Attached files
| file | filename |
|---|---|
| EX-23.1 - EX-23.1 - VIVUS INC | a2202317zex-23_1.htm |
| EX-31.2 - EX-31.2 - VIVUS INC | a2202317zex-31_2.htm |
| EX-10.7 - EX-10.7 - VIVUS INC | a2202317zex-10_7.htm |
| EX-31.1 - EX-31.1 - VIVUS INC | a2202317zex-31_1.htm |
| EX-32.1 - EX-32.1 - VIVUS INC | a2202317zex-32_1.htm |
| EX-21.1 - EX-21.1 - VIVUS INC | a2202317zex-21_1.htm |
| EX-10.28 - EX-10.28 - VIVUS INC | a2202317zex-10_28.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
ý |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the fiscal year ended December 31, 2010 |
||
OR |
||
o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 (NO FEE REQUIRED) |
|
For the transition period from to |
||
Commission File Number 001-33389
VIVUS, INC.
(Exact name of Registrant as specified in its charter)
| Delaware (State or other jurisdiction of incorporation or organization) |
94-3136179 (IRS employer identification number) |
|
1172 Castro Street Mountain View, California (Address of principal executive office) |
94040 (Zip Code) |
Registrant's telephone number, including area code: (650) 934-5200
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class | Name of Each Exchange on Which Registered | |
|---|---|---|
| Common Stock, $.001 Par Value (Title of class) |
The NASDAQ Global Select Market | |
| Preferred Share Purchase Rights (Title of class) |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ý No o
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by check mark whether the Registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of Registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ý | Accelerated filer o | Non-accelerated filer o (Do not check if a smaller reporting company) |
Smaller reporting company o |
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No ý
The aggregate market value of the common equity held by non-affiliates of the Registrant as of June 30, 2010 totaled approximately $774,287,837 based on the closing stock price as reported by the NASDAQ Global Market.
As of February 18, 2011, there were 81,888,089 shares of the Registrant's common stock, $0.001 par value per share, outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
| Document Description | 10-K part | |
|---|---|---|
Portions of the Registrant's notice of annual meeting of stockholders and proxy statement to be filed pursuant to Regulation 14A within 120 days after Registrant's fiscal year end of December 31, 2010 are incorporated by reference into Part III of this report. |
III, ITEMS 10, 11, 12, 13, 14 |
VIVUS, INC.
FISCAL 2010 FORM 10-K
INDEX
2
PART I
FORWARD-LOOKING STATEMENTS
This Form 10-K contains "forward-looking" statements that involve risks and uncertainties. These statements typically may be identified by the use of forward-looking words or phrases such as "may," "will," "believe," "expect," "intend," "anticipate," "predict," "should," "planned," "continue," "likely," "opportunity," "estimated," and "potential," the negative use of these words or other similar words. All forward-looking statements included in this document are based on our current expectations, and we assume no obligation to update any such forward-looking statements. The Private Securities Litigation Reform Act of 1995 provides a "safe harbor" for such forward-looking statements. In order to comply with the terms of the safe harbor, we note that a variety of factors could cause actual results and experiences to differ materially from the anticipated results or other expectations expressed in such forward-looking statements. On October 28, 2010, we received a Complete Response Letter, or CRL, regarding the New Drug Application, or NDA, for QNEXA® as a treatment for obesity. The CRL stated that in its current form, the NDA for QNEXA was not approvable. The CRL included the following areas: clinical, labeling, Risk Evaluation and Mitigation Strategy, or REMS, safety update, and drug scheduling. In the clinical section of the CRL, the FDA requested a comprehensive assessment of topiramate's and QNEXA's teratogenic potential including a detailed plan and strategy to evaluate and mitigate the potential teratogenic risks in women of childbearing potential taking the drug for the treatment of obesity. In addition, the FDA asked us to provide evidence that the elevation in heart rate (mean 1.6 beats per minute on the top dose) associated with QNEXA does not increase the risk for major adverse cardiovascular events. The FDA requested that we formally submit the results from the completed SEQUEL study (OB-305), a 52-week extension study for a subset of 675 patients who completed the previously reported 56-week CONQUER study. The FDA reserved the right to comment further on proposed labeling. On REMS, the FDA requested that a discussion of an already-submitted REMS plan be continued after we have submitted the written response. The agency also requested a safety update of any new adverse events be submitted to the NDA. Finally, the FDA stated that if approved, QNEXA would be a Schedule IV drug due to the phentermine component. On January 19, 2011, we held an End-of-Review meeting with the FDA to discuss the items contained in the CRL and the information we plan to include in the resubmission of the NDA for QNEXA. In anticipation of the meeting, we had provided a briefing document that included analyses integrating existing non-clinical and clinical data to provide a comprehensive assessment of the teratogenic potential of topiramate. In addition, we provided several new analyses to demonstrate that QNEXA does not increase the risk for major cardiovascular events, which analyses included cardiovascular data from our SEQUEL (OB-305) and Sleep Apnea (OB-204) studies. We also provided a synopsis of the final study report for the SEQUEL study. No new clinical studies were requested in the CRL. At the meeting, presentations were made on the comprehensive assessment of the teratogenic potential of topiramate and QNEXA, and evidence was presented that the increase in heart rate of 1.6 beats per minute does not increase the risk for major adverse cardiovascular events. The discussion also included elements of our proposed REMS program for QNEXA. The FDA chose to focus the meeting on the discussion of teratogenic potential for topiramate, specifically the incidence of oral clefts observed in the North American AED Pregnancy Registry and in the UK Epilepsy and Pregnancy Registry. As part of this meeting, the FDA requested that we assess the feasibility of performing a retrospective observational study utilizing existing electronic healthcare databases to review fetal outcomes including the historical incidence of congenital malformations with an interest in oral cleft and low birth weight (less than 2,500 gm) in the offspring of women who received prophylaxis treatment with 100 mg of topiramate for migraine during pregnancy, or the Feasibility Assessment. Although no other requests for additional information or studies were made by the FDA at the meeting or in the CRL, there can be no assurance that the FDA will not request or require us to provide additional information or undertake additional studies in connection with the QNEXA NDA. In the QNEXA studies, which included 15 offspring from women exposed to QNEXA or topiramate, there were no reports of any congenital malformations or low birth weight. The timing of the planned resubmission of the QNEXA NDA will be determined after agreement with the FDA is reached on the Feasibility Assessment and whether a retrospective observational study would be needed. We expect to reach agreement with the FDA, and if
3
deemed feasible, initiate the retrospective observational study on fetal outcomes within the next two months. It is our goal to resubmit the NDA for QNEXA to the FDA by the end of 2011. The risks and uncertainties that may affect the operations, performance, development, and results of our business include but are not limited to: (1) our ability to complete the Feasibility Assessment made by the FDA during our End-of-Review meeting held January 19, 2011. As no additional guidance was given by the FDA as to the potential parameters of the study it may be difficult to interpret the definition of feasibility. In addition, we do not know if we will be able to determine if enough live births to mothers exposed to 100 mg of topiramate exist, the condition of their records and the records of the infants, the ability to properly link records of the mother to those of the infant, the ability to determine other important infant health factors including but not limited to the health status of the mother and the diagnosis for which she was prescribed topiramate, concomitant use of medications, smoking status during pregnancy, use of alcohol or illegal substances during pregnancy, mothers weight, nutrition during pregnancy and folic acid supplementation, family history and any other information necessary to determine the nature of any malformations, in particular oral cleft. If the FDA does not agree with our assessment, we may appeal the decision or amend the NDA to exclude women of childbearing potential; (2) the FDA's interpretation of the data we submit relating to cardiovascular safety; (3) the FDA's interpretation of the data from our SEQUEL study (OB-305) and Sleep Apnea study (OB-204); (4) that we may be required to conduct additional clinical trials; (5) impact on future sales based on contraindications contained in the label and extent of the REMS program; (6) our history of losses and variable quarterly results; (7) substantial competition; (8) risks related to the failure to protect our intellectual property and litigation in which we may become involved; (9) uncertainties of government or third party payer reimbursement; (10) our reliance on sole source suppliers; (11) our limited sales and marketing efforts and our reliance on third parties; (12) failure to continue to develop innovative investigational drug candidates and drugs; (13) risks related to the failure to obtain United States Food and Drug Administration, or FDA, or foreign authority clearances or approvals and noncompliance with FDA regulations; (14) our ability to demonstrate through clinical testing the safety and effectiveness of our investigational drug candidates; (15) our dependence on the performance of our collaborative partners; (16) the timing of initiation and completion of clinical trials and submissions to the FDA; (17) the volatility and liquidity of the financial markets; (18) our liquidity and capital resources; (19) our expected future revenues, operations and expenditures; and (20) other factors that are described from time to time in our periodic filings with the Securities and Exchange Commission, or the SEC, including those set forth in this filing as "Item 1A. Risk Factors."
Overview
VIVUS, Inc. is a biopharmaceutical company, incorporated in 1991 as a California corporation and reincorporated in 1996 as a Delaware corporation, dedicated to the development and commercialization of therapeutic drugs for large underserved markets, including obesity and related morbidities, such as sleep apnea and diabetes and men's sexual health. With respect to obesity, it is estimated that the potential worldwide pharmaceutical market for obesity could approach $5 billion annually. Annual sales of approved drugs for diabetes currently exceed $10 billion. There are currently no approved pharmaceutical therapies for sleep apnea; however, the sales of devices and related consumables used to treat sleep apnea exceed $2 billion annually. The indications targeted by our investigational drug candidate as a treatment for erectile dysfunction represent a projected market greater than $1 billion annually.
Currently, we have one investigational drug candidate, QNEXA, which has been submitted for approval as a treatment for weight loss in the U.S. and the European Union. In the U.S., we received a CRL for QNEXA in October 2010, which contained requests for additional information. We held an End-of-Review meeting with the FDA on January 19, 2011 to discuss its requests and our planned resubmission of the NDA for QNEXA. The FDA has requested that we perform the Feasibility Assessment, which is currently in process. We also have investigational drug candidates in various stages
4
of clinical development for various indications related to obesity, erectile dysfunction and other diseases.
Recent Developments
On January 19, 2011, we held an End-of-Review meeting with the FDA to discuss our planned response to the CRL received on October 28, 2010, regarding the New Drug Application for QNEXA as a treatment for obesity. The CRL stated that in its current form, the NDA for QNEXA was not approvable. The CRL included the following areas: clinical, labeling, REMS, safety update, and drug scheduling. In the clinical section of the CRL, the FDA requested a comprehensive assessment of topiramate's and QNEXA's teratogenic potential including a detailed plan and strategy to evaluate and mitigate the potential teratogenic risks in women of childbearing potential taking the drug for the treatment of obesity. In addition, the FDA asked us to provide evidence that the elevation in heart rate (mean 1.6 beats per minute on the top dose) associated with QNEXA does not increase the risk for major adverse cardiovascular events. The FDA requested that we formally submit the results from the completed SEQUEL study (OB-305), a 52-week extension study for a subset of 675 patients who completed the previously reported 56-week CONQUER study. The FDA reserved the right to comment further on proposed labeling. On REMS, the FDA requested that a discussion of an already-submitted REMS plan be continued after we have submitted the written response. The agency also requested a safety update of any new adverse events be submitted to the NDA. Finally, the FDA stated that if approved, QNEXA would be a Schedule IV drug due to the phentermine component. In anticipation of the meeting held with the FDA on January 19, 2011, we had provided a briefing document that included analyses integrating existing non-clinical and clinical data to provide a comprehensive assessment of the teratogenic potential of topiramate. In addition, we provided several new analyses to demonstrate that QNEXA does not increase the risk for major cardiovascular events, which analyses included cardiovascular data from our SEQUEL (OB-305) and Sleep Apnea (OB-204) studies. We also provided a synopsis of the final study report for the SEQUEL study. No new clinical studies were requested in the CRL. At the meeting, presentations were made on the comprehensive assessment of the teratogenic potential of topiramate and QNEXA, and evidence was presented that the increase in heart rate of 1.6 beats per minute does not increase the risk for major adverse cardiovascular events. The discussion also included elements of our proposed REMS program for QNEXA. The FDA chose to focus the meeting on the discussion of teratogenic potential for topiramate. As part of this meeting, the FDA requested that we complete the Feasibility Assessment. Although no other requests for additional information or studies were made by the FDA at the meeting or in the CRL, there can be no assurance that the FDA will not request or require us to provide additional information or undertake additional studies in connection with the QNEXA NDA. In the QNEXA studies, which included 15 births from women exposed to QNEXA or topiramate, there were no reports of any congenital malformations or low birth weight. The timing of the planned resubmission of the QNEXA NDA will be determined after agreement with the FDA is reached on the Feasibility Assessment and whether a retrospective study would be needed. We expect to reach agreement with the FDA, and if deemed feasible, initiate the retrospective observational study on fetal outcomes within the next two months. It is our goal to resubmit the NDA for QNEXA to the FDA by the end of 2011.
On October 1, 2010, we entered into a definitive Asset Purchase Agreement with Meda AB, or Meda, to sell certain rights and assets related to MUSE, transurethral alprostadil, for the treatment of erectile dysfunction, or the MUSE Transaction. Meda has been our European distributor of MUSE since 2002. The assets sold in the MUSE Transaction include the U.S. and foreign MUSE patents, existing inventory, and the manufacturing facility located in Lakewood, New Jersey. We retained all of the liabilities associated with the pre-closing operations and products of the MUSE business and the accounts receivables for pre-closing MUSE sales. The transaction closed on November 5, 2010. Prior to the closing of the MUSE Transaction, we regained all of the rights to MUSE and avanafil held by Deerfield Management Company, L.P., and affiliates, and Crown Bank, N.A., or Crown.
5
On October 15, 2010, in preparation for the closing of the MUSE Transaction and in accordance with the terms of the agreements with Crown, we paid $4.8 million to Crown in satisfaction of all obligations owed to them under these agreements. As a result, the security interests and Certificate of Deposit held by Crown were terminated in our favor. On October 21, 2010, we exercised the Option under the Option and Put Agreement with the Deerfield Affiliates and the Deerfield Sub, dated April 3, 2008, and an Amended and Restated Option and Put Agreement dated March 16, 2009, or the OPA, and we paid an aggregate amount totaling $27.1 million, which consisted of the Base Option Price of $25 million, less the Option Premium Adjustment of $2 million, plus the Cash Adjustment of $2.8 million and the Royalty Adjustment of $1.3 million. These payments satisfied all of the financial obligations under the Funding and Royalty Agreement, or FARA, and the OPA. As a result, all of the outstanding shares and the $2.8 million of cash of the Deerfield Sub are owned by us, all of the outstanding loans owed by the Deerfield Sub have been repaid and the security interests in the collateral related to MUSE and avanafil held by the Deerfield Sub and the Deerfield Affiliates as part of the FARA and OPA were terminated. In December 2010, the Deerfield Sub was dissolved.
Under the terms of the MUSE Transaction, we received an upfront payment of $22 million upon the closing, on November 5, 2010, and are eligible to receive an additional $1.5 million based on future sales of MUSE, provided that certain sales milestones are reached. Meda is now responsible for the manufacturing, selling and marketing of MUSE. Meda also assumed all post-closing expenses and liabilities associated with MUSE. We have agreed not to develop, manufacture or sell any transurethral erectile dysfunction drugs for a period of three years following the closing of the MUSE Transaction. The assets and liabilities and results of operations associated with MUSE have been reported as discontinued operations for all periods presented.
On December 17, 2010, we filed a Marketing Authorization Application, or MAA, with the European Medicines Agency, or EMA, for QNEXA Controlled-Release Capsules in the European Union, or EU. The proposed indication in the EU is for the treatment of obesity, including weight loss and maintenance of weight loss, and should be used in conjunction with a mildly hypocaloric diet. If approved in the EU, QNEXA could be recommended for obese adult patients (BMI ³ 30 kg/m2), or overweight patients (BMI ³ 27 kg/m2) with weight-related co-morbidities such as hypertension, type 2 diabetes, dyslipidemia, or central adiposity (abdominal obesity). In Europe, approximately 150 million adults are considered overweight or obese, and the prevalence is rising rapidly. According to EMA guidelines for medicinal products used in weight control, a demonstration of weight loss of at least 10% of baseline weight, which is at least statistically greater than that associated with placebo, is considered to be a valid primary efficacy criterion. We believe QNEXA has met this efficacy criterion set by the EMA for obesity therapies. In addition, the mean weight loss for the mid- and top-dose of QNEXA at the end of two years was 10.4% and 11.4%, respectively, which met the efficacy benchmark set by the EMA for obesity therapies. These results were shown to be sustained over a two-year period and were associated with significant improvements in weight-related co-morbidities such as hypertension, dyslipidemia and diabetes. The EMA filing is comprised of data from over 4,500 overweight or obese patients with a broad range of weight-related co-morbidities. Two-year, double-blind data from SEQUEL (OB-305) were also included in the filing to demonstrate durability of treatment response and long-term safety. The EMA's review of QNEXA will follow their centralized marketing authorization procedure. If approved, QNEXA could receive marketing authorization in all 27 EU member countries. The MAA was officially validated for central procedure on January 19, 2011.
Our Future
Our goal is to build a successful biopharmaceutical company through the development and commercialization of innovative proprietary drugs. We intend to achieve this by:
- •
- seeking approval for QNEXA for the treatment of obesity in the U.S. and the European Union;
- •
- establishing internal capabilities or strategic relationships with marketing partners to maximize sales potential for our drugs that require significant commercial support; and
6
- •
- capitalizing on our clinical and regulatory expertise and experience to advance the development of investigational drug candidates in our pipeline.
It is our objective to become a leader in the development and commercialization of drugs for large underserved markets. We believe we have strong intellectual property supporting several opportunities in obesity and related disorders, such as sleep apnea and diabetes, and men's sexual health. Our future growth depends on our ability to further develop and obtain regulatory approval of our investigational drug candidates for indications that we have studied, or plan to study, as well as in-licensing and product line extensions.
We have funded operations primarily through private and public offerings of our common stock, through the sale of the rights to Evamist and through product sales of MUSE (alprostadil). We expect to generate future net losses due to increases in operating expenses as our various investigational drug candidates are advanced through the various stages of clinical development and for pre-commercialization activities. In connection with the sale of Evamist, to date we have received an aggregate of $150 million. The sale of Evamist was a unique transaction. An initial $10.0 million was paid at closing and $140 million was paid upon the FDA's approval of the Evamist NDA. These payments were non-refundable and were originally recorded as deferred revenue and were recognized as license and other revenue ratably over a 21.5-month period, from August 1, 2007 to May 15, 2009. All of the revenue deferred from the Evamist sale has been recognized. As of December 31, 2010, we have incurred a cumulative deficit of $300.1 million and expect to incur operating losses in future years.
Our Investigational Drug Candidates
Our investigational drug pipeline includes two late-stage clinical investigational drug candidates. One of these investigational drug candidates, QNEXA, has completed Phase 3 clinical trials for obesity and Phase 2 clinical trials for diabetes and obstructive sleep apnea. We submitted an NDA to the FDA for QNEXA in December 2009. On October 28, 2010, we received a CRL from the FDA regarding the QNEXA NDA stating that the NDA could not be approved in its present form. We are currently working with the FDA on their request for information contained in the CRL. In the EU, the MAA for QNEXA is currently under review through the centralized procedure. Avanafil has completed three pivotal Phase 3 trials for erectile dysfunction. Our investigational drug candidates are summarized as follows:
Drug
|
Indication | Status | Commercial rights | |||
|---|---|---|---|---|---|---|
QNEXA (phentermine and topiramate CR) |
Obesity | Phase 3 studies completed; NDA submitted; CRL received | Worldwide | |||
QNEXA (phentermine and topiramate CR) |
Obstructive Sleep Apnea | Phase 2 study completed | Worldwide | |||
QNEXA (phentermine and topiramate CR) |
Diabetes | Phase 2 study completed | Worldwide | |||
Avanafil (PDE5 inhibitor) |
Erectile dysfunction | Phase 3 completed; NDA preparation and MAA submission in progress | Worldwide license from Mitsubishi Tanabe Pharma Corporation (ex. certain Asian markets) |
QNEXA for Obesity
Obesity is a chronic disease condition that affects millions of people and often requires long-term or invasive treatment to promote and sustain weight loss. In the National Health and Nutrition
7
Examination Survey, or NHANES, conducted for 2007-2008, 68% of adults in the U.S. (72.3% of men and 64.1% of women) were classified as overweight, defined as a body mass index, or BMI >25, and 33.8% were obese (BMI >30). Data from NHANES also found that almost 17% of school children were obese and almost 32% were overweight for the 2007-2008 time period. The percentage of American men and women classified as overweight and obese has more than doubled since 1962. Researchers fear that the percentage of American adults that are obese could climb as high as 43% in the next 10 years. Obesity is the second leading cause of preventable death in the U.S.. According to a study performed by the Centers for Disease Control and Prevention, or CDC, as reported in the Journal of the American Medical Association, an estimated 112,000 excess deaths a year in the U.S. are attributable to obesity. Additionally, Americans spend more than $30 billion annually on weight-loss products and services.
QNEXA is our proprietary oral investigational drug candidate for the treatment of obesity, incorporating low doses of active ingredients from two previously approved drugs, phentermine and topiramate. We believe that by combining these compounds, QNEXA targets excessive appetite and high threshold for satiety, or the feeling of being full, the two main mechanisms that impact eating behavior. QNEXA is a once-a-day capsule containing a proprietary formulation of controlled release phentermine and topiramate. Our first U.S patent on QNEXA (U.S. 7,056,890 B2) and our EU patent on QNEXA (EU EP 1187603) both expire in 2020.
EQUIP (OB-302) AND CONQUER (0B-303) One-Year Phase 3 Studies
The QNEXA development program included two large Phase 3 randomized, double-blind, placebo-controlled, 3-arm, prospective studies across 93 centers comparing QNEXA to placebo over a 56-week treatment period. All Phase 3 studies utilized our once-a-day formulation of QNEXA, which at top-dose contains 15 mg phentermine and 92 mg of a proprietary controlled release formulation of topiramate. The Phase 3 studies were designed to prospectively demonstrate the safety and efficacy of QNEXA in obese and overweight patients with different baseline characteristics. The co-primary endpoints for these studies evaluated the differences between treatments in mean percent weight loss from baseline to the end of the treatment period and the differences between treatments in the percentage of patients achieving weight loss of 5% or more. Patients were asked to follow a hypocaloric diet representing a 500-calorie/day deficit and were advised to implement a simple lifestyle modification program.
The first year-long Phase 3 study, known as EQUIP, enrolled 1,267 morbidly obese patients (1,050 females and 217 males) with a BMI that equaled or exceeded 35 kg/m2 with or without controlled co-morbidities. The average baseline BMI of the study population was 42.1 kg/m2 and baseline weight was 256 pounds. Patients had a 4-week dose titration period followed by 52 weeks of treatment, with patients randomized to receive once-a-day treatment with low-dose QNEXA, full-dose QNEXA or placebo. Weight loss results from the study are summarized as follows:
| |
ITT-LOCF | Completers | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
EQUIP (OB-302) 56 weeks
|
Placebo (n=498) |
QNEXA low-dose (n=234) |
QNEXA top-dose (n=498) |
Placebo (n=241) |
QNEXA low-dose (n=138) |
QNEXA top-dose (n=301) |
|||||||||||||
Mean weight loss (%) |
1.6 | % | 5.1 | %* | 11 | % | 2.5 | % | 7 | %* | 14.7 | %* | |||||||
Greater than or equal to 5% weight loss rate |
17 | % | 45 | %* | 67 | %* | 26 | % | 59 | %* | 84 | %* | |||||||
ITT-LOCF: Intent-to-treat with last observation carried forward
- *
- p<0.0001 vs. placebo
The EQUIP study met the co-primary endpoints by demonstrating that patients treated with top-dose and low-dose QNEXA had an average weight loss of 11% and 5.1%, respectively, as compared to weight loss of 1.6% in the placebo group (ITT-LOCF p<0.0001). Average weight loss was 37 pounds and 18 pounds with top-dose QNEXA and low-dose QNEXA, respectively, as compared to 6
8
pounds in the placebo group. The proportion of patients losing 5% or more of their initial body weight was 67% for top-dose, 45% for low-dose and 17% for placebo (ITT-LOCF p<0.0001).
The most common drug-related adverse events reported in the EQUIP study for the top-dose, low-dose and placebo group were tingling of the extremities, dry mouth, altered taste, headache and constipation. A significantly greater proportion of patients completed the study on QNEXA as compared to placebo patients. Overall average completion rates were 59%, 57% and 47% for patients taking top-dose QNEXA, low-dose QNEXA and placebo, respectively.
The second year-long Phase 3 trial, known as CONQUER, enrolled 2,487 overweight and obese adult patients (1,737 females and 750 males) with BMI's from 27 kg/m2 to 45 kg/m2 and at least two co-morbid conditions, such as hypertension, dyslipidemia and type 2 diabetes. The average baseline BMI of the study population was 36.6 kg/m2 and baseline weight was 227 pounds. Patients had a 4-week dose titration period followed by 52 weeks of treatment, with patients randomized to receive once-a-day treatment with top-dose QNEXA, mid-dose QNEXA or placebo. Weight loss results from the study are summarized as follows:
| |
ITT-LOCF | Completers | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
CONQUER (OB 303) 56 weeks
|
Placebo (n=979) |
QNEXA mid-dose (n=488) |
QNEXA top-dose (n=981) |
Placebo (n=564) |
QNEXA mid-dose (n=344) |
QNEXA top-dose (n=634) |
|||||||||||||
Mean weight loss (%) |
1.8 | % | 8.4 | %* | 10.4 | %* | 2.4 | %* | 10.5 | %* | 13.2 | %* | |||||||
Greater than or equal to 5% weight loss rate |
21 | % | 62 | %* | 70 | %* | 26 | % | 75 | %* | 85 | %* | |||||||
- *
- p<0.0001 vs. placebo
The CONQUER study also met the co-primary endpoints by demonstrating that patients treated with top-dose and mid-dose QNEXA had an average weight loss of 10.4% and 8.4%, respectively, as compared to weight loss of 1.8% in the placebo group (ITT-LOCF p<0.0001). Average weight loss was 30 pounds and 24 pounds with top-dose QNEXA and mid-dose QNEXA, respectively, as compared to 6 pounds in the placebo group. The proportion of patients losing 5% or more of their initial body weight was 70% for top-dose, 62% for mid-dose and 21% for placebo (ITT-LOCF p<0.0001).
The most common drug-related adverse events reported in the CONQUER study for the top-dose, mid-dose, and placebo group were tingling of the extremities, dry mouth, altered taste, headache and constipation. A significantly greater proportion of patients completed the study on QNEXA as compared to placebo patients. Overall average completion rates were 64%, 69%, and 57% for patients taking top-dose QNEXA, mid-dose QNEXA and placebo, respectively.
SEQUEL (0B-305) one-year extension study
We also conducted a one-year extension study of a subset of patients who completed the 56-week CONQUER study. The SEQUEL study was a double-blind, placebo-controlled, 3-arm, prospective study across 36 centers comparing QNEXA to placebo over an additional 52-week treatment period for a total treatment duration of 108 weeks, or two years. Patients in SEQUEL continued in a blinded fashion to receive the same treatment they were receiving when they completed the CONQUER study. The co-primary endpoints for this study were the differences between treatments in mean weight loss and percent weight loss from start of the OB-303 study (baseline) to the end of the treatment period (two-years). Secondary endpoints include the differences between treatments in the percentage of patients achieving weight loss of 5% and 10% and the change in waist circumference. Patients were asked to continue a hypocaloric diet representing a 500-calorie/day deficit and were advised to implement a simple lifestyle modification program.
SEQUEL included 675 obese or overweight patients, all of whom had two or more weight related co-morbidities and an average baseline BMI of 36.1. There was no titration necessary for patients rolling over to OB-305 as they continued to receive blinded treatment. The total study period was
9
108 weeks. The purpose of this study was to provide long-term safety and efficacy data to support the MAA filing in Europe.
Patients in the study taking the top dose of QNEXA achieved and maintained average weight loss through two years of 26 pounds (ITT-LOCF). Consistent with the first-year experience, QNEXA therapy was well tolerated, with no new or unexpected adverse events. The most common side effects seen were constipation, tingling, dry mouth, altered taste and insomnia.
Weight loss with QNEXA in SEQUEL was associated with statistically significant improvements in weight-related co-morbidities such as hypertension, dyslipidemia and diabetes. Among patients without diabetes at baseline, the incidence of new onset of type 2 diabetes was reduced by 54% and 76% (mid- and top-dose, respectively) as compared to placebo.
Specific SEQUEL findings include the following primary endpoints: Patients taking top- and mid-dose QNEXA achieved and maintained weight loss over two years of 11.4% and 10.4% of their initial body weight, respectively, as compared to placebo-treated patients with 2.5% weight loss (ITT-LOCF, p<0.0001). A majority of all patients taking QNEXA exceeded 10% weight loss, the goal established by the National Institutes of Health, or NIH, to decrease the severity of obesity-associated risk factors. The percentage of patients achieving categorical weight loss of at least 5%, 10% and 15% on both QNEXA doses was statistically significant compared to placebo:
Categorical Weight Loss (ITT-LOCF)
|
5% | 10% | 15% | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Top-dose |
79 | %* | 54 | %* | 32 | %* | ||||
Mid-dose |
75 | %* | 50 | %* | 24 | %* | ||||
Placebo |
30 | % | 12 | % | 7 | % | ||||
- *
- p<0.0001 vs placebo
Treatment-emergent serious adverse event rates in SEQUEL were low (top-dose = 4.1%; mid-dose = 2.6%) and similar to placebo (4%), with no drug-related serious adverse events reported.
The completion rate in SEQUEL was approximately 83% for both QNEXA doses and 86% for the placebo group. Discontinuations due to adverse events were 3.9% and 4.1% for the mid- and top-dose, respectively, and 2.6% for the placebo group; with no single adverse event leading to discontinuation in more than 1% of patients. Additionally, SEQUEL data confirms previous safety findings, with no evidence of suicidality and no reports of suicidal attempts or behavior. Depression assessments, as measured by the PHQ-9 clinical depression scale, improved from baseline for all treatment groups. The incidence of targeted medical events for sleep disorders, depression, anxiety, cardiac disorders and cognitive disorders in SEQUEL was lower than observed during the one-year CONQUER study. Similar to previously presented data, effects of QNEXA in SEQUEL on heart rate were small and seen in conjunction with improvements in blood pressure from baseline. There were no clinically relevant decreases of serum bicarbonate in QNEXA -treated patients compared to placebo in year two of SEQUEL.
Across the entire QNEXA development program (4,323 patients), including the two-year data in SEQUEL, serious cardiovascular and neurovascular adverse event rates in patients taking QNEXA were similar to placebo with a relative risk of 0.54 (95% CI: 0.29-0.98). No teratogenic effects were observed across the entire development program in patients taking QNEXA.
The primary efficacy endpoint for Phase 3 weight loss trials in the U.S., as recommended by the FDA, is at least a 5% mean reduction in baseline body weight compared to placebo or at least 35% of patients losing 5% or more of their baseline body weight. In Europe, the Committee for Medicinal Products for Human Use of the European Medicines Agency has recommended that demonstration of significant weight loss of at least 10% of baseline weight is considered to be a valid primary endpoint for anti-obesity drugs. The FDA and foreign authorities require pivotal obesity studies to be conducted for at least one year. Although the results of our pivotal Phase 3 obesity trials met these current
10
guidelines for efficacy, there can be no assurance that these results will be acceptable to the FDA or the EMA.
We completed a "Thorough QT," or TQT, prolongation study evaluating patients taking QNEXA. The QT interval represents the time for both ventricular depolarization and repolarization to occur in the heart, and therefore roughly estimates the duration of an average ventricular action potential. If abnormally prolonged or shortened, there is a risk of developing ventricular arrhythmias. The study was completed with no drug-related signal for QT prolongation. We also conducted studies evaluating cognitive and psychomotor functions in patients taking QNEXA. Patients underwent complex and extensive cognitive and psychomotor testing using validated, FDA recognized testing methodologies. There was no clinically relevant change in overall cognitive function or effect on psychomotor skills seen in patients taking QNEXA.
We have entered into a Master Services Agreement and related Task Orders with Medpace, Inc., or Medpace, pursuant to which Medpace will perform certain clinical research services in connection with the clinical trials for QNEXA and work related to the preparation of the NDA for avanafil. Our aggregate payment obligations under the agreement for services entered into during 2007 through 2010, out of pocket expenses and pass through costs total approximately $80.2 million, of which we have paid approximately $75.5 million through December 31, 2010. We have agreed to defend and indemnify Medpace against third party claims arising from the services other than claims resulting from Medpace's negligence, willful misconduct, violation of law or material breach of the Master Service Agreement or a Task Order. We can terminate the agreement at any time without cause. Medpace may terminate the agreement following our material breach of the agreement that remains uncured.
QNEXA for Obstructive Sleep Apnea
Obstructive sleep apnea, or OSA, is a condition in which patients momentarily pause or stop breathing altogether while sleeping. The pauses in breathing can occur frequently throughout the course of sleep. Sleep apnea is often undiagnosed and can lead to severe health problems and even death if left untreated. It is estimated that about 18 million people in the U.S. have obstructive sleep apnea. Currently, there are no approved pharmacologic treatments for OSA. Modafinil is approved for the treatment of residual daytime sleepiness associated with OSA, but does not specifically treat the sleep apnea condition.
In January 2010, we announced positive results from a Phase 2 study evaluating the safety and efficacy of QNEXA for the treatment of OSA. This Phase 2 study (OB-204) was a single-center, randomized, double-blind, placebo-controlled parallel group trial including 45 obese men and women (BMI 30 to 40 kg/m2 inclusive), 30 to 65 years of age with OSA (apnea-hypopnea index, or AHI, greater than or equal to 15 at baseline), who had not been treated with, or who were not compliant with continuous positive airway pressure, or CPAP, within three months of screening. Patients were randomized to placebo or top-dose QNEXA. CPAP is the current standard of care treatment for the majority of patients with moderate or severe OSA, defined as an apnea-hypopnea index, or AHI, of 15 or more events per hour. Although CPAP is reported to be effective in treating OSA when properly and consistently used, compliance (as defined by use for at least 4 hours per night, on at least 70% of nights) may be as low as 50-60%.
11
In the OB-204 study, patients underwent a four-week dose titration followed by 24 weeks of additional treatment. All patients were also provided with a lifestyle modification program focusing on diet and exercise. Overnight polysomnography in a sleep laboratory was performed at baseline, Week 8 and Week 28. The primary endpoint was the change in AHI between baseline and Week 28; secondary endpoints included weight loss, improvement in overnight oxygen saturation and reduction in blood pressure.
The study demonstrated statistically significant improvement in AHI in patients with OSA treated with QNEXA for 28 weeks. QNEXA-treated patients also experienced significant weight loss, improvements in blood pressure, and overnight blood oxygen saturation.
Highlights of the study include:
- •
- Patients treated with QNEXA for 28 weeks had a 69% reduction in the AHI;
- •
- QNEXA treatment reduced the number of apnea-hypopnea events from a mean of 46 events per hour of sleep to
14—compared to placebo patients with a reduction from a mean 44 events per hour of sleep to 27 (ITT-LOCF p<0.001 active vs. placebo);
- •
- QNEXA treated patients lost 10.2% body weight, or 23.8 lbs in 28 weeks—compared to 4.3% for placebo
patients, or 10.4 lbs, (ITT-LOCF p<0.001 active vs. placebo);
- •
- Systolic blood pressure was reduced by 15 mm Hg in the QNEXA group from a mean of 138 mm Hg at baseline
(ITT-LOCF p<0.04 active vs. placebo); and
- •
- Mean overnight blood oxygen saturation was significantly improved in QNEXA patients (p<0.014 active vs. placebo).
Sleep apnea is one of the leading co-morbidities associated with obesity and research has shown that weight loss can improve OSA. QNEXA treatment was well-tolerated with no serious adverse events reported in the QNEXA arm; the most common side effects were dry mouth, altered taste and sinus infection.
QNEXA for Diabetes
Diabetes is a significant worldwide disease. Based on the fourth edition of the Diabetes Atlas published in 2009, the International Diabetes Federation estimated that in 2008 there were 285 million people with diabetes worldwide, with 27 million of those people living in the U.S. Diabetes, mostly type 2 diabetes, was projected to reach 6.6% of the world's adult population in 2010, with almost 70% of the total in developing countries. Based on the National Diabetes Fact Sheet, 2011, the CDC estimates that nearly 26 million people in the U.S. have diabetes, mostly type 2 diabetes, and that 79 million people have pre-diabetes, a condition that puts people at increased risk of diabetes. Type 2 diabetes is characterized by inadequate response to insulin and/or inadequate secretion of insulin as blood glucose levels rise. Currently approved therapies for type 2 diabetes are directed toward correcting the body's inadequate response with oral or injectable medications, or directly modifying insulin levels through injection of insulin or insulin analogs.
The currently approved oral medications for type 2 diabetes include insulin releasers such as glyburide, insulin sensitizers such as Actos and Avandia, inhibitors of glucose production by the liver such as metformin, DPP-IV inhibitors like Januvia, as well as Precose and Glyset, which slow the uptake of glucose from the intestine. The worldwide market for diabetes medications was estimated at $24 billion in 2007, according to IMS Health. However, it is estimated that a significant portion of type 2 diabetics fail oral medications and require injected insulin therapy. Current oral medications for type 2 diabetes have a number of common drug-related side effects, including hypoglycemia, weight gain and edema. Numerous pharmaceutical and biotechnology companies are seeking to develop insulin sensitizers, novel insulin formulations and other therapeutics to improve the treatment of diabetes.
12
Previous clinical studies of topiramate, a component of QNEXA, in type 2 diabetics resulted in a clinically meaningful reduction of hemoglobin A1c, a measure used to determine treatment efficacy of anti-diabetic agents.
In December 2008, we announced the results of our DM-230 diabetes study, a 56-week, Phase 2 clinical trial in 130 type 2 diabetics conducted at ten sites in the U.S.. Patients treated with QNEXA had a reduction in hemoglobin A1c of 1.6%, from 8.8% to 7.2%, as compared to 1.1% from 8.5% to 7.4% in the placebo group (ITT LOCF p=0.0381) at 56 weeks. Patients in the study were actively managed according to American Diabetes Association, or ADA, standards of care with respect to diabetes medications and lifestyle modification. For patients treated with placebo, increases in the number and doses of concurrent anti-diabetic medications were required to bring about the observed reduction in HbA1c. By contrast, concurrent anti-diabetic medications were reduced over the course of the trial in patients treated with QNEXA (p<0.05).
Fasting plasma glucose levels were reduced in patients treated with QNEXA from 176 mg/dL to 133 mg/dL, as compared to a decrease from 171 mg/dL to 145 mg/dL for the placebo group (p=0.02). Over 56 weeks, patients treated with QNEXA also lost 9.4% of their baseline body weight, or 20.5 pounds, as compared to 2.7%, or 6.1 pounds, for the placebo group (p<0.0001). Sixty-five percent of the QNEXA patients lost at least 5% of their body weight, as compared to 24% in the placebo group (p<0.001), and 37% of the QNEXA patients lost at least 10% of their body weight, as compared to 9% of patients in the placebo group (p<0.001). Patients treated with QNEXA had reductions in blood pressure, triglycerides and waist circumference. Both treatment groups had a study completion rate of greater than 90%.
The most common drug-related side effects reported were tingling, constipation and nausea. Patients on antidepressants such as SSRI's or SNRI's were allowed to participate in the studies. Patients were monitored for depression and suicidality using the PHQ-9 questionnaire, a validated mental health assessment tool agreed to by the FDA for use in our studies. Patients treated with QNEXA demonstrated greater improvements in PHQ-9 scores from baseline to the end of the study than patients in the placebo group.
Despite a mean baseline HbA1c level of 8.8%, 53% of the patients treated with QNEXA were able to achieve the ADA recommended goal of 7% or lower, versus 40% of the patients in the placebo arm (p<0.05). The incidence of hypoglycemia in the treatment and placebo arms was similar (12% and 9%, respectively). Patients in the QNEXA arm experienced no treatment-related serious adverse events.
The DM-230 Phase 2 study enrolled 130 patients, who completed OB-202, our Phase 2 study for the treatment of obesity, at 10 study sites to continue in a blinded fashion as previously randomized for an additional 28 weeks. The results of the DM-230 study included assessments from the start of the OB-202 study through the end of the DM-230 study in this population, for a total treatment period of 56 weeks. We also studied the effect of QNEXA on well-controlled diabetics as part of OB-303. The results were consistent and supportive of the Phase 2 results.
QNEXA for Other Indications
We believe QNEXA may be helpful in treating other obesity-related diseases including nonalcoholic steatohepatitis or its precursor, nonalcoholic fatty liver disease, also known as fatty liver disease. QNEXA may also be helpful in treating hyperlipidemia, or an elevation of lipids (fats) in the bloodstream. These lipids include cholesterol, cholesterol esters (compounds), phospholipids and triglycerides.
13
Avanafil for Erectile Dysfunction
Erectile dysfunction, or ED, is defined as the inability to attain or maintain an erection sufficient for intercourse. ED was reported by 52% of men between the ages of 40 to 70 in the Massachusetts Male Aging Study, with the incidence increasing with age. Erectile dysfunction, frequently associated with vascular problems, is particularly common in men with diabetes and in those who have had a radical prostatectomy for prostate cancer. PDE5 inhibitors such as sildenafil (Viagra), vardenafil (Levitra) and tadalafil (Cialis), which inhibit the breakdown of cyclic guanosine monophosphate, have been shown to be effective oral treatments for ED.
The worldwide sales in 2010 of PDE5 inhibitor products for the treatment of ED were in excess of $4.1 billion, including approximately $1.9 billion in sales of Viagra, approximately $1.7 billion in sales of Cialis and over $500 million in estimated sales of Levitra. Based on the aging population and the desire to maintain an active sexual lifestyle, we believe the market for PDE5 inhibitors will continue to grow.
Avanafil is an oral PDE5 inhibitor investigational drug candidate that we licensed from Tanabe Seiyaku Co., Ltd., or Tanabe, in 2001. In October 2007, Tanabe and Mitsubishi Pharma Corporation completed their merger and announced their name change to Mitsubishi Tanabe Pharma Corporation, or MTPC. Our U.S. patent on avanafil (U.S. 6,656,935) expires in 2020.
We have exclusive worldwide development and commercialization rights for avanafil with the exception of certain Asian markets.
Pre-clinical and clinical data suggest that avanafil:
- •
- is highly selective to PDE5, which we believe may result in a favorable side effect profile; and
- •
- is fast-acting as compared to the current commercially available PDE5 inhibitors based on a shorter Tmax, or time to maximum plasma concentration.
In November 2009, we announced results from the first of several pivotal Phase 3 studies of avanafil. The first study, REVIVE (TA-301), was a randomized, double-blind, placebo-controlled Phase 3 study of avanafil in 646 men. Participants in the study had ED for at least six months; 72% of study participants had tried at least one other ED treatment. Patients underwent a four-week, non-treatment run-in period followed by 12 weeks of treatment with one of three doses of avanafil: 50 mg, 100 mg and 200 mg or placebo. Patients were instructed to attempt sexual intercourse 30 minutes after taking avanafil, with no restrictions on food or alcohol consumption. The primary endpoints of the study were improvement in erectile function as measured by the Sexual Encounter Profile, or SEP, and improvement in the International Index of Erectile Function, or IIEF, score; secondary endpoints included patient satisfaction with erections and with sexual experience. This Phase 3 study was conducted under a Special Protocol Assessment, or SPA, with the FDA.
The REVIVE study met all primary endpoints across the three doses studied by demonstrating statistically significant improvement in erectile function as measured by the SEP and improvement in the IIEF score. Highlights of the study include:
- •
- Nearly 80% of sexual attempts among patients on the 200 mg dose of avanafil had erections sufficient for
intercourse (SEP2);
- •
- Full efficacy, as measured by successful intercourse (SEP3), was reported by some avanafil patients on all three dose
levels in 15 minutes or less;
- •
- Full efficacy was maintained for all doses across multiple time points from 15 minutes to beyond six hours based on diary
reports in some patients;
- •
- All FDA-defined primary endpoints were met across all three doses of avanafil;
- •
- Avanafil was well tolerated as demonstrated by a high retention rate (85%);
14
- •
- There were no reported drug-related serious adverse events in the study; and
- •
- Avanafil patients reported low rates of common PDE5i side effects (headache, flushing and upset stomach).
Patients on all three dose levels achieved a dose-related overall improvement in erectile function, as measured by improvement in the IIEF. IIEF scores range from 0-30 and measure the severity of erectile dysfunction as follows: severe dysfunction is less than or equal to 10; moderate is 11-16; and mild/minimal is 17-25. IIEF results of the study were:
| |
Baseline | End of Treatment |
|||||
|---|---|---|---|---|---|---|---|
Placebo |
12.4 | 15.3 | |||||
Avanafil 50 mg |
12.7 | 18.1 | |||||
Avanafil 100 mg |
12.6 | 20.9 | |||||
Avanafil 200 mg |
12.7 | 22.2 | |||||
(p</=0.001 vs. placebo)
Patients on avanafil had erections sufficient for vaginal penetration as measured by the Sexual Encounter Profile question number 2 (SEP2):
| |
Baseline | End of Treatment |
|||||
|---|---|---|---|---|---|---|---|
Placebo |
47 | % | 54 | % | |||
Avanafil 50 mg |
45 | % | 64 | % | |||
Avanafil 100 mg |
46 | % | 74 | % | |||
Avanafil 200 mg |
48 | % | 77 | % | |||
(p<0.001 vs. placebo)
Patients taking avanafil experienced successful intercourse as measured by the Sexual Encounter Profile question 3 (SEP3):
| |
Baseline | End of Treatment |
|||||
|---|---|---|---|---|---|---|---|
Placebo |
13 | % | 27 | % | |||
Avanafil 50 mg |
13 | % | 41 | % | |||
Avanafil 100 mg |
14 | % | 57 | % | |||
Avanafil 200 mg |
12 | % | 57 | % | |||
(p<0.001 vs. placebo)
The most commonly reported side effects in patients taking avanafil (all doses combined) included headache (7% vs. 1.2% placebo), flushing (4.6% vs. 0% placebo) and nasal congestion (2.3% vs. 1.2%). There were no reports of visual disturbances such as "blue vision."
In January 2010, we announced new data from an analysis of REVIVE TA-301. Patients who attempted intercourse within 15 minutes of dosing were successful 67%, 69% and 72% of the time on 50, 100 and 200 mg of avanafil, respectively, as compared to 29% of the patients on placebo (p<0.05).
We completed a Phase 1 "Thorough QT", or TQT, study evaluating 100 mg and 800 mg of avanafil compared to placebo and a known positive control. The study was successfully completed with no concern associated with QT prolongation.
15
In June 2010, we announced results from the Phase 3 REVIVE-Diabetes (TA-302) study, which evaluated the safety and efficacy of avanafil for the treatment of erectile dysfunction, or ED, in men with type 1 and type 2 diabetes. The REVIVE-Diabetes study met all three primary endpoints across the two doses studied by demonstrating statistically significant improvement in erectile function as measured by the Sexual Encounter Profile, or SEP, and improvement in the International Index of Erectile Function, or IIEF score. The study also demonstrated a favorable side effect profile and successful intercourse (as measured by SEP 3) in as early as 15 minutes and beyond six hours after dosing, without any restrictions for food or alcohol intake.
Highlights of the REVIVE-Diabetes study include:
- •
- More than 60% of patients on the 200 mg dose of avanafil had erections sufficient for intercourse (SEP2) at the end
of treatment;
- •
- Patients treated with 100 mg and 200 mg of avanafil improved their ability to have successful intercourse
three- and four-fold, respectively, from the start of treatment;
- •
- Treatment with avanafil improved erectile function in a dose-dependent manner with significant increases in
the IIEF scores from the beginning of treatment through the end of treatment. Erectile function scores increased 41% and 45% for patients on the 100 mg and 200 mg doses, respectively, as
compared to the placebo group with an increase of 17%; and
- •
- The most commonly reported side effects in patients taking avanafil included headache, nasopharyngitis, flushing, sinus congestion, sinusitis and dyspepsia. There were no drug-related serious adverse events in the study.
The REVIVE-Diabetes study was a randomized, double-blind, placebo-controlled efficacy and safety study that evaluated two doses of avanafil in men with diabetes and a history of ED. The results of the phase 3 study showed:
Patients achieved an overall statistically significant improvement in erectile function, as measured by the IIEF. EF-Domain scores range from 0-30 and measure the severity of erectile dysfunction as follows: severe dysfunction is less than or equal to 10; moderate is 11-16; and mild/minimal is 17-25. Results of the study were:
| |
Baseline | End of Treatment |
|||||
|---|---|---|---|---|---|---|---|
Placebo |
11.3 | 13.2 | |||||
Avanafil 100 mg |
11.2 | 15.8 | * | ||||
Avanafil 200 mg |
11.9 | 17.3 | ** | ||||
- *
- (p=0.002
100mg vs. placebo change from baseline)
- **
- (p<0.001 200mg vs. placebo change from baseline)
Patients on avanafil had erections sufficient for penetration as measured by the SEP question 2:
| |
Baseline | End of Treatment |
|||||
|---|---|---|---|---|---|---|---|
Placebo |
36 | % | 42 | % | |||
Avanafil 100 mg |
32 | % | 54 | %* | |||
Avanafil 200 mg |
42 | % | 63 | %* | |||
- *
- (p<0.001 active vs. placebo change from baseline)
16
Patients taking avanafil experienced successful intercourse as measured by the SEP question 3:
| |
Baseline | End of Treatment |
|||||
|---|---|---|---|---|---|---|---|
Placebo |
10 | % | 20 | % | |||
Avanafil 100 mg |
8 | % | 34 | %* | |||
Avanafil 200 mg |
8 | % | 40 | %* | |||
- *
- (p<0.001 active vs. placebo change from baseline)
The most commonly reported side effects in patients taking avanafil (all doses combined) included headache (7.8% vs. 1.5% placebo), nasopharyngitis (3.1% vs. 4.6% placebo), flushing (2.7% vs. 0% placebo), sinus congestion (1.9% vs. 0.8% placebo), sinusitis (1.9% vs. 0% placebo), and dyspepsia (1.6% vs. 0% placebo). No drug related serious adverse events were reported. The discontinuation rates for all patients enrolled were 15.4% placebo, 15.5% 100 mg, and 13% 200 mg.
REVIVE-Diabetes (TA-302) was a randomized, double-blind, placebo-controlled phase 3 study of avanafil in 390 men with ED as a result of their diabetes. On average patients had ED for at least six years and had diabetes for over 11 years. Seventy-six percent of study participants had tried at least one other ED treatment. Patients underwent a four-week, non-treatment run-in period followed by 12 weeks of treatment with one of two doses of avanafil: 100 mg and 200 mg, or placebo. Patients were instructed to attempt sexual intercourse approximately 30 minutes after taking the drug with no restrictions on food or alcohol consumption. The primary endpoints of the study were improvement in erectile function as measured by the SEP and improvement in the EF-Domain of the IIEF score; secondary endpoints included patient satisfaction with erections and with sexual experience.
The Phase 3 program includes two additional studies. REVIVE-RP (TA-303) has completed enrollment with 298 patients with ED following a radical prostatectomy. Patients undergo a four-week run-in period followed by 12 weeks of treatment. Patients are randomized to placebo or one of two dose levels of active drug. The primary endpoints of the study will be the same as those used in TA-301, namely, improvement in erectile function as measured by the Sexual Encounter Profile and the IIEF score. Patients are instructed to attempt sexual intercourse 30 minutes after taking avanafil, with minimal restrictions on food or alcohol consumption. REVIVE-RP will study two doses of avanafil: 100 mg and 200 mg.
In December 2010, we announced the positive results of the long term safety study, TA-314. TA-314 was conducted over one year in approximately 675 patients across 40 U.S. centers. Patients completing either the 12-week REVIVE or REVIVE-Diabetes studies were eligible to participate in TA-314. The study met all primary endpoints by demonstrating improvement from baseline in erectile function as measured by the Sexual Encounter Profile (both SEP2 and SEP3) and improvement in the International Index of Erectile Function, or IIEF. In the study, patients treated with avanafil who attempted sexual intercourse (SEP3) within the first 15 minutes of dosing had success rates of 80%. TA-314 confirms the longer term safety and efficacy results observed in the previously reported placebo-controlled Phase 3 studies of avanafil in patients with ED. With the completion of this study, we continue to anticipate the completion of the NDA filing for avanafil in the second quarter of 2011.
Highlights of the study include:
- •
- Eighty percent (80%) of sexual attempts among patients on avanafil had erections sufficient for intercourse (SEP2);
- •
- Sixty-seven percent (67%) of patients taking avanafil experienced successful intercourse (SEP3);
- •
- Successful intercourse was achieved as early as 15 minutes after dosing;
17
- •
- Avanafil was well tolerated as evidenced by a low rate of discontinuations due to adverse events (2.8%);
- •
- The most common side effects reported were headache (5.6%), flushing (3.5%), nasopharyngitis (3.4%) and nasal congestion
(2.1%); and
- •
- There were no drug-related serious adverse events reported in the study.
Patients achieved an overall improvement in erectile function, as measured by the erectile function, or EF, domain score of the IIEF. EF domain scores range from 0-30 and measure erectile function as follows: severe dysfunction is less than or equal to 10; moderate dysfunction is 11-16; mild/minimal dysfunction is 17-25; with normal function in the range of 26-30. At baseline, patients in the study had a mean EF domain score of 12.3 (high moderate). At the end of treatment, patients in the study had a mean EF domain score of 22.6, representing a change from baseline of 10.3.
Enrollment in the TA-303 study was slower than anticipated but was completed late in 2010. We believe in part this was due to the fact that physicians are routinely prescribing PDE5 inhibitors to their patients shortly following prostate surgery for penile rehabilitation, and patients do not wish to discontinue this rehabilitation therapy for the purpose of enrolling into a double-blind, placebo-controlled study such as TA-303. In October 2010, we held a pre-NDA meeting with the FDA and discussed our NDA filing for avanafil without the inclusion of TA-303. The FDA agreed that we may submit the NDA for avanafil prior to the completion of the TA-303 study. It is our intent to submit the results of the TA-303 study to the FDA once it is complete. We have been informed by the FDA that the submission of the TA-303 study results subsequent to our NDA filing will not impact the timing of their decision concerning the approvability of avanafil for other populations.
We have entered into a Master Services Agreement and related Task Orders with Quintiles, Inc., or Quintiles, pursuant to which Quintiles will perform certain clinical research services in connection with the clinical trials for avanafil. Our aggregate payment obligations entered into during 2008 through 2010 under the agreement for services, out of pocket expenses and pass through costs total approximately $29.2 million, of which we have paid approximately $25 million through December 31, 2010. We have agreed to defend and indemnify Quintiles against third party claims arising from the services other than claims resulting from Quintiles's negligence, willful misconduct, violation of law or material breach of the Master Service Agreement or a Task Order. We can terminate the agreement at any time without cause. Quintiles may terminate the agreement following our material breach of the agreement that remains uncured.
MUSE for Erectile Dysfunction
In 1997, we commercially launched MUSE in the U.S.. MUSE was the first minimally invasive therapy for erectile dysfunction approved by the FDA. On October 1, 2010, we entered into a definitive Asset Purchase Agreement with Meda to sell certain rights and assets related to MUSE, the MUSE Transaction. The assets sold include the U.S. and foreign MUSE patents, existing inventory, and the manufacturing facility located in Lakewood, New Jersey. We retained all of the liabilities associated with the pre-closing operations of the MUSE business and the accounts receivables for pre-closing MUSE sales. The transaction closed on November 5, 2010. Prior to the closing of the MUSE Transaction, we regained all of the rights to MUSE and avanafil held by Deerfield Management Company, L.P., and affiliates, and Crown Bank, N.A., or Crown. On October 15, 2010, we repaid the Crown loan in satisfaction of all obligations owed to them. As a result, the security interests and Certificate of Deposit held by Crown were terminated in our favor. On October 21, 2010, we exercised the Option under the Deerfield OPA, and satisfied all of the financial obligations under the FARA and OPA. As a result, the security interests in the collateral related to MUSE and avanafil held by the Deerfield Sub and the Deerfield Affiliates as part of the FARA and OPA were terminated.
18
Under the terms of the MUSE Transaction, we received an upfront payment of $22 million upon the closing and are eligible to receive an additional $1.5 million based on future sales of MUSE, provided that certain sales milestones are reached. Meda is now responsible for the manufacturing, selling and marketing of MUSE. Meda also assumed all post-closing expenses and liabilities associated with MUSE. We have agreed not to develop, manufacture or sell any transurethral erectile dysfunction drugs for a period of three years following the closing of the MUSE Transaction.
Other Programs
We have licensed and intend to continue to license from third parties the rights to other investigational drug candidates to treat various diseases and medical conditions. We also sponsor early stage clinical trials at various research institutions and intend to conduct early stage proof of concept studies on our own. We expect to continue to use our expertise in designing clinical trials, formulation and investigational drug candidate development to commercialize pharmaceuticals for unmet medical needs or for disease states that are underserved by currently approved drugs. We intend to develop products with a proprietary position or that complement our other products currently under development.
Sale of Evamist to K-V Pharmaceutical Company
On March 30, 2007, we entered into a definitive agreement with K-V Pharmaceutical Company, or K-V, to transfer our assets and grant a sublicense of our rights under the Evamist Agreement to K-V, or the K-V Transaction. The closing of the K-V Transaction occurred on May 15, 2007. Under the terms of the K-V Transaction, upon the closing, we received an upfront payment of $10 million. On July 27, 2007, we received FDA approval of the NDA for Evamist. On August 1, 2007, we transferred and assigned the Evamist FDA submissions, and all files related thereto to K-V, and on August 8, 2007, K-V paid us the additional $140 million milestone payment due upon FDA approval of the Evamist NDA. In August 2008, we assigned all of our rights and obligations under the Evamist license agreement to K-V. In connection with the K-V Transaction, in order to obtain MTPC's blanket release of liens against our assets including the Evamist assets and intellectual property, we repaid the MTPC line of credit.
In May 2006, we announced positive results from the pivotal Phase 3 clinical trial of Evamist. The study showed a statistically significant reduction in the number and severity of moderate and severe hot flashes. We submitted the NDA for Evamist to the FDA in the third quarter of 2006 and made a $1 million clinical development milestone payment to Acrux in October 2006 under the terms of our licensing agreement related to this submission. Upon approval of the NDA for Evamist, a $3 million product approval milestone became due and was paid to Acrux in August 2007. Under the terms of the K-V Transaction, K-V paid $1.5 million of this $3 million milestone.
Deerfield Financing
On April 3, 2008, we entered into several agreements with Deerfield Management Company, L.P., or Deerfield, a healthcare investment fund, and its affiliates, Deerfield Private Design Fund L.P. and Deerfield Private Design International, L.P. (collectively, the Deerfield Affiliates). Certain of the agreements were amended and restated on March 16, 2009, which included the addition of Deerfield PDI Financing L.P. as a Deerfield Affiliate. Under the agreements, Deerfield and its affiliates provided us with $30 million in funding, consisting of $20 million from the Funding and Royalty Agreement, or FARA, entered into with a newly incorporated subsidiary of Deerfield, or the Deerfield Sub, and $10 million from the sale of our common stock. We received all of the required payments under the FARA, or the Funding Payments. Under the FARA, we paid royalties on the net sales of MUSE to the Deerfield Sub. The FARA had a term of 10 years.
19
We entered into the Option and Put Agreement with the Deerfield Affiliates and the Deerfield Sub, dated April 3, 2008, and an Amended and Restated Option and Put Agreement dated March 16, 2009, or the OPA. Pursuant to the OPA, the Deerfield Affiliates granted us an option to purchase all of the outstanding shares of common stock of the Deerfield Sub from the Deerfield Affiliates, referred to as the Option. Our obligation to pay royalties terminated upon the exercise of the Option. The base consideration for the Option exercise, or Base Option Price, was $25 million, less $2 million we paid upon closing, as the Option was exercised on or prior to the third anniversary of the execution of the OPA. The aggregate consideration payable by us upon exercise of the Option, or the Option Purchase Price, was equal to the sum of the Base Option Price, plus: (i) the cash and cash equivalents held by the Deerfield Sub at the date of the closing of the resulting sale of the common stock of the Deerfield Sub, or the Cash Adjustment; (ii) accrued and unpaid royalties, or the Royalty Adjustment; and minus (i) the Option premium of $2 million that was paid at the closing of the transaction, or the Option Premium Adjustment ; (ii) accrued but unpaid taxes; (iii) unpaid Funding Payments; (iv) loans payable by the Deerfield Sub, or the Loan Balance Adjustment; and (v) any other outstanding liabilities of the Deerfield Sub, or the Adjusted Option Purchase Price.
In preparation for the closing of the MUSE Transaction and in accordance with the terms of the OPA, we exercised the Option, and on October 21, 2010 we paid an aggregate amount totaling $27.1 million, which consisted of the Base Option Price of $25 million, less the Option Premium Adjustment of $2 million, plus the Cash Adjustment of $2.8 million and the Royalty Adjustment of $1.3 million. These payments satisfied all of the financial obligations under the FARA and OPA. As a result, all of the outstanding shares and the $2.8 million of cash of the Deerfield Sub are owned by us, all of the outstanding loans owed by the Deerfield Sub have been repaid, and the security interests in the collateral related to MUSE and avanafil held by the Deerfield Sub and the Deerfield Affiliates as part of the FARA and OPA were terminated. In December 2010, the Deerfield Sub was dissolved. The payoff of the Deerfield loan resulted in a loss on the early extinguishment of debt of $6 million which was recognized in the fourth quarter of 2010.
Government Regulations
FDA Regulation
Prescription pharmaceutical products are subject to extensive pre- and post-marketing regulation by the FDA, including regulations that govern the testing, manufacturing, safety, efficacy, labeling, storage, record-keeping, advertising and promotion of the products under the Federal Food, Drug and Cosmetic Act, and by comparable agencies in most foreign countries.
The activities required before a pharmaceutical agent may be marketed in the U.S. begin with pre-clinical testing. Pre-clinical tests include laboratory evaluation of potential products and animal studies to assess the potential safety and efficacy of the product and its formulations. The results of these studies and other information must be submitted to the FDA as part of an Investigational New Drug, or IND, application, which must be reviewed and approved by the FDA before proposed clinical testing can begin. Clinical trials involve the administration of the investigational new drug to healthy volunteers or to patients under the supervision of a qualified principal investigator. Clinical trials must be conducted in accordance with Good Clinical Practices under protocols that detail the objectives of the study, the parameters to be used to monitor safety and the efficacy criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND application. Further, each clinical study must be conducted under the auspices of an independent institutional review board. The institutional review board will consider, among other things, ethical factors and the safety of human patients.
Typically, human clinical trials are conducted in three phases that may overlap. In Phase 1, clinical trials are conducted with a small number of patients to determine the early safety profile and pharmacology of the new therapy. In Phase 2, clinical trials are conducted with groups of patients afflicted with a specific disease or medical condition in order to determine preliminary efficacy, optimal
20
dosages and expanded evidence of safety. In Phase 3, large scale, multicenter clinical trials are conducted with patients afflicted with a target disease or medical condition in order to provide substantial evidence of efficacy and safety required by the FDA and others.
The results of the pre-clinical and clinical testing, together with chemistry and manufacturing information, are submitted to the FDA in the form of a New Drug Application, or NDA, for a pharmaceutical product in order to obtain approval to commence commercial sales. In responding to an NDA, the FDA may grant marketing approvals, may request additional information or further research or studies, or may deny the application if it determines that the application does not satisfy its regulatory approval criteria. FDA approval for a pharmaceutical product may not be granted on a timely basis, if at all, or if granted may not cover all the clinical indications for which approval is sought or may contain significant limitations in the form of warnings, precautions or contraindications with respect to conditions of use.
Satisfaction of FDA premarket approval requirements for new drugs typically takes several years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or targeted disease. Government regulation may delay or prevent marketing of potential products for a considerable period of time and may impose costly procedures upon our activities. Success in early stage clinical trials or with prior versions of products does not assure success in later stage clinical trials. Data obtained from clinical activities are not always conclusive and may be susceptible to varying interpretations that could delay, limit or prevent regulatory approval.
Once approved, the FDA may withdraw the product approval if compliance with post-marketing regulatory standards is not maintained or if problems occur after the product reaches the marketplace. In addition, the FDA may require post-marketing studies, referred to as Phase 4 studies, to monitor the effect of an approved product, and may limit further marketing of the product based on the results of these post-market studies. The FDA has broad post-market regulatory and enforcement powers, including the ability to levy fines and civil penalties, suspend or delay issuance of approvals, seize or recall products, or withdraw approvals. Additionally, the Food and Drug Amendment Act of 2007 requires all clinical trials we conduct for our investigational drug candidates, both before and after approval, and the results of those trials when available, to be included in a clinical trials registry database that is available and accessible to the public via the Internet. Our failure to properly participate in the clinical trial database registry would subject us to significant civil penalties.
Facilities used to manufacture drugs are subject to periodic inspection by the FDA, and other authorities where applicable, and must comply with the FDA's cGMP regulations. Failure to comply with the statutory and regulatory requirements subjects the manufacturer to possible legal or regulatory action, such as suspension of manufacturing, seizure of product or voluntary recall of a product. Certain adverse experiences with the product must be reported to the FDA and could result in the imposition of market restriction through labeling changes or product removal. Product approvals may be withdrawn if compliance with regulatory requirements is not maintained or if problems concerning safety or efficacy of the product occur following approval.
With respect to post-market product advertising and promotion, the FDA imposes a number of complex regulations on entities that advertise and promote pharmaceuticals, which include, among other things, standards and regulations relating to direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities, and promotional activities involving the Internet. The FDA has very broad enforcement authority. Failure to abide by these regulations can result in penalties including the issuance of a warning letter directing the entity to correct deviations from FDA standards, adverse publicity, a requirement that future advertising and promotional materials be pre-cleared by the FDA, and state and federal civil and criminal investigations and prosecutions.
We are subject to various laws and regulations regarding laboratory practices, the experimental use of animals, and the use and disposal of hazardous or potentially hazardous substances in connection
21
with our research. In each of these areas, as noted above, the government has broad regulatory and enforcement powers, including the ability to levy fines and civil penalties, suspend or delay issuance of approvals, seize or recall products, and withdraw approvals, any one or more of which could have a material adverse effect upon us.
Other Government Regulations
In addition to laws and regulations enforced by the FDA, we are also subject to regulation under National Institutes of Health guidelines as well as under the Controlled Substances Act, the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act and other present and potential future federal, state or local laws and regulations, as our research and development may involve the controlled use of hazardous materials, chemicals, viruses and various radioactive compounds.
In addition to regulations in the U.S., we are subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our investigational drug candidates. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
Corporate Collaborations and Licenses from Third Parties
Mitsubishi Tanabe Pharma Corporation
In January 2001, we entered into an exclusive development, license and supply agreement with Tanabe Seiyaku Co., Ltd., or Tanabe, now Mitsubishi Tanabe Pharma Corporation, or MTPC, and hereinafter collectively referred to as MTPC, for the development and commercialization of avanafil, a PDE5 inhibitor compound for the oral and local treatment of male and female sexual dysfunction. Under the terms of the agreement, MTPC agreed to grant an exclusive license to us for products containing avanafil outside of Japan, North Korea, South Korea, China, Taiwan, Singapore, Indonesia, Malaysia, Thailand, Vietnam and the Philippines. We agreed to grant MTPC an exclusive, royalty-free license within those countries for oral products that we develop containing avanafil. In addition, we agreed to grant MTPC an exclusive option to obtain an exclusive, royalty-bearing license within those countries for non-oral products that we develop containing avanafil. MTPC agreed to manufacture and supply us with avanafil for use in clinical trials, which will be our primary responsibility.
We have paid upfront licensing fees of $5 million to MTPC and have agreed to make additional payments upon the completion of certain development, regulatory and sales milestones. During the first quarter of 2004, we initiated a Phase 2 clinical trial with avanafil, which triggered one of the clinical development milestone criteria noted above. In 2006, we paid MTPC $2 million in connection with this milestone. We have further agreed to pay royalties on net sales of products containing avanafil. No payments were made under this agreement with MTPC in the years ended December 31, 2007 and 2008; however, we paid MTPC $4 million in January 2009 following the enrollment in December 2008 of the first patient in the first Phase 3 clinical studies. We expect to make other substantial payments to MTPC in accordance with our agreements with MTPC as we continue to develop and, if approved for sale, commercialize avanafil for the oral treatment of male sexual dysfunction. Such potential future milestone payments total $15 million and include payments upon: the first submission of an NDA, which is expected in the second quarter of 2011; obtainment of the first regulatory approval in the U.S. and any major European country, and achievement of $250 million or more in calendar year sales.
The term of the MTPC agreement is based on a country-by-country and on a product-by-product basis. The term shall continue until the later of (i) 10 years after the date of the first sale for a particular product, or (ii) the expiration of the last-to-expire patents within the MTPC patents covering
22
such product in such country. In the event that our investigational drug candidate is deemed to be (i) insufficiently effective or insufficiently safe relative to other PDE5 inhibitor compounds based on published information, or (ii) not economically feasible to develop due to unforeseen regulatory hurdles or costs as measured by standards common in the pharmaceutical industry for this type of product, we have the right to terminate the agreement with MTPC with respect to such investigational drug candidate.
Other
On October 16, 2001, we entered into an assignment agreement, or the Assignment Agreement, with Thomas Najarian, M.D. for a combination of pharmaceutical agents for the treatment of obesity and other disorders, or the Combination Therapy, that has since been the focus of our investigational drug candidate development program for QNEXA for the treatment of obesity, obstructive sleep apnea and diabetes. The Combination Therapy and all related patent applications, or the Patents, were transferred to us with worldwide rights to develop and commercialize the Combination Therapy and exploit the Patents. Pursuant to the Assignment Agreement, we have paid a total of $220,000 to Dr. Najarian through December 31, 2010 and have issued him options to purchase 40,000 shares of our common stock. We are obligated under the terms of the Assignment Agreement to make a milestone payment of $1 million and issue an option to purchase 20,000 shares of our common stock to Dr. Najarian upon marketing approval by the FDA of a drug for the treatment of obesity that is based upon the Combination Therapy and Patents. This assignment will require us to pay royalties on worldwide net sales of a drug for the treatment of obesity that is based upon the Combination Therapy and Patents until the last-to-expire of the assigned Patents. To the extent that we decide not to commercially exploit the Patents, the Assignment Agreement will terminate and the Combination Therapy and Patents will be assigned back to Dr. Najarian. In 2006, Dr. Najarian joined us as a part-time employee and currently serves as a Principal Scientist.
Patents and Proprietary Technology
We are the exclusive licensee of 34 patents and 11 published patent applications in the U.S. and Canada. We intend to develop, maintain and secure intellectual property rights and to aggressively defend and pursue new patents to expand upon our current patent base. Our portfolio of patents as it primarily relates to our investigational drugs is summarized as follows:
QNEXA |
||||
U.S. Patent No. 7,056,890 |
Expiring 06/14/2020 | |||
U.S. Patent No. 7,553,818 |
Expiring 06/14/2020 | |||
U.S. Patent No. 7,659,256 |
Expiring 06/14/2020 | |||
U.S. Patent No. 7,674,776 |
Expiring 06/14/2020 | |||
U.S. Patent Publication No. 2008/0255093 A1 |
Pending | |||
U.S. Patent Publication No. 2008/0312163 A1 |
Pending | |||
U.S. Patent Publication No. 2009/0304789 A1 |
Pending | |||
U.S. Patent Publication No. 2009/0304785 A1 |
Pending | |||
U.S. Patent Publication No. 2010/0105765 A1 |
Pending | |||
U.S. Patent Publication No. 2010/0215739 A1 |
Pending | |||
Canadian Patent No. 2,377,330 |
Expiring 06/14/2020 | |||
Canadian Patent Publication No. 2,691,991 A1 |
Pending | |||
Canadian Patent Publication No. 2,692,042 A1 |
Pending | |||
Canadian Patent Publication No. 2,727,313 A1 |
Pending | |||
Canadian Patent Publication No. 2,727,319 A1 |
Pending | |||
Canadian Patent Publication No. 2,686,633 A1 |
Pending | |||
AVANAFIL |
23
U.S. Patent No. 6,656,935 |
Expiring 09/13/2020 | |||
U.S. Patent No. 6,797,709 |
Expiring 09/13/2020 | |||
Canadian Patent No. 2,383,466 |
Expiring 09/13/2020 | |||
ERECTILE DYSFUNCTION |
||||
U.S. Patent No. 5,482,039 |
Expiring 03/25/2014 | |||
U.S. Patent No. 5,769,088 |
Expiring 03/25/2014 | |||
U.S. Patent No. 5,773,020 |
Expiring 04/25/2010 | |||
U.S. Patent No. 5,820,587 |
Expiring 03/14/2015 | |||
U.S. Patent No. 5,849,803 |
Expiring 12/15/2015 | |||
U.S. Patent No. 5,910,316 |
Expiring 06/08/2016 | |||
U.S. Patent No. 5,922,341 |
Expiring 10/28/2017 | |||
U.S. Patent No. 5,925,629 |
Expiring 10/28/2017 | |||
U.S. Patent No. 6,037,346 |
Expiring 10/28/2017 | |||
U.S. Patent No. 6,093,181 |
Expiring 07/25/2017 | |||
U.S. Patent No. 6,113,939 |
Expiring 09/05/2017 | |||
U.S. Patent No. 6,127,363 |
Expiring 10/28/2017 | |||
U.S. Patent No. 6,156,753 |
Expiring 10/28/2017 | |||
U.S. Patent No. 6,403,597 |
Expiring 10/28/2017 | |||
U.S. Patent No. 6,495,154 |
Expiring 11/21/2020 | |||
U.S. Patent No. 6,548,490 |
Expiring 10/28/2017 | |||
U.S. Patent No. 6,946,141 |
Expiring 11/21/2020 | |||
Canadian Patent No. 2,305,394 |
Expiring 10/28/2018 |
QNEXA contains two active substances, phentermine and topiramate, which have already been approved individually for use in medicinal products in the European Union. There is no approved product containing a fixed dose combination of phentermine and topiramate. We have been advised that QNEXA is considered a reference product under the EU rules governing medicinal products for the purpose of granting a fresh and independent period of regulatory data protection.
Since a new fixed combination product is considered a reference product within the meaning of Article 10(2)(a) of Directive 2001/83, it is entitled to a fresh period of regulatory data protection. According to Article 10(1) coupled with Article 10(2)(b) of Directive 2001/83/EC for nationally authorized products, this period of data protection is set at 10 years. Within eight years of those 10 years, a generic applicant is not permitted to cross refer to the preclinical and clinical trial data relating to the reference product. Even if the generic product is authorized, it cannot be placed on the market until the full 10-year regulatory data protection has expired. This 10-year data protection may be extended cumulatively to a maximum period of 11 years if during the first eight years of those 10 years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. This essentially is the formula of 8+2+1 for regulatory data protection applied in the EU. An identical formula for regulatory data protection is set out in Article 14(11) of Regulation (EC) 726/2004 for all centrally authorized products. If we are successful in obtaining a new indication, which is considered by the regulatory authorities to bring about a significant clinical benefit like diabetes or sleep apnea with QNEXA in the EU, we could, under the current EU rules, enjoy a total period of 11 years of data exclusivity.
The following U.S. patents and their foreign counterparts were transferred to Meda AB of Solna, Sweden, in connection with the previously reported closing of the sale of our MUSE assets November 5, 2010: 5,242,391; 5,474.535; 5,843,961; 5,886,039; and 5,942,512.
In connection with the previously reported termination of the Development and Commercialization Agreement for Luramist with FemPharm Pty Ltd., a wholly owned subsidiary of Acrux Limited, our
24
rights under the following U.S. patents, applications, and their foreign counterparts reverted to the licensor: 6,299,900; 6,818,226; 6,923,983; 7,438,203; 2006/0280783 A1; and 2008/0152597 A1.
We also hold foreign counterparts, patents and patent applications in major foreign jurisdictions related to our U.S. patents. We have developed and acquired exclusive rights to patented technology in support of our development and commercialization of our investigational drug candidates, and we rely on trade secrets and proprietary technologies in developing potential drugs. We continue to place significant emphasis on securing global intellectual property rights and are aggressively pursuing new patents to expand upon our strong foundation for commercializing investigational drug candidates in development.
Competition
Competition in the pharmaceutical and medical products industries is intense and is characterized by costly and extensive research efforts and rapid technological progress. We are aware of several pharmaceutical companies also actively engaged in the development of therapies for the treatment of obesity, diabetes and sexual health and medical device companies for the treatment of sleep apnea. Many of these companies have substantially greater research and development capabilities as well as substantially greater marketing, financial and human resources than we do. In addition, many of these companies have significantly greater experience than us in undertaking pre-clinical testing, human clinical trials and other regulatory approval procedures. Our competitors may develop technologies and products that are more effective than those we are currently marketing or researching and developing. Such developments could render our investigational drug candidates less competitive or possibly obsolete. We are also competing with respect to marketing capabilities and manufacturing efficiency, areas in which we have limited experience. Mergers, acquisitions, joint ventures and similar events may also significantly change the competition.
Current approved anti-obesity drugs include Xenical (orlistat), marketed by Roche, and the over-the-counter version, alli, marketed by GlaxoSmithKline, and phentermine, which is available from several generic manufacturers. Orlistat works by inhibiting lipase, thus preventing digestion and absorption of dietary fat in the gastrointestinal tract. Meridia (sibutramine) was marketed by Abbott Laboratories; however, in October 2010, Abbott Laboratories announced that it will withdraw Meridia in the U.S. at the FDA's request. The FDA requested the withdrawal because they believed Meridia's risks were not justified compared with the modest weight loss that patients achieved on the drug. In January 2010, the EMA suspended the marketing authorization of sibutramine in Europe. The impact on QNEXA of the withdrawal of sibutramine is unknown at this time. There are several drugs in development for obesity including an investigational drug candidate, liraglutide in Phase 3 clinical trials being developed by Novo Nordisk A/S and several other investigational drug candidates in Phase 2 clinical trials. Arena Pharmaceuticals, Inc. and Orexigen Therapeutics, Inc. have each submitted an NDA with the FDA for their investigational obesity drug candidates. Neither of these investigational drug candidates was approved by the FDA citing concerns around carcinogenicity in the case of Arena and the need to complete a pre-approval cardiovascular outcomes study in the case of Orexigen. The future of the Arena and Orexigen compounds is uncertain at this time.
Many of these drugs are or will be marketed by pharmaceutical companies with substantially greater resources than us. In addition, a number of generic pharmaceutical drugs are prescribed for obesity, predominantly phentermine. Phentermine is sold at much lower prices than we intend to charge for QNEXA, if approved. The availability of a branded prescription drugs, generic drugs and over-the-counter drugs could limit the demand for, and the price we are able to charge for QNEXA, if approved.
There are also surgical approaches to treat severe obesity that are becoming increasingly accepted and could become competitors against our investigational drug candidate, QNEXA. Two of the most well established surgical procedures are gastric bypass surgery and adjustable gastric banding. In
25
February 2011, the FDA approved the use of a lap band in patients with a BMI of 35 without co-morbidities and 30 with co-morbidities. The lowering of the BMI requirement will make more overweight and obese patients eligible for lap band surgery. The potential impact on QNEXA and/or other weight loss pharmacotherapy is unknown. In addition, other potential approaches that utilize various implantable devices or surgical tools are in development. Some of these approaches are in late stage development and may be approved for marketing. If approved, the companies that market these drugs may have substantially greater resources than we have.
Significant competitive therapies exist for avanafil in the form of oral medications marketed by Pfizer, Inc. under the name Viagra®, Cialis®, marketed by Eli Lilly and Company, and Levitra®, which is co-marketed by GlaxoSmithKline plc and Schering-Plough Corporation in the U.S. In 2010, the European Commission approved a new formulation of Levitra, or vardenafil HCI. Levitra 10mg oral-disintegrating tablet, or ODT, will be the first ED medication available in an ODT form. In Europe, the launch roll-out was expected in November 2010. The launch in the EU has not yet occurred. In the U.S., the medication was approved in June 2010 and will be marketed by GlaxoSmithKline and Merck & Co., Inc. under the tradename STAXYN™. Consistent with the oral version, the ODT formulation instructs men to take the medication 60 minutes prior to attempting sexual activity. However, the ODT version will be positioned as an "anytime, anywhere" treatment for ED. We are uncertain how the launch of ODT vardenafil will impact the future commercial potential of avanafil.
There are currently three PDE5 inhibitors approved in the U.S. for the treatment of erectile dysfunction: sildenafil, vardenafil, and tadalafil. Worldwide sales of these drugs were in excess of $4.1 billion in 2010. As the patents for the three major PDE5 inhibitors currently being sold expire, generic PDE5s will enter the marketplace. Generic PDE5s would likely be sold at lower prices and may reduce the demand for avanafil and the prices we intend to charge for avanafil, if approved. Additional PDE5 inhibitors are in various stages of development by other companies. Warner-Chilcott plc has licensed the U.S. rights to udenafil, a PDE5i from Dong-A. Warner-Chilcott continues the Phase 3 development of this compound for ED. Other treatments for ED exist, such as transurethral alprostadil, needle injection therapies, vacuum constriction devices and penile implants, and the manufacturers of these products will most likely continue to develop or improve these therapies.
New developments, including the development of other drug technologies and methods of preventing the incidence of disease, occur in the pharmaceutical and medical technology industries at a rapid pace. These developments may render our investigational drug candidates obsolete or noncompetitive. Compared to us, many of our potential competitors have substantially greater:
- •
- research and development resources, including personnel and technology;
- •
- regulatory experience;
- •
- investigational drug candidate development and clinical trial experience;
- •
- experience and expertise in exploitation and protection of intellectual property rights; and
- •
- access to strategic partners and capital resources.
As a result of these factors, our competitors may obtain regulatory approval of their products more rapidly than we or may obtain patent protection or other intellectual property rights that limit our ability to develop or commercialize our investigational drug candidates. Our competitors may also develop drugs or surgical approaches that are more effective, more useful and less costly than ours and may also be more successful in manufacturing and marketing their products. In addition, our competitors may be more effective in commercializing their products. We currently outsource our manufacturing and therefore rely on third parties for that competitive expertise. There can be no assurance that we will be able to develop or contract for these capabilities on acceptable economic terms, or at all.
26
Research and Development
We incurred $40 million in 2010, $70.9 million in 2009 and $77 million in 2008 in research and development expenses, primarily to discover and develop our investigational drug candidates in obesity, sleep apnea and diabetes treatment, to restore sexual function in men, to license from third parties the rights to investigational drug candidates to treat various sexual and nonsexual disorders and to sponsor early stage clinical trials at various research institutions.
Employees
As of February 18, 2011, we had 43 employees located at our corporate headquarters in Mountain View, California and other U.S. locations. None of our current employees are represented by a labor union or are the subject of a collective bargaining agreement. We believe that our relations with our employees are good and we have never experienced a work stoppage at any of our facilities.
Insurance
We maintain product liability insurance for our clinical trials. Insurance coverage is becoming increasingly expensive and no assurance can be given that we will be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. There can also be no assurance that we will be able to obtain commercially reasonable product liability insurance for any future drugs approved for marketing.
Available Information
Our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and amendments to reports filed pursuant to Section 13(a) and 15(d) of the Securities Exchange Act of 1934, as amended, are available on our website at www.vivus.com, when such reports are available on the Securities and Exchange Commission, or SEC, website. Copies of our annual report will be made available, free of charge, upon written request.
The public may read and copy any materials filed by VIVUS with the SEC at the SEC's Public Reference Room at 100 F Street, NE, Washington, DC 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains an Internet site that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC at http://www.sec.gov. The contents of these websites are not incorporated into this filing. Further, VIVUS' references to the URLs for these websites are intended to be inactive textual references only.
In addition, information regarding our code of ethics and the charters of our Audit, Compensation, and Nominating and Governance Committees are available free of charge on our website listed above, or in print upon written request.
Set forth below and elsewhere in this Annual Report on Form 10-K and in other documents we file with the Securities and Exchange Commission, or SEC, are risks and uncertainties that could cause actual results to differ materially from the results contemplated by the forward-looking statements contained in this Annual Report on Form 10-K. These are not the only risks and uncertainties facing VIVUS. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also impair our business operations.
27
Risks Relating to our Investigational Drug Candidate Development Efforts
We face significant risks in our investigational drug candidate development efforts.
The process of developing new drugs and/or therapeutic products is inherently complex, unpredictable, time-consuming, expensive and uncertain. We must make long-term investments and commit significant resources before knowing whether our development programs will result in drugs that will receive regulatory approval and achieve market acceptance. Investigational drug candidates that appear to be promising at all stages of development may not reach the market for a number of reasons that may not be predictable based on results and data of the non-clinical and clinical program. Investigational drug candidates may be found ineffective or may cause harmful side effects during clinical trials, may take longer to progress through clinical trials than had been anticipated, may not be able to achieve the pre-defined clinical endpoints due to statistical anomalies even though clinical benefit may have been achieved, may fail to receive necessary regulatory approvals, may prove impracticable to manufacture in commercial quantities at reasonable cost and with acceptable quality, or may fail to achieve market acceptance.
We are largely dependent on the success of our two investigational drug candidates: QNEXA, for treatment of obesity, and avanafil, for treatment of erectile dysfunction, and cannot be certain that either investigational drug candidate will receive timely regulatory approval, if at all, or be successfully commercialized.
We currently have only a limited number of investigational drug candidates in clinical development, and our business depends on their successful development and, if approved, commercialization. The research, testing, manufacturing, labeling, approval, sale, marketing and distribution of drug products are subject to extensive regulation by the U.S. Food and Drug Administration, or FDA, and other regulatory authorities in the U.S. and other countries, which regulations differ by agency and country. We are not permitted to market our investigational drug candidates in the U.S. until we receive approval of a new drug application, or NDA, from the FDA, or in any foreign countries until we receive the requisite approval from the regulatory authorities of such countries.
Regulatory approval of an NDA or NDA supplement is not guaranteed. Despite the time and expense exerted, failure can occur at any stage, and we could encounter problems that cause us to abandon clinical trials or to repeat or perform additional pre-clinical studies and clinical trials. The number of pre-clinical studies and clinical trials that will be required for FDA approval varies depending on the investigational drug candidate, the disease or condition that the investigational drug candidate is designed to target and the regulations applicable to any particular investigational drug candidate. The FDA may also ask for retrospective observational study or trials. A retrospective observational study or trial, also referred to as a historical study or trial, is a study in which the medical records of groups of individuals with certain characteristics are assessed for a particular outcome. In summary, in a retrospective observational study, all the events—exposure and subsequent development of the event, have already occurred in the past. We merely collect the data now, and establish the risk of developing an event if exposed to a particular risk factor. On the other hand, a prospective study is typically conducted by starting with two groups at the current point, and following up in the future for occurrence of an event, if any.
To date, all of the clinical studies we have performed on QNEXA are prospective studies. The FDA has asked for the feasibility of conducting a retrospective observational study of the historical incidence of fetal outcomes including congenital malformations with an interest in oral cleft and low birth weight in the offspring of women who received prophylaxis treatment with 100 mg of topiramate for migraine during pregnancy. We are currently assessing the feasibility of such a study. The FDA may require us to perform this study prior to the approval of QNEXA. This requirement may delay the approval of QNEXA and have a material adverse impact on our finances and future sales of QNEXA.
28
The FDA can delay, limit or deny approval of an investigational drug candidate for many reasons, including:
- •
- the FDA may not deem an investigational drug candidate safe and effective;
- •
- the FDA position can change or be adversely impacted due to unexpected or unpredictable external circumstances;
- •
- the FDA may not find the data from pre-clinical, clinical or retrospective observational studies or trials
sufficient to support approval;
- •
- the FDA may require additional pre-clinical, clinical or retrospective observational studies or trials;
- •
- the FDA may not accept our stability data for commercial product;
- •
- the FDA may not approve of our third-party manufacturers' processes and facilities;
- •
- the FDA may not accept future NDA submissions from us including amendments of the existing NDA for QNEXA or the filing of
an NDA for avanafil due to, among other reasons, the formatting of the submission; or
- •
- the FDA may change its approval policies, adopt new regulations or provide new guidance with significant requirements not currently included or considered by us when seeking NDA approval.
On October 28, 2010, we received a Complete Response Letter, or CRL, regarding the New Drug Application, or NDA, for QNEXA as a treatment for obesity. The CRL stated that in its current form, the NDA for QNEXA was not approvable. The CRL included the following areas: clinical, labeling, Risk Evaluation and Mitigation Strategy, or REMS, safety update, and drug scheduling. In the clinical section of the CRL, the FDA requested a comprehensive assessment of topiramate's and QNEXA's teratogenic potential including a detailed plan and strategy to evaluate and mitigate the potential teratogenic risks in women of childbearing potential taking the drug for the treatment of obesity. In addition, the FDA asked us to provide evidence that the elevation in heart rate (mean 1.6 beats per minute on the top dose) associated with QNEXA does not increase the risk for major adverse cardiovascular events. The FDA requested that we formally submit the results from the completed SEQUEL study (OB-305), a 52-week extension study for a subset of 675 patients who completed the previously reported 56-week CONQUER study. The FDA reserved the right to comment further on proposed labeling. On REMS, the FDA requested that a discussion of an already-submitted REMS plan be continued after we have submitted the written response. The agency also requested a safety update of any new adverse events be submitted to the NDA. Finally, the FDA stated that if approved, QNEXA would be a Schedule IV drug due to the phentermine component. On January 19, 2011, we held an End-of-Review meeting with the FDA to discuss the items contained in the CRL and the information we plan to include in the resubmission of the NDA for QNEXA. In anticipation of the meeting, we had provided a briefing document that included analyses integrating existing non-clinical and clinical data to provide a comprehensive assessment of the teratogenic potential of topiramate. In addition, we provided several new analyses to demonstrate that QNEXA does not increase the risk for major cardiovascular events, which analyses included cardiovascular data from our SEQUEL (OB-305) and Sleep Apnea (OB-204) studies. We also provided a synopsis of the final study report for the SEQUEL study. No new clinical studies were requested in the CRL. At the meeting, presentations were made on the comprehensive assessment of the teratogenic potential of topiramate and QNEXA, and evidence was presented that the increase in heart rate of 1.6 beats per minute does not increase the risk for major adverse cardiovascular events. The discussion also included elements of our proposed REMS program for QNEXA. The FDA chose to focus the meeting on the discussion of teratogenic potential for topiramate, specifically the incidence of oral clefts observed in the North American AED Pregnancy Registry and in the UK Epilepsy and Pregnancy Registry. As part of this meeting, the FDA requested that we complete the Feasibility Assessment. Although no other requests for additional
29
information or studies were made by the FDA at the meeting or in the CRL, there can be no assurance that the FDA will not request or require us to provide additional information or undertake additional studies in connection with the QNEXA NDA. In the QNEXA studies, which included 15 offspring from women exposed to QNEXA or topiramate, there were no reports of any congenital malformations or low birth weight. The timing of the planned resubmission of the QNEXA NDA will be determined after agreement with the FDA is reached on the Feasibility Assessment and whether a retrospective observational study would be needed. We expect to reach agreement with the FDA, and if deemed feasible, initiate the retrospective observational study on fetal outcomes within the next two months. It is our goal to resubmit the NDA for QNEXA to the FDA by the end of 2011.
There are no guarantees that our response to the FDA's request to perform the Feasibility Assessment will be sufficient to satisfy the FDA's safety concerns, that the FDA will not require us to conduct additional clinical or retrospective observational studies, that the FDA will accept our resubmission or that QNEXA will receive regulatory approval for any indication or prove to be commercially successful.
Our next most advanced investigational drug candidate, avanafil, has completed all of the large, pivotal Phase 3 clinical trials for efficacy and safety that are required to submit the NDA for approval. As part of our pre-NDA meeting with the FDA, it was determined that the completion of TA-303, a study of avanafil in patients who have undergone a radical prostatectomy, would not be required prior to submission of the avanafil NDA. Recruitment for the study was completed in December 2010. The final study report for TA-303 may be included as part of a supplemental filing to the avanafil NDA at a later time. All of the other avanafil Phase 3 studies have been completed and we anticipate filing the NDA in the second quarter of 2011. Notwithstanding our belief that the data collected from our three Phase 3 trials of QNEXA is promising, and even if we believe that data collected from our pre-clinical studies and clinical trials of avanafil and our other investigational drug candidates are promising and that our information and procedures regarding chemistry, manufacturing and controls are sufficient, our data may not be sufficient to support approval by the FDA or any other foreign regulatory authority.
In addition, we believe that the regulatory review of NDAs for investigational drug candidates intended for widespread use by a large proportion of the general population is becoming increasingly focused on safety. In this regard, it is likely that some of our investigational drug candidates, including QNEXA and avanafil, will be subject to increased scrutiny to show adequate safety than would investigational drug candidates for more acute or life-threatening diseases, such as cancer or HIV. In 2010, the FDA notified healthcare professionals that the review of additional data from a post-approval study of sibutramine indicates an increased risk of heart attack and stroke in patients with a history of cardiovascular disease using sibutramine. Meridia (sibutramine) was marketed by Abbott Laboratories; however, in October 2010, Abbott Laboratories withdrew Meridia in the U.S. at the FDA's request.
The European Medicines Agency also completed a review of the safety and effectiveness of sibutramine. The Agency's Committee for Medicinal Products for Human Use, or CHMP, has concluded that the benefits of sibutramine do not outweigh its risks, and that all marketing authorizations for medicines containing sibutramine should be suspended throughout Europe. We are unable to determine the impact on QNEXA, if any, of the recent actions in the U.S. and Europe with regards to sibutramine. Recently, Orexigen Therapeutics, manufacturers of Contrave, an investigational drug for the treatment of obesity, received a CRL from the FDA requesting that a long-term, randomized, placebo-controlled cardiovascular outcomes study be completed prior to refiling the NDA. To date, the FDA has not requested that we perform any additional studies, including cardiovascular outcome studies pre-approval, other than the request to perform the Feasibility Assessment. Cardiovascular outcomes studies can take several years, cost millions of dollars and may result in showing an increased risk for major adverse cardiovascular events for patients undergoing drug treatment. If any regulatory agency were to require additional studies, including studies to address cardiovascular events, the impact on the timing of approval and, if approved, commercialization of
30
QNEXA, avanafil or any of our investigational drug candidates could be delayed or adversely impacted. Even if approved, investigational drug candidates may not be approved for all indications requested and such approval may be subject to limitations on the indicated uses for which the drug may be marketed and may have restricted access programs. Our business and reputation may be harmed by any failure or significant delay in receiving regulatory approval for the sale of any drugs resulting from our investigational drug candidates. As a result, we cannot predict when or whether regulatory approval will be obtained for any of our investigational drug candidates currently under development.
The results of pre-clinical studies and completed clinical trials are not necessarily predictive of future results, and our current investigational drug candidates may not have favorable results in later studies or trials.
Pre-clinical studies and Phase 1 and Phase 2 clinical trials are not primarily designed to test the efficacy of an investigational drug candidate in the general population, but rather to test initial safety, to study pharmacokinetics and pharmacodynamics, to study limited efficacy in a small number of study subjects in a selected disease population, and to identify and attempt to understand the investigational drug candidate's side effects at various doses and dosing schedules. Success in pre-clinical studies or completed clinical trials does not ensure that later studies or trials, including continuing pre-clinical studies and large-scale clinical trials, will be successful nor does it necessarily predict future results. Favorable results in early studies or trials may not be repeated in later studies or trials, and investigational drug candidates in later stage trials may fail to show acceptable safety and efficacy despite having progressed through earlier trials. In addition, the placebo rate in larger studies may be higher than expected.
We have previously announced results from two pivotal Phase 3 clinical trials for safety and efficacy of our most advanced investigational drug candidate QNEXA, as a treatment for obesity, and we may continue to analyze the data collected in these trials. In addition, the briefing documents prepared by us and the FDA ahead of the Advisory Committee meeting, as well as materials used during that meeting, have been made available to the public. The meeting materials contain information not previously disclosed to the public. Top-line results of an extension study, OB-305, were released in the third quarter of 2010. Consequently, it is possible that further analysis of this information and other data on QNEXA may yield information or suggest conclusions not yet known that may negatively impact our ability to obtain regulatory approval for QNEXA as a treatment for obesity or, if approved, market acceptance.
Our other investigational drug candidate, avanafil, has successfully completed all of the large, pivotal Phase 3 trials for safety and efficacy that are required for submission for approval by the FDA and other worldwide regulatory authorities. Pre-clinical data and the limited clinical results that we have obtained for our investigational drug candidates may not predict results from studies in larger numbers of patients in multiple sites drawn from more diverse populations treated for longer periods of time. The smaller and shorter clinical trials also may not predict the ability of these investigational drugs to achieve or sustain the desired effects in the broad intended population or to do so safely. We may also decide to not conduct additional Phase 2 studies prior to the initiation of pivotal Phase 3 studies. In addition, we may elect to enter into pivotal Phase 3 studies with a new formulation, dosage, delivery system or choose to study different populations than had been studied in previous clinical trials.
QNEXA is our proprietary capsule formulation investigational drug candidate containing the active ingredients phentermine and topiramate. Phentermine was approved for the short-term treatment of obesity by the FDA in 1959. Topiramate is approved for seizures (1996) and migraine prevention (2004). Published studies on topiramate reported that topiramate treatment produced weight loss. By combining topiramate with phentermine, QNEXA attempts to simultaneously address excessive appetite and a high threshold for satiety, the two main mechanisms believed to impact eating behavior. Although we believe QNEXA affects the two major causes of overeating, excessive hunger and the
31
inability to feel satisfied, we may not be correct in our assessment of the impact the combination of these two ingredients may have on weight loss or their mechanism of action. Earlier studies with QNEXA were completed using a twice-a-day dose. The twice-a-day dose and timing of the administration of the immediate-release active ingredients was determined by the inventor through the treatment of patients in his private practice. We used a once-a-day, controlled-release formulation in our completed Phase 3 studies of QNEXA. We have completed various pharmacokinetic studies of the once-a-day formulations to characterize the pharmacokinetic profile of the once-a-day, controlled-release formulation of QNEXA. The FDA has also asked us to study the effects of a lower dose of QNEXA, which we did in the Phase 3 obesity trials. To date, the FDA has not raised any issues or concerns around the formulation of QNEXA. While we believe we can adequately address any future the issues raised by the FDA, there can be no assurance that we will be successful in obtaining or maintaining regulatory approval for QNEXA.
We may be required to demonstrate through large, long-term outcome trials that our investigational drug candidates are safe and effective for use in a broad population prior to obtaining regulatory approval. We are considering a cardiovascular outcome study post-approval for QNEXA. In addition, we are proposing the use of the pregnancy registry for QNEXA patients post-approval. We will also implement as necessary a patient access program to ensure only qualified patients receive QNEXA and will implement a Risk Evaluation and Mitigation Strategy, or REMS, to ensure that the benefits of QNEXA outweigh its risks. REMS for QNEXA will measure and monitor physician and patient compliance. There is typically a high rate of attrition from the failure of investigational drug candidates proceeding through clinical trials. If any of our investigational drug candidates fail to demonstrate sufficient safety and efficacy in any clinical trial, we will experience potentially significant delays in, or may decide to abandon development of, that investigational drug candidate. If we abandon or are delayed in our development efforts related to any of our investigational drug candidates, we may not be able to generate sufficient revenues to continue our operations and clinical studies at the current level or become profitable. Our reputation in the industry and in the investment community would likely be significantly damaged. It may not be possible for us to raise funds in the public or private markets, and our stock price would likely decrease significantly.
If the results of current or future pre-clinical studies, clinical testing and/or clinical trials indicate that our proposed investigational drug candidates are not safe or effective for human use, our business will suffer.
Unfavorable results from ongoing pre-clinical studies, clinical testing and/or clinical trials could result in delays, modifications or abandonment of ongoing or future clinical trials or development programs. A number of companies in the pharmaceutical industry have suffered significant setbacks in late stage clinical trials, even after promising results in mid to late-stage trials. Clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals. Negative or inconclusive results or adverse medical events during a clinical trial could cause a clinical trial to be delayed, repeated, modified or terminated. In addition, failure to design appropriate clinical trial protocols could result in the test or control group experiencing a disproportionate number of adverse events and could cause a clinical trial to be delayed, repeated, modified or terminated.
All of the investigational drug candidates that we are currently developing require extensive pre-clinical and/or clinical testing before we can submit any application for regulatory approval. Before obtaining regulatory approvals for the commercial sale of any of our investigational drug candidates, we must demonstrate with substantial evidence through pre-clinical testing and/or clinical trials that our investigational drug candidates are safe and effective in humans for the indication being studied. We may also be asked to complete retrospective observational studies. The FDA has recently requested that we complete the Feasibility Assessment. Retrospective observational studies can be complex and involve multiple data sources and electronic records that may not contain all the required information. We may be required to obtain consents from patients to further investigate findings in a retrospective observational study. In addition, the sample size selected may not be adequate to perform statistical
32
analysis or may not satisfy the requirements for approval. Conducting clinical trials and retrospective observational studies are complex, lengthy, expensive and uncertain processes. Completion of clinical trials and retrospective observational studies and approval by the FDA may take several years or more. Our ability to complete clinical trials and retrospective observational studies may be delayed, suspended or terminated by many factors, including, but not limited to:
- •
- inability to obtain or manufacture sufficient quantities of investigational drug candidates for use in clinical trials;
- •
- inability of the manufactured product to meet stability requirements;
- •
- failure to receive approval by the FDA of our clinical trial and retrospective observational studies protocols;
- •
- changes in clinical trial and retrospective observational studies protocols or analysis plans made by us or imposed by the
FDA;
- •
- poor safety or effectiveness of our investigational drug candidates;
- •
- slower than expected rate of and higher than expected cost of patient recruitment;
- •
- retaining patients who have initiated a clinical trial but may be prone to withdraw due to side effects from the therapy,
lack of efficacy or personal issues, or who are lost to further follow-up;
- •
- delay or failure to reach agreement on acceptable clinical trial agreement terms or clinical trial protocols with
prospective sites or investigators, or in the case of retrospective observational studies, failure to reach agreement with investigators, data providers, the institutional review board, or IRB,
patients, programmers and others on any aspect of a study;
- •
- delays in identifying and reaching agreement on acceptable terms with prospective clinical trial sites;
- •
- unfavorable results from ongoing clinical trials and pre-clinical studies;
- •
- uncertainty regarding proper dosing;
- •
- difficulty or inability to achieve bioequivalence between commercial formulations and clinical trial formulations;
- •
- failure of our clinical research organizations to comply with all regulatory and contractual requirements or otherwise
perform their services in a timely or acceptable manner;
- •
- scheduling conflicts with participating clinicians and clinical institutions;
- •
- termination of clinical trials by one or more clinical trial sites;
- •
- inability or unwillingness of medical investigators to follow our clinical protocols;
- •
- inability to adequately follow patients after treatment;
- •
- insufficient data to support regulatory approval;
- •
- collecting, reviewing and analyzing our clinical trial data;
- •
- unforeseen safety issues;
- •
- unforeseen issues with formulation or stability of investigational drug candidates;
- •
- obtaining IRB approval to conduct a clinical trial at a prospective site;
- •
- government or regulatory delays;
- •
- inability to raise the necessary cash to start or complete the trials;
- •
- inability to obtain access to historical records and information necessary to complete a retrospective observational study; or
33
- •
- inability to obtain consents from patients which, in the case of retrospective observational studies, can be several years after diagnosis.
Many of these factors may also ultimately lead to denial of regulatory approval of our investigational drug candidates. If we experience delays, suspensions or terminations and retrospective observational studies in our clinical trials for a particular investigational drug candidate, the commercial prospects for that investigational drug candidate will be harmed, and we may be unable to raise additional funds on favorable terms, if at all, or generate product revenues from that investigational drug candidate or revenues would be delayed, our reputation in the industry and in the investment community would likely be significantly damaged, and our stock price would likely decrease significantly. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials and retrospective observational studies may also ultimately lead to the denial of regulatory approval of an investigational drug candidate.
Our bioequivalence studies may fail to demonstrate acceptable comparability between formulations of investigational drug candidates used in our Phase 3 clinical trials and new formulations, if any, of investigational drug candidates we might choose to launch commercially, or choose to commercialize later, after launch.
We may choose to develop a new formulation of any or all of our investigational drug candidates that may be different from the formulation used in our Phase 3 clinical trials. If changes are made, or if a new formulation is used, we will need to demonstrate comparable bioequivalence between the formulation used in our Phase 3 clinical trials and the new formulation, should we choose to launch or later commercialize this new formulation. If we are unable to demonstrate that the formulation used in our Phase 3 clinical trials is bioequivalent to the new formulation we intend to launch or later commercialize, then we may be required to conduct additional clinical trials or repeat some or all of our Phase 3 clinical trials for our investigational drug candidate, or we may need to develop an alternative commercial formulation for the investigational drug candidate that is bioequivalent. As a result, our ability to obtain approval of the investigational drug candidate, if any, may be delayed. We have performed a bioequivalence study on a new formulation of QNEXA that we intend to launch, if approved, which was determined to be equivalent to the formulation used in the Phase 3 trials.
Association with fen-phen could lead to increased scrutiny of our investigational drug candidate, QNEXA.
One of the active ingredients in QNEXA, phentermine, had previously been used in combination with fenfluramine and dexfenfluramine. Phentermine is the most commonly prescribed anti-obesity drug. As phentermine is an older drug, no new efficacy trials have been conducted, with the exception of several trials on the combination of phentermine and fenfluramine in the early and mid 1990s and the EQUATE Phase 3 study that contained two phentermine arms. The combination of fenfluramine or PONDIMIN, or fen, and phentermine, or phen, was known as fen-phen. Fenfluramine received FDA approval in 1973 for the short-term treatment of obesity. Together, phentermine and fenfluramine were used by doctors to treat obesity. The FDA never approved the fen-phen combination; however, since the FDA approved fenfluramine, doctors were able to prescribe it as needed. The use of these drugs together for treatment of obesity was considered an off-label and unapproved use. In 1992, a published study cited fen-phen as a more effective method than dieting or exercise in reducing the weight of the chronically obese.
Neither combination, however, was ever tested for safety. By the summer of 1997, the Mayo Clinic reported 24 cases of heart valve disease in patients that had taken the fen-phen combination. The cluster of unusual cases of heart valve disease in fen-phen users suggested a correlation between fen-phen use and heart valve disease. On July 8, 1997, the FDA issued a Public Health Advisory to report the Mayo findings. The FDA continued to receive additional reports of heart valve disease, including reports from patients who had taken only fenfluramine or dexfenfluramine. Further
34
evaluations of patients taking fenfluramine or dexfenfluramine showed that approximately 30% had abnormal valve findings. This figure was much higher than expected for abnormal test results and suggested fenfluramine and dexfenfluramine as the likely causes of Primary Pulmonary Hypertension, or PPH, and valvular heart disease.
In September 1997, the FDA requested drug manufacturers to voluntarily withdraw fenfluramine and dexfenfluramine. At the same time, the FDA recommended that patients using either fenfluramine or dexfenfluramine stop taking them. The FDA did not, however, request the withdrawal of phentermine. Although studies to date have not demonstrated that phentermine causes PPH and valvular heart disease, when used in larger populations, there can be no assurance that QNEXA will not demonstrate rare, but significant cardiovascular or other detrimental side effects when used by the general population. Moreover, the adverse clinical history of fen-phen and dexfen-phen combinations for obesity may result in increased FDA regulatory scrutiny of the safety or the risk/benefit profile of QNEXA and may raise potential adverse publicity in the marketplace, which could affect clinical enrollment or ultimately market acceptance if QNEXA is approved for commercial sale.
Benfluorex is an anorectic and hypolipidemic agent that is structurally related to fenfluramine. Clinical studies have shown it may improve glycemic control and decrease insulin resistance in people with poorly controlled type 2 diabetes. On December 18, 2009, the European Medicines Agency recommended the withdrawal of all medicines containing benfluorex in the European Union because their risks, particularly the risk of heart valve disease (fenfluramine-like cardiovascular side effects), are greater than their benefits. In France, the medication had been marketed as Mediator by Servier as an adjuvant antidiabetic. It was on the market between 1976 and 2009 and is thought to have caused between 500 and 2,000 deaths. The drug was also used in Portugal and Cyprus. We are unable to determine the impact of the benfluorex withdrawal, if any, on the MAA for QNEXA.
Our investigational drug candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval or limit the commercial profile of an approved label.
Side effects caused by our investigational drug candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restricted label or the delay or denial of regulatory approval by the FDA or other regulatory authorities. The most common side effects reported in the first Phase 3 study of avanafil were headache, flushing and nasal congestion. The most common side effects reported in our Phase 3 trials of QNEXA were paresthesia (tingling of the extremities), dry mouth, altered taste, headache and constipation. In addition, the constituent drugs of QNEXA each has its own side effect profile that is included in its current product label. If QNEXA is approved by the FDA, we would anticipate that the label would, at a minimum, include the side effect profiles of each of the constituent drugs. We also expect the label to include warnings on use in pregnant, nursing mothers or women of childbearing potential. While the constituent drugs that make up QNEXA have post-marketing safety records and while we have tested these constituent drugs in combination in our clinical trials of QNEXA, to date, the combination of these constituent drugs has not received regulatory approval. While we believe our Phase 3 QNEXA clinical trials have generated sufficient safety and efficacy data, the approvability and eventual labeling of QNEXA will be determined by the FDA. For example, in 2007, the Endocrinologic and Metabolic Drugs Advisory Committee convened by the FDA reviewed another company's investigational obesity drug candidate, rimonabant, and voted not to recommend approval of the investigational drug candidate to the FDA, based on concerns regarding the safety profile of that investigational drug candidate in particular, depression, suicidality and seizures. Recently, two other obesity medications have also been rejected by the FDA citing safety concerns.
Phentermine and topiramate are each separately approved for sale by the FDA and have been on the market for many years. In general, significant adverse events and side effects observed in pre-clinical, clinical and post-marketing studies are included in the full prescribing information or label
35
for each drug. The label for TOPAMAX contains reports of side effects, warnings and precautions including metabolic acidosis, acute myopia and secondary angle closure glaucoma, decreased sweating and hyperthermia, cognitive-related dysfunction, psychiatric and behavioral disturbances including one completed suicide in a patient during a bipolar trial, somnolence and fatigue, sudden unexplained death in epileptics, kidney stones, paresthesia, various drug interactions, congenital malformations and low birth weight in infants born to mothers exposed to topiramate during pregnancy. The label for ADIPEX, a popular branded form of phentermine, contains warnings and precautions including recommendation against co-administration of phentermine with other drugs for weight loss. Adverse side effects include, among other things, pulmonary hypertension, valvular heart disease, drug abuse and dependence, overstimulation, restlessness, dizziness, insomnia, euphoria, dysphoria, tremor, headache, dryness of the mouth, diarrhea, constipation, impotence and changes in libido. The warnings and precautions for both of these drugs are updated often.
Previously published studies suggest that the administration of topiramate alone, in conjunction with diet and a behavioral modification program, results in weight reduction in obese patients. The most prominent side effect seen in the published studies was paresthesia (tingling of the extremities), experienced by 42% to 59% of patients. Dropouts due to paresthesia were 5% or less. In the EQUATE, EQUIP and CONQUER Phase 3 obesity studies, tingling was experienced in 23%, 19% and 21%, respectively, of the patients on the top-dose of QNEXA. In the Phase 2 diabetes study, paresthesia was experienced by 17% of the patients. The other common adverse events reported in the published topiramate monotherapy studies were also central nervous system, or CNS, related including fatigue, difficulty with attention, memory and concentration, and depression. In our obesity and diabetes studies, these CNS-related side effects were low, but they were higher than placebo. The pharmaceutical company performing research of topiramate alone for the treatment of obesity announced they had discontinued their development program including their controlled- release formulation.
The FDA has also issued an alert on the use of antiepileptic drugs and a potential risk of increased suicidal ideation. As part of our Phase 3 obesity trials for QNEXA, we prospectively assessed the potential risk of suicidal tendencies. The results of the extensive assessments performed in our Phase 3 trials for QNEXA indicated no signal for suicidal behavior or ideation. On July 10, 2008, the FDA held a Joint Meeting of the Peripheral and Central Nervous System Drugs Advisory Committee and the Psychopharmacologic Drugs Advisory Committee. The advisory committee and representatives from the Pediatric Advisory Committee, and the Drug Safety and Risk Management Advisory Committee considered the results of FDA's analysis of suicidality (both suicidal ideation and behavior) from placebo-controlled clinical studies of 11 antiepileptic drugs. One of the drugs included in the discussion was topiramate (marketed as TOPAMAX, Ortho-McNeil-Janssen Pharmaceuticals Inc.). The FDA discussed with the committee, in light of the results, whether any additional actions were necessary. The committee recognized that there was an increased risk of suicidality and recommended to the FDA that additional information should be provided to patients regarding the risks and benefits of antiepileptic drugs; however, the committee strongly recommended against a black box warning to be applied to antiepileptic drugs. In December 2008, the FDA asked the manufacturers of the antiepileptic drugs included in the analysis to add warnings about suicidality to the labels and to issue a medication guide covering the results of the meta-analysis. In April 2009, the FDA approved these new labels. We anticipate that the label for QNEXA, if approved, will, at a minimum, contain the similar suicidality warnings to those contained in the topiramate label.
The preliminary experience from an observational registration study conducted in the United Kingdom on women with epilepsy who became pregnant, published in the July 22, 2008 edition of Neurology, stated that the major congenital malformations, or MCM, rate observed in the study among infants born to women who were taking topiramate and other antiepileptics during their pregnancy raised some concerns. The UK Epilepsy and Pregnancy Register is a voluntary registry in the United Kingdom that collects information in order to gather and publish information on the relative safety of
36
antiepileptic drugs in this population. The results of the study were updated and published in the October edition of the Journal of Neurology, Neurosurgery and Psychiatry. In the study, 245 pregnancies were followed, of which 14 of 162, or 8.6%, had an MCM on polytherapy and three of 83, or 3.6%, had an MCM on topiramate monotherapy. The MCMs included oral clefts and hypospadias. It has been reported that prenatal exposure to certain antiepileptic drugs increases the risk of MCM from a background risk of between 1% and 2% to between 4% and 9%.
Pregnant women or women who planned on becoming pregnant were not eligible to participate in the QNEXA clinical trials. Women of childbearing potential were advised to use and agreed to use two forms of birth control during the study. Patients who became pregnant during the study period were required to immediately discontinue study medication. In our studies, we had 15 births from women exposed to QNEXA or topiramate. They were taken off the study medication and followed through to delivery. While we did not observe any congenital malformations or low birth weight (less than 2,500 gm) in those pregnancies, we anticipate the labeling for QNEXA, if approved, will contain a warning against use by women who are or are considering becoming pregnant. We might also be required to list QNEXA as a "Category X" drug. Drugs in Category X are designated as such when studies in animals or humans have demonstrated fetal abnormalities and/or there is positive evidence of human fetal risk based on adverse reaction data from investigational or marketing experience, and the risks involved in use of the drug in pregnant women clearly outweigh potential benefits. The FDA may also require us to establish a post-approval pregnancy exposure registry. The goal of pregnancy exposure registries is to provide clinically relevant human data that can be used in a product's labeling to provide medical care providers with useful information for treating or counseling patients who are pregnant or anticipating pregnancy.
In the U.S., the label for topiramate has been modified and includes warnings for pregnant mothers. The warnings suggest that topiramate may cause serious adverse fetal effects, based on pregnancy registry and non-clinical data. There are no adequate and well-controlled studies using topiramate in pregnant women. Topiramate should be used during pregnancy only if the potential benefit outweighs the potential risk to the fetus. Pregnancy registry data suggest that there may be an association between the use of topiramate during pregnancy and congenital malformations (e.g., craniofacial defects, such as cleft lip/palate, hypospadias, and anomalies involving various body systems). This has been reported with topiramate monotherapy and topiramate as part of a polytherapy regimen. Compared with a reference group not taking antiepileptic drugs, registry data for topiramate monotherapy showed a higher prevalence of low birth weight; however, the dose of topiramate and the duration of use during pregnancy were not reported. A causal relationship has not been established. Several factors affect infant birth weight including maternal weight, maternal health, concomitant drug use, smoking or alcohol use during pregnancy and use of prenatal vitamins. In the QNEXA clinical studies, none of the infants to mothers exposed to QNEXA or topiramate during pregnancy acheived low birth weight.
Two antiepileptic pregnancy registries have reported post-marketing reports of major congenital malformations in infants whose mothers were epileptic and exposed to topiramate. The dose and duration of topiramate from the North American Registry are unknown. In the UK registry, for the two oral clefts reported in infants, the maternal topiramate exposure was 200 mg and 600 mg. These two registries suggest the MCM rate is higher than compared to various control groups. These reports have been included in the label for topiramate and, if QNEXA is approved, we may be required to include these reports in the label for QNEXA. At the request of the FDA, we are currently conducting the Feasibility Assessment. If deemed feasible and the retrospective observational study is completed, the results may be included in the QNEXA label, if approved. In addition, if the risk to women of childbearing potential is deemed unacceptable, QNEXA may not be approved or any such approval may be limited to men and women of non-childbearing potential. If approval of QNEXA is limited to men and women of non-child bearing potential, future sales of QNEXA, if approved, would be adversely affected.
37
Patients in the year long QNEXA studies had a mean elevation in heart rate at the end of the 56-week studies of 1.3, 0.6 and 1.6 beats per minute on the low, mid and top dose as compared to no change in the placebo group. Patients also had a decrease in systolic blood pressure of 3.3, 5.2 and 5.2 mmHg on the low, mid and top dose as compared to a decrease of 2.1 mmHg in the placebo group. The clinical relevance of the increase in heart rate at these levels is unknown. In the CRL, the FDA asked us to provide evidence that the elevations in heart rate associated with QNEXA do not increase the risk for major adverse cardiovascular events. We have provided evidence from existing information and data analyses to show that the increase in heart rate associated with QNEXA does not increase the risk for major cardiovascular events; however, there can be no assurance that the FDA will accept or agree with the evidence we provide, that the FDA will not require us to conduct long-term cardiovascular outcomes studies or other clinical studies prior to approval, or that we will be able to rule out, to the FDA's satisfaction, that the elevations in heart rate associated with QNEXA do not increase the risk for major adverse cardiovascular events now or in the future.
In the Phase 3 EQUIP and CONQUER studies, there was no difference between QNEXA (0.4%) and placebo (0.4%) drug-related serious adverse events. In the Phase 3 EQUATE study, there were no reported drug-related serious adverse events. In the Phase 3 avanafil studies, there were no drug related serious adverse events. If our trials are not successful or are perceived as not successful by the FDA, physicians, analysts, investors, the media or the public in general our business, financial condition and results of operations will be materially harmed.
If any of our investigational drug candidates receives marketing approval and we, or others, identify unknown side effects caused by the drug, a number of potentially significant negative consequences could result, including:
- •
- regulatory authorities may withdraw their approval of the drug;
- •
- regulatory authorities may require the addition of labeling statements, such as a "black box" warning with QNEXA or a
contraindication;
- •
- we may be required to create a Medication Guide outlining the risks of such side effects for distribution to patients;
- •
- we may be required to change the way the drug is administered, conduct additional clinical trials or change the labeling
of the drug;
- •
- we could be asked to formulate a Risk Evaluation Mitigation Strategy, or REMS, that could include a program of
post-marketing surveillance or restricted distribution for physicians who prescribe our drugs and patients being treated with our drugs;
- •
- prescribing physicians may be requested to complete certain education programs on the drugs and their intended use;
- •
- patient access to the drug may be limited to certain populations and patients may be requested to provide certain
information prior to receiving their new or refill prescription;
- •
- patients who are contraindicated by the label may obtain the drug and experience adverse side effects;
- •
- we could be sued and held liable for injury to individuals exposed to or taking our drugs; and
- •
- our reputation may suffer.
Any of these events could prevent us from achieving or maintaining market acceptance of QNEXA and could substantially increase the costs of commercializing our investigational drug candidates.
38
Our investigational drug candidate, QNEXA, is a combination of drugs approved individually by the FDA that are commercially available and marketed by other companies. As a result, our drug may be subject to substitution with individual drugs contained in the QNEXA formulation and immediate competition.
Each of the approved drugs that are combined to produce our investigational drug candidate, QNEXA, is commercially available at prices lower than the price at which we would seek to market QNEXA, if approved. We cannot be sure that physicians will view QNEXA as sufficiently superior to a treatment regime of QNEXA's individual active pharmaceutical ingredients as to justify the significantly higher cost we expect to seek for QNEXA, and they may prescribe the individual generic drugs already approved and marketed by other companies instead of our combination drug. Although our U.S. and European patents contain composition, product formulation and method-of-use claims that we believe protect QNEXA, those patents may be ineffective to protect against physicians prescribing the individual drugs marketed by other companies instead of our combination drug. Our patents and pending patent applications do not prevent physicians from prescribing the generic constituents of our investigational drug candidates. Phentermine and topiramate are currently available in generic form, although the doses used in QNEXA are currently not available and no controlled or sustained release formulation of topiramate exists. We believe that a practitioner seeking safe and effective therapy is not likely to prescribe such off-label generics in place of QNEXA because the dosage strengths, pharmacokinetic profiles and titration regimens recommended for QNEXA are not available using existing generic preparations of immediate release, or IR, phentermine or topiramate. However, to the extent that the price of QNEXA is significantly higher than the prices of the individual components as marketed by other companies, physicians may have a greater incentive to write prescriptions for the individual components instead of for our combination drug, and this may limit how we price or market QNEXA. Similar concerns could also limit the reimbursement amounts private health insurers or government agencies in the U.S. are prepared to pay for QNEXA, which could also limit market and patient acceptance of our drug, and could negatively impact our revenues. A physician could seek to prescribe off-label generics in place of QNEXA. Off-label use occurs when a drug that is approved by the FDA for one indication is legally prescribed by physicians for a different indication not approved by the FDA. Topiramate, one of the ingredients in QNEXA, is not approved for obesity treatment.
With regard to off-label substitution at the pharmacy level, we cannot be certain that pharmacists and/or pharmacy benefit managers will not seek prescriber authorization to substitute generics in place of QNEXA, which could significantly diminish its market potential. Wide scale generic substitution by physicians and at the pharmacy level could have substantial negative consequences to our business.
In many regions and countries where we may plan to market QNEXA, including Europe and Canada, the pricing of prescription drugs is controlled by the government or regulatory agencies. The government or regulatory agencies in these countries could determine that the pricing for QNEXA should be based on prices for its active pharmaceutical ingredients when sold separately, rather than allowing us to market QNEXA at a premium as a new drug.
We may choose or be required by regulatory authorities to restrict distribution of QNEXA to specialty pharmacies after physicians and patients register to ensure a safe and secure launch. Our success in distributing our investigational drug candidate in this manner could be limited, which could have an adverse effect on our business, financial condition, results of operations and cash flow.
The FDA and other regulatory agencies will likely require more extensive or expensive trials for our combination investigational drug candidate, QNEXA, than may be required for single agent pharmaceuticals.
To obtain regulatory approval for QNEXA, we are required to show that each active pharmaceutical ingredient in our investigational drug candidate makes a contribution to the combined investigational drug candidate's claimed effects and that the dosage of each component, including amount, frequency and duration, is such that the combination is safe and more effective than each of
39
the components. As a result, we were required to include in our clinical trial protocols an evaluation of each component drug as well as for the component drug in combination. This required us to conduct more extensive and more expensive clinical trials than would be the case for many single agent pharmaceuticals. The need to conduct such trials could make it more difficult and costly to obtain regulatory approval of QNEXA than of a new drug containing only a single active pharmaceutical ingredient. The OB-301, or EQUATE, Phase 3 trial was designed to meet the combination guidelines set by the FDA. The EQUATE study contained separate component arms as well as the combination. We believe the results of the EQUATE study meet FDA guidelines for combination therapy studies; however, there can be no assurance that we have satisfied the combination requirements to the FDA's satisfaction or that further testing of the combination will not be required. The EQUATE study also contained a mid-dose of QNEXA containing 7.5 mg of phentermine and 46 mg of topiramate CR. The mid-dose was also included in the CONQUER, or OB-303, study. We did not complete a component study for the low-dose. We have filed for approval of all three doses. The number of patients on the low-dose in OB-302 or the mid-dose in the OB-303 study may not be sufficient for approval. We have no assurance that any of the doses of QNEXA will be approved or that additional pre-clinical and clinical testing may not be needed prior to approval. In addition, if the FDA does not approve the top-dose of QNEXA, there is no assurance that they would approve the mid-dose or any other dose of QNEXA. In the CRL received on October 28, 2010, there were no deficiencies noted relating to the combination guidelines or exposure numbers by dose; however, until approval, there can be no assurance that these items will not be subject to further review and comment by the FDA.
We have in-licensed all or a portion of the rights to our investigational drug candidates from third parties. If we default on any of our material obligations under those licenses, we could lose rights to our investigational drug candidates.
We have in-licensed and otherwise contracted for rights to our investigational drug candidates, and we may enter into similar licenses in the future to supplement our investigational drug candidate pipeline. Under the relevant agreements, we are subject to commercialization, development, supply, sublicensing, royalty, insurance and other obligations. If we fail to comply with any of these requirements, or otherwise breach these license agreements, the licensor may have the right to terminate the license in whole or to terminate the exclusive nature of the license. Loss of any of these licenses or the exclusive rights provided therein could harm our financial condition and operating results.
In particular, the rights to avanafil were licensed from Tanabe Seiyaku Co., Ltd., or Tanabe, in 2001. In October 2007, Tanabe and Mitsubishi Pharma Corporation completed their merger and announced their name change to Mitsubishi Tanabe Pharma Corporation, or MTPC. We are in discussion with MTPC about certain aspects of the license agreement relating to the manufacture and supply of products. Failure to reach agreement with MTPC may have an adverse impact on the commercial future of avanafil. The rights to QNEXA were licensed from Dr. Najarian in 2001. We believe we are in compliance with all the material terms of our current agreements; however, there can be no assurance that this compliance will continue or that the licensors would not have a differing interpretation of the material terms of the agreements. If the license agreements were terminated early or if the terms of the license were contested for any reason, it would have a material adverse impact on our ability to commercialize products subject to these agreements, our ability to raise funds to finance our operations, our stock price and our overall financial condition. The monetary and disruption costs of any disputes involving our collaborative agreements could be significant despite rulings in our favor.
For example, VIVUS and Acrux Limited, through its wholly owned subsidiary FemPharm Pty Ltd., or Acrux, were parties to the Testosterone Development and Commercialization Agreement dated February 12, 2004, or the Testosterone Agreement. The Testosterone Agreement covered our investigational drug candidate, Luramist, which was licensed from Acrux under the Testosterone
40
Agreement. On November 5, 2007, Acrux made a demand for arbitration under the Testosterone Agreement regarding certain claims related to Luramist. Acrux's demand sought a reversion of all rights assigned to us related to Luramist, monetary damages and the payment of a milestone payment for Luramist under the Testosterone Agreement and declaratory relief. We asserted counterclaims against Acrux in the arbitration and sought the enforcement of our rights under the Testosterone Agreement. The arbitration hearing concluded on January 23, 2009, and on April 6, 2009 the panel of arbitrators, or the Panel, issued its Interim Arbitration Award finding in favor of the Company that we were in compliance with the Testosterone Agreement and denying all of the relief sought by Acrux in its demand. The Panel found that we had used diligent, commercially reasonable efforts to develop Luramist. The Panel further ruled in our favor on our counterclaim that Acrux had breached the Testosterone Agreement by failing to provide certain know-how and certain improvements in the formulation and delivery device for Luramist. The Panel denied the Acrux claim for additional milestone payments. The Panel ordered Acrux to turn over certain information to us that was previously withheld in violation of the Testosterone Agreement by Acrux. After the parties failed to agree on a new Outside Date by which we were to commence our first Phase 3 trial for Luramist, the Panel reset the Outside Date of April 30, 2006 to April 1, 2010 to reflect the regulatory environment. On March 30, 2010, we provided written notice to Acrux of our intent to terminate the Testosterone Agreement. On April 6, 2010, in connection with Acrux's request for further briefing on the issue of damages in light of the our termination of the Testosterone Agreement, the Panel ordered the parties to enter into settlement discussions and to report back to the Panel no later than May 17, 2010 on whether a settlement had been reached. On May 6, 2010, the parties agreed to the terms of a settlement agreement and mutual release, or the Settlement Agreement, resolving any and all claims or potential claims in the arbitration and that may have or could have arisen from any case whatsoever, other than certain rights and obligations that survive the termination of the Testosterone Agreement or are required by the Settlement Agreement. Pursuant to the Settlement Agreement, we have transferred Luramist-related assets to Acrux, including clinical trial material, batch release documents, inventory of applicators, FDA correspondence, intellectual property and know-how and trademarks. In addition, we have ceased our clinical study program for Luramist as part of the settlement. The parties have not exchanged cash payments as a result of the settlement and termination of the Testosterone Agreement. The Panel retains jurisdiction over the matter to enforce the terms of the Settlement Agreement. Although we have now returned the rights to Luramist to Acrux and resolved the arbitration, there can be no assurance that Acrux will not pursue legal action against us for any of our continued obligations under the settlement or the provisions of the Testosterone Agreement that survive the termination. The monetary and disruption costs of this arbitration have been significant despite the favorable rulings by the Panel.
While we may be entitled to future milestone payments under existing contractual arrangements, we may not receive these payments.
Certain of our contractual arrangements include future milestone payments to us based upon the other party achieving defined sales targets. Meeting those milestone targets is dependent on the performance of the other party to the contractual arrangement and we have little, or no, control over those outcomes. We have no assurance any of those milestone targets will be achieved and that the milestones will be paid to us.
For example, on March 30, 2007, we entered into a definitive agreement with K-V Pharmaceutical Company, or K-V, to transfer the assets and grant a sublicense of our rights under our licensing agreement with Acrux related to Evamist, a metered-dose transdermal spray for the treatment of menopause symptoms, to K-V. Under the terms of this agreement, we are also eligible to receive certain one-time payments of up to $30 million based on K-V achieving certain annual net sales thresholds for Evamist. In January 2009, K-V and certain of its subsidiaries announced a voluntary recall of most of its prescription drugs. In addition, K-V voluntarily suspended the manufacturing and shipping of all of its
41
products. Subsequent to the recall, K-V announced plans to reduce its workforce by 700 employees. Evamist is not manufactured by K-V and was not subject to the recall. In July 2009, K-V announced that it had hired a firm that specializes in restructurings and bankruptcies. Given the uncertainties with K-V, it is difficult to determine the extent of the adverse impact on Evamist. Although we are entitled to additional milestone payments from future sales of Evamist by K-V, at the present time we do not anticipate receiving any additional milestone payments from sales of Evamist.
On October 1, 2010, we entered into a definitive Asset Purchase Agreement with Meda AB, or Meda, to sell certain rights and assets related to MUSE. The transaction closed on November 5, 2010. Under the terms of the transaction, we are eligible to receive an additional $1.5 million based on future sales of MUSE, provided that certain sales milestones are reached. There can be no guarantee that these future sales milestones will be achieved or that we will receive this additional compensation related to the sale of MUSE.
We are dependent upon collaborative arrangements and strategic alliances.
We are, and in the future expect to be, dependent upon collaborative arrangements or strategic alliances to complete the development and commercialization of some of our investigational drug candidates, particularly after the Phase 2 stage of clinical testing. These arrangements may place the development of our investigational drug candidates outside of our control, may require us to relinquish certain rights or pay royalties, or may otherwise be on terms unfavorable to us. In October 2007, Tanabe and Mitsubishi Pharma Corporation completed their merger and announced their name change to Mitsubishi Tanabe Pharma Corporation, or MTPC. The rights and obligation of our license agreement with Tanabe have been transferred to MTPC. It is unclear at this time what effect, if any, the merger has had on our agreement with MTPC. There can be no guarantee that the merger will not have an adverse material effect on the performance by MTPC under our agreement, which in turn could lead to a material adverse effect on our business, financial condition and results of operations.
We may be unable to enter into favorable agreements with third parties, which could delay or impair our ability to develop and commercialize our investigational drug candidates and could increase our costs of development and commercialization. Dependence on collaborative arrangements or strategic alliances will subject us to a number of risks, including the risk that:
- •
- we may not be able to control the amount, timing and quality of resources that our collaborators may devote to the
investigational drug candidates;
- •
- our collaborators may experience financial, regulatory or operational difficulties;
- •
- our collaborators may be required to disclose our confidential information or may fail to protect our confidential
information;
- •
- we may be required to relinquish important rights such as marketing and distribution rights;
- •
- business combinations or significant changes in a collaborator's business strategy may adversely affect a collaborator's
willingness or ability to satisfactorily complete its obligations to meet our requirements under any arrangement;
- •
- legal disputes or disagreements may occur with our collaborative partners;
- •
- a collaborator could independently move forward with a competing investigational drug candidate developed either
independently or in collaboration with others, including our competitors; and
- •
- collaborative arrangements are often terminated or allowed to expire, which would delay the development and may increase the cost of developing our investigational drug candidates.
42
Although we may terminate an existing collaborative arrangement or other agreement, we could be subject to continued costs or obligations thereunder.
The majority of our agreements with third parties contain termination clauses that provide for cancellation or termination of the agreement under certain circumstances. We may decide to terminate our agreements with third parties for business or other reasons at any time. For example, we have entered into contractual agreements for services in anticipation of the commercial launch of QNEXA. Although we may terminate these or any other agreements, we could be subject to continued costs or obligations under the terminated agreements.
We face significant governmental regulation during our investigational drug candidate development activities.
The research, testing, manufacturing, selling and marketing of investigational drug candidates and approved pharmaceuticals is subject to extensive regulations by the FDA and other regulatory agencies in the U.S. and other countries. We cannot predict with certainty if or when we might submit for regulatory review our investigational drug candidates currently under development, except for QNEXA, for which the NDA was submitted to the FDA on December 29, 2009 and for which we are currently in the process of resubmitting an NDA amendment. Even if submitted, the FDA can suspend or modify clinical studies at any time if the agency believes that the patients participating in such studies are being exposed to unacceptable health risks.
Regulatory approval is never guaranteed, and the approval process typically takes several years and is extremely expensive. The FDA has substantial discretion in the drug approval process. Despite the time and expense involved, failure can occur at any stage.
In July 2008, an FDA advisory committee discussed the role of cardiovascular outcomes assessment in the pre-approval and post-approval settings for drugs and biologics developed for the treatment of type 2 diabetes mellitus. The advisory committee recommended that sponsors conduct a long-term cardiovascular trial or to provide other equivalent evidence to rule out an unacceptable cardiovascular risk. The FDA has since published a guidance document in December 2008 for the evaluation of cardiovascular risk in new antidiabetic therapies specifically for the treatment of type 2 diabetes. In general, the FDA recommends that sponsors should compare the incidence of important cardiovascular events occurring with the antidiabetic investigational agent to the incidence of the same type of events with the control group to estimate the relative risk of the investigational antidiabetic agent. This may be accomplished by either conducting an integrated analysis (meta-analysis) of the Phase 2 and Phase 3 studies, if the investigational drug was in late-stage development at the time the December 2008 guidance was published, or conduct a single, large, prospective long-term cardiovascular safety outcomes study prior to NDA submission. A long-term cardiovascular study would take several years to complete and would require financial and personnel resources that may be beyond our current capabilities. QNEXA, in development for diabetes is subject to this recommendation. The FDA, however, has neither required a meta-analysis of the QNEXA Phase 2 and 3 data, nor a prospective long-term cardiovascular safety outcomes study to be performed for QNEXA as a treatment for obesity. There can be no assurance, however, that the FDA would not in the future require us to perform a cardiovascular safety outcomes study, pre- or post-approval, for QNEXA as a treatment for obesity. Previously, the FDA notified healthcare professionals that the review of data from a post-approval outcomes trial of an anti-obesity agent, subutramine, indicates an increased risk of heart attack and stroke in patients with a history of cardiovascular disease. Based on the serious nature of the review findings, the FDA requested, and the manufacturer agreed, to add a new contraindication to the sibutramine drug label stating that sibutramine is not to be used in patients with a history of cardiovascular disease. In 2010, subutramine was withdrawn from the market in the U.S. and in the EU. Recently, Orexigen Therapeutics, the developer of Contrave, received a CRL and reported that the CRL stated that before the NDA could be approved they must conduct a randomized, double-blind, placebo-controlled trial of sufficient size and duration to demonstrate that the risk of major
43
adverse cardiovascular events in overweight and obese subjects treated with naltrexone/bupropion does not adversely affect the drug's benefit-risk profile. We have no insight as to the cost, design or timing of this required study. We have no reason to believe QNEXA would be subject to the same requirements. If we are required to complete a long-term cardiovascular safety outcomes study for QNEXA, the ultimate approval may be delayed for several years and the overall cost of the program will significantly increase.
In June 2007, an FDA advisory committee recommended against approval of rimonabant, an oral obesity treatment that targets the CB1 receptor system. Rimonabant was a centrally acting drug that reduced patients' desire to eat being developed by another company. The advisory committee expressed concerns about the impact of the drug on depressed patients and also expressed concerns about patients having thoughts about suicide. In addition, concerns about rimonabant's mechanism of action and interference with the CB1 receptor pathway were also voiced. The company withdrew its NDA for rimonabant shortly after the advisory committee meeting. Although the active ingredients in QNEXA have been previously approved by FDA at higher doses for other indications, it is a centrally acting drug that may increase the risk of psychiatric side effects such as depression and/or suicidal ideation.
We are not permitted to market any of our investigational drug candidates in the U.S. until we receive approval from the FDA. As a consequence, any failure to obtain or delay in obtaining FDA approval for our investigational drug candidates would delay or prevent our ability to generate revenue from our investigational drug candidates, which would adversely affect our financial results and our business.
Our applications for regulatory approval could be delayed or denied due to problems with studies conducted before we licensed some of our investigational drug candidates from third parties.
We currently license some of our investigational drug candidates from third parties. Our present development programs involving these investigational drug candidates rely in part upon previous development work conducted by third parties over whom we had no control and before we licensed the investigational drug candidates. In order to receive regulatory approval of an investigational drug candidate, we must present to the FDA for its review all relevant data and information obtained during research and development, including research conducted prior to our license of the investigational drug candidate. Although we are not currently aware of any such problems, any problems that emerge with research and testing conducted prior to our licensing an investigational drug candidate may affect future results or our ability to document prior research and to conduct clinical trials, which could delay, limit or prevent regulatory approval for our investigational drug candidates.
Following regulatory approval of any investigational drug candidates, we will be subject to ongoing regulatory obligations and restrictions, which may result in significant expense and limit our ability to commercialize our potential drugs.
If one of our investigational drug candidates is approved by the FDA or by another regulatory authority for a territory outside of the U.S., we will be required to comply with extensive regulations for drug manufacturing, labeling, packaging, adverse event reporting, storage, distribution, advertising, promotion and record keeping. Regulatory approvals may also be subject to significant limitations on the indicated uses or marketing of the investigational drug candidates or to whom and how we may distribute our products. Even if U.S. regulatory approval is obtained, the FDA may still impose significant restrictions on a drug's indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies. For example, the label ultimately approved for QNEXA, if any, may include restrictions on use, including restrictions based on childbearing potential or pregnancy status, level of obesity and duration of treatment or a boxed warning related to concerns regarding antidepressants, antiepileptics or otherwise. The FDA may also require the distribution of a Medication Guide to patients outlining the increased risk of suicidal thinking or behavior in children and
44
adolescents or other populations. The FDA could also require a registry to track the patients utilizing the drug or implement a risk evaluation mitigation strategy, or REMS, that could restrict access to the drug.
Potentially costly post-marketing clinical studies may be required as a condition of approval to further substantiate safety or efficacy, or to investigate specific issues of interest to the regulatory authority.
Manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with current good manufacturing practices, or cGMP, regulations, which include requirements relating to quality control and quality assurance as well as the corresponding maintenance of records and documentation. Further, regulatory agencies must approve these manufacturing facilities before they can be used to manufacture our future approved drugs, if any, and these facilities are subject to ongoing regulatory inspections. In addition, regulatory agencies subject a drug, its manufacturer and the manufacturer's facilities to continual review and inspections. The subsequent discovery of previously unknown problems with a drug, including adverse events of unanticipated severity or frequency, or problems with the facility where the drug is manufactured, may result in restrictions on the marketing of that drug, up to and including withdrawal of the drug from the market. If our manufacturing facilities or those of our suppliers fail to comply with applicable regulatory requirements, it could result in regulatory action and additional costs to us. Failure to comply with applicable FDA and other regulatory requirements may, either before or after product approval, if any, subject our company to administrative or judicially imposed sanctions, including:
- •
- issuance of Form 483 notices, warning letters and adverse publicity by the FDA or other regulatory agencies;
- •
- imposition of fines and other civil penalties due to product liability or other issues;
- •
- criminal prosecutions;
- •
- injunctions, suspensions or revocations of regulatory approvals;
- •
- suspension of any ongoing clinical trials;
- •
- total or partial suspension of manufacturing;
- •
- delays in commercialization;
- •
- refusal by the FDA to approve pending applications or supplements to approved applications filed by us or our
collaborators;
- •
- refusals to permit drugs to be imported into or exported from the U.S.;
- •
- restrictions on operations, including costly new manufacturing requirements; and
- •
- product recalls or seizures.
In addition, the law or regulatory policies governing pharmaceuticals may change. New statutory requirements may be enacted or additional regulations may be enacted that could prevent or delay regulatory approval of our investigational drug candidates. Contract Manufacturing Organizations, or CMOs, and their vendors or suppliers may also face changes in regulatory requirements from governmental agencies in the U.S. and other countries. We cannot predict the likelihood, nature, extent or effects of government regulation that may arise from future legislation or administrative action, either in the U.S. or elsewhere. If we are not able to maintain regulatory compliance, we might not be permitted to market any future approved drugs and our business could suffer.
45
Even if we receive regulatory approval to commercialize our investigational drug candidates, our ability to generate revenues from any resulting drugs will be subject to a variety of risks, many of which are out of our control.
Even if our investigational drug candidates obtain regulatory approval, those drugs may not gain market acceptance among physicians, patients, healthcare payers or the medical community. The indication may be limited to a subset of the population or we may implement a distribution system and patient access program that is limited. Coverage and reimbursement of our investigational drug candidates by third-party payers, including government payers, generally is also necessary for optimal commercial success. We believe that the degree of market acceptance and our ability to generate revenues from such drugs will depend on a number of factors, including:
- •
- timing of market introduction of competitive drugs;
- •
- efficacy and safety of our investigational drug candidates;
- •
- prevalence and severity of any side effects;
- •
- potential or perceived advantages or disadvantages over alternative treatments including generics;
- •
- the relative convenience and ease of administration and dosing schedule;
- •
- strength of sales, marketing and distribution support;
- •
- price of any future drugs, if approved, both in absolute terms and relative to alternative treatments;
- •
- the effectiveness of our or any future collaborators' sales and marketing strategies;
- •
- the effect of current and future healthcare laws on our investigational drug candidates;
- •
- availability of coverage and reimbursement from government and other third-party payers;
- •
- patient access programs that require patients to provide certain information prior to receiving new and refill
prescriptions;
- •
- requirements for prescribing physicians to complete certain educational programs for prescribing drugs;
- •
- the willingness of patients to pay out of pocket in the absence of government or third-party coverage; and
- •
- product labeling or product insert requirements of the FDA or other regulatory authorities.
If approved, our investigational drug candidates may fail to achieve market acceptance or generate significant revenue to achieve or sustain profitability. In addition, our efforts to educate the medical community and third-party payers on the benefits of our investigational drug candidates may require significant resources and may never be successful.
We have limited sales and marketing experience and resources and we may not be able to effectively market and sell our investigational drug candidates, if approved, in the U.S. and/or internationally without a global pharmaceutical partner.
We are developing QNEXA, our investigational drug candidate for the treatment of obesity, for large markets traditionally served by general and family practitioners and internists. Generalist physicians number in the several hundred thousands in the U.S.. Traditional pharmaceutical companies employ groups of sales representatives numbering in the thousands to call on this large generalist physician population. In order to adequately address these physician groups, we must establish sales and marketing collaborations or co-promotion arrangements or expend significant resources to develop our own sales and marketing presence. We currently have no resources and may not be successful in
46
developing our own sales and marketing presence or establishing sales and marketing collaborations or co-promotion arrangements on acceptable terms, if at all. We may also decide to forego any form of collaboration and develop sales and marketing capabilities on our own. We also face competition in our search for collaborators, co-promoters and sales force personnel. We may rely on third parties to develop or commercialize our investigational drug candidates. These third parties may fail to develop or effectively commercialize our investigational drug candidates because they cannot obtain the necessary regulatory approvals, decide to pursue a competitive potential product that may be developed outside of the collaboration or fail to devote the resources necessary to realize the full commercial potential of our investigational drug candidates.
Even if our investigational drug candidates receive regulatory approval in the U.S., we may never receive approval for or commercialize our drugs outside of the U.S..
To market any of our investigational drug candidates outside of the U.S., we and our partners must comply with numerous and varying regulatory requirements of other countries. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from that required to obtain FDA approval. The regulatory approval process in other countries may include all of the risks associated with FDA approval as well as additional, presently unanticipated, risks. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others. Failure to obtain regulatory approval in other countries or any delay or setback in obtaining such approval could have the same adverse effects detailed above regarding FDA approval in the U.S.. As described above, such effects include the risks that our investigational drug candidates may not be approved for all indications requested, which could limit the uses of our investigational drug candidates and have an adverse effect on their commercial potential or require costly, post-marketing follow-up studies. On December 17, 2010, we filed a Marketing Authorization Application, or MAA, with the European Medicines Agency, or EMA, for QNEXA Controlled-Release Capsules in the European Union, or EU. The proposed indication in the EU is for the treatment of obesity, including weight loss and maintenance of weight loss, and should be used in conjunction with a mildly hypocaloric diet. If approved in the EU, QNEXA could be recommended for obese adult patients (BMI ³ 30 kg/m2), or overweight patients (BMI ³ 27 kg/m2) with weight-related co-morbidities such as hypertension, type 2 diabetes, dyslipidemia, or central adiposity (abdominal obesity). The EMA's review of QNEXA will follow their centralized marketing authorization procedure. If approved, QNEXA could receive marketing authorization in all 27 EU member countries. The MAA was officially validated for central procedure on January 19, 2011.
We rely on third parties to conduct pre-clinical clinical and retrospective observational trials and studies for our investigational drug candidates and those third parties may not perform satisfactorily.
We do not have the ability to conduct pre-clinical, clinical or retrospective observational studies for our investigational drug candidates without the assistance of third parties who conduct the studies on our behalf. These third parties are usually toxicology facilities, safety monitoring companies, epidemiologists, clinical investigators and clinical sites and clinical research organizations, or CROs, which have significant resources and experience in the conduct of pre-clinical and clinical studies. The toxicology facilities conduct the pre-clinical safety studies as well as all associated tasks connected with these studies. Safety monitoring companies collect reported adverse events that are reported from patients and healthcare providers during clinical trials. Clinical investigators and clinical sites enroll patients and conduct clinical testing according to clinical protocols. Epidemiologists employ programmers and have access to electronic databases that allow them to perform historical studies. The CROs typically review data generated by clinical investigators, perform project management, data management, statistical analysis, and other reporting functions. We intend to use several different
47
facilities and CROs for all of our pre-clinical and clinical studies. We have contracted with a safety monitoring company that we intend to use for all of our clinical trials. If these third party toxicology facilities, the safety monitoring company, clinical investigators, clinical sites or CROs do not successfully carry out their contractual duties or meet expected timelines, we may not be able to obtain regulatory approvals for our investigational drug candidates on a timely basis, if at all, and we may not be able to successfully commercialize these investigational drug candidates. If these third party toxicology facilities, the safety monitoring company, clinical investigators, epidemiologists, clinical sites or CROs do not perform satisfactorily, we may not be able to locate acceptable replacement third parties or enter into favorable agreements with these third parties, if at all. These third parties may also fail economically, which would impact our ability to obtain and utilize the results of the studies performed by these third parties.
We rely on third parties and collaborative partners to manufacture sufficient quantities of compounds within product specifications as required by regulatory agencies for use in our pre-clinical and clinical trials and future commercial operations and an interruption to this service may harm our business.
We do not have the ability to manufacture the materials we use in our pre-clinical and clinical trials and future commercial operations. Rather, we rely on various third parties to manufacture these materials and there may be long lead times to obtain materials. There can be no assurance that we will be able to identify, qualify and obtain prior regulatory approval for additional sources of clinical materials. If interruptions in this supply occur for any reason, including a decision by the third parties to discontinue manufacturing, technical difficulties, labor disputes, natural or other disasters, or a failure of the third parties to follow regulations, we may not be able to obtain regulatory approvals for our investigational drug candidates and may not be able to successfully commercialize these investigational drug candidates.
We or our third-party manufacturers and collaborative partners may encounter delays and problems in manufacturing our investigational drug candidates or approved drugs for a variety of reasons, including accidents during operation, failure of equipment, delays in receiving materials, natural or other disasters, political or governmental changes, or other factors inherent in operating complex manufacturing facilities. Supply chain management is difficult. Commercially available starting materials, reagents and excipients may become scarce or more expensive to procure, and we may not be able to obtain favorable terms in agreements with subcontractors. We or our third-party manufacturers may not be able to operate our respective manufacturing facilities in a cost-effective manner or in a time frame that is consistent with our expected future manufacturing needs. If we or our third-party manufacturers cease or interrupt production or if our third-party manufacturers and other service providers fail to supply materials, products or services to us for any reason, such interruption could delay progress on our programs, or interrupt the commercial supply, with the potential for additional costs and lost revenues. If this were to occur, we may also need to seek alternative means to fulfill our manufacturing needs.
We have completed the development of a once-a-day formulation of QNEXA for the treatment of obesity. The contract manufacturer we selected to develop a once-a-day formulation supplied the entire product for the Phase 3 program. In addition, this contract manufacturer is our sole source of clinical and commercial supplies for QNEXA. While this contract manufacturer has significant experience in commercial scale up manufacturing, there is no assurance that they will be successful with the commercial scale up of QNEXA, which could have a material adverse impact on our development plan, market price of our common stock and financial condition.
In the case of avanafil, we rely on MTPC to supply the active pharmaceutical ingredient, or API, and the finished goods. The MTPC manufacturing site where the materials are made for avanafil will be subject to a pre-approval inspection by the FDA. We are informed that this particular manufacturing site has never made any materials or products that have been approved by the FDA. This site has not
48
been subject to a pre-approval inspection in the past nor has it ever manufactured commercial quantities of avanafil. If MTPC is unable to receive and maintain approval from the FDA or manufacture avanafil API in sufficient quantities to meet projected demand, the approval and future sales of avanafil will be adversely effected, which in turn could have a detrimental impact on our financial results.
We rely on third parties to maintain appropriate levels of confidentiality of the data compiled during clinical, pre-clinical and retrospective observational studies and trials.
We seek to maintain the confidential nature of our confidential information through contractual provisions in our agreements with third parties, including our agreements with clinical research organizations, or CROs, that manage our clinical studies for our investigational drug candidates. These CROs may fail to comply with their obligations of confidentiality or may be required as a matter of law to disclose our confidential information. As the success of our clinical studies depends in large part on our confidential information remaining confidential prior to, during and after a clinical study, any disclosure could have a material adverse effect on the outcome of a clinical study, our business, financial condition and results of operations.
Risks Relating to our Operations
If we, or our suppliers, fail to comply with FDA and other regulatory agency regulations relating to our commercial manufacturing operations, we may be prevented from manufacturing our approved drugs, if any, or may be required to undertake significant expenditures to become compliant with such regulations.
After regulatory approval for a product is obtained, the product is subject to continual regulatory review. Manufacturing, labeling and promotional activities are continually regulated by the FDA and equivalent foreign regulatory agencies. For example, our third party manufacturers are required to maintain satisfactory compliance with current Good Manufacturing Practices, or cGMP. If these manufacturers fail to comply with applicable regulatory requirements, our ability to manufacture, market and distribute our drugs may be adversely affected. In addition, the FDA could issue warning letters or could require the seizure or recall of products. The FDA could also impose civil penalties or require the closure of our contract manufacturing facility until cGMP compliance is satisfactorily achieved.
We obtain the necessary raw materials and components for the manufacture of QNEXA and avanafil as well as certain services, such as analytical testing packaging and labeling, from third parties. We currently contract with suppliers and service providers, including foreign manufacturers. We and these suppliers and service providers are required to follow cGMP requirements and are subject to routine and unannounced inspections by the FDA and by state and foreign regulatory agencies for compliance with cGMP requirements and other applicable regulations. Upon inspection of these facilities, the FDA or foreign regulatory agencies may find the manufacturing process or facilities are not in compliance with cGMP requirements and other regulations.
Failure to achieve satisfactory cGMP compliance as confirmed by routine and unannounced inspections could have a material adverse effect on our ability to continue to manufacture and distribute our commercial drugs, if approved, and, in the most serious case, result in the issuance of a regulatory warning letter or seizure or recall of products, injunction and/or civil penalties or closure of our manufacturing facility until cGMP compliance is achieved.
If we fail to comply with healthcare regulations, we could face substantial penalties and our business, operations and financial condition could be adversely affected.
Even though we do not and will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third party payers, certain federal and state healthcare laws and regulations pertaining to fraud, abuse and patients' rights are and will be applicable to our business.
49
We are subject to healthcare fraud, abuse and patient privacy regulation by both the federal government and the states in which we conduct our business. The regulations that may affect our ability to operate include, but are not limited to:
- •
- the federal healthcare program Anti-Kickback Law, which prohibits, among other things, persons from
soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual, for an item or service or the purchasing or ordering of a good or service, for
which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs;
- •
- federal false claims laws, which prohibit, among other things, individuals or entities from knowingly presenting, or
causing to be presented, claims for payment from Medicare, Medicaid, or other third party payers that are false or fraudulent;
- •
- the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which prohibits executing a scheme to
defraud any healthcare benefit program or making false statements relating to healthcare matters and which also imposes certain requirements relating to the privacy, security and transmission of
individually identifiable health information; and
- •
- state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by any third party payer, including commercial insurers, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion of our drugs, if approved, from medical healthcare programs like Medicare and Medicaid, and the curtailment or restructuring of our operations. Any penalties, damages, fines, curtailment or restructuring of our operations could adversely affect our ability to operate our business and our financial results. Although compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, the risks cannot be entirely eliminated. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management's attention from the operation of our business. Moreover, achieving and sustaining compliance with applicable federal and state privacy, security and fraud laws may prove costly.
Any pre-marketing and marketing activities for our investigational drug candidates are subject to continued governmental regulation.
Prior to and after product approval by the FDA, any pre-marketing and marketing activities will be subject to FDA and other regulatory review. Certain activities undertaken prior to approval may be considered pre-approval promotion. Pre-approval promotion of investigational drug candidates is prohibited by FDA regulations. Failure to comply with these regulations may result in delays in the ultimate approval of our investigational drug candidates. After approval, if products are marketed in contradiction with FDA mandates, the FDA may issue warning letters that require specific remedial measures to be taken, as well as an immediate cessation of the impermissible conduct resulting in adverse publicity. The FDA may also order that all future promotional materials receive prior agency review and approval before use. Certain states have also adopted regulations and reporting requirements surrounding the promotion of pharmaceuticals. QNEXA, if approved, would be subject to these regulations. Failure to comply with state requirements may affect our ability to promote or sell pharmaceuticals drugs in certain states. This in turn could have a material adverse impact on our financial results and financial condition.
50
We must continue to monitor the use of our drugs, if approved, and may be required to complete post-approval studies mandated by the FDA.
Even if we receive regulatory approval of our investigational drug candidates, such approval may involve limitations on the indicated uses or marketing claims we may make for our drugs and distribution channels. The FDA may also require us to commit to perform lengthy post-approval studies, for which we would have to expend significant additional resources, which could have an adverse effect on our operating results, financial condition and stock price. Failure to comply with the applicable regulatory requirements can result in, among other things, civil penalties, suspensions of regulatory approvals, product recalls, operating restrictions and criminal prosecution. The restriction, suspension or revocation of regulatory approvals or any other failure to comply with regulatory requirements could have a material adverse effect on our business, financial condition, results of operations and stock price.
Sales of any future drugs are subject to continued governmental regulation, as well as our ability to accurately forecast demand and our ability to produce sufficient quantities to meet demand.
Sales of any future drugs both inside and outside the U.S. will be subject to regulatory requirements governing marketing approval. These requirements vary widely from country to country and could delay the introduction of our proposed drugs in those countries. After the FDA and international regulatory authorities approve a product, we must manufacture sufficient volumes to meet market demand. This is a process that requires accurate forecasting of market demand. There is no guarantee that there will be market demand for any future drugs or that we will be able to successfully manufacture or adequately support sales of any future drugs.
We have limited sales and marketing capabilities in the U.S..
If we are unable to establish capabilities to sell, market and distribute our investigational drug candidates, either by developing our own capabilities or entering into agreements with others, we will not be able to successfully launch our investigational drug candidates upon FDA approval. We cannot guarantee that we will be able to hire the qualified sales and marketing personnel we need. We may not be able to enter into any marketing or distribution agreements with third party providers on acceptable terms, if at all. In that event, our ability to generate revenues will be adversely affected.
The markets in which we operate are highly competitive and we may be unable to compete successfully against new entrants or established companies.
Competition in the pharmaceutical and medical products industries is intense and is characterized by costly and extensive research efforts and rapid technological progress. We are aware of several pharmaceutical companies also actively engaged in the development of therapies for the treatment of obesity, diabetes and sexual health and medical device companies for the treatment of sleep apnea. Many of these companies have substantially greater research and development capabilities as well as substantially greater marketing, financial and human resources than we do. In addition, many of these companies have significantly greater experience than us in undertaking pre-clinical testing, human clinical trials and other regulatory approval procedures. Our competitors may develop technologies and products that are more effective than those we are currently marketing or researching and developing. Such developments could render our drugs, if approved, or our investigational drug candidates less competitive or possibly obsolete. We are also competing with respect to marketing capabilities and manufacturing efficiency, areas in which we have limited experience. Mergers, acquisitions, joint ventures and similar events may also significantly change the competition.
Current approved anti-obesity drugs include Xenical (orlistat), marketed by Roche, the over-the-counter version, alli, marketed by GlaxoSmithKline, and phentermine, which is available from several generic manufacturers. Orlistat works by inhibiting lipase, thus preventing digestion and
51
absorption of dietary fat in the gastrointestinal tract. Meridia (sibutramine) was previously marketed by Abbott Laboratories; however, in October 2010, Abbott Laboratories withdrew Meridia in the U.S. at the FDA's request. The FDA requested the withdrawal because they believed Meridia's risks were not justified compared with the modest weight loss that patients achieved on the drug. In January 2010, the EMA suspended the marketing authorization of sibutramine in Europe. The impact on QNEXA of the withdrawal of sibutramine is unknown at this time. There are several drugs in development for obesity including an investigational drug candidate, liraglutide in Phase 3 clinical trials being developed by Novo Nordisk A/S and several other investigational drug candidates in Phase 2 clinical trials. Arena Pharmaceuticals, Inc. and Orexigen Therapeutics, Inc. have each submitted an NDA to the FDA for their investigational obesity drug candidates. Neither of these investigational drug candidates was approved by the FDA citing concerns around carcinogenicity in the case of Arena and the need to complete a pre-approval cardiovascular outcomes study in the case of Orexigen. The future of the Arena and Orexigen compounds is uncertain at this time.
Many of these drugs, if approved, are or will be marketed by pharmaceutical companies with substantially greater resources than us. In addition, a number of generic pharmaceutical drugs are prescribed for obesity, predominantly phentermine. Phentermine is sold at much lower prices than we intend to charge for our investigational drug candidate, QNEXA, if approved. The availability of a branded prescription drug, generic drugs and over-the-counter drugs could limit the demand for, and the price we are able to charge for QNEXA, if approved.
There are also surgical approaches to treat severe obesity that are becoming increasingly accepted and could become competitors against our investigational drug candidate, QNEXA. Two of the most well established surgical procedures are gastric bypass surgery and adjustable gastric banding. In February 2011, the FDA approved the use of a lap band in patients with a BMI of 35 without co-morbidities and 30 with co-morbidities. The lowering of the BMI requirement will make more overweight and obese patients eligible for lap band surgery. The potential impact on QNEXA and/or other weight loss pharmacotherapy is unknown. In addition, other potential approaches that utilize various implantable devices or surgical tools are in development. Some of these approaches are in late stage development and may be approved for marketing. If approved, the companies that market these drugs may have substantially greater resources than we have.
Significant competitive therapies exist for avanafil in the form of oral medications marketed by Pfizer, Inc. under the name Viagra®, Cialis®, marketed by Eli Lilly and Company, and Levitra®, which is co-marketed by GlaxoSmithKline plc and Schering-Plough Corporation in the U.S. In 2010, the European Commission approved a new formulation of Levitra (vardenafil HCI). Levitra 10mg oral-disintegrating tablet, or ODT, will be the first erectile dysfunction, or ED, medication available in an ODT form. In Europe, the launch roll-out was expected in November 2010. The launch in the EU has not yet occurred. In the U.S., the medication was approved in June 2010 and will be marketed by GlaxoSmithKline and Merck & Co., Inc. under the tradename STAXYN™. Consistent with the oral version, the ODT formulation instructs men to take the medication 60 minutes prior to attempting sexual activity. However, the ODT version will be positioned as an "anytime, anywhere" treatment for ED. We are uncertain how the launch of ODT vardenafil will impact the future commercial potential of avanafil.
There are currently three PDE5 inhibitors approved for the treatment of erectile dysfunction in the U.S.: sildenafil, vardenafil, and tadalafil. Worldwide sales of these products were in excess of $4.1 billion in 2010. As the patents for the three major PDE5 inhibitors currently being sold expire, generic PDE5s will enter the marketplace. Generic PDE5s would likely be sold at lower prices and may reduce the demand for avanafil and the prices we intend to charge for avanafil, if approved. Additional PDE5 inhibitors are in various stages of development by other companies. Warner-Chilcott plc has licensed the U.S. rights to udenafil, a PDE5i from Dong-A. Warner-Chilcott continues the Phase 3 development of this compound for ED. Other treatments for ED exist, such as needle injection
52
therapies, vacuum constriction devices and penile implants, and the manufacturers of these products will most likely continue to develop or improve these therapies.
New developments, including the development of other drug technologies and methods of preventing the incidence of disease, occur in the pharmaceutical and medical technology industries at a rapid pace. These developments may render our investigational drug candidates obsolete or noncompetitive. Compared to us, many of our potential competitors have substantially greater:
- •
- research and development resources, including personnel and technology;
- •
- regulatory experience;
- •
- investigational drug candidate development and clinical trial experience;
- •
- experience and expertise in exploitation of intellectual property rights; and
- •
- access to strategic partners and capital resources.
As a result of these factors, our competitors may obtain regulatory approval of their products more rapidly than we or may obtain patent protection or other intellectual property rights that limit our ability to develop or commercialize our investigational drug candidates. Our competitors may also develop drugs or surgical approaches that are more effective, more useful and less costly than ours and may also be more successful in manufacturing and marketing their products. In addition, our competitors may be more effective in commercializing their products. We currently outsource our manufacturing and therefore rely on third parties for that competitive expertise. There can be no assurance that we will be able to develop or contract for these capabilities on acceptable economic terms, or at all. Recently, Orexigen Therapeutics, manufacturers of Contrave, an investigational drug for the treatment of obesity, received a CRL from the FDA requesting that a long-term, randomized, placebo-controlled cardiovascular outcomes study be completed prior to refiling the NDA. To date, the FDA has not requested that we perform any additional studies, including cardiovascular outcome studies pre-approval, other than the Feasibility Assessment. Cardiovascular outcomes studies can take several years, cost millions of dollars and may result in showing an increased risk for major adverse cardiovascular events for patients in the treatment arm. If any regulatory agency were to require additional studies, including studies to address cardiovascular events, the impact on the timing of approval and, if approved, commercialization of QNEXA, avanafil or any of our investigational drug candidates, could be delayed or adversely impacted.
If our raw material supplier fails to supply us with the active pharmaceutical ingredients, or APIs, for our investigational drug candidates, for which availability is limited, we may experience delays in our investigational drug candidate development and commercialization.
We currently do not have supply agreements in place for phentermine or topiramate, the APIs used in our investigational drug candidate, QNEXA, nor do we have a supply agreement for the commercial manufacture of QNEXA, if approved. There can be no guarantees that we will be able to enter into such agreements under reasonable terms, if at all. We cannot guarantee that should we be successful in entering into such agreements we will be able to obtain the necessary prior regulatory approvals for these suppliers. MTPC manufactures the API for avanafil. To date, MTPC has never manufactured commercial quantities of API. In addition, we are informed that the avanafil manufacturing site has never been inspected by the FDA nor have they ever manufactured any materials that have been included in an FDA approved product. If MTPC is unable to produce the API for avanafil or if their facility does not receive and maintain approval from the FDA, the launch of avanafil could be delayed. Any potential delays with the commercial timeline for avanafil could have a material adverse impact on our future financial condition and could impact our ability to enter into a collaboration for the commercialization of avanafil.
53
We depend upon consultants and outside contractors extensively in important roles within our company.
We outsource many key functions of our business and therefore rely on a substantial number of consultants, and we will need to be able to effectively manage these consultants to ensure that they successfully carry out their contractual obligations and meet expected deadlines. However, if we are unable to effectively manage our outsourced activities or if the quality or accuracy of the services provided by consultants is compromised for any reason, our clinical trials or other development activities may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for our investigational drug candidates or otherwise advance our business. There can be no assurance that we will be able to manage our existing consultants or find other competent outside contractors and consultants on commercially reasonable terms, or at all.
If we fail to retain our key personnel and hire, train and retain qualified employees, we may not be able to compete effectively, which could result in reduced revenues or delays in the development of our investigational drug candidates .
Our success is highly dependent upon the skills of a limited number of key management personnel. To reach our business objectives, we will need to retain and hire qualified personnel in the areas of manufacturing, sales and marketing, research and development, regulatory and legal affairs, clinical trial design, execution and analysis, and pre-clinical testing. There can be no assurance that we will be able to hire or retain such personnel, as we must compete with other companies, academic institutions, government entities and other agencies. The loss of any of our key personnel or the failure to attract or retain necessary new employees could have an adverse effect on our research, investigational drug candidate development and business operations.
Allegations of discrimination, wrongful termination or other employment matters, regardless of merit, could negatively affect our operations by causing us to allocate additional monetary and personnel resources to these issues.
In the ordinary course of business we may become involved in lawsuits and subject to various claims from current and former employees including wrongful termination, sexual discrimination, retaliation, hostile work environment and other employment-related matters. We were party to a lawsuit involving a former employee which lawsuit was dismissed without prejudice to the plaintiff to refile certain of his state law claims. We have also been named as a potential defendant in a complaint filed by a former employee. We have investigated each of the former employee claims and believe the allegations have no merit and that we have meritorious defenses to any such allegations. Due to the current economic downturn, former employees may be more likely to file employment-related claims. Employment-related claims also appear more likely following a poor performance review. Although there may be no merit to such claims or legal matters, we may be required to allocate additional monetary and personnel resources to defend against these type of allegations.
Any adverse changes in reimbursement procedures by government and other third party payers may limit our ability to market and sell any future drugs, if approved or limit our product revenues and delay profitability.
In the U.S. and abroad, sales of pharmaceutical drugs are dependent, in part, on the availability of reimbursement to the consumer from third party payers, such as government and private insurance plans. Third party payers are increasingly challenging the prices charged for medical products and services. Some third party payer benefit packages restrict reimbursement or do not provide coverage for specific drugs or drug classes.
In addition, certain healthcare providers are moving towards a managed care system in which such providers contract to provide comprehensive healthcare services, including prescription drugs, for a fixed cost per person. We are unable to predict the reimbursement policies employed by third party healthcare payers.
54
The healthcare industry in the U.S. and abroad is undergoing fundamental changes that are the result of political, economic and regulatory influences. The levels of revenue and profitability of pharmaceutical companies may be affected by the continuing efforts of governmental and third party payers to contain or reduce healthcare costs through various means. Reforms that have been and may be considered include mandated basic healthcare benefits, controls on healthcare spending through limitations on the increase in private health insurance premiums and the types of drugs eligible for reimbursement and Medicare and Medicaid spending, the creation of large insurance purchasing groups and fundamental changes to the healthcare delivery system. These proposals include measures that would limit or prohibit payments for some medical treatments or subject the pricing of drugs to government control and regulations changing the rebates we are required to provide. These changes could impact our ability to maximize revenues in the Federal marketplace. In addition, healthcare reform legislation could affect the prices of our investigational drug candidates, if approved, under certain healthcare programs. These proposals include expanding the 340B drug pricing program to allow additional types of healthcare providers to purchase drugs at significant discounts and to require those discounts on inpatient drugs as well, increasing the minimum Medicaid drug rebate percentage, expanding Medicaid rebate liability to drugs purchased under Medicaid managed care contracts, increasing the Medicaid rebate on new formulations of existing drugs, and requiring Medicaid rebates to be paid on drugs provided to certain enrollees in the Medicare Part D prescription drug benefit. Due to uncertainties regarding the outcome of healthcare reform initiatives and their enactment and implementation, we cannot predict which, if any, of the reform proposals will be adopted or the effect such adoption may have on us. There can be no assurance that future healthcare legislation or other changes in the administration or interpretation of government healthcare or third party reimbursement programs will not have a material adverse effect on us. Healthcare reform is also under consideration in other countries where we intend to market QNEXA, if approved.
We expect to experience pricing pressures in connection with the sale of our investigational drug candidates, if approved, due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative proposals. If we fail to successfully secure and maintain reimbursement coverage for our investigational drug candidates or are significantly delayed in doing so, we will have difficulty achieving market acceptance of our investigational drug candidates and our business will be harmed. Congress has recently enacted healthcare reform and may enact further reform, which could adversely affect the pharmaceutical industry as a whole, and therefore could have a material adverse effect on our business.
Both of the active ingredients in QNEXA, phentermine and topiramate are available as generics. Based on the research we have completed to date, we are unable to determine whether QNEXA, if approved, will be subject to reimbursement or at what level reimbursement may occur. The exact doses of the active ingredients in the final formulation of QNEXA will be different than those currently available. State pharmacy laws prohibit pharmacists from substituting drugs with differing doses and formulations. The safety and efficacy of QNEXA is highly dependent on the titration, dosing and formulation, which we believe could not be easily duplicated, if at all, with the use of generic substitutes. However, there can be no assurance that we will be able to provide for optimal reimbursement of QNEXA as a treatment for obesity or any other indication, if approved, from third party payers or the U.S. government. Furthermore, there can be no assurance that healthcare providers would not actively seek to provide patients with generic versions of the active ingredients in QNEXA in order to treat obesity at a potential lower cost.
55
Federal legislation may increase the pressure to reduce prices of pharmaceutical drugs paid for by Medicare, which could adversely affect our future revenues, if any.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or MMA, expanded Medicare coverage for drug purchases by the elderly and disabled beginning in 2006. Under the MMA, private insurance plans subsidized by the government offer prescription drug coverage to Medicare beneficiaries who elect to enroll in their plans. Although almost all prescription drugs are potentially available to plan enrollees, the plans are allowed to use formularies, preferred drug lists and similar mechanisms to favor selected drugs and limit access to other drugs except in certain circumstances. The price of a drug as negotiated between the manufacturer and a plan is a factor that the plan can consider in determining its availability to enrollees.
As a result, we expect that there will be increased pressure to reduce prices for drugs to obtain favorable status for them under the plans offering prescription drug coverage to Medicare beneficiaries. This pressure could decrease the coverage and price that we receive for our investigational drug candidates, if approved, in the future and could seriously harm our business. It is possible that our investigational drug candidate, QNEXA, if approved, could be particularly subject to price reduction initiatives because it is based on combinations of lower priced existing drugs.
In addition, some members of Congress advocate that the federal government should negotiate directly with manufacturers for lower prices for drugs in the Medicare program, rather than rely on private plans. If the law were changed to allow or require such direct negotiation, there could be additional reductions in the coverage of and prices that we receive for any future approved drugs.
Federal legislation and actions by state and local governments may permit re-importation of drugs from foreign countries into the U.S., including foreign countries where the drugs are sold at lower prices than in the U.S., which could adversely affect our operating results and our overall financial condition.
We may face competition for our investigational drug candidates, if approved, from lower priced products from foreign countries that have placed price controls on pharmaceutical products. The Medicare Prescription Drug Improvement and Modernization Act of 2003 contains provisions that may change U.S. importation laws and expand consumers' ability to import lower priced versions of our investigational drug candidates and competing products from Canada, where there are government price controls. These changes to U.S. importation laws will not take effect unless and until the Secretary of Health and Human Services certifies that the changes will lead to substantial savings for consumers and will not create a public health safety issue. The Secretary of Health and Human Services has not yet announced any plans to make this required certification. As directed by Congress, a task force on drug importation conducted a comprehensive study regarding the circumstances under which drug importation could be safely conducted and the consequences of importation on the health, medical costs and development of new medicines for U.S. consumers. The task force issued its report in December 2004, finding that there are significant safety and economic issues that must be addressed before importation of prescription drugs is permitted. In addition, a number of federal legislative proposals have been made to implement the changes to the U.S. importation laws without any certification, and to broaden permissible imports in other ways. Even if the changes do not take effect, and other changes are not enacted, imports from Canada and elsewhere may continue to increase due to market and political forces, and the limited enforcement resources of the FDA, the U.S. Customs Service and other government agencies. For example, Pub. L. No. 109-295, which was signed into law in October 2006 and provides appropriations for the Department of Homeland Security for the 2007 fiscal year, expressly prohibits the U.S. Customs Service from using funds to prevent individuals from importing from Canada less than a 90-day supply of a prescription drug for personal use, when the drug otherwise complies with the Federal Food, Drug, and Cosmetic Act. Further, several states and local governments have implemented importation schemes for their citizens and, in the absence of federal action to curtail such activities, we expect other states and local governments to launch
56
importation efforts. The importation of foreign products that compete with our future approved drugs, if any, could negatively impact our financial condition.
Defending against claims relating to improper handling, storage or disposal of hazardous materials could be time consuming and expensive.
Our research and development involves the controlled use of hazardous materials and our operations produce hazardous waste products. We cannot eliminate the risk of accidental contamination or discharge and any resultant injury from those materials. Various laws and regulations govern the use, manufacture, storage, handling and disposal of hazardous materials. We may be sued for any injury or contamination that results from our use or the use by third parties of these materials. Compliance with environmental laws and regulations may be expensive, and current or future environmental regulations may impair our research, development and production efforts.
Our business and operations would suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems and those of our CROs, safety monitoring company and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, accidents, terrorism, war and telecommunication and electrical failures. While we have not experienced any such system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our investigational drug candidate development programs and drug manufacturing operations. For example, the loss of clinical trial data from completed or ongoing clinical trials for our investigational drug candidates could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach was to result in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development of our investigational drug candidates could be delayed. With the sale of our facility in New Jersey, we currently do not have a backup location to restore our information systems in the event of failure or disaster in our Mountain View, California facility. We are in the process of securing a backup facility; however, there can be no assurance that we will be able to locate and implement a backup process on a timely basis or that we will be able to restore our information network in the event of a failure at the Mountain View facility. If we are unable to restore our information systems in the event of a systems failure, our communications, daily operations and the ability to develop our investigational drug candidates would be severely affected.
Natural disasters or resource shortages could disrupt our investigational drug candidate development efforts and adversely affect results.
Our ongoing or planned clinical trials could be delayed or disrupted indefinitely upon the occurrence of a natural disaster. For example, in 2005, our clinical trials in the New Orleans area were interrupted by Hurricane Katrina. In addition, our offices are located in the San Francisco Bay Area near known earthquake fault zones and are therefore vulnerable to damage from earthquakes. In October 1989, a major earthquake in our area caused significant property damage and a number of fatalities. Our supplier of avanafil is located in Japan near known earthquake fault zones and is vulnerable to damage from earthquakes. We are also vulnerable to damage from other disasters, such as power loss, fire, floods and similar events. If a significant disaster occurs, our ability to continue our operations could be seriously impaired and we may not have adequate insurance to cover any resulting losses. Any significant unrecoverable losses could seriously impair our operations and financial conditions.
57
Risks Relating to our Intellectual Property
We may be sued for infringing the intellectual property rights of others or others may infringe on our intellectual property rights.
There can be no assurance that our investigational drug candidates do not or will not infringe on the patent or proprietary rights of others. In addition, third parties may already own or may obtain patents in the future and claim that use of our technologies infringes these patents. We could incur substantial costs and diversion of the time and attention of management and technical personnel in defending ourselves against any such claims. Furthermore, parties making claims against us may be able to obtain injunctive or other equitable relief that could effectively block our ability to further develop, commercialize and sell any future approved drugs, and such claims could result in the award of substantial damages against us. In the event of a successful claim of infringement against us, we may be required to pay damages and obtain one or more licenses from third parties. We may not be able to obtain these licenses at a reasonable cost, if at all. In that case, we could encounter delays in product introductions while we attempt to develop alternative investigational drug candidates or be required to cease commercializing any affected future approved drugs and our operating results would be harmed.
We believe the Supreme Court ruling in KSR International Co. vs. Teleflex, Inc. raised the standards for patentability and ease the ability to show that a patent is obvious. This ruling will make it more difficult to obtain patents for combination pharmaceutical drugs. At the present time, we are unable to predict the impact, if any, that this ruling will have on our current or future patents and patent applications. If we are unable to defend the patents currently issued on our investigational drug candidates, or to obtain new patents for any reason, our ability to commercialize any future approved drugs would be at risk.
Our inability to adequately protect our proprietary technologies could harm our competitive position and have a material adverse effect on our business.
We hold various patents and patent applications in the U.S. and abroad targeting obesity and morbidities related to obesity, including sleep apnea and diabetes, and male sexual health among other indications. QNEXA is our investigational drug candidate involving low doses of phentermine and topiramate. On June 6, 2006, the initial U.S. patent was issued by the USPTO. This patent contains composition, product, and other claims that should protect QNEXA, if approved, as a proprietary product for the treatment of obesity. The term of this patent extends into 2020. In January 2009, the European Patent Office granted European patent No. 1,187,603, which broadly covers QNEXA and its use as a weight loss treatment. The patent extends the intellectual property protection of QNEXA beyond the already issued patents in the U.S. and abroad. Patents have also been granted on QNEXA in Canada and Australia. We are in the process of prosecuting additional patent applications in these and other countries as well, to obtain significant foreign patent coverage for both QNEXA and future generations of QNEXA. Furthermore, we have filed additional patent applications in the U.S. to expand the coverage that will be provided by U.S. Patent No. 7,056,890 B2. On March 9, 2010, U.S. Patent No. 7,674,776 issued with method, composition, and dosage form claims, including claims drawn to a method for treating Syndrome X, a common multisymptomatic disorder often found in obese patients, and to a method for treating side effects of obesity such as sleep apnea. On February 9, 2010, U.S. Patent No. 7,659,256 issued significantly broadening both the method and composition-of-matter protection afforded QNEXA by our initial U.S. Patent No. 7,056,890 B2. On June 30, 2009, U.S. Patent No. 7,553,818 was issued drawn to a method for effecting weight loss by co-administration of varying doses of phentermine and topiramate. This patent also expands on the initial coverage provided by our U.S. Patent No. 7,056,890 B2. Each of these U.S. patents for QNEXA expires in 2020. The primary focus of the patent applications is on combination therapy using a sympathomimetic agent (such as phentermine) and an anticonvulsant (such as topiramate) for the treatment of obesity and other related disorders. We are aware of issued patents for the use of topiramate alone or in combination for
58
obesity. We have worked closely with our patent counsel to put together a cogent patent strategy and are building a strong patent portfolio in an attempt to obtain exclusivity over the life of the patents.
The procedures for obtaining a patent in the U.S. and in most foreign countries are complex. These procedures require an analysis of the scientific technology related to the invention and many sophisticated legal issues. Obtaining patent rights outside the U.S. often requires the translation of highly technical documents and an improper translation could lead to the loss of, or otherwise jeopardize, the patent protection of our inventions. Ensuring adequate quality of translators and foreign patent attorneys is often very challenging. Consequently, the process for having our pending patent applications issue as patents will be difficult, complex and time consuming. We do not know when, or if, we will obtain additional patents for our technologies, or if the scope of the patents obtained will be sufficient to protect our investigational drug candidates, or be considered sufficient by parties reviewing our patent positions pursuant to a potential licensing or financing transaction.
In addition, other entities may challenge the validity or enforceability of our patents and patent applications in litigation or administrative proceedings. Even the issuance of a patent is not conclusive as to its validity or enforceability. We cannot make assurances as to how much protection, if any, will be given to our patents if we attempt to enforce them or they are challenged. It is possible that a competitor or a generic pharmaceutical provider may successfully challenge our patents and those challenges may result in reduction or elimination of our patents' coverage.
The USPTO has over the last few years tried to enact and/or has proposed changes in the rules governing (i) the duties of patent applicants to disclose information that relates to their applications, (ii) the ability of patent applicants to file unlimited numbers of patent applications and patent claims that concern closely related inventions and/or different aspects of the same invention, and (iii) the manner in which the USPTO will decide whether to require patent applicants to separate closely related inventions into separate patent applications. Some of these rule changes are being challenged in the courts. It is unclear which of these rule changes, if any, will be allowed by the courts and which of them will continue to be pursued. In addition, the U.S. Congress is considering changes to federal patent laws on several issues including, but not limited to: (i) the information can be used to determine whether an invention is not new and, therefore, not patentable, (ii) the limits on the independent administrative rulemaking authority of the USPTO, (iii) the duties of patent applicants to disclose information that relates to their applications, (iv) whether, under what circumstances, and how many times a third party can challenge an issued U.S. patent before the USPTO, (v) whether and under what circumstances patent applicants can lose their ability to enforce their patents in the U.S. based on their failure to disclose certain information relating to their inventions, and (vi) how damages for patent infringement may be reduced based by a number of factors, including the similarity of a patented invention to preexisting technologies.
We believe that the U.S. is by far the largest single market for pharmaceuticals in the world. Because of the critical nature of patent rights to the pharmaceutical industry, changes in U.S. patent rules and laws could have a profound effect on our future profits. Several of the patent rule and law changes that are being considered could significantly weaken patent protections in the U.S. in general. They may also have a disproportionately large negative impact on the biotechnology and pharmaceutical industries in particular, as well as tilt the balance of market control and distribution of profits between the manufacturers of patented pharmaceutical products and the manufacturers of generic pharmaceutical products towards the generics manufacturers. At present there is considerable uncertainty as to which patent rules and laws will be changed and whether changes to the patent rules will ultimately be enforced or struck down by the courts.
Our existing patents and any future patents we obtain may not be sufficiently broad to prevent others from practicing our technologies or from developing competing products. Others may independently develop similar or alternative technologies or design around our patented technologies
59
or products. These companies would then be able to develop, manufacture and sell products that compete directly with our products. In that case, our revenues and operating results would decline.
We seek to protect our confidential information by entering into confidentiality agreements with employees, collaborators, CROs, consultants and potential investors. Nevertheless, employees, collaborators, consultants or potential investors may still disclose or misuse our confidential information, and we may not be able to meaningfully protect our trade secrets. In addition, others may independently develop substantially equivalent information or techniques or otherwise gain access to our trade secrets. Disclosure or misuse of our confidential information would harm our competitive position and could cause our revenues and operating results to decline.
A dispute regarding the infringement or misappropriation of our proprietary rights or the proprietary rights of others could be costly and result in delays or termination of our future research, development, manufacturing and sales activities.
Our commercial success also depends upon our ability to develop and manufacture our investigational drug candidates and market and sell any future approved drugs and conduct our research and development activities without infringing or misappropriating the proprietary rights of others. There are many patents and patent applications filed, and that may be filed, by others relating to drug discovery and development programs that could be determined to be similar, identical or superior to ours or our licensors or collaborators. We may be exposed to future litigation by others based on claims that our investigational drug candidates, technologies or activities infringe the intellectual property rights of others. Numerous U.S. and foreign issued patents and pending patent applications owned by others also exist in the therapeutic areas in, and for the therapeutic targets for, which we are developing investigational drug candidates. There are also numerous issued patents and patent applications to chemical compounds or synthetic processes that may be necessary or useful to use in our research, development, manufacturing or commercialization activities. These could materially affect our ability to develop our investigational drug candidates or manufacture, import or sell any future approved drugs, and our activities, or those of our licensors or collaborators, could be determined to infringe these patents. Because patent applications can take many years to issue, there may be currently pending applications, unknown to us, which may later result in issued patents that our investigational drug candidates or technologies may infringe. There also may be existing patents, of which we are not aware, that our investigational drug candidates or technologies may infringe. Further, there may be issued patents or pending patent applications in fields relevant to our business, of which we are or may become aware, that we believe (i) are invalid or we do not infringe; (ii) relate to immaterial portions of our overall drug discovery, development, manufacturing and commercialization efforts; or (iii) in the case of pending patent applications, the resulting patent would not be granted or, if granted, would not likely be enforced in a manner that would materially impact such efforts. We cannot assure you that others holding any of these patents or patent applications will not assert infringement claims against us for damages or seek to enjoin our activities. We also cannot assure you that, in the event of litigation, we will be able to successfully assert any belief we may have as to non-infringement, invalidity or immateriality, or that any infringement claims will be resolved in our favor.
In addition, others may infringe or misappropriate our proprietary rights, and we may have to institute costly legal action to protect our intellectual property rights. We may not be able to afford the costs of enforcing or defending our intellectual property rights against others.
There could also be significant litigation and other administrative proceedings in our industry that affect us regarding patent and other intellectual property rights. Any legal action or administrative
60
action against us, or our collaborators, claiming damages or seeking to enjoin commercial activities relating to our drug discovery, development, manufacturing and commercialization activities could:
- •
- require us, or our collaborators, to obtain a license to continue to use, manufacture or market the affected drugs,
methods or processes, which may not be available on commercially reasonable terms, if at all;
- •
- prevent us from importing, making, using, selling or offering to sell the subject matter claimed in patents held by others
and subject us to potential liability for damages;
- •
- consume a substantial portion of our managerial, scientific and financial resources; or
- •
- be costly, regardless of the outcome.
Furthermore, because of the substantial amount of pre-trial document and witness discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, during the course of this kind of litigation, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the trading price of our common stock.
We cannot protect our intellectual property rights throughout the world.
Filing, prosecuting, defending and enforcing patents on all of our drug discovery technologies and all of our potential investigational drug candidates throughout the world would be prohibitively expensive. Competitors may use our technologies to develop their own drugs in jurisdictions where we have not obtained patent protection. These drugs may compete with our investigational drug candidates and may not be covered by any of our patent claims or other intellectual property rights. The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the U.S., and many companies have encountered significant problems in protecting and defending such rights in foreign jurisdictions. Many countries, including certain countries in Europe, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties (for example, the patent owner has failed to "work" the invention in that country or the third party has patented improvements). In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of the patent. Compulsory licensing of life-saving drugs is also becoming increasingly popular in developing countries either through direct legislation or international initiatives. Such compulsory licenses could be extended to include some of our investigational drug candidates, which could limit our potential revenue opportunities. Moreover, the legal systems of certain countries, particularly certain developing countries, do not favor the aggressive enforcement of patents and other intellectual property protection, particularly those relating to biotechnology and/or pharmaceuticals, which makes it difficult for us to stop the infringement of our patents. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
We may face additional competition outside of the U.S. as a result of a lack of patent enforcement in foreign countries and off-label use of other dosage forms of the generic components in our investigational drug candidates.
While we have filed patent applications in many countries outside the U.S., and have obtained some patent coverage for certain of our investigational drug candidates in certain foreign countries, we do not currently have widespread patent protection for QNEXA outside the U.S. and have no protection in many foreign jurisdictions. Even if international patent applications ultimately issue or
61
receive approval, it is likely that the scope of protection provided by such patents will be different from, and possibly less than, the scope provided by our corresponding U.S. patents. The success of our international market opportunity is dependent upon the enforcement of patent rights in various other countries. A number of countries in which we have filed or intend to file patent applications have a history of weak enforcement and/or compulsory licensing of intellectual property rights. Even if we have patents issued in these jurisdictions, there can be no assurance that our patent rights will be sufficient to prevent generic competition or unauthorized use.
We may face competition from the off-label use of other dosage forms of the generic components in our investigational drug candidates. In addition, others may attempt to commercialize our investigational drug candidate combinations in countries or other markets where we do not have patent protection for all of our investigational drug candidates. In particular, it is possible that patients will seek to acquire the generic IR components of our investigational drug candidate QNEXA (phentermine and topiramate). The off-label use of the generic IR components in the U.S. or the importation of the generic IR components from foreign markets could adversely affect the commercial potential for our investigational drug candidates and adversely affect our overall business and financial results.
We may be subject to claims that we, or our employees, have wrongfully used or disclosed alleged trade secrets of their former employers.
We employ individuals who were previously employed at other pharmaceutical companies, including our competitors or potential competitors. Although we have no knowledge of any pending or overtly threatened claims, we may be subject to claims that these employees, or we, have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
We may be unable to in-license intellectual property rights or technology necessary to develop and commercialize our investigational drug candidates.
Depending on its ultimate formulation and method of use, before we can develop, clinically test, make, use, or sell a particular investigational drug candidate, we may need to obtain a license from one or more third parties who have patent or other intellectual property rights covering components of our investigational drug candidate or its method of use. There can be no assurance that such licenses will be available on commercially reasonable terms, or at all. If a third party does not offer us a necessary license or offers a license only on terms that are unattractive or unacceptable to us, we might be unable to develop and commercialize one or more of our investigational drug candidates.
Our failure to successfully acquire, develop and market additional investigational drug candidates or approved drugs would impair our ability to grow.
As part of our growth strategy, we may acquire, in-license, develop and/or market additional products and investigational drug candidates. Because our internal research capabilities are limited, we may be dependent upon pharmaceutical and biotechnology companies, academic scientists and other researchers to sell or license products or technology to us. The success of this strategy depends partly upon our ability to identify, select and acquire promising pharmaceutical investigational drug candidates and products.
The process of proposing, negotiating and implementing a license or acquisition of an investigational drug candidate or approved product is lengthy and complex. Other companies, including some with substantially greater financial, marketing and sales resources, may compete with us for the license or acquisition of investigational drug candidates and approved products. We have limited
62
resources to identify and execute the acquisition or in-licensing of third-party products, businesses and technologies and integrate them into our current infrastructure. Moreover, we may devote resources to potential acquisitions or in-licensing opportunities that are never completed, or we may fail to realize the anticipated benefits of such efforts. We may not be able to acquire the rights to additional investigational drug candidates on terms that we find acceptable, or at all.
In addition, future acquisitions may entail numerous operational and financial risks, including:
- •
- exposure to unknown liabilities;
- •
- disruption of our business and diversion of our management's time and attention to develop acquired products or
technologies;
- •
- incurrence of substantial debt or dilutive issuances of securities to pay for acquisitions;
- •
- higher than expected acquisition, integration and maintenance costs;
- •
- increased amortization expenses;
- •
- difficulty and cost in combining the operations and personnel of any acquired businesses with our operations and
personnel;
- •
- impairment of relationships with key suppliers or customers of any acquired businesses due to changes in management and
ownership; and
- •
- inability to retain key employees of any acquired businesses.
Further, any investigational drug candidate that we acquire may require additional development efforts prior to commercial sale, including extensive clinical testing and approval by the FDA and applicable foreign regulatory authorities. All investigational drug candidates are prone to risks of failure typical of pharmaceutical investigational drug candidate development, including the possibility that an investigational drug candidate will not be shown to be sufficiently safe and effective for approval by regulatory authorities. In addition, we cannot provide assurance that any drugs that we develop or approved products that we may acquire will be commercialized profitably or achieve market acceptance.
We may participate in new partnerships and other strategic transactions that could impact our liquidity, increase our expenses and present significant distractions to our management.
From time to time we consider strategic transactions, such as out-licensing or in-licensing of compounds or technologies, acquisitions of companies and asset purchases. Additional potential transactions we may consider include a variety of different business arrangements, including strategic partnerships, joint ventures, spin-offs, restructurings, divestitures, business combinations and investments. In addition, another entity may pursue us as an acquisition target. Any such transactions may require us to incur non-recurring or other charges, may increase our near and long-term expenditures and may pose significant integration challenges, require additional expertise or disrupt our management or business, which could harm our operations and financial results.
As part of an effort to enter into significant transactions, we conduct business, legal and financial due diligence with the goal of identifying and evaluating material risks involved in the transaction. Despite our efforts, we ultimately may be unsuccessful in ascertaining or evaluating all such risks and, as a result, might not realize the intended advantages of the transaction. If we fail to realize the expected benefits from any transaction we may consummate, whether as a result of unidentified risks, integration difficulties, regulatory setbacks or other events, our business, results of operations and financial condition could be adversely affected.
63
Setbacks and consolidation in the pharmaceutical and biotechnology industries, and our or our collaborators' inability to obtain third-party coverage and adequate reimbursement, could make partnering more difficult and diminish our revenues.
Setbacks in the pharmaceutical and biotechnology industries, such as those caused by safety concerns relating to high-profile drugs like Avandia, Vioxx and Celebrex, or investigational drug candidates, as well as competition from generic drugs, litigation, and industry consolidation, may have an adverse effect on us. For example, pharmaceutical companies may be less willing to enter into new collaborations or continue existing collaborations if they are integrating a new operation as a result of a merger or acquisition or if their therapeutic areas of focus change following a merger. Moreover, our and our collaborators' ability to commercialize any of our investigational drug candidates that may be approved will depend in part on government regulation and the availability of coverage and adequate reimbursement from third-party payers, including private health insurers and government payers, such as the Medicaid and Medicare programs, increases in government-run, single-payer health insurance plans and compulsory licenses of drugs. Government and third-party payers are increasingly attempting to contain healthcare costs by limiting coverage and reimbursement levels for new drugs. Given the continuing discussion regarding the cost of healthcare, managed care, universal healthcare coverage and other healthcare issues, we cannot predict with certainty what additional healthcare initiatives, if any, will be implemented or the effect any future legislation or regulation will have on our business. These efforts may limit our commercial opportunities by reducing the amount a potential collaborator is willing to pay to license our programs or investigational drug candidates in the future due to a reduction in the potential revenues from drug sales. Moreover, legislation and regulations affecting the pricing of pharmaceuticals may change before regulatory agencies approve our investigational drug candidates for marketing. Adoption of such legislation and regulations could further limit pricing approvals for, and reimbursement of, drugs. A government or third-party payer decision not to approve pricing for, or provide adequate coverage and reimbursements of, our investigational drug candidates could limit market acceptance of such drugs, if approved.
We will need to obtain FDA approval of our proposed product names and any failure or delay associated with such approval may adversely impact our business.
Any name we intend to use for our investigational drug candidates will require approval from the FDA, regardless of whether we have secured a formal trademark registration from the USPTO. The FDA typically conducts a rigorous review of proposed product names, including an evaluation of potential for confusion with other product names. The FDA may also object to a product name if it believes the name inappropriately implies medical claims. If the FDA objects to one of our proposed product names, we will be required to adopt an alternative name for that investigational drug candidate. If we adopt an alternative name, we would lose the benefit of our existing trademark applications and may be required to expend significant additional resources in an effort to identify a suitable product name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA, which could cause delays that would adversely impact our business. We may be unable to build a successful brand identity for a new trademark in a timely manner or at all, which would limit our ability to commercialize our investigational drug candidates.
64
Risks Relating to our Financial Position and Need for Financing
We require additional capital for our future operating plans, including, if approved, the commercial launch of QNEXA in the United States, and we may not be able to secure the requisite additional funding on acceptable terms, or at all, which would force us to delay, reduce or eliminate investigational drug candidate development programs or commercialization efforts.
We expect that our existing capital resources combined with future anticipated cash flows will be sufficient to support our operating activities at least into 2012. However, we anticipate that we will be required to obtain additional financing to fund the development of our research and development pipeline in future periods as well as to support the possible launch of any approved products. Our future capital requirements will depend upon numerous factors, including:
- •
- the timing and substance of our response to the FDA's request to perform the Feasibility Assessment and, if deemed
feasible, the cost, timing and outcome of such study and the other items included in the complete response letter;
- •
- the FDA's interpretation of the data we submit relating to cardiovascular safety;
- •
- the FDA's interpretation of the data from our SEQUEL study (OB-305) and Sleep Apnea study
(OB-204);
- •
- whether or not the FDA requires us to perform additional clinical studies for QNEXA in support of the NDA;
- •
- the cost and time required to set up a distribution system and a REMS program for QNEXA that is suitable to address any
FDA concerns;
- •
- the progress and costs of our research and development programs;
- •
- the scope, timing and results of pre-clinical, clinical and retrospective observational studies and trials;
- •
- the cost of access to electronic records and databases that allow for retrospective observational studies;
- •
- patient recruitment and enrollment in planned and future clinical trials;
- •
- the costs involved in seeking regulatory approvals for our investigational drug candidates;
- •
- the costs involved in filing and pursuing patent applications, defending and enforcing patent claims;
- •
- the establishment of collaborations, sublicenses and strategic alliances and the related costs, including milestone
payments;
- •
- the costs involved in establishing a commercial operation and in launching a product without a partner;
- •
- the cost of manufacturing and commercialization activities and arrangements;
- •
- the results of operations;
- •
- the cost, timing and outcome of regulatory reviews;
- •
- the rate of technological advances;
- •
- ongoing determinations of the potential commercial success of our investigational drug candidates under development;
- •
- the state of the economy and financing environment;
65
- •
- the level of resources devoted to any future sales and marketing capabilities;
- •
- the regulatory approval environment and regulatory hurdles for safety assessment for new investigational drug candidates;
- •
- the cost, timing and outcome of litigations;
- •
- the healthcare reimbursement system or the impact of healthcare reform, if any, imposed by the federal government; and
- •
- the activities of competitors.
Future capital requirements will also depend on the extent to which we acquire or invest in additional complementary businesses, products and technologies. We currently have no commitments or agreements relating to any of these types of transactions.
We have substantially less money than we need to develop our investigational drug compounds into commercially available drugs. It takes many years and potentially hundreds of millions of dollars to successfully develop a pre-clinical or early clinical compound into an approved and commercially marketed drug, and our efforts may not result in any marketed drugs. We may need additional funds or a partner to bring our most advanced investigational drug candidate, QNEXA, to market, if ever.
To obtain additional capital when needed, we will evaluate alternative financing sources, including, but not limited to, the issuance of equity or debt securities, corporate alliances, joint ventures and licensing agreements. However, there can be no assurance that funding will be available on favorable terms, if at all. We are continually evaluating our existing portfolio and we may choose to divest, sell or spin-off one or more of our investigational drug candidates at any time. We cannot assure you that we will successfully develop our investigational drug candidates under development or, if successfully developed or approved, that our drugs will generate revenues sufficient to enable us to earn a profit. If we are unable to obtain additional capital, management may be required to explore alternatives to reduce cash used by operating activities, including the termination of research and development efforts that may appear to be promising to us, the sale of certain assets and the reduction in overall operating activities. If adequate funds are not available, we may be required to delay, reduce the scope of or eliminate one or more of our development programs or our commercialization efforts.
Raising additional funds by issuing securities will cause dilution to existing stockholders and raising funds through lending and licensing arrangements may restrict our operations or require us to relinquish proprietary rights.
To the extent that we raise additional capital by issuing equity securities, our existing stockholders' ownership will be diluted. We have financed our operations, and we expect to continue to finance our operations, primarily by issuing and selling our common stock. In light of our need for additional financing, we may issue additional shares of common stock that could dilute your ownership in our company and may include terms that give new investors rights that are superior to yours. For example, on September 23, 2009, we sold 10,350,000 shares of our common stock through an underwriting agreement at a price of $10.50 per share resulting in gross proceeds to us of $108.7 million. Moreover, any issuances by us of equity securities may be at or below the prevailing market price of our common stock and in any event may have a dilutive impact on your ownership interest, which could cause the market price of our common stock to decline. To raise additional capital, we may chose to issue additional securities at any time and at any price.
We may also raise additional funds through the incurrence of debt, and the holders of any debt we may issue would have rights superior to your rights in the event we are not successful and are forced to seek the protection of bankruptcy laws. In addition, debt financing typically contains covenants that restrict operating activities. For example, our loan with Crown Bank, N.A., was secured by the land and
66
buildings, among other assets, located at our former principal manufacturing facility and a $700,000 Certificate of Deposit held by Crown. Additionally, our intellectual property and all of the accounts receivable, inventory and equipment arising out of or relating to MUSE and avanafil were pledged as collateral for the Deerfield transaction, which also contained a variety of covenants, including requiring us to use commercially reasonable efforts to preserve our intellectual property, manufacture, promote and sell MUSE, and develop avanafil. We have divested MUSE and the related assets and repaid all debt obligations secured by these assets. Any future debt financing we enter into may involve similar or more onerous covenants that restrict our operations.
If we raise additional funds through collaboration, licensing or other similar arrangements, it may be necessary to relinquish potentially valuable rights to our current investigational drug candidates, potential products or proprietary technologies, or grant licenses on terms that are not favorable to us. If adequate funds are not available, our ability to achieve profitability or to respond to competitive pressures would be significantly limited and we may be required to delay, significantly curtail or eliminate the development of one or more of our investigational drug candidates.
The current global economic environment poses severe challenges to our business strategy, which relies on access to capital from the markets and our collaborators.
The global economy, including credit markets and the financial services industry, has been experiencing a period of substantial turmoil and uncertainty. These conditions have generally made equity and debt financing more difficult to obtain, and may negatively impact our ability to complete financing transactions. The duration and severity of these conditions is uncertain, as is the extent to which they may adversely affect our business and the business of current and prospective collaborators and vendors. If the global economy does not improve or worsens, we may be unable to secure additional funding to sustain our operations or to find suitable partners to advance our internal programs, even if we receive positive results from our research and development or business development efforts.
Our investment in the clinical development and manufacture of a commercial supply of QNEXA may not result in any benefit to us if QNEXA is not approved for commercial sale.
We have invested significant resources in the clinical development of QNEXA. We are planning for and may invest significant resources now in preparation for marketing approval and planning for manufacture of commercial supply and sales and marketing. Our engagement in these resource-intensive activities puts significant investment at risk if we do not obtain regulatory approval and successfully commercialize QNEXA in the U.S. On October 28, 2010, we received a Complete Response Letter, or CRL, regarding the New Drug Application, or NDA, for QNEXA as a treatment for obesity. The CRL stated that in its current form, the NDA for QNEXA was not approvable. The CRL included the following areas: clinical, labeling, REMS, safety update, and drug scheduling. In the clinical section of the CRL, the FDA requested a comprehensive assessment of topiramate's and QNEXA's teratogenic potential including a detailed plan and strategy to evaluate and mitigate the potential teratogenic risks in women of childbearing potential taking the drug for the treatment of obesity. In addition, the FDA asked us to provide evidence that the elevation in heart rate (mean 1.6 beats per minute on the top dose) associated with QNEXA does not increase the risk for major adverse cardiovascular events. The FDA requested that we formally submit the results from the completed SEQUEL study (OB-305), a 52-week extension study for a subset of 675 patients who completed the previously reported 56-week CONQUER study. The FDA reserved the right to comment further on proposed labeling. On REMS, the FDA requested that a discussion of an already-submitted REMS plan be continued after we have submitted the written response. The agency also requested a safety update of any new adverse events be submitted to the NDA. Finally, the FDA stated that if approved, QNEXA would be a Schedule IV drug due to the phentermine component. On
67
January 19, 2011, we held an End-of-Review meeting with the FDA to discuss the items contained in the CRL and the information we plan to include in the resubmission of the NDA for QNEXA. In anticipation of the meeting, we had provided a briefing document that included analyses integrating existing non-clinical and clinical data to provide a comprehensive assessment of the teratogenic potential of topiramate. In addition, we provided several new analyses to demonstrate that QNEXA does not increase the risk for major cardiovascular events, which analyses included cardiovascular data from our SEQUEL (OB-305) and Sleep Apnea (OB-204) studies. We also provided a synopsis of the final study report for the SEQUEL study. No new clinical studies were requested in the CRL; however, in the event that any of the FDA concerns are not alleviated, additional clinical studies may be required. At the meeting, presentations were made on the comprehensive assessment of the teratogenic potential of topiramate and QNEXA, and evidence was presented that the increase in heart rate of 1.6 beats per minute does not increase the risk for major adverse cardiovascular events. The discussion also included elements of our proposed REMS program for QNEXA. The FDA chose to focus the meeting on the discussion of teratogenic potential for topiramate, specifically the incidence of oral clefts observed in the North American AED Pregnancy Registry and in the UK Epilepsy and Pregnancy Registry. As part of this meeting, the FDA requested that we complete the Feasibility Assessment. Although no other requests for additional information or studies were made by the FDA at the meeting or in the CRL, there can be no assurance that the FDA will not request or require us to provide additional information or undertake additional studies in connection with the QNEXA NDA. In the QNEXA studies, which included 15 offspring from women exposed to QNEXA or topiramate, there were no reports of any congenital malformations or low birth weight. The timing of the planned resubmission of the QNEXA NDA will be determined after agreement with the FDA is reached on the Feasibility Assessment and whether a retrospective observational study would be needed. We expect to reach agreement with the FDA, and if deemed feasible, initiate the retrospective observational study on fetal outcomes within the next two months. It is our goal to resubmit the NDA for QNEXA to the FDA by the end of 2011.
There is no assurance that our development of QNEXA will lead successfully to regulatory approval, or that obtaining regulatory approval will lead to commercial success. If QNEXA is not approved for commercial sale or if its development is delayed for any reason, our full investment in QNEXA may be at risk, we may be forced to write-off existing inventory, face significant costs to dispose of unusable inventory, and our business and financial condition would be materially adversely affected.
The investment of our cash balance and our available-for-sale securities are subject to risks which may cause losses and affect the liquidity of these investments.
At December 31, 2010, we had $37.2 million in cash and cash equivalents and $102 million in available-for-sale securities. While at December 31, 2010 our excess cash balances were invested in money market and U.S. Treasury securities, our investment policy as approved by the Board of Directors, also provides for investments in debt securities of U.S. government agencies, corporate debt securities and asset-backed securities. The investment policy has the primary investment objectives of preservation of principal; however, there may be times when certain of the securities in our portfolio will fall below the credit ratings required in the policy. If those securities are downgraded or impaired we would experience losses in the value of our portfolio which would have an adverse effect on our results of operations, liquidity and financial condition. An investment in money market mutual funds is not insured or guaranteed by the Federal Deposit Insurance Corporation or any other government agency. Although money market mutual funds seek to preserve the value of the investment at $1 per share, it is possible to lose money by investing in money market mutual funds.
68
The holders of our stock and other securities may take actions that are contrary to your interests, including selling their stock.
A small number of our stockholders hold a significant amount of our outstanding stock. These stockholders may support competing transactions and have interests that are different from yours. Sales of a large number of shares of our stock by these large stockholders or other stockholders within a short period of time could adversely affect our stock price.
Capital appreciation, if any, of our common stock will be your sole source of gain on an investment in our stock for the foreseeable future.
We have paid no cash dividends on any of our classes of capital stock to date and we currently intend to retain our future earnings, if any, to fund the development and growth of our business. We do not anticipate paying any cash dividends on our common stock in the foreseeable future. As a result, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future.
Our involvement in securities-related class action litigation could divert our resources and management's attention and harm our business.
The stock markets have from time to time experienced significant price and volume fluctuations that have affected the market prices for the common stock of pharmaceutical companies. These broad market fluctuations may cause the market price of our common stock to decline. In the past, securities-related class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biotechnology and biopharmaceutical companies often experience significant stock price volatility in connection with their investigational drug candidate development programs and the FDA's review of their NDAs. We are a defendant in a federal securities class action lawsuit and federal and consolidated state shareholder derivative lawsuits. These securities-related class action lawsuits generally allege that the Company and its officers misled the investing public regarding the safety and efficacy of QNEXA and the prospects for the FDA's approval of the QNEXA NDA as a treatment for obesity. Securities-related class action litigation often is expensive and diverts management's attention and our financial resources, which could adversely affect our business.
We have an accumulated deficit of $300.1 million as of December 31, 2010 and we may continue to incur substantial operating losses for the future.
We have generated a cumulative net loss of $300.1 million for the period from our inception through December 31, 2010, and we anticipate losses in future years due to continued investment in our research and development programs. There can be no assurance that we will be able to achieve or maintain profitability or that we will be successful in the future.
Our ability to utilize our net operating loss carryforwards to offset future taxable income may be limited.
As of December 31, 2010, we had approximately $234.5 million and $54.3 million of net operating loss, or NOL, carryforwards with which to offset our future taxable income for federal and state income tax reporting purposes, respectively. We used $121.2 million federal and $32.2 million state NOLs to offset our year ended December 31, 2007 federal and state taxable income, which included the $150 million in gain recognized from the Evamist sale. The Internal Revenue Code of 1986, as amended, contains provisions that may limit the net operating loss and credit carryforwards available for use in any given period upon the occurrence of certain events, including significant change in ownership interest. Should this occur, our future ability to use NOLs to offset taxable earnings would be limited in accordance with the Internal Revenue Code.
69
We may have exposure to additional tax liabilities that could negatively impact our income tax provision, net income, and cash flow.
We are subject to income taxes and other taxes in both the U.S. and the foreign jurisdictions in which we currently operate or have historically operated. The determination of our worldwide provision for income taxes and current and deferred tax assets and liabilities requires judgment and estimation. In the ordinary course of our business, there are many transactions and calculations where the ultimate tax determination is uncertain. We are subject to regular review and audit by U.S. tax authorities as well as subject to the prospective and retrospective effects of changing tax regulations and legislation. Although we believe our tax estimates are reasonable, the ultimate tax outcome may materially differ from the tax amounts recorded in our consolidated financial statements and may materially affect our income tax provision, net income, or cash flows in the period or periods for which such determination and settlement is made.
If we become subject to product liability claims, we may be required to pay damages that exceed our insurance coverage.
Past sales of our previously owned commercial product, MUSE, and our clinical trials expose us to a significant risk of product liability claims. In addition, pharmaceutical products are subject to heightened risk for product liability claims due to inherent side effects. We identified potential side effects in the patient package insert and physician insert, both of which were distributed with MUSE. While we believe that we are reasonably insured against these risks, we may not be able to obtain insurance in amounts or scope sufficient to provide us with adequate coverage against all potential liabilities. A product liability claim in excess of, or excluded from, our insurance coverage would have to be paid out of cash reserves and could have a material adverse effect upon our business, financial condition and results of operations. Product liability insurance is expensive, difficult to maintain, and current or increased coverage may not be available on acceptable terms, if at all.
In addition, we develop, test and manufacture investigational drug candidates that are used by humans. We face an inherent risk of product liability exposure related to the testing of our investigational drug candidates in clinical trials. An individual may bring a liability claim against us if one of our investigational drug candidates or future approved drugs, if any, causes, or merely appears to have caused, an injury. If we cannot successfully defend ourselves against a product liability claim, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
- •
- injury to our reputation;
- •
- withdrawal of clinical trial patients;
- •
- costs of related litigation;
- •
- substantial monetary awards to patients or other claimants; and
- •
- the inability to commercialize our investigational drug candidates.
Damages awarded in a product liability action could be substantial and could have a negative impact on our financial condition. Whether or not we were ultimately successful in product liability litigation, such litigation would consume substantial amounts of our financial and managerial resources, and might result in adverse publicity, all of which would impair our business.
70
Risks Relating to an Investment in our Common Stock
Our stock price has been and may continue to be volatile.
The market price of our common stock has been volatile and is likely to continue to be so. The market price of our common stock may fluctuate due to factors including, but not limited to:
- •
- the timing and substance of our response to the FDA's request to perform a feasibility study on a retrospective
observational study of infants born to women who received prophylaxis treatment with 100 mg of topiramate for migraine during pregnancy, and if deemed feasible the cost, timing and outcome of such
study and the other items included in the complete response letter;
- •
- the FDA's interpretation of the data we submit relating to cardiovascular safety;
- •
- the FDA's interpretation of the data from our SEQUEL study (OB-305) and Sleep Apnea study
(OB-204);
- •
- whether or not the FDA requires us to conduct additional clinical studies for QNEXA;
- •
- the cost and time required to set up a distribution system and a REMS program for QNEXA that is suitable to address any
FDA concerns;
- •
- results within the clinical trial programs for QNEXA and avanafil or other results or decisions affecting the development
of our investigational drug candidates;
- •
- announcements of technological innovations or new products by us or our competitors;
- •
- announcements of Phase 3 data of other anti-obesity compounds in development;
- •
- announcements by licensors of our technology;
- •
- actual or anticipated fluctuations in our financial results;
- •
- our ability to obtain needed financing;
- •
- sales by insiders;
- •
- economic conditions in the U.S. and abroad;
- •
- the volatility and liquidity of the financial markets;
- •
- comments by or changes in assessments of us or financial estimates by security analysts;
- •
- adverse regulatory actions or decisions;
- •
- any loss of key management;
- •
- the results of our clinical trials relative to those of our competitors;
- •
- deviations in our operating results from the estimates of securities analysts or other analyst comments;
- •
- discussions about us or our stock price by the financial and scientific press and in online investor communities;
- •
- developments or disputes concerning patents or other proprietary rights;
- •
- licensing, product, patent or securities litigation; and
- •
- public concern as to the safety and efficacy of investigational drug candidates or any future approved drugs developed by us.
These factors and fluctuations, as well as political and other market conditions, may adversely affect the market price of our common stock. Securities related class action litigation is often brought
71
against a company following periods of volatility in the market price of its securities. We are currently a defendant in a number of federal and state securities-related class action lawsuits and may be the target of similar litigation in the future. Securities related litigation, whether with or without merit, could result in substantial costs and divert management's attention and financial resources, which could harm our business and financial condition, as well as the market price of our common stock.
Additionally, volatility or a lack of positive performance in our stock price may adversely affect our ability to retain or recruit key employees, all of whom have been or will be granted stock options as an important part of their compensation packages.
We may be unable to receive the full sales consideration from the sale of the MUSE assets to Meda AB.
Under the terms of the Asset Purchase Agreement we entered into with Meda AB, or Meda, to sell certain of the assets related to the MUSE business to Meda, or the MUSE Transaction, we received $22 million upon the closing of the MUSE Transaction. The MUSE Transaction closed on November 5, 2010. We may also receive a one-time payment of $1.5 million should Meda achieve a $50 million sales milestone for MUSE in any one calendar year during any of the three full calendar years following the date of the Purchase Agreement.
Should Meda fail to achieve the sales milestone for MUSE, the potential milestone payment under the Purchase Agreement with Meda could be delayed or not occur at all.
Volatility in the stock prices of other companies may contribute to volatility in our stock price.
The stock market in general, and the NASDAQ Global Market and the market for life sciences companies in particular, have experienced significant price and volume fluctuations. Further, there has been particular volatility in the market prices of securities of early stage and development stage life sciences companies. These broad market and industry factors may seriously harm the market price of our common stock, regardless of our operating performance.
Our stock price could decline significantly based on the results and timing of prospective, retrospective observational and pre-clinical studies of, and decisions affecting, our most advanced investigational drug candidates.
The results and timing of prospective and retrospective trials and pre-clinical studies can affect our stock price. Pre-clinical studies include experiments performed in test tubes, in animals, or in cells or tissues from humans or animals. These studies include all drug studies except those conducted in human patients, and may occur before or after initiation of clinical trials for a particular compound. Results of prospective and retrospective trials and pre-clinical studies of QNEXA, avanafil or our other investigational drug candidates may not be viewed favorably by us or third parties, including investors, analysts, potential collaborators, the academic and medical community, and regulators. The same may be true of how we design the development programs of our most advanced investigational drug candidates and regulatory decisions (including by us or regulatory authorities) affecting those development programs. Biotechnology and biopharmaceutical company stock prices have declined significantly when such results and decisions were unfavorable or perceived negatively or when an investigational drug candidate did not otherwise meet expectations.
Our share ownership is concentrated, and our officers, directors and principal stockholders acting collectively can exert significant control over matters requiring stockholder approval.
Due to their combined stock holdings, our officers, directors and principal stockholders (stockholders holding greater than 5% of our common stock) acting collectively may have the ability to exercise significant influence over matters requiring stockholder approval including the election of directors and approval of significant corporate transactions. In addition, this concentration of ownership
72
may delay or prevent a change in control of our company and may make some transactions more difficult or impossible to complete without the support of these stockholders.
Our operating results may fluctuate from quarter to quarter and this fluctuation may cause our stock price to decline.
Our quarterly operating results have fluctuated in the past and are likely to fluctuate in the future. Factors contributing to these fluctuations include, among other items, the timing and enrollment rates of clinical trials for our investigational drug candidates, timing of milestone payments, the timing of recognition of deferred revenue, and our need for clinical and pre-commercialization supplies. Thus, quarter-to-quarter comparisons of our operating results are not indicative of what we might expect in the future. As a result, in some future quarters our operating results may not meet the expectations of securities analysts and investors, which could result in a decline in the price of our stock.
Future sales of our common stock may depress our stock price.
Sales of our stock by our executive officers and directors, or the perception that such sales may occur, could adversely affect the market price of our stock. We have also registered all common stock that we may issue under our employee benefits plans. As a result, these shares can be freely sold in the public market upon issuance, subject to restrictions under the securities laws. Some of our executive officers have adopted trading plans under SEC Rule 10b5-1 to dispose of a portion of their stock. Any of our executive officers or directors may adopt such trading plans in the future. If any of these events cause a large number of our shares to be sold in the public market, the sales could reduce the trading price of our common stock and impede our ability to raise future capital.
There may not be an active, liquid trading market for our common stock.
There is no guarantee that an active trading market for our common stock will be maintained on the NASDAQ Global Market. Investors may not be able to sell their shares quickly or at the latest market price if trading in our stock is not active.
Our charter documents and Delaware law could make an acquisition of our company difficult, even if an acquisition may benefit our stockholders.
Our Board of Directors has adopted a Preferred Shares Rights Plan. The Preferred Shares Rights Plan has the effect of causing substantial dilution to a person or group that attempts to acquire us on terms not approved by our Board of Directors. The existence of the Preferred Shares Rights Plan could limit the price that certain investors might be willing to pay in the future for shares of our common stock and could discourage, delay or prevent a merger or acquisition that a stockholder may consider favorable.
Some provisions of our Amended and Restated Certificate of Incorporation and Bylaws could delay or prevent a change in control of our company. Some of these provisions:
- •
- authorize the issuance of preferred stock by the Board of Directors without prior stockholder approval, commonly referred
to as "blank check" preferred stock, with rights senior to those of common stock;
- •
- prohibit stockholder actions by written consent;
- •
- specify procedures for director nominations by stockholders and submission of other proposals for consideration at
stockholder meetings; and
- •
- eliminate cumulative voting in the election of directors.
73
In addition, we are governed by the provisions of Section 203 of Delaware General Corporate Law. These provisions may prohibit large stockholders, in particular those owning 15% or more of our outstanding voting stock, from merging or combining with us. These and other provisions in our charter documents could reduce the price that investors might be willing to pay for shares of our common stock in the future and result in the market price being lower than it would be without these provisions.
Changes in financial accounting standards related to share-based payments are expected to continue to have a significant effect on our reported results.
On January 1, 2006, we adopted the revised statement of Financial Accounting Standards No. SFAS 123R, Share-Based Payment, as codified in FASB ASC topic 718, Compensation—Stock Compensation, or ASC 718, which requires that we record compensation expense in the statement of operations for share-based payments, such as employee stock options, using the fair value method. The adoption of this new standard is expected to continue to have a significant effect on our reported earnings, although it will not affect our cash flows, and could adversely impact our ability to provide accurate guidance on our future reported financial results due to the variability of the factors used to estimate the values of share-based payments. If factors change and we employ different assumptions or different valuation methods in the application of SFAS 123R in future periods, the compensation expense that we record under SFAS 123R may differ significantly from what we have recorded in the current period, which could negatively affect our stock price.
Compliance with changing regulation of corporate governance and public disclosure may result in additional expenses.
Changing laws, regulations and standards relating to corporate governance and public disclosure, including the Sarbanes-Oxley Act of 2002, new SEC regulations and NASDAQ Global Market rules, are creating significant obligations and uncertainty of compliance for companies such as ours. These new or changed laws, regulations and standards are subject to varying interpretations in many cases due to their lack of specificity, and as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies, which could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. We are committed to maintaining high standards of corporate governance and public disclosure. As a result, our efforts to comply with evolving laws, regulations and standards have resulted in, and are likely to continue to result in, increased general and administrative expenses and management time related to compliance activities. In particular, our efforts to comply with Section 404 of the Sarbanes-Oxley Act of 2002 and the related regulations regarding our required assessment of our internal controls over financial reporting and our external auditors' review and audit of our internal control over financial reporting has required the commitment of significant financial and managerial resources. We expect these efforts to require the continued commitment of significant resources. If we fail to comply with new or changed laws, regulations and standards, our reputation may be harmed and we might be subject to sanctions or investigation by regulatory authorities, such as the SEC. Any such action could adversely affect our financial results and the market price of our common stock.
Item 1B. Unresolved Staff Comments
None.
In November 2006, we entered into a 30-month lease for our corporate headquarters located in Mountain View, California. The lease commenced on February 1, 2007. The base monthly rent was set at $1.85 per square foot or $26,000 per month. The lease expired on July 31, 2009. On December 16, 2008, we entered into a first amendment to this lease. Under the terms of the amended lease, we
74
continue to lease the office space for our corporate headquarters for a two-year period commencing on August 1, 2009 and expiring on July 31, 2011. The base monthly rent was set at $1.64 per square foot or $23,000 per month. The amended lease allowed us one option to extend the term of the lease for one year from the expiration of the lease. On November 12, 2009, we entered into a second amendment to this lease. The second amendment commenced on January 1, 2010, expires on July 31, 2011 and expands the leased space. The base rent for the expansion space is set at $2.25 per square foot or $8,500 per month. The option to extend the term of the amended lease for one year from the expiration of the lease applied to this expansion space as well. In December 2010, we entered into a third amendment to this lease. The third amendment extended the lease term for the original premises and the expansion space for a period of twelve months commencing August 1, 2011 and terminating July 31, 2012. Under the third amendment, the base rent for the original space will be set at $1.69 per square foot, or $24,000 per month, and the base rent for the expansion space will be set at $2.31 per square foot, or $8,700 per month. The amended lease allows us one additional option to extend the term of the lease for one year from the expiration of the lease. The option to extend the term of the amended lease for one year from the expiration of the lease applies to the expansion space as well.
In general, our existing facilities are in good condition and adequate for all present and near term uses.
In the normal course of business, the Company receives claims and makes inquiries regarding patent infringement and other related legal matters. The Company believes that it has meritorious claims and defenses and intends to pursue any such matters vigorously.
Acrux Dispute
The Company and Acrux Limited, through its wholly owned subsidiary FemPharm Pty Ltd., or Acrux, were parties to the Testosterone Development and Commercialization Agreement dated February 12, 2004, or the Testosterone Agreement. The Testosterone Agreement covered the Company's investigational drug candidate, Luramist, which was licensed from Acrux under the Testosterone Agreement. On November 5, 2007, Acrux made a demand for arbitration under the Testosterone Agreement regarding certain claims related to Luramist. Acrux's demand sought a reversion of all rights assigned to the Company related to Luramist, monetary damages and the payment of a milestone payment for Luramist under the Testosterone Agreement and declaratory relief. The Company asserted counterclaims against Acrux in the arbitration and sought the enforcement of the Company's rights under the Testosterone Agreement. The arbitration hearing concluded on January 23, 2009, and on April 6, 2009 the panel of arbitrators, or the Panel, issued its Interim Arbitration Award finding in favor of the Company that it was in compliance with the Testosterone Agreement and denying all of the relief sought by Acrux in its demand. The Panel found that the Company was not in breach of the Luramist license agreement and that the Company had used diligent, commercially reasonable efforts to develop Luramist. The Panel further ruled in favor of the Company on its counterclaim that Acrux had breached the Luramist license agreement by failing to provide certain know-how and certain improvements in the formulation and delivery device for Luramist. The Panel denied the Acrux claim for additional milestone payments. The Panel ordered Acrux to turn over certain information to the Company that was previously withheld in violation of the agreement by Acrux. After the parties failed to agree on a new Outside Date by which the Company was to commence its first Phase 3 trial for Luramist, the Panel reset the Outside Date of April 30, 2006 to April 1, 2010 to reflect the regulatory environment.
On March 30, 2010, the Company provided written notice to Acrux of its intent to terminate the Testosterone Agreement. On April 6, 2010, in connection with Acrux's request for further briefing on the issue of damages in light of the Company's termination of the Testosterone Agreement, the Panel
75
ordered the parties to enter into settlement discussions. On May 6, 2010, the parties agreed to the terms of a settlement agreement and mutual release, or the Settlement Agreement, resolving any and all claims or potential claims in the arbitration and that may have or could have arisen from any case whatsoever, other than certain rights and obligations that survive the termination of the Testosterone Agreement or are required by the Settlement Agreement. Pursuant to the Settlement Agreement, the Company transferred Luramist-related assets to Acrux, including clinical trial material, batch release documents, inventory of applicators, FDA correspondence, intellectual property and know-how and trademarks. In addition, the Company ceased its clinical study program for Luramist as part of the settlement. The parties did not exchange cash payments as a result of the settlement and termination of the Testosterone Agreement. The Panel retains jurisdiction over the matter to enforce the terms of the Settlement Agreement.
Securities Related Class Action Lawsuits
A federal securities class action lawsuit captioned Kovtun v. Vivus, Inc., et al. is pending in the U.S. District Court, Northern District of California, which asserts claims for violations of Section 10(b) and 20(a) of the federal securities laws, purportedly relating to statements allegedly made by the Company in connection with its New Drug Application, or NDA, for QNEXA as a treatment for obesity. The essential factual allegation is that the Company and its officers misled the investing public regarding the prospects for QNEXA's NDA approval, and the drug's efficacy and safety. On February 2, 2011, the court granted a stipulation and order appointing a lead plaintiff and a lead counsel for the class. On February 3, 2011, the court granted a stipulation and order requiring the lead plaintiff to file any amendment to the operative complaint no later than 60 days from February 2, 2011, with the defendants' answer or motion to dismiss to be filed no later than 60 days after plaintiffs file the amended complaint. In the event the defendants file a motion to dismiss, plaintiffs' opposition to such motion must be filed no later than 60 days after the filing of the motion, with defendants' reply to the opposition being filed no later than 45 days after the filing of plaintiffs' opposition. Discovery is stayed in the Kovtun matter pending resolution of the defendants' motion to dismiss.
Also pending in the U.S. District Court, Northern District of California and formally related to the Kovtun matter is a shareholder derivative action captioned Turberg v. Logan, et al., which restates the allegations in Kovtun by claiming that certain of the Company's officers and directors caused or allowed the Company to violate the federal securities laws by issuing material misrepresentations to the investing public. On February 7, 2011, the court granted a stipulation and order requiring plaintiffs to file and serve any amendment to the complaint no later than 60 days from the filing of the consolidated amended complaint in the Kovtun matter, with the defendants' answer or motion to dismiss to be filed no later than 60 days after the court enters an order ruling on defendants' motion to dismiss in the Kovtun matter. In the event the defendants file a motion to dismiss in the Turberg matter, the plaintiffs will have 60 days to oppose the motion and the defendants will have 45 days to reply to the plaintiffs' opposition. The court order stayed all discovery in the Turberg matter pending resolution of the defendants' motion to dismiss.
Additionally, three separate shareholder derivative suits are pending in California Superior Court, Santa Clara County. The allegations in these shareholder derivative suits are nearly identical to those in the Turberg federal shareholder derivative action. The Company has informal agreement from plaintiffs' counsel that defendants (the members of the Company's Board of Directors) do not have to respond to the current complaints, and the Company intends to either agree with plaintiffs to stay these matters or will move the court to do so. On February 3, 2011, the court granted a stipulation and order consolidating the three suits into the action captioned Wilkinson v. Wilson, et al. and appointing a lead plaintiff and a lead counsel for the shareholder derivative actions. The court order requires the plaintiffs to file and serve a consolidated complaint no later than 60 days from the court's order and
76
the parties to meet and confer on a briefing schedule for the defendants' motion to dismiss the consolidated complaint no later than 30 days from the service of the consolidated complaint.
The Company believes that the allegations in the various federal and state actions have no merit and that the Company has meritorious defenses to the claims stated in such actions. The Company intends to vigorously defend itself in the various actions. Although there may be no merit to such allegations or claims, the Company will be required to allocate monetary and personnel resources to defend itself. The Company believes the disposition of the current lawsuits and claims is not likely to have a material effect on our financial condition or liquidity.
In the ordinary course of business the Company may become involved in lawsuits and subject to various claims from current and former employees including wrongful termination, sexual discrimination and employment matters. The Company is currently a party to a lawsuit involving a former employee. The Company has also been named as a potential defendant in a complaint filed by a former employee. The Company has investigated each of the claims and believes the allegations have no merit and that the Company has meritorious defenses to such charges. Due to the current economic downturn, employees may be more likely to file employment-related claims. Employment-related claims also may be more likely following a poor performance review. Although there may be no merit to such claims or legal matters, the Company may be required to allocate additional monetary and personnel resources to defend against these type of allegations. The Company believes the disposition of the current lawsuit and claims is not likely to have a material effect on our financial condition or liquidity.
The Company is not aware of any other asserted or unasserted claims against it where an unfavorable resolution would have an adverse material impact on the operations or financial position of the Company.
Item 4. (Removed and Reserved).
77
Item 5. Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
VIVUS' common stock trades publicly on the NASDAQ Global Select Market under the symbol "VVUS." The following table sets forth for the periods indicated the quarterly high and low sales prices of our common stock as reported on the NASDAQ Global Select Market.
| |
Three Months Ended | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
March 31 | June 30 | September 30 | December 31 | ||||||||||
2010 |
||||||||||||||
High |
$ | 10.54 | $ | 13.20 | $ | 12.44 | $ | 9.78 | ||||||
Low |
8.24 | 8.71 | 4.96 | 5.94 | ||||||||||
2009 |
||||||||||||||
High |
$ | 5.55 | $ | 6.20 | $ | 12.88 | $ | 10.85 | ||||||
Low |
2.72 | 3.61 | 5.55 | 7.07 | ||||||||||
Stockholders
As of February 18, 2011, there were 81,888,089 shares of outstanding common stock that were held by 3,549 shareholders of record and no outstanding shares of preferred stock. On February 18, 2011, the last reported sales price of our common stock on the NASDAQ Global Select Market was $7.78 per share.
Dividends
We have not paid any dividends since our inception and we do not intend to declare or pay any dividends on our common stock in the foreseeable future. Declaration or payment of future dividends, if any, will be at the discretion of our Board of Directors after taking into account various factors, including VIVUS' financial condition, operating results and current and anticipated cash needs.
Stock Options
Our stock option plans are part of a broad-based, long-term retention program that is intended to attract and retain talented employees and directors and align stockholder and employee interests.
Pursuant to our 2001 Stock Option Plan, or the 2001 Plan, which was approved by the stockholders at the annual meeting held on June 5, 2002, we may grant incentive or non-statutory stock options or stock purchase rights, or SPRs. The 2001 Plan allows us to grant incentive stock options to employees at not less than 100% of the fair market value of the stock (110% of fair market value for individuals who control more than 10% of our stock) at the date of grant, as determined by the Board of Directors. The 2001 Plan allows us to grant non-statutory stock options to employees, directors and consultants at a price to be determined by the Board of Directors. The term of the option is determined by the Board of Directors on the date of grant but shall not be longer than 10 years. The 2001 Plan allows us to grant SPRs to employees and consultants. Sales of stock under SPRs are made pursuant to restricted stock purchase agreements containing provisions established by the Board of Directors. We have a right, but not the obligation, to repurchase the shares at the original sale price, which expires at a rate to be determined by the Board of Directors. As of December 31, 2010, no SPRs had been granted under the 2001 Plan.
On July 12, 2006, the Board of Directors adopted an amendment to the 2001 Plan to add the ability to issue Restricted Stock Units, or RSUs, under the 2001 Plan. In contrast to restricted stock awards, the RSUs represent an obligation of VIVUS to issue unrestricted shares of common stock or
78
cash to the grantee only when and to the extent that the vesting criteria of the award are satisfied. As in the case of restricted stock awards, vesting criteria for RSUs can be based on time or other conditions specified by the Board or an authorized committee of the Board. However, until vesting occurs, the grantee is not entitled to any stockholder rights with respect to the unvested shares. Upon vesting of an RSU, the recipient receives one share of VIVUS stock for each vested restricted stock unit or a cash payment for the value thereof. VIVUS, in its sole discretion, may pay earned RSUs in cash, shares, or a combination thereof. Shares represented by RSUs that are fully paid in cash again will be available for grant under the Plan. We issue new shares for settlement of vested restricted stock units and exercises of stock options. We do not have a policy of purchasing our shares relating to our share-based programs.
On March 29, 2010, the Company's Board of Directors terminated the 2001 Plan. In addition, the Board of Directors adopted and approved a new 2010 Equity Incentive Plan, or the 2010 Plan, with 32,000 shares remaining reserved and unissued under the 2001 Plan, subject to the approval of the Company's stockholders. The 2001 Plan, however, continues to govern awards previously granted under it. On June 25, 2010, the Company's stockholders approved the 2010 Plan at the Company's 2010 Annual Meeting of Stockholders. The 2010 Plan provides for the grant of stock options, stock appreciation rights, restricted stock, restricted stock units, performance shares and performance units to employees, directors and consultants, to be granted from time to time as determined by the Board of Directors, the Compensation Committee of the Board of Directors, or its designees. The 2010 Plan's share reserve which the stockholders approved is 8,400,000 shares, plus any shares reserved but not issued pursuant to awards under the 2001 Plan as of the date of stockholder approval, plus any shares subject to outstanding awards under the 2001 Plan that expire or otherwise terminate without having been exercised in full, or are forfeited to or repurchased by the Company, up to a maximum of 8,111,273 shares (which was the number of shares subject to outstanding options under the 2001 Plan as of March 11, 2010).
On April 30, 2010, the Company's Board of Directors granted an option to purchase 400,000 shares of the Company's common stock, or the Inducement Grant, to Michael P. Miller, the Company's new Senior Vice President and Chief Commercial Officer. The Inducement Grant was granted outside of the Company's 2010 Plan and without stockholder approval pursuant to NASDAQ Listing Rule 5635(c)(4) and is subject to the terms and conditions of the Stand-Alone Stock Option Agreement between the Company and Michael P. Miller.
Additional information regarding our stock option plans and plan activity for fiscal 2010, 2009 and 2008 is provided in our consolidated financial statements. See Note 8: "Stock Option and Purchase Plans."
Information regarding equity compensation plans is incorporated by reference from Item 12 of this report.
79
Stock Performance Graph
The following graph shows a comparison of total stockholder return for holders of our common stock from December 31, 2005 through December 31, 2010 compared with the NASDAQ Stock Market (U.S.) Index and the RDG SmallCap Pharmaceutical Index. Total stockholder return assumes $100 invested at the beginning of the period in our common stock, the stock represented in the NASDAQ Stock Market (U.S.) Index and the stock represented by the RDG SmallCap Pharmaceutical Index, respectively. This graph is presented pursuant to SEC rules. We believe that while total stockholder return can be an important indicator of corporate performance, the stock prices of smallcap pharmaceutical stocks like VIVUS are subject to a number of market-related factors other than company performance, such as competitive announcements, mergers and acquisitions in the industry, the general state of the economy, and the performance of other medical technology stocks.
COMPARISON OF 5 YEAR CUMULATIVE TOTAL RETURN*
Among Vivus, Inc., the NASDAQ Composite Index
and the RDG SmallCap Pharmaceutical Index
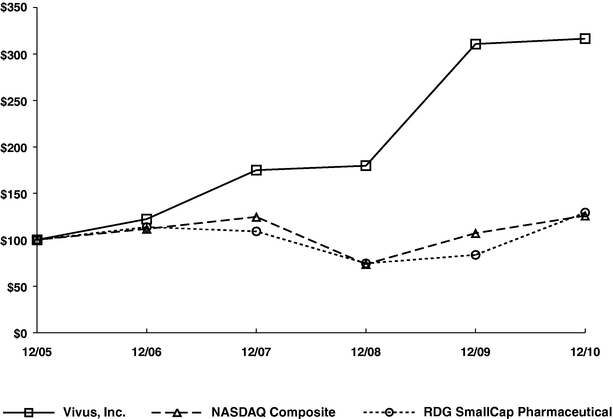
- *
- $100 invested on 12/31/05 in stock or index, including reinvestment of dividends. Fiscal year ending December 31.
80
Item 6. Selected Financial Data
The following selected financial data have been derived from our audited financial statements. The information set forth below is not necessarily indicative of the results of future operations and should be read in conjunction with "Management's Discussion and Analysis of Financial Condition and Results of Operations" and the financial statements and notes thereto included elsewhere in this Annual Report on Form 10-K. The selected data is not intended to replace the financial statements.
Selected Financial Data
(In thousands, except per share)
Selected Annual Financial Data
| |
Year Ended December 31 | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | 2007 | 2006 | |||||||||||||
Income Statement Data: |
||||||||||||||||||
License and other revenue |
$ | — | $ | 31,395 | $ | 83,721 | $ | 34,884 | $ | — | ||||||||
Operating expenses: |
||||||||||||||||||
Research and development |
39,971 | 70,940 | 76,673 | 26,240 | 12,366 | |||||||||||||
General and administrative |
25,656 | 13,870 | 12,253 | 9,981 | 7,534 | |||||||||||||
Total operating expenses |
65,627 | 84,810 | 88,926 | 36,221 | 19,900 | |||||||||||||
Loss from operations |
(65,627 | ) | (53,415 | ) | (5,205 | ) | (1,337 | ) | (19,900 | ) | ||||||||
Interest and other income (expense) |
||||||||||||||||||
Interest and other income |
468 | 1,998 | 4,406 | 4,672 | 1,562 | |||||||||||||
Interest expense |
(4,308 | ) | (3,693 | ) | (659 | ) | (45 | ) | (143 | ) | ||||||||
Other-than-temporary loss on impaired securities |
— | (654 | ) | (7,689 | ) | — | — | |||||||||||
Loss on early extinguishment of debt |
(5,958 | ) | — | — | — | — | ||||||||||||
Total interest and other income (expense) |
(9,798 | ) | (2,349 | ) | (3,942 | ) | 4,627 | 1,419 | ||||||||||
Income (loss) from continuing operations before income taxes |
(75,425 | ) | (55,764 | ) | (9,147 | ) | 3,290 | (18,481 | ) | |||||||||
Benefit (provision) for income taxes |
(9 | ) | 2,379 | 7 | (4,883 | ) | (1 | ) | ||||||||||
Net loss from continuing operations |
(75,434 | ) | (53,385 | ) | (9,140 | ) | (1,593 | ) | (18,482 | ) | ||||||||
Net income (loss) from discontinued operations, net of income taxes |
9,369 | (906 | ) | (800 | ) | (791 | ) | (3,142 | ) | |||||||||
Net loss |
$ | (66,065 | ) | $ | (54,291 | ) | $ | (9,940 | ) | $ | (2,384 | ) | $ | (21,624 | ) | |||
Basic and diluted net income (loss) per share: |
||||||||||||||||||
Continuing operations |
$ | (0.93 | ) | $ | (0.74 | ) | $ | (0.15 | ) | $ | (0.03 | ) | $ | (0.38 | ) | |||
Discontinued operations |
$ | 0.11 | $ | (0.01 | ) | $ | (0.01 | ) | $ | (0.01 | ) | $ | (0.07 | ) | ||||
Net loss per share |
$ | (0.82 | ) | $ | (0.75 | ) | $ | (0.16 | ) | $ | (0.04 | ) | $ | (0.45 | ) | |||
Shares used in per share computation: |
||||||||||||||||||
Basic |
81,017 | 72,779 | 63,724 | 58,522 | 48,103 | |||||||||||||
Diluted |
83,821 | 72,779 | 63,724 | 58,522 | 48,103 | |||||||||||||
Balance Sheet Data (at year end): |
||||||||||||||||||
Working capital |
$ | 131,781 | $ | 200,852 | $ | 134,880 | $ | 90,230 | $ | 57,564 | ||||||||
Total assets |
$ | 144,286 | $ | 230,032 | $ | 207,622 | $ | 199,289 | $ | 78,214 | ||||||||
Long-term debt |
$ | — | $ | 19,998 | $ | 11,177 | $ | 5,062 | $ | 11,488 | ||||||||
Accumulated deficit |
$ | (300,125 | ) | $ | (234,060 | ) | $ | (179,769 | ) | $ | (169,829 | ) | $ | (168,651 | ) | |||
Stockholders' equity |
$ | 132,002 | $ | 186,726 | $ | 131,213 | $ | 60,167 | $ | 53,140 | ||||||||
81
Item 7. Management's Discussion and Analysis of Financial Conditions and Results of Operations
Forward Looking Statement
This Management's Discussion and Analysis of Financial Conditions and Results of Operations and other parts of this Form 10-K contain "forward-looking" statements that involve risks and uncertainties. These statements typically may be identified by the use of forward-looking words or phrases such as "may," "will," "believe," "expect," "intend," "anticipate," "predict," "should," "planned," "continue," "likely," "opportunity," "estimated," and "potential," the negative use of these words or other similar words. All forward-looking statements included in this document are based on our current expectations, and we assume no obligation to update any such forward-looking statements. The Private Securities Litigation Reform Act of 1995 provides a "safe harbor" for such forward-looking statements. In order to comply with the terms of the safe harbor, we note that a variety of factors could cause actual results and experiences to differ materially from the anticipated results or other expectations expressed in such forward-looking statements. On October 28, 2010, we received a Complete Response Letter, or CRL, regarding the New Drug Application, or NDA, for QNEXA® as a treatment for obesity. The CRL stated that in its current form, the NDA for QNEXA was not approvable. The CRL included the following areas: clinical, labeling, Risk Evaluation and Mitigation Strategy, or REMS, safety update, and drug scheduling. In the clinical section of the CRL, the FDA requested a comprehensive assessment of topiramate's and QNEXA's teratogenic potential including a detailed plan and strategy to evaluate and mitigate the potential teratogenic risks in women of childbearing potential taking the drug for the treatment of obesity. In addition, the FDA asked us to provide evidence that the elevation in heart rate (mean 1.6 beats per minute on the top dose) associated with QNEXA does not increase the risk for major adverse cardiovascular events. The FDA requested that we formally submit the results from the completed SEQUEL study (OB-305), a 52-week extension study for a subset of 675 patients who completed the previously reported 56-week CONQUER study. The FDA reserved the right to comment further on proposed labeling. On REMS, the FDA requested that a discussion of an already-submitted REMS plan be continued after we have submitted the written response. The agency also requested a safety update of any new adverse events be submitted to the NDA. Finally, the FDA stated that if approved, QNEXA would be a Schedule IV drug due to the phentermine component. On January 19, 2011, we held an End-of-Review meeting with the FDA to discuss the items contained in the CRL and the information we plan to include in the resubmission of the NDA for QNEXA. In anticipation of the meeting, we had provided a briefing document that included analyses integrating existing non-clinical and clinical data to provide a comprehensive assessment of the teratogenic potential of topiramate. In addition, we provided several new analyses to demonstrate that QNEXA does not increase the risk for major cardiovascular events, which analyses included cardiovascular data from our SEQUEL (OB-305) and Sleep Apnea (OB-204) studies. We also provided a synopsis of the final study report for the SEQUEL study. No new clinical studies were requested in the CRL. At the meeting, presentations were made on the comprehensive assessment of the teratogenic potential of topiramate and QNEXA, and evidence was presented that the increase in heart rate of 1.6 beats per minute does not increase the risk for major adverse cardiovascular events. The discussion also included elements of our proposed REMS program for QNEXA. The FDA chose to focus the meeting on the discussion of teratogenic potential for topiramate, specifically the incidence of oral clefts observed in the North American AED Pregnancy Registry and in the UK Epilepsy and Pregnancy Registry. As part of this meeting, the FDA requested that we assess the feasibility of performing a retrospective observational study utilizing existing electronic healthcare databases to determine fetal outcomes including congenital malformations with an interest in oral cleft and low birth weight (less than 2,500 gm) in the offspring of women who received prophylaxis treatment with 100 mg of topiramate for migraine during pregnancy, or the Feasibility Assessment. Although no other requests for additional information or studies were made by the FDA at the meeting or in the CRL, there can be no assurance that the FDA will not request or require us to provide additional information or undertake additional studies in connection with the QNEXA NDA. In the QNEXA studies, which included 15 offspring from women exposed to QNEXA or topiramate, there were no reports of any congenital malformations or low birth weight. The timing of the planned resubmission of the QNEXA NDA will be determined after agreement
82
with the FDA is reached on the Feasibility Assessment and whether a retrospective observational study would be needed. We expect to reach agreement with the FDA, and if deemed feasible, initiate the retrospective observational study on fetal outcomes within the next two months. It is our goal to resubmit the NDA for QNEXA to the FDA by the end of 2011. The risks and uncertainties that may affect the operations, performance, development, and results of our business include but are not limited to: (1) our ability to complete the Feasibility Assessment made by the FDA during our End-of-Review meeting held January 19, 2011. As no additional guidance was given by the FDA as to the potential parameters of the study it may be difficult to interpret the definition of feasibility. In addition we do not know if we will be able to determine if enough live births to mothers exposed to 100 mg of topiramate exist, the condition of their records and the records of the infants, the ability to properly link records of the mother to those of the infant, the ability to determine other important infant health factors including but not limited to the health status of the mother and the diagnosis for which she was prescribed topiramate, concomitant use of medications, smoking status during pregnancy, use of alcohol or illegal substances during pregnancy, mothers weight, nutrition during pregnancy and folic acid supplementation, family history and any other information necessary to determine the nature of any malformations, in particular oral cleft. If the FDA does not agree with our assessment we may appeal the decision or amend the NDA to exclude women of childbearing potential; (2) the FDA's interpretation of the data we submit relating to cardiovascular safety; (3) the FDA's interpretation of the data from our SEQUEL study (OB-305) and Sleep Apnea study (OB-204); (4) that we may be required to conduct additional clinical trials; (5) impact on future sales based on contraindications contained in the label and extent of the REMS program; (6) our history of losses and variable quarterly results; (7) substantial competition; (8) risks related to the failure to protect our intellectual property and litigation in which we may become involved; (9) uncertainties of government or third party payer reimbursement; (10) our reliance on sole source suppliers; (11) our limited sales and marketing efforts and our reliance on third parties; (12) failure to continue to develop innovative investigational drug candidates and drugs; (13) risks related to the failure to obtain United States Food and Drug Administration, or FDA, or foreign authority clearances or approvals and noncompliance with FDA regulations; (14) our ability to demonstrate through clinical testing the safety and effectiveness of our investigational drug candidates; (15) our dependence on the performance of our collaborative partners; (16) the timing of initiation and completion of clinical trials and submissions to the FDA; (17) the volatility and liquidity of the financial markets; (18) our liquidity and capital resources; (19) our expected future revenues, operations and expenditures; and (20) other factors that are described from time to time in our periodic filings with the Securities and Exchange Commission, or the SEC, including those set forth in this filing as "Item 1A. Risk Factors."
All percentage amounts and ratios were calculated using the underlying data in thousands. Operating results for the year ended December 31, 2010, are not necessarily indicative of the results that may be expected for future fiscal years. The following discussion and analysis should be read in conjunction with our historical financial statements and the notes to those financial statements that are included in Item 8 of Part II of this Form 10-K.
Overview
VIVUS, Inc. is a biopharmaceutical company, incorporated in 1991 as a California corporation and reincorporated in 1996 as a Delaware corporation, dedicated to the development and commercialization of therapeutic drugs for large underserved markets. Currently, we have one investigational drug candidate, QNEXA, which has been submitted for approval as a treatment for weight loss in the U.S. and European Union. In the U.S., we received a Complete Response Letter, or CRL, for QNEXA in October 2010, which contained requests for additional information. On January 19, 2011, we held an End-of-Review meeting with the FDA to discuss its requests and our planned resubmission of the NDA for QNEXA. The FDA has requested that we perform the Feasibility Assessment, which is currently in process. We also have investigational drug candidates in various stages of clinical development, and we are focused on market opportunities in obesity and related morbidities, such as sleep apnea and diabetes, and men's sexual health. With respect to obesity, it is estimated that the potential
83
pharmaceutical market for obesity could approach $5 billion annually. Annual sales of approved drugs for diabetes currently exceed $10 billion. There are currently no approved pharmaceutical therapies for sleep apnea; however, the sales of devices and related consumables used to treat sleep apnea exceed $2 billion annually. The indications targeted by our investigational drug candidate as a treatment for erectile dysfunction represent a projected market greater than $4 billion annually.
Recent Developments
As mentioned above, on January 19, 2011, we held an End-of-Review meeting with the FDA to discuss our planned response to the CRL received on October 28, 2010, regarding the New Drug Application for QNEXA as a treatment for obesity. The CRL stated that in its current form, the NDA for QNEXA was not approvable. The CRL included the following areas: clinical, labeling, REMS, safety update, and drug scheduling. In the clinical section of the CRL, the FDA requested a comprehensive assessment of topiramate's and QNEXA's teratogenic potential including a detailed plan and strategy to evaluate and mitigate the potential teratogenic risks in women of childbearing potential taking the drug for the treatment of obesity. In addition, the FDA asked us to provide evidence that the elevation in heart rate (mean 1.6 beats per minute on the top dose) associated with QNEXA does not increase the risk for major adverse cardiovascular events. The FDA requested that we formally submit the results from the completed SEQUEL study (OB-305), a 52-week extension study for a subset of 675 patients who completed the previously reported 56-week CONQUER study. The FDA reserved the right to comment further on proposed labeling. On REMS, the FDA requested that a discussion of an already-submitted REMS plan be continued after we have submitted the written response. The agency also requested a safety update of any new adverse events be submitted to the NDA. Finally, the FDA stated that if approved, QNEXA would be a Schedule IV drug due to the phentermine component. In anticipation of the meeting held with the FDA on January 19, 2011, we had provided a briefing document that included analyses integrating existing non-clinical and clinical data to provide a comprehensive assessment of the teratogenic potential of topiramate. In addition, we provided several new analyses to demonstrate that QNEXA does not increase the risk for major cardiovascular events, which analyses included cardiovascular data from our SEQUEL (OB-305) and Sleep Apnea (OB-204) studies. We also provided a synopsis of the final study report for the SEQUEL study. No new clinical studies were requested in the CRL. At the meeting, presentations were made on the comprehensive assessment of the teratogenic potential of topiramate and QNEXA, and evidence was presented that the increase in heart rate of 1.6 beats per minute does not increase the risk for major adverse cardiovascular events. The discussion also included elements of our proposed REMS program for QNEXA. The FDA chose to focus the meeting on the discussion of teratogenic potential for topiramate, specifically the incidence of oral clefts observed in the North American AED Pregnancy Registry and in the UK Epilepsy and Pregnancy Registry. As part of this meeting, the FDA requested that we complete the Feasibility Assessment. Although no other requests for additional information or studies were made by the FDA at the meeting or in the CRL, there can be no assurance that the FDA will not request or require us to provide additional information or undertake additional studies in connection with the QNEXA NDA. In the QNEXA studies, which included 15 births from women exposed to QNEXA or topiramate, there were no reports of any congenital malformations or low birth weight. The timing of the planned resubmission of the QNEXA NDA will be determined after agreement with the FDA is reached on the Feasibility Assessment and whether a retrospective observational study would be needed. We expect to reach agreement with the FDA, and if deemed feasible, initiate the retrospective observational study on fetal outcomes within the next two months. It is our goal to resubmit the NDA for QNEXA to the FDA by the end of 2011.
On October 1, 2010, we entered into a definitive Asset Purchase Agreement with Meda AB, or Meda, to sell certain rights and assets related to MUSE, transurethral alprostadil, for the treatment of erectile dysfunction, or the MUSE Transaction. Meda has been our European distributor of MUSE since 2002. The assets sold in the MUSE Transaction include the U.S. and foreign MUSE patents,
84
existing inventory and the manufacturing facility located in Lakewood, New Jersey. We retained all of the liabilities associated with the pre-closing operations and products of the MUSE business and the accounts receivables for pre-closing MUSE sales. The transaction closed on November 5, 2010. Prior to the closing of the MUSE Transaction, we regained all of the rights to MUSE and avanafil held by Deerfield Management Company, L.P., and affiliates, and Crown Bank, N.A., or Crown.
On October 15, 2010, in preparation for the closing of the MUSE Transaction and in accordance with the terms of the agreements with Crown, we paid $4.8 million to Crown in satisfaction of all obligations owed to them under these agreements. As a result, the security interests and Certificate of Deposit held by Crown were terminated in our favor. On October 21, 2010, we exercised the Option under the Deerfield OPA, and we paid an aggregate amount totaling $27.1 million, which consisted of the Base Option Price of $25 million, less the Option Premium Adjustment of $2 million, plus the Cash Adjustment of $2.8 million and the Royalty Adjustment of $1.3 million. These payments satisfied all of the financial obligations under the FARA and OPA. As a result, all of the outstanding shares and the $2.8 million of cash of the Deerfield Sub are owned by us, all of the outstanding loans owed by the Deerfield Sub have been repaid and the security interests in the collateral related to MUSE and avanafil held by the Deerfield Sub and the Deerfield Affiliates as part of the FARA and OPA were terminated. In December 2010, the Deerfield Sub was dissolved.
Under the terms of the MUSE Transaction, we received an upfront payment of $22 million upon the closing, on November 5, 2010, and are eligible to receive an additional $1.5 million based on future sales of MUSE, provided that certain sales milestones are reached. Meda is now responsible for the manufacturing, selling and marketing of MUSE. Meda also assumed all post-closing expenses and liabilities associated with MUSE. We have agreed not to develop, manufacture or sell any transurethral erectile dysfunction drugs for a period of three years following the closing of the MUSE Transaction. The assets and liabilities and results of operations associated with MUSE have been reported as discontinued operations for all periods presented.
On December 17, 2010, we filed a Marketing Authorization Application, or MAA, with the European Medicines Agency, or EMA, for QNEXA Controlled-Release Capsules in the European Union, or EU. The proposed indication in the EU is for the treatment of obesity, including weight loss and maintenance of weight loss, and should be used in conjunction with a mildly hypocaloric diet. If approved in the EU, QNEXA could be recommended for obese adult patients (BMI ³ 30 kg/m2), or overweight patients (BMI ³ 27 kg/m2) with weight-related co-morbidities such as hypertension, type 2 diabetes, dyslipidemia, or central adiposity (abdominal obesity). In Europe, approximately 150 million adults are considered overweight or obese, and the prevalence is rising rapidly. According to EMA guidelines for medicinal products used in weight control, a demonstration of weight loss of at least 10% of baseline weight, which is at least greater than that associated with placebo, is considered to be a valid primary efficacy criterion. We believe QNEXA has met this efficacy criterion set by the EMA for obesity therapies. In addition, the mean weight loss for the mid- and top-dose of QNEXA at the end of two years was 10.4% and 11.4%, respectively, which met the efficacy benchmark set by the EMA for obesity therapies. These results were shown to be sustained over a two-year period and were associated with significant improvements in weight-related co-morbidities such as hypertension, dyslipidemia and diabetes. The EMA filing is comprised of data from over 4,500 overweight or obese patients with a broad range of weight-related co-morbidities. Two-year, double-blind data from SEQUEL (OB-305) were also included in the filing to demonstrate durability of treatment response and long-term safety. The EMA's review of QNEXA will follow their centralized marketing authorization procedure. If approved, QNEXA could receive marketing authorization in all 27 EU member countries. The MAA was officially validated for central procedure on January 19, 2011.
85
Our Future
Our goal is to build a successful biopharmaceutical company through the development and commercialization of innovative proprietary drugs. We intend to achieve this by:
- •
- seeking approval for QNEXA for the treatment of obesity in the U.S. and the E.U.;
- •
- establishing internal capabilities or strategic relationships with marketing partners to maximize sales potential for our
drugs that require significant commercial support; and
- •
- capitalizing on our clinical and regulatory expertise and experience to advance the development of investigational drug candidates in our pipeline.
It is our objective to become a leader in the development and commercialization of drugs for large underserved markets. We believe we have strong intellectual property supporting several opportunities in obesity and related disorders, such as sleep apnea and diabetes, and men's sexual health. Our future growth depends on our ability to further develop and obtain regulatory approval of our investigational drug candidates for indications that we have studied, or plan to study, as well as in-licensing and product line extensions.
We have funded operations primarily through private and public offerings of our common stock, through the sale of the rights to Evamist and through product sales of MUSE (alprostadil). We expect to generate future net losses due to increases in operating expenses as our various investigational drug candidates are advanced through the various stages of clinical development and for pre-commercialization activities. In connection with the sale of Evamist, to date we have received an aggregate of $150 million. The sale of Evamist was a unique transaction. An initial $10 million was paid at closing and $140 million was paid upon the FDA's approval of the Evamist NDA. These payments were non-refundable and were originally recorded as deferred revenue and were recognized as license and other revenue ratably over a 21.5-month period, from August 1, 2007 to May 15, 2009. All of the revenue deferred from the Evamist sale has been recognized. As of December 31, 2010, we have incurred a cumulative deficit of $300.1 million and expect to incur operating losses in future years.
Critical Accounting Policies and Estimates
The discussion and analysis of our financial condition and results of operations are based upon our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the U.S.. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosure of contingent assets and liabilities. On an ongoing basis, we evaluate our estimates, including those related to available-for-sale securities, research and development expenses, income taxes, inventories, contingencies and litigation and stock-based compensation. We base our estimates on historical experience, information received from third parties and on various other assumptions that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
We believe the following critical accounting policies affect our more significant judgments and estimates used in the preparation of our consolidated financial statements:
Revenue Recognition
License and Other Revenue: We recognize license revenue in accordance with the SEC's Staff Accounting Bulletin No. 104, Revenue Recognition, as codified in the Financial Accounting Standards Board's, or FASB, Accounting Standards Codification, or ASC, topic 605, Revenue Recognition, or ASC 605. When evaluating multiple element arrangements, we consider whether the components of the
86
arrangement represent separate units of accounting as defined in Emerging Issues Task Force Issue No. 00-21, Revenue Arrangements with Multiple Deliverables, or EITF 00-21, as codified in FASB ASC topic 605, subtopic 25 Multiple Element Arrangements, or ASC 605-25. In accordance with EITF 00-21, we recognize revenue for delivered elements only when the delivered element has stand-alone value and we have objective and reliable evidence of fair value for each undelivered element. If the fair value of any undelivered element included in a multiple element arrangement cannot be objectively determined, revenue is deferred until all elements are delivered and services have been performed, or until fair value can objectively be determined for any remaining undelivered elements, or such elements are insignificant. Application of this standard requires subjective determinations and requires management to make judgments about the fair value of the individual elements and whether such elements are separable from the other aspects of the contractual relationship.
Revenue from non-refundable, upfront license fees where we have continuing involvement is recognized ratably over the development or agreement period. Revenue associated with performance milestones is recognized based upon the achievement of the milestones, as defined in the respective agreements.
On May 15, 2007, we closed our transaction with K-V Pharmaceutical Company, or K-V, for the sale of our investigational drug candidate, Evamist, a metered-dose transdermal spray for the treatment of menopause symptoms. At the time of the sale, Evamist was an investigational product and was not yet approved by the FDA for marketing. The sale transaction contained multiple deliverables, including: the delivery at closing of the Evamist assets (mainly raw material inventory and certain fixed assets); a grant of a sublicense of our rights under a license related to Evamist, and a license to the MDTS applicator; the delivery upon receipt of regulatory approval of Evamist, along with all regulatory submissions; and, lastly, the delivery after FDA approval of certain transition services and a license to improvements to the MDTS applicator. We received approval from the FDA to market Evamist on July 27, 2007, or FDA Approval, and on August 1, 2007, we transferred and assigned the Evamist FDA submissions and all files related thereto to K-V. In August 2008, we assigned all of our rights and obligations under the Evamist license agreement to K-V. We received an upfront payment of $10 million in May 2007 upon the closing and received an additional $140 million milestone payment in August 2007 upon FDA Approval. These payments are non-refundable. We evaluated this multiple deliverable arrangement to determine whether the deliverables were divided into separate units of accounting. Upon FDA Approval, the two remaining deliverables were the transition services to be performed under the Transition Services Agreement, or TSA, and a license to improvements to the MDTS applicator, or Improvement License, during the two-year period commencing with the closing, or May 15, 2007, and ending on May 15, 2009. We were able to establish fair value for the TSA. As it relates to the Improvement License, no specific value was assigned in the agreement. We had no obligation to develop improvements to the MDTS applicator and had no plans to expend significant resources in this endeavor. However, we did not have objective, reliable evidence of fair value or evidence of inconsequential value to the customer of the Improvement License. Accordingly, the delivered items, together with the undelivered items, were bundled together and were treated as one unit of accounting. As a result, the initial $10 million paid at closing and the $140 million paid upon FDA Approval were recorded as deferred revenue and have been recognized as license revenue ratably over the 21.5-month term of the Improvement License, from August 1, 2007 to May 15, 2009. The revenue related to the transaction recognized for years ended December 31, 2009, 2008 and 2007 was $31.4 million, $83.7 million and $34.9 million, respectively.
In September 2009, the FASB issued Accounting Standards Update, or ASU, No. 2009-13, Revenue Recognition (Topic 605): Multiple-Deliverable Revenue Arrangements. ASU No. 2009-13 amends the guidance for measurement and separation of deliverables in multiple element arrangements under EITF Issue 00-21, as codified in FASB ASC 605-25, and significantly increases the related disclosure requirements. Under this new guidance, which will be effective for us beginning January 1, 2011, our accounting for the revenue from the sale of the Evamist transaction may have been different.
87
Research and Development Expenses
Research and development, or R&D, expenses include license fees, related compensation, consultants' fees, facilities costs, administrative expenses related to R&D activities and clinical trial costs at other companies and research institutions under agreements that are generally cancelable, among other related R&D costs. We also record accruals for estimated ongoing clinical trial costs. Clinical trial costs represent costs incurred by clinical research organizations, or CROs, and clinical sites and include advertising for clinical trials and patient recruitment costs. These costs are recorded as a component of R&D expenses and are expensed as incurred. Under our agreements, progress payments are typically made to investigators, clinical sites and CROs. We analyze the progress of the clinical trials, including levels of patient enrollment, invoices received and contracted costs when evaluating the adequacy of accrued liabilities. Significant judgments and estimates must be made and used in determining the accrued balance in any accounting period. Actual results could differ from those estimates under different assumptions. Revisions are charged to expense in the period in which the facts that give rise to the revision become known.
Income Taxes
We make certain estimates and judgments in determining income tax expense for financial statement purposes. These estimates and judgments occur in the calculation of certain tax assets and liabilities, which arise from differences in the timing of recognition of revenue and expense for tax and financial statement purposes.
As part of the process of preparing our consolidated financial statements, we are required to estimate our income taxes in each of the jurisdictions in which we operate. This process involves us estimating our current tax exposure under the most recent tax laws and assessing temporary differences resulting from differing treatment of items for tax and accounting purposes. These differences result in deferred tax assets and liabilities, which are included in our consolidated balance sheets.
We assess the likelihood that we will be able to recover our deferred tax assets. We consider all available evidence, both positive and negative, including historical levels of income, expectations and risks associated with estimates of future taxable income and ongoing prudent and feasible tax planning strategies in assessing the need for a valuation allowance. If it is not more likely than not that we will recover our deferred tax assets, we will increase our provision for taxes by recording a valuation allowance against the deferred tax assets that we estimate will not ultimately be recoverable. As a result of our analysis of all available evidence, both positive and negative, as of December 31, 2010, it was considered more likely than not that the Company's deferred tax assets would not be realized. However, should there be a change in our ability to recover our deferred tax assets; we would recognize a benefit to our tax provision in the period in which we determine that it is more likely than not that we will recover our deferred tax assets.
Inventories
Inventories are valued at the lower of cost (first in, first out) or market. We record inventory reserves for estimated obsolescence, unmarketable or excess inventory equal to the difference between the cost of inventory and the estimated market value based upon assumptions about future demand and market conditions. If actual market conditions are less favorable than those projected by management, inventory write-downs may be required.
Cash and Cash Equivalents
The Company considers highly liquid investments with maturities from the date of purchase of three months or less to be cash equivalents. At December 31, 2010, all cash equivalents are invested in money market funds and U.S. Treasury securities. These accounts are recorded at fair value.
88
Cash with restrictions for a period of greater than 12 months is classified as restricted cash, a non-current asset.
Available-for-Sale Securities
We focus on liquidity and capital preservation in our investments in available-for-sale securities. Our investment policy, as approved by the Audit Committee of the Board of Directors, allows us to invest our excess cash balances in money market and marketable securities, primarily U.S. Treasury securities and debt securities of U.S. government agencies, corporate debt securities and asset-backed securities. We periodically evaluate our investments to determine if impairment charges are required.
We determine the appropriate classification of marketable securities at the time of purchase and reevaluate such designation at each balance sheet date. Our marketable securities have been classified and accounted for as available-for-sale. We may or may not hold securities with stated maturities greater than 12 months until maturity. In response to changes in the availability of and the yield on alternative investments as well as liquidity requirements, we may sell these securities prior to their stated maturities. As these securities are viewed by us as available to support current operations securities with maturities beyond 12 months are classified as current assets.
Securities are carried at fair value, with the unrealized gains and losses, net of taxes, reported as a component of stockholders' equity, unless the decline in value is deemed to be other-than-temporary and we intend to sell such securities before recovering their costs, in which case such securities are written down to fair value and the loss is charged to other-than-temporary loss on impaired securities. We evaluate our investment securities for other-than-temporary declines based on quantitative and qualitative factors. Any realized gains or losses on the sale of marketable securities are determined on a specific identification method, and such gains and losses are reflected as a component of interest income.
SFAS No. 115, Accounting for Certain Investments in Debt and Equity Securities, FSP SFAS 115-2 and SFAS 124-4, Recognition and Presentation of Other-than-Temporary Impairments ("FSP 115-2/SFAS 124-2") and SAB Topic 5M, Accounting for Non-current Marketable Equity Securities, as codified in FASB ASC topic 320, Investments—Debt and Equity Securities, or ASC 320, provide guidance on determining when an investment is other-than-temporarily impaired. FSP 115-2/124-2 is effective for all periods ending after June 15, 2009 and provides additional guidance designed to create a greater clarity and consistency in accounting for and presenting impairment losses on securities. For securities that are deemed to be other-than-temporarily impaired, the security is adjusted to fair value and the resulting losses are recognized in other-than-temporary loss on impaired securities in the consolidated statements of operations.
During our quarterly impairment assessments, we determined that a decline in value of certain securities was other-than-temporary. Accordingly, we recorded other-than-temporary impairment adjustments of $654,000 in the year ended December 31, 2009. We included this non-cash impairment charge in other-than-temporary loss on impaired securities in the consolidated statements of operations. These securities covered a number of industries.
Contingencies and Litigation
We are periodically involved in disputes and litigation related to a variety of matters. When it is probable that we will experience a loss, and that loss is quantifiable, we record appropriate reserves. We record legal fees and costs as an expense when incurred.
Share-Based Payments
We follow the fair value method of accounting for share-based compensation arrangements in accordance with SFAS 123R, Share-Based Payment, as codified in FASB ASC topic 718, Compensation—
89
Stock Compensation, or ASC 718. We adopted SFAS 123R effective January 1, 2006 using the modified prospective method of transition. Under SFAS 123R, the estimated fair value of share-based compensation, including stock options and restricted stock units granted under our Stock Option Plan and purchases of common stock by employees at a discount to market price under the Employee Stock Purchase Plan, or the ESPP, is recognized as compensation expense. Compensation expense for purchases under the ESPP is recognized based on the estimated fair value of the common stock purchase rights during each offering period and the percentage of the purchase discount.
We recorded $7.4 million, $4.8 million and $4.7 million of share-based compensation expense from continuing and discontinued operations for the years ended December 31, 2010, 2009 and 2008, respectively. Share-based compensation expense is allocated among research and development, general and administrative expenses and discontinued operations based on the function of the related employee. This charge had no impact on our cash flows for the periods presented.
We use the Black-Scholes option pricing model to estimate the fair value of the share-based awards as of the grant date. The Black-Scholes model, by its design, is highly complex, and dependent upon key data inputs estimated by management. The primary data inputs with the greatest degree of judgment are the estimated lives of the share-based awards and the estimated volatility of our stock price. The Black-Scholes model is highly sensitive to changes in these two data inputs. The expected term of the options represents the period of time that options granted are expected to be outstanding and is derived by analyzing the historical experience of similar awards, giving consideration to the contractual terms of the stock-based awards, vesting schedules and expectations of future employee behavior. We determine expected volatility using the historical method, which is based on the daily historical trading data of our common stock over the expected term of the option. Management selected the historical method primarily because we have not identified a more reliable or appropriate method to predict future volatility. For more information about ASC 718, see Note 8: "Stock Option and Purchase Plans" to the notes to the consolidated financial statements included in this Form 10-K.
Fair Value
On January 1, 2008, we adopted SFAS No. 157 Fair Value Measurements, as codified in FASB ASC 820, Fair Value Measurements and Disclosures, or ASC 820, and effective October 10, 2008, we adopted FSP No. SFAS 157-3, Determining the Fair Value of a Financial Asset When the Market for That Asset Is Not Active, except as it applies to the nonfinancial assets and nonfinancial liabilities subject to FSP 157-2. On January 1, 2009, we adopted SFAS No 157 with respect to non-financial assets and non-financial liabilities. On June 15, 2009 we adopted FSP 157-4, Determining Fair Value When the Volume and Level of Activity for the Assets or Liabilities Have Significantly Decreased and Identifying Transactions That Are Not Orderly. Adoption of the provisions of these standards did not have a material effect on our financial position.
Financial Instruments Measured at Fair Value. Our cash and cash equivalents and available-for-sale financial instruments are carried at fair value and we make estimates regarding valuation of these assets measured at fair value in preparing the consolidated financial statements.
Fair Value Measurement—Definition and Hierarchy. SFAS No. 157 defines fair value as the price that would be received to sell an asset or paid to transfer a liability (i.e., the "exit price") in an orderly transaction between market participants at the measurement date.
Valuation Technique. SFAS No. 157 establishes a hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the observable inputs be used when available. Observable inputs are inputs that market participants would use in pricing the asset or liability developed based on market data obtained from sources independent of VIVUS. Unobservable inputs are inputs that reflect our assumptions about the assumptions market participants would use in pricing the asset or liability developed based on the best
90
information available in the circumstances. SFAS No. 157 prescribes three valuation techniques that shall be used to measure fair value as follows:
- 1.
- Market
Approach—uses prices or other relevant information generated by market transactions involving identical or comparable assets or
liabilities.
- 2.
- Income
Approach—uses valuation techniques to convert future cash flow amounts to a single present value amount (discounted).
- 3.
- Cost Approach—the amount that currently would be required to replace the service capacity of an asset (i.e., current replacement cost).
One or a combination of the approaches above can be used to calculate fair value, whichever results in the most representative fair value.
In addition to the three valuation techniques, SFAS No. 157 prescribes a fair value hierarchy in order to increase consistency and comparability in fair value measurements and related disclosures. The hierarchy is broken down into three levels based on the reliability of inputs as follows:
- •
- Level 1—Valuations based on quoted prices in active markets for identical assets. Since valuations are based on quoted prices that are readily and regularly available in an active market, valuation of these products does not entail a significant degree of judgment.
These types of instruments primarily consist of financial instruments whose value is based on quoted market prices such as cash, money market funds and U.S. Treasury securities that are actively traded. Management judgment was required to determine our policy that defines the levels at which sufficient volume and frequency of transactions is met for a market to be considered active.
- •
- Level 2—Valuations based on quoted prices in markets that are not active or for which all significant inputs are observable, directly or indirectly. Quoted prices for similar instruments in active markets, quoted prices for identical or similar instruments in markets that are not active, or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
The types of instruments valued based on other observable inputs include debt securities of U.S. government agencies, corporate bonds, mortgage-backed and asset-backed products. Substantially all of these assumptions are observable in the marketplace, can be derived from observable data or are supported by observable levels at which transactions are executed in the marketplace.
- •
- Level 3—Valuations based on inputs that are unobservable and significant to the overall fair value measurement.
These types of instruments have included certain corporate bonds, mortgage-backed securities and asset-backed securities. We have no Level 3 securities as of December 31, 2010 or 2009. Level 3 is comprised of unobservable inputs that are supported by little or no market activity. These instruments are considered Level 3 when their fair values are determined using pricing models, discounted cash flows or similar techniques and at least one significant model assumption or input is unobservable. Level 3 may still include some observable inputs such as yield spreads derived from markets with limited activity. Level 3 financial assets include securities for which there is limited market activity such that the determination of fair value requires significant judgment or estimation. The availability of observable inputs can vary from product to product and is affected by a wide variety of factors, including, for example, the type of product, whether the product is new and not yet established in the marketplace, and other characteristics particular to the transaction. To the extent that valuation is based on models or inputs that are less observable or unobservable in the market, the determination of fair value requires more judgment. Accordingly, the degree of judgment exercised by us in determining fair value is greatest for instruments categorized in Level 3. In certain cases, the inputs used to measure fair value may fall into different levels of the fair value hierarchy. In such cases, for disclosure purposes
91
the level in the fair value hierarchy within which the fair value measurement in its entirety falls is determined based on the lowest level input that is significant to the fair value measurement in its entirety.
Fair Value Measurements
As of December 31, 2010, our cash and cash equivalents and available-for-sale securities measured at fair value on a recurring basis totaled $139.2 million.
All of our cash and cash equivalents and available-for-sale securities are in cash, money market instruments and U.S. Treasury securities at December 31, 2010, and these are classified as Level 1. The valuation techniques used to measure the fair values of these financial instruments were derived from quoted market prices, as substantially all of these instruments have maturity dates, if any, within one year from the date of purchase and active markets for these instruments exists.
Deerfield Financing
On April 3, 2008, we entered into several agreements with Deerfield Management Company, L.P., or Deerfield, a healthcare investment fund, and its affiliates, Deerfield Private Design Fund L.P. and Deerfield Private Design International, L.P. (collectively, the Deerfield Affiliates). Certain of the agreements were amended and restated on March 16, 2009, which included the addition of Deerfield PDI Financing L.P. as a Deerfield Affiliate. Under the agreements, Deerfield and its affiliates provided us with $30 million in funding, consisting of $20 million from the Funding and Royalty Agreement, or FARA, entered into with a newly incorporated subsidiary of Deerfield, the Deerfield Sub, and $10 million from the sale of our common stock. Under the FARA, we paid royalties on the net sales of MUSE to the Deerfield Sub. The FARA had a term of 10 years.
We entered into the Option and Put Agreement with the Deerfield Affiliates and the Deerfield Sub, dated April 3, 2008, and an Amended and Restated Option and Put Agreement dated March 16, 2009, or the OPA. Pursuant to the OPA, the Deerfield Affiliates granted us an option to purchase all of the outstanding shares of common stock of the Deerfield Sub from the Deerfield Affiliates, referred to as the Option. Our obligation to pay royalties terminated upon the exercise of the Option. The base consideration for the Option exercise, or Base Option Price, was $25 million, less $2 million we paid upon closing, as the Option was exercised on or prior to the third anniversary of the execution of the OPA. The aggregate consideration payable by us upon exercise of the Option, or the Option Purchase Price, was equal to the sum of the Base Option Price, plus: (i) the cash and cash equivalents held by the Deerfield Sub at the date of the closing of the resulting sale of the common stock of the Deerfield Sub, or the Cash Adjustment; (ii) accrued and unpaid royalties, or the Royalty Adjustment; and minus (i) the Option premium of $2 million that was paid at the closing of the transaction, or the Option Premium Adjustment ; (ii) accrued but unpaid taxes; (iii) unpaid Funding Payments; (iv) loans payable by the Deerfield Sub, or the Loan Balance Adjustment; and (v) any other outstanding liabilities of the Deerfield Sub, or the Adjusted Option Purchase Price.
In preparation for the closing of the MUSE Transaction and in accordance with the terms of the OPA, we exercised the Option, and on October 21, 2010 we paid an aggregate amount totaling $27.1 million, which consisted of the Base Option Price of $25 million, less the Option Premium Adjustment of $2 million, plus the Cash Adjustment of $2.8 million and the Royalty Adjustment of $1.3 million. These payments satisfied all of the financial obligations under the FARA and OPA. As a result, all of the outstanding shares and the $2.8 million of cash of the Deerfield Sub are owned by us, all of the outstanding loans owed by the Deerfield Sub have been repaid and the security interests in the collateral related to MUSE and avanafil held by the Deerfield Sub and the Deerfield Affiliates as part of the FARA and OPA were terminated. In December 2010, the Deerfield Sub was dissolved. The payoff of the Deerfield loan resulted in a loss on the early extinguishment of debt of $6 million which was recognized in the fourth quarter of 2010.
92
RESULTS OF OPERATIONS
Executive Overview
For the year ended December 31, 2010, we reported a net loss of $66.1 million or $0.82 net loss per share, as compared to a net loss of $54.3 million, or $0.75 net loss per share during the same period in 2009. The net loss from continuing operations was $75.4 million, or $0.93 net loss per share and the net income from discontinued operations was $9.4 million, or $0.11 net income per share. The increased net loss from continuing operations in the year ended December 31, 2010, as compared to the year ended December 31, 2009, results from the completion of the recognition of the Evamist deferred revenue in 2009, where we recognized $31.4 million in deferred revenue, partially offset by decreased operating expenses in 2010. The decrease in operating expenses in 2010 as compared to 2009 was primarily attributable to reduced research and development spending on QNEXA for obesity, partially offset by increased general and administrative expense primarily due to QNEXA pre-commercialization expenses and an increase in non-cash share-based compensation expense. In addition, in 2009, we had a $2.4 million benefit for income taxes primarily due to a carryback claim of losses generated in 2008 which allowed us to obtain a refund for Federal income taxes which we paid in 2007.
On October 28, 2010, we received a Complete Response Letter, or CRL, from the FDA regarding the QNEXA NDA. The FDA issued the CRL to communicate its decision that the NDA could not be approved in its present form. On January 19, 2011, we held An End-of-Review meeting with the FDA to discuss the items contained in the CRL and the information we plan to include in the resubmission. In anticipation of the meeting, we had provided a briefing document that included analyses integrating existing non-clinical and clinical data to provide a comprehensive assessment of the teratogenic potential of topiramate. In addition, we provided several new analyses to demonstrate that QNEXA does not increase the risk for major cardiovascular events, which analyses included cardiovascular data from our SEQUEL (OB-305) and Sleep Apnea (OB-204) studies. We also provided a synopsis of the final study report for the SEQUEL study. No new clinical studies were requested in the CRL. At the meeting, presentations were made on the comprehensive assessment of the teratogenic potential of topiramate and QNEXA, and evidence was presented that the increase in heart rate of 1.6 beats per minute does not increase the risk for major adverse cardiovascular events. The discussion also included elements of our proposed REMS program for QNEXA. The FDA chose to focus the meeting on the discussion of teratogenic potential for topiramate, specifically the incidence of oral clefts observed in the North American AED Pregnancy Registry and in the UK Epilepsy and Pregnancy Registry. As part of this meeting the FDA requested that we complete the Feasibility Assessment. Although no other requests for additional information or studies were made by the FDA at the meeting or in the CRL, there can be no assurance that the FDA will not request or require us to provide additional information or undertake additional studies in connection with the QNEXA NDA. In the QNEXA studies, which included 15 births from women exposed to QNEXA or topiramate, there were no reports of any congenital malformations or low birth weight. The timing of the planned resubmission of the QNEXA NDA will be determined after agreement with the FDA is reached on the Feasibility Assessment and whether a retrospective observational study would be needed. We expect to reach agreement with the FDA, and if deemed feasible, initiate the retrospective observational study on fetal outcomes within the next two months. It is our goal to resubmit the NDA for QNEXA to the FDA by the end of 2011.
On October 1, 2010, we entered into a definitive Asset Purchase Agreement with Meda to sell certain rights and assets related to MUSE, transurethral alprostadil, for the treatment of erectile dysfunction, or the MUSE Transaction. Meda has been our European distributor of MUSE since 2002. The assets sold in the MUSE Transaction include the U.S. and foreign MUSE patents, existing inventory, and the manufacturing facility located in Lakewood, New Jersey. We retained all of the liabilities associated with the pre-closing operations and products of the MUSE business and the
93
accounts receivables for pre-closing MUSE sales. The transaction closed on November 5, 2010. The sale of MUSE will allow us to focus on the approval and commercialization of QNEXA and the development of avanafil. Prior to the closing of the MUSE Transaction, we regained all of the rights to MUSE and avanafil held by Deerfield Management Company, L.P., and affiliates, and Crown Bank, N.A., or Crown. On October 15, 2010, in preparation for the closing of the MUSE Transaction and in accordance with the terms of the agreements with Crown, we paid $4.8 million to Crown in satisfaction of all obligations owed to them under these agreements. As a result, the security interests and Certificate of Deposit held by Crown were terminated in our favor. Under the terms of the MUSE Transaction, we received an upfront payment of $22 million upon the closing and are eligible to receive an additional $1.5 million based on future sales of MUSE, provided that certain sales milestones are reached. Upon the closing of the MUSE Transaction, Meda is now responsible for the manufacturing, selling and marketing of MUSE. Meda also assumed all post-closing expenses and liabilities associated with MUSE. We have agreed not to develop, manufacture or sell any transurethral erectile dysfunction drugs for a period of three years following the closing of the MUSE Transaction. The assets and liabilities and results of operations associated with MUSE have been reported as discontinued operations for all periods presented.
On April 3, 2008, we entered into several agreements with Deerfield Management Company, L.P., or Deerfield, a healthcare investment fund, and its affiliates, Deerfield Private Design Fund L.P. and Deerfield Private Design International, L.P. (collectively, the Deerfield Affiliates). Certain of the agreements were amended and restated on March 16, 2009, which included the addition of Deerfield PDI Financing L.P. as a Deerfield Affiliate. Under the agreements, Deerfield and its affiliates provided us with $30 million in funding, consisting of $20 million from the Funding and Royalty Agreement, or FARA, entered into with a newly incorporated subsidiary of Deerfield, or the Deerfield Sub, and $10 million from the sale of our common stock. We received all of the required payments under the FARA, or the Funding Payments. Under the FARA, we paid royalties on the net sales of MUSE to the Deerfield Sub. The FARA had a term of 10 years.
We entered into the Option and Put Agreement with the Deerfield Affiliates and the Deerfield Sub, dated April 3, 2008, and an Amended and Restated Option and Put Agreement dated March 16, 2009, or the OPA. Pursuant to the OPA, the Deerfield Affiliates granted us an option to purchase all of the outstanding shares of common stock of the Deerfield Sub from the Deerfield Affiliates, referred to as the Option. Our obligation to pay royalties terminated upon the exercise of the Option. The base consideration for the Option exercise, or Base Option Price, was $25 million, less $2 million we paid upon closing, as the Option was exercised on or prior to the third anniversary of the execution of the OPA. The aggregate consideration payable by us upon exercise of the Option, or the Option Purchase Price, was equal to the sum of the Base Option Price, plus: (i) the cash and cash equivalents held by the Deerfield Sub at the date of the closing of the resulting sale of the common stock of the Deerfield Sub, or the Cash Adjustment; (ii) accrued and unpaid royalties, or the Royalty Adjustment; and minus (i) the Option premium of $2 million that was paid at the closing of the transaction, or the Option Premium Adjustment ; (ii) accrued but unpaid taxes; (iii) unpaid Funding Payments; (iv) loans payable by the Deerfield Sub, or the Loan Balance Adjustment; and (v) any other outstanding liabilities of the Deerfield Sub, or the Adjusted Option Purchase Price.
In preparation for the closing of the MUSE Transaction and in accordance with the terms of the OPA, we exercised the Option, and on October 21, 2010 we paid an aggregate amount totaling $27.1 million, which consisted of the Base Option Price of $25 million, less the Option Premium Adjustment of $2 million, plus the Cash Adjustment of $2.8 million and the Royalty Adjustment of $1.3 million. These payments satisfied all of the financial obligations under the FARA and OPA. As a result, all of the outstanding shares and the $2.8 million of cash of the Deerfield Sub are owned by us, all of the outstanding loans owed by the Deerfield Sub have been repaid and the security interests in the collateral related to MUSE and avanafil held by the Deerfield Sub and the Deerfield Affiliates as
94
part of the FARA and OPA were terminated. The payoff of the Deerfield loan resulted in a loss on the early extinguishment of debt of $6 million which was recognized in the fourth quarter of 2010. In December 2010, the Deerfield Sub was dissolved.
In connection with the sale of Evamist, we received $150 million. The sale of Evamist was a unique transaction. An initial $10 million was paid at closing and $140 million was paid upon FDA approval of Evamist. These payments were non-refundable and were recorded as deferred revenue and have been recognized as license and other revenue ratably over a 21.5-month period, from August 1, 2007 to May 15, 2009, which was the remaining term of a license to improvements to the MDTS applicator. No improvements to the MDTS applicator were made during this period. As compared to revenues from product sales, license and other revenue was significant on a quarterly basis during this 21.5-month period. Since the $150 million was received and we had no related contingencies, the recognition of revenue and the corresponding reduction of deferred revenue related to the Evamist sale had no impact on our cash flows from operations during this period. The revenue related to the Evamist transaction recognized in 2009 was $31.4 million.
We may have continued losses in future years, depending on the timing of our research and development expenditures, and we plan to continue to invest in clinical development of our current research and investigational drug candidates to bring those potential drugs to market.
Continuing operations
Revenue
| |
|
|
|
% Change (Decrease) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Years Ended December 31, | |||||||||||||||
| |
2010 vs 2009 | 2009 vs 2008 | ||||||||||||||
| |
2010 | 2009 | 2008 | |||||||||||||
| |
(In thousands, except percentages) |
|||||||||||||||
License and other revenue |
$ | — | $ | 31,395 | $ | 83,721 | (100 | )% | (63 | )% | ||||||
On March 30, 2007, we announced that we had entered into a definitive agreement with K-V, to transfer our assets and grant a sublicense of our rights under the Evamist Agreement to K-V, or the K-V Transaction. In August 2008, we assigned all of our rights and obligations under the Evamist license agreement to K-V. The closing of the K-V Transaction occurred on May 15, 2007 and on July 27, 2007 we received FDA approval of the Evamist NDA. An initial $10 million was paid at closing and $140 million was paid upon FDA approval. These payments were recorded as deferred revenue and have been recognized as revenue ratably over the 21.5-month term of the Improvement License, from August 1, 2007 to May 15, 2009. In the years ended December 31, 2010, 2009 and 2008, we recognized $0, $31.4 million and $83.7 million, respectively, in deferred revenue related to Evamist.
Research and development
| |
|
|
|
% Change (Decrease) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Years Ended December 31, | |||||||||||||||
| |
2010 vs 2009 | 2009 vs 2008 | ||||||||||||||
| |
2010 | 2009 | 2008 | |||||||||||||
| |
(In thousands, except percentages) |
|||||||||||||||
Research and development |
$ | 39,971 | $ | 70,940 | $ | 76,673 | (44 | )% | (7 | )% | ||||||
Research and development expenses in the year ended December 31, 2010 decreased $31 million, or 44%, to $40 million, as compared to $70.9 million in the same period last year. This decrease was the net result of decreased spending for QNEXA for obesity of $28.5 million, decreased spending for avanafil of $3.4 million and a net decrease in other research and development spending of $278,000, partially offset by $1.2 million in increased spending related to the filing of the Marketing Authorization Application for QNEXA in the European Union in the year ended December 31, 2010,
95
as compared to the same period last year. In the year ended December 31, 2010, we incurred $7.8 million for services provided by one clinical research organization on the QNEXA Phase 3 studies, which represented 20% of our research and development expenses for the period. Separately, we incurred another $8.2 million for services provided by another clinical research organization on the avanafil Phase 3 studies, which represented another 20% of our research and development expenses for the year ended December 31, 2010.
We anticipate that our research and development expenses in 2011 will decline significantly from costs incurred in 2010. The current remaining contractual obligation for payments to our primary contract research organization, or CRO, for the Phase 3 avanafil trials totals $3.2 million, which includes amounts in accrued research and clinical expenses as of December 31, 2010. The current remaining contractual obligation for payments to our primary CRO for the Phase 3 QNEXA for obesity trials and work related to the preparation of the NDA for avanafil totals $1.8 million, which also includes amounts in accrued research and clinical expenses as of December 31, 2010. There are likely to be additional research and development expenses related to avanafil and our other investigational drug candidates under development. Our research and development expenses may fluctuate from period to period due to the timing and scope of our development activities and the results of clinical and pre-clinical studies. Regardless, if we are successful in obtaining FDA regulatory approval for any new investigational drug candidates being developed through our research and development efforts, we do not expect to recognize revenue from sales of such new drugs, if any, for at least a year or more due to the length of time required to develop investigational drug candidates into commercially viable products.
Research and development expenses in the year ended December 31, 2009 decreased $5.7 million, or 7%, to $70.9 million, as compared to $76.7 million in the year ended December 31, 2008. This decrease was the net result of decreased spending for QNEXA for obesity of $12.2 million, QNEXA for diabetes of $3.3 million, decreased non-cash share-based compensation expense of $554,000, and a net decrease in other research and development spending of $377,000, partially offset by increased avanafil program spending of $10.7 million in the year ended December 31, 2009, as compared to the year ended December 31, 2008. In the years ended December 31, 2009 and 2008, we incurred $19.1 million and $44.7 million, respectively, for services provided by one clinical research organization on the QNEXA Phase 3 studies, which represented 27% and 58% of our research and development expenses for the period. We incurred another $17 million for services provided by another clinical research organization on the avanafil Phase 3 studies, which represented 24% of our research and development expenses for the year ended December 31, 2009. In the year ended December 31, 2008, we incurred $6.2 million for clinical supplies and formulation work performed by our sole-source manufacturer, which represented 8% of our total research and development expenses.
General and administrative
| |
|
|
|
% Change Increase |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Years Ended December 31, | |||||||||||||||
| |
2010 vs 2009 | 2009 vs 2008 | ||||||||||||||
| |
2010 | 2009 | 2008 | |||||||||||||
| |
(In thousands, except percentages) |
|||||||||||||||
General and administrative |
$ | 25,656 | $ | 13,870 | $ | 12,253 | 85 | % | 13 | % | ||||||
General and administrative expenses in the year ended December 31, 2010 increased $11.8 million, or 85% to $25.7 million as compared to the same period in 2009. In the year ended December 31, 2010, the increase is primarily due to QNEXA pre-commercialization expenses of $9.2 million and net increases of $2.2 million in non-cash share-based compensation expense and $359,000 in other general and administrative expenses as compared to the year ended December 31, 2009.
96
General and administrative expenses in the year ended December 31, 2009 increased $1.6 million, or 13% to $13.9 million as compared to the same period in 2008. In the year ended December 31, 2009, the increase is primarily due to QNEXA pre-commercialization expenses of $1.2 million and net increases of $422,000 in other general and administrative expenses as compared to the year ended December 31, 2008.
On January 19, 2011, we held an End-of-Review meeting with the FDA to discuss its requests in the CRL for QNEXA and our planned resubmission of the NDA for QNEXA. As part of the meeting, the FDA has requested that we perform the Feasibility Assessment, which is currently in process. As a result, we may evaluate our spending on the pre-commercial activities for QNEXA. Ultimately, our general and administrative expenses in 2011 will depend upon the timing and outcome of the FDA's decision on QNEXA, although at this time we do not anticipate an increase in these expenses as compared to 2010.
Interest income and expense
Interest and other income, net for the year ended December 31, 2010 was $468,000 as compared to $2 million for the year ended December 31, 2009. The decrease in interest and other income in the year ended December 31, 2010 as compared to the same period last year was largely due to lower cash balances and lower interest rates, year-over-year, on our cash, cash equivalents and available-for-sale securities balances resulting from the policy to invest primarily in U.S. government securities, which earn lower rates of interest. In 2009, we recognized a net realized gain of $1.1 million related to the sale of securities.
Interest and other income, net for the year ended December 31, 2009 was $2 million as compared to $4.4 million for the year ended December 31, 2008. The decrease in interest income in the year ended December 31, 2009 as compared to the same period last year was largely due to lower interest rates, year-over-year, on our cash, cash equivalents and available-for-sale securities balances resulting from a change in policy to invest primarily in U.S. government securities, which earn lower rates of interest. As mentioned above, in 2009, we recognized a net realized gain of $1.1 million related to the sale of securities.
Interest expense for the year ended December 31, 2010 was $4.3 million as compared to $3.7 million during the same period last year. The net increase in interest expense in the year ended December 31, 2010 as compared to the same period in 2009 is primarily due to the higher Deerfield loan balances in 2010.
Interest expense for the year ended December 31, 2009 was $3.7 million as compared to $659,000 during the same period last year. The net increase in interest expense in the year ended December 31, 2009 as compared to the same period in 2008 is primarily due to increased interest expense on the Deerfield financing.
We recognized an other-than-temporary loss on impaired securities of $0, $654,000 and $7.7 million for the years ended December 31, 2010, 2009 and 2008, respectively. The other-than-temporary loss on impaired available-for-sale securities in the year ended December 31, 2008 included a $2.4 million loss related to our investment in corporate bonds issued by Lehman Brothers Holdings Inc., or Lehman (or their respective subsidiaries, as appropriate). On September 15, 2008, Lehman filed for bankruptcy protection under Chapter 11 of the U.S. Bankruptcy Code. Accordingly, recovery of the full value of our Lehman bonds, if any, was deemed remote and we recognized an other-than-temporary impairment loss in the third quarter of 2008. The $7.7 million other-than-temporary loss primarily represented unrealized impairment losses recorded on securities that were classified as available-for-sale securities on our consolidated balance sheet as of December 31, 2008. The majority of the other-than-temporary losses on impaired securities were
97
recorded on securities obtained in late 2007 through the redemption-in-kind distribution from the Bank of America Columbia Strategic Cash Portfolio.
We recognized a $6 million loss on the early extinguishment of debt in connection with the payoff of the Deerfield loan in the year ended December 31, 2010.
Benefit (provision) for income taxes
In the year ended December 31, 2010, we recorded a provision for income taxes of $9,000. Our income tax return for the year ended December 31, 2007 is currently under examination by the California Franchise Tax Board. Based on the progress of the audit to date, we believe adjustments may be made in early years that will reduce tax attributes available to offset tax due in 2007. Therefore, we increased our unrecognized tax benefits to $7,000 for the year ended December 31, 2010. We recognize interest and penalties accrued on any unrecognized tax benefits as a component of our provision for income taxes. During the year ended December 31, 2010, $1,000 of interest on the unrecognized tax benefits was recorded.
We recorded a benefit for income taxes for the year ended December 31, 2009 of $2.4 million primarily due to our filing a carryback claim of losses generated in 2008, pursuant to the IRS Revenue Procedure 2009-52. The Revenue Procedure was issued in the fourth quarter of 2009 and allows losses generated in 2008 or 2009 to be carried back for up to five years. The provision for income taxes for the year ended December 31, 2008 was $7,000. The provision for income taxes for the year ended December 31, 2008 relates to state income taxes.
Discontinued operations
Revenue—discontinued operations.
| |
|
|
|
% Change Increase/(Decrease) |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Years Ended December 31, | ||||||||||||||||
| |
2010 vs 2009 | 2009 vs 2008 | |||||||||||||||
| |
2010 | 2009 | 2008 | ||||||||||||||
| |
(In thousands, except percentages) |
||||||||||||||||
United States product, net |
$ | 9,626 | $ | 15,836 | $ | 14,974 | (39 | )% | 6 | % | |||||||
International product |
2,425 | 2,347 | 3,076 | 3 | % | (24 | )% | ||||||||||
License and other revenue |
1,260 | 463 | 462 | 172 | % | 0 | % | ||||||||||
Total revenues |
$ | 13,311 | $ | 18,646 | $ | 18,512 | (29 | )% | 1 | % | |||||||
Product revenues for the year ended December 31, 2010 and December 31, 2009 totaled $12.1 million and $18.2 million, respectively. U.S. product revenues in the year ended December 31, 2010 decreased as compared to the same period in 2009. The decrease in U.S. product revenues as well as the increase in license and other revenue as compared to the year ended December 31, 2009 was primarily due to the sale of MUSE on November 5, 2010. In the fourth quarter of 2010, we recognized all of the remaining deferred license revenue from our international distributors.
International product revenues increased in the year ended December 31, 2010, as compared to the year ended December 31, 2009, primarily due to the timing of customer orders and a slight reduction in the international pricing reserve in the year ended December 31, 2010.
Worldwide product revenues from the sales of MUSE were $18.2 million in 2009, an increase of $133,000, or 1%, from the worldwide sales of MUSE in 2008. U.S. product revenues increased in 2009 as compared to the prior year period primarily due to higher prices for MUSE and a modest increase in units shipped.
The decrease in international revenue in 2009 as compared to 2008 was principally due to decreases in units shipped based upon the timing of orders from our international partners and to a
98
reduction in transfer prices resulting from the difference in currency exchange rates. Also contributing to the reduction in international revenues in 2009 was the allowance of $116,000 for certain out-of-specification production identified in the first quarter of 2009.
Cost of goods sold and manufacturing—discontinued operations.
| |
|
|
|
% Change Increase/(Decrease) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Years Ended December 31, | |||||||||||||||
| |
2010 vs 2009 | 2009 vs 2008 | ||||||||||||||
| |
2010 | 2009 | 2008 | |||||||||||||
| |
(In thousands, except percentages) |
|||||||||||||||
Cost of goods sold and manufacturing |
$ | 9,785 | $ | 11,950 | $ | 11,956 | (18 | )% | 0 | % | ||||||
Cost of goods sold and manufacturing, or cost of goods sold, in the year ended December 31, 2010 decreased $2.2 million, or 18%, to $9.8 million, as compared to $12 million in the year ended December 31, 2009. This decrease is primarily due to a decrease in product shipped in the year ended December 31, 2010 as compared to the year ended December 31, 2009 due to the sale of MUSE in the fourth quarter 2010.
Cost of goods sold was $12 million in the years ended December 31, 2009 and 2008. While cost of goods sold remained constant in 2009, there was a nominal reduction in units shipped as compared to 2008. Costs of goods sold for 2009 included an increase in quality control related expenses of $413,000 and an increase in inventory reserves of $487,000 for raw material that exceeded its shelf life, which were partially offset by recoveries from the supplier and insurance proceeds totaling $409,000 for the loss on non-conformance of raw materials that occurred in 2009. In 2008, we incurred inventory disposal costs of $444,000 due to the non-conformance of certain raw materials.
Research and development—discontinued operations.
| |
|
|
|
% Change Increase/(Decrease) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Years Ended December 31, | |||||||||||||||
| |
2010 vs 2009 | 2009 vs 2008 | ||||||||||||||
| |
2010 | 2009 | 2008 | |||||||||||||
| |
(In thousands, except percentages) |
|||||||||||||||
Research and development |
$ | 2,304 | $ | 135 | $ | 323 | 1,607 | % | (58 | )% | ||||||
Research and development expenses increased $2.2 million in the year ended December 31, 2010, or 1,607%, to $2.3 million, as compared to $135,000 for the year ended December 31, 2009. This increase is primarily due to the write-off of $2.2 million in MUSE PDUFA fee refund receivables from the FDA, which were determined to be uncollectible in the year ended December 31, 2010. Research and development expenses decreased $188,000 in the year ended December 31, 2009, or 58%, to $135,000, as compared to $323,000 for the year ended December 31, 2008.
Selling, general and administrative—discontinued operations.
| |
|
|
|
% Change Increase/(Decrease) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Years Ended December 31, | |||||||||||||||
| |
2010 vs 2009 | 2009 vs 2008 | ||||||||||||||
| |
2010 | 2009 | 2008 | |||||||||||||
| |
(In thousands, except percentages) |
|||||||||||||||
Selling, general and administrative |
$ | 5,124 | $ | 7,163 | $ | 6,651 | (28 | )% | 8 | % | ||||||
Selling, general and administrative expenses in the year ended December 31, 2010 decreased $2 million, or 28%, to $5.1 million, as compared to the year ended December 31, 2009. In the year ended December 31, 2010, the decrease is primarily due to the sale of MUSE to Meda on November 5, 2010, which resulted in decreased marketing expenses of $974,000, sales expenses of $828,000 and other net decreases in selling, general and administrative expenses of $238,000 as compared to the year ended December 31, 2009.
99
Selling, general and administrative expenses in the year ended December 31, 2009 increased $512,000, or 8%, to $7.2 million as compared to the year ended December 31, 2008. In the year ended December 31, 2009, the increase is primarily due to incremental increases of $714,000 in sales expenses (including $299,000 in MUSE related wholesaler distribution fees and increased compensation expense of $290,000), partially offset by a $201,000 incremental decrease in marketing expenses as compared to the year ended December 31, 2008.
Interest income and expense—discontinued operations.
Interest and other income, net for the years ended December 31, 2010, 2009 and 2008 was $10,000, $21,000 and $33,000, respectively. Interest income in all periods is a result of fluctuations in investment yields on the Crown Bank restricted cash certificate of deposit.
Interest expense for the years ended December 31, 2010, 2009 and 2008 was $384,000, $386,000 and $405,000, respectively. The net decrease in interest expense from year to year is primarily due to principal paydowns of the outstanding balance on the Crown Bank loan. The Crown Bank loan was paid in full in connection with the sale of MUSE to Meda in the fourth quarter 2010.
Gain on disposal of discontinued operations.
We recognized a $13.7 million gain in the fourth quarter of 2010 due to the sale of MUSE to Meda.
Benefit (provision) for income taxes—discontinued operations.
Provision for income taxes for the year ended December 31, 2010 was $29,000. The provision for income taxes for the year ended December 31, 2010 relates to state income taxes.
We recorded a benefit for income taxes for the year ended December 31, 2009 of $61,000 primarily due to our filing a carryback claim of losses generated in 2008, pursuant to the IRS Revenue Procedure 2009-52. The Revenue Procedure was issued in the fourth quarter of 2009 and allows losses generated in 2008 or 2009 to be carried back for up to five years.
Provision for income taxes for the year ended December 31, 2008 was $10,000. The provision for income taxes for the year ended December 31, 2008 relates to state income taxes.
Liquidity and Capital Resources
Continuing Operations
Cash. Unrestricted cash, cash equivalents and available-for-sale securities totaled $139.2 million at December 31, 2010, as compared to $206.8 million at December 31, 2009. The decrease in cash, cash equivalents and available-for-sale securities of $67.6 million is primarily the result of cash used for operating and financing activities. Included in these amounts is $3.9 million in net proceeds from common stock option exercises and ESPP purchases.
Since inception, we have financed operations primarily from the issuance of equity securities. Through December 31, 2010, we raised $407.6 million from financing activities, received $150 million from the sale of Evamist and had an accumulated deficit of $300.1 million at December 31, 2010.
At December 31, 2010, we had $37.2 million in cash and cash equivalents and $102 million in available-for-sale securities. We invest our excess cash balances in money market and marketable securities, primarily U.S. Treasury securities and debt securities of U.S. government agencies, corporate debt securities and asset-backed securities, in accordance with our investment policy. At December 31, 2010, all of our cash equivalents and available-for-sale securities were invested in either U.S. government securities or money market funds that invest only in U.S. Treasury securities. The
100
investment policy has the primary investment objectives of preservation of principal; however, there may be times when certain of the securities in our portfolio will fall below the credit ratings required in the policy. If those securities are downgraded or impaired, we would experience realized or unrealized losses in the value of our portfolio, which would have an adverse effect on our results of operations, liquidity and financial condition.
Investment securities are exposed to various risks, such as interest rate, market and credit. Due to the level of risk associated with certain investment securities and the level of uncertainty related to changes in the value of investment securities, it is possible that changes in these risk factors in the near term could have an adverse material impact on our results of operations or shareholders' equity.
Liabilities. Total liabilities were $12.3 million at December 31, 2010; $31 million lower than at December 31, 2009. The change in total liabilities includes a $15.3 million decrease in notes payable due to the pay off of the Deerfield loan in the fourth quarter 2010.
Operating Activities. Our operating activities used $68 million, $93.1 million and $64.3 million during the years ended December 31, 2010, 2009 and 2008, respectively. During the year ended December 31, 2010, our net operating loss of $75.4 million was offset by $6.4 million in non-cash share-based compensation expense, $6 million in loss on early extinguishment of the Deerfield loan, $138,000 in depreciation expense, a $2.7 million decrease in prepaid expenses and other assets, a $521,000 increase in accrued and other liabilities and a $477,000 increase in accrued employee compensation and benefits. These positive cash flows to our net operating loss were in turn offset by a $3.2 million increase in inventories due to the purchase of QNEXA raw material inventory and a $5.7 million decrease in accounts payable.
During the year ended December 31, 2009, our net operating loss of $53.4 million was offset by $3.9 million in non-cash share-based compensation expense, a $654,000 other-than-temporary loss on impaired securities, a $737,000 increase in accrued employee compensation and benefits and a $1.3 million increase in accrued and other liabilities, primarily due to an increase in accrued interest payable on the Deerfield loan. These positive cash flows to our net operating loss were in turn offset by the recognition of $31.4 million in revenue due to the amortization of deferred license revenue from the receipt of $150 million from K-V for the sale of Evamist, a $7.3 million decrease in accounts payable due to the timing of payments, a $2.6 million increase in prepaid expenses (primarily due to a $2.3 million receivable for continuing operations from the IRS related to carryback claims on our amended 2007 federal income tax return), a $1.1 million net realized gain on other-than temporarily-impaired securities and a $4 million decrease in accrued research and clinical expenses, primarily due to the winding down of the QNEXA for obesity development efforts.
During the year ended December 31, 2008, our net operating loss of $9.1 million was offset by a $7.7 million other-than-temporary loss on impaired securities, $3.9 million in non-cash share based compensation expense, a $5 million increase in accrued research and clinical expenses primarily due to the QNEXA for obesity development effort, a $8.2 million increase in accounts payable due to the timing of payments, a $2.1 million decrease in prepaid and other assets, a $553,000 increase in accrued employee compensation and benefits and a $979,000 loss on investments. These positive cash flows to our net operating loss were in turn offset by the recognition of $83.7 million in revenue due to the amortization of deferred license revenue from the receipt of $150 million from K-V for the sale of Evamist.
Investing Activities. Our investing activities provided $64.9 million, used $43.3 million and provided $10.1 million in cash during the years ended December 31, 2010, 2009 and 2008, respectively. The fluctuations from period to period are due primarily to the timing of purchases, sales and maturity of investment securities.
101
Financing Activities. Financing activities used cash of $19.1 million and provided cash of $114.3 million and $82.1 million during the years ended December 31, 2010, 2009 and 2008, respectively. In 2010, cash used in financing activities included $23 million to payoff the Deerfield loan (which includes a $6 million loss on early extinguishment of the loan). This amount was partially offset by $3.6 million in net proceeds from the exercise of stock options and $304,000 from ESPP purchases. In 2009, the cash provided by financing activities included $102.7 million in net proceeds from the underwritten public offering of our common stock, $10 million in cash from the Deerfield financing and $2.7 million from stock option exercises and ESPP purchases, partially offset by $1 million in principal payments under our notes payable. In 2008, the cash provided by financing activities included cash receipts from the Deerfield financing including $9.7 million in net proceeds from the issuance of common stock and $7.6 million from the Deerfield FARA, net proceeds of $63.7 million from the registered direct offering of our common stock, $2.2 million in proceeds from the exercise of stock options and $275,000 from the sale of common stock through our ESPP partially offset by $1.3 million in principal payments under our notes payable.
Discontinued Operations
Accounts receivable (net of allowance for doubtful accounts) at December 31, 2010 was $6,000, as compared to $7.3 million at December 31, 2009. As of February 18, 2011, we had collected 100% of the accounts receivable outstanding at December 31, 2010.
Cash provided by operating activities in the year ended December 31, 2010 was $2.2 million. In the year ended December 31, 2010, the $9.4 million net income was offset by a $13.7 million gain on the sale of MUSE. These offsets to our net operating income were in turn offset by positive cash flows including a $7.3 million decrease in accounts receivable (due to the collection of the December 2009 outstanding receivables), $915,000 in depreciation expense and $970,000 in non-cash share-based compensation expense.
Cash used for operating activities in the year ended December 31, 2009 was $3 million. In the year ended December 31, 2009, the $906,000 net loss was partially offset by $1.1 million in depreciation expense, $864,000 in non-cash share-based compensation expense, and a $487,000 provision for obsolete inventory due to alprostadil that has exceeded its shelf life. These positive cash flows to our net operating loss were in turn offset by a $3.1 million increase in accounts receivable (due to timing of the December 2009 buy-in as compared to the prior year) and a $1.4 million decrease in accounts payable, primarily due to the timing of payments.
Cash provided by operating activities in the year ended December 31, 2008 was $767,000. Included in this amount is a $1.2 million increase in accounts payable, primarily due to the timing of payments, $1.1 million in depreciation expense and $769,000 in non-cash share-based compensation expense.
Our investing activities provided $21.5 million and used $171,000 and $264,000 in cash during the years ended December 31, 2010, 2009 and 2008, respectively. In the year ended December 31, 2010, cash provided by investing activities included $21.6 million in cash proceeds from the sale of MUSE to Meda. Also included in these amounts in each year are purchases of property and equipment. Financing activities used $4.9 million, $145,000 and $130,000 during the years ended December 31, 2010, 2009 and 2008, respectively. These amounts represent principal payments made under our Crown Bank loan in all periods. In the year ended December 31, 2010, we paid off the loan to Crown Bank.
On December 22, 2005, we purchased from our landlord our principal manufacturing facility, which was previously leased, for $7.1 million. The purchase price was funded in part by $3.3 million, which was being held by the landlord as cash collateral for renovations to the facility upon the termination of the lease and the remainder with cash. On January 4, 2006, we obtained a $5.4 million loan from Crown Bank, N.A., or Crown. The land and buildings, among other assets, located at our principal manufacturing facility and a $700,000 Certificate of Deposit held by Crown served as
102
collateral for the Crown loan. The loan was payable over a 10-year term. The interest rate was adjusted annually to a fixed rate for the year equal to the prime rate plus 1%, with a floor of 7.5%. Principal and interest were payable monthly based upon a 20-year amortization schedule and were adjusted annually at the time of the interest rate reset. All remaining principal was due on February 1, 2016. The interest rate was 7.5% for 2010 and 2009. At December 31, 2009 and 2008, the manufacturing facility and restricted cash are reported as assets of discontinued operations on the consolidated balance sheets. On October 15, 2010, in preparation for the closing of the MUSE Transaction and in accordance with the terms of the agreements with Crown, we paid $4.8 million to Crown in satisfaction of all obligations owed to them under these agreements. As a result, the security interests and Certificate of Deposit held by Crown were terminated in our favor.
On October 1, 2010, we entered into a definitive Asset Purchase Agreement with Meda, to sell certain rights and assets related to MUSE, transurethral alprostadil, for the treatment of erectile dysfunction, or the MUSE Transaction. Meda has been our European distributor of MUSE since 2002. The assets sold in the MUSE Transaction include the U.S. and foreign MUSE patents, existing inventory, and the manufacturing facility located in Lakewood, New Jersey. We retained all of the liabilities associated with the pre-closing operations and products of the MUSE business and the accounts receivables for pre-closing MUSE sales. The transaction closed on November 5, 2010. Prior to the closing of the MUSE Transaction we regained all of the rights to MUSE and avanafil held by Deerfield Management Company, L.P., and affiliates, and Crown Bank, N.A.
Under the terms of the MUSE Transaction, we received an upfront payment of $22 million upon the closing and are eligible to receive an additional $1.5 million based on future sales of MUSE, provided that certain sales milestones are reached. Meda is now responsible for the manufacturing, selling and marketing of MUSE. Meda also assumed all post-closing expenses and liabilities associated with MUSE. We have agreed not to develop, manufacture or sell any transurethral erectile dysfunction drugs for a period of three years following the closing of the MUSE Transaction. The assets and liabilities and results of operations associated with MUSE have been reported as discontinued operations for all periods presented.
On April 3, 2008, we entered into several agreements with Deerfield Management Company, L.P., or Deerfield, a healthcare investment fund, and its affiliates, Deerfield Private Design Fund L.P. and Deerfield Private Design International, L.P. (collectively, the Deerfield Affiliates). Certain of the agreements were amended and restated on March 16, 2009, which included the addition of Deerfield PDI Financing L.P. as a Deerfield Affiliate. Under the agreements, Deerfield and its affiliates provided us with $30 million in funding, consisting of $20 million from the Funding and Royalty Agreement, or FARA, entered into with a newly incorporated subsidiary of Deerfield, or the Deerfield Sub, and $10 million from the sale of our common stock. We received all of the required payments under the FARA, or the Funding Payments. Under the FARA, we paid royalties on the net sales of MUSE to the Deerfield Sub. The FARA had a term of 10 years.
We entered into the Option and Put Agreement with the Deerfield Affiliates and the Deerfield Sub, dated April 3, 2008, and an Amended and Restated Option and Put Agreement dated March 16, 2009, or the OPA. Pursuant to the OPA, the Deerfield Affiliates granted us an option to purchase all of the outstanding shares of common stock of the Deerfield Sub from the Deerfield Affiliates, referred to as the Option. Our obligation to pay royalties terminated upon the exercise of the Option. The base consideration for the Option exercise, or Base Option Price, was $25 million, less $2 million we paid upon closing, as the Option was exercised on or prior to the third anniversary of the execution of the OPA. The aggregate consideration payable by us upon exercise of the Option, or the Option Purchase Price, was equal to the sum of the Base Option Price, plus: (i) the cash and cash equivalents held by the Deerfield Sub at the date of the closing of the resulting sale of the common stock of the Deerfield Sub, or the Cash Adjustment; (ii) accrued and unpaid royalties, or the Royalty Adjustment; and minus (i) the Option premium of $2 million that was paid at the closing of the transaction, or the Option
103
Premium Adjustment ; (ii) accrued but unpaid taxes; (iii) unpaid Funding Payments; (iv) loans payable by the Deerfield Sub, or the Loan Balance Adjustment; and (v) any other outstanding liabilities of the Deerfield Sub, or the Adjusted Option Purchase Price.
On October 21, 2010, in preparation for the closing of the MUSE transaction, we exercised the Option under the Deerfield OPA, and we paid an aggregate amount totaling $27.1 million, which consisted of the Base Option Price of $25 million, less the Option Premium Adjustment of $2 million, plus the addition of the Cash Adjustment of $2.8 million and the Royalty Adjustment of $1.3 million. These payments satisfied all of the financial obligations under the FARA and OPA. As a result, all of the outstanding shares and the $2.8 million of cash of the Deerfield Sub are owned by us, all of the outstanding loans owed by the Deerfield Sub have been repaid and the security interests in the collateral related to MUSE and avanafil held by the Deerfield Sub and the Deerfield Affiliates as part of the FARA and OPA were terminated. In December 2010, the Deerfield Sub was dissolved.
On August 6, 2008, we sold $65 million of our common stock in a registered direct offering. Under the terms of the financing, we sold 8,365,508 shares of our common stock at a price of $7.77 per share. On August 5, 2008, the Company filed a prospectus supplement with the SEC relating to this registered direct offering under the existing shelf Registration Statement (File Number 333-150649).
On September 17, 2009, we entered into an underwriting agreement, or the Underwriting Agreement, with J.P. Morgan Securities Inc., as representative of the several underwriters named therein, or the Underwriters, relating to the public offering and sale of 9,000,000 shares of our common stock. Pursuant to the Underwriting Agreement, the Underwriters agreed to purchase, subject to customary closing conditions, 9,000,000 shares of our common stock. We also granted the Underwriters a 30-day option to purchase up to 1,350,000 additional shares of common stock on the same terms and conditions as set forth above to cover over-allotments, which the Underwriters exercised in full. The 10,350,000 shares were sold at a price to the public of $10.50 per share which resulted in approximately $108.7 million in gross proceeds before deducting underwriting discounts and commissions and other offering expenses. The transaction closed on September 23, 2009. The offering was made pursuant to the effective shelf registration statement on Form S-3 (Registration No. 333-161948), including the prospectus dated September 16, 2009 contained therein, as supplemented.
On February 16, 2010, we filed a Form S-8 (File Number 333-164921) with the SEC registering 1,000,000 shares of common stock, par value $0.001 per share, under the 2001 Stock Option Plan, as amended.
On July 14, 2010, we filed a Form S-8 (File Number 333-168106) with the SEC registering 16,615,199 shares of common stock, par value $0.001 per share, to be issued pursuant to the 2010 Equity Incentive Plan, and registering 400,000 shares of common stock, par value $0.001 per share, to be issued pursuant to the Stand-Alone Stock Option Agreement with Michael P. Miller.
The funding necessary to execute our business strategies is subject to numerous uncertainties, which may adversely affect our liquidity and capital resources. Completion of clinical trials and approval by the FDA may take several years or more, but the length of time generally varies substantially according to the type, complexity, novelty and intended use of an investigational drug candidate. It is also important to note that if an investigational drug candidate is identified, the further development of that candidate can be halted or abandoned at any time due to a number of factors. These factors include, but are not limited to, funding constraints, lack of efficacy or safety or change in market demand.
The nature and efforts required to develop our investigational drug candidates into commercially viable drugs include research to identify a clinical candidate, pre-clinical development, clinical testing, FDA approval and commercialization. This process is very costly and can take in excess of 10 years to complete for each investigational drug candidate. The duration and the cost of clinical trials may vary
104
significantly over the life of a project as a result of matters arising during the clinical studies, including, among others, the following:
- •
- we or the FDA may suspend trials;
- •
- we may discover that an investigational drug candidate may cause harmful side effects or is not effective;
- •
- patient recruitment may be slower than expected; and
- •
- patients may drop out of the trials.
For each of our investigational drug candidate development programs, we periodically assess the scientific progress and the merits of the programs to determine if continued research and development is economically viable. Certain of our programs were terminated due to the lack of scientific progress and lack of prospects for ultimate commercialization. As such, the ultimate timeline and costs to commercialize a drug cannot be accurately estimated.
Our investigational drug candidates have not yet achieved FDA regulatory approval, which is required before we can market them as therapeutic products. In order to achieve regulatory approval, the FDA must conclude that our clinical data establish substantial evidence of safety and efficacy. The results from pre-clinical testing and early clinical trials may not be predictive of results in later clinical trials. It is possible for a candidate to show promising results in early clinical trials, but subsequently fail to demonstrate safety and efficacy data necessary to obtain regulatory approvals.
As a result of the uncertainties discussed above, among others, the duration and completion of our investigational drug candidate development programs are difficult to estimate and are subject to considerable variation. Our inability to complete our research and investigational drug candidate development programs in a timely manner or our failure to enter into collaborative agreements, when appropriate, could significantly increase our capital requirements and could adversely impact our liquidity. These uncertainties could force us to seek additional, external sources of financing from time to time in order to continue with our business strategy. Our inability to raise capital, or to do so on terms reasonably acceptable to us, would jeopardize the future success of our business.
We may also be required to make further substantial expenditures if unforeseen difficulties arise in other areas of our business. In particular, our future capital and additional funding requirements will depend upon or be impacted by numerous factors, including:
- •
- the timing and substance of our response to the FDA's request to perform a feasibility study on a retrospective
observational study of congenital malformations with an interest in oral cleft and low birth weight in infants born to women who received prophylaxis treatment with 100 mg of topiramate for migraine
during pregnancy, and if deemed feasible the cost, timing and outcome of such study and the other items included in the complete response letter;
- •
- the FDA's interpretation of the data we submit relating to cardiovascular safety;
- •
- the FDA's interpretation of the data from our SEQUEL study (OB-305) and Sleep Apnea (OB-204);
- •
- whether or not the FDA requires us to perform additional clinical studies for QNEXA;
- •
- the cost and time required to set up a distribution system and REMS program for QNEXA that is suitable to address any FDA
concerns;
- •
- the progress and costs of our research and development programs;
- •
- the scope, timing and results of pre-clinical, clinical and retrospective observational studies and trials;
105
- •
- the cost of access to electronic records and databases that allow for retrospective observational studies;
- •
- patient recruitment and enrollment in planned and future clinical trials;
- •
- the costs involved in seeking regulatory approvals for our investigational drug candidates;
- •
- the costs involved in filing and pursuing patent applications and enforcing patent claims;
- •
- the establishment of collaborations, sublicenses and strategic alliances and the related costs, including milestone
payments;
- •
- the costs involved in establishing a commercial operation and in launching a product without a partner;
- •
- the cost of manufacturing and commercialization activities and arrangements;
- •
- the results of operations;
- •
- the cost, timing and outcome of regulatory reviews;
- •
- the rate of technological advances;
- •
- ongoing determinations of the potential commercial success of our investigational drug candidates under development;
- •
- the state of the economy and financing environment;
- •
- the regulatory approval environment and regulatory hurdles for safety assessment for new products;
- •
- the cost, timing and outcome of litigations;
- •
- the healthcare reimbursement system or the impact of healthcare reform, if any, imposed by the U.S. federal government;
- •
- the level of resources devoted to sales and marketing capabilities;
- •
- perceptions and interpretations of QNEXA or the data by outside analysts or others; and
- •
- the activities of competitors.
We anticipate that our existing capital resources combined with anticipated future cash flows will be sufficient to support our operating needs at least into 2012. However, we anticipate that we may require additional funding to continue our research and investigational drug candidate development programs, to conduct pre-clinical studies and trials, to fund operating expenses, to pursue regulatory approvals for our investigational drug candidates, to finance the costs involved in filing and prosecuting patent applications and enforcing or defending our patent claims, if any, and we may require additional funding to establish additional or new manufacturing and marketing capabilities in the future, to manufacture quantities of our investigational drug candidates for approval and commercialization, or to launch a product. In particular, we expect to make other substantial payments to the inventor of QNEXA pending approval by the FDA and to MTPC, in accordance with our agreements with MTPC in connection with the licensing of avanafil. These payments are based on certain development, regulatory and sales milestones. In addition, we are required to make royalty payments on any future product sales. Similar to the transaction with Evamist, we may consider selling or licensing any of our investigational drug candidates in development in order to raise additional funding. We may seek to access the public or private equity markets at any time. The sale of additional equity securities would result in additional dilution to our stockholders. We may also seek additional funding through strategic alliances, acquisitions of companies with cash balances and other financing mechanisms. We cannot assure you that adequate funding will be available on terms acceptable to us, if at all. If adequate funds are not available, we may be required to curtail significantly one or more of our investigational drug
106
candidate development programs or obtain funds through arrangements with collaborators or others. This may require us to relinquish rights to certain of our technologies or investigational drug candidates and to pay royalties on future product sales. To the extent that we are unable to obtain third party funding for such expenses, we expect that increased expenses may result in future losses from operations. We are continually evaluating our existing portfolio and we may choose to divest or sell one or more of our investigational drug candidates at any time. We cannot assure you that we will successfully develop our products under development or that our products, if approved for sale, will generate revenues sufficient to enable us to earn a profit.
Contractual Obligations
The following table summarizes our contractual obligations at December 31, 2010 excluding amounts already recorded on our consolidated balance sheet as accounts payable, and the effect such obligations are expected to have on our liquidity and cash flow in future fiscal years. This table includes our enforceable and legally binding obligations and future commitments, as well as obligations related to all contracts that we are likely to continue, regardless of the fact that they were cancelable as of December 31, 2010. These do not include milestones and assumes non-termination of agreements. These obligations, commitments and supporting arrangements represent payments based on current operating plans, which are subject to change:
| |
Payments Due by Period | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Contractual obligations
|
Total | 2011 | 2012-2014 | 2015-2016 | Thereafter | |||||||||||
| |
(in thousands) |
|||||||||||||||
Continuing Operations: |
||||||||||||||||
Operating leases |
$ | 1,091 | $ | 685 | $ | 406 | $ | — | $ | — | ||||||
Other agreements |
6,605 | 5,085 | 1,520 | — | — | |||||||||||
Clinical trials |
6,699 | 6,699 | — | — | — | |||||||||||
Total contractual obligations |
$ | 14,395 | $ | 12,469 | $ | 1,926 | $ | — | $ | — | ||||||
Continuing Operations:
Operating Leases
In November 2006, we entered into a 30-month lease for our corporate headquarters located in Mountain View, California. The lease commenced on February 1, 2007. The base monthly rent was set at $1.85 per square foot, or $26,000 per month. The lease expired on July 31, 2009. On December 16, 2008, we entered into a first amendment to this lease. Under the terms of the amended lease, we continue to lease the office space for our corporate headquarters for a two-year period commencing on August 1, 2009 and expiring on July 31, 2011. The base monthly rent was set at $1.64 per square foot, or $23,000 per month. The amended lease allowed us one option to extend the term of the lease for one year from the expiration of the lease. On November 12, 2009, we entered into a second amendment to this lease. The second amendment commenced on January 1, 2010, expires on July 31, 2011 and expands the leased space. The base rent for the expansion space was set at $2.25 per square foot, or $8,500 per month. The option to extend the term of the amended lease for one year from the expiration of the lease applies to this expansion space as well. In December 2010, we entered into a third amendment to this lease. The third amendment extended the lease term for the original premises and the expansion space for a period of twelve months commencing August 1, 2011 and terminating July 31, 2012. Under the third amendment, the base rent for the original space will be set at $1.69 per square foot, or $24,000 per month and the base rent for the expansion space will be set at $2.31 per square foot, or $8,700 per month. The amended lease allows us one additional option to extend the term of the lease for one year from the expiration of the lease. The option to extend the term of the amended lease for one year from the expiration of the lease applies to the expansion space as well.
107
Other Agreements
Purchase obligations consist of agreements to purchase goods or services that are enforceable and legally binding on us and that specify all significant terms, including: fixed or minimum quantities to be purchased; fixed, minimum or variable price provisions; and the approximate timing of the transaction. These include obligations for research and development, general and administrative services, and media/market research contracts, as well as obligations related to those contracts that we are likely to continue, regardless of the fact that they were cancelable as of December 31, 2010.
We have remaining commitments under various general and administrative services agreements totaling $3 million at December 31, 2010, including $1.5 million related to Leland F. Wilson's Employment Agreement (see paragraph below). We have also entered into various agreements with research consultants and other contractors to perform regulatory services, drug research and testing and, at December 31 2010, our remaining commitment under these agreements totaled $3.6 million.
On December 19, 2007, the Compensation Committee of the Board of Directors of the Company approved an employment agreement, or the Employment Agreement, with Leland F. Wilson, the Company's Chief Executive Officer. The Employment Agreement includes salary, incentive compensation, retirement benefits and length of employment, among other items, as agreed to with Mr. Wilson. The Employment Agreement had an initial term of two years commencing on the effective date, June 1, 2007, or the Effective Date. On January 23, 2009, the Compensation Committee approved an amendment to the Employment Agreement, or the Amendment, which amends the Employment Agreement. Pursuant to the Amendment, the initial term of the Employment Agreement was increased from two to three years commencing on June 1, 2007 and other relevant dates were also extended to reflect the three-year initial term. On January 21, 2011, the Compensation Committee approved the second amendment to Mr. Wilson's Employment Agreement. Pursuant to the second amendment, the initial term of the Employment Agreement is increased to four years commencing on June 1, 2007.
Clinical Trials
We have entered into various agreements with clinical consultants, investigators, clinical suppliers and clinical research organizations to perform clinical trial management and clinical studies on our behalf and, at December 31, 2010, our remaining commitment under these agreements totaled $6.7 million, which includes nearly all of the accrued research and clinical expenses of $2.6 million in the consolidated balance sheet as of December 31, 2010. We make payments to these providers based upon the number of patients enrolled and the length of their participation in the trials. These obligations, however, are contingent on future events, e.g. the rate of patient accrual in our clinical trials. This amount represents the remaining contractual amounts due under various contracts, although all of these contracts could be cancelled by us, in which case we would only be liable to the vendors for work performed to the date of cancellation.
Additional Payments
We have entered into development, license and supply agreements that contain provisions for payments upon completion of certain development, regulatory and sales milestones. Due to the uncertainty concerning when and if these milestones may be completed or other payments are due, we have not included these potential future obligations in the above table.
Mitsubishi Tanabe Pharma Corporation
In January 2001, we entered into an exclusive development, license and supply agreement with Tanabe Seiyaku Co., Ltd., or Tanabe, now Mitsubishi Tanabe Pharma Corporation, or MTPC, and hereinafter collectively referred to as MTPC, for the development and commercialization of avanafil, a PDE5 inhibitor compound for the oral and local treatment of male and female sexual dysfunction.
108
Under the terms of the agreement, MTPC agreed to grant an exclusive license to us for products containing avanafil outside of Japan, North Korea, South Korea, China, Taiwan, Singapore, Indonesia, Malaysia, Thailand, Vietnam and the Philippines. We agreed to grant MTPC an exclusive, royalty-free license within those countries for oral products that we develop containing avanafil. In addition, we agreed to grant MTPC an exclusive option to obtain an exclusive, royalty-bearing license within those countries for non-oral products that we develop containing avanafil. MTPC agreed to manufacture and supply us with avanafil for use in clinical trials, which will be our primary responsibility.
We have paid upfront licensing fees of $5 million to MTPC and have agreed to make additional payments upon the completion of certain development, regulatory and sales milestones. During the first quarter of 2004, we initiated a Phase 2 clinical trial with avanafil, which triggered one of the clinical development milestone criteria noted above. In 2006, we paid MTPC $2 million in connection with this milestone. We have further agreed to pay royalties on net sales of products containing avanafil. No payments were made under this agreement with MTPC in the years ended December 31, 2007 and 2008; however, we paid MTPC $4 million in January 2009 following the enrollment in December 2008 of the first patient in the first Phase 3 clinical studies. We expect to make other substantial payments to MTPC in accordance with our agreements with MTPC as we continue to develop and, if approved for sale, commercialize avanafil for the oral treatment of male sexual dysfunction. Such potential future milestone payments total $15 million in the aggregate and include payments upon: the first submission of an NDA, expected in the second quarter of 2011; obtainment of the first regulatory approval in the U.S. and any major European country; and achievement of $250 million or more in calendar year sales.
The term of the MTPC agreement is based on a country-by-country and on a product-by-product basis. The term shall continue until the later of (i) 10 years after the date of the first sale for a particular product, or (ii) the expiration of the last-to-expire patents within the MTPC patents covering such product in such country. In the event that our product is deemed to be (i) insufficiently effective or insufficiently safe relative to other PDE5 inhibitor compounds based on published information, or (ii) not economically feasible to develop due to unforeseen regulatory hurdles or costs as measured by standards common in the pharmaceutical industry for this type of product, we have the right to terminate the agreement with MTPC with respect to such product.
Other
On October 16, 2001, we entered into an assignment agreement, or the Assignment Agreement, with Thomas Najarian, M.D. for a combination of pharmaceutical agents for the treatment of obesity and other disorders, or the Combination Therapy, that has since been the focus of our investigational drug candidate development program for QNEXA for the treatment of obesity, obstructive sleep apnea and diabetes. The Combination Therapy and all related patent applications, or the Patents, were transferred to us with worldwide rights to develop and commercialize the Combination Therapy and exploit the Patents. Pursuant to the Assignment Agreement, we have paid a total of $220,000 to Dr. Najarian through December 31, 2010 and have issued him options to purchase 40,000 shares of our common stock. We are obligated under the terms of the Assignment Agreement to make a milestone payment of $1 million and issue an option to purchase 20,000 shares of our common stock to Dr. Najarian upon marketing approval by the FDA of a product for the treatment of obesity that is based upon the Combination Therapy and Patents. The Assignment Agreement will require us to pay royalties on worldwide net sales of a product for the treatment of obesity that is based upon the Combination Therapy and Patents until the last-to-expire of the assigned Patents. To the extent that we decide not to commercially exploit the Patents, the Assignment Agreement will terminate and the Combination Therapy and Patents will be assigned back to Dr. Najarian. In 2006, Dr. Najarian joined us as a part-time employee and currently serves as a Principal Scientist.
109
Off-Balance Sheet Arrangements
We have not entered into any off-balance sheet financing arrangements and have not established any special purpose entities. We have not guaranteed any debt or commitments of other entities or entered into any options on non-financial assets.
Indemnifications
In the normal course of business, the Company provides indemnifications of varying scope to customers against claims of intellectual property infringement made by third parties arising from the use of its products and to its clinical research organizations and investigators sites against liabilities incurred in connection with any third-party claim arising from the work performed on behalf of the Company, among others. Historically, costs related to these indemnification provisions have not been significant and we are unable to estimate the maximum potential impact of these indemnification provisions on our future results of operations.
Pursuant to the terms of the Asset Purchase Agreement for the sale of the Evamist product to K-V, the Company made certain representations and warranties concerning its rights and assets related to Evamist and the Company's authority to enter into and consummate the transaction. The Company also made certain covenants that survive the closing date of the transaction, including a covenant not to operate a business that competes, in the U.S., and its territories and protectorates, with the Evamist product.
Pursuant to the terms of the Asset Purchase Agreement the Company entered into with Meda AB, or Meda, to sell certain of the assets related to the MUSE business to Meda, or the MUSE Transaction, the Company agreed to indemnify Meda in connection with the representations and warranties that it made concerning its rights, liabilities and assets related to the MUSE business and its authority to enter into and consummate the MUSE Transaction. The Company also made certain covenants in the Asset Purchase Agreement which survive the closing of the MUSE Transaction, including a three year covenant not to develop, manufacture, promote or commercialize a trans-urethral erectile dysfunction drug.
To the extent permitted under Delaware law, we have agreements whereby we indemnify our officers and directors for certain events or occurrences while the officer or director is, or was, serving at our request in such capacity. The indemnification period covers all pertinent events and occurrences during the officer's or director's lifetime. The maximum potential amount of future payments we could be required to make under these indemnification agreements is unlimited; however, we have director and officer insurance coverage that reduces our exposure and enables us to recover a portion of any future amounts paid. We believe the estimated fair value of these indemnification agreements in excess of applicable insurance coverage is minimal.
Recent Accounting Pronouncements
In December 2010, the FASB issued ASU 2010-27, Fees Paid to the Federal Government by Pharmaceutical Manufacturers, (amendments to FASB ASC Topic 720, Other Expenses. The objective of this Update is to address questions concerning how pharmaceutical manufacturers should recognize and classify in their income statements fees mandated by the Patient Protection and Affordable Care Act as amended by the Health Care and Education Reconciliation Act (the Acts). The amendments in this Update are effective for calendar years beginning after December 31, 2010, when the fee initially becomes effective. The adoption of this statement effective January 1, 2011 did not have a material effect on our consolidated financial statements.
In January 2010, the FASB issued ASU 2010-06, Fair Value Measurements Disclosures, which amends Subtopic 820-10 of the FASB Accounting Standards Codification to require new disclosures for
110
fair value measurements and provides clarification for existing disclosures requirements. More specifically, this update will require (a) an entity to disclose separately the amounts of significant transfers in and out of Levels 1 and 2 fair value measurements and to describe the reasons for the transfers; and (b) information about purchases, sales, issuances and settlements to be presented separately (i.e. present the activity on a gross basis rather than net) in the reconciliation for fair value measurements using significant unobservable inputs (Level 3 inputs). This update clarifies existing disclosure requirements for the level of disaggregation used for classes of assets and liabilities measured at fair value and requires disclosures about the valuation techniques and inputs used to measure fair value for both recurring and nonrecurring fair value measurements using Level 2 and Level 3 inputs. The adoption of this statement effective January 1, 2010 did not materially expand our consolidated financial statement footnote disclosures.
In October 2009, the FASB issued ASU 2009-13, Multiple-Deliverable Revenue Arrangements, (amendments to FASB ASC Topic 605, Revenue Recognition), or ASU 2009-13, and ASU 2009-14, Certain Arrangements That Include Software Elements, (amendments to FASB ASC Topic 985, Software), or ASU 2009-14. ASU 2009-13 requires entities to allocate revenue in an arrangement using estimated selling prices of the delivered goods and services based on a selling price hierarchy. The amendments eliminate the residual method of revenue allocation and require revenue to be allocated using the relative selling price method. ASU 2009-14 removes tangible products from the scope of software revenue guidance and provides guidance on determining whether software deliverables in an arrangement that includes a tangible product are covered by the scope of the software revenue guidance. ASU 2009-13 and ASU 2009-14 should be applied on a prospective basis for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010, with early adoption permitted. The adoption of these statements effective January 1, 2011 did not have a material effect on our consolidated financial statements.
In June 2009, the FASB issued SFAS No. 167, Amendments to FASB Interpretation No. 46(R), as codified in ASC 810. This statement amends the consolidation guidance applicable to variable interest entities and the definition of a variable interest entity, and requires enhanced disclosures to provide more information about an enterprise's involvement in a variable interest entity. This statement also requires ongoing assessments of whether an enterprise is the primary beneficiary of a variable interest entity. This statement is effective for our fiscal year beginning January 1, 2010. The adoption of this statement did not have a material effect on our consolidated financial statements.
Dividend Policy
We have not paid any dividends since our inception and do not intend to declare or pay any dividends on our common stock in the foreseeable future. Declaration or payment of future dividends, if any, will be at the discretion of our Board of Directors after taking into account various factors, including our financial condition, operating results and current and anticipated cash needs.
Cautionary Note on Forward-Looking Statements
Our business is subject to significant risks, including but not limited to, the risks inherent in our research and development activities, including the successful completion of clinical trials, the lengthy, expensive and uncertain process of seeking regulatory approvals, uncertainties associated both with the potential infringement of patents and other intellectual property rights of third parties, and with obtaining and enforcing our own patents and patent rights, uncertainties regarding government reforms and of product pricing and reimbursement levels, technological change, competition, manufacturing uncertainties and dependence on third parties. Even if our investigational drug candidates appear promising at an early stage of development, they may not reach the market for numerous reasons. Such reasons include the possibilities that the drug will be ineffective or unsafe during clinical trials, will fail to receive necessary regulatory approvals, will be difficult to manufacture on a large scale, will be
111
uneconomical to market or will be precluded from commercialization by proprietary rights of third parties. For more information about the risks we face, see "Item 1.A. Risk Factors" included in this report.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
The Securities and Exchange Commission's rule related to market risk disclosure requires that we describe and quantify our potential losses from market risk sensitive instruments attributable to reasonably possible market changes. Market risk sensitive instruments include all financial or commodity instruments and other financial instruments that are sensitive to future changes in interest rates, currency exchange rates, commodity prices or other market factors.
Market and Interest Rate Risk
Our cash, cash equivalents and available-for-sale securities as of December 31, 2010 consisted primarily of money market funds and U.S. Treasury securities. Our cash is invested in accordance with an investment policy approved by our Board of Directors that specifies the categories (money market funds, U.S. Treasury securities and debt securities of U.S. government agencies, corporate bonds, asset-backed securities, and other securities), allocations, and ratings of securities we may consider for investment. Currently, we have focused on investing in U.S. Treasuries until market conditions improve.
Our primary exposure to market risk is interest income sensitivity, which is affected by changes in the general level of U.S. interest rates, particularly because the majority of our investments are in short-term marketable debt securities. The primary objective of our investment activities is to preserve principal. Some of the securities that we invest in may be subject to market risk. This means that a change in prevailing interest rates may cause the value of the investment to fluctuate. For example, if we purchase a security that was issued with a fixed interest rate and the prevailing interest rate later rises, the value of our investment may decline. A hypothetical 100 basis point increase in interest rates would reduce the fair value of our available-for-sale securities at December 31, 2010 by approximately $343,000. In general, money market funds are not subject to market risk because the interest paid on such funds fluctuates with the prevailing interest rate.
There is ongoing concern in the credit markets regarding the value of a variety of mortgage-backed and auction rate securities and the resultant effect on various securities markets. In addition, continuing concerns over inflation, energy costs, geopolitical issues, the availability and cost of credit, the U.S. mortgage market and a declining residential real estate market in the U.S. have contributed to increased volatility and diminished expectations for the economy and the markets going forward. These factors combined with volatile oil prices, declining business and consumer confidence and increased unemployment, have precipitated an economic recession and fears of a possible depression. Domestic and international equity markets continue to experience heightened volatility and turmoil. These events and the continuing market upheavals may have an adverse effect on us. In the event of a continuing market downturn, our results of operations could be adversely affected by those factors in many ways including making it more difficult for us to raise funds if necessary and may cause stock price volatility. Our investment policy, as approved by our Board of Directors, allows us to invest in cash, cash equivalents and available-for-sale securities that are not federally insured. Given the current economic instability, we cannot provide assurance that we will not experience losses on these investments.
112
Item 8. Financial Statements and Supplementary Data
1. Index to Consolidated Financial Statements
The following financial statements are filed as part of this Report:
113
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The
Board of Directors and Stockholders of
VIVUS, Inc.
We have audited the accompanying consolidated balance sheets of VIVUS, Inc. as of December 31, 2010 and 2009, and the related consolidated statements of operations, stockholders' equity and cash flows for each of the three years in the period ended December 31, 2010. Our audits also included the financial statement schedule listed in Item 15(a)(2). These financial statements and schedule are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements audited by us present fairly, in all material respects, the consolidated financial position of VIVUS, Inc. at December 31, 2010 and 2009, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2010, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly in all material respects the information set forth therein.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), VIVUS, Inc.'s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated February 25, 2011 expressed an unqualified opinion thereon.
/s/ ODENBERG, ULLAKKO, MURANISHI & CO. LLP
San Francisco, CA
February 25, 2011
114
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The
Board of Directors and Stockholders of
VIVUS, Inc.
We have audited VIVUS, Inc.'s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). VIVUS, Inc.'s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in Management's Annual Report on Internal Control Over Financial Reporting included in Item 9A. Our responsibility is to express an opinion on the Company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, VIVUS, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of VIVUS, Inc. as of December 31, 2010 and 2009, and the related consolidated statements of operations, stockholders' equity and cash flows for each of the three years in the period ended December 31, 2010 and our report dated February 25, 2011 expressed an unqualified opinion thereon.
/s/ ODENBERG, ULLAKKO, MURANISHI & CO. LLP
San Francisco, CA
February 25, 2011
115
VIVUS, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except par value)
| |
December 31 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | |||||||
ASSETS |
|||||||||
Current assets: |
|||||||||
Cash and cash equivalents |
$ | 37,216 | $ | 40,533 | |||||
Available-for-sale securities |
101,970 | 166,241 | |||||||
Inventories |
3,225 | — | |||||||
Prepaid expenses and other assets |
1,648 | 4,106 | |||||||
Current assets of discontinued operations |
6 | 12,482 | |||||||
Total current assets |
144,065 | 223,362 | |||||||
Property and equipment, net |
221 | 259 | |||||||
Non-current assets of discontinued operations |
— | 6,411 | |||||||
Total assets |
$ | 144,286 | $ | 230,032 | |||||
LIABILITIES AND STOCKHOLDERS' EQUITY |
|||||||||
Current liabilities: |
|||||||||
Accounts payable |
$ | 2,395 | $ | 8,082 | |||||
Accrued research and clinical expenses |
2,625 | 2,426 | |||||||
Accrued employee compensation and benefits |
2,820 | 2,343 | |||||||
Accrued and other liabilities |
932 | 2,000 | |||||||
Current liabilities of discontinued operations |
3,512 | 7,659 | |||||||
Total current liabilities |
12,284 | 22,510 | |||||||
Notes payable—net of current portion |
— | 15,255 | |||||||
Non-current liabilities of discontinued operations |
— | 5,541 | |||||||
Total liabilities |
12,284 | 43,306 | |||||||
Commitments and contingencies |
|||||||||
Stockholders' equity: |
|||||||||
Preferred stock; $1.00 par value; 5,000 shares authorized; no shares issued and outstanding at December 31, 2010 and 2009 |
— | — | |||||||
Common stock; $.001 par value; 200,000 shares authorized at December 31, 2010 and 2009; 81,568 and 80,607 shares issued and outstanding at December 31, 2010 and December 31, 2009, respectively |
82 | 81 | |||||||
Additional paid-in capital |
432,041 | 420,708 | |||||||
Accumulated other comprehensive income (loss) |
4 | (3 | ) | ||||||
Accumulated deficit |
(300,125 | ) | (234,060 | ) | |||||
Total stockholders' equity |
132,002 | 186,726 | |||||||
Total liabilities and stockholders' equity |
$ | 144,286 | $ | 230,032 | |||||
See accompanying notes to consolidated financial statements.
116
VIVUS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except per share data)
| |
Years Ended December 31 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | |||||||||
Revenue: |
||||||||||||
License and other revenue |
$ | — | $ | 31,395 | $ | 83,721 | ||||||
Operating expenses: |
||||||||||||
Research and development |
39,971 | 70,940 | 76,673 | |||||||||
General and administrative |
25,656 | 13,870 | 12,253 | |||||||||
Total operating expenses |
65,627 | 84,810 | 88,926 | |||||||||
Loss from operations |
(65,627 | ) | (53,415 | ) | (5,205 | ) | ||||||
Interest and other income (expense): |
||||||||||||
Interest and other income, net |
468 | 1,998 | 4,406 | |||||||||
Interest expense |
(4,308 | ) | (3,693 | ) | (659 | ) | ||||||
Other-than-temporary loss on impaired securities |
— | (654 | ) | (7,689 | ) | |||||||
Loss on early extinguishment of debt |
(5,958 | ) | — | — | ||||||||
Total interest and other income (expense) |
(9,798 | ) | (2,349 | ) | (3,942 | ) | ||||||
Loss from continuing operations before income taxes |
(75,425 | ) | (55,764 | ) | (9,147 | ) | ||||||
Benefit (provision) for income taxes |
(9 | ) | 2,379 | 7 | ||||||||
Net loss from continuing operations |
(75,434 | ) | (53,385 | ) | (9,140 | ) | ||||||
Discontinued operations: |
||||||||||||
Income (loss) before income taxes |
9,398 | (967 | ) | (790 | ) | |||||||
Benefit (provision) for income taxes |
(29 | ) | 61 | (10 | ) | |||||||
Net income (loss) from discontinued operations |
9,369 | (906 | ) | (800 | ) | |||||||
Net loss |
$ | (66,065 | ) | $ | (54,291 | ) | $ | (9,940 | ) | |||
Basic and diluted net income (loss) per share: |
||||||||||||
Continuing operations |
$ | (0.93 | ) | $ | (0.74 | ) | $ | (0.15 | ) | |||
Discontinued operations |
0.11 | (0.01 | ) | (0.01 | ) | |||||||
Net loss per share |
$ | (0.82 | ) | $ | (0.75 | ) | $ | (0.16 | ) | |||
Shares used in per share computation: |
||||||||||||
Basic |
81,017 | 72,779 | 63,724 | |||||||||
Diluted |
83,821 | 72,779 | 63,724 | |||||||||
See accompanying notes to consolidated financial statements.
117
VIVUS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
(In thousands)
| |
Common Stock | |
Accumulated Other Comprehensive Income (Loss) |
|
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Additional Paid-In Capital |
Accumulated Deficit |
|
|||||||||||||||||
| |
Shares | Amount | Total | |||||||||||||||||
Balances, December 31, 2007 |
58,873 | $ | 59 | $ | 230,005 | $ | (68 | ) | $ | (169,829 | ) | $ | 60,167 | |||||||
Sale of common stock through employee stock purchase plan |
64 | — | 275 | — | — | 275 | ||||||||||||||
Exercise of common stock options for cash |
738 | 1 | 2,182 | — | — | 2,183 | ||||||||||||||
Share-based compensation expense |
— | — | 4,718 | — | — | 4,718 | ||||||||||||||
Proceeds from private placement of common stock |
9,992 | 10 | 74,990 | — | — | 75,000 | ||||||||||||||
Issue costs for private placement of common stock |
— | — | (1,612 | ) | — | — | (1,612 | ) | ||||||||||||
Net unrealized gain on securities |
— | — | — | 422 | — | 422 | ||||||||||||||
Net loss |
— | — | — | — | (9,940 | ) | (9,940 | ) | ||||||||||||
Balances, December 31, 2008 |
69,667 | 70 | 310,558 | 354 | (179,769 | ) | 131,213 | |||||||||||||
Sale of common stock through employee stock purchase plan |
99 | — | 361 | — | — | 361 | ||||||||||||||
Exercise of common stock options for cash |
491 | 1 | 2,349 | — | — | 2,350 | ||||||||||||||
Share-based compensation expense |
— | — | 4,791 | — | — | 4,791 | ||||||||||||||
Proceeds from underwritten public offering of common stock |
10,350 | 10 | 108,665 | — | — | 108,675 | ||||||||||||||
Issue costs for underwritten public offering of common stock |
— | — | (6,016 | ) | — | — | (6,016 | ) | ||||||||||||
Net unrealized loss on securities |
— | — | — | (357 | ) | — | (357 | ) | ||||||||||||
Net loss |
— | — | — | — | (54,291 | ) | (54,291 | ) | ||||||||||||
Balances, December 31, 2009 |
80,607 | 81 | 420,708 | (3 | ) | (234,060 | ) | 186,726 | ||||||||||||
Sale of common stock through employee stock purchase plan |
48 | — | 304 | — | — | 304 | ||||||||||||||
Exercise of common stock options for cash |
913 | 1 | 3,616 | — | — | 3,617 | ||||||||||||||
Share-based compensation expense |
— | — | 7,413 | — | — | 7,413 | ||||||||||||||
Net unrealized gain on securities |
— | — | — | 7 | — | 7 | ||||||||||||||
Net loss |
— | — | — | — | (66,065 | ) | (66,065 | ) | ||||||||||||
Balances, December 31, 2010 |
81,568 | $ | 82 | $ | 432,041 | $ | 4 | $ | (300,125 | ) | $ | 132,002 | ||||||||
See accompanying notes to consolidated financial statements.
118
VIVUS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| |
Years Ended December 31 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | ||||||||||
Cash flows from operating activities: |
|||||||||||||
Net loss from continuing operations |
$ | (75,434 | ) | $ | (53,385 | ) | $ | (9,140 | ) | ||||
Adjustments to reconcile net loss from continuing operations to net cash used for operating activities from continuing operations: |
|||||||||||||
Depreciation |
138 | 75 | 63 | ||||||||||
Net realized (gain)/loss on investments |
(5 | ) | (1,085 | ) | 979 | ||||||||
Other-than-temporary loss on impaired securities |
— | 654 | 7,689 | ||||||||||
Share-based compensation expense |
6,443 | 3,927 | 3,949 | ||||||||||
Loss on early extinguishment of debt |
5,958 | — | — | ||||||||||
Changes in assets and liabilities: |
|||||||||||||
Inventories |
(3,225 | ) | — | — | |||||||||
Prepaid expenses and other assets |
2,658 | (2,582 | ) | 2,097 | |||||||||
Accounts payable |
(5,687 | ) | (7,289 | ) | 8,225 | ||||||||
Accrued research and clinical expenses |
199 | (4,009 | ) | 4,953 | |||||||||
Accrued employee compensation and benefits |
477 | 737 | 553 | ||||||||||
Deferred revenue |
— | (31,395 | ) | (83,721 | ) | ||||||||
Accrued and other liabilities |
521 | 1,302 | 18 | ||||||||||
Net cash used for operating activities from continuing operations |
(67,957 | ) | (93,050 | ) | (64,335 | ) | |||||||
Net cash provided by (used for) operating activities from discontinued operations |
2,195 | (3,033 | ) | 767 | |||||||||
Net cash used for operating activities |
(65,762 | ) | (96,083 | ) | (63,568 | ) | |||||||
Cash flows from investing activities: |
|||||||||||||
Property and equipment purchases |
(105 | ) | (237 | ) | (186 | ) | |||||||
Release of restricted cash |
700 | — | — | ||||||||||
Investment purchases |
(209,759 | ) | (220,606 | ) | (123,381 | ) | |||||||
Proceeds from sale/maturity of securities |
274,042 | 177,572 | 133,674 | ||||||||||
Net cash provided by (used for) investing activities from continuing operations |
64,878 | (43,271 | ) | 10,107 | |||||||||
Net cash provided by (used for) investing activities from discontinued operations |
21,546 | (171 | ) | (264 | ) | ||||||||
Net cash provided by (used for) investing activities |
86,424 | (43,442 | ) | 9,843 | |||||||||
Cash flows from financing activities: |
|||||||||||||
Proceeds from notes payable |
— | 10,000 | 7,556 | ||||||||||
Payments of notes payable |
(23,000 | ) | (1,022 | ) | (1,278 | ) | |||||||
Exercise of common stock options |
3,617 | 2,350 | 2,183 | ||||||||||
Sale of common stock through employee stock purchase plan |
304 | 361 | 275 | ||||||||||
Net proceeds from issuance of common stock |
— | 102,659 | 73,388 | ||||||||||
Net cash (used for) provided by financing activities from continuing operations |
(19,079 | ) | 114,348 | 82,124 | |||||||||
Net cash used for financing activities from discontinued operations |
(4,900 | ) | (145 | ) | (130 | ) | |||||||
Net cash (used for) provided by financing activities |
(23,979 | ) | 114,203 | 81,994 | |||||||||
Net increase (decrease) in cash and cash equivalents |
(3,317 | ) | (25,322 | ) | 28,269 | ||||||||
Cash and cash equivalents: |
|||||||||||||
Beginning of year |
40,533 | 65,855 | 37,586 | ||||||||||
End of year |
$ | 37,216 | $ | 40,533 | $ | 65,855 | |||||||
Supplemental cash flow disclosure: |
|||||||||||||
Interest paid |
$ | 6,030 | $ | 2,861 | $ | 831 | |||||||
Income taxes paid |
$ | 41 | $ | 16 | $ | 64 | |||||||
Non-cash investing and financing activities: |
|||||||||||||
Unrealized gain (loss) on securities |
$ | 7 | $ | (357 | ) | $ | 422 | ||||||
See accompanying notes to consolidated financial statements.
119
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note 1. Business and Significant Accounting Policies
Business
VIVUS is a biopharmaceutical company, incorporated in 1991, developing innovative, next-generation therapies to address unmet needs in obesity and related morbidities, including sleep apnea and diabetes, and sexual health. The Company's lead drug in clinical development, QNEXA®, has completed Phase 3 clinical trials for the treatment of obesity and a New Drug Application, or NDA, was submitted to the Food and Drug Administration, or FDA, in December 2009. QNEXA is also in Phase 2 clinical development for the treatment of sleep apnea and type 2 diabetes. In the area of sexual health, VIVUS is in Phase 3 development with avanafil, a PDE5 inhibitor for the treatment of erectile dysfunction. On November 5, 2010, the Company completed the sale of MUSE® (alprostadil), a first-generation therapy for the treatment of erectile dysfunction, or ED. Current and investigational drug candidates in development will encompass patented proprietary formulations and novel delivery systems. Investigational drug candidates may be developed by seeking new indications for previously approved pharmaceutical drugs.
At December 31, 2010, the Company's accumulated deficit was approximately $300.1 million. Based on current plans, management expects to incur further losses for the foreseeable future. Management believes that the Company's cash, cash equivalents, and available-for-sale securities at December 31, 2010 will be sufficient to meet the Company's obligations into 2012. Until the Company can generate sufficient levels of cash from its operations, the Company expects to continue to finance its future cash needs primarily through proceeds from equity or debt financing, loans and collaborative agreements with corporate partners. The Company operates in a single segment, the development and commercialization of novel therapeutic products.
Significant Accounting Policies
Reclassifications
Certain prior year amounts in the consolidated financial statements have been reclassified to conform to the current year's presentation. On November 5, 2010, the Company completed the sale of MUSE®. As discussed in Note 3: "Discontinued Operations," the results of operations, the assets and the liabilities related to MUSE have been reported as discontinued operations in accordance with FASB ASC topic 205, Discontinued Operations, or ASC 205. Accordingly, the assets, liabilities and results of operations related to MUSE from prior periods have been reclassified to discontinued operations.
Principles of Consolidation
The consolidated financial statements include the accounts of VIVUS, Inc., and its wholly owned subsidiaries: VIVUS Real Estate LLC, VIVUS International Limited, VIVUS U.K. Limited and VIVUS B.V. Limited. All significant intercompany transactions and balances have been eliminated in consolidation. On December 31, 2005, VIVUS U.K. Limited became a dormant company. On March 20, 2008, VIVUS International Limited was dissolved. The Company acquired 100% of the outstanding shares of Deerfield ED Corp., a Delaware corporation, on November 5, 2010. Deerfield ED Corp. was dissolved on December 9, 2010.
120
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 1. Business and Significant Accounting Policies (Continued)
Use of Estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosures of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Cash and Cash Equivalents
The Company considers highly liquid investments with maturities from the date of purchase of three months or less to be cash equivalents. At December 31, 2010, all cash equivalents are invested in money market funds and U.S. Treasury securities. These accounts are recorded at fair value.
Cash with restrictions for a period of greater than 12 months is classified as restricted cash, a non-current asset.
Available-for-Sale Securities
The Company focuses on liquidity and capital preservation in its investments in available-for-sale securities. The Company's investment policy, as approved by the Audit Committee of the Board of Directors, allows it to invest its excess cash balances in money market and marketable securities, primarily U.S. Treasury securities and debt securities of U.S. government agencies, corporate debt securities and asset-backed securities in accordance with its investment policy. The Company periodically evaluates its investments to determine if impairment charges are required.
The Company determines the appropriate classification of marketable securities at the time of purchase and reevaluates such designation at each balance sheet date. Marketable securities have been classified and accounted for as available-for-sale. The Company may or may not hold securities with stated maturities greater than 12 months until maturity. In response to changes in the availability of and the yield on alternative investments as well as liquidity requirements, the Company may sell these securities prior to their stated maturities. As these securities are viewed by the Company as available to support current operations, securities with maturities beyond 12 months are classified as current assets.
Securities are carried at fair value, with the unrealized gains and losses, net of taxes, reported as a component of stockholders' equity, unless the decline in value is deemed to be other-than-temporary and the Company intends to sell such securities before recovering their costs, in which case such securities are written down to fair value and the loss is charged to other-than-temporary loss on impaired securities. The Company evaluates its investment securities for other-than-temporary declines based on quantitative and qualitative factors. Any realized gains or losses on the sale of marketable securities are determined on a specific identification method, and such gains and losses are reflected as a component of interest income.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist primarily of cash, cash equivalents and available-for-sale-securities. The Company has established guidelines to limit its exposure to credit risk by placing investments with a number of high credit
121
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 1. Business and Significant Accounting Policies (Continued)
quality institutions, in U.S. Treasury securities or diversifying its investment portfolio and placing investments with maturities that maintain safety and liquidity within the Company's liquidity needs.
Inventories
Inventories are valued at the lower of cost (first in, first out) or market. The Company records inventory reserves for estimated obsolescence, unmarketable or excess inventory equal to the difference between the cost of inventory and the estimated market value based upon assumptions about future demand and market conditions. If actual market conditions are less favorable than those projected by the Company, inventory write-downs may be required.
Prepaid Expenses and Other Assets
Prepaid expenses and other assets generally consist of deposits, other receivables and prepayments for future services. Prepayments are expensed when the services are received.
Property and Equipment
Property and equipment is stated at cost and includes leasehold improvements, computers and software and furniture and fixtures. For financial reporting, depreciation is computed using the straight-line method over estimated useful lives of two to seven years for computers and software and furniture and fixtures. Leasehold improvements are amortized using the straight-line method over the shorter of the expected lease term or the estimated useful lives. Expenditures for repairs and maintenance, which do not extend the useful life of the property and equipment, are expensed as incurred. Upon retirement, the asset cost and related accumulated depreciation are relieved from the accompanying consolidated balance sheets. Gains and losses associated with dispositions are reflected as a component of other income, net in the accompanying consolidated statements of operations.
Long-lived assets, such as property and equipment, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to an estimate of undiscounted future cash flows expected to be generated by the asset. If the carrying amount of the asset exceeds its estimated future cash flows, an impairment charge is recognized by the amount by which the carrying amount of the asset exceeds the fair value of the asset. Assets to be disposed of would be separately presented in the consolidated balance sheet and reported at the lower of the carrying amount or fair value less costs to sell, and are no longer depreciated. The assets and liabilities of a disposed group classified as held for sale would be presented separately in the appropriate asset and liability sections of the consolidated balance sheet. The Company believes the future cash flows to be received from the long-lived assets will exceed the assets' carrying value, and accordingly the Company has not recognized any impairment losses through December 31, 2010.
Fair Value
On January 1, 2008, the Company adopted SFAS No. 157 Fair Value Measurements, as codified in FASB ASC 820, Fair Value Measurements and Disclosures, or ASC 820, and effective October 10, 2008, the Company adopted FSP No. SFAS 157-3, Determining the Fair Value of a Financial Asset When the Market for That Asset Is Not Active, except as it applies to the nonfinancial assets and nonfinancial liabilities subject to FSP 157-2. On January 1, 2009, the Company adopted SFAS No 157 with respect
122
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 1. Business and Significant Accounting Policies (Continued)
to non-financial assets and non-financial liabilities. On June 15, 2009, the Company adopted FSP 157-4, Determining Fair Value When the Volume and Level of Activity for the Assets or Liabilities Have Significantly Decreased and Identifying Transactions That Are Not Orderly. Adoption of the provisions of these standards did not have a material effect on the Company's financial position.
Financial Instruments Measured at Fair Value. Cash and cash equivalents and available-for-sale financial instruments are carried at fair value and the Company makes estimates regarding valuation of these assets measured at fair value in preparing the consolidated financial statements.
Fair Value Measurement—Definition and Hierarchy. SFAS No. 157 defines fair value as the price that would be received to sell an asset or paid to transfer a liability (i.e., the "exit price") in an orderly transaction between market participants at the measurement date.
Valuation Technique. SFAS No. 157 establishes a hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the observable inputs be used when available. Observable inputs are inputs that market participants would use in pricing the asset or liability developed based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company's assumptions about the assumptions market participants would use in pricing the asset or liability developed based on the best information available in the circumstances. SFAS No. 157 prescribes three valuation techniques that shall be used to measure fair value as follows:
- 1.
- Market
Approach—uses prices or other relevant information generated by market transactions involving identical or comparable assets or
liabilities.
- 2.
- Income
Approach—uses valuation techniques to convert future cash flow amounts to a single present value amount (discounted).
- 3.
- Cost Approach—the amount that currently would be required to replace the service capacity of an asset (i.e., current replacement cost).
One or a combination of the approaches above can be used to calculate fair value, whichever results in the most representative fair value.
In addition to the three valuation techniques, SFAS No. 157 prescribes a fair value hierarchy in order to increase consistency and comparability in fair value measurements and related disclosures. The hierarchy is broken down into three levels based on the reliability of inputs as follows:
- •
- Level 1—Valuations based on quoted prices in active markets for identical assets. Since valuations are based on quoted prices that are readily and regularly available in an active market, valuation of these products does not entail a significant degree of judgment.
These types of instruments primarily consist of financial instruments whose value is based on quoted market prices such as cash, money market funds and U.S. Treasury securities that are actively traded. Management judgment was required to determine the Company's policy that defines the levels at which sufficient volume and frequency of transactions is met for a market to be considered active.
- •
- Level 2—Valuations based on quoted prices in markets that are not active or for which all significant inputs are observable, directly or indirectly. Quoted prices for similar instruments in active markets, quoted prices for identical or similar instruments in markets that are not active,
123
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 1. Business and Significant Accounting Policies (Continued)
or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
The types of instruments valued based on other observable inputs include debt securities of U.S. government agencies, corporate bonds, mortgage-backed and asset-backed products. Substantially all of these assumptions are observable in the marketplace, can be derived from observable data or are supported by observable levels at which transactions are executed in the marketplace.
- •
- Level 3—Valuations based on inputs that are unobservable and significant to the overall fair value measurement.
These types of instruments have included certain corporate bonds, mortgage-backed securities and asset-backed securities. The Company had no Level 3 securities at December 31, 2010. Level 3 is comprised of unobservable inputs that are supported by little or no market activity. These instruments are considered Level 3 when their fair values are determined using pricing models, discounted cash flows or similar techniques and at least one significant model assumption or input is unobservable. Level 3 may still include some observable inputs such as yield spreads derived from markets with limited activity. Level 3 financial assets include securities for which there is limited market activity such that the determination of fair value requires significant judgment or estimation.
The availability of observable inputs can vary from product to product and is affected by a wide variety of factors, including, for example, the type of product, whether the product is new and not yet established in the marketplace, and other characteristics particular to the transaction. To the extent that valuation is based on models or inputs that are less observable or unobservable in the market, the determination of fair value requires more judgment. Accordingly, the degree of judgment exercised by the Company in determining fair value is greatest for instruments categorized in Level 3. In certain cases, the inputs used to measure fair value may fall into different levels of the fair value hierarchy. In such cases, for disclosure purposes, the level in the fair value hierarchy within which the fair value measurement in its entirety falls is determined based on the lowest level input that is significant to the fair value measurement in its entirety.
Fair Value Measurements
As of December 31, 2010, the Company's cash and cash equivalents and available-for-sale securities measured at fair value on a recurring basis totaled $139.2 million. All of the Company's cash and cash equivalents and available-for-sale securities are cash, money market instruments and U.S. Treasury securities and these are classified as Level 1. The valuation techniques used to measure the fair values of these financial instruments were derived from quoted market prices, as substantially all of these instruments have maturity dates, if any, within one year from the date of purchase and active markets for these instruments exists.
Revenue Recognition
License and Other Revenue
The Company recognizes license revenue in accordance with the Securities and Exchange Commission's, or SEC's, Staff Accounting Bulletin No. 104, Revenue Recognition, and Emerging Issues Task Force, or EITF, Issue 00-21, Revenue Arrangements with Multiple Deliverables, as codified in FASB ASC topic 605, Revenue Recognition, or ASC 605. Revenue arrangements with multiple deliverables are
124
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 1. Business and Significant Accounting Policies (Continued)
divided into separate units of accounting if certain criteria are met, including whether the delivered item has standalone value to the customer, and whether there is objective, reliable evidence of the fair value of the undelivered items. Consideration received is allocated among the separate units of accounting based on their relative fair values, and the applicable revenue recognition criteria are identified and applied to each of the units.
Revenue from non-refundable, upfront license fees where the Company has continuing involvement is recognized ratably over the development or agreement period. Revenue associated with performance milestones is recognized based upon the achievement of the milestones, as defined in the respective agreements.
Sale of Evamist product
On May 15, 2007, the Company closed its transaction with K-V Pharmaceutical Company, or K-V, for the sale of its investigational drug candidate, Evamist. At the time of the sale, Evamist was an investigational drug candidate and was not yet approved by the Food and Drug Administration, or FDA, for marketing. The sale transaction contained multiple deliverables, including: the delivery at closing of the Evamist assets; a grant of a sublicense of the Company's rights under a license agreement related to Evamist, and a license to the metered-dose transdermal spray, or MDTS, applicator; the delivery upon receipt of regulatory approval of the approved drug along with all regulatory submissions; and, lastly, the delivery after FDA approval of certain transition services and a license to improvements to the MDTS applicator. The Company received approval from the FDA to market Evamist on July 27, 2007, or FDA Approval, and, on August 1, 2007, the Company transferred and assigned the Evamist FDA submissions, and all files related thereto, to K-V. The Company received an upfront payment of $10 million upon the closing and received an additional $140 million milestone payment in August 2007 upon FDA Approval. These payments are non-refundable. In August 2008, the Company assigned all of its rights and obligations under the Evamist license agreement to K-V.
Upon FDA Approval, the two remaining deliverables were the transition services to be performed under the Transition Services Agreement, or TSA, and a license to improvements to the MDTS applicator during the two-year period commencing with the closing, or May 15, 2007, and ending on May 15, 2009. The Company was able to establish fair value for the TSA. Given the unique nature of the license to improvements, the Company was unable to obtain objective, reliable evidence of its fair value.
Accordingly, the delivered items, together with the undelivered items, were treated as one unit of accounting. Since the deliverables were treated as a single unit of accounting, the total cash received, $150 million, was recognized as revenue on a pro-rata basis over the term of the last deliverable, which in this case was the license to improvements that expired on May 15, 2009. As a result, the initial $10 million paid at closing and the $140 million paid upon FDA Approval were recorded as deferred revenue and have been recognized as revenue ratably over the remaining 21.5-month term of the license to improvements, from August 1, 2007 to May 15, 2009. All of the revenue deferred from the Evamist sale has been recognized.
The Company is also eligible to receive milestone payments of up to $30 million based upon sales of Evamist through the term of the agreements. Revenue associated with performance milestones will be recognized based upon the achievement of the milestones, as defined in the respective agreements.
125
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 1. Business and Significant Accounting Policies (Continued)
Under the terms of the transaction, K-V reimbursed the Company for $1.5 million of the $3 million milestone payment paid by the Company to Acrux upon FDA Approval of the NDA.
Research and Development Expenses and Accruals
Research and development, or R&D, expenses include license fees, related compensation, consultants' fees, facilities costs, administrative expenses related to R&D activities and clinical trial costs at other companies and research institutions under agreements that are generally cancelable, among other related R&D costs. The Company also records accruals for estimated ongoing clinical trial costs. Clinical trial costs represent costs incurred by clinical research organizations, or CROs, and clinical sites and include advertising for clinical trials and patient recruitment costs. These costs are recorded as a component of R&D expenses and are expensed as incurred. Under the Company's agreements, progress payments are typically made to investigators, clinical sites and CROs. The Company analyzes the progress of the clinical trials, including levels of patient enrollment, invoices received and contracted costs when evaluating the adequacy of accrued liabilities. Significant judgments and estimates must be made and used in determining the accrued balance in any accounting period. Actual results could differ from those estimates under different assumptions. Revisions are charged to expense in the period in which the facts that give rise to the revision become known.
Share-Based Payments
The Company follows the fair value method of accounting for share-based compensation arrangements in accordance with SFAS 123R, Share-Based Payment, as codified in FASB ASC topic 718, Compensation—Stock Compensation, or ASC 718. Compensation expense is recognized, using a fair-value based method, for all costs related to share-based payments including stock options and restricted stock units and stock issued under the employee stock purchase plan. The Company estimates the fair value of share-based payment awards on the date of the grant using an option-pricing model. The Company adopted SFAS 123R effective January 1, 2006 using the modified prospective method of transition. The Company adopted the simplified method to calculate the beginning balance of the additional paid-in capital, or APIC, pool of excess tax benefits, and to determine the subsequent effect on the APIC pool and consolidated statements of cash flows of the tax effects of employee stock-based compensation awards. See Note 8: "Stock Option and Purchase Plans" for further discussion of the Company's stock-based compensation plans.
Income Taxes
The Company makes certain estimates and judgments in determining income tax expense for financial statement purposes. These estimates and judgments occur in the calculation of certain tax assets and liabilities, which arise from differences in the timing of recognition of revenue and expense for tax and financial statement purposes.
As part of the process of preparing the Company's consolidated financial statements, the Company is required to estimate its income taxes in each of the jurisdictions in which the Company operates. This process involves the Company estimating its current tax exposure under the most recent tax laws and assessing temporary differences resulting from differing treatment of items for tax and accounting purposes. These differences result in deferred tax assets and liabilities, which are included in the Company's consolidated balance sheets.
126
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 1. Business and Significant Accounting Policies (Continued)
The Company assesses the likelihood that it will be able to recover its deferred tax assets. The Company considers all available evidence, both positive and negative, including historical levels of income, expectations and risks associated with estimates of future taxable income and ongoing prudent and feasible tax planning strategies in assessing the need for a valuation allowance. If it is not more likely than not that the Company will recover its deferred tax assets, the Company will increase its provision for taxes by recording a valuation allowance against the deferred tax assets that the Company estimates will not ultimately be recoverable. As a result of the Company's analysis of all available evidence, both positive and negative, as of December 31, 2010, it was considered more likely than not that the Company's deferred tax assets would not be realized. However, should there be a change in the Company's ability to recover its deferred tax assets, the Company would recognize a benefit to its tax provision in the period in which the Company determines that it is more likely than not that it will recover its deferred tax assets.
Contingencies and Litigation
The Company is periodically involved in disputes and litigation related to a variety of matters. When it is probable that the Company will experience a loss, and that loss is quantifiable, the Company records appropriate reserves. The Company records legal fees and costs as an expense when incurred.
License Agreements
The Company has obtained rights to patented technologies under several licensing agreements. Non-refundable licensing payments made on technologies that are yet to be proven are expensed to research and development.
Net Income (Loss) Per Share
The Company computes basic net income (loss) per share applicable to common shareholders based on the weighted average number of common shares outstanding during the period. Diluted net income (loss) per share is based on the weighted average number of common and common equivalent shares, which represent shares that may be issued in the future upon the exercise of outstanding stock options. Common share equivalents are excluded from the computation in periods in which they have an anti-dilutive effect. Stock options for which the price exceeds the average market price over the period have an anti-dilutive effect on net income per share and, accordingly, are excluded from the calculation. When there is a net loss, other potentially dilutive common equivalent shares are not included in the calculation of net loss per share since their inclusion would be anti-dilutive.
127
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 1. Business and Significant Accounting Policies (Continued)
The computation of basic and diluted net income (loss) per share from continuing and discontinued operations for the years ended December 31, 2010, 2009 and 2008 are as follows:
| |
2010 | 2009 | 2008 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
(In thousands, except per share data) |
|||||||||
Net loss |
$ | (66,065 | ) | $ | (54,291 | ) | $ | (9,940 | ) | |
Net loss per share—basic |
$ | (.82 | ) | $ | (.75 | ) | $ | (.16 | ) | |
Effect of dilutive securities |
— | — | — | |||||||
Net loss per share—diluted |
$ | (.82 | ) | $ | (.75 | ) | $ | (.16 | ) | |
Shares used in the computation of net loss per share—basic |
81,017 | 72,779 | 63,724 | |||||||
Effect of dilutive securities |
2,804 | — | — | |||||||
Diluted shares |
83,821 | 72,779 | 63,724 | |||||||
As the Company recognized a net loss from continuing operations and net income from discontinued operations for the year ended December 31, 2010, 4,383,624 and 1,579,660 potentially dilutive options outstanding were not included in the computation of diluted net loss from continuing operations and diluted net income from discontinued options, respectively, because the effect would have been anti-dilutive. In the years ended December 31, 2009 and 2008, all potential common equivalent shares were excluded for these periods as they were anti-dilutive. Potentially dilutive options outstanding of 3,333,108 and 3,282,348 at December 31, 2009 and 2008, respectively, were not included in the computation of diluted net loss per share for the Company because the effect would be anti-dilutive.
Recent Accounting Requirements
In December 2010, the FASB issued ASU 2010-27, Fees Paid to the Federal Government by Pharmaceutical Manufacturers, (amendments to FASB ASC Topic 720, Other Expenses. The objective of this update is to address questions concerning how pharmaceutical manufacturers should recognize and classify in their income statements fees mandated by the Patient Protection and Affordable Care Act as amended by the Health Care and Education Reconciliation Act (the Acts). The amendments in this update are effective for calendar years beginning after December 31, 2010, when the fee initially becomes effective. The adoption of this statement effective January 1, 2011 did not have a material effect on the Company's consolidated financial statements.
In January 2010, the FASB issued ASU 2010-06, Fair Value Measurements Disclosures, which amends Subtopic 820-10 of the FASB Accounting Standards Codification to require new disclosures for fair value measurements and provides clarification for existing disclosures requirements. More specifically, this update will require (a) an entity to disclose separately the amounts of significant transfers in and out of Levels 1 and 2 fair value measurements and to describe the reasons for the transfers; and (b) information about purchases, sales, issuances and settlements to be presented separately (i.e. present the activity on a gross basis rather than net) in the reconciliation for fair value measurements using significant unobservable inputs (Level 3 inputs). This update clarifies existing disclosure requirements for the level of disaggregation used for classes of assets and liabilities measured
128
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 1. Business and Significant Accounting Policies (Continued)
at fair value and requires disclosures about the valuation techniques and inputs used to measure fair value for both recurring and nonrecurring fair value measurements using Level 2 and Level 3 inputs. The adoption of this statement effective January 1, 2010 did not materially expand the Company's consolidated financial statement footnote disclosures.
In October 2009, the FASB issued ASU 2009-13, Multiple-Deliverable Revenue Arrangements, (amendments to FASB ASC Topic 605, Revenue Recognition), or ASU 2009-13, and ASU 2009-14, Certain Arrangements That Include Software Elements, (amendments to FASB ASC Topic 985, Software), or ASU 2009-14. ASU 2009-13 requires entities to allocate revenue in an arrangement using estimated selling prices of the delivered goods and services based on a selling price hierarchy. The amendments eliminate the residual method of revenue allocation and require revenue to be allocated using the relative selling price method. ASU 2009-14 removes tangible products from the scope of software revenue guidance and provides guidance on determining whether software deliverables in an arrangement that includes a tangible product are covered by the scope of the software revenue guidance. ASU 2009-13 and ASU 2009-14 should be applied on a prospective basis for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010, with early adoption permitted. The adoption of this statement effective January 1, 2011 did not have a material effect on the Company's consolidated financial statements.
In June 2009, the FASB issued SFAS No. 167, Amendments to FASB Interpretation No. 46(R), as codified in ASC 810. This statement amends the consolidation guidance applicable to variable interest entities and the definition of a variable interest entity, and requires enhanced disclosures to provide more information about an enterprise's involvement in a variable interest entity. This statement also requires ongoing assessments of whether an enterprise is the primary beneficiary of a variable interest entity. This statement is effective for our fiscal year beginning January 1, 2010. The adoption of this statement did not have a material effect on the Company's consolidated financial statements.
Note 2. Cash, Cash Equivalents and Available-for-Sale Securities
The fair value and the amortized cost of cash, cash equivalents, and available-for-sale securities by major security type at December 31, 2010 and 2009 are presented in the tables that follow.
As of December 31, 2010 (in thousands):
Cash and cash equivalents
|
Amortized Cost |
Estimated Fair Value |
Gross Unrealized Gains |
Gross Unrealized Losses |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Cash and money market funds |
$ | 37,216 | $ | 37,216 | $ | — | $ | — | ||||||
Total cash and cash equivalents |
$ | 37,216 | $ | 37,216 | $ | — | $ | — | ||||||
Available-for-sale securities
|
Amortized Cost |
Estimated Fair Value |
Gross Unrealized Gains |
Gross Unrealized Losses |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
U.S. Treasury securities |
$ | 101,966 | $ | 101,970 | $ | 12 | $ | (8 | ) | |||||
Total available-for-sale securities |
$ | 101,966 | $ | 101,970 | $ | 12 | $ | (8 | ) | |||||
129
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 2. Cash, Cash Equivalents and Available-for-Sale Securities (Continued)
As of December 31, 2009 (in thousands):
Cash and cash equivalents
|
Amortized Cost |
Estimated Fair Value |
Gross Unrealized Gains |
Gross Unrealized Losses |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Cash and money market funds |
$ | 38,525 | $ | 38,525 | $ | — | $ | — | ||||||
U.S. Treasury securities |
2,009 | 2,008 | — | (1 | ) | |||||||||
Total cash and cash equivalents |
$ | 40,534 | $ | 40,533 | $ | — | $ | (1 | ) | |||||
Available-for-sale securities
|
Amortized Cost |
Estimated Fair Value |
Gross Unrealized Gains |
Gross Unrealized Losses |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
U.S. Treasury securities |
$ | 166,243 | $ | 166,241 | $ | 34 | $ | (36 | ) | |||||
Total available-for-sale securities |
$ | 166,243 | $ | 166,241 | $ | 34 | $ | (36 | ) | |||||
The following table summarizes the Company's available-for-sale securities by the contractual maturity date as of December 31, 2010 (in thousands):
| |
Amortized Cost |
Estimated Fair Value |
|||||
|---|---|---|---|---|---|---|---|
Due within one year |
$ | 101,966 | $ | 101,970 | |||
|
$ | 101,966 | $ | 101,970 | |||
The following table summarizes the net realized gains (losses) on available-for-sale securities for the periods presented (in thousands):
| |
Years Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | |||||||
Realized gains |
$ | 5 | $ | 1,637 | $ | 394 | ||||
Realized losses |
— | (552 | ) | (1,373 | ) | |||||
Net realized gains (losses) |
$ | 5 | $ | 1,085 | $ | (979 | ) | |||
During the year ended December 31, 2010, the Company sold a total of $28.5 million of fixed income securities, which resulted in net realized gains of $5,000. During the year ended December 31, 2009, the Company sold and received paydowns totaling $24 million of fixed income securities, which resulted in net realized gains of $1.1 million. During the year ended December 31, 2008, the Company sold and received paydowns totaling $85.2 million of fixed income securities which resulted in net realized losses of $979,000. In the ordinary course of business, the Company may sell securities at a loss for a number of reasons, including, but not limited to: (i) changes in the investment environment; (ii) expectation that the fair value could deteriorate further; (iii) desire to reduce exposure to an issuer or an industry; (iv) changes in credit quality; or (v) changes in expected cash flow.
130
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 2. Cash, Cash Equivalents and Available-for-Sale Securities (Continued)
At December 31, 2010, the Company had the following available-for-sale securities that were in an unrealized loss position (in thousands):
| |
Less Than 12 Months | |||||||
|---|---|---|---|---|---|---|---|---|
December 31, 2010
|
Gross Unrealized Losses |
Estimated Fair Value |
||||||
U.S. Treasury securities |
$ | (8 | ) | $ | 42,822 | |||
Total |
$ | (8 | ) | $ | 42,822 | |||
At December 31, 2009, the Company had the following cash equivalent and available-for-sale securities that were in an unrealized loss position (in thousands):
| |
Less Than 12 Months | |||||||
|---|---|---|---|---|---|---|---|---|
December 31, 2009
|
Gross Unrealized Losses |
Estimated Fair Value |
||||||
U.S. Treasury securities |
$ | (37 | ) | $ | 79,709 | |||
Total |
$ | (37 | ) | $ | 79,709 | |||
The gross unrealized losses reported above for December 31, 2010 and 2009 were primarily caused by general fluctuations in market interest rates from the respective purchase date of these securities through the end of those periods. The gross unrealized loss of $37,000 at December 31, 2009 is attributable to the Company's holding in 32 individual securities from one issuer, the U.S. Treasury.
As the Company presently does not intend to sell its debt securities and believes it will not likely be required to sell the securities that are in an unrealized loss position before recovery of their amortized cost, the Company does not consider these securities to be other-than-temporarily impaired.
As of December 31, 2010 and 2009, the temporary unrealized gains (losses) on cash, cash equivalents and available-for-sale securities, net of tax, of $4,000 and $(3,000), respectively, were included in accumulated other comprehensive income (loss) in the accompanying consolidated balance sheets.
SFAS No. 115, Accounting for Certain Investments in Debt and Equity Securities, FSP SFAS 115-2 and SFAS 124-4, Recognition and Presentation of Other-than-Temporary Impairments ("FSP 115-2/SFAS 124-2") and SAB Topic 5M, Accounting for Non-current Marketable Equity Securities, as codified in FASB ASC topic 320-10, Investments—Debt and Equity Securities, or ASC 320-10, provides guidance on determining when an investment is other-than-temporarily impaired. Investments are reviewed quarterly for indicators of other-than-temporary impairment. Effective for all periods ending after June 15, 2009, it provides additional guidance designed to create a greater clarity and consistency in accounting for and presenting impairment losses on securities. In reviewing its non-U.S. Government available-for-sale securities, the Company concluded that it intends to sell the debt securities before recovering their costs. Therefore, in accordance with the above recent guidance, the Company recognized an "other-than-temporary" impairment of $654,000 on these securities during the year ended December 31, 2009. The Company included this non-cash impairment charge in other-than-temporary loss on impaired securities in the consolidated statements of operations and other
131
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 2. Cash, Cash Equivalents and Available-for-Sale Securities (Continued)
comprehensive loss. These securities covered a number of industries. At December 31, 2010, all available-for-sale securities were invested in U.S. Treasuries.
During the Company's quarterly impairment assessments in 2008, the Company determined that a decline in value of certain securities was other-than-temporary. Accordingly, the Company recorded other-than-temporary impairment adjustments of $7.7 million in the year ended December 31, 2008. The Company included this non-cash impairment charge in other-than-temporary loss on impaired securities in the consolidated statements of operations. Included in the charge taken in 2008 was $2.4 million related to corporate bonds issued by Lehman Brothers Holdings Inc., or Lehman (or their respective subsidiaries, as appropriate). In addition, other-than-temporary impairments recognized in 2008 included impairments on investments for which the Company determined that the impairment was other-than-temporary due to credit downgrades and/or the Company's intent and ability to hold the investment to maturity. These securities covered a number of industries.
Fair Value Measurements
Effective January 1, 2008, the Company adopted SFAS No. 157, Fair Value Measurements, as codified in FASB ASC 820, Fair Value Measurements and Disclosures, or ASC 820, which defines fair value, establishes a framework for measuring fair value in generally accepted accounting principles and expands disclosures about fair value measurements. Broadly, the framework clarifies that fair value is an exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is a market-based measurement that should be determined based on assumptions that market participants would use in pricing an asset or liability.
As a basis for considering such assumptions, this statement establishes a three-tier value hierarchy, which prioritizes the inputs used in measuring fair value as follows: (Level 1) observable inputs such as quoted prices in active markets; (Level 2) inputs other than the quoted prices in active markets that are observable either directly or indirectly; and (Level 3) unobservable inputs in which there is little or no market data, which require the Company to develop its own assumptions. This hierarchy requires the Company to use observable market data, when available, and to minimize the use of unobservable inputs when determining fair value. On a recurring basis, the Company measures its marketable securities at fair value.
The following fair value hierarchy tables present information about the Company's assets (cash and cash equivalents, available-for-sale securities) measured at fair value on a recurring basis as of December 31, 2010 (in thousands):
| |
Basis of Fair Value Measurements | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Balance at December 31, 2010 |
Level 1 | Level 2 | Level 3 | ||||||||||
Cash and cash equivalents: |
||||||||||||||
Cash and money market funds |
$ | 37,216 | $ | 37,216 | $ | — | $ | — | ||||||
Total cash and cash equivalents |
$ | 37,216 | $ | 37,216 | $ | — | $ | — | ||||||
132
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 2. Cash, Cash Equivalents and Available-for-Sale Securities (Continued)
| |
Basis of Fair Value Measurements | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Balance at December 31, 2010 |
Level 1 | Level 2 | Level 3 | ||||||||||
Available-for-sale securities: |
||||||||||||||
U.S. Treasury securities |
$ | 101,970 | $ | 101,970 | $ | — | $ | — | ||||||
Total available-for-sale securities |
$ | 101,970 | $ | 101,970 | $ | — | $ | — | ||||||
Reported as: |
||||||||||||||
Cash and cash equivalents |
$ | 37,216 | ||||||||||||
Available-for-sale securities |
101,970 | |||||||||||||
Total |
$ | 139,186 | ||||||||||||
The following fair value hierarchy tables present information about the Company's assets (cash and cash equivalents and available-for-sale securities) measured at fair value on a recurring basis as of December 31, 2009 (in thousands):
| |
Basis of Fair Value Measurements | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Balance at December 31, 2009 |
Level 1 | Level 2 | Level 3 | ||||||||||
Cash and cash equivalents: |
||||||||||||||
Cash and money market funds |
$ | 38,525 | $ | 38,525 | $ | — | $ | — | ||||||
U.S. Treasury securities |
2,008 | 2,008 | — | — | ||||||||||
Total cash and cash equivalents |
$ | 40,533 | $ | 40,533 | $ | — | $ | — | ||||||
| |
Basis of Fair Value Measurements | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Balance at December 31, 2009 |
Level 1 | Level 2 | Level 3 | ||||||||||
Available-for-sale securities: |
||||||||||||||
U.S. Treasury securities |
$ | 166,241 | $ | 166,241 | $ | — | $ | — | ||||||
Total available-for-sale securities |
$ | 166,241 | $ | 166,241 | $ | — | $ | — | ||||||
Reported as: |
||||||||||||||
Cash and cash equivalents |
$ | 40,533 | ||||||||||||
Available-for-sale securities |
166,241 | |||||||||||||
Total |
$ | 206,774 | ||||||||||||
133
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 2. Cash, Cash Equivalents and Available-for-Sale Securities (Continued)
Fair values are based on quoted market prices, where available. These fair values are obtained primarily from third party pricing services, which generally use Level 1 or Level 2 inputs for the determination of fair value in accordance with ASC 820. Third party pricing services normally derive the security prices through recently reported trades for identical or similar securities making adjustments through the reporting date based upon available market observable information. For securities not actively traded, the third party pricing services may use quoted market prices of comparable instruments or discounted cash flow analyses, incorporating inputs that are currently observable in the markets for similar securities. Inputs that are often used in the valuation methodologies include, but are not limited to, benchmark yields, broker quotes, credit spreads, default rates and prepayment speeds. The Company performs a review of the prices received from third parties to determine whether the prices are reasonable estimates of fair value.
The Company generally obtains one price for each investment security. The Company performs a review to assess if the evaluated prices represent a reasonable estimate of their fair value. This process involves quantitative and qualitative analysis by the Company. Examples of procedures performed include, but are not limited to, initial and ongoing review of pricing service methodologies, review of the prices received from the pricing service, and comparison of prices for certain securities with different appropriate price sources for reasonableness. As a result of this analysis, if the Company determines there is a more appropriate fair value based upon available market data, which happens infrequently, the price of a security is adjusted accordingly. The pricing service provides information to indicate which securities were priced using market observable inputs so that the Company can properly categorize its financial assets in the fair value hierarchy.
As of December 31, 2010, the Company does not have any liabilities that are measured at fair value on a recurring basis.
Certain assets and liabilities are measured at fair value on a nonrecurring basis; that is, the instruments are not measured at fair value on an ongoing basis but are subject to fair value adjustments only in certain circumstances (for example, when there is evidence of impairment). There were no assets or liabilities measured at fair value on a nonrecurring basis during the year ended December 31, 2010.
Note 3. Discontinued Operations
On October 1, 2010, the Company entered into a definitive Asset Purchase Agreement with Meda AB, or Meda, to sell certain rights and assets related to MUSE, transurethral alprostadil, for the treatment of erectile dysfunction, or the MUSE Transaction. Meda has been the Company's European distributor of MUSE since 2002. The assets sold in the MUSE Transaction include the U.S. and foreign MUSE patents, existing inventory, and the manufacturing facility located in Lakewood, New Jersey. The Company retained all of the liabilities associated with the pre-closing operations and products of the MUSE business and the accounts receivables for pre-closing MUSE sales. The transaction closed on November 5, 2010. Prior to the closing of the MUSE Transaction, the Company terminated all of the rights to MUSE and avanafil held by Deerfield Management Company, L.P. and affiliates and by Crown Bank, N. A. as collateral to the Company's note payable (see Note 6: "Notes Payable"). Under the terms of the MUSE Transaction, the Company received an upfront payment of $22 million upon the closing and is eligible to receive an additional $1.5 million based on future sales of MUSE, provided that certain sales milestones are reached. Meda is now responsible for the manufacturing,
134
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 3. Discontinued Operations (Continued)
selling and marketing of MUSE. Meda also assumed all post-closing expenses and liabilities associated with MUSE. The Company has agreed not to develop, manufacture or sell any transurethral erectile dysfunction drugs for a period of three years following the closing of the MUSE Transaction.
The sale of the MUSE product and certain related assets has been reported as discontinued operations in the consolidated statements of operations for all periods presented, since (i) the MUSE product and related assets have identifiable cash flows that are largely independent of the cash flows of other groups of assets and liabilities, (ii) the Company will have no significant continuing involvement with the product after the close of the transaction, and (iii) the cash milestone payment to be received upon achievement of certain sales levels is considered an indirect cash flow. The assets and liabilities related to the MUSE operations are reported as assets and liabilities of discontinued operations in the consolidated balance sheets for all periods presented.
The following table presents the major classes of assets and liabilities that have been presented as assets and liabilities of discontinued operations in the consolidated balance sheets (in thousands):
| |
December 31, 2010 |
December 31, 2009 |
||||||
|---|---|---|---|---|---|---|---|---|
ASSETS |
||||||||
Cash and cash equivalents |
$ | — | $ | 217 | ||||
Trade accounts receivable, net |
6 | 7,259 | ||||||
Inventories, net |
— | 2,702 | ||||||
Prepaid expenses and other assets |
— | 2,304 | ||||||
Total current assets of discontinued operations |
6 | 12,482 | ||||||
Property and equipment, net |
— | 5,711 | ||||||
Restricted cash |
— | 700 | ||||||
Total non-current assets of discontinued operations |
— | 6,411 | ||||||
Total assets of discontinued operations |
$ | 6 | $ | 18,893 | ||||
| |
December 31, 2010 |
December 31, 2009 |
||||||
|---|---|---|---|---|---|---|---|---|
LIABILITIES |
||||||||
Accounts payable |
$ | 211 | $ | 403 | ||||
Accrued product returns |
2,598 | 3,026 | ||||||
Accrued chargeback reserve |
472 | 1,617 | ||||||
Accrued employee compensation and benefits |
47 | 729 | ||||||
Accrued and other liabilities |
184 | 1,884 | ||||||
Total current liabilities of discontinued operations |
3,512 | 7,659 | ||||||
Note payable Crown Bank, N.A., net of current portion |
— | 4,743 | ||||||
Deferred revenue |
— | 798 | ||||||
Total non-current liabilities of discontinued operations |
— | 5,541 | ||||||
Total liabilities of discontinued operations |
$ | 3,512 | $ | 13,200 | ||||
135
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 3. Discontinued Operations (Continued)
The following table presents summarized results of operations for the discontinued operations presented in the consolidated statements of operations (in thousands):
| |
Years Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | |||||||
Total revenues |
$ | 13,311 | $ | 18,646 | $ | 18,512 | ||||
Loss from operations |
$ | (3,902 | ) | $ | (602 | ) | $ | (418 | ) | |
Income (loss) before provision (benefit) for income taxes |
$ | 9,398 | $ | (967 | ) | $ | (790 | ) | ||
Net income (loss) from discontinued operations |
$ | 9,369 | $ | (906 | ) | $ | (800 | ) | ||
Note 4. Prepaid Expenses and Other Assets
Prepaid expenses and other assets as of December 31, 2010 and 2009, respectively, consist of (in thousands):
| |
2010 | 2009 | ||||||
|---|---|---|---|---|---|---|---|---|
Refundable federal income taxes |
$ | 141 | $ | 2,381 | ||||
Prepaid clinical studies |
5 | 520 | ||||||
Interest receivable |
553 | 472 | ||||||
Prepaid insurance |
594 | 429 | ||||||
Other prepaid expenses and assets |
355 | 304 | ||||||
Prepaid expenses and other assets |
$ | 1,648 | $ | 4,106 | ||||
Note 5. Property and Equipment
Property and equipment as of December 31, 2010 and 2009, respectively, consist of (in thousands):
| |
2010 | 2009 | |||||
|---|---|---|---|---|---|---|---|
Computers and software |
$ | 541 | $ | 522 | |||
Furniture and fixtures |
461 | 412 | |||||
Leasehold improvements |
272 | 240 | |||||
|
1,274 | 1,174 | |||||
Accumulated depreciation |
(1,053 | ) | (915 | ) | |||
Property and equipment, net |
$ | 221 | $ | 259 | |||
For the years ended December 31, 2010, 2009 and 2008, depreciation expense was $138,000, $75,000 and $63,000, respectively.
136
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 6. Notes Payable
Deerfield Financing
On April 3, 2008, the Company entered into several agreements with Deerfield Management Company, L.P., or Deerfield, a healthcare investment fund, and its affiliates, Deerfield Private Design Fund L.P. and Deerfield Private Design International, L.P. (collectively, the Deerfield Affiliates). Certain of the agreements were amended and restated on March 16, 2009, which included the addition of Deerfield PDI Financing L.P. as a Deerfield Affiliate. Under the agreements, Deerfield and its affiliates agreed to provide $30 million in funding to the Company. The $30 million in funding consisted of $20 million from a Funding and Royalty Agreement, or FARA, entered into with a newly incorporated subsidiary of Deerfield, or the Deerfield Sub, and $10 million from the sale of the Company's common stock under a securities purchase agreement. Under the FARA, the Deerfield Sub made $3.3 million payments to the Company in April, September and December 2008 and February, June and September 2009, constituting all of the required payments under the FARA. The Company paid royalties on the net sales of MUSE and if approved, on future sales of avanafil, an investigational drug candidate, to the Deerfield Sub. The term of the FARA was 10 years. The FARA included covenants requiring the Company to use commercially reasonable efforts to preserve its intellectual property, manufacture, promote and sell MUSE, and develop avanafil.
The agreements also provided the Company with an option to purchase, and the Deerfield Affiliates with an option to compel the Company to purchase, or put right, the Deerfield Sub holding the royalty rights. If the Company exercised its right to purchase the Deerfield Sub, the net price would be $23 million if exercised before April 3, 2011, or $26 million if exercised after April 3, 2011 but before April 3, 2012 (the purchase prices are subject to other adjustments as defined in the agreement). After April 3, 2011, the Deerfield Affiliates could have exercised the right to compel the Company to purchase the Deerfield Sub at a price of $17 million. This price could have increased up to $26 million, and the timing of the sale of the shares could have been accelerated under certain conditions including a change-in-control, sale of MUSE or avanafil, sale of major assets and the sale of securities in a transaction or a series of related transactions by the Company that exceed 20% of the Company's outstanding common stock at the date the Option and Put Agreement was signed if at the time of the sale the Company's market capitalization is below $300 million (each, a Major Transaction). Under these conditions, the cost of the shares of the Deerfield Sub would have been $23 million on or before April 3, 2011 and $26 million from April 3, 2011 through April 3, 2018. The sale of the shares of the Deerfield Sub could also have been accelerated if the Company's cash, cash equivalents and available for sale securities fell below $15 million or the Company's market capitalization fell below $50 million. The purchase prices under the put right were subject to other adjustments as defined in the agreements. If either party exercised its option, any further royalty payments would be effectively terminated. In exchange for the option right, the Company paid $2 million to the Deerfield Affiliates. The Company's intellectual property and all of the accounts receivable, inventory and machinery and equipment arising out of or relating to MUSE and avanafil were collateral for this transaction. At December 31, 2009 and 2008, substantially all of the accounts receivable, inventory and machinery and equipment, reported as assets of discontinued operations (See Note 3: "Discontinued Operations") on the Company's consolidated balance sheet related to MUSE and served as collateral for this transaction.
In preparation for the closing of the MUSE Transaction and in accordance with the terms of the OPA, the Company exercised the option right and on October 21, 2010, it paid $27.1 million in satisfaction of all of its financial obligations under the FARA and OPA. The gross amount paid
137
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 6. Notes Payable (Continued)
consisted of the Base Option Price of $25 million less the $2 million Option Premium Adjustment, or $23 million, plus the Cash Adjustment of $2.8 million and the Royalty Adjustment of $1.3 million. The Royalty Adjustment was calculated based upon royalties on MUSE sales not yet paid to the Deerfield Sub at the time of Option Closing. The Cash Adjustment was the total amount of cash remaining in the Deerfield Sub at time of Option Closing. As a result, all of the outstanding shares of the Deerfield Sub were acquired by the Company, the royalty rights to MUSE and avanafil were terminated and the notes payable of the Deerfield Sub were cancelled. In addition, the $2.8 million of cash held by the Deerfield Sub is now owned by the Company. All the security interests in the collateral related to MUSE and avanafil held by the Deerfield Sub and the Deerfield Affiliates as part of the FARA and OPA were terminated. In December 2010, the Deerfield Sub was dissolved. The payoff of the Deerfield loan resulted in a loss on the early extinguishment of debt of $6 million which was recognized in the fourth quarter of 2010.
The Company evaluated the Deerfield financing in accordance with FASB Financial Interpretation No., or FIN, 46(R), Consolidation of Variable Interest Entities, or FIN 46R, as codified in FASB ASC topic 810, Consolidation, or ASC 810, and determined that the Deerfield Sub may constitute a Variable Interest Entity, or VIE; however, the Company also determined that it was not the primary beneficiary of this VIE and therefore concluded that the Company was not required to consolidate the Deerfield Sub. In December 2010, the Deerfield Sub was dissolved.
In accordance with Emerging Issues Task Force (EITF) Issue 88-18, Sale of Future Revenues, as codified in FASB ASC 605, the FARA transaction was in substance a financing arrangement, or loan, that was repaid by the Company. The minimum repayment amount was $17 million, the amount of the unconditional put option held by the Deerfield Affiliates, plus royalties paid during the term of the agreement on sales of MUSE and, if approved, avanafil. Accordingly, the Company recorded the advances from the Deerfield Affiliates, net of the $2 million option right payment and related fees and expenses, as a loan. The Company received all of the required advances under the financing arrangement. Per the agreement, the loan amount would be lower than the contractual amounts owed if the Company exercised its call option of $23 million to $26 million, or if the Deerfield Affiliates required the Company to purchase the shares as a result of a "Major Transaction". Using the interest method under APB Opinion No. 21, Interest on Receivables and Payables, as codified in FASB ASC topic 835, Interest, subtopic 30, Imputation of Interest or ASC 835-30, interest expense on the loan was calculated and recognized over three years, which was the estimated term of the loan based on the earliest date that the Deerfield Affiliates could require the Company to repay the amounts advanced. The Deerfield Affiliates received quarterly payments based on net sales of MUSE. The initial imputed effective annual interest rate on the financing was approximately 32% as calculated based upon quarterly advances under the FARA, up to a loan balance of $17 million, offset by the estimated quarterly royalty payments to the Deerfield Affiliates. The imputed interest rate was revised to 31% at December 31, 2009 and 33% at December 31, 2008 based on the actual royalty payments made and the timing of payments and advances in 2009 and 2008, respectively. The imputed effective interest rate was utilized for purposes of calculating the interest expense only and did not reflect the amount of royalty paid to the Deerfield Affiliates on a quarterly basis. Quarterly royalty payments were based on a percentage of net MUSE sales at a rate substantially lower than the imputed effective interest rate used to calculate interest expense.
138
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 6. Notes Payable (Continued)
Crown Bank N.A. Loan
On January 4, 2006, VIVUS, Inc. and Vivus Real Estate LLC, a wholly owned subsidiary of VIVUS, Inc., jointly, the Company, entered into a Term Loan Agreement and a Commercial Mortgage Note, or the Agreements, with Crown Bank N. A., or Crown, secured by the land and buildings, among other assets, located at 735 Airport Road and 745 Airport Road in Lakewood, New Jersey, or the Facility. The Facility was the Company's former principal manufacturing facility, which the Company purchased on December 22, 2005. Under the Agreements, the Company borrowed $5.4 million on January 4, 2006 from Crown payable over a 10-year term. The interest rate was adjusted annually to a fixed rate for the year equal to the prime rate plus 1%, with a floor of 7.5%. Principal and interest were payable monthly based upon a 20-year amortization schedule and were adjusted annually at the time of the interest rate reset. All remaining principal was due on February 1, 2016. The interest rate was 7.5% for the year ended December 31, 2010 and 2009, respectively. Because the interest rate was variable, and based on a market rate, the carrying value of the debt approximates fair value. The Agreements contained prepayment penalties, and a requirement to maintain a depository account at Crown with a minimum collected balance of $100,000 which, if not maintained, would result in an automatic increase in the interest rate on the note of one-half percent (0.5%). The Facility, assignment of rents and leases on the Facility, and a $700,000 Certificate of Deposit held by Crown, classified as restricted cash, served as collateral for these Agreements. At December 31, 2009, the Facility and restricted cash were reported as assets of discontinued operations, (see Note 3: "Discontinued Operations"). On October 15, 2010, in preparation for the closing of the MUSE Transaction and in accordance with the terms of the Agreements, the Company paid $4.8 million to Crown in satisfaction of all obligations owed to Crown under the Agreements. As a result, the security interests and Certificate of Deposit held by Crown were terminated in favor of the Company. The Crown Bank N.A. Loan is reported as a liability of discontinued operations in the consolidated balance sheets for all periods presented (see Note 3: "Discontinued Operations").
Total long-term notes payable consist of the following (in thousands):
| |
December 31, 2010 | December 31, 2009 | ||||||
|---|---|---|---|---|---|---|---|---|
Deerfield loan |
$ | — | $ | 15,255 | ||||
Less current portion |
— | — | ||||||
Total long-term notes payable |
$ | — | $ | 15,255 | ||||
Note 7. Stockholders' Equity
Common Stock
The Company is authorized to issue 200 million shares of common stock. As of December 31, 2010 and 2009, there were 81,568,082 and 80,607,481 shares, respectively, issued and outstanding.
On April 3, 2008, the Company entered into several agreements with the Deerfield Affiliates (see Note 6: Notes Payable). Under the agreements, Deerfield and its affiliates agreed to provide $30 million in funding to the Company. The $30 million in funding consists of $20 million from a FARA and $10 million from the sale of the Company's common stock under a securities purchase agreement. At the closing on April 15, 2008, under the securities purchase agreement, the Deerfield Affiliates purchased 1,626,017 shares of the Company's common stock for an aggregate purchase price
139
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 7. Stockholders' Equity (Continued)
of $10 million and the Company paid to the Deerfield Affiliates a $500,000 fee and reimbursed approximately $200,000 in certain expenses incurred in this transaction, registered under the shelf Registration Statement (File Number 333-135793) filed with the SEC on July 14, 2006. The number of shares was determined based on the volume weighted average price on the NASDAQ Global Market of the Company's common stock on the three days prior to the execution of the securities purchase agreement dated as of April 3, 2008.
On August 6, 2008, the Company sold $65 million of its common stock in a registered direct offering. Under the terms of the financing, the Company sold 8,365,508 shares of its common stock at a price of $7.77 per share. On August 5, 2008, the Company filed a prospectus supplement with the SEC relating to this registered direct offering under the existing shelf Registration Statement (File Number 333-150649).
On March 9, 2009, the Company filed a Form S-8 (File Number 333-157787) with the SEC registering 1,000,000 shares of common stock, par value $0.001 per share, under the 2001 Stock Option Plan, as amended.
On September 17, 2009, the Company entered into an underwriting agreement, or the Underwriting Agreement, with J.P. Morgan Securities Inc., as representative of the several underwriters named therein, or the Underwriters, relating to the public offering and sale of 9,000,000 shares of the Company's common stock. Pursuant to the Underwriting Agreement, the Underwriters agreed to purchase, subject to customary closing conditions, 9,000,000 shares of the Company's common stock. The Company also granted the Underwriters a 30-day option to purchase up to 1,350,000 additional shares of common stock on the same terms and conditions as set forth above to cover over-allotments, which the Underwriters exercised in full. The 10,350,000 shares were sold at a price to the public of $10.50 per share which resulted in approximately $108.7 million in gross proceeds to the Company before deducting underwriting discounts and commissions and other offering expenses. The transaction closed on September 23, 2009. The offering was made pursuant to the Company's effective shelf registration statement on Form S-3 (Registration No. 333-161948) including the prospectus dated September 16, 2009 contained therein, as supplemented.
On February 16, 2010, the Company filed a Form S-8 (File Number 333-164921) with the SEC registering 1,000,000 shares of common stock, par value $0.001 per share, under the 2001 Stock Option Plan, as amended.
On July 14, 2010, the Company filed a Form S-8 (File Number 333-168106) with the SEC registering 16,615,199 shares of common stock, par value $0.001 per share, to be issued pursuant to the 2010 Equity Incentive Plan, and registering 400,000 shares of common stock, par value $0.001 per share, to be issued pursuant to the Stand-Alone Stock Option Agreement with Michael P. Miller.
Preferred Stock
The Company is authorized to issue 5 million shares of undesignated preferred stock with a par value of $1.00 per share. As of December 31, 2010 and 2009, there were no preferred shares issued or outstanding. The Company may issue shares of preferred stock in the future, without stockholder approval, upon such terms as the Company's management and Board of Directors may determine.
140
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 7. Stockholders' Equity (Continued)
Stockholder Rights Plan
On March 26, 2007, the Board of Directors of the Company adopted a Stockholder Rights Plan, or the Rights Plan, and amended its bylaws. Under the Rights Plan, the Company will issue a dividend of one right for each share of its common stock held by stockholders of record as of the close of business on April 13, 2007.
The Rights Plan is designed to guard against partial tender offers and other coercive tactics to gain control of the Company without offering a fair and adequate price and terms to all of the Company's stockholders. The Rights Plan is intended to provide the Board of Directors with sufficient time to consider any and all alternatives to such an action and is similar to plans adopted by many other publicly traded companies. The Rights Plan was not adopted in response to any efforts to acquire the Company and the Company is not aware of any such efforts.
Each right will initially entitle stockholders to purchase a fractional share of the Company's preferred stock for $26.00. However, the rights are not immediately exercisable and will become exercisable only upon the occurrence of certain events. If a person or group acquires, or announces a tender or exchange offer that would result in the acquisition of 15% or more of the Company's common stock while the Stockholder Rights Plan remains in place, then, unless the rights are redeemed by the Company for $.001 per right, the rights will become exercisable by all rights holders except the acquiring person or group for the Company's shares or shares of the third party acquirer having a value of twice the right's then-current exercise price. The Rights will expire on the earliest of (i) April 13, 2017 (the final expiration date), or (ii) redemption or exchange of the Rights.
Note 8. Stock Option and Purchase Plans
Stock Option Plan
Under the 2001 Stock Option Plan, or the 2001 Plan, which was approved by the stockholders at the annual meeting held on June 5, 2002, the Company may grant incentive or non-statutory stock options or stock purchase rights, or SPRs. The maximum aggregate number of shares that may be granted under the 2001 Plan is 1,000,000 shares plus (a) any shares that have been reserved but not issued under the Company's 1991 Incentive Stock Option Plan, or the 1991 Plan; (b) any shares returned to the 1991 Plan as a result of termination of options or repurchase of shares issued under the 1991 Plan; and (c) an annual increase to be added on the first day of the Company's fiscal year beginning 2003, equal to the lesser of (i) 1,000,000 shares, (ii) 2.5% of the outstanding shares on such date, or (iii) a lesser amount determined by the Board. The 2001 Plan allows the Company to grant incentive stock options to employees at not less than 100% of the fair market value of the stock (110% of fair market value for individuals who control more than 10% of the Company stock) at the date of grant, as determined by the Board of Directors.
The 2001 Plan allows the Company to grant non-statutory stock options to employees, directors and consultants at a price to be determined by the Board of Directors. The term of the option is determined by the Board of Directors on the date of grant but shall not be longer than 10 years. Options under this plan generally vest over four years, and all options expire after 10 years. The 2001 Plan allows the Company to grant SPRs to employees and consultants. Sales of stock under SPRs are made pursuant to restricted stock purchase agreements containing provisions established by the Board of Directors. The Company has a right, but not the obligation, to repurchase the shares at the original
141
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 8. Stock Option and Purchase Plans (Continued)
sale price, which expires at a rate to be determined by the Board of Directors. As of December 31, 2009, no SPRs have been granted under the 2001 Plan.
Under the 2001 Plan, non-employee directors will receive an option to purchase 32,000 shares of common stock when they join the Board of Directors. These options vest 25% after one year and 25% annually thereafter. Each non-employee director shall automatically receive an option to purchase 8,000 shares of the Company's common stock annually upon their reelection and these options are fully exercisable ratably over eight months. Non-employee directors are also eligible to receive additional stock option grants.
On March 29, 2010, the Company's Board of Directors terminated the 2001 Plan. In addition, the Board of Directors adopted and approved a new 2010 Equity Incentive Plan, or the 2010 Plan, with 32,000 shares remaining reserved and unissued under the 2001 Plan, subject to the approval of the Company's stockholders. The 2001 Plan, however, continues to govern awards previously granted under it. On June 25, 2010, the Company's stockholders approved the 2010 Plan at the Company's 2010 Annual Meeting of Stockholders. The 2010 Plan provides for the grant of stock options, stock appreciation rights, restricted stock, restricted stock units, performance shares and performance units to employees, directors and consultants, to be granted from time to time as determined by the Board of Directors, the Compensation Committee of the Board of Directors, or its designees. The term of the option is determined by the Board of Directors on the date of grant but shall not be longer than 10 years. Options under this plan generally vest over four years, and all options expire after 10 years. The 2010 Plan's share reserve, which the stockholders approved, is 8,400,000 shares, plus any shares reserved but not issued pursuant to awards under the 2001 Plan as of the date of stockholder approval, or 99,975 shares, plus any shares subject to outstanding awards under the 2001 Plan that expire or otherwise terminate without having been exercised in full, or are forfeited to or repurchased by the Company, up to a maximum of 8,111,273 shares (which was the number of shares subject to outstanding options under the 2001 Plan as of March 11, 2010). Awards exercisable for 274,750 shares were granted in the year ended December 31, 2010 pursuant to the 2010 Plan.
On April 30, 2010, the Company's Board of Directors granted an option to purchase 400,000 shares of the Company's common stock, or the Inducement Grant, to Michael P. Miller, the Company's new Senior Vice President and Chief Commercial Officer. The Inducement Grant was granted outside of the Company's 2010 Plan and without stockholder approval pursuant to NASDAQ Listing Rule 5635(c)(4) and is subject to the terms and conditions of the Stand-Alone Stock Option Agreement between the Company and Michael P. Miller.
142
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 8. Stock Option and Purchase Plans (Continued)
A summary of stock option award activity under these plans is as follows:
| |
Years Ended December 31, | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | ||||||||||||||||
| |
Number of Shares |
Weighted- Average Exercise Price |
Number of Shares |
Weighted- Average Exercise Price |
Number of Shares |
Weighted- Average Exercise Price |
|||||||||||||
Balance at beginning of year |
7,553,776 | $ | 4.74 | 6,107,304 | $ | 4.80 | 5,348,501 | $ | 4.25 | ||||||||||
Options: |
|||||||||||||||||||
Granted |
1,729,135 | $ | 9.37 | 2,132,382 | $ | 4.62 | 1,557,058 | $ | 6.05 | ||||||||||
Exercised |
(982,594 | ) | $ | 4.34 | (491,134 | ) | $ | 4.79 | (698,262 | ) | $ | 3.40 | |||||||
Cancelled |
(381,304 | ) | $ | 6.66 | (194,776 | ) | $ | 5.01 | (99,993 | ) | $ | 4.52 | |||||||
Balance at end of year |
7,919,013 | $ | 5.71 | 7,553,776 | $ | 4.74 | 6,107,304 | $ | 4.80 | ||||||||||
Exercisable at end of year |
5,171,827 | $ | 4.82 | 4,493,391 | $ | 4.63 | 3,739,766 | $ | 4.52 | ||||||||||
Weighted average grant-date fair value of options granted during the year |
$ | 5.74 | $ | 2.72 | $ | 3.33 | |||||||||||||
Restricted Stock Units
On July 12, 2006, the Board of Directors adopted an amendment to the 2001 Plan to add the ability to issue Restricted Stock Units, or RSUs, under the 2001 Plan. In contrast to restricted stock awards, the RSUs represent an obligation of the Company to issue unrestricted shares of common stock or cash to the grantee only when and to the extent that the vesting criteria of the award are satisfied. As in the case of restricted stock awards, vesting criteria for RSUs can be based on time or other conditions specified by the Board or an authorized committee of the Board. However, until vesting occurs, the grantee is not entitled to any stockholder rights with respect to the unvested shares. Upon vesting of an RSU, the recipient receives one share of VIVUS stock for each vested restricted stock unit or a cash payment for the value thereof. The Company, in its sole discretion, may pay earned RSUs in cash, shares, or a combination thereof. Shares represented by RSUs that are fully paid in cash again will be available for grant under the Plan. The Company issues new shares for settlement of vested restricted stock units and exercises of stock options. The Company does not have a policy of purchasing its shares relating to its share-based programs.
There were no restricted stock units outstanding as of December 31, 2010.
143
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 8. Stock Option and Purchase Plans (Continued)
Summary of Stock Options
At December 31, 2010, stock options were outstanding and exercisable as follows:
| Options Outstanding | Options Exercisable | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Range of Exercise Prices
|
Number Outstanding at December 31, 2010 |
Weighted- Average Remaining Contractual Life |
Weighted- Average Exercise Price |
Number Exercisable December 31, 2010 |
Weighted- Average Exercise Price |
||||||||||
$2.95 - $4.23 |
2,924,866 | 6.1 years | $ | 3.96 | 2,147,755 | $ | 3.87 | ||||||||
$4.25 - $6.05 |
2,938,682 | 5.8 years | $ | 5.13 | 2,570,273 | $ | 5.04 | ||||||||
$6.10 - $10.19 |
2,055,465 | 8.2 years | $ | 9.03 | 453,799 | $ | 8.09 | ||||||||
$2.95 - $10.19 |
7,919,013 | 6.5 years | $ | 5.71 | 5,171,827 | $ | 4.82 | ||||||||
The aggregate intrinsic value of outstanding options as of December 31, 2010 was $29.5 million, of which $23.5 million related to exercisable options.
At December 31, 2010, 8,511,624 options remain available for grant. On January 21, 2011, the Company granted 1,265,790 of these options. During the year ended December 31, 2010, in accordance with the terms of the 2010 Plan, the Company transferred a net total of 285,649 expired plan shares to the 2010 Plan.
Stock Purchase Plan
Under the 1994 Employee Stock Purchase Plan, or the Stock Purchase Plan, the Company reserved 800,000 shares of common stock for issuance to employees pursuant to the Stock Purchase Plan, under which eligible employees may authorize payroll deductions of up to 10% of their base compensation (as defined) to purchase common stock at a price equal to 85% of the lower of the fair market value as of the beginning or the end of the offering period.
At the annual meeting held on June 4, 2003, the stockholders approved amendments to the Stock Purchase Plan to (i) extend the original term of the Stock Purchase Plan by an additional 10 years such that the Stock Purchase Plan will now expire in April 2014 (subject to earlier termination as described in the Stock Purchase Plan) and (ii) increase the number of shares of common stock reserved for issuance under the Stock Purchase Plan by 600,000 shares to a new total of 1,400,000 (collectively referred to herein as the 1994 Purchase Plan Amendments).
As of December 31, 2010, 1,327,628 shares have been issued to employees and there are 72,372 shares available for issuance under the Employee Stock Purchase Plan. The weighted average fair value of shares issued under the Stock Purchase Plan in 2010, 2009 and 2008 was $3.60, $2.34 and $1.76 per share, respectively.
Share-based Compensation Expense
The Company accounts for share-based compensation in accordance with SFAS No. 123R, Share-Based Payment, as codified in FASB ASC topic 718, Compensation—Stock Compensation, or ASC 718, which was adopted January 1, 2006, utilizing the modified prospective transition method.
144
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 8. Stock Option and Purchase Plans (Continued)
Total estimated share-based compensation expense, related to all of the Company's share-based awards, recognized for the years ended December 31, 2010, 2009 and 2008 was comprised as follows (in thousands, except per share data):
| |
2010 | 2009 | 2008 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Continuing Operations: |
||||||||||
Research and development |
$ | 1,204 | $ | 921 | $ | 1,476 | ||||
General and administrative |
5,240 | 3,006 | 2,473 | |||||||
|
6,444 | 3,927 | 3,949 | |||||||
Discontinued Operations: |
||||||||||
Cost of goods sold and manufacturing |
810 | 688 | 602 | |||||||
Selling, general and administrative |
159 | 176 | 167 | |||||||
|
969 | 864 | 769 | |||||||
Share-based compensation expense before taxes |
7,413 | 4,791 | 4,718 | |||||||
Related income tax benefits |
— | — | — | |||||||
Share-based compensation expense, net of taxes |
$ | 7,413 | $ | 4,791 | $ | 4,718 | ||||
Net share-based compensation expense, per common share: |
||||||||||
Basic and diluted |
$ | 0.09 | $ | 0.07 | $ | 0.07 | ||||
Stock-based compensation expense for continuing and discontinued operations for the year ended December 31, 2010 included $7.3 million related to stock options and $141,000 related to the employee stock purchase plan, net of the estimated forfeitures. Stock-based compensation expense for continuing and discontinued operations for the year ended December 31, 2009 included $4.6 million related to stock options and $181,000 related to the employee stock purchase plan, net of the estimated forfeitures.
As of December 31, 2010, unrecognized estimated compensation expense totaled $5.9 million related to non-vested stock options and $81,000 related to the employee stock purchase plan. The weighted average remaining requisite service period of the non-vested options was 1.2 years and of the employee stock purchase plan was 4.5 months.
Valuation Assumptions
The fair value of each option award is estimated on the grant date using a Black-Scholes option-pricing model that uses the weighted average assumptions noted in the following table. Prior to January 1, 2008, the Company calculated the estimated life of stock options granted using a "simplified" method, which is based on the average of the vesting term and the term of the option, as a result of guidance from the SEC, as contained in Staff Accounting Bulletin No. 107 permitting the initial use of this method. Effective January 1, 2008, the expected term, which represents the period of time that options granted are expected to be outstanding, is derived by analyzing the historical experience of similar awards, giving consideration to the contractual terms of the stock-based awards, vesting schedules and expectations of future employee behavior. Expected volatilities are estimated using the historical share price performance over the expected term of the option. The Company also considers other factors such as its planned clinical trials and other company activities that may affect
145
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 8. Stock Option and Purchase Plans (Continued)
the volatility of VIVUS' stock in the future but determined that at this time, the historical volatility was more indicative of expected future stock price volatility. The risk-free interest rate for the period matching the expected term of the option is based on the U.S. Treasury yield curve in effect at the time of the grant. The Black-Scholes Model also requires a single expected dividend yield as an input. The Company does not anticipate paying any dividends in the near future. The Company develops pre-vesting forfeiture assumptions based on an analysis of historical data.
The following table sets forth information about the weighted-average assumptions used for options granted in the years ended December 31, 2010, 2009 and 2008:
| |
2010 | 2009 | 2008 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Expected life (in years) |
5.82 | 5.67 | 5.58 | |||||||
Volatility |
67.72 | % | 65.49 | % | 59.92 | % | ||||
Risk-free interest rate |
2.59 | % | 2.14 | % | 2.73 | % | ||||
Dividend yield |
— | — | — | |||||||
Note 9. Agreements
In 2001, VIVUS entered into a Development, Licensing and Supply Agreement with Tanabe for the development of avanafil, an oral PDE5 inhibitor investigational drug candidate for the treatment of erectile dysfunction. In October 2007, Tanabe and Mitsubishi Pharma Corporation completed their merger and announced their name change to Mitsubishi Tanabe Pharma Corporation, or MTPC. Under the terms of the 2001 Development, Licensing and Supply Agreement with Tanabe, the Company paid a $2 million license fee obligation to Tanabe in the year ended December 31, 2006. No payments were made under this agreement with MTPC in the year ended December 31, 2008; however, the Company paid MTPC $4 million in January 2009 following the enrollment in December 2008 of the first patient in the first Phase 3 clinical study. The Company expects to make other substantial payments to MTPC in accordance with its agreements with MTPC as the Company continues to develop and, if approved for sale, commercialize avanafil for the oral treatment of male sexual dysfunction. Such potential future milestone payments total $15 million in the aggregate and include payments upon: the first submission of an NDA, expected in the second quarter of 2011; obtainment of the first regulatory approval in the U.S. and any major European country; and achievement of $250 million or more in calendar year sales.
The term of the MTPC agreement is based on a country-by-country and on a product-by-product basis. The term shall continue until the later of (i) 10 years after the date of the first sale for a particular product, or (ii) the expiration of the last-to-expire patents within the MTPC patents covering such product in such country. In the event that the Company's product is deemed to be (i) insufficiently effective or insufficiently safe relative to other PDE5 inhibitor compounds based on published information, or (ii) not economically feasible to develop due to unforeseen regulatory hurdles or costs as measured by standards common in the pharmaceutical industry for this type of product, the Company has the right to terminate the agreement with MTPC with respect to such product.
On October 16, 2001, the Company entered into an assignment agreement, or the Assignment Agreement, with Thomas Najarian, M.D. for a combination of pharmaceutical agents for the treatment of obesity and other disorders, or the Combination Therapy, that has since been the focus of our investigational drug candidate development program for QNEXA for the treatment of obesity, obstructive sleep apnea and diabetes. The Combination Therapy and all related patent applications, or
146
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 9. Agreements (Continued)
the Patents, were transferred to the Company with worldwide rights to develop and commercialize the Combination Therapy and exploit the Patents. Pursuant to the Assignment Agreement, the Company has paid a total of $220,000 to Dr. Najarian through December 31, 2010 and has issued him options to purchase 40,000 shares of our common stock. The Company is obligated under the terms of the Assignment Agreement to make a milestone payment of $1 million and issue an option to purchase 20,000 shares of VIVUS' common stock to Dr. Najarian upon marketing approval by the FDA of a product for the treatment of obesity that is based upon the Combination Therapy and Patents. The Assignment Agreement will require the Company to pay royalties on worldwide net sales of a product for the treatment of obesity that is based upon the Combination Therapy and Patents until the last-to-expire of the assigned Patents. To the extent that the Company decides not to commercially exploit the Patents, the Assignment Agreement will terminate and the Combination Therapy and Patents will be assigned back to Dr. Najarian. In 2006, Dr. Najarian joined the Company as a part-time employee and currently serves as a Principal Scientist.
Note 10. Interest and Other Income, net
The components of interest and other income, net were as follows (in thousands):
| |
Years Ended December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | ||||||||
Interest income |
$ | 219 | $ | 913 | $ | 5,385 | |||||
Realized gains (losses) on marketable securities, net |
5 | 1,085 | (979 | ) | |||||||
Therapeutic discovery grant |
244 | — | — | ||||||||
Interest and other income, net |
$ | 468 | $ | 1,998 | $ | 4,406 | |||||
Note 11. Comprehensive Income (Loss)
Comprehensive income (loss) consisted of the following (in thousands):
| |
Years Ended December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | ||||||||
Net loss |
$ | (66,065 | ) | $ | (54,291 | ) | $ | (9,940 | ) | ||
Unrealized gain (loss) on securities, net of taxes |
7 | (357 | ) | 422 | |||||||
Total comprehensive loss |
$ | (66,058 | ) | $ | (54,648 | ) | $ | (9,518 | ) | ||
Note 12. Commitments
Lease Commitments
In November 2006, the Company entered into a 30-month lease for its corporate headquarters located in Mountain View, California. The lease commenced on February 1, 2007. The base monthly rent was set at $1.85 per square foot or $26,000 per month. The lease expired on July 31, 2009. On December 16, 2008, the Company entered into a first amendment to this lease. Under the terms of the amended lease, it continues to lease the office space for its corporate headquarters for a two-year period commencing on August 1, 2009 and expiring on July 31, 2011. The base monthly rent was set at
147
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 12. Commitments (Continued)
$1.64 per square foot or $23,000 per month. The amended lease allowed the Company one option to extend the term of the lease for one year from the expiration of the lease. On November 12, 2009, the Company entered into a second amendment to this lease. The second amendment commenced on January 1, 2010, expires on July 31, 2011 and expands the leased space. The base rent for the expansion space was set at $2.25 per square foot or $8,500 per month. The option to extend the term of the amended lease for one year from the expiration of the lease applies to this expansion space as well. In December 2010, the Company entered into a third amendment to this lease. The third amendment extended the lease term for the original premises and the expansion space for a period of twelve months commencing August 1, 2011 and terminating July 31, 2012. Under the third amendment, the base rent for the original space will be set at $1.69 per square foot or $24,000 per month and the base rent for the expansion space will be set at $2.31 per square foot or $8,700 per month. The amended lease allows the Company one additional option to extend the term of the lease for one year from the expiration of the lease. The option to extend the term of the amended lease for one year from the expiration of the lease applies to this expansion space as well.
Future minimum lease payments under operating leases are as follows (in thousands):
2011 |
$ | 685 | |||
2012 |
406 | ||||
Total |
$ | 1,091 | |||
Rent expense under operating leases totaled $676,000, $531,000 and $549,000 for the years ended December 31, 2010, 2009 and 2008, respectively.
Other Agreements
The Company has entered into various agreements with clinical consultants and clinical research organizations to perform clinical studies on its behalf and at December 31, 2010, its remaining commitment under these agreements totaled $6.6 million. The Company has remaining commitments under various general and administrative services agreements totaling $3 million at December 31, 2010, including $1.5 million related to Leland F. Wilson's Employment Agreement (see paragraph below). The Company has also entered into various agreements with research consultants and other contractors to perform regulatory services, drug research and testing and, at December 31, 2010, its remaining commitment under these agreements totaled $3.6 million.
On December 19, 2007, the Compensation Committee of the Board of Directors of the Company approved an employment agreement, or the Employment Agreement, with Leland F. Wilson, the Company's President and Chief Executive Officer. The Employment Agreement includes salary, incentive compensation, retirement benefits and length of employment, among other items, as agreed to with Mr. Wilson. The Employment Agreement had an initial term of two years commencing on the effective date, June 1, 2007, or the Effective Date. On January 23, 2009, the Compensation Committee approved an amendment to the Employment Agreement, or the Amendment, which amends the Employment Agreement. Pursuant to the Amendment, the initial term of the Employment Agreement was increased from two to three years commencing on June 1, 2007 and other relevant dates were also extended to reflect the three-year initial term. On January 21, 2011, the Compensation Committee approved the second amendment to Mr. Wilson's Employment Agreement. Pursuant to the second
148
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 12. Commitments (Continued)
amendment, the initial term of the Employment Agreement is increased to four years commencing on June 1, 2007.
Indemnifications
In the normal course of business, the Company provides indemnifications of varying scope to certain customers against claims of intellectual property infringement made by third parties arising from the use of its products and to its clinical research organizations and investigator sites against liabilities incurred in connection with any third-party claim arising from the work performed on behalf of the Company, among others. Historically, costs related to these indemnification provisions have not been significant and the Company is unable to estimate the maximum potential impact of these indemnification provisions on its future results of operations.
Pursuant to the terms of the Asset Purchase Agreement for the sale of the Evamist product to K-V, the Company made certain representations and warranties concerning its rights and assets related to Evamist and the Company's authority to enter into and consummate the transaction. The Company also made certain covenants that survive the closing date of the transaction, including a covenant not to operate a business that competes, in the U.S., and its territories and protectorates, with the Evamist product. See Note 16: "Legal Matters" for further information regarding Acrux.
Pursuant to the terms of the Asset Purchase Agreement, (see Note 3: "Discontinued Operations"), the Company entered into with Meda AB, or Meda, to sell certain of the assets related to the MUSE business to Meda, or the MUSE Transaction, the Company agreed to indemnify Meda in connection with the representations and warranties that it made concerning its rights, liabilities and assets related to the MUSE business and its authority to enter into and consummate the MUSE Transaction. The Company also made certain covenants in the Asset Purchase Agreement which survive the closing of the MUSE Transaction, including a three year covenant not to develop, manufacture, promote or commercialize a trans-urethral erectile dysfunction drug.
To the extent permitted under Delaware law, the Company has agreements whereby it indemnifies its officers and directors for certain events or occurrences while the officer or director is, or was, serving at the Company's request in such capacity. The indemnification period covers all pertinent events and occurrences during the officer's or director's lifetime. The maximum potential amount of future payments the Company could be required to make under these indemnification agreements is unlimited; however, the Company maintains director and officer insurance coverage that reduces its exposure and enables the Company to recover a portion of any future amounts paid. The Company believes the estimated fair value of these indemnification agreements in excess of applicable insurance coverage is minimal.
149
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 13. Income Taxes
Deferred income taxes result from differences in the recognition of expenses for tax and financial reporting purposes, as well as operating loss and tax credit carryforwards. Significant components of the Company's deferred income tax assets as of December 31, 2010 and 2009 are as follows (in thousands):
| |
2010 | 2009 | |||||||
|---|---|---|---|---|---|---|---|---|---|
Deferred tax assets: |
|||||||||
Net operating loss carry forwards |
$ | 85,518 | $ | 63,836 | |||||
Research and development credit carry forwards |
14,120 | 12,251 | |||||||
Inventory reserve |
— | 603 | |||||||
Accruals and other |
10,073 | 8,021 | |||||||
Depreciation |
3,393 | 5,816 | |||||||
Deferred revenue |
— | 483 | |||||||
|
113,104 | 91,010 | |||||||
Valuation allowance |
(113,104 | ) | (91,010 | ) | |||||
Total |
$ | — | $ | — | |||||
The Company makes certain estimates and judgments in determining income tax expense for financial statement purposes. These estimates and judgments occur in the calculation of certain tax assets and liabilities, which arise from differences in the timing of recognition of revenue and expense for tax and financial statement purposes.
As part of the process of preparing its consolidated financial statements, the Company is required to estimate its income taxes in each of the jurisdictions in which it operates. This process involves estimating its current tax exposure under the most recent tax laws and assessing temporary differences resulting from differing treatment of items for tax and accounting purposes. The differences result in deferred tax assets and liabilities, which are included in the Company's consolidated balance sheets.
The Company assesses the likelihood that it will be able to recover its deferred tax assets. The Company considers all available evidence, both positive and negative, including historical levels of income, expectations and risks associated with estimates of future taxable income and ongoing prudent and feasible tax planning strategies in assessing the need for a valuation allowance. If it is not more likely than not that the Company will recover its deferred tax assets, the Company will increase its provision for taxes by recording a valuation allowance against the deferred tax assets that the Company estimates will not ultimately be recoverable. As a result of the Company's analysis of all available evidence, both positive and negative, as of December 31, 2010, it was considered more likely than not that the Company's deferred tax assets would not be realized. However, should there be a change in the Company's ability to recover its deferred tax assets, the Company would recognize a benefit to its tax provision in the period in which the Company determines that it is more likely than not that it can recover its deferred tax assets.
The net increase in the valuation allowance for the years ended December 31, 2010, 2009 and 2008 was $22.1 million, $23.7 million and $4.1 million, respectively. As of December 31, 2010, the Company had no significant deferred tax liabilities.
For federal and state income tax reporting purposes, respective net operating loss, or NOL, carryforwards of approximately $234.5 million and $54.3 million are available to reduce future taxable income, if any. ASC 718 prohibits recognition of a deferred income tax asset for excess tax benefits due
150
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 13. Income Taxes (Continued)
to stock option exercises that have not yet been realized through a reduction in income taxes payable. Post-adoption of ASC 718, the unrecognized deferred tax benefits totaled $2.8 million, of which $81,000 have been accounted for as a credit to additional paid-in capital, as they have been realized through a reduction in income taxes payable. For federal and state income tax reporting purposes, respective credit carryforwards of approximately $13.2 million and $2.9 million are available to reduce future taxable income, if any. These net operating loss and tax credit carryforwards, except for the California research and development credit, expire on various dates through 2030. The California research and development credits do not expire. The Internal Revenue Code of 1986, as amended, contains provisions that may limit the net operating loss and credit carryforwards available for use in any given period upon the occurrence of certain events, including a significant change in ownership interest.
The (benefit)/provision for income taxes is based upon (loss)/income from continuing operations before (benefit)/provision for income taxes as follows, for the years ended December 31, 2010, 2009 and 2008 (in thousands):
| |
2010 | 2009 | 2008 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Loss before income taxes: |
|||||||||||
Domestic |
$ | (75,425 | ) | $ | (55,764 | ) | $ | (9,128 | ) | ||
International |
— | — | 19 | ||||||||
Loss before taxes |
$ | (75,425 | ) | $ | (55,764 | ) | $ | (9,147 | ) | ||
The (benefit)/provision for income taxes consists of the following components for the years ended December 31, 2010, 2009 and 2008 (in thousands):
Continuing Operations:
| |
2010 | 2009 | 2008 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Current |
|||||||||||
Federal |
$ | — | $ | (2,380 | ) | $ | (8 | ) | |||
State |
9 | 1 | 1 | ||||||||
Foreign |
— | — | — | ||||||||
Total current (benefit)/provision for income taxes |
$ | 9 | $ | (2,379 | ) | $ | (7 | ) | |||
Deferred |
|||||||||||
Federal |
$ | — | $ | — | $ | — | |||||
State |
— | — | — | ||||||||
Foreign |
— | — | — | ||||||||
Total deferred benefit for income taxes |
$ | — | $ | — | $ | — | |||||
Total (benefit)/provision for income taxes from continuing operations |
$ | 9 | $ | (2,379 | ) | $ | (7 | ) | |||
151
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 13. Income Taxes (Continued)
Discontinued Operations:
| |
2010 | 2009 | 2008 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Total (benefit)/provision for income taxes from discontinued operations |
$ | 29 | $ | (61 | ) | $ | 10 | |||
A reconciliation between the U.S. federal statutory tax rate and the Company's effective tax rate from continuing operations is as follows, for the years ended December 31, 2010, 2009 and 2008:
| |
2010 | 2009 | 2008 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Tax at U.S. federal statutory rate |
(35 | )% | (35 | )% | (35 | )% | |||||
Change in valuation allowance |
35 | 39 | 35 | ||||||||
Permanent items |
1 | 1 | 10 | ||||||||
Extinguishment of debt |
3 | — | — | ||||||||
Tax credits |
(4 | ) | (5 | ) | (10 | ) | |||||
Refund of prior taxes |
— | (4 | ) | — | |||||||
Effective tax rate |
0 | % | (4 | )% | 0 | % | |||||
Effective January 1, 2007, the Company adopted Financial Accounting Standards Interpretation, or FIN, No. 48, Accounting for Uncertainty in Income Taxes—an interpretation of FASB Statement No. 109, as codified in FASB ASC topic 740, Income Taxes, or ASC 740, which prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of uncertain tax positions taken or expected to be taken in a company's income tax return, and also provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosure, and transition. ASC 740-10 utilizes a two-step approach for evaluating uncertain tax positions accounted for in accordance with SFAS No. 109, Accounting for Income Taxes, also as codified in ASC 740. Step one, Recognition, requires a company to determine if the weight of available evidence indicates that a tax position is more likely than not to be sustained upon audit, including resolution of related appeals or litigation processes, if any. Step two, Measurement, is based on the largest amount of benefit, which is more likely than not to be realized on ultimate settlement.
The total gross unrecognized tax benefits as of December 31, 2010 is $1.2 million and relates to state tax exposures, of which $7,000 would affect the effective tax rate if recognized.
A reconciliation of the beginning and ending amount of unrecognized tax benefits in 2010 and 2009 is as follows (in thousands):
| |
2010 | 2009 | 2008 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Unrecognized tax benefits as of January 1 |
$ | — | $ | — | $ | — | ||||
Gross increase for tax positions of prior years |
1,171 | — | — | |||||||
Gross decrease for tax positions of prior years |
— | — | — | |||||||
Gross increase for tax positions of current year |
— | — | — | |||||||
Gross decrease for tax positions of current year |
— | — | — | |||||||
Settlements |
— | — | — | |||||||
Lapse of statute of limitations |
— | — | — | |||||||
Unrecognized tax benefits balance at December 31 |
$ | 1,171 | $ | — | $ | — | ||||
152
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 13. Income Taxes (Continued)
The total unrecognized tax benefits as of December 31, 2010 include approximately $1.2 million of unrecognized tax benefits that have been netted against the related deferred tax assets. The remaining balance recorded on the Company's consolidated balance sheets in 2010 and 2009 is as follows (in thousands):
| |
2010 | 2009 | |||||
|---|---|---|---|---|---|---|---|
Total unrecognized tax benefits balance |
$ | 1,171 | $ | — | |||
Amounts netted against deferred tax assets |
(1,164 | ) | — | ||||
Unrecognized tax benefits recorded on consolidated balance sheets |
$ | 7 | $ | — | |||
The Company recognizes interest and penalties accrued on any unrecognized tax benefits as a component of its provision for income taxes. As of January 1, 2010, the Company had no accrual for payment of interest and penalties related to unrecognized tax benefits. To the extent accrued interest and penalties do not ultimately become payable, amounts accrued will be reduced and reflected as a reduction of the overall income tax provision in the period that such determination is made. During 2010, $1,000 of interest was recognized.
Although the Company files U.S. federal, various state, and foreign tax returns, the Company's only major tax jurisdictions are the U.S., California and New Jersey. The Company's income tax return for the year ended December 31, 2007 is currently under examination by the California Franchise Tax Board. Based on the progress of the audit to date, the Company believes adjustments may be made in early years that will reduce tax attributes available to offset tax due in 2007. Therefore, the Company has increased their unrecognized tax benefits to $7,000 for the year ended December 31, 2010.
The Company's income tax return for the years ended December 31, 2007 and 2008 are currently under examination by the Internal Revenue Service. Because the Company used net operating loss carryforwards and other tax attributes to offset its taxable income in on its 2007 income tax returns for U.S. Federal and California, such attributes can be adjusted by these taxing authorities until the statute closes on the year in which such attributes were utilized. Tax years 1991 to 2009 remain subject to examination by the appropriate governmental agencies due to tax loss carryovers from those years.
The Company is in various stages of the examination process in connection with all of its tax audits and it is difficult to determine when these examinations will be settled. It is reasonably possible that over the next twelve-month period the Company may experience an increase or decrease in its unrecognized tax benefits. It is not possible to determine either the magnitude or range of any increase or decrease at this time.
Note 14. Concentration of Suppliers
The Company relies on third party sole-source manufacturers to produce its clinical trial materials, components and raw materials. Third party manufacturers may not be able to meet the Company's needs with respect to timing, quantity or quality. Several of the Company's manufacturers are sole-source manufacturers where no alternative suppliers exist. For the years ended December 31, 2010, 2009 and 2008, the Company incurred $7.8 million, $19.1 million and $44.7 million, respectively, for services provided by one clinical research organization on the QNEXA Phase 3 studies, which represented 20%, 27% and 58%, respectively, of the Company's total research and development expenses. Separately, the Company incurred another $8.2 million and $17 million for services provided
153
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 14. Concentration of Suppliers (Continued)
by another clinical research organization on the avanafil Phase 3 studies, which represented 20% and 24% of the Company's total research and development expenses for the years ended December 31, 2010 and 2009, respectively. For the years ended December 31, 2010, 2009 and 2008, the Company incurred $4.9 million, $7.8 million and $6.2 million, respectively, for clinical supplies and formulation work performed by the Company's sole-source manufacturer, which represented 12%, 11% and 8%, respectively, of the Company's total research and development expenses.
Note 15. 401(k) Plan
All of the Company's full-time employees are eligible to participate in the VIVUS 401(k) Plan. The employer-matching portion of the 401(k) plan began on July 1, 2000. Employer-matching contributions for the years ended December 31, 2010, 2009 and 2008 were as follows (in thousands):
| |
2010 | 2009 | 2008 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Continuing Operations: |
||||||||||
Research and development |
$ | 90 | $ | 92 | $ | 63 | ||||
General and administrative |
105 | 63 | 49 | |||||||
|
195 | 155 | 112 | |||||||
Discontinued Operations: |
||||||||||
Cost of goods sold and manufacturing |
121 | 122 | 101 | |||||||
Selling, general and administrative |
37 | 40 | 33 | |||||||
|
158 | 162 | 134 | |||||||
Total 401(k) Plan employer-matching contributions |
$ | 353 | $ | 317 | $ | 246 | ||||
Note 16. Legal Matters
In the normal course of business, the Company receives claims and makes inquiries regarding patent infringement and other related legal matters. The Company believes that it has meritorious claims and defenses and intends to pursue any such matters vigorously.
Acrux Dispute
The Company and Acrux Limited, through its wholly owned subsidiary FemPharm Pty Ltd., or Acrux, were parties to the Testosterone Development and Commercialization Agreement dated February 12, 2004, or the Testosterone Agreement. The Testosterone Agreement covers the Company's investigational product candidate, Luramist, which was licensed from Acrux under the Testosterone Agreement. On November 5, 2007, Acrux made a demand for arbitration under the Testosterone Agreement regarding certain claims related to Luramist. Acrux's demand sought a reversion of all rights assigned to the Company related to Luramist, monetary damages and the payment of a milestone payment for Luramist under the Testosterone Agreement and declaratory relief. The Company asserted counterclaims against Acrux in the arbitration and sought the enforcement of the Company's rights under the Testosterone Agreement. The arbitration hearing concluded on January 23, 2009, and on April 6, 2009 the panel of arbitrators, or the Panel, issued its Interim Arbitration Award finding in favor of the Company that it was in compliance with the Testosterone Agreement and denying all of the relief sought by Acrux in its demand. The Panel found that the Company was not in breach of the
154
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 16. Legal Matters (Continued)
Luramist license agreement and that the Company had used diligent, commercially reasonable efforts to develop Luramist. The Panel further ruled in favor of the Company on its counterclaim that Acrux had breached the Luramist license agreement by failing to provide certain know-how and certain improvements in the formulation and delivery device for Luramist. The Panel denied the Acrux claim for additional milestone payments. The Panel ordered Acrux to turn over certain information to the Company that was previously withheld in violation of the agreement by Acrux. After the parties failed to agree on a new Outside Date by which the Company was to commence its first Phase 3 trial for Luramist, the Panel reset the Outside Date of April 30, 2006 to April 1, 2010 to reflect the regulatory environment.
On March 30, 2010, the Company provided written notice to Acrux of its intent to terminate the Testosterone Agreement. On April 6, 2010, in connection with Acrux's request for further briefing on the issue of damages in light of the Company's termination of the Testosterone Agreement, the Panel ordered the parties to enter into settlement discussions. On May 6, 2010, the parties agreed to the terms of a settlement agreement and mutual release, or the Settlement Agreement, resolving any and all claims or potential claims in the arbitration and that may have or could have arisen from any case whatsoever, other than certain rights and obligations that survive the termination of the Testosterone Agreement or are required by the Settlement Agreement. Pursuant to the Settlement Agreement, the Company transferred Luramist-related assets to Acrux, including clinical trial material, batch release documents, inventory of applicators, FDA correspondence, intellectual property and know-how and trademarks. In addition, the Company ceased its clinical study program for Luramist as part of the settlement. The parties did not exchange cash payments as a result of the settlement and termination of the Testosterone Agreement. The Panel retains jurisdiction over the matter to enforce the terms of the Settlement Agreement.
Securities Related Class Action Lawsuits
A federal securities class action lawsuit captioned Kovtun v. Vivus, Inc., et al. is pending in the U.S. District Court, Northern District of California, which asserts claims for violations of Section 10(b) and 20(a) of the federal securities laws, purportedly relating to statements allegedly made by the Company in connection with its New Drug Application, or NDA, for QNEXA as a treatment for obesity. The essential factual allegation is that the Company and its officers misled the investing public regarding the prospects for QNEXA's NDA approval, and the drug's efficacy and safety. On February 2, 2011, the court granted a stipulation and order appointing a lead plaintiff and a lead counsel for the class. On February 3, 2011, the court granted a stipulation and order requiring the lead plaintiff to file any amendment to the operative complaint no later than 60 days from February 2, 2011, with the defendants' answer or motion to dismiss to be filed no later than 60 days after plaintiffs file the amended complaint. In the event the defendants file a motion to dismiss, plaintiffs' opposition to such motion must be filed no later than 60 days after the filing of the motion, with defendants' reply to the opposition being filed no later than 45 days after the filing of plaintiffs' opposition. Discovery is stayed in the Kovtun matter pending resolution of the defendants' motion to dismiss.
Also pending in the U.S. District Court, Northern District of California and formally related to the Kovtun matter is a shareholder derivative action captioned Turberg v. Logan, et al., which restates the allegations in Kovtun by claiming that certain of the Company's officers and directors caused or allowed the Company to violate the federal securities laws by issuing material misrepresentations to the investing public. On February 7, 2011, the court granted a stipulation and order requiring plaintiffs to
155
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 16. Legal Matters (Continued)
file and serve any amendment to the complaint no later than 60 days from the filing of the consolidated amended complaint in the Kovtun matter, with the defendants' answer or motion to dismiss to be filed no later than 60 days after the court enters an order ruling on defendants' motion to dismiss in the Kovtun matter. In the event the defendants file a motion to dismiss in the Turberg matter, the plaintiffs will have 60 days to oppose the motion and the defendants will have 45 days to reply to the plaintiffs' opposition. The court order stayed all discovery in the Turberg matter pending resolution of the defendants' motion to dismiss.
Additionally, three separate shareholder derivative suits are pending in California Superior Court, Santa Clara County. The allegations in these shareholder derivative suits are nearly identical to those in the Turberg federal shareholder derivative action. On February 3, 2011, the court granted a stipulation and order consolidating the three suits into the action captioned Wilkinson v. Wilson, et al. and appointing a lead plaintiff and a lead counsel for the shareholder derivative actions. The court order requires the plaintiffs to file and serve a consolidated complaint no later than 60 days from the court's order and the parties to meet and confer on a briefing schedule for the defendants' motion to dismiss the consolidated complaint no later than 30 days from the service of the consolidated complaint.
The Company believes that the allegations in the various federal and state actions have no merit and that the Company has meritorious defenses to the claims stated in such actions. The Company intends to vigorously defend itself in the various actions. Although there may be no merit to such allegations or claims, the Company will be required to allocate monetary and personnel resources to defend itself. The Company believes the disposition of the current lawsuits and claims is not likely to have a material effect on its financial condition or liquidity.
In the ordinary course of business the Company may become involved in lawsuits and subject to various claims from current and former employees including wrongful termination, sexual discrimination and employment matters. The Company is currently a party to a lawsuit involving a former employee. The Company has also been named as a potential defendant in a complaint filed by a former employee. The Company has investigated each of the claims and believes the allegations have no merit and that the Company has meritorious defenses to such charges. Due to the current economic downturn, employees may be more likely to file employment-related claims. Employment-related claims also may be more likely following a poor performance review. Although there may be no merit to such claims or legal matters, the Company may be required to allocate additional monetary and personnel resources to defend against these type of allegations. The Company believes the disposition of the current lawsuit and claims is not likely to have a material effect on its financial condition or liquidity.
The Company is not aware of any other asserted or unasserted claims against it where an unfavorable resolution would have an adverse material impact on the operations or financial position of the Company.
Note 17. Related Party Transactions
Mario M. Rosati, one of the Company's directors until June 13, 2008, is also a member of Wilson Sonsini Goodrich & Rosati, Professional Corporation, which has served as the Company's outside corporate counsel since its formation and has received compensation at normal commercial rates for these services. During the year ended December 31, 2008, the Company paid $758,000 to Wilson Sonsini Goodrich and Rosati for legal services. On April 17, 2008, Mr. Rosati notified the Company of
156
VIVUS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 17. Related Party Transactions (Continued)
his decision not to stand for re-election at the Company's 2008 annual meeting held on June 13, 2008 and is no longer a member of the Company's Board of Directors as of that date.
Note 18. Subsequent Events (Unaudited)
On January 19, 2011, the Company held an End-of-Review meeting with the FDA to discuss its planned response to the Complete Response Letter, or CRL, received on October 28, 2010, regarding the New Drug Application, or NDA, for QNEXA as a treatment for obesity and the Company's planned resubmission of the NDA for QNEXA. The CRL stated that in its current form, the NDA for QNEXA was not approvable. The FDA has requested that the Company assess the feasibility of performing a retrospective observational study utilizing existing databases to review fetal outcomes including the historical incidence of congenital malformations with an interest in oral cleft and low birth weight (less than 2,500 gm) in the offspring of women who received prophylaxis treatment with 100mg of topiramate for migraine during pregnancy, or the Feasibility Assessment, which is currently in process.
Note 19. Selected Financial Data (Unaudited)
Selected Quarterly Financial Data (in thousands)
| |
Quarter Ended, | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
March 31 | June 30 | September 30 | December 31 | |||||||||||
2010 |
|||||||||||||||
Revenue |
$ | — | $ | — | $ | — | $ | — | |||||||
Operating expenses |
$ | 15,375 | $ | 20,326 | $ | 16,815 | $ | 13,111 | |||||||
Net loss from continuing operations |
$ | (16,609 | ) | $ | (21,570 | ) | $ | (18,136 | ) | $ | (19,119 | ) | |||
Net income (loss) from discontinued operations |
$ | (2,209 | ) | $ | (1,187 | ) | $ | 157 | $ | 12,608 | |||||
Basic and diluted net income (loss) per share: |
|||||||||||||||
Continuing operations |
$ | (0.21 | ) | $ | (0.27 | ) | $ | (0.22 | ) | $ | (0.23 | ) | |||
Discontinued operations |
$ | (0.02 | ) | $ | (0.01 | ) | $ | 0.00 | $ | 0.15 | |||||
2009 |
|||||||||||||||
Revenue |
$ | 20,930 | $ | 10,465 | $ | — | $ | — | |||||||
Operating expenses |
$ | 24,043 | $ | 23,065 | $ | 20,496 | $ | 17,206 | |||||||
Net loss from continuing operations |
$ | (3,980 | ) | $ | (12,748 | ) | $ | (21,114 | ) | $ | (15,543 | ) | |||
Net income (loss) from discontinued operations |
$ | (2,829 | ) | $ | (456 | ) | $ | 48 | $ | 2,331 | |||||
Basic and diluted net income (loss) per share: |
|||||||||||||||
Continuing operations |
$ | (0.06 | ) | $ | (0.18 | ) | $ | (0.30 | ) | $ | (0.19 | ) | |||
Discontinued operations |
$ | (0.04 | ) | $ | (0.01 | ) | $ | 0.00 | $ | 0.03 | |||||
157
The financial statement Schedule II—VALUATION AND QUALIFYING ACCOUNTS is filed as part of the Form 10-K.
VIVUS, Inc.
SCHEDULE II—VALUATION AND QUALIFYING ACCOUNTS
(in thousands)
Each of the following valuation and qualifying accounts are reported as assets and liabilities of discontinued operations in the consolidated balance sheets for all periods presented.
| |
Balance at Beginning of Period |
Charged to Operations |
Charges Utilized |
Balance at End of Period |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Allowance for Doubtful Accounts |
||||||||||||||
Fiscal year ended December 31, 2008 |
$ | 29 | $ | (6 | ) | $ | — | $ | 23 | |||||
Fiscal year ended December 31, 2009 |
23 | 45 | (41 | ) | 27 | |||||||||
Fiscal year ended December 31, 2010 |
27 | (7 | ) | (20 | ) | — | ||||||||
Inventory Reserve |
||||||||||||||
Fiscal year ended December 31, 2008 |
1,664 | 213 | (432 | )(1) | 1,445 | |||||||||
Fiscal year ended December 31, 2009 |
1,445 | 224 | (95 | )(2) | 1,574 | |||||||||
Fiscal year ended December 31, 2010 |
1,574 | (353 | ) | (1,221 | )(3) | — | ||||||||
Accrued Product Returns |
||||||||||||||
Fiscal year ended December 31, 2008 |
2,498 | 1,314 | (947 | ) | 2,865 | |||||||||
Fiscal year ended December 31, 2009 |
2,865 | 1,425 | (1,264 | ) | 3,026 | |||||||||
Fiscal year ended December 31, 2010 |
3,026 | 906 | (1,334 | ) | 2,598 | |||||||||
Accrued Chargebacks Reserve |
||||||||||||||
Fiscal year ended December 31, 2008 |
1,314 | 4,081 | (4,016 | ) | 1,379 | |||||||||
Fiscal year ended December 31, 2009 |
1,379 | 4,538 | (4,300 | ) | 1,617 | |||||||||
Fiscal year ended December 31, 2010 |
$ | 1,617 | $ | 3,103 | $ | (4,248 | ) | $ | 472 | |||||
- (1)
- The
Company used $67,000 of its fully reserved component parts inventory in production. The fully reserved inventory is charged to cost of goods sold at a
zero basis when used, which has a favorable impact on gross profit.
- (2)
- The
Company used $97,000 of its fully reserved component parts inventory in production. The fully reserved inventory is charged to cost of goods sold at a
zero basis when used, which has a favorable impact on gross profit.
- (3)
- The Company used $98,000 of its fully reserved component parts inventory and $367,000 of its fully reserved raw materials inventory in production. The fully reserved inventory is charged to cost of goods sold at a zero basis when used, which has a favorable impact on gross profit.
158
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
(a.) Evaluation of disclosure controls and procedures. We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the timelines specified in the Securities and Exchange Commission's rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can only provide reasonable assurance of achieving the desired control objectives, and in reaching a reasonable level of assurance, management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
As required by SEC Rule 13a-15(b), the Company carried out an evaluation, under the supervision and with the participation of the Company's management, including the Company's Chief Executive Officer and the Company's Chief Financial Officer, of the effectiveness of the design and operation of the Company's disclosure controls and procedures as of the end of the year covered by this report. Based on the foregoing, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level.
(b.) Changes in internal controls. During the fourth quarter of 2010, we sold certain of our rights and assets related to MUSE, our approved treatment for erectile dysfunction. Most of the employees relating to MUSE in New Jersey were hired by the purchaser of MUSE. Certain functions that were previously performed by employees in New Jersey, such as payroll and benefits processing, human resources, information systems, among other functions have been transferred to personnel at our headquarters in Mountain View, California. As a result, the responsibilities for certain internal controls were modified, or transferred to personnel in Mountain View to maintain our existing internal controls over financial reporting. We have assessed the internal controls over the key processes affected by the sale of MUSE and concluded that we have maintained adequate internal control over financial reporting.
Management's Annual Report on Internal Control Over Financial Reporting
Internal control over financial reporting refers to the process designed by, or under the supervision of, our Chief Executive Officer and Chief Financial Officer, and effected by our Board of Directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that:
- (1)
- Pertain
to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets;
- (2)
- Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorization of our management and directors; and
159
- (3)
- Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisitions, use or disposition of our assets that could have a material effect on the financial statements.
Internal control over financial reporting cannot provide absolute assurance of achieving financial reporting objectives because of its inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk. Management is responsible for establishing and maintaining adequate internal control over financial reporting for the company.
Management has used the framework set forth in the report entitled Internal Control-Integrated Framework published by the Committee of Sponsoring Organizations of the Treadway Commission, known as COSO, to evaluate the effectiveness of the Company's internal control over financial reporting. Based on this assessment, management has concluded that our internal control over financial reporting was effective as of December 31, 2010. Odenberg Ullakko Muranishi & Co. LLP, the independent registered public accounting firm that audited the consolidated financial statements included in the Annual Report on Form 10-K, has issued an attestation report on the effectiveness of our internal control over financial reporting as of December 31, 2010. This report, which expresses an unqualified opinion on the effectiveness of our internal controls over financial reporting as of December 31, 2010, is included herein.
There has been no change in our internal controls over financial reporting during our most recent fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal controls over financial reporting.
None.
160
Item 10. Directors, Executive Officers and Corporate Governance
The information required by this item is hereby incorporated by reference from the information under the captions "Election of Directors" and "Executive Officers" contained in the Company's definitive Proxy Statement, to be filed with the Securities and Exchange Commission no later than 120 days from the end of the Company's last fiscal year in connection with the solicitation of proxies for its 2011 Annual Meeting of Stockholders. The information required by Section 16(a) is incorporated by reference from the information under the caption "Compliance with Section 16(a) of the Securities Exchange Act of 1934" in the Proxy Statement.
The Company has adopted a code of ethics that applies to its Chief Executive Officer, Chief Financial Officer, and to all of its other officers, directors, employees and agents. The code of ethics is available at the Corporate Governance section of the Investor Relations page on the Company's website at www.vivus.com. The Company intends to disclose future amendments to, or waivers from, certain provision of its code of ethics on the above website within five business days following the date of such amendment or waiver.
Item 11. Executive Compensation
The information required by this item is incorporated by reference from the information under the caption "Executive Officer Compensation" in the Company's Proxy Statement referred to in Item 10 above.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Equity Compensation Plans Approved by Stockholders
Information about our equity compensation plans at December 31, 2010 that were approved by our stockholders was as follows:
Plan Category
|
Number of Shares to be issued Upon Exercise of Outstanding Options and Rights(a) |
Weighted Average Exercise Price of Outstanding Options |
Number of Shares Remaining Available for Future Issuance(c) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
Equity compensation plans approved by stockholders |
7,519,013 | $ | 5.47 | 8,583,996 | ||||||
Equity compensation plans not approved by stockholders(b) |
400,000 | $ | 10.19 | — | ||||||
Total |
7,919,013 | $ | 5.71 | 8,583,996 | ||||||
- (a)
- Consists
of three plans: our 1994 Stock Option Plan, our 2001 Stock Option Plan and our 2010 Stock Option Plan.
- (b)
- On
April 30, 2010, the Company's Board of Directors granted an option to purchase 400,000 shares of the Company's common stock, or the Inducement
Grant, to Michael P. Miller, the Company's new Senior Vice President and Chief Commercial Officer. The Inducement Grant was granted outside of the Company's 2010 Plan and without stockholder approval
pursuant to NASDAQ Listing Rule 5635(c)(4) and is subject to the terms and conditions of the Stand-Alone Stock Option Agreement between the Company and Michael P. Miller.
- (c)
- Includes 8,511,624 shares for the 2010 Stock Option Plan and 72,372 shares for the 1994 Employee Stock Purchase Plan.
161
The information required by this item is incorporated by reference from the information under the caption "Security Ownership of Certain Beneficial Owners and Management" in the Company's Proxy Statement referred to in Item 10 above.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this item is incorporated by reference from the information under the caption "Certain Relationships and Related Transactions" in the Company's Proxy Statement referred to in Item 10 above.
Item 14. Principal Accounting Fees and Services
The information required by this item is incorporated by reference from the information under the caption "Ratification of Appointment of Independent Auditors" in the Company's Proxy Statement referred to in Item 10 above.
162
Item 15. Exhibits and Financial Statement Schedules
(a) Documents filed as part of this report.
1. Financial Statements
The following Financial Statements of VIVUS, Inc. and Reports of Independent Registered Public Accounting Firm have been filed as part of this Form 10-K. See index to Financial Statements under Item 8, above:
2. Financial Statement Schedules
The following financial statement schedule of VIVUS, Inc. as set forth on page 158 is filed as part of this Form 10-K and should be read in conjunction with the Financial Statements of VIVUS, Inc. incorporated by reference herein:
Schedule II—Valuation and Qualifying Accounts
All other schedules are omitted because they are not applicable or the required information is shown in the Financial Statements or the notes thereto.
3. Exhibits Refer to Item 15(b) immediately below.
(b) The list of Exhibits required by Item 601 of Regulation S-K are listed in the Exhibit Index immediately preceding the exhibits and are incorporated herein.
163
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized:
| VIVUS, INC., a Delaware Corporation |
|||
By: |
/s/ LELAND F. WILSON Leland F. Wilson Chief Executive Officer (Principal Executive Officer) |
||
Date: February 28, 2011
164
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints each of Leland F. Wilson and Timothy E. Morris as his attorney-in-fact for him, in any and all capacities, to sign each amendment to this Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that said attorney-in-fact or his substitute or substitutes may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated:
Signature
|
Title
|
Date
|
||
|---|---|---|---|---|
| /s/ LELAND F. WILSON Leland F. Wilson |
Chief Executive Officer (Principal Executive Officer) and Director | February 28, 2011 | ||
/s/ MARK B. LOGAN Mark B. Logan |
Chairman of the Board and Director |
February 28, 2011 |
||
/s/ PETER Y. TAM Peter Y. Tam |
President and Director |
February 28, 2011 |
||
/s/ TIMOTHY E. MORRIS Timothy E. Morris |
Senior Vice President of Finance and Chief Financial Officer (Principal Financial Officer) |
February 28, 2011 |
||
/s/ LEE B. PERRY Lee B. Perry |
Vice President and Chief Accounting Officer (Principal Accounting Officer) |
February 28, 2011 |
||
/s/ CHARLES J. CASAMENTO Charles J. Casamento |
Director |
February 28, 2011 |
||
/s/ LINDA M. DAIRIKI SHORTLIFFE, M.D. Linda M. Dairiki Shortliffe, M.D. |
Director |
February 28, 2011 |
165
VIVUS, INC.
REPORT ON FORM 10-K FOR
THE YEAR ENDED DECEMBER 31, 2010
| Exhibit Number |
Description | ||
|---|---|---|---|
| 2.1 | (1)† | Asset Purchase Agreement between the Registrant and K-V Pharmaceutical Company dated as of March 30, 2007 | |
| 3.1 | (2) | Amended and Restated Certificate of Incorporation of the Registrant | |
| 3.2 | (3) | Amended and Restated Bylaws of the Registrant | |
| 3.3 | (4) | Amended and Restated Certificate of Designation of Rights, Preferences and Privileges of Series A Participating Preferred Stock of the Registrant | |
| 4.1 | (5) | Specimen Common Stock Certificate of the Registrant | |
| 4.2 | (6) | Preferred Stock Rights Agreement dated as of March 27, 2007 between the Registrant and Computershare Investor Services, LLC | |
| 10.1 | (7)* | Form of Indemnification Agreement by and among the Registrant and the Directors and Officers of the Registrant | |
| 10.2 | (8)* | 1991 Incentive Stock Plan and Form of Agreement, as amended | |
| 10.3 | (9)* | 1994 Employee Stock Purchase Plan and Form of Subscription Agreement | |
| 10.4 | (10)* | 2001 Stock Option Plan and Form of Agreement thereunder | |
| 10.5 | (11)* | 2001 Stock Option Plan, as amended on July 12, 2006 | |
| 10.6 | (12)* | Form of Notice of Grant and Restricted Stock Unit Agreement under the VIVUS, Inc. 2001 Stock Option Plan | |
| 10.7 | 2010 Equity Incentive Plan and form of agreement thereunder | ||
| 10.8 | (13)* | Stand-Alone Stock Option Agreement with Michael P. Miller dated as of April 30, 2010 | |
| 10.9 | (14)* | Employment Agreement dated December 20, 2007 between the Registrant and Leland F. Wilson | |
| 10.10 | (15)* | First Amendment to the Employment Agreement dated December 20, 2007 by and between the Registrant and Leland F. Wilson | |
| 10.11 | (16)* | Second Amendment to the Employment Agreement dated December 20, 2007 by and between the Registrant and Leland F. Wilson | |
| 10.12 | (17)* | Form of Change of Control and Severance Agreement by and between the Registrant and Certain of its Executive Officers | |
| 10.13 | (18)* | Form of Change of Control and Severance Agreement dated April 30, 2010 by and between the Registrant and Michael P. Miller | |
| 10.14 | (19)† | Agreement effective as of December 28, 2000 between the Registrant and Tanabe Seiyaku Co., Ltd. | |
| 10.15 | (20) | Amendment No. 1 effective as of January 9, 2004 to the Agreement effective as of December 28, 2000 between the Registrant and Tanabe Seiyaku Co., Ltd. | |
166
| Exhibit Number |
Description | ||
|---|---|---|---|
| 10.16 | (21) | Termination and Release executed by Tanabe Holding America, Inc. dated May 1, 2007 | |
| 10.17 | (22)† | Settlement and Modification Agreement dated July 12, 2001 between ASIVI, LLC, AndroSolutions, Inc., Gary W. Neal and the Registrant | |
| 10.18 | (23)†† | Assignment Agreement between Thomas Najarian, M.D. and the Registrant dated October 16, 2001 | |
| 10.19 | (24)† | Testosterone Development and Commercialization Agreement dated as of February 7, 2004 between the Registrant, Fempharm Pty Ltd. and Acrux DDS Pty Ltd. | |
| 10.20 | (25)† | Estradiol Development and Commercialization Agreement dated as of February 12, 2004 between the Registrant, Fempharm Pty Ltd. and Acrux DDS Pty Ltd. | |
| 10.21 | (26)† | Master Services Agreement dated as of September 12, 2007 between the Registrant and Medpace, Inc. | |
| 10.22 | (27)† | Exhibit A: Medpace Task Order Number: 06 dated as of December 15, 2008 pursuant to that certain Master Services Agreement, between the Registrant and Medpace, Inc., dated as of September 12, 2007 | |
| 10.23 | (28)†† | Asset Purchase Agreement dated October 1, 2010 between the Registrant, MEDA AB and Vivus Real Estate, LLC | |
| 10.24 | (29)†† | Transition Services Agreement dated November 5, 2010 between the Registrant and MEDA AB | |
| 10.25 | (30) | Lease Agreement effective November 1, 2006 by and between the Registrant and Castro Mountain View, LLC, Thomas A. Lynch, Trudy Molina Flores, Trustee of the Jolen Flores and Trudy Molina Flores Joint Living Trust dated April 3, 2001, E William and Charlotte Duerkson, The Duerkson Family Trust dated February 16, 1999, The Dutton Family Trust dated September 16, 1993, The Noel S. Schuurman Trust, The Duarte Family Partners, L.P., The Marie Antoinette Clough Revocable Living Trust dated January 11, 1989, Blue Oak Properties, Inc., and CP6CC, LLC | |
| 10.26 | (31) | First Amendment to Lease effective November 18, 2008 between Castro Mountain View, LLC, CP6CC, LLC and the Registrant | |
| 10.27 | (32) | Second Amendment to Lease effective November 12, 2009 between Castro Mountain View, LLC, CP6CC, LLC and the Registrant | |
| 10.28 | Third Amendment to Lease effective December 3, 2010 between Castro Mountain View LLC, CP6CC, LLC and the Registrant | ||
| 21.1 | Subsidiaries of the Registrant | ||
| 23.1 | Consent of Odenberg, Ullakko, Muranishi & Co. LLP, Independent Registered Public Accounting Firm | ||
| 24.1 | Power of Attorney (See signature page) | ||
| 31.1 | Certification of Chief Executive Officer pursuant to Rules 13a-14 and 15d-14 promulgated under the Securities Exchange Act of 1934, as amended | ||
| 31.2 | Certification of Chief Financial Officer pursuant to Rules 13a-14 and 15d-14 promulgated under the Securities Exchange Act of 1934, as amended | ||
167
| Exhibit Number |
Description | ||
|---|---|---|---|
| 32.1 | Certification of Chief Executive Officer and Chief Financial Officer pursuant to Section 18 U.S.C. 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | ||
- †
- Confidential
treatment granted.
- ††
- Confidential
portions of this exhibit have been redacted and filed separately with the Commission pursuant to a confidential
treatment request in accordance with Rule 24b-2 of the Securities Exchange Act of 1934, as amended.
- *
- Indicates
management contract or compensatory plan or arrangement.
- (1)
- Incorporated
by reference to the same numbered exhibit filed with the Registrant's Current Report on Form 8-K filed with the Commission
on May 21, 2007.
- (2)
- Incorporated
by reference to Exhibit 3.2 filed with the Registrant's Annual Report on Form 10-K for the fiscal year ended
December 31, 1996.
- (3)
- Incorporated
by reference to the same numbered exhibit filed with the Registrant's Current Report on Form 8-K filed with the Commission
on April 30, 2010.
- (4)
- Incorporated
by reference to the same numbered exhibit filed with the Registrant's Registration Statement on Form 8-A filed with the
Commission on March 28, 2007.
- (5)
- Incorporated
by reference to the same numbered exhibit filed with the Registrant's Annual Report on Form 10-K/A for the fiscal year ended
December 31, 1996 filed with the Commission on April 16, 1997.
- (6)
- Incorporated
by reference to Exhibit 4.1 filed with the Registrant's Registration Statement on Form 8-A filed with the Commission
on March 28, 2007.
- (7)
- Incorporated
by reference to Exhibit 10.11 filed with the Registrant's Form 8-B filed with the Commission on June 25, 1996.
- (8)
- Incorporated
by reference to the same numbered exhibit filed with the Registrant's Registration Statement on Form S-1
No. 33-90390, as amended.
- (9)
- Incorporated
by reference to the same numbered exhibit filed with the Registrant's Registration Statement on Form S-1
No. 33-75698, as amended.
- (10)
- Incorporated
by reference to the Exhibit 10.44 filed with the Registrant's Registration Statement on Form S-8 filed with the
Commission on November 15, 2001.
- (11)
- Incorporated
by reference to Exhibit 10.1 filed with the Registrant's Current Report on Form 8-K filed with the Commission on
July 13, 2006.
- (12)
- Incorporated
by reference to Exhibit 10.2 filed with the Registrant's Current Report on Form 8-K filed with the Commission on
July 13, 2006.
- (13)
- Incorporated
by reference to Exhibit 10.1 filed with the Registrant's Current Report on Form 8-K filed with the Commission on
May 6, 2010.
- (14)
- Incorporated
by reference to Exhibit 10.63 filed with the Registrant's Current Report on Form 8-K filed with the Commission on
December 24, 2007.
- (15)
- Incorporated
by reference to Exhibit 10.1 filed with the Registrant's Current Report on Form 8-K filed with the Commission on
January 29, 2009.
- (16)
- Incorporated by reference to Exhibit 10.1 filed with the Registrant's Current Report on Form 8-K filed with the Commission on January 26, 2011.
168
- (17)
- Incorporated
by reference to Exhibit 10.64 filed with the Registrant's Current Report on Form 8-K filed with the Commission on
December 24, 2007.
- (18)
- Incorporated
by reference to Exhibit 10.1 filed with the Registrant's Current Report on Form 8-K filed with the Commission on
April 30, 2010.
- (19)
- Incorporated
by reference to Exhibit 10.42 filed with the Registrant's Annual Report on Form 10-K for the fiscal year ended
December 31, 2000.
- (20)
- Incorporated
by reference to Exhibit 10.42A filed with the Registrant's Quarterly Report on Form 10-Q for the quarter ended
March 31, 2004.
- (21)
- Incorporated
by reference to Exhibit 10.61 filed with the Registrant's Current Report on Form 8-K filed with the Commission on
May 4, 2007.
- (22)
- Incorporated
by reference to Exhibit 10.43 filed with the Registrant's Quarterly Report on Form 10-Q for the quarter ended
June 30, 2001.
- (23)
- Incorporated
by reference to Exhibit 10.79 filed with the Registrant's Annual Report on Form 10-K for the fiscal year ended
December 31, 2009.
- (24)
- Incorporated
by reference to Exhibit 10.50 filed with the Registrant's Quarterly Report on Form 10-Q for the quarter ended
March 31, 2004.
- (25)
- Incorporated
by reference to Exhibit 10.51 filed with the Registrant's Quarterly Report on Form 10-Q for the quarter ended
March 31, 2004.
- (26)
- Incorporated
by reference to Exhibit 10.62 filed with the Registrant's Current Report on Form 8-K filed with the Commission on
September 18, 2007.
- (27)
- Incorporated
by reference to Exhibit 10.1 filed with the Registrant's Current Report on Form 8-K filed with the Commission on
December 24, 2008.
- (28)
- Incorporated
by reference to Exhibit 10.1 filed with the Registrant's Current Report on Form 8-K filed with the Commission on
November 10, 2010.
- (29)
- Incorporated
by reference to Exhibit 10.1 filed with the Registrant's Current Report on Form 8-K filed with the Commission on
November 8, 2010.
- (30)
- Incorporated
by reference to Exhibit 10.60 filed with the Registrant's Current Report on Form 8-K filed with the Commission on
November 7, 2006.
- (31)
- Incorporated
by reference to Exhibit 10.1 filed with the Registrant's Current Report on Form 8-K filed with the Commission on
December 18, 2008.
- (32)
- Incorporated by reference to Exhibit 10.78 filed with the Registrant's Annual Report on Form 10-K for the fiscal year ended December 31, 2009.
169