Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
ANNUAL AND TRANSITION REPORTS
PURSUANT TO SECTIONS 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2010
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission File No. 000-31135
INSPIRE PHARMACEUTICALS, INC.
(Exact Name of Registrant as Specified in Its Charter)
| Delaware | 04-3209022 | |
| (State or Other Jurisdiction of Incorporation or Organization) | (I.R.S. Employer Identification No.) | |
| 8081 Arco Corporate Drive, Suite 400, Raleigh, North Carolina | 27617 | |
| (Address of Principal Executive Offices) | (Zip Code) | |
(919) 941-9777
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class |
Name of Each Exchange on Which Registered | |
| Common Stock, $.001 par value | The Nasdaq Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
(Title of Class)
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the Registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one)
| Large accelerated filer ¨ |
Accelerated filer x | |
| Non-accelerated filer ¨ (do not check if a smaller reporting company) |
Smaller reporting company ¨ |
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No x
State the aggregate market value of the voting and non-voting common equity held by non-affiliates computed by reference to the price at which the common equity was last sold, or the average bid and asked price of such common equity, as of the last business day of the registrant’s most recently completed second fiscal quarter. $297,177,125.
Indicate the number of shares outstanding of each of the Registrant’s classes of common stock, as of January 31, 2011.
| Class |
Number of Shares | |
| Common Stock, $.001 par value |
83,172,132 |
Documents incorporated by reference
| Document Description |
10-K Part III | |
| Portions of the Registrant’s proxy statement to be filed pursuant to Regulation 14A within 120 days after Registrant’s fiscal year end of December 31, 2010 are incorporated by reference into Part III of this report. | Items 10, 11, 12, 13, 14 |
Table of Contents
2010 FORM 10-K ANNUAL REPORT
TABLE OF CONTENTS
| Page | ||||||
| PART I. |
||||||
| Item 1. |
1 | |||||
| Item 1A. |
19 | |||||
| Item 1B. |
39 | |||||
| Item 2. |
39 | |||||
| Item 3. |
39 | |||||
| Item 4. |
39 | |||||
| PART II |
||||||
| Item 5. |
40 | |||||
| Item 6. |
42 | |||||
| Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
43 | ||||
| Item 7A. |
61 | |||||
| Item 8. |
62 | |||||
| Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
63 | ||||
| Item 9A. |
63 | |||||
| Item 9B. |
64 | |||||
| PART III |
||||||
| Item 10. |
65 | |||||
| Item 11. |
65 | |||||
| Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
65 | ||||
| Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
65 | ||||
| Item 14. |
65 | |||||
| PART IV |
||||||
| Item 15. |
66 | |||||
| 67 | ||||||
We own or have rights to various trademarks, copyrights and trade names used in our business. AzaSite® is a trademark owned by InSite Vision Incorporated. Restasis®, Elestat® and ProlacriaTM are trademarks owned by Allergan, Inc. DiquasTM is a trademark owned by Santen Pharmaceutical Co., Ltd. This report also includes trademarks, service marks and trade names of other companies.
Table of Contents
PART I
Overview
We are a specialty pharmaceutical company focused on developing and commercializing ophthalmic products. Our strategy is to create a sustainable portfolio of ophthalmic products by leveraging our commercial capabilities and pipeline assets and pursuing corporate development and licensing opportunities. Our specialty eye care sales force generates revenue from the promotion of AzaSite (azithromycin ophthalmic solution) 1% for bacterial conjunctivitis and the co-promotion of Elestat (epinastine HCl ophthalmic solution) 0.05% for allergic conjunctivitis. We receive royalties based on net sales of Restasis (cyclosporine ophthalmic emulsion) 0.05% for dry eye and expect to begin receiving royalties in 2011 based on net sales of Diquas Ophthalmic Solution 3% (diquafosol tetrasodium) for dry eye in Japan.
| PRODUCTS AND PRODUCT CANDIDATES |
THERAPEUTIC AREA/ |
CONTRACTUAL PARTNER (1) |
CURRENT STATUS | |||
| Products |
||||||
| AzaSite |
Bacterial conjunctivitis | InSite Vision | Promoting | |||
| Elestat |
Allergic conjunctivitis | Allergan | Co-promoting | |||
| Restasis |
Dry eye disease | Allergan | Receiving royalty | |||
| Diquas |
Dry eye disease | Santen Pharmaceutical | Receiving royalty (2) | |||
| Product Candidates |
||||||
| Denufosol tetrasodium |
Cystic fibrosis | None | Phase 3 (3) | |||
| Prolacria (diquafosol tetrasodium) |
Dry eye disease | None | Phase 3 (4) | |||
| DE-089 (diquafosol tetrasodium) |
Dry eye disease | Santen Pharmaceutical | Phase 3 (5) | |||
| AzaSite |
Blepharitis | InSite Vision | Phase 2 | |||
| INS115644, INS117548 |
Glaucoma | Wisconsin Alumni Research Foundation | Phase 1 | |||
| (1) | See “Agreements” for a detailed description of our agreements with these contractual partners. |
| (2) | In December 2010, Diquas received pricing approval and was launched for sale in Japan by Santen Pharmaceutical Co., Ltd. |
| (3) | In January 2011, we announced that our Phase 3 clinical trial (TIGER-2) did not meet its primary or key secondary endpoints. In February 2011, we announced our decision to discontinue our development of denufosol. |
| (4) | In January 2010, we announced that our Phase 3 clinical trial (Trial 03-113) did not meet its primary or secondary endpoints. We are not currently proceeding with the clinical development of Prolacria. |
| (5) | Santen is conducting Phase 3 clinical trials with DE-089 in China. |
We were incorporated as a Delaware corporation in October 1993 and commenced operations in March 1995. Our corporate headquarters are located in Raleigh, North Carolina.
1
Table of Contents
PRODUCTS
AzaSite
AzaSite (azithromycin ophthalmic solution) 1% is a topical anti-infective, in which azithromycin is formulated into an ophthalmic solution utilizing DuraSite®, a novel ocular drug delivery system. Azithromycin is a semi-synthetic antibiotic that is derived from erythromycin and, since 1992, has been available via oral administration by Pfizer Inc. under the trade name Zithromax®. In April 2007, AzaSite was approved by the U.S. Food and Drug Administration, or FDA, for the treatment of bacterial conjunctivitis in adults and children one year of age and older.
In February 2007, we entered into a license agreement with InSite Vision Incorporated, or InSite Vision, pursuant to which we acquired exclusive rights to commercialize AzaSite for use in the treatment of human ocular or ophthalmic indications. The license agreement grants us exclusive rights to develop, make, use, market, commercialize and sell AzaSite in the United States and Canada. We are obligated to pay InSite Vision royalties on net sales of AzaSite in the United States and Canada. See “—Agreements—InSite Vision Incorporated.”
In August 2007, we launched AzaSite in the United States and are promoting it to eye care specialists. The manufacture and sale of AzaSite is protected in the United States under use and formulation patents, the latest of which expires in March 2019. In addition, the sale of AzaSite is also protected in the United States under a use patent that we sub-licensed from Pfizer that expires in November 2018.
Market Opportunity. The U.S. single agent ocular antibiotic market was approximately $576 million and total prescriptions for all branded products in the U.S. ocular antibiotic market were approximately 15 million for the 12 months ended December 31, 2010, according to data compiled from IMS Health.
Elestat
Elestat (epinastine HCl ophthalmic solution) 0.05% is a topical antihistamine developed by Allergan, Inc., or Allergan, for the prevention of ocular itching associated with allergic conjunctivitis. Elestat was approved by the FDA in October 2003 and is indicated for adults and children at least three years old.
We receive co-promotion revenue from Allergan on its U.S. net sales of Elestat. When a generic form of Elestat or an over-the-counter form of epinastine ophthalmic solution is introduced into the market, our agreement with Allergan to co-promote Elestat will no longer be in effect, and our revenues from sales of Elestat will be nominal. See “—Agreements—Allergan, Inc.—Elestat.”
Subject to applicable law, competitors are permitted to submit to the FDA an Abbreviated New Drug Application, or ANDA, for a generic version of Elestat, due to the expiration of the marketing exclusivity period for Elestat provided under the Hatch-Waxman Act on October 15, 2008. We are aware that the following companies have filed an ANDA for a generic version of Elestat: Apotex, Inc., Cypress Pharmaceutical, Inc., Paddock Laboratories, Inc., PharmaForce, Inc. and Sandoz, Inc. The date of submission of the first ANDA filing to the FDA Office of Generic Drugs was October 14, 2008, according to the FDA’s website (www.fda.gov). Also, according to the FDA’s website, Apotex, Inc., PharmaForce, Inc. and Sandoz, Inc. have each received tentative approval for their respective epinastine hydrochloride ophthalmic solution.
We plan to continue co-promoting and receiving co-promotion revenues on Elestat sales during the FDA’s review period of these ANDAs. We do not know when the FDA will complete its review of the ANDAs relating to a generic version of Elestat and we do not expect to be notified by any party prior to any approval. Based upon our assessment, we expect that a generic form of epinastine may be launched at any time. See “Risk Factors—“When a generic form of Elestat or an over-the-counter form of epinastine ophthalmic solution is introduced into the market, which we expect may occur at any time, our agreement with Allergan to co-promote Elestat will no longer be in effect, and our revenues from sales of Elestat will be nominal”—for further discussion of the risk related to the ANDA filings.
2
Table of Contents
Restasis
Restasis (cyclosporine ophthalmic emulsion) 0.05% is the first approved prescription product in the United States for the treatment of dry eye disease. It is indicated to increase tear production in adults and children at least 16 years old whose tear production is presumed to be suppressed due to ocular inflammation associated with keratoconjunctivitis sicca, or dry eye disease. Restasis was approved by the FDA in December 2002, and Allergan launched the product in the United States in April 2003.
In June 2001, we entered into an agreement with Allergan to develop and commercialize our product candidate, Prolacria (diquafosol tetrasodium), for the treatment of dry eye disease. The agreement also provided that Allergan pay us royalties on net sales of Restasis. In August 2010, we amended and restated our agreement with Allergan. Under the amended and restated agreement, which runs through December 31, 2020, we are entitled to receive revenues at one global rate based on net sales of Restasis and any other human ophthalmic formulation of cyclosporine owned or controlled by Allergan, with no requirement to co-promote Restasis. Effective January 1, 2011, the worldwide rate stepped down from the 2010 rate by three percentage points. The rate will step down a further 0.25 percentage point in 2013 and a final 0.50 percentage point in 2014, remaining at this level through the end of the term in 2020. See “—Agreements—Allergan, Inc.—Restasis and Prolacria.”
The manufacture and sale of Restasis is protected in the United States by a formulation patent that expires in May 2014.
Market Opportunity. Dry eye disease is associated with aging, environmental factors, autoimmune disorders and various medications. For the years ended December 31, 2010, 2009 and 2008, Allergan recorded approximately $621 million, $523 million and $444 million, respectively, of revenue from net sales of Restasis.
Diquas
Diquas ophthalmic solution 3% (diquafosol tetrasodium) has been developed by Santen Pharmaceutical Co., Ltd., or Santen, as a treatment for dry eye.
In December 1998, we entered into an agreement with Santen that allows Santen to develop diquafosol tetrasodium for the therapeutic treatment of ocular surface diseases, such as dry eye disease, in Japan and nine other Asian countries, and provides for certain milestone payments to be paid to us upon achievement of development milestones by Santen. In December 2010, we received a milestone payment of $1.25 million as Diquas received pricing approval and was launched for sale in Japan. We are entitled to receive royalty payments based upon a tiered rate on net sales of Diquas in Japan, with a minimum rate in the high single digits and a maximum rate in the low double digits. See “—Agreements—Santen Pharmaceutical Co., Ltd.”
The manufacture and sale of Diquas is protected in Japan under patents that have claims to the drug substance, the formulation, and method of use that expire in February 2018, subject to any applicable patent restoration that may extend protection up to an additional five years from the date of expiration of the applicable patent, if any, for which restoration is sought.
Product Risks
For a more detailed discussion of the risks associated with these products, please see Item 1.—Risk Factors below.
3
Table of Contents
PRODUCT CANDIDATES
Denufosol tetrasodium for the treatment of cystic fibrosis
Overview. Denufosol tetrasodium is an ion channel regulator that was being developed to address the underlying ion transport defect in the lungs of patients with cystic fibrosis. Denufosol was designed to enhance airway hydration and mucociliary clearance through receptor-mediated mechanisms that increase chloride secretion, inhibit sodium absorption and increase ciliary beat frequency. The manufacture and sale of denufosol tetrasodium is protected in the United States under patents that have claims to the drug substance, the formulation, and method of use that expire in February 2017, subject to any applicable patent restoration that may extend protection up to an additional five years from the date of expiration of the applicable patent, if any, for which restoration is sought.
Development Status. In January 2011, we announced the top-line results from our second Phase 3 clinical trial, TIGER-2, with denufosol tetrasodium for the treatment of cystic fibrosis. The trial did not achieve statistical significance for its primary efficacy endpoint, which was change from baseline in FEV1 (Forced Expiratory Volume in One Second) at the Week 48 Endpoint (48 weeks or last observation carried forward). Patients receiving denufosol in the 466-patient, double-blind, placebo-controlled clinical trial had an improvement of 40 mL, compared to 32 mL for the patients receiving placebo (p = 0.742). Additionally, there were no statistically significant differences between denufosol and placebo for three key secondary endpoints, which were (i) rate of change in percent predicted FEV1 over 48 weeks; (ii) change from baseline in FEF25%-75% (Forced Expiratory Flow) at the Week 48 Endpoint; and (iii) time to first pulmonary exacerbation. Based on our assessment of the full data set from the TIGER-2 Phase 3 trial of denufosol, the open-label denufosol-only trial (Trial 08-114) data and our current corporate resources, we decided to discontinue our development of denufosol.
Prolacria (diquafosol tetrasodium) for the treatment of dry eye disease
Overview. Diquafosol tetrasodium is a dinucleotide which functions as an agonist at the P2Y2 receptor and was being developed for the treatment of dry eye disease. Prolacria, the proposed U.S. tradename for diquafosol tetrasodium ophthalmic solution 2%, is designed to stimulate the release of three components of natural tears—mucin, lipids and fluid. The manufacture and sale of Prolacria is protected in the United States under drug substance and formulation patents that expire in July 2016, as well as under use patents that expire in February 2017, subject to any applicable patent restoration that may extend protection up to an additional five years from the date of expiration of the applicable patent, if any, for which restoration is sought.
Under our amended and restated agreement with Allergan, we have sole control over any future Prolacria development and commercialization. We are not currently proceeding with the clinical development of Prolacria. In the event we resume the Prolacria clinical development program and receive regulatory approval for a Prolacria product in a particular country, we will have the option to offer Prolacria commercialization rights to Allergan for such country upon the original commercial terms previously agreed to between us and Allergan. If we choose not to offer Allergan Prolacria commercialization rights with respect to a country, we will receive all the commercialization revenues related to Prolacria in such country and our rights to receive royalties from Allergan based on net sales of Restasis products in such country will terminate. See “—Agreements—Allergan, Inc.—Restasis and Prolacria.”
Development Status. In June 2003, we filed a New Drug Application, or NDA, with the FDA for Prolacria for the treatment of dry eye disease. We received approvable letters from the FDA in December 2003 and December 2005. In January 2010, we announced that an additional Phase 3 clinical trial (Trial 03-113) did not meet its primary endpoint (p = 0.526) or its secondary endpoint (p = 0.368). We are not currently proceeding with the clinical development of Prolacria.
4
Table of Contents
DE-089 (diquafosol tetrasodium) for the treatment of dry eye disease
Our agreement with Santen allows Santen to develop diquafosol tetrasodium for the therapeutic treatment of ocular surface diseases, such as dry eye disease, in Japan, the People’s Republic of China and eight other Asian countries. Santen is conducting Phase 3 clinical trials with DE-089 in China. We are eligible to receive a single digit royalty on net sales of DE-089 in China, if it is ultimately approved and commercialized in China.
AzaSite for the treatment of blepharitis
Overview. Blepharitis is an ocular disease characterized by inflammation of the lid margin that is common, complex, and has a multi-factorial etiology. Blepharitis coexists with other common ocular surface conditions and we believe is often under-diagnosed and misdiagnosed in general clinical practice. Blepharitis can be subdivided into two categories: anterior and posterior blepharitis. Although they are distinct diseases, they can overlap. Anterior blepharitis is generally associated with the presence of bacteria, lid debris and/or sebaceous gland activity and is most often an acute disease. Posterior blepharitis is almost always associated with dysfunctional meibomian glands or altered meibomian gland secretions and is generally considered a chronic disease.
Our market research and input from eye care specialists suggests that blepharitis is an under-diagnosed and under-treated disease. Survey data published in The Ocular Surface and funded by Inspire indicated that 15% of adults reported having at least one of the three symptoms that clinicians associate with anterior blepharitis at least half of the time in the previous 12 months. Based on the overall U.S. adult population of 232 million, this implies potentially as many as 34 million adults might have suffered from some form of blepharitis over such time frame. Currently, there are no FDA-approved prescription pharmaceutical products indicated for the treatment of this disease. Patients currently manage the acute and often chronic effects of blepharitis with the use of warm compresses, lid hygiene, topical antibiotic ointments and, when exacerbated, with topical steroids or oral antibiotics.
Development Status. During 2008, we conducted a series of Phase 4 clinical trials with AzaSite evaluating the safety and efficacy of AzaSite in ocular conditions, such as blepharitis. In late 2008, we sought input from numerous medical experts and evaluated our Phase 4 data along with market research on the prevalence and awareness of the disease to evaluate AzaSite’s potential opportunity as it relates to the treatment of blepharitis. In addition, we had preliminary discussions with the FDA on potential regulatory pathways. Based on preliminary information gathered, we decided to pursue a Phase 2 program to study AzaSite for the treatment of blepharitis.
In May 2009, we initiated Phase 2 work which consisted of two randomized, vehicle-controlled clinical trials that enrolled approximately 600 patients with anterior blepharitis. Trial 044-101 included a two-week treatment period with a two-week follow-up period and Trial 044-102 included a four-week treatment period with a four-week follow-up period. Patients were randomized to AzaSite or the DuraSite vehicle and received one drop in each eye twice-a-day for the first two days, then one drop in each eye daily for the remainder of the treatment period. All patients in the trials performed lid hygiene using commercially available lid scrubs once daily for the duration of the trials.
In March 2010, we announced the results of these two trials. In the four-week trial, improvements for AzaSite compared to vehicle were achieved for a number of blepharitis signs and symptoms at various time points with p-values £ 0.05, but statistical significance was not achieved for the primary endpoint of mean lid margin hyperemia. In the two-week trial, there were no statistically significant improvements for AzaSite compared to vehicle, including for the primary endpoint of clearing of lid debris. In both trials, the AzaSite treatment group and the vehicle treatment group showed statistically significant improvements relative to baseline for all measured signs and symptoms of blepharitis. Additionally, AzaSite was well-tolerated in both trials.
5
Table of Contents
In December 2010, we initiated an exploratory Phase 2, double-masked, vehicle-controlled clinical trial (Trial 044-103) comparing AzaSite to vehicle for four weeks of treatment with an eight-week follow-up period in up to 300 blepharitis patients at approximately 25 U.S. sites. We will be assessing the patient population, length of dosing and measurements of multiple signs and symptoms of blepharitis, including utilizing photographic methods and a centralized reading center. We expect information from this trial in the second half of 2011.
Given the limited data available and the early stage of development of this program, we are currently unable to reasonably project the future dates and costs associated with clinical trials or a prospective NDA filing for this program.
Glaucoma product candidates
Overview. In November 2004, we licensed technology for use in developing and commercializing new treatments for glaucoma from Wisconsin Alumni Research Foundation, or WARF. See “—Agreements—Wisconsin Alumni Research Foundation.”
Development Status. In 2007, we initiated a Phase 1 proof-of-concept placebo-controlled, dose-ranging clinical trial (Trial 032-101) for INS115644, the first compound in a series of compounds, in glaucoma patients to evaluate the safety and tolerability of INS115644, as well as changes in IOP. In 2008, we initiated a Phase 1 proof-of-concept placebo-controlled, dose-ranging clinical trial (Trial 037-101) for a second compound, referred to as INS117548, to evaluate the safety, tolerability and IOP-lowering effects of INS117548 in approximately 80 subjects with early stage glaucoma or ocular hypertension.
In September 2009, we announced top-line results of these trials. In Trial 032-101 with INS115644, a Latrunculin B formulation, we observed dose-dependent IOP-lowering effects and the compound was well-tolerated. Trial 037-101 with INS117548, which is one of a series of Rho kinase molecules we have developed, showed mild IOP-lowering effects but had some dose-related tolerability issues, specifically with ocular discomfort such as burning and stinging. We are in the process of evaluating next steps for the overall glaucoma program, based on our clinical and preclinical data, expert opinions and resource availability.
Due to the uncertainty of the future of this program, and given the limited data available and the early stage of development of this program, we are currently unable to reasonably project the future dates and costs associated with clinical trials or a prospective NDA filing for either of these product candidates.
Product Candidates Risks
For a discussion of the risks associated with our development programs, please see Item 1.—Risk Factors below.
Research & Development Expenses
Additional information about the costs and expenses associated with all of our research and development programs is discussed below in “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Results of Operations—Years Ended December 31, 2010, 2009 and 2008—Costs and Expenses.”
Agreements
Allergan, Inc.—Elestat
In December 2003, we entered into an agreement with Allergan to co-promote Elestat in the United States. Under the agreement, we have the responsibility for promoting and marketing Elestat to ophthalmologists, optometrists and allergists in the United States and paying the associated costs. We receive co-promotion revenue from Allergan on its U.S. net sales of Elestat. Allergan records sales of Elestat and is responsible for supply chain management, managed health care, customer order processing and regulatory compliance.
6
Table of Contents
The Elestat co-promotion agreement provides that unless earlier terminated, the term of such agreement will be in effect until the earlier of (i) the approval and launch of the first generic epinastine product after expiration of the FDA exclusivity period covering Elestat in the United States, or (ii) the approval and launch of the first over-the-counter epinastine product after expiration of the listing of Elestat in the FDA’s Orange Book. As stated earlier, a generic form of epinastine may be launched at any time. Following the termination of such co-promotion agreement, we will no longer have rights to co-promote Elestat. We will be entitled to receive post-termination payments from Allergan, based on any remaining net sales of Elestat for a period of 36 months. During the three successive 12-month periods immediately following the termination of the agreement, Allergan will be obligated to pay to us 20%, 15% and 10%, respectively, on any net sales of Elestat in the United States. See “—Products—Elestat” for a discussion of the filing of ANDAs by various pharmaceutical companies relating to generic forms of Elestat.
Either Allergan or we may terminate the agreement in the event of a material breach of the agreement by the other or in the event of the other’s insolvency. Allergan can terminate the agreement upon a change of control where we become an affiliate of a direct competitor of Allergan as that term is defined in the agreement. We can terminate the agreement in the event that Elestat is withdrawn from the market for more than 90 days.
Allergan, Inc.—Restasis and Prolacria
In August 2010, we and Allergan entered into an Amended and Restated License, Development and Marketing Agreement (the “Restated Agreement”) to our License, Development and Marketing Agreement, dated as of June 22, 2001, as previously amended (the “Original Agreement”).
Pursuant to the terms of the Original Agreement, we granted to Allergan certain rights relating to the development and commercialization of Inspire’s diquafosol tetrasodium products, including Prolacria, throughout the world, except in Japan and nine other Asian countries (the “Territory”), and, as partial consideration therefore, we received certain rights relating to the commercialization of Restasis (and any other human ophthalmic formulations of cyclosporine owned by Allergan), including the right to receive revenue based on net sales of Restasis (and such other products, if any) in the Territory.
Restasis Rights and Obligations
Under the Original Agreement, we were entitled to receive from Allergan net sales payments on Restasis and any other cyclosporine product, on a country-by-country basis for a term equal to the later of (i) the applicable patent term covering such product in such country, and (ii) 10 years from commercial launch of such product. Pursuant to the Restated Agreement, Allergan is obligated to make applicable net sales payments on Restasis and any other human ophthalmic formulations of cyclosporine owned or controlled by Allergan until December 31, 2020.
Under the terms of the Restated Agreement, we will be entitled to receive revenue payments based on one worldwide rate that will apply to all sales of Restasis products (including Restasis and any other human ophthalmic formulations of cyclosporine owned or controlled by Allergan) in the Territory. Effective January 1, 2011, the worldwide rate stepped down from the 2010 rate by three percentage points. The rate will step down a further 0.25 percentage point in 2013 and a final 0.50 percentage point in 2014, remaining at this level through the end of the term in 2020. We are not required to co-promote Restasis in order to receive this revenue stream.
Diquafosol Rights and Obligations
Pursuant to the terms of the Restated Agreement, we have sole control over the development of diquafosol products, including the ability to determine in our sole and absolute discretion whether or not to conduct any development activities at all. In furtherance of the foregoing, under the Restated Agreement, we have the right to control the regulatory filings and intellectual property relating to diquafosol products. At this time, we are not planning to proceed with further clinical development of Prolacria.
7
Table of Contents
In the event we resume development and receive regulatory approval for a diquafosol product in a particular country in the Territory, we will have the option for a period of 60 days from receipt of such regulatory approval to offer commercialization rights to Allergan in such country. Allergan may either accept or reject the commercialization rights relating to the product in such country. If Allergan accepts such commercialization rights, we and Allergan will enter into an Exclusive License Agreement. In accordance with the terms of such Exclusive License Agreement, Allergan will be obligated to commercialize the product in such country and to make milestone and other payments based on net sales to Inspire on substantially the same terms as set forth in the Original Agreement with respect to diquafosol products. Allergan has the option to terminate such Exclusive License Agreement at the end of 2020, in which case all rights to diquafosol thereunder would revert back to us.
If Allergan rejects the offered diquafosol commercialization rights with respect to a country, we will retain such commercialization rights with respect to diquafosol, and we will also continue to be entitled to receive revenue payment based on net sales of Restasis products in such country. In the event that we choose not to offer diquafosol commercialization rights to Allergan with respect to a country, we will retain all commercialization rights to diquafosol in such country, and once the product is launched in such country, our rights to receive revenue payments from Allergan based on net sales of Restasis products in such country will terminate.
Cystic Fibrosis Foundation Therapeutics, Inc.
In October 2002, we entered into a study funding agreement with the Cystic Fibrosis Foundation Therapeutics, Inc., or CFFT, a non-profit drug development affiliate of the Cystic Fibrosis Foundation, for the funding of one Phase 2 clinical trial for denufosol for the treatment of cystic fibrosis. Under the agreement, the CFFT provided the majority of funding of external costs for one Phase 2 clinical trial of denufosol, which we completed in April 2004, in exchange for post-commercialization development and sales milestone payments. The agreement will immediately terminate if our ongoing development efforts of denufosol for the treatment of cystic fibrosis fail to yield statistically significant results. We, in our sole discretion, may elect to cease development efforts following such failure. In addition, either the CFFT or we may terminate the agreement if the other materially breaches the agreement.
InSite Vision Incorporated
In February 2007, we entered into a license agreement with InSite Vision pursuant to which we acquired exclusive rights to commercialize AzaSite, as well as other potential topical anti-infective products containing azithromycin as the sole active ingredient for use in the treatment of human ocular or ophthalmic indications. The license agreement also grants us exclusive rights to develop, make, use, market, commercialize and sell each product in the United States and Canada. We are currently responsible for all regulatory obligations and strategies relating to the further development and commercialization of a product in the United States and Canada.
Pursuant to the license agreement, we paid InSite Vision an upfront license fee of $13.0 million and an additional $19.0 million milestone related to FDA approval of AzaSite. In addition, we paid a 20% royalty for the first two years of commercialization and in July 2009 began paying a 25% royalty on net sales of AzaSite in the United States and Canada, which will continue for the duration of the agreement. We are obligated to pay royalties under the agreement for the longer of (i) 11 years from the launch of the subject product and (ii) the period during which a valid claim under a patent licensed from InSite Vision covers a subject product. Under the terms of the agreement, our obligation to pay royalties to InSite Vision is subject to pre-determined minimum annual royalty payments. The determination of whether or not we will owe any such payments is based upon the amount of royalties accrued over a 12-month royalty period. There are five successive 12-month minimum royalty periods, the third of which commenced on October 1, 2010.
Contemporaneously with the license agreement, InSite Vision entered into an exclusive license agreement with Pfizer for certain Pfizer patent rights relating to the treatment of ocular infection with azithromycin for certain products. Under the terms of our license agreement with InSite Vision, we obtained from InSite Vision a
8
Table of Contents
sublicense to such Pfizer patent rights, in addition to the license to the InSite Vision patent rights, subject to certain limitations. Also, Inspire and Pfizer entered into a related agreement that provides for the continuation of our sublicense rights under the Pfizer patent rights upon a termination of the license agreement between InSite Vision and Pfizer. The agreement between us and Pfizer also provides an opportunity to cure any breaches by InSite Vision of the license agreement between InSite Vision and Pfizer and the opportunity to maintain and enforce such Pfizer patent rights under certain circumstances.
Santen Pharmaceutical Co., Ltd.
In December 1998, we entered into a Development, License and Supply Agreement with Santen. The terms of the agreement allow Santen to develop diquafosol tetrasodium for the therapeutic treatment of ocular surface diseases, such as dry eye disease, in Japan, China, South Korea, the Philippines, Thailand, Vietnam, Taiwan, Singapore, Malaysia and Indonesia (the “Santen Territory”). The agreement provides for certain payments to be paid to Inspire upon achievement of development milestones by Santen and for royalties relating to the net sales of products containing diquafosol tetrasodium developed by Santen. Santen is responsible for all development, regulatory submissions, filings and approvals, and all marketing of potential products in its territory.
Under the terms of the agreement, Santen has developed a formulation of diquafosol, known as Diquas ophthalmic solution 3%, which received regulatory approval from the Japanese Ministry of Health in April 2010 and received pricing approval and was launched in December 2010.
In June 2010, we amended the agreement to relieve Inspire of its manufacturing obligations with respect to the supply of diquafosol tetrasodium active pharmaceutical ingredients, or API, and to grant to Santen expanded rights allowing it to manufacture, or have manufactured, the API throughout the world for use in products in the Santen Territory. In connection therewith, the licensed technology was expanded to include know-how relating to manufacturing. Royalty rates relating to net sales of products, including Diquas, in Japan were amended to a tiered royalty range with a minimum rate in the high single digits and a maximum rate in the low double digits. In addition, the royalty rate with respect to net sales, if any, of any product approved for sale in the Santen Territory other than Japan were amended to a single digit royalty rate.
Under the terms of the agreement, we received an up-front equity investment of $1.5 million in exchange for shares of our preferred stock in December 1998, that were subsequently converted into shares of our common stock. We have received total milestone payments of $4.25 million based on the achievement of certain regulatory work, the completion of Phase 3 clinical testing of diquafosol tetrasodium in Japan, and Japanese approval of Diquas. There are no further milestones to be earned under the agreement.
The agreement will terminate when all patents licensed under the agreement have expired. Either Santen or we may terminate the agreement if the other materially breaches the agreement. In addition, we have the right to terminate the agreement at any time if we determine, subject to a coordinating committee’s review and arbitration, that Santen has not made reasonably sufficient progress in the development or commercialization of potential products. If Santen breaches the agreement, or if we terminate the agreement because Santen has not made sufficient progress, Santen’s license will terminate. Santen will provide us with all data and information relating to our products, and will assign or permit us to cross-reference all regulatory filings and approvals; provided, however, that Santen is not required to provide us with any data or information arising after the agreement which relate solely to the manufacture of the API.
Wisconsin Alumni Research Foundation
In November 2004, we licensed technology for use in developing and commercializing new treatments for glaucoma from WARF. Under the terms of the agreement, we are obligated to make contingent payments of up to an aggregate of $1.8 million upon the achievement of development milestones and to pay royalties on sales of any regulatory approved product utilizing the licensed patents.
9
Table of Contents
We will design and fund all future research, development, testing, regulatory filings and potential marketing activities related to any product candidate under development or product developed from the license. Unless terminated earlier, the agreement will expire on a country-by-country basis upon the expiration of the patents in such country. The U.S. government may have limited rights in some of this patented technology. WARF may terminate the license if we fail to make timely payment of any amount due to WARF under the agreement or commit a material breach of any material covenant contained in the agreement, subject to our right to cure.
Research and Development
During the years ended December 31, 2010, 2009 and 2008, our research and development expenses were $46.1 million, $51.1 million and $44.6 million, respectively. In February 2009, we eliminated our early preclinical and molecule discovery activities and refocused our resources on the development of existing later-stage clinical programs and commercially available products. In February 2011, we implemented a strategic corporate restructuring focusing activities on our eye care business and discontinuing our pulmonary therapeutic area. This restructuring resulted in a substantial reduction of resources devoted to future research and development.
We collaborate with external contract research organizations that allow us to perform development activities, including toxicology, pharmacokinetics, toxicokinetics, and other studies required for NDA regulatory submissions.
For more information about our research and development costs and expenses, see “Management’s Discussion and Analysis of Financial Conditions and Results of Operations—Research and Development Expenses.”
Sales and Marketing
We currently employ approximately 90 territory managers to provide us with national U.S. sales coverage for AzaSite and Elestat. We also have marketing, managed care, training and operation groups to support our commercialization efforts. Our sales and marketing organization focuses its promotional activities for AzaSite and Elestat on eye care specialists.
We believe our commercial operations provides us with the foundation to leverage opportunities to market and sell other products we are developing, or products that we may in-license or otherwise acquire, and to maximize their commercial value in the United States.
Compliance
We conduct our business in an ethical, fair, honest and lawful manner. We act responsibly, respectfully and with integrity in our relationships with patients, health care professionals, providers, governments, regulatory entities, customers, stockholders, suppliers and vendors.
We have designated a Chief Compliance Officer who reports to the Executive Vice President and Chief Administrative and Legal Officer, and the Chairperson of the Audit Committee of the Board of Directors. Among other duties, this officer oversees compliance training, education, auditing and monitoring; enforces disciplinary guidelines for any infractions of our Comprehensive Compliance Program; implements new policies and procedures; responds to any detected issues; and undertakes corrective action procedures. The Chief Compliance Officer provides updates to senior management, the Audit Committee of the Board of Directors, and to the full Board of Directors. Our controls address compliance matters relating to requirements and entities that govern public pharmaceutical companies including, but not limited to the following: federal and state law, such as the Sarbanes-Oxley Act of 2002 and the U.S. Foreign Corrupt Practices Act of 1977; NASDAQ listing requirements; the Financial Industry Regulatory Authority; the Securities and Exchange Commission; the Food and Drug Administration; the United States Department of Health and Human Services; and the Office of Inspector
10
Table of Contents
General, along with voluntary industry standards developed by the Pharmaceutical Research and Manufacturers of America. Our standard operating procedures are designed to provide a framework for corporate governance in accordance with ethical standards and legal best practices. Our codes and policies that have been implemented include, but are not limited to, Code of Conduct and Business Ethics; Whistleblower Policy; and Code of Conduct: Promotional Interactions with Health Care Professionals.
Manufacturing and Supply
We rely on single source manufacturers for our commercial products. Allergan is responsible for the manufacture of both Restasis and Elestat and relies on single source manufacturers for the API in both products. We rely on InSite Vision for supply of the API for AzaSite, which InSite Vision obtains from a single source manufacturer. We are responsible for the remaining finished product manufacturing of AzaSite, for which we rely on a single source manufacturer. Additionally, we rely upon a single third party to provide distribution services for AzaSite. See “Risk Factors—Reliance on a single party to manufacture and supply either finished product or the bulk active pharmaceutical ingredients for a product or product candidates could adversely affect us.”
We conduct qualification and routine audits of our contract manufacturers. These contract manufacturers are identified in our regulatory agency filings, such as with the FDA, and are subject to regulatory agency inspections. We also attempt to stay informed on the financial condition of contract manufacturers and their status with regulatory agencies.
The manufacture of our products and product candidates is based, in part, on technology that we believe to be proprietary to our contract manufacturers or our collaborative partners. Such manufacturers may not abide by the limitations or confidentiality restrictions in agreements with us. In addition, any such manufacturer may develop process technology related to the manufacture of our compounds that such supplier owns either independently or jointly with us. This would increase our reliance on such manufacturer or require us to obtain a license from such manufacturer in order to have our products manufactured using such technology.
Catalent Pharma Solutions
In September 2007, we entered into a long term manufacturing services agreement with Catalent Pharma Solutions, LLC, or Catalent, for the manufacture of the finished product AzaSite, pursuant to which Catalent agreed to manufacture AzaSite to Inspire’s specifications for a period of six years. Under the agreement, we agreed to purchase from Catalent on an annual basis a specified minimum number of units of AzaSite for the first four years at a per unit price that is specified in the contract.
Either party may terminate the agreement upon 60 days’ prior written notice if the other party materially breaches the agreement. However, if we fail to make payments to Catalent within 15 days after such payments are due, Catalent may terminate the agreement or Catalent may cease performing under the agreement until all of the outstanding payments are brought current. We may terminate the agreement if a force majeure event prevents Catalent from fully performing its obligations under the agreement for a period of 120 days. In addition, following the conclusion of the third contract year, the agreement may be terminated on 12 months’ notice by us or on 24 months’ notice by Catalent.
InSite Vision
In February 2007, we entered into a supply agreement with InSite Vision for the active pharmaceutical ingredient, azithromycin. Previously, InSite Vision entered into a third-party supply agreement for the production of azithromycin. Under the supply agreement, InSite Vision agreed to supply our requirements of azithromycin, pursuant to certain forecasting and ordering procedures. The initial term of the supply agreement is until 2012, subject to certain customary termination provisions, such as termination for material breach of the agreement. Either we or InSite Vision may terminate the supply agreement upon 180 days’ notice to the other party. After
11
Table of Contents
2012, the supply agreement automatically renews for successive three-year periods unless terminated pursuant to such termination provisions. The supply agreement requires that InSite Vision produce for us a specified stockpile of azithromycin.
Novasep
On December 16, 2010, we entered into a commercial supply agreement with Finorga S.A.S., a corporation organized and existing under the laws of France and a wholly-owned subsidiary of Groupe Novasep S.A.S. (hereinafter referred to as “Novasep”), for the active pharmaceutical ingredient, denufosol. Under the supply agreement, we are not obligated to purchase any particular volumes of denufosol from Novasep.
Yamasa Corporation
Effective September 25, 2009, we entered into a Technology License Agreement for the Manufacture of Denufosol with Yamasa Corporation. During our denufosol development program, Yamasa has manufactured all of the denufosol used by Inspire in its related clinical trials. The purpose of the technology agreement was to facilitate the transfer of the current denufosol manufacturing technology, including intellectual property, to Novasep.
Pursuant to the terms and conditions of this technology agreement, Yamasa granted to Inspire an exclusive, worldwide, royalty-free right and license to use, make, have made and sell denufosol and any pharmaceutical formulation containing denufosol as an active pharmaceutical ingredient, under certain intellectual property developed by Yamasa. During the term of this technology agreement, Inspire may designate a single manufacturer (in addition to Yamasa) to use the rights licensed from Yamasa at any given time.
In consideration of the grant of rights under the technology agreement, in October 2009, Inspire paid Yamasa three hundred million Japanese Yen (¥ 300,000,000), which was approximately $3.3 million at the prevailing exchange rate. Additionally, Inspire was obligated to pay Yamasa (i) three hundred million Yen (¥ 300,000,000) within thirty (30) days after the receipt of a process validation report by December 31, 2010 following the successful completion of three validation batches; and (ii) four hundred million Yen (¥ 400,000,000) within thirty (30) days after both (a) the acceptance of a pre-approval inspection of denufosol at Yamasa by the FDA, and (b) the approval of a New Drug Application of a formulated denufosol drug product at Inspire by the FDA.
In February 2011, we entered into an amended and restated technology agreement with Yamasa. The amended and restated technology agreement relieves Inspire of any restrictions on its ability to manufacture, or have manufactured, denufosol API. Yamasa reserves no rights to manufacture, or have manufactured, denufosol API. In addition, the seven hundred million Yen (¥ 700,000,000) of potential milestone payments, mentioned above, have been eliminated and there are no further milestones to be earned under the amended and restated technology agreement. In addition, certain of the patents and patent applications licensed to Inspire under the technology agreement were assigned to Inspire.
Patents and Proprietary Rights
We believe that the proprietary protection of our product candidates, processes and know-how is important to the success of our business. We file and prosecute patents covering our proprietary technology and, if warranted, will defend our patents and proprietary technology. We seek trademark protection in the United States and foreign countries, as appropriate. We also rely upon trade secrets, know-how, continuing technological innovation and licensing opportunities to develop and maintain our competitive position.
As of January 31, 2011, our patent estate included approximately 80 U.S. patents that we own or co-own and approximately 20 U.S. patents that we have licensed, as well as over 250 counterpart patents in countries other than the United States. Our issued patents and pending patent applications in the United States include
12
Table of Contents
composition of matter coverage on a number of different structural families of compounds as well as related formulation and use coverage. The actual protection afforded by a patent varies from country to country and depends upon many factors, including the type of patent, the scope of its coverage and the availability of legal remedies in a particular country.
Below is a summary of certain patent information, as of January 31, 2011, relating to our commercial products as well as product candidates that are in Phase 3 development. The information relating to the products listed below reflects those patents listed in the FDA’s Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations, with respect to such product. The information relating to Prolacria represents those patents listed in our NDA filed with respect to such candidate and the information regarding denufosol tetrasodium reflects the U.S. patents that we own and consider to be particularly important to the protection of such candidate. In addition to the patents reflected in the table, for some of these product candidates we have other patents that cover a particular form or composition or relate to manufacturing methods, as well as pending patent applications. These issued patents and any patents issued in relation to a pending applications, could provide additional or a longer period of protection.
| PRODUCTS AND PRODUCT CANDIDATES |
NUMBER OF SPECIFIED PATENTS IN U.S. |
TYPES OF PATENTS |
PATENT OWNER IN |
RANGE OF
U.S. | ||||||
| Products |
||||||||||
| AzaSite |
5 | Use and Formulation Patents | InSite Vision/Pfizer 1 | November 2018 – March 2019 | ||||||
| Elestat |
N/A2 | N/A 2 | N/A | N/A | ||||||
| Diquas |
N/A3 | N/A 3 | N/A | N/A | ||||||
| Restasis |
1 | Formulation | Allergan | May 2014 | ||||||
| Product Candidates in |
||||||||||
| Denufosol tetrasodium | 6 | Drug substance, Formulation and Use | Inspire | February 2017 4 | ||||||
| Prolacria (diquafosol tetrasodium) |
8 | Drug substance, Formulation and Use | Inspire | July 2016 – February 2017 4 | ||||||
| (1) | In-licensed to Inspire |
| (2) | See “Product—Elestat” for a discussion of the expiration of market exclusivity under the Hatch-Waxman Act. |
| (3) | Diquas is commercialized in Japan and accordingly is not covered by any U.S. patents. |
| (4) | Subject to any applicable patent restoration that may extend protection up to an additional five years from the date of expiration of the applicable patent, if any, for which restoration is sought. |
We seek patent protection for our proprietary technology and products in the United States and Canada and in key commercial European and Asia/Pacific countries and other major commercial sectors of the world, as appropriate. We have in-licensed patents related to AzaSite in Canada, and have received patents related to denufosol tetrasodium and diquafosol tetrasodium in Canada, Europe, Australia, New Zealand and other Asia/Pacific countries. See “Agreements—Allergan, Inc.—Restasis and Prolacria” and “Agreements—Santen Pharmaceutical Co., Ltd.” for additional discussion of responsibilities for diquafosol tetrasodium development and potential commercialization outside of the United States
13
Table of Contents
Competition
Many pharmaceutical and biotechnology companies engage in research and development to commercialize products to treat allergic conjunctivitis, bacterial conjunctivitis, dry eye disease, blepharitis, glaucoma, and other ocular diseases. We compete with these companies for funding, access to licenses, personnel, third-party collaborators and product development. However, most large pharmaceutical and biotechnology companies have significantly larger intellectual property estates, substantially greater financial, marketing, sales, distribution and technical resources and greater capabilities and experience in preclinical and clinical development, sales, marketing, manufacturing and regulatory affairs than we do. The introduction of new products or the development of new processes by competitors or new information about existing products may result in price reductions or product replacements, even for products protected by patents.
The following treatments compete, or may compete, with our products and product candidates, as applicable:
Allergic Conjunctivitis. There are multiple therapies available to treat or prevent allergic conjunctivitis. The primary products that Elestat competes with are Patanol® and PatadayTM, both by Alcon, Inc.; LastacaftTM by Allergan; BepreveTM by ISTA Pharmaceuticals, Inc.; Zaditor® by Novartis and its related generic; and Optivar® by Meda Pharmaceuticals and its related generic. Patanol and Pataday currently have the majority of the prescriptions in the allergic conjunctivitis market.
Bacterial Conjunctivitis. The current prescription ocular anti-infective treatments for bacterial conjunctivitis that compete with AzaSite include single agent ocular antibiotics Vigamox® and Ciloxan®, both by Alcon; Zymar®, Zymaxid® and Ocuflox®, all by Allergan; Quixin® and Iquix®, both by Vistakon Pharmaceuticals, LLC; and Besivance® by Bausch & Lomb, Inc. In addition, there are several generics used to treat bacterial conjunctivitis, which include erythromycin, gentamycin and tobramycin.
Dry Eye Disease. The current prescription and non-prescription treatments for dry eye disease include Restasis by Allergan; Lacrisert® by Aton Pharma, a Division of Valeant Pharmaceuticals International; and numerous over the counter artificial tear solutions and lubricant eye drops. We are aware of several companies that are developing products for the treatment of dry eye disease.
Governmental Regulation
The research, development, testing, manufacture, promotion, marketing and distribution of human therapeutic products are extensively regulated by governmental authorities in the United States and other countries. The FDA regulates drug products in the United States and similar regulatory agencies exist in other countries. The steps ordinarily required before a new drug may be marketed in the United States, which are similar to steps required in most other countries, include:
| • | Preclinical laboratory tests, preclinical studies in animals and formulation studies and the submission to the FDA of an Investigational New Drug Application, or IND, prior to beginning clinical trials for a new drug; |
| • | Adequate and well-controlled clinical trials to establish the safety and efficacy of the drug for each indication; |
| • | The submission of an NDA to the FDA; and |
| • | FDA review and approval of the NDA before any commercial sale or shipment of the drug. |
Preclinical tests include laboratory evaluation of product toxicity and formulation, as well as animal studies. The results of preclinical testing are submitted to the FDA as part of an IND. A 30-day waiting period after the filing of each IND is required before the commencement of clinical testing in humans. At any time during this 30-day period or later, the FDA may place a clinical hold and halt proposed or ongoing clinical trials for any one
14
Table of Contents
of several conditions that are set out in regulations, and the clinical trial may not resume until the FDA withdraws its hold on the clinical trials. Moreover, positive results of preclinical tests will not necessarily indicate positive results in clinical trials.
Clinical trials to support NDAs are typically conducted in three sequential phases, but the phases may overlap.
Phase 1—During Phase 1, the initial introduction of the drug into healthy volunteers, the drug is tested to assess metabolism, pharmacokinetics and pharmacological actions and safety, including side effects associated with increasing doses.
Phase 2—Phase 2 usually involves studies in a limited patient population to: (1) assess the efficacy of the drug in specific, targeted indications; (2) assess dosage tolerance and optimal dosage; and (3) identify possible adverse effects and safety risks.
Phase 3—If a compound is found to be potentially effective and to have an acceptable safety profile in Phase 2 evaluations, Phase 3 clinical trials, also called pivotal studies, major studies or advanced clinical trials, are undertaken to further demonstrate clinical efficacy and to further test for safety within an expanded patient population at geographically dispersed clinical trial sites. In general, the FDA requires that at least two adequate and well-controlled Phase 3 clinical trials be conducted.
After successful completion of the required clinical testing, generally an NDA is submitted. The FDA may request additional information before accepting an NDA for filing, in which case the application must be resubmitted with the additional information. Once the submission has been accepted for filing, the FDA has 180 days to review the application and respond to the applicant. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA may refer the NDA to an appropriate advisory committee for review, evaluation and recommendation as to scientific issues relevant to whether the application should be approved, but the FDA is not bound by the recommendation of an advisory committee.
Based on its review of the NDA and associated support, such as the results from inspections of manufacturing and clinical sites, the FDA will either approve or refuse to approve the NDA, unless the FDA evaluation is inconclusive, in which case the FDA will issue a “complete response letter.” The complete response letter replaced the FDA’s “approvable” and “non-approvable” letters on August 11, 2008. A complete response letter will state that the FDA cannot approve the NDA in its present form and, usually, will describe all of the specific deficiencies that the FDA has identified in the application. The complete response letter, when possible, will include the FDA’s recommended actions to place the application in a condition for approval. Deficiencies can be minor (e.g., labeling changes) or major (e.g., requiring additional clinical trials). A complete response letter may also be issued before the FDA conducts the required facility inspection and/or reviews labeling, leaving the possibility that additional deficiencies in the original NDA could be subsequently cited. An applicant receiving a complete response letter is permitted to resubmit the NDA addressing the identified deficiencies (in which case a new two or six month review cycle will begin), or withdraw the NDA. The FDA may consider a failure to take action within one year of a complete response letter to be a request to withdraw, unless the applicant has requested an extension of time in which to resubmit. If the FDA approves an NDA, the marketing of the drug product will be limited to the particular disease states and conditions of use that are described in the product label.
We and all of our contract manufacturers are also required to comply with the applicable FDA current Good Manufacturing Practice, or cGMP, regulations during clinical development and to ensure that the product can be consistently manufactured to meet the specifications submitted in an NDA. The cGMP regulations include requirements relating to product quality as well as the corresponding maintenance of records and documentation. Manufacturing facilities must be approved by the FDA before they can be used to manufacture our products. Based on an inspection, the FDA determines whether manufacturing facilities are in compliance with applicable regulations. Manufacturing facilities in non-U.S. countries that are utilized to manufacture drugs for distribution into the United States are subject to inspection by the FDA. Additionally, failure to comply with local regulatory requirements could affect production and availability of product in relevant markets.
15
Table of Contents
We must also comply with multiple governmental requirements and best practices associated with the marketing, sale and distribution of our products and product samples. These include, but are not limited to, compliance with federal and state reporting laws; review, approval and distribution of product promotional materials; review and monitoring of promotional and educational programs; interactions with health care providers; repackaging and labeling requirements; and distribution of product samples.
With regard to AzaSite, we are responsible for monitoring the safety of the product, reporting adverse events, and taking corrective actions as necessary. In addition, we enter into contracts with managed care organizations for both private and government programs, including Medicare Part D and also directly with state and federal governments for certain programs, including Medicaid programs.
Outside the United States, our ability to market our products will also depend on our receipt of marketing authorizations from the appropriate regulatory authorities, as well as the efforts of our collaborative partners to obtain authorizations. The requirements governing the conduct of clinical trials and marketing authorization vary widely from country to country. At present, foreign marketing authorizations are applied for at a national level, although within the European Union procedures are available to companies seeking to market a product in more than one member state. If the regulatory authority is satisfied that adequate evidence of safety, quality and efficacy has been presented, a marketing authorization will be granted. Foreign regulatory approval processes, including those in Europe and Japan, involve risks similar to those associated with obtaining FDA marketing approval.
Health Care Reform Measures and Third-Party Reimbursement
Sales of prescription drugs depend significantly on the availability of reimbursement to the consumer and/or provider from third-party payors, such as government health programs and private insurance plans. These third-party payors frequently require that drug companies provide predetermined discounts or rebates from list prices, and they are increasingly challenging the prices for medical products and services. Third-party payors may not consider our products to be cost effective and may not reimburse the consumer sufficiently to allow us, and/or our collaborators, to sell our products on a profitable basis. In addition, budgetary concerns at the federal and state levels and an increasing emphasis on managed care in the United States continue to increase pressure on drug pricing. Additional legislative or regulatory proposals or changes in managed care practices may be adopted that may have an adverse effect on our business and our financial condition, including our profits.
In both the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes to the health care system adopted in recent years that could affect our ability to sell our products profitably, and additional changes could be adopted in the future. For instance, in the United States, the Medicare Prescription Drug, Improvement and Modernization Act of 2003 established a voluntary Medicare outpatient prescription drug benefit under Part D of the Social Security Act. The program, which went into effect January 1, 2006, is administered by the Centers for Medicare & Medicaid Services, or CMS, within the Department of Health and Human Services, or HHS, and is implemented and operated by private sector Part D plan sponsors. Under the Part D program, each participating drug plan is permitted by regulation to develop and establish its own unique drug formulary that may exclude certain drugs from coverage and impose prior authorization and other coverage restrictions, and negotiate payment levels with drug manufacturers that may be lower than reimbursement levels available through private health plans or other payers. Moreover, beneficiary co-insurance requirements can vary, influencing which products are recommended by physicians and selected by patients. CMS has issued extensive regulations and other subregulatory guidance documents to assist Part D plan sponsors with implementing the new benefit. Moreover, the HHS Office of Inspector General has issued regulations and other guidance in connection with the program. The federal government continues to issue guidance and regulations regarding the obligations of Part D sponsors on an ongoing basis.
Allergan is responsible for Medicare Part D program activities relating to Restasis and Elestat and has contracted with Part D plan sponsors to cover such drugs under the Part D benefit. We are responsible for contracting with Part D plan sponsors with respect to AzaSite. There is no assurance that any drug that we
16
Table of Contents
co-promote or sell will be covered by drug plans participating under the Medicare Part D program or, if covered, what the terms of any such coverage will be, or that the drugs will be reimbursed at amounts that reflect current or historical payment levels. Our results of operations could be materially adversely affected by coverage or reimbursement changes resulting from the Medicare prescription drug program, including changes in Part D formularies or prices negotiated with Part D drug plans. To the extent that private insurers or managed care programs follow Medicare coverage and payment developments, the adverse effects of lower Medicare payments may be magnified by private insurers adopting similar lower payment. In addition, health care reform legislation in the United States and in foreign countries, along with other federal or state prescription drug reimbursement and coverage policies, may be enacted or adopted in the future that could further lower payment for our products.
More recently, in March 2010, Congress passed significant health reform legislation, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, “ACA”). The legislation is designed to expand access to affordable health insurance through subsidies, Medicaid expansion, and insurance market reforms (including the development of new health benefit exchanges), financed in part through reduced federal health care spending and various taxes and fees.
Among other things, the ACA makes a number of significant changes affecting pharmaceutical manufacturers. With regard to Medicaid drug pricing, the ACA provides for increases to the minimum Medicaid rebate percentages from 15.1% to 23.1%, increases in “additional rebates” for new formulations of brand name drugs, the establishment of a maximum rebate amount, and the extension of Medicaid rebates to Medicaid managed care organization utilization. In addition, the ACA broadens the definition of “average manufacturer price,” or AMP, which in turn may have the effect of increasing Medicaid rebate and Public Health Service section 340B drug discount program payment obligations. The ACA also provides that federal upper limits for multiple source drugs available for purchase by retail community pharmacies on a nationwide basis must be set at no less than 175% of the weighted average (based on utilization) of the most recently reported monthly AMP, using a smoothing process. The payments included in the calculation of AMP were further modified by the Education Jobs and Medicaid Assistance Act, which was enacted on August 10, 2010.
The ACA also includes provisions impacting the Medicare Part D program. For instance, the ACA requires drug manufacturers to provide a 50% discount on brand-name prescriptions filled in the Medicare Part D “coverage gap” beginning in 2011. The legislation also provides a $250 payment to Part D beneficiaries who reach the coverage gap during 2010, and mandates the gradual elimination of the coverage gap, beginning in 2011 and finishing in 2020. Moreover, the ACA reduces Part D premium subsidies for higher-income beneficiaries, expands medication therapy management requirements, and makes a number of other revisions to Part D program requirements. In addition, the ACA: extends the 340B drug discount program to additional entities, expands oversight of the 340B program, and increases manufacturer reporting requirements; creates an Independent Payment Advisory Board to develop recommendations to reduce Medicare spending under certain circumstances, with such recommendations to go into effect automatically unless Congress adopts alternative savings; encourages comparative effectiveness research; expands public disclosure requirements regarding drug manufacturer financial arrangements with physicians and teaching hospitals; and establishes a regulatory pathway for generic versions of biologics. Further, beginning in 2011, manufacturers and importers of branded prescription drugs and biologics will be assessed an annual fee ($2.5 billion in 2011 and increasing amounts in subsequent years up to $4.1 billion in 2018 and $2.8 billion annually thereafter), with individual company allocations to be determined by the Secretary of the Treasury, based generally on market share. The IRS recently extended the deadline for filing sales information, and has not yet made preliminary fee calculations for individual manufacturers.
While the Obama Administration has issued guidance on certain aspects of implementation of the ACA, details on many other ACA policies have yet to be finalized. Although the ACA provisions implemented to date have not had a material adverse impact on our operations or profitability, there can be no assurances that the prescription drug user fees and other ACA provisions impacting prescription drug policy will not adversely impact reimbursement and/or coverage of our products or otherwise impact our financial position.
17
Table of Contents
In addition to federal health reform legislation, a number of states have adopted or are considering measures to contain state health care costs, institute other health care coverage and delivery reforms, and require disclosure of certain manufacturer relationships with providers. The adoption of cost-control and other reform proposals on a state-by-state basis could impact our reimbursement levels and require us to develop state-specific marketing and sales approaches.
Employees
As of January 31, 2011, we had approximately 240 full-time and part-time employees. In February 2011, we implemented a strategic corporate restructuring focusing activities on our eye care business and discontinuing our pulmonary therapeutic area. This restructuring resulted in a workforce reduction of approximately 65 positions or approximately 27% of our full-time employees, primarily affecting functions in research and development; manufacturing and technical operations; and general and administrative. Our future success will depend in large part upon our ability to attract and retain highly qualified personnel. Our employees are not represented by any collective bargaining agreements, and we have never experienced a work stoppage. Employees are required to execute confidentiality and assignment of intellectual property agreements.
Available Information
Our Internet site is located at www.inspirepharm.com. Copies of our reports filed pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, including annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and any amendments to those reports may be accessed from our website, free of charge, as soon as reasonably practicable after we electronically file such reports with, or furnish such reports to, the Securities and Exchange Commission, or SEC. Please note that the information contained on our website is not incorporated by reference into our reports that are filed with the SEC. The public may read or copy any materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street NE, Room 1580, Washington D.C. 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains an Internet site that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. The address of that site is www.sec.gov.
18
Table of Contents
RISK FACTORS
An investment in our common stock involves a substantial risk of loss. You should carefully read this entire report and should give particular attention to the following risk factors. You should recognize that other significant risks may arise in the future, which we cannot foresee at this time. Also, the risks that we now foresee might affect us to a greater or different degree than expected. There are a number of important factors that could cause our actual results to differ materially from those indicated by any forward-looking statements in this report. These factors include, without limitation, the risk factors listed below and other factors presented throughout this report and any other reports filed by us with the SEC.
Risks Related to Product Commercialization
Failure to adequately market and commercialize AzaSite will negatively impact our revenues.
The commercial success of AzaSite will depend on a number of factors, including:
| • | Acceptance by patients and physicians; |
| • | Effectiveness of our sales and marketing efforts; |
| • | Ability to differentiate AzaSite relative to our competitors’ products; |
| • | Ability to further develop clinical information to support AzaSite; |
| • | Market satisfaction with existing alternative therapies; |
| • | Perceived efficacy relative to other available therapies; |
| • | Disease prevalence; |
| • | Cost of treatment; |
| • | Pricing and availability of alternative products, including generic or over-the-counter products; |
| • | Marketing and sales activities of competitors; |
| • | Shifts in the medical community to new treatment paradigms or standards of care; |
| • | Relative convenience and ease of administration; |
| • | Adequate production and supply of commercial product and samples; and |
| • | Our ability to enter into managed care and governmental agreements on favorable terms. |
We are responsible for all aspects of the commercialization of this product, including the determination of formularies upon which AzaSite is listed, manufacturing, distribution, marketing and sales. The determination of formularies upon which AzaSite is listed, the discounts and pricing under such formularies, as well as the amount of time it takes for us to obtain favorable formulary status under various plans, will impact our commercialization efforts. Additionally, inclusion on certain formularies requires significant price concessions through rebate programs that impact the level of revenue that we receive. The need to give price concessions can be particularly acute where competing products are listed on the same formulary, such as the area of bacterial conjunctivitis. If AzaSite is not successfully commercialized, our revenues will be limited.
Under our agreement with InSite Vision, we are obligated to make pre-determined minimum annual royalty payments to InSite Vision. To the extent annual royalty payments actually paid to InSite Vision on our sales of AzaSite are less than the minimum annual royalty amounts established under our agreement with InSite Vision, we are obligated to pay the difference. In the event we are required to make annual minimum royalty payments,
19
Table of Contents
our profits with respect to AzaSite, if any, will be decreased or any losses with respect to the product will be increased. Such circumstances may result in us ceasing our commercialization of AzaSite and terminating our agreement with InSite Vision. Based on actual net sales from AzaSite during the fourth quarter of 2010 and our expectation of net sales from AzaSite in 2011, we anticipate incurring a shortfall of the minimum royalty due to InSite Vision at the end of September 30, 2011.
Under our agreement with the manufacturer of AzaSite, we are required to purchase a minimum number of units of AzaSite annually, regardless of our ability to sell AzaSite. If we are unable to sell the quantities of AzaSite that we are required to purchase, our inventory of the product will increase and the shelf life of the inventory will be adversely impacted. In such circumstances, we may be required to make price concessions to sell short-dated product or write-off and dispose of expired product, which may have an adverse affect on our AzaSite profitability.
If Restasis is not successfully commercialized by Allergan, our revenues will be negatively impacted.
Allergan is responsible for commercializing Restasis. Accordingly, our revenues on the net sales of Restasis are dependent on the actions and success of Allergan, over whom we have no control.
The manufacture and sale of Restasis is protected in the United States by a formulation patent that expires in May 2014. While a formulation patent may afford certain limited protection, a competitor may attempt to gain FDA approval for a cyclosporine product using a different formulation. Furthermore, following the expiration of the formulation patent in 2014, a generic form of Restasis could be introduced into the market. If and when Restasis experiences competition from a cyclosporine product, including generics, our revenues attributable to Restasis may be significantly impacted.
Other factors that could affect the commercialization of Restasis include:
| • | Extent and effectiveness of Allergan’s sales and marketing efforts; |
| • | Satisfaction with existing alternative therapies, including generic or over-the-counter products; |
| • | Perceived efficacy relative to other available therapies; |
| • | Changes in, or the levels of, third-party reimbursement of product costs; |
| • | Coverage and reimbursement under Medicare Part D, state government sponsored plans and commercial plans; |
| • | Cost of treatment; |
| • | Development and FDA approval of competing dry eye products; and |
| • | Shifts in the medical community to new treatment paradigms or standards of care. |
When a generic form of Elestat or an over-the-counter form of epinastine ophthalmic solution is introduced into the market, which we expect may occur at any time, our agreement with Allergan to co-promote Elestat will no longer be in effect, and our revenues from sales of Elestat will be nominal.
Our Elestat co-promotion agreement with Allergan provides that unless earlier terminated, the term of such agreement will be in effect until the earlier of (i) the approval and launch of the first generic epinastine product after expiration of the FDA exclusivity period covering Elestat in the United States, or (ii) the approval and launch of the first over-the-counter epinastine product after expiration of the listing of Elestat in the FDA publication Approved Drug Products with Therapeutic Equivalence (commonly called the “Orange Book”). Following the termination of the co-promotion agreement, we will no longer have the right to co-promote Elestat. We will be entitled to receive post-termination payments from Allergan, based on any remaining net sales of Elestat for a period of 36 months. During the three successive 12-month periods immediately following the termination of the agreement, Allergan will be obligated to pay to us 20%, 15% and 10%, respectively, on any net sales of Elestat in the United States.
20
Table of Contents
Subject to applicable law, competitors are permitted to submit to the FDA an ANDA for a generic version of Elestat, due to the expiration of the marketing exclusivity period for Elestat provided under the Hatch-Waxman Act on October 15, 2008. We are aware that the following companies have filed an ANDA for a generic version of Elestat: Apotex, Inc., Cypress Pharmaceutical, Inc., Paddock Laboratories, Inc., PharmaForce, Inc. and Sandoz, Inc. The date of submission of the first ANDA filing to the FDA Office of Generic Drugs was October 14, 2008, according to the FDA’s website (www.fda.gov). Also, according to the FDA’s website, Apotex, Inc., PharmaForce, Inc. and Sandoz, Inc. have each received tentative approval for their respective epinastine hydrochloride ophthalmic solution.
The FDA’s review of an ANDA is a confidential process between the FDA and the applicable ANDA filer. We do not expect to be informed by the FDA, any ANDA filer or any other party regarding the status or timing of the review relating to any of the ANDA filings pertaining to a generic form of Elestat. The FDA may complete its review of the filed ANDAs at any time. As a result, we expect to be required to stop the co-promotion of Elestat with little, if any, advance notice. We expect that a generic form of epinastine may be launched at any time. The loss of co-promotion revenue from Elestat will significantly impact our results of operations and cash flows.
If we do not successfully market and promote Elestat, our revenues will be negatively impacted.
Notwithstanding the expected termination of the Elestat agreement upon the launch of a generic form of Elestat, our present revenues depend, in part, upon the continued acceptance of Elestat by eye care professionals, allergists and patients. Other factors that could affect the commercialization of Elestat include:
| • | Satisfaction with existing alternative therapies, including therapies requiring only one dose per day; |
| • | Decreases in the size of the market for topical allergic conjunctivitis products; |
| • | Extent and effectiveness of our sales and marketing efforts; |
| • | Changes in, or the levels of, third-party reimbursement of product costs; |
| • | Coverage and reimbursement under Medicare Part D, state government sponsored plans and commercial plans; |
| • | Pricing and availability of alternative products, including generic or over-the-counter products; and |
| • | Marketing and sales activities of competitors. |
We rely on third parties to distribute and sell our products and those third parties may not perform.
We are dependent on third parties to perform or assist us in the distribution or sale of AzaSite, and are dependent on third parties, primarily Allergan, for the distribution and sale of Elestat. We rely on the services of a single source, third-party distributor to deliver AzaSite to our customers. In addition to the physical storage and distribution of AzaSite, this third-party distributor maintains and provides us with information and data with regard to our AzaSite inventory, orders, billings and receivables, chargebacks and returns, among others, on which our accounting estimates are based. If third parties do not successfully carry out their contractual duties in maximizing the commercial potential of our products, we may be required to hire or expand our own staff and sales force to compete successfully, which may not be possible. If third parties or Allergan do not perform, or assist us in performing these functions, or if there is a delay or interruption in the distribution of our products, it could have an adverse effect on product revenue, accounting estimates and our overall operations.
We depend on three pharmaceutical wholesalers for the vast majority of our AzaSite sales in the United States, and the loss of any of these wholesalers could negatively impact our revenues.
The prescription drug wholesaling industry in the United States is highly concentrated, with a vast majority of all sales made by three major full-line companies: Cardinal Health, McKesson Corporation and AmerisourceBergen. Greater than 85% of our AzaSite revenues come from sales to these three companies. The loss of any of these wholesalers could have a negative impact on our commercialization of AzaSite.
21
Table of Contents
It is also possible that these wholesalers, or others, could decide to change their policies and fees in the future. This could result in or cause us to incur higher product distribution costs, lower margins or the need to find alternative methods of distributing our products. Such alternative methods may not be economically or administratively feasible.
If Diquas is not successfully commercialized by Santen in Japan, we may not receive any royalty revenues with respect to the sales of such product.
In December 2010, Santen launched Diquas in Japan. Santen is solely responsible for commercializing Diquas. Accordingly, royalty revenues on the net sales of Diquas are dependent on the successful commercialization efforts of Santen, over whom we have no control. Factors that could affect the commercialization of Diquas in Japan include:
| • | The production of sufficient quantities of the product in order to facilitate the launch of the product and adequately supply the market; |
| • | Perceived efficacy relative to other available therapies; |
| • | Development and regulatory approval of competing dry eye products; |
| • | The extent and effectiveness of Santen’s sales and marketing efforts; and |
| • | Satisfaction with existing alternative therapies, including generic or over-the-counter products. |
Risks Related to Manufacture and Supply
If we are unable to contract with third parties for the manufacture of active pharmaceutical ingredients required for preclinical testing, for the manufacture of drug products for clinical trials or for the large-scale manufacture of any approved products, we may be unable to develop or commercialize our drug products.
The manufacturing of sufficient quantities of new products or product candidates is a time-consuming and complex process. We have no experience or capabilities to conduct the manufacture of any of our products or product candidates. In order to successfully commercialize AzaSite and continue to develop our product candidates, we need to contract or otherwise arrange for the necessary manufacturing services. There are a limited number of manufacturers that operate under the FDA’s cGMP regulations capable of manufacturing for us or our collaborators. We depend upon third parties for the manufacture of API, finished drug products for clinical trials, and for the manufacture of AzaSite. We expect to depend upon third parties for the large-scale manufacture of commercial quantities of any other approved product. This dependence may adversely affect our ability to develop and deliver such products on a timely and competitive basis. Similarly, our dependence on our partners to arrange for their own supplies of finished drug products may adversely affect our operations and revenues. If we, or our partners, are unable to engage or retain third-party manufacturers on a long-term basis or on commercially acceptable terms, our products may not be commercialized as planned, and the development of our product candidates could be delayed.
Reliance on a single party to manufacture and supply either finished product or the bulk active pharmaceutical ingredients for a product or product candidates could adversely affect us.
Under our agreements with Allergan, Allergan is responsible for the manufacture and supply of Restasis and Elestat. Allergan relies upon an arrangement with a single third party for the manufacture and supply of APIs for each of Restasis and Elestat. Allergan then completes the manufacturing process to yield finished product.
Under our supply agreement with InSite Vision, InSite Vision is responsible for supplying us with azithromycin, the API used in AzaSite. InSite Vision, in turn, relies upon an arrangement with a single third party for the manufacture and supply of such API. We are responsible for producing the finished product form of
22
Table of Contents
AzaSite, which is currently manufactured by a single party. There can be no assurance that such manufacturer will be able to continue to produce sufficient quantities of finished product in a timely manner to support the commercialization of AzaSite.
In the event a third-party manufacturer is unable to supply Allergan or InSite Vision (as the case may be), if such supply is unreasonably delayed, or if Allergan or our finished product contract supplier are unable to complete the manufacturing cycle, sales of the applicable product could be adversely impacted, which would result in a reduction in any applicable product revenue. In addition, if Allergan or the third-party manufacturers do not maintain cGMP compliance, the FDA could require corrective actions or take enforcement actions that could affect production and availability of the applicable product, thus adversely affecting sales.
It would be time consuming and costly to identify and qualify new sources for manufacture of APIs or finished products. If our vendors were to terminate our arrangement or fail to meet our supply needs we might be forced to delay our development programs and/or be unable to supply products to the market, which could delay or reduce revenues and result in loss of market share.
Risks Related to Product Development
If the FDA does not conclude that our product candidates meet statutory requirements for safety and efficacy, we will be unable to obtain regulatory approval for marketing in the United States.
We have to conduct significant development activities, non-clinical and clinical tests and obtain regulatory approval before our product candidates can be commercialized. Product candidates that may appear to be promising at early stages of development may not successfully reach the market for a number of reasons. The results of preclinical and clinical testing of our product candidates under development may not necessarily indicate the results that will be obtained from later or more extensive testing. Additionally, companies in the pharmaceutical and biotechnology industries, including us, have suffered significant setbacks in advanced clinical trials, even after obtaining promising results in earlier clinical trials. Our ongoing clinical trials might be delayed or halted for various reasons, including:
| • | The measure of efficacy of the drug is not statistically significant compared to placebo; |
| • | Patients experience severe side effects or serious adverse events during treatment; |
| • | Patients die during the clinical trial because their disease is too advanced or because they experience medical problems that may or may not relate to the drug being studied; |
| • | Patients do not enroll in the clinical trials at the rate we expect; |
| • | We decide to modify the drug or the clinical trial protocol during testing; |
| • | Our commercial partners, or future commercial partners, delay, amend or change our development plan or strategy; and |
| • | We allocate our limited financial and other resources to other clinical and preclinical programs. |
Changes in regulatory policy or new regulations as well as clinical investigator misconduct could also result in delays or rejection of our applications for approval of our product candidates. Clinical investigator misconduct that raises questions about the integrity of data in one or more applications (e.g., fraud, bribery, omission of a material fact, gross negligence) could be used by the FDA as grounds to suspend substantive scientific review of any or all pending marketing applications until the data in question have successfully undergone a validity assessment. Product candidates that fail validity assessments must be withdrawn from FDA review or, if the drug is an approved, marketed product, such product must be removed from the market.
Additionally, the introduction of our products in foreign markets will subject us to foreign regulatory clearances, the receipt of which may be unpredictable, uncertain and may impose substantial additional costs and
23
Table of Contents
burdens which we or our partners in such foreign markets may be unwilling or unable to fund. As with the FDA, foreign regulatory authorities must be satisfied that adequate evidence of safety and efficacy of the product has been presented before marketing authorization is granted. The foreign regulatory approval process includes all of the risks associated with obtaining FDA marketing approval. Approval by the FDA does not ensure approval by other regulatory authorities, nor does approval by any foreign regulatory authority ensure approval by the FDA.
Since there may be no regulatory precedents for our clinical candidates, conducting clinical trials and obtaining regulatory approval may be difficult, expensive and prolonged, which would delay any commercialization of our products.
To complete successful clinical trials, our product candidates must demonstrate safety and provide substantial evidence of efficacy. The FDA generally evaluates efficacy based on the statistical significance of a product candidate meeting predetermined clinical endpoints. The design of clinical trials to establish meaningful endpoints is done in collaboration with the FDA prior to the commencement of clinical trials. We establish these endpoints based on guidance from the FDA, including FDA guidance documents applicable to establishing the efficacy, safety and tolerability measures required for approval of products. Depending upon the disease our product candidate is designed to treat, the FDA may not have established guidelines for the design of our clinical trials and may take longer than average to consider our product candidates for approval. The FDA could change its view on clinical trial design and establishment of appropriate standards for efficacy, safety and tolerability and require a change in clinical trial design, additional data or even further clinical trials before granting approval of our product candidates. We could encounter delays and increased expenses in our clinical trials if the FDA concludes that the endpoints established for a clinical trial do not adequately predict a clinical benefit.
We have initiated a Phase 2 development program to evaluate the use of AzaSite for the treatment of blepharitis. The FDA has not published guidelines on the approval of a product for the treatment of blepharitis. Furthermore, to date, no prescription product has been approved by the FDA for the treatment of blepharitis. The FDA may require that we evaluate the product in relation to different primary and/or secondary clinical endpoints than those being used presently. This may require us to undertake additional Phase 2 clinical trials, which could lead to increased costs and program delays.
We may need to develop alternate dosing regimens for our product candidates.
In order to achieve broad market acceptance of our product candidates, we may need to develop, alone or with others, alternate dosing regimens and methods for administering our products. If the number of doses, or the method of dosing, is not convenient, patients may not use our product. Furthermore, if patients use our products at a dosing level that is less than the dosing level tested in our clinical trials, the drug may not be efficacious or may be less efficacious. In such cases, the patient may look for alternative therapies.
Clinical trials may take longer to complete and cost more than we expect, which would adversely affect our ability to commercialize product candidates and achieve profitability.
The number and type of studies that may be required by the FDA, or other regulatory authorities, for a particular compound are based on the compound’s clinical profile compared to existing therapies for the targeted patient population. While all new compounds require standard regulated phases of testing, the actual type and scope of testing can vary significantly among different product candidates and as a result, creates additional complexity when estimating program costs. Factors that affect the costs of a clinical trial include:
| • | The number of patients required to participate in clinical trials to demonstrate statistical significance for a drug’s safety and efficacy and the number and geographical location of clinical trial sites necessary to enroll such patients; |
24
Table of Contents
| • | The time required to enroll the targeted number of patients in clinical trials, which may vary depending on the size and availability of the relevant patient population, the nature of the protocol, the proximity of patients to clinical sites, the eligibility criteria for the clinical trial, the perceived benefit to the clinical trial participants and approval of the Institutional Review Board; |
| • | Appropriate identification of optimal treatment regimens; |
| • | Adequate supplies of drug product; |
| • | Monitoring and auditing; and |
| • | The number and type of required laboratory tests supporting clinical trials. |
Delays in patient enrollment can result in increased costs and longer development times. Even if we successfully complete clinical trials, we may not receive regulatory approval for the product candidate. In addition, if the FDA or foreign regulatory authorities require additional clinical trials, we could face increased costs and significant development delays.
Additionally, ongoing development programs and associated costs are subject to frequent, significant and unpredictable changes due to a number of factors, including:
| • | Data collected in preclinical or clinical trials may prompt significant changes, delays or enhancements to an ongoing development program; |
| • | Commercial partners and the underlying contractual agreements may require additional or more involved clinical or preclinical activities; |
| • | The FDA or other regulatory authorities may direct the sponsor to change or enhance its ongoing development program based on developments in the testing of similar compounds or related compounds; |
| • | Unexpected regulatory requirements, changes in regulatory policy or review standards, or interim reviews by regulatory agencies may cause delays or changes to development programs; |
| • | Unexpected and serious events, reactions, or experiences of clinical trial participants may cause delays or changes to the development programs and could result in a clinical hold, which would stop the clinical trial until the FDA determined that the trial or development program could be resumed; and |
| • | Anticipated manufacturing costs may change significantly due to necessary changes in manufacturing processes, variances from anticipated manufacturing process yields or changes in the cost and/or availability of starting materials, and other costs to ensure the manufacturing facility is in compliance with cGMP requirements and is capable of consistently producing the product candidate in accordance with established specifications submitted to the FDA. |
The occurrence of any of these factors may result in significant disparities in total costs required to complete the respective development programs.
If we fail to reach milestones or to make annual minimum payments or otherwise breach our obligations under our license agreements, our licensors may terminate our agreements with them and seek additional remedies.
If we fail to meet payment obligations, performance milestones relating to the timing of regulatory filings, development and commercial diligence obligations, fail to make milestone payments in accordance with applicable provisions, or fail to pay the minimum annual payments under our respective licenses, our licensors may terminate the applicable license. As a result, our development of the respective product candidate or commercialization of the product would cease.
25
Table of Contents
Risks Related to Governmental Regulation
Failure to comply with all applicable regulations, including those that require us to obtain and maintain governmental approvals for our product candidates, may result in fines, corrective actions, administrative sanctions and restrictions, including the withdrawal of a product from the market.
Pharmaceutical companies are subject to significant regulation by a number of local, state, and federal governmental agencies, including the FDA. Such regulations and their authorizing statutes are amended from time to time. There are laws and regulations that govern areas including financial controls, clinical trials, testing, manufacturing, labeling, safety, packaging, shipping, distribution, post-approval safety surveillance, marketing, and promotion of pharmaceuticals, including those governing interactions with prescribers and health care professionals in a position to prescribe, recommend, or arrange for the provision of our products. Failure to comply with applicable regulatory requirements could, among other things, result in warning letters, fines, corrective actions, administrative sanctions, suspensions or delays of product manufacture or distribution or both, product recalls, delays in marketing activities and sales, withdrawal of marketing approvals, and civil or criminal sanctions including seizure of product, court-ordered injunctions, and possible exclusion from eligibility for federal government contracts and reimbursement of our products by Medicare, Medicaid, and other third-party payors. Senior management and the executives of an FDA-regulated company, such as ours, can be individually criminally prosecuted for violations of the Food, Drug, and Cosmetic Act and, unlike typical criminal actions, the government is not required to show an intent to violate the law. Successful criminal prosecutions can lead to debarment from providing any services to a drug company in any capacity. If this occurred, the reputation of the company could suffer, sales could decrease, formularies could refuse to include our drug products, and/or our revenues could be reduced and result in loss of market share.
After initial regulatory approval, the manufacturing and marketing of drugs, including our products, are subject to continuing FDA and foreign regulatory requirements and oversight. The FDA requires drug manufacturers and distributors to monitor the safety of a drug after it is approved and marketed. We are required to document and investigate reports of adverse events and report serious adverse events to the FDA. Additionally, the FDA encourages health professionals to report significant adverse events associated with products. The FDA may require additional clinical studies, known as Phase 4 studies, to evaluate product safety effects. In addition to studies required by the FDA after approval, we may conduct our own Phase 4 studies to explore the use of the approved drug product for treatment of new indications or to broaden our knowledge of the product. The subsequent discovery of previously unknown problems with a product’s safety or efficacy as a result of these studies or as reported in their prescribed use may result in the implementation of an FDA-required risk evaluation and mitigation strategy known as REMS, restrictions through labeling changes, or withdrawal of the product from the market.
The FDA periodically inspects drug manufacturing facilities to ensure compliance with applicable cGMP regulations. Failure to comply with statutory and regulatory requirements subjects the manufacturer to possible legal or regulatory action, such as suspension of manufacturing, seizure of product or court-ordered injunction, which could include mandatory recall of a product, and consent decrees that could be in place for up to five years or more until the government is satisfied with the company’s corrective and preventive actions to ensure compliance. Under a consent decree, a company is typically required to submit to third-party inspections, additional FDA reporting requirements, additional corrective actions, limitations on doing business in certain states, and disgorgement of any profits as a result of the wrongful conduct alleged by the government, all of which could significantly affect the cost of doing business and the company’s reputation, reduce revenues, and result in a loss of market share. Before taking such actions, the FDA may first issue one or more notices of compliance deficiencies. Such notices include inspectional observations on Form FDA-483, warning letters, and other untitled written correspondence; however, the FDA may also take action without such notice in situations of egregious noncompliance or where public safety is at risk. The FDA may also request us to take actions voluntarily or we may initiate actions ourselves such as recalls or suspension of manufacturing to ensure compliance with cGMP regulations.
26
Table of Contents
Additional authority to take post-approval actions was given to the FDA under the FDA Amendments Act of 2007. The FDA is authorized to revisit and change its prior determinations if new information raises questions about our product’s safety profile. The FDA is authorized to impose additional post-marketing requirements, such as development of a risk evaluation and mitigation strategy which could include additional monitoring of post-approval adverse events, providing additional patient safety information, and adding warnings, precautions, and other safety information to the labeling. Failure to implement such requirements could result in corrective actions, fines, withdrawal of marketing approval, or any combination of such actions.
In its regulation of drug product advertising and promotional activities, the FDA may issue correspondence to pharmaceutical companies alleging that their advertising, promotional materials or activities are false or misleading and requesting corrective actions to remove and mitigate claims made in such materials. Pharmaceutical advertising and promotional activity must be true, fairly balanced between benefits and risks, provide adequate risk information, and be within the labeled indications. Drug manufacturers are prohibited from promoting unapproved, investigational drug products, and any approved and marketed drug product for a use that is not on the approved labeling; however, healthcare professionals are free to use the product for any use that, in the judgment of the healthcare professional, may be appropriate for any individual patient. The FDA and the HHS Office of the Inspector General (“OIG”) have the power to impose a wide array of sanctions on companies for advertising practices that are found to be false or misleading, do not include adequate risk information, or promote an unapproved or off-label use and, if we were to receive correspondence from the FDA or the OIG alleging such practices, it may be necessary for us to:
| • | Incur substantial expenses, including fines, penalties, legal fees and costs to conform to the FDA’s limits on such promotion; |
| • | Change our methods of marketing, promoting and selling products; |
| • | Take corrective action, which could include placing advertisements or sending letters to physicians correcting statements made in previous advertisements or promotions; or |
| • | Disrupt the distribution of products and halt or suspend sales until we are in compliance with the FDA’s interpretation of applicable laws and regulations. |
Medicare prescription drug coverage legislation and legislative or regulatory reform of the health care system may affect our or our partners’ ability to sell products profitably.
In both the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes to the health care system that could affect our ability to sell our products profitably. In the United States, the Medicare Prescription Drug, Improvement and Modernization Act of 2003 established a voluntary Medicare outpatient prescription drug benefit under Part D of the Social Security Act. The program, which went into effect January 1, 2006, is administered by the Centers for Medicare & Medicaid Services within the HHS and is implemented and operated by private sector Part D plan sponsors. Each participating drug plan is permitted by regulation to develop and establish its own unique drug formulary that may exclude certain drugs from coverage and impose prior authorization and other coverage restrictions, and negotiate payment levels with drug manufacturers that may be lower than reimbursement levels available through private health plans or other payers. Moreover, beneficiary co-insurance requirements can vary, which could influence which products are recommended by physicians and selected by patients. The federal government continues to issue guidance and regulations regarding the obligations of Part D sponsors under the program, and a number of changes to the Part D program were included in recently-enacted health reform legislation, as discussed below.
Allergan is responsible for Medicare Part D program activities relating to Restasis and Elestat and has contracted with Part D plan sponsors to cover such drugs under the Part D benefit. We are responsible for contracting with Part D plan sponsors with respect to AzaSite. There is no assurance that any drug that we co-promote or sell will be covered by drug plans participating under the Medicare Part D program or, if covered, what the terms of any such coverage will be, or that the drugs will be reimbursed at amounts that reflect current or historical payment levels. Our results of operations could be materially adversely affected by the
27
Table of Contents
reimbursement changes associated with the Medicare prescription drug program or by changes in the formularies or price negotiations with Part D drug plans. To the extent that private insurers or managed care programs follow Medicare coverage and payment developments, the adverse effects of lower Medicare payment may be magnified by private insurers adopting similar lower payment.
Our products also can be impacted by state and federal legislative and regulatory changes in Medicaid reimbursement policy and in mandated levels of Medicaid drug rebates paid by pharmaceutical manufacturers. Many states are facing serious budgetary pressures that could lead to adoption of additional cost-containment measures, including provisions aimed at reducing Medicaid drug prices. Medicaid drug pricing policies also have been subject to periodic revision by Congress, including as part of federal health reform efforts, as discussed below. Further, on February 3, 2011, the HHS announced that it is undertaking a national survey of pharmacies to create a national database of actual acquisition costs that states can use to determine state-specific pharmaceutical reimbursement rates; the data will be available to states later this year. There can be no assurances that new state and federal policies will not lower Medicaid reimbursement levels for our products.
In March 2010, Congress passed significant health reform legislation, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, “ACA”). The legislation is designed to expand access to affordable health insurance through subsidies, Medicaid expansion, and insurance market reforms (including the development of new health benefit exchanges), financed in part through reduced federal health care spending and various taxes and fees.
Among many other things, the ACA makes a number of significant changes affecting pharmaceutical manufacturers. With regard to Medicaid drug pricing, the ACA provides for increases to the minimum Medicaid rebate percentages from 15.1% to 23.1%, increased “additional rebates” for new formulations of brand name drugs, the establishment of a maximum rebate amount, and the extension of Medicaid rebates to Medicaid managed care organization utilization. In addition, the ACA broadens the definition of “average manufacturer price,” or AMP, which in turn may have the effect of increasing Medicaid rebate and Public Health Service section 340B drug discount program payment obligations. The ACA also provides that federal upper limits for multiple source drugs available for purchase by retail community pharmacies on a nationwide basis must be set at no less than 175% of the weighted average (based on utilization) of the most recently reported monthly AMP, using a smoothing process. The payments included in the calculation of AMP were further modified by the Education Jobs and Medicaid Assistance Act , which was enacted on August 10, 2010.
With regard to the Medicare Part D program, the ACA also requires drug manufacturers to provide a 50% discount on brand-name prescriptions filled in the Medicare Part D “coverage gap” beginning in 2011. The legislation also provided a $250 payment to Part D beneficiaries who reach the coverage gap during 2010, and mandates the gradual elimination of the coverage gap, beginning in 2011 and finishing in 2020. Moreover, the ACA reduces Part D premium subsidies for higher-income beneficiaries, expands medication therapy management requirements, and makes a number of other revisions to Part D program requirements. In addition, the ACA: extends the 340B program to additional entities, expands oversight of the 340B program, and increases manufacturer reporting requirements; creates an Independent Payment Advisory Board to develop recommendations to reduce Medicare spending under certain circumstances, with such recommendations to go into effect automatically unless Congress adopts alternative savings; expands public disclosure requirements regarding drug manufacturer financial arrangements with physicians; and establishes a licensure pathway for generic versions of biologics. Further, beginning in 2011, manufacturers and importers of branded prescription drugs and biologics will be assessed an annual fee ($2.5 billion in 2011 and increasing amounts in subsequent years up to $4.1 billion in 2018 and $2.8 billion annually thereafter), with individual company allocations to be determined by the Secretary of the Treasury, based generally on market share. The IRS recently extended the deadline for filing sales information, and has not yet made preliminary fee calculations for individual manufacturers. There can be no assurances that implementation of the new health reform legislation will not have an adverse impact on revenues, as well as reimbursement and/or coverage of our products.
28
Table of Contents
In addition to federal health reform legislation, a number of states have adopted or are considering measures to contain state health care costs and institute other health care coverage and delivery reform, which could impact our reimbursement levels and require us to develop state-specific marketing and sales approaches. Further, a number of states require disclosure of financial arrangements with physicians, marketing expenditures, and the like, and other states may do so in the future; our failure to comply with these as well as the federal disclosure requirements could have an adverse impact.
Congress also has enacted the American Recovery and Reinvestment Act, which included a major expansion of federal efforts to compare the effectiveness of different medical treatments, including pharmaceuticals, which eventually could impact Medicare and private payer coverage and payment policies. This comparative effectiveness initiative was expanded by the ACA, and can include studies regarding the comparative clinical effectiveness of drugs. The federal government may consider additional proposals that could lead to coverage or reimbursement constraints and restrictions on access to certain products. We cannot predict the outcome of such initiatives, and it is difficult to predict the impact on our business of future legislative and regulatory changes.
We are subject to “fraud and abuse” and similar government laws and regulations, and a failure to comply with such laws and regulations, or an investigation into our compliance with such laws and regulations, or a failure to prevail in any litigation related to noncompliance, could harm our business.
We are subject to multiple state and federal laws pertaining to health care fraud and abuse. Pharmaceutical pricing, sales, and marketing programs and arrangements, and related business practices in the health care industry generally are under increasing scrutiny from federal and state regulatory, investigative, prosecutorial, and administrative entities. Many health care laws, including the federal and state anti-kickback laws and federal and state statutory and common law false claims laws, have been construed broadly by the courts and permit government entities to exercise considerable discretion. Further, the ACA contains a number of provisions strengthening the scope of the federal laws, including making a violation under the federal anti-kickback law a predicate action for violation of the federal False Claims Act and expanding the authority for Medicaid exclusions . The ACA also provides that a person need not have actual knowledge of the anti-kickback law, among others, or a specific intent to commit a violation of the law, to be found in violation of it. The ACA further applies the False Claims Act to payments made through new health benefits exchanges if the payments include any federal funds. In the event that government entities believed that wrongdoing had occurred, such entities could institute civil, administrative, or criminal proceedings which, if instituted and resolved unfavorably, could subject us to substantial fines, penalties, and injunctive and administrative remedies, including exclusion from government reimbursement programs. The adverse outcome of a government investigation also could prompt other government entities to commence investigations, or cause those entities or private parties to bring civil actions against us. We cannot predict whether investigations or enforcement actions would affect our marketing or sales practices. Any such result could have a material adverse impact on our results of operations, cash flows, financial condition, and our business. Such investigations and enforcement actions could be costly, divert management’s attention from our business, and result in damage to our reputation. We cannot guarantee that measures that we have taken to prevent violations, including our corporate compliance program, will protect us from future violations, lawsuits or investigations by governmental entities or private whistleblowers. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant negative impact on our business, including the imposition of significant fines or other sanctions.
Failure to adequately ensure compliance with applicable laws and regulations may subject us to whistleblower and government actions.
In recent years, pharmaceutical companies have been the targets of extensive whistleblower actions in which the person bringing the action alleges violations of the federal civil False Claims Act or its state equivalent, including allegations that manufacturers aided and abetted in the submission of false claims. These actions have
29
Table of Contents
focused on such areas as pricing practices, off-label product promotion, manufacturing deficiencies, sales and marketing practices, and improper relationships with physicians and other health care professionals, among others. The ACA eliminates a number of jurisdictional bars to bringing a false claims action, and therefore may make it easier for whistleblowers to bring successful actions. If our relationships with health care professionals and/or our promotional or other activities are found not to comply with applicable laws, regulations or guidelines, we may be subject to warnings from, or investigative or enforcement action by, regulatory and other federal or state governmental authorities. The potential ramifications are far-reaching if there are areas identified as out of compliance by regulatory agencies and governmental authorities including, but not limited to, significant financial penalties, manufacturing and clinical trial consent decrees, commercialization restrictions, exclusion from government programs, product recalls or seizures, the imposition of corporate integrity agreements and deferred prosecution agreements, or other restrictions and litigation. Furthermore, there can be no assurance that we will not be subject to a whistleblower or other state or federal investigative or enforcement action at some time in the future. Even an unsuccessful challenge to our operations or activities could prove costly and divert management’s attention.
Risks Associated with Our Business and Industry
If we do not receive timely regulatory approvals of our product candidates and successfully launch such products, we may need substantial additional funds to support our expanding capital requirements.
We have used substantial amounts of cash to fund our research and development and commercial activities. Our operating expenses were approximately $142.2 million and approximately $129.8 million for the years ended December 31, 2010 and 2009, respectively. Our cash, cash equivalents and investments totaled approximately $94.3 million on December 31, 2010. Based on current operating plans, we expect our cash and investments to provide liquidity beyond 2011.
We expect that our capital and operating expenditures will continue to exceed our revenue over the next several years as we conduct our research and development, licensing, acquisition and/or commercial activities. Many factors will influence our future capital requirements, including:
| • | The number, breadth and progress of our research and development programs; |
| • | The level of activities relating to commercialization of our products; |
| • | The ability to attract collaborators for our products and establish and maintain those relationships; |
| • | The receipt or payment of milestone payments under our current collaborative agreements and any future collaborations; |
| • | Progress by our collaborators with respect to the development of product candidates; |
| • | Competing technological and market developments; |
| • | The timing and terms of any business development activities; |
| • | The costs involved in defending any litigation claims against us; |
| • | The costs involved in responding to government, the Financial Industry Regulatory Authority, Inc., or other applicable investigations against us; and |
| • | The costs involved in enforcing patent claims and other intellectual property rights. |
In addition, our capital requirements will depend upon:
| • | The level of net sales generated for AzaSite; |
| • | The receipt of revenue from Allergan on net sales of Restasis; |
30
Table of Contents
| • | The timing of the introduction of a generic form of Elestat; |
| • | The receipt of revenue from wholesalers and other customers on net sales of AzaSite; |
| • | The ability to obtain approval from the FDA for our product candidates; and |
| • | Payments from existing and future collaborators. |
In the event that we do not receive timely regulatory approvals, we may need substantial additional funds to support our on-going operations, including business development, product commercialization and co-promotion efforts as well as continue our development activities associated with our product candidates. We may seek such additional funding through public or private equity offerings and debt financings. Additional financing may not be available when needed. If available, such financing may not be on terms favorable to us or our stockholders. Our stockholders’ ownership will be diluted if we raise additional capital by issuing equity securities. If we are required to raise funds through future collaborations and licensing arrangements, we may have to give up rights to our technologies or product candidates or grant licenses on unfavorable terms. If adequate funds are not available, we would have to scale back or terminate research programs and product development and we may not be able to successfully commercialize any product candidate.
Our co-promotion and royalty revenues are based, in part, upon Allergan’s revenue recognition policy and other accounting policies over which we have limited or no control.
We recognize co-promotion revenue based on Allergan’s net sales of Elestat and royalty revenue based on Allergan’s net sales of Restasis, as defined in the agreements and as reported to us by Allergan. Accordingly, our co-promotion and royalty revenues are based upon Allergan’s revenue recognition policy and other accounting policies, over which we have limited or no control, and the underlying terms of our agreements. Allergan’s filings with the SEC indicate that Allergan maintains disclosure controls and procedures in accordance with applicable laws, which are designed to provide reasonable assurance that the information required to be reported by Allergan in its Exchange Act filings is reported timely and in accordance with applicable laws, rules and regulations. We are not entitled to review Allergan’s disclosure controls and procedures. All of our co-promotion and royalty revenues are currently derived from Allergan’s net sales of Restasis and Elestat as reported to us by Allergan. We are unable to provide complete assurance that Allergan will not revise reported revenue amounts in the future. If Allergan’s reported revenue amounts were inaccurate, it could have a material impact on our financial statements, including financial statements for prior periods.
Revenues in future periods could vary significantly and may not cover our operating expenses.
Our revenues may fluctuate from period to period due in part to:
| • | The timing of the introduction of a generic form of Elestat; |
| • | Fluctuations in future sales of AzaSite, Restasis and Elestat due to competition, the intensity of an allergy season, disease prevalence, manufacturing difficulties, reimbursement and pricing under commercial or government plans, seasonality, or other factors that affect the sales of a product; |
| • | Deductions from gross sales relating to estimates of sales returns, credits and allowances, normal trade and cash discounts, managed care sales rebates and other allocated costs; |
| • | The duration of market exclusivity of AzaSite and Restasis; |
| • | The timing of approvals, if any, for other possible future products; |
| • | The progress toward and the achievement of developmental milestones by us or our partners; |
| • | The initiation of new contractual arrangements with other companies; and |
| • | The failure or refusal of a collaborative partner to pay royalties or milestone payments. |
31
Table of Contents
Inventory levels of AzaSite held by wholesalers can also cause our operating results to fluctuate unexpectedly. Although we attempt to monitor wholesaler inventory of our products, we rely upon information provided by third parties to quantify the inventory levels maintained by wholesalers. In addition, we and the wholesalers may not be effective in matching inventory levels to end-user demand. Significant differences between actual and estimated inventory levels and product demand may result in (1) inadequate or excessive (i) inventory production, (ii) product supply in distribution channels, or (iii) product availability at the retail level, and (2) unexpected increases or decreases in orders from our major customers. Any of these events may cause our revenues to fluctuate significantly from quarter to quarter, and in some cases may cause our operating results for a particular quarter to be below expectations.
If we continue to incur operating losses for a period longer than anticipated, or in an amount greater than anticipated, we may be unable to continue our operations.
We have experienced significant losses since inception. We incurred net losses of approximately $35.4 million and approximately $40.0 million for the years ended December 31, 2010 and 2009. As of December 31, 2010, our accumulated deficit was approximately $435.9 million. We currently expect to incur operating losses over the next several years. We expect that losses will fluctuate from quarter to quarter. Such fluctuations will be affected by the timing and level of the following:
| • | Commercialization activities to support AzaSite and Elestat; |
| • | Revenues from Restasis; |
| • | Regulatory approvals of our product candidates; |
| • | Patient demand for our products and any licensed products; |
| • | Payments to and from licensors and corporate partners; |
| • | Research and development activities; |
| • | Investments in new technologies and product candidates; and |
| • | The costs involved in defending any litigation claims against, or government investigations of, us. |
To achieve and sustain profitable operations, we must, alone or with others, develop successfully, obtain regulatory approval for, manufacture, introduce, market and sell our products. The time frame necessary to achieve market success is long and uncertain. We may not generate sufficient product revenues to become profitable or to sustain profitability. If the time required to achieve profitability is longer than we anticipate, we may not be able to continue our operations.
Recent stock market and credit market conditions have been extremely volatile and unpredictable. It is difficult to predict whether these conditions will continue or worsen, and, if so, whether the conditions would impact us and whether such impact could be material.
We have exposure to many different industries and counterparties, including commercial banks, investment banks and customers (which include wholesalers, managed care organizations and governments) that may be unstable or may become unstable in the current economic environment. Any such instability may impact these parties’ ability to fulfill contractual obligations to us or they might limit or place burdensome conditions upon future transactions with us. Customers may also reduce spending during times of economic uncertainty. Also, it is possible that suppliers may be negatively impacted. If such events were to occur, there could be a resulting material and adverse impact on our operations and results of operations.
We may decide to access the equity or debt markets to meet capital or liquidity needs. However, the recent constriction and volatility in these markets may restrict our future flexibility to do so when such needs arise. Further, recent economic conditions have resulted in severe downward pressure on the stock and credit markets, which could reduce the return available on invested corporate cash, which if severe and sustained could have a material and adverse impact on our results of operations and cash flows.
32
Table of Contents
Our dependence on collaborative relationships may lead to delays in product development, lost revenues and disputes over rights to technology.
Our business strategy depends to some extent upon the formation of research collaborations, licensing and/or marketing arrangements. We currently have collaboration agreements with several companies, including Allergan, InSite Vision and Santen. The termination of any collaboration will result in the loss of any unmet development or commercial milestone payments, may lead to delays in product development and disputes over technology rights, and may reduce our ability to enter into collaborations with other potential partners. In the event we breach an agreement with a collaborator, the collaborator is entitled to terminate our agreement with them in the event we do not cure the breach within a specified period of time, which is typically 60 or 90 days from the notice date. With respect to the Allergan collaboration, in the event we become an affiliate of a third party that manufactures, markets or sells any then currently promoted prescription ophthalmic product, Allergan will have the right to terminate our Elestat co-promotion agreement, which right must be exercised within 3 months of the occurrence of such event. If we do not maintain our current collaborations, or establish additional research and development collaborations or licensing arrangements, it will be difficult to develop and commercialize potential products. Any future collaborations or licensing arrangements may not be on terms favorable to us.
Our current or any future collaborations or licensing arrangements ultimately may not be successful. Under our current strategy, and for the foreseeable future, we do not expect to develop or market products outside North America without a collaborative partner or outside our therapeutic areas of focus.
It may be necessary in the future for us to obtain additional licenses to avoid infringement of third-party patents. Additionally, we may enter into license arrangements with other third parties as we build our product portfolio. We do not know the terms on which such licenses may be available, if at all.
We will continue to depend on collaborators and/or contractors for research and development, manufacturing and marketing of our potential products. Our agreements with collaborators typically allow them some discretion in electing whether to pursue such activities. If any collaborator were to breach or terminate its agreement with us or otherwise fail to conduct collaborative activities in a timely and successful manner, the clinical development or commercialization of product candidates or research programs would be delayed or terminated. Any delay or termination in clinical development or commercialization would delay or eliminate potential product revenues relating to our product candidates.
Disputes may arise in the future over the ownership of rights to any technology developed with collaborators. These and other possible disagreements between us and our collaborators could lead to delays in the collaborative development or commercialization of products. Such disagreements could also result in litigation or require arbitration to resolve.
Failure to hire and retain key personnel may hinder our product development programs and our business efforts.
We depend on the principal members of our management and scientific staff, including Adrian Adams, our President and Chief Executive Officer and a director, and Thomas R. Staab, II, our Chief Financial Officer and Treasurer. If these people leave us, we may have difficulty conducting our operations. Our future success will depend in part on our ability to attract, hire or appoint, and retain additional personnel skilled or experienced in the pharmaceutical industry. There is significant competition for such qualified personnel and we may not be able to attract and retain such personnel.
We may not be able to successfully compete with other biotechnology companies and pharmaceutical companies.
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. There are many companies seeking to develop products for the same
33
Table of Contents
indications that we are working on. Our competitors in the United States and elsewhere are numerous and include, among others, major multinational pharmaceutical and chemical companies and specialized biotechnology firms.
Most of these competitors have greater resources than we do, including greater financial resources, larger research and development staffs and more experienced marketing and manufacturing organizations. In addition, most of our competitors have greater experience than we do in conducting clinical trials and obtaining FDA and other regulatory approvals. Accordingly, our competitors may succeed in obtaining FDA or other regulatory approvals for product candidates more rapidly than we do. Companies that complete clinical trials, obtain required regulatory approvals, and commence commercial sale of their drugs before we do may achieve a significant competitive advantage, including patent and FDA marketing exclusivity rights that would delay our ability to market products. Drugs resulting from our research and development efforts, or from our joint efforts with our collaborative partners, may not compete successfully with competitors’ existing products or products under development.
Our competitors may also develop technologies and drugs that are safer, more effective, or less costly than any we are developing or which would render our technology and future drugs obsolete and non-competitive.
If our intellectual property protection is inadequate, the development and any possible sales of our product candidates could suffer or competitors could force our products completely out of the market.
Our business and competitive position depends on our ability to continue to develop and protect our products and processes, proprietary methods and technology and to prevent others from infringing on our patents, trademarks and other intellectual property rights. Upon the expiration or loss of patent protection for a product, we or our applicable collaborative partners may lose a significant portion of sales of that product in a short period of time as other companies manufacture generic forms of the previously protected product or manufacture similar products at lower cost, without having had to incur significant research and development costs in formulating the product. Therefore, our future financial success may depend in part on our and our partners obtaining patent protection for technologies incorporated into our products. We cannot be assured that such patents will be issued, or that any existing or future patents will be of commercial benefit. In addition, it is impossible to anticipate the breadth or degree of protection that any such patents will afford, and we cannot be assured that any such patents will not be successfully challenged in the future. If we are unsuccessful in obtaining or preserving patent protection, or if any of our products rely on unpatented proprietary technology, we cannot be assured that others will not commercialize products substantially identical to those products. We also believe that the protection of our trademarks is an important factor in product recognition and in our ability to maintain or increase market share. If we do not adequately protect our rights in our various trademarks from infringement, their value to us could be lost or diminished, seriously impairing our competitive position. Moreover, the laws of certain foreign countries do not protect our intellectual property rights to the same extent as the laws of the United States.
Certain of our patents are use patents containing claims covering methods of treating disorders and diseases by administering therapeutic chemical compounds. Use patents may provide limited protection for commercial efforts in the United States, but may afford a lesser degree of protection, if any, in other countries due to their patent laws. Besides our use patents, we have patents and patent applications covering compositions (new chemical compounds), pharmaceutical formulations and processes for manufacturing our new chemical compounds. Many of the chemical compounds included in the claims of our use patents and process applications were known in the scientific community prior to our patent applications. None of our composition patents or patent applications covers these previously known chemical compounds, which are in the public domain. As a result, competitors may be able to commercialize products that use the same previously known chemical compounds used by us for the treatment of disorders and diseases not covered by our use patents. Such competitors’ activities may reduce our revenues.
34
Table of Contents
Third parties may challenge, invalidate or circumvent our patents and patent applications relating to our products, product candidates or technologies at any time. If we must defend a patent suit, or if we choose to initiate a suit to have a third-party patent declared invalid, or allege infringement of our patents, we may need to make considerable expenditures of money and expend considerable management time in litigation. We believe that there is significant litigation in the pharmaceutical and biotechnology industry regarding patent and other intellectual property rights. A patent does not provide the patent holder with freedom to operate in a way that infringes the patent rights of others. We may be accused of patent infringement at any time. A judgment against us in a patent infringement action could cause us to pay monetary damages, require us to obtain licenses, or prevent us from manufacturing or marketing the affected products. In addition, we may need to initiate litigation to enforce our proprietary rights against others. Initiation of litigation may result in considerable expenditures of money and management time and may result in our patents being declared invalid. Further, we may need to participate in interference proceedings in the U.S. Patent and Trademark Office, or USPTO, to determine the priority of invention of any of our technologies.
Our ability to develop sufficient patent rights in our pharmaceutical, biopharmaceutical and biotechnology products to support commercialization efforts is uncertain and involves complex legal and factual questions. For instance, the USPTO examiners may not allow our claims in examining our patent applications. If we have to appeal a decision to the USPTO’s Appeals Board for a final determination of patentability, we could incur significant legal fees. Lengthy and uncertain patent prosecutions, including the utilization of the appeals process, can add uncertainty, delay and expense to the process of obtaining intellectual property rights for our products, and as such may add delay and uncertainty to the development program for any such product.
Use of our products may result in product liability claims for which we may not have adequate insurance coverage.
Manufacturing, marketing and sale of our products or conducting clinical trials of our product candidates may expose us to liability claims from the use of those products and product candidates. Product liability claims could result in the imposition of substantial liability on us, a recall of products, or a change in the indications for which they may be used. Although we carry product liability insurance and clinical trial liability insurance, we, or our collaborators, may not maintain sufficient insurance to cover these potential claims. We do not have the financial resources to self-insure and it is unlikely that we will have these financial resources in the foreseeable future. If we are unable to protect against potential product liability claims adequately, we may find it difficult or impossible to continue to commercialize our products or the product candidates we develop. If claims or losses exceed our liability insurance coverage, there could be a resulting material adverse effect on our business.
Insurance coverage is increasingly more costly and difficult to obtain or maintain.
While we currently have insurance for our business, property, directors and officers, and our products, insurance is increasingly more costly and narrower in scope, and we may be required to assume more risk in the future. If we are subject to claims or suffer a loss or damage in excess of our insurance coverage, we will be required to share that risk in excess of our insurance limits. If we are subject to claims or suffer a loss or damage that is outside of our insurance coverage, we may incur significant uninsured costs associated with loss or damage that could have an adverse effect on our operations and financial position. Furthermore, any claims made on our insurance policies may impact our ability to obtain or maintain insurance coverage at reasonable costs or at all.
35
Table of Contents
Risks Related to Our Stock
Our common stock price has been volatile and your investment in our stock may decline in value.
The market price of our common stock has been volatile. These fluctuations create a greater risk of capital losses for our stockholders as compared to less volatile stocks. Factors that have caused volatility and could cause additional volatility in the market price of our common stock include among others:
| • | Announcements made by us concerning results of clinical trials with our product candidates; |
| • | Announcements regarding the commercialization of AzaSite; |
| • | Announcements regarding potential FDA approval (or lack thereof) of any of our product candidates; |
| • | Market acceptance and market share of AzaSite, Restasis and Elestat; |
| • | The timing of the introduction of a generic form of Elestat; |
| • | Duration of market exclusivity of AzaSite and Restasis; |
| • | Volatility in other securities including pharmaceutical and biotechnology securities; |
| • | Changes in government regulations; |
| • | Regulatory actions and/or investigations; |
| • | Changes in the development priorities of our collaborators that result in changes to, or termination of, our agreements with such collaborators; |
| • | Developments concerning proprietary rights including patents by us or our competitors; |
| • | Variations in our operating results; |
| • | FDA approval of other treatments for the same indication as any one of our product candidates; |
| • | Business development activities; and |
| • | Litigation. |
Extreme price and volume fluctuations occur in the stock market and in particular market sectors from time to time that may affect the prices of biotechnology companies. These extreme fluctuations are sometimes unrelated to the actual performance of the affected companies.
Warburg is able to exercise substantial control over our business.
Warburg Pincus Private Equity IX, L.P., or Warburg, holds 22,907,488 shares of our common stock, which represented approximately 28% of our outstanding common stock as of January 31, 2011. Warburg and its affiliates may acquire the lesser of: (x) 32.5% of our voting securities on a fully diluted basis and (y) 34.9% of our then outstanding voting securities, without triggering the provisions of our stockholder rights plan. Warburg has the right to designate one person for election to our Board of Directors for so long as Warburg owns a significant percentage of our securities. Pursuant to this right, Jonathan S. Leff serves as a Class C member of the Board of Directors. Pursuant to a Securities Purchase Agreement, dated July 17, 2007, for so long as Warburg owns at least 10% of the shares of common stock issued upon the exchange of Exchangeable Preferred Stock it acquired in July 2007, it will have subscription rights to acquire a pro rata amount of future issuances of equity securities by us, subject to certain exceptions. As a result of the foregoing, Warburg is able to exercise substantial influence over our business, policies and practices.
36
Table of Contents
Our existing principal stockholders hold a substantial amount of our common stock and may be able to influence significant corporate decisions, which may conflict with the interest of other stockholders.
As of January 31, 2011, our current 5% and greater stockholders (which includes Warburg) and their affiliates beneficially owned approximately 45% of our outstanding common stock. These stockholders, if they act together, may be able to influence the outcome of matters requiring approval of the stockholders, including the election of our directors and other corporate actions such as:
| • | a merger or corporate combination with or into another company; |
| • | a sale of substantially all of our assets; and |
| • | amendments to our certificate of incorporation. |
The decisions of these individual stockholders may conflict with our interests or those of our other stockholders.
Future sales and issuances of securities may dilute the ownership interests of our current stockholders and cause our stock price to decline.
Future sales of our common stock by current stockholders into the public market could cause the market price of our stock to fall. As of January 31, 2011, there were 83,172,132 shares of our common stock outstanding. We may from time to time issue securities in relation to a license arrangement, collaboration, merger or acquisition.
In addition, on October 6, 2010, we filed with the SEC a shelf registration statement on Form S-3. This shelf registration statement has been declared effective and allows us to sell up to $150 million of securities, including common stock, preferred stock, debt securities, depositary shares and securities warrants, from time to time at prices and on terms to be determined at the time of sale.
As of January 31, 2011, there were 14,392,736 stock options and restricted stock units outstanding and 6,611,750 shares available for issuance under our Amended and Restated 2010 Equity Compensation Plan and equity compensation grants outside such plan. The shares underlying existing stock options and restricted stock units and possible future stock options, stock appreciation rights and stock awards have been registered pursuant to registration statements on Form S-8.
If some or all of such shares are sold or otherwise issued into the public market over a short period of time, our current stockholders’ ownership interests may be diluted and the value of all publicly traded shares is likely to decline, as the market may not be able to absorb those shares at then-current market prices. Additionally, such sales and issuances may make it more difficult for us to sell equity securities or equity-related securities in the future at a time and price that our management deems acceptable, or at all.
Our Rights Agreement, the provisions of our Change in Control Severance Benefit Plans, the anti-takeover provisions in our Amended and Restated Certificate of Incorporation, as amended, and Amended and Restated Bylaws, standstill agreements, and our right to issue preferred stock, may discourage a third party from making a take-over offer that could be beneficial to us and our stockholders and may make it difficult for stockholders to replace our Board of Directors and effect a change in our management if they desire to do so.
In October 2002, we entered into a Rights Agreement with Computershare Trust Company. The Rights Agreement could discourage, delay or prevent a person or group from acquiring 15% or more of our common stock. The Rights Agreement provides that if a person acquires 15% or more of our common stock without the approval of our Board of Directors, all other stockholders will have the right to purchase securities from us at a price that is less than its fair market value, which would substantially reduce the value of our common stock owned by the acquiring person. As a result, our Board of Directors has significant discretion to approve or disapprove a person’s efforts to acquire 15% or more of our common stock. In connection with the transaction with Warburg, we and Computershare entered into a First Amendment to Rights Agreement which provides that
37
Table of Contents
Warburg and its affiliates will be exempt from the definition of an “Acquiring Person” under the Rights Agreement, unless Warburg or certain of its affiliates becomes the beneficial owner of the lesser of: (x) 32.5% of our voting securities on a fully diluted basis and (y) 34.9% of our then outstanding voting securities. In addition to Warburg’s ability to exercise substantial control over our business, the First Amendment to Rights Agreement could further discourage, delay or prevent a person or group from acquiring 15% or more of our common stock. As part of the same transaction with Warburg, we entered into a standstill agreement, dated July 20, 2007, pursuant to which Warburg and certain of its affiliates agreed for three years not to increase their holdings of our common stock beyond the levels described above in the First Amendment to Rights Agreement. On August 4, 2009, we amended the standstill agreement to extend its term until August 4, 2012.
Our employees are covered under Change in Control Severance Benefit Plans which provide severance benefits in the event they are terminated after a change in control. In addition, the Company’s Executive Employment Agreements with Mr. Adams, our President and Chief Executive Officer, and Mr. Koven, our Executive Vice President and Chief Administrative and Legal Officer, provide for severance benefits in the event of a change of control. These arrangements would increase the acquisition costs to a purchasing company that triggers the change in control provisions and as a result, may discourage, delay or prevent a change in control.
Our Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws contain provisions which could discourage, delay or prevent a third party from acquiring shares of our common stock or replacing members of our Board of Directors. Our Amended and Restated Certificate of Incorporation allows our Board of Directors to issue shares of preferred stock. Our Board of Directors can determine the price, rights, preferences and privileges of those shares without any further vote or action by the stockholders. As a result, our Board of Directors could make it difficult for a third party to acquire a majority of our outstanding voting stock. Since management is appointed by the Board of Directors, any inability to effect a change in the Board of Directors may result in the entrenchment of management.
Our Amended and Restated Certificate of Incorporation also provides that the members of the Board will be divided into three classes. Each year, the terms of approximately one-third of the directors will expire. Our Amended and Restated Bylaws include director nomination procedures and do not permit our stockholders to call a special meeting of stockholders. The staggering of directors’ terms of office, the director nomination procedures and the inability of stockholders to call a special meeting may make it difficult for stockholders to remove or replace the Board of Directors should they desire to do so. The director nomination requirements include a provision that requires stockholders give advance notice to our Secretary of any nominations for director or other business to be brought by stockholders at any stockholders’ meeting. Our directors may be removed from our Board of Directors only for cause. These provisions may discourage, delay or prevent changes of control or management, either by third parties or by stockholders seeking to change control or management.
In the ordinary course of our business, from time to time we discuss possible collaborations, licenses and other transactions with various third parties, including pharmaceutical companies and biotechnology companies. When we deem it appropriate, we enter into standstill agreements with such third parties in relation to the discussions. These standstill agreements, several of which may be in force from time-to-time, typically prohibit such parties from acquiring our securities for a period of time.
We are also subject to the anti-takeover provisions of Section 203 of the Delaware General Corporation Law. Under these provisions, if anyone becomes an “interested stockholder,” we may not enter a “business combination” with that person for three years without special approval, which could discourage a third party from making a take-over offer and could delay or prevent a change of control. For purposes of Section 203, “interested stockholder” means, generally, someone owning 15% or more of our outstanding voting stock or an affiliate of ours that owned 15% or more of our outstanding voting stock during the past three years, subject to certain exceptions as described in Section 203. In connection with our prior sale of stock to Warburg, we agreed to waive Warburg’s acquisition of securities from the provisions of Section 203 of the Delaware General Corporation Law.
38
Table of Contents
FORWARD LOOKING STATEMENTS
This annual report on Form 10-K, including the documents that we incorporate by reference, contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended, that are subject to the “safe harbor” created by those sections. Any statements about our expectations, beliefs, plans, objectives, assumptions or future events or performance are not historical facts and may be forward-looking. These statements are often, but not always, made through the use of words or phrases such as “anticipate,” “estimate,” “plan,” “project,” “continuing,” “believe,” “expect,” “future” and “intend” and similar expressions to identify forward-looking statements. There are a number of important factors that could cause our actual results to differ materially from those indicated by any forward-looking statements, including, without limitation, the risk factors listed above and those relating to product development, revenue and earnings expectations, intellectual property rights and litigation, competitive products, results of clinical trials, the need for additional research and testing, delays in manufacturing, funding and the timing and content of decisions made by regulatory authorities, including the FDA and other factors presented throughout this annual report and any other documents filed by us with the SEC.
In light of these assumptions, risks and uncertainties, the results and events discussed in the forward-looking statements contained in this annual report on Form 10-K or in any document incorporated by reference might not occur. Stockholders are cautioned not to place undue reliance on the forward-looking statements, which speak only of the date of this report or the date of the document incorporated by reference in this document. We are not under any obligation, and we expressly disclaim any obligation, to update or alter any forward-looking statements, whether as a result of new information, future events or otherwise. All subsequent forward-looking statements attributable to us or to any person acting on our behalf are expressly qualified in their entirety by the cautionary statements contained or referred to in this section.
Item 1B. Unresolved Staff Comments
None.
Effective December 27, 2010, we began leasing administrative facilities that comprise approximately 43,278 square feet in Raleigh, North Carolina. The lease expires in December 31, 2017 and has two renewable periods of three years each. We believe our facilities are adequate to meet our current operational needs. In addition, we lease approximately 500 square feet of administrative space as a sales office in Dallas, Texas.
Not applicable.
Item 4. [Removed and Reserved].
39
Table of Contents
PART II
Item 5. Market for the Company’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Our common stock has been traded on the Nasdaq National Market, and later the Nasdaq Global Market, under the symbol “ISPH” since August 3, 2000. The following table sets forth, for the calendar periods indicated, the range of high and low sale prices for our common stock on the Nasdaq Global Market:
| 2009 |
High | Low | ||||||
| First Quarter |
$ | 4.21 | $ | 2.59 | ||||
| Second Quarter |
$ | 5.70 | $ | 3.52 | ||||
| Third Quarter |
$ | 6.54 | $ | 4.33 | ||||
| Fourth Quarter |
$ | 6.07 | $ | 4.36 | ||||
| 2010 |
High | Low | ||||||
| First Quarter |
$ | 7.07 | $ | 4.95 | ||||
| Second Quarter |
$ | 7.69 | $ | 4.83 | ||||
| Third Quarter |
$ | 6.04 | $ | 4.42 | ||||
| Fourth Quarter |
$ | 8.74 | $ | 5.82 | ||||
As of January 31, 2011, there were 52 record stockholders and approximately 7,400 beneficial stockholders of our common stock. As of January 31, 2011, the last sale price reported on the Nasdaq Global Market for our common stock was $3.95 per share.
We have not paid or declared dividends on our common stock since our inception and do not plan to pay dividends on our common stock in the foreseeable future. Any earnings that we may realize will be retained to finance our growth.
See “Part III—Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” for certain equity compensation plan information.
40
Table of Contents
RELATIVE STOCK PERFORMANCE
The graph below compares the cumulative 5-year total stockholder return on our common stock with the cumulative total returns of the NASDAQ Composite index and the NASDAQ Biotechnology index. The graph tracks the performance of a $100 investment in our common stock and in each of the indexes (with the reinvestment of all dividends) from December 31, 2005 to December 31, 2010.
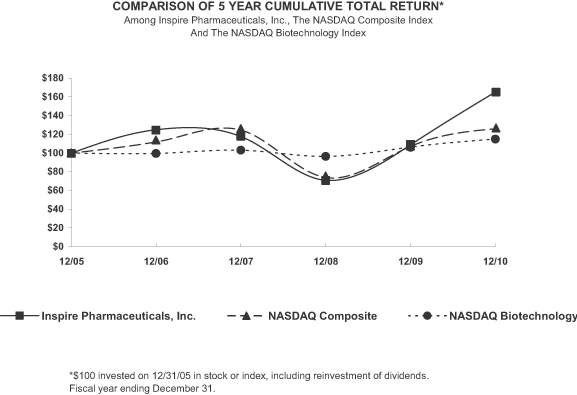
| Cumulative Total Returns | ||||||||||||||||||||||||
| 12/31/05 | 12/31/06 | 12/31/07 | 12/31/08 | 12/31/09 | 12/31/10 | |||||||||||||||||||
| Inspire Pharmaceuticals, Inc. |
$ | 100.00 | 125.00 | 117.72 | 70.87 | 108.66 | 165.35 | |||||||||||||||||
| NASDAQ Composite |
100.00 | 111.74 | 124.67 | 73.77 | 107.12 | 125.93 | ||||||||||||||||||
| NASDAQ Biotechnology |
100.00 | 99.71 | 103.09 | 96.34 | 106.49 | 114.80 | ||||||||||||||||||
The comparisons in the graph are required by the SEC and are not intended to forecast or be indicative of possible future performance of our common stock.
41
Table of Contents
Item 6. Selected Financial Data.
The selected statement of operations data and balance sheet data with respect to the years ended December 31, 2010, 2009, 2008, 2007 and 2006 set forth below are derived from our financial statements. The selected financial data set forth below should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” contained in Item 7 below, and our financial statements and the notes thereto appended to this annual report. Historical results are not necessarily indicative of our future results.
| (in thousands, except per share amounts) | ||||||||||||||||||||
| Year Ended December 31, | ||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| Statement of Operations Data: |
||||||||||||||||||||
| Revenue |
$ | 106,399 | $ | 92,159 | $ | 70,498 | $ | 48,665 | $ | 37,059 | ||||||||||
| Operating expenses: |
||||||||||||||||||||
| Cost of sales |
14,915 | 11,271 | 6,412 | 1,622 | — | |||||||||||||||
| Research and development |
46,149 | 51,134 | 44,637 | 53,391 | 42,537 | |||||||||||||||
| Selling and marketing |
50,151 | 49,304 | 54,568 | 45,543 | 25,265 | |||||||||||||||
| General and administrative |
30,939 | 16,053 | 14,540 | 13,986 | 15,880 | |||||||||||||||
| Restructuring |
— | 2,014 | — | — | — | |||||||||||||||
| Total operating expenses |
142,154 | 129,776 | 120,157 | 114,542 | 83,682 | |||||||||||||||
| Loss from operations |
(35,755 | ) | (37,617 | ) | (49,659 | ) | (65,877 | ) | (46,623 | ) | ||||||||||
| Other income/(expense), net |
308 | (2,359 | ) | (1,944 | ) | 2,137 | 4,508 | |||||||||||||
| Net loss |
$ | (35,447 | ) | $ | (39,976 | ) | $ | (51,603 | ) | $ | (63,740 | ) | $ | (42,115 | ) | |||||
| Non-cash deemed dividend related to beneficial conversion feature of exchangeable preferred stock |
— | — | — | (8,285 | ) | — | ||||||||||||||
| Net loss attributable to common stockholders |
$ | (35,447 | ) | $ | (39,976 | ) | $ | (51,603 | ) | $ | (72,025 | ) | $ | (42,115 | ) | |||||
| Net loss per common share—basic and diluted |
$ | (0.43 | ) | $ | (0.60 | ) | $ | (0.91 | ) | $ | (1.61 | ) | $ | (1.00 | ) | |||||
| Common shares used in computing weighted average common shares outstanding—basic and diluted |
82,733 | 66,797 | 56,609 | 44,763 | 42,227 | |||||||||||||||
| (in thousands) | ||||||||||||||||||||
| December 31, | ||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| Balance Sheet Data: |
||||||||||||||||||||
| Cash, cash equivalents and investments |
$ | 94,251 | $ | 129,099 | $ | 72,966 | $ | 139,724 | $ | 102,281 | ||||||||||
| Trade receivables, net |
22,442 | 22,682 | 16,544 | 12,974 | 8,245 | |||||||||||||||
| Inventories, net |
776 | 1,717 | 689 | 1,280 | — | |||||||||||||||
| Working capital |
72,491 | 85,412 | 52,512 | 107,651 | 89,655 | |||||||||||||||
| Total assets |
142,499 | 178,770 | 114,224 | 180,503 | 116,699 | |||||||||||||||
| Deferred revenue |
— | — | — | 371 | — | |||||||||||||||
| Debt obligations, including current portion(1) |
— | 25,175 | 43,605 | 57,701 | 21,357 | |||||||||||||||
| Total stockholders’ equity |
95,238 | 119,168 | 44,387 | 91,693 | 78,371 | |||||||||||||||
| Shares of common stock outstanding |
83,159 | 82,346 | 56,672 | 56,501 | 42,238 | |||||||||||||||
| (1) | Includes capital leases as of December 31, 2008, 2007 and 2006. |
42
Table of Contents
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
Cautionary Statement
The discussion herein contains forward-looking statements regarding our financial condition and our results of operations that are based upon our financial statements, which have been prepared in accordance with accounting principles generally accepted within the United States, as well as projections for the future. The preparation of these financial statements requires our management to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosure of contingent assets and liabilities. We evaluate our estimates on an ongoing basis. Our estimates are based on historical experience and on various other assumptions that are believed to be reasonable under the circumstances. The results of our estimates form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources.
We operate in a highly competitive environment that involves a number of risks, some of which are beyond our control. We are subject to risks common to specialty pharmaceutical companies, including risks inherent in our development and commercialization efforts, clinical trials, uncertainty of regulatory actions and marketing approvals, reliance on collaborative partners, enforcement of patent and proprietary rights, the need for future capital, competition associated with products, potential competition associated with our product candidates and retention of key employees. In order for any of our product candidates to be commercialized, it will be necessary for us, or our collaborative partners, to conduct clinical trials, demonstrate efficacy and safety of the product candidate to the satisfaction of regulatory authorities, obtain marketing approval, enter into manufacturing, distribution and marketing arrangements, and obtain market acceptance and adequate reimbursement from government and private insurers. We cannot provide assurance that we will generate significant revenues or achieve and sustain profitability in the future. In addition, we can provide no assurance that we will have sufficient funding to meet our future capital requirements. Statements contained in Management’s Discussion and Analysis of Financial Condition and Results of Operations and elsewhere in this report which are not historical facts are, or may constitute, forward-looking statements. Forward-looking statements involve known and unknown risks that could cause our actual results to differ materially from expected results. The most significant known risks are discussed in the section entitled “Risk Factors.” Although we believe the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements.
Our revenues are difficult to predict and depend on numerous factors. The effectiveness of our ability and the ability of third parties on which we rely to help us manufacture, distribute and market AzaSite; physician and patient acceptance of AzaSite; competitor response to AzaSite; as well as discounts, pricing and coverage on governmental and commercial formularies, and the recently passed healthcare reform legislation are all factors, among others, that will impact the level of revenue recorded for AzaSite in subsequent periods. Our co-promotion and royalty revenues are based upon Allergan’s revenue recognition policy and other accounting policies, over which we have limited or no control, and on the underlying terms of our agreements. Our co-promotion and royalty revenues are impacted by the number of governmental and commercial formularies upon which Restasis and Elestat are listed, the discounts and pricing under such formularies, as well as the estimated and actual amount of rebates, all of which are managed by Allergan. Other factors that are difficult to predict and that impact our co-promotion and royalty revenues are the extent and effectiveness of Allergan’s sales and marketing efforts, our sales and marketing efforts, timing of a generic epinastine launch, coverage and reimbursement under Medicare Part D and Medicaid programs, the recently passed healthcare reform legislation and the sales and marketing activities of competitors. Additionally, our ability to receive revenues on future sales of AzaSite, Restasis and Elestat are dependent upon the duration of market exclusivity and the strength of patent protection. Revenues related to development activities are dependent upon the progress toward and the achievement of developmental milestones by us or our collaborative partners.
Our operating expenses are also difficult to predict and depend on several factors. Cost of sales related to AzaSite contain variable and fixed cost components. Research and development expenses, drug manufacturing,
43
Table of Contents
and clinical research activities, depend on the ongoing requirements of our development programs, availability of capital and direction from regulatory agencies, which are difficult to predict. In addition, we have incurred and expect to continue to incur significant selling and marketing expenses to commercialize our products. Management may be able to control the timing and level of research and development, selling and marketing and general and administrative expenses, but many of these expenditures will occur irrespective of our actions due to contractually committed activities and/or payments.
As a result of these factors, we believe that period to period comparisons are not necessarily meaningful and you should not rely on them as an indication of future performance. Due to all of the foregoing factors, it is possible that our operating results will be below the expectations of market analysts and investors. In such event, the prevailing market price of our common stock could be materially adversely affected.
Overview
We are a specialty pharmaceutical company focused on developing and commercializing ophthalmic products. Our strategy is to create a sustainable portfolio of ophthalmic products by leveraging our commercial capabilities and pipeline assets and pursuing corporate development and licensing opportunities. Our specialty eye care sales force generates revenue from the promotion of AzaSite and the co-promotion of Elestat. We receive royalties based on net sales of Restasis and expect to begin receiving royalties in 2011 based on net sales of Diquas in Japan.
In February 2007, we signed an exclusive licensing agreement with InSite Vision for the U.S. and Canadian commercialization rights of AzaSite for the treatment of bacterial conjunctivitis. In April 2007, AzaSite was approved by the FDA for the treatment of bacterial conjunctivitis in adults and children one year of age and older. In August 2007, we launched AzaSite in the United States and are promoting it to eye care specialists.
In 2004, we launched Elestat for the treatment of allergic conjunctivitis.
In August 2010, we amended and restated our agreement with Allergan relating to Restasis and Prolacria. Under the amended and restated agreement, which runs through December 31, 2020, we are entitled to receive revenues at one global rate based on net sales of Restasis and any other human ophthalmic formulation of cyclosporine owned or controlled by Allergan, with no requirement to co-promote Restasis. Under agreements with Allergan, we receive revenue based upon Allergan’s net sales of these products.
See Part I—Item 1. Business of this report for a full discussion of our agreements with InSite Vision, Allergan and other significant agreements, as well our other product candidates in development.
We have incurred significant operating losses since our inception and, as of December 31, 2010, we had an accumulated deficit of $435.9 million. Revenue from sales of AzaSite, Restasis and Elestat did not exceed our total operating expenses in 2010. We expect to incur operating losses for the next several years. We have financed our operations through the sale of equity securities, including private sales of preferred stock and public offerings of common stock, debt, and with revenue from corporate partnerships, including co-promotion and royalty revenue. We operate as a single business segment and do not have any foreign operations; however, we do receive royalties based on net sales outside of the United States.
Critical Accounting Policies and Estimates
The accompanying discussion and analysis of our financial condition and results of operations are based upon our financial statements and the related disclosures, which have been prepared in accordance with generally accepted accounting principles. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues, expenses and related disclosure of contingent assets and liabilities. We evaluate our estimates, judgments and the policies underlying these estimates on a periodic basis, as situations change and regularly discuss financial events, policies, and issues with
44
Table of Contents
members of our audit committee and our independent registered public accounting firm. In addition, recognition of revenue from product co-promotion and earned royalties is affected by certain estimates and judgments made by Allergan on which we rely when recording this revenue. We routinely evaluate our estimates and policies regarding revenue recognition, product returns, rebates and incentives, inventory and manufacturing, taxes, stock-based compensation, research and development, marketing and other expenses and any associated liabilities.
We believe the following policies to be the most critical to an understanding of our financial condition and results of operations because they require us to make estimates and judgments about matters that are inherently uncertain.
Revenue Recognition
We record all of our revenue from: (1) sales of AzaSite; (2) product co-promotion activities and earned royalties; and (3) collaborative research agreements, when realized or realizable and earned. Revenue is realized or realizable and earned when all of the following criteria are met: (i) persuasive evidence of an arrangement exists; (ii) delivery has occurred or services have been rendered; (iii) the seller’s price to the buyer is fixed or determinable; and (iv) collectibility is reasonably assured.
Product Revenues
We recognize revenue for sales of AzaSite when title and substantially all the risks and rewards of ownership have transferred to the customer, which generally occurs on the date of shipment to wholesalers and distributors. In the United States, we sell AzaSite to wholesalers and distributors, who, in turn, sell to pharmacies and federal, state and commercial health care organizations.
Sales deductions consist of statutory rebates to state Medicaid, Medicare and other government agencies, contractual rebates with commercial managed care organizations, wholesaler chargebacks, sales discounts (including trade discounts and distribution service fees), allowances for coupon and voucher programs and product returns. These deductions are recorded as reductions to revenue from AzaSite in the same period as the related sales with estimates of future utilization derived from historical experience adjusted to reflect known changes in the factors that impact such reserves.
We utilize data from external sources to help us estimate our gross-to-net sales adjustments as they relate to the recognition of revenue for AzaSite sold. External sourced data includes, but is not limited to, information obtained from certain wholesalers with respect to their inventory levels and sell-through to customers, targeted surveys as well as data from IMS Health, a third-party supplier of market research data to the pharmaceutical industry. We also utilize this data to help estimate and identify prescription trends and patient demand, as well as product levels in the supply chain.
We account for these sales deductions in accordance with the Financial Accounting Standards Board, or FASB, authoritative guidance on revenue recognition when consideration is given by a vendor to a customer as well as when the right of return exists.
We have categorized and described more fully, the following significant sales deductions, all of which involve estimates and judgments which we consider to be critical accounting estimates, and require us to use information from external sources.
Rebates and Chargebacks
Statutory rebates to state Medicaid agencies and Medicare and contractual rebates to commercial managed care organizations are based on statutory or negotiated discounts to AzaSite’s selling price. As it can take up to
45
Table of Contents
nine months or more for information to be received on actual usage of AzaSite in managed care and Medicaid and other governmental programs as well as on the total discounts to be reimbursed, we maintain reserves for amounts payable under these programs relating to AzaSite sales.
Chargebacks claimed by wholesalers are based on the differentials between product acquisition prices paid by wholesalers and lower government contract pricing paid by eligible customers covered under federally qualified programs.
The amount of the reserve for rebates and chargebacks is based on multiple qualitative and quantitative factors, including the historical and projected utilization levels, historical payment experience, changes in statutory laws and interpretations as well as contractual terms, product pricing (both normal selling prices and statutory or negotiated prices), changes in prescription demand patterns and utilization of our product through private or public benefit plans, and the levels of AzaSite inventory in both the wholesale and retail distribution channel. Other factors that we may consider, if determined relevant, would include price changes from competitors and introductions of generics and/or competitive new products. We acquire prescription utilization data from IMS Health, a third-party supplier of market research data to the pharmaceutical industry. We apply these multiple factors, the quantitative historical data along with other qualitative aspects, such as management’s judgment regarding future utilization trends, to the respective period’s sales of AzaSite to determine the rebate accrual and related expense. We update our estimates and assumptions each period and record any necessary adjustments to our reserves. Settlements of rebates and chargebacks typically occur within nine months from point of sale. To the extent actual rebates and chargebacks differ from our estimates, additional reserves may be required or reserves may need to be reversed, either of which would impact current period product revenue. As of December 31, 2010 and 2009, reserves for rebates and chargebacks were $9.1 million and $3.5 million, respectively.
Discounts and Other Sales Incentives
Discounts and other sales incentives consist of the following:
| • | Prompt pay discounts—Prompt payment discounts are offered to all wholesalers in return for payment within 30 days following the invoice date. We record sales of AzaSite net of the discount amount based on historical experience. We adjust the reserve at the end of each reporting period to approximate the percentage discount applicable to the outstanding gross accounts receivable balances. |
| • | Inventory Management Agreement (“IMA”) Fees—Per contractual agreements with our largest wholesalers, we provide an IMA fee based on a percentage of their purchases of AzaSite. The IMA fee rates are set forth in the individual contracts. We track sales to these wholesalers each period and accrue a liability relating to the unpaid portion of these fees by applying the contractual rates to such sales. |
| • | Product coupons and vouchers—Product coupons and vouchers, made available by us online or through pharmacies and prescribing physicians, offer patients the ability to receive free or discounted product. We use a third-party administrator to coordinate program activities and who invoices us on a periodic basis for the cost of coupons and vouchers redeemed in the period. We base our estimates on the historical coupon and voucher redemption rate of similar programs. |
As of December 31, 2010 and 2009, reserves for discounts and other sales incentives were $1.2 million and $889,000, respectively.
Product Returns
At the time of sale of AzaSite, we record product return allowances based on our estimate of the portion of sales that will be returned by our customers in the future. The return allowances are established in accordance with our returned goods policy. Our returned goods policy generally allows for returns of AzaSite within an
46
Table of Contents
18-month period, from six months prior to the expiration date and up to 12 months following the expiration date, but may differ from customer to customer, depending on certain factors. Future estimated returns of AzaSite are based primarily on the return data for comparative products and our own historical experience with AzaSite. Historical return data on AzaSite is analyzed on a specific production lot basis. In determining our return allowance we also consider other relevant factors, including:
| • | Levels of inventory in the distribution channel and any significant changes to these levels; |
| • | Estimated expiration date or remaining shelf life of inventory in the distribution channel; |
| • | Current and projected demand of AzaSite that could be impacted by introductions of generic products and/or introductions of competitive new products; and |
| • | Competitive product shortages, recalls and/or discontinuances. |
Our estimates of the level of AzaSite inventory in the distribution channel is based on inventory data provided by wholesalers and third-party prescription data. As of December 31, 2010 and 2009, reserves for returns of AzaSite were $2.6 million and $1.5 million, respectively.
The following table reflects the gross to net sales accruals and the balance in the related allowance accounts for the years ended December 31, 2010, 2009 and 2008.
| Balance at Beginning of Year |
Related to Current Period Sales |
Related to Prior Period Sales |
Credits/ Payments |
Balance at End of Year |
||||||||||||||||
| Year Ended December 31, 2010 |
||||||||||||||||||||
| Reserve for Rebates and Chargebacks |
$ | 3,488,000 | $ | 14,945,000 | $ | (58,000 | ) | $ | (9,290,000 | ) | $ | 9,085,000 | ||||||||
| Reserve for Discounts and Other Sales Incentives |
889,000 | 4,871,000 | — | (4,601,000 | ) | 1,159,000 | ||||||||||||||
| Allowance for Returns |
1,523,000 | 1,625,000 | 4,000 | (562,000 | ) | 2,590,000 | ||||||||||||||
| Total Sales Deductions Accruals |
$ | 5,900,000 | $ | 21,441,000 | $ | (54,000 | ) | $ | (14,453,000 | ) | $ | 12,834,000 | ||||||||
| Year Ended December 31, 2009 |
||||||||||||||||||||
| Reserve for Rebates and Chargebacks |
$ | 783,000 | $ | 6,015,000 | $ | 188,000 | $ | (3,498,000 | ) | $ | 3,488,000 | |||||||||
| Reserve for Discounts and Other Sales Incentives |
519,000 | 3,328,000 | — | (2,958,000 | ) | 889,000 | ||||||||||||||
| Allowance for Returns |
701,000 | 1,259,000 | 301,000 | (738,000 | ) | 1,523,000 | ||||||||||||||
| Total Sales Deductions Accruals |
$ | 2,003,000 | $ | 10,602,000 | $ | 489,000 | $ | (7,194,000 | ) | $ | 5,900,000 | |||||||||
| Year Ended December 31, 2008 |
||||||||||||||||||||
| Reserve for Rebates and Chargebacks |
$ | 149,000 | $ | 1,501,000 | $ | (7,000 | ) | $ | (860,000 | ) | $ | 783,000 | ||||||||
| Reserve for Discounts and Other Sales Incentives |
89,000 | 1,394,000 | — | (964,000 | ) | 519,000 | ||||||||||||||
| Allowance for Returns |
95,000 | 661,000 | 16,000 | (71,000 | ) | 701,000 | ||||||||||||||
| Total Sales Deductions Accruals |
$ | 333,000 | $ | 3,556,000 | $ | 9,000 | $ | (1,895,000 | ) | $ | 2,003,000 | |||||||||
Product Co-promotion and Royalty Revenues
We recognize co-promotion revenue based on net sales of Elestat and royalty revenue based on net sales of Restasis, as defined in the applicable agreements, and as reported to us by Allergan. Our co-promotion and royalty revenues are based upon Allergan’s revenue recognition policy and other accounting policies over which we have limited or no control and on the underlying terms of our agreements. Allergan recognizes revenue from product sales when goods are shipped and title and risk of loss transfers to the customer. The agreements provide for gross sales to be reduced by estimates of sales returns, credits and allowances, normal trade and cash discounts, managed care sales rebates and other allocated costs as defined in the agreements, all of which are
47
Table of Contents
determined by Allergan and are outside our control. We record a percentage of Allergan’s reported net sales to us for Elestat and Restasis, as co-promotion revenue and royalty revenue, respectively. We receive monthly net sales information from Allergan and perform analytical reviews and trend analyses using prescription information that we receive from IMS Health. In addition, we exercise our audit rights under the contractual agreements with Allergan to annually perform an examination of Allergan’s sales records of both Restasis and Elestat. We make no adjustments to the amounts reported to us by Allergan other than reductions in net sales to reflect the incentive programs managed by us. We offer and manage certain incentive programs associated with Elestat, which are utilized by us in addition to those programs managed by Allergan. We reduce co-promotion revenue from net sales of Elestat by estimating the portion of sales that are subject to these incentive programs based on information reported to us by our third-party administrator of the incentive programs. The rebates associated with the programs we manage represent an insignificant amount, as compared to the rebate and discount programs administered by Allergan and as compared to our aggregate co-promotion and royalty revenue. Prior to January 1, 2010, under the co-promotion agreement for Elestat, we were obligated to meet predetermined minimum calendar year net sales target levels. If the annual minimum was not achieved, we recorded revenues using a reduced percentage of net sales based upon our level of achievement of the predetermined calendar year net sales target levels. Amounts receivable from Allergan in excess of recorded co-promotion revenue were recorded as deferred revenue. Calendar year 2009 was the last year in which there was a minimum annual net sales target level for Elestat under the co-promotion agreement. Accordingly, effective January 1, 2010, all co-promotion revenue from net sales of Elestat is recognized in the same period in which the sales occur.
Collaborative Research and Development Revenues
We recognize revenue under our collaborative research and development agreements when we or our collaborative partner have met a contractual milestone triggering a payment to us. We are entitled to receive milestone payments under our collaborative research and development agreements based upon the achievement of agreed upon development events that are substantively at-risk by our collaborative partners or us. This collaborative research and development revenue is recognized upon the achievement and acknowledgement of our collaborative partner of a development event, which is generally at the date payment is received from the collaborative partner or is reasonably assured. Accordingly, our revenue recognized under our collaborative research and development agreements may fluctuate significantly from period to period. In each of the years ended December 31, 2010 and 2008, we recognized $1.25 million of collaborative research and development revenue from Santen related to its development and approval of Diquas in accordance with our development, license and supply agreement.
Inventories
Our inventories are related to AzaSite and are valued at the lower of cost or market using the first-in, first-out (i.e., FIFO) method. Cost includes materials, labor, overhead, shipping and handling costs. Our inventories are subject to expiration dating. We regularly evaluate the carrying value of our inventories and provide valuation reserves for any estimated obsolete, short-dated or unmarketable inventories. In addition, we may experience spoilage of our raw materials, which primarily consist of API. Our determination that a valuation reserve might be required, in addition to the quantification of such reserve, requires us to utilize significant judgment. We base our analysis, in part, on the level of inventories on hand in relation to our estimated forecast of product demand, production requirements for forecasted product demand, expected market conditions and the expiration dates or remaining shelf life of inventories. As of December 31, 2010 and 2009, we had net reserves of $50,000 and $25,000, respectively.
Taxes
We account for uncertain tax positions in accordance with FASB authoritative guidance regarding the accounting for taxes. Significant management judgment is required in determining our provision for income taxes, deferred tax assets and liabilities and any valuation allowance recorded against net deferred tax assets. We
48
Table of Contents
have recorded a valuation allowance against all potential tax assets due to uncertainties in our ability to utilize deferred tax assets, primarily consisting of certain net operating losses carried forward, before they expire. The valuation allowance is based on estimates of taxable income in each of the jurisdictions in which we operate and the period over which our deferred tax assets will be recoverable.
Liabilities
We generally enter into contractual agreements with third-party vendors who provide research and development, manufacturing, and other services in the ordinary course of business. Some of these contracts are subject to milestone-based invoicing and services are completed over an extended period of time. We record liabilities under these contractual commitments when we determine an obligation has been incurred, regardless of the timing of the invoice. We monitor all significant research and development, manufacturing, sales and marketing and other service activities and the progression of work related to these activities. We estimate the underlying obligation for each activity based upon our estimate of the amount of work performed and compare the estimated obligation against the amount that has been invoiced. Because of the nature of certain contracts and related delay in the contract’s invoicing, the obligation to these vendors may be based upon management’s estimate of the underlying obligation. We record the larger of our estimated obligation or invoiced amounts for completed service. In all cases, actual results may differ from our estimate.
Stock-Based Compensation Expense
We recognize stock-based compensation expense in accordance with FASB authoritative guidance regarding the accounting for share-based payments, which requires us to measure compensation cost for share-based payment awards at fair value and recognize compensation expense over the service period for awards expected to vest. We utilize the Black-Scholes option-pricing model to value our awards and recognize compensation expense on a straight-line basis over the vesting periods of our awards. We consider many factors when estimating expected forfeitures, including types of awards, employee class, and historical experience. Our expected volatility is determined based on our own historical volatility. The estimation of share-based payment awards that will ultimately vest requires judgment, and to the extent actual results or updated estimates differ from our current estimates, such amounts will be recorded as a cumulative adjustment in the period estimates are revised. Significant management judgment is also required in determining estimates of future stock price volatility and forfeitures to be used in the valuation of the options. Actual results, and future changes in estimates, may differ substantially from our current estimates.
Impact of Inflation
We do not believe that our operating results have been materially impacted by inflation during the past three years. However, we cannot be assured that our operating results will not be adversely affected by inflation in the future. We will continually seek to mitigate the adverse effects of inflation on the costs of goods and services that we use through improved operating efficiencies and cost containment and periodic price increases for our product.
Results of Operations
Years Ended December 31, 2010, 2009 and 2008
Revenues
Total revenues were approximately $106.4 million for the year ended December 31, 2010, as compared to approximately $92.2 million in 2009 and approximately $70.5 million in 2008. The increase in 2010 revenue of approximately $14.2 million, or 15%, as compared to 2009, was primarily due to an increase in product revenue from net sales of AzaSite and an increase in royalty revenue from net sales of Restasis, partially offset by a reduction in co-promotion revenue from net sales of Elestat. The increase in 2009 revenue of approximately $21.7 million, or 31%, as compared to 2008, was primarily due to an increase in product revenue from net sales of AzaSite, as well as increased royalty revenue from net sales of Restasis. In addition, total revenues for both the
49
Table of Contents
2010 and 2008 period included the recognition of development milestones of $1.25 million from Santen related to its development and approval of Diquas in accordance with our development, license and supply agreement, as previously discussed in this report.
Product Sales, net
Product sales of AzaSite, net of rebates and discounts, for the year ended December 31, 2010 were approximately $42.7 million, as compared to approximately $35.0 million in 2009 and approximately $18.3 million in 2008. The increase in 2010 revenue for AzaSite of approximately $7.7 million, or 22%, as compared to 2009, was primarily due to increased patient usage of AzaSite and an increase in prescribers of AzaSite, as evidenced by an increase in prescriptions year-over-year, as well as price increases for the product between the periods. Additionally, approximately $1.0 million of revenue from net sales of AzaSite in 2010, as compared to approximately $3.0 million to $4.0 million in 2009, was associated with hospital usage of AzaSite during a temporary supply shortage of erythromycin ophthalmic ointment, as discussed below. The erythromycin ophthalmic ointment supply shortage began in the third quarter of 2009 and was resolved in the first quarter of 2010.
The increase in 2009 revenue for AzaSite of approximately $16.7 million, or 91%, as compared to 2008, was primarily due to increased patient and physician usage of AzaSite, evidenced by an increase in prescriptions year-over-year, as well as a price increase for the product between the periods. Approximately $3.0 million to $4.0 million of revenue from sales of AzaSite for the year ended December 31, 2009 was associated with hospital usage of AzaSite as a substitute therapy during a temporary supply shortage of erythromycin ophthalmic ointment (0.5%), as discussed below.
In September 2009, erythromycin ophthalmic ointment was placed on the FDA Drug Shortages website. Erythromycin ophthalmic ointment is a macrolide antibiotic routinely used in neonates for prophylaxis of ophthalmia neonatorum, a form of bacterial conjunctivitis that may be contracted by newborns during delivery. Due to this shortage, the Centers for Disease Control and Prevention (CDC) asked healthcare professionals to reserve erythromycin supplies for neonatal use and also recommended the use of AzaSite as an acceptable substitute for neonatal prophylaxis use when erythromycin was not available.
The increase in AzaSite revenues for the year ended December 31, 2010, as compared to 2009, was partially offset by an increase in gross-to-net sales deductions. Total sales deductions as a percentage of gross revenues increased approximately 9%. The impact of the net rate increase as a percentage of gross revenues was approximately $5.9 million in additional provisions for the year ended December 31, 2010. The increase was primarily attributable to an approximate 10% increase in rebates and discounts due to (1) an increase in the number of formularies that now list AzaSite and (2) the price concessions required to secure this coverage under Medicare and commercial managed care organizations. In addition, we initiated several new and more significant mail-in rebate and voucher programs which increased our rebates over 2009. These increases were partially offset by a slight decline in accrual rates for product return allowances. Our reserves related to rebates and discounts for the year ended December 31, 2010 were not significantly impacted by the new U.S. healthcare reform legislation passed in March 2010 and those provisions of the legislation that became effective during the first quarter of 2010. We expect future sales of AzaSite to be negatively impacted by the provisions of the new legislation that become effective in 2011.
For the year ended December 31, 2010, based on prescription data from IMS Health, there were approximately 719,000 prescriptions written for AzaSite, representing approximately 5% of all prescriptions in the single agent ocular antibiotic market, defined as both branded and generic single agent ocular antibiotics. In comparison, approximately 529,000 prescriptions were written for AzaSite in 2009, excluding hospital usage, representing approximately 4% of all prescriptions in the single agent ocular antibiotic market. For the year ended December 31, 2010, the average monthly prescriptions per sales force associate and the average monthly prescriptions per targeted physician increased 36% and 30%, respectively, as compared to 2009. Since launch, actual units of AzaSite dispensed have been slightly higher than the number of prescriptions as reported by IMS
50
Table of Contents
Health due to the issuance of multiple unit prescriptions by some physicians. For the year ended December 31, 2010, the single agent ocular antibiotic market, in terms of prescriptions, increased approximately 1% from the prior year.
Our average market share in our primary call audience of eye care specialists, mainly ophthalmologists and optometrists, was approximately 11% for the year ended December 31, 2010, as compared to approximately 9% in 2009. In addition, our average market share in our target call audience of eye care specialists was approximately 20% for the year ended December 31, 2010, as compared to approximately 13% in 2009.
Product Co-Promotion and Royalty
Total co-promotion and royalty revenue for the year ended December 31, 2010 was approximately $62.5 million, as compared to approximately $57.2 million in 2009 and approximately $50.9 million in 2008.
Our royalty revenue from net sales of Restasis for the year ended December 31, 2010 was approximately $45.6 million, as compared to approximately $38.4 million in 2009 and approximately $32.8 million in 2008. In August 2010, we amended and restated our agreement with Allergan. Under the amended and restated agreement, which runs through December 31, 2020, we are entitled to receive revenues at one global rate based on net sales of Restasis and any other human ophthalmic formulation of cyclosporine owned or controlled by Allergan, with no requirement to co-promote Restasis. The royalty rate for Restasis in the United States remained unchanged for 2010. Effective January 1, 2011, the annual global rate steps down from the 2010 rate by three percentage points. The rate will step down a further 0.25 percentage point in 2013 and a final 0.50 percentage point in 2014, remaining at this level through the end of the term in 2020. For the years ended December 31, 2010, 2009 and 2008, Allergan recorded revenue from net sales of Restasis of approximately $621 million, $523 million and $444 million, respectively.
The increase in both 2010 and 2009 royalty revenue for Restasis, as compared to 2009 and 2008, respectively, was primarily due to increased patient usage of Restasis, as evidenced by an increase in prescriptions year-over-year. In addition, there were annual price increases in the first quarters of 2010 and 2009.
Co-promotion revenue from net sales of Elestat for the year ended December 31, 2010, was approximately $16.9 million, as compared to approximately $18.8 million in 2009 and approximately $18.1 million in 2008. The decrease in co-promotion revenue from net sales of Elestat of $1.9 million, or 10%, as compared to 2009, was primarily due to a lower share of the total U.S. allergic conjunctivitis market, partially offset by an annual price increase. The 2009 increase in co-promotion revenue from net sales of Elestat of $615,000, or 3%, as compared to 2008, was primarily due to an annual price increase, partially offset by a slight decrease in the total U.S. allergic conjunctivitis market in 2009. The 2010 and 2009 annual price increases for Elestat were effective in the first quarters of both years.
Elestat is a seasonal product with product demand mirroring seasonal trends for topical allergic conjunctivitis products. Typically, demand is highest during the Spring months followed by moderate demand in the Summer and Fall months. The lowest demand is during the Winter months. Based upon national prescription data from IMS Health, for the years ended December 31, 2010, 2009 and 2008, Elestat prescriptions, as a percentage of the total U.S. allergic conjunctivitis market, represented approximately 6%, 7% and 7%, respectively, of the total U.S. allergic conjunctivitis market. Based upon monthly data from IMS Health, for the year ended December 31, 2010, the total U.S. allergic conjunctivitis market, in terms of prescriptions, increased approximately 3% and for the years ended December 31, 2009 and 2008, decreased approximately 1% and 6%, respectively.
Under our agreement with Allergan related to our co-promotion of Elestat, prior to 2010, we were obligated to meet predetermined minimum calendar year net sales target levels, which increased annually. We were entitled to an escalating percentage of net sales of Elestat based upon predetermined calendar year net sales target levels. During a fiscal year, we recognized product co-promotion revenue associated with targeted net
51
Table of Contents
sales levels for Elestat achieved during that time period and deferred revenue in excess of the sales level achieved. Under the co-promotion agreement with Allergan, calendar year 2009 was the last year that our co-promotion revenues of Elestat were subject to annual minimum target levels.
Subject to applicable law, competitors are permitted to submit to the FDA an ANDA for a generic version of Elestat, due to the expiration of the marketing exclusivity period for Elestat provided under the Hatch-Waxman Act on October 15, 2008. We are aware that the following companies have filed an ANDA for a generic version of Elestat: Apotex, Inc., Cypress Pharmaceutical, Inc., Paddock Laboratories, Inc., PharmaForce, Inc. and Sandoz, Inc. The date of submission of the first ANDA filing to the FDA Office of Generic Drugs was October 14, 2008, according to the FDA’s website (www.fda.gov). Also, according to the FDA’s website, Apotex, Inc., PharmaForce, Inc. and Sandoz, Inc. have each received tentative approval for their respective epinastine hydrochloride ophthalmic solution.
The Elestat co-promotion agreement provides that unless earlier terminated, the term of such agreement will be in effect until the earlier of (i) the approval and launch of the first generic epinastine product after expiration of the FDA exclusivity period covering Elestat in the United States, or (ii) the approval and launch of the first over-the-counter epinastine product after expiration of the listing of Elestat in the FDA’s Orange Book. Following the termination of such co-promotion agreement, we will no longer have the right to co-promote Elestat. We will be entitled to receive post-termination payments from Allergan, based on any remaining net sales of Elestat for a period of 36 months. During the three successive 12-month periods immediately following the termination of the agreement, Allergan will be obligated to pay to us 20%, 15% and 10%, respectively, on any net sales of Elestat in the United States. We expect any revenue from net sales of Elestat received during this 36-month post-termination period to be nominal compared to revenue in years where we were actively co-promoting the product. We plan to continue co-promoting and receiving co-promotion revenues on Elestat sales during the FDA’s review period of these ANDAs. We do not know when the FDA will complete its review but we expect that a generic form of epinastine may be launched at any time. Loss of our co-promotion revenue from Elestat will significantly impact our results of operations and cash flows.
Collaborative Research and Development
In December 2010, Santen received pricing approval for Diquas Ophthalmic Solution 3% (diquafosol tetrasodium), for which we received a milestone payment of $1.25 million. There are no future milestones to be earned under the amended agreement with Santen. We are entitled to receive royalty payments based upon a tiered rate on net sales of Diquas in Japan, which Santen launched in December 2010, with a minimum rate in the high single digits and a maximum rate in the low double digits. In May 2008, Santen completed its Phase 3 clinical testing of diquafosol tetrasodium in Japan for which we received a milestone payment of $1.25 million.
Our future revenue will depend on various factors including the effectiveness of our commercialization of AzaSite and continued commercial success and duration of commercial exclusivity of Restasis and Elestat. In addition to the foregoing, pricing, rebates, discounts and returns for all products; the effect of competing products; coverage and reimbursement under commercial or government plans; and the recently passed healthcare reform legislation will impact future revenues. If Allergan significantly under-estimates or over-estimates rebate amounts, there could be a material effect on our revenue. In addition to the continuing sales of AzaSite, Restasis and Elestat, our future revenue will also depend on Santen’s ability to successfully commercialize Diquas, our ability to enter into additional collaboration agreements, and to achieve milestones under existing or future collaboration agreements, as well as whether we obtain regulatory approvals for our product candidates.
Cost of Sales
Cost of sales related to the sales of AzaSite were approximately $14.9 million for the year ended December 31, 2010, as compared to approximately $11.3 million in 2009 and approximately $6.4 million in 2008. The increase in cost of sales of $3.6 million, or 32%, as compared to 2009, was primarily due to increased sales volume of AzaSite, which resulted in increased royalties, as well as an increase in the royalty rate paid to
52
Table of Contents
InSite Vision. In July 2009, our royalty rate to InSite Vision on net sales of AzaSite increased from 20% to 25% in accordance with the terms of our licensing agreement, and will remain at 25% for the remaining term of the licensing agreement. Additionally, during the year ended December 31, 2010 we incurred approximately $450,000 of costs, including a reserve of $253,000, associated with the spoilage of API used to manufacture AzaSite and related unanticipated manufacturing activities.
Under the terms of the license agreement with InSite Vision, our obligation to pay royalties to InSite Vision is subject to pre-determined minimum annual royalty payments. The determination of whether or not we will owe any such payments is based upon the amount of royalties accrued over a 12-month royalty period. There are five successive 12-month minimum royalty periods, the third of which commenced on October 1, 2010. Based on actual net sales from AzaSite during the fourth quarter of 2010 and our expectation of net sales from AzaSite in 2011, we anticipate incurring a shortfall of the minimum royalty due at the end of September 30, 2011. As a result, beginning with the fourth quarter of 2010, we have started to accrue additional royalty expense based on our anticipated shortfall from the minimum royalty. The amount of additional royalty expensed during the fourth quarter of 2010 as it relates to the shortfall from the minimum royalty due was approximately $229,000. The total shortfall, if any, does not have to be paid until the end of the 12-month minimum royalty period.
The increase in cost of sales in 2009 of $4.9 million, or 76%, as compared to 2008, was primarily due to increased sales volume of AzaSite, which has resulted in increased royalties, as well as an increase in the royalty rate, paid to InSite Vision, as described earlier.
Cost of sales expense consists of variable and fixed cost components. Variable cost components include royalties to InSite Vision on net sales of AzaSite, the cost of AzaSite inventory sold, distribution, shipping and logistic service charges from our third-party logistics provider, and changes to our inventory reserve for overstocking, short-dated or otherwise unmarketable product or unusable materials. Fixed cost components are primarily the amortization of the $19.0 million approval milestone that we paid InSite Vision as part of our licensing agreement. This approval milestone is being amortized ratably on a straight-line basis through the term of the underlying patent coverage for AzaSite, which expires in March 2019.
Certain costs included in cost of sales are subject to annual increases for which we have limited control. We expect that cost of sales will increase in relation to, but not proportionately to, the increases in revenue from sales of AzaSite.
Costs and Expenses
Research and Development Expenses
Research and development expenses were approximately $46.1 million for the year ended December 31, 2010, as compared to approximately $51.1 million in 2009 and approximately $44.6 million in 2008.
The decrease in research and development expenses of approximately $5.0 million, or 10%, for the year ended December 31, 2010, as compared to 2009, was primarily due to the substantial completion of both our Phase 3 trial activities associated with our product candidate, Prolacria, for the treatment of dry eye and our Phase 2 trials researching AzaSite for the treatment of blepharitis in the first quarter of 2010, as well as completion of Phase 1 trial activities related to our glaucoma program in the third quarter of 2009. Additionally, we experienced cost savings due to our elimination of preclinical and drug discovery activities as a result of a restructuring that occurred in the first quarter of 2009. These decreases in expenses were partially offset by increased costs associated with our cystic fibrosis development program, including pre-launch development activities with Novasep associated with developing an additional and/or alternative commercial supplier of denufosol.
53
Table of Contents
The increase in research and development expenses of approximately $6.5 million, or 15%, for the year ended December 31, 2009, as compared to 2008, was primarily due to increased costs associated with our cystic fibrosis program, including the recognition of an approximate $3.3 million milestone payable to Yamasa Corporation upon entering into an agreement to facilitate the transfer of denufosol manufacturing technology, including intellectual property, to Novasep and thus enable a two-supplier strategy for denufosol. The remaining increase in research and development expenses was due to the initiation of a Phase 2 program for AzaSite for the treatment of blepharitis.
These increases in research and development expenses for the year ended December 31, 2009, as compared to 2008, were partially offset by reduced spending on our glaucoma program as a result of the completion of Phase 1 trial activities, the discontinuation of our program for the development of epinastine nasal spray for allergic rhinitis in 2008, and cost savings from our restructuring activities in the first quarter of 2009. See Part I—Item 1. Business of this report for additional information regarding various development programs.
Research and development expenses include all direct and indirect costs, including salaries for our research and development personnel, consulting fees, clinical trial costs, including the development and manufacture of drug product for clinical trials, sponsored research costs, clinical trial insurance, upfront license fees and milestone payments relating to research and development as well as other fees and costs related to the development of product candidates. Research and development expenses vary according to the number of programs in clinical development and the stage of development of our clinical programs. Later stage clinical programs tend to cost more than earlier stage programs due to the length of the clinical trials and the number of patients enrolled in later stage clinical trials.
Our future research and development expenses will depend on the results and magnitude or scope of our clinical activities and requirements imposed by regulatory agencies. Year over year spending on active development programs can vary due to the differing levels and stages of development activity, the timing of certain expenses and other factors. Accordingly, our research and development expenses may fluctuate significantly from period to period. In addition, if we in-license or out-license rights to product candidates, our research and development expenses may fluctuate significantly from prior periods.
Our research and development expenses for the years ended December 31, 2010, 2009 and 2008 and from the respective project’s inception are shown below and includes the percentage of overall research and development expenditures for the years listed.
| (In thousands) Year ended December 31, |
Cumulative from Inception to December 31, 2010 |
% | ||||||||||||||||||||||||||||||
| 2010 | % | 2009 | % | 2008 | % | |||||||||||||||||||||||||||
| Denufosol tetrasodium for cystic fibrosis (1) |
$ | 36,433 | 79 | $ | 29,590 | 58 | $ | 18,633 | 42 | $ | 130,803 | 32 | ||||||||||||||||||||
| AzaSite for blepharitis |
2,937 | 6 | 6,297 | 12 | 83 | — | 9,317 | 2 | ||||||||||||||||||||||||
| AzaSite (2) |
2,204 | 5 | 2,311 | 5 | 1,621 | 4 | 20,734 | 5 | ||||||||||||||||||||||||
| Prolacria (diquafosol tetrasodium) for dry eye disease |
902 | 2 | 6,621 | 13 | 7,632 | 17 | 58,474 | 14 | ||||||||||||||||||||||||
| INS115644 and INS117548 for glaucoma and related research and development |
265 | 1 | 1,555 | 3 | 5,552 | 12 | 16,712 | 4 | ||||||||||||||||||||||||
| Other research, preclinical and unallocated development costs (3) |
3,408 | 7 | 4,760 | 9 | 11,116 | 25 | 174,084 | 43 | ||||||||||||||||||||||||
| Total |
$ | 46,149 | 100 | $ | 51,134 | 100 | $ | 44,637 | 100 | $ | 410,124 | 100 | ||||||||||||||||||||
| (1) | Includes the expensing of an approximate $3.3 million milestone in September 2009. |
| (2) | Expenses in 2010 include costs associated with new bottle design and development activities. |
| (3) | Prior to February 2009, other research, preclinical and development costs represent all unallocated research and development costs or those costs allocated to preclinical programs as well as costs of discontinued and/or inactive programs. In February 2009, we restructured our operations eliminating our preclinical and molecule discovery activities. |
54
Table of Contents
Selling and Marketing Expenses
Selling and marketing expenses were approximately $50.2 million for the year ended December 31, 2010, as compared to approximately $49.3 million in 2009 and approximately $54.6 million in 2008.
The increase in selling and marketing expenses of approximately $847,000, or 2%, for the year ended December 31, 2010, as compared to 2009, was primarily due to a general increase in personnel related expenses, as well as the timing of certain marketing activities.
The decrease in selling and marketing expenses of approximately $5.3 million, or 10%, for the year ended December 31, 2009, as compared to 2008, was due to an overall reduction in promotional and marketing activities, including Phase 4 clinical trial activities, partially offset by a general increase in personnel related expenses, including stock-based compensation expense.
Our commercial organization currently focuses its promotional efforts on approximately 10,000 eye care specialists. Our selling and marketing expenses include all direct costs associated with the commercial organization, which include our sales force and marketing programs. Our sales force expenses include salaries, training and educational program costs, product sample costs, fleet management and travel. Our marketing and promotion expenses include product management, promotion, advertising, public relations, Phase 4 clinical trial costs, physician training and continuing medical education and administrative expenses. We adjust the timing, magnitude and targeting of our advertising, promotional, Phase 4 clinical trials and other commercial activities for our products based on seasonal trends and other factors, and accordingly, these costs can fluctuate from period to period.
Future selling and marketing expenses will depend on the level of our future commercialization activities. We expect selling and marketing expenses will increase in periods that immediately precede and follow product launches. In addition, if we in-license or out-license rights to products, our selling and marketing expenses may fluctuate significantly from prior periods.
General and Administrative Expenses
General and administrative costs were approximately $30.9 million for the year ended December 31, 2010, as compared to approximately $16.1 million in 2009 and approximately $14.5 million in 2008.
The increase in general and administrative expenses of approximately $14.8 million, or 93%, for the year ended December 31, 2010, as compared to 2009, was primarily due to an increase in executive compensation expense, the majority of which consisted of non-cash stock based compensation expense, associated with recent changes in management. For the year ended December 31, 2010, executive compensation increased approximately $8.1 million, including non-cash stock based compensation expense, which increased by approximately $5.0 million, as compared to 2009. In addition, there was an increase in consulting expenses and other fees of approximately $4.1 million, primarily associated with business and corporate development, compliance activities and the CEO transition, for the year ended December 31, 2010, as compared to 2009. General and administrative expenses for the prior year period were reduced by a reimbursement of legal expenses of approximately $875,000 from our insurance carrier as discussed below.
The increase in general and administrative expenses of approximately $1.5 million, or 10%, for the year ended December 31, 2009, as compared to 2008, was primarily due to an increase in consulting and legal expenses, as well as an increase in personnel related expenses and stock-based compensation. These increases were partially offset by a final reimbursement of legal fees in 2009 received from our insurance provider of approximately $875,000 related to the stockholder litigation and SEC investigation referenced below.
On September 30, 2008, the SEC approved a non-monetary settlement of the previously announced investigation of Inspire and two of our prior officers by the SEC staff relating to our disclosures regarding a Phase 3 clinical trial of our dry eye product candidate, Prolacria.
55
Table of Contents
On July 26, 2007, the United States District Court for the Middle District of North Carolina granted Inspire’s and the other defendants’ motion and dismissed the Consolidated Class Action Complaint with prejudice. On December 12, 2008, the Fourth Circuit of the United States Court of Appeals issued an opinion affirming the judgment of the District Court.
Our general and administrative expenses consist primarily of personnel, facility and related costs for general corporate functions, including business development, finance, accounting, legal, human resources, quality/compliance, facilities and information systems.
Future general and administrative expenses will depend on the level and extent of support required to conduct our future research and development, commercialization, business development, and corporate activities.
Restructurings
In March 2009, we announced a restructuring of our operations to eliminate preclinical and drug discovery activities and refocus our resources on the development of existing later-stage clinical programs and commercially available products. Significant components of the restructuring charge were one-time termination benefits for employees impacted by the restructuring, estimated costs to write-down idle lab equipment to net realizable value, losses associated with leased lab space that was vacated, and costs to satisfy contract commitments related to activities and programs no longer associated with our supported programs and on-going operations. In connection with the restructuring, we recorded restructuring charges of approximately $2.0 million for the year ended December 31, 2009.
On January 3, 2011, we announced that the top-line results from our second Phase 3 clinical trial, TIGER-2, with denufosol tetrasodium for the treatment of cystic fibrosis trial did not achieve statistical significance. Based upon the overall data relating to the program, we have decided to discontinue development of denufosol tetrasodium and change our strategic approach to solely focus on our eye care business as announced on February 17, 2011. As a result, we expect to recognize a restructuring charge of approximately $10 million to $13 million in the first quarter of 2011, primarily comprised of costs to (i) restructure our research and development organization and eliminate our infrastructure associated with the pulmonary therapeutic area; (ii) complete all necessary remaining contractual activities associated with TIGER-2 and the open-label denufosol-only clinical trial as well as other denufosol activities; and (iii) record an impairment charge associated with the equipment that was developed and purchased in anticipation of FDA approval and costs related to idle facilities. In addition, we expect to incur one-time termination benefits associated with a reduction of approximately 27% of our full-time employee base as a result of eliminating our pulmonary therapeutic area. See Note 19 “Subsequent Events” to our financial statements for further discussion.
Other Income (Expense)
For the year ended December 31, 2010, we incurred other income, net of approximately $308,000, as compared to approximately $2.4 million in other expense, net in 2009 and approximately $1.9 million in other expense, net in 2008.
The increase in other income, net of approximately $2.7 million for the year ended December 31, 2010, as compared to 2009, was due to a decrease in interest expense. The decrease in interest expense is the result of a lower average outstanding principal balance of our term loan facility during the year ended December 31, 2010, as compared to 2009. In December 2010, we repaid the remaining principal balance of our term loan facility. In addition, in December 2010, we recognized $978,000 awarded to us under the Federal Government’s Qualifying Therapeutic Discovery Project in other income.
The increase in other expense, net of approximately $415,000 for the year ended December 31, 2009, as compared to 2008, was due to decreased interest income partially offset by a decrease in interest expense.
56
Table of Contents
Interest income was negatively impacted due to lower average cash and investment balances combined with a lower rate of return during 2009, as compared to 2008. The decrease in interest expense was the result of a lower average outstanding principal balance of our term loan facility during 2009, as compared to 2008.
Other income/(expense) fluctuates from year to year depending on the level of interest income earned on variable cash and investment balances, realized gains and losses on investments due to changes in fair market value and interest expense on debt. Future other income/(expense) will depend on our future cash and investment balances, the return and change in fair market value on these investments, as well as levels of debt and the associated interest rates.
Liquidity and Capital Resources
We have financed our operations primarily through the sale of equity securities, including private sales of preferred stock and public offerings of common stock and, to a lesser extent, through a term loan facility. We also currently receive product revenue from net sales of AzaSite, royalty revenue from net sales of Restasis, and co-promotion revenue from net sales of Elestat. We do not expect our revenue to exceed our operating expenses in 2011.
As of December 31, 2010, we had net working capital of approximately $72.5 million, a decrease of approximately $12.9 million from approximately $85.4 million at December 31, 2009. The decrease in working capital was principally due to the funding of normal operating expenses associated with commercialization activities and the development of our product candidates, as well as principal and interest payments on our term loan facility. Our principal sources of liquidity at December 31, 2010 were approximately $42.2 million in cash and cash equivalents, approximately $51.2 million in investments, which are considered available-for-sale, and approximately $22.4 million in trade receivables.
Net cash used in operating activities was approximately $6.7 million for the year ended December 31, 2010, as compared to approximately $33.5 million in 2009 and approximately $51.5 million in 2008. The decrease in net cash used in operating activities in 2010, as compared to 2009, was primarily attributable to a reduction in our net loss for the period, increases in non-cash stock-based compensation expense and an increase in accounts payable at December 31, 2010, primarily associated with denufosol activities. The decrease in net cash used in operating activities in 2009, as compared to 2008, was primarily attributable to a reduction in our net loss for the period. Our annual net losses since 2007 have trended downward, primarily driven by annual increases in net product sales of AzaSite and royalty revenue from Restasis. However, we expect 2011 revenues to decrease from 2010 levels due to the anticipated introduction of a generic form of epinastine ophthalmic solution into the market and the 3% reduction in our royalty rate for Restasis in accordance with the terms of the amended and restated agreement with Allergan.
Net cash provided by investing activities was approximately $20.7 million for the year ended December 31, 2010, as compared to net cash used in investing activities of approximately $63.7 million in 2009, and net cash provided by investing activities of approximately $22.1 million in 2008. Increases in net cash provided by investing activities are generally the result of the conversion of our investments in available-for-securities to cash to enable us to fund our ongoing operating activities. Increases in net cash used in investing activities generally occur when we have raised cash either through the sale of equity securities or through borrowings and have invested the proceeds in available-for-sale securities.
In August 2009, we completed a public offering of 25,555,555 shares of our common stock at a price of $4.50 per share for gross proceeds of $115 million. Net proceeds were $109 million, after deducting underwriting discounts and offering expenses.
Net cash used in financing activities was approximately $24.0 million in 2010, as compared to net cash provided by financing activities of approximately $91.0 million in 2009, and net cash used in financing activities of approximately $14.0 million in 2008. Net cash used in financing activities have primarily involved the
57
Table of Contents
scheduled repayment of our term loan facility. Payments on notes payable and capital lease obligations have been approximately $25.2 million, $18.4 million and $14.1 million for the years ended December 31, 2010, 2009 and 2008, respectively. Net cash provided by financing activities in 2009 was primarily related to net cash proceeds from our common stock sale in 2009, as mentioned above.
In December 2006, we entered into a loan and security agreement in order to obtain debt financing of up to $40.0 million to fund in-licensing opportunities and related development. In June 2007, we amended the loan and security agreement to enable us to draw upon a new supplemental term loan facility in the amount of $20.0 million. We had borrowed the full $60.0 million under the term loan facility. In December 2010, we repaid the remaining principal balance of the term loan facility. See Note 9 “Debt” to our financial statements for further discussion regarding the term loan facility.
At December 31, 2010, our cash and investments totaled approximately $94.3 million, for which all but $815,000 of these investments provide short-term liquidity and can be readily liquidated into cash to fund our ongoing operations. These investments, in conjunction with the restructuring activities announced in February 2011 which significantly reduce our expected future operating expenses, will allow us to adequately fund our operations in 2011 and for the next several years with the potential ability to achieve cash flow positive operations in that time period. Based upon our current trends and planned operations, we project our 2011 cash utilization to be in the range of $15 million to $22 million. Our future working capital requirements, including the need for additional working capital, will be largely determined by the commercial success of our products, the successful and timely completion of our blepharitis development program, and the in-licensing or acquisition of any additional products that we may identify in the future.
More specifically, our working capital requirements will be dependant on: the number, magnitude, scope and timing of our development programs; regulatory approval of our product candidates; the commercial potential and success of our product candidates; the loss of commercial exclusivity of any of our products; the loss of revenue from our products due to competition or loss of market share; the level of ongoing costs related to the commercialization of AzaSite and Elestat; the cost, timing and outcome of regulatory reviews, regulatory investigations, and changes in regulatory requirements; the costs of obtaining patent protection for our product candidates; the timing and terms of business development activities; the rate of technological advances relevant to our operations; the timing, method and cost of the commercialization of our product candidates; the efficiency of manufacturing processes developed on our behalf by third parties; the level of required administrative support for our daily operations; and our ability to successfully restructure our operations, among other factors.
2011 Financial Guidance
In February 2011, we announced a corporate restructuring as a result of the failed TIGER-2 clinical trial and discontinuation of our cystic fibrosis program. The restructuring includes a workforce reduction of approximately 65 positions or approximately 27% of our full-time employees. Following the restructuring, we expect to reduce 2011 non-cost of sales operating expenses by more than $40 million, excluding restructuring charges.
Based upon current trends and assumptions, and our planned strategic direction and development operations, we expect to record the following for 2011:
| • | Aggregate revenue in the range of $80-$90 million. |
| • | Total operating expenses in the range of $110-$125 million, which includes projected stock-based compensation costs of approximately $7-$12 million and restructuring costs: |
| • | Cost of sales in the range of $18-$21 million; |
| • | Research and development expenses in the range of $13-$18 million; |
58
Table of Contents
| • | Selling and marketing expenses in the range of $45-$50 million; |
| • | General and administrative expenses in the range of $22-$26 million; and |
| • | Restructuring expenses of $10-$13 million. |
| • | Operating cash utilization in the range of $15-$22 million. |
Our ability to remain within our operating expense target range is subject to multiple factors, including unanticipated or additional development costs, including the in-licensing or acquisition of additional products in the future, unanticipated or additional costs to successfully commercialize our products, the commercial success of our current products and other factors described under the Risk Factors located elsewhere in this report.
Contractual Obligations and Commitments
In the normal course of business, we enter into various agreements that create contractual obligations and commitments that may require future cash payments. Contractual obligations at December 31, 2010 included operating leases of $8.5 million and purchase obligations and other commitments, as further described below.
As part of our drug development strategy, we outsource significant amounts of our clinical development program activities as well as the manufacture of drug substance used in those programs. In addition, we have manufacturing, promotion and clinical responsibilities and activities associated with the commercialization of AzaSite. Based on these requirements and activities, we have entered into contractual commitments or purchase obligations with various clinical research organizations, promotion and advertising agencies, manufacturers of active pharmaceutical ingredients and drug product for clinical and commercial use as well as with other service providers. These financial commitments, which totaled approximately $15.7 million as of December 31, 2010, are reflected as purchase obligations in the table below and include both cancelable and non-cancelable arrangements. Since many of these commitment amounts are dependent upon variable components of the agreements, actual payments and the timing of those payments may differ from management’s estimates.
We are obligated to pay royalties to InSite Vision as part of our in-licensing agreement for AzaSite. Under the terms of the agreement, our obligation to pay royalties to InSite Vision is subject to pre-determined minimum annual royalty payments. The determination of whether or not we will owe any such payments is based upon the amount of royalties accrued over a 12-month royalty period. There are five successive 12-month minimum royalty periods, the first of which commenced on October 1, 2008. The minimum royalties escalate each year. Remaining minimum royalties as of December 31, 2010 total $51.0 million. Based on actual net sales from AzaSite during the fourth quarter of 2010 and our expectation of net sales from AzaSite in 2011, we anticipate incurring a shortfall of the minimum royalty due to InSite Vision at the end of September 30, 2011.
59
Table of Contents
The table below reflects contractual and potential obligations as of December 31, 2010. Some of the amounts we include in this table are based on management’s estimate and assumptions about these obligations, including their duration, the possibility of renewal, anticipated actions by third parties, and other factors as previously described. In addition, based upon our decision to discontinue the cystic fibrosis program following the results of the TIGER-2 clinical trial and the restructuring activities announced in February 2011, the majority of the potential development milestone obligations listed in the table will no longer apply following the restructuring. See Note 19 “Subsequent Events” to our financial statements for further discussion. Because these estimates and assumptions are necessarily subjective, the obligations we will actually pay in future periods may vary from those reflected in the table:
| (In thousands) Payment due by Period as of December 31, 2010 |
||||||||||||||||||||
| Contractual and Potential Obligations |
Total | Less than 1 year |
1-3 years |
3-5 years |
More than 5 years |
|||||||||||||||
| Operating Lease Obligations (1) |
$ | 8,516 | $ | 1,463 | $ | 3,152 | $ | 1,936 | $ | 1,965 | ||||||||||
| Purchase Obligations (2) |
15,742 | 15,742 | — | — | — | |||||||||||||||
| Development Milestone Obligations (3)(4) |
21,857 | 3,693 | — | — | 18,164 | |||||||||||||||
| Minimum Royalties and Sales Milestone Obligations (3)(4) |
55,000 | 15,000 | 36,000 | — | 4,000 | |||||||||||||||
| Total |
$ | 101,115 | $ | 35,898 | $ | 39,152 | $ | 1,936 | $ | 24,129 | ||||||||||
| (1) | Includes estimated payments of $1.8 million for the cancelable portion of operating leases, primarily our fleet vehicles under a master lease agreement. See Note 14 “Commitments and Contingencies” to our financial statements for a full discussion. |
| (2) | Purchase obligations reflect all contractual obligations, including amounts that are cancelable, under legally enforceable contracts with contract terms that are both fixed and determinable. These amounts exclude obligations for goods and services that already have been incurred and are reflected on our Balance Sheet as of December 31, 2010. |
| (3) | Development and sales milestone obligations represent potential amounts payable by us contingent on a number of factors, including the progress of our research and development programs, our ability to obtain regulatory approvals, and the commercial success of our approved products. Approximately $1.9 million of “other long-term liabilities” as recorded on our Balance Sheet as of December 31, 2010, is included in Development Milestone Obligations. |
| (4) | On January 3, 2011, we announced that the top-line results from our second Phase 3 clinical trial, TIGER-2, with denufosol tetrasodium for the treatment of cystic fibrosis did not achieve statistical significance. Based upon the overall data relating to the program, we have decided to discontinue development of denufosol tetrasodium. In February 2011, we amended our agreement with Yamasa and are no longer obligated with regards to potential development milestones of approximately $8.6 million. In addition, it is unlikely that other potential development milestone obligations of approximately $11.5 million and sales milestones of $4.0 million, which relate to the denufosol program will continue as potential obligations and ultimately be paid. See Note 19 “Subsequent Events” to our financial statements for further discussion. |
Impact of Recently Issued Accounting Pronouncements
In April 2010, the FASB issued authoritative guidance on defining a milestone and determining when it may be appropriate to apply the milestone method of revenue recognition for research or development transactions. Consideration that is contingent on achievement of a milestone in its entirety may be recognized as revenue in the period in which the milestone is achieved only if the milestone is judged to meet certain criteria to be considered substantive. Milestones should be considered substantive in their entirety and may not be bifurcated. An arrangement may contain both substantive and nonsubstantive milestones, and each milestone should be evaluated individually to determine if it is substantive. The guidance is effective on a prospective basis
60
Table of Contents
for milestones achieved in fiscal years, and interim periods within those years, beginning on or after June 15, 2010, with early adoption permitted. We do not expect the adoption of this guidance to have a material impact on our consolidated financial statements.
Healthcare Reform
In March 2010, Congress passed significant health reform legislation, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, “Affordable Care Act” or “ACA”). The legislation is designed to expand access to affordable health insurance through subsidies, Medicaid expansion, and insurance market reforms (including the development of new health benefit exchanges), financed in part through reduced federal health care spending. Among many other things, the ACA makes a number of significant changes affecting pharmaceutical manufacturers.
Although many provisions of the ACA do not take effect immediately, several provisions became effective in the first quarter of 2010. For instance, the ACA provides for increases to the minimum Medicaid rebate percentages from 15.1% to 23.1%, increased “additional rebates” for new formulations of brand name drugs, the establishment of a maximum rebate amount, and the extension of Medicaid rebates to Medicaid managed care organization utilization. In addition, the ACA broadens the definition of “average manufacturer price” effective October 1, 2010, which in turn may have the effect of increasing Medicaid rebate and Public Health Service section 340B drug discount program payment obligations.
Beginning in 2011, the ACA requires that drug manufacturers provide a 50% discount to Medicare beneficiaries whose prescription drug costs cause them to be subject to the Medicare Part D coverage gap. Also, beginning in 2011, we will be assessed our share of a new fee assessed on all branded prescription drug and biologics manufacturers and importers. This fee will be calculated based upon each organization’s percentage share of total branded prescription drug sales to U.S. government programs (such as Medicare, Medicaid and VA and PHS discount programs) made during the previous year.
The Obama Administration has issued guidance on certain aspects of implementation, including the manufacturer discount program for certain Medicare Part D drugs, which we are assessing. However, details on many other ACA policies have yet to be finalized, including how the annual fee on branded prescription drugs will be calculated and allocated in 2011. We do not expect the provisions that became effective in the first quarter of 2010 to have a significant financial impact on our 2010 revenues from AzaSite sales. We expect future AzaSite revenues to be negatively impacted beginning in 2011 but are unable to adequately assess at this time the extent of the impact of provisions that have not yet gone into effect until additional operational details are available. There can be no assurance that the prescription drug user fees and other ACA provisions yet to be implemented will not impact our financial position.
Off Balance Sheet Arrangements
As of December 31, 2010, we were not a party to any off-balance sheet arrangements.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
Interest Rate Risk
We are subject to interest rate risk on our investment portfolio and borrowings under our term loan facility.
We invest in marketable securities in accordance with our investment policy. The primary objectives of our investment policy are to preserve capital, maintain proper liquidity to meet operating needs and maximize yields. Our investment policy specifies credit quality standards for our investments and limits the amount of credit exposure to any single issue, issuer or type of investment. Our investment portfolio may consist of a variety of
61
Table of Contents
securities, including U.S. government and agency securities, money market and mutual fund investments, municipal and corporate bonds and commercial paper and asset or mortgage-backed securities, among others. As of December 31, 2010, cash equivalents consisted of $6.2 million in a money market account, $21.2 million in money market funds and $6.3 million in corporate bonds, commercial paper and U.S. Government agency securities. Our investment portfolio as of December 31, 2010 consisted of corporate bonds, commercial paper, U.S. Government and agency securities, and an international government security which collectively had an average maturity of less than 12 months, using the stated maturity. All of our cash, cash equivalents and investments are maintained at two banking institutions.
Our investment exposure to market risk for changes in interest rates relates to the increase or decrease in the amount of interest income we can earn on our portfolio, changes in the market value due to changes in interest rates and other market factors as well as the increase or decrease in any realized gains and losses. Our investment portfolio includes only marketable securities and instruments with active secondary or resale markets to help ensure portfolio liquidity and we have implemented guidelines limiting the duration of investments. At December 31, 2010, our portfolio of available-for-sale investments consisted of approximately $47.5 million of investments maturing within one year and approximately $3.7 million of investments maturing after one year but within 24 months. In general, securities with longer maturities are subject to greater interest-rate risk than those with shorter maturities. A hypothetical 100 basis point drop in interest rates along the entire interest rate yield curve would not significantly affect the fair value of our interest sensitive financial instruments. We generally have the ability to hold our fixed-income investments to maturity and therefore do not expect that our operating results, financial position or cash flows will be materially impacted due to a sudden change in interest rates. However, our future investment income may fall short of expectations due to changes in interest rates, or we may suffer losses in principal if forced to sell securities which have declined in market value due to changes in interest rates or other factors, such as changes in credit risk related to the securities’ issuers.
We do not use interest rate derivative instruments to manage exposure to interest rate changes. We ensure the safety and preservation of invested principal funds by limiting default risk, market risk and reinvestment risk. We reduce default risk by investing in investment grade securities.
Investment Risk
In addition to our normal investment portfolio, we have an investment in Parion Sciences, Inc. of $200,000 as of December 31, 2010. This investment is in the form of unregistered common stock and is subject to higher investment risk than our normal investment portfolio due to the lack of an active resale market for the Parion Sciences, Inc. securities.
Foreign Currency Exchange Risk
The majority of our transactions occur in U.S. dollars and we do not have subsidiaries or investments in foreign countries. We receive royalties on Restasis and Diquas that are based upon sales outside the United States; however, we are compensated for these sales in royalty payments that are made in U.S. dollars. Therefore, we are not subject to significant foreign currency exchange risk. We do, however, have foreign currency exposure with regard to the purchase of active pharmaceutical ingredients as they relate to AzaSite, which is manufactured by a foreign-based company and is payable in Euros. We have established policies and procedures for assessing market and foreign exchange risk. As of December 31, 2010, we did not have any foreign currency hedges.
Item 8. Financial Statements and Supplementary Data.
The financial statements required to be filed pursuant to this Item 8 are appended to this Annual Report on Form 10-K. A list of the financial statements filed herewith is found at “Index to Financial Statements” on page F-1.
62
Table of Contents
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
Not applicable.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
Our management is responsible for establishing and maintaining an adequate system of internal control over our financial reporting. The design, monitoring and revision of the system of internal accounting controls involves, among other items, management’s judgments with respect to the relative cost and expected benefits of specific control measures. The effectiveness of the control system is supported by the selection, retention and training of qualified personnel and an organizational structure that provides an appropriate division of responsibility and formalized procedures. The system of internal accounting controls is periodically reviewed and modified in response to changing conditions. Internal audit consultants regularly monitor the adequacy and effectiveness of internal accounting controls. In addition to the system of internal accounting controls, management maintains corporate policy guidelines that help monitor proper overall business conduct, possible conflicts of interest, compliance with laws and confidentiality of proprietary information. Our Chief Executive Officer and Chief Financial Officer have reviewed and evaluated the effectiveness of our disclosure controls and procedures as defined in Rules 13a-15(e) and 15d-15(e) of the Securities Exchange Act of 1934 as of the end of the period covered by this report. Based on that evaluation, the Chief Executive Officer and Chief Financial Officer have concluded that our current disclosure controls and procedures are effective.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over our financial reporting as defined in Rules 13a-15(f) and 15d-15(f) of the Securities Exchange Act of 1934, and for performing an assessment of the effectiveness of internal control over financial reporting as of December 31, 2010. Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our system of internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Management conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on this evaluation, management, including our principal executive officer and principal financial officer, concluded that our internal control over financial reporting was effective as of December 31, 2010. The effectiveness of our internal control over financial reporting as of December 31, 2010 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report which is presented in this Annual Report on Form 10-K.
Changes in Internal Control Over Financial Reporting
No change in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) occurred during the year ended December 31, 2010 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
63
Table of Contents
Audit Committee Oversight
The Audit Committee of our Board of Directors, consisting solely of independent directors, appoints the independent registered public accounting firm and receives and reviews the reports submitted by them. The Audit Committee meets several times during the year with management, the internal auditors and the independent registered public accounting firm to discuss audit activities, internal controls and financial reporting matters. The internal auditors and the independent registered public accounting firm have full and free access to the Audit Committee.
Not applicable.
64
Table of Contents
PART III
Item 10. Directors, Executive Officers and Corporate Governance.
The information required by this item is incorporated by reference to the material responsive to this item contained in our Proxy Statement to be filed in connection with our 2011 Annual Meeting of Stockholders.
Item 11. Executive Compensation.
The information required by this item is incorporated by reference to the material responsive to this item contained in our Proxy Statement to be filed in connection with our 2011 Annual Meeting of Stockholders.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The following table sets forth certain information with respect to securities authorized for issuance under equity incentive plans as of December 31, 2010.
Equity Compensation Plan Information
| Plan category |
(a) Number of securities to be issued upon exercise of outstanding options, warrants and rights |
(b) Weighted-average exercise price of outstanding options, warrants and rights |
(c) Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column(a)) |
|||||||||
| Equity compensation plans approved by security holders |
12,318,198 | $ | 7.00 | 7,645,791 | ||||||||
| Equity compensation plans not approved by security holders |
1,687,500 | $ | 5.93 | 0 | ||||||||
| Total |
14,005,698 | $ | 6.94 | 7,645,791 | ||||||||
The additional information required by this item is incorporated by reference to the material responsive to this item contained in our Proxy Statement to be filed in connection with our 2011 Annual Meeting of Stockholders.
Item 13. Certain Relationships and Related Transactions, and Director Independence.
The information required by this item is incorporated by reference to the material responsive to this item contained in our Proxy Statement to be filed in connection with our 2011 Annual Meeting of Stockholders.
Item 14. Principal Accountant Fees and Services.
The information required by this item is incorporated by reference to the material responsive to this item contained in our Proxy Statement to be filed in connection with our 2011 Annual Meeting of Stockholders.
65
Table of Contents
PART IV
Item 15. Exhibits and Financial Statements Schedules.
(a) The following documents are included as part of this Annual Report on Form 10-K:
| Page | ||||
| F-2 | ||||
| F-3 | ||||
| F-4 | ||||
| F-5 | ||||
| F-6 | ||||
| F-7 | ||||
2. Financial Statement Schedule:
Schedule of Valuation and Qualifying Accounts
(in thousands)
| Additions | ||||||||||||||||||||
| Beginning Balance |
Charged to Costs and Expenses |
Charged to Other Accounts |
Deductions from Allowances |
Ending Balance |
||||||||||||||||
| Year ended December 31, 2008 |
||||||||||||||||||||
| Allowance for rebates, chargebacks and other sales incentives |
$ | 238 | $ | 2,888 | $ | — | $ | (1,824 | ) | $ | 1,302 | |||||||||
| Allowance for returns |
95 | 677 | — | (71 | ) | 701 | ||||||||||||||
| Allowance for uncollectible accounts |
10 | — | — | (3 | ) | 7 | ||||||||||||||
| Inventory allowance |
125 | 105 | — | (220 | ) | 10 | ||||||||||||||
| Valuation allowance for income taxes |
133,209 | 25,321 | — | — | 158,530 | |||||||||||||||
| Year ended December 31, 2009 |
||||||||||||||||||||
| Allowance for rebates, chargebacks and other sales incentives |
$ | 1,302 | $ | 9,531 | $ | — | $ | (6,456 | ) | $ | 4,377 | |||||||||
| Allowance for returns |
701 | 1,560 | — | (738 | ) | 1,523 | ||||||||||||||
| Allowance for uncollectible accounts |
7 | 43 | — | — | 50 | |||||||||||||||
| Inventory allowance |
10 | 15 | — | — | 25 | |||||||||||||||
| Valuation allowance for income taxes |
158,530 | 19,629 | — | — | 178,159 | |||||||||||||||
| Year ended December 31, 2010 |
||||||||||||||||||||
| Allowance for rebates, chargebacks and other sales incentives |
$ | 4,377 | $ | 19,758 | $ | — | $ | (13,891 | ) | $ | 10,244 | |||||||||
| Allowance for returns |
1,523 | 1,629 | — | (562 | ) | 2,590 | ||||||||||||||
| Allowance for uncollectible accounts |
50 | — | — | — | 50 | |||||||||||||||
| Inventory allowance |
25 | 253 | — | (228 | ) | 50 | ||||||||||||||
| Valuation allowance for income taxes |
178,159 | 16,970 | — | — | 195,129 | |||||||||||||||
For additional information regarding the Company’s reserves and allowances for rebates, chargebacks, discounts and returns, see—“Management’s Discussion and Analysis of Financial Condition and Results of Operations—Revenue Recognition.”
All other schedules are omitted as the information required is inapplicable or the information is presented in the financial statements.
3. Exhibits:
See the Exhibit Index located at the end of this document.
66
Table of Contents
Pursuant to the requirements of Section 13 of 15(d) the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Inspire Pharmaceuticals, Inc. | ||
| By: |
/s/ ADRIAN ADAMS | |
| Adrian Adams President & Chief Executive Officer and Director | ||
| Date: February 25, 2011 | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ ADRIAN ADAMS Adrian Adams |
President & Chief Executive Officer (principal executive officer) and Director |
February 25, 2011 | ||
| /s/ THOMAS R. STAAB, II Thomas R. Staab, II |
Chief Financial Officer & Treasurer (principal financial officer and principal accounting officer) |
February 25, 2011 | ||
| /s/ KENNETH B. LEE, JR. Kenneth B. Lee, Jr. |
Chairman of the Board of Directors |
February 25, 2011 | ||
| /s/ GEORGE ABERCROMBIE George Abercrombie |
Director |
February 25, 2011 | ||
| /s/ KIP A. FREY Kip A. Frey |
Director |
February 25, 2011 | ||
| /s/ ALAN F. HOLMER Alan F. Holmer |
Director |
February 25, 2011 | ||
| /s/ NANCY J. HUTSON Nancy J. Hutson |
Director |
February 25, 2011 | ||
| /s/ JONATHAN S. LEFF Jonathan S. Leff |
Director |
February 25, 2011 | ||
| /s/ RICHARD S. KENT Richard S. Kent |
Director |
February 25, 2011 | ||
67
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
| Page(s) | ||||
| F-2 | ||||
| F-3 | ||||
| F-4 | ||||
| F-5 | ||||
| F-6 | ||||
| F-7 | ||||
F-1
Table of Contents
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders
of Inspire Pharmaceuticals, Inc.
In our opinion, the financial statements listed in the index appearing under Item 15(a)1 present fairly, in all material respects, the financial position of Inspire Pharmaceuticals, Inc. at December 31, 2010 and December 31, 2009, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2010 in conformity with accounting principles generally accepted in the United States of America. In addition, in our opinion, the financial statement schedule listed in the index appearing under Item 15(a)2 presents fairly, in all material respects, the information set forth therein when read in conjunction with the related financial statements. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for these financial statements and financial statement schedule, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in Management’s Report on Internal Control over Financial Reporting under Item 9A. Our responsibility is to express opinions on these financial statements, on the financial statement schedule, and on the Company’s internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP
Raleigh, North Carolina
February 24, 2011
F-2
Table of Contents
BALANCE SHEETS
(in thousands, except per share amounts)
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 42,204 | $ | 52,256 | ||||
| Investments |
47,510 | 54,367 | ||||||
| Trade receivables, net |
22,442 | 22,682 | ||||||
| Prepaid expenses and other receivables |
3,792 | 5,479 | ||||||
| Inventories, net |
776 | 1,717 | ||||||
| Other assets |
— | 153 | ||||||
| Total current assets |
116,724 | 136,654 | ||||||
| Property and equipment, net |
7,896 | 4,429 | ||||||
| Assets held-for-sale |
— | 402 | ||||||
| Investments |
3,922 | 21,861 | ||||||
| Restricted deposits |
615 | 615 | ||||||
| Intangibles, net |
13,154 | 14,748 | ||||||
| Other assets |
188 | 61 | ||||||
| Total assets |
$ | 142,499 | $ | 178,770 | ||||
| Liabilities and Stockholders’ Equity |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 12,766 | $ | 10,056 | ||||
| Accrued expenses |
31,467 | 21,246 | ||||||
| Short-term debt and capital leases |
— | 19,940 | ||||||
| Total current liabilities |
44,233 | 51,242 | ||||||
| Long-term debt and capital leases |
— | 5,235 | ||||||
| Other long-term liabilities |
3,028 | 3,125 | ||||||
| Total liabilities |
47,261 | 59,602 | ||||||
| Commitments and contingencies (Note 14) |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.001 par value, 1,860 shares authorized; no shares issued and outstanding |
— | — | ||||||
| Common stock, $0.001 par value, 200,000 shares authorized; 83,159 and 82,346 shares issued and outstanding, respectively |
83 | 82 | ||||||
| Additional paid-in capital |
530,983 | 519,462 | ||||||
| Accumulated other comprehensive income |
63 | 68 | ||||||
| Accumulated deficit |
(435,891 | ) | (400,444 | ) | ||||
| Total stockholders’ equity |
95,238 | 119,168 | ||||||
| Total liabilities and stockholders’ equity |
$ | 142,499 | $ | 178,770 | ||||
The accompanying notes are an integral part of these financial statements.
F-3
Table of Contents
STATEMENTS OF OPERATIONS
(in thousands, except per share amounts)
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Revenues: |
||||||||||||
| Product sales, net |
$ | 42,671 | $ | 34,961 | $ | 18,349 | ||||||
| Product co-promotion and royalty |
62,478 | 57,198 | 50,899 | |||||||||
| Collaborative research and development |
1,250 | — | 1,250 | |||||||||
| Total revenue |
106,399 | 92,159 | 70,498 | |||||||||
| Operating expenses: |
||||||||||||
| Cost of sales |
14,915 | 11,271 | 6,412 | |||||||||
| Research and development |
46,149 | 51,134 | 44,637 | |||||||||
| Selling and marketing |
50,151 | 49,304 | 54,568 | |||||||||
| General and administrative |
30,939 | 16,053 | 14,540 | |||||||||
| Restructuring |
— | 2,014 | — | |||||||||
| Total operating expenses |
142,154 | 129,776 | 120,157 | |||||||||
| Loss from operations |
(35,755 | ) | (37,617 | ) | (49,659 | ) | ||||||
| Other income/(expense): |
||||||||||||
| Interest income |
594 | 668 | 2,642 | |||||||||
| Interest expense |
(1,269 | ) | (3,027 | ) | (4,586 | ) | ||||||
| Other |
983 | — | — | |||||||||
| Other income/(expense), net |
308 | (2,359 | ) | (1,944 | ) | |||||||
| Net loss |
$ | (35,447 | ) | $ | (39,976 | ) | $ | (51,603 | ) | |||
| Basic and diluted net loss per common share |
$ | (0.43 | ) | $ | (0.60 | ) | $ | (0.91 | ) | |||
| Weighted average common shares used in computing basic and diluted net loss per common share |
82,733 | 66,797 | 56,609 | |||||||||
The accompanying notes are an integral part of these financial statements.
F-4
Table of Contents
STATEMENTS OF CASH FLOWS
(in thousands)
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Cash flows from operating activities: |
||||||||||||
| Net loss |
$ | (35,447 | ) | $ | (39,976 | ) | $ | (51,603 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Amortization expense |
1,752 | 1,952 | 2,134 | |||||||||
| Depreciation of property and equipment |
1,060 | 755 | 939 | |||||||||
| Asset impairment |
218 | 484 | — | |||||||||
| Loss/(gain) on disposal of property and equipment, net |
38 | (90 | ) | 7 | ||||||||
| Loss/(gain) on investments |
(5 | ) | — | — | ||||||||
| Allowance for doubtful accounts |
18 | 43 | (3 | ) | ||||||||
| Inventory reserve |
253 | 15 | 105 | |||||||||
| Stock-based compensation expense |
10,382 | 5,063 | 4,443 | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Trade receivables |
222 | (6,181 | ) | (3,567 | ) | |||||||
| Prepaid expenses and other receivables |
1,553 | (1,413 | ) | 345 | ||||||||
| Inventories |
688 | (1,043 | ) | 486 | ||||||||
| Other assets |
(132 | ) | — | (40 | ) | |||||||
| Accounts payable |
3,178 | (970 | ) | (3,675 | ) | |||||||
| Accrued expenses and other liabilities |
8,257 | 7,860 | (671 | ) | ||||||||
| Deferred rent |
1,298 | — | — | |||||||||
| Deferred revenue |
— | — | (371 | ) | ||||||||
| Net cash used in operating activities |
(6,667 | ) | (33,501 | ) | (51,471 | ) | ||||||
| Cash flows from investing activities: |
||||||||||||
| Purchase of investments |
(43,108 | ) | (94,049 | ) | (7,574 | ) | ||||||
| Proceeds from sale of investments |
67,906 | 31,945 | 30,694 | |||||||||
| Purchase of property and equipment |
(4,498 | ) | (1,791 | ) | (1,045 | ) | ||||||
| Proceeds from sale of property and equipment |
350 | 161 | — | |||||||||
| Net cash provided by/(used in) investing activities |
20,650 | (63,734 | ) | 22,075 | ||||||||
| Cash flows from financing activities: |
||||||||||||
| Proceeds from the sale of common stock |
— | 109,000 | — | |||||||||
| Proceeds from exercise of common stock options |
1,994 | 433 | 88 | |||||||||
| Payments related to net settlement of employee stock-based awards |
(854 | ) | — | — | ||||||||
| Payments on notes payable and capital lease obligations |
(25,175 | ) | (18,430 | ) | (14,096 | ) | ||||||
| Net cash provided by/(used in) financing activities |
(24,035 | ) | 91,003 | (14,008 | ) | |||||||
| Decrease in cash and cash equivalents |
(10,052 | ) | (6,232 | ) | (43,404 | ) | ||||||
| Cash and cash equivalents, beginning of year |
52,256 | 58,488 | 101,892 | |||||||||
| Cash and cash equivalents, end of year |
$ | 42,204 | $ | 52,256 | $ | 58,488 | ||||||
| Supplemental disclosure of cash flow information: |
||||||||||||
| Cash paid for interest |
$ | 1,280 | $ | 2,794 | $ | 4,257 | ||||||
| Supplemental disclosure of non-cash investing information: |
||||||||||||
| Purchase of equipment included in accounts payable and accrued expenses |
$ | 1,641 | $ | 1,409 | $ | — | ||||||
The accompanying notes are an integral part of these financial statements.
F-5
Table of Contents
STATEMENTS OF STOCKHOLDERS’ EQUITY
(in thousands)
| Common Stock | Additional Paid-in Capital |
Accumulated Other Comprehensive Income/(loss) |
Accumulated Deficit |
Stockholders’ Equity |
||||||||||||||||||||
| Number of Shares |
Amount | |||||||||||||||||||||||
| Balance at December 31, 2007 |
56,501 | $ | 57 | $ | 400,460 | $ | 41 | $ | (308,865 | ) | $ | 91,693 | ||||||||||||
| Issuance of common stock—stock compensation plans |
171 | — | 88 | — | — | 88 | ||||||||||||||||||
| Unrealized loss on investments |
— | — | — | (234 | ) | — | (234 | ) | ||||||||||||||||
| Stock-based compensation |
— | — | 4,443 | — | — | 4,443 | ||||||||||||||||||
| Net loss |
— | — | — | — | (51,603 | ) | (51,603 | ) | ||||||||||||||||
| Balance at December 31, 2008 |
56,672 | 57 | 404,991 | (193 | ) | (360,468 | ) | 44,387 | ||||||||||||||||
| Issuance of common stock—stock compensation plans |
118 | — | 433 | — | — | 433 | ||||||||||||||||||
| Common Stock Offering |
25,556 | 25 | 108,975 | — | 109,000 | |||||||||||||||||||
| Unrealized gain on investments |
— | — | — | 261 | — | 261 | ||||||||||||||||||
| Stock-based compensation |
— | — | 5,063 | — | — | 5,063 | ||||||||||||||||||
| Net loss |
— | — | — | — | (39,976 | ) | (39,976 | ) | ||||||||||||||||
| Balance at December 31, 2009 |
82,346 | 82 | 519,462 | 68 | (400,444 | ) | 119,168 | |||||||||||||||||
| Issuance of common stock—stock compensation plans |
813 | 1 | 1,993 | — | — | 1,994 | ||||||||||||||||||
| Unrealized loss on investments |
— | — | — | (5 | ) | — | (5 | ) | ||||||||||||||||
| Stock-based compensation |
— | — | 10,382 | — | — | 10,382 | ||||||||||||||||||
| Payments related to net settlement of employee stock-based awards |
— | — | (854 | ) | — | — | (854 | ) | ||||||||||||||||
| Net loss |
— | — | — | — | (35,447 | ) | (35,447 | ) | ||||||||||||||||
| Balance at December 31, 2010 |
83,159 | $ | 83 | $ | 530,983 | $ | 63 | $ | (435,891 | ) | $ | 95,238 | ||||||||||||
The accompanying notes are an integral part of these financial statements.
F-6
Table of Contents
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share amounts)
1. Organization
Inspire Pharmaceuticals, Inc. (the “Company” or “Inspire”) was incorporated in October 1993 and commenced operations in March 1995. Inspire is located in Raleigh, North Carolina.
Inspire is a specialty pharmaceutical company focused on developing and commercializing ophthalmic products. The Company’s specialty eye care sales force generates revenue from the promotion of AzaSite (azithromycin ophthalmic solution) 1% for bacterial conjunctivitis and the co-promotion of Elestat (epinastine HCl ophthalmic solution) 0.05% for allergic conjunctivitis. The Company receives royalties based on net sales of Restasis (cyclosporine ophthalmic emulsion) 0.05% for dry eye and expects to begin receiving royalties in 2011 based on net sales of Diquas Ophthalmic Solution 3% (diquafosol tetrasodium) for dry eye in Japan.
Inspire has incurred losses and negative cash flows from operations since inception. Based on current operating plans, the Company expects it has sufficient liquidity to continue its planned operations beyond 2011. The Company’s liquidity needs will largely be determined by the commercial success of its products, key development and regulatory events and whether it successfully in-licenses or acquires additional products. The Company will continue to incur operating losses until revenues reach a level sufficient to support ongoing operations.
2. Summary of Significant Accounting Policies and Concentrations of Risk
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ materially from those estimates and assumptions.
Cash, Cash Equivalents, Interest and Other Receivables
The Company considers all highly-liquid investments with a maturity of three months or less at the date of purchase to be cash equivalents. The carrying values of cash, cash equivalents, interest and other receivables approximate their fair value due to the short-term nature of these items.
Trade Receivables
The Company’s trade receivables consist of co-promotion revenue based on net sales of Elestat and royalty revenue based on net sales of Restasis, both of which are earned from Allergan, Inc. (“Allergan”) and product revenue from sales of AzaSite. The Company is required to estimate the amount of trade receivables which ultimately will be uncollectible. The Company calculates an estimate of uncollectible accounts based on a review of specific customer balances, as well as a consideration of other industry and economic environment factors.
Investments
The Company invests in high-credit quality investments in accordance with its investment policy which minimizes the possibility of loss. Per its policy, the Company is able to invest in marketable debt securities that may consist of U.S. government and government agency securities, money market and mutual fund investments, municipal and corporate notes and bonds, commercial paper and asset or mortgage-backed securities, among
F-7
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
others. The Company’s investment policy requires it to purchase high-quality marketable securities with a maximum individual maturity of three years and requires an average portfolio maturity of no more than one year. Investments with original maturities at date of purchase beyond three months and which mature at or less than 12 months from the balance sheet date are classified as current. Investments with a maturity beyond 12 months from the balance sheet date are classified as long-term. Investments in marketable debt securities are classified as available-for-sale and are carried at fair value with unrealized gains and losses recognized in other comprehensive income (loss). Realized gains and losses are reflected in other income/(expense), net in the accompanying Statement of Operations and are determined using the specific identification method and transactions are recorded on a settlement date basis. Marketable and non-marketable equity investments are evaluated periodically for impairment. If it is determined that a decline of any investment is other than temporary, then the investment would be written down to fair value and the write-down would be included in the Company’s operating results.
The Company has an equity investment in Parion Sciences, Inc., a non-public entity for which its fair value is not readily determinable. For this investment in which the Company does not have significant influence and owns less than 5%, the investment is carried at cost and is subject to a write-down for impairment whenever events or changes in circumstances indicate that the carrying value may not be recoverable. As of December 31, 2010 and 2009, this investment’s recorded value was $200.
Property and Equipment
Property and equipment is primarily comprised of manufacturing and computer equipment, leasehold improvements, furniture and software which are recorded at cost and depreciated using the straight-line method over their estimated useful lives which range from three to seven years.
In March 2010, the Company and Finorga S.A.S. (“Novasep”) entered into a Technical Transfer & Development Services Agreement for the purpose of enabling Novasep to become a qualified manufacturer of the active pharmaceutical ingredients (“API”) for denufosol. Novasep is responsible for procuring, installing at one of its facilities, qualifying and maintaining certain equipment to be used for the manufacture of denufosol API. The manufacturing equipment is owned by the Company and will be used by Novasep for the manufacture of denufosol API. As of December 31, 2010, the Company had $3,844 of capitalized equipment, net, to be used for the manufacture of denufosol API.
The carrying values of property and equipment are periodically reviewed to determine if the facts and circumstances suggest that a potential impairment may have occurred. The review includes a determination of the carrying values of assets based on an analysis of undiscounted cash flows over the remaining depreciation period. If the review indicates that carrying values may not be recoverable, the Company will reduce the carrying values to the estimated fair value. See Note 7 “Property and Equipment” and Note 19 “Subsequent Events” for additional information.
Assets Held-For-Sale
In the fourth quarter of 2009, the Company began disposing of its laboratory equipment in connection with the corporate restructuring that occurred in the first quarter of 2009. The assets held-for-sale are reported at the lower of the carrying value or fair value, less costs to sell, and are no longer being depreciated. See Note 3 “Fair Value” and Note 6 “Restructuring” for additional information.
F-8
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
Restricted Deposits
Restricted deposits consist of cash and cash equivalents which collateralize letters of credit that are required under the terms of certain agreements to which the Company is involved. Restricted deposits are classified as current or long-term based upon the expected release date of such restriction. The carrying amount of these restricted deposits approximates fair value.
Intangible Assets
Costs associated with obtaining patents on the Company’s product candidates and license initiation and preservation fees, including milestone payments by the Company to its licensors, are evaluated based on the stage of development of the related product candidate and whether the underlying product candidate has an alternative use. Costs of these types incurred for product candidates not yet approved by the U.S. Food and Drug Administration (“FDA”) and for which no alternative future use exists are recorded as an expense. In the event a product candidate has been approved by the FDA or an alternative future use exists for a product candidate, patent and license costs are capitalized and amortized over the expected life of the related product candidate. Milestone payments to the Company’s collaborators are recognized when the underlying requirement is met.
Upon FDA approval of AzaSite in April 2007, the Company paid a $19,000 milestone payment to InSite Vision Incorporated (“InSite Vision”). The $19,000 is being amortized ratably on a straight-line basis through the term of the underlying patent coverage for AzaSite, or March 2019, which represents the expected period of commercial exclusivity. As of December 31, 2010 and 2009, the Company had $5,846 and $4,252, respectively, in accumulated amortization related to this milestone payment.
The carrying values of intangible assets are periodically reviewed to determine if the facts and circumstances suggest that a potential impairment may have occurred. The review includes a determination of the carrying values of intangible assets based on an analysis of undiscounted cash flows over the remaining amortization period. If the review indicates that carrying values may not be recoverable, the Company would record an impairment charge to reduce the carrying values to the estimated fair value. The Company had no impairments of its intangible assets for the years ended December 31, 2010, 2009 and 2008.
Deferred Rent
During 2010, the Company entered into a lease for its new headquarters to be located in Raleigh, NC. The lease term was effective on December 27, 2010 and ends on December 31, 2017. The lease also entitled the Company to receive a tenant improvement allowance of $1,298. The tenant improvement allowance was recorded as a deferred rent liability, which is included in other long-term liabilities on the accompanying balance sheet and will be amortized as a reduction of rent expense over the non-cancelable term of the lease.
The lease also provides for fixed non-contingent rent escalation and a rent free period. The Company will recognize rent expense on a straight line basis over the lease term. The difference between rent expense recorded and amounts paid under lease agreements will be recorded as deferred rent.
Revenue Recognition
The Company records all of its revenue from: (1) sales of AzaSite; (2) product co-promotion activities and earned royalties; and (3) collaborative research agreements, when realized or realizable and earned. Revenue is realized or realizable and earned when all of the following criteria are met: (i) persuasive evidence of an arrangement exists; (ii) delivery has occurred or services have been rendered; (iii) the seller’s price to the buyer is fixed or determinable; and (iv) collectibility is reasonably assured.
F-9
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
Product Revenues
The Company recognizes revenue for sales of AzaSite when title and substantially all the risks and rewards of ownership have transferred to the customer, which generally occurs on the date of shipment to wholesalers and distributors. In the United States, the Company sells AzaSite to wholesalers and distributors, who, in turn, sell to pharmacies and federal, state and commercial health care organizations.
Sales deductions consist of statutory rebates to state Medicaid, Medicare and other government agencies, contractual rebates with commercial managed care organizations, wholesaler chargebacks, sales discounts (including trade discounts and distribution service fees), allowances for coupon and voucher programs and product returns. These deductions are recorded as reductions to revenue from AzaSite in the same period as the related sales with estimates of future utilization derived from historical experience adjusted to reflect known changes in the factors that impact such reserves.
The Company utilizes data from external sources to help it estimate gross-to-net sales adjustments as they relate to the recognition of revenue for AzaSite sold. External sourced data includes, but is not limited to, information obtained from certain wholesalers with respect to their inventory levels and sell-through to customers, targeted surveys as well as data from IMS Health, a third-party supplier of market research data to the pharmaceutical industry. The Company also utilizes this data to help estimate and identify prescription trends and patient demand, as well as product levels in the supply chain.
The Company accounts for these sales deductions in accordance with the Financial Accounting Standards Board (“FASB”) authoritative guidance on revenue recognition when consideration is given by a vendor to a customer as well as when the right of return exists.
The Company has categorized and described more fully the following significant sales deductions, all of which involve estimates and judgments, which the Company considers to be critical accounting estimates, and requires it to use information from external sources.
Rebates and Chargebacks
Statutory rebates to state Medicaid agencies and Medicare and contractual rebates to commercial managed care organizations are based on statutory or negotiated discounts to AzaSite’s selling price. As it can take up to nine months or more for information to be received on actual usage of AzaSite in managed care and Medicaid and other governmental programs as well as on the total discounts to be reimbursed, the Company maintains reserves for amounts payable under these programs relating to AzaSite sales.
Chargebacks claimed by wholesalers are based on the differentials between product acquisition prices paid by wholesalers and lower government contract pricing paid by eligible customers covered under federally qualified programs.
The amount of the reserve for rebates and chargebacks is based on multiple qualitative and quantitative factors, including the historical and projected utilization levels, historical payment experience, changes in statutory laws and interpretations as well as contractual terms, product pricing (both normal selling prices and statutory or negotiated prices), changes in prescription demand patterns and utilization of the Company’s product through private or public benefit plans, and the levels of AzaSite inventory in both the wholesale and retail distribution channel. Other factors that the Company may consider, if determined relevant, would include price changes from competitors and introductions of generics and/or competitive new products. The Company acquires
F-10
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
prescription utilization data from IMS Health, a third-party supplier of market research data to the pharmaceutical industry. The Company applies these multiple factors, the quantitative historical data along with other qualitative aspects, such as management’s judgment regarding future utilization trends, to the respective period’s sales of AzaSite to determine the rebate accrual and related expense. The Company updates its estimates and assumptions each period and records any necessary adjustments to its reserves. Settlements of rebates and chargebacks typically occur within nine months from point of sale. To the extent actual rebates and chargebacks differ from the Company’s estimates, additional reserves may be required or reserves may need to be reversed, either of which would impact current period product revenue. As of December 31, 2010 and 2009, reserves for rebates and chargebacks were $9,085 and $3,488, respectively.
Discounts and Other Sales Incentives
Discounts and other sales incentives consist of the following:
| • | Prompt pay discounts—Prompt payment discounts are offered to all wholesalers in return for payment within 30 days following the invoice date. The Company records sales of AzaSite net of the discount amount based on historical experience. The Company adjusts the reserve at the end of each reporting period to approximate the percentage discount applicable to the outstanding gross accounts receivable balances. |
| • | Inventory Management Agreement (“IMA”) Fees—Per contractual agreements with the Company’s largest wholesalers, the Company provides an IMA fee based on a percentage of their purchases of AzaSite. The IMA fee rates are set forth in the individual contracts. The Company tracks sales to these wholesalers each period and accrues a liability relating to the unpaid portion of these fees by applying the contractual rates to such sales. |
| • | Product coupons and vouchers—Product coupons and vouchers, made available by us online or through pharmacies and prescribing physicians, offer patients the ability to receive free or discounted product. The Company uses a third-party administrator who coordinates program activities and invoices on a periodic basis for the cost of coupons and vouchers redeemed in the period. The Company bases its estimates on the historical coupon and voucher redemption rate of similar programs. |
As of December 31, 2010 and 2009, the Company’s reserves for discounts and other sales incentives were $1,159 and $889, respectively.
Product Returns
At the time of sale of AzaSite, the Company records product return allowances based on its estimate of the portion of sales that will be returned by its customers in the future. The return allowances are established in accordance with the Company’s returned goods policy. The Company’s returned goods policy generally allows for returns of AzaSite within an 18-month period, from six months prior to the expiration date and up to 12 months following the expiration date, but may differ from customer to customer, depending on certain factors. Future estimated returns of AzaSite are based primarily on the return data for comparative products and the Company’s own historical experience with AzaSite. Historical return data on AzaSite is analyzed on a specific production lot basis. In determining the Company’s return allowance, the Company also considers other relevant factors, including:
| • | Levels of inventory in the distribution channel and any significant changes to these levels; |
| • | Estimated expiration date or remaining shelf life of inventory in the distribution channel; |
F-11
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
| • | Current and projected demand of AzaSite that could be impacted by introductions of generic products and/or introductions of competitive new products; and |
| • | Competitive product shortages, recalls and/or discontinuances. |
The Company’s estimates of the level of AzaSite inventory in the distribution channel is based on inventory data provided by wholesalers and third-party prescription data. As of December 31, 2010 and 2009, reserves for returns of AzaSite were $2,590 and $1,523, respectively.
Product Co-promotion and Royalty Revenues
The Company recognizes co-promotion revenue based on net sales of Elestat and royalty revenue based on net sales of Restasis, as defined in the applicable agreements, and as reported to Inspire by Allergan, Inc. (“Allergan”). The Company’s co-promotion and royalty revenues are based upon Allergan’s revenue recognition policy and other accounting policies over which the Company has limited or no control and on the underlying terms of the agreements. Allergan recognizes revenue from product sales when goods are shipped and title and risk of loss transfers to the customer. The agreements provide for gross sales to be reduced by estimates of sales returns, credits and allowances, normal trade and cash discounts, managed care sales rebates and other allocated costs as defined in the agreements, all of which are determined by Allergan and are outside the Company’s control. The Company records a percentage of Allergan’s reported net sales to Inspire for Elestat and Restasis, as co-promotion revenue and royalty revenue, respectively. The Company receives monthly sales information from Allergan and performs analytical reviews and trend analyses using prescription information that it receives from IMS Health. In addition, the Company exercises its audit rights under the contractual agreements with Allergan to annually perform an examination of Allergan’s sales records of both Restasis and Elestat. The Company makes no adjustments to the amounts reported to it by Allergan other than reductions in net sales to reflect the incentive programs managed by the Company. The Company offers and manages certain incentive programs associated with Elestat, which are utilized by it in addition to those programs managed by Allergan. The Company reduces co-promotion revenue from net sales of Elestat by estimating the portion of sales that are subject to these incentive programs based on information reported to it by a third-party administrator of the incentive program. The rebates associated with the programs that the Company manages represent an insignificant amount, as compared to the rebate and discount programs administered by Allergan and as compared to the Company’s aggregate co-promotion and royalty revenue. Prior to January 1, 2010, under the co-promotion agreement for Elestat, the Company was obligated to meet predetermined minimum calendar year net sales target levels. If the annual minimum was not achieved, the Company recorded revenues using a reduced percentage of net sales based upon its level of achievement of the predetermined calendar year net sales target levels. Amounts receivable from Allergan in excess of recorded co-promotion revenue were recorded as deferred revenue. Calendar year 2009 was the last year in which there was a minimum annual net sales target level for Elestat under the co-promotion agreement. Accordingly, effective January 1, 2010, all co-promotion revenue from net sales of Elestat is recognized in the same period in which the sales occur.
Collaborative Research and Development Revenues
The Company recognizes revenue under its collaborative research and development agreements when the Company or its collaborative partner have met a contractual milestone triggering a payment to the Company. The Company is entitled to receive milestone payments under its collaborative research and development agreements based upon the achievement of agreed upon development events that are substantively at-risk by its collaborative partners or the Company. This collaborative research and development revenue is recognized upon the achievement and acknowledgement of the Company’s collaborative partner of a development event, which is
F-12
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
generally at the date payment is received from the collaborative partner or is reasonably assured. Accordingly, the Company’s revenue recognized under its collaborative research and development agreements may fluctuate significantly from period to period.
Research and Development
Research and development expenses include all direct costs and indirect development costs related to the development of the Company’s portfolio of product candidates. These expenses include: salaries for research and development personnel, consulting fees, clinical trial costs, including the development and manufacture of drug product for clinical trials, sponsored research costs, clinical trial insurance, up-front license fees and milestone payments relating to research and development as well as other fees and costs related to the development of product candidates. These costs have been charged to operating expense as incurred. License milestone payments to the Company’s licensors are recognized as expense when the underlying requirement is met or service has been provided.
Income Taxes
The Company accounts for income taxes using the liability method which requires the recognition of deferred tax assets or liabilities for the temporary differences between financial reporting and tax bases of the Company’s assets and liabilities and for tax carryforwards at enacted statutory tax rates in effect for the years in which the differences are expected to reverse. The effect on deferred taxes of a change in tax rates is recognized in income in the period that includes the enactment date. The Company accounts for uncertain tax positions in accordance with FASB authoritative guidance regarding the accounting for taxes. Significant management judgment is required in determining the provision for income taxes, deferred tax assets and liabilities and any valuation allowance recorded against net deferred tax assets. The Company has recorded a valuation allowance against all potential tax assets due to uncertainties in the Company’s ability to utilize the deferred tax assets, primarily consisting of certain net operating losses carried forward, before they expire. The valuation allowance is based on estimates of taxable income in each of the jurisdictions in which the Company operates and the period over which the deferred tax assets will be recoverable.
Stock-Based Compensation
The Company recognizes stock-based compensation expense in accordance with FASB authoritative guidance regarding the accounting for share-based payments, which requires that share-based payments be measured at fair value and recognized as compensation expense over the service period in which the awards are expected to vest. The Company utilizes the Black-Scholes option-pricing model to value its awards and recognizes compensation expense on a straight-line basis over the vesting periods of the awards. The Company considers many factors when estimating expected forfeitures, including types of awards, employee class, and historical experience. Expected volatility is determined based on the Company’s own historical volatility. The estimation of share-based payment awards that will ultimately vest requires judgment, and to the extent actual results or updated estimates differ from the Company’s current estimates, such amounts will be recorded as a cumulative adjustment in the period estimates are revised. Significant management judgment is required in determining estimates of future stock price volatility and forfeitures to be used in the valuation of the awards. Actual results, and future changes in estimates, may differ substantially from current estimates.
In accordance with SEC authoritative guidance, if a company concludes that its historical share option experience does not provide a reasonable basis upon which to estimate expected term, it may use the simplified
F-13
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
method to estimate the expected term of its options. The Company has utilized the simplified method to estimate expected term for share-based payment awards issued in the years ended December 31, 2010, 2009 and 2008. See Note 11 “Stock-Based Compensation” for additional information.
Other Income
During the year ended December 31, 2010, the Company was awarded $978 under the Federal Government’s Qualifying Therapeutic Discovery Project.
Net Loss Per Common Share
Basic net loss per common share is computed by dividing net loss attributable to common stockholders by the weighted average number of common shares outstanding during the period. Diluted net loss per common share is computed by dividing net loss attributable to common stockholders by the weighted average number of common shares outstanding and dilutive potential common shares then outstanding. Dilutive potential common shares consist of shares issuable upon the exercise of stock options and restricted stock units that are paid in shares of the Company’s stock upon conversion. The calculation of diluted earnings per share for the years ended December 31, 2010, 2009 and 2008 does not include 1,310, 261 and 183, respectively, of potential common shares, as their impact would be antidilutive.
Comprehensive Loss
Accumulated other comprehensive loss is comprised of unrealized gains and losses on marketable securities and is disclosed as a component of stockholders’ equity. At December 31, 2010 and 2009, the Company had $63 and $68, respectively, of unrealized gains on its investments.
Comprehensive loss consists of the following components for the years ended December 31,:
| 2010 | 2009 | 2008 | ||||||||||
| Net loss |
$ | (35,447 | ) | $ | (39,976 | ) | $ | (51,603 | ) | |||
| Adjustment for realized gain in other income/(expense), net |
(5 | ) | — | — | ||||||||
| Change in unrealized gain/(losses) on investments |
— | 261 | (234 | ) | ||||||||
| Total comprehensive loss |
$ | (35,452 | ) | $ | (39,715 | ) | $ | (51,837 | ) | |||
Advertising
The Company engages in general and direct-response advertising when promoting and marketing AzaSite and Elestat. These advertising costs are expensed as the costs are incurred. Advertising and product promotion expenses were $9,463, $9,666 and $12,314 for the years ended December 31, 2010, 2009 and 2008, respectively.
Significant Customers and Risk
The Company relies primarily on three pharmaceutical wholesalers to purchase and supply the majority of AzaSite at the retail level. These three pharmaceutical wholesalers accounted for greater than 85% of all AzaSite product sales in the years ended December 31, 2010, 2009 and 2008, and accounted for approximately 27% of the Company’s outstanding trade receivables as of December 31, 2010 and 2009. The loss of one or more of these wholesalers as a customer could negatively impact the commercialization of AzaSite. All co-promotion and
F-14
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
royalty revenues recognized and recorded were from one collaborative partner, Allergan. The Company is entitled to receive co-promotion revenue from net sales of Elestat and royalty revenue from net sales of Restasis under the terms of its applicable agreements with Allergan, and accordingly, all trade receivables for these two products are solely due from Allergan and accounted for 71% and 72% of the Company’s outstanding trade receivables as of December 31, 2010 and 2009, respectively. Due to the nature of these agreements, Allergan has significant influence over the commercial success of Restasis and Elestat.
Risk from Generic Competition
The Company’s revenues are subject to risk due to generic product entrants. The Elestat co-promotion agreement provides that unless earlier terminated, the term of such agreement will be in effect until the earlier of (i) the approval and launch of the first generic epinastine product after expiration of the FDA exclusivity period covering Elestat in the United States, or (ii) the approval and launch of the first over-the-counter epinastine product after expiration of the listing of Elestat in the FDA’s Orange Book. Following the termination of such co-promotion agreement, the Company will no longer have rights to co-promote Elestat. The Company will be entitled to receive post-termination payments from Allergan, based on any remaining net sales of Elestat for a period of 36 months.
The Company is aware that the following companies have filed an Abbreviated New Drug Application (“ANDA”) for a generic version of Elestat: Apotex, Inc., Cypress Pharmaceutical, Inc., Paddock Laboratories, Inc., PharmaForce, Inc. and Sandoz, Inc. The date of submission of the first ANDA filing to the FDA Office of Generic Drugs was October 14, 2008, according to the FDA’s website (www.fda.gov). Also, according to the FDA’s website, Apotex, Inc., PharmaForce, Inc. and Sandoz, Inc. have each received tentative approval for their respective epinastine hydrochloride ophthalmic solution. The Company does not know when the FDA will complete its review of these ANDAs, but it expects that a generic form of epinastine may be launched at any time.
The manufacture and sale of Restasis is protected in the United States by a formulation patent that expires in May 2014. While a formulation patent may afford certain limited protection, a competitor may attempt to gain FDA approval for a cyclosporine product using a different formulation. Furthermore, following the expiration of the formulation patent in 2014, a generic form of Restasis could be introduced into the market. If and when Restasis experiences competition from a cyclosporine product, including a generic cyclosporine product, the Company’s revenues attributable to Restasis may be significantly impacted.
Credit Risk
Cash equivalents and investments are financial instruments which potentially subject the Company to concentration of risk to the extent recorded on the balance sheet. The Company deposits excess cash with major financial institutions in the United States. Balances may exceed the amount of insurance provided on such deposits. Management of the Company believes it has established guidelines for investment of its excess cash relative to diversification and maturities that maintain safety and liquidity. To minimize the exposure due to adverse shifts in interest rates, the Company maintains a portfolio of investments with an average maturity of 12 months or less.
Risks from Third Party Manufacturing and Distribution Concentration
The Company relies on single source manufacturers for its commercial products. Allergan is responsible for the manufacture of both Restasis and Elestat and relies on single source manufacturers for the API in both
F-15
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
products. The Company relies on InSite Vision for the supply of the API for AzaSite, which InSite Vision obtains from a single source manufacturer. The Company is responsible for the remaining finished product manufacturing of AzaSite, for which it relies on a single source manufacturer. Additionally, the Company relies upon a single third party to provide warehousing and distribution services for AzaSite.
Delays in the manufacture or distribution of any product could adversely impact the marketing and sale of the Company’s products. Furthermore, the Company has no control over the manufacturing or the overall product supply chain of Restasis and Elestat.
Recent Accounting Pronouncements
In April 2010, the FASB issued authoritative guidance on defining a milestone and determining when it may be appropriate to apply the milestone method of revenue recognition for research or development transactions. Consideration that is contingent on achievement of a milestone in its entirety may be recognized as revenue in the period in which the milestone is achieved only if the milestone is judged to meet certain criteria to be considered substantive. Milestones should be considered substantive in their entirety and may not be bifurcated. An arrangement may contain both substantive and non substantive milestones, and each milestone should be evaluated individually to determine if it is substantive. The guidance is effective on a prospective basis for milestones achieved in fiscal years, and interim periods within those years, beginning on or after June 15, 2010, with early adoption permitted. The Company does not expect the adoption of this guidance to have a material impact on its consolidated financial statements.
3. Fair Value
The Company’s financial assets recorded at fair value on a recurring basis have been categorized based upon a fair value hierarchy. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities and the lowest priority to unobservable inputs. Specifically, Level 1 fair values are defined as observable inputs such as quoted prices in active markets; Level 2 fair values are defined as inputs other than quoted prices in active markets that are either directly or indirectly observable. These include quoted prices for similar assets or liabilities in active markets; quoted prices for identical or similar assets or liabilities in markets that are not active; and inputs to valuation models or other pricing methodologies that do not require significant judgment; Level 3 fair values are defined as inputs for which little or no market data exists, therefore requiring an entity to develop its own assumptions.
F-16
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
The following fair value hierarchy tables present information about the Company’s financial assets measured at fair value on a recurring basis as of December 31, 2010 and 2009:
| Total Balance | Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
||||||||||
| As of December 31, 2010: |
||||||||||||
| Cash equivalents |
$ | 27,433 | $ | 21,185 | $ | 6,248 | ||||||
| Investments—Available-for-sale securities: |
||||||||||||
| U.S. Treasury |
3,012 | 3,012 | — | |||||||||
| U.S. Government agencies |
9,535 | — | 9,535 | |||||||||
| Corporate bonds and commercial paper |
36,679 | — | 36,679 | |||||||||
| International Government |
2,006 | — | 2,006 | |||||||||
| Total |
$ | 78,665 | $ | 24,197 | $ | 54,468 | ||||||
| As of December 31, 2009: |
||||||||||||
| Cash equivalents |
$ | 41,052 | $ | 36,053 | $ | 4,999 | ||||||
| Investments—Available-for-sale securities: |
||||||||||||
| U.S. Treasury |
5,980 | 5,980 | — | |||||||||
| U.S. Government agencies |
12,658 | — | 12,658 | |||||||||
| Corporate bonds and commercial paper |
44,778 | — | 44,778 | |||||||||
| Negotiated certificates of deposit |
12,612 | — | 12,612 | |||||||||
| Total |
$ | 117,080 | $ | 42,033 | $ | 75,047 | ||||||
Level 1 cash equivalents consist of investments concentrated in money market funds which are primarily invested in U.S. Treasury securities. Level 2 cash equivalents and available-for-sale securities consist of investments in U.S. government agency securities, corporate bonds and commercial paper, international government securities and negotiated certificates of deposit. The fair value of these securities is derived using a market approach such as pricing models that rely on relevant observable market data including interest rates, yield curves, recently reported trades of comparable securities, and benchmark securities.
The Company’s investment policy dictates that investments in money market instruments are limited to those that have a rating of at least A-1 and P-1 according to Standard & Poor’s and Moody’s Investor Services, respectively. Likewise, for investments made in corporate obligations, the Company’s investment policy requires ratings of at least A and A-2 according to Standard & Poor’s and Moody’s Investor Services. The Company does not have any direct investments in auction-rate securities or securities that are collateralized by assets that include mortgages or subprime debt.
Level 3 Fair Valued Assets
As of December 31, 2009, the Company had laboratory equipment and related assets classified as held-for-sale that are fair valued on a non-recurring basis and are considered Level 3 valuations. During 2010, the Company recorded impairment charges of $218 to reduce the value of the laboratory equipment and related assets held-for-sale associated with the restructuring that occurred in 2009 to $0. The Company had previously recorded an impairment charge of $484 in the first quarter of 2009 to reflect the estimated fair value of the idle assets, based on internally established estimates and the selling prices of similar assets. The Company began marketing and disposing of all its laboratory equipment in the fourth quarter of 2009. As of December 31, 2010, the Company had substantially completed the sale of its laboratory equipment.
F-17
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
4. Investments
A summary of the fair market value of the Company’s investments by classification, as well as contractual maturities of marketable debt securities, is as follows:
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Available-for-sale debt securities: |
||||||||
| Less than one year |
$ | 47,510 | $ | 54,367 | ||||
| After one year through five years |
3,722 | 21,661 | ||||||
| Total available-for-sale debt securities |
$ | 51,232 | $ | 76,028 | ||||
| Preferred stock |
200 | 200 | ||||||
| Total Investments |
$ | 51,432 | $ | 76,228 | ||||
The following is a summary of the Company’s marketable debt securities which are classified as available-for-sale as of December 31, 2010 and 2009:
| Cost | Gross Unrealized Gain |
Gross Unrealized Loss |
Fair Value |
|||||||||||||
| As of December 31, 2010: |
||||||||||||||||
| Corporate bonds and commercial paper |
$ | 36,630 | $ | 54 | $ | (5 | ) | $ | 36,679 | |||||||
| U.S. Treasury |
3,012 | — | — | 3,012 | ||||||||||||
| U.S. Government agencies |
9,518 | 17 | — | 9,535 | ||||||||||||
| International Government |
2,007 | — | (1 | ) | 2,006 | |||||||||||
| Total |
$ | 51,167 | $ | 71 | $ | (6 | ) | $ | 51,232 | |||||||
| As of December 31, 2009: |
||||||||||||||||
| Corporate bonds and commercial paper |
$ | 44,702 | $ | 88 | $ | (12 | ) | $ | 44,778 | |||||||
| U.S. Treasury |
5,981 | — | (1 | ) | 5,980 | |||||||||||
| U.S. Government agencies |
12,664 | — | (6 | ) | 12,658 | |||||||||||
| Negotiated certificates of deposit |
12,612 | — | — | 12,612 | ||||||||||||
| Total |
$ | 75,959 | $ | 88 | $ | (19 | ) | $ | 76,028 | |||||||
The following table shows the gross unrealized losses and fair value of the Company’s investments in marketable debt securities with unrealized losses that are deemed to be temporarily impaired, aggregated by length of time that the individual securities have been in a continuous unrealized loss position as of December 31, 2010 and 2009:
| Less than 12 months | ||||||||
| Fair Value | Unrealized Loss | |||||||
| As of December 31, 2010: |
||||||||
| Corporate bonds and commercial paper |
$ | 12,319 | $ | (5 | ) | |||
| International Government |
2,006 | (1 | ) | |||||
| Total |
$ | 14,325 | $ | (6 | ) | |||
| As of December 31, 2009: |
||||||||
| Corporate bonds |
$ | 20,586 | $ | (12 | ) | |||
| U.S. Treasury |
5,980 | (1 | ) | |||||
| U.S. Government agencies |
9,158 | (6 | ) | |||||
| Total |
$ | 35,724 | $ | (19 | ) | |||
F-18
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
The contractual terms of these investments do not permit the issuer to settle the securities at a price less than the amortized cost of the investment. Because the Company has the ability and intent to hold its investments until a recovery of fair value, which may be at maturity, the Company does not consider its investments to be other-than-temporarily impaired at December 31, 2010. The Company did not have any investments in marketable debt securities that have been in a continuous unrealized loss position for 12 months or greater as of December 31, 2010 and 2009. In addition to the unrealized losses on its available-for-sale securities, as detailed above, the Company had an unrealized loss of $2 on a U.S. Government agency security that was classified as a cash equivalent as of December 31, 2010. The Company recognized a gain of $5 on the sale of a debt security during 2010 which was recorded in other income/(expense) net in the accompanying Statement of Operations. There were no realized gains or losses on the Company’s available-for-sale securities for the years ended December 31, 2009 and 2008.
As of December 31, 2010 and 2009, the Company had available-for-sale marketable debt securities with maturities of one year or less of $47,510 and $54,367, respectively. As of December 31, 2010 and 2009, the Company had available-for-sale marketable debt securities with maturities after one year through five years of $3,722 and $21,661, respectively.
5. Inventories
The Company’s inventories are related to AzaSite and are valued at the lower of cost or market using the first-in, first-out (i.e., FIFO) method. Cost includes materials, labor, overhead, shipping and handling costs. The Company’s inventories are subject to expiration dating and the Company has reserved for potential overstocking. The Company’s inventories consisted of the following:
| As of | ||||||||
| December 31, 2010 |
December 31, 2009 |
|||||||
| Finished Goods |
$ | 230 | $ | 808 | ||||
| Work-in-Process |
152 | 96 | ||||||
| Raw Materials |
444 | 838 | ||||||
| Total Inventories |
826 | 1,742 | ||||||
| Less Valuation Reserve |
(50 | ) | (25 | ) | ||||
| Total Inventories, net |
$ | 776 | $ | 1,717 | ||||
During the year ended December 31, 2010, the Company recorded a valuation reserve of approximately $253 primarily associated with spoilage of a portion of its azithromycin inventory, which is the API used to manufacture AzaSite and associated manufacturing costs. The Company subsequently wrote-off approximately $228 of API during the year ended December 31, 2010. During the years ended December 31, 2009 and 2008, the Company recorded valuation reserves of $15 and $105, respectively, for potential overstocking and short-dated product.
6. Restructuring
The Company restructured its operations during the first quarter of 2009, eliminating preclinical and drug discovery activities and refocusing its resources on the development of existing later-stage clinical programs and commercially available products. In connection with the restructuring, the Company recorded restructuring charges of $2,014 for both the year ended December 31, 2009. The Company recorded its restructuring activities in accordance with FASB authoritative guidance regarding the accounting for the impairment and disposal of long-lived assets and the accounting for costs associated with exit or disposal activities.
F-19
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
For the year ended December 31, 2009, the Company incurred the following restructuring charges:
| • | Employee separation costs of $1,072 consisted of one-time termination benefits, primarily severance costs, associated with the reduction in the Company’s workforce. |
| • | Asset impairments of $484 consisted of property and equipment, primarily lab equipment, that was used for discovery and preclinical research activities. The Company performed an impairment analysis and determined that the carrying value of the idle assets exceeded their fair value and recorded an impairment charge of $484. Fair value was based on internally established estimates and the selling prices of similar assets. |
| • | Contract charges of $203 consisted of costs associated with contractual commitments and work performed subsequent to the restructuring related to programs the Company no longer supported as part of its planned ongoing research and development activities. |
| • | Facility related charges of $255 consisted of estimated losses associated with leased lab space at the Company’s Durham, North Carolina headquarters that the Company no longer used in its operations. The unoccupied leased space is approximately 14,000 square feet and the lease term is through January 2011. The Company used a discounted cash-flow analysis to calculate the amount of this liability. The probability-weighted discounted cash-flow analysis is based on management’s assumptions and estimates of its ongoing lease obligations, including contractual rental commitments, build-out commitments and building operating costs, estimates of income from subleases, and market conditions for similar rental properties in its geographic area. The estimated cash flows were discounted using a credit-adjusted risk free rate of approximately 15%. The Company incurred approximately $45 of accretion expense over the term of the lease. |
As of December 31, 2009, the activities associated with the restructuring were substantially complete and the liabilities associated with employee separation costs and contractual commitments were fully paid. During 2010, the Company made all payments under the existing Durham, NC facility lease and recognized the remaining accretion expense. As a result, there were no outstanding liabilities associated with the 2009 restructuring as of December 31, 2010.
The following table sets forth activity in the restructuring liability for the years ended December 31, 2010 and 2009:
| Employee separation costs |
Facilities related charges |
Other restructuring charges |
Total | |||||||||||||
| Balance at December 31, 2008 |
$ | — | $ | — | $ | — | $ | — | ||||||||
| Accruals |
1,072 | 255 | 203 | 1,530 | ||||||||||||
| Payments |
(1,072 | ) | (140 | ) | (203 | ) | (1,415 | ) | ||||||||
| Balance at December 31, 2009 |
— | 115 | — | 115 | ||||||||||||
| Payments |
— | (115 | ) | — | (115 | ) | ||||||||||
| Balance at December 31, 2010 |
$ | — | $ | — | $ | — | $ | — | ||||||||
See Note 19 “Subsequent Events” for information regarding an additional restructuring in 2011.
F-20
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
7. Property and Equipment
Property and equipment consist of the following:
| Useful Life (Years) | December 31, | |||||||||
| 2010 | 2009 | |||||||||
| Equipment |
5 - 7 | $ | 7,071 | $ | 6,339 | |||||
| Leasehold improvements |
Lesser of lease term or useful life | 1,399 | 2,236 | |||||||
| Software |
5 | 1,326 | 1,165 | |||||||
| Furniture and fixtures |
7 | 1,068 | 836 | |||||||
| Computer hardware |
3 | 543 | 578 | |||||||
| 11,407 | 11,154 | |||||||||
| Less accumulated depreciation |
(3,511 | ) | (6,725 | ) | ||||||
| Property and equipment, net |
$ | 7,896 | $ | 4,429 | ||||||
As of December 31, 2010, the Company had vacated its previous headquarters located in Durham, NC and relocated to its new headquarters in Raleigh, NC. The Company wrote-off substantially all of the leasehold improvements and related accumulated depreciation associated with the Durham location, resulting in a loss of approximately $9. In addition, the Company received a tenant improvement allowance of $1,298 in connection with its lease of the new headquarters. This amount has been capitalized as leasehold improvements and will be depreciated over the new lease term.
As of December 31, 2010, the Company had $3,844 of capitalized equipment, net, to be used for the manufacture of denufosol API. See Note 19 “Subsequent Events” for additional information.
8. Accrued Expenses
Accrued expenses are comprised of the following:
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Allowances for discounts, rebates, chargebacks and returns |
$ | 12,834 | $ | 5,900 | ||||
| Compensation and benefits |
7,878 | 7,574 | ||||||
| Development costs |
5,917 | 4,605 | ||||||
| Fixed assets and equipment |
927 | 12 | ||||||
| Selling and marketing costs |
853 | 906 | ||||||
| Inventory |
638 | 893 | ||||||
| Professional fees |
464 | 314 | ||||||
| Duties and taxes |
279 | 226 | ||||||
| Accrued royalty |
229 | — | ||||||
| Other |
1,448 | 816 | ||||||
| $ | 31,467 | $ | 21,246 | |||||
As of December 31, 2010 and 2009, the carrying value of accrued expenses approximates fair value due to their short-term settlement.
F-21
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
9. Debt
In December 2006, the Company entered into a loan and security agreement with two participating financial institutions, which provided a term loan facility to the Company in an aggregate amount of $40,000. In June 2007, the Company amended the loan and security agreement with the two participating financial institutions to enable the Company to draw upon a new supplemental term loan facility in the amount $20,000, effectively increasing the total term loan facility to $60,000. The Company borrowed the full $60,000 available under the term loan facility. The interest rates associated with each of the borrowings under the facility ranged from approximately 7.6% to 8.0%.
The final maturity date for all loan advances under the original term loan facility and the supplemental term loan facility is March 2011. Interest accrues on the unpaid principal amount of each loan advance at a per annum rate equal to the five-year U.S. Treasury note yield plus a predetermined percentage at the time each advance is made. Repayment of each advance is made according to a schedule of six monthly installments of interest-only followed by equal monthly installments of principal and interest until the maturity date. The Company has the right to prepay the principal of any advance in minimum incremental amounts of $1,000. Each of the loan advances were subject to a 2% final payment, for a total of $1,200.
In December 2010, the Company repaid all of the remaining principal balance of the term loan facility, without penalty, including the final payment of $1,200 and any accrued and unpaid interest.
10. Stockholders’ Equity
Sales of Common Stock
In August 2009, the Company completed a public offering of 25,556 shares of its common stock at a price of $4.50 per share, which included the underwriter’s over-allotment allocation of an additional 3,333 shares, for gross proceeds of $115,000. Net proceeds were $109,000, after deducting underwriting discounts and estimated offering expenses, including reimbursing Warburg Pincus Private Equity IX, L.P. (“Warburg”), a related party, for $500 of expenses incurred. Warburg purchased $40,000, or 8,889 shares, of the common stock offering for total holdings of 22,907 shares of the Company’s common stock.
The holders of the Company’s common stock are entitled to receive dividends from time to time as may be declared by the Board of Directors, but a common stock dividend has never been declared, nor is a dividend payment expected in the near-term. The holders of shares of common stock are entitled to one vote for each share held with respect to all matters voted on by the stockholders of the Company.
Rights Agreement
In October 2002, the Company entered into a Rights Agreement with Computershare Trust Company. The Rights Agreement provides for a dividend of one preferred stock purchase right for each outstanding share of the
F-22
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
Company’s common stock. Each right entitles a stockholder, after the rights become exercisable, to buy 1/1,000th of a share of Inspire’s Series H Preferred Stock at an exercise price of $50. Each right will become exercisable following the tenth day after an acquiring person or group acquires, or announces its intention to acquire, 15% or more of the common stock. The Company will be entitled to redeem the rights at $0.001 per right at any time on or before the close of business on the tenth day following acquisition by a person or group of 15% or more of the common stock. Under the Rights Agreement, if a person acquires 15% or more of the common stock without the approval of the Company’s Board of Directors, all other stockholders will have the right to purchase securities from the Company at a price that is less than its fair market value, which would substantially reduce the value of the common stock owned by the acquiring person. As a result, the rights will cause substantial dilution to a person or group that attempts to acquire the Company on terms not approved by the Company’s Board of Directors, except pursuant to an offer conditioned on a substantial number of Rights being acquired. The rights should not interfere with any merger or other business combination approved by the Board of Directors since the rights may be redeemed by the Company at the redemption price of $0.001 prior to the occurrence of a distribution date. In connection with the transaction with Warburg, the Company and Computershare entered into a First Amendment to Rights Agreement dated July 17, 2007. The First Amendment to Rights Agreement provides that Warburg and its affiliates will be exempt from the definition of an “Acquiring Person” under the Rights Agreement, unless Warburg or certain of its affiliates becomes the beneficial owner of the lesser of: (x) 32.5% of the Company’s voting securities on a fully diluted basis and (y) 34.9% of the Company’s then outstanding voting securities.
11. Stock-Based Compensation
The Company accounts for stock-based compensation in accordance with FASB authoritative guidance regarding share-based payments. Total stock-based compensation was allocated as follows:
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Research and development |
$ | 1,387 | $ | 1,295 | $ | 1,319 | ||||||
| Selling and marketing |
2,035 | 1,849 | 1,402 | |||||||||
| General and administrative |
6,960 | 1,919 | 1,722 | |||||||||
| Total stock-based compensation expense |
$ | 10,382 | $ | 5,063 | $ | 4,443 | ||||||
Equity Compensation Plans
The Company’s stock-based compensation plan, the Amended and Restated 2010 Equity Compensation Plan (the “2010 Plan”), allows for the granting of both incentive and non-qualified stock options, stock appreciation rights, restricted stock and restricted stock units to directors, officers, employees and consultants. At December 31, 2010, there were 7,646 shares available for grant as options, deferred stock units (“DSUs”), restricted stock units (“RSUs”) or other forms of share-based payments under the 2010 Plan.
The Board of Directors, or an appropriate committee of the Board of Directors, determines the terms of all options and other equity arrangements under both plans. The maximum term for any option grant under the 2010 Plan is seven years from the date of the grant. Options granted to executive employees vest 25% upon completion of one full year of employment from date of grant and on a monthly basis over the following three years of their employment and have a term of seven years. Options granted to non-executive employees vest 33% upon completion of one full year of employment from date of grant and on a monthly basis over the following two
F-23
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
years of their employment and have a term of five years. Options granted to members of the Board of Directors vest on a quarterly basis over one year and have a term of seven years. The vesting period typically begins on the date of hire for new employees and on the date of grant for existing employees.
CEO Transition
In February 2010, the Company’s CEO, Christy Shaffer, resigned from the Company and Adrian Adams was hired as CEO. As part of this transition, certain existing stock options and DSUs previously granted to Dr. Shaffer were modified. In addition, Dr. Shaffer was also granted 100 stock options which vested immediately and a stock award for 100 shares of the Company’s common stock. The modification of the existing options and the granting of the new awards to Dr. Shaffer resulted in stock-based compensation expense of $2,230 during the year ended December 31, 2010.
As part of his hiring, Mr. Adams was granted 350 stock options. On the date of grant, 25% of these options vested immediately and the remaining 75% will vest ratably over the 36 months following the first anniversary of the effective date of Mr. Adams’ employment. In addition, Mr. Adams was granted an award of 650 RSUs. On the date of grant, 25% of these RSUs became non-forfeitable or vested and the remaining 75% will become non-forfeitable ratably over the 36 months following the first anniversary of the effective date of his employment. The total value of these awards to Mr. Adams was $5,199, of which $1,300 was expensed during the year ended December 31, 2010.
Additional Sign-On Grants
On May 10, 2010, the Company hired Andrew I. Koven as Executive Vice President and Chief Administrative and Legal Officer. As part of his hiring, Mr. Koven was granted 200 stock options. On the date of grant, 25% of these options vested immediately and the remaining 75% will vest ratably over the 36 months following the first anniversary of the effective date of Mr. Koven’s employment. In addition, Mr. Koven was granted an award of 500 RSUs. On the date of grant, 50% of these RSUs became non-forfeitable or vested and the remaining 50% will become non-forfeitable ratably over the 36 months following the first anniversary of the effective date of his employment. The total value of these awards to Mr. Koven was $3,725, of which $1,686 was expensed during the year ended December 31, 2010.
Basis for Fair Value Estimate of Share-Based Payments
Stock Options
The Company uses its own historical volatility to estimate its future volatility. Actual volatility, and future changes in estimated volatility, may differ substantially from the Company’s current estimates.
The Company utilizes the simplified method of calculating the expected life of options for grants made to its employees under the 2010 Plan due to the lack of adequate historical data with regard to exercise activity on options. For options granted to directors under the 2010 Plan, the Company uses the contractual term of seven years as the expected life of options. The Company will continue with these assumptions in determining the expected life of options under the 2010 Plan until such time that adequate historical data is available. The Company estimates the forfeiture rate based on its historical experience. These estimates will be revised in future periods if actual forfeitures differ from the estimate. Changes in forfeiture estimates impact compensation cost in the period in which the change in estimate occurs.
F-24
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
The table below presents the weighted average expected life of options granted during the period indicated. The risk-free rate of the stock options is based on the U.S. Treasury yield curve in effect at the time of grant, which corresponds with the expected term of the option granted. The fair value of share-based payments, granted during the period indicated, was estimated using the Black-Scholes option pricing model with the following assumptions and weighted average fair values as follows:
| Stock Options for Year Ended December 31, |
||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Risk-free interest rate |
1.47 | % | 1.73 | % | 2.43 | % | ||||||
| Dividend yield |
0 | % | 0 | % | 0 | % | ||||||
| Expected volatility |
69 | % | 71 | % | 65 | % | ||||||
| Expected life of options (years) |
3.9 | 3.9 | 3.9 | |||||||||
| Weighted average fair value of grants (per option) |
$ | 3.12 | $ | 2.34 | $ | 2.01 | ||||||
The following table summarizes the stock option activity for the year ended December 31, 2010:
| Number of Shares |
Weighted Average Exercise Price (per share) |
Weighted Average Remaining Contractual Term (in Years) |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding at December 31, 2009 |
11,001 | $ | 7.47 | 3.5 | $ | 6,503 | ||||||||||
| Granted |
3,262 | 6.02 | ||||||||||||||
| Exercised |
(439 | ) | (4.54 | ) | ||||||||||||
| Forfeited/cancelled/expired |
(985 | ) | (10.94 | ) | ||||||||||||
| Outstanding at December 31, 2010 |
12,839 | $ | 6.94 | 3.1 | $ | 32,183 | ||||||||||
| Vested and exercisable at December 31, 2010 |
8,741 | $ | 7.60 | 2.4 | $ | 20,349 | ||||||||||
| Expected to vest at December 31, 2010 |
12,666 | $ | 6.95 | 3.1 | $ | 31,733 | ||||||||||
Total intrinsic value of stock options exercised for the years ended December 31, 2010, 2009 and 2008 was $1,163, $174 and $534, respectively. Due to the Company’s net loss position, no windfall tax benefits have been realized during the year ended December 31, 2010. As of December 31, 2010, approximately $10,852 of total unrecognized compensation cost related to unvested stock options is expected to be recognized over a weighted-average period of 2.4 years.
The following table summarizes information concerning options outstanding at December 31, 2010:
| Options Outstanding |
Weighted Average Exercise Price (per share) |
Weighted Average Remaining Contractual Term (in Years) |
Options Exercisable |
|||||||||||||
| Exercise Price range (per share): |
||||||||||||||||
| $ 2.25 - $ 4.44 |
2,724 | $ | 3.83 | 2.7 | 2,048 | |||||||||||
| $ 4.47 - $ 5.25 |
2,963 | 4.95 | 3.7 | 1,684 | ||||||||||||
| $ 5.27 - $ 6.35 |
2,941 | 6.02 | 3.5 | 1,823 | ||||||||||||
| $ 6.36 - $11.65 |
2,582 | 7.80 | 2.8 | 1,557 | ||||||||||||
| $12.25 - $20.30 |
1,629 | 16.02 | 2.7 | 1,629 | ||||||||||||
| 12,839 | $ | 6.94 | 3.1 | 8,741 | ||||||||||||
F-25
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
Deferred Restricted Stock Units
In July 2006, the Compensation Committee authorized the issuance of DSUs to each of the Company’s then executive officers. The value of DSUs granted was based on the closing market price of the Company’s common stock on the date of grant and is amortized on a straight-line basis over the five year requisite service period. A total of 195 DSUs were granted and had a total fair value at the date of grant of $811. The DSUs vest 20% annually over five years from the date of grant. Any DSUs that have not vested at the time of termination of service to the Company are forfeited. The DSUs do not have voting rights, and the shares underlying the DSUs are not considered issued and outstanding until conversion. Any vested units will convert into an equivalent number of shares of common stock upon termination of employment with the Company.
The following table summarizes the DSU activity for the year ended December 31, 2010:
| Number of Shares |
Weighted Average Grant Date Fair Value (per share) |
|||||||
| Nonvested Awards at December 31, 2009 |
70 | $ | 4.16 | |||||
| Vested |
(45 | ) | (4.16 | ) | ||||
| Forfeited |
(9 | ) | (4.16 | ) | ||||
| Nonvested Awards at December 31, 2010 |
16 | $ | 4.16 | |||||
| Vested and deferred awards at December 31, 2010 |
150 | $ | 4.16 | |||||
During the year ended December 31, 2010, approximately 45 DSUs with an aggregate fair value of $214 became vested. DSUs outstanding at December 31, 2010 had a weighted-average remaining contractual life of 0.6 years and a total aggregate intrinsic value of $1,394.
Time-Based Restricted Stock Units
Beginning in 2010, the Compensation Committee began approving grants of RSUs to the Company’s executive officers and to certain other employees. In general, the RSUs vest on an annual basis over 4 years. Any RSUs that have not vested at the time of termination of service to the Company are forfeited. The RSUs do not have voting rights, and the shares underlying the RSUs are not considered issued and outstanding until vested and released. The Company also began granting RSUs to its Board of Directors which vest annually over one year.
The following table summarizes the time-based RSU activity for the year ended December 31, 2010:
| Number of Shares |
Weighted Average Grant Date Fair Value (per share) |
|||||||
| Outstanding Awards at December 31, 2009 |
— | $ | — | |||||
| Granted |
1,418 | 5.95 | ||||||
| Vested |
(414 | ) | (6.05 | ) | ||||
| Forfeited |
(3 | ) | (6.29 | ) | ||||
| Outstanding and nonvested Awards at December 31, 2010 |
1,001 | $ | 5.91 | |||||
F-26
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
During the year ended December 31, 2010, approximately 414 RSUs with an aggregate fair value of $2,507 became vested. RSUs outstanding at December 31, 2010 had a weighted-average remaining contractual life of 1.7 years and a total aggregate intrinsic value of $8,405. RSUs outstanding and expected to vest at December 31, 2010 had a weighted-average remaining contractual life of 1.7 years and a total aggregate intrinsic value of $7,984.
Stock Awards
As discussed earlier, during the year ended December 31, 2010, 100 stock awards were granted to Dr. Shaffer as part of the CEO transition. The 100 stock awards were issued, fully vested upon grant, with an aggregate fair value of $627.
12. Income Taxes
The Company had no federal, state or foreign income tax expense for the years ended December 31, 2010, 2009 and 2008.
Significant components of the Company’s deferred tax assets and liabilities consist of the following:
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Current deferred tax assets: |
||||||||
| Compensation related items |
$ | 2,887 | $ | 307 | ||||
| Accrued expenses and other |
1,362 | 508 | ||||||
| Noncurrent deferred tax assets: |
||||||||
| Accrued expenses and other |
879 | 861 | ||||||
| Domestic net operating loss carryforwards |
128,798 | 125,234 | ||||||
| Research and development credits |
48,009 | 38,660 | ||||||
| Property, equipment and intangible assets |
7,047 | 7,666 | ||||||
| Stock-based compensation |
5,752 | 4,255 | ||||||
| Contributions |
392 | 434 | ||||||
| Investments |
3 | 234 | ||||||
| Total deferred tax assets |
195,129 | 178,159 | ||||||
| Valuation allowance |
(195,129 | ) | (178,159 | ) | ||||
| Deferred tax assets |
$ | — | $ | — | ||||
At December 31, 2010 and 2009, the Company provided a full valuation allowance against its net deferred tax assets since realization of these benefits could not be reasonably assured. The valuation allowance has increased $16,970, $19,629 and $25,321 for the years ended December 31, 2010, 2009 and 2008, respectively. The increase in the valuation allowance of $16,970 during the year ended December 31, 2010 resulted primarily from the generation of additional net operating loss carryforwards and research and development credits, partially offset by a reduction in deferred tax assets related to 2010 and prior years of $2,405 that are unlikely to be realized.
As of December 31, 2010, the Company had federal and state net operating loss carryforwards of $319,605 and $377,614, respectively. The net operating loss carryforwards expire in various amounts starting in 2009 and 2010 for federal and state tax purposes, respectively. Net operating loss carryforwards that expired in 2009 and 2010 and that are set to expire in 2011 are $308, $2,443 and $5,450, respectively. The utilization of the federal net operating loss carryforwards may be subject to limitation under the rules regarding a change in stock
F-27
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
ownership as determined by the Internal Revenue Code. If the Company’s utilization of its net operating loss carryforwards is limited and the Company has taxable income which exceeds the permissible yearly net operating loss carryforward, the Company would incur a federal income tax liability even though its net operating loss carryforwards exceed its taxable income. Additionally, as of December 31, 2010 and 2009, the Company had federal research and development and orphan drug credit carryforwards of $48,009 and $38,660, respectively. The research and development carryforwards and orphan drug carryforwards begin to expire in varying amounts starting in 2011 and 2013, respectively. There are no orphan drug carryforwards set to expire in 2011 and the carryforwards related to research and development set to expire in 2011 are insignificant.
On January 1, 2007, the Company adopted the provisions of FASB authoritative guidance related to accounting for uncertainty in income taxes. The guidance prescribes a recognition threshold and a measurement attribute for the financial statement recognition and measurement of tax positions taken or expected to be taken in a tax return. For those benefits to be recognized, a tax position must be more-likely-than-not to be sustained upon examination by taxing authorities. The amount recognized is measured as the largest amount of benefit that is greater than 50 percent likely of being realized upon ultimate settlement. The Company does not expect its unrecognized tax benefits to change significantly over the next 12 months.
A reconciliation of the beginning and ending amount of unrecognized tax benefits is as follows:
| 2010 | 2009 | 2008 | ||||||||||
| Balance at January 1 |
$ | 10,923 | $ | 8,388 | $ | 6,651 | ||||||
| Additions to current year tax positions |
2,428 | 2,442 | 1,689 | |||||||||
| Additions to tax positions of prior years |
161 | 93 | 48 | |||||||||
| Reductions for tax provisions of prior years |
(184 | ) | — | — | ||||||||
| Balance at December 31 |
$ | 13,328 | $ | 10,923 | $ | 8,388 | ||||||
All of the Company’s unrecognized tax benefits of $13,328 as of December 31, 2010, would, if recognized, reduce the Company’s effective tax rate; however, currently all of the Company’s deferred tax assets are subject to a full valuation allowance. The Company has no current pending or open tax examinations or audits. The Company is subject to tax examinations by U.S. Federal and state and local authorities for tax years subsequent to 2004. However, the net operating loss carryforwards and various research and development credits dating back to 1993 are open to adjustment by the taxing authorities.
Taxes computed at the statutory federal income tax rate of 35% are reconciled to the provision for income taxes as follows:
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| U.S. Federal tax at statutory rate |
$ | (12,406 | ) | $ | (13,992 | ) | $ | (20,685 | ) | |||
| State taxes (net of Federal benefit) |
(1,449 | ) | (1,619 | ) | (2,140 | ) | ||||||
| Change in valuation reserve |
16,970 | 19,629 | 25,321 | |||||||||
| Research and development credit |
(9,348 | ) | (9,660 | ) | (6,395 | ) | ||||||
| Net operating loss expiration |
(2,443 | ) | (308 | ) | (22 | ) | ||||||
| Reversal of the benefit booked in prior years |
3,298 | 415 | 29 | |||||||||
| Nondeductible expenses due to credits |
(323 | ) | 56 | (3 | ) | |||||||
| Other nondeductible expenses |
5,701 | 5,479 | 3,895 | |||||||||
| Provision for income taxes |
$ | — | $ | — | $ | — | ||||||
F-28
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
13. Collaboration Agreements
Allergan, Inc.
Elestat
In December 2003, the Company entered into an agreement with Allergan to co-promote Elestat in the United States. Under the agreement, Inspire has the responsibility for promoting and marketing Elestat to ophthalmologists, optometrists and allergists in the United States and paying the associated costs. Inspire receives co-promotion revenue from Allergan on its U.S. net sales of Elestat. Allergan records sales of Elestat and is responsible for supply chain management, managed health care, customer order processing and regulatory compliance.
The Elestat co-promotion agreement provides that unless earlier terminated, the term of such agreement will be in effect until the earlier of (i) the approval and launch of the first generic epinastine product after expiration of the FDA exclusivity period covering Elestat in the United States, or (ii) the approval and launch of the first over-the-counter epinastine product after expiration of the listing of Elestat in the FDA’s Orange Book. As stated earlier, a generic form of epinastine may be launched at any time. Following the termination of such co-promotion agreement, Inspire will no longer have rights to co-promote Elestat. Inspire will be entitled to receive post-termination payments from Allergan, based on any remaining net sales of Elestat for a period of 36 months. During the three successive 12-month periods immediately following the termination of the agreement, Allergan will be obligated to pay to Inspire 20%, 15% and 10%, respectively, on any net sales of Elestat in the United States.
Several companies have filed an ANDA for a generic version of Elestat. The date of submission of the first ANDA filing to the FDA Office of Generic Drugs was October 14, 2008, according to the FDA’s website (www.fda.gov). Also, according to the FDA’s website, Apotex, Inc., PharmaForce, Inc. and Sandoz, Inc. have each received tentative approval for their respective epinastine hydrochloride ophthalmic solution.
Either Allergan or Inspire may terminate the agreement in the event of a material breach of the agreement by the other or in the event of the other’s insolvency. Allergan can terminate the agreement upon a change of control where Inspire becomes an affiliate of a direct competitor of Allergan as that term is defined in the agreement. Inspire can terminate the agreement in the event that Elestat is withdrawn from the market for more than 90 days.
Restasis and Prolacria
In June 2001, the Company entered into a joint license, development and marketing agreement with Allergan to develop and commercialize the Company’s product candidate, Prolacria. The agreement also provided the Company with revenue on net sales of Allergan’s Restasis. The agreement was amended and restated in August 2010. Under the amended and restated agreement, which runs through December 31, 2020, the Company is entitled to receive revenues at one global rate based on net sales of Restasis and any other human ophthalmic formulation of cyclosporine owned or controlled by Allergan, with no requirement to co-promote Restasis. Effective January 1, 2011, the worldwide rate steps down from the 2010 rate by three percentage points. The rate will step down a further 0.25 percentage point in 2013 and a final 0.50 percentage point in 2014, remaining at this level through the end of the term in 2020.
Additionally, the Company has sole control over any future Prolacria development and commercialization. The Company is not currently proceeding with the clinical development of Prolacria. In the event the Company
F-29
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
resumes the Prolacria clinical development program and receives regulatory approval for a Prolacria product in a particular country, the Company will have the option to offer Prolacria commercialization rights to Allergan for such country upon the original commercial terms previously agreed upon. If the Company chooses not to offer Allergan Prolacria commercialization rights with respect to a country, the Company will receive all the commercialization revenues related to Prolacria in such country and its rights to receive royalties from Allergan based on net sales of Restasis products in such country will terminate.
Unless earlier terminated pursuant to other terms of the agreement, the agreement will terminate on December 31, 2020 and, therefore, Allergan will be obligated to make applicable net sales payments on Restasis and any other human ophthalmic formulations of cyclosporine owned or controlled by Allergan until December 31, 2020.
Cystic Fibrosis Foundation Therapeutics, Inc.
In October 2002, the Company entered into a study funding agreement with the Cystic Fibrosis Foundation Therapeutics, Inc. (“CFFT”), whereby the majority of the expenses for one Phase 2 clinical trial for denufosol for the treatment of cystic fibrosis were funded by the CFFT, but the Company also recorded the corresponding expenses and liabilities as the CFFT incurred these costs. This clinical trial was completed in 2004. If the Company receives FDA approval for denufosol for the treatment of cystic fibrosis, the Company will be obligated to pay a development milestone, and possibly a sales milestone, to the CFFT. The aggregate potential milestones under this agreement are approximately $15,488. The agreement will immediately terminate if the Company’s ongoing development efforts of denufosol for the treatment of cystic fibrosis fail to yield statistically significant results. The Company, in its sole discretion, may elect to cease development efforts following such failure. In addition, either the CFFT or the Company may terminate the agreement if the other materially breaches the agreement.
The Company has recorded a $1,915 liability associated with this agreement in other long-term liabilities in the accompanying Balance Sheets as of December 31, 2010 and 2009. If the agreement terminates upon denufosol’s failure to yield statistically significant results in a trial or denufosol does not receive FDA approval for the treatment of cystic fibrosis, the Company will have no financial obligation to the CFFT, including the Phase 2 clinical trial costs the CFFT funded on the Company’s behalf. See Note 19 “Subsequent Events” for additional information.
InSite Vision Incorporated.
In February 2007, the Company entered into a license agreement with InSite Vision pursuant to which Inspire acquired exclusive rights to commercialize AzaSite, as well as other potential topical anti-infective products containing azithromycin as the sole active ingredient for use in the treatment of human ocular or ophthalmic indications. The license agreement also grants Inspire exclusive rights to develop, make, use, market, commercialize and sell each product in the United States and Canada. Inspire is currently responsible for all regulatory obligations and strategies relating to the further development and commercialization of a product in the United States and Canada.
Pursuant to the license agreement, the Company paid an upfront licensing fee of $13,000. The Company paid an additional $19,000 milestone payment upon regulatory approval of AzaSite by the FDA. In addition, the Company paid a 20% royalty for the first two years of commercialization and in July 2009 began paying a 25% royalty on net sales of AzaSite in the United States and Canada, which will continue for the duration of the agreement. The Company is obligated to pay royalties under the agreement for the longer of (i) 11 years from the
F-30
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
launch of the subject product and (ii) the period during which a valid claim under a patent licensed from InSite Vision covers a subject product. Under the terms of the agreement, the Company’s obligation to pay royalties to InSite Vision is subject to pre-determined minimum annual royalty payments. The determination of whether or not the Company will owe any such payments is based upon the amount of royalties accrued over a 12-month royalty period. There are five successive 12-month minimum royalty periods, the third of which commenced on October 1, 2010. The Company launched AzaSite in August 2007, and began paying royalties to InSite Vision in the fourth quarter of 2007.
Santen Pharmaceuticals Co., Ltd.
In December 1998, the Company entered into a development, license and supply agreement with Santen Pharmaceutical Co., Ltd (“Santen”). The terms of the agreement allow Santen to develop diquafosol tetrasodium for the therapeutic treatment of ocular surface diseases, such as dry eye disease, in Japan, China, South Korea, the Philippines, Thailand, Vietnam, Taiwan, Singapore, Malaysia and Indonesia (the “Santen Territory”). The agreement provides for certain payments to be paid to Inspire upon achievement of development milestones by Santen and for royalties relating to the net sales of products containing diquafosol tetrasodium developed by Santen. Santen is responsible for all development, regulatory submissions, filings and approvals, and all marketing of potential products in its territory.
Under the terms of the agreement, Santen has developed a formulation of diquafosol, known as Diquas ophthalmic solution 3%, which received regulatory approval from the Japanese Ministry of Health in April 2010 and received pricing approval and was launched in December 2010.
In June 2010, the Company amended the agreement to relieve Inspire of its manufacturing obligations with respect to the supply of diquafosol tetrasodium API, and to grant to Santen expanded rights allowing it to manufacture, or have manufactured, the API throughout the world for use in products in the Santen Territory. In connection therewith, the licensed technology was expanded to include know-how relating to manufacturing. Royalty rates relating to net sales of products, including Diquas, in Japan were amended to a tiered royalty range with a minimum rate in the high single digits and a maximum rate in the low double digits. In addition, the royalty rate with respect to net sales, if any, of any product approved for sale in the Santen Territory other than Japan were amended to a single digit royalty rate.
Under the terms of the agreement, Inspire has received a total of $1,500 in equity and $4,250 in milestone payments, including $1,250 milestone payments received in both May 2008 and in December 2010. There are no further milestones to be earned under the amendment.
The agreement will terminate when all patents licensed under the agreement have expired. Either Santen or the Company may terminate the agreement if the other materially breaches the agreement. In addition, the Company has the right to terminate the agreement at any time if it determines, subject to a coordinating committee’s review and arbitration, that Santen has not made reasonably sufficient progress in the development or commercialization of potential products. If Santen breaches the agreement, or if the Company terminates the agreement because Santen has not made sufficient progress, Santen’s license will terminate. Santen will provide the Company with all data and information relating to the Company’s products, and will assign or permit the Company to cross-reference all regulatory filings and approvals; provided, however, that Santen is not required to provide the Company with any data or information arising after the agreement which relate solely to the manufacture of the API.
F-31
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
Wisconsin Alumni Research Foundation
In November 2004, the Company licensed several patents for use in developing and commercializing new treatments for glaucoma from the Wisconsin Alumni Research Foundation (“WARF”). The Company is obligated for contingent payments of up to an aggregate of $1,750 upon the achievement of development milestones, and royalties on sales of any regulatory approved product utilizing the licensed patents.
Inspire will design and fund all future research, development, testing, regulatory filings and potential marketing activities related to any product candidate under development or product developed from the license. Unless terminated earlier, the agreement will expire on a country-by-country basis upon the expiration of the patents in such country. The U.S. government may have limited rights in some of this patented technology. WARF may terminate the license if Inspire fails to make timely payment of any amount due to WARF under the agreement or commit a material breach of any material covenant contained in the agreement, subject to the right to cure.
14. Commitments and Contingencies
Operating Leases
Total rent expense for operating leases during 2010, 2009 and 2008 was $1,907, $1,869 and $2,098, respectively. Future minimum lease payments under non-cancelable operating leases as of December 31, 2010 are as follows:
| Year Ending December 31, |
Operating Leases |
|||
| 2011 |
$ | 817 | ||
| 2012 |
1,089 | |||
| 2013 |
981 | |||
| 2014 |
944 | |||
| 2015 |
957 | |||
| Thereafter |
1,965 | |||
| Total minimum lease payments |
$ | 6,753 | ||
During 2010, the Company entered into a non-cancelable lease for its new headquarters located in Raleigh, NC. The lease term is seven years with two renewable periods of three years each and was effective December 27, 2010. The lease also provides for fixed non-contingent rent escalations as well as a rent free period. The Company will recognize rent expense on a straight-line basis over the non-cancelable lease term. The difference between rent expense recorded and amounts paid under lease agreements will be recorded as deferred rent.
The Company has entered into non-cancelable operating leases for its fleet of vehicles and office equipment that extend through 2013 and are subject to voluntary renewal options. The Company leases vehicles for its commercial organization under a Master Lease Agreement that allows for individual vehicle leases to be cancelable after one year. The Master Lease Agreement requires the Company to maintain a Standby Letter of Credit in the amount of $515 during the term of the lease. The vehicle Master Lease Agreement also requires that the vehicles under lease serve as collateral for the obligation.
F-32
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
Other Commitments
The Company has entered into contractual commitments or purchase obligations with various clinical research organizations, promotion and advertising agencies, manufacturers of active pharmaceutical ingredients and drug product for clinical and commercial use as well as with other service providers. These financial commitments, which include both cancelable and non-cancelable arrangements, totaled approximately $15,742 as of December 31, 2010. Since many of these commitment amounts are dependent upon variable components of the agreements, actual payments and the timing of those payments may differ from management’s estimates. In addition, the Company is obligated to pay royalties to InSite Vision as part of its license agreement for AzaSite. Under the terms of the agreement, the Company’s obligation to pay royalties to InSite Vision is subject to pre-determined minimum annual royalty payments. The determination of whether or not the Company will owe any such payments is based upon the amount of royalties accrued over a 12-month royalty period. There are five successive 12-month minimum royalty periods, the first of which commenced on October 1, 2008. The minimum royalties escalate each year. Remaining minimum royalties as of December 31, 2010 total $51,000.
Contingencies
As of December 31, 2010, the Company’s existing license, collaboration and sponsored research agreements may require cash payments contingent upon the occurrence of certain future events. In the aggregate, these agreements may require payments of up to $21,857 assuming the achievement of all development milestones and up to an additional $4,000 assuming the achievement of all sales milestones. Amounts payable by the Company under these agreements are uncertain and are contingent on a number of factors, including the progress of its research and development programs, its ability to obtain regulatory approvals, the commercial success of its approved products and future annual product sales levels. See Note 19 “Subsequent Events” for additional information. The Company is also obligated to pay royalties on net sales, if any, of certain product candidates currently in its portfolio.
15. Related Party Transactions
In February 2009, the Company entered into an agreement with Clinipace, Inc. (“Clinipace”) for the provision of various data management and biostatistics services to support two Phase 2 clinical trials of AzaSite for the treatment of blepharitis. Under this agreement, the Company paid Clinipace $365 upon execution and is obligated to pay an additional $171 per month for 12 months commencing in March 2009. In addition, Clinipace has performed similar services in support of other development programs of the Company in 2010, 2009 and 2008, considered insignificant. The Company had expenses associated with Clinipace activities in 2010, 2009 and 2008, of $410, $2,137 and $24, respectively. Kenneth B. Lee, Jr., the Chairman of the Company’s Board of Directors, is a general partner of Hatteras Venture Partners, LLC, which has a significant ownership in Clinipace. Christy Shaffer, the Company’s President and Chief Executive Officer during 2009 and a portion of 2010, also serves as a director of Clinipace. Neither Mr. Lee nor Dr. Shaffer have a personal interest in any amounts paid by the Company to Clinipace.
In August 2009, the Company completed a public offering of its common stock. Warburg participated in the public offering and acquired an additional 8,889 shares of common stock for total holdings of 22,907 shares of the Company’s common stock, which represented approximately 28% of the Company’s outstanding stock as of December 31, 2009. As part of the offering, the Company reimbursed Warburg for $500 of expenses incurred, related to their participation in the common stock sale. Prior to the sale of common stock in this offering, Warburg owned approximately 14,019, or 25%, of the then outstanding common stock of the Company. Jonathan S. Leff, serves as a member of the Company’s Board of Directors and as a member of its Corporate Governance
F-33
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
Committee. Since January 2000, he has served as a General Partner of Warburg, Pincus & Company, which is the managing partner of Warburg Pincus LLC, and as a Member and Managing Director of Warburg Pincus LLC.
16. Employee Benefit Plan
The Company adopted a 401(k) Profit Sharing Plan (“the 401(k) Plan”) covering all qualified employees on August 1, 1995. Participants may elect a salary reduction of 1% or more up to the IRS allowed maximum as a tax-deferred contribution to the 401(k) Plan. The 401(k) Plan permits discretionary employer contributions. If employer discretionary contributions are implemented, participants will begin vesting 100% immediately in such contributions. In 2010, 2009 and 2008, the Company elected a safe harbor contribution at 3.0% of annual compensation. These safe harbor contributions totaled $1,008, $935 and $938 for the years ended December 31, 2010, 2009 and 2008, respectively.
17. Revenue by Product Line
The Company operates its business as one operating segment. The Company derives all of its product revenue for AzaSite and all its co-promotion revenue for Elestat from product sales in the United States. Approximately 4%, 2% and 2% of royalty revenue for Restasis in fiscal years 2010, 2009 and 2008, respectively, was derived from product sales outside the United States.
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Product Sales: |
||||||||||||
| AzaSite |
$ | 42,671 | $ | 34,961 | $ | 18,349 | ||||||
| Co-Promotion and Royalty Sales: |
||||||||||||
| Restasis |
45,614 | 38,445 | 32,761 | |||||||||
| Elestat |
16,864 | 18,753 | 18,138 | |||||||||
| Total |
$ | 105,149 | $ | 92,159 | $ | 69,248 | ||||||
18. Quarterly Financial Data (unaudited)
| 2010 | ||||||||||||||||||||
| First | Second | Third | Fourth | Total | ||||||||||||||||
| Revenue |
$ | 22,068 | $ | 27,272 | $ | 26,732 | $ | 30,327 | $ | 106,399 | ||||||||||
| Cost of sales |
3,015 | 3,694 | 3,689 | 4,517 | 14,915 | |||||||||||||||
| Net loss |
(14,787 | ) | (8,848 | ) | (7,558 | ) | (4,254 | ) | (35,447 | ) | ||||||||||
| Net loss per common share—basic and diluted |
$ | (0.18 | ) | $ | (0.11 | ) | $ | (0.09 | ) | $ | (0.05 | ) | $ | (0.43 | ) | |||||
| 2009 | ||||||||||||||||||||
| Revenue |
$ | 14,331 | $ | 23,051 | $ | 25,168 | $ | 29,609 | $ | 92,159 | ||||||||||
| Cost of sales |
1,961 | 2,284 | 3,032 | 3,994 | 11,271 | |||||||||||||||
| Net loss |
(19,407 | ) | (9,511 | ) | (8,493 | ) | (2,565 | ) | (39,976 | ) | ||||||||||
| Net loss per common share—basic and diluted |
$ | (0.34 | ) | $ | (0.17 | ) | $ | (0.12 | ) | $ | (0.03 | ) | $ | (0.60 | ) | |||||
F-34
Table of Contents
INSPIRE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
(in thousands, except per share amounts)
19. Subsequent Events
On January 3, 2011, the Company announced that the top-line results from its second Phase 3 clinical trial, TIGER-2, with denufosol tetrasodium for the treatment of cystic fibrosis did not achieve statistical significance for its primary efficacy endpoint, which was change from baseline in FEV1 (Forced Expiratory Volume in One Second) at the Week 48 Endpoint (48 weeks or last observation carried forward). Patients receiving denufosol in the 466-patient, double-blind, placebo-controlled clinical trial had an improvement of 40 mL, compared to 32 mL for the patients receiving placebo (p=0.742). Additionally, there were no statistically significant differences between denufosol and placebo for three key secondary endpoints, which were (i) rate of change in percent predicted FEV1 over 48 weeks; (ii) change from baseline in FEF25%-75% (Forced Expiratory Flow) at the Week 48 Endpoint; and (iii) time to first pulmonary exacerbation. Based upon the overall data relating to the program, the Company decided to discontinue development of denufosol tetrasodium.
On February 17, 2011, the Company announced a strategic corporate restructuring, discontinuing its pulmonary therapeutic area. The corporate restructuring includes a workforce reduction of approximately 65 positions, primarily effecting functions in research and development; manufacturing and technical operations; and general and administrative. As a result, the Company expects to recognize a restructuring charge in the first quarter of 2011 of approximately $10,000 to $13,000 which includes severance costs, termination of ongoing denufosol contracts and activities, the write-off of impaired assets and idle facility charges.
Also in February 2011, the Company entered into an amended and restated technology agreement with Yamasa. The amended and restated technology agreement relieves the Company of any restrictions on its ability to manufacture, or have manufactured, denufosol API. Yamasa reserves no rights to manufacture, or have manufactured, denufosol API. In addition, the seven hundred million Yen (¥ 700,000,000) of potential milestone payments have been eliminated and there are no further milestones to be earned under the amended and restated technology agreement.
Furthermore, the Company expects that it will have no further obligations under the CFFT study funding agreement, including any development and sales milestones as they relate to the denufosol program and expects to reverse the $1,915 liability included in other long-term liabilities on the accompanying Balance Sheet as of December 31, 2010.
F-35
Table of Contents
Exhibit Index
| Exhibit Number |
Description | |
| 3.1 | Amended and Restated Certificate of Incorporation (Incorporated by reference to Exhibit 99.2 to the Company’s Current Report on Form 8-K filed on June 8, 2010). | |
| 3.2 | Amended and Restated Bylaws (Incorporated by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed on December 18, 2007). | |
| 4.1 | Specimen Common Stock Certificate (Incorporated by reference to Exhibit 4.1 to the Company’s registration statement on Form S-1 (Registration No. 333-31174) which became effective on August 3, 2000). | |
| 4.2 | Rights Agreement, dated as of October 21, 2002, between the Company and Computershare Trust Company, which includes the form of Certificate of Designation of Series H Preferred Stock of Inspire Pharmaceuticals, Inc. as Exhibit “A”, the form of Rights Certificate as Exhibit “B” and the Summary of Rights to Purchase Preferred Stock as Exhibit “C” (Incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on October 22, 2002). | |
| 4.3 | First Amendment to Rights Agreement, dated July 17, 2007, by and between Inspire Pharmaceuticals, Inc. and Computershare Trust Company (Incorporated by reference to Exhibit 4.2 to the Company’s Current Report on Form 8-K filed on July 23, 2007). | |
| 10.1* | Development, License and Supply Agreement between Inspire Pharmaceuticals, Inc. and Santen Pharmaceutical Co., Ltd., dated as of December 16, 1998 (Incorporated by reference to Exhibit 10.15 to the Company’s registration statement on Form S-1 (Registration No. 333-31174) which became effective on August 3, 2000). | |
| 10.2† | Employee Confidentiality, Invention Assignment and Non-Compete Agreement between Inspire Pharmaceuticals, Inc. and Benjamin R. Yerxa dated February 4, 2000 (Incorporated by reference to Exhibit 10.26 to the Company’s registration statement on Form S-1 (Registration No. 333-31174) which became effective on August 3, 2000). | |
| 10.3† | Employee Confidentiality, Invention Assignment and Non-Compete Agreement between Inspire Pharmaceuticals, Inc. and Christy L. Shaffer dated February 10, 2000 (Incorporated by reference to Exhibit 10.28 to the Company’s registration statement on Form S-1 (Registration No. 333-31174) which became effective on August 3, 2000). | |
| 10.4† | Employee Confidentiality, Invention Assignment and Non-Compete Agreement between Inspire Pharmaceuticals, Inc. and Joseph Schachle dated April 3, 2001 (Incorporated by reference to Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q filed on August 10, 2001). | |
| 10.5* | Elestat (Epinastine) Co-Promotion Agreement, entered into as of December 8, 2003, by and between Allergan Sales, LLC and Inspire Pharmaceuticals, Inc. (Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on July 1, 2004). | |
| 10.6† | Employee Confidentiality, Invention Assignment and Non-Compete Agreement between Inspire Pharmaceuticals, Inc. and Thomas R. Staab, II, dated May 16, 2003 (Incorporated by reference to Exhibit 10.35 to the Company’s Annual Report on Form 10-K filed March 12, 2004). | |
| 10.7 | Master Lease Agreement between GE Capital Fleet Services and Inspire Pharmaceuticals, Inc., dated as of November 18, 2003, and related documentation (Incorporated by reference to Exhibit 10.38 to the Company’s Annual Report on Form 10-K filed March 12, 2004). | |
| 10.8† | Employee Confidentiality, Invention Assignment and Non-Compete Agreement between the Company and R. Kim Brazzell, dated August 5, 2004 (Incorporated by reference to Exhibit 10.4 to the Company’s Quarterly Report on Form 10-Q filed November 9, 2004). | |
Table of Contents
| Exhibit Number |
Description | |
| 10.9* | Exclusive License Agreement between Inspire Pharmaceuticals, Inc. and the Wisconsin Alumni Research Foundation, effective November 2, 2004 (Incorporated by reference to Exhibit 10.44 to the Company’s Annual Report on Form 10-K filed March 11, 2005). | |
| 10.10† | Form of Incentive Stock Option Grant Agreement (Incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on June 16, 2005). | |
| 10.11† | Form of Nonqualified Stock Option Grant Agreement (Incorporated by reference to Exhibit 10.3 to the Company’s Current Report on Form 8-K filed on June 16, 2005). | |
| 10.12† | Form of Director’s Nonqualified Stock Option Grant Agreement (Incorporated by reference to Exhibit 10.4 to the Company’s Current Report on Form 8-K filed on June 16, 2005). | |
| 10.13† | Form of Restricted Stock Unit Agreement (Incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed on November 7, 2006). | |
| 10.14† | Employee Confidentiality, Invention Assignment and Non-Compete Agreement between Inspire Pharmaceuticals, Inc. and Joseph M. Spagnardi, dated May 10, 2005 (Incorporated by reference to Exhibit 10.43 to the Company’s Annual Report on Form 10-K filed on March 16, 2007). | |
| 10.15* | License Agreement by and between Inspire Pharmaceuticals, Inc. and InSite Vision Incorporated, dated as of February 15, 2007 (Incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed on May 10, 2007). | |
| 10.16* | Supply Agreement by and between Inspire Pharmaceuticals, Inc. and InSite Vision Incorporated, dated as of February 15, 2007 (Incorporated by reference to Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q filed on May 10, 2007). | |
| 10.17 | Trademark License Agreement by and between Inspire Pharmaceuticals, Inc. and InSite Vision Incorporated, dated as of February 15, 2007 (Incorporated by reference to Exhibit 10.3 to the Company’s Quarterly Report on Form 10-Q filed on May 10, 2007). | |
| 10.18 | Side Letter by and between Inspire Pharmaceuticals, Inc., InSite Vision Incorporated and Pfizer Inc., dated as of February 15, 2007 (Incorporated by reference to Exhibit 10.4 to the Company’s Quarterly Report on Form 10-Q filed on May 10, 2007). | |
| 10.19† | Form of Nonqualified Stock Option Grant Agreement (Incorporated by reference to Exhibit 10.6 to the Company’s Quarterly Report on Form 10-Q filed on May 10, 2007). | |
| 10.20† | Form of Director’s Nonqualified Stock Option Grant Agreement (Incorporated by reference to Exhibit 10.7 to the Company’s Quarterly Report on Form 10-Q filed on May 10, 2007). | |
| 10.21† | Executive Officer Annual Cash Bonus Plan (Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on June 13, 2007). | |
| 10.22 | Standstill Agreement, dated July 20, 2007, among Inspire Pharmaceuticals, Inc., Warburg Pincus Private Equity IX, L.P., Warburg Pincus IX, LLC, Warburg Pincus Partners, LLC and Warburg Pincus & Co. (Incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on July 23, 2007). | |
| 10.23* | Manufacturing Services Agreement, dated September 11, 2007, by and between Inspire Pharmaceuticals, Inc. and Catalent Pharma Solutions, LLC (Incorporated by reference to Exhibit 10.3 to the Company’s Quarterly Report on Form 10-Q filed on November 9, 2007). | |
| 10.24† | Inspire Pharmaceuticals, Inc. Executive Change in Control Severance Benefit Plan, Amended and Restated as of July 8, 2009 (Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on July 13, 2009). | |
Table of Contents
| Exhibit Number |
Description | |
| 10.25† | Inspire Pharmaceuticals, Inc. Change in Control Severance Benefit Plan, Amended and Restated as of July 8, 2009 (Incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on July 13, 2009). | |
| 10.26† | Form of 1995 Stock Plan Nonqualified Stock Option Grant Agreement (Incorporated by reference to Exhibit 10.4 to the Company’s Current Report on Form 8-K filed on July 13, 2009). | |
| 10.27† | Form of Restricted Stock Unit Agreement Under the Amended and Restated 2005 Equity Compensation Plan (Incorporated by reference to Exhibit 10.5 to the Company’s Current Report on Form 8-K filed on July 13, 2009). | |
| 10.28† | Form of Inspire Pharmaceuticals, Inc. 1995 Stock Plan Director’s Nonqualified Stock Option Grant Agreement (Incorporated by reference to Exhibit 10.6 to the Company’s Current Report on Form 8-K filed on July 13, 2009). | |
| 10.29† | Form of Inspire Pharmaceuticals, Inc. 2005 Equity Compensation Plan Director’s Nonqualified Stock Option Grant Agreement (Incorporated by reference to Exhibit 10.7 to the Company’s Current Report on Form 8-K filed on July 13, 2009). | |
| 10.30 | Amendment No. 1 to Standstill Agreement, dated August 4, 2009, between Inspire Pharmaceuticals, Inc. and Warburg Pincus Private Equity IX, L.P. (Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on August 10, 2009). | |
| 10.31† | Executive Incentive Compensation Recovery (“Clawback”) Policy, dated December 18, 2009. Incorporated by reference to Exhibit 10.53 to the Company’s Annual Report on Form 10-K filed on March 15, 2010). | |
| 10.32† | Amended and Restated Equity Compensation Grant Policy, dated December 18, 2009. Incorporated by reference to Exhibit 10.54 to the Company’s Annual Report on Form 10-K filed on March 15, 2010). | |
| 10.33† | Executive Employment Agreement, between Inspire Pharmaceuticals, Inc. and Adrian Adams, made as of February 18, 2010 (Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on February 24, 2010). | |
| 10.34† | Stock Option Agreement by and between Inspire Pharmaceuticals, Inc. and Adrian Adams, effective as of February 22, 2010 (Incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on February 24, 2010). | |
| 10.35† | Separation of Employment and Consulting Agreement between Inspire Pharmaceuticals, Inc. and Dr. Christy L. Shaffer, Ph.D., made as of February 19, 2010 (Incorporated by reference to Exhibit 10.3 to the Company’s Current Report on Form 8-K filed on February 24, 2010). | |
| 10.36† | Form of Restricted Stock Unit Agreement by and between Inspire Pharmaceuticals, Inc. and Adrian Adams, effective as of March 18, 2010 (Incorporated by reference to Exhibit 99.3 to the Company’s Registration Statement on Form S-8 filed on March 18, 2010). | |
| 10.37† | Form of Restricted Stock Unit Grant Agreement (for Directors) (Incorporated by reference to Exhibit 10.1 to the company’s Quarterly Report on Form 10-Q filed on May 4, 2010). | |
| 10.38† | Form of Restricted Stock Unit Grant Agreement (for Officers) (Incorporated by reference to Exhibit 10.2 to the company’s Quarterly Report on Form 10-Q filed on May 4, 2010). | |
| 10.39* | Technical Transfer & Development Services Agreement, between Inspire Pharmaceuticals, Inc. and Finorga S.A.S., acting in its own name and on behalf of Novasep Process, dated as of March 26, 2010 (Incorporated by reference to Exhibit 10.3 to the company’s Quarterly Report on Form 10-Q filed on May 4, 2010). | |
Table of Contents
| Exhibit Number |
Description | |
| 10.40† | Executive Employment Agreement, between Inspire Pharmaceuticals, Inc. and Andrew I. Koven, made as of April 2, 2010 (Incorporated by reference to Exhibit 10.4 to the Company’s Quarterly Report on Form 10-Q filed on May 4, 2010). | |
| 10.41† | Stock Option Agreement by and between Inspire Pharmaceuticals, Inc. and Andrew I. Koven, effective as of May 10, 2010 (Incorporated by reference to Exhibit 99.2 to the Company’s Registration Statement on Form S-8 filed on May 19, 2010). | |
| 10.42† | Form of Restricted Stock Unit Agreement by and between Inspire Pharmaceuticals, Inc. and Andrew I. Koven, effective as of May 19, 2010 (Incorporated by reference to Exhibit 99.3 to the Company’s Registration Statement on Form S-8 filed on May 19, 2010). | |
| 10.43† | Amended and Restated 2010 Equity Compensation Plan (Incorporated by reference to Exhibit 99.1 to the Company’s Current Report on Form 8-K filed on June 8, 2010). | |
| 10.44* | First Amendment to Development, License and Supply Agreement, dated June 8, 2010, by and between Inspire Pharmaceuticals, Inc. and Santen Pharmaceutical Co. Ltd. (Incorporate by reference to Exhibit 10.5 to the Company’s Quarterly Report on Form 10-Q filed on August 6, 2010) | |
| 10.45 | Lease Agreement between Inspire Pharmaceuticals, Inc. and Brier Creek Corporate Center Associates Limited Partnership dated as of June 28, 2010 (Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on July 1, 2010). | |
| 10.46† | Separation of Employment and Consulting Agreement by and between Inspire Pharmaceuticals, Inc. and Benjamin R. Yerxa, dated as of June 29, 2010 (Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on July 1, 2010).) | |
| 10.47† | Executive Employment Agreement, between Inspire Pharmaceuticals, Inc. and R. Kim Brazzell, dated as of July 7, 2010 (Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on July 13, 2010). | |
| 10.48† | Executive Employment Agreement, between Inspire Pharmaceuticals, Inc. and Joseph K. Schachle, dated as of July 7, 2010 (Incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on July 13, 2010). | |
| 10.49† | Executive Employment Agreement, between Inspire Pharmaceuticals, Inc. and Joseph M. Spagnardi, dated as of July 7, 2010 (Incorporated by reference to Exhibit 10.3 to the Company’s Current Report on Form 8-K filed on July 13, 2010). | |
| 10.50† | Executive Employment Agreement, between Inspire Pharmaceuticals, Inc. and Thomas R. Staab, II, dated as of July 7, 2010 (Incorporated by reference to Exhibit 10.4 to the Company’s Current Report on Form 8-K filed on July 13, 2010). | |
| 10.51† | Executive Employment Agreement between Inspire Pharmaceuticals, Inc. and Charles A. Johnson, dated August 12, 2010 (Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on August 13, 2010). | |
| 10.52* | Amended and Restated License, Development and Marketing Agreement, dated August 19, 2010, between Inspire Pharmaceuticals, Inc. and Allergan, Inc., Allergan Sales, LLC and Allergan Pharmaceuticals Holdings (Ireland) Ltd. (Incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed on November 5, 2010) | |
| 10.53† | Stock Option Agreement between Inspire Pharmaceuticals, Inc. and Charles A. Johnson, dated September 15, 2010 (Incorporated by reference to Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q filed on November 5, 2010). | |
Table of Contents
| Exhibit Number |
Description | |
| 10.54† | Restricted Stock Unit Agreement between Inspire Pharmaceuticals, Inc. and Charles A. Johnson, dated September 15, 2010. (Incorporated by reference to Exhibit 10.3 to the Company’s Quarterly Report on Form 10-Q filed on November 5, 2010). | |
| 10.55* | API Commercial Supply Agreement (Denufosol), dated as of December 14, 2010, by and between Inspire Pharmaceuticals, Inc. and Finorga S.A.S. (Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on December 22, 2010) | |
| 10.56 | Parent Guarantee from Groupe Novasep S.A.S. concerning the terms of the API Commercial Supply Agreement (Denufosol), dated as of December 14, 2010, by and between Inspire Pharmaceuticals, Inc. and Finorga S.A.S. (Incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on December 22, 2010) | |
| 10.57† | Amended and Restated Director Compensation Policy, dated November 17, 2010. | |
| 23.1 | Consent of PricewaterhouseCoopers LLP, independent registered public accounting firm. | |
| 31.1 | Certification of the Chief Executive Officer pursuant to Rule 13a-14(a)/15d-14(a) of the Securities Exchange Act of 1934. | |
| 31.2 | Certification of the Chief Financial Officer pursuant to Rule 13a-14(a)/15d-14(a) of the Securities Exchange Act of 1934. | |
| 32.1 | Certification of the Chief Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 32.2 | Certification of the Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| * | Confidential treatment has been granted with respect to a portion of this Exhibit. |
| † | Denotes a management contract or compensation plan or arrangement required to be filed as an exhibit pursuant to Item 15(c) of this Form 10-K. |