Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
(Mark One)
| þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For The Fiscal Year Ended December 31, 2010
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Transition Period from to
Commission File 000-30039
ADOLOR CORPORATION
(Exact name of registrant as specified in its charter)
| Delaware | 31-1429198 | |
| (State or other jurisdiction of incorporation or organization) |
(IRS Employer Identification Number) | |
| 700 Pennsylvania Drive, Exton, Pennsylvania | 19341 | |
| (Address of principal executive offices) | (Zip code) | |
(484) 595-1500
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| (Title of each class) | (Name of each exchange on which registered) | |
| Common Stock, $0.0001 par value | NASDAQ | |
| Series A Junior Participating Preferred Stock Purchase Rights |
None |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. Yes þ No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. þ
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. Check one:
Large accelerated filer ¨ Accelerated filer ¨ Non-accelerated filer ¨ Smaller reporting company þ
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No þ
The aggregate market value of common stock held by non-affiliates of the registrant was $49.2 million as of June 30, 2010, the last business day of the registrant’s most recently completed second fiscal quarter. Such aggregate market value was computed by reference to the closing price of the common stock as reported on the NASDAQ Global Market on June 30, 2010. For purposes of determining this amount only, the registrant has defined affiliates as including (a) the executive officers of the registrant as of June 30, 2010 and (b) all directors of the registrant as of June 30, 2010.
The number of shares of the registrant’s common stock outstanding as of February 18, 2011 was 46,427,507.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive proxy statement (the “Definitive Proxy Statement”) to be filed with the Securities and Exchange Commission in connection with the Company’s Annual Meeting of Stockholders for the fiscal year ended December 31, 2010 are incorporated by reference into Items 10, 11, 12, 13 and 14 of Part III of this Form 10-K.
Table of Contents
ADOLOR CORPORATION
FORM 10-K
December 31, 2010
| Page No. | ||||||
| 1 | ||||||
| PART I | ||||||
| Item 1. |
3 | |||||
| Item 1A. |
15 | |||||
| Item 1B. |
25 | |||||
| Item 2. |
25 | |||||
| Item 3. |
26 | |||||
| Item 4. |
26 | |||||
| PART II | ||||||
| Item 5. |
27 | |||||
| Item 6. |
29 | |||||
| Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
29 | ||||
| Item 7A. |
42 | |||||
| Item 8. |
43 | |||||
| Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
71 | ||||
| Item 9A. |
71 | |||||
| Item 9B. |
71 | |||||
| PART III | ||||||
| Item 10. |
72 | |||||
| Item 11. |
72 | |||||
| Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
72 | ||||
| Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
72 | ||||
| Item 14. |
72 | |||||
| PART IV | ||||||
| Item 15. |
73 | |||||
| 79 | ||||||
Table of Contents
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
In addition to historical facts or statements of current condition, this report and the documents into which this report is and will be incorporated contain forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. Forward-looking statements contained in this report or incorporated herein by reference constitute our expectations or forecasts of future events as of the date this report was filed with the Securities and Exchange Commission and are not statements of historical fact. You can identify these statements by the fact that they do not relate strictly to historical or current facts. Such statements may include words such as “anticipate,” “will,” “estimate,” “expect,” “project,” “intend,” “should,” “plan,” “believe,” “hope” and other words and terms of similar meaning in connection with any discussion of, among other things, sales, future operating or financial performance, strategic initiatives and business strategies, regulatory or competitive environments, our intellectual property and product development. In particular, these forward-looking statements include, among others, statements about:
| • | our dependence on sales of ENTEREG® (alvimopan) in the United States and the commercial prospects and future marketing efforts for this product; |
| • | our dependence on our current collaboration with Glaxo Group Limited and our ability to attract future partners to finance development and commercialization of our product candidates; |
| • | our anticipated progress in our research and development of potential pharmaceutical products, including our ongoing or planned clinical trials, the status, timing, costs and results of such trials, the ability to secure regulatory approval for our product candidates and the likelihood or timing of revenues from these products, if any; |
| • | the scope and duration of our intellectual property protection for our products and product candidates, our ability to adequately protect our technologies and enforce our intellectual property rights and the future expiration of patent and/or regulatory exclusivity on alvimopan; |
| • | our anticipated operating losses and cash requirements, projections regarding the levels of our cash, cash equivalents and investments and our ability to raise additional funds in light of our current and projected level of operations; and |
| • | other statements regarding matters that are not historical facts or statements of current condition. |
Any or all of our forward-looking statements in this report and in the documents that we have referred you to may turn out to be wrong. They can be affected by inaccurate assumptions we might make or by known or unknown risks and uncertainties. Therefore, you should not place undue reliance on any such forward-looking statements. The factors that could cause actual results to differ from those expressed or implied by our forward-looking statements include, among others:
| • | the acceptance of ENTEREG by hospital formularies and pharmacies, physicians and patients in the marketplace; |
| • | setbacks with respect to research programs, clinical trials or manufacturing or commercial activities; |
| • | the timing and unpredictability of regulatory actions; |
| • | our ability to develop and commercialize new products effectively; |
| • | unanticipated cash requirements to support current operations, to expand our business or for capital expenditures; |
| • | the inability to adequately protect our key intellectual property rights; |
| • | the loss of key management or scientific personnel; |
| • | the activities of our competitors; |
1
Table of Contents
| • | regulatory, legal or other setbacks with respect to our operations or business; |
| • | market conditions in the capital markets and the biopharmaceutical industry that make raising capital or consummating acquisitions difficult, expensive or both; and |
| • | enactment of new government laws, regulations, court decisions, regulatory interpretations or other initiatives that are adverse to us or our interests. |
We do not intend to update publicly any forward-looking statement, whether as a result of new information, future events or otherwise, except as required by law. We discuss in more detail the risks that we anticipate in Part I, Item 1A of this report. This discussion is permitted by the Private Securities Litigation Reform Act of 1995.
2
Table of Contents
PART I
| ITEM 1. | BUSINESS |
OVERVIEW
Adolor Corporation is a biopharmaceutical company focused on the discovery, development and commercialization of novel prescription pain and pain-management products. In May 2008, the U.S. Food and Drug Administration (FDA) approved our first product, ENTEREG® (alvimopan). ENTEREG is indicated to accelerate upper and lower gastrointestinal (GI) recovery following partial large or small bowel resection surgery with primary anastomosis. Delayed GI recovery following bowel resection surgery can postpone hospital discharge until its resolution, resulting in an increased cost burden on healthcare providers. In June 2008, we launched ENTEREG in the United States in collaboration with Glaxo Group Limited (Glaxo).
We have a number of product candidates in various stages of clinical and preclinical development. We are conducting two Phase 2 clinical trials of ADL5945 to treat opioid-induced constipation (OIC), a condition that often results from long-term use of opioid analgesics in the management of chronic pain conditions. In addition, we are continuing limited development efforts on ADL7445, a second peripherally-acting mu opioid receptor antagonist, which is considered a back-up compound in our OIC program. We have completed Phase 1 clinical evaluation of ADL6906 (beloxepin), a compound with a novel and potentially differentiating pharmacological profile for treating chronic pain. Our preclinical pipeline includes novel, selective centrally-acting mu opioid receptor antagonists (CAMORs) currently in development for the treatment of l-DOPA-induced dyskinesia (LID) associated with the management of Parkinson’s disease.
For the year ended December 31, 2010, our total revenues and net loss were $43.3 million and $27.3 million, respectively. Net sales of ENTEREG for the year ended December 31, 2010 were $25.4 million. We will need net sales of ENTEREG to increase significantly beyond current levels before we will be able to achieve profitability and positive cash flow from operations. Ultimately, we may never generate sufficient revenues from ENTEREG for us to reach profitability, generate positive cash flow or sustain, on an ongoing basis, our current or projected levels of operations. In addition, our success is highly dependent on the results of our Phase 2 clinical trials with respect to ADL5945, our most advanced product candidate. There is no assurance that these studies will yield positive results, and even if the results are positive, it is unlikely that we will have sufficient available resources to fund a Phase 3 clinical program for ADL5945 without either securing a collaboration partner or raising proceeds in the capital markets.
We are a Delaware corporation with our principal executive offices located at 700 Pennsylvania Drive, Exton, Pennsylvania, 19341. Our telephone number is (484) 595-1500 and our web site address is www.adolor.com. We include our web site address in this Annual Report on Form 10-K only as an inactive textual reference and do not intend it to be an active link to our web site. The information on our website is not incorporated by reference in this Annual Report on Form 10-K.
Our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and all amendments to those reports, as well as other documents we file with the U.S. Securities and Exchange Commission (SEC), are available free of charge through the Investor Insights section of our web site as soon as reasonably practicable after such material is electronically filed with or furnished to the SEC. The public can obtain documents that we file with the SEC at www.sec.gov.
ENTEREG
ENTEREG (alvimopan) is a small molecule, peripherally-acting mu opioid receptor antagonist intended to block the adverse side effects of opioid analgesics on the GI tract without affecting their beneficial analgesic
3
Table of Contents
effects. ENTEREG was approved by the FDA on May 20, 2008 and together with our partner, Glaxo, we launched the product in the United States in mid-2008. ENTEREG is specifically indicated to accelerate upper and lower GI recovery following partial large or small bowel resection surgery with primary anastomosis.
Market Opportunity and Commercialization
The impairment of GI motility after intra-abdominal surgery can be associated with abdominal distension and bloating, persistent abdominal pain, nausea and vomiting, variable reduction of bowel sounds, delayed passage of or an inability to pass flatus (gas) or stool and an inability to tolerate oral intake or progress to a solid diet. Delayed GI recovery following bowel resection surgery can postpone hospital discharge until its resolution, resulting in an increased cost burden on healthcare providers.
There are over 4,000 hospitals in the United States that perform bowel resection surgeries, with approximately 1,600 hospitals collectively performing approximately 80% of such surgeries. ENTEREG is detailed by Glaxo’s national hospital-based sales organization, and Adolor is co-promoting ENTEREG in certain hospitals with a field force that numbers approximately 25 persons.
Most hospitals have committees that meet periodically to determine which pharmaceutical products to add to its formulary, or list of accepted drugs. Once a drug is on formulary, it is easier for a physician within a hospital or hospital group to prescribe the drug. As such, we consider hospital formulary approval to be critical for ENTEREG to become a commercial success.
In April 2007, Glaxo and we announced the results of Study 014, a Phase 3 long-term safety study of alvimopan in patients taking opioids for chronic non-cancer pain and experiencing opioid bowel dysfunction (OBD). Results from Study 014 showed a higher number of myocardial infarctions reported by patients treated long-term (1 year) with alvimopan compared to placebo. As a result, ENTEREG was approved subject to a Risk Evaluation and Mitigation Strategy (REMS), and the product labeling carries a boxed warning that ENTEREG is available only for short-term (15 doses) use in hospitalized patients, consistent with the approved indication. The REMS is designed to maintain the benefits associated with short-term use in the bowel resection population and prevent long-term, outpatient use.
Under the REMS, ENTEREG is available only to hospitals that perform bowel resection surgeries and that are enrolled in the ENTEREG Access Support and Education (E.A.S.E.®) Program. Hospitals can enroll in the E.A.S.E Program if they acknowledge that they: have received the E.A.S.E. Program educational materials and provided such materials to healthcare practitioners who are responsible for the ordering, dispensing or administration of ENTEREG, have systems in place to limit the use of ENTEREG to no more than 15 doses per patient for administration in the hospital only, will not dispense ENTEREG for outpatient use and will not transfer ENTEREG to an unregistered hospital. Upon enrollment, hospitals can order ENTEREG through wholesalers and on receipt and verification of the order, Glaxo will drop-ship ENTEREG directly to the hospital pharmacy.
The FDA recently completed a post-marketing drug safety evaluation (PDSE) of ENTEREG to determine if there are any new serious adverse events not previously identified during the development of ENTEREG, known side effects reported in unusual numbers or potential new safety concerns now that ENTEREG is being used in the general population. The PDSE identified and reviewed cases of GI perforation and myocardial infarction in patients taking ENTEREG. Cases of GI perforation were complicated by the patients’ underlying surgical condition, making a causal attribution to ENTEREG difficult. Cases of myocardial infarction were determined to be related to underlying coronary artery disease, age or other causes, rather than to ENTEREG. As a result of the PDSE, the FDA did not require any changes to the ENTEREG label.
As required by the FDA as a post-approval requirement for ENTEREG, we began a Phase 4 clinical trial in 2009 intended to evaluate the safety and efficacy of ENTEREG in patients undergoing radical cystectomy.
4
Table of Contents
Radical cystectomy involves extensive resection of abdominal and pelvic structures and these patients are at high risk for delayed GI recovery. The trial is expected to enroll approximately 300 patients and enrollment is expected to be completed in late 2011. In addition, consistent with FDA regulation and the ENTEREG approval letter, two pediatric Phase 4 clinical trials are required. The first of these studies is expected to be initiated during 2013.
Glaxo Collaboration and Other License Agreements
In April 2002, we entered into our collaboration agreement with Glaxo for the exclusive worldwide development and commercialization of ENTEREG for certain indications. At that time, Glaxo paid us a non-refundable and non-creditable signing fee of $50 million, which is being recognized as revenue over a period of fourteen years, the estimated performance period under the collaboration agreement. Since 2002, we have received and recognized $30 million of milestone revenue, including $20 million received following FDA approval of ENTEREG in May 2008. Under the Glaxo agreement, we have a profit-sharing arrangement pursuant to which we receive 45% and Glaxo receives 55% of profits and losses, as defined, through June of 2011, after which the profit split is 50% each. Profits and losses are calculated as net sales of ENTEREG in the United States less certain agreed-upon costs, subject to certain adjustments.
The term of the collaboration agreement for ENTEREG in the United States is ten years from the first commercial sale of ENTEREG, which occurred in June 2008. Glaxo has the right, upon notice, to terminate its rights and obligations with respect to ENTEREG. Glaxo also has the right to terminate the collaboration agreement for breach of the agreement by us or for safety-related reasons as defined in the collaboration agreement. In September 2008, Glaxo returned to us all worldwide rights to alvimopan for the treatment of chronic OIC; in December 2009, they returned to us the rights to alvimopan outside of the United States for the treatment of delayed GI recovery. We currently do not have any plans to seek approval to market alvimopan for OIC or, outside of the United States, for delayed GI recovery.
Under the collaboration agreement, certain external expenses incurred in the United States by each company are reimbursed by the other party pursuant to contractually agreed percentages. Monies reimbursable to Glaxo by us for profit-share and certain marketing amounts are recorded gross as selling, general and administrative expense on our consolidated statements of operations. Contract reimbursement amounts owed to us by Glaxo for research and development activities are recorded gross on our consolidated statements of operations as contract revenues. We also have a distribution agreement with Glaxo under which it performs certain distribution and contracting services for ENTEREG on our behalf for a fee. Pursuant to that agreement, we have the option to assume responsibility for such distribution and contracting services beginning in June 2011.
In November 1996, Roberts Laboratories Inc. (Roberts) licensed from Eli Lilly and Company (Lilly) certain intellectual property rights relating to ENTEREG. In June 1998, we entered into an option and license agreement with Roberts under which we sublicensed these rights from Roberts. In December 2000, Shire U.S. Inc. became the successor in interests to Roberts under our option and license agreement with Roberts. Our obligations to pay, in the aggregate, single-digit royalties on net sales of ENTEREG to Shire and Lilly is expected to expire on the date of the last to expire of the licensed Lilly patents.
Intellectual Property Position and Regulatory Exclusivity
ENTEREG is protected by a U.S. composition of matter patent licensed to us from Lilly that expires on March 29, 2011. The U.S. Patent and Trademark Office (PTO) has issued a Notice of Final Determination that this patent is eligible for a five year patent term extension; it is expected that the PTO will issue shortly a Certificate of Extension evidencing the extension of this patent to March 29, 2016. The product also is protected by a U.S. patent claiming the use of ENTEREG that expires in 2020 and a second composition of matter patent that expires in 2013; in addition, there are five other patents and patent applications covering the product.
In addition to the patents related to ENTEREG, alvimopan is classified as a new chemical entity (NCE) under the Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman
5
Table of Contents
Act. The Hatch-Waxman Act provides five years of NCE marketing exclusivity to the first applicant to gain approval of a New Drug Application (NDA) for a product that does not contain an active ingredient found in any other approved product. We have five years of NCE exclusivity for ENTEREG that expires on May 20, 2013. The NCE exclusivity prevents the submission to the FDA of any Abbreviated New Drug Application (ANDA) for any pharmaceutical product containing alvimopan until May 2013 (or May 2012 if the ANDA applicant certifies that the patents covering ENTEREG are invalid or will not be infringed by the ANDA product).
Manufacturing and Product Supply
We have two approved suppliers under our NDA of the active pharmaceutical ingredient (API) in ENTEREG: Piramal Healthcare (Canada) Limited (formerly Torcan Chemical Ltd.) and Central Glass Germany GmbH (formerly Girindus AG). We also have two approved manufacturers under our NDA of ENTEREG finished capsules: Pharmaceutics International, Inc. and Pharmaceutical Manufacturers Research Services Inc. We seek to maintain inventories of finished products to protect against supply disruptions. Any future change in manufacturers or manufacturing process requires regulatory approval.
Competition
Currently, ENTEREG is the only FDA-approved product indicated for the acceleration of GI recovery following bowel resection surgery. We are aware of other products in various stages of clinical development for this condition. For example, we are aware of molecules in development by Tranzyme Pharma, Helsinn Therapeutics and other companies that could compete with ENTEREG at some point in the future.
Customers
Our principal customers are wholesale drug distributors. These customers comprise a significant part of the distribution network for all pharmaceutical products in the United States. Three large wholesale drug distributors, Cardinal Health, Inc., McKesson Corporation and AmerisourceBergen Corporation, comprise a significant share of this network. These three wholesale customers, in the aggregate, accounted for 93% of our total product sales for the year ended December 31, 2010. Under the E.A.S.E. Program, hospital orders are processed through wholesalers; however, ENTEREG is drop-shipped from Glaxo’s warehouse directly to registered hospitals.
DEVELOPMENT PROGRAMS
Opioid-induced Constipation (OIC) Program
We are developing two molecules, ADL5945 and ADL7445, to treat OIC. These compounds are small molecule, potent, peripherally-acting mu opioid receptor antagonists intended to block the adverse effects of opioid analgesics on the GI tract without affecting analgesia.
Market Opportunity
Mu opioid receptors in the GI tract (characterized as peripheral mu opioid receptors as they reside outside the central nervous system) regulate functions such as motility, secretion and absorption. Stimulation of these GI mu opioid receptors by morphine, or other opioid analgesics, disrupts normal gut motility resulting in non-propulsive contractions of the bowel wall, ultimately delaying transit time of intestinal contents. This is the primary mechanism underlying OIC.
Clinical Development
During 2010, we conducted our initial clinical evaluation of both ADL5945 and ADL7445. The single dose and multiple-ascending dose studies of ADL5945 and the single-ascending dose and multiple-ascending dose
6
Table of Contents
studies of ADL7445 enrolled both healthy volunteers and chronic non-cancer pain patients on long-term opioid therapy with OIC. At target therapeutic doses, both compounds were well-tolerated and, in the patients with OIC, produced greater increases (over baseline) in weekly average number of spontaneous bowel movements (SBMs) as compared with placebo. The most commonly reported adverse events were dose-dependent and primarily GI related, including abdominal cramping and nausea, the majority of which were of mild severity. There were no serious adverse events reported.
In October 2010, we initiated a Phase 2 study of ADL5945 in chronic non-cancer pain patients suffering from OIC. The study will evaluate two doses of ADL5945 (0.10 mg and 0.25 mg given twice daily) versus placebo over a 4-week, double-blind treatment period, with approximately 120 patients expected to enroll (n = 40/treatment group). In January 2011, we initiated a second Phase 2 study of ADL5945 (0.25 mg) in patients suffering from OIC. The study design is similar to the Phase 2 study initiated in October 2010; however, once-daily dosing will be evaluated with approximately 80 patients (n = 40/treatment group) expected to enroll. The primary endpoint of the studies will be the change from baseline in the weekly average number of SBMs over the 4-week treatment period. Results from these studies are expected in the third quarter of 2011.
Limited development efforts also continue on ADL7445, which is considered a back-up compound in our OIC program.
License Agreement
In September 2009, we acquired from Lilly the exclusive worldwide rights to ADL5945 for an up-front payment of $2.0 million. We also will be required to pay milestones, which are contingent upon achievement of pre-defined, late-stage clinical and regulatory events and achievement of certain sales targets, and royalties on net sales of the product.
Intellectual Property Position
Under the Lilly license agreement, we have an exclusive, world-wide license to patents covering the composition of ADL5945 in the United States and various foreign countries. The U.S. patent is projected to expire in 2025. We also have pending patent applications in the United States and certain foreign countries claiming the use of ADL5945 for the treatment of OIC. For ADL7445, we own two issued U.S. patents covering the composition of this compound that also are projected to expire in 2025; in addition, we have pending patent applications in the United States and certain foreign countries covering, among other things, the use of this compound for the treatment of OIC.
Competition
There are no FDA-approved products indicated for OIC. We are aware of other products in various stages of clinical development for this condition. For example, we are aware of molecules in development by Progenics Pharmaceuticals, Inc. in collaboration with Salix Pharmaceuticals Inc., AstraZeneca PLC in collaboration with Nektar Therapeutics, Theravance Inc. and Alkermes, Inc. that could compete with ADL5945 and ADL7445 at some point in the future.
ADL6906 (beloxepin) for Chronic Pain
Serotonin (5-HT) and norepinephrine (NE) in the brain and spinal cord are believed to help regulate the perception of pain. Reduced levels of endogenous 5-HT and NE activity at both spinal and supraspinal levels are believed to contribute substantially to chronic pain. It is suggested that the dual 5-HT/NE reuptake inhibitors (SNRIs) attenuate pain by increasing levels of NE and 5-HT post-synaptically, leading to activation of the descending inhibitory pain pathway. Combining drugs that act at different receptors and on different pain mechanisms is commonly used in clinical practice to treat various pain conditions. As a result, there is interest in identifying novel analgesics interacting specifically with multiple pain targets.
7
Table of Contents
We are developing ADL6906 (beloxepin), a dual NE reuptake inhibitor and 5-HT2 receptor antagonist, giving ADL6906 a novel and potentially differentiating pharmacological profile for treating chronic pain. ADL6906 is active in several preclinical models of pain after oral administration. Preclinical studies conducted by Adolor also suggest that the analgesic efficacy of ADL6906 is the result of a complementary effect between the dual mechanisms of action of NE reuptake inhibition and 5-HT2 receptor antagonism.
We recently completed a Phase 1 multiple ascending dose study of ADL6906 that evaluated the safety, tolerability and pharmacokinetics of ADL6906. ADL6906 was generally well-tolerated. We currently are evaluating the initiation of Phase 2 testing of ADL6906, either independently or in collaboration with a potential partner.
CAMOR Program for Parkinson’s Disease
In the United States, it is estimated that one million people are living with Parkinson’s disease, and about 50,000 new cases are reported annually. Symptoms of Parkinson’s disease include muscle rigidity or stiffness, slowing of normal movements, tremors and postural instability. The standard of care for the treatment of Parkinson’s disease is the use of l-DOPA to increase levels of dopamine in the brain. However, within 5 to 10 years of starting on l-DOPA, the majority of patients develop a debilitating side effect known as l-DOPA-induced dyskinesia (LID), characterized by erratic, uncontrollable movements often involving the arms and head. Currently, there is no FDA-approved treatment for LID.
Scientific evidence suggests that increased opioid peptide transmission in the basal ganglia might underlie dyskinesia after chronic l-DOPA treatment and that opioid antagonists, therefore, might be useful as adjuncts to l-DOPA therapy for Parkinson’s disease. We have discovered a family of novel, selective centrally-acting mu opioid receptor antagonists (CAMORs). These compounds have been shown to be highly efficacious, after oral administration, in well-validated, non-human primate preclinical models of LID. During the fourth quarter of 2010, the Michael J. Fox Foundation for Parkinson’s Research awarded us a second round of funding to support our development of CAMORs for the treatment of LID associated with Parkinson’s disease. The $0.4 million award will be paid over a period of 18 months.
Delta Opioid Receptor Agonist Program
We previously collaborated with Pfizer Inc. (Pfizer) for the development and commercialization of the delta opioid receptor agonist compounds ADL5859 and ADL5747 for the treatment of pain.
During June 2010, Pfizer and we completed a Phase 2a clinical trial of ADL5859 and ADL5747 in patients with osteoarthritis (OA). The trial was a randomized, double-blind, placebo-controlled, multi-center trial designed to evaluate the safety, tolerability and clinical activity of ADL5859 and ADL5747 in patients with OA of the knee. The study enrolled over 400 patients. Top-line results of the study showed a high placebo response, with neither oxycodone CR nor ADL5859 nor ADL5747 demonstrating a statistically-significant improvement in pain intensity over placebo.
In December 2010, Pfizer and we announced that we were discontinuing further development of ADL5859 and ADL5747. Based on the clinical evaluation of ADL5859 and ADL5747 in multiple indications, the companies concluded that there was not compelling evidence to continue the development program. In December 2010, Pfizer delivered to us a notice of termination of our collaboration agreement, which will be effective as of March 2011.
INTELLECTUAL PROPERTY
We seek United States and international patent protection for important and strategic components of our technology. There can be no assurance that any of our patents will guarantee protection or market exclusivity for
8
Table of Contents
our products and product candidates. We also rely on trade secret protection for certain of our confidential and proprietary information, and we use license agreements both to access external technologies and assets and to convey certain intellectual property rights to others. Our success will be dependent in part on our ability to obtain commercially valuable patent claims, protect our intellectual property rights and operate without infringing upon the proprietary rights of others.
The patent positions of pharmaceutical and biotechnology companies, including ours, are generally uncertain and involve complex legal and factual questions. Our business could be negatively impacted by any of the following:
| • | the claims of any patents that are issued may not provide meaningful protection, may not provide a basis for commercially viable products or may not provide us with any competitive advantages; |
| • | our patents may be challenged by third parties; |
| • | others may have patents that relate to our technology or business that may prevent us from marketing our product candidates unless we are able to obtain a license to those patents; |
| • | the pending patent applications to which we have rights may not result in issued patents; and |
| • | we may not be successful in developing additional proprietary technologies that are patentable. |
In addition, patent law relating to the scope of claims in the field in which we operate is still evolving. The degree of future protection for some of our rights, therefore, is uncertain. Furthermore, others may independently develop similar or alternative technologies, duplicate any of our technologies and, if patents are licensed or issued to us, design around the patented technologies licensed to or developed by us. In addition, we could incur substantial costs in litigation if we have to defend ourselves in patent suits brought by third parties or if we initiate such suits.
With respect to proprietary know-how that is not patentable and for processes for which patents are difficult to enforce, we rely on trade secret protection and confidentiality agreements to protect our interests. While we require all employees, consultants and potential business partners to enter into confidentiality agreements, we may not be able to protect adequately our trade secrets or other proprietary information. Others may independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets.
COMPETITION
In addition to the competition we may face for ENTEREG and ADL5945 as discussed above, we face intense competition and rapid technological change in the pharmaceutical marketplace. Large and small companies, academic institutions and other public and private research organizations conduct research, seek patent protection and establish collaborative arrangements for product development in competition with us. Products developed by any of these entities may compete directly with those we may develop or sell. In addition, many of the companies and institutions that compete with us have substantially greater capital resources, research and development staffs and facilities than we have, and substantially greater experience in conducting clinical trials, obtaining regulatory approvals and manufacturing and marketing pharmaceutical products. These entities represent significant competition for us. In addition, competitors who are developing prescription pain and pain-management products might succeed in developing technologies and products that are more effective than any that we develop or sell or that would render our technology and products obsolete or noncompetitive. Competition and innovation from these or other sources potentially could negatively affect sales of our products or make them obsolete. Advances in current treatment methods also may adversely affect the market for such products. In addition, we may be at a competitive marketing disadvantage against companies that have broader product lines and whose sales personnel are able to offer more complementary products than we can. Any failure to maintain our competitive position could adversely affect our business and results of operations.
9
Table of Contents
Our competitive position also will depend upon our ability to attract and retain qualified personnel, obtain patent protection or otherwise develop proprietary products or processes and secure sufficient capital resources for the typically substantial period between technological conception and commercial sales. Competitive disadvantages in any of these factors could materially harm our business and financial condition.
MANUFACTURING
We presently do not maintain our own manufacturing facilities. We maintain an internal manufacturing organization to manage our relationships with third parties for the manufacture and supply of products for preclinical, clinical and commercial purposes. We have contracted with certain of these third-party manufacturers for the supply of certain amounts of material to meet our needs for commercial and developmental purposes. Commercial and development quantities of any products that we develop will need to be manufactured in facilities, and by processes, that comply with FDA regulations and other federal, state and local regulations.
GOVERNMENT REGULATION
Approval Process
In the United States, pharmaceutical and diagnostic products intended for use in humans are subject to rigorous FDA regulation. The process of completing clinical trials and obtaining FDA approval for a new drug is likely to take a number of years and require the expenditure of substantial resources. There can be no assurance that any of our product candidates will receive FDA approval.
The process of drug development is complex and lengthy and the activities undertaken before a new pharmaceutical product may be marketed in the United States include:
| • | discovery research; |
| • | preclinical studies; |
| • | submission to the FDA of an Investigational New Drug Application (IND), which must become effective before human clinical trials commence; |
| • | adequate and well-controlled human clinical trials to establish the safety and efficacy of the product candidate; |
| • | submission to the FDA of a NDA; and |
| • | FDA approval of the NDA prior to any commercial sale of the product. |
Preclinical studies include laboratory evaluation of product chemistry and formulation, as well as in vivo studies and other studies to assess the potential safety and efficacy of the product candidate. The results of preclinical studies are then submitted to the FDA as a part of an IND and are reviewed by the FDA prior to the commencement of human clinical trials. Unless the FDA objects to, or otherwise responds to, an IND submission, the IND becomes effective 30 days following its receipt by the FDA.
Human clinical trials are typically conducted in three sequential phases that may overlap:
| • | Phase 1: The drug candidate is initially introduced into healthy human volunteers or patients and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and clinical pharmacology. In addition, it is sometimes possible to conduct a preliminary evaluation of efficacy in Phase 1 trials. |
| • | Phase 2: This phase involves studies in a limited patient population to identify possible adverse effects and safety risks, to evaluate the efficacy of the product for specific targeted diseases and to determine optimal dosage and tolerance. |
10
Table of Contents
| • | Phase 3: When Phase 2 evaluations demonstrate that a dose range of the product candidate is effective and has an acceptable safety profile, Phase 3 trials are undertaken to further evaluate dosage and clinical efficacy, to further test for safety in an expanded patient population at geographically dispersed clinical study sites and to determine the overall risk-benefit ratio of the drug candidate and to provide an adequate basis for product labeling. |
Each trial is conducted in accordance with certain standards under protocols that detail the objectives of the study, the parameters to be used to monitor safety and the efficacy criteria to be evaluated. In the United States, each protocol must be submitted to the FDA as part of the NDA. Further, one or more independent Institutional Review Boards (IRBs) must evaluate each clinical study. The IRB considers, among other things, ethical factors, the safety of the study, the adequacy of informed consent by human subjects and the possible liability of the institution.
Data from preclinical studies and clinical trials are submitted to the FDA in an NDA for marketing approval. Preparing an NDA involves considerable data collection, verification, analyses and expense and there can be no assurance that the FDA will accept the application or grant an approval on a timely basis, if at all. The marketing or sale of pharmaceuticals in the United States may not begin without FDA approval. The approval process is affected by a number of factors, including primarily the safety and efficacy demonstrated in clinical trials and the severity of the disease. The FDA may deny an application if, in its sole discretion, it determines that applicable regulatory criteria have not been satisfied or if, in its judgment, additional testing or information is required to ensure the efficacy and safety of the product. One of the conditions for initial marketing approval, as well as continued post-approval marketing, is that a prospective manufacturer’s quality control and manufacturing procedures conform to the current Good Manufacturing Practices (cGMPs) regulations of the FDA. In complying with these regulations, a manufacturer must continue to expend time, money and effort in the area of production, quality control and quality assurance to ensure full compliance. Manufacturing establishments, both foreign and domestic, are subject to inspections by or under the authority of the FDA and by other federal, state, local or foreign agencies. Discovery of previously unknown problems with a product or manufacturer may result in restrictions on such product or manufacturer, including withdrawal of the product from the market.
Post-marketing Regulations
After FDA approval has been obtained, further studies, including Phase 4 post-marketing studies, may be required to provide additional data on safety, to validate surrogate efficacy endpoints or for other reasons, and the failure of such studies can result in a range of regulatory actions, including withdrawal of the product from the market. Further studies are required to gain approval for the use of a product as a treatment for clinical indications other than those for which the product was initially approved. As discussed above, we began a Phase 4 clinical trial in 2009 intended to evaluate the safety and efficacy of ENTEREG in patients undergoing radical cystectomy. In addition, our FDA approval letter also requires two pediatric Phase 4 clinical trials of ENTEREG. The first of these studies is expected to be initiated during 2013.
Results of post-marketing programs may limit or expand the further marketing of the products. Further, if there are any modifications to the drug, including any change in indication, manufacturing process, labeling or manufacturing facility, it may be necessary to submit an application seeking approval of such changes to the FDA. Also, the FDA can place restrictions on approval and marketing utilizing its authority under applicable regulations. For example, ENTEREG was approved subject to a REMS under which the product is available only to hospitals that perform bowel resection surgeries and are enrolled in the E.A.S.E. Program.
Additionally, the FDA regulates the labeling, storage, record keeping, advertising and promotion of prescription pharmaceuticals. Marketed products also are subject to continued regulatory oversight by the Office of Medical Policy Division of Drug Marketing, Advertising, and Communications, and the failure to comply with applicable regulations could result in marketing restrictions, financial penalties and/or other sanctions. Certain products approved by the FDA may only be marketed if the promotional materials advertising such products
11
Table of Contents
carry a so-called boxed warning. ENTEREG has a boxed warning that alerts prescribers to the restriction on ENTEREG imposed by the REMS – in particular, that no more than 15 doses of ENTEREG should be prescribed to any patient and ENTEREG should only be used in the hospital and not in an outpatient population.
The Controlled Substances Act imposes various registration, record-keeping and reporting requirements, procurement and manufacturing quotas, labeling and packaging requirements, security controls and a restriction on prescription refills on certain pharmaceutical products. A principal factor in determining the particular requirements of this act, if any, applicable to a product is its actual or potential abuse profile. A pharmaceutical product may be listed as a Schedule I, II, III, IV or V substance, with Schedule I substances considered to present the highest risk of substance abuse and Schedule V substances the lowest. Because of the potential for abuse, drugs that are able to activate mu opioid receptors in the brain (morphine-like opioid analgesics) are regulated, or scheduled, under the Controlled Substances Act.
Since we sell ENTEREG to the Department of Veterans Affairs (VA) and other governmental agencies, we are subject to additional legal requirements arising out of our status as a government contractor. In addition, we also are subject to healthcare anti-kickback statutes and false claims statutes as well as regulation under the Occupational Safety and Health Act, the Environmental Protection Act, the Clean Air Act and other federal, state and local regulations. Failure to comply with these requirements could result, among other things, in suspension of regulatory approval, recalls, injunctions, product seizures, non-coverage of our products under government healthcare programs or civil or criminal sanctions.
Hatch-Waxman Act
Under the Hatch-Waxman Act, Congress created an abbreviated FDA review process for generic versions of pioneer (brand name) drug products. The law also provides incentives by awarding, in certain circumstances, non-patent marketing exclusivities to pioneer drug manufacturers. Newly approved drug products and changes to the conditions of use of approved products may benefit from periods of non-patent marketing exclusivity in addition to any patent protection the drug product may have. The Hatch-Waxman Act provides five years of NCE marketing exclusivity to the first applicant to gain approval of an NDA for a product that does not contain an active ingredient found in any other approved product.
The Hatch-Waxman Act also provides three years of exclusivity for applications containing the results of new clinical investigations (other than bioavailability studies) essential to the FDA’s approval of new uses of approved products, such as new indications, dosage forms, strengths or conditions of use.
The Hatch-Waxman Act requires NDA applicants and NDA holders to provide certain patent information for listing in “Approved Drug Products with Therapeutic Equivalence Evaluations,” also known as the “Orange Book.” Applicants under rules relating to ANDA or applications pursuant to Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act must then certify regarding each of the FDA Orange Book-listed patents for the reference product(s). A certification that each listed patent is invalid or will not be infringed by the marketing of the applicant’s product is called a “Paragraph IV certification.” If the ANDA or 505(b)(2) applicant provides such a notification of patent invalidity or noninfringement, then the FDA may accept the ANDA or 505(b)(2) application four years after approval of the NDA.
If a Paragraph IV certification is filed and the ANDA has been accepted as a reviewable filing by the FDA, the ANDA or 505(b)(2) applicant must then within 30 days provide notice to the NDA holder and patent owner stating that the application has been submitted and providing the factual and legal basis for the applicant’s opinion that the patent is invalid or not infringed. If the NDA holder or patent owner files suit against the ANDA or 505(b)(2) applicant for patent infringement within 45 days of receiving notice of the Paragraph IV certification, a one-time 30-month stay of the FDA’s ability to approve the ANDA or 505(b)(2) application is triggered. The 30-month stay begins at the end of the NDA holder’s five years of data exclusivity or, if data exclusivity has expired, on the date that the patent holder is notified. The FDA may approve the proposed
12
Table of Contents
product before the expiration of the 30-month stay if a court finds the patent invalid or not infringed, or if the court shortens the period because the parties have failed to cooperate in expediting the litigation.
Foreign Regulation
Whether or not FDA approval has been obtained, approval of a product by regulatory authorities in foreign countries must be obtained prior to the commencement of commercial sales of the product in such countries. The requirements governing the conduct of clinical trials and product approvals vary widely from country to country, and the time required for approval may be longer or shorter than that required for FDA approval. Although there are procedures for unified filings for most European countries, in general, each country also has its own additional procedures and requirements, especially related to pricing of new pharmaceuticals. Further, the FDA and other federal agencies regulate the export of products produced in the United States and, in some circumstances, may prohibit or restrict the export even if such products are approved for sale in other countries.
LEGAL MATTERS
None.
EMPLOYEES
As of December 31, 2010, we had 75 full-time employees. Most of our senior management and professional employees have had prior experience in pharmaceutical or biotechnology companies. None of our employees is covered by collective bargaining agreements. We believe that our relations with our employees are good.
EXECUTIVE OFFICERS OF THE REGISTRANT
The names, ages and positions held by our executive officers as of the filing date of this Annual Report on Form 10-K are as follows:
| Name |
Age | Title | ||||
| Michael R. Dougherty |
53 | President, Chief Executive Officer and Director | ||||
| John M. Limongelli, Esq. |
41 | Senior Vice President, General Counsel and Secretary | ||||
| George R. Maurer |
55 | Senior Vice President, Manufacturing and Pharmaceutical Technologies | ||||
| Stephen W. Webster |
49 | Senior Vice President, Finance and Chief Financial Officer | ||||
Michael R. Dougherty. Mr. Dougherty was named as our President and Chief Executive Officer and appointed as a member of the Board of Directors on December 14, 2006. Mr. Dougherty served as our Senior Vice President, Chief Operating Officer and Treasurer from October 31, 2005 until December 14, 2006. From April 2003 until October 31, 2005, he was Chief Financial Officer. He joined us as our Senior Vice President of Commercial Operations in November 2002. From November 2000 to November 2002, Mr. Dougherty was President and Chief Operating Officer of Genomics Collaborative, Inc., a privately-held functional genomics company. Previously, Mr. Dougherty served as President and Chief Executive Officer at Genaera Corporation, formerly Magainin Pharmaceuticals Inc., a publicly-traded biotechnology company, and as Senior Vice President and Chief Financial Officer at Centocor, Inc., a publicly-traded biotechnology company. Mr. Dougherty received a B.S. from Villanova University. Mr. Dougherty has served on the Board of Directors of ViroPharma Incorporated since January 2004.
John M. Limongelli, Esq. Mr. Limongelli joined us as our Senior Vice President, General Counsel and Secretary in September 2008. From July 2002 to September 2008, Mr. Limongelli held several roles of increasing responsibility with Cephalon, Inc., most recently serving as Vice President and Associate General Counsel. Prior to joining Cephalon in 2002, Mr. Limongelli was an associate with the law firm of Morgan, Lewis & Bockius, LLP, in Philadelphia. From 1995 to 1997, Mr. Limongelli worked as a financial analyst for Bell Atlantic Corp., and from 1991 to 1995 he was an auditor with KPMG LLP. Mr. Limongelli obtained both his Juris Doctor and Masters in Business Administration from Temple University.
13
Table of Contents
George R. Maurer. Mr. Maurer was named as our Senior Vice President, Manufacturing and Pharmaceutical Technologies, in January 2009. Mr. Maurer joined Adolor in 2002 as Senior Director, Commercial Manufacturing, and then served as Vice President, Commercial Manufacturing from 2005 to January 2009. Prior to joining Adolor, Mr. Maurer was Director, Commercial Manufacturing at ViroPharma Incorporated. Prior to ViroPharma, for 22 years Mr. Maurer held positions of increasing responsibility with Schering-Plough Corporation, including directing production planning of manufacturing functions supporting domestic sales and management of manufacturing operations in Puerto Rico and New Jersey. Mr. Maurer earned his B.S. in Biology from Rensselaer Polytechnic Institute.
Stephen W. Webster. Mr. Webster joined us as our Senior Vice President, Finance and Chief Financial Officer in June 2008. From 2007 until joining Adolor, Mr. Webster was Managing Director, Investment Banking Division, Health Care Group for Broadpoint Capital (formerly First Albany Capital). From 2000 to 2006, he was with Neuronyx, Inc., a development-stage biotechnology company he co-founded, as President from 2000 to 2006 and Chief Executive Officer from 2003 to 2006. From 1987 to 2000, he served in positions of increased responsibility including as Director, Investment Banking Division, Health Care Group for PaineWebber Incorporated. He received his A.B. in Economics cum laude from Dartmouth College in 1983 and his Master of Business Administration in Finance from The Wharton School, The University of Pennsylvania in 1987. Mr. Webster serves on the Board of Directors of HearUSA Inc. and Pennsylvania Bio.
14
Table of Contents
| ITEM 1A. | RISK FACTORS |
You should carefully consider the risks described below, in addition to the other information contained in this Annual Report on Form 10-K, before making an investment decision. Our business, financial condition or results of operations could be harmed by any of these risks. The risks and uncertainties described below are not the only ones we face. Additional risks not presently known to us or other factors not perceived by us to present significant risks to our business at this time also may impair our business operations.
Our OIC program may not lead to successful drug candidates or commercially successful products.
Our future success is highly dependent on the results of our clinical development efforts, particularly with respect to our OIC program where we are testing ADL5945 in Phase 2 clinical trials. While earlier studies of ADL5945 supported the initiation of the Phase 2 studies, there is no assurance that these studies will yield positive results. If the results are not positive, we may need to discontinue development of our OIC program, which currently is our most advanced clinical program. Even if the results are positive, it is unlikely that we will have sufficient available resources to fund a Phase 3 clinical program in OIC without either securing a partner for this program or raising proceeds in the capital markets. We cannot be certain that such a partnership or capital raising transaction would be available on terms acceptable to us, if at all, particularly in light of current economic and capital market conditions. If we are able to successfully develop ADL5945, we still face significant commercial hurdles. There are a number of other products in various stages of clinical development for OIC that could have a material adverse impact on our ability to successfully market and sell ADL5945.
Ultimately, drug development is a highly uncertain process; our OIC product candidates may not be safe or effective and we may not be successful in our clinical development programs. Development of our OIC product candidates may not lead to commercially successful products.
We are and will continue to be highly dependent on the commercial success of ENTEREG, and we may be unable to attain profitability and/or positive cash flow from operations based solely on product sales of ENTEREG.
In May 2008, the FDA approved ENTEREG for use in accelerating GI recovery following partial large or small bowel resection surgery with primary anastomosis. ENTEREG is indicated for in-hospital use only and is available only to hospitals that have been registered under our E.A.S.E. Program. The commercial success of ENTEREG depends on several factors, including the following:
| • | the acceptance of ENTEREG in the medical community and the marketplace, particularly with respect to whether healthcare professionals and hospitals view ENTEREG as safe and effective for its labeled indication, whether the product carries with it an acceptable benefit-to-risk profile and whether the product offers sufficient pharmacoeconomic benefit to hospitals in light of its cost; |
| • | the number of hospitals that register for the E.A.S.E. Program; |
| • | the ability to obtain formulary acceptance of ENTEREG at registered hospitals; |
| • | the effectiveness of Glaxo’s and our sales and marketing efforts; |
| • | the occurrence of any side effects, adverse reactions or misuse (or any unfavorable publicity relating thereto) stemming from the use of ENTEREG; and |
| • | the development of competing products or therapies that accelerate GI recovery following bowel resection surgery. |
Ultimately, we may never generate sufficient revenues from ENTEREG for us to reach profitability, generate positive cash flow or sustain, on an ongoing basis, our current or projected levels of operations.
15
Table of Contents
We expect to continue to incur significant operating losses over the next several years and without additional funding, we likely will be unable to continue our operations.
We have generated operating losses in each year since we began operations in November 1994, and as of December 31, 2010, our accumulated deficit was $530.3 million and our cash, cash equivalents and short-term investments were $46.6 million. Our losses have resulted principally from costs incurred in research and development, including clinical trials, and from selling, general and administrative expenses associated with our operations. Notwithstanding our restructurings in June 2009 and July 2010, we expect to continue to generate substantial losses and utilize substantial amounts of cash, cash equivalents and short-term investments for the foreseeable future, driven primarily by expenditures related to research and development and sales and marketing activities. We believe that our existing cash, cash equivalents and short-term investments are adequate to fund our operations through mid-2012 based upon the level of research and development and marketing and administrative activities we believe will be necessary to achieve our strategic objectives. We cannot be sure that we will ever achieve significant product revenue from ENTEREG or any of our other product candidates sufficient for us to generate positive cash flows from operations. Sales of ENTEREG will need to increase significantly beyond current levels before we will be able to achieve profitability and/or positive cash flows from operations. Even if we are able to achieve profitability, we may be unable to maintain profitability on a continuing basis or at a level sufficient to support our current or projected levels of continuing investment. We anticipate that we will need to obtain funding to support our operational needs in the future, and we cannot be certain that such funding will be available on terms acceptable to us, if at all, particularly in light of current economic and capital market conditions. Any capital raising necessary to continue our operations may be through one or a combination of approaches, which could include public or private financing, issuances of equity or debt, sale or partnering of one or more of our development programs or other strategic initiatives. Any public or private financings involving issuances of common stock or other classes of our equity would likely dilute existing stockholders’ percentage ownership. If we are unable to obtain funding to support operations, we will be forced to curtail our operations and we may be less likely to develop products successfully.
Restrictions on ENTEREG may have the effect of limiting the commercial prospects for the product.
In April 2007, Glaxo and we announced the results of Study 014, a Phase 3 long-term safety study of alvimopan in patients taking opioids for chronic non-cancer pain and experiencing OBD. Results from Study 014 showed a higher number of myocardial infarctions reported by patients treated long-term (1 year) with alvimopan compared to placebo. As a result, ENTEREG was approved in its labeled indication by the FDA subject to a Risk Evaluation and Mitigation Strategy (REMS). The FDA has determined that a REMS is necessary to ensure that the benefits of ENTEREG outweigh the risks. The REMS is subject to modification by the FDA at anytime and it is possible that the FDA could require changes to the REMS or other restrictions that would make it even more difficult to market and sell ENTEREG.
Our ENTEREG product labeling carries a boxed warning that ENTEREG is available only for short-term (15 doses) use in hospitalized patients. The REMS and the boxed warning may make it more difficult to market and sell ENTEREG. We are not permitted to market and sell ENTEREG to hospitals that do not register in the E.A.S.E. Program. Hospitals may be unwilling or unable to comply with the requirements for registration in the E.A.S.E. Program. For example, hospitals may not have systems, order sets, protocols or other measures in place to limit the use of ENTEREG to no more than 15 doses per patient and to ensure in-hospital use only. Hospitals also may not have controls in place to ensure that ENTEREG will neither be dispensed for outpatient use nor be transferred to unregistered hospitals. In such cases, we will be unable to register these hospitals in the E.A.S.E. Program.
Selling a pharmaceutical product in the hospital setting presents certain challenges. Hospitals differ widely and each hospital’s or hospital group’s prescribing is influenced by a list of accepted drugs called a formulary. Most hospitals have a committee, often called a pharmacy and therapeutics (P&T) committee, which meets periodically to determine which pharmaceutical products to add to the formulary. Many factors are assessed by
16
Table of Contents
such committees, including the cost of the drug and its pharmacoeconomic profile. Once a pharmaceutical is on formulary, it is easier for a physician within a hospital or hospital group to prescribe the drug. Hospital formulary approval is critical if ENTEREG is to become a commercial success and we cannot assure you that a sufficient number of hospitals will include ENTEREG on their formulary.
Notwithstanding success in registering hospitals in the E.A.S.E. Program and having those registered hospitals include ENTEREG on their formulary, there can be no assurance that such hospitals will order ENTEREG in meaningful amounts, if at all.
Our stock price has been highly volatile, and your investment in our stock could decline significantly in value.
The market price for our common stock has been, and may continue to be in the future, highly volatile. For example, during the period January 1, 2008 through December 31, 2010, the price of our common stock reached a low of $1.00 per share on July 7, 2010 and a high of $6.09 per share on May 21, 2008.
The market price for our common stock is highly dependent on, among other things, the performance of ENTEREG in the market, the success of our product development efforts, the amount of our available cash and investments and our level of cash utilization. Negative announcements, including, among others:
| • | disappointing sales of ENTEREG; |
| • | negative clinical trial results or adverse regulatory decisions for our product candidates; |
| • | disappointing developments concerning our collaborations; |
| • | adverse period-to-period fluctuations in our sales or operating results or financial results that fall below the market’s expectations; or |
| • | legal challenges, disputes and/or other adverse developments impacting patents or other proprietary rights that protect our products, |
could trigger a significant decline in the price of our common stock. In addition, external events, such as news concerning economic or market conditions in the general economy or within our industry, the activities of our competitors or our customers, changes in U.S. or foreign government regulations impacting our industry or the movement of capital into or out of our industry, also are likely to affect the price of our common stock, regardless of our operating performance. Further declines in our stock price or our failure to meet other conditions required by the NASDAQ Global Market could impact our continued listing on the NASDAQ Global Market.
Our success is highly dependent on the efforts of our collaborators and third-party contractors and on our ability to secure future partnerships.
Because we have limited resources, we seek to enter into, and in the past we have entered into, agreements with other pharmaceutical companies and third-party contractors. These agreements may call for our partners to control or play a significant role in, among other things:
| • | the development of a product candidate, including, among other things, toxicology, formulation and clinical research efforts; |
| • | the design and execution of clinical studies; |
| • | the process of obtaining regulatory approval to market the product; and |
| • | the manufacturing, marketing and selling of any approved product. |
17
Table of Contents
In each of these areas, our partners may not support fully our research and commercial interests since our programs may compete for time, attention and resources with the internal programs of our corporate collaborators. As such, we cannot be sure that our corporate collaborators will share our perspectives on the relative importance of our program, that they will commit sufficient resources to our program to move it forward effectively or that the program will advance as rapidly as it might if we had retained complete control of all research, development, regulatory and commercialization decisions.
We are highly dependent on the efforts and expertise of Glaxo in the distribution, marketing and selling of ENTEREG. Presently, ENTEREG is detailed primarily by Glaxo’s national hospital-based sales organization. We are co-promoting ENTEREG in certain hospitals with a field force that numbers approximately 25 persons.
Any failure by our partners to perform their obligations or any decision by our partners to terminate these agreements could negatively impact our ability to successfully develop, obtain regulatory approvals for and commercialize our product and product candidates, which would likely materially impact our financial condition, results of operations and outlook. In addition, any termination of our collaboration agreements will terminate the funding we may receive under the relevant collaboration agreement and may materially and adversely impact our ability to fund further development efforts and our progress in our development programs. In December 2010, Pfizer and we announced that we were discontinuing further development of our delta opioid receptor agonist compounds, ADL5859 and ADL5747. We had been developing such compounds under an exclusive worldwide license and collaboration agreement; however, clinical evaluation in multiple indications had not yielded compelling evidence for either party to continue the development program. Pfizer delivered to us a notice of termination of the collaboration agreement, which will be effective as of March 2011.
In the future, we will likely seek to enter into other collaborative arrangements for the development, marketing, sale and/or distribution of certain of our product candidates, including in our OIC program, which may require us to share profits or revenues. Despite our efforts, we may be unable to secure additional collaborative licensing or other arrangements that are necessary for us to further develop and commercialize our product candidates. Moreover, we may not realize the contemplated benefits from such collaborative licensing or other arrangements. These arrangements may place responsibility on our collaborative partners for preclinical testing, human clinical trials, the preparation and submission of applications for regulatory approval, or for marketing, sales and distribution support for product commercialization. These arrangements also may require us to transfer certain material rights or issue our equity securities to corporate partners, licensees or others. Moreover, we may not derive any revenues or profits from these arrangements.
The results and timing of future clinical trials cannot be predicted and future setbacks may materially and adversely affect our business.
We must demonstrate through preclinical testing and clinical trials that a product candidate is safe and efficacious. The results from preclinical testing and early clinical trials may not be predictive of results obtained in subsequent clinical trials, and we cannot be sure that these clinical trials will demonstrate the safety and efficacy necessary to obtain regulatory approval for any product candidates.
Many companies in the biotechnology and pharmaceutical industries have suffered significant setbacks in advanced clinical trials. Product candidates that appear to be promising at earlier stages of development may not reach the market or be marketed successfully for a number of reasons, including, but not limited to, the following:
| • | researchers may find during later preclinical testing or clinical trials that the product candidate is ineffective or has harmful side effects; |
| • | new information about the mechanisms by which a drug candidate works may adversely affect its development; |
18
Table of Contents
| • | one or more competing products may be approved for the same or a similar condition, raising the hurdles to approval of the product candidate; |
| • | the product candidate may fail to receive necessary regulatory approval or clearance; or |
| • | competitors may market equivalent or superior products. |
The completion of clinical trials of our product candidates may be delayed by many factors, one of which is the rate of enrollment of patients. Neither we nor our collaborators can control the rate at which patients present themselves for enrollment, and we cannot be sure that the rate of patient enrollment will be consistent with our expectations or be sufficient to enable clinical trials of our product candidates to be completed in a timely manner. In addition, we contract with third parties to conduct our clinical trials, and are subject to the risk that these third parties fail to perform their obligations properly and in compliance with applicable FDA and other governmental regulations. Any significant delays in, or termination of, clinical trials of our product candidates may have a material adverse effect on our business and the price of our common stock.
We cannot be sure that we will be permitted by regulatory authorities to undertake additional clinical trials for any of our product candidates, or that if such trials are conducted, any of our product candidates will prove to be safe and efficacious or will receive regulatory approvals. In addition, we, or a regulatory authority, may suspend ongoing clinical trials at any time if the subjects participating in the trial are exposed to unacceptable health risks or if the regulatory agency finds deficiencies in the conduct of the trials. Any delays in or termination of these clinical trial efforts could have a material and adverse effect on our business.
We have no commercial manufacturing capability and infrastructure, and we rely on third parties to manufacture our product and product candidates in sufficient quantities, at an acceptable cost and in compliance with regulatory requirements.
We depend on Piramal Healthcare (Canada) Limited (formerly Torcan Chemical Ltd.) and Central Glass Germany GmbH (formerly Girindus AG) as the approved suppliers under our NDA of the active pharmaceutical ingredient in ENTEREG. We also depend on Pharmaceutics International, Inc. and Pharmaceutical Manufacturers Research Services Inc. as the approved manufacturers under our NDA of ENTEREG finished capsules. These suppliers have been inspected by the FDA but are subject to periodic re-inspection. In the event that these vendors cannot supply material for any reason, FDA approval is required to change or add new suppliers and manufacturers and obtaining such approval could result in delays that disrupt the supply of the product. While we seek to maintain inventories of the active pharmaceutical ingredient and finished products to protect against supply disruptions, if they were to occur they could materially and adversely affect the commercial success of ENTEREG.
Completion of our clinical trials and commercialization of our product candidates require access to, or development of, facilities to manufacture a sufficient supply of our product candidates. We have depended, and expect to continue to depend, on third parties for the manufacture of our product candidates for preclinical, clinical and commercial purposes. While we have established relationships with a number of different third-party manufacturing service providers, we may not be able to contract for the manufacture of sufficient quantities of the products we develop, or meet our needs for preclinical or clinical development. Our products may be in competition with other products for manufacturing capacity of third parties and suitable alternatives may be unavailable. Consequently, our products may be subject to manufacturing delays if outside contractors give their own or other products greater priority than ours. It is difficult, time consuming and expensive to change contract manufacturers for pharmaceutical products. Our dependence upon others for the manufacture of our products may adversely affect our future profit margin and our ability to commercialize products, if additional products are approved, on a timely and competitive basis.
To receive regulatory approval for any future product, contract manufacturers will need to be identified that can obtain FDA approval of their manufacturing facilities used to manufacture that product, and there is a risk
19
Table of Contents
that such approval may not be obtained. In an NDA, we are required to submit information and data regarding chemistry, manufacturing and controls which satisfy the FDA that our contract manufacturers are able to make that product in accordance with cGMPs. Under cGMPs, our manufacturers and we will be required to manufacture our products and maintain records in a prescribed manner with respect to manufacturing, testing and quality control activities. We are dependent on our third-party manufacturers to comply with these regulations in the manufacture of our products and these parties may have difficulties complying with cGMPs. The failure of any third-party manufacturer to comply with applicable government regulations could cause us substantial harm by delaying or preventing regulatory approval and marketing of our products.
It is difficult and costly to protect our intellectual property rights, and we cannot ensure the protection of these rights; we may be sued by others for infringing their intellectual property.
Our commercial success will depend, in part, on obtaining patent protection for new technologies, products and processes and successfully defending these patents against third-party challenges. To that end, we file applications for patents covering the compositions, uses and proprietary processes for the manufacture of our product and product candidates. The patent positions of pharmaceutical and biotechnology companies can be highly uncertain and involve complex legal, scientific and factual questions. Accordingly, we cannot predict the breadth of claims allowed in our patents or those of our collaborators. The patents and patent applications relating to our products, product candidates and technologies may be challenged, invalidated or circumvented by third parties and might not protect us against competitors with similar products or technology. Patent disputes in our industry are frequent and can preclude commercialization of products. If we ultimately engage in and lose any such disputes, we could be subject to competition and/or significant liabilities, we could be required to enter into third-party licenses or we could be required to cease using the technology or product in dispute. In addition, even if such licenses are available, the terms of any license requested by a third party could be unacceptable to us.
Others have filed, and in the future are likely to file, patent applications covering products and technologies that are similar, identical or competitive to ours, or important to our business. We cannot be certain that any patent application owned by a third party will not have priority over patent applications filed or in-licensed by us, or that we or our licensors will not be involved in interference proceedings before U.S. or foreign patent offices.
Although no third party has asserted a claim of infringement against us, others may hold proprietary rights that could prevent our product candidates from being marketed unless we can obtain a license to those proprietary rights. Any patent-related legal action against our collaborators or us claiming damages and seeking to enjoin commercial activities relating to our products and processes could subject us to potential liability for damages and require our collaborators or us to obtain a license to continue to manufacture or market the affected products and processes. We cannot predict whether our collaborators or we would prevail in any such actions or that any license required under any of these patents would be made available on commercially acceptable terms, if at all.
There has been, and we believe that there will continue to be, significant litigation in the industry regarding patent and other intellectual property rights. The results of patent litigation among third parties has caused, and may continue to cause, changes to the ways patents are interpreted, enforced and/or challenged. These changes may adversely affect our ability to protect our products. If we become involved in litigation, it could consume substantial managerial and financial resources.
We also rely on trade secrets to protect technology in cases when we believe patent protection is not appropriate or obtainable. However, trade secrets are difficult to protect. While we require employees, academic collaborators, consultants and other contractors to enter into confidentiality agreements, we may not be able to adequately protect our trade secrets or other proprietary information. Our research collaborators and scientific advisors have rights to publish data and information in which we have rights. If we cannot maintain the confidentiality of our technology and other confidential information in connection with our collaborators and advisors, our ability to receive patent protection or protect our proprietary information may be imperiled.
20
Table of Contents
We are a party to various license agreements that give us rights to use specified technologies in our research and development processes. If we are not able to continue to license such technology on commercially reasonable terms, our product development and research may be delayed. In addition, we generally do not fully control the prosecution of patents relating to in-licensed technology (or technology subject to a collaboration) and, accordingly, are unable to exercise the same degree of control over this intellectual property as we exercise over our internally developed technology.
Our long-term success depends upon our ability to discover, license and develop, receive regulatory approval for and commercialize our product candidates.
Our product candidates require governmental approvals prior to commercialization. Because these product candidates are in development, we face the substantial risks of failure inherent in developing drugs based on new technologies. Our product candidates must satisfy rigorous standards of safety and efficacy before the FDA and foreign regulatory authorities will approve them for commercial use. There can be no assurance that these standards will remain consistent over time, further complicating our ability to obtain marketing approvals for our product candidates. To satisfy these standards, we will need to conduct significant additional research, preclinical testing and clinical trials.
Preclinical testing and clinical development are long, expensive and highly uncertain processes. Failure can occur at any stage of testing. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful. Based on results at any stage of clinical trials, we may decide to discontinue development of our product candidates. Even if we obtain approval and begin marketing a product, ongoing clinical trials, including for other indications, may result in additional information that could affect our ability or decision to continue marketing the product. Even if we receive regulatory approval for our product candidates, we must comply with applicable FDA post-marketing regulations governing manufacturing, promotion, labeling, risk management and reporting of adverse events and other information, as well as other regulatory requirements. Failure to comply with applicable regulatory requirements could subject us to criminal penalties, civil penalties, recall or seizure of products, withdrawal of marketing approval, total or partial suspension of production or injunction, as well as other regulatory actions against our product or us.
In each of 2009 and 2010 we undertook restructurings to reduce costs and realign our workforce and operations based on current business objectives. During 2011, we will be funding Phase 2 studies of ADL5945 in OIC and our ongoing Phase 4 study of ENTEREG in patients undergoing radical cystectomy. While we also expect to continue to conduct research, preclinical studies and process development activities on our other product candidates, the level of such expenditures will be significantly reduced compared to previous years, which may limit or delay our ability to discover new products or develop our product candidates and may increase the risk that our long-term business objectives will not be met.
Despite our limited resources, we intend to explore opportunities to expand our product portfolio by acquiring or in-licensing product candidates. Although we conduct extensive evaluations of product candidate opportunities as part of our due diligence efforts, there can be no assurance that our product candidate development efforts related thereto will be successful or that we will not become aware of issues or complications that will cause us to alter, delay or terminate our product candidate development efforts.
If we market products in a manner that violates healthcare fraud and abuse laws, or if we violate government price reporting laws, we may be subject to civil or criminal penalties.
In addition to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal healthcare fraud and abuse laws have been applied in recent years to restrict certain marketing practices in the pharmaceutical industry. These laws include false claims statutes and anti-kickback statutes. Because of the breadth of these laws and the narrowness of the safe harbors, it is possible that some of our business activities could be subject to challenge under one or more of these laws.
21
Table of Contents
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government or knowingly making, or causing to be made, a false statement to get a false claim paid. The federal healthcare program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce, or in return for, purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Although there are several statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchasing or recommending may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Our practices may not in all cases meet all of the criteria for safe harbor protection from anti-kickback liability.
Over the past several years, numerous pharmaceutical and other healthcare companies have been prosecuted under these laws for a variety of alleged promotional and marketing activities, such as: allegedly providing free trips, free goods, sham consulting fees and grants and other monetary benefits to prescribers; reporting to pricing services inflated average wholesale prices that were then used by federal programs to set reimbursement rates; engaging in off-label promotion that caused claims to be submitted to Medicaid for non-covered, off-label uses; and submitting inflated best price information to the Medicaid Rebate Program to reduce liability for Medicaid rebates. Most states also have statutes or regulations similar to the federal anti-kickback law and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. Sanctions under these federal and state laws may include civil monetary penalties, exclusion of a manufacturer’s products from reimbursement under government programs, criminal fines and imprisonment.
We are required to submit pricing data to the federal government as a condition of selling ENTEREG to healthcare facilities of the VA, Department of Defense (DoD), Public Health Service and U.S. Coast Guard (collectively, the VA Network). These price reports are used to determine the amount of discount that must be provided to the VA Network. Pharmaceutical manufacturers have been prosecuted under false claims laws for knowingly submitting inaccurate pricing information to the government to reduce their liability for providing discounts. The rules governing the calculation of these reported prices are complex. We depend upon Glaxo to calculate these prices for ENTEREG, and it is possible that the methodologies we are using for calculating these prices could be challenged under false claims laws or other laws. If this were to occur, we could face substantial liability.
We face product liability risks, which may have a negative effect on our financial performance.
The administration of drugs to humans, whether in clinical trials or commercially, can result in product liability claims regardless of whether or not the drugs are actually at fault for causing an injury. The product labeling for ENTEREG includes a boxed warning that ENTEREG is available only for short-term (15 doses) use in hospitalized patients. The product label informs physicians that there were more reports of myocardial infarctions in patients treated with alvimopan 0.5 milligrams twice daily compared with placebo-treated patients in a 12-month study of patients treated with opioids for chronic pain. ENTEREG also is marketed and sold under a REMS.
If ENTEREG is used more widely or if physicians prescribe the product for conditions other than its labeled indication, the likelihood of an adverse drug reaction, unintended side effect or incident of misuse may increase. Product liability claims can be expensive to defend and may result in large judgments or settlements against us, which could have a negative effect on our financial performance or condition. We maintain product liability insurance and self-insurance retentions in amounts we believe to be commercially reasonable, but which may not cover the potential liability associated with significant product liability claims. Even if a product liability claim is not successful, the adverse publicity and time and expense of defending such a claim may harm the commercial
22
Table of Contents
prospects for the product or otherwise interfere with our business. Regardless of whether we face product liability lawsuits, we may be required to disclose adverse events associated with ENTEREG. Although ENTEREG may not be the cause of these adverse events, these disclosures could nevertheless have a material adverse effect on our sales of ENTEREG and our financial condition.
Additionally, we enter into various agreements where we indemnify third parties such as manufacturers and investigators for certain product liability claims related to our products. These indemnification obligations may require us to pay significant sums of money for claims that are covered by these indemnifications.
If competitors develop and market products that are more effective, have fewer side effects, are less expensive than our product or product candidates or offer other advantages, our commercial opportunities will be limited.
Other companies have product candidates in development to treat the conditions we are seeking to treat and they may develop effective and commercially successful products. Our competitors may succeed in developing products that are more effective than those that we may develop, that have fewer side effects, that are less expensive or that they market before we market any products we may develop.
We are aware of other products in various stages of clinical development for the acceleration of GI recovery following bowel resection surgery, OIC and other manifestations of opioid bowel dysfunction. For the acceleration of GI recovery following bowel resection surgery, we are aware of molecules in development by Tranzyme Pharma, Helsinn Therapeutics and other companies that could compete with ENTEREG at some point in the future. For OIC, we are aware of molecules in development by Progenics Pharmaceuticals, Inc. in collaboration with Salix Pharmaceuticals Inc., AstraZeneca PLC in collaboration with Nektar Therapeutics, Theravance Inc. and Alkermes, Inc. that could compete with our OIC product candidates. There are additional competitive products being developed that could have a material adverse effect on our ability to successfully market and sell our product and product candidates.
Our competitors include fully-integrated pharmaceutical companies and biotechnology companies, universities and public and private research institutions. Many of the organizations competing with us have substantially greater capital resources, larger research and development staffs and facilities, greater experience in drug development and in obtaining regulatory approvals, and greater manufacturing and marketing capabilities than we do. These organizations also compete with us to:
| • | attract partners for acquisitions, joint ventures or other collaborations; |
| • | license proprietary technology; and |
| • | attract qualified personnel. |
Reductions in the number of bowel resection surgeries or in the use of opioid analgesics would reduce the potential market for ENTEREG and any products developed to treat OIC.
ENTEREG is indicated for the acceleration of gastrointestinal recovery following partial small or large bowel resection surgery with primary anastomosis. If medical advances or surgical techniques are developed that reduce the need for these surgeries, the potential market for ENTEREG would be similarly reduced. Moreover, if the use of drugs or techniques that reduce the requirement for mu opioid analgesics becomes more widespread, the market for ENTEREG and our OIC product candidates would decrease. Various techniques to reduce the use of opioids are being utilized in an attempt to reduce the impact of opioid side effects. The use of local anesthetics in epidural catheters during and after surgery with the continuation of the epidural into the postoperative period can reduce or eliminate the use of opioids. Non-steroidal anti-inflammatory agents also may reduce total opioid requirements. Continuous infusion of local anesthetic into a wound or near major nerves can reduce the use of opioids in limited types of procedures and pain states. Novel analgesics that act at receptors other than mu opioid
23
Table of Contents
receptors also are under development. Many companies have developed and are developing analgesic products that compete with opioids or which, if approved, would compete with opioids. If these analgesics significantly reduce the use of opioids, it would have a negative impact on the potential market for ENTEREG and our OIC product candidates.
Our ability to generate product sales could diminish if we fail to obtain acceptable prices or an adequate level of reimbursement for our products from third-party payors.
Our ability to successfully commercialize pharmaceutical products, alone or with collaborators, may depend in part on the extent to which reimbursement is available from government and health administration authorities or private health insurers and third-party payors.
The continuing efforts of government and third-party payors to contain or reduce the costs of healthcare through various means may limit our commercial opportunity. Increasing emphasis on managed care in the United States will continue to put pressure on the pricing of in-patient hospital procedures and pharmaceutical products. Significant uncertainty exists as to the reimbursement status of newly approved healthcare products. Cost control initiatives could adversely affect the commercialization of our products, decrease the price received for any products in the future and may impede patients’ ability to obtain reimbursement under their insurance programs for our products.
In the case of partial large or small bowel resection surgery, a hospital typically will be reimbursed a fixed fee for the procedure by a private health insurer or Medicare. Pharmaceutical products such as ENTEREG that may be used in connection with the surgery are not separately reimbursed and, therefore, a hospital must assess the cost of ENTEREG relative to its benefits. Current and future efforts to limit the level of reimbursement for in-patient hospital procedures could cause a hospital to decide to not use ENTEREG or to discontinue use of the product.
We are or may become involved in legal proceedings that, if adversely adjudicated or settled, could materially impact our financial condition.
As a public biopharmaceutical company, we are or may become a party to litigation in the ordinary course of our business, including, among others, matters alleging product liability, patent or other intellectual property rights infringement, patent invalidity, violations of securities laws, employment discrimination or breach of commercial contract. In general, litigation claims can be expensive and time consuming to bring or defend against and could result in settlements or damages that could significantly impact our results of operations and financial condition.
Our business could suffer if we are unable to attract, retain and motivate skilled personnel and cultivate key academic collaborations.
We are a small company, and our success depends on our continued ability to attract, retain and motivate highly-qualified management and scientific personnel. Moreover, we may not be successful in attracting qualified individuals to replace employees or to expand our business should the need arise. If we lose the services of any of our existing personnel or if we fail to appropriately motivate them, it could impede significantly the achievement of our objectives. We do not know if we will be able to attract, retain or motivate personnel or maintain important academic, scientific and commercial relationships. We do not maintain key man life insurance on any of our employees.
If we engage in an acquisition, reorganization or business combination, we will incur a variety of risks that could adversely affect our business operations or our stockholders.
From time to time we have considered, and we will continue to consider in the future, strategic business initiatives intended to further the development of our business. These initiatives may include acquiring
24
Table of Contents
businesses, technologies or products or entering into a business combination with another company. If we do pursue such a strategy, we could, among other things:
| • | issue equity securities that would dilute our current stockholders’ percentage ownership; |
| • | incur substantial debt that may place strains on our operations; |
| • | spend substantial operational, financial and management resources in integrating new businesses, technologies and products; |
| • | assume substantial actual or contingent liabilities; or |
| • | merge with, or otherwise enter into a business combination with, another company in which our stockholders would receive cash or shares of the other company or a combination of both on terms that our stockholders may not deem desirable. |
We are not in a position to predict what, if any, collaborations, alliances or other transactions may result or how, when or if these activities would have a material effect on us or the development of our business.
Certain provisions of both our charter documents and Delaware law, our adoption of a stockholder rights plan and certain limitations within our collaboration agreements may make an acquisition of us, which may be beneficial to our stockholders, more difficult.
Provisions of our amended and restated certificate of incorporation and restated bylaws, as well as provisions of Delaware law, could make it more difficult for a third party to acquire us, even if doing so would benefit our stockholders.
Authorized shares of our common stock and preferred stock are available for future issuance without stockholder approval. The existence of unissued common stock and preferred stock may enable our Board of Directors to issue shares to persons friendly to current management or to issue preferred stock with terms that could render more difficult or discourage a third-party attempt to obtain control of us by means of a merger, tender offer, proxy contest or otherwise, which would protect the continuity of our management. In addition, we adopted a stockholder rights plan, the effect of which may be to make an acquisition of us more difficult.
Our amended and restated certificate of incorporation provides for our Board of Directors to be divided into three classes, with the term of one such class expiring each year, and we have eliminated the ability of our stockholders to consent in writing to the taking of any action pursuant to Section 228 of the Delaware General Corporation Law.
Under our collaboration agreements with Glaxo and Pfizer, there are certain limitations on the ability of Glaxo and Pfizer to acquire our securities. These limitations make it more difficult for Glaxo or Pfizer to acquire us, even if such an acquisition would benefit our stockholders. The limitations do not prevent Glaxo or Pfizer, among other things, from acquiring our securities in certain circumstances following initiation by a third party of an unsolicited tender offer to purchase more than a certain percentage of any class of our publicly traded securities. As a result of the termination of our collaboration agreement with Pfizer effective March 2011, the limitations on Pfizer’s ability to acquire our securities will terminate effective March 2013.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None.
| ITEM 2. | PROPERTIES |
We lease our corporate headquarters in Exton, Pennsylvania, under a ten-year lease agreement expiring in May 2013. The lease includes a renewal option for two consecutive additional five-year periods and we have a
25
Table of Contents
purchase option exercisable at the tenth year of the lease term. The building has approximately 80,000 square feet of space consisting of approximately 30,000 square feet of administrative office space, 25,000 square feet of laboratory space and 25,000 square feet of unfinished space. We have subleased approximately 10,000 square feet of laboratory and office space, with a term that expires in April 2013.
| ITEM 3. | LEGAL PROCEEDINGS |
None.
| ITEM 4. | (REMOVED AND RESERVED) |
26
Table of Contents
PART II
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Our common stock is traded on the NASDAQ Global Market under the symbol “ADLR.” The price range per share reflected in the table below is the highest and lowest per share sales price for our stock as reported by the NASDAQ Global Market during each quarter of the two most recent years.
| High | Low | |||||||
| 2010 |
||||||||
| First Quarter |
$ | 2.20 | $ | 1.43 | ||||
| Second Quarter |
2.24 | 1.04 | ||||||
| Third Quarter |
1.22 | 1.00 | ||||||
| Fourth Quarter |
1.37 | 1.05 | ||||||
| 2009 |
||||||||
| First Quarter |
$ | 2.55 | $ | 1.61 | ||||
| Second Quarter |
2.38 | 1.45 | ||||||
| Third Quarter |
2.04 | 1.44 | ||||||
| Fourth Quarter |
1.68 | 1.33 | ||||||
As of February 18, 2011, there were 114 holders of record of our common stock. This does not reflect beneficial stockholders who hold their stock in nominee or “street” name through various brokerage firms. On February 18, 2011, the last reported sale price of our common stock as reported on the NASDAQ Global Market was $1.44 per share.
We have never declared or paid cash dividends on our capital stock, and we do not intend to pay cash dividends in the foreseeable future. We plan to retain earnings, if any, for use in the operation of our business and to fund future growth.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table gives information about our common stock that may be issued upon the exercise of stock options, warrants and rights under all of our existing equity compensation plans as of December 31, 2010, including the Amended and Restated 2003 Stock-Based Incentive Compensation Plan (the 2003 Plan). There were no shares available for future grants under the 1994 Amended and Restated Equity Compensation Plan, which expired in February 2010.
Equity Compensation Plan Information
| Plan Category |
(a) Number of Securities to be Issued Upon Exercise of Outstanding Options, Warrants and Rights |
(b) Weighted Average Exercise Price of Outstanding Options, Warrants and Rights |
(c) Number of Securities Remaining Available for Future Issuance (Excludes Securities Reflected in Column (a)) |
|||||||||
| Equity compensation plans approved by stockholders |
5,147,418 | (1) | $ | 5.78 | 878,743 | (2) | ||||||
| Equity compensation plans not approved by stockholders |
— | — | ||||||||||
| Total |
5,147,418 | $ | 5.78 | 878,743 | ||||||||
| (1) | Does not include awards covering 1,919,343 shares of unvested restricted and deferred stock awards that are outstanding as of December 31, 2010. |
| (2) | Consists of shares available for grant by us under our 2003 Plan. |
27
Table of Contents
Performance Graph
The following graph compares the cumulative five-year total return on our common stock relative to the cumulative total returns of the NASDAQ Composite Index and the NASDAQ Biotechnology Index. An investment of $100 (with reinvestment of all dividends) is assumed to have been made in our common stock and in each of the indexes on December 31, 2005 and its relative performance is tracked through December 31, 2010.
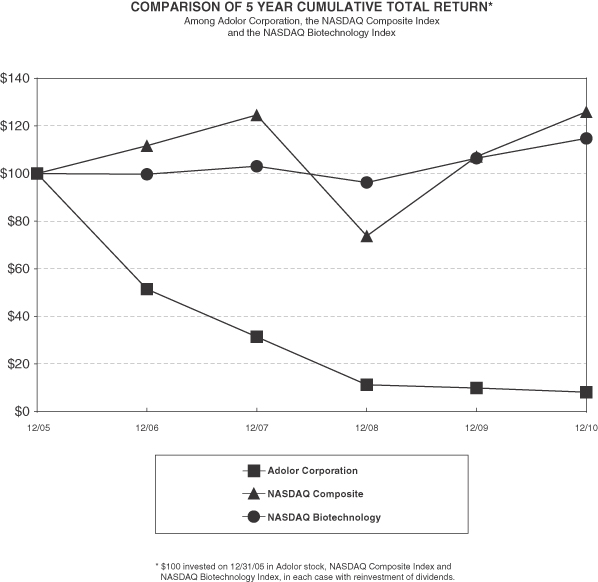
| As of December 31, | ||||||||||||||||||||||||
| 2005 | 2006 | 2007 | 2008 | 2009 | 2010 | |||||||||||||||||||
| Adolor Corporation |
100.00 | 51.51 | 31.51 | 11.37 | 10.00 | 8.29 | ||||||||||||||||||
| NASDAQ Composite Index |
100.00 | 111.74 | 124.67 | 73.77 | 107.12 | 125.93 | ||||||||||||||||||
| NASDAQ Biotechnology Index |
100.00 | 99.71 | 103.09 | 96.34 | 106.49 | 114.80 | ||||||||||||||||||
The stock price performance included in this graph is not necessarily indicative of future stock price performance.
28
Table of Contents
| ITEM 6. | SELECTED FINANCIAL DATA |
The following selected consolidated financial data should be read in conjunction with our consolidated financial statements and the related notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included in Part II, Item 7 in this Annual Report on Form 10-K. The Consolidated Statements of Operations data for the years ended December 31, 2010, 2009 and 2008 and the Consolidated Balance Sheet data as of December 31, 2010 and 2009 are derived from our audited consolidated financial statements which are included elsewhere in this Annual Report on Form 10-K. The Consolidated Statements of Operations data for the years ended December 31, 2007 and 2006 and the Consolidated Balance Sheet data as of December 31, 2008, 2007 and 2006 are derived from audited consolidated financial statements not included in this Annual Report on Form 10-K. Historical results are not necessarily indicative of the results to be expected in the future.
| Year ended December 31, | ||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||||||
| Consolidated Statements of Operations |
||||||||||||||||||||
| Product sales, net |
$ | 25,386 | $ | 14,609 | $ | 1,248 | $ | — | $ | — | ||||||||||
| Contract revenues |
17,916 | 22,752 | 48,208 | 9,120 | 15,087 | |||||||||||||||
| Total revenues, net |
43,302 | 37,361 | 49,456 | 9,120 | 15,087 | |||||||||||||||
| Operating expenses incurred: |
||||||||||||||||||||
| Cost of product sales |
2,876 | 1,515 | 204 | — | — | |||||||||||||||
| Research and development |
33,210 | 43,930 | 52,664 | 41,610 | 56,387 | |||||||||||||||
| Selling, general and administrative |
34,053 | 36,948 | 31,115 | 23,970 | 35,458 | |||||||||||||||
| Restructuring charge |
1,919 | 3,933 | — | — | 2,504 | |||||||||||||||
| Total operating expenses |
72,058 | 86,326 | 83,983 | 65,580 | 94,349 | |||||||||||||||
| Loss from operations |
(28,756 | ) | (48,965 | ) | (34,527 | ) | (56,460 | ) | (79,262 | ) | ||||||||||
| Interest income and other income, net |
1,482 | 1,051 | 4,405 | 8,017 | 9,524 | |||||||||||||||
| Net loss |
$ | (27,274 | ) | $ | (47,914 | ) | $ | (30,122 | ) | $ | (48,443 | ) | $ | (69,738 | ) | |||||
| Basic and diluted net loss per share |
$ | (0.59 | ) | $ | (1.03 | ) | $ | (0.65 | ) | $ | (1.05 | ) | $ | (1.56 | ) | |||||
| Shares used in computing basic and diluted net loss per share |
46,339 | 46,296 | 46,158 | 45,933 | 44,731 | |||||||||||||||
| As of December 31, | ||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Consolidated Balance Sheet |
||||||||||||||||||||
| Cash, cash equivalents and short-term investments |
$ | 46,587 | $ | 83,206 | $ | 131,910 | $ | 167,189 | $ | 185,562 | ||||||||||
| Working capital |
38,345 | 66,989 | 112,250 | 147,543 | 173,130 | |||||||||||||||
| Total assets |
52,758 | 91,459 | 144,427 | 178,677 | 200,598 | |||||||||||||||
| Accumulated deficit |
(530,255 | ) | (502,981 | ) | (455,067 | ) | (424,944 | ) | (376,502 | ) | ||||||||||
| Total stockholders’ equity |
19,658 | 44,054 | 88,619 | 112,353 | 153,181 | |||||||||||||||
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
Management’s Discussion and Analysis of Financial Condition and Results of Operations (MD&A) is intended to provide information to assist you in better understanding and evaluating our financial condition and results of operations. We encourage you to read this MD&A in conjunction with our consolidated financial statements included in Part II, Item 8 of this Annual Report on Form 10-K and the “Risk Factors” contained in Part I, Item 1A of this Annual Report on Form 10-K.
29
Table of Contents
EXECUTIVE SUMMARY
We are a biopharmaceutical company focused on the discovery, development and commercialization of novel prescription pain and pain-management products. In May 2008, the U.S. Food and Drug Administration (FDA) approved our first product, ENTEREG® (alvimopan). ENTEREG is indicated to accelerate upper and lower gastrointestinal (GI) recovery following partial large or small bowel resection surgery with primary anastomosis. Delayed GI recovery following bowel resection surgery can postpone hospital discharge until its resolution, resulting in an increased cost burden on healthcare providers.
We have a number of product candidates in various stages of clinical and preclinical development. We are conducting two Phase 2 clinical trials of ADL5945 to treat opioid-induced constipation (OIC), a condition that often results from long-term use of opioid analgesics in the management of chronic pain conditions. In addition, we are continuing limited development efforts on ADL7445, a second peripherally-acting mu opioid receptor antagonist, which is considered a back-up compound in our OIC program. We have completed Phase 1 clinical evaluation of ADL6906 (beloxepin), a compound with a novel and potentially differentiating pharmacological profile for treating chronic pain. Our preclinical pipeline includes novel, selective centrally-acting mu opioid receptor antagonists (CAMORs) currently in development for the treatment of l-DOPA-induced dyskinesia (LID) associated with the management of Parkinson’s disease.
For the year ended December 31, 2010, our total revenues and net loss were $43.3 million and $27.3 million, respectively. Net sales of ENTEREG for the year ended December 31, 2010 were $25.4 million. We will need net sales of ENTEREG to increase significantly beyond current levels before we will be able to achieve profitability and positive cash flow from operations. Ultimately, we may never generate sufficient revenues from ENTEREG for us to reach profitability, generate positive cash flow or sustain, on an ongoing basis, our current or projected levels of operations. In addition, our success is highly dependent on the results of our Phase 2 clinical trials with respect to ADL5945, our most advanced product candidate. There is no assurance that these studies will yield positive results, and even if the results are positive, it is unlikely that we will have sufficient available resources to fund a Phase 3 clinical program for ADL5945 without either securing a collaboration partner or raising proceeds in the capital markets.
On July 15, 2010, we announced a restructuring plan to reduce costs and realign our workforce and operations based on our current business objectives. We recorded a restructuring charge of $1.9 million for the year ended December 31, 2010, which consisted of employee severance and benefits-related costs.
ENTEREG
Together with our partner, Glaxo Group Limited (Glaxo), we launched ENTEREG in the United States in mid-2008. ENTEREG is detailed primarily by Glaxo’s national hospital-based sales organization. In certain hospitals, we co-promote ENTEREG with a field force that numbers approximately 25 persons. ENTEREG was approved by the FDA subject to a Risk Evaluation and Mitigation Strategy under which the product is available only to hospitals that perform bowel resections and are enrolled in the ENTEREG Access Support and Education (E.A.S.E.®) Program.
Under our agreement with Glaxo, we have a profit-sharing arrangement pursuant to which we receive 45% and Glaxo receives 55% of profits and losses, as defined, through June of 2011, after which the profit split is 50% each. Profits and losses are calculated as net sales of ENTEREG in the United States less certain agreed-upon costs, subject to certain adjustments.
Opioid-induced Constipation (OIC) Program
Peripheral mu opioid receptors in the GI tract regulate functions such as motility, secretion and absorption. Stimulation of these GI mu opioid receptors by morphine, or other opioid analgesics, disrupts normal gut motility ultimately delaying transit time of intestinal contents.
30
Table of Contents
We are developing two molecules, ADL5945 and ADL7445, to treat OIC. These compounds are small molecule, potent, peripherally-acting mu opioid receptor antagonists intended to block the adverse effects of opioid analgesics on the GI tract without affecting analgesia. During 2010, we conducted our initial clinical evaluation of both ADL5945 and ADL7445. The single dose and multiple-ascending dose studies of ADL5945 and the single-ascending dose and multiple-ascending dose studies of ADL7445 enrolled both healthy volunteers and chronic non-cancer pain patients on long-term opioid therapy with OIC. At target therapeutic doses, both compounds were well-tolerated and, in the patients with OIC, produced greater increases (over baseline) in weekly average number of spontaneous bowel movements as compared with placebo.
We currently are conducting Phase 2 clinical evaluation of ADL5945 in patients suffering from OIC. Limited development efforts also continue on ADL7445, which is considered a back-up compound in our OIC program.
Other Development Programs
ADL6906 (beloxepin) for Chronic Pain
We are developing ADL6906 (beloxepin), a dual NE reuptake inhibitor and 5-HT2 receptor antagonist, giving ADL6906 a novel and potentially differentiating pharmacological profile for treating chronic pain. ADL6906 is active in several preclinical models of pain after oral administration. We recently completed a Phase 1 multiple ascending dose study of ADL6906 that evaluated the safety, tolerability and pharmacokinetics of ADL6906 and we currently are evaluating the initiation of Phase 2 testing of ADL6906, either independently or in collaboration with a potential partner.
CAMOR Program for Parkinson’s Disease
Scientific evidence suggests that increased opioid peptide transmission in the basal ganglia might underlie dyskinesia after chronic l-DOPA treatment and that opioid antagonists might, therefore, be useful as adjuncts to l-DOPA therapy for Parkinson’s disease. We have discovered a family of novel, selective CAMORs that have been shown to be highly efficacious, after oral administration, in well-validated, non-human primate preclinical models of l-DOPA-induced dyskinesia (LID). During the fourth quarter of 2010, the Michael J. Fox Foundation for Parkinson’s Research awarded us a second round of funding to support our development of CAMORs for the treatment of LID associated with Parkinson’s disease. The $0.4 million award will be paid over a period of 18 months.
Delta Opioid Receptor Agonist Program
We previously collaborated with Pfizer Inc. (Pfizer) for the development and commercialization of the delta opioid receptor agonist compounds ADL5859 and ADL5747 for the treatment of pain.
In December 2010, Pfizer and we announced that we were discontinuing further development of ADL5859 and ADL5747. Based on the clinical evaluation of ADL5859 and ADL5747 in multiple indications, the companies concluded that there was not compelling evidence to continue the development program. In December 2010, Pfizer delivered to us a notice of termination of our collaboration agreement, which will be effective as of March 2011.
The following discussion is included to describe our consolidated financial position and results of operations as of December 31, 2010 and 2009 and for each of the years in the three-year period ended December 31, 2010. The consolidated financial statements and notes thereto contain detailed information that should be referred to in conjunction with this discussion.
LIQUIDITY AND CAPITAL RESOURCES
Cash, cash equivalents and short-term investments were $46.6 million at December 31, 2010 and $83.2 million at December 31, 2009, representing 88% and 91%, respectively, of our total assets. We invest excess
31
Table of Contents
cash in U.S. Treasury obligations. Our working capital, which is calculated as current assets less current liabilities, was $38.3 million at December 31, 2010 compared to $67.0 million at December 31, 2009. The decrease in cash, cash equivalents, short-term investments and working capital was primarily from the use of cash to fund our operations during the year ended December 31, 2010.
The following is a summary of selected cash flow information for the years ended December 31, 2010, 2009 and 2008:
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Net loss |
$ | (27,274,068 | ) | $ | (47,914,314 | ) | $ | (30,122,260 | ) | |||
| Adjustments for non-cash operating items |
(10,115,451 | ) | (3,507,038 | ) | (6,226,735 | ) | ||||||
| (37,389,519 | ) | (51,421,352 | ) | (36,348,995 | ) | |||||||
| Net change in assets and liabilities |
704,929 | 6,037,551 | 3,815,612 | |||||||||
| Net cash used in operating activities |
$ | (36,684,590 | ) | $ | (45,383,801 | ) | $ | (32,533,383 | ) | |||
| Net cash provided by investing activities |
$ | 34,100,552 | $ | 45,062,321 | $ | 24,770,394 | ||||||
| Net cash used in financing activities |
$ | (26,301 | ) | $ | — | $ | (277,669 | ) | ||||
Net Cash Used in Operating Activities
Net operating cash outflows of $36.7 million, $45.4 million and $32.5 million for the years ended December 31, 2010, 2009 and 2008, respectively, resulted primarily from research and development expenditures associated with our product candidates and selling, general and administrative expenses. For the year ended December 31, 2009, we received $9.3 million related to the acceleration of payments owed by Glaxo to us under the terms of Amendment No. 4 of the Glaxo collaboration agreement. In 2008, we received a $20.0 million milestone payment from Glaxo following FDA approval of ENTEREG.
Net Cash Provided by Investing Activities
Net cash provided by investing activities relates to purchases and maturities of investment securities, expenditures for property and equipment and in-process research and development, and proceeds from the sale of equipment. Capital expenditures are primarily for the purchase of laboratory equipment, furniture and fixtures, office equipment and leasehold improvements associated with our leased facility.
Net cash provided by investing activities was $34.1 million for the year ended December 31, 2010 as compared to $45.1 million for the year ended December 31, 2009. The decrease in cash provided by investing activities for the year ended December 31, 2010 as compared to the prior year period is primarily attributable to a decrease of $12.8 million in the net maturities of available-for-sale securities, offset partially by $2.3 million of payments made during the year ended December 31, 2009 related to the purchase of in-process research and development.
Net cash provided by investing activities was $45.1 million for the year ended December 31, 2009 as compared to $24.8 million for the year ended December 31, 2008. The increase in cash provided by investing activities in 2009 is primarily attributable to an increase of $21.0 million in the net maturities of available-for-sale securities and cash proceeds of $1.0 million received from the disposal of equipment, offset partially by $2.3 million of payments made during 2009 related to the purchase of in-process research and development.
We expect to fund a significant portion of our future operations through the sale or maturities of investments in our portfolio, which consists of U.S. Treasury obligations.
32
Table of Contents
Contractual Commitments
The following table summarizes our obligations to make future payments under current contracts:
| Payments Due by Period | ||||||||||||||||||||
| Contractual obligations |
Total | 2011 | 2012 and 2013 | 2014 and 2015 | 2016 and Thereafter |
|||||||||||||||
| Operating leases |
$ | 3,013,000 | $ | 1,262,000 | $ | 1,751,000 | $ | — | $ | — | ||||||||||
| Purchase obligations |
845,000 | 773,000 | 72,000 | — | — | |||||||||||||||
| Total |
$ | 3,858,000 | $ | 2,035,000 | $ | 1,823,000 | $ | — | $ | — | ||||||||||
Contractual commitments in the table above represent future cash obligations that are legally binding and enforceable under agreements with third parties. These amounts relate to future minimum payments for the manufacture of ENTEREG, the use of software and operating leases for office and laboratory space. The table summarizes our significant contractual commitments as of December 31, 2010 and the effects such commitments are expected to have on our liquidity and cash flows in future periods.
Excluded from the table above are contingent liabilities associated with various agreements into which we have entered for services with third-party vendors, including agreements to conduct clinical trials, to manufacture product candidates and for consulting and other contracted services. These contingent liabilities require future performance by the third party in order for payment to be executed, and we accrue the costs of these agreements based on estimates of work completed to date. We estimate that approximately $16.2 million will be payable in future periods under the arrangements in place at December 31, 2010. Of this amount, approximately $2.4 million has been accrued for work estimated to have been completed as of December 31, 2010 and approximately $13.8 million relates to future performance under these arrangements.
In addition to the above, we have committed to make potential future royalty and milestone payments to third parties as part of our in-licensing agreements. Payments generally become due and payable only upon the achievement of certain developmental, regulatory and/or commercial milestones. Because the achievement of these milestones is neither probable nor reasonably estimable, we have not recorded a liability on our consolidated balance sheet for any such contingencies. As of December 31, 2010, the maximum potential milestone payments due under current contractual agreements are $106.5 million.
Glaxo Collaboration Agreement
Under the terms of the Glaxo agreement, we are obligated to partially reimburse Glaxo for certain third-party expenses incurred by Glaxo related to ENTEREG, pursuant to an agreed upon development plan and budget. We also incur certain third-party expenses related to ENTEREG, pursuant to an agreed upon development plan and budget, a portion of which are reimbursable to us by Glaxo. We record these expenses gross on our consolidated statement of operations as incurred.
In addition, we and Glaxo have a profit-sharing arrangement pursuant to which we receive 45% and Glaxo receives 55% of profits and losses, as defined, through June of 2011, after which the profit split is 50% each. Profits and losses are calculated as net sales of ENTEREG in the United States less certain agreed-upon costs, subject to certain adjustments. We record these profit-sharing expenses as selling, general and administrative expense gross on our consolidated statement of operations as incurred.
License Agreements
In November 1996, Roberts Laboratories, Inc. (Roberts) licensed from Lilly certain intellectual property rights relating to ENTEREG. In June 1998, we entered into an option and license agreement with Roberts under which we sublicensed these rights from Roberts. In December 2000, Shire U.S. Inc. (Shire) became the successor
33
Table of Contents
in interests to Roberts under our option and license agreement with Roberts. The Company has made license and milestone payments under this agreement totaling $2.5 million. Our obligations to pay royalties to Shire and Lilly are expected to expire on the date of the last to expire of the licensed Lilly patents.
In September 2009, we acquired from Lilly the exclusive worldwide rights to ADL5945 for an up-front payment of $2.0 million. We also will be required to pay milestones, which are contingent upon achievement of pre-defined, late-stage clinical and regulatory events and achievement of certain sales targets, and royalties on net sales of the product.
Outlook
We expect to use our cash, cash equivalents and short-term investments to fund our operations. Since inception, we have experienced significant operating losses and negative operating cash flow and have funded our operations primarily from the proceeds received from the sale of our equity securities and amounts received under collaboration agreements. Our accumulated deficit at December 31, 2010 was $530.3 million and we expect to continue to incur substantial losses for at least the next several years.
We may never generate significant product sales, achieve profitable operations or generate positive cash flows from operations and, even if profitable operations are achieved, they may not be sustained on a continuing basis or sufficient to support our current or projected levels of investment in our research and development programs and our other operations. At this time, we cannot accurately assess a number of factors that will influence the levels of future product sales, such as the degree of market acceptance, patent protection and exclusivity of ENTEREG, the impact of competition, the effectiveness of our sales and marketing efforts and the outcome of our current efforts to develop, receive approval for and successfully launch other product candidates. However, at current expenditure levels, we will need ENTEREG sales to increase significantly beyond current levels before we will be able to achieve profitability and positive cash flows from operations.
We expect to continue to incur significant levels of research and development expenditures related to our clinical product candidates. During 2011, we will be funding Phase 2 studies of ADL5945 in OIC and our ongoing Phase 4 study of ENTEREG in patients undergoing radical cystectomy. We also expect to continue to conduct research, preclinical studies and process development activities on our other product candidates, although as a result of our restructurings in 2009 and 2010, the level of such expenditures will be significantly reduced compared to previous years. Should these programs advance to later stages of development, it is likely that expenses related to these efforts will increase over time.
We believe that our existing cash, cash equivalents and short-term investments are adequate to fund our operations through mid-2012 based upon the level of research and development and marketing and administrative activities we believe will be necessary to achieve our strategic objectives. We anticipate that we will need to obtain funding to support our operational needs in the future, and we cannot be certain that funding will be available on terms acceptable to us, or at all.
RESULTS OF OPERATIONS
This section should be read in conjunction with the discussion above under “Liquidity and Capital Resources.”
34
Table of Contents
Revenues
The following table sets forth revenues for the years ended December 31, 2010, 2009 and 2008:
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Product sales, net |
$ | 25,386,285 | $ | 14,609,022 | $ | 1,247,271 | ||||||
| Contract revenues |
17,916,233 | 22,751,584 | 48,208,224 | |||||||||
| Total revenues, net |
$ | 43,302,518 | $ | 37,360,606 | $ | 49,455,495 | ||||||
Product Sales, Net
Net product sales are derived solely from ENTEREG. ENTEREG was approved by the FDA in May 2008 and product shipments to hospitals began in June 2008. For a discussion of our revenue recognition policy, see “Critical Accounting Policies and Estimates – Revenue Recognition” section of MD&A.
Net sales of ENTEREG were $25.4 million and $14.6 million for the years ended December 31, 2010 and 2009, respectively. The increase in net product sales during 2010 as compared to 2009 was driven primarily by an increase in the number of hospitals ordering ENTEREG and increased penetration within existing hospital customers.
Net sales of ENTEREG were $14.6 million and $1.2 million for the years ended December 31, 2009 and 2008, respectively. The increase in net product sales during 2009 resulted primarily from a full year of sales as well as an increase in the number of hospitals ordering ENTEREG. In addition, net product sales for the year ended December 31, 2009 include $0.9 million for product that was shipped to registered hospitals in 2008, but not recognized as revenue at the time of shipment under our previous revenue recognition policy.
Contract Revenues
Contract revenues are derived from our collaboration agreements with Glaxo and Pfizer and include milestone payments, cost reimbursement, amortization of up-front license fees and other revenue. Contract revenues were $17.9 million, $22.8 million and $48.2 million in the years ended December 31, 2010, 2009 and 2008, respectively. Contract revenues decreased in 2010 compared to 2009 primarily as a result of reduced reimbursements under the Glaxo and Pfizer collaboration agreements.
Contract revenues decreased in 2009 compared to 2008 due primarily to a $20.0 million milestone payment received from Glaxo during 2008 in conjunction with the FDA approval of ENTEREG. In addition, contract revenues from Pfizer for the year ended December 31, 2009 decreased due to a reduction in delta program costs, which resulted in lower cost reimbursement, as well as reduced amortization of deferred licensing fees due to extensions of the estimated performance period in the third quarter of 2008 and the first quarter of 2009. These decreases were partially offset by increased revenue that was recognized related to $9.3 million of payments received from Glaxo during the year ended December 31, 2009 under the terms of Amendment No. 4 to the collaboration agreement. Of the $9.3 million received, $8.4 million is being recognized as revenue on a straight-line basis over the estimated remaining performance period under the collaboration agreement, which extends to March 2016, and the remaining $0.9 million was recognized as revenue in 2009.
35
Table of Contents
Operating Expenses
The following table sets forth operating expenses for the years ended December 31, 2010, 2009 and 2008:
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Cost of product sales |
$ | 2,876,503 | $ | 1,515,073 | $ | 203,972 | ||||||
| Research and development |
33,210,404 | 43,930,303 | 52,664,213 | |||||||||
| Selling, general and administrative |
34,053,247 | 36,947,749 | 31,114,718 | |||||||||
| Restructuring charge |
1,918,701 | 3,932,582 | — | |||||||||
| Total operating expenses |
$ | 72,058,855 | $ | 86,325,707 | $ | 83,982,903 | ||||||
Cost of Product Sales
Cost of product sales was $2.9 million, $1.5 million and $0.2 million for the years ended December 31, 2010, 2009 and 2008, respectively, and consisted of royalty payments under certain alvimopan license agreements, FDA fees and manufacturing costs. The increase year-over-year was due to increased sales of ENTEREG. Cost of product sales as a percentage of net product sales were 11%, 10% and 16% for the years ended December 31, 2010, 2009 and 2008, respectively.
Costs associated with the manufacture of alvimopan prior to FDA approval of ENTEREG in May 2008 were expensed to research and development. As a result, at December 31, 2010, we have inventory related to alvimopan that carries a zero-cost and is not reflected on the December 31, 2010 consolidated balance sheet. To the extent that this inventory is sold, our cost of product sales will not reflect all costs associated with such product manufacture, and our gross margins will be favorably impacted. We currently are unable to estimate the timing of the impact to future profitability resulting from the sell-through of any inventory manufactured after FDA approval of ENTEREG.
Research and Development Expenses
Our research and development expenses can be identified as internal or external expenses. External expenses include expenses incurred with clinical research organizations, contract manufacturers and other third-party vendors. Internal expenses include expenses such as personnel, laboratory and overhead expenses.
Research and development expenses were $33.2 million, $43.9 million and $52.7 million for the years ended December 31, 2010, 2009 and 2008, respectively, and consisted of the following:
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| External research and development expenses: |
||||||||||||
| OIC program |
$ | 8,102,939 | $ | 6,906,846 | $ | 1,704,907 | ||||||
| ENTEREG |
3,946,351 | 4,638,899 | 5,040,590 | |||||||||
| Delta agonist program |
5,915,845 | 7,050,948 | 10,499,302 | |||||||||
| Other programs |
2,058,329 | 4,625,961 | 9,971,473 | |||||||||
| Total external research and development expenses |
20,023,464 | 23,222,654 | 27,216,272 | |||||||||
| Total internal research and development expenses |
13,186,940 | 20,707,649 | 25,447,941 | |||||||||
| Total research and development expenses |
$ | 33,210,404 | $ | 43,930,303 | $ | 52,664,213 | ||||||
We report all expenses gross within our consolidated statements of operations and, as such, the above table does not reflect any cost reimbursements from our collaboration partners.
36
Table of Contents
Total research and development expenses decreased during 2010 as compared to 2009 primarily due to reductions in expenses associated with our restructurings in June 2009 and July 2010 and lower expenses in other programs. In addition, our delta program expenses decreased due to lower levels of clinical activity for ADL5859 and ADL5747 during 2010. These decreases were partially offset by higher OIC program costs, which resulted from our initial clinical evaluation of ADL5945 and ADL7445 and the initiation of Phase 2 clinical testing for ADL5945 during 2010.
Total research and development expenses decreased for the year ended December 31, 2009 as compared to the year ended December 31, 2008 primarily due to lower costs of clinical studies incurred during 2009 in our delta agonist program, lower expenses in other programs and a reduction in headcount and depreciation expenses resulting from our June 2009 restructuring. These decreases were partially offset by higher expenses in our OIC program, including a $2.0 million payment to Lilly for the rights to ADL5945 in September 2009 which was expensed as in-process research and development.
There are significant risks and uncertainties inherent in the preclinical and clinical studies associated with each of our research and development programs. These studies may yield varying results that could delay, limit or prevent the advancement of a program through the various stages of product development and significantly impact the costs to be incurred, and time involved, in bringing a program to completion. As a result, the cost to complete such programs, as well as the period in which net cash inflows from significant programs are expected to commence, are not reasonably estimable.
Selling, General and Administrative Expenses
Our selling, general and administrative expenses were $34.1 million, $36.9 million and $31.1 million for the years ended December 31, 2010, 2009 and 2008, respectively. Included in selling, general and administrative expenses for 2010 and 2009 were $7.9 million and $2.1 million, respectively, of profit-sharing expenses under the Glaxo collaboration agreement. The higher profit-sharing expenses were driven by improved profitability resulting primarily from increased sales of ENTEREG. Remaining selling, general and administrative expenses decreased by $8.6 million in 2010 compared to 2009 primarily due to a reduction of $2.8 million in general and administrative expenses as a result of our June 2009 and July 2010 restructurings and lower marketing expenses of $4.9 million year-over-year.
The increase in 2009 compared to 2008 was primarily driven by higher sales and marketing expenses associated with ENTEREG, including the expansion of our sales force and marketing activities. These higher sales and marketing expenses were partially offset by lower general and administrative expenses year-over-year. Selling, general and administrative expenses for the year ended December 31, 2009 also include $0.5 million of profit-sharing expense that was recognized during 2009 as a result of our change in revenue recognition policy. Prior to 2009, ENTEREG profit-sharing expenses that were based on net product shipments were deferred until the corresponding net product shipments were recognized as revenue.
Restructuring Charges
As a result of our July 2010 restructuring, we recorded a restructuring charge of $1.9 million for the year ended December 31, 2010, which consisted of employee severance and benefits-related costs. As a result of our restructuring in June 2009, we recorded a charge of $3.9 million for the year ended December 31, 2009, which consisted of $2.1 million of employee severance and benefits-related costs and a $1.8 million non-cash impairment charge primarily related to leasehold improvements and laboratory equipment used for activities which were eliminated pursuant to our restructuring. There were no restructuring charges for the year ended December 31, 2008.
37
Table of Contents
Interest Income and Other Income, Net
The following table sets forth interest income and other income, net, for the years ended December 31, 2010, 2009 and 2008:
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Interest income |
$ | 188,202 | $ | 1,050,787 | $ | 4,314,658 | ||||||
| Other income, net |
1,294,067 | — | 90,490 | |||||||||
| Total interest income and other income, net |
$ | 1,482,269 | $ | 1,050,787 | $ | 4,405,148 | ||||||
Interest Income
Our interest income was $0.2 million, $1.1 million and $4.3 million for the years ended December 31, 2010, 2009 and 2008, respectively. The decreases year-over-year were due to lower investment balances resulting primarily from the use of cash in operating activities and lower prevailing interest rates in each successive year.
Other Income, Net
Other income, net, was $1.3 million and $0.1 million for the years ended December 31, 2010 and 2008, respectively. In November 2010, we received four grants totaling $1.0 million under the Qualified Therapeutic Discovery Project Grants Program. The Qualified Therapeutic Discovery Project Grants Program was included in the healthcare reform legislation, and established a one-time pool of $1 billion for grants to small biotechnology companies developing novel therapeutics which show potential to: (a) result in new therapies that either treat areas of unmet medical need, or prevent, detect, or treat chronic or acute diseases and conditions; (b) reduce long-term health care costs in the United States; or (c) significantly advance the goal of curing cancer within a the 30-year period. In addition, we recognized $0.2 million of Michael J. Fox Foundation grant income during 2010 in conjunction with the development of our CAMOR program and $0.1 million of sublease income related to laboratory and office space at our corporate office.
Other income, net, in 2008 primarily represents cash received from the sale of certain Pennsylvania research and development tax credits. There was no other income for the year ended December 31, 2009.
Income Taxes
As of December 31, 2010, we had $445.4 million of Federal net operating loss carryforwards and $404.8 million of state net operating loss carryforwards, which are potentially available to offset future taxable income. The state net operating loss carryforwards begin expiring during 2019 and the Federal net operating loss carryforwards began expiring in 2010. In addition, the utilization of the state net operating loss carryforwards is subject to annual limitation. At December 31, 2010, we also had $12.8 million of Federal and $0.3 million of state research and development tax credit carryforwards, which begin expiring in 2011, and are available to reduce Federal and state income taxes.
The Tax Reform Act of 1986 (the Act) provides for a limitation on the annual use of net operating loss and research and development tax credit carryforwards (following certain ownership changes, as defined by the Act) that could significantly limit our ability to utilize these carryforwards. We may have experienced various ownership changes, as defined by the Act, as a result of past financings. Additionally, because United States and certain state tax laws limit the time during which these carryforwards may be applied against future taxes, we may not be able to take full advantage of these attributes for Federal and state income tax purposes.
CRITICAL ACCOUNTING POLICIES AND ESTIMATES
Our MD&A discusses our consolidated financial statements, which we have prepared in accordance with U.S. generally accepted accounting principles (GAAP). In preparing our consolidated financial statements, we
38
Table of Contents
must make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. We develop and periodically change these estimates and assumptions based on historical experience and on various other factors that we believe are reasonable under the circumstances. Actual results may differ from these estimates under different assumptions or conditions.
Our significant accounting policies are described in Note 2 to our consolidated financial statements for the year ended December 31, 2010 included in Part II, Item 8 of this Annual Report on Form 10-K. In addition, the Securities and Exchange Commission (SEC) defines critical accounting policies as those that are, in management’s view, most important to the portrayal of the company’s financial condition and results of operations and most demanding of their judgment. Management considers the following policies to be critical to an understanding of our consolidated financial statements and the uncertainties associated with the complex judgments made by us that could impact our consolidated results of operations, financial position and cash flows.
Revenue Recognition
We recognize revenue in accordance with SEC Staff Accounting Bulletin (SAB) No. 101, Revenue Recognition in Financial Statements (SAB 101), as amended by SAB No. 104, Revenue Recognition (SAB 104) and Financial Accounting Standards Board (FASB) Accounting Standards Codification (ASC) 605, Revenue Recognition (ASC 605).
Product Sales, Net
ENTEREG was approved by the FDA in May 2008 and product shipments to hospitals began in June 2008. Hospital orders are processed through wholesalers; however, ENTEREG is drop-shipped from Glaxo directly to an ordering hospital registered under the E.A.S.E. Program. Wholesalers remit payment to Glaxo and, on a monthly basis, Glaxo remits the net proceeds to us. In accordance with SAB 101 and SAB 104, we recognize revenue from product sales when the following four revenue recognition criteria are met: persuasive evidence of an arrangement exists, delivery has occurred, the selling price is fixed or determinable and collectability is reasonably assured. Prior to the fourth quarter of 2009, we deferred recognition of revenue associated with the first shipment of ENTEREG to each hospital customer as we were not able to reasonably estimate future returns. When an existing customer placed a new order, we recognized product sales on the previous shipment for an amount equal to the lesser of (a) the previous shipment or (b) the new order. During the fourth quarter of 2009, we began to recognize net product sales on the shipment of product to the registered hospital upon our determination that we had sufficient historical experience to reasonably estimate future returns of ENTEREG. In the future, actual returns could exceed our estimates of expected returns, which would result in a reduction to our net product sales. As a result of the change in our revenue recognition policy in the fourth quarter of 2009, net product sales for the year ended December 31, 2009 include $0.9 million for product that was shipped to registered hospitals in 2008, but not recognized as revenue at the time of shipment. We also recognized $0.5 million of selling, general and administrative expenses during 2009 that had been deferred under our previous revenue recognition policy. The change in our revenue recognition policy resulted in a decrease to net loss of $0.4 million, or $0.01 per share, for the year ended December 31, 2009.
We record product sales net of prompt payment discounts, returns, group purchasing organization fees, chargebacks and other discounts as reported to us by Glaxo. Calculating these allowances requires significant estimates and judgments and while we undertake certain procedures to review the reasonableness of the information, we cannot obtain absolute assurance over the accuracy of such information provided by Glaxo.
Contract Revenues
Contract revenues, which include revenues from collaborative agreements, consist primarily of milestone payments, cost reimbursement, amortization of up-front license fees and other revenue under such agreements.
39
Table of Contents
Non-refundable up-front license fees are recognized as revenue if we have a contractual right to receive such payment, the contract price is fixed or determinable, the collection of the resulting receivable is reasonably assured and we have no further performance obligations under the license agreement. Multiple element arrangements, such as license and development arrangements, are analyzed to determine whether the deliverables, which often include a license and performance obligations, can be separated for revenue recognition purposes. When the license is considered to either (i) not have standalone value or (ii) have standalone value but the fair value of any of the undelivered performance obligations could not be determined, the up-front license payment is recognized as revenue ratably over the estimated period of when our performance obligations are to be performed. We estimate our performance period based on the specific terms of each collaborative agreement and subsequently adjust the performance periods, if appropriate, based upon available facts and circumstances. During the third quarter of 2008, the Pfizer performance period was extended by six months to August 2010 based on the status of the development programs. The effect of the third quarter 2008 change in estimate was an increase to net loss of $1.2 million, or $0.03 per share, for the year ended December 31, 2008. During the first quarter of 2009, the performance period was further extended by four months to December 2010 based on the status of the development programs. The effect of the first quarter 2009 change in estimate was an increase to net loss of $1.6 million, or $0.03 per share, for the year ended December 31, 2009. In December 2010, Pfizer and we announced that we were discontinuing further development of ADL5859 and ADL5747. Based on the clinical evaluation of ADL5859 and ADL5747 in multiple indications, the companies concluded that there was not compelling evidence to continue the development program. Pfizer delivered to us a notice of termination of our collaboration agreement, which will be effective as of March 2011. As we had completed substantially all performance under the collaboration agreement as of December 31, 2010, the performance period was not extended beyond December 2010.
Our collaboration agreements also contain substantive milestone payments. Substantive milestone payments are considered to be performance payments that are recognized upon achievement of the milestone only if all of the following conditions are met: (i) the milestone payment is non-refundable; (ii) achievement of the milestone involves a degree of risk and was not reasonably assured at the inception of the arrangement; (iii) substantive effort is involved in achieving the milestone; (iv) the amount of the milestone payment is reasonable in relation to the effort expended or the risk associated with achievement of the milestone; and (v) a reasonable amount of time passes between the up-front license payment and the first milestone payment as well as between each subsequent milestone payment. Determination as to whether a milestone meets the aforementioned conditions involves the judgment of management. If any of these conditions are not met, the resulting payment would not be considered a substantive milestone, and such payment would therefore be recognized as revenue as such performance obligations are performed in accordance with the policies described above.
Reimbursement of costs is recognized as revenue provided the provisions of ASC 605 are met, the amounts are determinable and collection of the related receivable is reasonably assured.
Amounts received prior to satisfying the above revenue recognition criteria for contract revenues are recorded as deferred revenue in the accompanying consolidated balance sheets. Amounts not expected to be recognized within one year of the balance sheet date are classified as non-current deferred revenue. The estimate of the classification of deferred revenue as current or non-current is based upon management’s estimate of its performance period based on the specific terms of each collaborative agreement. These estimates may change in the future and such changes to estimates would be accounted for prospectively and would result in a change in the amount of revenue recognized in future periods.
Research and Development Expenses
We have entered into contracts with third parties to conduct certain research and development activities including preclinical, clinical and manufacturing activities. We accrue expenses related to such contracts based upon an estimate of the amounts due for work completed under the contracts. Factors considered in preparing such estimates include the number of subjects enrolled in studies and other criteria relating to the progress of efforts by our vendors.
40
Table of Contents
Inventories
Inventories are stated at the lower of cost or market, as determined on a first-in, first-out, or FIFO, basis. On a quarterly basis, we analyze our inventory levels to determine if there is inventory that is expected to expire prior to being sold, inventory that has a cost basis in excess of its expected net realizable value or inventory in excess of expected sales requirements. Based on our review, we will determine if a write-down of inventory is necessary, and if so, will record the charge to cost of product sales. Expired inventory is disposed of and the related costs are written off to cost of product sales. If actual market conditions are less favorable than those projected by management, additional inventory write-downs may be required. Since inception, we have not recorded any write-downs of inventory.
Valuation of Equipment and Leasehold Improvements
Our equipment and leasehold improvements have been recorded at cost and are being depreciated on a straight-line basis over the estimated useful life of those assets or the lease term, whichever is shorter. We periodically assess our equipment and leasehold improvements for impairment in accordance with ASC 360, Property, Plant and Equipment. We review long-lived assets, specifically equipment and leasehold improvements, for impairment whenever events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable. If indicators of impairment exist, impairment loss would be tested for if estimated undiscounted future cash flows expected to result from the use of the asset and its eventual disposition is less than its carrying amount. The impairment charge, if any, is determined based on the excess of the carrying value of the asset over its fair value, with fair value determined based on an estimate of discounted future cash flows or other appropriate measures of fair value. During the year ended December 31, 2009, we recorded a $1.8 million non-cash impairment charge primarily related to leasehold improvements and laboratory equipment used for activities that were eliminated pursuant to our June 2009 restructuring.
Equity-based Compensation
We follow the fair value method of accounting for stock-based awards granted to employees and directors, which requires the recognition of compensation expense in our consolidated statement of operations. Under the fair value method, stock-based compensation cost is measured at the grant date based on the fair value of the award and is generally recognized as expense on a straight-line basis over the service period, which represents the vesting period. For equity-based awards with performance conditions, we recognize compensation cost if and when we conclude that it is probable that the performance condition will be achieved. For any equity-based awards that do not vest on a monthly basis, the Company estimates a forfeiture rate based on its historical experience of pre-vesting forfeitures.
We calculate the fair value of our stock options using the Black-Scholes option pricing model. The Black-Scholes option-pricing model requires several inputs, including volatility and the expected life of our options. Our estimate of volatility is based upon the historical volatility experienced in our stock price. To the extent volatility of our stock price increases in the future, our estimates of the fair value of stock options granted in the future could increase, thereby increasing stock-based compensation expense in future periods. The expected life of our stock options represents the period of time that option awards are expected to be outstanding giving consideration to vesting schedules and our historical exercise patterns, which we believe are relevant indicators of future exercise patterns. We calculate the fair value of our deferred and restricted stock based upon the market value of the underlying common stock on the date of the grant.
Recent Accounting Pronouncements
In December 2010, the FASB ratified a consensus of the Emerging Issues Task Force related to an annual fee to be paid to the federal government by pharmaceutical manufacturers as mandated by the Patient Protection and Affordable Care Act as amended by the Health Care and Education Reconciliation Act. This consensus requires the liability related to the annual fee to be estimated and recorded in full upon the first qualifying sale
41
Table of Contents
with a corresponding deferred cost that is amortized to expense generally using a straight-line method of allocation over the calendar year that it is payable. This guidance is effective for calendar years beginning after December 31, 2010, when the fee initially became effective. We are currently evaluating the effect that this guidance will have on our consolidated financial statements.
In January 2010, the FASB issued ASU 2010-06, Fair Value Measurements and Disclosures (Topic 820): Improving Disclosures about Fair Value Measurements (ASU 2010-06), which amends the existing fair value measurement and disclosure guidance currently included in Accounting Standards Codification (ASC) Topic 820, Fair Value Measurements and Disclosures, to require additional disclosures regarding fair value measurements. Specifically, ASU 2010-06 requires entities to disclose the amounts of significant transfers between Level 1 and Level 2 of the fair value hierarchy and the reasons for these transfers, the reasons for any transfer in or out of Level 3 and information in the reconciliation of recurring Level 3 measurements about purchases, sales, issuances and settlements on a gross basis. In addition, ASU 2010-06 also clarifies the requirement for entities to disclose information about both the valuation techniques and inputs used in estimating Level 2 and Level 3 fair value measurements. ASU 2010-06 is effective for interim and annual reporting periods beginning after December 15, 2009, except for additional disclosures related to Level 3 fair value measurements, which are effective for fiscal years beginning after December 15, 2010. The adoption of ASU 2010-06 did not impact our consolidated financial statements or results of operations.
In September 2009, the FASB issued ASU 2009-13, Revenue Recognition (Topic 605): Multiple-Deliverable Revenue Arrangements (ASU 2009-13), which requires companies to allocate revenue in arrangements involving multiple deliverables based on the estimated selling price of each deliverable when such deliverables are not sold separately either by the company or other vendors. ASU 2009-13 eliminates the requirement that all undelivered elements must have objective and reliable evidence of fair value before a company can recognize the portion of the overall arrangement fee that is attributable to items that already have been delivered. As a result, the new guidance may allow some companies to recognize revenue on transactions that involve multiple deliverables earlier than under current requirements. ASU 2009-13 is effective for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010. Early adoption is permitted at the beginning of a company’s fiscal year. We do not expect that the adoption of ASU 2009-13 will have a material impact on our consolidated financial statements.
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
Our investment assets consist of U.S. Treasury obligations. The market value of such investments fluctuates with current market interest rates. In general, as rates increase, the market value of a debt instrument would be expected to decrease. The opposite also is true. To minimize such market risk, we have in the past held and, to the extent possible, will continue in the future to hold, such debt instruments to maturity at which time the debt instrument will be redeemed at its stated, or face, value. Due to the short duration and nature of these instruments, we do not believe that we have a material exposure to interest rate risk related to our investment portfolio. The investment portfolio at December 31, 2010 totaled $42.0 million, and the weighted-average yield-to-maturity was approximately 0.2% with maturities of investments ranging up to 12 months.
42
Table of Contents
| ITEM 8. | CONSOLIDATED FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
43
Table of Contents
Management’s Report on Consolidated Financial Statements
Our management is responsible for the preparation, integrity and fair presentation of information in our consolidated financial statements, including estimates and judgments. The consolidated financial statements presented in this Annual Report on Form 10-K have been prepared in accordance with accounting principles generally accepted in the United States. Our management believes the consolidated financial statements and other financial information included in this Annual Report on Form 10-K fairly present, in all material respects, our consolidated financial condition, results of operations and cash flows as of and for the periods presented in this Annual Report on Form 10-K. The consolidated financial statements have been audited by KPMG LLP, an independent registered public accounting firm, as stated in their report, which is included herein.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Our internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of consolidated financial statements for external purposes in accordance with accounting principles generally accepted in the United States.
Our internal control over financial reporting includes those policies and procedures that:
| • | pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect our transactions and dispositions of our assets; |
| • | provide reasonable assurance that our transactions are recorded as necessary to permit preparation of our consolidated financial statements in accordance with accounting principles generally accepted in the United States and that our receipts and expenditures are being made only in accordance with authorization of our management and our directors; and |
| • | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the consolidated financial statements. |
Because of its inherent limitations, internal control over financial reporting can provide only reasonable assurance and may not prevent or detect misstatements. Also, projections of any evaluation of the effectiveness of such controls in future periods are subject to the risk that the controls may become inadequate because of changes in conditions or that the degree of compliance with the policies and procedures may deteriorate.
Our management conducted an assessment of the effectiveness of internal control over financial reporting based on the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this assessment, our management concluded that, as of December 31, 2010, our internal control over financial reporting was effective to provide reasonable assurance regarding the reliability of financial reporting and the preparation of consolidated financial statements for external purposes in accordance with accounting principles generally accepted in the United States.
44
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Adolor Corporation:
We have audited the accompanying consolidated balance sheets of Adolor Corporation and subsidiary as of December 31, 2010 and 2009, and the related consolidated statements of operations, comprehensive loss, stockholders’ equity, and cash flows for each of the years in the three-year period ended December 31, 2010. In connection with our audits of the consolidated financial statements, we also have audited the financial statement schedule, Schedule II—Valuation and Qualifying Accounts. These consolidated financial statements and financial statement schedule are the responsibility of Adolor Corporation’s management. Our responsibility is to express an opinion on these consolidated financial statements and financial statement schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the consolidated financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Adolor Corporation and subsidiary as of December 31, 2010 and 2009, and the results of their operations and their cash flows for each of the years in the three-year period ended December 31, 2010, in conformity with U.S. generally accepted accounting principles. Also in our opinion, the related consolidated financial statement schedule, when considered in relation to the basic consolidated financial statements taken as a whole, presents fairly, in all material respects, the information set forth therein.
/s/ KPMG LLP
Philadelphia, Pennsylvania
February 24, 2011
45
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
Consolidated Balance Sheets
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 4,603,327 | $ | 7,213,666 | ||||
| Short-term investments |
41,983,210 | 75,992,310 | ||||||
| Accounts receivable |
3,105,493 | 3,816,205 | ||||||
| Inventory |
933,857 | 1,006,809 | ||||||
| Prepaid expenses and other current assets |
1,560,725 | 2,462,668 | ||||||
| Total current assets |
52,186,612 | 90,491,658 | ||||||
| Equipment and leasehold improvements, net |
464,052 | 860,776 | ||||||
| Other assets |
107,000 | 107,000 | ||||||
| Total assets |
$ | 52,757,664 | $ | 91,459,434 | ||||
| Liabilities and Stockholders’ Equity |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 272,607 | $ | 1,742,362 | ||||
| Accrued expenses |
8,923,508 | 8,421,570 | ||||||
| Deferred revenue and rent—current |
4,645,674 | 13,338,404 | ||||||
| Total current liabilities |
13,841,789 | 23,502,336 | ||||||
| Deferred revenue and rent—non-current |
19,257,551 | 23,903,425 | ||||||
| Total liabilities |
33,099,340 | 47,405,761 | ||||||
| Commitments (Note 7) |
||||||||
| Stockholders’ equity: |
||||||||
| Series A Junior Participating preferred stock, $0.01 par value; 35,000 shares authorized; none issued and outstanding |
— | — | ||||||
| Preferred stock, $0.01 par value; 1,000,000 shares authorized; none issued and outstanding |
— | — | ||||||
| Common stock, $0.0001 par value; 99,000,000 shares authorized; 46,433,663 and 46,333,735 shares issued; 46,414,759 and 46,333,735 shares outstanding at December 31, 2010 and 2009, respectively |
4,643 | 4,633 | ||||||
| Additional paid-in capital |
549,929,447 | 546,995,528 | ||||||
| Treasury stock, at cost, 18,904 shares at December 31, 2010 |
(26,301 | ) | — | |||||
| Unrealized gains on available for sale securities |
5,660 | 34,569 | ||||||
| Accumulated deficit |
(530,255,125 | ) | (502,981,057 | ) | ||||
| Total stockholders’ equity |
19,658,324 | 44,053,673 | ||||||
| Total liabilities and stockholders’ equity |
$ | 52,757,664 | $ | 91,459,434 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
46
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
Consolidated Statements of Operations
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Revenues: |
||||||||||||
| Product sales, net |
$ | 25,386,285 | $ | 14,609,022 | $ | 1,247,271 | ||||||
| Contract revenues |
17,916,233 | 22,751,584 | 48,208,224 | |||||||||
| Total revenues, net |
43,302,518 | 37,360,606 | 49,455,495 | |||||||||
| Operating expenses incurred: |
||||||||||||
| Cost of product sales |
2,876,503 | 1,515,073 | 203,972 | |||||||||
| Research and development |
33,210,404 | 43,930,303 | 52,664,213 | |||||||||
| Selling, general and administrative |
34,053,247 | 36,947,749 | 31,114,718 | |||||||||
| Restructuring charge |
1,918,701 | 3,932,582 | — | |||||||||
| Total operating expenses |
72,058,855 | 86,325,707 | 83,982,903 | |||||||||
| Loss from operations |
(28,756,337 | ) | (48,965,101 | ) | (34,527,408 | ) | ||||||
| Interest income |
188,202 | 1,050,787 | 4,314,658 | |||||||||
| Other income, net |
1,294,067 | — | 90,490 | |||||||||
| Net loss |
$ | (27,274,068 | ) | $ | (47,914,314 | ) | $ | (30,122,260 | ) | |||
| Basic and diluted net loss per share |
$ | (0.59 | ) | $ | (1.03 | ) | $ | (0.65 | ) | |||
| Shares used in computing basic and diluted net loss per share |
46,338,538 | 46,296,235 | 46,158,458 | |||||||||
The accompanying notes are an integral part of these consolidated financial statements.
47
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
Consolidated Statements of Comprehensive Loss
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Net loss |
$ | (27,274,068 | ) | $ | (47,914,314 | ) | $ | (30,122,260 | ) | |||
| Other comprehensive income (loss): |
||||||||||||
| Unrealized gains (losses) on available for sale securities, net |
(28,909 | ) | (636,888 | ) | 271,666 | |||||||
| Comprehensive loss |
$ | (27,302,977 | ) | $ | (48,551,202 | ) | $ | (29,850,594 | ) | |||
The accompanying notes are an integral part of these consolidated financial statements.
48
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
Consolidated Statements of Stockholders’ Equity
| Common stock | Additional paid-in capital |
Treasury stock | Unrealized gains on available for sale securities |
Accumulated deficit |
Total stockholders’ equity |
|||||||||||||||||||||||||||
| Number of shares |
Amount | Number of shares |
Amount | |||||||||||||||||||||||||||||
| Balance, January 1, 2008 |
46,027,003 | $ | 4,603 | $ | 536,893,567 | — | $ | — | $ | 399,791 | $ | (424,944,483 | ) | $ | 112,353,478 | |||||||||||||||||
| Vesting of deferred stock |
343,109 | 34 | (34 | ) | — | — | — | — | — | |||||||||||||||||||||||
| Stock issued to directors |
10,881 | 1 | (1 | ) | — | — | — | — | — | |||||||||||||||||||||||
| Forfeiture of restricted stock |
(5,000 | ) | — | — | — | — | — | — | — | |||||||||||||||||||||||
| Purchase and retirement of deferred stock |
(81,326 | ) | (8 | ) | (425,230 | ) | — | — | — | — | (425,238 | ) | ||||||||||||||||||||
| Stock-based compensation expense |
— | — | 6,393,347 | — | — | — | — | 6,393,347 | ||||||||||||||||||||||||
| Exercise of stock options |
39,068 | 3 | 147,566 | — | — | — | — | 147,569 | ||||||||||||||||||||||||
| Unrealized gains on available for sale securities, net |
— | — | — | — | — | 271,666 | — | 271,666 | ||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | (30,122,260 | ) | (30,122,260 | ) | ||||||||||||||||||||||
| Balance, December 31, 2008 |
46,333,735 | 4,633 | 543,009,215 | — | — | 671,457 | (455,066,743 | ) | 88,618,562 | |||||||||||||||||||||||
| Stock-based compensation expense |
— | — | 3,986,313 | — | — | — | — | 3,986,313 | ||||||||||||||||||||||||
| Unrealized losses on available for sale securities, net |
— | — | — | — | — | (636,888 | ) | — | (636,888 | ) | ||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | (47,914,314 | ) | (47,914,314 | ) | ||||||||||||||||||||||
| Balance, December 31, 2009 |
46,333,735 | 4,633 | 546,995,528 | — | — | 34,569 | (502,981,057 | ) | 44,053,673 | |||||||||||||||||||||||
| Vesting of deferred stock |
99,928 | 10 | (10 | ) | — | — | — | — | — | |||||||||||||||||||||||
| Stock-based compensation expense |
— | — | 2,933,929 | — | — | — | — | 2,933,929 | ||||||||||||||||||||||||
| Treasury stock acquired |
— | — | — | 18,904 | (26,301 | ) | — | — | (26,301 | ) | ||||||||||||||||||||||
| Unrealized losses on available for sale securities, net |
— | — | — | — | — | (28,909 | ) | — | (28,909 | ) | ||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | (27,274,068 | ) | (27,274,068 | ) | ||||||||||||||||||||||
| Balance, December 31, 2010 |
46,433,663 | $ | 4,643 | $ | 549,929,447 | 18,904 | $ | (26,301 | ) | $ | 5,660 | $ | (530,255,125 | ) | $ | 19,658,324 | ||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
49
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
Consolidated Statements of Cash Flows
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Net cash flows from operating activities: |
||||||||||||
| Net loss |
$ | (27,274,068 | ) | $ | (47,914,314 | ) | $ | (30,122,260 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Stock-based compensation expense |
2,946,590 | 3,827,591 | 6,363,827 | |||||||||
| Amortization of premiums and discounts on short-term investments |
(103,217 | ) | 866,922 | 1,677,269 | ||||||||
| Amortization of deferred revenue and rent |
(13,338,404 | ) | (13,462,122 | ) | (16,432,499 | ) | ||||||
| Depreciation and amortization expense |
379,580 | 1,155,031 | 2,164,668 | |||||||||
| Non-cash restructuring charge |
— | 1,805,540 | — | |||||||||
| Acquired in-process research and development |
— | 2,300,000 | — | |||||||||
| Changes in assets and liabilities: |
||||||||||||
| Accounts receivable |
710,712 | (227,270 | ) | (1,996,926 | ) | |||||||
| Inventory |
60,091 | (851,327 | ) | — | ||||||||
| Prepaid expenses and other current assets |
901,943 | 1,867,953 | (394,400 | ) | ||||||||
| Other assets |
— | 5,414 | 52,511 | |||||||||
| Accounts payable |
(1,469,755 | ) | (567,968 | ) | 1,435,336 | |||||||
| Accrued expenses |
501,938 | (2,339,826 | ) | 4,444,666 | ||||||||
| Customer deposits |
— | (274,425 | ) | 274,425 | ||||||||
| Deferred revenue |
— | 8,425,000 | — | |||||||||
| Net cash used in operating activities |
(36,684,590 | ) | (45,383,801 | ) | (32,533,383 | ) | ||||||
| Net cash flows from investing activities: |
||||||||||||
| Purchases of equipment and leasehold improvements |
(48,991 | ) | (502,370 | ) | (1,062,638 | ) | ||||||
| Proceeds from disposal of equipment |
66,135 | 985,751 | — | |||||||||
| Purchase of in-process research and development |
— | (2,300,000 | ) | — | ||||||||
| Purchases of short-term investments |
(48,916,592 | ) | (96,615,060 | ) | (156,153,968 | ) | ||||||
| Maturities of short-term investments |
83,000,000 | 143,494,000 | 181,987,000 | |||||||||
| Net cash provided by investing activities |
34,100,552 | 45,062,321 | 24,770,394 | |||||||||
| Net cash flows from financing activities: |
||||||||||||
| Net proceeds from exercise of stock options |
— | — | 147,569 | |||||||||
| Payment of withholding taxes related to deferred stock |
(26,301 | ) | — | (425,238 | ) | |||||||
| Net cash used in financing activities |
(26,301 | ) | — | (277,669 | ) | |||||||
| Net decrease in cash and cash equivalents |
(2,610,339 | ) | (321,480 | ) | (8,040,658 | ) | ||||||
| Cash and cash equivalents at beginning of year |
7,213,666 | 7,535,146 | 15,575,804 | |||||||||
| Cash and cash equivalents at end of year |
$ | 4,603,327 | $ | 7,213,666 | $ | 7,535,146 | ||||||
| Supplemental disclosure of cash flow information: |
||||||||||||
| Cash received from sale of Pennsylvania research and development tax credits |
$ | — | $ | — | $ | 93,656 | ||||||
| Supplemental disclosure of non-cash investing activities: |
||||||||||||
| Unrealized gains (losses) on available for sale securities, net |
$ | (28,909 | ) | $ | (636,888 | ) | $ | 271,666 | ||||
| Change in accrued expenses related to purchases of equipment |
$ | — | $ | (179,663 | ) | $ | (207,577 | ) | ||||
The accompanying notes are an integral part of these consolidated financial statements.
50
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
| 1. | ORGANIZATION AND BUSINESS ACTIVITIES |
Adolor Corporation (the Company) is a biopharmaceutical company focused on the discovery, development and commercialization of novel prescription pain and pain-management products. In May 2008, the U.S. Food and Drug Administration (FDA) approved the Company’s first product, ENTEREG® (alvimopan). ENTEREG is indicated to accelerate upper and lower gastrointestinal (GI) recovery following partial large or small bowel resection surgery with primary anastomosis. Delayed GI recovery following bowel resection surgery can postpone hospital discharge until its resolution, resulting in an increased cost burden on healthcare providers. In June 2008, the Company launched ENTEREG in the United States in collaboration with Glaxo Group Limited (Glaxo).
The Company has a number of product candidates in various stages of clinical and preclinical development. The Company is conducting two Phase 2 clinical trials of ADL5945 to treat opioid-induced constipation (OIC), a condition that often results from long-term use of opioid analgesics in the management of chronic pain conditions. In addition, the Company is continuing limited development efforts on ADL7445, a second peripherally-acting mu opioid receptor antagonist, which is considered a back-up compound in the Company’s OIC program. The Company has completed Phase 1 clinical evaluation of ADL6906 (beloxepin), a compound with a novel and potentially differentiating pharmacological profile for treating chronic pain. Adolor’s preclinical pipeline includes novel, selective centrally-acting mu opioid receptor antagonists (CAMORs) currently in development for the treatment of l-DOPA-induced dyskinesia associated with Parkinson’s disease.
| 2. | BASIS OF ACCOUNTING AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES |
Basis of Presentation and Consolidation
The accompanying consolidated financial statements include the accounts of Adolor and its wholly-owned subsidiary. All intercompany balances and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues and expenses and related disclosure of contingent assets and liabilities. The estimates made are principally in the areas of revenue recognition, research and development accruals and stock option expenses. Management bases its estimates on historical experience and various assumptions that are believed to be reasonable under the circumstances. Actual results may differ from those estimates.
Concentration of Risk
Financial instruments that potentially expose the Company to concentrations of credit risk consist primarily of cash equivalents, short-term investments and receivables. The Company invests its excess cash in accordance with a policy objective that seeks both liquidity and safety of principal. The policy limits investments to instruments issued by the U.S. government and commercial institutions with strong investment grade credit ratings and places restrictions on maturity terms and concentrations by type and issuer.
For the years ended December 31, 2010, 2009 and 2008, three wholesale customers, Cardinal Health, Inc., McKesson Corporation and AmerisourceBergen Corporation, accounted for 93%, 92% and 93%, respectively, of the Company’s total product sales. Through its distribution services agreement with Glaxo, the Company and Glaxo have established credit rating guidelines relative to sales of ENTEREG to wholesalers.
51
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
For the years ended December 31, 2010, 2009 and 2008, all of the Company’s contract revenues resulted from its collaboration agreements with Glaxo and Pfizer. Any failure by the Company’s collaborators to perform their obligations or any decision by its collaborators to terminate these agreements under the terms provided for in their respective agreements could negatively impact the Company’s ability to successfully develop, obtain regulatory approvals for and commercialize its products and product candidates, which would likely materially impact its financial condition and results of operations. In addition, any termination of the Company’s collaboration agreements will terminate the funding it receives under the relevant collaboration agreement and may materially and adversely impact the Company’s ability to fund further development efforts. In December 2010, Pfizer and the Company announced that they were discontinuing further development of ADL5859 and ADL5747. Pfizer delivered to the Company a notice of termination of the collaboration agreement, which will be effective as of March 2011.
The Company depends on Piramal Healthcare (Canada) Limited (formerly Torcan Chemical Ltd.) and Central Glass Germany GmbH (formerly Girindus AG) as the approved suppliers under its New Drug Application (NDA) of the active pharmaceutical ingredient in ENTEREG. The Company also depends on Pharmaceutics International, Inc. and Pharmaceutical Manufacturers Research Services Inc. to manufacture ENTEREG finished capsules. The Company seeks to maintain inventories of finished products to protect against supply disruptions. Any future change in manufacturers or manufacturing process would require regulatory approval.
Fair Value of Financial Instruments
The carrying amounts of the Company’s financial instruments, which include cash equivalents, short-term investments, accounts receivable and accounts payable, approximate their fair values due to the short-term maturities of these instruments.
Cash Equivalents and Short-term Investments
The Company considers all highly liquid investments with a maturity of three months or less when purchased to be cash equivalents. The Company has $3.2 million in short-term money market accounts as of December 31, 2010.
The Company’s entire portfolio of short-term investments is currently classified as available for sale and is stated at fair value as determined by quoted market values. Investments are comprised of U.S. Treasury obligations. All investments are short-term and are classified as current assets. Net unrealized gains and losses are included as a separate component of stockholders’ equity and changes in such amounts are included in comprehensive loss. For purposes of determining realized gains and losses, the cost of short-term investments sold is based upon specific identification. The Company has not experienced any other-than-temporary losses.
Inventory
Inventories are stated at the lower of cost or market, as determined on a first-in, first-out, or FIFO, basis. The Company periodically reviews its inventory for excess or obsolete inventory, and will write-down the excess or obsolete inventory value to its net realizable value, if applicable. Costs associated with the manufacture of alvimopan prior to the FDA approval of ENTEREG were expensed to research and development when incurred. Costs associated with manufacturing the Company’s clinical and preclinical candidates are expensed to research and development when incurred.
52
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
Equipment and Leasehold Improvements
Purchases of laboratory, computer and office equipment, furniture and fixtures and leasehold improvements are recorded at cost. Depreciation and amortization is calculated using the straight-line method over the shorter of the estimated useful lives of the assets, generally three to seven years, or the lease term, if applicable. Expenditures for repairs and maintenance are charged to expense as incurred. When assets are retired or otherwise disposed, the assets and related allowances for depreciation and amortization are eliminated from the accounts and any resulting gain or loss is reflected in operating expenses.
Valuation of Equipment and Leasehold Improvements
The Company periodically assesses its equipment and leasehold improvement assets for impairment in accordance with Financial Accounting Standards Board (FASB) Accounting Standards Codification (ASC) 360, Property, Plant and Equipment. The Company reviews long-lived assets, specifically equipment and leasehold improvements, for impairment whenever events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable. If indicators of impairment exist, impairment loss would be tested for if the estimated undiscounted future cash flows expected to result from the use of the asset and its eventual disposition is less than its carrying amount. The impairment charge, if any, is determined based on the excess of the carrying value of the asset over its fair value, with fair value determined based on an estimate of discounted future cash flows or other appropriate measures of fair value.
Revenue Recognition
The Company recognizes revenue in accordance with Securities and Exchange Commission (SEC) Staff Accounting Bulletin (SAB) No. 101, Revenue Recognition in Financial Statements (SAB 101), as amended by SAB No. 104, Revenue Recognition (SAB 104) and ASC 605, Revenue Recognition (ASC 605).
Product Sales, Net
ENTEREG was approved by the FDA in May 2008 and product shipments to hospitals began in June 2008. In accordance with SAB 101 and SAB 104, the Company recognizes revenue from product sales when the following four revenue recognition criteria are met: persuasive evidence of an arrangement exists, delivery has occurred, the selling price is fixed or determinable, and collectability is reasonably assured. Prior to the fourth quarter of 2009, the Company deferred recognition of revenue associated with the first shipment of ENTEREG to each hospital customer as it was not able to reasonably estimate future returns. When an existing customer placed a new order, the Company recognized product sales on the previous shipment for an amount equal to the lesser of (a) the previous shipment or (b) the new order. During the fourth quarter of 2009, the Company began to recognize net product sales upon the shipment of product to the registered hospital, as the Company had developed sufficient historical experience to reasonably estimate future returns of ENTEREG. As a result of the change in the Company’s revenue recognition policy, net product sales for the year ended December 31, 2009 include $0.9 million for product that was shipped in 2008 to registered hospitals, but not recognized as revenue at the time of shipment. The Company also recognized $0.5 million of selling, general and administrative expenses during 2009 that had been deferred under the Company’s previous revenue recognition policy. The change in the Company’s revenue recognition policy resulted in a decrease to net loss of $0.4 million, or $0.01 per share, for the year ended December 31, 2009.
The Company records product sales net of prompt payment discounts, returns, group purchasing organization fees, chargebacks and other discounts as reported to it by Glaxo. Calculating these allowances requires significant estimates and judgments and while the Company undertakes certain procedures to review the
53
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
reasonableness of the information, it cannot obtain absolute assurance over the accuracy of such information provided by Glaxo. For the years ended December 31, 2010, 2009 and 2008, provisions for these charges, which were offset against product sales, were $1.9 million, $1.2 million and $0.1 million, respectively.
Contract Revenues
Contract revenues consist primarily of milestone payments, cost reimbursement, amortization of up-front license fees and other revenue under collaboration agreements. Non-refundable up-front license fees are recognized as revenue if the Company has a contractual right to receive such payment, the contract price is fixed or determinable, the collection of the resulting receivable is reasonably assured and the Company has no further performance obligations under the license agreement. Multiple element arrangements, such as license and development arrangements, are analyzed to determine whether the deliverables, which often include a license and performance obligations, can be separated for revenue recognition purposes. When the license is considered to either (i) not have standalone value or (ii) have standalone value but the fair value of any of the undelivered performance obligations can not be determined, the up-front license payment is recognized as revenue ratably over the estimated period of when the Company’s performance obligations are to be performed. The Company estimates its performance period based on the specific terms of each collaborative agreement and, as appropriate, will adjust the performance periods based upon available facts and circumstances.
The Company’s collaboration agreements also contain substantive milestone payments. Substantive milestone payments are considered to be performance payments that are recognized upon achievement of the milestone only if all of the following conditions are met: (i) the milestone payment is non-refundable; (ii) achievement of the milestone involves a degree of risk and was not reasonably assured at the inception of the arrangement; (iii) substantive effort is involved in achieving the milestone; (iv) the amount of the milestone payment is reasonable in relation to the effort expended or the risk associated with achievement of the milestone; and (v) a reasonable amount of time passes between the up-front license payment and the first milestone payment as well as between each subsequent milestone payment. Determination as to whether a milestone meets the aforementioned conditions involves the judgment of management. If any of these conditions are not met, the resulting payment would not be considered a substantive milestone, and such payment would therefore be recognized as revenue as such performance obligations are performed in accordance with the policies described above.
Reimbursement of costs is recognized as revenue provided the provisions of ASC 605 are met, the amounts are determinable and collection of the related receivable is reasonably assured.
Amounts received prior to satisfying the above revenue recognition criteria for contract revenues are recorded as deferred revenue in the accompanying consolidated balance sheets. Amounts not expected to be recognized within one year of the balance sheet date are classified as non-current deferred revenue. The estimate of the classification of deferred revenue as current or non-current is based upon management’s estimate of its performance period based on the specific terms of each collaborative agreement. These estimates may change in the future and such changes to estimates would be accounted for prospectively and would result in a change in the amount of revenue recognized in future periods.
Grant income
Grants received are recognized as income when the related work is performed and the qualifying research and development costs are incurred and are presented as other income, net, on the Company’s consolidated statement of operations. In November 2010, the Company received four grants totaling $1.0 million under the Qualified Therapeutic Discovery Project Grants Program. The Qualified Therapeutic Discovery Project Grants
54
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
Program was included in the healthcare reform legislation and established a one-time pool of $1 billion for grants to small biotechnology companies developing novel therapeutics which show potential to: (a) result in new therapies that either treat areas of unmet medical need, or prevent, detect, or treat chronic or acute diseases and conditions; (b) reduce long-term health care costs in the United States; or (c) significantly advance the goal of curing cancer within a the 30-year period. In addition, the Company recognized $0.2 million of grant income during 2010 related to a Michael J. Fox Foundation grant made to support the development of the Company’s CAMOR program.
Research and Development Expenses
Research and product development expenses are charged to expense as incurred. Research and development expenses include, among other costs, salaries and other personnel-related costs, costs to conduct clinical trials, costs to manufacture drug candidates and clinical supplies, laboratory supplies costs and in-process research and development.
Advertising Costs
Advertising costs are charged to expense as incurred and are included in selling, general and administrative expense within the consolidated statements of operations. Advertising costs, which include promotional and other marketing expenses, were $4.7 million, $8.3 million and $6.4 million for the years ended December 31, 2010, 2009 and 2008, respectively.
Accounting for Income Taxes
The Company provides for income taxes under the asset and liability method. Deferred income tax assets and liabilities are determined based on differences between the financial statement reporting and tax bases of assets and liabilities and net operating loss and credit carryforwards, and are measured using the enacted tax rates and laws that will be in effect when such items are expected to reverse. Deferred income tax assets are reduced, as necessary, by a valuation allowance when management determines it is more likely than not that some or all of the tax benefits will not be realized. The effect on deferred income tax assets and liabilities of a change in tax rates is recognized in the period that such tax rate changes are enacted.
Collaborative Arrangements
The Company enters into collaborative arrangements with pharmaceutical companies to develop and commercialize certain of its products and product candidates. ASC 808-10, Collaborative Arrangements, defines a collaborative arrangement as one in which the participants are actively involved and are exposed to significant risks and rewards that depend on the ultimate commercial success of the endeavor. For the years ended December 31, 2010, 2009 and 2008, the Company had collaborative arrangements with Glaxo for the development and commercialization of ENTEREG and Pfizer for the development of its delta opioid receptor agonist compounds. As part of these collaborative arrangements, the Company earns license fees, milestone payments and /or other revenue, which are recorded as contract revenues on the Company’s consolidated statement of operations. Under the guidelines of ASC 605-45-45, Overall Considerations of Reporting Revenue Gross as a Principal vs. Net as an Agent, the Company has determined that it is the principal in the Glaxo and Pfizer collaborative arrangements and, therefore, presents revenues and costs incurred with third parties in connection with collaborative arrangements gross on its consolidated statement of operations. As a result, sales of ENTEREG under the Glaxo collaborative arrangement are presented gross as net product sales and cost reimbursement amounts owed to the Company by Glaxo and Pfizer for certain third-party research and development activities are recorded gross as contract revenues. Monies reimbursable to Glaxo and Pfizer by the
55
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
Company for certain third-party research and development and marketing expenses, as well as for Glaxo profit-share amounts, are recorded gross as research and development expense or selling, general and administrative expense, as applicable, on the Company’s consolidated statements of operations.
Segment Information
The Company is operated as one business and is managed by a single management team that reports to the chief executive officer. The Company does not operate separate lines of business or separate business entities with respect to any of its product candidates. Accordingly, the Company does not prepare discrete financial information with respect to separate product areas or by location, and does not have separately reportable segments.
Net Loss per Common Share
Basic net loss per common share is computed by dividing net loss by the weighted average number of common shares outstanding. Diluted net loss per common share is computed by dividing net loss by the weighted average number of common shares and dilutive potential common share equivalents then outstanding. Potential common shares consist of shares issuable upon the exercise of stock options and unvested deferred and restricted stock awards calculated using the treasury stock method. Because the inclusion of potential common stock would be anti-dilutive for all periods presented as a result of the net losses, diluted net loss per share is the same as basic net loss per share.
The following table sets forth the potential common stock excluded from the calculation of net loss per share because its inclusion would be anti-dilutive:
| December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Options to purchase common stock |
5,147,418 | 6,137,302 | 5,520,472 | |||||||||
| Unvested deferred and restricted common stock |
1,919,343 | 932,687 | 62,500 | |||||||||
Stock-Based Compensation
Stock-based compensation cost is measured at the grant date based on the fair value of the award and is generally recognized as expense on a straight-line basis over the service period, which represents the vesting period. For equity-based awards with performance conditions, the Company recognizes compensation cost if and when it concludes that it is probable that the performance condition will be achieved. The Company calculates the fair value of its stock options using the Black-Scholes option pricing model and the fair value of its deferred and restricted stock is based upon the market value of the underlying common stock on the date of the grant. For any equity-based awards that do not vest on a monthly basis, the Company estimates a forfeiture rate based on its historical experience of pre-vesting forfeitures.
Recent Accounting Pronouncements
In December 2010, the FASB ratified a consensus of the Emerging Issues Task Force related to an annual fee to be paid to the federal government by pharmaceutical manufacturers as mandated by the Patient Protection and Affordable Care Act as amended by the Health Care and Education Reconciliation Act. This consensus requires the liability related to the annual fee to be estimated and recorded in full upon the first qualifying sale with a corresponding deferred cost that is amortized to expense generally using a straight-line method of
56
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
allocation over the calendar year that it is payable. This guidance is effective for calendar years beginning after December 31, 2010, when the fee initially became effective. The Company is currently evaluating the effect that this guidance will have on its consolidated financial statements.
In January 2010, the FASB issued ASU 2010-06, Fair Value Measurements and Disclosures (Topic 820): Improving Disclosures about Fair Value Measurements (ASU 2010-06), which amends the existing fair value measurement and disclosure guidance currently included in Accounting Standards Codification (ASC) Topic 820, Fair Value Measurements and Disclosures, to require additional disclosures regarding fair value measurements. Specifically, ASU 2010-06 requires entities to disclose the amounts of significant transfers between Level 1 and Level 2 of the fair value hierarchy and the reasons for these transfers, the reasons for any transfer in or out of Level 3 and information in the reconciliation of recurring Level 3 measurements about purchases, sales, issuances and settlements on a gross basis. In addition, ASU 2010-06 also clarifies the requirement for entities to disclose information about both the valuation techniques and inputs used in estimating Level 2 and Level 3 fair value measurements. ASU 2010-06 is effective for interim and annual reporting periods beginning after December 15, 2009, except for additional disclosures related to Level 3 fair value measurements, which are effective for fiscal years beginning after December 15, 2010. The adoption of ASU 2010-06 did not impact the Company’s consolidated financial statements or results of operations.
In September 2009, the FASB issued ASU 2009-13, Revenue Recognition (Topic 605): Multiple-Deliverable Revenue Arrangements (ASU 2009-13), which requires companies to allocate revenue in arrangements involving multiple deliverables based on the estimated selling price of each deliverable when such deliverables are not sold separately either by the company or other vendors. ASU 2009-13 eliminates the requirement that all undelivered elements must have objective and reliable evidence of fair value before a company can recognize the portion of the overall arrangement fee that is attributable to items that already have been delivered. As a result, the new guidance may allow some companies to recognize revenue on transactions that involve multiple deliverables earlier than under current requirements. ASU 2009-13 is effective for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010. Early adoption is permitted at the beginning of a company’s fiscal year. The Company does not expect that the adoption of ASU 2009-13 will have a material impact on its consolidated financial statements.
| 3. | SHORT-TERM INVESTMENTS |
Short-term investments consist of investment-grade, fixed-income securities with original maturities of greater than three months. All investments are classified as available for sale and are considered current assets as the contractual maturities of the Company’s short-term investments are all less than one year.
The following summarizes the short-term investments at December 31, 2010 and 2009:
| Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Fair Value | |||||||||||||
| U.S. Government obligations at December 31, 2010 |
$ | 41,977,550 | $ | 7,301 | $ | (1,641 | ) | $ | 41,983,210 | |||||||
| U.S. Government obligations at December 31, 2009 |
$ | 75,957,741 | $ | 40,944 | $ | (6,375 | ) | $ | 75,992,310 | |||||||
ASC 820, Fair Value Measurements and Disclosures, establishes a valuation hierarchy for disclosure of the inputs to valuations techniques used to measure fair value. This hierarchy prioritizes the inputs into three broad levels. Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities. Level 2
57
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
inputs include quoted prices for identical or similar assets and liabilities that are not active, quoted prices for similar assets and liabilities in active markets or inputs that are observable for the asset or liability, either directly or indirectly through market corroboration, for substantially the full term of the financial instrument. Level 3 inputs are unobservable inputs based on management’s own assumptions used to measure assets and liabilities at fair value. The classification of a financial asset or liability within the hierarchy is determined based on the lowest level input that is significant to the fair value measurement.
The following table provides the Company’s assets and liabilities carried at fair value measured on a recurring basis as of December 31, 2010 and 2009:
| Fair Value Measurements Using | ||||||||||||||||
| Total Carrying Value |
Quoted
Prices in Active Markets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
|||||||||||||
| U.S. Government obligations at December 31, 2010 |
$ | 41,983,210 | $ | 41,983,210 | $ | — | $ | — | ||||||||
| U.S. Government obligations at December 31, 2009 |
$ | 75,992,310 | $ | 75,992,310 | $ | — | $ | — | ||||||||
| 4. | INVENTORY |
As of December 31, 2010 and 2009, inventory consisted of the following:
| December 31, 2010 |
December 31, 2009 |
|||||||
| Raw materials |
$ | 67,648 | $ | — | ||||
| Work-in-process |
157,536 | 527,036 | ||||||
| Finished goods |
708,673 | 479,773 | ||||||
| Total inventory |
$ | 933,857 | $ | 1,006,809 | ||||
The above inventory was manufactured subsequent to the approval of ENTEREG by the FDA. As of December 31, 2010 and 2009, $0.6 million and $0.1 million, respectively, of ENTEREG finished goods inventory held at Glaxo warehouses was classified as “inventory consigned to others.” Costs associated with the manufacture of alvimopan prior to the FDA approval of ENTEREG were expensed to research and development when incurred. As a result, at December 31, 2010 and 2009, the Company has inventory related to alvimopan, including raw material, which carries a zero-cost and is not reflected on the consolidated balance sheets at December 31, 2010 and 2009.
58
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
| 5. | EQUIPMENT AND LEASEHOLD IMPROVEMENTS |
Equipment and leasehold improvements consist of the following:
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Laboratory, computer and office equipment |
$ | 4,154,363 | $ | 6,275,413 | ||||
| Furniture, fixtures and leasehold improvements |
7,460,938 | 7,591,281 | ||||||
| 11,615,301 | 13,866,694 | |||||||
| Less accumulated depreciation and amortization: |
||||||||
| Laboratory, computer and office equipment |
(3,974,396 | ) | (5,944,967 | ) | ||||
| Furniture, fixtures and leasehold improvements |
(7,176,853 | ) | (7,060,951 | ) | ||||
| (11,151,249 | ) | (13,005,918 | ) | |||||
| $ | 464,052 | $ | 860,776 | |||||
Depreciation and amortization expense related to equipment and leasehold improvements was $0.4 million, $1.2 million and $2.2 million for the years ended December 31, 2010, 2009 and 2008, respectively. During the year ended December 31, 2009, the Company recorded a $1.8 million non-cash impairment charge primarily related to leasehold improvements and laboratory equipment used for activities which were eliminated pursuant to the Company’s June 2009 restructuring (see Note 12).
| 6. | ACCRUED EXPENSES |
Accrued expenses consist of the following:
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Clinical development costs |
$ | 1,211,386 | $ | 920,679 | ||||
| Manufacturing costs |
625,244 | 644,277 | ||||||
| Sales and marketing costs |
153,090 | 692,176 | ||||||
| Consulting and other costs |
1,327,295 | 2,614,231 | ||||||
| Collaboration agreement expenses |
3,255,176 | 1,313,098 | ||||||
| Professional fees |
450,777 | 200,840 | ||||||
| Personnel related costs |
1,900,540 | 2,036,269 | ||||||
| $ | 8,923,508 | $ | 8,421,570 | |||||
59
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
| 7. | COMMITMENTS |
Future minimum lease payments under non-cancelable operating leases for equipment and office and laboratory space are as follows:
| Year Ending December 31, |
Operating | Operating Sublease Rental Income |
||||||
| 2011 |
$ | 1,262,000 | $ | (355,000 | ) | |||
| 2012 |
1,243,000 | (349,000 | ) | |||||
| 2013 |
508,000 | (116,000 | ) | |||||
| 2014 |
— | — | ||||||
| 2015 |
— | — | ||||||
| 2016 and beyond |
— | — | ||||||
| $ | 3,013,000 | $ | (820,000 | ) | ||||
Rent expense was $1.1 million, $1.1 million and $1.0 million for the years ended December 31, 2010, 2009 and 2008, respectively. In December 2002, the Company signed a lease agreement for office and laboratory space that expires in May 2013. The lease includes a renewal option for two consecutive additional five-year periods and the Company has a purchase option exercisable at the tenth year of the lease term. The Company recognizes rent expense on a straight-line basis over the life of the lease, and as a result, rent escalation expense is deferred on the consolidated balance sheet. During 2010, the Company subleased certain laboratory and office space, with a term that expires in April 2013.
Other Commitments
The Company has committed to make future minimum payments to third parties for the manufacture of ENTEREG and the use of software. These obligations are legally binding and enforceable under agreements with third parties, and as of December 31, 2010, these minimum purchase commitments totaled $0.8 million.
The Company also has contingent liabilities associated with various agreements that it has entered into for services with third-party vendors, including agreements to conduct clinical trials, to manufacture product candidates and for consulting and other contracted services. These contingent liabilities require future performance by the third party in order for payment to be executed, and the Company accrues the costs of these agreements based on estimates of work completed to date. The Company estimates that $16.2 million will be payable in future periods under the arrangements in place at December 31, 2010. Of this amount, $2.4 million has been accrued for work estimated to have been completed as of December 31, 2010 and $13.8 million relates to future performance under these arrangements.
In addition to the above, the Company has committed to make potential future milestone payments to third parties as part of its in-licensing agreements. Payments generally become due and payable only upon the achievement of certain developmental, regulatory and/or commercial milestones. Because the achievement of these milestones is neither probable nor reasonably estimable, the Company has not recorded a liability on its consolidated balance sheet for any such contingencies. As of December 31, 2010, the maximum potential milestone payments due under current contractual agreements are $106.5 million.
Glaxo Collaboration Agreement
Under the terms of the Glaxo agreement, the Company is obligated to partially reimburse Glaxo for certain third-party expenses incurred by Glaxo related to ENTEREG, pursuant to an agreed upon development plan and
60
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
budget. The Company also incurs certain third-party expenses related to ENTEREG, pursuant to an agreed upon development plan and budget, a portion of which are reimbursable to the Company by Glaxo. The Company records these expenses gross on its consolidated statement of operations as incurred.
In addition, the Company and Glaxo have a profit-sharing arrangement pursuant to which the Company receives 45% and Glaxo receives 55% of profits and losses, as defined, through June of 2011, after which the profit split is 50% each. Profits and losses are calculated as net sales of ENTEREG in the United States less certain agreed-upon costs, subject to certain adjustments. The Company records these profit-sharing expenses as selling, general and administrative expenses gross on its consolidated statement of operations as incurred.
Pfizer Collaboration Agreement
Under the terms of the Pfizer agreement, which will terminate effective March 2011, the Company is obligated to partially reimburse Pfizer for third-party expenses incurred by Pfizer in the development of certain delta agonist compounds in support of regulatory filings in the United States, pursuant to an agreed upon development plan and budget. The Company also incurs expenses in the development of certain delta agonist compounds, pursuant to an agreed upon development plan and budget, a portion of which are reimbursable to the Company by Pfizer. The Company records these expenses gross on its consolidated statement of operations as incurred.
License Agreements
With regard to the Company’s commercial product, ENTEREG, the Company has commitments to Roberts Laboratories, Inc. (Roberts) and Eli Lilly and Company (Lilly). In November 1996, Roberts licensed from Lilly certain intellectual property rights relating to ENTEREG. In June 1998, the Company entered into an Option and License Agreement with Roberts under which the Company sublicensed these rights from Roberts. In December 2000, Shire U.S. Inc. (Shire) became the successor in interests to Roberts under the Company’s option and license agreement with Roberts. The Company has made license and milestone payments under this agreement totaling $2.5 million. In addition to the license and milestone payments, the Company is obligated under the agreement to pay, in the aggregate, single-digit royalties on commercial sales of ENTEREG. For the years ended December 31, 2010, 2009 and 2008, the Company incurred $2.0 million, $1.2 million and $0.1 million, respectively, of royalty expense related to sales of ENTEREG. The Company’s obligations to pay royalties to Shire and Lilly are expected to expire on the date of the last to expire of the licensed Lilly patents.
In August 2002, the Company entered into a separate exclusive license agreement with Lilly under which the Company obtained an exclusive license to six issued U.S. patents and related foreign equivalents and know-how relating to peripherally selective opioid antagonists. The Company paid Lilly $4.0 million upon signing the agreement and is subject to additional clinical and regulatory milestone payments and royalty payments to Lilly on sales, if any, of new products utilizing the licensed technology. Under this license agreement, the Company also paid Lilly $4.0 million upon acceptance for review of the Company’s ENTEREG NDA by the FDA, which payment was made in the third quarter of 2004. However, there are no ongoing royalties on commercial sales of ENTEREG due to Lilly under this agreement.
In September 2009, the Company acquired from Lilly the exclusive worldwide rights to ADL5945, a clinical-stage opioid receptor antagonist. Under the terms of the agreement, the Company paid Lilly $2.0 million. As a result of requiring additional research and development efforts and regulatory approval in order to commercialize this product candidate, the $2.0 million was recognized as acquired in-process research and development and recorded as research and development expense for the year ended December 31, 2009. In
61
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
addition to the $2.0 million payment, the Company will be required to pay milestones, which are contingent upon achievement of pre-defined, late-stage clinical and regulatory events and achievement of certain sales targets, and royalties on net sales of the product.
The Company charges to expense research and development milestone payments that are required to be made upon the occurrence of future events prior to receipt of applicable regulatory approval.
| 8. | STOCKHOLDERS’ EQUITY |
Shareholder Rights Plan
The Company’s Board of Directors adopted a Shareholder Rights Plan (the Plan) in February 2001, which was renewed and extended in January 2011. Under the Plan, preferred stock purchase rights (each, a Right) were distributed as a dividend at the rate of one Right for each share of common stock outstanding as of the close of business on February 20, 2001, and automatically attach to shares issued thereafter. Each Right entitles the holder to purchase one ten-thousandth of a share of newly created Series A Junior Participating preferred stock of the Company at an exercise price of $18.00 (the Exercise Price) per Right. In general, the Rights will be exercisable if a person or group (Acquiring Person) becomes the beneficial owner of 20% or more of the outstanding common stock of the Company or announces a tender offer for 20% or more of the common stock of the Company. When the Rights become exercisable, a holder, other than the Acquiring Person, will have the right to receive, upon exercise, common stock having a value equal to two times the Exercise Price of the Right. The Board of Directors will in general be entitled to redeem the Rights for $.00001 per Right at any time prior to the occurrence of the stock acquisition events described above. If not redeemed, the Rights will expire on January 31, 2021.
Standstill Arrangements
The Glaxo collaboration agreement generally provides that during its term, Glaxo will not, directly or indirectly, alone or in concert with others: (i) acquire, or agree to acquire any shares of the Company’s common stock or any securities exercisable for or convertible into the Company’s common stock; (ii) make, or in any way participate in, any solicitation of proxies to vote the Company’s common stock; or (iii) acquire or agree to acquire any of the Company’s tangible or intangible assets not offered for sale by the Company. However, Glaxo may, under certain circumstances, acquire equity securities of the Company set forth in the agreement, including following the initiation by a third party of an unsolicited tender offer to purchase the Company or in connection with stock splits or recapitalizations or on exercise of pre-emptive rights afforded to the Company’s stockholders generally.
The Pfizer license and collaboration agreement generally provides that for a period lasting until the earlier of (a) a number of years as defined in the agreement and (b) a period of time following the effective date of termination, Pfizer will not directly or indirectly, and will not encourage others to: (i) acquire, or agree to acquire securities of the Company; (ii) make, or in any way participate in, any solicitation of proxies to vote the Company’s common stock; or (iii) acquire or agree to acquire any of the assets, tangible or intangible, of the Company. The agreement sets forth certain circumstances under which Pfizer may acquire securities of the Company or be released from the standstill arrangement. As a result of the termination of the Company’s collaboration agreement with Pfizer effective March 2011, the limitations on Pfizer’s ability to acquire the Company’s securities will terminate effective March 2013.
Equity Compensation Plans
The Company has established equity compensation plans for its employees and directors and certain other individuals. The equity plans are administered by the Compensation Committee of the Company’s Board of
62
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
Directors and consist of the Company’s 1994 Amended and Restated Equity Compensation Plan, as amended (the 1994 Plan), which expired in February 2010, and 2003 Amended and Restated Stock-Based Incentive Compensation Plan (the 2003 Plan), together known as the Plans. The 2003 Plan allows for the granting of incentive and nonqualified stock options and deferred and restricted stock awards to employees, directors, consultants and contractors. In aggregate, 12,950,000 shares of the Company’s common stock are authorized to be issued under the Plans. As of December 31, 2010, there were 878,743 shares available for future grants under the 2003 Plan and the Company has reserved approximately 7,900,000 shares of common stock for the exercise of stock options and vesting of deferred and restricted stock awards granted or to be granted under the Plans. There were no shares available for future grants under the 1994 Plan as of December 31, 2010.
The following aggregate stock-based compensation expense resulting from stock options, restricted stock awards and deferred stock awards was included in the Company’s consolidated statements of operations:
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Cost of goods sold |
$ | 50,969 | $ | 36,270 | $ | — | ||||||
| Research and development |
891,748 | 1,502,160 | 2,966,272 | |||||||||
| Selling, general and administrative |
2,003,873 | 2,289,161 | 3,397,555 | |||||||||
| Total stock-based compensation expense |
$ | 2,946,590 | $ | 3,827,591 | $ | 6,363,827 | ||||||
During the years ended December 31, 2010 and 2009, the Company capitalized approximately $40,000 and $0.2 million, respectively, of employee stock-based compensation to inventory. There were no recognized tax benefits related to stock compensation during the years ended December 31, 2010, 2009 or 2008, as any benefit was offset by the Company’s full valuation allowance on its net deferred tax asset. In addition, the Company has not recognized any windfall tax benefit as the resulting deduction has not been realized through a reduction of income taxes payable.
Stock Options
Stock options generally vest over four years from the date of grant and stock options are exercisable generally for a period of ten years from the date of grant. Upon the exercise of stock options, new shares of the Company’s common stock are issued. The Company uses the Black-Scholes option pricing model to calculate the fair value of stock options on the date of grant, and option awards are generally granted with an exercise price equal to the closing market price of the Company’s stock at the date of grant. Expected volatility for the expected life of the option is based upon historical volatility and the expected life of the stock options represents the period of time that option awards are expected to be outstanding giving consideration to vesting schedules and the Company’s historical exercise patterns. The risk-free interest rate is calculated using the U.S. Treasury yield curves in effect at the time of grant, for the period equal to the expected life of the options. The Company has not paid any dividends in the past and does not plan to pay any dividends in the foreseeable future. The fair value of stock options granted to employees and directors was estimated using the following weighted-average assumptions for the years ended December 31, 2010, 2009 and 2008:
| 2010 | 2009 | 2008 | ||||||||||
| Expected dividend yield |
— | — | — | |||||||||
| Expected stock price volatility |
80.5 | % | 84.4 | % | 81.2 | % | ||||||
| Risk-free interest rate |
2.17 | % | 2.03 | % | 3.11 | % | ||||||
| Expected life (in years) |
5.0 | 5.0 | 5.0 | |||||||||
63
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
The following table summarizes the aggregate stock option activity for the year ended December 31, 2010:
| Options | Weighted Average Exercise Price |
Weighted Average Remaining Contractual Life (years) |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding at January 1, 2010 |
6,137,302 | $ | 5.79 | |||||||||||||
| Granted |
214,400 | 1.62 | ||||||||||||||
| Exercised |
— | — | ||||||||||||||
| Forfeited |
(515,553 | ) | 2.65 | |||||||||||||
| Expired |
(688,731 | ) | 6.93 | |||||||||||||
| Outstanding at December 31, 2010 |
5,147,418 | $ | 5.78 | 6.8 | $ | — | ||||||||||
| Exercisable at December 31, 2010 |
3,579,951 | $ | 7.43 | 6.1 | $ | — | ||||||||||
Intrinsic value in the above table was calculated as the difference between the Company’s closing stock price on the last trading day of 2010 and the exercise price, multiplied by the number of options. For any of the Company’s outstanding stock options with an exercise price equal to or greater than the Company’s closing stock price on December 31, 2010, the intrinsic value was considered to be zero.
On May 12, 2009, the Company’s stockholders approved a stock option exchange program under which eligible employees (which excluded executive officers and directors) were given the opportunity to exchange some or all of their outstanding options with exercise prices of $7.00 per share or higher for a lesser number of new options with an exercise price per share equal to the fair market value on the exchange date. The program commenced on July 22, 2009 and expired on August 19, 2009. Under the program, the Company accepted for cancellation eligible options to purchase 367,413 shares of the Company’s common stock and the Company issued new options to purchase up to 37,575 shares of the Company’s common stock under the Plans in exchange for the options surrendered. The new options have an exercise price of $1.57 per share, the closing price of the Company’s common stock on August 20, 2009, and vest on a semi-annual basis over a period of one or two years. The exchange was treated as a modification and the incremental expense associated with the exchange was immaterial.
As of December 31, 2010, total unrecognized compensation cost related to outstanding stock options was $1.8 million, which will be amortized over the weighted average remaining service period of 2.0 years. There were no in-the-money options exercisable as of December 31, 2010. For the year ended December 31, 2008, the Company received net proceeds of $0.1 million from the exercise of stock options. There were no exercises of stock options during the years ended December 31, 2010 and 2009.
The weighted-average grant date fair value of the options issued during the years ended December 31, 2010, 2009 and 2008 was $1.05, $1.09 and $2.90 per share, respectively. The intrinsic value of stock options exercised for the year ended December 31, 2008 was $43,000.
64
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
A summary of options outstanding and exercisable by price range at December 31, 2010, is as follows:
| Options Outstanding | Options Exercisable | |||||||||||||||||||
| Range of Exercise Prices |
Number of Shares |
Weighted Average Remaining Option Life |
Weighted Average Exercise Price (per share) |
Number of Shares |
Weighted Average Exercise Price (per share) |
|||||||||||||||
| $ 1.19—2.79 |
2,080,723 | 8.6 | $ | 1.63 | 784,795 | $ | 1.76 | |||||||||||||
| $ 2.80—5.59 |
1,376,318 | 7.0 | $ | 4.16 | 1,109,041 | $ | 4.15 | |||||||||||||
| $ 5.60—8.39 |
629,383 | 6.0 | $ | 7.99 | 625,121 | $ | 8.00 | |||||||||||||
| $ 8.40—11.19 |
240,517 | 4.3 | $ | 9.46 | 240,517 | $ | 9.46 | |||||||||||||
| $11.20—13.99 |
191,957 | 2.6 | $ | 12.97 | 191,957 | $ | 12.97 | |||||||||||||
| $14.00—16.79 |
404,520 | 3.7 | $ | 14.86 | 404,520 | $ | 14.86 | |||||||||||||
| $16.80—19.59 |
34,000 | 2.1 | $ | 18.16 | 34,000 | $ | 18.16 | |||||||||||||
| $19.60—22.39 |
100,000 | 2.5 | $ | 21.03 | 100,000 | $ | 21.03 | |||||||||||||
| $22.40—25.31 |
90,000 | 5.4 | $ | 23.22 | 90,000 | $ | 23.22 | |||||||||||||
| 5,147,418 | 6.8 | $ | 5.78 | 3,579,951 | $ | 7.43 | ||||||||||||||
Deferred and Restricted Stock Awards
Deferred and restricted stock awards generally vest when either a performance condition is satisfied or ratably over one to four years from the date of grant. The Company granted deferred and restricted stock awards to employees and non-employee directors as follows: 1,434,827 awards during 2010; 935,901 awards during 2009; and 179,169 awards during 2008. Of the awards granted during 2010, 277,500 shares were granted to the Company’s officers and vest upon the achievement of certain product sales and development targets within a specified period of time. The Company will recognize compensation cost for these awards if and when it concludes that it is probable that the performance condition will be achieved. The remaining awards consisted of 1,120,350 employee deferred stock awards that vest in full in September 2012 and 36,977 non-employee director deferred stock awards that vest in full in May 2011. During the year ended December 31, 2010, 130,000 performance shares, which were granted to certain of the Company’s executive officers in 2009, expired due to the Company not achieving certain ENTEREG sales targets within a specified period of time. During 2010, 99,928 deferred stock awards vested under the terms of employee and non-employee director grant agreements. In connection with the vesting of these restricted stock awards, 81,024 shares of the Company’s common stock were issued to employees and non-employee directors and 18,904 shares were surrendered to the Company in satisfaction of minimum tax withholding obligations. The surrendered shares were recorded as treasury stock on the Company’s consolidated balance sheet. During 2008, 380,609 deferred and restricted stock awards vested upon FDA approval of ENTEREG. In connection with the vesting of deferred and restricted stock awards during the year ended December 31, 2008, approximately 81,000 shares, with an aggregate fair value of $0.4 million, were withheld and retired in satisfaction of minimum tax withholding obligations. The following table summarizes deferred and restricted stock awards for the year ended December 31, 2010:
| Number of Shares |
Weighted Average Grant Date Fair Value (Per Share) |
Aggregate Intrinsic Value |
||||||||||
| Nonvested at January 1, 2010 |
932,687 | $ | 1.87 | |||||||||
| Granted |
1,434,827 | 1.09 | ||||||||||
| Vested |
(99,928 | ) | 2.25 | |||||||||
| Forfeited |
(348,243 | ) | 1.68 | |||||||||
| Nonvested at December 31, 2010 |
1,919,343 | $ | 1.30 | $ | 2,322,405 | |||||||
65
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
As of December 31, 2010, total unrecognized compensation cost related to unvested deferred and restricted stock awards was $1.8 million, which will be expensed over the applicable vesting term or upon the achievement, if any, of certain performance conditions. The weighted-average grant date fair value of deferred and restricted stock issued during the years ended December 31, 2010, 2009 and 2008 was $1.09, $1.51 and $4.43 per share, respectively. The fair value of deferred and restricted stock awards that vested during the years ended December 31, 2010 and 2008 was $0.2 million and $2.1 million, respectively. There were no deferred or restricted stock awards that vested during the year ended December 31, 2009.
| 9. | INCOME TAXES |
No federal and state taxes are payable as of December 31, 2010 and 2009.
As of December 31, 2010, the Company had $445.4 million of Federal and $404.8 million of state net operating loss carryforwards potentially available to offset future taxable income. The Federal and state net operating loss carryforwards will expire as follows:
| Federal | State | |||||||
| 2011 |
$ | 1,079,000 | — | |||||
| 2012 |
1,867,000 | — | ||||||
| 2013 |
— | — | ||||||
| 2014 |
— | — | ||||||
| 2015 |
— | — | ||||||
| 2016 |
— | — | ||||||
| 2017 |
— | — | ||||||
| Thereafter |
442,462,000 | 404,765,000 | ||||||
| $ | 445,408,000 | $ | 404,765,000 | |||||
Federal and state net operating loss carryforwards described above do not reflect a portion of the benefit related to certain stock option exercises as prescribed by ASC 718, Compensation—Stock Compensation. At December 31, 2010, the Company also has $12.8 million of Federal and $0.3 million of state research and development tax credit carryforwards, which begin expiring in 2011, and are available to reduce Federal and state income taxes.
The Tax Reform Act of 1986 (the Act) provides for a limitation on the annual use of net operating loss and research and development tax credit carryforwards following certain ownership changes (as defined by the Act). Because the Company may have experienced various ownership changes, as defined by the Act, as a result of past financings and its initial public offering, the Company’s ability to utilize the above mentioned Federal and state net operating loss and credit carryforwards in any given year may be limited. Federal and state tax law limits the time during which carryforwards may be applied against future taxes and Pennsylvania tax law limits the utilization of state net operating loss carryforwards to $3.0 million annually.
Significant components of the Company’s deferred tax assets and liabilities are shown below. At December 31, 2010, a valuation allowance of $216.3 million has been recognized to offset the deferred tax asset. A valuation allowance to reduce the deferred tax asset is required if, based on the weight of the evidence, it is more likely than not that some, or all, of the deferred tax assets will not be realized. Realization of the Company’s deferred tax asset is dependent upon generating future taxable income and given the uncertainty of future profitability, management has determined that a valuation allowance is necessary. The change in the deferred tax asset valuation allowance for the years ended December 31, 2010, 2009 and 2008 was $8.6 million,
66
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
$18.0 million and $12.8 million, respectively, and such change reduced the statutory Federal tax benefit at a rate of 35% to no tax benefit or provision in the consolidated statement of operations.
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Deferred tax assets: |
||||||||
| Net operating losses |
$ | 182,124,000 | $ | 165,636,000 | ||||
| Capitalized research and development costs |
4,137,000 | 7,757,000 | ||||||
| Tax credit carryforwards |
13,012,000 | 12,392,000 | ||||||
| Deferred revenue |
10,386,000 | 15,342,000 | ||||||
| Accrued expenses and other |
6,678,000 | 6,606,000 | ||||||
| Total deferred tax assets |
216,337,000 | 207,733,000 | ||||||
| Less valuation allowance |
(216,335,000 | ) | (207,719,000 | ) | ||||
| Net deferred tax assets |
2,000 | 14,000 | ||||||
| Deferred tax liability |
(2,000 | ) | (14,000 | ) | ||||
| Net deferred tax |
$ | — | $ | — | ||||
In addition, other income, net, of $0.1 million for the year ended December 31, 2008 represents cash received from the sale of certain Pennsylvania research and development tax credits.
| 10. | CONTRACT REVENUES |
Contract revenues consist of the following:
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Amortization of deferred revenue |
$ | 13,166,677 | $ | 13,290,394 | $ | 16,269,972 | ||||||
| Cost reimbursement under collaboration agreements |
4,749,556 | 8,513,270 | 11,938,252 | |||||||||
| Milestone revenue |
— | — | 20,000,000 | |||||||||
| Other |
— | 947,920 | — | |||||||||
| Total contract revenues |
$ | 17,916,233 | $ | 22,751,584 | $ | 48,208,224 | ||||||
In April 2002, the Company entered into a collaboration agreement with Glaxo for the exclusive worldwide development and commercialization of ENTEREG for certain indications. Under the terms of the agreement, Glaxo paid the Company a non-refundable and non-creditable signing fee of $50.0 million. The $50.0 million signing fee was recorded as deferred revenue and is being recognized as revenue on a straight-line basis over the estimated performance period under the collaboration agreement, which extends to March 2016. Revenue related to the Glaxo collaboration agreement of $3.3 million was recognized in each of the years ended December 31, 2010, 2009 and 2008. During the year ended December 31, 2008, the Company recorded $20.0 million in milestone revenue under the Glaxo collaboration agreement following FDA approval of ENTEREG in May 2008.
During the first quarter of 2009, the Company and Glaxo entered into Amendment No. 4 to the collaboration agreement, under which Glaxo paid the Company $8.4 million during 2009. The $8.4 million was recorded as deferred revenue and is being recognized as revenue on a straight-line basis over the estimated remaining performance period under the collaboration agreement. Revenue related thereto of $1.2 million and $1.0 million was recognized in the years ended December 31, 2010 and 2009, respectively. Under the terms of Amendment No. 4, the Company also received a $0.9 million payment from Glaxo in 2009 that it recognized as revenue.
67
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
Certain external expenses incurred in the United States by each party are reimbursed pursuant to contractually agreed percentages. Reimbursement amounts owed to the Company by Glaxo are recorded gross in the consolidated statements of operations as contract revenues. The Company recorded collaboration agreement reimbursements from Glaxo of $1.4 million, $4.4 million and $5.6 million for the years ended December 31, 2010, 2009 and 2008, respectively. As of December 31, 2010 and 2009, $0.2 million and $0.8 million, respectively, was receivable from Glaxo for reimbursement of expenses incurred by the Company pursuant to the collaboration agreement.
In December 2007, the Company entered into a collaboration agreement with Pfizer for the exclusive worldwide development and commercialization of ADL5859 and ADL5747. Under the terms of the agreement, Pfizer paid the Company an up-front payment of $30.0 million and reimbursed $1.9 million of Phase 2a development costs incurred by the Company prior to entering into the collaboration agreement. The $31.9 million up-front fee was recorded as deferred revenue and was recognized as revenue on a straight-line basis over the estimated performance period under the collaboration agreement. During the third quarter of 2008, the performance period was extended by six months to August 2010 based on the status of the development programs. The effect of the third quarter 2008 change in estimate was an increase to net loss of $1.2 million, or $0.03 per share, for the year ended December 31, 2008. During the first quarter of 2009, the performance period was further extended by four months to December 2010 based on the status of the development programs. The effect of the first quarter 2009 change in estimate was an increase to net loss of $1.6 million, or $0.03 per share, for the year ended December 31, 2009. The Company recorded revenue related to the deferred license fees under the collaboration agreement with Pfizer of $8.7 million, $9.0 million and $13.0 million for the years ended December 31, 2010, 2009 and 2008, respectively.
Certain external expenses for research and development and marketing activities incurred in the United States by each party are reimbursed pursuant to contractually agreed percentages. Reimbursement amounts owed to the Company by Pfizer are recorded gross on the consolidated statements of operations as contract revenues. The Company recorded collaboration cost reimbursement from Pfizer of $3.3 million, $4.1 million and $6.3 million for the years ended December 31, 2010, 2009 and 2008, respectively. As of December 31, 2010 and 2009, $0.3 million and $1.3 million, respectively, was receivable from Pfizer for reimbursement of expenses incurred by the Company pursuant to the collaboration agreement.
In December 2010, Pfizer and the Company announced that they were discontinuing further development of ADL5859 and ADL5747. Based on the clinical evaluation of ADL5859 and ADL5747 in multiple indications, the companies concluded that there is not compelling evidence to continue the development program. Pfizer delivered to the Company a notice of termination of their collaboration agreement, which will be effective as of March 2011. As the Company had completed substantially all performance under the collaboration agreement as of December 31, 2010, the performance period was not extended beyond December 2010.
| 11. | 401(k) PROFIT SHARING PLAN |
The Company maintains a 401(k) Profit Sharing Plan (the 401(k) Plan) available to all employees meeting certain eligibility criteria. The 401(k) Plan permits participants to contribute up to 100% of their salary, not to exceed the limits established by the Internal Revenue Code. All contributions made by participants into the participant’s account vest immediately. For the years ended December 31, 2010, 2009 and 2008, the Company made contributions to the 401(k) Plan of $0.3 million, $0.4 million and $0.2 million, respectively. The Company’s common stock is not, and never has been, an investment option for 401(k) Plan participants.
68
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
| 12. | RESTRUCTURING |
In July 2010, the Company announced a restructuring plan to reduce costs and realign its workforce and operations based on its current business objectives. The restructuring resulted in the reduction of 30 positions, or approximately 27% of the Company’s workforce, as well as other cost saving initiatives. Terminated employees were provided with severance payments, continued benefits for a specified period of time and outplacement assistance. The Company recorded a restructuring charge of $1.9 million for the year ended December 31, 2010, which consisted of employee severance and benefit-related costs. The Company has completed all restructuring activities and recognized all anticipated restructuring charges as of December 31, 2010 in connection with this restructuring plan.
In June 2009, the Company announced a restructuring that resulted in the reduction of 45 positions, or 28% of the Company’s workforce, as well as other cost saving initiatives. The Company reduced and restructured its discovery group to focus on late-stage, preclinical compounds, with fewer resources dedicated to early-stage programs. Employees directly affected by the restructuring were provided with severance payments, continued benefits for a specified period of time and outplacement assistance. The Company recorded a restructuring charge of $3.9 million for the year ended December 31, 2009, which consisted of $2.1 million of employee severance and benefit-related costs and $1.8 million of a non-cash impairment charge primarily related to leasehold improvements and laboratory equipment used for activities which were eliminated pursuant to the Company’s restructuring. The Company received proceeds of $1.0 million related to the sale of these impaired assets during the year ended December 31, 2009.
The following table summarizes activity related to the Company’s restructurings:
| Employee Termination Costs |
Asset Write-downs |
Total | ||||||||||
| Balance at January 1, 2009 |
$ | — | $ | — | $ | — | ||||||
| Restructuring charge |
2,127,042 | 1,805,540 | 3,932,582 | |||||||||
| Cash payments |
(2,125,287 | ) | — | (2,125,287 | ) | |||||||
| Non-cash impairment |
— | (1,805,540 | ) | (1,805,540 | ) | |||||||
| Balance at December 31, 2009 |
1,755 | — | 1,755 | |||||||||
| Restructuring charge |
1,918,701 | — | 1,918,701 | |||||||||
| Cash payments |
(1,898,477 | ) | — | (1,898,477 | ) | |||||||
| Balance at December 31, 2010 |
$ | 21,979 | $ | — | $ | 21,979 | ||||||
69
Table of Contents
ADOLOR CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(continued)
| 13. | UNAUDITED QUARTERLY INFORMATION |
The table below summarizes the unaudited results of operations for each quarter of 2010 and 2009:
| Quarter Ended | ||||||||||||||||
| March 31 | June 30 | September 30 | December 31 | |||||||||||||
| Fiscal 2010 |
||||||||||||||||
| Revenue |
$ | 10,668,087 | $ | 10,946,293 | $ | 10,673,609 | $ | 11,014,529 | ||||||||
| Loss from operations |
$ | (9,667,234 | ) | $ | (8,328,508 | ) | $ | (6,151,873 | ) | $ | (4,608,722 | ) | ||||
| Net loss |
$ | (9,599,539 | ) | $ | (8,277,050 | ) | $ | (6,061,851 | ) | $ | (3,335,628 | ) | ||||
| Basic and diluted net loss per share |
$ | (0.21 | ) | $ | (0.18 | ) | $ | (0.13 | ) | $ | (0.07 | ) | ||||
| Fiscal 2009 |
||||||||||||||||
| Revenue |
$ | 6,679,660 | $ | 9,063,938 | $ | 8,662,941 | $ | 12,954,067 | ||||||||
| Loss from operations |
$ | (13,673,941 | ) | $ | (16,850,298 | ) | $ | (11,288,914 | ) | $ | (7,151,948 | ) | ||||
| Net loss |
$ | (13,187,340 | ) | $ | (16,541,847 | ) | $ | (11,129,072 | ) | $ | (7,056,055 | ) | ||||
| Basic and diluted net loss per share |
$ | (0.28 | ) | $ | (0.36 | ) | $ | (0.24 | ) | $ | (0.15 | ) | ||||
During the fourth quarter of 2009, the Company began to recognize net product sales upon the shipment of product to the registered hospital, as the Company had developed sufficient historical experience to reasonably estimate future returns of ENTEREG. As a result of the change in the Company’s revenue recognition policy, net product sales for the quarter ended December 31, 2009 include $2.6 million for product that was shipped to registered hospitals through September 30, 2009, but not recognized as revenue at the time of shipment. The Company also recognized $1.4 million of selling, general and administrative expenses during the quarter ended December 31, 2009 that had been deferred under the Company’s previous revenue recognition policy. The change in the Company’s revenue recognition policy resulted in a decrease to net loss of $1.2 million, or $0.03 per share, for the quarter ended December 31, 2009.
70
Table of Contents
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
| ITEM 9A. | CONTROLS AND PROCEDURES |
(a) Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our President and Chief Executive Officer and our Senior Vice President, Finance and Chief Financial Officer (the principal financial and accounting officer), evaluated the effectiveness of our disclosure controls and procedures (as defined in Rule 13a-15(e) of the Securities Exchange Act of 1934) as of the end of the period covered by this Annual Report on Form 10-K. Based on that evaluation, the President and Chief Executive Officer and the Senior Vice President, Finance and Chief Financial Officer concluded that our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K have been designed and are functioning effectively to provide reasonable assurance that the information required to be disclosed by us in reports filed under the Securities Exchange Act of 1934 is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. We believe that a control system, no matter how well designed and operated, cannot provide absolute assurance that the objectives of the control system are met, and no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within a company have been detected.
(b) Management’s Annual Report on Internal Control over Financial Reporting
Management’s Report on Internal Control over Financial Reporting is included in Part II, Item 8 of this Annual Report on Form 10-K and incorporated into this Item 9A by reference.
(c) Attestation Report of the Registered Public Accounting Firm
This Annual Report on Form 10-K does not include an attestation report of our registered public accounting firm regarding internal control over financial reporting as required by Section 404(c) of the Sarbanes Oxley Act of 2002. Management’s report was not subject to attestation by our registered public accounting firm pursuant to Section 989G(a) of the Dodd-Frank Wall Street Reform and Consumer Protection Act, which permits us to provide only management’s report in this Annual Report on Form 10-K.
(d) Changes in Internal Control over Financial Reporting
There have been no changes in our internal control over financial reporting during our most recent fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| ITEM 9B. | OTHER INFORMATION |
None.
71
Table of Contents
PART III
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
Directors
The information required by this Item 10 with respect to our Directors is incorporated herein by reference to the information contained under the caption “Proposal 1—Election of Directors” in our definitive proxy statement related to the 2011 annual meeting of stockholders, to be filed within 120 days after the end of the year covered by this Annual Report on Form 10-K.
Executive Officers
The information concerning our executive officers required by this Item 10 is provided under the caption “Executive Officers of the Registrant” in Part I, Item 1 of this Annual Report on Form 10-K.
Section 16(a) Beneficial Ownership Reporting Compliance
The information required by this Item 10 concerning Section 16(a) beneficial ownership reporting compliance by our directors and executive officers is incorporated herein by reference to the information contained under the caption “Section 16(a) Beneficial Ownership Reporting Compliance” in our definitive proxy statement related to the 2011 annual meeting of stockholders, to be filed within 120 days after the end of the year covered by this Annual Report on Form 10-K.
Code of Ethics
The information required by this Item 10 concerning our code of ethics is incorporated herein by reference to the information contained under the caption “Governance of the Company—Does the Company have a ‘Code of Ethics’?” in our definitive proxy statement related to the 2011 annual meeting of stockholders, to be filed within 120 days after the end of the year covered by this Annual Report on Form 10-K.
| ITEM 11. | EXECUTIVE COMPENSATION |
The information required by this Item 11 is incorporated herein by reference to the information contained in our definitive proxy statement related to the 2011 annual meeting of stockholders, to be filed within 120 days after the end of the year covered by this Annual Report on Form 10-K.
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required by this Item 12 is incorporated herein by reference to the information contained in our definitive proxy statement related to the 2011 annual meeting of stockholders, to be filed within 120 days after the end of the year covered by this Annual Report on Form 10-K.
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information required by this Item 13 is incorporated herein by reference to the information contained in our definitive proxy statement related to the 2011 annual meeting of stockholders, to be filed within 120 days after the end of the year covered by this Annual Report on Form 10-K.
| ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The information required by this Item 14 is incorporated herein by reference to the information contained in our definitive proxy statement related to the 2011 annual meeting of stockholders, to be filed within 120 days after the end of the year covered by this Annual Report on Form 10-K.
72
Table of Contents
PART IV
| ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES |
(a) DOCUMENTS FILED AS PART OF THIS REPORT
The following is a list of our consolidated financial statements and supplementary data included in this Annual Report on Form 10-K under Item 8 of Part II hereof:
| 1. | CONSOLIDATED FINANCIAL STATEMENTS AND SUPPLEMENTAL DATA |
Report of Management.
Report of Independent Registered Public Accounting Firm.
Consolidated Balance Sheets as of December 31, 2010 and 2009.
Consolidated Statements of Operations for the years ended December 31, 2010, 2009 and 2008.
Consolidated Statements of Comprehensive Loss for the years ended December 31, 2010, 2009 and 2008.
Consolidated Statements of Stockholders’ Equity for the years ended December 31, 2010, 2009 and 2008.
Consolidated Statements of Cash Flows for the years ended December 31, 2010, 2009 and 2008.
Notes to Consolidated Financial Statements.
| 2. | CONSOLIDATED FINANCIAL STATEMENT SCHEDULE |
Schedule II—Valuation and Qualifying Accounts.
Schedules, other than those listed above, are omitted because they are not applicable or are not required, or because the required information is included in the consolidated financial statements or notes thereto.
73
Table of Contents
(b) Exhibits
The following is a list of exhibits filed as part of this Annual Report on Form 10-K. When so indicated by footnote, exhibits that were previously filed are incorporated by reference. For exhibits incorporated by reference, the location of the exhibit in the previous filing is indicated.
| Exhibit Number |
Description | |
| 3.1 | Amended and Restated Certificate of Incorporation of Adolor, filed as Exhibit 3.1 to the Company’s Quarterly Report on Form 10-Q filed on May 15, 2001. | |
| 3.2 | Amended and Restated Bylaws of the Company, dated February 24, 2009, filed as Exhibit 3.1 to the Company’s Current Report on Form 8-K filed on February 24, 2009. | |
| 4.1 | Amended and Restated Rights Agreement, dated as of January 31, 2011, between Adolor and StockTrans, a Broadridge Company, which includes the Form of Certificate of Designation, Preferences and Rights of Series A Junior Participating Preferred Stock (Exhibit A), the Form of Rights Certificate (Exhibit B) and the Form of Summary of Rights (Exhibit C), filed as Exhibit 1 to the Company’s Registration Statement on Form 8-A/A (Amendment No. 2) filed on January 31, 2011. | |
| 4.2 | Form of Common Stock Certificate, filed as Exhibit 4.14 to Amendment No. 3 to the Company’s Registration Statement on Form S-1 filed on March 21, 2000. | |
| †10.1 | Amended and Restated 1994 Equity Compensation Plan (effective as of May 12, 2009), filed as Exhibit 10.2 to the Company’s Current Report on Form 8-K/A filed on July 20, 2009. | |
| †10.2 | Adolor Corporation Amended and Restated 2003 Stock-Based Incentive Compensation Plan (effective as of May 12, 2009), filed as Exhibit 10.3 to the Company’s Current Report on Form 8-K filed on May 14, 2009. | |
| †10.3 | Amended and Restated Adolor Corporation Incentive Compensation Plan, effective as of January 2009, filed as Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on January 12, 2009. | |
| †10.4 | Adolor Corporation Incentive Compensation Plan, as amended and restated February 22, 2011, filed as Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on February 23, 2011. | |
| †10.5 | Adolor Corporation Executive Severance Pay Program (for executives appointed prior to December 31, 2008, filed as Exhibit 10.8 to the Company’s Annual Report on Form 10-K filed on February 26, 2010). | |
| †10.6 | Adolor Corporation Executive Severance Pay Program (for executives appointed after December 31, 2008, filed as Exhibit 10.9 to the Company’s Annual Report on Form 10-K filed on February 26, 2010). | |
| †10.7 | Letter Agreement between the Company and Michael R. Dougherty, dated October 24, 2002, filed as Exhibit 10.15 to the Company’s Annual Report on Form 10-K filed on March 18, 2003. | |
| †10.8 | Amendment dated January 26, 2004 to Letter Agreement between the Company and Michael R. Dougherty, dated October 24, 2002, filed as Exhibit 10.6 to the Company’s Annual Report on Form 10-K filed on March 4, 2004. | |
| †10.9 | Letter Agreement between the Company and Michael R. Dougherty dated December 14, 2006 amending letter agreement dated October 24, 2002, as amended January 26, 2004, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on December 14, 2006. | |
| †10.10 | Amendment dated February 21, 2008 to Letter Agreement between the Company and Michael R. Dougherty dated October 24, 2002, as amended January 26, 2004, as further amended December 14, 2006, filed as Exhibit 10.20 to the Company’s Annual Report on Form 10-K filed on February 29, 2008. | |
74
Table of Contents
| Exhibit Number |
Description | |
| †10.11 | Amendment dated December 31, 2008 to Letter Agreement between the Company and Michael R. Dougherty dated October 24, 2002, as amended January 26, 2004, as further amended December 14, 2006, as further amended February 21, 2008, filed as Exhibit 10.11 to the Company’s Annual Report on Form 10-K filed on February 26, 2009. | |
| †10.12 | Letter Agreement between the Company and Stephen Webster, M.B.A. dated June 13, 2008, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on June 13, 2008. | |
| †10.13 | Letter Agreement between the Company and John M. Limongelli, Esq., dated August 15, 2008, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on September 15, 2008. | |
| †10.14 | Letter Agreement between the Company and George R. Maurer dated January 6, 2009, filed as Exhibit 10.19 to the Company’s Annual Report on Form 10-K filed on February 26, 2010. | |
| †10.15 | Stock Award Letter Agreement between the Company and Michael R. Dougherty dated December 14, 2006, filed as Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on December 14, 2006. | |
| †10.16 | Performance Stock Option Award between the Company and Michael R. Dougherty dated January 8, 2008, filed as Exhibit 10.22 to the Company’s Annual Report on Form 10-K filed February 29, 2008. | |
| †10.17 | Summary of Oral Agreement for Payment of Services between Adolor Corporation and its Board of Directors (effective as of May 2009), filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on May 14, 2009. | |
| †10.18 | Form of Stock Option Agreement for members of the Board of Directors of Adolor Corporation, filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed on July 31, 2006. | |
| †10.19 | Form of Stock Option Agreement (monthly vesting), filed as Exhibit 10.26 to the Company’s Annual Report on Form 10-K filed on March 1, 2005. | |
| †10.20 | Form of Stock Option Agreement (annual vesting; effective 2009) , filed as Exhibit 10.26 to the Company’s Annual Report on Form 10-K filed on February 26, 2010. | |
| †10.21 | Form of Deferred Stock Agreement with Annual Vestings, filed as Exhibit 10.27 to the Company’s Annual Report on Form 10-K filed on February 26, 2010. | |
| †10.22 | Form of Deferred Stock Agreement with Vestings at year 2 and 4 (effective December 2009) , filed as Exhibit 10.28 to the Company’s Annual Report on Form 10-K filed on February 26, 2010. | |
| †10.23 | Form of Performance Deferred Stock Award (January 2008), filed as Exhibit 10.29 to the Company’s Annual Report on Form 10-K filed on February 29, 2008. | |
| *†10.24 | Form of Performance-Based Deferred Stock Award dated September 9, 2010. | |
| *†10.25 | Form of Deferred Stock Agreement dated September 9, 2010. | |
| 10.40 | Option and License Agreement between Adolor and Roberts Laboratories, Inc., dated June 10, 1998, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on September 3, 2008. (1) | |
| 10.41 | Collaboration Agreement dated as of April 14, 2002, by and between the Company and Glaxo Group Limited, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K/A filed on December 22, 2005. (1) | |
75
Table of Contents
| Exhibit Number |
Description | |
| 10.42 | Amendment No. 1, dated as of June 22, 2004, to the Collaboration Agreement by and between Glaxo Group Limited and the Company, filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed on August 4, 2004. | |
| 10.43 | Amendment No. 2, dated as of December 22, 2004, to the Collaboration Agreement by and between Glaxo Group Limited and the Company, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K/A filed on February 25, 2005. (1) | |
| 10.44 | Amendment No. 3, dated as of July 23, 2008, to the Collaboration Agreement by and between Glaxo Group Limited and the Company, filed as Exhibit 10.4 to the Company’s Quarterly Report on Form 10-Q filed on October 28, 2008. (1) | |
| 10.45 | Amendment No. 4, dated as of January 30, 2009, to the Collaboration Agreement by and between Glaxo Group Limited and the Company, filed as Exhibit 10.3 to the Company’s Quarterly Report on Form 10-Q filed on April 30, 2009. (1) | |
| 10.46 | Amendment No. 5, effective as of December 16, 2009, to the Collaboration Agreement by and between Glaxo Group Limited and the Company, filed as Exhibit 10.46 to the Company’s Annual Report on Form 10-K filed on February 26, 2010. | |
| 10.47 | Notice of Termination For GI Products Received from Glaxo Group Limited dated August 29, 2008, filed as Exhibit 10.5 to the Company’s Quarterly Report on Form 10-Q filed on October 28, 2008. | |
| 10.48 | Distribution Services Agreement between SmithKline Beecham Corporation and the Company, dated June 29, 2004, filed as Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q filed on August 4, 2004. (1) | |
| 10.49 | Amendment No. 1 to Distribution Services Agreement by and between the Company and GlaxoSmithKline LLC (formerly SmithKline Beecham Corporation), dated December 16, 2009, filed as Exhibit 10.49 to the Company’s Annual Report on Form 10-K filed on February 26, 2010. | |
| 10.50 | API Compound Supply Agreement between the Company and Torcan Chemical Ltd., dated July 13, 2004, filed as Exhibit 10.3 to the Company’s Quarterly Report on Form 10-Q filed on August 4, 2004. (1) | |
| 10.51 | API Compound Supply Agreement Between the Company and Girindus AG (now Central Glass Germany), dated July 6, 2004, filed as Exhibit 10.4 to the Company’s Quarterly Report on Form 10-Q filed on August 4, 2004. (1) | |
| 10.52 | Drug Product Supply Agreement between the Company and Pharmaceutics International, Inc., dated July 1, 2004, filed as Exhibit 10.5 to the Company’s Quarterly Report on Form 10-Q filed on August 4, 2004. (1) | |
| 10.53 | License Agreement between Adolor Corporation and Eli Lilly and Company, dated August 8, 2002, filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed on November 1, 2002. (1) | |
| 10.54 | License and Collaboration Agreement between the Company and Pfizer Inc. dated December 4, 2007, filed as Exhibit 10.14 to the Company’s Annual Report on Form 10-K filed on February 29, 2008. (1) | |
| *10.55 | Notice of Termination from Pfizer Inc. dated December 16, 2010 to the License and Collaboration Agreement between the Company and Pfizer Inc. dated December 4, 2007. | |
76
Table of Contents
| Exhibit Number |
Description | |
| 10.56 | Amended and Restated Build to Suit Lease between the Company and 700 Pennsylvania Drive Associates, dated February 27, 2003, filed as Exhibit 10.4 to the Company’s Annual Report on Form 10-K filed on March 18, 2003. | |
| *21.1 | Subsidiaries of Adolor Corporation | |
| *23.1 | Consent of KPMG LLP. | |
| *31.1 | Certification Pursuant to Rule 13a-14(a)/15d-14(a) of the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| *31.2 | Certification Pursuant to Rule 13a-14(a)/15d-14(a) of the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| *32.1 | Certification Pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| *32.2 | Certification Pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| * | Filed or furnished herewith. |
| † | Compensation plans and arrangements for executives and others. |
| (1) | Portions of the Exhibit have been omitted and have been filed separately pursuant to an application for confidential treatment granted by the Securities and Exchange Commission. |
77
Table of Contents
ADOLOR CORPORATION
SCHEDULE II—VALUATION AND QUALIFYING ACCOUNTS
| Year ended December 31, |
Balance at Beginning of the Year |
Additions (Deductions) (1) |
Other Additions (Deductions) (2) |
Balance at End of the Year |
||||||||||||
| Income tax valuation allowance: |
||||||||||||||||
| 2010 |
$ | 207,719,000 | $ | 8,616,000 | $ | — | $ | 216,335,000 | ||||||||
| 2009 |
$ | 189,682,000 | $ | 18,037,000 | $ | — | $ | 207,719,000 | ||||||||
| 2008 |
176,897,000 | 12,785,000 | — | 189,682,000 | ||||||||||||
| (1) | Amounts represent increases to the valuation allowance. |
| (2) | Amounts represent utilization and adjustments of balance sheet reserve accounts. |
78
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Security Exchange Act of 1934, the Registrant has duly caused this Annual Report to be signed on its behalf by the undersigned, thereunto duly authorized.
Date: February 24, 2011
| ADOLOR CORPORATION | ||
| By: | /S/ MICHAEL R. DOUGHERTY | |
| Name: | Michael R. Dougherty | |
| Title: | President, Chief Executive Officer and Director | |
Pursuant to the requirements of the Securities Exchange Act of 1934, this Annual Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /S/ MICHAEL R. DOUGHERTY Michael R. Dougherty |
President, Chief Executive Officer and Director (Principal Executive Officer) |
February 24, 2011 | ||
| /S/ STEPHEN W. WEBSTER Stephen W. Webster |
Senior Vice President, Finance and Chief Financial Officer (Principal Financial and Accounting Officer) | February 24, 2011 | ||
| /S/ ARMANDO ANIDO Armando Anido |
Director | February 24, 2011 | ||
| /S/ GEORGES GEMAYEL Georges Gemayel |
Director | February 24, 2011 | ||
| /S/ PAUL GODDARD Paul Goddard |
Director | February 24, 2011 | ||
| /S/ GEORGE V. HAGER, JR. George V. Hager, Jr. |
Director | February 24, 2011 | ||
| /S/ DAVID M. MADDEN David M. Madden |
Director | February 24, 2011 | ||
| /S/ GUIDO MAGNI Guido Magni |
Director | February 24, 2011 | ||
| /S/ CLAUDE H. NASH Claude H. Nash |
Director | February 24, 2011 | ||
| /S/ DONALD E. NICKELSON Donald E. Nickelson |
Director | February 24, 2011 | ||
79