UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| |
|
|
| þ |
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES
EXCHANGE ACT OF 1934 |
For the fiscal year ended June 30, 2010
OR
| |
|
|
| o |
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES
EXCHANGE ACT OF 1934 |
Commission file number: 000-51772
CARDICA, INC.
(Exact name of registrant as specified in its charter)
| |
|
|
| Delaware
|
|
94-3287832 |
| (State or other jurisdiction of
|
|
(I.R.S. Employer |
| Incorporation or Organization)
|
|
Identification No.) |
900 Saginaw Drive
Redwood City, California 94063
(650) 364-9975
(Address, including zip code, and telephone number, including area code, of principal executive offices)
Securities registered pursuant to Section 12(b) of the Act:
| |
|
|
| Title of each class
|
|
Name of each exchange on which registered |
| Common Stock, $0.001 par value per share
|
|
The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule
405 of the Securities Act.
Yes o No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section
13 or Section 15(d) of the Act.
Yes o No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by
Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for
such shorter period that the registrant was required to file such reports), and (2) has been
subject to such filing requirements for the past 90 days.
Yes þ No o
Indicate by check mark whether the registrant has submitted electronically and posted on its
corporate Web site, if any, every Interactive Data File required to be submitted and posted
pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months
(or for such shorter period that the registrant was required to submit and post such files).
Yes o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§
229.405 of this chapter) is not contained herein, and will not be contained, to the best of
registrant’s knowledge, in definitive proxy or information statements incorporated by reference in
Part III of this Form 10-K or any amendment to this Form 10-K. þ
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a
non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated
filer”, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
(Check one):
| |
|
|
|
|
|
|
| Large accelerated filer o
|
|
Accelerated filer o
|
|
Non-accelerated filer o
(Do not check if a smaller reporting company)
|
|
Smaller reporting company þ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of
the Act). Yes o No þ
The aggregate market value of the voting stock held by non-affiliates of the registrant as of
December 31, 2009 was approximately $14,865,255 (based on the closing sales price of the
registrant’s common stock as reported by the NASDAQ Global Market, on December 31, 2009).
The number of shares of common stock outstanding as of September 9, 2010 was 25,263,499.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s Proxy Statement for its 2010 Annual Meeting of Stockholders to be
filed with the Securities and Exchange Commission within 120 days after the registrant’s fiscal
year ended June 30, 2010 are incorporated by reference in Part III, Items 10-14 of this Annual
Report on Form 10-K.
Cardica, inc.
ANNUAL REPORT ON FORM 10-K
For the Year Ended June 30, 2010
TABLE OF CONTENTS
This Annual Report on Form 10-K contains forward-looking statements within the meaning of
Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange
Act of 1934, as amended, which are subject to the “safe harbor” created by those sections.
Forward-looking statements are based on our management’s beliefs and assumptions and on information
currently available to our management. In some cases, you can identify forward-looking statements
by terms such as “may,” “will,” “should,” “could,” “would,” “expect,” “plan,” “anticipate,”
“believe,” “estimate,” “project,” “predict,” “potential” and similar expressions intended to
identify forward-looking statements. These statements involve known and unknown risks,
uncertainties and other factors which may cause our actual results, performance, time frames or
achievements to be materially different from any future results, performance, time frames or
achievements expressed or implied by the forward-looking statements. We discuss many of these
risks, uncertainties and other factors in this Annual Report on Form 10-K in greater detail under
the heading “Risk Factors.” Given these risks, uncertainties and other factors, you should not
place undue reliance on these forward-looking statements. Also, these forward-looking statements
represent our estimates and assumptions only as of the date of this filing. You should read this
Annual Report on Form 10-K completely and with the understanding that our actual future results may
be materially different from what we expect. We hereby qualify our forward-looking statements by
our cautionary statements. Except as required by law, we assume no obligation to update these
forward-looking statements publicly, or to update the reasons actual results could differ
materially from those anticipated in these forward-looking statements, even if new information
becomes available in the future.
Item 1. Business
Overview
Historically, our business focused on the design, manufacture and marketing of proprietary
automated anastomotic systems used by cardiac surgeons to perform coronary bypass surgery. We have
re-focused our business on the development of an endoscopic microcutter product line intended for
use by general, thoracic, gynecologic, bariatric and urologic surgeons. Unless and until a
microcutter product is developed and cleared for marketing in the United States or elsewhere, or we
enter into an arrangement with a development and commercialization partner that provides us with
development revenue, we will have ongoing costs related to development of this potential product
line without related revenue.
Our C-Port® Distal Anastomosis Systems, or C-Port systems, are sold in the United
States and Europe. The C-Port systems are used to perform a distal anastomosis, which is the
connection between a bypass graft vessel and the target artery. As of June 30, 2010, more than
10,600 C-Port systems had been sold in the United States and Europe. Our PAS-Port®
Proximal Anastomosis System, or PAS-Port system, is sold in the United States, Europe and Japan.
The PAS-Port system is used to perform a proximal anastomosis, which is the connection between a
graft vessel, typically a saphenous vein, and the aorta or other source of blood. As of June 30,
2010, more than 19,500 PAS-Port systems had been sold in the United States, Europe and Japan. In
addition to our commercialized cardiac surgery products, we are developing the Cardica Microcutter
ES8, a multi-fire endolinear microcutter device based on our proprietary “staple-on-a-strip”
technology, which would expand our commercial opportunity into additional surgical markets. We are
using the technical and intellectual property foundation for the Cardica Microcutter ES8 as the
technology platform for our planned future development of a broad range of surgical stapling
devices in a variety of configurations and formats. We recently entered into a license agreement
with Intuitive Surgical Operations, Inc., or Intuitive Surgical, a subsidiary of Intuitive
Surgical, Inc., providing Intuitive Surgical with a worldwide, exclusive license to
our intellectual property, which relates to tissue cutting, stapling and clip appliers, for use in
the robotics field.
We use independent distributors and manufacturers’ representatives to support a small core
direct sales team for our C-Port and PAS-Port systems in the United States to contain sales costs
while continuing to serve our customers and potential customers for our automated anastomosis
product line. We have shifted our development efforts to focus on the Cardica Microcutter ES8 and
other potential products in this anticipated product line.
1
Our Strategy
Our goals are to develop and market endoscopic microcutter products intended for use by
general, thoracic, gynecologic, bariatric and urologic surgeons and to become the leading provider
of automated anastomotic systems for coronary artery bypass grafting, or CABG, procedures and
closure devices for other surgical procedures. Other existing technologies either do not enable or
are less compatible with less invasive and minimally invasive surgery. Because less invasive
surgery has many advantages relative to patient outcomes, our strategy involves developing and,
ultimately, marketing and selling devices that enable or facilitate less invasive and minimally
invasive procedures, which in turn may help enlarge the market for these types of surgeries.
The principal elements of our strategy to achieve our vision and goals include:
| |
• |
|
Developing our endoscopic microcutter. We have begun development of
the Cardica Microcutter ES8, a multi-fire endolinear microcutter
device based on our proprietary “staple-on-a-strip” technology, which
would expand our commercial opportunity into additional surgical
markets. Our microcutter technology is designed to allow the
connecting, stapling and cutting of tissue similar to currently
marketed competitive endolinear stapling products. The innovative
features we plan to incorporate into this new product line are an
ability to deploy multiple successive rows of staples without
replacing cartridges, a significant reduction in tool shaft diameter
and the ability to increase the amount of articulation of the
end-effector. Our planned introduction of the microcutter would
enable more minimally invasive procedures in general, bariatric,
thoracic, gynecologic and urologic surgeries, such as single port
surgery. |
| |
• |
|
Leveraging the Cardica Microcutter ES8 technology to develop a broad
range of surgical stapling devices. We believe that our technology for
the Cardica Microcutter ES8, which comprises extensive technological
innovations, can be adapted for a variety of surgical stapling
devices. The product applications of our technology that we plan to
develop include stapling devices that have incremental benefits, such
as smaller size, a flexible instrument shaft, greater degrees of
articulation and potentially larger staples. |
| |
• |
|
Obtaining regulatory clearance and commencing marketing of the Cardica
Microcutter ES8. We anticipate that we will complete product
development for the Cardica Microcutter ES8 in early calendar year
2011 and apply for regulatory clearance through the 510(k) process
with the FDA based on the results of our animal trials. |
| |
• |
|
Driving market adoption of the C-Port and PAS-Port systems. We intend
to drive commercial adoption of our C-Port systems, our PAS-Port
system and any future anastomosis products by marketing them as
integrated anastomotic tools for use in both on- and off-pump CABG
procedures. |
| |
• |
|
Establishing a strong proprietary position. As of June 30, 2010, we
had 82 issued U.S. patents, 68 additional patent applications in the
United States, six issued foreign patents and another 11 patent
applications filed in selected international markets. We plan to
continue to invest in building our intellectual property portfolio. |
Cardiac Industry Background
Coronary Artery Disease
According to the American Heart Association, approximately 17.6 million people in the United
States have coronary artery disease, and approximately 425,400 people in the United States die each
year as a result of the disease. Coronary artery disease, sometimes referred to as atherosclerosis,
is a degenerative disease resulting from the deposit of cholesterol and other fatty materials on
the interior walls of blood vessels, forming a build-up known as plaque. The accumulation of
plaque, usually over decades, causes the vessel to become inelastic and progressively narrows the
interior of the artery, impairing its ability to supply blood and oxygen to the heart muscle. When
there is insufficient blood flow to the heart muscle, an injury may occur, often resulting in chest
pain, or angina, a heart attack or even death. Coronary artery disease is caused by aging and is
exacerbated by dietary and environmental factors, as well as by genetic predisposition. As a
patient ages, the disease will typically advance and become more diffuse, compromising the coronary
artery system more globally and occluding more small-diameter vessels.
2
Current Treatment Alternatives for Coronary Artery Disease
Physicians and patients may select among a variety of treatments to address coronary artery
disease, with the selection often depending upon the stage and severity of the disease and the age
of the patient. In addition to changes in patient lifestyle, such as smoking cessation, weight
reduction, diet changes and exercise programs, the principal existing treatments for coronary
artery disease include the following:
Medical Treatment with Pharmaceuticals
Before the advent of interventional cardiology or bypass surgery, medical treatment with
pharmaceuticals was the only form of therapy available to patients with coronary artery disease. In
patients with less severe disease, pharmaceuticals remain the primary treatment approach and
include drugs such as platelet adhesion inhibitors or drugs that reduce the blood cholesterol or
triglyceride levels. The objective for medical treatment with pharmaceutical agents is to reduce
the incidence, progression or exacerbation of coronary artery disease and its associated symptoms.
For more serious disease, however, pharmacological therapy alone is often inadequate.
Interventional Cardiology Techniques
Coronary Angioplasty. Percutaneous transluminal coronary angioplasty, commonly referred to as
balloon angioplasty, is a surgical procedure that involves the dilation of the obstructed artery
with a balloon catheter. To perform an angioplasty, the surgeon maneuvers a flexible balloon
catheter to the site of the blockage in the coronary artery, inflates the balloon, compressing the
plaque and stretching the artery wall to create a larger channel for blood flow. The balloon is
then deflated and removed. Angioplasty is generally successful in increasing immediate blood flow
and, relative to current surgical procedures, offers the benefits of shorter periods of
hospitalization, quicker recovery times, reduced patient discomfort and lower cost. However,
angioplasty does not always provide prolonged efficacy: independent studies indicate that 25% to
40% of vessels treated with balloon angioplasty return to their pre-treatment, narrowed size, a
process known as restenosis, within six months following the procedure. Restenosis is primarily the
result of cell proliferation in response to the “injury” caused by the angioplasty procedure.
Stents. High rates of restenosis following treatment by balloon angioplasty led to the
introduction of stents, mesh-like metallic tubes that are placed within the narrowed portion of the
coronary vessel to hold the vessel open after the angioplasty balloon has been removed. Although
clinical outcomes for procedures using stents reflect an improvement over balloon angioplasty
alone, the effectiveness of stents is still limited by restenosis, which for bare metal stents
occurs in about 10% to 35% of cases within six months of the procedure.
Some manufacturers have introduced drug-eluting stents, which incorporate, on the surface of
the stent, specially formulated, slow-release drugs designed to prevent restenosis. According to
published studies, currently marketed drug-eluting stents have been shown in clinical trials to
reduce the rate of restenosis, within the first nine months after placement, to less than 10%.
Market adoption of drug-eluting stents has been rapid, and industry observers had predicted that
drug-eluting stents would capture approximately 90% of the stent market within three years.
However, some studies have been presented that associate drug eluting stents with late stage
thrombosis, or clotting, which can be an adverse event. Drug eluting stents are still widely used,
with a current market share relative to total stent usage in the range of 70-80%.
Despite the advancements and market success of drug-eluting stents and angioplasty therapies,
these interventional procedures may be less effective than CABG procedures in addressing diffuse
progressive coronary artery disease. In this advanced stage of coronary artery disease,
intervention is required for multiple vessels, many of which are less than two millimeters in
internal diameter, a diameter currently unsuited for angioplasty and stenting. In addition, stents
have been shown to be difficult to place in patients with coronary lesions in sections with vessel
branches and in patients with narrowings in the left main coronary artery.
3
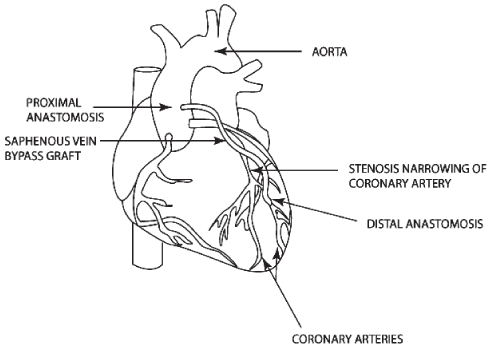
Bypass Surgery. CABG involves the construction of an alternative path to bypass a narrowed or
occluded coronary artery and restore blood flow from the aorta to an area past the occlusion. This
procedure can be accomplished using either veins or arteries as bypass grafts. Veins are typically
harvested from the patient’s leg (saphenous vein), while arteries are taken from either the
patient’s arm (radial artery) or chest wall (mammary artery). For vein grafts and radial arteries,
one end of the harvested vessel is then generally attached to the aorta for blood inflow, and the
opposite end is attached to the target coronary vessel. If a mammary artery is used as the
bypass graft, it must be dissected from the chest wall, leaving one end in place, while the
opposite end is attached to the target vessel, providing uninterrupted blood flow from the arterial
circulation. Once in place, these grafts provide sufficient blood flow to bypass the narrowed or
occluded portion of the coronary artery. (See Figure Below).
Although CABG surgery is generally a highly invasive and even traumatic procedure, an
independent study comparing CABG and implantation of conventional stents has shown that CABG is the
more effective treatment for coronary artery disease, achieving the best long-term patient outcomes
as measured by survival rate and need for intervention. Studies have shown that following CABG,
grafts can remain patent, or open, and functional for as long as 10 years in approximately 50% of
venous grafts and approximately 90% of arterial grafts. In addition, CABG procedures can be used to
treat diffuse, end-stage coronary artery disease states that are often not amenable to treatment by
angioplasty or stents.
According to an independent analysis by Medtech Insight, a division of Windhover Information,
entitled “U.S. Surgical Procedure Volumes,” dated February 2007, an estimated 257,000 CABG
procedures were performed in 2007 in the United States, as compared to approximately 260,000
procedures in 2006. We believe that the decrease in CABG procedures is primarily attributable to
the increase in other interventional cardiology procedures, including the increased use of
drug-eluting stents. The average CABG surgery requires approximately three bypass grafts per
patient, and a majority of grafts require an anastomotic connection at both ends of the graft.
Assuming an average of approximately five anastomoses per CABG procedure, we estimate that
approximately 1.8 million of these blood vessel connections are performed in connection with CABG
procedures annually in the United States. We believe approximately two-thirds of the procedures are
performed using veins as the bypass graft. A similar number of CABG procedures with similar
grafting frequency are performed outside of the US.
4
Types of CABG Procedures
There are currently three types of CABG procedures, two of which are commonly performed:
Conventional On-Pump CABG Procedures. Conventional on-pump CABG procedures are particularly
invasive and traumatic to the patient, typically requiring the surgeon to open the patient’s chest
cavity by splitting the sternum and to place the patient on a pump to circulate the blood
throughout the body. Redirecting the blood flow to a pump enables the surgeon to clamp the aorta
and stop the heart, which results in a motionless and bloodless field in which the surgeon can
perform the difficult and tedious task of manually suturing the small vessels to one another. The
absence of blood flow and motion are important factors in ensuring precision and providing positive
clinical outcomes; however, the use of a pump for circulation exposes the patient’s blood to
foreign surfaces, which has been shown to increase the incidence of bleeding and short-term
neurocognitive defects. Additionally, stopping the heart may result in impairment or damage to the
heart muscle. Moreover, clamping of the aorta has been shown in clinical studies to cause the
release of particles into the blood stream that may produce blockages in other parts of the body,
such as the brain. Blockages in the brain can lead to neurological damage, including strokes.
Clamping the aorta also carries the risk of injury to the vessel wall with later bleeding
complications. Notwithstanding these potential problems, the majority of CABG procedures performed
today use this on-pump technique.
Off-Pump CABG Procedures. In 1995, a new method of performing CABG procedures was introduced
that avoids the use of external pumps, requiring the surgeon to perform the anastomosis while the
heart is beating. The clinical literature suggests that this procedure, termed off-pump coronary
artery bypass, or OPCAB, offers several benefits as compared to on-pump CABG procedures, including
reductions in bleeding, kidney dysfunction, short-term neurocognitive dysfunction and length of
hospital stay. OPCAB procedures currently represent approximately 25% of all CABG procedures
performed in the United States.
Notwithstanding these advantages, the technical challenges inherent in OPCAB have impeded its
widespread adoption. Because the patient’s heart is beating during the procedure, the surgeon is
required to perform the delicate anastomosis on a target vessel, which could be as small as one
millimeter in internal diameter, while the vessel is moving with each heart contraction. The
technical demands of the procedure, together with the longer learning curve required to achieve
surgical proficiency, may also initially adversely affect long-term graft patency and completion of
revascularization. In addition, surgeons will still typically be required to place a partially
occluding clamp on the ascending aorta to hand suture the proximal vein graft anastomosis. As a
result, even in OPCAB procedures, patients still face the risk of the serious adverse effects
associated with the application of aortic clamps.
Minimally Invasive Endoscopic Procedures. Recently, a very small number of CABG procedures
have been performed using minimally invasive endoscopic procedures to reduce patient trauma. These
procedures are known as totally endoscopic coronary artery bypass, or TECAB, and typically involve
the use of Intuitive Surgical’s da Vinci surgical robot system. In this approach, the sternum is
left intact and the surgery is performed through small access ports. The anastomoses are performed
on selected, readily reachable vessels using special surgical instruments or the da Vinci robot
system, and this procedure requires special surgical skills. Although endoscopic procedures offer
the promise of faster post-operative patient recovery times, rapid ambulation, long-term graft
patency and a low incidence of adverse outcomes, in the past there were a number of challenges to
wide-scale realization of that potential, including the absence of a method to enable surgeons to
perform reproducible and effective anastomoses that can be rapidly deployed through small
incisions. While many patients may be eligible for minimally invasive endoscopic techniques, the
TECAB procedures are currently performed in less than 1% of all CABG patients.
Surgical Techniques for Anastomoses
The current method of performing anastomoses, which surgeons generally view as the most
critical aspect of CABG procedures, typically employs tedious and time-consuming hand-sewn
placement of individual stitches with a continuous suture to connect the bypass graft to the aorta
or coronary vessels. Conventional anastomosis can require ten to 25 minutes to suture, depending
upon the size and disease state of the vessels. Proper vessel alignment and suture tension among
the many individually placed fine stitches are critical for optimal bypass graft blood flow and
function. Furthermore, long-term clinical outcomes may be improved if the anastomosis is
“compliant,” that is, if its shape and size can adapt to changes in flow and blood pressure by
placement of many single sutures rather than one continuous suture. However, most surgeons prefer
the use of a continuous suture because placement of individual sutures may be more technically
challenging and time-consuming. Whether the surgeon elects to operate on the patient on- or
off-pump, a hand-sewn proximal anastomosis generally requires clamping of the aorta and therefore
carries with it the risk of neurological damage and other serious adverse effects. Recently, new
technology
has been introduced that allows the surgeon to perform hand-sewn proximal anastomoses to the aorta
without clamping of the aorta. These facilitating devices temporarily cover the opening in the
aortic wall from the inside while the surgeon places the stitches to create the anastomosis and are
removed after the anastomosis has been completed to allow blood flow into the bypass graft. We
believe these systems, in their current implementations, are not suitable for endoscopic bypass
surgery.
5
The laborious and time-consuming nature of manually applied sutures and the limitations
associated with their use, together with advances occurring in coronary surgical procedures, have
fueled the need for easy-to-use, fast and highly reliable automated systems to expedite and
standardize the performance of anastomoses in CABG procedures. Although a number of companies have
attempted to develop automated systems to perform anastomoses, to date, Cardica is the only company
with FDA clearance to market distal and proximal anastomosis devices in the United States, and only
one other non-automated system for use in performing a proximal anastomosis is currently
commercially available in the United States.
Microcutter Industry Background
Market
Laparoscopic surgery is a type of minimally invasive surgery in which a small incision is made
in the abdominal wall through which an endoscope, an instrument usually consisting of a fiber-optic
tube connected to a viewing device, is inserted to permit structures within the abdomen and pelvis
to be seen. A number of different tubes or instruments can be introduced through the same opening,
which enables performing a number of surgical procedures without the need for a large surgical
incision. The advantages of laparoscopic surgery include a shorter post-operative recovery period
with less pain, shorter lengths of stay in the hospital, decreases in post-operative complications
and a quicker return to routine activities compared to traditional open surgical procedures.
Laparoscopic surgery was originally used by gynecologists for the diagnosis of diseases of the
ovary and uterus. Removal of the gall bladder by laparoscopic techniques was introduced in the late
1980s. Smaller surgical instruments and improvements in endoscopic fiber-optic and video cameras
have expanded the use of laparoscopic surgery to surgical procedures involving the appendix,
stomach, lungs, colon, uterus and other organs and procedures.
The use of disposable devices closing and/or cutting in both traditional and laparoscopic
surgical procedures has been broadly adopted clinically in a number of surgical specialties
including colorectal, bariatric, gynecologic, urologic and thoracic surgery. The world-wide
laparoscopic surgery products market is estimated at $3.6 billion, with the cutter and stapler
segment representing approximately $1.3 billion. Based on a 2010 Millennium Research Group report,
55-70% of the worldwide laparoscopic stapling-cutting closure product revenue is generated in the
United States market.
We estimate there are approximately 1.4 million surgical procedures per year in the United
States involving bariatric and general, thoracic, gynecologic and urologic surgery, involving, we
estimate, over 4 million staple cartridge deployments, 3 million of which we believe are deployed
in laparoscopic procedures.
Current Devices for Surgical Stapling
Current, conventional surgical stapling technology generally involves:
| |
• |
|
deploying multiple U-shaped wires against a deforming surface, called an anvil, to
reshape the wires into B-shaped wires and thereby connecting or sealing tissue; |
| |
• |
|
deploying multiple rows of staples, usually two to three rows per side, with a tissue
dividing cut between the rows; |
| |
• |
|
individually placing sets of staples in reloadable cartridges, designed for single use; |
| |
• |
|
using a deployment tool, consisting of a handle and shaft (with a minimum diameter of 12
millimeters), that is reusable within a single surgical procedure; and |
| |
• |
|
using cartridges that can be loaded, following each deployment, into a receptacle at the
end of the deployment tool. |
6
Unlike many other surgical instruments and devices, there have been few significant
innovations in surgical stapling technology over the past ten years.
Our Cardiac Solutions
We design, manufacture and market proprietary automated anastomotic systems used by surgeons
to perform anastomoses during on- or off-pump CABG procedures. We believe that by enabling
consistent and reliable anastomoses of the vessels at this most critical step in CABG surgery
through a fast, automated process, our products can improve the quality and consistency of these
anastomoses, which we believe will ultimately contribute to improved patient outcomes. We have
designed our products to meet the needs of surgeons, including:
| |
• |
|
Physiological features. Our clips use medical grade stainless steel
that is identical to that used in conventional coronary stents, which
is known to be compatible with the human body (in the absence of
allergies to certain components of medical grade stainless steel). Our
products minimize trauma to both the graft and target vessel during
loading and deployment, thereby reducing the risk of scar formation
and associated narrowings or occlusions. Additionally, our PAS-Port
system can be used without clamping the aorta, which has been shown to
be a cause of adverse events, including neurological complications. In
addition, our C-Port system creates compliant anastomoses, which
potentially allow the shape and size of the anastomosis to adapt to
changes in flow and blood pressure. |
| |
• |
|
Handling features. Our anastomotic systems can create anastomoses more
rapidly than hand suturing, resulting in a surgical procedure that can
be performed more quickly. For example the PAS-Port system can be
set-up and deployed in approximately three minutes compared with
approximately ten to 25 minutes for a hand-sewn anastomosis. In
addition, the system is easy to use, typically requiring only a few
hours of training to become technically proficient in the technique.
The C-Port system is compatible with coronary arteries as small as 1.3
millimeters in internal diameter, which is typically the lower limit
of target vessels considered to be candidates for revascularization.
The C-Port system can also be deployed at various angles, allowing
access to all coronary targets during both on- and off-pump
procedures. Both the C-Port system and the PAS-Port system are
designed as integrated products, where all steps necessary to create
an anastomosis are performed by a single tool, with one user
interface. The need for target vessel preparation is minimal for the
PAS-Port system, a feature that is especially important in patients
undergoing a second or third coronary bypass procedure with the
presence of significant scarring in and around the heart and aorta. |
| |
• |
|
Standardized results. Our products enable consistent, reproducible
anastomoses, largely independent of surgical technique and skill set,
using a wide range in quality of graft tissues. In comparison with
hand-sewn sutures, our systems offer mechanically-governed
repeatability and reduced procedural complexity. |
| |
• |
|
Reduced costs. Because our products can help to expedite the CABG
procedure, we believe that they may contribute to reduced operating
room time and a reduction in associated expenses, partially offset by
the increased direct cost of our products compared to current
alternatives, such as sutures. Additionally, our C-Port system creates
anastomoses rapidly and does not require the interruption of blood
flow. This may reduce some of the technical challenges inherent in
performing anastomosis in off-pump procedures, which may advance
adoption of the off-pump approach. By helping more surgeons perform
off-pump CABG, the need for a costly pump may also be reduced or
eliminated, thereby potentially reducing the total direct costs of the
procedure. The C-Port Flex A allows the surgeon to perform coronary
revascularization through small openings in the chest wall, thereby
reducing the trauma and morbidity associated with the CABG procedure,
which therefore may help reduce costs by reducing the time to patient
discharge. Finally, to the extent complications such as strokes or
injury to the heart muscle decrease, post-operative costs of a CABG
procedure may be significantly reduced. |
7
Microcutter Product Development
Based upon much of the technology we developed for our cardiac surgery anastomosis products,
we have begun development of a new product line of multi-fire endolinear stapling devices, a
product line we have termed the microcutter. We believe that our endoscopic microcutter design
potentially will address many of the limitations in currently available stapling products and would
provide surgeons with a smaller and more effective stapling and cutting device for more minimally
invasive surgical procedures, including:
| |
• |
|
Staple Design and Formation. Our microcutter would utilize our innovative three
dimensional, or 3D, staple design, which we engineered in connection with our vascular
anastomotic products, that in vascular applications allows single rows of staples to
effectively prevent blood leakage at physiological blood pressures. These 3D staples allow
for a large contact surface between staple and tissue, which dramatically improves sealing
while significantly reducing the likelihood of the staple cutting through tissue. These 3D
staples are guided into their final shape by the anvil rather than forced to buckle, which
reduces the forming forces and helps to eliminate malformed staples. The 3D design with a
rectangular cross-section significantly increases staple stiffness compared to round wire,
resulting in a much stronger final form that is significantly more resistant to unbending
or yielding. |
| |
• |
|
Device Size. By changing the technology used to form the staple, our microcutter is
being designed to have a smaller-sized end-effector and tool shaft. Depending upon the
chosen staple line length and staple height, the microcutter’s outer diameter could be as
small as five millimeters. Due to its smaller size, our microcutter should enable
procedures requiring minimal access, such as robot-assisted surgery and the rapidly
emerging area of single incision laparoscopic surgery, or SILS. |
| |
• |
|
“Staple-On-A-Strip” Technology. We have further advanced our 3D staple technology in
connection with the microcutter by introducing an innovative design in which 3D staples are
stamped from sheet metal and left connected to a metal band that is then loaded into the
device. This differs from conventional technology where individual staples are typically
loaded into cartridge bays. We believe that our “staple-on-a-strip” technology will enable
tighter spacing between individual staples, which improves sealing performance. |
| |
• |
|
True Multi-Fire Capability. Our “staple-on-a-strip” technology is being designed to
allow the surgeon to conduct multiple deployments within a single procedure, without the
need to remove the stapler from the tissue site or having to replace the staple cartridge.
Conventional stapling technology requires a tedious, repetitive 13-step process after each
deployment in which the stapler is first clamped and then removed from the body cavity.
Our true multi-fire capability would reduce this multi-step process to one simple step:
following a deployment the device is reset by activating a simple slider. |
| |
• |
|
Low Deployment Forces. We are designing our microcutter products to deploy staples with
significantly lower deployment forces. Reduced deployment forces potentially gives the
user more control during deployment. Additionally, our compact staple mechanism would allow
more design space to be dedicated to the anvil, which helps to ensure favorable tissue
compression. These features combine to result in excellent staple formation. |
| |
• |
|
Articulation, Rotation and Handling. End-effector articulation and rotation clearly
improve tissue access and ease of use, and both are expected by surgeons in stapling
devices. Our microcutter’s design incorporates an end-effector that in one product format
we are planning would be angled up to 80 degrees, as compared to the 45 degrees of maximum
articulation achieved with the vast majority of currently marketed linear stapling
technologies. In addition, our microcutter is being designed to enable 360-degree rotation
of the end-effector. Our microcutter also would be a truly single-hand operated handle for
articulating staplers: 360 degree rotation and 80 degree articulation would be accomplished
by rotating a single knob at the end of the handle. |
8
Our Cardiac Products
We currently market four proprietary products to perform anastomoses, the C-Port xA system,
C-Port Flex A system, C-Port X-CHANGE system and the PAS-Port system. The C-Port systems automate a
distal anastomosis between the graft vessel and target artery. The C-Port xA system was developed
to use veins and arteries as the bypass graft vessel and received 510(k) clearance in November
2006. A new generation of the C-Port xA system, the C-Port Flex A system, designed to further
enable minimally invasive CABG surgery, received 510(k) clearance from the FDA in March 2007, and
the C-Port X-CHANGE system, a reloadable cartridge-based system, received 510(k) clearance from the
FDA in December 2007. Each of our C-Port systems has received the CE Mark for sales
in Europe. As of June 30, 2010, we had sold an aggregate of nearly 10,600 units of all the
versions of our C-Port systems. The PAS-Port system automates the performance of a proximal
anastomosis between a graft vessel, typically a saphenous vein, and the aorta. The PAS-Port system
received 510(k) clearance from the FDA in September 2008 following successful completion of a
prospective, international, randomized study. Our PAS-Port system also has received the CE Mark.
The PAS-Port system is cleared or approved for sale and marketed in the United States, Europe and
Japan. As of June 30, 2010, over 19,500 PAS-Port systems had been sold, primarily in Japan and the
United States. Total product sales of our C-Port and PAS-Port systems were $3.8 million, $6.8
million and $4.9 million for fiscal years ended June 30, 2010, 2009 and 2008, respectively. Total
product sales represent 95%, 69% and 65% of total revenue for fiscal years ended June 30, 2010,
2009 and 2008, respectively.
C-Port® Distal Anastomosis Systems
C-Port® xA Anastomosis System
Our C-Port xA Distal Anastomosis System, which may be used in either on- or off-pump CABG
procedures, is designed to perform an end-to-side distal anastomosis by attaching the end of a
bypass graft to a coronary artery downstream of an occlusion or narrowing. The C-Port xA system is
inserted in a small incision in the coronary artery with a bypass graft vessel attached to the
device. The C-Port xA system is actuated by depressing a trigger which activates a manifold powered
by a cylinder of compressed carbon dioxide to provide smooth actuation. Miniature stainless steel
staples are deployed to securely attach the bypass graft to the coronary artery and at the same
time a miniature knife completes an opening inside the coronary artery to complete the bypass.
After deployment, the C-Port system is removed from the coronary artery and the entry incision is
closed typically with a single stitch. Our C-Port xA system is effective in creating compliant
anastomoses in vessels as small as 1.3 millimeters in internal diameter. In addition, the C-Port xA
system has been designed to:
| |
• |
|
perform an end-to-side anastomosis without interruption of native
coronary blood flow, which is not possible in a conventional hand-sewn
anastomosis during off-pump surgery without the use of a temporarily
placed vascular shunt; |
| |
• |
|
achieve nearly complete alignment of the natural blood lining surfaces
of the coronary artery and the bypass graft to minimize scarring and
potential occlusion of the anastomosis; |
| |
• |
|
minimize the amount of foreign material in the blood stream that may
cause clotting and subsequent graft failure; and |
| |
• |
|
suitable for all grafts typically used in CABG procedures with wall
thicknesses of less than or equal to 1.4 millimeters. |
C-Port® Flex A Anastomosis System
The C-Port Flex A system includes modifications to the C-Port xA system that are designed to
enable automated anastomoses to be performed as part of minimally invasive and robot-facilitated
CABG procedures. The C-Port Flex A system includes all of the features and benefits of the C-Port
xA system and has a flexible, rather than rigid, shaft. The flexible shaft is designed to allow the
working end of the device that creates the anastomosis to be inserted through a 14-millimeter
diameter port to access the chest cavity and heart. The device is designed to be loaded with the
bypass graft vessel inside or outside the chest cavity and deployed to create the anastomosis to
the coronary artery. This product is designed to enable technology for completion of robotically
assisted, including endoscopic, CABG surgery through four or five relatively small incisions
between the ribs. Avoiding both the incision through the sternum and the use of the pump should
significantly reduce patient trauma and accelerate post-operative recovery.
9
C-Port® X-CHANGE System
The C-Port X-CHANGE system, the most recent offering in the C-Port product line, is a
cartridge-based reloadable C-Port system. The C-Port X-CHANGE system includes modifications to the
C-Port xA system that are designed to enable multiple automated anastomoses to be performed using
the same handle with up to three separate cartridges. The C-Port X-CHANGE system provides for a
lower cost per deployment for multiple deployments in one CABG procedure.
PAS-Port® Proximal Anastomosis System
Our PAS-Port system is a fully automated device used to perform an end-to-side proximal
anastomosis between a saphenous vein and the aorta. To complete a proximal anastomosis, the cardiac
surgeon simply loads the bypass graft vessel into the PAS-Port system, places the end of the
delivery device against the aorta and turns the knob on the opposite end of the delivery tool. The
device first creates an opening in the aorta and subsequently securely attaches the bypass graft to
the aortic wall, using a medical grade stainless steel implant that is formed into its final shape
by the delivery tool. The innovative design of the PAS-Port system allows the surgeon to load the
bypass graft and rapidly complete the anastomosis, typically in approximately three minutes, with
little or no injury to the bypass graft vessel or the aorta.
An important advantage of our PAS-Port system is that, in contrast to conventional hand-sewn
proximal anastomoses, the vascular connections created can be performed without clamping the aorta,
potentially avoiding associated risks, such as neurological complications. Surgeons use our
PAS-Port system in conventional CABG procedures and in OPCAB. Similar to hand-sewn anastomosis,
anastomoses completed using our PAS-Port system occasionally requires additional stitches
intra-operatively to obtain hemostasis (absence of bleeding in the anastomosis site). These
additional stitches may be required intra-operatively in an individual anastomosis depending on the
quality of the target and graft vessels, adequacy of target site preparation and quality of the
loading of the graft to the deployment cartridge.
Product Development Programs
Microcutter
We intend to launch a full range of surgical stapling devices that cover the needs of general,
bariatric, thoracic, urologic and gynecologic surgeons. The first of these products would be the
Cardica Microcutter ES8, which is currently under development. We are currently conducting acute
and chronic animal trials to validate the performance of the staple line of the Cardica Microcutter
ES8. The ability of the staple line to achieve hemostasis and to achieve acceptable sealing are two
of the key measures in the animal trials. We anticipate that we will complete product development
for the Cardica Microcutter ES8 in early calendar year 2011 and apply for regulatory clearance
through the 510(k) process with the FDA based on the results of our animal trials. See
“-Government Regulation-510(k) Clearance Pathway” below. Assuming that we receive clearance from
the FDA under the 510(k) process, we plan to initially launch the Cardica Microcutter ES8 in the
United States to a limited number of targeted clinical sites. We plan to learn from these sites
the time and training required to achieve routine clinical adoption of the Cardica Microcutter ES8.
We would base a broader launch of the Cardica Microcutter ES8 on our experience from this initial
product introduction.
In addition, we plan to secure a CE Mark and gain our “first in man” clinical experience for
the Cardica Microcutter ES8 in Europe. See “Government Regulation-International Regulation”
below.
We anticipate that our microcutter product line would ultimately include products that provide
staple line lengths from 30 to 60 millimeters, come in shaft diameters ranging from five to twelve
millimeters, accommodate staple heights from 2 to 5.3 millimeters and articulate up to 80 degrees.
Depending on the specific product application, we anticipate that some of these products will have
true multi-fire capability, while others will be cartridge-based. In all instances the
true-multi-fire or cartridge design would be combined with our unique staple design, including the
“staple-on-a-strip” technology. In the true multi-fire design, we anticipate that each device will
provide a number of deployments that is a function of shaft length and desired staple line length,
ranging from six to 12 deployments in one device. In addition, we plan to expand the microcutter
product line by introducing products with flexible shafts to facilitate minimally invasive
procedures.
10
Cardica Hybrid Technology
Because our microcutter would be significantly smaller than other endostaplers and would
incorporate our true multi-fire technology, we anticipate that it would allow different product
platforms to be combined into one product. One of these potential combined products would be a
hybrid of the Cardica Microcutter ES8 and would combine both bi-polar, or thermal, tissue sealing
technology and true multi-fire endostapling. With this potential product, the surgeon would be
able to switch between tissue sealing and stapling, depending on the structures encountered during
tissue dissection, enabling the surgeon to quickly advance through tissue without the need to
switch products. We may also explore other potential forms of hybrid technology that would include
the use of different staple sizes within one product, which would allow the surgeon to have
procedure-specific products that deliver varying staple sizes, as required within a procedure, in
one product. Finally, based upon our C-Port Flex A system technology, we plan to develop a
flexible microcutter. We believe that, due to its anticipated small shaft diameter (as small as
five millimeters) and its flexibility, this potential product would offer surgeons new capability
to perform single incision laparoscopic surgery and intraluminal resections.
License Agreement and Collaborations
Our product research and development efforts are focused on building innovative devices that
enhance our current products or leverage our core competency in mechanical micro-clip formation for
applications in the robotics field and endoscopic CABG and other medical fields. To date, we have
entered into a license agreement with Intuitive Surgical and two agreements, neither of which
remains active, with Cook Incorporated, or Cook, to apply our proprietary technology to solve other
medical needs.
Agreement with Intuitive Surgical
On August 16, 2010, we entered into a License Agreement with Intuitive Surgical pursuant to
which we granted to Intuitive Surgical a worldwide, sublicenseable, exclusive license to use our
intellectual property in the robotics field in diagnostic or therapeutic medical procedures,
excluding vascular anastomosis applications, or the License Agreement. In consideration for this
license, we received an upfront license fee of $9 million. We will also be eligible to receive a
milestone payment if sales of any products incorporating our patent rights achieve a specified
level of net sales within a specified period after the date of the License Agreement and will also
be eligible to receive single-digit royalties on sales by Intuitive Surgical, its affiliates or its
sublicensees of specified stapler and clip applier products covered by our patent rights as well as
on sales of certain other products covered by our patent rights that may be developed in the
future. Each party has the right to terminate the License Agreement in the event of the other
party’s uncured material breach or bankruptcy. Following any termination of the License Agreement,
the licenses granted to Intuitive Surgical will continue, and except in the case of termination for
our uncured material breach or insolvency, Intuitive Surgical’s payment obligations will continue
as well.
Regulatory Status
Other than our PAS-Port system and C-Port systems, all of our products are in a pre-clinical
development stage.
Cardiac Product Sales and Marketing
United States
Our initial products focus on the needs of cardiovascular surgeons worldwide. We have changed
our sales approach in the United States. Rather than building a direct sales force, we are
building a network of independent medical device manufacturers’ representatives and distributors to
sell our products domestically. We are targeting manufacturers’ representatives and distributors
who carry other cardiac surgery products, are clinically knowledgeable and are capable of training
cardiac surgeons on the use of our products and proctoring initial cases in the operating room. We
manage this network of manufacturers’ representatives and distributors with a direct sales force of
two sales representatives across the United States. We anticipate that these manufacturers’
representatives
and distributors will target, as our direct sales force has, selected influential surgeons in high
volume cardiac surgery centers in the United States to sell our C-Port and PAS-Port systems.
11
International
We currently distribute our PAS-Port system in Japan through our exclusive distributor,
Century Medical, Inc., or Century. For the fiscal years ended June 30, 2010, 2009 and 2008, sales
to Century comprised approximately 22%, 10% and 13%, respectively, of our total revenue and
approximately 23%, 15% and 20%, respectively, of our product sales. As of June 30, 2010, Century
had trained over 300 Japanese cardiac surgeons in over 200 hospitals. Century has a direct sales
organization of approximately 16 representatives who are responsible for the development of the
anastomotic device market and directly contact cardiac surgeons. Century provides clinical training
and support for end-users in Japan. We provide Century with promotional support, ongoing clinical
training, representation at trade shows and guidance in Century’s sales and marketing efforts. Our
agreement with Century expires in July 2014, but automatically renews for an additional five-year
term if Century meets certain sales milestones. Either party may terminate this agreement if the
other party defaults in performance of material obligations and such default is not cured within a
specified period or if the other party becomes insolvent or subject to bankruptcy proceedings. In
addition, we may terminate the agreement within 90 days following a change of control by payment of
a specified termination fee. For the specifics of our revenue by geographic location please see
Note 1, Concentrations of Credit Risk and Certain Other Risks, located in Notes to Financial
Statements.
Total product sales of our C-Port and PAS-Port systems were $3.8 million, $6.8 million and
$4.9 million, for fiscal years ended June 30, 2010, 2009 and 2008, respectively. Total product
sales represent 95%, 69% and 65% of total revenues for fiscal years ended June 30, 2010, 2009 and
2008, respectively.
We are continuing to sell to selected international customers and will continue to evaluate
further opportunities to expand our distribution network in Europe and in other parts of the world
where the healthcare economics are conducive to the introduction and adoption of new medical device
technologies.
Competition
Cardiac Products
The market for medical devices used in the treatment of coronary artery disease is intensely
competitive, subject to rapid change, and significantly affected by new product introductions and
other market activities of industry participants. We believe the principal competitive factors in
the market for medical devices used in the treatment of coronary artery disease include:
| |
• |
|
improved patient outcomes; |
| |
• |
|
access to and acceptance by leading physicians; |
| |
• |
|
product quality and reliability; |
| |
• |
|
device cost-effectiveness; |
| |
• |
|
physician relationships; and |
| |
• |
|
sales and marketing capabilities. |
12
There are numerous potential competitors in the medical device, biotechnology and
pharmaceutical industries, such as Maquet Cardiovascular LLC, formerly the cardiac surgery division
of Boston Scientific Corporation, Edwards Lifesciences Corporation, Johnson & Johnson, Inc.,
Abbott Laboratories, which acquired an additional
division of Guidant Corporation, Medtronic, Inc. and St. Jude Medical, that are targeting the
treatment of coronary artery disease broadly. Each of these companies has significantly greater
financial, clinical, manufacturing, marketing, distribution and technical resources and experience
than we have. In addition, new companies have been, and are likely to continue to be, formed to
pursue opportunities in our market.
The landscape of active competitors in the market for anastomotic solutions is currently
limited. Several companies market systems designed to facilitate or stabilize proximal anastomoses,
such as Maquet Cardiovascular’s Heartstring Aortic Occluder and Novare Surgical Systems’ Enclose
anastomotic assist device. St. Jude Medical previously had a commercially available proximal
anastomotic system that was marketed both in the United States and Europe; however, St. Jude
Medical voluntarily withdrew this product from the market in 2004. Johnson & Johnson obtained FDA
clearance for a proximal system that was developed by Bypass Inc. but has divested the division
that was originally responsible for selling this product, and this proximal anastomosis product is
now not available for cardiac surgeons in the United States or abroad.
Our C-Port systems are the only automated anastomosis devices for distal anastomosis cleared
for marketing in the United States. The only currently marketed facilitating device for distal
anastomosis is the U-Clip, which substitutes clips for sutures, but still requires manual
application of typically 12 to 14 individually placed clips per anastomosis by the surgeon.
Currently, the vast majority of anastomoses are performed with sutures and, for the
foreseeable future, sutures will continue to be the principal competitor for alternative
anastomotic solutions. The direct cost of sutures used for anastomoses in CABG procedures is far
less expensive than the direct cost of automated anastomotic systems, and surgeons, who have been
using sutures for their entire careers, have been reluctant to consider alternative technologies,
despite potential advantages.
In addition, cardiovascular diseases may also be treated by other methods that do not require
anastomoses, including interventional techniques such as balloon angioplasty and use of
drug-eluting stents, pharmaceuticals, atherectomy catheters and lasers. Further, technological
advances with other therapies for cardiovascular disease such as drugs, local gene therapy or
future innovations in cardiac surgery techniques could make other methods of treating this disease
safer, more effective or less expensive than CABG procedures.
Microcutter
The Cardica Microcutter ES8, if it receives FDA clearance and is successfully launched, would
compete in the market for stapling and cutting devices within laparoscopic stapling and sealing
devices currently marketed in the United States. We believe the principal competitive factors in
the market for laparascopic staplers include:
| |
• |
|
product quality and reliability; |
| |
• |
|
device cost-effectiveness; |
| |
• |
|
degree of articulation; |
| |
• |
|
physician relationships; and |
| |
• |
|
sales and marketing capabilities. |
Two large competitors, Ethicon Endo-Surgery, part of Johnson & Johnson, and Covidien currently
control over 80% of this market. Other large competitors in the laparoscopic device market include
Stryker Endoscopy and Olympus, which acquired another competitor, Gyrus Medical. Ethicon
Endo-Surgery and Covidien, which recently acquired a small competitor, Power Medical, each have
large direct sales forces in the United States and have been
the largest participants in the market for single use disposable laparoscopic stapling devices for
many years. Competing against large established competitors with significant resources may make
establishing a market for any products that we develop difficult.
13
Manufacturing
Our manufacturing operations, sterile products manufacturing, assembly, packaging, storage and
shipping, as well as our research and development laboratories and administrative activities all
take place at our headquarters facility. We believe that our current facilities will be sufficient
to meet our manufacturing needs for at least the next two years.
We believe our manufacturing operations are in compliance with regulations mandated by the FDA
and the European Union. Our facility is ISO 13485:2003 certified. In connection with our CE mark
approval and compliance with European quality standards, our facility was initially certified in
June 2002 and has been inspected annually thereafter.
There are a number of critical components and sub-assemblies required for manufacturing the
C-Port and PAS-Port systems that we purchase from third-party suppliers. The vendors for these
materials are qualified through stringent evaluation and monitoring of their performance over time.
We audit our critical component manufacturers on a regular basis and at varied intervals based on
the nature and complexity of the components they provide and the risk associated with the
components’ failure.
We use or rely upon sole source suppliers for certain components and services used in
manufacturing our products, and we utilize materials and components supplied by third parties, with
which we do not have any long-term contracts. In recent years, many suppliers have ceased supplying
materials for use in implantable medical devices. We cannot quickly establish additional or
replacement suppliers for certain components or materials, due to both the complex nature of the
manufacturing processes employed by our suppliers and the time and effort that may be required to
obtain FDA clearance or other regulatory approval to use materials from alternative suppliers. Any
significant supply interruption or capacity constraints affecting our facilities or those of our
suppliers would affect our ability to manufacture and distribute our products.
Third-Party Reimbursement
Sales of medical products are increasingly dependent in part on the availability of
reimbursement from third-party payors such as government and private insurance plans. Currently,
payors provide coverage and reimbursement for CABG procedures only when they are medically
necessary. Our cardiac surgery technology will be used concomitantly in CABG procedures. Cardica
cardiac surgery technologies bring added direct costs to medical providers and may not be
reimbursed separately by third-party payors at rates sufficient to allow us to sell our products on
a competitive and profitable basis.
We believe the majority of bypass graft patients in the United States will be Medicare
beneficiaries. Further, private payors often consider Medicare’s coverage and payment decisions
when developing their own policies. The Centers for Medicare & Medicaid Services, or CMS, is the
agency within the Department of Health and Human Services that administers Medicare and will be
responsible for reimbursement decisions for the Cardica cardiac surgery devices when used to treat
Medicare beneficiaries during CABG surgery.
Once a device has received approval or clearance for marketing by the FDA, there is no
assurance that Medicare will cover the device and related services. In some cases, CMS may place
certain restrictions on the circumstances in which coverage will be available. In making such
coverage determinations, CMS considers, among other things, peer-reviewed publications concerning
the effectiveness of the technology, the opinions of medical specialty societies, input from the
FDA, the National Institutes of Health, and other government agencies.
In general, Medicare makes a predetermined, fixed payment amount for its beneficiaries
receiving covered inpatient services in acute care hospitals. This payment methodology is part of
the inpatient prospective payment system, or IPPS. For acute care hospitals, under IPPS, payment
for an inpatient stay is based on diagnosis-related groups, or DRGs, which include reimbursement
for all covered medical services and medical products that are
provided during a hospital stay. Additionally, a relative weight is calculated for each individual
DRG which represents the average resources required to care for cases in that particular DRG
relative to the average resources required to treat cases in all DRGs. Generally, DRG relative
weights are adjusted annually to reflect changes in medical practice in a budget neutral manner.
14
CMS has made no decisions with respect to DRG assignment when patients undergo CABG procedures
in which our cardiac surgery products would be used, and there can be no assurance that the DRG to
which such patients will be assigned will result in Medicare payment levels that are considered by
hospitals to be adequate to support purchase of our products.
Under current CMS reimbursement policies, CMS offers a process to obtain add-on payment for a
new medical technology when the existing DRG prospective payment rate is inadequate. To obtain
add-on payment, a technology must be considered “new,” demonstrate substantial improvement in care
and exceed certain payment thresholds. Add-on payments are made for no less than two years and no
more than three years. We must demonstrate the safety and effectiveness of our cardiac surgery
technology to the FDA in addition to CMS requirements before add-on payments can be made. Further,
Medicare coverage is based on our ability to demonstrate the treatment is “reasonable and
necessary” for Medicare beneficiaries. The process involved in applying for additional
reimbursement for new medical technologies from CMS is lengthy and expensive. In November 2006, CMS
denied our request for an add-on payment. According to CMS, we met the “new” criteria and exceeded
the payment threshold but did not in their view demonstrate substantial improvement in care. Our
cardiac surgery products may not be awarded additional or separate reimbursement in the foreseeable
future, if at all. Moreover, many private payors look to CMS in setting their reimbursement
policies and payment amounts. If CMS or other agencies limit coverage and decrease or limit
reimbursement payments for hospitals and physicians, this may affect coverage and reimbursement
determinations by many private payors.
For classification of physician services, the American Medical Association, referred to as the
AMA, has developed a coding system known as the Current Procedural Terminology, or CPT. CPT codes
are established by the AMA and adopted by the Medicare program in the Healthcare Common Procedure
Coding System, to describe and develop payment amounts for physician services. Physician services
are reimbursed by Medicare based on a physician fee schedule whereby payment is based generally on
the number of “relative value units” assigned by CMS to the service furnished by the physician. No
decision has been made concerning whether existing CPT codes would be appropriate for use in coding
anastomosis procedures when our products are used or if new CPT codes and payment are required. We
cannot assure you that codes used for submitting claims for anastomosis procedures using our
products will result in incremental payment to physicians. CPT codes are used by many other
third-party payors in addition to Medicare. Failure by physicians to receive what they consider to
be adequate reimbursement for anastomosis procedures in which our products are used could have a
material adverse effect on our business, financial condition and results of operations.
Research and Development
As of June 30, 2010, we had 12 employees in our research and development department. Future
research and development efforts will involve development of the microcutter in a variety of
formats that accommodate different staple sizes and staple line lengths and different tool form
factors, such as flexible versus rigid shafts, and combining stapling with sealing devices. We are
also continuing development of the C-Port X-CHANGE II system, a cartridge based device with
enhanced ease of use features and lower cost of goods than existing C-Port systems. We are also
exploring the development of other products that can be derived from our core technology platform
and intellectual property. Research and development expenses for fiscal years ended June 30, 2010,
2009, and 2008 were $5.4 million, $8.2 million and $8.6 million respectively. We expect research
and development expenses to be higher in absolute dollar terms in fiscal year 2011 based on
multiple microcutter development activities.
Patents and Intellectual Property
We believe our competitive position will depend significantly upon our ability to protect our
intellectual property. Our policy is to seek to protect our proprietary position by, among other
methods, filing U.S. and foreign patent applications related to our technology, inventions and
improvements that are important to the development of our business. As of June 30, 2010, we had 82
issued U.S. patents, 68 additional U.S. patent applications, six issued
foreign patents and another 11 patent applications filed in select international markets. Our
issued patents expire between 2018 and 2028.
15
We also rely upon trade secrets, technical know-how and continuing technological innovation to
develop and maintain our competitive position. We typically require our employees, consultants and
advisors to execute confidentiality and assignment of inventions agreements in connection with
their employment, consulting or advisory relationships with us. There can be no assurance, however,
that these agreements will not be breached or that we will have adequate remedies for any breach.
Furthermore, no assurance can be given that competitors will not independently develop
substantially equivalent proprietary information and techniques or otherwise gain access to our
proprietary technology, or that we can meaningfully protect our rights in unpatented proprietary
technology.
Patent applications in the United States and in foreign countries are maintained in secrecy
for a period of time after filing, which results in a delay between the actual discoveries and the
filing of related patent applications and the time when discoveries are published in scientific and
patent literature. Patents issued and patent applications filed relating to medical devices are
numerous, and there can be no assurance that current and potential competitors and other third
parties have not filed or in the future will not file applications for, or have not received or in
the future will not receive, patents or obtain additional proprietary rights relating to products,
devices or processes used or proposed to be used by us. We are aware of patents issued to third
parties that contain subject matter related to our technology. We believe that the technologies we
employ in our products and systems do not infringe the valid claims of any such patents. There can
be no assurance, however, that third parties will not seek to assert that our devices and systems
infringe their patents or seek to expand their patent claims to cover aspects of our products and
systems.
The medical device industry, in general, and the industry segment that includes products for
the treatment of cardiovascular disease in particular, has been characterized by substantial
litigation regarding patents and other intellectual property rights. Any such claims, regardless of
their merit, could be time-consuming and expensive to respond to and could divert our technical and
management personnel. We may be involved in litigation to defend against claims of infringement by
other patent holders, to enforce patents issued to us, or to protect our trade secrets. If any
relevant claims of third-party patents are upheld as valid and enforceable in any litigation or
administrative proceeding, we could be prevented from practicing the subject matter claimed in such
patents, or would be required to obtain licenses from the patent owners of each such patent, or to
redesign our products, devices or processes to avoid infringement. There can be no assurance that
such licenses would be available or, if available, would be available on terms acceptable to us or
that we would be successful in any attempt to redesign our products or processes to avoid
infringement. Accordingly, an adverse determination in a judicial or administrative proceeding or
failure to obtain necessary licenses could prevent us from manufacturing and selling our products,
which would have a material adverse effect on our business, financial condition and results of
operations. We intend to vigorously protect and defend our intellectual property. Costly and
time-consuming litigation brought by us may be necessary to enforce patents issued to us, to
protect trade secrets or know-how owned by us or to determine the enforceability, scope and
validity of the proprietary rights of others. See “Risk Factors.”
Government Regulation
The FDA and other regulatory bodies extensively regulate the research, development,
manufacture, labeling, distribution and marketing of our products. Our current products are
regulated by the FDA as medical devices, and we are required to obtain review and clearance or
approval from the FDA prior to commercializing our devices in the United States.
FDA regulations govern nearly all of the activities that we perform, or that are performed on
our behalf, to ensure that medical products distributed domestically or exported internationally
are safe and effective for their intended uses. The activities that the FDA regulates include the
following:
| |
• |
|
product design, development and manufacture; |
| |
• |
|
product safety, testing, labeling and storage; |
| |
• |
|
pre-clinical testing in animals and in the laboratory; |
16
| |
• |
|
clinical investigations in humans; |
| |
• |
|
marketing applications, such as 510(k) notifications and Premarket Approval, or PMA, applications; |
| |
• |
|
record keeping and document retention procedures; |
| |
• |
|
advertising and promotion; |
| |
• |
|
product marketing, distribution and recalls; and |
| |
• |
|
post-marketing surveillance and medical device reporting, including reporting of deaths, serious
injuries, device malfunctions or other adverse events. |
FDA’s Premarket Clearance and Approval Requirements. Unless an exemption applies, each medical
device distributed commercially in the United States will require either prior 510(k) clearance or
PMA from the FDA. The FDA classifies medical devices into one of three classes. Class I devices are
subject to only general controls, such as establishment registration and device listing, labeling,
medical devices reporting, and prohibitions against adulteration and misbranding. Class II medical
devices generally require prior 510(k) clearance before they may be commercially marketed in the
United States. The FDA will clear marketing of a medical device through the 510(k) process if the
FDA is satisfied that the new product has been demonstrated to be substantially equivalent to
another legally marketed device, or predicate, device, and otherwise meets the FDA’s requirements.
Class II devices are also subject to general controls and may be subject to performance standards
and other special controls. Devices deemed by the FDA to pose the greatest risk, such as
life-sustaining, life-supporting or implantable devices, or devices deemed not substantially
equivalent to a predicate device, are placed in Class III, generally requiring submission of a PMA
supported by clinical trial data.
510(k) Clearance Pathway. To obtain 510(k) clearance, we must submit a notification to the FDA
demonstrating that our proposed device is substantially equivalent to a predicate device, i.e., a
device that was in commercial distribution before May 28, 1976, a device that has been reclassified
from Class III to Class I or Class II, or a 510(k)-cleared device. The FDA’s 510(k) clearance
process generally takes from three to 12 months from the date the application is submitted, but can
take significantly longer. If the FDA determines that the device, or its intended use, is not
substantially equivalent to a previously-cleared device or use, the device is automatically placed
into Class III, requiring the submission of a PMA. Any modification to a 510(k)-cleared device that
would constitute a major change in its intended use, design or manufacture, requires a new 510(k)
clearance and may even, in some circumstances, require a PMA, if the change raises complex or novel
scientific issues. The FDA requires every manufacturer to make the determination regarding the need
for a new 510(k) submission in the first instance, but the FDA may review any manufacturer’s
decision. If the FDA disagrees with a manufacturer’s determination, the FDA can require the
manufacturer to cease marketing and/or recall the device until 510(k) clearance or PMA is obtained.
If the FDA requires us to seek 510(k) clearance or PMAs for any modifications, we may be required
to cease marketing and/or recall the modified device, if already in distribution, until 510(k)
clearance or PMA is obtained and we could be subject to significant regulatory fines or penalties.
The FDA has undertaken a systematic review of the 510(k) clearance process and is currently
soliciting public comments on a preliminary report that outlines ten major recommendations that
could adversely impact the timing and data burden required to obtain 510(k) clearance. The
recommendations include but are not limited to restrictions on the use of certain devices for use
as predicate devices in determining substantial equivalence, creation of a new administrative
device classification within the Class II regulatory framework that will provide clarity on the
need for clinical data for moderate risk devices that are not subject to the PMA regulations,
inclusion of manufacturing process information not previously required in 510(k) premarket
notifications, inclusion of more complete safety and efficacy information and pre-clearance
facility inspections. Furthermore, our products could be subject to voluntary recall if we or the
FDA determines, for any reason, that our products pose a risk of injury or are otherwise defective.
Moreover, the FDA can order a mandatory recall if there is a reasonable probability that our device
would cause serious adverse health consequences or death. Delays in receipt or failure to receive
clearances or approvals, the loss of previously received clearances or approvals, or the failure to
comply with existing or future regulatory requirements could reduce our sales, profitability and
future growth prospects.
17
Premarket Approval Pathway. A PMA must be submitted to the FDA if the device cannot be cleared
through the 510(k) process. The PMA process is much more demanding than the 510(k) notification
process. A PMA must be supported by extensive data, including but not limited to data obtained from
preclinical or clinical studies or relating to manufacturing and labeling to demonstrate to the
FDA’s satisfaction the safety and effectiveness of the device.
After a PMA is complete, the FDA begins an in-depth review of the submitted information, which
generally takes between one and three years, but may take significantly longer. During this review
period, the FDA will typically request additional information or clarification of the information
already provided. Also, an advisory panel of experts from outside the FDA may be convened to review
and evaluate the application and provide recommendations to the FDA as to the approvability of the
device. The FDA may or may not accept the panel’s recommendation. In addition, the FDA will conduct
a pre-approval inspection of the manufacturing facility to ensure compliance with Quality System
Regulation, or QSR. New PMA applications or PMA supplements are required for significant
modifications to the device, including, for example, certain types of modifications to the device’s
indication for use, manufacturing process, labeling and design. PMA supplements often require
submission of the same type of information as a PMA application, except that the supplement is
limited to information needed to support any changes from the device covered by the original PMA
application and may not require as extensive clinical data or the convening of an advisory panel.
Clinical Trials. Clinical trials are generally required to support a PMA application and are
sometimes required for 510(k) clearance. To perform a clinical trial in the United States for a
significant risk device, prior submission of an application for an Investigational Device
Exemption, or IDE, to the FDA is required. An IDE amendment must also be submitted before
initiating a new clinical study under an existing IDE, such as initiating a pivotal trial following
the conclusion of a feasibility trial. The IDE application must be supported by appropriate data,
such as animal and laboratory testing results, and any available data on human clinical experience,
showing that it is safe to test the device in humans and that the testing protocol is
scientifically sound. The animal and laboratory testing must meet the FDA’s good laboratory
practice requirements.
The IDE and any IDE supplement for a new trial must be approved in advance by the FDA for a
specific number of patients. Clinical trials conducted in the United States for significant risk
devices may not begin until the IDE application or IDE supplement is approved by the FDA and the
appropriate institutional review boards, or IRBs, overseeing the welfare of the research subjects
and responsible for that particular clinical trial. If the product is considered a non-significant
risk device under FDA regulations, only the patients’ informed consent and IRB approval are
required. Under its regulations, the agency responds to an IDE or an IDE amendment for a new trial
within 30 days. The FDA may approve the IDE or amendment, grant an approval with certain
conditions, or identify deficiencies and request additional information. It is common for the FDA
to require additional information before approving an IDE or amendment for a new trial, and thus
final FDA approval on a submission may require more than the initial 30 days. The FDA may also
require that a small-scale feasibility study be conducted before a pivotal trial may commence. In a
feasibility trial, the FDA limits the number of patients, sites and investigators that may
participate. Feasibility trials are typically structured to obtain information on safety and to
help determine how large a pivotal trial should be to obtain statistically significant results.
Clinical trials are subject to extensive recordkeeping and reporting requirements. Our
clinical trials must be conducted under the oversight of an IRB for the relevant clinical trial
sites and must comply with FDA regulations, including but not limited to those relating to good
clinical practices. We are also required to obtain the patients’ informed consent in form and
substance that complies with both FDA requirements and state and federal privacy and human subject
protection regulations. We, the FDA or the IRB may suspend a clinical trial at any time for various
reasons, including a belief that the risks to study subjects outweigh the anticipated benefits.
Even if a trial is completed, the results of clinical testing may not adequately demonstrate the
safety and efficacy of the device or may otherwise not be sufficient to obtain FDA approval to
market the product in the United States. Similarly, in Europe the clinical study must be approved
by a local ethics committee and in some cases, including studies with high-risk devices, by the
ministry of health in the applicable country.
18
We intend to seek 510(k) clearance for the Cardica Microcutter ES8. The statutory review time
for a traditional 510(k) is ninety (90) days, assuming that no major deficiencies are identified.
Once 510(k) clearance is obtained for an initial Cardica microcutter device, that Cardica
microcutter could then serve as the predicate device for
subsequent iterations and product line extensions. Given the long history of safe and effective use
of surgical staples, 510(k) clearances for linear stapling devices have relied heavily on bench and
animal data to confirm the design and performance characteristics. There is precedent for the FDA’s
acceptance of bench and animal data to support 510(k) clearance for significant technological
innovations, including material changes, design changes, changes in principles of operations and
changes to the indications for use. However, the FDA may at any time mandate that human clinical
data are required to support a substantial equivalence determination for any of our proposed
products, thereby delaying clearance until such time as any required clinical data has been
collected and subsequently reviewed by the FDA.
Any products that we develop that require regulatory clearance or approval, including the
Cardica Microcutter ES8, may not be cleared or approved on the timing that we anticipate, if at
all. We cannot assure that any new products or any product enhancements that we develop will be
subject to the shorter 510(k) clearance process instead of the more lengthy PMA requirements.
Pervasive and Continuing Regulation. There are numerous regulatory requirements governing the
approval and marketing of a product. These include:
| |
• |
|
product listing and establishment registration, which helps facilitate FDA
inspections and other regulatory action; |
| |
• |
|
QSR, which requires manufacturers, including third-party manufacturers, to follow
stringent design, testing, control, documentation and other quality assurance
procedures during all aspects of the manufacturing process; |
| |
• |
|
labeling regulations and FDA prohibitions against the promotion of products for
uncleared, unapproved or off-label use or indication; |
| |
• |
|
clearance or approval of product modifications that could significantly affect
safety or efficacy or that would constitute a major change in intended use; |
| |
• |
|
medical device reporting regulations, which require that manufacturers comply with
FDA requirements to report if their device may have caused or contributed to an
adverse event, a death or serious injury or malfunctioned in a way that would
likely cause or contribute to a death or serious injury if the malfunction were to
recur; |
| |
• |
|
post-approval restrictions or conditions, including post-approval study commitments; |
| |
• |
|
post-market surveillance regulations, which apply when necessary to protect the
public health or to provide additional safety and effectiveness data for the
device; and |
| |
• |
|
notices of correction or removal and recall regulations. |
Advertising and promotion of medical devices are also regulated by the Federal Trade
Commission and by state regulatory and enforcement authorities. Recently, some promotional
activities for FDA-regulated products have been the subject of enforcement action brought under
healthcare reimbursement laws and consumer protection statutes. In addition, under the federal
Lanham Act and similar state laws, competitors and others can initiate litigation relating to
advertising claims.
We have registered with the FDA as a medical device manufacturer. The FDA has broad
post-market and regulatory enforcement powers. We are subject to unannounced inspections by the FDA
to determine our compliance with the QSR, and other regulations, and these inspections may include
the manufacturing facilities of our suppliers.
Failure by us or by our suppliers to comply with applicable regulatory requirements can result
in enforcement action by the FDA or state authorities, which may include any of the following
sanctions:
| |
• |
|
warning letters, fines, injunctions, consent decrees and civil penalties; |
| |
• |
|
customer notifications, repair, replacement, refunds, recall or seizure of our products; |
| |
• |
|
operating restrictions, partial suspension or total shutdown of production; |
| |
• |
|
delay in processing marketing applications for new products or modifications to existing products; |
19
| |
• |
|
mandatory product recalls; |
| |
• |
|
withdrawing approvals that have already been granted; and |
Fraud and Abuse and False Claims. We are directly and indirectly subject to various federal
and state laws governing our relationship with healthcare providers and pertaining to healthcare
fraud and abuse, including anti-kickback laws. In particular, the federal healthcare program
Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, offering,
receiving or providing remuneration, directly or indirectly, in exchange for or to induce either
the referral of an individual, or the furnishing, arranging for or recommending a good or service,
for which payment may be made in whole or part under federal healthcare programs, such as the
Medicare and Medicaid programs. Penalties for violations include criminal penalties and civil
sanctions such as fines, imprisonment and possible exclusion from Medicare, Medicaid and other
federal healthcare programs. The Anti-Kickback Statute is broad and prohibits many arrangements and
practices that are lawful in businesses outside of the healthcare industry. In implementing the
statute, the Office of Inspector General of the U.S. Department of Health and Services, or OIG, has
issued a series of regulations, known as the “safe harbors.” These safe harbors set forth
provisions that, if all their applicable requirements are met, will assure healthcare providers and
other parties that they will not be prosecuted under the Anti-Kickback Statute. The failure of a
transaction or arrangement to fit precisely within one or more safe harbors does not necessarily
mean that it is illegal or that prosecution will be pursued. However, conduct and business
arrangements that do not fully satisfy each applicable element of a safe harbor may result in
increased scrutiny by government enforcement authorities, such as the OIG.
The Federal False Claims Act imposes civil liability on any person or entity who submits, or
causes the submission of a false or fraudulent claim to the United States Government. Damages under
the Federal False Claims Act can be significant and consist of the imposition of fines and
penalties. The Federal False Claims Act also allows a private individual or entity with knowledge
of past or present fraud on the federal government to sue on behalf of the government to recover
the civil penalties and treble damages. The U.S. Department of Justice on behalf of the government
has successfully enforced the Federal False Claims Act against pharmaceutical manufacturers.
Federal suits have alleged that pharmaceutical manufacturers whose marketing and promotional
practices were found to have included the off-label promotion of drugs or the payment of prohibited
kickbacks to doctors violated the Federal False Claims Act on the grounds that these prohibited
activities resulted in the submission of claims to federal and state healthcare entitlement
programs such as Medicaid, resulting in the payment of claims by Medicaid for the off-label use of
the drug that was not a use of the drug otherwise covered by Medicaid. Such manufacturers have
entered into settlements with the federal government under which they paid amounts and entered into
corporate integrity agreements that require, among other things, substantial reporting and remedial
actions.
The Federal authorities, and state equivalents, may likewise seek to enforce the False Claims
Act against medical device manufacturers. We believe that our marketing practices are not in
violation of the Federal False Claims Act or state equivalents, but we cannot assure you that the
federal authorities will not take action against us and, if such action were successful, we could
be required to pay significant fines and penalties and change our marketing practices. Such
enforcement could have a significant adverse effect on our ability to operate.
We engage in a variety of activities that are subject to these laws and that have come under
particular scrutiny in recent years by federal and state regulators and law enforcement entities.
These activities have included, consulting arrangements with cardiothoracic surgeons, grants for
training and other education, grants for research, and other interactions with doctors.
International Regulation. International sales of medical devices are subject to foreign
governmental regulations, which vary substantially from country to country. The time required to
obtain certification or approval by a foreign country may be longer or shorter than that required
for FDA clearance or approval, and the requirements may differ.
20
The primary regulatory body in Europe is the European Union, which has adopted numerous
directives and has promulgated voluntary standards regulating the design, manufacture and labeling
of and clinical trials and adverse
event reporting for medical devices. In March 2010, amendments to the European Union Medical
Devices Directive came into force that could potentially impact the ability to obtain and maintain
CE marking for medical devices put on the market in the participating EU and EFTA countries.
Elements of the amendments include the provision of evidence of clinical efficacy regardless of
device classification (this can include supporting information from the literature, prospective
clinical data or a combination of both), consideration of the applicability of the machinery and
personal protective equipment directives, active postmarket surveillance up to and including
prospective, postmarket registries, and increased scrutiny of third party subcontractors. Devices
that comply with the requirements of a relevant directive will be entitled to bear CE conformity
marking, indicating that the device conforms with the essential requirements of the applicable
directives and, accordingly, can be commercially distributed throughout the member states of the
European Union and other countries that comply with or mirror these directives. The method for
assessing conformity varies depending upon the type and class of the product, but normally involves
a combination of self-assessment by the manufacturer and a third-party assessment by a notified
body, which is an independent and neutral institution appointed by a country to conduct the
conformity assessment. This third-party assessment may consist of an audit of the manufacturer’s
quality system and specific testing of the manufacturer’s device. Such an assessment is required
for a manufacturer to commercially distribute the product throughout these countries. International
Standards Organization, or ISO, 9001 and ISO 13845 certifications are voluntary standards.
Compliance establishes the presumption of conformity with the essential requirements for the CE
Mark. We have the authorization to affix the CE Mark to the PAS-Port and C-Port devices and to
commercialize the devices in the European Union for coronary artery bypass grafting. In addition,
we plan to secure a CE Mark and gain our “first in man” clinical experience for the Cardica
Microcutter ES8 in Europe.
In Japan, medical devices must be approved prior to importation and commercial sale by the
Ministry of Health, Labor and Welfare, or MHLW. Manufacturers of medical devices outside of Japan
are required to utilize a contractually bound In-Country Caretaker, or ICC, to submit an
application for device approval to the MHLW. The MHLW evaluates each device for safety and
efficacy. As part of its approval process, the MHLW may require that the product be tested in
Japanese laboratories. The approval process for products such as our existing anastomotic products
is typically 13 to 14 months. Other medical devices may require a longer review period for
approval. Once approved, the manufacturer may import the device into Japan for sale by the
manufacturer’s contractually bound importer or distributor.
After a device is approved for importation and commercial sale in Japan, the MHLW continues to
monitor sales of approved products for compliance with labeling regulations, which prohibit
promotion of devices for unapproved uses and reporting regulations, which require reporting of
product malfunctions, including serious injury or death caused by any approved device. Failure to
comply with applicable regulatory requirements can result in enforcement action by the MHLW, which
may include fines, injunctions, and civil penalties, recall or seizure of our products, operating
restrictions, partial suspension or total shutdown of sales in Japan, or criminal prosecution.
We have received approval from the MHLW to distribute our PAS-Port system in Japan. We will be
required to submit applications with respect to all new products and product enhancements for
review and approval by the MHLW. Our contract with Century, our distributor in Japan, has a
multi-year term and is renewable for additional multi-year terms upon mutual agreement of the
parties.
In addition to MHLW oversight, the regulation of medical devices in Japan is also governed by
the Japanese Pharmaceutical Affairs Law, or PAL. PAL was substantially revised in July 2002, and
the new provisions were implemented in stages through April 2005. Revised provisions of the
approval and licensing system of medical devices in Japan, which constitutes the core of import
regulations, came into effect on April 1, 2005. The revised law changes class categorizations of
medical devices in relation to risk, introduces a third-party certification system, strengthens
safety countermeasures for biologically derived products, and reinforces safety countermeasures at
the time of resale or rental. The revised law also abolishes the ICC system and replaces it with
the “primary distributor” system. Under the PAL in effect prior to April 1, 2005, manufacturers of
medical devices outside of Japan were required to utilize a Marketing Authorization Holder (MAH) to
obtain on their behalf approval of each product by the MHLW prior to the sale or distribution of
their products in Japan. Under the revised PAL, manufacturers outside of Japan must now appoint a
“primary distributor” located in Japan that holds a primary distributor license for medical devices
to provide primary distribution services, including conducting quality assurance and safety control
tasks for each product at the time an application for the approval of each such product is
submitted to the MHLW. Century Medical serves as the “primary distributor” for Cardica. As an
interim measure, an ICC licensed under the
PAL in effect prior to April 1, 2005 will be deemed to be the primary distributor under the revised
PAL if that ICC had a license to import and distribute the relevant medical devices that was
applied for and obtained under the old PAL. We are unable at this time to determine the impact of
such changes on our approved products or future products. We do not anticipate that these changes
will have a material impact on our existing level of third-party reimbursement for sales of our
products in Japan.
21
Employees
As of June 30, 2010, we had 34 employees, including 8 employees in manufacturing, 3 employees
in sales and marketing, 4 employees in clinical, regulatory and quality assurance, 7 employees in
general and administrative and 12 employees in research and development. We believe that our future
success will depend upon our continued ability to attract, hire and retain qualified personnel.
None of our employees is represented by a labor union or party to a collective bargaining
agreement, and we believe our employee relations are good.
Corporate Information
We were incorporated in Delaware in October 1997 as Vascular Innovations, Inc. and changed our
name to Cardica, Inc. in November 2001. Our principal executive offices are located at 900 Saginaw
Drive, Redwood City, California 94063 and our telephone number is (650) 364-9975. We make our
periodic and current reports available, free of charge, on our website as soon as practicable after
such material is electronically filed with the Securities and Exchange Commission. Our website
address is www.cardica.com and the reports are filed under “SEC Filings”, on the Investors/Media
portion of our website.
22
Executive Officers of the Registrant
The following table sets forth certain information concerning our executive officers
as of August 31, 2010:
| |
|
|
|
|
|
|
| Name |
|
Age |
|
Position |
Bernard A. Hausen, M.D., Ph.D.
|
|
|
50 |
|
|
President, Chief Executive Officer,
Chief Medical Officer and Director |
Robert Y. Newell
|
|
|
62 |
|
|
Vice President, Finance and Chief Financial Officer |
Frederick M. Bauer
|
|
|
57 |
|
|
Vice President, Operations |
Bryan D. Knodel, Ph.D.
|
|
|
50 |
|
|
Vice President, Research and Development |
Bernard A. Hausen, M.D., Ph.D. has been our President and Chief Executive Officer since
December 2000. Dr. Hausen co-founded the Company in October 1997 and has served as a director and
our Chief Medical Officer since inception. Dr. Hausen received a medical degree from Hannover
Medical School in Germany in 1988 and was trained there as a general and cardiothoracic surgeon.
Upon completion of his training, he received a Ph.D. degree in Medical Physiology in 1999. From
1996 to 2000, he was employed as a Senior Research Scientist in the Laboratory for Transplantation
Immunology of the Department of Cardiothoracic Surgery at Stanford University. Until Dr. Hausen
became our full-time employee in October of 2000, he remained responsible for all surgery-related
research in that laboratory.
Robert Y. Newell has been our Vice President, Finance and Chief Financial Officer since March
2003 and was Vice President, Finance and Operations, from July 2005 to July 2008. From January 2000
to February 2003 he was Vice President, Finance and Chief Financial Officer for Omnicell, Inc., a
hospital supply and medication management company. Mr. Newell holds a B.A. degree in Mathematics
from the College of William & Mary and an M.B.A. degree from the Harvard Business School.
Frederick M. Bauer joined Cardica as our Vice President of Operations in July 2008. From
August 2005 to June 2008, he was President and Owner of 3RLatex, LLC, a containment, transportation and
recycling company for the construction industry and from November 2002 to November 2005, he was
general manager of Amazon Environmental, a latex paint recycling company. From October 1996 to
November 2001, he was Vice President Operations for the Cardiac Surgery division and Vice President
Operations for the Perfusion Systems division of Medtronic, Inc., a medical device company. He also
held a number of operations and engineering executive positions with Baxter Healthcare
International, a healthcare company, from 1981 to 1996. He currently serves as a member of the
board of the Orange County ARC, a non-profit servicing 700 developmentally disabled adults.
Mr. Bauer holds a B.S. degree in Civil Engineering from the University of Detroit Mercy.
Bryan D. Knodel, Ph.D. joined Cardica as our Vice President of Research and Development in
July 2005. Since January 1998, he has been president of Bryan D. Knodel, Inc., a consulting firm
specializing in medical device design and product development. From April 2001 until June 2005, Mr.
Knodel consulted for us in product development. From 1992 to 1997, he was a principal engineer with
Ethicon Endo-Surgery, a Johnson & Johnson company developing medical devices for less invasive
surgery. Mr. Knodel holds B.S., M.S. and Ph.D. degrees in Mechanical Engineering from the
University of Illinois.
23
Item 1A. Risk Factors
We have identified the following risks and uncertainties that may have a material adverse effect on
our business, financial condition or results of operations. The risks described below are not the
only ones we face. Additional risks not presently known to us or that we currently believe are
immaterial may also significantly impair our business operations.
Risks Related to Our Finances and Capital Requirements
We need to generate higher product sales to become and remain profitable.
Our ability to become and remain profitable depends upon our ability to generate higher
product sales. Our ability to generate significantly higher revenue depends upon a number of
factors, including:
| |
• |
|
achievement of broad acceptance for our current products or future products that we may commercialize; |
| |
• |
|
achievement of U.S. regulatory clearance or approval for additional products; and |
| |
• |
|
successful sales, manufacturing, marketing and distribution of our products. |
Sales of our products and development activities generated only $4.0 million, $9.9 million and
$7.6 million of revenue for fiscal years ended June 30, 2010, 2009 and 2008, respectively. We do
not anticipate that we will generate significantly higher product sales for the foreseeable future.
Sales of our C-Port and PAS-Port systems have not met the levels that we had anticipated, and to
date our systems have had limited commercial adoption. Failure to obtain broader commercial
adoption of our systems will continue to negatively impact our financial results and financial
position and may require us to delay, further reduce the scope of or eliminate our
commercialization efforts with respect to one or more of our products or one or more of our
research and development projects.
We need substantial additional funding and may be unable to raise capital, which would force us to
delay, reduce or eliminate our research and development programs or commercialization efforts and
could cause us to cease operations.
Our development efforts have consumed substantial capital to date. Including the cash received
in August 2010 in connection with the License Agreement with Intuitive Surgical and related equity
investment, we believe that our existing cash and cash equivalents, along with the
cash that we expect to generate from operations, will be sufficient to meet our anticipated cash
needs to enable us to conduct our business substantially as currently conducted through September
30, 2011. We have based our estimate on assumptions that may prove to be wrong, including
assumptions with respect to the level of revenue from product sales, and we could exhaust our
available financial resources sooner than we currently expect. While our cash resources would
permit us to continue through September 30, 2011, we would need to further reduce expenses in
advance of that date in the event that we are unable to complete a financing, strategic or
commercial transaction to ensure that we have sufficient capital to meet our obligations and
continue on a path designed to create and preserve stockholder value.
The sufficiency of our current cash resources and our need for additional capital, and the
timing thereof, will depend upon numerous factors. These factors include, but are not limited to,
the following:
| |
• |
|
the extent of our ongoing research and development programs and related costs, including costs
related to the development of the Cardica Microcutter ES8 and additional potential products in our
anticipated microcutter product line; |
| |
• |
|
our ability to enter into additional license, development and/or collaboration agreements with
respect to our technology, and the terms thereof; |
| |
• |
|
market acceptance and adoption of our current products or future products that we may commercialize; |
24
| |
• |
|
costs associated with our sales and marketing initiatives and manufacturing activities; |
| |
• |
|
costs and timing of obtaining and maintaining FDA and other regulatory clearances and approvals for
our products and potential additional products; |
| |
• |
|
securing, maintaining and enforcing intellectual property rights and the costs thereof; and |
| |
• |
|
the effects of competing technological and market developments. |
Because we do not anticipate that we will generate sufficient product sales to achieve
profitability for the foreseeable future, if at all, we need to raise substantial additional
capital to finance our operations in the future. To raise capital, we may seek to sell additional
equity or debt securities, obtain a credit facility or enter into product development, license or
distribution agreements with third parties or divest one or more of our commercialized products or
products in development. The sale of additional equity or convertible debt securities could result
in significant dilution to our stockholders, particularly in light of the prices at which our
common stock has been recently trading. If additional funds are raised through the issuance of debt
securities, these securities could have rights senior to those associated with our common stock and
could contain covenants that would restrict our operations. Any product development, licensing,
distribution or sale agreements that we enter into may require us to relinquish valuable rights,
including with respect to commercialized products or products in development that we would
otherwise seek to commercialize or develop ourselves. We may not be able to obtain sufficient
additional funding or enter into a strategic transaction in a timely manner. Our need to raise
capital may require us to accept terms that may harm our business or be disadvantageous to our
current stockholders. If adequate funds are not available or revenues from product sales do not
increase, we would be required to further reduce our workforce, delay, reduce the scope of or
eliminate our commercialization efforts with respect to one or more of our products or one or more
of our research and development programs in advance of September 30, 2011 to ensure that we have
sufficient capital to meet our obligations and continue on a path designed to create and preserve
stockholder value. Failure to raise additional capital may result in our ceasing to be publicly
traded or ceasing operations.
We have a history of net losses, which we expect to continue for the foreseeable future, and we are
unable to predict the extent of future losses or when we will become profitable, if at all.
We have incurred net losses since our inception in October 1997. As of June 30, 2010, our
accumulated deficit was approximately $120.3 million. We expect to incur substantial additional
losses until we can achieve significant commercial sales of our products, which depend upon a
number of factors, including increased commercial sales of our C-Port and PAS-Port systems in the
United States and receipt of regulatory clearance or approval and market adoption of our additional
proposed products in the United States. We commenced commercial sales of the C-Port system in
Europe in 2004 and in the United States in 2006 and of the PAS-Port system in Europe in 2003, in
Japan in 2004 and in the United States in September 2008.
Our cost of product sales was 98% and 79% of our net product sales for fiscal years ended June
30, 2010 and 2009, respectively. We expect high cost of product sales to continue for the
foreseeable future. If, over the long term, we are unable to reduce our cost of producing goods and
expenses relative to our net revenue, we may not achieve profitability even if we are able to
generate significant sales of the C-Port and PAS-Port systems. Our failure to achieve and sustain
profitability would negatively impact the market price of our common stock.
Creditors may have rights to our assets that are senior to our stockholders.
On August 17, 2010, we paid off the remaining $1.4 million principal balance of our note
payable to Century Medical, Inc. and, therefore, no longer have any outstanding debt obligations.
However, future arrangements with creditors may allow these creditors to liquidate our assets,
which may include our intellectual property rights, if we are in default or breach of our debt
obligations for a continued period of time. The proceeds of any sale or liquidation of our assets
under these circumstances would be applied first to any of our debt obligations that would have
priority over any of our capital stock. After satisfaction of our debt obligations, we may have
little or no proceeds left under these circumstances to distribute to the holders of our capital
stock.
25
Our quarterly operating results and stock price may fluctuate significantly.
We expect our operating results to be subject to quarterly fluctuations. The revenue we
generate, if any, and our operating results will be affected by numerous factors, many of which are
beyond our control, including:
| |
• |
|
the extent of our ongoing research and development programs and related costs, including
costs related to the development of the Cardica Microcutter ES8 and additional potential
products; |
| |
• |
|
our ability to enter into additional license, development and/or collaboration agreements
with respect to our technology, and the terms thereof; |
| |
• |
|
market acceptance and adoption of our products; |
| |
• |
|
costs associated with our sales and marketing initiatives and manufacturing activities; |
| |
• |
|
costs and timing of obtaining and maintaining FDA and other regulatory clearances and
approvals for our products and potential additional products; |
| |
• |
|
securing, maintaining and enforcing intellectual property rights and the costs thereof; and |
| |
• |
|
the effects of competing technological and market developments. |
Quarterly fluctuations in our operating results may, in turn, cause the price of our stock to
fluctuate substantially.
Risks Related to Our Business
We are dependent upon the success of our current products to generate revenue in the near term, and
we have U.S. regulatory clearance for our C-Port and PAS-Port systems only. We cannot be certain
that the C-Port and PAS-Port systems can be successfully commercialized in the United States. If we
are unable to successfully commercialize our products in the United States, our ability to generate
higher revenue will be significantly delayed or halted, and our business will be harmed.
We have expended significant time, money and effort in the development of our current
commercial products, the C-Port systems and the PAS-Port system. If we are not successful in
commercializing our C-Port and PAS-Port systems, we may never generate substantial revenue, our
business, financial condition and results of operations would be materially and adversely affected,
and we may be forced to cease operations. We commenced sales of our C-Port xA system in December
2006 (after introduction of our original C-Port system in January 2006), our C-Port Flex A in April
2007 and our C-Port X-CHANGE in December 2007. We commenced U.S. sales of our PAS-Port system in
September 2008. We anticipate that our ability to increase our revenue significantly will depend on
the continued adoption of our current PAS-Port and C-Port systems in the United States and
commercialization of additional potential products, including the Cardica Microcutter ES8 in
particular.
We may not be successful in our efforts to expand our product portfolio, and our failure to do so
could cause our business and prospects to suffer.
We intend to use our knowledge and expertise in anastomotic technologies to discover, develop
and commercialize new applications in additional markets. In particular, we are developing the
Cardica Microcutter ES8, an endoscopic microcutter intended for use by general, thoracic,
gynecologic, bariatric and urologic surgeons, and other potential products in our anticipated
microcutter product line. We have not yet commenced human testing of this device. Significant
additional research and development and financial resources will be required to develop the Cardica
Microcutter ES8 into a commercially viable product and to obtain necessary regulatory approvals to
commercialize the device. We cannot assure you that our development efforts will be successful or
that they will be completed within our publicly stated anticipated timelines, and we may never be
successful in developing a viable
product for the markets intended to be addressed by the Cardica Microcutter ES8 or other potential
microcutter products. Our failure to successfully develop the Cardica Microcutter ES8 and/or other
microcutter products would have a material adverse effect on our business, growth prospects and
ability to raise additional capital.
26
Our products may never gain any significant degree of market acceptance, and a lack of market
acceptance would have a material adverse effect on our business.
To date, our anastomoses products have not gained, and we cannot assure you that our
anastomoses products or any other products that we may develop will gain, any significant degree of
market acceptance among physicians or patients. We believe that recommendations by physicians will
be essential for market acceptance of our products; however, we cannot assure you that significant
recommendations will be obtained. Physicians will not recommend our products unless they conclude,
based on clinical data and other factors, that the products represent a safe and acceptable
alternative to other available options. In particular, physicians may elect not to recommend using
our anastomoses products in surgical procedures until such time, if ever, as we successfully
demonstrate with long-term data that our products result in patency rates comparable to or better
than those achieved with hand-sewn anastomoses, and we resolve any technical limitations that may
arise.
Assuming that we receive FDA clearance for one or more of our microcutter products, a number
of factors will influence our ability to gain clinical adoption. In many surgical specialties, the
use of laparoscopic and open surgical stapling devices is routine in clinical practice and an
accepted standard of care. Two large companies, Johnson & Johnson and Covidien, dominate the market
for surgical stapling devices. For our products to be clinically adopted, they must show benefits
that are significant enough for surgeons to communicate their preference and to overcome any
constraints on their hospitals’ ability to purchase competing products, such as purchasing
contracts, to buy one of our stapling products to replace a competing device. In addition to this
obstacle, our microcutter products must demonstrate the degree of reliability that surgeons have
experienced with products that they have been using for years. Market acceptance of our products
also depends on our ability to demonstrate consistent quality and safety of our products. Our
anticipated initial lack of human clinical data with respect to the use of any microcutter products
that we may commercialize is likely to negatively impact the rate and extent of clinical adoption
of the products. Any future recalls may impact physicians’ and hospitals’ perception of our
products.
Widespread use of our products will require the training of numerous physicians, and the time
required to complete training could result in a delay or dampening of market acceptance. Even if
the safety and efficacy of our products is established, physicians may elect not to use our
products for a number of reasons beyond our control, including inadequate or no reimbursement from
health care payors, physicians’ reluctance to use products that have not been proven through time
in the market, the introduction of competing devices by our competitors and pricing for our
products. Failure of our products to achieve any significant market acceptance would have a
material adverse effect on our business, financial condition and results of operations.
Our PAS-Port and C-Port systems, as well as our other future products, may still face future
development and regulatory difficulties.
Even though the current generations of the C-Port and PAS-Port systems have received U.S.
regulatory clearance, the FDA may still impose significant restrictions on the indicated uses or
marketing of these products or ongoing requirements for potentially costly post-clearance studies.
Any of our future products, including the Cardica Microcutter ES8 and any future generations of the
C-Port systems, may not obtain regulatory clearances or approvals required for marketing or may
face these types of restrictions or requirements, particularly as the FDA is in the process of
revising its 510(k) clearance system to, in certain cases, require human clinical data and to
prohibit the combination of multiple predicate devices as the basis for a 510(k). The process of
obtaining regulatory clearances or approvals to market a medical device, particularly from the FDA,
can be costly and time consuming, and there can be no assurance that such clearances or approvals
will be granted on a timely basis, if at all. The FDA permits commercial distribution of most new
medical devices only after the device has received 510(k) clearance or is the subject of an
approved pre-market approval application, or PMA. The FDA will clear the marketing of a medical
device through the 510(k) process if it is demonstrated that the new product has the same intended
use, is substantially equivalent to
27
another legally marketed device, including a 510(k)-cleared
product, and otherwise meets
the FDA’s requirements. We intend to seek 510(k) clearance for the Cardica Microcutter ES8. The PMA
approval process is more costly, lengthy and uncertain than the 510(k) clearance process and
requires the development and submission of clinical studies supporting the safety and effectiveness
of the device. Product modifications may also require the submission of a new 510(k) clearance or
the approval of a PMA before the modified product can be marketed. Any products that we develop
that require regulatory clearance or approval, including the Cardica Microcutter ES8, may not be
approved on the timelines that we currently anticipate, if approved at all. We cannot assure that
any new products or any product enhancements that we develop will be subject to the shorter 510(k)
clearance process instead of the more lengthy PMA requirements. Regulatory agencies subject a
product, its manufacturer and the manufacturer’s facilities to continual review, regulation and
periodic inspections. If a regulatory agency discovers previously unknown problems with a product,
including adverse events of unanticipated severity or frequency, or problems with the facility
where the product is manufactured, a regulatory agency may impose restrictions on that product, our
collaborators or us, including requiring withdrawal of the product from the market. Our products
will also be subject to ongoing FDA requirements for the labeling, packaging, storage, advertising,
promotion, record-keeping and submission of safety and other post-market information on the
product. If our products fail to comply with applicable regulatory requirements, a regulatory
agency may impose any of the following sanctions:
| |
• |
|
warning letters, fines, injunctions, consent decrees and civil penalties; |
| |
• |
|
customer notifications, repair, replacement, refunds, recall or seizure of our products; |
| |
• |
|
operating restrictions, partial suspension or total shutdown of production; |
| |
• |
|
delay in processing marketing applications for new products or modifications to existing products; |
| |
• |
|
withdrawing approvals that have already been granted; and |
To market any products internationally, we must establish and comply with numerous and varying
regulatory requirements of other countries regarding safety and efficacy. Approval procedures vary
among countries and can involve additional product testing and additional administrative review
periods. The time required to obtain approval in other countries might differ from that required to
obtain FDA clearance or approval. The regulatory approval process in other countries may include
all of the risks detailed above regarding FDA clearance or approval. Regulatory approval in one
country does not ensure regulatory approval in another, but a failure or delay in obtaining
regulatory approval in one country may negatively impact the regulatory process in others. Failure
to obtain regulatory approval in other countries or any delay or setback in obtaining such approval
could have the same adverse effects detailed above regarding FDA clearance or approval,including
the risk that our products may not be approved for use under all of the circumstances requested,
which could limit the uses of our products and adversely impact potential product sales, and that
such clearance or approval may require costly, post-marketing follow-up studies. If we fail to
comply with applicable foreign regulatory requirements, we may be subject to fines, suspension or
withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions
and criminal prosecution.
If we do not achieve our projected development goals in the time frames we announce and expect, the
commercialization of our product candidates may be delayed and, as a result, our stock price may
decline.
From time to time, we may estimate and publicly announce the timing anticipated for the
accomplishment of various clinical, regulatory and other product development goals, which we
sometimes refer to as milestones. These milestones may include submissions for and receipt of
clearances or approvals from regulatory authorities, other clinical and regulatory events or the
launch of new products. These estimates are based on a variety of assumptions. The actual timing of
these milestones can vary dramatically compared to our estimates, in some cases for reasons beyond
our control. In particular, our estimate of the timing of completion of development of the Cardica
Microcutter ES8 and submission to the FDA for 510(k) clearance is subject to a number of risks
related to development and regulatory matters that are discussed in this Annual Report. If we do
not meet milestones as
publicly announced, the commercialization of our products may be delayed and, as a result, our
stock price may decline.
28
Our manufacturing facilities, and those of our suppliers, must comply with applicable regulatory
requirements. Failure of our manufacturing facilities to comply with quality requirements would
harm our business and our results of operations.
Our manufacturing facilities and processes are subject to periodic inspections and audits by
various U.S. federal, U.S. state and foreign regulatory agencies. For example, our facilities have
been inspected by State of California regulatory authorities pursuant to granting a California
Device Manufacturing License and by the FDA. Additionally, to market products in Europe, we are
required to maintain ISO 13485:2003 certification and are subject to periodic surveillance audits.
We are currently ISO 13485:2003 certified; however, our failure to maintain necessary regulatory
compliance and permits for our manufacturing facilities could prevent us from manufacturing and
selling our products.
Additionally, our manufacturing processes and, in some cases, those of our suppliers, are
required to comply with the FDA’s Quality System Regulation, or QSR, which covers the procedures
and documentation of the design, testing, production, control, quality assurance, labeling,
packaging, storage and shipping of our products, including the PAS-Port and C-Port systems. We are
also subject to similar state requirements and licenses. In addition, we must engage in extensive
record keeping and reporting and must make available our manufacturing facilities and records for
periodic inspections by governmental agencies, including FDA, state authorities and comparable
agencies in other countries. If we are given notice of significant violations in a QSR inspection,
our operations could be disrupted and our manufacturing interrupted. Failure to take adequate
corrective action in response to an adverse QSR inspection could result in, among other things, a
shut-down of our manufacturing operations, significant fines, suspension of product distribution or
other operating restrictions, seizures or recalls of our devices and criminal prosecutions, any of
which would cause our business to suffer. Furthermore, our key component suppliers may not
currently be or may not continue to be in compliance with applicable regulatory requirements, which
may result in manufacturing delays for our products and cause our revenue to decline.
We may also be required to recall our products due to manufacturing supply defects. If we
issue recalls of our products in the future, our revenue and business could be harmed.
If we are unable to establish sales and marketing capabilities or enter into and maintain
arrangements with third parties to market and sell our products, our business may be harmed.
We have limited experience as a company in the sale, marketing and distribution of our
products. Century Medical is responsible for marketing and commercialization of the PAS-Port system
in Japan. To promote our current and future products in the United States and Europe, we must
develop our sales, marketing and distribution capabilities or make arrangements with third parties
to perform these services. Competition for qualified sales personnel is intense. Developing a sales
force is expensive and time consuming and could delay any product launch. We may be unable to
establish and manage an effective sales force in a timely or cost-effective manner, if at all, and
any sales force we do establish may not be capable of generating sufficient demand for our
products. We have entered into arrangements with third parties to perform sales and marketing
services, which may result in lower product sales than if we directly marketed and sold our
products. We expect to rely on third-party distributors for substantially all of our international
sales. If we are unable to establish adequate sales and marketing capabilities, independently or
with others, we may not be able to generate significant revenue and may not become profitable. For
our microcutter products, we will have to compete for sales and acceptance of our products against
two large companies, Johnson & Johnson and Covidien, with large sales forces and dominant market
positions.
Lack of third-party coverage and reimbursement for our products could delay or limit their
adoption.
We may experience limited sales growth resulting from limitations on reimbursements made to
purchasers of our products by third-party payors, and we cannot assure you that our sales will not
be impeded and our business harmed if third-party payors fail to provide reimbursement that
hospitals view as adequate.
29
In the United States, our products will be purchased primarily by medical institutions, which
then bill various third-party payors, such as the Centers for Medicare & Medicaid Services, or CMS,
which administer the Medicare program, and other government programs and private insurance plans,
for the health care services provided to their patients. The process involved in applying for
coverage and incremental reimbursement from CMS is lengthy and expensive. Under current CMS
reimbursement policies, CMS offers a process to obtain add-on payment for a new medical technology
when the existing Diagnosis-Related Group, or DRG, prospective payment rate is inadequate. To
obtain add-on payment, a technology must be considered “new,” demonstrate substantial improvement
in care and exceed certain payment thresholds. Add-on payments are made for no less than two years
and no more than three years. We must demonstrate the safety and effectiveness of our technology to
the FDA in addition to CMS requirements before add-on payments can be made. Further, Medicare
coverage is based on our ability to demonstrate the treatment is “reasonable and necessary” for
Medicare beneficiaries. In November 2006, CMS denied our request for an add-on payment with respect
to our C-Port systems. According to CMS, we met the “new” criteria and exceeded the payment
threshold but did not in their view demonstrate substantial improvement in care. Even if our
products receive FDA and other regulatory clearance or approval, they may not be granted coverage
and reimbursement in the foreseeable future, if at all. Moreover, many private payors look to CMS
in setting their reimbursement policies and amounts. If CMS or other agencies limit coverage or
decrease or limit reimbursement payments for doctors and hospitals, this may affect coverage and
reimbursement determinations by many private payors.
We cannot assure you that CMS will provide coverage and reimbursement for our products. If a
medical device does not receive incremental reimbursement from CMS, then a medical institution
would have to absorb the cost of our products as part of the cost of the procedure in which the
products are used. Acute care hospitals are now generally reimbursed by CMS for inpatient operating
costs under a Medicare hospital inpatient prospective payment system. Under the Medicare hospital
inpatient prospective payment system, acute care hospitals receive a fixed payment amount for each
covered hospitalized patient based upon the DRG to which the inpatient stay is assigned, regardless
of the actual cost of the services provided. At this time, we do not know the extent to which
medical institutions would consider insurers’ payment levels adequate to cover the cost of our
products. Failure by hospitals and physicians to receive an amount that they consider to be
adequate reimbursement for procedures in which our products are used could deter them from
purchasing our products and limit our revenue growth. In addition, pre-determined DRG payments may
decline over time, which could deter medical institutions from purchasing our products. If medical
institutions are unable to justify the costs of our products, they may refuse to purchase them,
which would significantly harm our business.
We have limited data regarding the safety and efficacy of the PAS-Port and C-Port systems. Any data
that is generated in the future may not be positive or consistent with our existing data, which
would affect market acceptance and the rate at which our devices are adopted.
The C-Port and PAS-Port systems are innovative products, and our success depends upon their
acceptance by the medical community as safe and effective. An important factor upon which the
efficacy of the C-Port and PAS-Port systems will be measured is long-term data regarding the
duration of patency, or openness, of the artery or the graft vessel. Equally important will be
physicians’ perceptions of the safety of our products. Our technology is relatively new in cardiac
bypass surgery, and the results of short-term clinical experience of the C-Port and PAS-Port
systems do not necessarily predict long-term clinical benefit. We believe that physicians will
compare long-term patency for the C-Port and PAS-Port devices against alternative procedures, such
as hand-sewn anastomoses. If the long-term rates of patency do not meet physicians’ expectations,
or if physicians find our devices unsafe, the C-Port and PAS-Port systems may not become widely
adopted and physicians may recommend alternative treatments for their patients. In addition, we
have recently commenced U.S. commercialization of our C-Port and PAS-Port systems. Any adverse
experiences of physicians using the C-Port and PAS-Port systems, or adverse outcomes to patients,
may deter physicians from using our products and negatively impact product adoption.
30
Our C-Port and PAS-Port systems were designed for use with venous grafts. Additionally, while
our indications for use of the C-Port system cleared by the FDA refer broadly to grafts, we have
studied the use of the C-Port systems only with venous grafts and not with arterial grafts. Using
the C-Port systems with arterial grafts may not yield patency rates or material adverse cardiac
event rates comparable to those found in our clinical trials using venous grafts, which could
negatively affect market acceptance of our C-Port systems. In addition, the clips and staples
deployed by our products are made of 316L medical-grade stainless steel, to which some patients are
allergic. These allergies, especially if not previously diagnosed or unknown, may result in adverse
reactions that negatively affect the patency of the anastomoses or the healing of the implants and
may therefore adversely affect outcomes, particularly when compared to anastomoses performed with
other materials, such as sutures. Additionally, in the event a surgeon, during the course of
surgery, determines that it is necessary to convert to a hand-sewn anastomosis and to remove an
anastomosis created by one of our products, the removal of the implants may result in more damage
to the target vessel (such as the aorta or coronary artery) than would typically be encountered
during removal of a hand-sewn anastomosis. Moreover, the removal may damage the target vessel to an
extent that could further complicate construction of a replacement hand-sewn or automated
anastomosis, which could be detrimental to patient outcome. These or other issues, if experienced,
could limit physician adoption of our products.
Even if the data collected from future clinical studies or clinical experience indicates
positive results, each physician’s actual experience with our devices outside the clinical study
setting may vary. Clinical studies conducted with the C-Port and PAS-Port systems have involved
procedures performed by physicians who are technically proficient, high-volume users of the C-Port
and PAS-Port systems. Consequently, both short- and long-term results reported in these studies may
be significantly more favorable than typical results of practicing physicians, which could
negatively impact rates of adoption of the C-Port and PAS-Port systems.
Any clinical trials that we may conduct may not begin on time, or at all, and may not be completed
on schedule, or at all.
The commencement or completion of any clinical trials that we may conduct may be delayed or
halted for numerous reasons, including, but not limited to, the following:
| |
• |
|
the FDA or other regulatory authorities suspend or place on hold a clinical trial, or do
not approve a clinical trial protocol or a clinical trial; |
| |
• |
|
the data and safety monitoring committee of a clinical trial recommends that a trial be
placed on hold or suspended; |
| |
• |
|
patients do not enroll in clinical trials at the rate we expect; |
| |
• |
|
patients are not followed-up at the rate we expect; |
| |
• |
|
clinical trial sites decide not to participate or cease participation in a clinical trial; |
| |
• |
|
patients experience adverse side effects or events related to our products; |
| |
• |
|
patients die or suffer adverse medical effects during a clinical trial for a variety of
reasons, which may not be related to our product candidates, including the advanced stage
of their disease and other medical problems; |
| |
• |
|
third-party clinical investigators do not perform our clinical trials on our anticipated
schedule or consistent with the clinical trial protocol and good clinical practices, or
other third-party organizations do not perform data collection and analysis in a timely
or accurate manner; |
| |
• |
|
regulatory inspections of our clinical trials or manufacturing facilities may, among
other things, require us to undertake corrective action or suspend or terminate our
clinical trials if investigators find us not to be in compliance with regulatory
requirements; |
| |
• |
|
third-party suppliers fail to provide us with critical components that conform to design
and performance specifications; |
| |
• |
|
the failure of our manufacturing processes to produce finished products that conform to
design and performance specifications; |
| |
• |
|
changes in governmental regulations or administrative actions; |
31
| |
• |
|
the interim results of the clinical trial are inconclusive or negative; |
| |
• |
|
pre-clinical or clinical data is interpreted by third parties in different ways; or |
| |
• |
|
our trial design, although approved, is inadequate to demonstrate safety and/or efficacy. |
Clinical trials sometimes experience delays related to outcomes experienced during the course
of the trials. For example, in our PAS-Port pivotal trial, we had an administrative hold of the
trial related to an adverse event, which lasted approximately 72 hours while the adverse event was
investigated. The data safety monitoring board subsequently concluded that there was no clear
evidence that our device had caused the adverse event, and enrollment continued. While this event
was resolved in a timely manner and did not result in any material delay in the trial, future
similar or other types of events could lead to more significant delays or other effects in future
trials.
Clinical trials may require the enrollment of large numbers of patients, and suitable patients
may be difficult to identify and recruit. Patient enrollment in clinical trials and completion of
patient follow-up in clinical trials depend on many factors, including the size of the patient
population, the nature of the trial protocol, the proximity of patients to clinical sites and the
eligibility criteria for the study and patient compliance. For example, patients may be discouraged
from enrolling in our clinical trials if the trial protocol requires them to undergo extensive
post-treatment procedures to assess the safety and effectiveness of our product candidates, or they
may be persuaded to participate in contemporaneous trials of competitive products. Delays in
patient enrollment or failure of patients to continue to participate in a study may cause an
increase in costs and delays or result in the failure of the trial.
Our clinical trial costs will increase if we have material delays in our clinical trials or if
we need to perform more or larger clinical trials than planned. Adverse events during a clinical
trial could cause us to repeat a trial, terminate a trial or cancel an entire program.
If the third parties on whom we rely to conduct our clinical trials do not perform as contractually
required or expected, we may not be able to obtain regulatory approval for or commercialize our
product candidates.
We do not have the ability to independently conduct clinical trials for our product
candidates, and we must rely on third parties, such as contract research organizations, medical
institutions, clinical investigators and contract laboratories, to conduct our clinical trials. In
addition, we rely on third parties to assist with our pre-clinical development of product
candidates. Furthermore, our third-party clinical trial investigators may be delayed in conducting
our clinical trials for reasons outside of their control, such as changes in regulations, delays in
enrollment, and the like. If these third parties do not successfully carry out their contractual
duties or regulatory obligations or meet expected deadlines, if these third parties need to be
replaced or if the quality or accuracy of the data they obtain is compromised due to the failure to
adhere to our clinical protocols or regulatory requirements or for other reasons, any clinical
trials that we may conduct may be extended, delayed, suspended or terminated, and we may not be
able to obtain regulatory approval for or successfully commercialize our product candidates on a
timely basis, if at all.
Because one customer accounts for a substantial portion of our product sales, the loss of this
significant customer would cause a substantial decline in our revenue.
We derive a substantial portion of our revenue from sales to Century Medical, our distributor
in Japan. The loss of Century Medical as a customer would cause a decrease in revenue and,
consequently, an increase in net loss. For fiscal years ended June 30, 2010 and 2009, sales to
Century Medical accounted for approximately 23% and 15%, respectively, of our total product sales.
We expect that Century Medical will continue to account for a substantial portion of our sales in
the near term. As a result, if we lose Century Medical as a customer, our revenue and net loss
would be adversely affected. In addition, customers that have accounted for significant revenue in
the past may not generate revenue in any future period. The failure to obtain new significant
customers or additional orders from existing customers will materially affect our operating
results.
32
If our competitors have products that are approved in advance of ours, marketed more effectively or
demonstrated to be safer or more effective than ours, our commercial opportunity will be reduced or
eliminated and our business will be harmed.
The market for anastomotic solutions and cardiac bypass products is competitive. Competitors
include a variety of public and private companies that currently offer or are developing cardiac
surgery products generally and automated anastomotic systems specifically that would compete
directly with ours.
We believe that the primary competitive factors in the market for medical devices used in the
treatment of coronary artery disease include:
| |
• |
|
improved patient outcomes; |
| |
• |
|
access to and acceptance by leading physicians; |
| |
• |
|
product quality and reliability; |
| |
• |
|
device cost-effectiveness; |
| |
• |
|
physician relationships; and |
| |
• |
|
sales and marketing capabilities. |
We may be unable to compete successfully on the basis of any one or more of these factors,
which could have a material adverse affect on our business, financial condition and results of
operations.
A number of different technologies exist or are under development for performing anastomoses,
including sutures, mechanical anastomotic devices, suture-based anastomotic devices and shunting
devices. Currently, substantially all anastomoses are performed with sutures and, for the
foreseeable future we believe that sutures will continue to be the principal alternative to our
anastomotic products. Sutures are far less expensive than our automated anastomotic products, and
other anastomotic devices may be less expensive than our own. Surgeons, who have been using sutures
for their entire careers, may be reluctant to consider alternative technologies, despite potential
advantages. Any resistance to change among practitioners could delay or hinder market acceptance of
our products, which would have a material adverse effect on our business.
Cardiovascular diseases may also be treated by other methods that do not require anastomoses,
including, interventional techniques such as balloon angioplasty with or without the use of stents,
pharmaceuticals, atherectomy catheters and lasers. Several of these alternative treatments are
widely accepted in the medical community and have a long history of use. In addition, technological
advances with other therapies for cardiovascular disease, such as drugs, or future innovations in
cardiac surgery techniques could make other methods of treating these diseases more effective or
lower cost than bypass procedures. For example, the number of bypass procedures in the United
States and other major markets has declined in recent years and is expected to decline in the years
ahead because competing treatments are, in many cases, far less invasive and provide acceptable
clinical outcomes. Many companies working on treatments that do not require anastomoses may have
significantly greater financial, manufacturing, marketing, distribution and technical resources and
experience than we have. Many of our competitors have significantly greater financial resources and
expertise in research and development, manufacturing, pre-clinical testing, clinical trials,
obtaining regulatory clearance or approval and marketing approved products than we do. Smaller or
early-stage companies may also prove to be significant competitors, particularly through
collaborative arrangements with large and established companies. Our competitors may succeed in
developing technologies and therapies that are more effective, better tolerated or less costly than
any that we are developing or
that would render our product candidates obsolete and noncompetitive. Our competitors may succeed
in obtaining clearance or approval from the FDA and foreign regulatory authorities for their
products sooner than we do for ours. We will also face competition from these third parties in
recruiting and retaining qualified scientific and management personnel, establishing clinical trial
sites and patient enrollment for clinical trials and in acquiring and in-licensing technologies and
products complementary to our programs or advantageous to our business.
33
The Cardica Microcutter ES8, if it receives FDA clearance and is successfully launched, would
compete in the market for laparoscopic stapling and cutting devices in the United States. We
believe the principal competitive factors in the market for laparascopic staplers include:
| |
• |
|
product quality and reliability; |
| |
• |
|
device cost-effectiveness; |
| |
• |
|
degree of articulation; |
| |
• |
|
physician relationships; and |
| |
• |
|
sales and marketing capabilities. |
Two large competitors, Ethicon Endo-Surgery, part of Johnson & Johnson, and Covidien currently
control over 80% of this market. Other large competitors in the laparoscopic device market include
Stryker Endoscopy and Olympus which acquired another competitor, Gyrus Medical. Ethicon
Endo-Surgery and Covidien, which recently acquired a small competitor, Power Medical, each have
large direct sales forces in the United States and have been the largest participants in the market
for single use disposable laparoscopic stapling devices for many years. Competing against large
established competitors with significant resources may make establishing a market for our products
difficult.
We are dependent upon a number of key suppliers, including single source suppliers, the loss of
which would materially harm our business.
We use or rely upon sole source suppliers for certain components and services used in
manufacturing our products, and we utilize materials and components supplied by third parties with
which we do not have any long-term contracts. In recent years, many suppliers have ceased supplying
materials for use in implantable medical devices. We cannot assure you that materials required by
us will not be restricted or that we will be able to obtain sufficient quantities of such materials
or services in the future. Moreover, the continued use by us of materials manufactured by third
parties could subject us to liability exposure. Because we do not have long-term contracts, none of
our suppliers is required to provide us with any guaranteed minimum production levels.
We cannot quickly replace suppliers or establish additional new suppliers for some of our
components, particularly due to both the complex nature of the manufacturing process used by our
suppliers and the time and effort that may be required to obtain FDA clearance or approval or other
regulatory approval to use materials from alternative suppliers. Any significant supply
interruption or capacity constraints affecting our facilities or those of our suppliers would have
a material adverse effect on our ability to manufacture our products and, therefore, a material
adverse effect on our business, financial condition and results of operations.
34
We have limited manufacturing experience and may encounter difficulties in increasing production
to provide an adequate supply to customers.
To date, our manufacturing activities have consisted primarily of producing moderate
quantities of our products for use in clinical studies and for commercial sales in Japan, Europe
and the United States. Production in increased commercial quantities will require us to expand our
manufacturing capabilities and to hire and train additional
personnel. We may encounter difficulties in increasing our manufacturing capacity and in
manufacturing larger commercial quantities, including:
| |
• |
|
maintaining product yields; |
| |
• |
|
maintaining quality control and assurance; |
| |
• |
|
providing component and service availability; |
| |
• |
|
maintaining adequate control policies and procedures; and |
| |
• |
|
hiring and retaining qualified personnel. |
Difficulties encountered in increasing our manufacturing could have a material adverse effect
on our business, financial condition and results of operations.
The manufacture of our products is a complex and costly operation involving a number of
separate processes and components. Any shipment delays could harm perception of our products and
have a material adverse impact on our results of operations.
In addition, the current unit costs for our products, based on limited manufacturing volumes,
are very high, and it will be necessary to achieve economies of scale to become profitable. Certain
of our manufacturing processes are labor intensive, and achieving significant cost reductions will
depend in part upon reducing the time required to complete these processes. We cannot assure you
that we will be able to achieve cost reductions in the manufacture of our products and, without
these cost reductions, our business may never achieve profitability.
We have considered, and will continue to consider as appropriate, manufacturing in-house
certain components currently provided by third parties, as well as implementing new production
processes. Manufacturing yields or costs may be adversely affected by the transition to in-house
production or to new production processes, when and if these efforts are undertaken, which would
materially and adversely affect our business, financial condition and results of operations.
If we fail to retain key personnel, or to retain our executive management team, we may be unable to
successfully develop or commercialize our products.
Our business and future operating results depend significantly on the continued contributions
of our key technical personnel and senior management, including those of our co-founder, CEO and
President, Bernard Hausen, M.D., Ph.D. These services and individuals would be difficult or
impossible to replace and none of these individuals is subject to a post-employment non-competition
agreement. While we are subject to certain severance obligations to Dr. Hausen, either he or we may
terminate his employment at any time and for any lawful reason or for no reason. Additionally,
although we have key-person life insurance in the amount of $3.0 million on the life of Dr. Hausen,
we cannot assure you that this amount would fully compensate us for the loss of Dr. Hausen’s
services. The loss of key employees, the failure of any key employee to perform or our inability to
attract and retain skilled employees, as needed, could materially adversely affect our business,
financial condition and results of operations.
As of June 30, 2010, we had 34 employees. Our business and future operating results depend
significantly on our ability to attract and retain qualified management, manufacturing, technical,
marketing, sales and support personnel for our operations. Competition for such personnel is
intense, and there can be no assurance that we will be successful in attracting or retaining such
personnel. We will need to maintain an appropriate level of managerial, operational, financial and
other resources to manage and fund our operations and clinical trials, continue our research and
development activities and commercialize our products, and we expect our recent reductions in force
will impair our ability to maintain or increase our product sales. It is possible that our
management and scientific personnel, systems and facilities currently in place may not be adequate
to maintain future operating activities, and we may be required to effect additional reductions in
force.
35
We may in the future be a party to patent litigation and administrative proceedings that could be
costly and could interfere with our ability to sell our products.
The medical device industry has been characterized by extensive litigation regarding patents
and other intellectual property rights, and companies in the industry have used intellectual
property litigation to gain a competitive advantage. We may become a party to patent infringement
claims and litigation or interference proceedings declared by the U.S. Patent and Trademark Office
to determine the priority of inventions. The defense and prosecution of these matters are both
costly and time consuming. Additionally, we may need to commence proceedings against others to
enforce our patents, to protect our trade secrets or know-how or to determine the enforceability,
scope and validity of the proprietary rights of others. These proceedings would result in
substantial expense to us and significant diversion of effort by our technical and management
personnel.
We are aware of patents issued to third parties that contain subject matter related to our
technology. We cannot assure you that these or other third parties will not assert that our
products and systems infringe the claims in their patents or seek to expand their patent claims to
cover aspects of our products and systems. An adverse determination in litigation or interference
proceedings to which we may become a party could subject us to significant liabilities or require
us to seek licenses. In addition, if we are found to willfully infringe third-party patents, we
could be required to pay treble damages in addition to other penalties. Although patent and
intellectual property disputes in the medical device area have often been settled through licensing
or similar arrangements, costs associated with these arrangements may be substantial and could
include ongoing royalties. We may be unable to obtain necessary licenses on satisfactory terms, if
at all. If we do not obtain necessary licenses, we may be required to redesign our products to
avoid infringement, and it may not be possible to do so effectively. Adverse determinations in a
judicial or administrative proceeding or failure to obtain necessary licenses could prevent us from
manufacturing and selling the C-Port or PAS-Port systems or any other product we may develop, which
would have a significant adverse impact on our business.
Intellectual property rights may not provide adequate protection, which may permit third parties to
compete against us more effectively.
We rely upon patents, trade secret laws and confidentiality agreements to protect our
technology and products. Our pending patent applications may not issue as patents or, if issued,
may not issue in a form that will be advantageous to us. Any patents we have obtained or will
obtain in the future might be invalidated or circumvented by third parties. If any challenges are
successful, competitors might be able to market products and use manufacturing processes that are
substantially similar to ours. We may not be able to prevent the unauthorized disclosure or use of
our technical knowledge or other trade secrets by consultants, vendors or former or current
employees, despite the existence generally of confidentiality agreements and other contractual
restrictions. Monitoring unauthorized use and disclosure of our intellectual property is difficult,
and we do not know whether the steps we have taken to protect our intellectual property will be
adequate. In addition, the laws of many foreign countries may not protect our intellectual property
rights to the same extent as the laws of the United States. To the extent that our intellectual
property protection is inadequate, we are exposed to a greater risk of direct competition. In
addition, competitors could purchase any of our products and attempt to replicate some or all of
the competitive advantages we derive from our development efforts or design around our protected
technology. If our intellectual property is not adequately protected against competitors’ products
and methods, our competitive position could be adversely affected, as could our business.
We also rely upon trade secrets, technical know-how and continuing technological innovation to
develop and maintain our competitive position. We require our employees, consultants and advisors
to execute appropriate confidentiality and assignment-of-inventions agreements with us. These
agreements typically provide that all materials and confidential information developed or made
known to the individual during the course of the individual’s relationship with us be kept
confidential and not disclosed to third parties except in specific circumstances and that all
inventions arising out of the individual’s relationship with us shall be our exclusive property.
These agreements may be breached, and in some instances, we may not have an appropriate remedy
available for breach of the agreements. Furthermore, our competitors may independently develop
substantially equivalent proprietary information and techniques, reverse engineer our information
and techniques, or otherwise gain access to our proprietary technology.
36
Our products face the risk of technological obsolescence, which, if realized, could have a material
adverse effect on our business.
The medical device industry is characterized by rapid and significant technological change.
There can be no assurance that third parties will not succeed in developing or marketing
technologies and products that are more effective than ours or that would render our technology and
products obsolete or noncompetitive. Additionally, new, less invasive surgical procedures and
medications could be developed that replace or reduce the importance of current procedures that use
or could use our products. Accordingly, our success will depend in part upon our ability to respond
quickly to medical and technological changes through the development and introduction of new
products. The relative speed with which we can develop products, complete clinical testing and
regulatory clearance or approval processes, train physicians in the use of our products, gain
reimbursement acceptance, and supply commercial quantities of products to the market are expected
to be important competitive factors. Product development involves a high degree of risk, and we
cannot assure you that our new product development efforts will result in any commercially
successful products. We have experienced delays in completing the development and commercialization
of our planned products, and there can be no assurance that these delays will not continue or recur
in the future. Any delays could result in a loss of market acceptance and market share.
We may be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws and
regulations and, if we are unable to fully comply with such laws, could face substantial penalties.
Our operations may be directly or indirectly affected by various broad state and federal
healthcare fraud and abuse laws, including the federal healthcare program Anti-Kickback Statute,
which prohibits any person from knowingly and willfully offering, paying, soliciting or receiving
remuneration, directly or indirectly, to induce or reward either the referral of an individual, or
the furnishing or arranging for an item or service, for which payment may be made under federal
healthcare programs, such as the Medicare and Medicaid programs. Foreign sales of our products are
also subject to similar fraud and abuse laws, including application of the U.S. Foreign Corrupt
Practices Act. If our operations, including any consulting arrangements we may enter into with
physicians who use our products, are found to be in violation of these laws, we or our officers may
be subject to civil or criminal penalties, including large monetary penalties, damages, fines,
imprisonment and exclusion from Medicare and Medicaid program participation. If enforcement action
were to occur, our business and financial condition would be harmed.
We could be exposed to significant product liability claims, which could be time consuming and
costly to defend, divert management attention, and adversely impact our ability to obtain and
maintain insurance coverage. The expense and potential unavailability of insurance coverage for our
company or our customers could adversely affect our ability to sell our products, which would
adversely affect our business.
The testing, manufacture, marketing, and sale of our products involve an inherent risk that
product liability claims will be asserted against us. Additionally, we are currently training
physicians in the United States on the use of our C-Port and PAS-Port systems. During training,
patients may be harmed, which could also lead to product liability claims. Product liability claims
or other claims related to our products, or their off-label use, regardless of their merits or
outcomes, could harm our reputation in the industry, reduce our product sales, lead to significant
legal fees, and result in the diversion of management’s attention from managing our business. As of
August 31, 2010, we were not aware of any existing product liability claims.
Although we maintain product liability insurance in the amount of $5,000,000, we may not have
sufficient insurance coverage to fully cover the costs of any claim or any ultimate damages we
might be required to pay. We may not be able to obtain insurance in amounts or scope sufficient to
provide us with adequate coverage against all potential liabilities. Any product liability claims
brought against us, with or without merit, could increase our product liability insurance rates or
prevent us from securing continuing coverage. Product liability claims in excess of our insurance
coverage would be paid out of cash reserves, harming our financial condition and adversely
affecting our operating results.
Some of our customers and prospective customers may have difficulty in procuring or
maintaining liability insurance to cover their operations and use of the C-Port or PAS-Port
systems. Medical malpractice carriers are withdrawing coverage in certain states or substantially
increasing premiums. If this trend continues or worsens, our
customers may discontinue using the C-Port or PAS-Port systems and potential customers may opt
against purchasing the C-Port or PAS-Port systems due to the cost or inability to procure insurance
coverage.
37
We sell our systems internationally and are subject to various risks relating to these
international activities, which could adversely affect our revenue.
To date, a substantial portion of our product sales has been attributable to sales in
international markets. By doing business in international markets, we are exposed to risks separate
and distinct from those we face in our domestic operations. Our international business may be
adversely affected by changing economic conditions in foreign countries. Because most of our sales
are currently denominated in U.S. dollars, if the value of the U.S. dollar increases relative to
foreign currencies, our products could become more costly to the international customer and,
therefore, less competitive in international markets, which could affect our results of operations.
Engaging in international business inherently involves a number of other difficulties and risks,
including:
| |
• |
|
export restrictions and controls relating to technology; |
| |
• |
|
the availability and level of reimbursement within prevailing foreign healthcare payment systems; |
| |
• |
|
pricing pressure that we may experience internationally; |
| |
• |
|
required compliance with existing and changing foreign regulatory requirements and laws; |
| |
• |
|
laws and business practices favoring local companies; |
| |
• |
|
difficulties in enforcing agreements and collecting receivables through certain foreign legal systems; |
| |
• |
|
political and economic instability; |
| |
• |
|
potentially adverse tax consequences, tariffs and other trade barriers; |
| |
• |
|
international terrorism and anti-American sentiment; |
| |
• |
|
difficulties and costs of staffing and managing foreign operations; and |
| |
• |
|
difficulties in enforcing intellectual property rights. |
Our exposure to each of these risks may increase our costs, impair our ability to market and
sell our products and require significant management attention. We cannot assure you that one or
more of these factors will not harm our business.
Our operations are currently conducted at a single location that may be at risk from earthquakes,
terror attacks or other disasters.
We currently conduct all of our manufacturing, development and management activities at a
single location in Redwood City, California, near known earthquake fault zones. We have taken
precautions to safeguard our facilities, including insurance, health and safety protocols, and
off-site storage of computer data. However, any future natural disaster, such as an earthquake, or
a terrorist attack, could cause substantial delays in our operations, damage or destroy our
equipment or inventory and cause us to incur additional expenses. A disaster could seriously harm
our business and results of operations. Our insurance does not cover earthquakes and floods and may
not be adequate to cover our losses in any particular case.
38
If we use hazardous materials in a manner that causes injury, we may be liable for damages.
Our research and development and manufacturing activities involve the use of hazardous
materials. Although we believe that our safety procedures for handling and disposing of these
materials comply with federal, state and local laws and regulations, we cannot entirely eliminate
the risk of accidental injury or contamination from the use, storage, handling or disposal of these
materials. We do not carry specific hazardous waste insurance coverage, and our property and
casualty and general liability insurance policies specifically exclude coverage for damages and
fines arising from hazardous waste exposure or contamination. Accordingly, in the event of
contamination or injury, we could be held liable for damages or penalized with fines in an amount
exceeding our resources, and our clinical trials or regulatory clearances or approvals could be
suspended or terminated.
We may be subject to fines, penalties or injunctions if we are determined to be promoting the use
of our products for unapproved or “off-label” uses.
In relation to our products that have received FDA clearance or approval, our promotional
materials and training methods regarding physicians need to comply with FDA and other applicable
laws and regulations. If the FDA determines that our promotional materials or training constitutes
promotion of an unapproved use, it could request that we modify our training or promotional
materials or subject us to regulatory enforcement actions, including the issuance of a warning
letter, injunction, seizure, civil fine and/or criminal penalties. It is also possible that other
federal, state or foreign enforcement authorities might take action if they consider our
promotional or training materials to constitute promotion of an unapproved use, which could result
in significant fines or penalties under other statutory authorities, such as laws prohibiting false
claims for reimbursement. In that event, our reputation could be damaged and adoption of our
products would be impaired.
Risks Related to Our Common Stock
We may not be able to maintain our listing on The NASDAQ Global Market, which would adversely
affect the price and liquidity of our common stock.
On June 25, 2010, we announced that we received a letter, dated June 21, 2010, from the
Listing Qualifications Department of The NASDAQ Stock Market notifying us that we did not comply
with the $50.0 million minimum market capitalization for continued listing on The NASDAQ Global
Market set forth in NASDAQ Marketplace Rule 5450(b)(2)(A) or the Minimum Market Capitalization
Standard. The NASDAQ Marketplace rules provide the Company with a grace period of 180 days, or
until December 20, 2010 to regain compliance with the listing standards. On September 16, 2010, we
received a letter from the Listing Qualifications Department of The NASDAQ Stock Market notifying
us that we had regained compliance with the Minimum Market Capitalization Standard. However, even
though we regained compliance with the listing requirements of The NASDAQ Global Market, there is
no assurance that in the future we will continue to satisfy such listing requirements, with the
result that our common stock may be delisted from that market.
If our stock is delisted from The NASDAQ Global Market, we may still meet the listing
requirements for the NASDAQ Capital Market, including the requirement to have a minimum of $1.0
million in stockholders’ equity. If we are unable to list on The NASDAQ Capital Market, it would
likely be more difficult to trade in or obtain accurate quotations as to the market price of our
common stock. Delisting of our common stock would materially and adversely affect the market price
and market liquidity of our common stock and our ability to raise necessary capital.
The price of our common stock may continue to be volatile, and the value of an investment in our
common stock may decline.
An active and liquid trading market for our common stock may not develop or be sustained.
Factors that could cause volatility in the market price of our common stock include, but are not
limited to:
| |
• |
|
completion of development of our microcutter products, and the timing thereof; |
| |
• |
|
market acceptance and adoption of our products; |
39
| |
• |
|
regulatory clearance or approvals of our products; |
| |
• |
|
volume and timing of orders for our products; |
| |
• |
|
changes in earnings estimates, investors’ perceptions, recommendations by securities
analysts or our failure to achieve analysts’ earning estimates; |
| |
• |
|
quarterly variations in our or our competitors’ results of operations; |
| |
• |
|
general market conditions and other factors unrelated to our operating performance or
the operating performance of our competitors; |
| |
• |
|
the announcement of new products or product enhancements by us or our competitors; |
| |
• |
|
announcements related to patents issued to us or our competitors and to litigation; and |
| |
• |
|
developments in our industry. |
In addition, the stock prices of many companies in the medical device industry have
experienced wide fluctuations that have often been unrelated to the operating performance of those
companies. These factors may materially and adversely affect the market price of our common stock.
The ownership of our common stock is highly concentrated, and your interests may conflict with the
interests of our existing stockholders.
Our executive officers and directors and their affiliates, together with other stockholders
that own 5% or more of our outstanding common stock, beneficially owned approximately 36% of our
outstanding common stock as of June 30, 2010. Accordingly, these stockholders have significant
influence over the outcome of corporate actions requiring stockholder approval and continue to have
significant influence over our operations. The interests of these stockholders may be different
than the interests of other stockholders on these matters. This concentration of ownership could
also have the effect of delaying or preventing a change in our control or otherwise discouraging a
potential acquirer from attempting to obtain control of us, which in turn could reduce the price of
our common stock.
Evolving regulation of corporate governance and public disclosure will result in additional
expenses and continuing uncertainty.
Changing laws, regulations and standards relating to corporate governance and public
disclosure, including the Sarbanes-Oxley Act of 2002, new SEC regulations and The Nasdaq Stock
Market rules are creating uncertainty for public companies. We are presently evaluating and
monitoring developments with respect to new and proposed rules and cannot predict or estimate the
amount of the additional compliance costs we may incur or the timing of such costs. These new or
changed laws, regulations and standards are subject to varying interpretations, in many cases due
to their lack of specificity, and as a result, their application in practice may evolve over time
as new guidance is provided by courts and regulatory and governing bodies. This could result in
continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing
revisions to disclosure and governance practices. Maintaining appropriate standards of corporate
governance and public disclosure will result in increased general and administrative expenses and a
diversion of management time and attention from product-generating and revenue-generating
activities to compliance activities. In addition, if we fail to comply with new or changed laws,
regulations and standards, regulatory authorities may initiate legal proceedings against us and our
business and reputation may be harmed.
40
Our future operating results may be below securities analysts’ or investors’ expectations, which
could cause our stock price to decline.
The revenue and income potential of our products and our business model are unproven, and we
may be unable to generate significant revenue or grow at the rate expected by securities analysts
or investors. In addition, our costs
may be higher than we, securities analysts or investors expect. If we fail to generate
sufficient revenue or our costs are higher than we expect, our results of operations will suffer,
which in turn could cause our stock price to decline. Our results of operations will depend upon
numerous factors, including:
| |
• |
|
completion of development of our microcutter products, and the timing thereof; |
| |
• |
|
FDA or other regulatory clearance or approval of our products; |
| |
• |
|
demand for our products; |
| |
• |
|
the performance of third-party contract manufacturers and component suppliers; |
| |
• |
|
our ability to develop sales and marketing capabilities; |
| |
• |
|
our ability to develop, introduce and market new or enhanced versions of our
products on a timely basis; and |
| |
• |
|
our ability to obtain and protect proprietary rights. |
Our operating results in any particular period may not be a reliable indication of our future
performance. In some future quarters, our operating results may be below the expectations of
securities analysts or investors. If this occurs, the price of our common stock will likely
decline.
Anti-takeover defenses that we have in place could prevent or frustrate attempts to change our
direction or management.
Provisions of our certificate of incorporation and bylaws and applicable provisions of
Delaware law may make it more difficult for or prevent a third party from acquiring control of us
without the approval of our board of directors. These provisions:
| |
• |
|
limit who may call a special meeting of stockholders; |
| |
• |
|
establish advance notice requirements for nominations for election to
our board of directors or for proposing matters that can be acted upon
at stockholder meetings; |
| |
• |
|
prohibit cumulative voting in the election of our directors, which
would otherwise permit less than a majority of stockholders to elect
directors; |
| |
• |
|
prohibit stockholder action by written consent, thereby requiring all
stockholder actions to be taken at a meeting of our stockholders; and |
| |
• |
|
provide our board of directors with the ability to designate the terms
of and issue a new series of preferred stock without stockholder
approval. |
In addition, Section 203 of the Delaware General Corporation Law generally prohibits us from
engaging in any business combination with certain persons who own 15% or more of our outstanding
voting stock or any of our associates or affiliates who at any time in the past three years have
owned 15% or more of our outstanding voting stock. These provisions may have the effect of
entrenching our management team and may deprive you of the opportunity to sell your shares to
potential acquirors at a premium over prevailing prices. This potential inability to obtain a
control premium could reduce the price of our common stock.
41
We may become involved in securities class action litigation that could divert management’s
attention and harm our business.
The stock market in general, the Nasdaq Global Market and the market for medical device
companies in particular, has experienced extreme price and volume fluctuations that have often been
unrelated or disproportionate to the operating performance of those companies. Further, the market
prices of securities of medical device companies have been particularly volatile. These broad
market and industry factors may materially harm the market price of our common stock, regardless of
our operating performance. In the past, following periods of volatility in the market price of a
particular company’s securities, securities class action litigation has often been brought against
that company. We may become involved in this type of litigation in the future. Litigation often is
expensive and diverts management’s attention and resources, which could materially harm our
financial condition and results of operations.
We have never paid dividends on our capital stock, and we do not anticipate paying any cash
dividends in the foreseeable future.
We have paid no cash dividends on any of our classes of capital stock to date, and we
currently intend to retain our future earnings to fund the development and growth of our business.
As a result, capital appreciation, if any, of our common stock will be the sole source of gain to
our stockholders for the foreseeable future.
Item 1B. Unresolved Staff Comments.
None.
Item 2. Properties
We currently lease approximately 30,000 square feet of office, manufacturing and laboratory
space in Redwood City, California. We believe that our existing facility should meet our needs for
at least the next 24 months. Our facility is subject to periodic inspections by state and federal
regulatory authorities.
Item 3. Legal Proceedings
We are not subject to any material legal proceeding.
Item 4. (Removed and Reserved)
42
PART II
|
|
|
| Item 5. |
|
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market for Common Equity
Our common stock began trading on the NASDAQ Global Market on February 3, 2006 under the
symbol “CRDC”. The table below sets forth the high and low intraday sales prices for our common
stock for the periods indicated:
| |
|
|
|
|
|
|
|
|
| |
|
High |
|
|
Low |
|
Fiscal year 2010 |
|
|
|
|
|
|
|
|
First Quarter ended September 30, 2009 |
|
$ |
3.19 |
|
|
$ |
1.15 |
|
Second Quarter ended December 31, 2009 |
|
$ |
1.63 |
|
|
$ |
1.00 |
|
Third Quarter ended March 31, 2010 |
|
$ |
2.44 |
|
|
$ |
1.16 |
|
Fourth Quarter ended June 30, 2010 |
|
$ |
2.85 |
|
|
$ |
1.41 |
|
|
|
|
|
|
|
|
|
|
Fiscal year 2009 |
|
|
|
|
|
|
|
|
First Quarter ended September 30, 2008 |
|
$ |
11.13 |
|
|
$ |
7.14 |
|
Second Quarter ended December 31, 2008 |
|
$ |
8.28 |
|
|
$ |
2.50 |
|
Third Quarter ended March 31, 2009 |
|
$ |
4.27 |
|
|
$ |
2.25 |
|
Fourth Quarter ended June 30, 2009 |
|
$ |
3.24 |
|
|
$ |
1.03 |
|
As of September 9, 2010, there were 108 holders of record of common stock. This number does
not include the number of persons whose shares are held by a nominee or in “street name” accounts
through brokers.
Dividend Policy
We have never declared or paid any dividends on our capital stock. We currently intend to
retain all of our future earnings, if any, to finance our operations and do not anticipate paying
any cash dividends on our capital stock in the foreseeable future.
Recent Sales of Unregistered Securities
On September 30, 2009, institutional and individual investors, including existing
stockholders, purchased approximately $10.2 million of the Company’s common stock and warrants to
purchase the Company’s common stock in a private placement (the “Private Placement”). The net
proceeds were approximately $9.9 million after offering expenses. Under the terms of the purchase
agreement with these investors, the Company sold 8,142,082 units at a purchase price of $1.2525 per
unit, with each unit consisting of one share of common stock and one warrant to purchase 0.50 of a
share of common stock, or 4,071,046 shares of the Company’s common stock. The warrants are
exercisable commencing on April 1, 2010 at $1.45 per share and will expire five years after the
date of issuance. There were no underwriters or placement agents involved with the Private
Placement, and no underwriting discounts or commissions or similar fees were payable in connection
with the Private Placement. The issuance was made in reliance upon exemptions from registration
pursuant to Section 4(2) under the Securities Act of 1933, as amended, (the “Securities Act”) and
Rule 506 promulgated thereunder, and was made without general solicitation or advertising. Each
investor represented that it was an accredited investor with access to information about us
sufficient to evaluate the investment and that the common stock and warrants were being acquired
without a view to distribution or resale in violation of the Securities Act.
On
August 16, 2010, we entered into a Stock Purchase Agreement with
Intuitive Surgical pursuant to
which Intuitive Surgical paid $3 million to purchase from us an aggregate of 1,249,541 newly-issued shares
of our common stock (the “Stock Issuance”). There were no underwriters or placement agents involved
with the Stock Issuance, and no underwriting discounts or commissions or similar fees were payable
in connection with the Stock Issuance. The shares were issued to Intuitive Surgical in reliance upon
exemptions from registration pursuant to Section 4(2) under the Securities Act
and Rule 506 promulgated thereunder, and was made without general solicitation or advertising.
Intuitive Surgical represented that it was an accredited investor with access to information about
us sufficient to evaluate the investment and that the common stock was being acquired without a
view to distribution or resale in violation of the Securities Act.
43
Issuer Purchases of Equity Securities
During the quarter ended June 30, 2010 we did not repurchase any equity securities.
Item 6. Selected Financial Data
The following selected financial data should be read in conjunction with “Management’s
Discussion and Analysis of Financial Condition and Results of Operations” and our financial
statements and notes to those statements included elsewhere in this report.
The following selected balance sheet data as of June 30, 2010 and 2009 and the statements of
operations data for each of the three fiscal years in the period ended June 30, 2010 have been
derived from our audited financial statements, which are included elsewhere in this annual report.
The selected balance sheet data as of June 30, 2008, 2007 and 2006 and the selected statements of
operations data for the fiscal years ended June 30, 2007 and 2006 have been derived from our
audited financial statements not included in this annual report. Historical results are not
necessarily indicative of the results to be expected in future periods.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Fiscal Year Ended June 30, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
|
2007 |
|
|
2006 |
|
| |
|
(in thousands, except per share data) |
|
Statements of Operations Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net revenue: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product sales, net |
|
$ |
3,764 |
|
|
$ |
6,798 |
|
|
$ |
4,934 |
|
|
$ |
2,103 |
|
|
$ |
1,028 |
|
Development revenue |
|
|
124 |
|
|
|
2,995 |
|
|
|
2,564 |
|
|
|
1,370 |
|
|
|
1,000 |
|
Royalty revenue
(includes amounts from
related party
of $67, $56 and$31 in
fiscal years 2008, 2007
and 2006, respectively) |
|
|
93 |
|
|
|
85 |
|
|
|
67 |
|
|
|
56 |
|
|
|
31 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total net revenue |
|
|
3,981 |
|
|
|
9,878 |
|
|
|
7,565 |
|
|
|
3,529 |
|
|
|
2,059 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating costs and expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cost of product sales |
|
|
3,687 |
|
|
|
5,341 |
|
|
|
4,808 |
|
|
|
2,880 |
|
|
|
2,102 |
|
Research and development |
|
|
5,437 |
|
|
|
8,217 |
|
|
|
8,609 |
|
|
|
7,014 |
|
|
|
6,459 |
|
Selling, general and
administrative |
|
|
5,734 |
|
|
|
13,632 |
|
|
|
13,175 |
|
|
|
9,057 |
|
|
|
5,645 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating costs and
expenses |
|
|
14,858 |
|
|
|
27,190 |
|
|
|
26,592 |
|
|
|
18,951 |
|
|
|
14,206 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations |
|
|
(10,877 |
) |
|
|
(17,312 |
) |
|
|
(19,027 |
) |
|
|
(15,422 |
) |
|
|
(12,147 |
) |
Interest income |
|
|
35 |
|
|
|
177 |
|
|
|
926 |
|
|
|
1,113 |
|
|
|
782 |
|
44
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Fiscal Year Ended June 30, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
|
2007 |
|
|
2006 |
|
| |
|
(in thousands, except per share data) |
|
Interest expense (includes
related-party interest expense
of $320 and $897 in fiscal
years 2007 and 2006,
respectively) |
|
|
(112 |
) |
|
|
(120 |
) |
|
|
(101 |
) |
|
|
(458 |
) |
|
|
(1,047 |
) |
Other income (expense), net |
|
|
(1 |
) |
|
|
(22 |
) |
|
|
6 |
|
|
|
2 |
|
|
|
(4 |
) |
Gain on early retirement of
notes payable to related-party |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,183 |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss before income tax
benefit |
|
|
(10,955 |
) |
|
|
(17,277 |
) |
|
|
(18,196 |
) |
|
|
(13,582 |
) |
|
|
(12,416 |
) |
Income tax benefit |
|
|
31 |
|
|
|
72 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(10,924 |
) |
|
$ |
(17,205 |
) |
|
$ |
(18,196 |
) |
|
$ |
(13,582 |
) |
|
$ |
(12,416 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per
common share |
|
$ |
(0.50 |
) |
|
$ |
(1.09 |
) |
|
$ |
(1.23 |
) |
|
$ |
(1.25 |
) |
|
$ |
(2.58 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Shares used in computing basic
and diluted net loss per
common share |
|
|
21,927 |
|
|
|
15,776 |
|
|
|
14,844 |
|
|
|
10,878 |
|
|
|
4,817 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
As of June 30, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
|
2007 |
|
|
2006 |
|
Balance Sheet Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash, cash equivalents and
short-term investments |
|
$ |
6,561 |
|
|
$ |
5,328 |
|
|
$ |
23,265 |
|
|
$ |
23,434 |
|
|
$ |
32,080 |
|
Working capital |
|
|
5,016 |
|
|
|
4,134 |
|
|
|
20,959 |
|
|
|
22,049 |
|
|
|
31,602 |
|
Total assets |
|
|
9,791 |
|
|
|
10,340 |
|
|
|
28,250 |
|
|
|
27,324 |
|
|
|
35,158 |
|
Short-term note payable |
|
|
1,400 |
|
|
|
2,000 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Long-term liabilities |
|
|
31 |
|
|
|
44 |
|
|
|
2,000 |
|
|
|
2,020 |
|
|
|
15,836 |
|
Total stockholders’ equity |
|
|
6,477 |
|
|
|
6,262 |
|
|
|
21,417 |
|
|
|
21,989 |
|
|
|
17,677 |
|
45
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion and analysis of our financial condition and results of operations should
be read in conjunction with our financial statements and the related notes to those statements
included elsewhere in this report. In addition to historical financial information, the following
discussion and analysis contains forward-looking statements that involve risks, uncertainties and
assumptions. Our actual results and timing of selected events may differ materially from those
anticipated in these forward-looking statements as a result of many factors, including those
discussed under “Risk Factors” and elsewhere in this Report.
Overview
Historically, our business focused on the design, manufacture and marketing of proprietary
automated anastomotic systems used by cardiac surgeons to perform coronary bypass surgery. We have
re-focused our business on the development of an endoscopic microcutter product line intended for
use by general, thoracic, gynecologic, bariatric and urologic surgeons. Unless and until a
microcutter product is developed and cleared for marketing in the United States or elsewhere, or we
enter into an arrangement with a development and commercialization partner that provides us with
development revenue, we will have ongoing costs related to the development of this potential
product line without related revenue.
Our C-Port® Distal Anastomosis Systems, or C-Port systems, are sold in the United
States and Europe. The C-Port systems are used to perform a distal anastomosis, which is the
connection between a bypass graft vessel and the target artery. As of June 30, 2010, more than
10,600 C-Port systems have been sold in the United States and Europe. We also currently sell our
PAS-Port® Proximal Anastomosis System, or PAS-Port system, in the United States, Europe
and Japan. The PAS-Port system is used to perform a proximal anastomosis, which is the connection
of a bypass graft vessel to the aorta or other source of blood. As of June 30, 2010, more than
19,500 PAS-Port systems had been sold in the United States, Europe and Japan. In addition to our
commercialized cardiac surgery products, we are developing the Cardica Microcutter ES8, a
multi-fire endolinear microcutter device based on our proprietary “staple-on-a-strip” technology,
which would expand our commercial opportunity into additional surgical markets.
We use independent distributors and manufacturers’ representatives to support a small core
direct sales team for our C-Port systems and PAS-Port system in the United States to contain sales
costs while continuing to serve our customers and potential customers for our automated anastomosis
product line. We have shifted our development efforts to focus on the Cardica Microcutter ES8 and
other potential products in this anticipated product line.
We manufacture our C-Port and PAS-Port systems with parts we manufacture and components
supplied by vendors, which we then assemble, test and package. For fiscal year 2010, we generated
net revenue of $4.0 million, including $124,000 of development revenue, and incurred a net loss of
$10.9 million.
Since our inception, we have incurred significant net losses, and we expect to continue to
incur net losses for the foreseeable future. To date, our C-Port and PAS-Port systems have had
limited commercial adoption, and sales have not met the levels that we had anticipated. Revenues
from product sales and milestone payments were not sufficient to support the operation of our
business as we had planned. If revenue from product sales does not increase, we may be required to
delay, further reduce the scope of or eliminate our commercialization efforts with respect to one
or more of our products or one or more of our research and development programs.
On August 16, 2010, we entered into a License Agreement with Intuitive Surgical pursuant to
which we granted to Intuitive Surgical a worldwide, sublicenseable, exclusive license to use our
intellectual property in the robotics field in diagnostic or therapeutic medical procedures, but
excluding vascular anastomosis applications (the “License Agreement”) for an upfront license fee of
$9 million. We will also be eligible to receive a milestone payment if sales of any products
incorporating our patent rights achieve a specified level of net sales within a specified period
after the date of the License Agreement and will also be eligible to receive single-digit royalties
on sales by Intuitive Surgical, its affiliates or its sublicensees of specified stapler and clip
applier products covered by our patent rights as well as on sales of certain other products covered
by our patent rights that may be developed in the future.
46
In
addition, on the same date, we entered into a Stock Purchase
Agreement with Intuitive Surgical
pursuant to which Intuitive Surgical paid $3 million to purchase from us an aggregate of 1,249,541
newly-issued shares of our common stock (the “Stock Issuance”).
On April 1, 2010, we entered into an amendment, or Note Agreement Amendment, to our
subordinated convertible note agreement, dated June 16, 2003 and as amended to date, with Century
Medical, Inc., or Century Medical, our distributor in Japan (referred to as the Note Agreement).
Under the terms of the Note Agreement Amendment, we made a principal payment of $600,000 to Century
Medical in April 2010, with the remaining $1.4 million principal amount owed to Century Medical
becoming due on June 17, 2011, which is one year later than the maturity date prior to the Note
Agreement Amendment. On August 17, 2010, we repaid the remaining $1.4 million principal balance and
interest due on the debt facility.
As of June 30, 2010, we had cash and cash equivalents of $6.6 million and total short-term
debt of $1.4 million. Including the cash received in August 2010 in connection with the License
Agreement with Intuitive Surgical and related equity investment, we believe that our
existing cash and cash equivalents, along with the cash that we expect to generate from operations,
will be sufficient to meet our anticipated cash needs to enable us to conduct our business
substantially as currently conducted through September 30, 2011. While our cash resources would
permit us to continue through September 30, 2011, we would need to further reduce expenses in
advance of that date in the event that we are unable to complete a financing, strategic or
commercial transaction to ensure that we have sufficient capital to meet our obligations and
continue on a path designed to create and preserve stockholder value. The sufficiency of our
current cash resources and our need for additional capital, and the timing thereof, will depend on
many factors, including primarily our ability to meet our microcutter product development
objectives and milestones, obtaining regulatory clearance for the Cardica Microcutter ES8 and
other microcutter products, the level of market acceptance of our microcutter products, the extent
of our sales and marketing efforts related to our products and the amount of revenue that we
receive from product sales, as well as other factors described in the “Liquidity and Capital
Resources” section below.
We may seek to sell additional equity or debt securities, obtain a credit facility, enter into
product development, license or distribution agreements with third parties or divest one or more of
our commercialized products or products in development. The sale of additional equity or
convertible debt securities could result in significant dilution to our stockholders, particularly
in light of the prices at which our common stock has been recently trading. In addition, if we
raise additional funds through the sale of equity securities, new investors could have rights
superior to our existing stockholders. If additional funds are raised through the issuance of debt
securities, these securities could have rights senior to those associated with our common stock and
could contain covenants that would restrict our operations. Any product development, licensing,
distribution or sale agreements that we enter into may require us to relinquish valuable rights,
including with respect to commercialized products or products in development that we would
otherwise seek to commercialize or develop ourselves. We may not be able to obtain sufficient
additional financing or enter into a strategic transaction in a timely manner. Our need to raise
capital may require us to accept terms that may harm our business or be disadvantageous to our
current stockholders.
Agreements with Cook Incorporated
In June 2007, we entered into, and in September 2007 and in June 2009 amended, a license,
development and commercialization agreement with Cook Incorporated, or Cook, to develop and
commercialize a specialized device, referred to as the PFO device, designed to close holes in the
heart from genetic heart defects known as patent foramen ovales, or PFOs. Under the agreement, Cook
funded certain development activities and we and Cook jointly developed the device. If developed,
Cook would receive an exclusive, worldwide, royalty-bearing license, with the right to grant
sublicenses, to make, have made, use, sell, offer for sale and import the PFO device. Under this
agreement, we received payments totaling $0, $1.0 million and $1.7 million in fiscal years ended
June 30, 2010, 2009 and 2008, respectively. We recorded as development revenue under the agreement
a total of $124,000, $1.4 million and $1.2 million in fiscal years ended June 30, 2010, 2009 and
2008, respectively. A total of $403,000 under this agreement has been recorded as deferred
development revenue on the balance sheet as of June 30, 2010. We are also entitled to receive from
Cook up to a total of an additional $275,000 in future payments if development milestones under the
agreement are achieved. Amounts paid but not yet earned on the project are recorded as deferred
revenue until such time as the related development expenses for certain project activities are
incurred. We
would also be entitled to receive a royalty based on Cook’s annual worldwide sales of the PFO
device, if any. On January 6, 2010, we and Cook mutually agreed to suspend work on the PFO project
and, accordingly, we do not anticipate receiving any additional payments or recording any
additional revenue related to this agreement in the foreseeable future.
47
On December 9, 2005, we entered into, and in September 2007 amended and in July 2009 amended
and partially terminated, an agreement with Cook to develop the Cook Vascular Closure Device. Under
the agreement, Cook funded certain development activities, and we and Cook jointly developed the
device, under the direction of a Development Committee that included representatives from each
party. Under the original agreement and the first amendment in September 2007, Cook received an
exclusive, worldwide, royalty-bearing license, with the right to grant sublicenses, to make, have
made, use, sell, offer for sale and import the Cook Vascular Closure Device for medical procedures
in any part of the body. Under this agreement, we received payments totaling approximately $5.3
million, including $0, $1.0 million and $1.5 million in fiscal years ended June 30, 2010, 2009 and
2008, respectively. We recorded as development revenue under the agreement a total of $0, $1.6
million and $1.4 million for fiscal years ended June 30, 2010, 2009 and 2008, respectively. In July
2009, we entered into a partial termination and second amendment of this agreement to terminate
Cook’s participation in the project and to provide to Cook a royalty on net sales of the Cook
Vascular Closure Device if Cardica successfully commercializes the product. The remaining deferred
revenue balance was recognized as revenue in the fourth quarter of fiscal 2009 as we had completed
all of our activities under the agreement and no amounts were refundable to Cook. In addition,
during the fiscal year ended June 30, 2009, we recognized a total of $251,000 of product sales to
Cook of the Cook Vascular Closure Device.
Deficiency letter from The NASDAQ Global Market
On June 25, 2010, we announced that we received a letter, dated June 21, 2010, from the
Listing Qualifications Department of The NASDAQ Stock Market notifying us that we did not comply
with the $50.0 million minimum market capitalization for continued listing on The NASDAQ Global
Market set forth in NASDAQ Marketplace Rule 5450(b)(2)(A) or the Minimum Market Capitalization
Standard. The NASDAQ Marketplace rules provide the Company with a grace period of 180 days, or
until December 20, 2010 to regain compliance with the listing standards. On September 16, 2010, we
received a letter from the Listing Qualifications Department of The NASDAQ Stock Market notifying
us that we had regained compliance with the Minimum Market Capitalization Standard. However, even
though we regained compliance with the listing requirements of The NASDAQ Global Market, there is
no assurance that in the future we will continue to satisfy such listing requirements, with the
result that our common stock may be delisted from that market.
If our stock is delisted from The NASDAQ Global Market, we may still meet the listing
requirements for the NASDAQ Capital Market, including the requirement to have a minimum of $1.0
million in stockholders’ equity. If we are unable to list on The NASDAQ Capital Market, it would
likely be more difficult to trade in or obtain accurate quotations as to the market price of our
common stock. Delisting of our common stock would materially and adversely affect the market price
and market liquidity of our common stock and our ability to raise necessary capital.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of our financial condition and results of operations
are based on our financial statements which have been prepared in accordance with accounting
principles generally accepted in the United States, or GAAP. The preparation of our financial
statements requires management to make estimates and assumptions that affect the amounts reported
in our financial statements and accompanying notes. Actual results could differ materially from
those estimates.
We believe that the following critical accounting policies are the most critical to an
understanding of our financial statements because they require us to make significant judgments and
estimates that are used in the preparation of our financial statements.
48
Revenue Recognition. We recognize revenue when four basic criteria are met: (1) persuasive
evidence of an
arrangement exists; (2) title has transferred; (3) the fee is fixed or determinable; and (4)
collectability is reasonably assured. We generally use contracts and customer purchase orders to
determine the existence of an arrangement. We use shipping documents and third-party proof of
delivery to verify that title has transferred. We assess whether the fee is fixed or determinable
based upon the terms of the agreement associated with the transaction. To determine whether
collection is probable, we assess a number of factors, including past transaction history with the
customer and the creditworthiness of the customer. If we determine that collection is not
reasonably assured, then the recognition of revenue is deferred until collection becomes reasonably
assured, which is generally upon receipt of payment.
We record product sales net of estimated product returns and discounts from the list prices
for our products. The amounts of product returns and the discount amounts have not been material to
date.
Revenue generated from development contracts is recognized when it is earned and
non-refundable upon receipt of milestone payments or upon incurrence of the related development
expenses in accordance with contractual terms, based on the actual costs incurred to date plus
overhead costs for certain project activities. Amounts paid but not yet earned on the project are
recorded as deferred revenue until such time as the related development expenses are incurred.
Inventory. We state our inventories at the lower of cost or market value on a first-in,
first-out basis. Inventory write-downs are established when conditions indicate that the net
realizable value could be less than cost due to physical deterioration, usage, obsolescence,
reductions in estimated future demand or reductions in selling prices. Inventory write-downs are
measured as the difference between the cost of inventory and estimated net realizable value.
Inventory write-downs are charged to cost of product sales and establish a lower cost basis for the
inventory. We balance the need to maintain strategic inventory levels with the risk of obsolescence
due to changing technology and the risk of lower customer demand levels. While we believe the
current value of inventories represents all known and estimated changes in demand, we have
experienced reduced demand for our C-Port systems and further unfavorable changes in market
conditions may result in a need for additional inventory write-downs that could adversely impact
our financial results.
Clinical Trial Accounting. Clinical trial costs are a component of research and development
expenses and include fees paid to participating hospitals and other service providers that conduct
clinical trial activities with patients on our behalf and the cost of clinical trial insurance. The
various costs of the trial are contractually based on the nature of the services, and we accrue the
costs as the services are provided. Accrued costs are based on estimates of the work completed
under the service agreements, patient enrollment and past experience with similar contracts. Our
estimate of the work completed and associated costs to be accrued, includes our assessment of
information received from our third-party service providers and the overall status of our clinical
trial activities. If we have incomplete or inaccurate information, we may underestimate costs
associated with various trials at a given point in time. Although our experience in estimating
these costs is limited, the difference between accrued expenses based on our estimates and actual
expenses have not been material to date.
Stock-Based Compensation. We account for employee and director share-based compensation plans,
including stock options and restricted stock units, or RSUs, pursuant to Accounting Standards
Codification, or ASC, 718 “Compensation — Stock Compensation”. Stock-based compensation cost is
measured on the grant date, based on fair value-based measurement of the award, and is recognized
as an expense over the requisite service period.
The Company selected the Black-Scholes option pricing model for determining the estimated fair
value-based measurements of share-based awards. The use of the Black-Scholes model requires the use
of assumptions including expected term, expected volatility, risk-free interest rate and expected
dividends. The expected term of options granted is determined using the “simplified” method. Under
this approach, the expected term is presumed to be the mid-point between the vesting date and the
end of the contractual term. Since the Company has limited historical data on volatility of its
stock, the expected volatility is based on the volatility of similar entities (referred to as
“guideline” companies). In evaluating similarity, we considered factors such as industry, stage of
life cycle, size, and financial leverage. The risk-free interest rate for the expected term of each
option is based on a risk-free zero-coupon spot interest rate at the time of grant. We have never
declared or paid any cash dividends and do not presently plan to pay cash dividends in the
foreseeable future. We estimate forfeitures in calculating the expense related to stock-based
compensation. We recognize stock-based compensation expense for options and restricted stock
awards using
the accelerated method over the requisite service period of the award, which generally equals
the vesting period of each grant. We recorded fair value-based stock-based compensation expense
under ASC 718 of $1.2 million, or $0.06 per share, $1.6 million, or $0.10 per share, and $1.4
million, or $0.09 per share for fiscal years ended June 30, 2010, 2009 and 2008, respectively.
Total compensation expense related to unvested awards not yet recognized is approximately $0.9
million at June 30, 2010 and is expected to be recognized over a weighted average period of 2.8
years.
49
Prior to the adoption of ASC 718, certain stock options were granted with exercise prices that
were below the estimated fair value of our common stock at the date of grant. We recorded employee
stock-based compensation expense associated with the amortization of deferred stock compensation
related to these awards of $21,000, $254,000 and $307,000 for fiscal years ended June 30, 2010,
2009 and 2008, respectively. There was no unamortized deferred stock compensation recorded as of
June 30, 2010.
Results of Operations
Comparison of Fiscal Years ended June 30, 2010 and 2009
Net Revenue. Net revenue decreased $5.9 million, or 60%, to $4.0 million in fiscal year 2010
compared to $9.9 million in fiscal year 2009.
Net product sales decreased $3.0 million, or 45%, to $3.8 million in fiscal year 2010 from
$6.8 million in fiscal year 2009. The decrease of product sales for the fiscal year ended June 30,
2010 was primarily attributable to both lower PAS-Port and C-Port systems sales in the United
States as we transitioned from a direct sales force approach to third party manufacturers’
representatives and distributors.
For fiscal years 2010 and 2009, sales to Century Medical, Inc., our distributor in Japan,
accounted for approximately 23% and 15%, respectively, of our total product sales.
Development revenue was $124,000 and $3.0 million in fiscal years 2010 and 2009, respectively.
The development revenue for the fiscal year 2010 was comprised of $124,000 for development
activities on the PFO project with Cook. Our revenue-generating development activities related to
the Cook Vascular Closure Device were completed in fiscal year 2009, and there was no revenue
recognized for the fiscal year ended June 30, 2010 related to that agreement. The 2009 total was
comprised of $1.4 million for development activities for the PFO device under a development
agreement with Cook that we entered into in June 2007, and $1.6 million for development activities
for the Cook Vascular Closure Device under a separate development agreement with Cook. On January
6, 2010, we and Cook mutually agreed to temporarily suspend work on the PFO project.
Cost of Product Sales. Cost of product sales consists primarily of material, labor and
overhead costs. Cost of product sales decreased $1.7 million, or 31%, to $3.7 million in fiscal
year 2010 from $5.3 million in fiscal year 2009.
The decrease in cost of product sales in fiscal year 2010 compared to fiscal year 2009
resulted primarily from decreased product sales, partially offset by the increased cost per unit of
our inventory due to decreased production volumes.
Our cost of product sales was 98% and 79% of our net product sales in fiscal years 2010 and
2009, respectively, due to higher overhead costs per unit sold resulting from lower production
volumes. We expect higher costs relative to product sales to continue for the foreseeable future.
Research and Development Expenses. Research and development expenses consist primarily of
personnel costs within our product development, regulatory and clinical groups and the costs of
clinical trials. Research and development expenses decreased $2.8 million, or 34%, to $5.4 million
in fiscal year 2010 from $8.2 million in fiscal year 2009.
The decrease in research and development expenses in fiscal year 2010 compared to fiscal year
2009 was attributable to a decrease in salaries and benefits of $1.6 million due primarily to a net
decrease in the number of
personnel, decreased prototype project materials of $259,000, lower non-cash stock-based
compensation expenses of $192,000 and lower clinical trial expenses of $1.1 million as a result of
completing the PAS-Port trials and European trials, partially offset by higher molds and tooling
expenses of $231,000 related to the microcutter program development activities.
50
We anticipate that research and development expenses will increase in absolute terms in fiscal
year 2011 as we begin to develop new applications of our technology, including the Cardica
Microcutter ES8 and additional microcutter products.
Selling, General and Administrative Expenses. Selling, general and administrative expenses
consist primarily of costs for administrative and sales and marketing personnel, intellectual
property and marketing expenses. Selling, general and administrative expenses decreased $7.9
million, or 58%, to $5.7 million in fiscal year 2010 from $13.6 million in fiscal year 2009.
The net decrease in selling, general and administrative expenses in fiscal year 2010 compared
to fiscal year 2009 was attributable to lower personnel, recruiting and travel expenses of
$5.5 million primarily due to a net decrease in personnel, a decrease in marketing activities of
$393,000 in the United States, a decrease in demonstration product expenses of $747,000 for the
training of physicians in the United States and a decrease in consulting and professional service
expenses of $858,000.
We expect selling, general and administrative expenses to increase slightly in absolute terms
in fiscal year 2011 as we increase the number of individuals in our sales force and the number of
manufacturers’ representatives and distributors that sell our products.
Interest Income. Interest income decreased $142,000, or 80%, to $35,000 for fiscal year 2010
from $177,000 for fiscal year 2009. The decrease in interest income in fiscal year 2010 was
primarily attributable to lower cash and short-term investment balances available for investing
during the period and lower overall market interest rates for the fiscal year.
Interest Expense. Interest expense decreased $8,000, or 7%, to $112,000 for fiscal year 2010
from $120,000 in fiscal year 2009. The decrease in interest expense in fiscal year 2010 reflected a
contractual interest rate of 6% per annum payable on our $1.4 million debt to Century Medical.
Income Tax Benefit. Under the Housing and Economic Recovery Act of 2008 and the American
Recovery and Reinvestment Act of 2009, or the Acts, signed into law in July 2008 and February 2009,
respectively, taxpayers can claim a refundable alternative minimum tax or research and development
credit if they forego bonus depreciation on certain qualified fixed assets placed in service
between April 2008 and December 2009. We computed and recognized credits based on fixed assets
placed into service in our fiscal years ended June 30, 2010 and 2009. We recorded income tax
benefits of $31,000 and $72,000 in fiscal year 2010 and 2009 for the U.S. federal refundable
credits as provided by the Acts.
Comparison of Fiscal Years ended June 30, 2009 and 2008
Net Revenue. Net revenue increased $2.3 million, or 31%, to $9.9 million in fiscal year 2009
compared to $7.6 million in fiscal year 2008.
Net product sales increased $1.9 million, or 38%, to $6.8 million in fiscal year 2009 from
$4.9 million in fiscal year 2008. The increase in product sales for the fiscal year ended June 30,
2009 was primarily the result of the introduction of our PAS-Port system in the United States.
Product sales for the fiscal year ended June 30, 2008 did not include any PAS-Port system sales in
the United States as the system was not cleared by the FDA until September 2008. However, in the
fourth quarter of fiscal year 2009, total revenue was $2.0 million compared to $2.8 million in the
fourth quarter of fiscal year 2008. The lower sales for the fourth quarter of fiscal year 2009 were
due primarily to lower product sales caused by reductions in force in April and May 2009 which
included a significant number of individuals in our direct sales force as well as our Vice
President of Sales and Marketing. In addition, in the third quarter of fiscal year 2009 we received
low orders from our distributor in Japan and did not receive a significant order in the fourth
quarter for Japan.
51
For fiscal years 2009 and 2008, sales to Century Medical, Inc., our distributor in Japan,
accounted for approximately 15% and 20%, respectively, of our total product sales.
Development revenue was $3.0 million and $2.6 million in fiscal years 2009 and 2008,
respectively. The 2009 total was comprised of $1.4 million for development activities for the PFO
device under a development agreement with Cook that we entered into in June 2007, and $1.6 million
for development activities for the Cook Vascular Closure Device under a separate development
agreement with Cook. The 2008 total was comprised of $1.2 million for development activities for
the PFO device under a development agreement with Cook that we entered into in June 2007, and $1.4
million for development activities for the Cook Vascular Closure Device under a separate
development agreement with Cook.
Cost of Product Sales. Cost of product sales consists primarily of material, labor and
overhead costs. Cost of product sales increased $533,000, or 11%, to $5.3 million in fiscal year
2009 from $4.8 million in fiscal year 2008.
The increase in cost of product sales in fiscal year 2009 compared to fiscal year 2008 was
primarily attributable to increased unit sales of all of our products worldwide, due primarily to
increased adoption of PAS-Port systems in the United States, of $513,000, an excess inventory
reserve on C-Port raw materials of $248,000, and higher production scrap expense of $112,000 for
the PAS-Port system; offset in part by lower warranty charges of $144,000, and decreased lower of
cost or market reserves of $162,000.
Our cost of product sales was 79% and 97% of our net product sales in fiscal years 2009 and
2008, respectively, due to lower overhead per unit resulting from higher production volumes.
Research and Development Expenses. Research and development expenses decreased $392,000, or
5%, to $8.2 million in fiscal year 2009 from $8.6 million in fiscal year 2008.
The net decrease in research and development expenses in fiscal year 2009 compared to fiscal
year 2008 was attributable to a decrease in salaries and benefits of $232,000 due primarily to a
net decrease in the number of personnel, decreased prototype project materials for the C-Port xV
and Cook projects of $414,000, lower non-cash stock-based compensation expenses of $84,000 and
lower clinical trial expense of $223,000 as a result of completing the PAS-Port trials, offset in
part by higher molds and tooling expenses of $475,000 related to retirement of certain assets for
the C-Port xV System, which is no longer under development since the C-Port X-CHANGE II System
performs a comparable function while offering additional features and has nearly caught up to the
C-Port xV System in development, and higher facilities costs of $71,000.
Selling, General and Administrative Expenses. Selling, general and administrative expenses
increased $457,000, or 3%, to $13.6 million in fiscal year 2009 from $13.2 million in fiscal year
2008.
The net increase in selling, general and administrative expenses in fiscal year 2009 compared
to fiscal year 2008 was attributable to higher sales and marketing expenses to support field sales
activities in the United States to sell C-Port and PAS-Port systems, including increased salaries
and benefits of $861,000, higher non-cash stock-based compensation expenses of $156,000, higher
recruiting fees of $111,000 due to the expansion of the sales force and higher product
demonstration and trade show expense of $129,000, offset in part by lower accounting and auditing
fees of $119,000 primarily related to our change in filing status to be a non-accelerated filer,
and lower legal expense of $658,000 due to lower litigation expense in fiscal 2009 based on a
settlement reached in fiscal 2008.
Interest Income. Interest income decreased $749,000, or 81%, to $177,000 for fiscal year 2009
from $926,000 for fiscal year 2008. The decrease in interest income in fiscal year 2009 was
primarily attributable to lower average investment balances available for investing during the
period and lower overall market interest rates for the fiscal year.
Interest Expense. Interest expense increased $19,000, or 19%, to $120,000 for fiscal year 2009
from $101,000 in fiscal year 2008. The increase in interest expense in fiscal year 2009 reflects a
higher contractual interest rate of 6% per annum payable on our $2.0 million debt to Century
Medical.
52
Income Tax Benefit. Under the Housing and Economic Recovery Act of 2008 and the American
Recovery and Reinvestment Act of 2009, or the Acts, signed into law in July 2008 and February 2009,
respectively, taxpayers can claim a refundable alternative minimum tax or research and development
credit if they forego bonus depreciation on certain qualified fixed assets placed in service
between April 2008 and December 2009. Under these Acts, we computed and recognized a credit based
on fixed assets placed into service in our fiscal year ended June 30, 2009. We recorded an income
tax benefit of $72,000 in fiscal year 2009 for the U.S. federal refundable credit as provided by
the Acts.
Income Taxes
Due to uncertainty surrounding the realization of our deferred tax assets through future
taxable income, we have provided a full valuation allowance and no benefit has been recognized for
our net operating losses and other deferred tax assets. Accordingly, deferred tax asset valuation
allowances have been established as of June 30, 2010 and 2009 to reflect these uncertainties. At
June 30, 2010, we had unrecognized tax benefits of $595,000, all of which would not currently affect our
effective tax rate if recognized due to our deferred tax assets being fully offset by a valuation
allowance.
As of June 30, 2010, we had net operating loss carry-forwards to reduce future taxable income,
if any, of approximately $104.5 million for federal income tax purposes and $75.2 million available
to reduce future taxable income, if any, for state income taxes. The net operating loss
carry-forwards begin to expire in the fiscal year 2013. We also had federal and state research and
development credit carry-forwards of approximately $0.6 million and $2.3 million, respectively, at
June 30, 2010. The federal credits begin to expire in fiscal year 2021 if not utilized. The state
credit carry-forwards have an unlimited carry-forward period and the State of Arizona credit begin
to expire in fiscal year 2024. We have completed a study of our tax attributes under Section 382 of
the Internal Revenue Code of 1986 which resulted in significant limitations on our net operating
loss and credit carry-forwards prior to utilization. The related reductions are reflected in the
carry-forward amounts discussed above.
Liquidity and Capital Resources
As of June 30, 2010, our accumulated deficit was $120.3 million and we had cash and cash
equivalents of $6.6 million and total short-term debt of $1.4 million. We currently invest our cash
and cash equivalents in money market funds. Since inception, we have financed our operations
primarily through private sales of convertible preferred stock, long-term notes payable and public
and private sales of common stock.
In June 2007, we received approximately $10.9 million in net proceeds from the sale of
2,301,337 shares of our common stock and warrants to purchase up to 575,347 shares of common stock
in a private placement. In November 2007, we received $11.5 million in net proceeds from the sale
of 1,500,000 shares of our common stock in a public offering. In December 2007, we received $3.8
million in net proceeds from the sale of an additional 481,170 shares of our common stock upon
exercise of the over-allotment option. In September 2009, we received approximately $9.9 million in
net proceeds from the sale of 8,142,082 shares of our common stock and warrants to purchase up to
4,071,046 shares of our common stock in a private placement.
We had a note payable that was originally issued under a Note Agreement entered into in
connection with our Japan Distribution Agreement with Century Medical in June 2003. We extended the
distribution agreement and restructured the $3.0 million note in March 2007, whereby $1.0 million
of the note was paid in April 2007 and the remaining $2.0 million had a June 2010 maturity date. On
April 1, 2010, we entered into the Note Agreement Amendment, under which we made a principal
payment of $600,000 to Century Medical in April 2010, with the remaining $1.4 million principal
amount owed to Century Medical becoming due on June 17, 2011, which is one year later than the
maturity date prior to the Note Agreement Amendment. The note bore interest at 5% per annum through
June 2008 and then increased to 6% per annum until maturity, and all interest due under our note to
Century Medical was payable quarterly. On August 17, 2010, we paid off the remaining $1.4 million
principal balance and interest due through August 17, 2010 so that we currently do not have any
outstanding notes payable.
Under the operating lease for our facility in Redwood City, California, we are required to
maintain a letter of credit with a restricted cash balance at our bank. A certificate of deposit of
$150,000 and $300,000 has been
recorded as restricted cash in our balance sheets at June 30, 2010 and 2009, respectively,
related to the letter of credit.
53
Summary cash flow data is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Fiscal Year Ended June 30, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
| |
|
(In thousands) |
|
Net cash used in operating activities |
|
$ |
(7,731 |
) |
|
$ |
(16,703 |
) |
|
$ |
(14,222 |
) |
Net cash (used in) provided by investing activities |
|
|
(365 |
) |
|
|
12,664 |
|
|
|
(6,613 |
) |
Net cash provided by financing activities |
|
|
9,329 |
|
|
|
146 |
|
|
|
15,517 |
|
Net cash used in operating activities for fiscal years 2010, 2009 and 2008 was $7.7 million,
$16.7 million, and $14.2 million, respectively. Our net use of cash for fiscal year 2010 was
primarily attributable to our net loss adjusted for non-cash stock-based compensation charges of
$1.2 million and approximately $836,000 of depreciation and amortization expenses, plus decreased
inventories of $764,000. Our net use of cash for fiscal year 2009 was primarily attributable to our
net loss, adjusted for non-cash stock-based compensation charges of $1.9 million, approximately
$614,000 for C-Port xV-related fixed assets that were retired and $917,000 of depreciation and
amortization, less increased inventories of $555,000, decreases in accounts payable, other accrued
liabilities and accrued compensation totaling $1.8 million and a decrease in deferred development
revenue of $958,000 mainly due to the completion of our activities under the Cook Vascular Closure
Device development and commercialization agreement. Our net use of cash for fiscal year 2008 was
primarily attributable to our net loss, adjusted for non-cash stock-based compensation charges of
$1.7 million, approximately $425,000 of our common stock issued for settlement of a patent
litigation and $944,000 of depreciation and amortization expenses, less higher accounts receivable
of $433,000 as a result of increased sales of our products in the United States offset in part by
increases in accounts payable, other accrued liabilities and accrued compensation totaling $1.1
million and an increase in deferred development revenue of $603,000 due to cash received from Cook.
Net cash used in investing activities was $365,000 for fiscal year 2010 and related to the
purchase of property and equipment. Net cash provided by investing activities was $12.7 million
for fiscal year 2009, resulting from net maturities of short-term investments of $14.1 million
required to fund our operating loss in fiscal year 2009 offset in part by $1.4 million used to
purchase property and equipment. Net cash used in investing activities was $6.6 million for
fiscal year 2008, resulting from net purchases of available-for-sale investments of $5.2 million
due to excess cash resources received from the sale of our common stock in November and December
2007, and $1.5 million used to purchase property and equipment.
Net cash provided by financing activities of $9.3 million for fiscal 2010 was due primarily to
$9.9 million of net proceeds received from sales of shares of common stock and warrants to purchase
shares of common stock in September 2009, offset in part by $600,000 of notes payable to Century
Medical that were repaid in April 2010. Net cash provided by financing activities of $146,000 for
fiscal 2009 was due to net proceeds received from exercises of options to purchase our common
stock. Net cash provided by financing activities of $15.5 million for fiscal 2008 was primarily due
to net proceeds received from sales of our common stock in November and December 2007.
On August 16, 2010, we entered into a License Agreement with Intuitive Surgical pursuant to
which we granted to Intuitive Surgical a worldwide, sublicenseable, exclusive license to use our
intellectual property in the robotics field in diagnostic or therapeutic medical procedures, but
excluding vascular anastomosis applications (the “License Agreement”) for an upfront license fee of
$9 million. We will also be eligible to receive a milestone payment if sales of any products
incorporating our patent rights achieve a specified level of net sales within a specified period
after the date of the License Agreement and will also be eligible to receive single-digit royalties
on sales by Intuitive Surgical, its affiliates or its sublicensees of specified stapler and clip
applier products covered by our patent rights as well as on sales of certain other products covered
by our patent rights that may be developed in the future.
In
addition, on the same date, we entered into a Stock Purchase
Agreement with Intuitive Surgical
pursuant to which Intuitive Surgical paid $3 million to purchase from us an aggregate of 1,249,541
newly-issued shares of our common stock (the “Stock Issuance”).
54
Including the cash received in August 2010 in connection with the License Agreement with
Intuitive Surgical and related equity investment by Intuitive, we believe that our existing cash
and cash equivalents, along with the cash that we expect to generate from operations, will be
sufficient to meet our anticipated cash needs to enable us to conduct our business substantially as
currently conducted through September 30, 2011.
Our estimates and our future capital requirements depend upon numerous factors. In addition,
we have based our estimates on assumptions that may prove to be wrong, including assumptions with
respect to the level of revenues from product sales, and we could exhaust our available financial
resources sooner than we currently expect. While our cash resources would permit us to continue
through September 30, 2011, we would need to further reduce expenses in advance of that date in the
event that we are unable to complete a financing, strategic or commercial transaction that
generates additional capital to ensure that we have sufficient capital to meet our obligations and
continue on a path designed to create and preserve stockholder value.
The sufficiency of our current cash resources and our need for additional capital, and the
timing thereof, will depend upon numerous factors. These factors include, but are not limited to,
the following:
| |
• |
|
the extent of our ongoing research and development programs and related costs,
including costs related to the development of the Cardica Microcutter ES8 and additional
potential products in our anticipated microcutter product line; |
| |
• |
|
our ability to enter into additional license, development and/or collaboration
agreements with respect to our technology, and the terms thereof; |
| |
• |
|
market acceptance and adoption of our current products or future products that we may
commercialize; |
| |
• |
|
costs associated with our sales and marketing initiatives and manufacturing activities; |
| |
• |
|
costs and timing of obtaining and maintaining FDA and other regulatory clearances and
approvals for our products and potential additional products; |
| |
• |
|
securing, maintaining and enforcing intellectual property rights and the costs thereof;
and |
| |
• |
|
the effects of competing technological and market developments |
Until we can generate significant continuing revenue, if ever, we expect to satisfy our future
cash needs through public or private equity offerings, debt financings or corporate collaboration
and licensing arrangements, as well as through interest income earned on cash balances. We cannot
be certain that additional funding of any kind will be available on acceptable terms, or at all.
The sale of additional equity or convertible debt securities could result in dilution to our
stockholders. If additional funds are raised through the issuance of securities, these securities
could have rights senior to those associated with our common stock and could contain covenants that
would restrict our operations. Any licensing or strategic agreements we enter into may require us
to relinquish valuable rights. If adequate funds are not available, we may be required to delay,
reduce the scope of or eliminate our commercialization efforts on one or more of our research and
development programs, cease operations or cease to be publicly traded.
55
Recent Accounting Pronouncements
Effective July 1, 2009, we adopted the Financial Accounting Standards Board, or FASB,
Accounting Standards Codification, or the Codification. The Codification is the source of
authoritative accounting principles recognized by the FASB to be applied by nongovernmental
entities in the preparation of financial statements in conformity with generally accepted
accounting principles, or GAAP, and became effective for interim periods and
fiscal years ending on or after September 15, 2009. The Codification explicitly recognizes
rules and interpretive releases of the Securities and Exchange Commission, or SEC, under federal
securities laws as authoritative GAAP for SEC registrants. We have updated all existing GAAP
references in our financial statements in accordance with the Codification. As the Codification was
not intended to change or alter existing GAAP, the adoption of the Codification did not have any
impact on the amounts included in our financial statements.
In October 2009, the FASB issued Accounting Standards Update, or ASU, No. 2009-13 which
addresses the accounting for multiple-element arrangements to potentially enable vendors to account
for products or services separately rather than as a combined unit. This guidance also modifies the
manner in which transaction consideration is allocated across the separately identified elements
and significantly expands the disclosure requirements for multiple-element revenue arrangements. We
will adopt ASU No. 2009-13 as of July 1, 2010 and apply it prospectively to arrangements entered
into or materially modified after the adoption date. We do not expect the adoption of ASU No.
2009-13 to have any impact on our results of operations or financial condition upon its required
adoption.
In April of 2010, ASC 605, Revenue Recognition, was amended to define a milestone and clarify
that the milestone method of revenue recognition is a valid application of the proportional
performance model when applied to research or development arrangements. Accordingly, a company can
make an accounting policy election to recognize a payment that is contingent upon the achievement
of a substantive milestone in its entirety in the period in which the milestone is achieved. This
guidance is effective for the Company as of July 1, 2010 and will be applied on a prospective
basis. The Company does not expect this guidance to have any impact on its results of operations
or financial condition upon its required adoption.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements, including structured finance, special
purpose or variable interest entities.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Not applicable.
56
Item 8. Financial Statements and Supplementary Data
The following tables set forth selected unaudited quarterly statement of operations data for
the eight most recent quarters. The information for each of these quarters has been prepared on the
same basis as the audited financial statements included in this report and, in the opinion of
management, includes all adjustments necessary for the fair presentation of the results of
operations for such periods. This data should be read in conjunction with the audited financial
statements and the related notes included in this report. These quarterly operating results are not
necessarily indicative of our operating results for any future period.
Quarterly Financial Data
Fiscal year 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
1st Quarter |
|
|
2nd Quarter |
|
|
3rd Quarter |
|
|
4th Quarter |
|
| |
|
(Unaudited, in thousands, except per share data) |
|
Total net revenue |
|
$ |
947 |
|
|
$ |
1,064 |
|
|
$ |
979 |
|
|
$ |
991 |
|
Gross profit (loss) on product sales |
|
|
(23 |
) |
|
|
324 |
|
|
|
(109 |
) |
|
|
(115 |
) |
Net loss |
|
|
(2,664 |
) |
|
|
(2,226 |
) |
|
|
(2,983 |
) |
|
|
(3,051 |
) |
Basic and diluted net loss per common share |
|
|
(0.17 |
) |
|
|
(0.09 |
) |
|
|
(0.12 |
) |
|
|
(0.13 |
) |
Shares used in computing basic and diluted
net loss per common share |
|
|
15,796 |
|
|
|
23,930 |
|
|
|
23,966 |
|
|
|
24,000 |
|
Fiscal year 2009:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
1st Quarter |
|
|
2nd Quarter |
|
|
3rd Quarter |
|
|
4th Quarter |
|
| |
|
(Unaudited, in thousands, except per share data) |
|
Total net revenue |
|
$ |
2,106 |
|
|
$ |
2,944 |
|
|
$ |
2,838 |
|
|
$ |
1,989 |
|
Gross profit (loss) on product sales |
|
|
450 |
|
|
|
532 |
|
|
|
551 |
|
|
|
(77 |
) |
Net loss |
|
|
(5,154 |
) |
|
|
(4,697 |
) |
|
|
(3,902 |
) |
|
|
(3,452 |
) |
Basic and diluted net loss per common share |
|
|
(0.33 |
) |
|
|
(0.30 |
) |
|
|
(0.25 |
) |
|
|
(0.22 |
) |
Shares used in computing basic and diluted
net loss per common share |
|
|
15,741 |
|
|
|
15,781 |
|
|
|
15,785 |
|
|
|
15,796 |
|
See Item 15, below, for our audited financial statements and related notes.
57
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Disclosure Controls and Procedures
Based on their evaluation, our Chief Executive Officer and Chief Financial Officer have
concluded that our disclosure controls and procedures (as defined in Rule 13a-15(e) and 15d-15(e)
of the Securities Exchange Act of 1934, as amended) were effective as of June 30, 2010.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over
financial reporting (as defined in Rule 13a-15(f) of the Securities Exchange Act of 1934). Under
the supervision and with the participation of our management, including our Chief Executive Officer
and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal
control over financial reporting as of June 30, 2010, based on the criteria set forth in Internal
Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway
Commission. Based on our evaluation under the criteria set forth in Internal Control — Integrated
Framework, our management concluded that our internal control over financial reporting was
effective as of June 30, 2010.
This annual report does not include an attestation report of our registered public accounting
firm regarding internal control over financial reporting. Management’s report was not subject to
attestation by our registered public accounting firm pursuant to Section 404(c) of the
Sarbanes-Oxley Act of 2002 and the rules of the Securities and Exchange Commission that permit us
to provide only management’s report in this annual report.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting during the quarter
ended June 30, 2010 that have materially affected, or are reasonably likely to materially affect,
our internal control over financial reporting.
Inherent Limitations on Effectiveness of Controls
A control system, no matter how well conceived and operated, can provide only reasonable, not
absolute, assurance that the objectives of the control system are met. Because of inherent
limitations in all control systems, no evaluation of controls can provide absolute assurance that
all control issues, if any, within an organization have been detected. Accordingly, our disclosure
controls and procedures are designed to provide reasonable, not absolute, assurance that the
objectives of our disclosure control system are met and, as set forth above, our principal
executive officer and principal financial officer have concluded, based on their evaluation as of
the end of the period covered by this report, that our disclosure controls and procedures were
effective to provide reasonable assurance that the objectives of our disclosure control system were
met.
Item 9B. Other Information
None.
58
PART III
Item 10. Directors, Executive Officers and Corporate Governance
Identification of Executive Officers and Directors
Reference is made to the information regarding executive officers appearing under the heading
“Business — Executive Officers of the Registrant” in Part I Item 1 of this Annual Report on Form
10-K, which information is hereby incorporated by reference. Reference is made to the information
regarding our directors and nominees for director appearing under the heading “Proposal 1 —
Election of Directors” in our proxy statement for our 2010 annual meeting of stockholders, or 2010
Proxy Statement, which information is hereby incorporated by reference.
Identification of Audit Committee and Audit Committee Financial Expert
Reference is made to the information regarding directors appearing under the headings
“Information Regarding the Board of Directors and Corporate Governance — Information Regarding
Committees of the Board of Directors” and “Information Regarding the Board of Directors and
Corporate Governance — Information Regarding Committees of the Board of Directors — Audit
Committee” in our 2010 Proxy Statement, which information is hereby
incorporated by reference.
Material Changes to Procedures for Recommending Directors
Reference is made to the information regarding directors appearing under the heading
“Information Regarding the Board of Directors and Corporate Governance” in our 2010 Proxy
Statement, which information is hereby incorporated by reference.
Compliance with Section 16(a) of the Exchange Act
Reference is made to the information appearing under the heading “Section 16(a) Beneficial
Ownership Reporting Compliance” in our 2010 Proxy Statement, which information is hereby
incorporated by reference.
Code of Conduct
Reference is made to the information appearing under the heading “Information Regarding the
Board of Directors and Corporate Governance — Code of Business Conduct and Ethics” in our 2010
Proxy Statement, which information is hereby incorporated by reference. A copy of our code of
business conduct and ethics can be found on our website, www.cardica.com in the section titled
“Investors/Media” under the subsection titled “Corporate Governance”. The contents of our website
are not a part of this Annual Report on Form 10-K.
Item 11. Executive Compensation
Reference is made to the information appearing under the headings “Executive Compensation”,
“Compensation Committee Interlocks and Insider Participation” and “Compensation Committee Report”
in our 2010 Proxy Statement, which information is hereby incorporated by reference.
59
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Security Ownership
The information required by this item will be set forth in our 2010 Proxy Statement under the
caption “Security Ownership of Certain Beneficial Owners and Management” and is incorporated herein
by reference.
Equity Compensation Plan Information
Information concerning our equity compensation plans will be set forth in our 2010 Proxy
Statement under the caption “Equity Compensation Plan Information” and is incorporated herein by
reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this item will be set forth in our 2010 Proxy Statement under the
captions “Transactions with Related Persons” and “Independence of the Board of Directors” and is
incorporated herein by reference.
Item 14. Principal Accountant Fees and Services
The information required by this item will be set forth in our 2010 Proxy Statement under the
caption “Principal Accountant Fees and Services” and is incorporated herein by reference.
60
PART IV
Item 15. Exhibits and Financial Statement Schedules
(a) Documents filed as part of this report
1. Financial Statements
Cardica, Inc.
Index to Financial Statements
61
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Cardica, Inc.
We have audited the accompanying balance sheets of Cardica, Inc. as of June 30, 2010 and 2009,
and the related statements of operations, stockholders’ equity and cash flows for each of the three
years in the period ended June 30, 2010. These financial statements are the responsibility of the
Company’s management. Our responsibility is to express an opinion on these financial statements
based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting
Oversight Board (United States). Those standards require that we plan and perform the audit to
obtain reasonable assurance about whether the financial statements are free of material
misstatement. We were not engaged to perform an audit of the Company’s internal control over
financial reporting. Our audits included consideration of internal control over financial
reporting as a basis for designing audit procedures that are appropriate in the circumstances, but
not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control
over financial reporting. Accordingly, we express no such opinion. An audit also includes
examining, on a test basis, evidence supporting the amounts and disclosures in the financial
statements, assessing the accounting principles used and significant estimates made by management,
and evaluating the overall financial statement presentation. We believe that our audits provide a
reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material
respects, the financial position of Cardica, Inc. at June 30, 2010 and 2009, and the results of its
operations and its cash flows for each of the three years in the period ended June 30, 2010, in
conformity with U.S. generally accepted accounting principles.
/s/ Ernst & Young LLP
Palo Alto, California
September 24, 2010
62
Cardica, Inc.
BALANCE SHEETS
(In thousands, except share and per share data)
| |
|
|
|
|
|
|
|
|
| |
|
June 30, |
|
| |
|
2010 |
|
|
2009 |
|
Assets |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Current assets |
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
6,561 |
|
|
$ |
5,328 |
|
Accounts receivable |
|
|
376 |
|
|
|
624 |
|
Inventories |
|
|
1,131 |
|
|
|
1,895 |
|
Prepaid expenses and other current assets |
|
|
231 |
|
|
|
321 |
|
|
|
|
|
|
|
|
Total current assets |
|
|
8,299 |
|
|
|
8,168 |
|
|
|
|
|
|
|
|
|
|
Property and equipment, net |
|
|
1,338 |
|
|
|
1,862 |
|
Restricted cash |
|
|
154 |
|
|
|
310 |
|
|
|
|
|
|
|
|
Total assets |
|
$ |
9,791 |
|
|
$ |
10,340 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Liabilities and stockholders’ equity |
|
|
|
|
|
|
|
|
Current liabilities |
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
496 |
|
|
$ |
551 |
|
Accrued compensation |
|
|
440 |
|
|
|
319 |
|
Other accrued liabilities |
|
|
517 |
|
|
|
637 |
|
Deferred development revenue |
|
|
403 |
|
|
|
527 |
|
Deferred rent |
|
|
27 |
|
|
|
— |
|
Note payable |
|
|
1,400 |
|
|
|
2,000 |
|
|
|
|
|
|
|
|
Total current liabilities |
|
|
3,283 |
|
|
|
4,034 |
|
|
|
|
|
|
|
|
|
|
Other non-current liabilities |
|
|
31 |
|
|
|
44 |
|
|
|
|
|
|
|
|
Total liabilities |
|
|
3,314 |
|
|
|
4,078 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Commitments and contingencies (Note 6) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stockholders’ equity |
|
|
|
|
|
|
|
|
Preferred stock, $0.001 par value: 5,000,000 shares authorized,
no shares issued and outstanding at June 30, 2010 and 2009 |
|
|
— |
|
|
|
— |
|
Common stock, $0.001 par value: 45,000,000 shares authorized,
24,005,813
and 15,825,549 shares issued and outstanding at June 30,
2010 and 2009,
respectively |
|
|
24 |
|
|
|
16 |
|
Additional paid-in capital |
|
|
127,381 |
|
|
|
116,272 |
|
Treasury stock at cost (66,227 shares at June 30, 2010 and 2009) |
|
|
(596 |
) |
|
|
(596 |
) |
Deferred stock-based compensation |
|
|
— |
|
|
|
(22 |
) |
Accumulated deficit |
|
|
(120,332 |
) |
|
|
(109,408 |
) |
|
|
|
|
|
|
|
Total stockholders’ equity |
|
|
6,477 |
|
|
|
6,262 |
|
|
|
|
|
|
|
|
Total liabilities and stockholders’ equity |
|
$ |
9,791 |
|
|
$ |
10,340 |
|
|
|
|
|
|
|
|
See accompanying notes to financial statements.
63
Cardica, Inc.
STATEMENTS OF OPERATIONS
(In thousands, except per share data)
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Fiscal Year Ended June 30, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net revenue |
|
|
|
|
|
|
|
|
|
|
|
|
Product sales, net |
|
$ |
3,764 |
|
|
$ |
6,798 |
|
|
$ |
4,934 |
|
Development revenue |
|
|
124 |
|
|
|
2,995 |
|
|
|
2,564 |
|
Royalty revenue (including $67 from a related party in
2008) |
|
|
93 |
|
|
|
85 |
|
|
|
67 |
|
|
|
|
|
|
|
|
|
|
|
Total net revenue |
|
|
3,981 |
|
|
|
9,878 |
|
|
|
7,565 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating costs and expenses |
|
|
|
|
|
|
|
|
|
|
|
|
Cost of product sales |
|
|
3,687 |
|
|
|
5,341 |
|
|
|
4,808 |
|
Research and development |
|
|
5,437 |
|
|
|
8,217 |
|
|
|
8,609 |
|
Selling, general and administrative |
|
|
5,734 |
|
|
|
13,632 |
|
|
|
13,175 |
|
|
|
|
|
|
|
|
|
|
|
Total operating costs and expenses |
|
|
14,858 |
|
|
|
27,190 |
|
|
|
26,592 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations |
|
|
(10,877 |
) |
|
|
(17,312 |
) |
|
|
(19,027 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
35 |
|
|
|
177 |
|
|
|
926 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest expense |
|
|
(112 |
) |
|
|
(120 |
) |
|
|
(101 |
) |
Other income (expense), net |
|
|
(1 |
) |
|
|
(22 |
) |
|
|
6 |
|
|
|
|
|
|
|
|
|
|
|
Net loss before income tax benefit |
|
|
(10,955 |
) |
|
|
(17,277 |
) |
|
|
(18,196 |
) |
Income tax benefit |
|
|
31 |
|
|
|
72 |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(10,924 |
) |
|
$ |
(17,205 |
) |
|
$ |
(18,196 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per common share |
|
$ |
(0.50 |
) |
|
$ |
(1.09 |
) |
|
$ |
(1.23 |
) |
|
|
|
|
|
|
|
|
|
|
Shares used in computing basic and diluted net loss per common
share |
|
|
21,927 |
|
|
|
15,776 |
|
|
|
14,844 |
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes to financial statements.
64
Cardica, Inc.
STATEMENTS OF STOCKHOLDERS’ EQUITY
(In thousands, except share data)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deferred |
|
|
Accumulated |
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
Additional |
|
|
|
|
|
|
Stock- |
|
|
other |
|
|
|
|
|
|
Total |
|
| |
|
Common Stock |
|
|
Paid-in |
|
|
Treasury |
|
|
Based |
|
|
comprehensive |
|
|
Accumulated |
|
|
Stockholders’ |
|
| |
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Stock |
|
|
Compensation |
|
|
loss |
|
|
Deficit |
|
|
Equity |
|
Balance at June 30, 2007 |
|
|
13,606,333 |
|
|
|
14 |
|
|
|
97,171 |
|
|
|
(596 |
) |
|
|
(591 |
) |
|
|
(2 |
) |
|
|
(74,007 |
) |
|
|
21,989 |
|
Issuance of common stock upon
exercise of employee stock
options for cash |
|
|
77,036 |
|
|
|
— |
|
|
|
167 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
167 |
|
Sale of common stock, net of
financing costs of $1,481 |
|
|
1,981,170 |
|
|
|
2 |
|
|
|
15,348 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
15,350 |
|
Issuance of stock options to
non-employees for services |
|
|
— |
|
|
|
— |
|
|
|
10 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
10 |
|
Issuance of common stock for
settlement of patent litigation |
|
|
60,000 |
|
|
|
— |
|
|
|
425 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
425 |
|
Issuance of shares pursuant to net
exercise of warrants |
|
|
7,666 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Issuance of restricted stock
awards |
|
|
52,450 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Stock-based compensation
expense |
|
|
— |
|
|
|
— |
|
|
|
1,365 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,365 |
|
Early exercise of stock options
no longer subject to repurchase |
|
|
— |
|
|
|
— |
|
|
|
10 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
10 |
|
Reversal of deferred stock-based
compensation for terminated
employees |
|
|
— |
|
|
|
— |
|
|
|
(2 |
) |
|
|
— |
|
|
|
2 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Amortization of deferred
stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
307 |
|
|
|
— |
|
|
|
— |
|
|
|
307 |
|
Comprehensive loss: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(18,196 |
) |
|
|
(18,196 |
) |
Net change in unrealized loss
on marketable securities |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(10 |
) |
|
|
— |
|
|
|
(10 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(18,206 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at June 30, 2008 |
|
|
15,784,655 |
|
|
|
16 |
|
|
|
114,494 |
|
|
|
(596 |
) |
|
|
(282 |
) |
|
|
(12 |
) |
|
|
(92,203 |
) |
|
|
21,417 |
|
65
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deferred |
|
|
Accumulated |
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
Additional |
|
|
|
|
|
|
Stock- |
|
|
other |
|
|
|
|
|
|
Total |
|
| |
|
Common Stock |
|
|
Paid-in |
|
|
Treasury |
|
|
Based |
|
|
comprehensive |
|
|
Accumulated |
|
|
Stockholders’ |
|
| |
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Stock |
|
|
Compensation |
|
|
loss |
|
|
Deficit |
|
|
Equity |
|
Issuance of common stock upon
exercise of employee stock
options for cash |
|
|
44,144 |
|
|
|
— |
|
|
|
146 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
146 |
|
Issuance of stock options to
non-employees for services |
|
|
— |
|
|
|
— |
|
|
|
10 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
10 |
|
Cancellation of restricted stock awards, net of
issuance of shares |
|
|
(3,250 |
) |
|
|
|
|
|
|
(2 |
) |
|
|
— |
|
|
|
2 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Stock-based compensation
expense |
|
|
— |
|
|
|
— |
|
|
|
1,629 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,629 |
|
Reversal of deferred stock-based
compensation for terminated
employees |
|
|
— |
|
|
|
— |
|
|
|
(5 |
) |
|
|
— |
|
|
|
5 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Amortization of deferred
stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
253 |
|
|
|
— |
|
|
|
— |
|
|
|
253 |
|
Comprehensive loss: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(17,205 |
) |
|
|
(17,205 |
) |
Net change in unrealized loss
on marketable securities |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
12 |
|
|
|
— |
|
|
|
12 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(17,193 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at June 30, 2009 |
|
|
15,825,549 |
|
|
|
16 |
|
|
|
116,272 |
|
|
|
(596 |
) |
|
|
(22 |
) |
|
|
— |
|
|
|
(109,408 |
) |
|
|
6,262 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of common stock upon
exercise of employee stock
options for cash |
|
|
8,257 |
|
|
|
— |
|
|
|
11 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
11 |
|
Issuance of common stock upon release of restricted
share units |
|
|
49,925 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Issuance of stock options to
non-employees for services |
|
|
— |
|
|
|
— |
|
|
|
31 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
31 |
|
Cancellation of restricted stock awards, net of
issuance of shares |
|
|
(20,000 |
) |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Sale of common stock, net of financing costs of $280 |
|
|
8,142,082 |
|
|
|
8 |
|
|
|
9,910 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
9,918 |
|
Stock-based compensation
expense |
|
|
— |
|
|
|
— |
|
|
|
1,158 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,158 |
|
Amortization of deferred
stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
(1 |
) |
|
|
— |
|
|
|
22 |
|
|
|
— |
|
|
|
— |
|
|
|
21 |
|
Comprehensive loss: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net and comprehensive loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(10,924 |
) |
|
|
(10,924 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(10,924 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at June 30, 2010 |
|
|
24,005,813 |
|
|
|
24 |
|
|
|
127,381 |
|
|
|
(596 |
) |
|
|
— |
|
|
|
— |
|
|
|
(120,332 |
) |
|
|
6,477 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes to financial statements.
66
Cardica, Inc.
STATEMENTS OF CASH FLOWS
(In thousands)
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Fiscal Year Ended June 30, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating activities |
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(10,924 |
) |
|
$ |
(17,205 |
) |
|
$ |
(18,196 |
) |
Adjustments to reconcile net loss to net cash used in operating activities |
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
836 |
|
|
|
917 |
|
|
|
944 |
|
Loss on disposal or retirement of property and equipment |
|
|
53 |
|
|
|
614 |
|
|
|
12 |
|
Amortization of deferred stock-based compensation expense |
|
|
21 |
|
|
|
253 |
|
|
|
307 |
|
Issuance of common stock to settle intellectual property litigation |
|
|
— |
|
|
|
— |
|
|
|
425 |
|
Stock-based compensation on grants of stock options to non-employees |
|
|
31 |
|
|
|
10 |
|
|
|
10 |
|
Stock-based compensation on grants of stock options to employees |
|
|
1,158 |
|
|
|
1,629 |
|
|
|
1,365 |
|
Changes in assets and liabilities
|
|
|
|
|
|
|
|
|
|
|
|
|
Accounts receivable |
|
|
248 |
|
|
|
92 |
|
|
|
(433 |
) |
Prepaid expenses and other current assets |
|
|
98 |
|
|
|
115 |
|
|
|
— |
|
Inventories |
|
|
764 |
|
|
|
(555 |
) |
|
|
(164 |
) |
Restricted Cash |
|
|
156 |
|
|
|
200 |
|
|
|
— |
|
Accounts payable and other accrued liabilities |
|
|
(175 |
) |
|
|
(1,129 |
) |
|
|
643 |
|
Accrued compensation |
|
|
121 |
|
|
|
(692 |
) |
|
|
495 |
|
Deferred rent |
|
|
6 |
|
|
|
17 |
|
|
|
(111 |
) |
Deferred development revenue |
|
|
(124 |
) |
|
|
(958 |
) |
|
|
603 |
|
Leasehold improvement obligation |
|
|
— |
|
|
|
(11 |
) |
|
|
(122 |
) |
|
|
|
|
|
|
|
|
|
|
Net cash used in operating activities |
|
|
(7,731 |
) |
|
|
(16,703 |
) |
|
|
(14,222 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Investing activities |
|
|
|
|
|
|
|
|
|
|
|
|
Purchases of property and equipment |
|
|
(365 |
) |
|
|
(1,392 |
) |
|
|
(1,454 |
) |
Purchases of short-term investments |
|
|
— |
|
|
|
(4,974 |
) |
|
|
(45,981 |
) |
Proceeds from maturities of short-term investments |
|
|
— |
|
|
|
19,030 |
|
|
|
40,822 |
|
|
|
|
|
|
|
|
|
|
|
Net cash (used in) provided by investing activities |
|
|
(365 |
) |
|
|
12,664 |
|
|
|
(6,613 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Financing activities |
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from sales of common stock, net of issuance costs |
|
|
9,918 |
|
|
|
— |
|
|
|
15,350 |
|
Payment of note payable |
|
|
(600 |
) |
|
|
— |
|
|
|
— |
|
Proceeds from issuance of common stock pursuant to the exercise of stock
options |
|
|
11 |
|
|
|
146 |
|
|
|
167 |
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by financing activities |
|
|
9,329 |
|
|
|
146 |
|
|
|
15,517 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net increase (decrease) in cash and cash equivalents |
|
|
1,233 |
|
|
|
(3,893 |
) |
|
|
(5,318 |
) |
Cash and cash equivalents at beginning of period |
|
|
5,328 |
|
|
|
9,221 |
|
|
|
14,539 |
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents at end of period |
|
$ |
6,561 |
|
|
$ |
5,328 |
|
|
$ |
9,221 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental disclosure of cash flow information
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid for interest |
|
$ |
118 |
|
|
$ |
117 |
|
|
$ |
101 |
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes to financial statements.
67
Cardica, Inc.
Notes to Financial Statements
Note 1. Organization and Summary of Significant Accounting Policies
Organization
Cardica, Inc. (the “Company”) was incorporated in the state of Delaware on October 15, 1997,
as Vascular Innovations, Inc. On November 26, 2001, the Company changed its name to Cardica, Inc.
The Company designs, manufactures and markets proprietary automated anastomotic systems used in
surgical procedures. The Company’s first product, the PAS-Port system, received the CE Mark for
sales in Europe in March 2003, regulatory approval for sales in Japan in January 2004 and 510(k)
clearance from the FDA on September 5, 2008. The Company’s second product, the C-Port system,
received the CE Mark for sales in Europe in April 2004 and 510(k) clearance in the United States in
November 2005. The C-Port xA system, a next generation C-Port system, received the CE Mark for
sales in Europe in July 2006 and 510(k) clearance in the U.S. in November 2006. The C-Port Flex A
system was cleared by the FDA in March 2007 and the C-Port X-CHANGE was cleared by the FDA in
December 2007.
Since its inception, the Company has incurred significant net losses and expects to continue
to incur net losses for the foreseeable future. To date, the Company’s C-Port and PAS-Port systems
have had limited commercial adoption. In addition to its commercialized cardiac surgery products,
the Company has re-focused its business on the development of an endoscopic microcutter product
line intended for use by general, thoracic, gynecologic, bariatric and urologic surgeons. The
Company is developing the Cardica Microcutter ES8, a multi-fire endolinear microcutter device based
on the Company’s proprietary “staple-on-a-strip” technology, which would expand the Company’s
commercial opportunity into additional surgical markets.
On August 16, 2010, the Company entered into a License Agreement with Intuitive Surgical pursuant to
which we granted to Intuitive Surgical a worldwide, sublicenseable, exclusive license to use our
intellectual property in the robotics field in diagnostic or therapeutic medical procedures, but
excluding vascular anastomosis applications (see Note 14, Subsequent Event).
Use of Estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting
principles (“GAAP”) generally requires management to make estimates and assumptions that affect the
amounts reported in the financial statements. Actual results could differ from these estimates.
Cash and Cash Equivalents
The Company’s cash and cash equivalents are maintained in checking, money market and mutual
fund investment accounts. The Company considers all highly liquid investments with maturities
remaining on the date of purchase of three months or less to be cash equivalents.
Available-for-Sale Securities
The Company held no investments in marketable securities as of June 30, 2010 and 2009. The
Company classifies its investments in marketable securities as available-for-sale. The cost of
securities sold is based on the specific-identification method. Interest on securities classified
as available-for-sale is included in interest income. Unrealized gains or losses on
available-for-sale securities are classified as other comprehensive income or loss and reported as
a separate component of stockholders’ equity until realized.
68
Restricted Cash
Under an operating lease for its facility in Redwood City, California, the Company is required
to maintain a letter of credit with a restricted cash balance at the Company’s bank. A certificate
of deposit for the amount of $150,000 and $300,000 at June 30, 2010 and 2009, respectively, has
been recorded as restricted cash in the accompanying balance sheets, related to the letter of
credit (see Note 5).
A certificate of deposit of $4,000 and $10,000 at June 30, 2010 and 2009, respectively, has
been recorded as restricted cash in the accompanying balance sheets related to the deposit on the
Company’s merchant credit card.
Concentrations of Credit Risk and Certain Other Risks
Financial instruments that potentially subject the Company to concentrations of credit risk
consist primarily of cash and cash equivalents, and accounts receivable. The Company places its
cash and cash equivalents with high-credit quality financial institutions. The Company is exposed
to credit risk in the event of default by the institutions holding the cash and cash equivalents
securities to the extent of the amounts recorded on the balance sheet.
The Company sells its products to hospitals in the U.S. and Europe and to distributors in
Japan and Saudi Arabia that resell the products to hospitals. The Company does not require
collateral to support credit sales. The Company has had no credit losses to date.
The following table illustrates total net revenue from the geographic location in which the
Company’s customers are located.
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Fiscal Year Ended June 30, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
United States |
|
|
63 |
% |
|
|
84 |
% |
|
|
82 |
% |
Japan |
|
|
23 |
% |
|
|
10 |
% |
|
|
13 |
% |
Europe |
|
|
13 |
% |
|
|
4 |
% |
|
|
4 |
% |
Rest of world |
|
|
1 |
% |
|
|
2 |
% |
|
|
1 |
% |
The following table illustrates the concentration of greater than 10% with any individual
customer.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Percent of Total Net |
|
|
Percent of Total |
|
| |
|
Revenue for |
|
|
Accounts Receivable |
|
| |
|
Fiscal Year Ended June 30, |
|
|
as of June 30, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
|
2010 |
|
|
2009 |
|
Century Medical |
|
|
22 |
% |
|
|
10 |
% |
|
|
13 |
% |
|
|
— |
|
|
|
10 |
% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cook Incorporated |
|
|
3 |
% |
|
|
30 |
% |
|
|
34 |
% |
|
|
— |
|
|
|
— |
|
The Company depends upon a number of key suppliers, including single source suppliers, the
loss of which would materially harm the Company’s business. Single source suppliers are relied upon
for certain components and services used in manufacturing the Company’s products. The Company does
not have long-term contracts with any of the suppliers; rather, purchase orders are submitted for
each order. Because long-term contracts do not exist, none of the suppliers are required to provide
the Company any guaranteed minimum quantities.
Inventories
Inventories are recorded at the lower of cost or market on a first-in, first-out basis. The
Company periodically assesses the recoverability of all inventories, including materials,
work-in-process and finished goods, to determine whether adjustments for impairment are required.
Inventory that is obsolete or in excess of forecasted usage is written down to its estimated net
realizable value based on assumptions about future demand and market conditions. Further reduced
demand may result in the need for additional
inventory write-downs in the near term. Inventory write-downs are charged to cost of product
sales and establish a lower cost basis for the inventory.
69
Property and Equipment
Property and equipment are stated at cost and depreciated on a straight-line basis over the
estimated useful lives of the related assets, which are generally three to five years. Amortization
of leasehold improvements is computed using the straight-line method over the shorter of the
remaining lease term or the estimated useful life of the related assets. Upon sale or retirement of
assets, the costs and related accumulated depreciation and amortization are removed from the
balance sheet and the resulting gain or loss is reflected in the statement of operations.
Impairment of Long-Lived Assets
The Company reviews long-lived assets, including property and equipment, for impairment
whenever events or changes in business circumstances indicate that the carrying amount of the
assets may not be fully recoverable. An impairment loss is recognized when estimated undiscounted
future cash flows expected to result from the use of the asset and its eventual disposition are
less than its carrying amount. Impairment, if any, is assessed using discounted cash flows. In the
year ended June 30, 2009, the Company recorded a loss of $475K related to the impairment of certain
assets specific to a product no longer under development. No other significant impairment losses
have been recorded through June 30, 2010.
Revenue Recognition
The Company recognizes revenue when four basic criteria are met: (1) persuasive evidence of an
arrangement exists; (2) title has transferred; (3) the fee is fixed or determinable; and (4)
collectability is reasonably assured. The Company uses contracts and customer purchase orders to
determine the existence of an arrangement. The Company uses shipping documents and third-party
proof of delivery to verify that title has transferred. The Company assesses whether the fee is
fixed or determinable based upon the terms of the agreement associated with the transaction. To
determine whether collection is probable, the Company assesses a number of factors, including past
transaction history with the customer and the creditworthiness of the customer. If the Company
determines that collection is not reasonably assured, then the recognition of revenue is deferred
until collection becomes reasonably assured, which is generally upon receipt of payment.
The Company records product sales net of estimated product returns and discounts from the list
prices for its products. The amounts of product returns and the discount amounts have not been
material to date. The Company includes shipping and handling costs in cost of product sales.
Each of the Company’s development contracts with Cook Incorporated is accounted for as a
separate arrangement. Revenue generated from development contracts is recognized when it is earned
and non-refundable upon receipt of milestone payments or upon incurrence of the related development
expenses in accordance with contractual terms, based on the actual costs incurred to date plus
overhead costs for certain project activities. Amounts paid but not yet earned on a project are
recorded as deferred revenue until such time as the related development expenses plus overhead
costs for certain project activities are incurred.
Research and Development
Research and development expenses consist of costs incurred for internally sponsored research
and development, direct expenses, research-related overhead expenses, and costs incurred on
development contracts. Research and development costs are charged to research and development
expenses as incurred.
70
Clinical Trials
The Company accrues and expenses costs for clinical trial activities performed by third
parties based upon estimates of the percentage of work completed over the life of the individual
study in accordance with agreements established with contract research organizations and clinical
trial sites. The Company determines the estimates through discussion with internal clinical
personnel and outside service providers as to progress or stage of completion of trials or services
and the agreed upon fee to be paid for such services. Costs of setting up clinical trial sites for
participation in the trials are expensed immediately as research and development expenses. Clinical
trial site costs related to patient enrollment are accrued as patients are entered into the trial.
Deferred Rent
Rent expense is recognized on a straight-line basis over the non-cancelable term of the
Company’s facility operating lease. The difference between the actual amounts paid and amounts
recorded as rent expense is recorded to deferred rent.
Income Taxes
The Company utilizes the liability method of accounting for income taxes. Under this method,
deferred tax assets and liabilities are determined based on differences between the financial
reporting and tax reporting bases of assets and liabilities and are measured using enacted tax
rates and laws that are expected to be in effect when the differences are expected to reverse.
Valuation allowances are established when necessary to reduce deferred tax assets to the amounts
expected to be realized.
The Company would classify interest and penalties related to uncertain tax positions in income
tax expense, if applicable. There was no interest expense or penalties related to unrecognized tax
benefits recorded through June 30, 2010.
Segments
The Company operates in one segment. Management uses one measurement of profitability and does
not segregate its business for internal reporting. All of the Company’s operations are in the
United States and all of its long-lived assets are maintained in the United States.
Comprehensive Loss
Comprehensive loss is comprised of net loss and unrealized holding gains and losses on
available-for-sale securities, if any, as follows (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Fiscal Year Ended June 30, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
Net loss |
|
$ |
(10,924 |
) |
|
$ |
(17,205 |
) |
|
$ |
(18,196 |
) |
Change in unrealized gain (loss) on investments |
|
|
— |
|
|
|
12 |
|
|
|
(10 |
) |
|
|
|
|
|
|
|
|
|
|
Comprehensive loss |
|
$ |
(10,924 |
) |
|
$ |
(17,193 |
) |
|
$ |
(18,206 |
) |
|
|
|
|
|
|
|
|
|
|
71
Net Loss per Share
Basic net loss per share is calculated by dividing the net loss by the weighted-average number
of common shares outstanding for the period less the weighted average unvested common shares
subject to repurchase and without consideration of potential common shares. Diluted net loss per
share is computed by dividing the net loss by the weighted-average number of common shares
outstanding for the period less the weighted average unvested common shares subject to repurchase
and dilutive potential common shares for the period determined using the treasury-stock method. For
purposes of this calculation, options and warrants to purchase stock and unvested restricted stock
awards are considered to be potential common shares and are only included in the calculation of
diluted net loss per share when their effect is dilutive (in thousands, except per share data).
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Fiscal Year Ended June 30, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
Numerator: |
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(10,924 |
) |
|
$ |
(17,205 |
) |
|
$ |
(18,196 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Denominator: |
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-average common shares outstanding |
|
|
21,935 |
|
|
|
15,815 |
|
|
|
14,893 |
|
Less: Weighted-average non-vested common shares
subject to repurchase |
|
|
— |
|
|
|
— |
|
|
|
(1 |
) |
Less: Weighted-average non-vested restricted stock awards |
|
|
(8 |
) |
|
|
(39 |
) |
|
|
(48 |
) |
|
|
|
|
|
|
|
|
|
|
Denominator for basic and diluted net loss per common share |
|
|
21,927 |
|
|
|
15,776 |
|
|
|
14,844 |
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per common share |
|
$ |
(0.50 |
) |
|
$ |
(1.09 |
) |
|
$ |
(1.23 |
) |
|
|
|
|
|
|
|
|
|
|
Outstanding securities not included in diluted net loss per common share calculation:
(in thousands)
| |
|
|
|
|
|
|
|
|
| |
|
As of June 30, |
|
| |
|
2010 |
|
|
2009 |
|
Options to purchase common stock |
|
|
3,062 |
|
|
|
1,446 |
|
Non-vested restricted stock units and awards |
|
|
46 |
|
|
|
229 |
|
Warrants |
|
|
4,706 |
|
|
|
648 |
|
|
|
|
|
|
|
|
|
|
|
7,814 |
|
|
|
2,323 |
|
|
|
|
|
|
|
|
Stock-Based Compensation
Stock-based compensation cost related to employee and director share-based compensation plans,
including stock options and restricted stock units, is measured on the grant date, based on the
fair value-based measurement of the award and is recognized as an expense over the requisite
service period. The Company recognizes compensation expense using the accelerated method.
The Company selected the Black-Scholes option pricing model for determining the estimated fair
value-based measurements of share-based awards. The use of the Black-Scholes model requires the use
of assumptions including expected term, expected volatility, risk-free interest rate and expected
dividends. The Company used the following assumptions in its fair value-based measurements:
| |
|
|
|
|
|
|
| |
|
Fiscal Year Ended June 30, |
| |
|
2010 |
|
2009 |
|
2008 |
Risk-free interest rate |
|
1.49% – 2.35% |
|
0.10% – 2.57% |
|
2.39% – 3.70% |
Dividend yield |
|
— |
|
— |
|
— |
Weighted-average expected life |
|
3.5 – 4.58 years |
|
0.25 – 4.5 years |
|
4.56 years |
Volatility |
|
68% – 81% |
|
53% – 136% |
|
58% |
Since the Company has limited historical data on volatility of its stock, the expected
volatility is based on the volatility of similar entities (referred to as “guideline” companies).
In evaluating similarity, the Company considered factors such as industry, stage of life cycle,
size, and financial leverage.
72
The expected term of options granted is determined using the “simplified” method. Under this
approach, the expected term is presumed to be the mid-point between the vesting date and the end of
the contractual term. The risk-free interest rate for the expected term of each option is based on
a risk-free zero-coupon spot interest rate at the time of grant. The Company recognizes the stock
compensation expense for option awards using the accelerated method over the requisite service
period of the award, which generally equals the vesting period of each grant. The Company has never
declared or paid any cash dividends and does not presently plan to pay cash dividends in the
foreseeable future. The Company estimates forfeitures in calculating the expense related to
stock-based compensation. The Company
recorded stock-based compensation expenses under ASC 718 of $1.2 million, or $0.06 per share,
$1.6 million, or $0.10 per share, and $1.4 million, or $0.09 per share for fiscal years ended June
30, 2010, 2009, and 2008, respectively. Total compensation expense related to unvested awards not
yet recognized is approximately $0.9 million at June 30, 2010 and is expected to be recognized over
a weighted average period of 2.8 years.
Included in the statement of operations are the following non-cash stock-based compensation
amounts for the periods reported, including non-employee stock based compensation expense and the
amortization of deferred compensation (in thousands).
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Fiscal Year Ended June 30, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
Cost of product sales |
|
$ |
178 |
|
|
$ |
187 |
|
|
$ |
51 |
|
Research and development |
|
|
329 |
|
|
|
522 |
|
|
|
605 |
|
Selling, general and administrative |
|
|
703 |
|
|
|
1,183 |
|
|
|
1,026 |
|
|
|
|
|
|
|
|
|
|
|
Total |
|
$ |
1,210 |
|
|
$ |
1,892 |
|
|
$ |
1,682 |
|
|
|
|
|
|
|
|
|
|
|
Recent Accounting Pronouncements
Effective July 1, 2009, the Company adopted the Financial Accounting Standards Board (the
“FASB”) Accounting Standards Codification (“ASC” or “Codification”). The Codification is the source
of authoritative accounting principles recognized by the FASB to be applied by nongovernmental
entities in the preparation of financial statements in conformity with GAAP and became effective
for interim periods and fiscal years ending on or after September 15, 2009. The Codification
explicitly recognizes rules and interpretive releases of the Securities and Exchange Commission
(“SEC”) under federal securities laws as authoritative GAAP for SEC registrants. The Company has
updated all existing GAAP references in these financial statements in accordance with the
Codification. As the Codification was not intended to change or alter existing GAAP, the adoption
of the Codification did not have any impact on the amounts included in the Company’s financial
statements.
In October 2009, the FASB issued Accounting Standards Update (“ASU”) No. 2009-13 which
addresses the accounting for multiple-element arrangements to potentially enable vendors to account
for products or services separately rather than as a combined unit. This guidance also modifies the
manner in which transaction consideration is allocated across the separately identified elements
and significantly expands the disclosure requirements for multiple-element revenue arrangements.
The Company will adopt this guidance as of July 1, 2010 and apply it prospectively to arrangements
entered into or materially modified after the adoption date. The Company does not expect the
adoption of ASU No. 2009-13 to have any impact on its results of operations or financial condition
upon its required adoption.
In April of 2010, ASC 605, “Revenue Recognition,” was amended to define a milestone and
clarify that the milestone method of revenue recognition is a valid application of the proportional
performance model when applied to research or development arrangements. Accordingly, a company can
make an accounting policy election to recognize a payment that is contingent upon the achievement
of a substantive milestone in its entirety in the period in which the milestone is achieved. This
guidance is effective for the Company as of July 1, 2010 and will be applied on a prospective
basis. The Company does not expect this guidance to have any impact on its results of operations
or financial condition upon its required adoption.
73
Note 2. Fair Value Measurements
ASC 820, “Fair Value Measurements and Disclosures,” defines fair value as the exchange price
that would be received for an asset or paid to transfer a liability (an exit price) in the
principal or most advantageous market for the asset or liability in an orderly transaction between
market participants at the measurement date. ASC 820 establishes a three-level fair value hierarchy
that prioritizes the inputs used to measure fair value. The three levels of inputs used to measure
fair value are as follows:
| |
|
|
Level 1 —
|
|
Quoted prices in active markets for identical assets or liabilities. |
|
|
|
Level 2 —
|
|
Observable inputs other than quoted prices included in Level 1,
such as quoted prices for similar assets and liabilities in
active markets; quoted prices for identical or similar assets
and liabilities in markets that are not active; or other inputs
that are observable or can be corroborated by observable market
data. |
|
|
|
Level 3 —
|
|
Unobservable inputs that are supported by little or no market
activity and that are significant to the fair value of the
assets or liabilities. |
The Company does not have any liabilities that are measured at fair value. All assets that
are measured at fair value have been segregated into the most appropriate level within the fair
value hierarchy based on the inputs used to determine the fair value at the measurement date. These
assets measured at fair value are summarized below (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
As of June 30, 2010 |
|
| |
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
|
Total |
|
Cash equivalents: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Money market funds |
|
$ |
511 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
511 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total assets at fair value |
|
$ |
511 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
511 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
As of June 30, 2009 |
|
| |
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
|
Total |
|
Cash equivalents: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Money market funds |
|
$ |
510 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
510 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total assets at fair value |
|
$ |
510 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
510 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash equivalents, consisting of funds held in money market instruments, are included in Level
1 as their fair value is based on market prices/quotes for identical assets in active markets.
As of June 30, 2010, the Company’s material financial assets and liabilities not carried at
fair value, including its trade accounts receivable, accounts payable, deferred development revenue
and note payable, are reported at their current carrying values which approximate fair value given
the short term nature of these instruments.
74
Note 3. Inventories
During the fiscal year ended June 30, 2009 the Company experienced reduced demand for the
Company’s C-Port systems and recorded a $248,000 write-down for excess and obsolete C-Port system
inventory at June 30, 2009. As of June 30, 2010, raw materials continue to include a write-down of
$246,000 related to this excess and obsolete C-Port inventory. Additionally, as of June 30, 2010,
the Company recorded a write-down of $189,000 primarily on its PAS-Port inventory to state the
finished goods inventory at the lower of cost or market due to higher product cost per unit
manufactured.
Inventories consisted of the following (in thousands):
| |
|
|
|
|
|
|
|
|
| |
|
June 30, |
|
| |
|
2010 |
|
|
2009 |
|
Raw materials |
|
$ |
250 |
|
|
$ |
256 |
|
Work in progress |
|
|
62 |
|
|
|
404 |
|
Finished goods |
|
|
819 |
|
|
|
1,235 |
|
|
|
|
|
|
|
|
|
|
$ |
1,131 |
|
|
$ |
1,895 |
|
|
|
|
|
|
|
|
Note 4. Property and Equipment
Property and equipment consisted of the following (in thousands):
| |
|
|
|
|
|
|
|
|
| |
|
June 30, |
|
| |
|
2010 |
|
|
2009 |
|
Computer hardware and software |
|
$ |
501 |
|
|
$ |
485 |
|
Office furniture and equipment |
|
|
220 |
|
|
|
220 |
|
Machinery and equipment |
|
|
5,545 |
|
|
|
5,122 |
|
Leasehold improvements |
|
|
567 |
|
|
|
567 |
|
Construction in process |
|
|
— |
|
|
|
149 |
|
|
|
|
|
|
|
|
|
|
|
6,833 |
|
|
|
6,543 |
|
Less: accumulated depreciation and amortization |
|
|
(5,495 |
) |
|
|
(4,681 |
) |
|
|
|
|
|
|
|
|
|
$ |
1,338 |
|
|
$ |
1,862 |
|
|
|
|
|
|
|
|
Note 5. Commitments and Contingencies
In April 2003, the Company entered into a non-cancelable operating lease for office
space that was scheduled to expire in July 2008. In December 2007, the Company entered into a
second amendment to its Office Lease Agreement extending the operating lease for its headquarters
through August 2011. Pursuant to the initial terms of the Operating Lease Agreement, the Company
obtained a letter of credit for $500,000 and, in order to obtain the letter of credit, the Company
placed cash funds in the amount of $500,000 in a certificate of deposit account. The security
deposit was reduced to $300,000 in the second amendment and further reduced to $150,000 in the
third amendment. The cash funds amount is restricted until the expiration of the lease agreement in
August 2011 and is recorded as non-current restricted cash.
Future minimum lease payments under the non-cancelable operating leases having initial
terms in excess of one year as of June 30, 2010, are as follows (in thousands):
| |
|
|
|
|
| |
|
Operating |
|
| Fiscal year ending June 30, |
|
Leases |
|
2011 |
|
|
858 |
|
2012 |
|
|
144 |
|
|
|
|
|
Total minimum lease payments |
|
$ |
1,002 |
|
|
|
|
|
75
Rent expense for fiscal years 2010, 2009 and 2008, was $832,000, $753,000 and $245,000,
respectively.
Note 6. License, Development and Commercialization Agreements
In June 2007, the Company entered into, and in September 2007 and in June 2009 amended, a
license, development and commercialization agreement with Cook Incorporated, or Cook, to develop
and commercialize a specialized device, referred to as the PFO device, designed to close holes in
the heart from genetic heart defects known as patent foramen ovales, or PFOs. Under the agreement,
Cook funded certain development activities and the Company and Cook jointly developed the device.
If developed, Cook would receive an exclusive, worldwide, royalty-bearing license, with the right
to grant sublicenses, to make, have made, use, sell, offer for sale and import the PFO device.
Under this agreement, the Company received payments of $1.0 million and $1.7 million in fiscal
years ended June 30, 2009 and 2008, respectively. The Company received no payments in fiscal 2010.
On January 6, 2010, the Company and Cook mutually agreed to suspend work on the PFO project. The
Company recorded as development revenue under the agreement a total of $124,000, $1.4 million and
$1.2 million in fiscal years ended June 30, 2010, 2009 and 2008, respectively. A total of $403,000
under this agreement has been recorded as deferred development revenue on the balance sheet as of
June 30, 2010. The Company is entitled to receive from Cook up to a total of an additional $275,000
in future payments if development milestones under the agreement are achieved. The Company is also
entitled to receive a royalty based on Cook’s annual worldwide sales of the PFO device, if any.
On December 9, 2005, the Company entered into, and in September 2007 amended and in July 2009
amended and partially terminated, an agreement with Cook to develop the Cook Vascular Closure
Device. Under the agreement, Cook funded certain development activities, and the Company and Cook
jointly developed the device, under the direction of a Development Committee that included
representatives from each party. Under the original agreement and the first amendment in September
2007, Cook received an exclusive, worldwide, royalty-bearing license, with the right to grant
sublicenses, to make, have made, use, sell, offer for sale and import the Cook Vascular Closure
Device for medical procedures in any part of the body. Under this agreement, the Company received
payments totaling approximately $5.3 million, including $0, $1.0 million and $1.5 million in fiscal
years ended June 30, 2010, 2009 and 2008, respectively. The Company recorded as development revenue
under the agreement a total of $0, $1.6 million and $1.4 million for fiscal years ended June 30,
2010, 2009, and 2008, respectively. In July 2009, the Company entered into a partial termination
and second amendment of this agreement to terminate Cook’s participation in the project and to
provide to Cook a royalty on net sales of the Cook Vascular Closure Device if Cardica successfully
commercializes the product. The remaining deferred revenue balance was recognized as revenue in the
fourth quarter of fiscal 2009 as all of the Company’s activities under this agreement had been
completed and no amounts were refundable to Cook. In addition, during fiscal year 2009, the Company
recognized a total of $251,000 of product sales to Cook of the Cook Vascular Closure Device.
Note 7. Related-Party Transactions
Financing Activities
In November 2007, the Company sold 1,500,000 shares of its common stock, and Guidant
Investment Corporation (“Guidant Investment”) sold 2,575,795 shares of the Company’s common stock,
in an underwritten public offering. The Company received net proceeds of approximately
$11.5 million. In December 2007, the Company received approximately $3.8 million in net proceeds
from the sale of an additional 481,170 shares of its common stock upon exercise of the
over-allotment option. The Company did not receive any funds from the sale of its common stock by
Guidant Investment. As of June 30, 2008, Guidant Investment was no longer a stockholder or related
party of the Company. In connection with the
sales of shares offered by the Company in 2007, Allen & Company LLC acted as a co-manager on
these transactions and received total fees of approximately $198,000.
76
In March 2009, the Company engaged Allen & Company LLC to help evaluate strategic
alternatives. A member of the Company’s Board of Directors, John Simon, is a Managing Director at
Allen & Company LLC. Allen & Company LLC received $200,000 in connection with this engagement.
Development and Supply Agreement
In December 2003, the Company entered into a Development and Supply Agreement with Guidant
Investment, a note holder and a related party for the development and commercialization of an
aortic cutter for Guidant, the Heartstring product. The agreement called for the Company to develop
and manufacture aortic cutters. Production of the aortic cutter has been outsourced by Guidant to a
third-party manufacturer, and the Company will receive royalties quarterly for each unit sold in
the future. During fiscal years ended June 30, 2010, 2009, and 2008, the Company received $93,000,
$85,000 and $67,000, respectively, of royalty revenue under this agreement. As of June 30, 2008,
Guidant Investment was no longer a related party of the Company.
Note 8. Note Payable
In June 2003, the Company entered into, and in March 2007 amended, a distribution agreement
with Century Medical, Inc. Also in June 2003, the Company issued a subordinated note to Century
Medical in the amount of $3.0 million due in June 2008 bearing 5% interest per annum. In March
2007, Century Medical and the Company restructured the note payable such that the note was no
longer subordinate, the Company paid $1.0 million in April 2007 and the remaining $2.0 million of
the note payable was due in June 2010.
On April 1, 2010, the Company entered into the Note Agreement Amendment, under which the
Company made a principal payment of $600,000 to Century Medical in April 2010, with the remaining
$1.4 million principal amount owed to Century Medical becoming due on June 17, 2011, which is one
year later than the maturity date prior to the Note Agreement Amendment. In the event that the
Company obtained at least $10 million in equity or debt financing during the period from April 1,
2010 through the new maturity date, the Company was required to make a payment of at least $400,000
of the then-outstanding principal within ten business days after the date on which the amount of
such aggregate financing was completed. In connection with the Note Agreement Amendment, the
Company entered into the Distribution Agreement Amendment, under which the Company agreed that,
during the time during which any amounts are outstanding under the Note Agreement, the Company will
not increase the price to Century Medical of the PAS-Port system being distributed by Century
Medical.
The note bore interest at 5% per annum through June 2008 and then increased to 6% per annum
until maturity. All interest due under the note to Century Medical was payable quarterly in arrears
on January 31, April 30, July 31, and October 31 of each year. The Company made interest payments
of $118,000, $117,000, and $101,000 in fiscal years ended June 30, 2010, 2009, and 2008,
respectively. The interest payable at June 30, 2010 and 2009 was $14,000 and $20,000, respectively,
and is included in other accrued liabilities in the accompanying balance sheets. Related to the
note, Century Medical had a continuing security interest in all of our personal property and
assets, including intellectual property, until the note was repaid. There were no covenants
associated with this debt. On August 17, 2010, we repaid the remaining $1.4 million principal
balance and interest due on the debt facility.
Note 9. Stockholders’ Equity
The total number of shares that the Company is authorized to issue is 50,000,000 shares, with
45,000,000 shares designated as common stock and 5,000,000 shares designated as preferred stock.
77
Private Placement Offering
On September 30, 2009, institutional and individual investors, including existing stockholders,
purchased approximately $10.2 million of the Company’s common stock and warrants to purchase the
Company’s common stock in a private placement (the “Private Placement”). The net proceeds were
approximately $9.9 million after offering expenses. Under the terms of the purchase agreement with
these investors, the Company sold 8,142,082 units at a purchase price of $1.2525 per unit, with
each unit consisting of one share of common stock and one warrant to purchase 0.50 of a share of
common stock, or 4,071,046 shares of the Company’s common stock. The warrants are exercisable
commencing on April 1, 2010 at $1.45 per share and will expire five years after the date of
issuance. There were no underwriters or placement agents involved with the Private Placement, and
no underwriting discounts or commissions or similar fees were payable in connection with the
Private Placement. The issuance was made in reliance upon exemptions from registration pursuant to
Section 4(2) under the Securities Act of 1933, as amended, (the “Securities Act”) and Rule 506
promulgated thereunder, and was made without general solicitation or advertising. Each investor
represented that it was an accredited investor with access to information about us sufficient to
evaluate the investment and that the common stock and warrants were being acquired without a view
to distribution or resale in violation of the Securities Act.
Public Offering
In November 2007, the Company sold 1,500,000 shares of its common stock, and Guidant
Investment sold 2,575,795 shares of the Company’s common stock, in a public offering for aggregate
gross proceeds to the Company of $12.7 million. After deducting the underwriters’ commissions and
discounts and other issuance costs, the Company received net proceeds of $11.5 million. The
Company did not receive any proceeds from the sale of its common stock by Guidant Investment.
In December 2007, the Company sold an additional 481,170 shares of its common stock to the
underwriters pursuant to the exercise of the over-allotment option. The Company received aggregate
gross proceeds of $4.1 million from the exercise of the underwriters’ over-allotment option. After
deducting the underwriters’ commission and related expenses, the Company received from the exercise
of the underwriters’ over-allotment option net proceeds of $3.8 million.
Common Stock
Holders of common stock are entitled to one vote per share on all matters to be voted upon by
the stockholders of the Company. Subject to the preferences that may be applicable to any
outstanding shares of preferred stock, the holders of common stock are entitled to receive ratably
such dividends, if any, as may be declared by the Board of Directors. No dividends have been
declared to date.
Preferred Stock
The Company has 5,000,000 shares of authorized preferred stock issuable in one or more series.
Upon issuance the Company can determine the rights, preferences, privileges and restrictions
thereof. These rights, preferences and privileges could include dividend rights, conversion rights,
voting rights, terms of redemption, liquidation preferences, sinking fund terms and the number of
shares constituting any series or the designation of such series, any or all of which may be
greater than the rights of common stock. The issuance of the preferred stock could adversely affect
the voting power of holders of common stock and the likelihood that such holders will receive
dividend payments and payments upon liquidation. In addition, the issuance of preferred stock could
have the effect of delaying, deferring or preventing a change in control of the Company or other
corporate action. There was no preferred stock outstanding as of June 30, 2010 or 2009.
78
Shares Reserved
Shares of common stock reserved for future issuance are as follows:
| |
|
|
|
|
| |
|
June 30, |
|
| |
|
2010 |
|
Stock options outstanding |
|
|
3,061,926 |
|
Shares available for grant under stock option plan |
|
|
794,135 |
|
Warrants for common stock |
|
|
4,706,410 |
|
|
|
|
|
|
|
|
8,562,471 |
|
|
|
|
|
Stock Options
In 1997, the Company adopted the 1997 Equity Incentive Plan (the “1997 Plan”). The 1997 Plan
provides for the granting of options to purchase common stock and the issuance of shares of common
stock, subject to Company repurchase rights, to directors, employees and consultants. Certain
options are immediately exercisable, at the discretion of the Board of Directors. Shares issued
pursuant to the exercise of an unvested option are subject to the Company’s right of repurchase
which lapses over periods specified by the board of directors, generally four years from the date
of grant. In February 2006, the Company terminated all remaining unissued shares under the 1997
Plan. Although the 1997 Plan terminated, all outstanding options thereunder will continue to be
governed by their existing terms.
In October 2005, the Company’s Board of Directors adopted, and in December 2005 the
stockholders approved, the 2005 Equity Incentive Plan, as amended (the “2005 Plan”). A total of
3,400,000 shares of common stock have been reserved for issuance under the 2005 Plan.
Stock awards granted under the 2005 Plan may either be incentive stock options, nonstatutory
stock options, stock bonuses or rights to acquire restricted stock. Incentive stock options may be
granted to employees with exercise prices of no less than the fair value of the common stock on the
date of grant, as determined by the Board of Directors, and nonstatutory options may be granted to
employees, directors or consultants at exercise prices of no less than the fair value. If, at the
time the Company grants an option, the optionee directly or by attribution owns stock possessing
more than 10% of the total combined voting power of all classes of stock of the Company, the option
price shall be at least 110% of the fair value and shall not be exercisable more than five years
after the date of grant. Options may be granted with vesting terms as determined by the Board of
Directors. Options expire no more than 10 years after the date of grant, or earlier if employment
is terminated.
Common stock options may include a provision whereby the holder, while an employee, director
or consultant, may elect at any time to exercise the option as to any part or all of the shares
subject to the option prior to the full vesting of the option. Any unvested shares so purchased are
subject to repurchase by the Company at its option and at a price equal to the original purchase
price of the stock. The Company does not consider the stock issued upon exercise of an unvested
stock option substantively exercised, and the cash paid for the exercise price is considered a
deposit or a prepayment of the exercise price that is recognized by the Company as a liability. As
the underlying shares vest, the deposit liability is reclassified as equity. As of June 30, 2010
and 2009, no such shares are subject to the Company’s right of repurchase and excluded from
stockholders’ equity.
79
Option activity under all Plans is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
Outstanding Options |
|
| |
|
|
|
|
|
|
|
|
|
Weighted-
Average |
|
| |
|
Shares |
|
|
|
|
|
|
Exercise |
|
| |
|
Available for |
|
|
Number of |
|
|
Price Per |
|
| |
|
Grant |
|
|
Shares |
|
|
Share |
|
Balance at June 30, 2007 |
|
|
117,967 |
|
|
|
1,316,006 |
|
|
$ |
4.67 |
|
Shares reserved |
|
|
500,000 |
|
|
|
— |
|
|
|
— |
|
Options granted |
|
|
(155,450 |
) |
|
|
155,450 |
|
|
|
7.61 |
|
Restricted stock awards |
|
|
(52,450 |
) |
|
|
— |
|
|
|
— |
|
Options exercised |
|
|
— |
|
|
|
(77,036 |
) |
|
|
2.19 |
|
Options forfeited |
|
|
60,288 |
|
|
|
(60,288 |
) |
|
|
5.98 |
|
|
|
|
|
|
|
|
|
|
|
|
Balance at June 30, 2008 |
|
|
470,355 |
|
|
|
1,334,132 |
|
|
|
5.10 |
|
Shares reserved |
|
|
500,000 |
|
|
|
— |
|
|
|
— |
|
Options granted |
|
|
(530,050 |
) |
|
|
530,050 |
|
|
|
8.03 |
|
Restricted stock awards |
|
|
(250,850 |
) |
|
|
— |
|
|
|
— |
|
Options exercised |
|
|
— |
|
|
|
(44,144 |
) |
|
|
3.33 |
|
Options forfeited |
|
|
373,889 |
|
|
|
(373,889 |
) |
|
|
7.09 |
|
Awards
forfeited |
|
|
55,250 |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
Balance at June 30, 2009 |
|
|
618,594 |
|
|
|
1,446,149 |
|
|
|
5.65 |
|
Shares reserved |
|
|
1,750,000 |
|
|
|
— |
|
|
|
— |
|
Options granted |
|
|
(2,217,785 |
) |
|
|
2,217,785 |
|
|
|
1.28 |
|
Restricted stock awards |
|
|
(66,425 |
) |
|
|
— |
|
|
|
— |
|
Options exercised |
|
|
— |
|
|
|
(8,257 |
) |
|
|
1.81 |
|
Options forfeited |
|
|
593,751 |
|
|
|
(593,751 |
) |
|
|
4.29 |
|
Awards
forfeited |
|
|
116,000 |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
Balance at June 30, 2010 |
|
|
794,135 |
|
|
|
3,061,926 |
|
|
$ |
2.63 |
|
|
|
|
|
|
|
|
|
|
|
|
The following table summarizes information about options outstanding, vested and
exercisable at June 30, 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Options Outstanding |
|
|
Options exercisable |
|
| |
|
|
|
|
|
Weighted- |
|
|
Weighted |
|
|
|
|
|
|
Weighted |
|
| |
|
|
|
|
|
Average |
|
|
Average |
|
|
|
|
|
|
Average |
|
| |
|
|
|
|
|
Remaining |
|
|
Exercise |
|
|
|
|
|
|
Exercise |
|
| |
|
Number of |
|
|
Contractual |
|
|
Price per |
|
|
Number of |
|
|
Price per |
|
| Exercise Prices |
|
Shares |
|
|
Life (years) |
|
|
Share |
|
|
Shares |
|
|
Share |
|
$1.12 |
|
|
1,257,202 |
|
|
|
6.36 |
|
|
$ |
1.12 |
|
|
|
211,725 |
|
|
$ |
1.12 |
|
$1.21 – $1.55 |
|
|
761,743 |
|
|
|
6.05 |
|
|
|
1.46 |
|
|
|
385,708 |
|
|
|
1.48 |
|
$1.71 – $4.53 |
|
|
512,674 |
|
|
|
5.28 |
|
|
|
2.57 |
|
|
|
432,220 |
|
|
|
2.69 |
|
$4.60 – $9.00 |
|
|
291,691 |
|
|
|
4.26 |
|
|
|
6.91 |
|
|
|
240,716 |
|
|
|
7.00 |
|
$9.20 |
|
|
223,618 |
|
|
|
5.10 |
|
|
|
9.20 |
|
|
|
107,520 |
|
|
|
9.20 |
|
$9.75 |
|
|
14,998 |
|
|
|
5.46 |
|
|
|
9.75 |
|
|
|
14,998 |
|
|
|
9.75 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total outstanding |
|
|
3,061,926 |
|
|
|
5.81 |
|
|
$ |
2.63 |
|
|
|
1,392,887 |
|
|
$ |
3.44 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options vested and
expected to vest |
|
|
2,795,805 |
|
|
|
5.76 |
|
|
$ |
2.72 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The weighted average remaining contractual life for all currently exercisable options as of
June 30, 2010 was 5.2 years. The aggregate intrinsic value as of June 30, 2010 of all outstanding
options was $788,000, options vested and expected to vest was $681,000 and options exercisable was
$170,000. The
aggregate intrinsic value as of June 30, 2009 of all outstanding options was $2,000, options
vested and expected to vest was $2,000 and options exercisable was $2,000.
The weighted-average estimated grant date fair value of options granted to employees and
directors during fiscal years 2010, 2009, and 2008 was $0.70, $3.69, and $3.25, respectively. The
intrinsic value of all options exercised during fiscal years 2010, 2009, and 2008 was $4,000,
$189,000, and $447,000, respectively. The fair value of all stock awards actually vesting in fiscal
years 2010, 2009, and 2008 was $594,000, $740,000, and $850,000, respectively.
80
Restricted Stock Units and Awards
The following table summarizes information about restricted stock activity.
| |
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
Weighted- |
|
| |
|
|
|
|
|
Average |
|
| |
|
|
|
|
|
Grant- |
|
| |
|
|
|
|
|
Date Fair |
|
| |
|
|
|
|
|
Value per |
|
| |
|
Shares |
|
|
Share |
|
Non-vested restricted stock at June 30, 2007 |
|
|
12,917 |
|
|
|
|
|
Awarded |
|
|
52,450 |
|
|
$ |
11.25 |
|
Vested |
|
|
(5,950 |
) |
|
|
|
|
Forfeited |
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
Non-vested restricted stock at June 30, 2008 |
|
|
59,417 |
|
|
|
|
|
Awarded |
|
|
250,850 |
|
|
$ |
3.62 |
|
Vested |
|
|
(26,167 |
) |
|
|
|
|
Forfeited |
|
|
(55,250 |
) |
|
|
|
|
|
|
|
|
|
|
|
Non-vested restricted stock at June 30, 2009 |
|
|
228,850 |
|
|
|
|
|
Awarded |
|
|
— |
|
|
|
|
|
Vested |
|
|
(66,425 |
) |
|
|
|
|
Forfeited |
|
|
(116,000 |
) |
|
|
|
|
|
|
|
|
|
|
|
Non-vested restricted stock at June 30, 2010 |
|
|
46,425 |
|
|
|
|
|
|
|
|
|
|
|
|
|
The fair value of each restricted stock award is estimated based upon the closing price of the
Company’s common stock on the grant date. Share-based compensation expense related to restricted
stock units and awards is recognized over the requisite service period as adjusted for estimated
forfeitures.
Warrants
The Company has outstanding warrants to purchase common stock that are all exercisable at June
30, 2010 as follows:
| |
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
Exercise |
|
|
|
|
| |
|
|
|
Price Per |
|
|
|
|
| Shares |
|
|
Share |
|
|
Expiration |
|
| |
4,071,046 |
|
|
$ |
1.45 |
|
|
September 2014 |
| |
575,347 |
|
|
|
5.65 |
|
|
June 2012 |
| |
60,017 |
|
|
|
11.58 |
|
|
October 2010 |
| |
|
|
|
|
|
|
|
|
|
| |
4,706,410 |
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
Note 10. Reductions in Force
During the fiscal year ended June 30, 2009, the Company reduced its workforce by 51 employees
in an effort to reduce the Company’s operating expenses. The total charge incurred during the
fiscal year ended June 30, 2009 in connection with this reduction in workforce was approximately
$847,000, of which
approximately $798,000 was paid as severance payments and the balance was for outplacement
services. The charges were included in research and development expense and selling, general and
administrative expense.
Note 11. Income Taxes
Under the Housing and Economic Recovery Act of 2008 and the American Recovery and Reinvestment
Act of 2009 ( the “Acts”), signed into law in July 2008 and February 2009, respectively, taxpayers
can claim refundable AMT or research and development credit if they forego bonus depreciation on
certain qualified fixed assets placed in service between April 2008 and December 2009. The Company
computed and recognized a credit based on fixed assets placed into service in the fiscal years
ended June 30, 2010 and 2009. The Company recorded an income tax benefit of $31,000 and $72,000 in
the fiscal years ended June 30, 2010 and 2009, respectively, for the U.S. federal refundable credit
as provided by the Acts.
81
Deferred income taxes reflect the net tax effects of net operating loss and tax
credit carryovers and temporary differences between the carrying amounts of assets and liabilities
for financial reporting purposes and the amounts used for income tax purposes. Significant
components of the Company’s deferred tax assets are as follows (in thousands):
| |
|
|
|
|
|
|
|
|
| |
|
June 30, |
|
| |
|
2010 |
|
|
2009 |
|
Net operating loss carry-forwards |
|
$ |
39,267 |
|
|
$ |
36,008 |
|
Research credits |
|
|
1,747 |
|
|
|
2,340 |
|
Capitalized research and development expenses |
|
|
77 |
|
|
|
107 |
|
Fixed asset depreciation |
|
|
365 |
|
|
|
291 |
|
Other |
|
|
795 |
|
|
|
462 |
|
|
|
|
|
|
|
|
Total deferred tax assets |
|
|
42,251 |
|
|
|
39,208 |
|
Valuation allowance |
|
|
(42,251 |
) |
|
|
(39,208 |
) |
|
|
|
|
|
|
|
Net deferred tax assets |
|
$ |
— |
|
|
$ |
— |
|
|
|
|
|
|
|
|
Realization of the deferred tax assets is dependent upon future taxable income, if any, the
amount and timing of which are uncertain. Accordingly, the net deferred tax assets have been fully
offset by a valuation allowance. The net valuation allowance increased by approximately $3.0
million, $6.3 million, and $3.8 million during fiscal years ended June 30, 2010, 2009, and 2008,
respectively.
As of June 30, 2010, the Company had federal net operating loss carry-forwards and research
credit carry-forwards of approximately $104.5 million and $0.6 million, respectively. The net
operating loss carry-forwards begin to expire in the fiscal year 2013. The research credit
carry-forwards begin to expire in the fiscal year 2021. Additionally,
the Company’s state net
operating loss carry-forwards of approximately $75.2 million begin to expire in the fiscal year
2013. The Company has state research credit carry-forwards of $2.3 million which have no expiration
date.
Included in the valuation allowance balance as of June 30, 2010 is $0.3 million related to the
exercise of stock options which are not reflected as an expense for financial reporting purposes.
Accordingly, any future reduction in the valuation allowance relating
to this amount will be credited
directly to equity and not reflected as an income tax benefit in the Statement of Operations.
82
The reconciliation of income tax benefits attributable to the net loss computed at the U.S.
federal statutory rates to the income tax benefit recorded (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Fiscal Year Ended June 30, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
Tax benefit at U.S. statutory rate |
|
$ |
(3,725 |
) |
|
$ |
(5,874 |
) |
|
$ |
(6,186 |
) |
Loss for which no tax benefit is currently recognizable |
|
|
3,428 |
|
|
|
5,201 |
|
|
|
5,726 |
|
Refundable research credits |
|
|
(31 |
) |
|
|
(72 |
) |
|
|
— |
|
Stock based
compensation |
|
|
279 |
|
|
|
616 |
|
|
|
404 |
|
Other, net |
|
|
18 |
|
|
|
57 |
|
|
|
56 |
|
|
|
|
|
|
|
|
|
|
|
|
|
$ |
(31 |
) |
|
$ |
(72 |
) |
|
$ |
— |
|
|
|
|
|
|
|
|
|
|
|
Utilization of the net operating loss carry-forwards and credit carry-forwards may be subject
to a substantial annual limitation due to the limitations set forth in Sections 382 and 383 of the
Internal Revenue Code of 1986, as amended, and similar state
provisions. In the fiscal year ended June 30, 2010, the Company concluded a
detailed analysis to determine whether an ownership change under Section 382 of the Internal
Revenue Code had occurred. The effect of an ownership change would be the imposition of an annual
limitation on the use of the net operating loss carry-forwards and credit carry-forwards
attributable to periods before the change. The Company concluded that approximately $4.9 million of
federal net operating loss carry-forwards, $1.5 million of federal
credit carry-forwards, $122,000 of California state credit carry-forwards and
approximately $19.5 million of California state net operating loss carry-forwards are significantly
limited to offset future income, if any. The reductions are reflected in the carry-forward amounts
included above.
At June 30, 2010, the Company had unrecognized tax benefits of $595,000, all of which would
not currently affect the Company’s effective tax rate if recognized due to the Company’s deferred
tax assets being fully offset by a valuation allowance. A reconciliation of the beginning and
ending amount of unrecognized tax benefits is as follows (in thousands):
| |
|
|
|
|
| |
|
Amount |
|
Balance at July 1, 2007 |
|
$ |
574 |
|
Additions based on tax positions related to current year |
|
|
72 |
|
Reductions for tax positions of prior year |
|
|
(7 |
) |
|
|
|
|
Balance at June 30, 2008 |
|
|
639 |
|
Additions based on tax positions related to current year |
|
|
106 |
|
Additions for tax positions of prior year
|
|
|
10 |
|
Reductions for tax positions of prior year |
|
|
(18 |
) |
|
|
|
|
Balance at June 30, 2009 |
|
|
737 |
|
Additions based on tax positions related to current year |
|
|
77 |
|
Reductions for tax positions of current year |
|
|
(11 |
) |
Reductions for tax positions of prior year
|
|
|
(208 |
) |
|
|
|
|
Balance at June 30, 2010 |
|
$ |
595 |
|
|
|
|
|
The Company would classify interest and penalties related to uncertain tax positions in income
tax expense, if applicable. There was no interest expense or penalties related to unrecognized tax
benefits recorded through June 30, 2010. The tax years 1998 through 2010 remain open to
examination by one or more major taxing jurisdictions to which the Company is subject.
The Company does not anticipate that total unrecognized tax benefits will significantly change
prior to June 30, 2011 other than in the normal course of business.
Note 12. Employee Benefit Plan
In January 2001, the Company adopted a 401(k) Profit Sharing Plan that allows voluntary
contributions by eligible employees. Employees may elect to contribute up to the maximum allowed
under the Internal Revenue Service regulations. The Company may make discretionary contributions as
determined by the Board of Directors. No amount was contributed by the Company to the plan during
fiscal years ended June 30, 2010, 2009 or 2008.
Note 13. Indemnification
From time to time, the Company enters into contracts that require the Company, upon the
occurrence of certain contingencies, to indemnify parties against third-party claims. These
contingent obligations primarily relate to (i) claims against the Company’s customers for violation
of third-party intellectual property rights caused by the Company’s products; (ii) claims resulting
from personal injury or property damage resulting from the Company’s activities or products;
(iii) claims by the Company’s office lessor arising out of the Company’s use of the premises; and
(iv) agreements with the Company’s officers and directors under which the Company may be required
to indemnify such persons for liabilities arising out of their activities on behalf of the Company.
Because the obligated amounts for these types of agreements usually are not explicitly stated, the
overall maximum potential amount of these obligations cannot be reasonably estimated. No
liabilities have been recorded for these obligations on the Company’s balance sheets as of June 30,
2010 or 2009 as there are no amounts currently estimable or probable of payment.
83
Note 14. Subsequent Event
On August 16, 2010, the Company entered into a License Agreement with Intuitive Surgical
Operations, Inc. (“Intuitive Surgical”), a subsidiary of Intuitive Surgical, Inc.,
pursuant to which the Company granted to Intuitive Surgical a worldwide, sublicenseable, exclusive
license to use the Company’s intellectual property in the robotics field in diagnostic or
therapeutic medical procedures, but excluding vascular anastomosis applications (the “License
Agreement”) for an upfront license fee of $9 million. The Company will also be eligible to receive
a milestone payment if sales of any products incorporating the Company’s patent rights achieve a
specified level of net sales within a specified period after the date of the License Agreement and
will also be eligible to receive single-digit royalties on sales by Intuitive Surgical, its
affiliates or its sublicensees of specified stapler and clip applier products covered by the
Company’s patent rights as well as on sales of certain other products covered by the Company’s
patent rights that may be developed in the future.
In addition, on the same date, the Company and Intuitive Surgical entered into a Stock Purchase
Agreement pursuant to which Intuitive Surgical paid $3 million to purchase from the Company an aggregate of
1,249,541 newly-issued shares of the Company’s common stock.
2. Financial Statement Schedules
All financial statement schedules are omitted because the information is not applicable or is
presented in the Financial Statements or Notes thereto.
84
3. The following exhibits are included herein or incorporated herein by reference:
(a) Exhibits
| |
|
|
|
|
| Exhibit |
|
|
| Number |
|
Description |
| |
|
|
|
|
| |
3.1 |
(1) |
|
Amended and Restated Certificate of Incorporation of the Registrant as currently in effect. |
| |
3.2 |
(6) |
|
Bylaws of the Registrant as currently in effect. |
| |
3.3 |
(1) |
|
Specimen Common Stock certificate of the Registrant. |
| |
4.1 |
(1) |
|
Warrant dated March 17, 2000 exercisable for 36,810 shares of common stock (on a pre-split
basis). |
| |
4.2 |
(1) |
|
Warrant dated October 31, 2002 exercisable for 180,052 shares of common stock (on a
pre-split basis). |
| |
4.3 |
(2) |
|
Form of Warrant dated June 2007. |
| |
4.4 |
(12) |
|
Form of Warrant dated September 2009. |
| |
10.1 |
(1) |
|
1997 Equity Incentive Plan and forms of related agreements and documents. + |
| |
10.2 |
(3) |
|
2005 Equity Incentive Plan and forms of related agreements and documents. + |
| |
10.3 |
(1) |
|
Office Lease Agreement dated April 25, 2003, and First Amendment to Office Lease Agreement
dated January 21, 2004. |
| |
10.4 |
(8) |
|
Second Amendment to Office Lease Agreement, executed and delivered on December effective
November 19, 2007. |
| |
10.5 |
(1) |
|
Distribution Agreement by and between Cardica, Inc. and Century Medical, Inc. dated
June 16, 2003.† |
| |
10.6 |
(4) |
|
First Amendment to Distribution Agreement, dated March 30, 2007, by and between Cardica,
Inc. and Century Medical, Inc.† |
| |
10.7 |
|
|
Amendment No. 2 to Distribution Agreement, dated June 13, 2007, by and between Cardica,
Inc. and Century Medical, Inc. † |
| |
10.8 |
|
|
Amendment No. 3 to Distribution Agreement, dated January 24, 2008, by and between Cardica,
Inc. and Century Medical, Inc. |
| |
10.9 |
(13) |
|
Amendment No. 4 to Distribution Agreement, dated April 1, 2010, by and between Cardica,
Inc. and Century Medical, Inc.† |
| |
10.10 |
(1) |
|
Subordinated Convertible Note Agreement with Century Medical, Inc. dated June 16, 2003,
and Amendment No. 1 thereto, dated August 6, 2003.† |
| |
10.11 |
(4) |
|
Amendment No. 2 to Subordinated Convertible Note Agreement, dated March 30, 2007, by and
between Cardica, Inc. and Century Medical, Inc. † |
| |
10.12 |
(13) |
|
Amendment No. 3 to Subordinated Convertible Note Agreement, dated April 1, 2010, by and
between Cardica, Inc. and Century Medical, Inc. † |
| |
10.13 |
(4) |
|
Amended and Restated Note issued pursuant to Amendment No. 2 to Subordinated Convertible
Note Agreement with Century Medical, Inc. |
| |
10.14 |
(2) |
|
Registration Rights Agreement, dated June 7, 2007, by and among Cardica, Inc., and the
purchasers listed on the signature pages thereto. |
| |
10.15 |
(16) |
|
Additional Compensation Information for named executive officers. + |
| |
10.16 |
|
|
Cardica, Inc. Non-Employee Director Compensation. + |
| |
10.17 |
(1) |
|
Benefit Agreement with Bernard Hausen, M.D., Ph.D.+ |
| |
10.18 |
(9) |
|
Letter Agreement with Frederic M. Bauer+ |
| |
10.19 |
(10) |
|
Cardica, Inc. Change in Control and Severance Benefit Plan. + |
| |
10.20 |
(11) |
|
Form of Restricted Stock Unit Grant Notice and Restricted Stock Unit Grant Agreement. + |
| |
10.21 |
(5) |
|
License, Development and Commercialization Agreement by and between the Company and Cook
Incorporated, dated June 12, 2007. |
| |
10.22 |
(7) |
|
Amendment to License, Development and Commercialization Agreement by and between Cardica,
Inc. and Cook Incorporated, dated September 19, 2007. |
| |
10.23 |
(15) |
|
Second Amendment to License, Development and Commercialization Agreement by and between
Cardica, Inc. and Cook Incorporated, dated June 19, 2009. |
| |
10.24 |
(12) |
|
Securities Purchase Agreement, dated September 29, 2009, by and among Cardica, Inc., and
purchasers listed on the signature pages thereto. |
| |
10.25 |
(12) |
|
Registration Rights Agreement, dated September 25, 2009, by and among Cardica, Inc., and
the purchasers listed on the signature pages thereto. |
85
| |
|
|
|
|
| Exhibit |
|
|
| Number |
|
Description |
| |
|
|
|
|
| |
10.26 |
(14) |
|
Stock Purchase Agreement, dated August 16, 2010, by and between Cardica, Inc., and
Intuitive Surgical Operations, Inc. |
| |
10.27 |
(14) |
|
Registration Rights Agreement, dated August 16, 2010, by and between Cardica, Inc., and
Intuitive Surgical Operations, Inc. |
| |
10.28 |
|
|
License Agreement, dated August 16, 2010, by and between Cardica, Inc., and Intuitive
Surgical Operations, Inc. † |
| |
21.1 |
(1) |
|
Subsidiaries of Registrant. |
| |
23.1 |
|
|
Consent of Independent Registered Public Accounting Firm. |
| |
24.1 |
|
|
Power of Attorney (included on signature page). |
| |
31.1 |
|
|
Certification of chief executive officer. |
| |
31.2 |
|
|
Certification of chief financial officer. |
| |
32.1 |
|
|
Section 1350 Certification. |
| |
|
|
| † |
|
Portions of this exhibit (indicated by asterisks) have been omitted
pursuant to a request for confidential treatment and this exhibit has
been filed separately with the SEC. |
| |
| + |
|
Indicates management contract or compensatory plan. |
| |
| (1) |
|
Filed as an exhibit to the Registrant’s Registration Statement on
Form S-1, file no. 333-129497, declared effective on February 2,
2006, as amended, and incorporated herein by reference. |
| |
| (2) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on June 13, 2007,
excluding Item 3.02 and incorporated herein by reference. |
| |
| (3) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on November 13, 2009 and
incorporated herein by reference. |
| |
| (4) |
|
Filed as exhibits to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on April 5, 2007 and
incorporated herein by reference. |
| |
| (5) |
|
Filed as exhibits to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on June 21, 2007,
excluding Items 3.01 and incorporated herein by reference. |
| |
| (6) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on August 19, 2008 and
incorporated herein by reference. |
| |
| (7) |
|
Filed as an exhibit to the Company’s Quarterly Report on Form 10-Q
for the quarterly period ended September 30, 2007 and incorporated
herein by reference. |
| |
| (8) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on December 5, 2007 and
incorporated herein by reference. |
| |
| (9) |
|
Filed as an exhibit to the Company’s Quarterly Report on Form 10-Q
filed with the Securities and Exchange Commission on November 7, 2008
and incorporated herein by reference. |
| |
| (10) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on February 18, 2009 and
incorporated herein by reference. |
| |
| (11) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on February 20, 2009 and
incorporated herein by reference. |
| |
| (12) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on September 29, 2009 and
incorporated herein by reference. |
| |
| (13) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on April 7, 2010 and
incorporated herein by reference. |
| |
| (14) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on August 20, 2010 and
incorporated herein by reference. |
| |
| (15) |
|
Filed as an exhibit to the Company’s Annual Report on Form 10-K filed
with the Securities and Exchange Commission on September 18, 2009 and
incorporated herein by reference. |
| |
| (16) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on August 18, 2009 and
incorporated herein by reference. |
(b) Financial Statement Schedules
None.
86
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly
caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| |
|
|
|
|
Cardica, Inc.
Registrant |
|
|
|
September 24, 2010
Date
|
|
/s/ Robert Y. Newell
Robert Y. Newell |
|
|
Chief Financial Officer |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes
and appoints Robert Y. Newell, as his true and lawful attorney-in-fact and agent, with full power
of substitution for him, and in his name in any and all capacities, to sign any and all amendments
to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents
in connection therewith, with the Securities and Exchange Commission, granting unto said
attorney-in-fact and agent, full power and authority to do and perform each and every act and thing
requisite and necessary to be done therewith, as fully to all intents and purposes as he might or
could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent, and
any of them or his or her substitute or substitutes, may lawfully do or cause to be done by virtue
hereof.
Pursuant to the requirements of the Securities Act of 1934, this report has been signed by the
following persons on behalf of the Registrant in the capacities indicated on the date set forth
below:
| |
|
|
|
|
| Name and Signature |
|
Title |
|
Date |
|
|
|
|
|
/s/ Bernard A. Hausen
Bernard A. Hausen, M.D., Ph.D.
|
|
President, Chief Executive
Officer and Director
(Principal Executive Officer)
|
|
September 24, 2010 |
|
|
|
|
|
/s/ Robert Y. Newell
Robert Y. Newell
|
|
Chief Financial Officer
(Principal Financial and
Accounting Officer)
|
|
September 24, 2010 |
|
|
|
|
|
/s/ Kevin T. Larkin
Kevin T. Larkin
|
|
Director
|
|
September 24, 2010 |
|
|
|
|
|
/s/ Richard P. Powers
Richard P. Powers
|
|
Director
|
|
September 24, 2010 |
|
|
|
|
|
/s/ Jeffrey L. Purvin
Jeffrey L. Purvin
|
|
Director
|
|
September 24, 2010 |
|
|
|
|
|
/s/ John Simon
John Simon, Ph.D.
|
|
Director
|
|
September 24, 2010 |
|
|
|
|
|
/s/ William H. Younger, Jr.
William H. Younger, Jr.
|
|
Director
|
|
September 24, 2010 |
87
| |
|
|
|
|
| Exhibit |
|
|
| Number |
|
Description |
| |
|
|
|
|
| |
3.1 |
(1) |
|
Amended and Restated Certificate of Incorporation of the Registrant as currently in effect. |
| |
3.2 |
(6) |
|
Bylaws of the Registrant as currently in effect. |
| |
3.3 |
(1) |
|
Specimen Common Stock certificate of the Registrant. |
| |
4.1 |
(1) |
|
Warrant dated March 17, 2000 exercisable for 36,810 shares of common stock (on a pre-split
basis). |
| |
4.2 |
(1) |
|
Warrant dated October 31, 2002 exercisable for 180,052 shares of common stock (on a
pre-split basis). |
| |
4.3 |
(2) |
|
Form of Warrant dated June 2007. |
| |
4.4 |
(12) |
|
Form of Warrant dated September 2009. |
| |
10.1 |
(1) |
|
1997 Equity Incentive Plan and forms of related agreements and documents. + |
| |
10.2 |
(3) |
|
2005 Equity Incentive Plan and forms of related agreements and documents. + |
| |
10.3 |
(1) |
|
Office Lease Agreement dated April 25, 2003, and First Amendment to Office Lease Agreement
dated January 21, 2004. |
| |
10.4 |
(8) |
|
Second Amendment to Office Lease Agreement, executed and delivered on December effective
November 19, 2007. |
| |
10.5 |
(1) |
|
Distribution Agreement by and between Cardica, Inc. and Century Medical, Inc. dated
June 16, 2003.† |
| |
10.6 |
(4) |
|
First Amendment to Distribution Agreement, dated March 30, 2007, by and between Cardica,
Inc. and Century Medical, Inc.† |
| |
10.7 |
|
|
Amendment No. 2 to Distribution Agreement, dated June 13, 2007, by and between Cardica,
Inc. and Century Medical, Inc. † |
| |
10.8 |
|
|
Amendment No. 3 to Distribution Agreement, dated January 24, 2008, by and between Cardica,
Inc. and Century Medical, Inc. |
| |
10.9 |
(13) |
|
Amendment No. 4 to Distribution Agreement, dated April 1, 2010, by and between Cardica,
Inc. and Century Medical, Inc.† |
| |
10.10 |
(1) |
|
Subordinated Convertible Note Agreement with Century Medical, Inc. dated June 16, 2003,
and Amendment No. 1 thereto, dated August 6, 2003.† |
| |
10.11 |
(4) |
|
Amendment No. 2 to Subordinated Convertible Note Agreement, dated March 30, 2007, by and
between Cardica, Inc. and Century Medical, Inc. † |
| |
10.12 |
(13) |
|
Amendment No. 3 to Subordinated Convertible Note Agreement, dated April 1, 2010, by and
between Cardica, Inc. and Century Medical, Inc. † |
| |
10.13 |
(4) |
|
Amended and Restated Note issued pursuant to Amendment No. 2 to Subordinated Convertible
Note Agreement with Century Medical, Inc. |
| |
10.14 |
(2) |
|
Registration Rights Agreement, dated June 7, 2007, by and among Cardica, Inc., and the
purchasers listed on the signature pages thereto. |
| |
10.15 |
(16) |
|
Additional Compensation Information for named executive officers. + |
| |
10.16 |
|
|
Cardica, Inc. Non-Employee Director Compensation. + |
| |
10.17 |
(1) |
|
Benefit Agreement with Bernard Hausen, M.D., Ph.D.+ |
| |
10.18 |
(9) |
|
Letter Agreement with Frederic M. Bauer+ |
| |
10.19 |
(10) |
|
Cardica, Inc. Change in Control and Severance Benefit Plan. + |
| |
10.20 |
(11) |
|
Form of Restricted Stock Unit Grant Notice and Restricted Stock Unit Grant Agreement. + |
| |
10.21 |
(5) |
|
License, Development and Commercialization Agreement by and between the Company and Cook
Incorporated, dated June 12, 2007. |
| |
10.22 |
(7) |
|
Amendment to License, Development and Commercialization Agreement by and between Cardica,
Inc. and Cook Incorporated, dated September 19, 2007. |
| |
10.23 |
(15) |
|
Second Amendment to License, Development and Commercialization Agreement by and between
Cardica, Inc. and Cook Incorporated, dated June 19, 2009. |
| |
10.24 |
(12) |
|
Securities Purchase Agreement, dated September 29, 2009, by and among Cardica, Inc., and
purchasers listed on the signature pages thereto. |
| |
10.25 |
(12) |
|
Registration Rights Agreement, dated September 25, 2009, by and among Cardica, Inc., and
the purchasers listed on the signature pages thereto. |
88
| |
|
|
|
|
| Exhibit |
|
|
| Number |
|
Description |
| |
| |
10.26 |
(14) |
|
Stock Purchase Agreement, dated August 16, 2010, by and between Cardica, Inc., and
Intuitive Surgical Operations, Inc. |
| |
10.27 |
(14) |
|
Registration Rights Agreement, dated August 16, 2010, by and between Cardica, Inc., and
Intuitive Surgical Operations, Inc. |
| |
10.28 |
|
|
License Agreement, dated August 16, 2010, by and between Cardica, Inc., and Intuitive
Surgical Operations, Inc. † |
| |
21.1 |
(1) |
|
Subsidiaries of Registrant. |
| |
23.1 |
|
|
Consent of Independent Registered Public Accounting Firm. |
| |
24.1 |
|
|
Power of Attorney (included on signature page). |
| |
31.1 |
|
|
Certification of chief executive officer. |
| |
31.2 |
|
|
Certification of chief financial officer. |
| |
32.1 |
|
|
Section 1350 Certification. |
| |
|
|
| † |
|
Portions of this exhibit (indicated by asterisks) have been omitted
pursuant to a request for confidential treatment and this exhibit has
been filed separately with the SEC. |
| |
| + |
|
Indicates management contract or compensatory plan. |
| |
| (1) |
|
Filed as an exhibit to the Registrant’s Registration Statement on
Form S-1, file no. 333-129497, declared effective on February 2,
2006, as amended, and incorporated herein by reference. |
| |
| (2) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on June 13, 2007,
excluding Item 3.02 and incorporated herein by reference. |
| |
| (3) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on November 13, 2009 and
incorporated herein by reference. |
| |
| (4) |
|
Filed as exhibits to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on April 5, 2007 and
incorporated herein by reference. |
| |
| (5) |
|
Filed as exhibits to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on June 21, 2007,
excluding Items 3.01 and incorporated herein by reference. |
| |
| (6) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on August 19, 2008 and
incorporated herein by reference. |
| |
| (7) |
|
Filed as an exhibit to the Company’s Quarterly Report on Form 10-Q
for the quarterly period ended September 30, 2007 and incorporated
herein by reference. |
| |
| (8) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on December 5, 2007 and
incorporated herein by reference. |
| |
| (9) |
|
Filed as an exhibit to the Company’s Quarterly Report on Form 10-Q
filed with the Securities and Exchange Commission on November 7, 2008
and incorporated herein by reference. |
| |
| (10) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on February 18, 2009 and
incorporated herein by reference. |
| |
| (11) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on February 20, 2009 and
incorporated herein by reference. |
| |
| (12) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on September 29, 2009 and
incorporated herein by reference. |
| |
| (13) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on April 7, 2010 and
incorporated herein by reference. |
| |
| (14) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on August 20, 2010 and
incorporated herein by reference. |
| |
| (15) |
|
Filed as an exhibit to the Company’s Annual Report on Form 10-K filed
with the Securities and Exchange Commission on September 18, 2009 and
incorporated herein by reference. |
| |
| (16) |
|
Filed as an exhibit to the Company’s Current Report on Form 8-K filed
with the Securities and Exchange Commission on August 18, 2009 and
incorporated herein by reference. |
89