Attached files
| file | filename |
|---|---|
| EX-31.2 - PHARMACYCLICS INC | ex312to10k07380_06302010.htm |
| EX-32.1 - PHARMACYCLICS INC | ex321to10k07380_06302010.htm |
| EX-23.1 - PHARMACYCLICS INC | ex231to10k07380_06302010.htm |
| EX-31.1 - PHARMACYCLICS INC | ex311to10k07380_06302010.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
______________
FORM 10-K
(Mark One)
|
x
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES
|
|
|
EXCHANGE ACT OF 1934
|
For the fiscal year ended June 30, 2010
|
¨
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
|
|
|
SECURITIES EXCHANGE ACT OF 1934
|
For the Transition Period from ________ to ________
Commission File Number 000-26658
|
PHARMACYCLICS, INC.
|
|
|
(Exact Name of Registrant as Specified in its Charter)
|
|
|
DELAWARE
|
94-3148201
|
|
(State or other jurisdiction of incorporation or organization)
|
(I.R.S. Employer Identification No.)
|
|
995 E. Arques Avenue, Sunnyvale, California
|
94085-4521
|
|
(Address of principal executive offices)
|
(Zip code)
|
|
Registrant’s telephone number, including area code: (408) 774-0330
|
|
Securities registered pursuant to Section 12(b) of the Act:
|
|
|
|
Title of each class
|
Name of each exchange on which registered
|
|
Common Stock, $0.0001 par value
|
NASDAQ Capital Market
|
Securities registered pursuant to Section 12 (g) of the Act: None
(Title of Class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. o Yes x No
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. o Yes x No
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. x Yes o No
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§229.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files) o Yes o No
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendments to this Form 10-K. x
Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. Check one:
|
Large Accelerated Filer o
|
Accelerated Filer x
|
|
Non-Accelerated Filer o
|
Smaller Reporting Company x
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). o Yes x No
The aggregate market value of the voting and non-voting stock held by non-affiliates of the Registrant was $116,760,649 based on the closing sale price of the Registrant's common stock on The NASDAQ Stock Market LLC on the last business day of the Registrant's most recently completed second fiscal quarter. Shares of the Registrant's common stock beneficially owned by each executive officer and director of the Registrant and by each person known by the Registrant to beneficially own 10% or more of its outstanding common stock have been excluded, in that such persons may be deemed to be affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes. The number of outstanding shares of the Registrant’s common stock as of August 31, 2010 was 59,249,483.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the following document are incorporated by reference into Part III of this Form 10-K: the Definitive Proxy Statement for the Registrant’s 2010 Annual Meeting of Stockholders which will be filed with the Securities and Exchange Commission within 120 days after the end of the Registrant’s fiscal year.
ANNUAL REPORT ON FORM 10-K
FOR THE FISCAL YEAR ENDED JUNE 30, 2010
TABLE OF CONTENTS
|
Page
|
||||
|
PART I
|
3 | |||
| 4 | ||||
| 29 | ||||
| 45 | ||||
| 46 | ||||
| 46 | ||||
| 46 | ||||
|
PART II
|
46 | |||
| 46 | ||||
| 47 | ||||
| 48 | ||||
| 62 | ||||
| 63 | ||||
| 99 | ||||
| 99 | ||||
| 100 | ||||
|
PART III
|
100 | |||
| 100 | ||||
| 101 | ||||
| 101 | ||||
| 101 | ||||
| 101 | ||||
|
PART IV
|
102 | |||
| 102 | ||||
| 103 | ||||
| 104 | ||||
Part I
Important Factors Regarding Forward-Looking Statements
This report contains forward-looking statements. These statements relate to future events or our future financial performance. In some cases, you can identify forward-looking statements by terminology such as “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “might,” “plan,” “possible,” “potential,” “predict,” “should” or “will” or the negative of such terms or other comparable terminology. In particular, forward-looking statements include:
|
|
·
|
statements about our future capital requirements and the sufficiency of our cash, cash equivalents, marketable securities and other financing proceeds to meet these requirements;
|
|
|
·
|
information concerning possible or assumed future results of operations, trends in financial results and business plans;
|
|
|
·
|
statements about our product development schedule;
|
|
|
·
|
statements about our expectations for and timing of regulatory approvals for any of our product candidates;
|
|
|
·
|
statements about the level of our expected costs and operating expenses;
|
|
|
·
|
statements about the potential results of ongoing or future clinical trials;
|
|
|
·
|
other statements about our plans, objectives, expectations and intentions; and
|
|
|
·
|
other statements that are not historical fact.
|
From time to time, we also may provide oral or written forward-looking statements in other materials we release to the public. Forward-looking statements are only predictions that provide our current expectations or forecasts of future events. Any or all of our forward-looking statements in this report and in any other public statements are subject to unknown risks, uncertainties and other factors may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by such forward-looking statements. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, performance or achievements. You should not place undue reliance on these forward-looking statements.
We undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise. You are advised, however, to consult any further disclosures we make on related subjects in our Quarterly Reports on Form 10-Q and Current Reports on Form 8-K. Also note that we provide a cautionary discussion of risks, uncertainties, assumptions and other factors relevant to our business under the caption Risk Factors and elsewhere in this report. These are risks that we think could cause our actual results to differ materially from expected or historical results.
Item1. Business
Company Overview
We are a clinical-stage biopharmaceutical company focused on developing and commercializing innovative small-molecule drugs for the treatment of cancer and immune mediated diseases. Our mission and goal is to build a viable biopharmaceutical company that designs, develops and commercializes novel therapies intended to improve quality of life, increase duration of life and resolve serious unmet medical healthcare needs; and to identify promising product candidates based on exceptional scientific development expertise, develop our products in a rapid, cost-efficient manner and pursue commercialization and/or development partners when and where appropriate. We exist to make a difference for the better and these are important times to do just that.
Presently, we have four product candidates in clinical development, a clinical development candidate in late-stage preclinical evaluation and several preclinical molecules in lead optimization. To date, substantially all of our resources have been dedicated to the research and development of our products, and we have not generated any commercial revenues from the sale of our products. We do not anticipate the generation of any product commercial revenues until we receive the necessary regulatory and marketing approvals to launch one of our products.
Our Pipeline
Most of our clinical development and product candidates are small-molecule enzyme inhibitors designed to target key biochemical pathways involved in human diseases with critical unmet needs. We currently have four proprietary drug candidates under clinical development, a clinical development candidate in late-stage preclinical evaluation and a several preclinical lead molecules. This includes: an inhibitor of Bruton’s tyrosine kinase (Btk) (PCI-32765) currently in a Phase Ia and Phase Ib clinical trials targeting indications in oncology; a Btk inhibitor (PCI-45292) in advanced preclinical development targeting autoimmune indications (with an initial targeted indication of rheumatoid arthritis); an inhibitor of Factor VIIa (PCI-27483) in a Phase II clinical trial in pancreatic cancer patients; a histone deacetylase inhibitor (PCI-24781) currently in a Phase II clinical trial in solid tumors and hematological malignancies and an HDAC8 inhibitor program which is currently in the lead optimization stage. Motexafin gadolinium (MGd) has completed accrual in a Phase I/II trial being conducted by the National Cancer Institute (NCI) in patients with newly diagnosed glioblastoma multiforme.
Status of Products Under Preclinical and Clinical Development
The table below summarizes our product candidates and their stage of development:
|
Product Candidates
|
Disease Indication
|
Development Status(1)
|
||
|
PCI-32765
Bruton’sTyrosine Kinase Inhibitor
|
B-cell lymphomas
|
Phase 1a/Ib – enrolling
|
||
|
PCI-45292
Bruton’s Tyrosine Kinase Inhibitor
|
Autoimmune diseases
|
Advanced preclinical development
IND submission – planned for late Q2 of calendar 2011
|
||
|
PCI-27483
Factor VIIa Inhibitor
|
Pancreatic cancer
|
Phase II – enrolling
|
||
|
PCI-24781
HDAC Inhibitor
|
Recurrent lymphomas
soft tissue sarcoma
|
Phase II – enrolling
Phase I/II – enrolling
|
||
|
HDAC8 Inhibitor Program
|
Cancer
|
Lead optimization and preclinical testing
|
||
|
Motexafin Gadolinium
|
Primary brain tumor(2)
|
Phase II – enrollment completed
|
|
(1)
|
“Phase I” means initial human clinical trials designed to establish the safety, dose tolerance, pharmacokinetics (i.e. absorption, metabolism, excretion), and pharmacodynamics (i.e. surrogate markers for efficacy) of a compound. “Phase II” means human clinical trials designed to establish safety, optimal dosage and preliminary activity of a compound in a patient population. “Preclinical” means the stage of drug development prior to human clinical trials in which a molecule is optimized for “drug like” properties and evaluated in laboratory animals for efficacy, pharmacokinetics, pharmacodynamics and safety. “IND” refers to an investigational new drug application filed with the FDA.
|
|
(2)
|
Study sponsored by the National Cancer Institute.
|
Our Drug Development Programs
Btk Inhibitors
Pharmacyclics is pioneering the development of orally bioavailable inhibitors of Bruton’s tyrosine kinase (Btk), a signaling molecule that is critically important for the activity of B cells (cells that can develop into antibody producing cells). When B cells are overactive, the immune system can produce antibodies that begin to attack the body’s own tissue, leading to autoimmune diseases. Also, B-cell lymphomas and leukemias, which are common blood cancers, result from mutations acquired during B-cell development that lead to uncontrolled B-cell proliferation. Both autoimmune diseases and B cell malignancies are thought to be driven by overactive signaling and activation of the B-cell antigen receptor, a process that is dependent on Btk.
Pharmacyclics has development programs based on two proprietary and chemically-distinct inhibitors. PCI-32765 has been optimized for oncology indications and has demonstrated clinical activity at well tolerated doses in a Phase Ia clinical trial in B-cell lymphomas. Specific cancer indications we are pursuing clinically include chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), and diffuse large B cell lymphoma (DLBCL). A second Btk inhibitor, PCI-45292, has been optimized for autoimmune diseases, and we anticipate filing an Investigational New Drug Application (IND) with the FDA in late second quarter of calendar 2011.
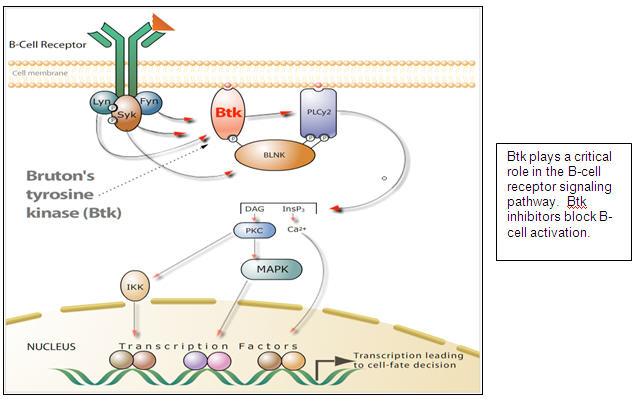
Genetic validation of Btk in mice and humans: In humans, mutations in the genes for Btk cause the rare genetic disease, X-linked agammaglobulinemia (XLA). Patients with XLA have very low or no B cells; therefore, they do not have very many antibodies. Other than the B-cell abnormalities, patients with XLA have no abnormalities in other organs or tissues. These patients are treated with antibody replacement therapy to protect them from infections. This suggests that a Btk-selective inhibitor would act specifically on B cells and have the potential for a wide safety margin.
PCI-32765 Btk Inhibitor Program for Oncology
PCI-32765 is a highly potent, selective, orally bioavailable, small-molecule inhibitor of Btk. A paper describing the preclinical efficacy of PCI-32765 in both lymphoma and autoimmune disease was recently published in the Proceedings of the National Academy of Science (Honigberg et al., Proc Natl Acad Sci USA, 2010; 107: 13075-80).
Mechanim of action: PCI-32765 binds irreversibly to the active site of Btk, thereby inhibiting the activity of Btk (IC50 of 0.5 nM). The inhibition of Btk by PCI-32765 has been confirmed by in vitro tests using B cells, which show that the activity of key cellular proteins “downstream” of the Btk signaling cascade are inhibited for about 18 hours. Importantly, as Btk is not found in T cells, in vitro application of PCI-32765 to T cells shows that PCI-32765 does not affect T-cell function. PCI-32765 is a highly selective inhibitor and does not appear to bind to other cellular proteins, with few exceptions, as strongly and as rapidly as it does to Btk. In humans, the levels of PCI-32765 in the blood are reduced by half within 1.5 to 2.5 hours. The unique combination of irreversible binding and rapid elimination reduces the likelihood of “off-target”effects of PCI-32765 on other cellular proteins. This has clinical relevance as often off-target effects contribute to the toxicity of drugs.
Clinical Development: Bruton's Tyrosine Kinase Inhibitor, PCI-32765, results from phase I trial were presented at an oral presentation at the American Society of Clinical Oncology (ASCO) annual meeting that took place in Chicago, Illinois in June 2010. The trial is an ongoing open-label, dose-escalation study of PCI-32765 in recurrent B cell malignancies treating a minimum of 6 patients per cohort. Five dose levels are being explored—1.25, 2.5, 5.0, 8.3 and 12.5 mg/kg/day. Each cycle of treatment consists of 28 consecutive days of once daily dosing followed by a 7-day rest period. An additional dose group at 8.3 mg/kg/day is also being explored using a 35-day cycle with no rest period ("continuous dosing" or "CD"). Patients were evaluated for dose limiting toxicities at the end of the first cycle and drug efficacy is being evaluated on an on-going basis every 2 cycles. Safety is being monitored throughout the trial. Data from patients receiving doses of 2.5 mg/kg/day or more have indicated: 1) PCI-32765 fully occupied the active site of the target enzyme Btk in peripheral blood cells with minimal variability, 2) PCI-32765 fully inhibited surrogate biomarkers for up to 24 hours postdose, and 3) PCI-32765 was well tolerated by patients.
Trial Results as of August 31, 2010: A total of 47 relapsed/refractory and progressing patients with a variety of B cell malignancies were enrolled in 6 dose groups (1.25, 2.5, 5.0, 8.3, 12.5 mg/kg/day of standard dosing -4 weeks on / 1 week off- and 8.3 mg/kg/day continuous dosing). Forty-one of the enrolled patients had an on-treatment tumor assessment after completing two cycles of therapy and are evaluable for efficacy as of August 31, 2010. Twenty-one of these evaluable patients had a complete or partial response as their best response and eleven patients had stable diseases. This equates to a response rate of 51% in the evaluable patients (69% in CLL, 75% in Mantle, 42% in Follicular, 33% in Marginal and 38% in DLBCL). On an intent-to-treat ("ITT") basis the overall response rate (ORR) was 45%.
As of August 31, 2010, 23 patients remain on study. Median duration of response has not yet been determined in this ongoing trial. We have submitted an abstract describing the duration of response to the American Society of Hematology (ASH) that will be held in Orlando, FL on December 4-7, 2010. ASH’s regulations require us not to disclose any information contained in the abstract prior to the meeting.
PCI-32765 appears to be well tolerated at all dose levels evaluated (1.25 to 12.5 mg/kg/day). Only 2 patients of the enrolled 47 patients have experienced a dose limiting toxicity (DLT) on this trial. One patient, with a history of allergies to prescription drugs developed an allergic hypersensitivity to PCI-32765. The other patient, who had a history of neutropenia, developed neutropenia while on treatment and required a delay in treatment of more than 7 days. No other dose limiting toxicities were observed. Thirteen of the 47 heavily pretreated patients had one or more serious adverse events, but only 2 were considered drug related by investigators.
|
N
|
Complete Response
|
Partial Response
|
Stable Disease
|
Progressive Disease
|
Not Evaluable*
|
Evaluable RR %
|
ORR %
|
||
|
ITT**
|
|||||||||
|
Chronic/Small Lymphocytic Leukemia (CLL/SLL)
|
15
|
1
|
8
|
4
|
0
|
2
|
69%
|
60%
|
|
|
(9/13)
|
(9/15)
|
||||||||
|
Mantle Cell (MCL)
|
4
|
2
|
1
|
1
|
0
|
75%
|
75%
|
||
|
(3/4)
|
(3/4)
|
||||||||
|
Diffuse Large B Cell Lymphoma (DLBCL)
|
8
|
3
|
1
|
4
|
38%
|
38%
|
|||
|
(3/8)
|
(3/8)
|
||||||||
|
Follicular Lymphoma (FL)
|
15
|
1
|
4
|
3
|
4
|
3
|
42%
|
33%
|
|
|
(5/12)
|
(5/15)
|
||||||||
|
Marginal
|
3
|
1
|
1
|
1
|
33%
|
33%
|
|||
|
(1/3)
|
(1/3)
|
||||||||
|
Malt
|
1
|
0
|
1
|
0%
|
0%
|
||||
|
(0/0)
|
(0/1)
|
||||||||
|
Waldenstrom
|
1
|
1
|
0
|
0%
|
0%
|
||||
|
(0/1)
|
(0/1)
|
||||||||
|
Total
|
47
|
4
|
17
|
11
|
9
|
6
|
51%
|
45%
|
|
|
(21/41)
|
(21/47)
|
||||||||
|
* Includes those patients who did not complete 2 cycles of therapy and withdrew from study before tumor assessment could be determined.
|
||||||||
|
** Overall response rate includes all "Intent To Treat" patients.
|
||||||||
|
Dose
|
# of
|
Evaluable # of Pts *
|
Clinical Findings as of 31 August 2010
|
|||||||
|
Cohort
|
(mg/kg)
|
Patients
|
RR % **
|
CR
|
PR
|
SD
|
PD
|
NE
|
||
|
I
|
1.25
|
7
|
7
|
29%
|
2
|
1
|
4
|
|||
|
II
|
2.5
|
9
|
7
|
57%
|
1
|
3
|
1
|
2
|
2
|
|
|
III
|
5
|
6
|
5
|
60%
|
1
|
2
|
1
|
1
|
1
|
|
|
IV
|
8.3
|
8
|
7
|
43%
|
1
|
2
|
3
|
1
|
1
|
|
|
Continuous
|
8.3
|
10
|
9
|
67%
|
1
|
5
|
2
|
1
|
1
|
|
|
V
|
12.5
|
7
|
6
|
50%
|
3
|
3
|
1
|
|||
|
Total
|
47
|
41
|
51%
|
4
|
17
|
11
|
9
|
6
|
||
|
10%
|
41%
|
27%
|
22%
|
15%
|
||||||
* Evaluable number of patients: Patients who completed two cycles of treatment and/or had at least one treatment tumor assessment done
** Definitions:
RR%: Response Rate of evaluable patients.
PR: partial response is defined as a > 50% tumor reduction and meets international working group criteria for CLL (2008) and Chesson 2007 criteria for NHL.
CR: is a complete response, as defined by the former criteria.
SD: is a stable disease (for additional information and full definition please see our website at www.pharmacyclics.com)
PD: is a progressive disease
NE: non-evaluable patients
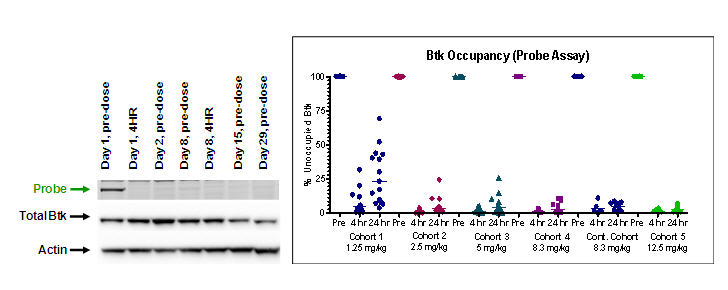
Pharmacodynamic Probe Assay: Pharmacyclics developed an assay to measure occupancy of Btk in PBMCs (described in Honigberg et al., Proc Natl Acad Sci USA, 2010; 107: 13075-80) using a cell-permeable fluorescently-labeled derivative of PCI-32765. This probe assay has demonstrated full occupancy of Btk by PCI-32765 at 4 and 24 hours post-dose beginning at the 2.5 mg/kg dose level, at AUC >200ng hr/mL. See figure below for an example of the probe assay data, as well as data from all patients dosed to date.
PCI-45292 for Autoimmune Disease
(including Rheumatoid Arthritis)
In animal models of rheumatoid arthritis, we observed that oral administration of PCI-32765 led to regression of established disease (Honigbert et al., Proc Natl Acad Sci USA, 2010; 107: 13075-80). This observation provided preclinical validation of Btk as a drug target in rheumatoid arthritis. Using the same chemical scaffold as PCI-32765, work was initiated on a second series of patented Btk inhibitors with the goal of optimizing the molecules for potency, pharmacokinetics, and safety. As a result of this lead optimization effort, PCI-45292 was chosen as the clinical development candidate. Compared to PCI-32765, PCI-45292 was shown to have increased selectivity for Btk inhibition, a reduced potential for off-target protein binding, and improved metabolic stability. Also, as shown in the figure below, PCI-45292 ameliorated inflammation in collagen-induced arthritis models at very low doses. Therefore we believe that PCI-45292 has the qualities to become a new oral disease modifying anti-rheumatic drug (DMARD).

PCI-45292 completely suppresses arthritic inflammation in a Collagen-induced Arthritis Model
Market: Pharmacyclics is not aware of any other competitors in clinical trials with other Btk inhibitors. The anti-B-cell biologics such as Rituxan® and Lymphostat B all have a distinction of massive B-cell depletion and lack of convenient oral dosing. The overall Non-Hodgkin’s Lymphoma market is projected to increase from $3.3 billion in 2007 to $4.7 billion in 2017 (3.6% a year). The market for rheumatoid arthritis (RA) therapies will show robust growth between 2009 and 2019; major market sales are projected at $11.1 billion in 2014 and $12.4 billion in 2019 (Decision Resource 2010).
Patents: PCI-32765, PCI-45292, and other Btk inhibitors (as compounds, in pharmaceutical compositions, PD markers, methods, and in uses for treating a variety of diseases) are covered by US patent applications (issued and pending) and PCT national phase patent applications in fifteen other jurisdictions, including Europe, Canada, Japan, China, India, South Korea, Australia, Brazil, etc. The projected expiration of global coverage is through Dec 2026 and beyond (without including patent term extensions in the various territories which can be up to five years).
Factor VIIa Inhibitor Program
Factor VII is a proteolytic enzyme that becomes activated (FVIIa) by binding to the cell surface protein tissue factor (TF). TF is overexpressed in many cancers including gastric, breast, colon, lung, prostate, ovarian, and pancreatic cancers. In these tumors, the FVIIa/TF complex induces intracellular signaling pathways by activating PAR-2. This in turn increases the expression of interleukin-8 (IL-8) and vascular endothelial growth factor (VEGF), two proteins that play an important role in tumor growth and metastases as well as angiogenesis. FVIIa also initiates the coagulation processes implicated in the high incidence of thromboembolic complications seen in cancer patients. Thromboembolic events are a major cause of death in patients with cancer, and anticoagulant treatment has been shown to improve survival in a variety of cancers (Klerk et al. JCO. 2005).
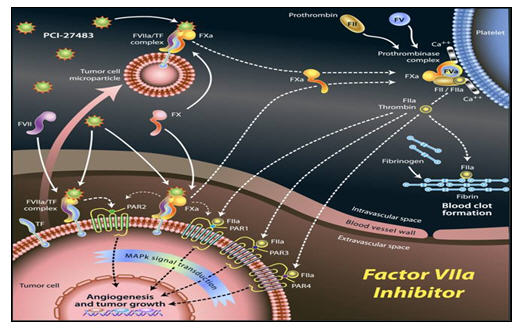
PCI-27483 Factor VIIa Inhibitor
Pharmacyclics’ Factor VIIa inhibitor PCI-27483 is a novel first-in-human small molecule inhibitor that selectively targets FVIIa. As an inhibitor of FVIIa, PCI-27483 has two potential mechanisms of action: 1) inhibition of intracellular signaling involved in tumor growth and metastases and 2) inhibition of early coagulation processes associated with thromboembolism.
PCI-27483 Anti-Tumor Effects in a Pancreatic Tumor Xenograft Model: Preclinical studies have shown PCI-27483 to significantly inhibit tumor growth in human pancreatic xenograft mice models, with or without gemcitabine.
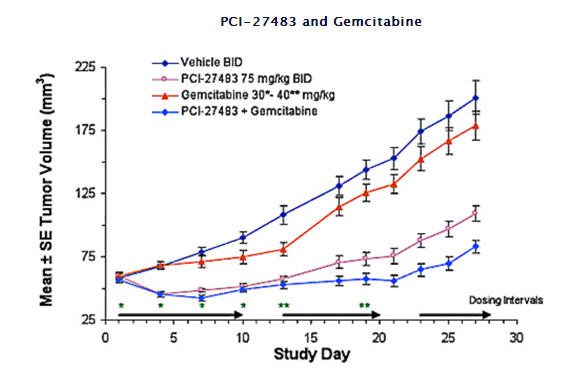
Tumor growth inhibition:
· 16.7% with gemcitabine alone
· 71.3% with PCI-27483 alone
· 89.7% with PCI-27483 plus gemcitabine
In cancer, the Factor VIIa:TF complex triggers a host of physiologic processes that facilitate tumor angiogenesis, growth, and metastases. Laboratory studies and animal models indicate that PCI-27483 inhibits tunor angiogenesis, growth and metastases.
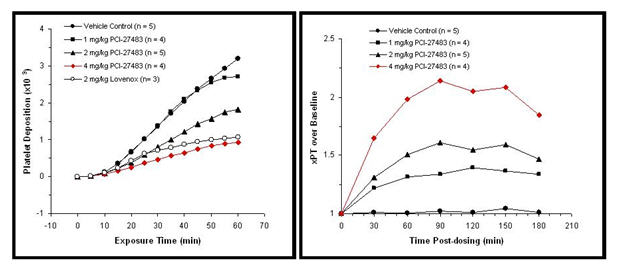
PCI-27483 Anti-Thrombotic Effects in an Arterial Thrombosis Model: The anti-thrombotic effects of PCI-27483 were determined in a baboon model of arterial thrombosis. In this model, PCI-27483 showed dose-dependent inhibition of thrombus formation, fibrin accumulation, and prothrombin time, and 4mg/kg PCI-27483 demonstrated comparable anti-coagulation effects as 2mg/kg Lovenox.
Clinical Development: Pancreatic cancer is one of the most common causes of death from cancer in the US and Europe. Despite the improvements in the diagnosis and treatment of cancer, patients with locally advanced or metastatic pancreatic cancer have a poor prognosis. Gemcitabine is the most active drug in the treatment of advanced pancreatic cancer; however, single agent gemcitabine treatment is associated with a median survival time of only 5.7 and 6.9 months. Recently a chemotherapy regimen including 5-FU/leucovorin, irinotecan and oxaliplatin was shown to increase median survival time to 10.5 months but was associated with a 48% incidence of grade 3/4 neutropenia (Conroy et al, J Clin Oncol (Meeting Abstracts) 2010; 28: 4010). Clearly, more effective and better tolerated therapies are needed.
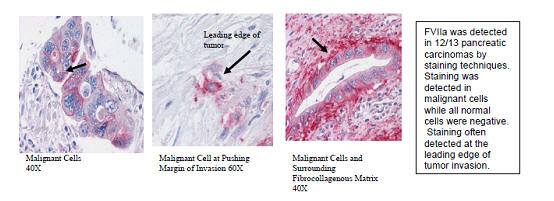
TF expression has been observed in 89% of pancreatic cancers, but not within the normal non-cancerous pancreas. Pancreatic cancer patients with high TF expression have a venous thromboembolism rate of 26.3% compared with 4.5% in patients with low TF expression. (Korana et. al. Clin Cancer Res. 2007;;13(10):2870-5). In addition of pancreatic, venous thromboembolism is a common complication of breast, ovarian, and brain tumors (Lee AYY and Levine MN. Circulation 2003;107:I-17). Among 66,000 patients with cancer admitted to US medical centers from 1995 to 2002, patients with pancreatic cancer had the highest risk of thromboembolic complications (12.1% per hospitalization) (Khorana et. al. J Clin Oncol 2006; 24: 484-90). TF expression occurs early in pancreatic cancer, thus PCI-27483 is being evaluated in recently diagnosed pancreatic patients to establish a clinical proof of concept.
We have completed our initial Phase I testing of PCI-27483 in healthy volunteers. The primary objective of the ascending dose study was to assess the pharmacodynamic and pharmacokinetic profiles of PCI-27483 following a single, subcutaneous injection. In addition, the safety and tolerability of PCI-27483 was evaluated. The drug was well tolerated and no adverse event was observed at any dose level. The International Normalized Ratio (INR) of prothrombin time, a laboratory test for coagulation, was used to measure pharmacodynamic effect at dose levels of 0.05, 0.20, 0.80 and 2.0 mg/kg. A mean peak INR of 2.7 was achieved without adverse effects at the highest dose level administered. The target INR range for oral anti-coagulants i.e. Coumadin, is between 2 and 3. The half-life of PCI-27483 was 9 to 10 hours, which compares favorably to the single-dose half-life of the low molecular weight heparin products Lovenox (4.5 hours) and Fragmin (3 to 5 hours).
In a multicenter Phase I/II study PCI-27483 is being evaluated for its safety and its potential to delay tumor progression and increase overall survival in patients with locally advanced or metastatic ductal adenocarcinoma of the pancreas. All patients enrolled in this study receive treatment with gemcitabine at a dosage of 1000 mg/m2 as weekly infusions on 3 out of every 4 weeks. In the Phase I portion of the study, ascending repeated doses of PCI-27483 were evaluated in 8 patients. PCI-27483 was administered twice daily by subcutaneous injection at dosages ranging from 0.8 to 1.5 mg/kg. The planned duration of treatment was 12 weeks. Patients with stable disease at the end to 12 weeks have the option to continue PCI-27483 treatment until disease progression. Pharmacodynamic responses to PCI-27483 are being monitored with periodic INR assessments performed 2 hours postdose. In the subsequent Phase II portion of the study, patients are being randomized into two groups to receive gemcitabine alone or gemcitabine plus PCI-27483. Twice daily dosing with PCI-27483 at a dosage of 1.2 mg/kg will proceed for 12 weeks with the option for continuation. Additional endpoints will include, levels of circulating tissue factor and frequency of venous thrombotic complications. The Phase II portion of the study began on August 9, 2010.
Market: Each year 230,000 individuals worldwide are diagnosed with pancreatic cancer (UK Cancer Research) (in the US more than 36,640 are diagnosed each year) (Decision Resources PatientBase 2010). Worldwide incidence of other cancers types that also have been shown to have high TF expression include: colon; ovarian; breast ; prostate, and lung cancer.
Patents: PCI-27483 is covered by US patent applications (issued and pending) and PCT national phase patent applications in fourteen other jurisdictions, including Europe, Canada, Japan, China, India, South Korea, Australia and Brazil. The projected expiration of this coverage is through Dec 2023 and beyond (without including patent term extensions in the various territories which can be up to five years).
Histone Deacetylase Inhibitor Program
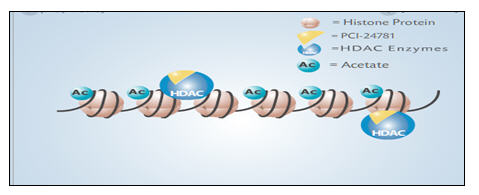
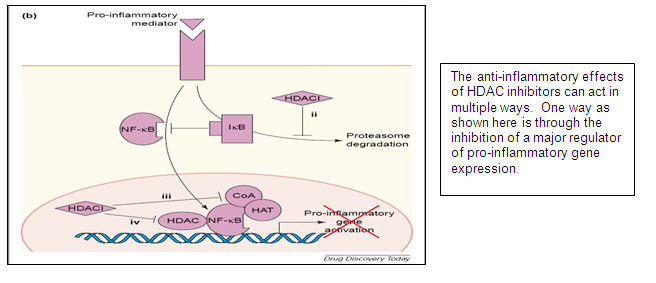
The human genome consists of a complex collection of genes which are turned on or off depending on the needs of the cell. Cancer is characterized by genome-wide changes in gene expression within the tumor. Turning off the expression of certain genes favors a tumor’s ability to multiply, to avoid apoptosis (i.e. programmed cell death) or to become resistant to chemotherapy. One of the ways in which genes are turned on or off is by means of chemical modification of histone proteins. Histone proteins are structural components of chromosomes, and form a scaffold upon which DNA, the genetic material, is arranged, see image below. Histone acetylation (i.e. the addition of an acetyl group to histones) alters the expression of genes involved in cell cycle control, cell division, and apoptosis. Histone deacetylation reverses histone acetylation by removing the acetyl groups. The process of histone deacetylation is controlled by a family of enzymes known as histone deacetylases (“HDAC”). HDAC inhibitors prevent deacetylation, leading to an increase in histone acetylation and an increased expression of certain genes. This effect limits the tumor’s ability to multiply, to avoid apoptosis or to become resistant to chemotherapy. HDAC inhibitors block cancer cell proliferation in vitro (i.e. in cultured cells) and cancer cell growth arrest is observed in vivo (i.e. in animals) at non-toxic concentrations.
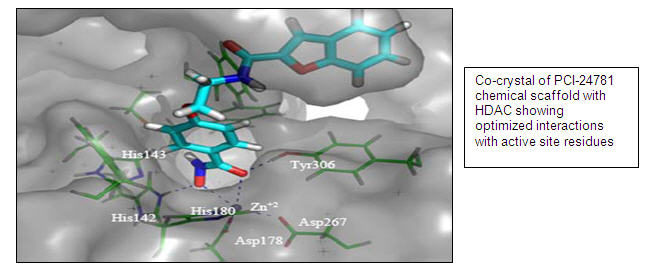
PCI-24781 (Pan HDAC Inhibitor)
PCI-24781 is a novel, potent, small-molecule inhibitor of HDAC enzymes with anti-tumor activity in vitro and in vivo (Buggy et al Mol Cancer Ther 2006; 5: 1309-17). PCI-24781 treatment leads to synergistic efficacy in tumor cells in combination with DNA-damaging agents such as radiation and chemotherapy agents. The mechanism of the synergy may involve inhibition of DNA repair. PCI-24781 has activity against primary human tumors from patients with colon, ovarian, lung and many hematological (i.e. blood related) cancers. We believe PCI-24781 has an improved safety profile compared to competitor drugs (e.g. Zolinza® or pamobinostat).
Clinical Development: Clinical development began with intravenous administration of PCI-24781 in an initial Phase I study, and progressed to two clinical studies by the oral route in 2007, both of which have completed Phase I enrollment. The first study employing an oral capsule formulation (PCYC-0402) is a Phase I, ascending dose study in patients with solid tumors. This study was conducted at four clinical centers (www.clinicaltrials.gov) and is now closed to enrollment. Single agent stable disease has been achieved in a number of solid tumors, with one patient with appendiceal cancer on treatment for over 16 months.
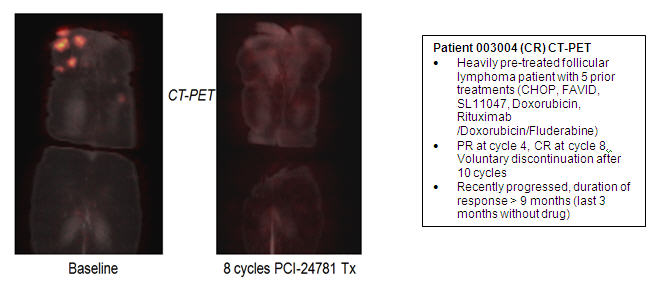
The second study by the oral route (PCYC-0403) is a Phase I/II trial in patients with recurrent lymphomas. The improved potency and pharmacokinetic aspects of PCI-24781 served as a basis for the ongoing proof of concept studies in Phase I/II in lymphoma. One complete response (Pt # 003004 in figure below), four partial responses and seven patients with stable disease have been observed, and these data were published as a poster at the American Society for Hematology (ASH) Annual Meeting in December 2009 (Evens et al., Blood 2009; 114: 2726, ASH 2009 Abstracts). Three of these patients are still on treatment for 19, 14 and 13 months, respectively. Thrombocytopenia (reduced platelet count) was the most commonly observed adverse event in this trial, and dose scheduling changes have been optimized to minimize this effect. Thrombocytopenia observed in PCI-24781 patients has been rapidly reversible, is likely related to the pharmacologic mechanism of action, and has been observed with a number of other HDAC inhibitors. To date there have been only two other non-hematological Grade 3 or 4 (dose-limiting) toxicities in this trial.
Based upon the responses observed in the Phase I arm, the Phase II portion of the trial PCYC-0403 has been commenced in two histologies, which are follicular and mantle cell lymphomas.
In solid tumors, a Phase I/II trial testing PCI-24781 in combination with doxorubicin in patients with soft tissue sarcoma was initiated in December 2009. This trial is co-sponsored by prominent investigators at Massachusetts General Hospital and Dana-Farber/Harvard Cancer Center, including Drs. George Demetri and Edwin Choy.
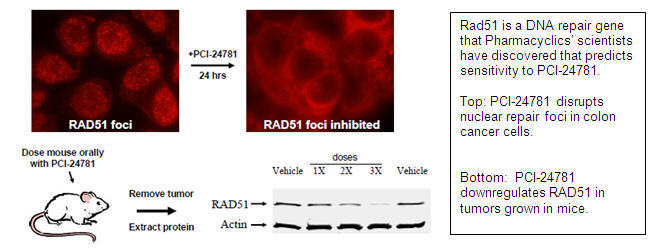
Proprietary Predictive Assays: Following chemotherapy or radiation treatment, some patients’ tumors may turn on certain genes as a strategy by the tumor to adapt to the therapy and become resistant to cell death. One example of a genetic change that occurs in many cancers is the activation of the DNA repair gene RAD51. In response to treatment with DNA-damaging chemotherapy or radiation, tumors will often turn on DNA repair genes, such as RAD51, as an adaptive strategy to help the tumor repair the DNA damage done by these agents. In pre-clinical models, PCI-24781 was able to turn off RAD51 (and other DNA repair genes), effectively blocking the ability of the tumor to repair its damaged DNA, sensitizing the tumor to chemotherapy and radiation. PCYC has patented the predictive use of the biomarker RAD51 which was found by Pharmacyclics’ scientists to potentially underlie resistance to therapy and may be used as a predictive measure of HDAC inhibitor activity that could be useful in the clinic. This research was published in the Proceedings of the National Academy of Sciences (Adimoolam et al., Proc Natl Acad Sci U S A. 2007; 104:19482-7. Epub 2007 Nov 27).
Thus PCI-24781 is effective at inhibiting repair of damaged DNA by downregulating RAD51, which is particularly essential for repair of double-strand breaks (DSB). It was demonstrated by Pharmacyclics that PCI-24781 effectively prevents DSB repair via one of the two major repair pathways, called the homologous recombination pathway, by modulation of RAD51. This allows PCI-24781 to synergize effectively with other agents that damage DNA, such as radiation (Banuelos et al., Clin Cancer Res, 2007; 13:6816-26) and chemotherapeutics i.e. doxorubicin (Adimoolam et al., Proc.Natl.Acad.Sci.U.S.A 2007;104:19482-87).
We showed recently that RAD51 is over expressed in a majority of human lymphoma samples and that pretreatment with PCI-24781 down regulates RAD51 and potentiates cell killing by subsequent addition of doxorubicin (Balasubramanian et al., Blood (ASH 2007 Abstracts); 110:1377). One of our collaborators, Dr. Dina Lev at MD Anderson Cancer Center, has shown that PCI-24781 can also synergize with doxorubicin in sarcoma, both in cells and in animal models (Lopez et al., Clin Cancer Res., 2009; 15:3472-83. Epub 2009 May 5). More recently, it was also shown that PCI-24781 can reverse resistance in a number of drug-resistant sarcoma cell lines (Yang et al., Cancer Chemother Pharmacol. 2010; May 12. [Epub ahead of print]). Accordingly, as mentioned above we are currently enrolling a Phase I/II trial of PCI-24781 in combination with doxorubicin for treating sarcoma with Dr. Edwin Choy at Massachusetts General Hospital and Dr. George Demetri at Dana-Farber Cancer Institute. These investigators are part of one of the leading consortiums in sarcoma in the world today. It is anticipated that clinical activity in this trial may pave the way to other indications for PCI-24781 in combination with doxorubicin, which is also used extensively in treatment of other cancers, including lymphoma, breast, lung, ovarian and liver cancer.
Market: Pan-HDAC inhibitors may have the potential for broad anti-cancer indications in hematologic and solid malignancies when used in combination with numerous chemotherapeutic drugs and radiation.
Patents: PCI-24781 and Pan-HDAC inhibitors patents; covering their composition, pharmaceutical formulation, methods of uses, biomarkers; are issued or pending with coverage through 2024 and beyond in US and fifteen other international territories including Canada, Europe, Japan, China, India and Brazil. All patents would be subject to patent term extensions due to development life cycles.
Competition: Merck’s vorinostat (Zolinza®) has been approved by the FDA for cutaneous T-cell lymphoma patients who have progressive, persistent or recurrent disease on or following failure of two systemic therapies, making the drug the first in its class to reach the market. Recently, a second HDAC inhibitor, Romidepsin (from Gloucester Pharmaceuticals, now Celgene) received US FDA approval, also for the same indication of Cutaneous T-cell Lymphoma (CTCL). A number of other structurally distinct HDAC inhibitors are currently in clinical trials including Novartis’ LBH-589, Spectrum Pharmaceuticals’ belinostat and the benzamide, SYND-275. HDAC inhibitors have exhibited clinical activity against a variety of human malignancies in initial clinical trials. For example, clinical improvements have been observed in patients with renal cell carcinoma, head and neck squamous carcinoma, mesothelioma, small-cell lung cancer, melanoma, papillary thyroid carcinoma and B- and T-cell lymphomas. Thrombocytopenia (a reduction in platelets, which are cells responsible for clotting blood) was identified as a dose-limiting toxicity for patients administered a number of these agents. Several of the competitors have reported cardiac toxicities such as Grade 3 QTc prolongation, arrhythmias and atrial fibrillation, in addition to fatigue, anorexia, infection, headache and nausea. Preliminary data suggests that oral PCI-24781 has a side-effect/toxicity profile better than its competitors with relatively few Grade 3/ 4 non-hematological toxicities and relatively low incidence of Grade 3/ 4 thrombocytopenia.
Partnering: In April 2009, we entered into a collaboration agreement with Les Laboratoires Servier (”Servier”) pursuant to which Pharmacyclics granted to Servier an exclusive license for its Pan-HDAC inhibitors, including PCI-24781, for territories throughout the world excluding the United States and its possessions. Under the terms of the agreement, Servier will pay a royalty to Pharmacyclics on sales outside of the United States. Pharmacyclics will continue to own all rights within the United States.
HDAC8-specific Inhibitor Program
Pharmacyclics’ scientists have been in the forefront of research into inhibitors for specific HDAC enzymes beginning with the cloning of the human HDAC8 in 2000 (Buggy et al., Biochem.J 2000;350(1):199-205). Since then, we were the first to publish the crystal structure of a human HDAC (HDAC8) in 2004 (Somoza et al., Structure 2004;12:1325-34), the first to publish the most selective inhibitor of human HDAC8 (PCI-34051) in 2008 (Balasubramanian et al., Leukemia 2008, 22:1026-34), and the first to discover a novel anti-inflammatory activity of a HDAC8 inhibitor (Balasubramanian et al., Blood [ASH Annual Meeting Abstracts], Nov 2008; 112: 2581; manuscript in preparation). Pharmacyclics has continued to strengthen its intellectual property position in HDAC8 inhibitors, with multiple patents on the gene, protein and a large selective inhibitor panel, and worldwide recognition of our efforts with seminar and poster presentations at major international conferences including the first HDAC inhibitors conference in 2007 and subsequent ones in 2008 and 2009, as well as AACR and ASH conferences. Our pre-eminent position in this field has also led to invited reviews (Isoform-specific histone deacetylase inhibitors: The next step? Balasubramanian et al., Cancer Lett 2009; 280:211-21) and workshops (Workshop on Epigenetic Cancer Therapy, Heidelberg, Germany, June 2010).
Using our unique knowledge of the crystal structure of HDAC8 complexed with multiple pan- and selective inhibitors, we had previously discovered a novel HDAC8 selective inhibitor, PCI-34051, which inhibits HDAC8 with a Ki of 10 nM (a measure of potency) with >200 fold selectivity over the other HDACs tested. Based on preclinical optimization efforts, we have identified two completely novel scaffolds that provided new leads with better pharmacokinetic (PK) properties while maintaining the HDAC8 selectivity and potency. HDAC8 inhibitors possess certain unique activities across a range of clinical indications, including T-cell malignancies, neuroblastoma and inflammation as indicated below.
T-cell lymphoma: We showed that HDAC8 selective inhibitor PCI-34051 induces growth arrest and apoptosis in cell lines derived from T-cell lymphomas and leukemias, but not in any other hematologic and most solid tumor cell lines (Balasubramanian et al., Leukemia 2008; 22:1026-1034). We have since shown that primary tumor cells obtained from patients suffering from cutaneous T-cell lymphoma (CTCL) can also be inhibited from proliferating and killed specifically with HDAC8 inhibitors.
Pediatric neuroblastoma: HDAC8, uniquely among all HDAC enzymes, is overexpressed in pediatric neuroblastoma tumors, and a high HDAC8 expression level is strongly associated with a poor prognosis (Oehme et al., Clin Cancer Res 2009, 15: 91-99). HDAC8-specific inhibitors induce growth inhibition of neuroblastoma cells and eventually lead to cell cycle arrest, and death or terminal differentiation into non-cancerous cells.
Inflammatory disease: We have discovered that PCI-34051 inhibits the secretion of many pro-inflammatory proteins from blood cells (Balasubramanian et al., Blood [ASH Annual Meeting Abstracts], Nov 2008;112:2581). PCI-34051 is particularly effective at modulating the proteins interleukin-1 beta (IL1b) and interleukin-18, both of which are associated with many autoimmune disorders. Anti-IL1b protein therapeutics have proven effective in treatment of RA and systemic juvenile RA (Pascual et al., J Exp.Med 2005;201:1479-1486). We have also shown that PCI-34051 is effective at reducing IL1b secretion from blood cells of patients with RA and psoriasis.
Motexafin Gadolinium (MGd)
MGd is a radiation and chemotherapy sensitizing agent with a novel mechanism of action. MGd is designed to accumulate selectively in cancer cells. Once inside cancer cells, MGd in combination with radiation induces apoptosis (programmed cell death) by disrupting redox-dependent pathways. MGd is also detectable by magnetic resonance imaging (MRI) and may allow for more precise tumor detection. The National Cancer Institute (NCI) is currently sponsoring one Phase I/II trial which have and continue to provide valuable developmental insights and directions.
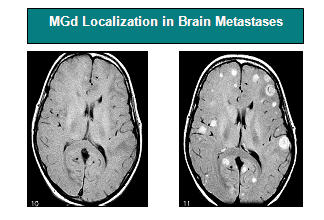
We are currently evaluating MGd in newly diagnosed glioblastoma multiforme (GBM), wherein proof-of-efficacy relies on extending survival time. GBM is the most common primary brain tumor in adults accounting for 40% of primary central nervous system tumors. Radiation increases median survival by approximately 12 months, addition of temozolomide increases this to 14.6 months (Stupp et al. N. Eng. J. Med. 2005), but despite numerous studies of other potential therapies, the outcome of newly diagnosed GBM has not changed beyond this. Previously, a NCI-sponsored study led by Dr. Judith Ford at UCLA (Int J Radiat Oncol Biol Phys. 2007;69(3):831-8), showed that in a case matched analysis, newly diagnosed GBM patients treated with MGd (n=31) and radiation therapy had a median survival of 16.1 months compared to the matched RTOG (Radiation Therapy Oncology Group) database patients with a median survival of 11.8 months. MGd has completed enrollment in a NCI-RTOG sponsored Phase II multi-center study in newly diagnosed GBM in combination with radiation therapy and temozolomide (www.clinicaltrials.gov; 113 patients study). The primary endpoint is survival and results are expected in 2011. The principal investigator, Dr. David G. Brachman, is heading this study at the Barrow Neurological Institute at St. Joseph’s Hospital in Phoenix, AZ. Previous studies in malignant gliomas headed by Dr. William Shapiro from the Barrow Institute have shown that the combination of MGd and temozolomide have no additional overlapping toxicities when used in combination.
Patents: MGd is protected by various US and international issued and pending patents through 2025, subject to patent term expensions. Certain patents in the MGd/texaphyrin portfolio will expire as early as October 2010. However, the MGd/texaphyrin program remains covered by additional global patent protection through 2025, with the potential for patent term extensions.
Our Business Strategy
The key elements of our business strategy include:
|
|
·
|
Focusing on creating novel, patentable, differentiated biopharmaceutical products. We are leveraging our expertise in chemistry, biology and clinical development to create multiple novel drug candidates.
|
|
|
·
|
Focusing on proprietary drugs that address large markets of unmet medical need for the treatment of oncology and immune mediated diseases. Although our versatile technology platform can be used to develop a wide range of pharmaceutical agents, we have focused most of our initial efforts in oncology and immune mediated diseases where we have established strength in preclinical and clinical development.
|
|
|
·
|
Utilize biomarkers and predictive pharmacodynamic assays wherever possible. Targeting the right drug to the right patient at the right time with the right dose has the potential to greatly expedite intelligent clinical development and reduce the time, cost and risk of clinical programs.
|
|
|
·
|
Provide major pharmaceutical companies access to validated drug candidates. Major pharmaceutical companies have a need for promising drug candidates, which still may require large clinical trials. We focus on satisfying this need for novel, best in class or first in class drugs. A partnership with Pharmacyclics may provide these companies the opportunity to leverage the innovation and excellence of a creative, focused and experienced scientific team.
|
|
|
·
|
Establish strategic alliances and collaborations. Except for the non US HDAC rights which we licensed to Servier, we own the worldwide rights to our multiple product candidates. When, as and if appropriate we maintain the option to establish strategic alliances and collaborations for the development and commercialization of our products.
|
|
|
·
|
Leverage development with outsourcing. We utilize outside vendors with expertise and capability in manufacturing and clinical development to more efficiently develop our multiple product candidates.
|
|
|
·
|
Create a large clinical pipeline. We improve our probability of success by taking multiple “shots on goal.”
|
We are subject to risks common to pharmaceutical companies developing products, including risks inherent in our research, development and commercialization efforts, preclinical testing, clinical trials, uncertainty of regulatory and marketing approvals, uncertainty of market acceptance of our products, history of and expectation of future operating losses, reliance on collaborative partners, enforcement of patent and proprietary rights, and the need for future capital. In order for a product to be commercialized, we must conduct preclinical tests and clinical trials, demonstrate efficacy and safety of our product candidates to the satisfaction of regulatory authorities, obtain marketing approval, enter into manufacturing, distribution and marketing arrangements, build U.S. commercial capability, obtain market acceptance and, in many cases, obtain adequate coverage of and reimbursement for our products from government and private insurers. We cannot provide assurance that we will generate revenues or achieve and sustain profitability in the future.
Collaboration and License Agreements, Acquired Products
Collaboration and License Agreement with Servier. In April 2009, we entered into a collaboration and license agreement with Servier to research, develop and commercialize PCI-24781, an orally active, novel, small molecule inhibitor of Pan HDAC enzymes. Servier is the leading independent pharmaceutical company in France. Under the terms of the agreement, Servier acquired the exclusive right to develop and commercialize the Pan HDAC inhibitor product worldwide except for the United States and its possessions. Pharmacyclics will continue to own all rights within the United States. In May 2009, Pharmacyclics received an upfront payment of $11,000,000 from Servier, less applicable withholding taxes of $550,000, for a net receipt of $10,450,000. The withholding tax paid to the French government will be reclaimed due to a recent revision in the Double Tax Treaty between the US and France.
Under the agreement, Pharmacyclics will also receive from Servier $4,000,000 for research collaboration over a twenty-four month period, paid in equal semi-annual installments, of which $2,000,000 had been received through June 30, 2010. Servier is solely responsible for conducting and paying for all development activities outside the United States. In addition, we could also receive from Servier up to approximately $24,500,000 upon the achievement of certain future milestones up to and including commercialization, as well as royalty payments.
The collaboration and license agreement continues until the later of the expiration of any patent rights licensed under the license agreement and the expiration of all periods of market exclusivity with respect to licensed compounds. Servier as well as Pharmacyclics can terminate the agreement under certain circumstances, including material breach and insolvency. Servier can terminate the agreement at any time due to safety or public health issues or after the second anniversary of the effective date of the agreement.
Celera Corporation. In April 2006, we acquired multiple small molecule drug candidates for the treatment of cancer and other diseases from Celera Genomics, an Applera Corporation business (now Celera Corporation). Under the terms of the agreement, we acquired Celera technology and intellectual property relating to drugs that target histone deacetylase (HDAC) enzymes, selective HDAC enzymes, a Factor VIIa inhibitor targeting a tumor signaling pathway involved in angiogenesis, tumor growth and metastases, and B-cell associated tyrosine kinase inhibitors potentially useful for the treatment of lymphomas.
Total consideration paid was $6,647,000 which consisted of 1,000,000 shares of our common stock, $2,000,000 of cash and $147,000 of transaction costs. We recorded an expense of $6,647,000 related to the consideration for the acquired drug candidates which had not yet reached technological feasibility and had no alternative future use due to the early stage of development and the significant regulatory requirements remaining. In May 2008, we amended our agreement with Celera pertaining to the potential sublicensing of its HDAC compounds. Under the amendment, Celera may receive a portion of any upfront licensing payments we receive from sublicensing an HDAC product and the total future potential milestone payments due to Celera were reduced from $144,000,000 to $104,000,000. In addition, Celera will also be entitled to royalty payments in the mid-to high single digits based on annual sales of any drugs commercialized from these programs.
The agreement with Celera was amended for the second time in March 2009. Pursuant to this amendment, the total future milestone payments to Celera were reduced to approximately $98,000,000, of which approximately $97,000,000 was outstanding at June 30, 2010. We currently cannot predict if or when any of the milestones will be achieved. Approximately 90% of this amount will become due upon regulatory approval for the drug programs in different geographic markets and with the achievement of certain net sales levels of any drugs commercialized from the HDAC program. We also reduced the US and ex-US royalties to mid-single digit level.
The Celera agreement was amended for the third time at the end of March 2009. That amendment changed the payment timeline of certain payments to Celera and also changed the obligations for us to pay royalties under certain conditions to Celera. In connection with this third amendment, we paid Celera $1,000,000 in April 2009. The amount was recorded as research and development expense in the quarter ended March 31, 2009, as the technology rights are being utilized in research and development and it is not clear that an alternative future use exists for such technology.
The University of Texas License. In 1991, we entered into a license agreement with the University of Texas under which we received the exclusive worldwide rights to develop and commercialize porphyrins, expanded porphyrins (e.g. Motexafin Gadolinium) and other porphyrin-like substances covered by their patents. In consideration for the license, we have paid a total of $300,000. We are obligated to pay royalties based on net sales of products that utilize the licensed technology. The term of the license agreement ends upon the last to expire of the patents covered by the license. We have royalty obligations under the license as long as valid and unexpired patents covering the licensed technology exist. Currently, the dates the last United States and ex-United States (international) patents covered by the agreement expire are 2020 and 2014, respectively. Under this agreement, we must be attempting to commercialize one or more products covered by the licensed technology. In the event we fail to attempt to commercialize one or more products covered by the licensed technology, the University of Texas may convert the exclusive license into a non-exclusive license.
Patents and Proprietary Technology
We believe our success depends in part upon our ability to protect and defend our proprietary technology and product candidates through patents and trade secret protection. We, therefore, aggressively pursue, prosecute, protect and defend patent applications, issued patents, trade secrets, and licensed patent and trade secret rights covering certain aspects of our technology. The evaluation of the patentability of United States and foreign patent applications can take years to complete and can involve considerable expense.
We have a number of patents and patent applications related to our compounds but we cannot be certain that issued patents will be enforceable or provide adequate protection or that pending patent applications will issue as patents. Even if patents are issued and maintained, these patents may not be of adequate scope to benefit us, or may be held invalid and unenforceable against third parties.
Our patents, patent applications, and licensed patent rights cover various compounds, pharmaceutical formulations and methods of use. Pharmacyclics owns or licenses rights to:
|
·
|
61 issued U.S. patents; and
|
|
·
|
36 other pending U.S. patent applications.
|
These issued U.S. patents expire at various times depending on product programs (see above program sections). In addition, Pharmacyclics owns or licenses approximately 89 issued foreign patents, 3 Patent Cooperation Treaty (“PCT”) patent applications, and more than 120 pending non-U.S. patent applications filed with the European Patent Office, and nationally in Canada, Japan, China, Australia and other countries.
All of these issued patents would be subject to potential patent term extensions in the U.S. and non-U.S. international territories (up to five years depending on territory).
We may be unsuccessful in prosecuting our patent applications or patents may not issue from our patent applications. Even if patents are issued and maintained, these patents may not be of adequate scope to benefit us, or may be held invalid and unenforceable against third parties.
We also rely upon trade secrets, technical know-how and continuing technological innovation to develop and maintain our competitive position. We require all of our employees, consultants, advisors and the like to execute appropriate confidentiality and assignment-of-inventions agreements. These agreements typically provide that all materials and confidential information developed or made known to the individual during the course of the individual's relationship with us is to be kept confidential and not disclosed to third parties, except in specific circumstances, and that all inventions arising out of the relationship with Pharmacyclics shall be our exclusive property.
Research and Development
The majority of our operating expenses to date have been related to research and development, or R&D. R&D expenses consist of independent R&D costs and costs associated with collaborative R&D. R&D expenses were $17,358,000 in fiscal 2010, $13,954,000 in fiscal 2009 and $18,180,000 in fiscal 2008.
Marketing and Sales
We currently are not directly pursuing marketing, sales, or distribution activities.
Manufacturing
Our chemical development and manufacturing group consists of 16 full-time employees, of which 5 hold Ph.D's. We use third parties to manufacture various components of our products under development. We have entered into several commercial supply agreements with manufacturers.
Competition
We face intense competition for each of our drug targets from pharmaceutical companies, universities, governmental entities and others in the development of therapeutic and diagnostic agents for the treatment of diseases which we target. See "Risk Factors — Risks Related to Our Industry – We face rapid technological change and intense competition."
In addition, see the section titled “Our Drug Development Programs” for further information on some of the competition for our drug programs.
Government Regulation and Product Approval
The FDA and comparable regulatory agencies in state and local jurisdictions and in foreign countries impose substantial requirements upon the clinical development,
manufacture and marketing of pharmaceutical products. These agencies and other federal, state and local entities regulate research and development activities and the testing, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising and promotion of our product candidates. Failure to comply with FDA requirements, both before and after product approval, may subject us to administrative or judicial sanctions, including but not limited to, FDA refusal to approve pending applications, warning letters, product recalls, product seizures, or total or partial suspension of production or distribution, fines, injunctions, or civil or criminal penalties.
The process required by the FDA before our products may be marketed in the U.S. generally involves the following:
|
|
·
|
completion of preclinical laboratory and animal tests;
|
|
|
·
|
submission of an Investigational New Drug (IND) application, which must become effective before clinical trials may begin;
|
|
|
·
|
performance of adequate and well-controlled human clinical trials to establish the safety and efficacy for each intended use;
|
|
|
·
|
submission to the FDA of a New Drug Application (NDA); and
|
|
|
·
|
satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the product candidate is made to assess compliance with the FDA’s current good manufacturing practice (cGMP) regulations.
|
The testing and approval process requires substantial time, effort, and financial resources; and we cannot be certain that any approval will be granted on a timely basis, if at all.
Preclinical tests include laboratory evaluation of the product, its chemistry, formulation and stability, as well as animal studies to assess the potential safety and efficacy of the product. We then submit the results of the preclinical tests, together with manufacturing information and analytical data, to the FDA as part of an IND, which must become effective before we may begin human clinical trials. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about the conduct of the trials as outlined in the IND, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can begin. Our submission of an IND may not result in FDA authorization to commence clinical trials. Further, an independent Institutional Review Board at the medical center proposing to conduct the clinical trials must review and approve any clinical study.
Human clinical trials are typically conducted in three sequential phases which may overlap:
|
|
·
|
Phase I: The drug is initially introduced into healthy human subjects or patients and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion.
|
|
|
·
|
Phase II: Involves studies in a limited patient population to identify possible adverse effects and safety risks, to evaluate preliminarily the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage.
|
|
|
·
|
Phase III: When Phase II evaluations demonstrate that a dosage range of the product may be effective and has an acceptable safety profile, Phase lll trials are undertaken to further evaluate dosage and clinical efficacy and to further test for safety in an expanded patient population at geographically dispersed clinical study sites.
|
In the case of products for severe or life-threatening diseases such as cancer, the initial human testing is often conducted in patients rather than in healthy volunteers. Since these patients already have the target disease, these studies may provide initial evidence of efficacy traditionally obtained in Phase II trials and thus these trials are frequently referred to as Phase I/II trials. We cannot be certain that we will successfully complete Phase I, Phase II or Phase III testing of our product candidates within any specific time period, if at all. Furthermore, the FDA, the relevant Institutional Review Board or the sponsor may suspend clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
The results of product development, preclinical studies and clinical studies are submitted to the FDA as part of a New Drug Application, or NDA, for approval of the marketing and commercial shipment of the product. The FDA may not accept the NDA for review if the applicable regulatory criteria are not satisfied or may require additional clinical data. Even if such data are accepted for filing, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. In addition, before approving an NDA, the FDA will inspect the facilities at which the product is manufactured and will not approve the product unless the facility is in substantial compliance with cGMP regulations. Once issued, the FDA may withdraw product approval if compliance with regulatory standards is not maintained or if problems occur after the product reaches the market. In addition, the FDA may require testing and surveillance programs to monitor the effect of approved products which have been commercialized, and the agency has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs.
Satisfaction of the above FDA requirements or similar requirements of state, local and foreign regulatory agencies typically takes several years and the actual time required may vary substantially, based upon the type, complexity and novelty of the pharmaceutical product. Government regulation may delay or prevent marketing of potential products for a considerable period of time and impose costly procedures upon our activities. We cannot be certain that the FDA or any other regulatory agency will grant approval for any of our products under development on a timely basis, if at all. Success in preclinical or early stage clinical trials does not assure success in later stage clinical trials. Data obtained from preclinical and clinical activities is not always conclusive and may be susceptible to varying interpretations which could delay, limit or prevent regulatory approval. Even if a product receives regulatory approval, the approval may be significantly limited to specific indications. Further, even after regulatory approval is obtained, later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Delays in obtaining, or failures to obtain regulatory approvals would have a material adverse effect on our business. Marketing our products abroad will require similar regulatory approvals and is subject to similar risks. In addition, we cannot predict what adverse governmental regulations may arise from future U.S. or foreign governmental action.
Any products manufactured or distributed by us pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including record-keeping requirements and reporting of adverse experiences with the drug. Drug manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with Good Manufacturing Practice regulations, which impose certain procedural and documentation requirements upon us and our third-party manufacturers. We cannot be certain that we or our present or future suppliers will be able to comply with the current Good Manufacturing Practice, or cGMP, regulations and other FDA regulatory requirements.
The FDA regulates drug labeling and promotion activities. The FDA has actively enforced regulations prohibiting the marketing of products for unapproved uses. The FDA will permit the promotion of a drug for an unapproved use in certain circumstances, but subject to very stringent requirements. We and our products are also subject to a variety of state laws and regulations in those states or localities where our products are or will be marketed. Any applicable state or local regulations may hinder our ability to market our products in those states or localities. We are also subject to numerous federal, state and local laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control, and disposal of hazardous or potentially hazardous substances. We may incur significant costs to comply with such laws and regulations now or in the future.
The FDA's policies may change and additional government regulations may be enacted which could prevent or delay regulatory approval of our potential products. Moreover, increased attention to the containment of health care costs in the U.S. and in foreign markets could result in new government regulations which could have a material adverse effect on our business. We cannot predict the likelihood, nature or extent of adverse governmental regulation which might arise from future legislative or administrative action, either in the U.S. or abroad.
Employees
As of June 30, 2010, we had fifty-eight employees, all of whom were full-time employees. Forty-two of our employees are engaged in research, development, preclinical and clinical testing, manufacturing, quality assurance and quality control and regulatory affairs and sixteen are in finance and administration. Sixteen of our employees have an M.D. or Ph.D. degree. Our future performance depends in significant part upon the continued service of our key scientific, technical and senior management personnel, none of whom is bound by an employment agreement requiring service for any defined period of time. The loss of the services of one or more of our key employees could harm our business. None of our employees are represented by a labor union. We consider our relations with our employees to be good.
Available Information
We were incorporated in Delaware in 1991 and commenced operations in 1992.
We file electronically with the Securities and Exchange Commission, or SEC, our annual reports on Form 10-K, quarterly interim reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934. We maintain a site on the worldwide web at www.pcyc.com; however, information found on our website is not incorporated by reference into this report. We make our SEC filings available free of charge on or through our website, including our annual report on Form 10-K, quarterly interim reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act of 1934 as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. Further, a copy of this annual report on Form 10-K is located at the Securities and Exchange Commission’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549. Information on the operation of the Public Reference Room can be obtained by calling the Securities and Exchange Commission at 1-800-SEC-0330. The Securities and Exchange Commission maintains a website that contains reports, proxy and information statements and other information regarding our filings at www.sec.gov.
In 2004, we adopted a code of ethics that applies to our officers, directors and employees, including our principal executive officer, principal financial officer and principal accounting officer. We have posted the text of our code of ethics on our website at www.pcyc.com in connection with “Investor” materials. In addition, we intend to promptly disclose (1) the nature of any amendment to our code of ethics that applies to our principal executive officer, principal financial officer, principal accounting officer, or persons performing similar functions and (2) the nature of any waiver, including an implicit waiver, from a provision of our code of ethics that is granted to one of these specified officers, the name of such person who is granted the waiver and the date of the waiver on our website in the future.
Item 1A. Risk Factors
An investment in our securities involves a high degree of risk. Anyone who is making an investment decision regarding our securities should carefully consider the following risk factors, as well as the other information contained or incorporated by reference in this report. The risks and uncertainties described below are those that we currently believe may materially affect our company or your investment. Other risks and uncertainties that we do not presently consider to be material, or of which we are not presently aware, may become important factors that adversely affect our security holders or us in the future. If any of the risks discussed below actually materialize, then our business, financial condition, operating results, cash flows and future prospects, or your investment in our securities, could be materially and adversely affected, resulting in a loss of all or part of your investment.
Risks Relating to Pharmacyclics
We will need substantial additional financing and we may have difficulty raising needed capital in the future.
We have expended and will continue to expend substantial funds to complete the research, development and clinical testing of our products. We are unable to entirely fund these efforts with our current financial resources. We may also raise additional funds through the public or private sale of securities, bank debt, collaborations or otherwise. If we are unable to secure additional funds, whether through additional partnership collaborations or sale of our securities, we will have to delay, reduce the scope of or discontinue one or more of our product development programs. Based upon the current status of our product development plans, we believe that our cash, cash equivalents and marketable securities, will be adequate to satisfy our capital needs for at least the next twelve months. We may, however, choose to raise additional funds before then. Our actual capital requirements will depend on many factors, including:
|
|
·
|
our ability to establish new partnership collaboration arrangements and the timing of such arrangements;
|
|
|
·
|
continued progress of our research and development programs;
|
|
|
·
|
our ability to establish and maintain collaborative arrangements with third parties;
|
|
|
·
|
progress with preclinical studies and clinical trials;
|
|
|
·
|
the time and costs involved in obtaining regulatory approval;
|
|
|
·
|
the costs involved in preparing, filing, prosecuting, maintaining and enforcing patent claims;
|
|
|
·
|
the amount and timing of capital equipment purchases; and
|
|
|
·
|
competing technological and market developments.
|
In addition, our ability to raise additional capital may be dependent upon our stock being quoted on the NASDAQ Capital Market. In the past, our stock price has fallen below the $1.00 minimum bid price requirement for continued listing set forth in NASDAQ Marketplace Rule 4450(a)(5). While we have since regained compliance with Marketplace Rule 4450(a)(5), we cannot assure you that our stock price will continue to remain above the required minimum bid price. If we do not remain in compliance with the $1.00 minimum bid price requirement or any other NASDAQ listing requirement, our stock may be delisted by NASDAQ.
We also expect to raise any necessary additional funds through the public or private sale of securities, bank debt financings, collaborative arrangements with corporate partners or other sources that may be highly dilutive or otherwise disadvantageous, to existing stockholders or subject us to restrictive covenants. In addition, in the event that additional funds are obtained through arrangements with collaborative partners or other sources, such arrangements may require us to relinquish rights to some of our technologies, product candidates or products under development that we would otherwise seek to develop or commercialize ourselves. Additional funds may not be available on acceptable terms, if at all. Our failure to raise capital when needed and on acceptable terms would require us to reduce our operating expenses, delay or reduce the scope of or eliminate one or more of our research or development programs and would limit our ability to respond to competitive pressures or unanticipated requirements and to continue operations. Any one of the foregoing would have a material adverse effect on our business, financial condition and results of operations.
Failure to obtain product approvals or comply with ongoing governmental regulations could adversely affect our business.
The manufacture and marketing of our products and our research and development activities are subject to extensive regulation for safety, efficacy and quality by numerous government authorities in the United States and abroad. Before receiving FDA approval to market a product, we will have to demonstrate to the satisfaction of the FDA that the product is safe and effective for the patient population and for the diseases that will be treated. Clinical trials, and the manufacturing and marketing of products, are subject to the rigorous testing and approval process of the FDA and equivalent foreign regulatory authorities. The Federal Food, Drug and Cosmetic Act and other federal, state and foreign statutes and regulations govern and influence the testing, manufacture, labeling, advertising, distribution and promotion of drugs and medical devices. As a result, clinical trials and regulatory approval can take a number of years to accomplish and require the expenditure of substantial resources. Data obtained from clinical trials are susceptible to varying interpretations that could delay, limit or prevent regulatory approvals.
In addition, we may encounter delays or rejections based upon additional government regulation from future legislation or administrative action or changes in FDA policy during the period of product development, clinical trials and FDA regulatory review. We may encounter similar delays in foreign countries. We may be unable to obtain requisite approvals from the FDA and foreign regulatory authorities and even if obtained, such approvals may not be received on a timely basis, or they may not cover the clinical uses that we specify.
Furthermore, regulatory approval may entail ongoing requirements for post-marketing studies. The manufacture and marketing of drugs are subject to continuing FDA and foreign regulatory review and later discovery of previously unknown problems with a product, manufacturer or facility may result in restrictions, including withdrawal of the product from the market. Any of the following events, if they were to occur, could delay or preclude us from further developing, marketing or realizing full commercial use of our products, which in turn would have a material adverse effect on our business, financial condition and results of operations:
|
|
·
|
failure to obtain and thereafter maintain requisite governmental approvals;
|
|
|
·
|
failure to obtain approvals for specific indications of our products under development; or
|
|
|
·
|
identification of serious and unanticipated adverse side effects in our products under development.
|
Any regulatory approval that we receive for a product candidate may be subject to limitations on the indicated uses for which the product may be marketed. In addition, if the FDA and/or foreign regulatory agencies approve any of our product candidates, the labeling, packaging, adverse event reporting, storage, advertising and promotion of the product will be subject to extensive regulatory requirements. We and the manufacturers of our product candidates must also comply with the applicable FDA Good Manufacturing Practice (“GMP”) regulations, which include quality control and quality assurance requirements as well as the corresponding maintenance of records and documentation. Manufacturing facilities are subject to ongoing periodic inspection by the FDA and corresponding state agencies, including unannounced inspections, and must be licensed before they can be used in commercial manufacturing of our products. We or our present or future suppliers may be unable to comply with the applicable GMP regulations and other FDA regulatory requirements. Failure of our suppliers to follow current GMP Practice or other regulatory requirements may lead to significant delays in the availability of products for commercial or clinical use and could subject us to fines, injunctions and civil penalties.
All of our product candidates are in development, and we cannot be certain that any of our products under development will be commercialized.
To be profitable, we must successfully research, develop, obtain regulatory approval for, manufacture, introduce, market and distribute our products under development. The time frame necessary to achieve these goals for any individual product is long and uncertain. Before we can sell any of our products under development, we must demonstrate to the satisfaction of the FDA and regulatory authorities in foreign markets through the submission of preclinical (animal) studies and clinical (human) trials that each product is safe and effective for human use for each targeted disease. We have conducted and plan to continue to conduct extensive and costly clinical trials to assess the safety and effectiveness of our potential products. We cannot be certain that we will be permitted to begin or continue our planned clinical trials for our potential products, or if permitted, that our potential products will prove to be safe and produce their intended effects.
The completion rate of our clinical trials depends upon, among other factors, the rate of patient enrollment, the adequacy of patient follow-up and the completion of required clinical evaluations. Many factors affect patient enrollment, including the size of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, competing clinical trials and new drugs or procedures used for the conditions we are investigating. Other companies are conducting clinical trials and have announced plans for future trials that are seeking or are likely to seek patients with the same diseases that we are studying. We may fail to obtain adequate levels of patient enrollment in our clinical trials. Delays in planned patient enrollment may result in increased costs, delays or termination of clinical trials, which could have a material adverse effect on us. Many factors can affect the adequacy of patient follow-up and completion of required clinical evaluations, including failure of patients to return for scheduled visits or failure of clinical sites to complete necessary documentation. Delays in or failure to obtain required clinical follow-up and completion of clinical evaluations could also have a material adverse effect on the timing and outcome of our clinical trials and product approvals.
Additionally, clinical trials require substantial administration and monitoring. We may fail to effectively oversee and monitor the various trials we have underway at any particular time which would result in increased costs or delays of our clinical trials.
Data already obtained from preclinical studies and clinical trials of our products under development do not necessarily predict the results that will be obtained from later preclinical studies and clinical trials. Moreover, data from clinical trials we are conducting are susceptible to varying interpretations that could delay, limit or prevent regulatory approval. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials, even after promising results in earlier trials. The failure to adequately demonstrate the safety and effectiveness of a product under development could limit or prevent regulatory approval of the potential product and would materially harm our business. Our clinical trials may not demonstrate the sufficient levels of safety and efficacy necessary to obtain the requisite regulatory approval or may not result in marketable products.
We have a history of operating losses and we expect to continue to have losses in the future.
We have incurred significant operating losses since our inception in 1991 and, as of June 30, 2010, had an accumulated deficit of approximately $377,922,000. We expect to continue to incur substantial additional operating losses until such time, if ever, as the commercialization of our products generates sufficient revenues to cover our expenses. All of our product candidates are in the early stages of development and the commercialization of those products will not occur, if at all, for at least the next several years. Our achieving profitability depends upon our ability, alone or with others, to successfully complete the development of our product candidates, and to obtain required regulatory approvals and to successfully manufacture and market our proposed product. While we have most recently generated $9,307,000 in fiscal 2010 revenue from collaborations, we have to date not generated significant revenue from either the licensing or commercial sale of our products.
Acceptance of our products in the marketplace is uncertain, and failure to achieve market acceptance will harm our business.
Even if approved for marketing, our products may not achieve market acceptance. The degree of market acceptance will depend upon a number of factors, including:
|
|
·
|
the receipt of regulatory approvals for the indications that we are studying, and the acceptance by physicians and patients of the clinical benefits that our products may offer;
|
|
|
·
|
the establishment and demonstration in the medical community of the safety, clinical efficacy and cost-effectiveness of our products and their potential advantages over existing therapeutic products;
|
|
|
·
|
marketing and distribution support;
|
|
|
·
|
the introduction, market penetration and pricing strategies of competing and future products;
|
|
|
·
|
coverage and reimbursement policies of governmental and other third- party payors such as insurance companies, health maintenance organizations and other plan administrators; and
|
|
|
·
|
physicians, patients, payors or the medical community in general may be unwilling to accept, purchase, utilize or recommend any of our products.
|
We may fail to adequately protect or enforce our intellectual property rights or secure rights to third-party patents.
Our success depends in part upon our ability to protect and defend our proprietary technology and product candidates through patents and trade secret protection. We, therefore, aggressively pursue, prosecute, protect and defend patent applications, issued patents, trade secrets, and licensed patent and trade secret rights covering certain aspects of our technology. The evaluation of the patentability of United States and foreign patent applications can take years to complete and can involve considerable expense.
We have a number of patents and patent applications related to our compounds but we cannot be certain that issued patents will be enforceable or provide adequate protection or that pending patent applications will issue as patents. Even if patents are issued and maintained, these patents may not be of adequate scope to benefit us, or may be held invalid and unenforceable against third parties.
We face risks and uncertainties related to our intellectual property rights. For example:
|
|
·
|
we may be unable to obtain or maintain patent or other intellectual property protection for any products or processes that we may develop;
|
|
|
·
|
third parties may obtain patents covering the manufacture, use or sale of these products, which may prevent us from commercializing any of our products under development globally or in certain regions; and
|
|
|
·
|
any future patents that we may obtain may not prevent other companies from competing with us by designing their products or conducting their activities so as to avoid the coverage of our patents.
|
The actual protection afforded by a patent varies depending on the product candidate and country and depends upon many factors, including the type of patent, the scope of its coverage, the availability of regulatory related extensions, the availability of legal remedies in a particular country and the validity and enforceability of the patents under existing and future laws. Our ability to maintain or enhance our proprietary position for our product candidates will depend on our success in obtaining effective claims and enforcing those claims once granted. Our issued patents and those that may issue in the future, or those licensed to us, may be challenged, invalidated or circumvented, and the rights granted under any issued patents may not provide us with proprietary protection or competitive advantages against competitors with similar products. Due to the extensive amount of time required for the development, testing and regulatory review of a potential product, it is possible that, before any of our products can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of the patent.
We also rely upon trade secrets, technical know-how and continuing technological innovation to develop and maintain our competitive position. Although we take steps to protect our proprietary rights and information, including the use of confidentiality and other agreements with our employees and consultants, and in our academic and commercial relationships, these steps may be inadequate, these agreements may be violated, or there may be no adequate remedy available for a violation. In addition, courts outside the United States are sometimes less willing to protect trade secrets. Furthermore, our competitors may independently develop substantially equivalent proprietary information and techniques, reverse engineer our information and techniques, or otherwise gain access to our proprietary technology. We may be unable to meaningfully protect our rights in unpatented proprietary technology.
We rely heavily on third parties for product and clinical development of our products.
We currently depend heavily and will depend heavily in the future on third parties for support in product development and clinical development of our products. The termination of a significant number of our existing collaborative arrangements, or our inability to establish and maintain collaborative arrangements could have a material adverse effect on our ability to complete clinical development of our products. Given our limited resources, it may be necessary to establish partnerships with other pharmaceutical companies that have greater financial and technical resources in order to successfully develop and commercialize our products. Although we recently entered into a global strategic alliance with Servier, the leading French independent pharmaceutical company, related to the research, development, and commercialization of Pharmacyclics’ PCI-24781, an orally active, novel, small molecule inhibitor of Pan HDAC enzymes, that is currently in Phase I/II clinical trials in the United States and being developed for the treatment of solid tumors and hematologic malignancies, there is no assurance that any additional partnerships can be obtained, and if obtained, may require us to relinquish product rights that could affect the financial success of these products.
We rely on contract clinical research organizations, or CROs, for various aspects of our clinical development activities including clinical trial monitoring, data collection, safety monitoring and data management. As a result, we have had and continue to have less control over the conduct of clinical trials, the timing and completion of the trials, the required reporting of adverse events and the management of data developed through the trial than would be the case if we were relying entirely upon our own staff. Although we rely on CROs to conduct some of our clinical trials, we are responsible for confirming that each of our clinical trials is conducted in accordance with the investigational plan and protocol. Moreover, the FDA and foreign regulatory agencies require us to comply with regulations and standards, commonly referred to as good clinical practices, for conducting, recording and reporting the results of clinical trials to assure that the data and results are credible and accurate and that the trial participants are adequately protected. Our reliance on third parties does not relieve us of these responsibilities and requirements.
Outside parties may have staffing difficulties, may undergo changes in priorities or may become financially distressed, adversely affecting their willingness or ability to conduct our trials. We may experience unexpected cost increases that are beyond our control. Any failure of such CROs to successfully accomplish clinical trial monitoring, data collection, safety monitoring and data management and the other services they provide for us in a timely manner and in compliance with regulatory requirements could have a material adverse effect on our ability to complete clinical development of our products and obtain regulatory approval. Problems with the timeliness or quality of the work of a CRO may lead us to seek to terminate the relationship and use an alternate service provider. However, making such changes may be costly and may delay our trials, and contractual restrictions may make such a change difficult or impossible. Additionally, it may be difficult to find a replacement organization that can conduct our trials in an acceptable manner and at an acceptable cost.
We lack the resources, capability and experience necessary to manufacture pharmaceuticals and thus rely heavily upon contract manufacturers.
We have no manufacturing facilities and we currently rely on third parties for manufacturing and storage activities related to all of our products in development. Our manufacturing strategy presents the following risks:
|
|
·
|
delays in scale-up to quantities needed for multiple clinical trials, or failure to manufacture such quantities to our specifications, or deliver such quantities on the dates we require, could cause delay or suspension of clinical trials, regulatory submissions and commercialization of our products in development;
|
|
|
·
|
there is no guarantee that the supply of clinical materials can be maintained during the clinical development of our product candidates;
|
|
|
·
|
our current and future manufacturers are subject to ongoing periodic unannounced inspections by the FDA and corresponding regulatory agencies for compliance with strictly enforced current Good Manufacturing Practice and similar foreign standards. Failure to pass these inspections could have a material adverse effect on our ability to produce our products to support our operations;
|
|
|
·
|
if we need to change to other commercial manufacturing contractors, there is no guarantee that we will be able to locate a suitable replacement contractor. The FDA and comparable foreign regulators must approve material manufactured by these contractors prior to our use. This would require new testing and compliance inspections. The new manufacturers would have to practice substantially equivalent processes for the production of our products;
|
|
|
·
|
our current manufacturers might not be able to fulfill our commercial needs, which would require us to seek new manufacturing arrangements and may result in substantial delays in meeting market demand; and
|
|
|
·
|
any disruption of the ability of our manufacturing contractors to supply necessary quantities of our products could have a material adverse effect on our ability to support our operations.
|
Any of these factors could delay clinical trials or commercialization of our products under development and entail higher costs.
We lack marketing, distribution and sales experience.
We have no experience marketing, selling or distributing drug products and currently lack the internal capability to do so. If any of our product candidates are approved by the FDA, we will need a drug sales force with technical expertise prior to the commercialization of any of our product candidates. We have no experience in developing, training or managing a sales force. We will incur substantial additional expenses in developing, training and managing such an organization. We may be unable to build such a sales force, the cost of establishing such a sales force may exceed any product revenues, or our direct marketing and sales efforts may be unsuccessful. In addition, we compete with many other companies that currently have extensive and well-funded marketing and sales operations. Our marketing and sales efforts may be unable to compete successfully against those of such other companies. We will need to enter into co-promotion or other licensing arrangements with larger pharmaceutical or biotechnology firms in order to increase the commercial success of our products. To the extent we enter into co-promotion or other licensing agreements, our product revenues are likely to be lower than if we directly marketed and sold our products, and some or all of the revenues we receive will depend upon the efforts of third parties, which may not be successful and may not be within our control. If we are unable to enter into co-promotion or other licensing agreements on acceptable terms or at all, we may not be able to successfully commercialize our existing and future product candidates. If we are not successful in commercializing our existing and future product candidates, either on our own or through collaborations with one or more third parties, our future product revenue will suffer and we may incur significant losses.
If we lose or are unable to hire and retain qualified personnel, then we may not be able to develop our products or processes and obtain the required regulatory approvals.
We are highly dependent on qualified scientific and management personnel. We will need to expand and effectively manage our managerial, operational, financial, development and other resources in order to successfully pursue our research, development and commercialization efforts for our existing and future product candidates. Our success depends on our continued ability to attract, retain and motivate highly qualified management and personnel with pre-clinical and clinical experience. We will need to hire additional personnel as we continue to expand our research and development and partnering activities.
We face intense competition from other companies and research and academic institutions for qualified personnel. We may not be able to attract or retain qualified management and scientific personnel in the future due to the intense competition for qualified personnel among biotechnology, pharmaceutical and other businesses, particularly in the San Francisco, California area. In September 2008, four members of our Board of Directors resigned and were replaced by four new members. At the same time of this change in our Board, our CEO and CFO resigned their positions and were replaced with Robert W. Duggan as CEO and Rainer (Ramses) Erdtmann as Vice President of Finance and Administration. We are highly dependent on these officers, and in fact Mr. Duggan has provided significant financing to us. If Mr. Duggan were to terminate his position with us, or we were to lose an additional executive officer, any of our senior scientists, a manager of one of our programs, or a significant number of any of our staff or are unable to hire and retain qualified personnel, then our ability to develop and commercialize our products and processes, raise additional capital or implement our business strategy may be adversely affected or prevented and our business may be harmed as a result.
Our business is subject to risks associated with international operations and collaborations.
The laws of foreign countries do not protect our intellectual property rights to the same extent as do the laws of the United States. In countries where we do not have and/or have not applied for patents on our products, we will be unable to prevent others from developing or selling similar products. In addition, in jurisdictions outside the United States where we acquire patent rights, we may be unable to prevent unlicensed parties from selling or importing products or technologies derived elsewhere using our patented technology.
Until we or our licensees obtain the required regulatory approvals for pharmaceuticals in any specific foreign country, we or our licensees will be unable to sell these products in that country. International regulatory authorities have imposed numerous and varying regulatory requirements and the approval procedures can involve additional testing. Approval by one regulatory authority does not ensure approval by any other regulatory authority.
We may be subject to damages resulting from claims that our employees or we have wrongfully used or disclosed alleged trade secrets of their former employers.
Many of our employees were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could hamper or prevent our ability to commercialize certain potential drugs, which could severely harm our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
We may need to implement additional finance and accounting systems, procedures and controls to satisfy reporting requirements.
As a public reporting company, we are required to comply with the Sarbanes-Oxley Act of 2002, including Section 404, and the related rules and regulations of the Securities and Exchange Commission, including expanded disclosures and accelerated reporting requirements and more complex accounting rules. Compliance with Section 404 and other requirements will increase our costs and require additional management resources. We may need to continue to implement additional finance and accounting systems, procedures and controls to satisfy reporting requirements. While we have been able to complete an unqualified assessment as to the adequacy of our internal control over financial reporting for our fiscal year ending June 30, 2010, there is no assurance that future assessments of the adequacy of our internal control over financial reporting will be unqualified. If we are unable to obtain future unqualified reports as to the effectiveness of our internal control over financial reporting, investors could lose confidence in the reliability of our internal control over financial reporting, which could adversely affect our stock price.
Our corporate compliance program cannot guarantee that we are in compliance with all potentially applicable regulations.
The development, manufacturing, pricing, sales, coverage and reimbursement of our products, together with our general operations, are subject to extensive regulation by federal, state and other authorities within the United States and numerous entities outside of the United States. While we have developed and instituted a corporate compliance program based on what we believe are the current best practices, we cannot provide any assurance that governmental authorities will find that our business practices comply with current or future administrative or judicial interpretations of potentially applicable laws and regulations. If we fail to comply with any of these laws and regulations, we could be subject to a range of regulatory actions, including suspension or termination of clinical trials, the failure to approve a product candidate, restrictions on our products or manufacturing processes, withdrawal of products from the market, significant fines, or other sanctions or litigation.
Our facility in California is located near an earthquake fault, and an earthquake or other types of natural disasters or resource shortages could disrupt our operations and adversely affect results.
Important documents and records, such as hard copies of our laboratory books and records for our drug candidates and compounds, are located in our corporate headquarters at a single location in Sunnyvale, California, which is near active earthquake zones. We do not have a formal business continuity or disaster recovery plan, and could therefore experience a significant business interruption in the event of a natural disaster, such as an earthquake, drought or flood, or localized extended outages of critical utilities or transportation systems. In addition, California from time to time has experienced shortages of water, electric power and natural gas. Future shortages and conservation measures could disrupt our operations and cause expense, thus adversely affecting our business and financial results.
Anti-takeover provisions in our charter documents and Delaware law could prevent or delay a change in control.
Our certificate of incorporation and bylaws may discourage, delay or prevent a merger or acquisition that a stockholder may consider favorable. In addition, provisions of the Delaware General Corporation Law also restrict certain business combinations with interested stockholders. These provisions are intended to encourage potential acquirers to negotiate with us and allow our board of directors the opportunity to consider alternative proposals in the interest of maximizing stockholder value. However, these prohibitions may also discourage acquisition proposals or delay or prevent a change in control, which could harm our stock price.
Risks Related to Our Industry
We face rapid technological change and intense competition.
The pharmaceutical industry is subject to rapid and substantial technological change. Therapies designed by other companies to treat the conditions that are the focus of our products are currently in clinical trials. Developments by others may render our products under development or technologies noncompetitive or obsolete, or we may be unable to keep pace with technological developments or other market factors. Technological competition in the industry from pharmaceutical and biotechnology companies, universities, governmental entities and others diversifying into the field is intense and is expected to increase. Many of these entities have significantly greater research and development capabilities than we do, as well as substantially more marketing, sales, manufacturing, financial and managerial resources. These entities represent significant competition for us. Acquisitions of, or investments in, competing pharmaceutical or biotechnology companies by large corporations could increase such competitors’ financial, marketing, manufacturing and other resources. In addition, we may experience competition from companies that have acquired or may acquire technology from universities and other research institutions. As these companies develop their technologies, they may develop proprietary positions that compete with our products. We are engaged in the development of novel therapeutic technologies. Our resources are limited and we may experience technical challenges inherent in such novel technologies.
Competitors have developed or are in the process of developing technologies that are, or in the future may be, the basis for competitive products. Some of these products may have an entirely different approach or means of accomplishing similar therapeutic effects than our products. Our competitors may develop products that are safer, more effective or less costly than our products and, therefore, present a serious competitive threat to our product offerings. Our competitors may price their products below ours, may receive better coverage and/or reimbursement or may have products that are more cost effective than ours.
The widespread acceptance of therapies that are alternatives to ours may limit market acceptance of our products even if commercialized. The diseases for which we are developing our therapeutic products can also be treated, in the case of cancer, by surgery, radiation, biologics and chemotherapy. These treatments are widely accepted in the medical community and have a long history of use. The established use of these competitive products may limit the potential for our products to receive widespread acceptance if commercialized.
The price of our common stock may be volatile.
The market prices for securities of biotechnology companies, including ours, have historically been highly volatile. Our stock, like that of many other companies, has from time to time experienced significant price and volume fluctuations sometimes unrelated to operating performance. For example, during the period beginning July 1, 2007 and ending August 31, 2010, the sales price for one share of our common stock reached a high of $8.60 per share and a low of $0.55 per share. The market price of our common stock may fluctuate significantly due to a variety of factors, including:
|
|
·
|
the progress and results of our preclinical testing, clinical trials, product development and partnering activities;
|
|
|
·
|
quarterly fluctuations in our financial results;
|
|
|
·
|
the development of technological innovations or new therapeutic products by us, our competitors or others;
|
|
|
·
|
changes in governmental regulation;
|
|
|
·
|
developments in patent or other proprietary rights by us, our competitors or others;
|
|
|
·
|
developments and/or announcements by us, our competitors or others;
|
|
|
·
|
litigation;
|
|
|
·
|
public concern as to the safety of products developed by us, our competitors or others;
|
|
|
·
|
departure of key personnel;
|
|
|
·
|
ability to manufacture our products to commercial standards;
|
|
|
·
|
changes in the structure of healthcare payment systems and the coverage and reimbursement policies of governmental and other third-party payors;
|
|
|
·
|
our ability to successfully commercialize our products if they are approved;
|
|
|
·
|
comments by securities analysts; and
|
|
|
·
|
general market conditions in our industry.
|
In addition, if any of the risks described in this section entitled “Risk Factors” actually occur, there could be a dramatic and material adverse impact on the market price of our common stock.
If our products are not accepted by the market or if users of our products are unable to obtain adequate coverage of and reimbursement for our products from government and other third-party payors, our revenues and profitability will suffer.
Our ability to commercialize our products successfully will depend in significant part on the extent to which appropriate coverage of and reimbursement for our products and related treatments are obtained from governmental authorities, private health insurers and other organizations, such as HMOs. Third-party payors are increasingly challenging the prices charged for medical products and services. We cannot provide any assurances that third- party payors will consider our products cost-effective or provide coverage of and reimbursement for our products, in whole or in part.
Uncertainty exists as to the coverage and reimbursement status of newly approved medical products and services and newly approved indications for existing products. Third-party payors may conclude that our products are less safe, less clinically effective, or less cost-effective than existing products, and third-party payors may not approve our products for coverage and reimbursement. If we are unable to obtain adequate coverage of and reimbursement for our products from third-party payors, physicians may limit how much or under what circumstances they will prescribe or administer them. Such reduction or limitation in use of our products could cause our sales to suffer. Even if third-party payors make reimbursement available, payment levels may not be sufficient to make the sale of our products profitable.
Also, the trend towards managed health care in the United States and the concurrent growth of organizations such as HMOs, which could control or significantly influence the purchase of medical services and products, may result in inadequate coverage of and reimbursement for our products. Many third- party payors, including in particular HMOs, are pursuing various ways to reduce pharmaceutical costs, including, for instance, the use of formularies. The market for our products depends on access to such formularies, which are lists of medications for which third-party payors provide reimbursement. These formularies are increasingly restricted, and pharmaceutical companies face significant competition in their efforts to place their products on formularies of HMOs and other third-party payors. This increased competition has led to a downward pricing pressure in the industry. The cost containment measures that third-party payors are instituting could have a material adverse effect on our ability to operate profitably.
Current health care laws and regulations, including the recently enacted health care reform, as well as future legislative or regulatory changes to the healthcare system, may affect our ability to sell our products profitably.
In the United States, there has been recent legislation, as well as legislative and regulatory proposals, changing the healthcare system in ways that may affect our future revenues and profitability, and the future revenues and profitability of our potential customers, suppliers and collaborative partners, and the availability of capital. Federal and state lawmakers regularly propose and, at times, enact legislation that would result in significant changes to the healthcare system and, in particular, that are intended to contain or reduce the costs of medical products and services.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or MMA, influenced the distribution and reimbursement of prescription drugs in the United States. One of the most important aspects of MMA was the creation of the Medicare Part D prescription drug benefit, effective on January 1, 2006. This program subsidizes Medicare beneficiaries for the costs of prescription drugs from private sector providers. Though the program is over four years old, its lasting effects have yet to be seen. One potential risk is that the program will create downward pricing pressure resulting from the enhanced purchasing power of private sector providers under the Part D.
The most significant recent health care legislation is the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or the “Healthcare Reform Act”, which President Obama signed into law in March 2010. This law substantially changes how health care is funded by the state and federal government as well as private insurers, and significantly impacts the pharmaceutical industry. Though the full effect of the Healthcare Reform Act on pharmaceutical companies has yet to be seen, the changes that may affect our business include those governing enrollment in federal healthcare programs, reimbursement changes, new governmental programs, and fraud and abuse enforcements. The Healthcare Reform Act takes effect in stages through 2018.
Certain aspects of the Health Care Reform Act are likely to adversely affect pharmaceutical manufacturers in particular. For example, in 2011, the Healthcare Reform Act will impose non-deductible annual flat fees on pharmaceutical manufacturers and importers based upon relative market share. Furthermore, as part of the Healthcare Reform Act closing a funding gap in the Medicare Part D prescription drug program, certain pharmaceutical manufacturers will be required to provide a 50% discount on drugs dispensed to beneficiaries within this funding gap.
There also have been and likely will continue to be legislative and regulatory proposals at the state and federal levels that could bring about significant changes to the Medicaid drug rebate program and other federal pharmaceutical pricing and rebate programs. Given these and other recent federal and state government initiatives directed at lowering the total cost of health care, federal and state lawmakers will likely continue to focus on health care reform, the cost of prescription pharmaceuticals and on the reform of the Medicare and Medicaid programs. These efforts could adversely affect our business by, among other possibilities, limiting the prices that can be charged for drugs we develop or the amount of reimbursement available for these products from governmental agencies or third-party payors, limiting the profits that pharmaceutical companies may earn on certain sales, increasing the tax obligations on pharmaceutical companies, increasing our rebate liability, or limiting our commercial opportunity. We cannot predict the impact on our business of any legislation or regulations that may be adopted in the future. Any cost containment measures and other healthcare system reforms that are adopted could have a material adverse effect on our ability to operate profitably.
We may need to change our business practices to comply with health care fraud and abuse regulations, and our failure to comply with such laws could adversely affect our business, financial condition and results of operations.
Our operations will be directly, or indirectly through our customers, subject to various state and federal fraud and abuse laws, including, without limitation, the federal Anti-Kickback Statute and False Claims Act. These laws may impact, among other things, our proposed sales, marketing and education programs.
The federal Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual, or the furnishing or arranging for a good or service, for which payment may be made under a federal healthcare program such as the Medicare and Medicaid programs. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the statute has been violated. The Anti-Kickback Statute is broad and prohibits many arrangements and practices that are lawful in businesses outside of the healthcare industry. Recognizing that the Anti-Kickback Statute is broad and may technically prohibit many innocuous or beneficial arrangements, Congress authorized the Department of Health and Human Services, Office of Inspector General (“OIG”) to issue a series of regulations, known as the “safe harbors.” These safe harbors set forth provisions that, if all their applicable requirements are met, will assure healthcare providers and other parties that they will not be prosecuted under the Anti-Kickback Statute. The failure of a transaction or arrangement to fit precisely within one or more safe harbors does not necessarily mean that it is illegal or that prosecution will be pursued. However, conduct and business arrangements that do not fully satisfy each applicable safe harbor may result in increased scrutiny by government enforcement authorities such as the OIG. Penalties for violations of the federal Anti-Kickback Statute include criminal penalties and civil sanctions such as fines, imprisonment and possible exclusion from Medicare, Medicaid and other federal healthcare programs. Many states have also adopted laws similar to the federal Anti-Kickback Statute, some of which apply to the referral of patients for healthcare items or services reimbursed by any source, not only the Medicare and Medicaid programs.
The federal False Claims Act prohibits persons from knowingly filing or causing to be filed a false claim to, or the knowing use of false statements to obtain payment from, the federal government. Suits filed under the False Claims Act, known as “qui tam” actions, can be brought by any individual on behalf of the government and such individuals, sometimes known as “relators” or, more commonly, as “whistleblowers,” may share in any amounts paid by the entity to the government in fines or settlement. The frequency of filing of qui tam actions has increased significantly in recent years, causing greater numbers of healthcare companies to have to defend a False Claim action. When an entity is determined to have violated the federal False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties of between $5,500 to $11,000 for each separate false claim. Various states have also enacted laws modeled after the federal False Claims Act.
In addition to the laws described above, the Health Insurance Portability and Accountability Act of 1996 created two new federal crimes: healthcare fraud and false statements relating to healthcare matters. The healthcare fraud statute prohibits knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private payors. A violation of this statute is a felony and may result in fines, imprisonment or exclusion from government sponsored programs. The false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. A violation of this statute is a felony and may result in fines or imprisonment.
If our operations are found to be in violation of any of the laws described above and other applicable state and federal fraud and abuse laws, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from government healthcare programs, and the curtailment or restructuring of our operations.
Our business exposes us to product liability claims.
The testing, manufacture, marketing and sale of our products involve an inherent risk that product liability claims will be asserted against us. We face the risk that the use of our products in human clinical trials will result in adverse effects. If we complete clinical testing for our products and receive regulatory approval to market our products, we will mark our products with warnings that identify the known potential adverse effects and the patients who should not receive our product. We cannot ensure that physicians and patients will comply with these warnings. In addition, unexpected adverse effects may occur even with use of our products that receive approval for commercial sale. Although we are insured against such risks in connection with clinical trials, our present product liability insurance may be inadequate. A successful product liability claim in excess of our insurance coverage could have a material adverse effect on our business, financial condition and results of operations. Any successful product liability claim may prevent us from obtaining adequate product liability insurance in the future on commercially desirable or reasonable terms. In addition, product liability coverage may cease to be available in sufficient amounts or at an acceptable cost. An inability to obtain sufficient insurance coverage at an acceptable cost or otherwise to protect against potential product liability claims could prevent or inhibit the commercialization of our pharmaceutical products. A product liability claim or recall would have a material adverse effect on our reputation, business, financial condition and results of operations.
Our business involves environmental risks.
In connection with our research and development activities and our manufacture of materials and products, we are subject to federal, state and local laws, rules, regulations and policies governing the use, generation, manufacture, storage, air emission, effluent discharge, handling and disposal of certain materials, biological specimens and wastes. Although we believe that we have complied with the applicable laws, regulations and policies in all material respects and have not been required to correct any material noncompliance, we may be required to incur significant costs to comply with environmental and health and safety regulations in the future. Our research and development involves the controlled use of hazardous materials, including but not limited to certain hazardous chemicals and radioactive materials. Although we believe that our safety procedures for handling and disposing of such materials comply with the standards prescribed by state and federal regulations, we cannot completely eliminate the risk of contamination or injury from these materials. In the event of such an occurrence, we could be held liable for any damages that result and any such liability could exceed our resources.
Item 1B. Unresolved Staff Comments
None.
Item 2. Properties
Our corporate offices are located in Sunnyvale, California, where, as of July 1, 2010, we lease approximately 32,000 square feet under a lease that expires in December 2011. Our facility includes administrative and research and development space. The lease is a non-cancelable operating lease. We are currently looking to lease additional space.
Item 3. Legal Proceedings
None.
Item 4. (Removed and Reserved)
PART II
Our common stock trades on the NASDAQ Stock Market under the symbol "PCYC." The following table sets forth, for the periods indicated, the high and low sales prices of our common stock.
|
HIGH
|
LOW
|
|||||||
|
FISCAL YEAR ENDED JUNE 30, 2010
|
||||||||
|
First Quarter
|
$ | 2.41 | $ | 1.13 | ||||
|
Second Quarter
|
3.40 | 1.71 | ||||||
|
Third Quarter
|
6.92 | 3.01 | ||||||
|
Fourth Quarter
|
8.60 | 5.45 | ||||||
|
FISCAL YEAR ENDED JUNE 30, 2009
|
||||||||
|
First Quarter
|
$ | 2.82 | $ | 1.42 | ||||
|
Second Quarter
|
2.25 | 0.66 | ||||||
|
Third Quarter
|
1.38 | 0.57 | ||||||
|
Fourth Quarter
|
1.47 | 0.99 | ||||||
As of August 31, 2010, there were 118 holders of record of our common stock. We have not paid cash dividends on our common stock since our inception and we do not anticipate paying any in the foreseeable future.
Sales of Unregistered Securities
Not Applicable.
Stock Repurchases in the Fourth Quarter
Not Applicable.
Securities Authorized for Issuance Under Equity Compensation Plans
See Item 12, “Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” for information with respect to our compensation plans under which equity securities are authorized for issuance.
Item 6. Selected Financial Data
The data set forth below should be read in conjunction with Management’s Discussion and Analysis of Financial Condition and Results of Operations and the financial statements and related notes included elsewhere herein.
|
Period
|
||||||||||||||||||||||||
|
from
|
||||||||||||||||||||||||
|
Inception
|
||||||||||||||||||||||||
|
(April 19, 1991)
|
||||||||||||||||||||||||
|
through
|
||||||||||||||||||||||||
|
Year Ended June 30,
|
June 30,
|
|||||||||||||||||||||||
|
2010
|
2009
|
2008
|
2007
|
2006
|
2010
|
|||||||||||||||||||
|
(in thousands, except per share amounts)
|
||||||||||||||||||||||||
|
STATEMENT OF OPERATIONS DATA:
|
||||||||||||||||||||||||
|
Revenues(1):
|
||||||||||||||||||||||||
|
License and milestone revenues
|
$ | 9,307 | $ | - | $ | - | $ | - | $ | - | $ | 17,162 | ||||||||||||
|
Grant and contract revenues
|
- | - | - | 126 | 181 | 6,154 | ||||||||||||||||||
|
Total revenues
|
9,307 | - | - | 126 | 181 | 23,316 | ||||||||||||||||||
|
Operating expenses:
|
||||||||||||||||||||||||
|
Research and development
|
17,358 | 13,954 | 18,180 | 21,115 | 25,737 | 343,234 | ||||||||||||||||||
|
General and administrative
|
7,561 | 8,474 | 7,332 | 7,403 | 11,919 | 92,176 | ||||||||||||||||||
|
Purchased in-process research
|
||||||||||||||||||||||||
|
and development
|
- | - | - | - | 6,647 | 6,647 | ||||||||||||||||||
|
Total operating expenses
|
24,919 | 22,428 | 25,512 | 28,518 | 44,303 | 442,057 | ||||||||||||||||||
|
Loss from operations
|
(15,612 | ) | (22,428 | ) | (25,512 | ) | (28,392 | ) | (44,122 | ) | (418,741 | ) | ||||||||||||
|
Interest income
|
81 | 137 | 1,206 | 2,175 | 1,964 | 43,025 | ||||||||||||||||||
|
Interest expense and other
|
||||||||||||||||||||||||
|
income (expense), net
|
(43 | ) | (606 | ) | 8 | - | - | (2,206 | ) | |||||||||||||||
|
Loss before provision
|
||||||||||||||||||||||||
|
for income taxes
|
(15,574 | ) | (22,897 | ) | (24,298 | ) | (26,217 | ) | (42,158 | ) | (377,922 | ) | ||||||||||||
|
Provision for income taxes
|
550 | (550 | ) | - | - | - | - | |||||||||||||||||
|
Net Loss
|
$ | (15,024 | ) | $ | (23,447 | ) | $ | (24,298 | ) | $ | (26,217 | ) | $ | (42,158 | ) | $ | (377,922 | ) | ||||||
|
Basic and diluted net
|
||||||||||||||||||||||||
|
loss per share(2)
|
$ | (0.31 | ) | $ | (0.88 | ) | $ | (0.93 | ) | $ | (1.08 | ) | $ | (2.12 | ) | |||||||||
|
Weighted average shares used to compute basic and
|
||||||||||||||||||||||||
|
diluted net loss per share
|
48,344 | 26,570 | 25,989 | 24,175 | 19,889 | |||||||||||||||||||
|
June 30,
|
||||||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||||||
|
(in thousands)
|
||||||||||||||||||||||||
|
BALANCE SHEET DATA:
|
||||||||||||||||||||||||
|
Cash, cash equivalents and marketable
|
||||||||||||||||||||||||
|
securities(3)
|
$ | 74,149 | $ | 16,326 | $ | 16,755 | $ | 38,762 | $ | 40,477 | ||||||||||||||
|
Total assets
|
76,820 | 18,301 | 18,367 | 41,095 | 42,729 | |||||||||||||||||||
|
Deferred revenue
|
6,099 | 11,628 | - | - | - | |||||||||||||||||||
|
Total liabilities
|
10,059 | 20,042 | 1,922 | 2,694 | 3,409 | |||||||||||||||||||
|
Deficit accumulated during
|
||||||||||||||||||||||||
|
development stage
|
(377,922 | ) | (362,898 | ) | (339,451 | ) | (315,153 | ) | (288,936 | ) | ||||||||||||||
|
Total stockholders' equity (deficit)
|
66,761 | (1,741 | ) | 16,445 | 38,401 | 39,320 | ||||||||||||||||||
|
(1) See Note 2 to the financial statements for a discussion of revenue recognition related to the Servier agreement.
|
||||||||||||||||||||||||
|
(2) See Note 1 to the financial statements for a description of the computation of basic and diluted net loss per share.
|
||||||||||||||||||||||||
|
(3) See Note 6 to the financial statements for a description of equity financings completed during fiscal 2010.
|
||||||||||||||||||||||||
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
In addition to historical information, this report contains predictions, estimates, assumptions and other forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended and Section 21E of the Securities Exchange Act of 1934, as amended. Actual results could differ materially from any future performance suggested in this report as a result of the risks, uncertainties and other factors described herein and elsewhere in this report, including those discussed in “Risk Factors.”
Company Overview
We are a clinical-stage biopharmaceutical company focused on developing and commercializing innovative small-molecule drugs for the treatment of cancer and immune mediated diseases. Our mission and goal is to build a viable biopharmaceutical company that designs, develops and commercializes novel therapies intended to improve quality of life, increase duration of life and resolve serious unmet medical healthcare needs; and to identify promising product candidates based on exceptional scientific development expertise, develop our products in a rapid, cost-efficient manner and pursue commercialization and/or development partners when and where appropriate. We exist to make a difference for the better and these are important times to do just that.
Presently, we have four product candidates in clinical development, a clinical development candidate in late-stage preclinical evaluation and several preclinical molecules in lead optimization. To date, substantially all of our resources have been dedicated to the research and development of our products, and we have not generated any commercial revenues from the sale of our products. We do not anticipate the generation of any product commercial revenues until we receive the necessary regulatory and marketing approvals to launch one of our products.
We have incurred significant operating losses since our inception in 1991, and as of June 30, 2010, had an accumulated deficit of approximately $377,922,000. The process of developing and commercializing our products requires significant research and development, preclinical testing and clinical trials, manufacturing arrangements as well as regulatory and marketing approvals. These activities, together with our general and administrative expenses, are expected to result in significant operating losses until the commercialization of our products, or partner collaborations, generate sufficient revenues to cover our expenses. We expect that losses will fluctuate from quarter to quarter and that such fluctuations may be substantial. Our achieving profitability depends upon our ability to successfully complete the development of our products, obtain required regulatory approvals and successfully manufacture and market our products.
Bruton’s Tyrosine Kinase (Btk) Inhibitor for oncology, PCI-32765 is an orally active small molecule inhibitor of Bruton’s tyrosine kinase (Btk) that is being developed by Pharmacyclics for the treatment of patients with B-cell lymphoma or leukemia (B-cells are white blood cells and are an important part of our immune system, B-cell lymphoma and leukemia are types of blood cancers). Btk plays a prominent role in the development as well as the survival of white blood cells, in particular B-cells. B-cell lymphocyte maturation is mediated by B-cell receptor (BCR) signal transduction and Btk is part of the BCR signaling pathway. Recent studies indicate that some large B-cell lymphomas have proteins that are activated and lie downstream of the BCR and that Btk inhibition can induce apoptosis (i.e. cell death) in these B-cells. BCR signaling is also thought to promote cancer cell expansion and survival in chronic lymphocytic leukemia, a prevalent blood cancer. In preclinical models, inhibition of Btk by PCI-32765 led to apoptosis in multiple malignant B-cell lines, and inhibited B-cell lymphoma progression in vivo.
A multicenter U.S. Phase I trial in B-cell lymphoma is currently enrolling patients. This Phase I trial is designed to determine the safety and tolerability of our Btk Inhibitor PCI-32765, and to evaluate effects of Btk inhibition (using pharmacodynamic assays) and tumor response.
We reported interim results of the trial as an oral presentation at the American Society for Clinical Oncology (ASCO) Annual Meeting on June 4-8, 2010 in Chicago, IL. Our abstract was selected among the few to be presented in an oral Session (Abstract Control Number: 54096, Permanent Abstract ID: 8012, Abstract Title: Btk inhibitor PCI-32765 monotherapy induces objective responses in patients with relapsed aggressive NHL: Evidence of antitumor activity from a phase I study).
Trial Results as of August 31, 2010: A total of 47 relapsed/refractory and progressing patients with a variety of B cell malignancies were enrolled in 6 dose groups (1.25, 2.5, 5.0, 8.3, 12.5 mg/kg/day of standard dosing [4 weeks on / 1 week off] and 8.3 mg/kg/day continuous dosing). Forty-one of the enrolled patients had an on-treatment tumor assessment after completing two cycles of therapy and are evaluable for efficacy as of August 31, 2010. Twenty-one of these evaluable patients had a complete or partial response as their best response and eleven patients had stable diseases. This equates to a response rate of 51% in the evaluable patients (69% in CLL, 75% in Mantle, 42% in Follicular, 33% in Marginal and 38% in DLBCL). On an intent-to-treat ("ITT") basis the overall response rate (ORR) was 45%.
Ongoing research continues to expand clinical indications for our Btk inhibitor PCI-32765 beyond blood cancers (i.e. hematologic tumors) and to support our ongoing clinical trials in lymphoma and CLL. PCI-32765 is a potent inhibitor of histamine release (also known as degranulation) from mast cells and basophils, which are specific immune cells. Largely based on the work of Dr. Gerard Evan and others at UCSF, mast cells have emerged as an important component of the proinflammatory microenvironment, which is crucial for the physical expansion and maintenance of tumor growth (Nature Medicine 13 (10), p. 1211-18). Dr. Evan’s lab tested our Btk Inhibitor PCI-32765 in his mouse pancreatic model. In the model, PCI-32765 completely blocked mast cell degranulation which led to tumor regression and vascular collapse. This work was presented at the 2010 American Association for Cancer Research (AACR) Annual Meeting as an oral presentation on April 20, 2010. With respect to research in lymphoma the NCI has made valuable discoveries indicating that many types of lymphoma are driven by chronic active B-cell receptor signaling, a process dependent upon Btk activity. These findings, which included experiments with PCI-32765, were published in the journal Nature (Davis et al. (2010) Vol. 463, p88-94).
Bruton’s Tyrosine Kinase (Btk) Inhibitor, PCI-45292 is an orally-active small molecule inhibitor of Bruton’s tyrosine kinase that is being developed by Pharmacyclics for the treatment of patients with autoimmune diseases. In our most recent quarter we identified PCI-45292 as a clinical development candidate. Most recently we have started IND-enabling preclinical safety studies. Pharmacyclics is intending to complete the in-life phase of these studies by the end of 2010 and is planning to file an IND (Investigational New Drug Application) in the late second quarter of 2011. Compared to PCI-32765, PCI-45292 was shown to have increased selectivity for Btk inhibition, a reduced potential for off-target protein binding, and improved metabolic stability. PCI-45292 ameliorated inflammation in collagen-induced arthritis models. Therefore we believe that PCI-45292 has the qualities to become a new oral disease modifying anti-rheumatic drug (DMARD).
Factor VIIa Inhibitor, PCI-27483 is a potent and highly selective small molecule inhibitor of coagulation (clotting) Factor VIIa. This drug targets Factor VIIa, the active form of Factor VII that arises from interaction with the cell surface membrane protein known as tissue factor (TF). In cancer, the Factor VIIa:TF complex is found in abundance in pancreatic, gastric, colon and other tumors, and triggers a host of patho-physiologic processes that facilitate tumor blood vessel formation (angiogenesis), growth and metastases. The Factor VIIa:TF complex is thought to be the cause of the increased propensity of cancer patients to develop thromboses, also known as blood clots. Laboratory studies and animal models indicate that our inhibitor of Factor VIIa blocks the growth of tumors that express TF.
We have completed a Phase I testing of Factor VIIa Inhibitor PCI-27483 in healthy volunteers. The primary objective of the ascending dose Phase I study was to assess the pharmacodynamic and pharmacokinetic profiles (i.e. the relationship between drug level and pharmacological response) of Factor VIIa Inhibitor PCI-27483 following a single, subcutaneous injection. In addition, the safety and tolerability of our Factor VIIa Inhibitor was evaluated. The drug was well tolerated and no adverse event was observed at any dose level. The International Normalized Ratio (INR) of prothrombin time, a simple laboratory test for coagulation, was used to measure pharmacodynamic effect at dose levels of 0.05, 0.20, 0.80 and 2.0 mg/kg. A mean peak INR of 2.7 was achieved without adverse effects at the highest dose level administered. The target INR range for oral anti-coagulants i.e. Coumadin, is between 2 and 3.
A multicenter Phase I/II study began with Factor VIIa Inhibitor PCI-27483 in the fourth quarter of calendar 2009. The study is enrolling patients with locally advanced or metastatic pancreatic cancer that are either receiving or are planned to receive gemcitabine therapy. In the Phase I portion of the study, ascending repeated doses of PCI-27483 were evaluated in 8 patients receiving concomitant gemcitabine therapy. PCI-27483 was administered twice daily by subcutaneous injection at dosages ranging from 0.8 to 1.5 mg/kg. The Phase II portion of the study has now started and patients are being randomized into two groups to receive either gemcitabine alone or gemcitabine plus PCI-27483. For both phases of the study, the planned duration of treatment with PCI-27483 is 12 weeks. Patients with stable disease at the end of 12 weeks have the option to continue PCI-27483 treatment until disease progression. The objectives of the study are to: a) assess the safety of Pharmacyclics Factor VIIa Inhibitor PCI-27483 at pharmacologically active dose levels; b) to assess potential inhibition of tumor progression and c) obtain initial information of the effects on the incidence of thromboembolic events.
Histone deacetylase (HDAC) Inhibitor, PCI-24781 is an orally-bioavailable histone deacetylase inhibitor that is currently in multiple clinical trials, including a Phase I trial in patients with advanced solid tumors, a Phase I/II trial in patients with recurrent lymphomas and a Phase I/II trial in sarcoma patients (in combination with doxorubicin, an anti tumor agent). Histone Deacetylases are enzymes in a cell responsible for turning off and on genes. PCI-24781 targets histone deacetylase (HDAC) enzymes and inhibits their function. We have shown that PCI-24781 impacts the cell by multiple mechanisms including a re-expression of tumor suppressors, disruption of DNA repair mechanisms, cell cycle inhibition and by generating an increase in reactive oxygen species, all of which may contribute to tumor cell toxicity. Previous clinical trials have demonstrated that our HDAC Inhibitor PCI-24781 has favorable systemic elimination properties when dosed orally, and inhibits the target enzymes. Clinical response or control of tumor growth has been recorded in the three single-agent clinical trials to date.
We announced the Phase I trial results of our HDAC Inhibitor PCI 24781, tested in heavily pretreated relapsed lymphoma patients (those who had a response but the cancer reoccurred) and/or refractory lymphoma patients (those who had an earlier treatment that did not have an effect on the cancer). The results of this trial were published at the 51st annual meeting of the American Society for Hematology in December of 2009. In a review of the clinical data at the end of the Phase I dose-escalation portion of the current lymphoma trial, we have found that PCI-24781 produced encouraging efficacy results with minimal toxicity when administered as a single agent. Among 16 lymphoma patients evaluated for disease control after the completion of at least 2 treatment cycles, we observed 1 complete response (follicular lymphoma), 4 partial responses (2 follicular lymphoma, 1 diffuse large B-cell lymphoma, 1 mantle cell lymphoma) and 7 patients with stable disease. Of the 4 follicular lymphoma patients evaluated in this trial, 3 had objective response and one manifested stable disease. The HDAC Inhibitor PCI-24781 has demonstrated a good safety profile in approximately 90 patients treated so far, with the main dose-limiting toxicity observed being a rapidly reversible thrombocytopenia (a decrease in platelets, which are blood cells necessary for clotting). This effect is common among HDAC Inhibitors and is considered a class effect, related to the pharmacologic mechanism of action. The duration and severity of the thrombocytopenia is being successfully managed using novel dose scheduling strategies that we have developed and tested in the clinic.
In preclinical models, we have identified synergy of the Pharmacyclics HDAC inhibitor PCI-24781 with several approved cancer therapeutics, and some of these combinations are being or will be tested in the clinic, including the use of PCI-24781 with doxorubicin in sarcoma patients (sarcoma is a cancer of soft tissues). This trial is co-sponsored by investigators at Massachusetts General Hospital and Dana-Farber / Harvard Cancer Center. During this trial we will also test the novel biomarker RAD51 that we have developed in collaboration with scientists at Stanford University (Adimoolam et al., Proc Natl Acad Sci USA 2007; 104:19482-7), which may be useful as predictive biomarker in clinical testing by improving patient selection.
In April 2009, we entered into a collaboration agreement with Servier pursuant to which Pharmacyclics granted to Servier an exclusive license for its pan-HDAC inhibitors, including PCI-24781, for territories throughout the world excluding the United States. Under the terms of the agreement, Servier acquired the exclusive right to develop and commercialize the pan-HDAC inhibitor product worldwide except for the United States and will pay a royalty to Pharmacyclics on sales outside of the United States. Pharmacyclics will continue to own all rights to develop and commercialize within the United States. Servier is committing resources to the clinical development of Pharmacyclics HDAC Inhibitor PCI-24781. During our fiscal year 2010 we signed a supply agreement and delivered to Servier 10 kg of PCI-24781 drug substance, to support their Phase I and Phase II clinical development program.
Histone deacetylase-8 (HDAC8) isoform-specific Inhibitor Program is at the preclinical lead optimization stage. Most recently, a poster presentation describing the lead optimization and preclinical development of our HDAC8 isoform-specific inhibitors, including their efficacy in certain cancer subtypes like T-cell leukemia and lymphoma and neuroblastoma, as well as their potential utility in autoimmune disease was presented on April 21, 2010 during the 101st annual meeting of the American Association for Cancer Research (AACR), in Washington, D.C.
Motexafin gadolinium (MGd, PCI-0120) is a radiation and chemotherapy sensitizing agent with a novel mechanism of action. MGd is designed to accumulate selectively in cancer cells. Once inside cancer cells, MGd in combination with radiation induces apoptosis (cell death) by disrupting the oxygen dependent pathways in a cell. MGd is also detectable by magnetic resonance imaging (MRI) and may allow for more precise tumor detection. The National Cancer Institute (NCI) is currently sponsoring a Phase I/II multi-center study in newly diagnosed patients with glioblastoma multiform (a type of primary brain cancer). In this study, which completed enrollment in July 2009, MGd is being administered in combination with radiation therapy and temozolomide, a chemotherapy for brain cancers. Previous studies in malignant brain cancers have shown that the combination of MGd and temozolomide have no overlapping toxicities when used in combination.
We are subject to risks common to pharmaceutical companies developing products, including risks inherent in our research, development and commercialization efforts, preclinical testing, clinical trials, uncertainty of regulatory and marketing approvals, uncertainty of market acceptance of our products, history of and expectation of future operating losses, reliance on collaborative partners, enforcement of patent and proprietary rights, and the need for future capital. In order for a product to be commercialized, we must conduct preclinical tests and clinical trials, demonstrate efficacy and safety of our product candidates to the satisfaction of regulatory authorities, obtain marketing approval, enter into manufacturing, distribution and marketing arrangements, build a U.S. commercial capability, obtain market acceptance and, in many cases, obtain adequate coverage of and reimbursement for our products from government and private insurers. We cannot provide assurance that we will generate revenues or achieve and sustain profitability in the future.
Critical Accounting Policies, Estimates and Judgments
This discussion and analysis of financial condition and results of operations is based upon our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosures. On an on-going basis, we evaluate our estimates, including those related to revenue recognition and clinical trial accruals. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results, however, may differ significantly from these estimates under different assumptions or conditions and may adversely affect the financial statements.
We believe the following critical accounting policies reflect the more significant judgments and estimates used in the preparation of our financial statements and accompanying notes.
Revenue Recognition
We recognize revenue when all four revenue recognition criteria have been met: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the fee is fixed or determinable; and collectability is reasonably assured. Revenue under our license and collaboration arrangements is recognized based on the performance requirements of the contract. Amounts received under such arrangements consist of up-front collaboration payments, periodic milestone payments and payments for research activities. Our collaborations with multiple elements are evaluated and are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value and whether there is verifiable objective and reliable evidence of the fair value of the undelivered items. The consideration we receive is combined and recognized as a single unit of accounting when criteria for separation are not met.
Up-front payments under agreements which include future performance requirements are recorded as deferred revenue and are recognized over the performance period. The performance period is estimated at the inception of the arrangement and is reevaluated at each reporting period. The reevaluation of the performance period may shorten or lengthen the period during which the deferred revenue is recognized. Revenues related to substantive, at-risk collaboration milestones are recognized upon achievement of the event specified in the underlying agreement. Revenues for research activities are recognized as the related research efforts are performed.
Research and Development Expenses and Accruals
Research and development expenses include personnel and facility-related expenses, outside contracted services including clinical trial costs, manufacturing and process development costs, research costs and other consulting services. Research and development costs are expensed as incurred.
Our cost accruals for clinical trials are based on estimates of the services received and efforts expended pursuant to contracts with clinical trial centers and clinical research organizations. In the normal course of business we contract with third parties to perform various clinical trial activities in the on-going development of potential products. The financial terms of these agreements are subject to negotiation and vary from contract to contract and may result in uneven payment flows. Payments under the contracts depend on factors such as the achievement of certain events, the successful enrollment of patients, the completion of portions of the clinical trial or similar conditions. The objective of our accrual policy is to match the recording of expenses in our financial statements to the actual services received and efforts expended. As such, expense accruals related to clinical trials are recognized based on our estimate of the degree of completion of the event or events specified in the specific clinical study or trial contract.
Share-Based Compensation
Share-based compensation cost for employee stock options is measured at the grant date based on the fair value of the award. The accounting grant date for employee stock options with performance obligations is the date on which the performance goals have been defined and a mutual understanding of the terms has been reached. Generally options with performance obligations vest over a four year period, with the goals set and agreed upon annually. Share-based compensation for non-employee stock options is re-estimated at each period-end through the vesting date.
The fair value of each stock option is estimated using the Black-Scholes option pricing model. Expected volatility is based on historical volatility data of our stock. The expected term of stock options granted is based on historical data and represents the period of time that stock options are expected to be outstanding. The expected term for each of our employee options, non-employee options and our Employee Stock Purchase Plan is calculated for and applied to one group of grants as we do not expect substantially different exercise or post-vesting termination behavior among our employee or non-employee population. The risk-free interest rate is based on a zero-coupon United States Treasury bond whose maturity period equals the expected term of the our options.
Options vest upon the passage of time or a combination of time and the achievement of certain performance obligations. The Compensation Committee of the Board of Directors will determine if the performance conditions have been met. Share-based compensation expense for the options with performance obligations is recorded when the company believes that the vesting of these options is probable.
Recent Accounting Pronouncements
In October 2009, the FASB issued two updates relating to revenue recognition. The first update eliminates the requirement that all undelivered elements in an arrangement with multiple deliverables have objective and reliable evidence of fair value before revenue can be recognized for items that have been delivered. The update also no longer allows use of the residual method when allocating consideration to deliverables. Instead, arrangement consideration is to be allocated to deliverables using the relative selling price method, applying a selling price hierarchy. Vendor specific objective evidence (VSOE) of selling price should be used if it exists. Otherwise, third party evidence (TPE) of selling price should be used. If neither VSOE nor TPE is available, the company’s best estimate of selling price should be used. The second update eliminates tangible products from the scope of software revenue recognition guidance when the tangible products contain software components and non-software components that function together to deliver the tangible products’ essential functionality. Both updates require expanded qualitative and quantitative disclosures and are effective for fiscal years beginning on or after June 15, 2010, with prospective application for new or materially modified arrangements or retrospective application permitted. Early adoption is permitted. The same transition method and period of adoption must be used for both updates. We are in the process of analyzing the impact of these updates.
In January 2010, the FASB issued ASU 2010-06, “Fair Value Measurements and Disclosures (ASC 820): Improving Disclosures about Fair Value Measurements.” This update will require (1) an entity to disclose separately the amounts of significant transfers in and out of Levels 1 and 2 fair value measurements and to describe the reasons for the transfers; and (2) information about purchases, sales, issuances and settlements to be presented separately (i.e., present the activity on a gross basis rather than net) in the reconciliation for fair value measurements using significant unobservable inputs (Level 3 inputs). This guidance clarifies existing disclosure requirements for the level of disaggregation used for classes of assets and liabilities measured at fair value and requires disclosures about the valuation techniques and inputs used to measure fair value for both recurring and nonrecurring fair value measurements using Level 2 and Level 3 inputs. The new disclosures and clarifications under this ASU are effective over a period of two fiscal years, for interim and annual reporting periods beginning after December 15, 2009 and after December 15, 2010. The updates for the first adoption date under ASU 2010-06 did not have a material impact on our financial statements. The adoption of the updates for the second date is not expected to have a material impact on our financial statements.
In March 2010, the FASB issued new guidance on the use of the milestone method of recognizing revenue for research and development arrangements under which consideration to be received by the vendor is contingent upon the achievement of certain milestones. The update provides guidance on the criteria that should be met for determining whether the milestone method of revenue recognition is appropriate. A vendor can recognize consideration in its entirety as revenue in the period in which the milestone is achieved only if the milestone meets all criteria to be considered substantive. Additional disclosures describing the consideration arrangement and the entity’s accounting policy for recognition of such milestone payments are also required. The new guidance is effective for fiscal years, and interim periods within such fiscal years, beginning on or after June 15, 2010, with early adoption permitted. The guidance may be applied prospectively to milestones achieved during the period of adoption or retrospectively for all prior periods. We do not believe the adoption of this guidance will have a significant impact on our financial statements.
Results of Operations
Revenues
The following table summarizes our revenue over the last three fiscal years:
|
2010
|
2009
|
2008
|
||||||||||
|
License and milestone revenues
|
$ | 9,307,000 | $ | - | $ | - | ||||||
We recorded $9,307,000 in revenue in the year ended June 30, 2010 associated with our collaboration and license agreement with Servier which was entered into in April 2009. Given that the deliverables under the collaboration agreement with Servier do not meet criteria in the accounting rules for separation (e.g., no separately identifiable fair value), the arrangement is being treated as a single unit-of-accounting for purposes of revenue recognition. We recognize the combined unit of accounting over the estimated period required to complete the research activities (two years), which coincides with the delivery period for all substantive obligations or “deliverables” associated with this agreement.
The collaboration and license agreement required us to enter into an agreement to supply drug product for Servier’s use in clinical trials. As the supply agreement is considered part of the arrangement we deferred recognition of all revenue under the Servier collaboration agreement until the supply agreement was completed and executed. We completed the drug supply agreement with Servier in the second quarter ended December 31, 2009 and therefore began recognizing revenue from the Servier collaboration agreement in fiscal 2010. Of the amount recognized in fiscal 2010, $1,211,000 represents the pro rata portion of revenue attributable to the period from April 2009 (i.e., the signing of the collaboration agreement) to June 30, 2009, had the supply agreement been completed in April 2009.
Of the total research and development collaboration revenues for the year ended June 30, 2010, $6,645,000 represents the amortization of the $11,000,000 upfront payment from Servier received in April 2009. The remaining revenue of $2,662,000 represents the pro-rata completion of services attributable to payments of $4,406,000 from Servier associated with research payments, our supply commitment and reimbursements of patent expenses received since establishing the collaboration.
Research and Development Expenses
The following table summarizes the period over period changes in our research and development (R&D) expenses over the last three fiscal years:
|
2010
|
Change
|
2009
|
Change
|
2008
|
|||||||||
|
R & D expenses
|
$
|
17,358,000
|
24%
|
$
|
13,954,000
|
-23%
|
|
$ |
18,180,000
|
The increase of 24% or $3,404,000 in research and development expenses for the year ended June 30, 2010 as compared to the year ended June 30, 2009, was primarily due to a $1,260,000 increase in stock compensation charges associated with increased non-employee option grants and expense associated with our Employee Stock Purchase Plan and increases of $934,000 in drug manufacturing costs associated with our HDAC, Btk and Factor VIIa programs, $835,000 in outside clinical trial costs, $496,000 in outside contracts and research, $358,000 in outside services and consulting expenses and $254,000 in outside preclinical costs. These increases were partially offset by a $1,000,000 expense incurred in the prior year associated with the amendment of our agreement with Celera Corporation.
R&D expenses in fiscal 2009 decreased by $4,226,000 compared to fiscal 2008 primarily due to a decrease of $1,736,000 in personnel costs due to lower headcount and a decrease of $1,517,000 in drug manufacturing costs and a decrease of $1,464,000 in outside preclinical costs associated with our HDAC, Btk and Factor VIIa programs, partially offset by an increase of $1,000,000 in expense associated with the amendment of our agreement with Celera Corporation and an increase of $342,000 in outside clinical trial costs.
Research and development costs are identified as either directly attributed to one of our research and development programs or as an indirect cost, with only direct costs being tracked by specific program. Direct costs consist of personnel costs directly associated with a program, preclinical study costs, clinical trial costs, and related clinical drug and device development and manufacturing costs, drug formulation costs, contract services and other research expenditures. Indirect costs consist of personnel costs not directly associated with a program, overhead and facility costs and other support service expenses. The following table summarizes our principal product development initiatives, including the related stages of development for each product, the direct costs attributable to each product and total indirect costs for each respective period. For a discussion of the risks and uncertainties associated with the timing and cost of completing a product development phase, see the Risk Factors discussed in this Annual Report.
Prior to fiscal 1999, we did not track our research and development expenses by specific program and for this reason we cannot accurately estimate our total historical costs on a specific program basis. Direct costs by program and indirect costs were as follows:
|
Related R & D Expenses
|
|||||||||||||||
|
Years ended June 30,
|
|||||||||||||||
|
Estimated
|
|||||||||||||||
|
Phase of
|
Completion
|
||||||||||||||
|
Product
|
Description
|
Development
|
of Phase
|
2010
|
2009
|
2008
|
|||||||||
|
HDAC Inhibitors
|
Cancer/autoimmune
|
Phase I/II
|
Unknown
|
$ | 2,596,000 | $ | 3,731,000 | $ | 3,429,000 | ||||||
|
Factor VIIa Inhibitor
|
Cancer
|
Phase II
|
Unknown
|
2,227,000 | 1,475,000 | 2,349,000 | |||||||||
|
Btk Inhibitors
|
Cancer/autoimmune
|
Phase Ia/1b
|
Unknown
|
6,565,000 | 3,075,000 | 3,402,000 | |||||||||
|
MGd
|
Cancer
|
Phase II
|
Unknown
|
174,000 | 964,000 | 2,413,000 | |||||||||
|
OTHER
|
- | - | - | ||||||||||||
|
Total direct costs
|
11,562,000 | 9,245,000 | 11,593,000 | ||||||||||||
|
Indirect costs
|
5,796,000 | 4,709,000 | 6,587,000 | ||||||||||||
|
Total research and
|
|||||||||||||||
|
development costs
|
$ | 17,358,000 | $ | 13,954,000 | $ | 18,180,000 | |||||||||
Research and development expenses increased $3,404,000, or 24% for the year ended June 30, 2010 compared to the year ended June 30, 2009 primarily due to the following:
|
|
·
|
HDAC program costs decreased $1,135,000, or 30% primarily due to a $1,000,000 payment in the prior year associated with the amendment of our license agreement with Celera.
|
|
|
·
|
Factor Vlla program costs increased $752,000, or 51% primarily due to increases of $467,000 in drug costs and $285,000 in outside clinical trial costs.
|
|
|
·
|
Btk program costs increased $3,490,000, or 113% primarily due to increases of $615,000 in personnel costs, $557,000 in drug costs, $1,049,000 in outside clinical trial costs, $266,000 in preclinical costs, $423,000 in outside services and consulting costs and other increases associated with the increased activity.
|
|
|
·
|
MGd program costs decreased $790,000, or 82% primarily due to decreases of $589,000 in personnel costs and $196,000 in consulting costs.
|
|
|
·
|
Indirect costs increased $1,087,000, or 23% primarily due to an increase of $963,000 in share-based compensation costs.
|
Research and development expenses decreased $4,226,000, or 23%, for the year ended June 30, 2009 compared to the year ended June 30, 2008 primarily due to the following:
|
|
·
|
HDAC program costs increased $302,000, or 9%, primarily due to a $1,000,000 payment associated with the amendment of our license agreement with Celera and a $746,000 increase in outside clinical trial costs partially offset by a $873,000 decrease in drug costs and a $450,000 decrease in personnel costs.
|
|
|
·
|
Factor VIIa programs costs decreased $874,000, or 37%, primarily due to a $640,000 decrease in pre-clinical study costs and a decrease of $510,000 in drug costs. However outside clinical trial costs increased by $325,000.
|
|
|
·
|
Btk program costs decreased $327,000, or 10%, primarily due to a $774,000 decrease in pre-clinical study costs, partially offset by an increase of $476,000 in personnel costs associated with an increase in clinical activity.
|
|
|
·
|
MGd program costs decreased $1,449,000, or 60%, primarily due to a decrease of $926,000 in outside clinical trial costs and a decrease of $691,000 in personnel costs, partially offset by a $206,000 increase in consulting costs associated with Phase III reporting requirements.
|
|
|
·
|
Indirect costs decreased $1,878,000, or 29%, primarily due to a decrease of $1,236,000 in personnel costs, a decrease of $353,000 in facility and related costs and a decrease of $148,000 in share-based compensation costs.
|
General and Administrative Expenses.
The following table summarizes the period over period changes in our general and administrative (G&A) expenses over the last three fiscal years.
|
2010
|
Change
|
2009
|
Change
|
2008
|
|||||||||
|
General and administrative expenses
|
$
|
7,561,000
|
-11%
|
$
|
8,474,000
|
16%
|
$
|
7,332,000
|
The decrease of 11% or $913,000 in general and administrative expenses for the year ended June 30, 2010, as compared to the year ended June 30, 2009, was primarily due to a non-cash decrease in share-based compensation of $1,363,000 and a $522,000 decrease in other payroll-related costs that were primarily due to the absence of significant severance costs in 2010. These decreases were partially offset by a fiscal 2010 increase of $819,000 in financial advisory expenses and $362,000 related to the provision of certain legal and patent outside services.
G&A expenses in fiscal 2009 increased by $1,142,000 compared to fiscal 2008 primarily due to share-based compensation expense of $1,795,000 and $740,000 of severance expenses associated with separation agreements entered into with our former CEO and CFO in September 2008, partially offset by lower personnel costs of $852,000 due to lower headcount and a reduction of $524,000 in non-severance related share-based compensation expense.
Interest and Other Income (Expense), Net.
The following table summarizes the period over period changes in our interest and other income, net, over the last three fiscal years.
|
2010
|
Change
|
2009
|
Change
|
2008
|
|||||||||
|
Interest income
|
$
|
81,000
|
-41%
|
$
|
137,000
|
-89%
|
|
$ |
1,206,000
|
||||
|
Interest expense
|
(43,000)
|
-93%
|
(618,000)
|
100%
|
-
|
||||||||
|
Gain (loss) on sale of investments
|
-
|
-100%
|
1,000
|
-86%
|
7,000
|
||||||||
|
Gain (loss) on sale of fixed assets
|
-
|
-100%
|
11,000
|
1000%
|
|
1,000
|
|||||||
|
Interest and other income (expense), net
|
38,000
|
108%
|
(469,000)
|
-139%
|
1,214,000
|
The increase of $507,000, or 108% in interest and other income (expense), net, for the year ended June 30, 2010, as compared to the year ended June 30, 2009, was primarily due to a decrease of $575,000 in interest expense due to settlement of the related party loans, partially offset by a $56,000 reduction of interest income due to lower average interest rates.
The decrease of $1,683,000, or 139% in interest and other income (expense), net, for the year ended June 30, 2009 compared to fiscal 2008 was primarily due to a decline in interest rates earned on our investments, $549,000 of non-cash interest expense associated with discounting $6,400,000 in related party loans to fair value and $69,000 of interest expense based on the stated interest rates of the notes.
Income Taxes.
In the year ended June 30, 2009, we recorded a $550,000 income tax provision as result of withholding taxes on the $11,000,000 upfront licensing payment received from Servier. In the year ended June 30, 2010, we recorded a $550,000 income tax receivable related to a tax credit resulting from a December 2009 tax treaty revision enacted between France and the United States that eliminates withholding taxes related to licensing agreements and provides that prior withholding taxes may be reclaimed.
At June 30, 2010, we had federal and state net operating loss carryforwards of approximately $141,100,000 and $86,500,000, respectively. The federal and state net operating loss carryforwards will begin to expire in 2011. Federal and state tax credit carryforwards of $3,500,000 and $9,200,000, respectively, are available to offset future taxable income. The federal tax credits will begin to expire in 2025. State research and development credits can be carried forward indefinitely.
Under the Tax Reform Act of 1986, the amounts of and the benefit from net operating losses and tax credit carry-forwards that can be carried forward may be impaired or limited in certain circumstances. These circumstances include, but are not limited to, a cumulative stock ownership change of greater than 50%, as defined, over a three year period. Such an annual limitation may result in the expiration of net operating losses before utilization. We have determined that a cumulative stock ownership change happened and we have estimated that a significant portion of our net operating losses for federal and state tax purposes, as well as some amount of our federal research credits, will not be available for use in future periods due to these limitation rules. As a result we have revised our 2009 deferred tax asset footnote by reducing the amount of previously reported net operating losses and credits for the estimated impairment amounts. The reduction to the 2009 deferred tax assets have no effect on our statement of operations, earnings per share, balance sheet, statement of cash flows or statement of stockholders’ equity (deficit) for any period presented.
A full valuation allowance has been established for our deferred tax assets since realization of such assets through the generation of future taxable income is uncertain.
Liquidity and Capital Resources
Our principal sources of working capital have been private and public equity financings and also proceeds from collaborative research and development agreements, as well as interest income. Since inception, we have used approximately $329,650,000 of cash for operating activities and approximately $16,471,000 of cash for the purchase of laboratory and office equipment, leasehold improvements and payments under capital lease agreements. As of June 30, 2010, we had $74,149,000 in cash, cash equivalents and marketable securities.
Net cash used in operating activities of $15,468,000 during the year ended June 30, 2010 resulted primarily from our net loss, a decrease in deferred revenue and an increase in prepaid and other assets, partially offset by share-based compensation expense and an increase in accounts payable. Net cash used in operating activities was $8,175,000 for the year ended June 30, 2009 and resulted primarily from the operating loss partially offset by an increase in deferred revenue and non-cash share-based compensation expense. Net cash used in operating activities was $22,105,000 for the year ended June 30, 2008 and resulted primarily from operating losses adjusted for non-cash compensation expense and changes in accounts payable, accrued liabilities, prepaid expenses and other assets.
Net cash used in investing activities of $21,810,000 in the year ended June 30, 2010 consisted primarily of purchases of marketable securities, partially offset by maturities of marketable securities. Net cash provided by investing activities of $2,657,000 and $22,361,000 in the years ended June 30, 2009 and 2008, respectively, primarily consisted of the net effect of purchases, maturities and sales of marketable securities.
Net cash provided by financing activities of $73,943,000 for the year ended June 30, 2010 consisted primarily of $21,720,000 in net proceeds from the sale of approximately 22.5 million shares of common stock in a rights offering completed in August 2009, net proceeds of $50,793,000 from the sale of approximately 8.1 million shares of common stock in a registered direct offering completed in June 2010 and approximately $1,744,000 in proceeds from the exercise of stock options and sale of stock under our employee stock purchase plan. Net cash provided by financing activities of $7,792,000 in the year ended June 30, 2009 consisted of proceeds from notes payable and the sale of common stock. Net cash provided by financing activities of $63,000 in the year ended June 30, 2008 primarily consisted of proceeds from the sale of common stock.
In April 2009, we signed a collaboration and license agreement with Servier. In May 2009, we received an upfront payment from Servier of $11,000,000 less applicable withholding taxes of $550,000, for a net payment of $10,450,000. The withholding tax paid to the French Government will be reclaimed due to a recent revision in the Double Tax Treaty between the US and France, (see Note 8).
In February 2009, we sold approximately 1.5 million shares of unregistered common stock at $0.93 per share for net proceeds of approximately $1,400,000.
In December 2008, we borrowed $5,000,000 from an affiliate of Robert W. Duggan. In March 2009, the loan amount was increased to $6,400,000. In August 2009, pursuant to the terms of the loans, we repaid the $6,400,000 loans outstanding at June 30, 2009 through the issuance of shares in the rights offering.
Our future contractual obligations at June 30, 2010 were as follows:
|
Operating
|
||||
|
Lease
|
||||
|
Commitments
|
||||
|
Less than 1 year
|
$ | 644 | ||
|
1-3 years
|
330 | |||
|
Total
|
$ | 974 | ||
In April 2006, we acquired multiple small molecule drug candidates for the treatment of cancer and other diseases from Celera Genomics, an Applera Corporation (now Celera Corporation) business. Future milestone payments we could be required to make under the agreement, as amended, could total as much as $97,000,000, although we currently cannot predict if or when any of the milestones will be achieved. In addition, Celera will also be entitled to royalty payments based on annual sales of drugs commercialized from these programs.
Based upon the current status of our product development plans, we believe that our existing cash, cash equivalents and marketable securities will be adequate to satisfy our capital needs through at least the next twelve months. We expect research and development expenses, as a result of on-going and future clinical trials, to consume a large portion of our existing cash resources. Changes in our research and development plans or other changes affecting our operating expenses may affect actual future consumption of existing cash resources as well. In any event, due to our extensive drug programs we will need to raise substantial additional capital to fund our operations in the future. We may seek partnership collaborations to help fund the development of our product candidates. We also expect to raise additional funds through the public or private sale of securities, bank debt, partnership collaboration or otherwise. If we are unable to secure additional funds, whether through partnership collaborations or sale of our securities, we will have to delay, reduce the scope of or discontinue one or more of our product development programs. Our actual capital requirements will depend on many factors, including the following:
|
|
·
|
our ability to establish and the scope of any new collaborations;
|
|
|
·
|
the progress and success of clinical trials of our product candidates; and
|
|
|
·
|
the costs and timing of obtaining regulatory approvals.
|
Our forecast of the period of time through which our financial resources will be adequate to support our operations is a forward-looking statement that involves risks and uncertainties, and actual results could vary materially. The factors described above will impact our future capital requirements and the adequacy of our available funds. If we are required to raise additional funds, we cannot be certain that such additional funding will be available on terms attractive to us, or at all. Furthermore, any additional equity financing may be highly dilutive, or otherwise disadvantageous, to existing stockholders and debt financing, if available, may involve restrictive covenants. Collaborative arrangements, if necessary to raise additional funds, may require us to relinquish rights to certain of our technologies, products or marketing territories. Our failure to raise capital when needed and on acceptable terms, would require us to reduce our operating expenses and would limit our ability to respond to competitive pressures or unanticipated requirements to develop our product candidates and to continue operations, any of which would have a material adverse effect on our business, financial condition and results of operations.
Off-Balance Sheet Arrangements
None.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Our exposure to interest rate risk relates primarily to our investment portfolio. Fixed rate securities may have their fair market value adversely impacted due to fluctuations in interest rates, while floating rate securities may produce less income than expected if interest rates fall. Due in part to these factors, our future investment income may fall short of expectations due to changes in interest rates or we may suffer losses in principal if forced to sell securities which have declined in market value due to changes in interest rates. The primary objective of our investment activities is to preserve principal while at the same time improving yields without significantly increasing risk. To achieve this objective, we invest in debt instruments of the U.S. Government and its agencies and high-quality corporate issuers, and, by policy, restrict our exposure to any single corporate issuer by imposing concentration limits. To minimize the exposure due to adverse shifts in interest rates, we maintain investments at an average maturity of generally less than two years. Assuming a hypothetical increase in interest rates of one percentage point, the fair value of our total investment portfolio as of June 30, 2010 would have potentially declined by approximately $89,000.
The table below presents the fair value of our marketable securities at June 30, 2010 and weighted-average interest rates by year of stated maturity for our investment portfolio (in thousands, except interest rates):
|
Fiscal Year
|
||||
|
2011
|
||||
|
Marketable securities
|
$ | 22,950 | ||
|
Weighted-average interest rate
|
0.50 | % | ||
Item 8. Financial Statements and Supplementary Data
INDEX TO FINANCIAL STATEMENTS
|
|
Page
|
|
Report of Independent Registered Public Accounting Firm
|
64
|
|
Balance Sheets
|
65 |
|
Statements of Operations
|
66 |
|
Statements of Cash Flows
|
67 |
|
Statements of Stockholders' Equity (Deficit)
|
68 |
|
Notes to Financial Statements
|
77 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of Pharmacyclics, Inc.:
In our opinion, the accompanying balance sheets and the related statements of operations, stockholders' equity (deficit) and cash flows present fairly, in all material respects, the financial position of Pharmacyclics, Inc. (the"Company") (a development stage enterprise) at June 30, 2010 and 2009, and the results of its operations and its cash flows for each of the three years in the period ended June 30, 2010 and, cumulatively, for the period from April 19, 1991 (date of inception) to June 30, 2010 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of June 30, 2010, based on criteria established in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company's management is responsible for these financial statements, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in Management's Annual Report on Internal Control Over Financial Reporting, appearing under Item 9A(b). Our responsibility is to express opinions on these financial statements and on the Company's internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP
San Jose, California
September 13, 2010
PHARMACYCLICS, INC.
(a development stage enterprise)
BALANCE SHEETS
(in thousands, except share and per share amounts)
|
June 30,
|
||||||||
|
2010
|
2009
|
|||||||
|
ASSETS
|
||||||||
|
Current Assets:
|
||||||||
|
Cash and cash equivalents
|
$ | 51,199 | $ | 14,534 | ||||
|
Marketable securities
|
22,950 | 1,792 | ||||||
|
Accounts receivable
|
194 | 632 | ||||||
|
Prepaid expenses and other current assets
|
1,702 | 583 | ||||||
|
Total current assets
|
76,045 | 17,541 | ||||||
|
Property and equipment, net
|
459 | 470 | ||||||
|
Other assets
|
316 | 290 | ||||||
|
Total assets
|
$ | 76,820 | $ | 18,301 | ||||
|
LIABILITIES AND STOCKHOLDERS' EQUITY (DEFICIT)
|
||||||||
|
Current liabilities:
|
||||||||
|
Accounts payable
|
$ | 2,856 | $ | 1,167 | ||||
|
Accrued liabilities
|
1,054 | 801 | ||||||
|
Notes payable to related party
|
- | 6,379 | ||||||
|
Deferred revenue - current portion (Note 2)
|
6,099 | 7,025 | ||||||
|
Total current liabilities
|
10,009 | 15,372 | ||||||
|
Deferred revenue - non-current portion (Note 2)
|
- | 4,603 | ||||||
|
Deferred rent
|
50 | 67 | ||||||
|
Total liabilities
|
10,059 | 20,042 | ||||||
|
Commitments (Notes 2 and 9)
|
||||||||
|
Stockholders' equity (deficit):
|
||||||||
|
Preferred stock, $0.0001 par value; 1,000,000 shares authorized at June 30, 2010 and 2009; no shares issued and outstanding
|
- | - | ||||||
|
Common stock, $0.0001 par value; 100,000,000 shares authorized at June 30, 2010 and 2009; shares issued and outstanding - 59,199,406 and 27,539,378 at June 30, 2010 and 2009
|
6 | 3 | ||||||
|
Additional paid-in capital
|
444,683 | 361,153 | ||||||
|
Accumulated other comprehensive income
|
(6 | ) | 1 | |||||
|
Deficit accumulated during development stage
|
(377,922 | ) | (362,898 | ) | ||||
|
Total stockholders' equity (deficit)
|
66,761 | (1,741 | ) | |||||
|
Total liabilities and stockholders' equity (deficit)
|
$ | 76,820 | $ | 18,301 | ||||
The accompanying notes are an integral part of these financial statements.
PHARMACYCLICS, INC.
(a development stage enterprise)
STATEMENTS OF OPERATIONS
(in thousands, except per share amounts)
|
Period
|
||||||||||||||||
|
from
|
||||||||||||||||
|
Inception
|
||||||||||||||||
|
(April 19, 1991)
|
||||||||||||||||
|
through
|
||||||||||||||||
|
Year Ended June 30,
|
June 30,
|
|||||||||||||||
|
2010
|
2009
|
2008
|
2010
|
|||||||||||||
|
Revenues(1):
|
||||||||||||||||
|
License and milestone revenues
|
$ | 9,307 | $ | - | $ | - | $ | 17,162 | ||||||||
|
Grant and contract revenues
|
- | - | - | 6,154 | ||||||||||||
|
Total revenues
|
9,307 | - | - | 23,316 | ||||||||||||
|
Operating expenses:
|
||||||||||||||||
|
Research and development *
|
17,358 | 13,954 | 18,180 | 343,234 | ||||||||||||
|
General and administrative *
|
7,561 | 8,474 | 7,332 | 92,176 | ||||||||||||
|
Purchased in-process research
|
||||||||||||||||
|
and development
|
- | - | - | 6,647 | ||||||||||||
|
Total operating expenses
|
24,919 | 22,428 | 25,512 | 442,057 | ||||||||||||
|
Loss from operations
|
(15,612 | ) | (22,428 | ) | (25,512 | ) | (418,741 | ) | ||||||||
|
Interest income
|
81 | 137 | 1,206 | 43,025 | ||||||||||||
|
Interest expense and other
|
||||||||||||||||
|
income (expense), net
|
(43 | ) | (606 | ) | 8 | (2,206 | ) | |||||||||
|
Loss before benefit (provision) for
|
||||||||||||||||
|
income taxes
|
(15,574 | ) | (22,897 | ) | (24,298 | ) | (377,922 | ) | ||||||||
|
Benefit (provision) for income taxes
|
550 | (550 | ) | - | - | |||||||||||
|
Net loss
|
$ | (15,024 | ) | $ | (23,447 | ) | $ | (24,298 | ) | $ | (377,922 | ) | ||||
|
Basic and diluted net loss per share
|
$ | (0.31 | ) | $ | (0.88 | ) | $ | (0.93 | ) | |||||||
|
Weighted average shares used to compute basic
|
||||||||||||||||
|
and diluted net loss per share
|
48,344 | 26,570 | 25,989 | |||||||||||||
|
*Includes non-cash share-based
|
||||||||||||||||
|
compensation of:
|
||||||||||||||||
|
Research and development
|
$ | 1,998 | $ | 738 | $ | 961 | $ | 8,712 | ||||||||
|
General and administrative
|
1,192 | 2,555 | 1,299 | 10,294 | ||||||||||||
|
(1) See Note 2 for a discussion of revenue recognition related to the Servier agreement.
|
||||||||||||||||
The accompanying notes are an integral part of these financial statements.
PHARMACYCLICS, INC.
(a development stage enterprise)
STATEMENTS OF CASH FLOWS
(in thousands)
|
Period
|
||||||||||||||||
|
from
|
||||||||||||||||
|
Inception
|
||||||||||||||||
|
(April 19, 1991)
|
||||||||||||||||
|
through
|
||||||||||||||||
|
Year Ended June 30,
|
June 30,
|
|||||||||||||||
|
Cash flows from operating activities:
|
2010
|
2009
|
2008
|
2010
|
||||||||||||
|
Net loss
|
$ | (15,024 | ) | $ | (23,447 | ) | $ | (24,298 | ) | $ | (377,922 | ) | ||||
|
Adjustments to reconcile net loss to net cash
|
||||||||||||||||
|
used in operating activities:
|
||||||||||||||||
|
Depreciation and amortization
|
235 | 299 | 348 | 15,519 | ||||||||||||
|
Amoritization of premium/discount on marketable securities, net
|
421 | (32 | ) | (196 | ) | 395 | ||||||||||
|
Amoritization of debt discount
|
21 | 549 | - | 570 | ||||||||||||
|
Purchased in-process research and development
|
- | - | - | 4,500 | ||||||||||||
|
Share-based compensation
|
3,190 | 3,293 | 2,260 | 19,006 | ||||||||||||
|
Common stock issued in exchange for services provided
|
- | 15 | - | 15 | ||||||||||||
|
Loss (gain) on sale of marketable securities
|
- | (1 | ) | (7 | ) | 50 | ||||||||||
|
Write-down (proceeds from sale) of fixed assets
|
- | (11 | ) | - | 370 | |||||||||||
|
Changes in assets and liabilities:
|
||||||||||||||||
|
Accounts receivable
|
438 | (632 | ) | - | (194 | ) | ||||||||||
|
Prepaid expenses and other assets
|
(1,145 | ) | 51 | 560 | (2,018 | ) | ||||||||||
|
Accounts payable
|
1,689 | 112 | (371 | ) | 2,856 | |||||||||||
|
Accrued liabilities
|
253 | 5 | (393 | ) | 1,054 | |||||||||||
|
Deferred revenue
|
(5,529 | ) | 11,628 | - | 6,099 | |||||||||||
|
Deferred rent
|
(17 | ) | (4 | ) | (8 | ) | 50 | |||||||||
|
Net cash used in operating activities
|
(15,468 | ) | (8,175 | ) | (22,105 | ) | (329,650 | ) | ||||||||
|
Cash flows from investing activities:
|
||||||||||||||||
|
Purchase of property and equipment
|
(224 | ) | (81 | ) | (187 | ) | (12,590 | ) | ||||||||
|
Proceeds from sale of property and equipment
|
- | 11 | - | 123 | ||||||||||||
|
Purchase of marketable securities
|
(36,595 | ) | (4,971 | ) | (5,446 | ) | (571,112 | ) | ||||||||
|
Proceeds from sales of marketable securities
|
- | 998 | 6,994 | 85,934 | ||||||||||||
|
Proceeds from maturities of marketable securities
|
15,009 | 6,700 | 21,000 | 461,777 | ||||||||||||
|
Net cash provided by (used in) investing activities
|
(21,810 | ) | 2,657 | 22,361 | (35,868 | ) | ||||||||||
|
Cash flows from financing activities:
|
||||||||||||||||
|
Issuance of common stock, net of issuance costs
|
72,606 | 1,388 | 63 | 382,912 | ||||||||||||
|
Exercise of stock options
|
1,651 | 4 | - | 8,086 | ||||||||||||
|
Proceeds from related party notes payable
|
- | 6,400 | - | 6,400 | ||||||||||||
|
Proceeds from notes payable
|
- | - | - | 3,000 | ||||||||||||
|
Issuance of convertible preferred stock, net of issuance costs
|
- | - | - | 20,514 | ||||||||||||
|
Payments under capital lease obligations
|
- | - | - | (3,881 | ) | |||||||||||
|
Repayment of notes payable
|
(314 | ) | - | - | (314 | ) | ||||||||||
|
Net cash provided by financing activities
|
73,943 | 7,792 | 63 | 416,717 | ||||||||||||
|
Increase in cash and cash equivalents
|
36,665 | 2,274 | 319 | 51,199 | ||||||||||||
|
Cash and cash equivalents at beginning of period
|
14,534 | 12,260 | 11,941 | - | ||||||||||||
|
Cash and cash equivalents at end of period
|
$ | 51,199 | $ | 14,534 | $ | 12,260 | $ | 51,199 | ||||||||
|
Supplemental Disclosures of Cash Flow Information:
|
||||||||||||||||
|
Interest paid
|
$ | 91 | $ | - | $ | - | $ | 1,360 | ||||||||
|
Supplemental disclosure of non-cash investing and financing activities:
|
||||||||||||||||
|
Settlement of related party notes payable by issuance of common stock
|
6,086 | - | - | 6,086 | ||||||||||||
|
Property and equipment acquired under capital lease obligations
|
- | - | - | 3,881 | ||||||||||||
|
Warrants issued
|
- | - | - | 49 | ||||||||||||
|
Conversion of notes payable and accrued interest into convertible preferred stock
|
- | - | - | 3,051 | ||||||||||||
The accompanying notes are an integral part of these financial statements.
PHARMACYCLICS, INC.
(a development stage enterprise)
STATEMENTS OF STOCKHOLDERS' EQUITY (DEFICIT)
For the period from inception (April 19, 1991) through June 30, 2010
(in thousands, except share and per share amounts)
|
Deficit
|
||||||||||||||||||||||||||||||||
|
Accumulated
|
Accumulated
|
|||||||||||||||||||||||||||||||
|
Convertible
|
Additional
|
other
|
During
|
|||||||||||||||||||||||||||||
|
Preferred Stock
|
Common Stock
|
Paid-in
|
Comprehensive
|
Development
|
||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Income (Loss)
|
Stage
|
Total
|
|||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||
|
Issuance of common stock for cash at $0.02 per share
|
- | $ | - | 400,000 | $ | - | $ | 6 | $ | - | $ | - | $ | 6 | ||||||||||||||||||
|
Balance at June 30, 1991
|
- | - | 400,000 | - | 6 | - | - | 6 | ||||||||||||||||||||||||
|
Issuance of common stock for cash at an average price of $0.02 per share
|
- | - | 97,111 | - | 2 | - | - | 2 | ||||||||||||||||||||||||
|
Issuance of convertible preferred stock for cash, net of issuance costs, at an average price of $1.32 per share
|
2,040,784 | - | - | - | 2,667 | - | - | 2,667 | ||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (523 | ) | (523 | ) | ||||||||||||||||||||||
|
Balance at June 30, 1992
|
2,040,784 | - | 497,111 | - | 2,675 | - | (523 | ) | 2,152 | |||||||||||||||||||||||
|
Issuance of common stock for cash at an average price of $0.06 per share
|
- | - | 49,000 | - | 3 | - | - | 3 | ||||||||||||||||||||||||
|
Issuance of convertible preferred stock for cash, net of issuance costs, at $4.88 per share
|
1,580,095 | - | - | - | 7,674 | - | - | 7,674 | ||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (3,580 | ) | (3,580 | ) | ||||||||||||||||||||||
|
Balance at June 30, 1993
|
3,620,879 | - | 546,111 | - | 10,352 | - | (4,103 | ) | 6,249 | |||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||
|
Issuance of common stock upon exercise of stock options at an average price of $0.12 per share
|
- | - | 324,188 | - | 38 | - | - | 38 | ||||||||||||||||||||||||
|
Issuance of convertible preferred stock for cash, net of issuance costs, at an average price of $8.63 per share
|
886,960 | - | - | - | 7,623 | - | - | 7,623 | ||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (5,141 | ) | (5,141 | ) | ||||||||||||||||||||||
|
Balance at June 30, 1994
|
4,507,839 | - | 870,299 | - | 18,013 | - | (9,244 | ) | 8,769 | |||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||
|
Issuance of common stock upon exercise of stock options at an average price of $0.24 per share
|
- | - | 38,403 | - | 9 | - | - | 9 | ||||||||||||||||||||||||
|
Issuance of warrants
|
- | - | - | - | 49 | - | - | 49 | ||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (10,479 | ) | (10,479 | ) | ||||||||||||||||||||||
|
Balance at June 30, 1995
|
4,507,839 | - | 908,702 | - | 18,071 | - | (19,723 | ) | (1,652 | ) | ||||||||||||||||||||||
The accompanying notes are an integral part of these financial statements
PHARMACYCLICS, INC.
(a development stage enterprise)
STATEMENTS OF STOCKHOLDERS' EQUITY (DEFICIT) (Continued)
For the period from inception (April 19, 1991) through June 30, 2010
(in thousands, except share and per share amounts)
|
Deficit
|
||||||||||||||||||||||||||||||||
|
Accumulated
|
Accumulated
|
|||||||||||||||||||||||||||||||
|
Convertible
|
Additional
|
other
|
During
|
|||||||||||||||||||||||||||||
|
Preferred Stock
|
Common Stock
|
Paid-in
|
Comprehensive
|
Development
|
||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Income (Loss)
|
Stage
|
Total
|
|||||||||||||||||||||||||
|
Issuance of convertible preferred stock for notes payable and accrued interest at an average of $8.63 per share
|
353,483 | - | - | - | 3,051 | - | - | 3,051 | ||||||||||||||||||||||||
|
Issuance of convertible preferred stock for cash, net of issuance costs, at an average price of $8.63 per share
|
295,649 | - | - | - | 2,550 | - | - | 2,550 | ||||||||||||||||||||||||
|
Issuance of common stock upon initial public offering, net of issuance costs, for cash at $12 per share
|
- | - | 2,383,450 | 1 | 26,042 | - | - | 26,043 | ||||||||||||||||||||||||
|
Conversion of convertible preferred stock into common stock
|
(5,156,971 | ) | - | 5,156,971 | - | - | - | - | - | |||||||||||||||||||||||
|
Issuance of common stock upon exercise of stock options at an average exercise price of $1.33 per share
|
- | - | 91,922 | - | 122 | - | - | 122 | ||||||||||||||||||||||||
|
Issuance of common stock upon exercise of purchase rights at an exercise price of $10.20 per share
|
- | - | 8,379 | - | 86 | - | - | 86 | ||||||||||||||||||||||||
|
Share-based compensation expense
|
- | - | - | - | 26 | - | - | 26 | ||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (8,235 | ) | (8,235 | ) | ||||||||||||||||||||||
|
Balance at June 30, 1996
|
- | - | 8,549,424 | 1 | 49,948 | - | (27,958 | ) | 21,991 | |||||||||||||||||||||||
|
Issuance of common stock, net of issuance costs, for cash at an average price of $16.93 per share
|
- | - | 1,442,190 | - | 24,420 | - | - | 24,420 | ||||||||||||||||||||||||
|
Issuance of common stock upon exercise of stock options at an average price of $2.74 per share
|
- | - | 96,283 | - | 264 | - | - | 264 | ||||||||||||||||||||||||
The accompanying notes are an integral part of these financial statements
PHARMACYCLICS, INC.
(a development stage enterprise)
STATEMENTS OF STOCKHOLDERS' EQUITY (DEFICIT) (Continued)
For the period from inception (April 19, 1991) through June 30, 2010
(in thousands, except share and per share amounts)
|
Deficit
|
||||||||||||||||||||||||||||||||
|
Accumulated
|
Accumulated
|
|||||||||||||||||||||||||||||||
|
Convertible
|
Additional
|
other
|
During
|
|||||||||||||||||||||||||||||
|
Preferred Stock
|
Common Stock
|
Paid-in
|
Comprehensive
|
Development
|
||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Income (Loss)
|
Stage
|
Total
|
|||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||
|
Issuance of common stock upon exercise of purchase rights at an exercise price of $10.51 per share
|
- | - | 14,557 | - | 153 | - | - | 153 | ||||||||||||||||||||||||
|
Share-based compensation expense
|
- | - | - | - | 126 | - | - | 126 | ||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (10,258 | ) | (10,258 | ) | ||||||||||||||||||||||
|
Balance at June 30, 1997
|
- | - | 10,102,454 | 1 | 74,911 | - | (38,216 | ) | 36,696 | |||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||
|
Issuance of common stock, net of issuance costs, for cash $21.75 per share
|
- | - | 2,012,500 | - | 40,796 | - | - | 40,796 | ||||||||||||||||||||||||
|
Issuance of common stock upon exercise of stock options at an average price of $6.57 per share
|
- | - | 88,933 | - | 584 | - | - | 584 | ||||||||||||||||||||||||
|
Issuance of common stock upon exercise of purchase rights at an exercise price of $14.36 per share
|
- | - | 10,372 | - | 149 | - | - | 149 | ||||||||||||||||||||||||
|
Issuance of common stock upon exercise of warrants
|
- | - | 80,033 | - | - | - | - | - | ||||||||||||||||||||||||
|
Share-based compensation expense
|
- | - | - | - | 91 | - | - | 91 | ||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (9,675 | ) | (9,675 | ) | ||||||||||||||||||||||
|
Balance at June 30, 1998
|
- | - | 12,294,292 | 1 | 116,531 | - | (47,891 | ) | 68,641 | |||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||
|
Issuance of common stock upon exercise of stock options at an average price of $5.10 per share
|
- | - | 75,275 | - | 384 | - | - | 384 | ||||||||||||||||||||||||
The accompanying notes are an integral part of these financial statements
PHARMACYCLICS, INC.
(a development stage enterprise)
STATEMENTS OF STOCKHOLDERS' EQUITY (DEFICIT) (Continued)
For the period from inception (April 19, 1991) through June 30, 2010
(in thousands, except share and per share amounts)
|
Accumulated
|
Accumulated
|
|||||||||||||||||||||||||||||||
|
Convertible
|
Additional
|
other
|
During
|
|||||||||||||||||||||||||||||
|
Preferred Stock
|
Common Stock
|
Paid-in
|
Comprehensive
|
Development
|
||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Income (Loss)
|
Stage
|
Total
|
|||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||
|
Issuance of common stock upon exercise of purchase rights at an exercise price of $12.77 per share
|
- | - | 13,643 | - | 174 | - | - | 174 | ||||||||||||||||||||||||
|
Issuance of common stock upon exercise of warrants
|
- | - | 45,661 | - | - | - | - | - | ||||||||||||||||||||||||
|
Share-based compensation expense
|
- | - | - | - | 89 | - | - | 89 | ||||||||||||||||||||||||
|
Change in unrealized gain (loss) on marketable securities
|
- | - | - | - | - | (85 | ) | - | (85 | ) | ||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (19,246 | ) | (19,246 | ) | ||||||||||||||||||||||
|
Total comprehensive loss
|
(19,331 | ) | ||||||||||||||||||||||||||||||
|
Balance at June 30, 1999
|
- | - | 12,428,871 | 1 | 117,178 | (85 | ) | (67,137 | ) | 49,957 | ||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||
|
Issuance of common stock upon exercise of stock options at an average price of $13.88 per share
|
- | - | 102,372 | - | 1,421 | - | - | 1,421 | ||||||||||||||||||||||||
|
Issuance of common stock upon exercise of purchase rights at an exercise price of $25.62 per share
|
- | - | 11,213 | - | 287 | - | - | 287 | ||||||||||||||||||||||||
|
Issuance of common stock, net of issuance costs, for cash at an average price of $44.36 per share
|
- | - | 3,465,000 | 1 | 153,711 | - | - | 153,712 | ||||||||||||||||||||||||
|
Share-based compensation expense
|
- | - | - | - | 88 | - | - | 88 | ||||||||||||||||||||||||
|
Change in unrealized gain (loss) on marketable securities
|
- | - | - | - | - | (421 | ) | - | (421 | ) | ||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (23,630 | ) | (23,630 | ) | ||||||||||||||||||||||
|
Total comprehensive loss
|
(24,051 | ) | ||||||||||||||||||||||||||||||
|
Balance at June 30, 2000
|
- | - | 16,007,456 | 2 | 272,685 | (506 | ) | (90,767 | ) | 181,414 | ||||||||||||||||||||||
The accompanying notes are an integral part of these financial statements
PHARMACYCLICS, INC.
(a development stage enterprise)
STATEMENTS OF STOCKHOLDERS' EQUITY (DEFICIT) (Continued)
For the period from inception (April 19, 1991) through June 30, 2010
(in thousands, except share and per share amounts)
|
Accumulated
|
Accumulated
|
|||||||||||||||||||||||||||||||
|
Convertible
|
Additional
|
other
|
During
|
|||||||||||||||||||||||||||||
|
Preferred Stock
|
Common Stock
|
Paid-in
|
Comprehensive
|
Development
|
||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Income (Loss)
|
Stage
|
Total
|
|||||||||||||||||||||||||
|
Issuance of common stock upon exercise of stock options at an average price of $16.17 per share
|
- | - | 93,528 | - | 1,512 | - | - | 1,512 | ||||||||||||||||||||||||
|
Issuance of common stock upon exercise of purchase rights at an exercise price of $27.89 per share
|
- | - | 15,386 | - | 429 | - | - | 429 | ||||||||||||||||||||||||
|
Share-based compensation expense
|
- | - | - | - | 326 | - | - | 326 | ||||||||||||||||||||||||
|
Change in unrealized gain (loss) on marketable securities
|
- | - | - | - | - | 1,599 | - | 1,599 | ||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (30,925 | ) | (30,925 | ) | ||||||||||||||||||||||
|
Total comprehensive loss
|
(29,326 | ) | ||||||||||||||||||||||||||||||
|
Balance at June 30, 2001
|
- | - | 16,116,370 | 2 | 274,952 | 1,093 | (121,692 | ) | 154,355 | |||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||
|
Issuance of common stock upon exercise of stock options at an average price of $13.93 per share
|
- | - | 13,257 | - | 183 | - | - | 183 | ||||||||||||||||||||||||
|
Issuance of common stock upon exercise of purchase rights at an exercise price of $8.32 per share
|
- | - | 58,169 | - | 484 | - | - | 484 | ||||||||||||||||||||||||
|
Share-based compensation expense
|
- | - | - | - | 91 | - | - | 91 | ||||||||||||||||||||||||
|
Change in unrealized gain (loss) on marketable securities
|
- | - | - | - | - | (930 | ) | - | (930 | ) | ||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (36,575 | ) | (36,575 | ) | ||||||||||||||||||||||
|
Total comprehensive loss
|
(37,505 | ) | ||||||||||||||||||||||||||||||
|
Balance at June 30, 2002
|
- | - | 16,187,796 | 2 | 275,710 | 163 | (158,267 | ) | 117,608 | |||||||||||||||||||||||
PHARMACYCLICS, INC.
(a development stage enterprise)
STATEMENTS OF STOCKHOLDERS' EQUITY (DEFICIT) (Continued)
For the period from inception (April 19, 1991) through June 30, 2010
(in thousands, except share and per share amounts)
|
Deficit
|
||||||||||||||||||||||||||||||||
|
Accumulated
|
Accumulated
|
|||||||||||||||||||||||||||||||
|
Convertible
|
Additional
|
other
|
During
|
|||||||||||||||||||||||||||||
|
Preferred Stock
|
Common Stock
|
Paid-in
|
Comprehensive
|
Development
|
||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Income (Loss)
|
Stage
|
Total
|
|||||||||||||||||||||||||
|
Issuance of common stock upon exercise of stock options at an average price of $1.03 per share
|
- | - | 3,397 | - | 3 | - | - | 3 | ||||||||||||||||||||||||
|
Issuance of common stock upon exercise of purchase rights at an exercise price of $2.64 per share
|
- | - | 38,908 | - | 103 | - | - | 103 | ||||||||||||||||||||||||
|
Share-based compensation expense
|
- | - | - | - | 13 | - | - | 13 | ||||||||||||||||||||||||
|
Change in unrealized gain (loss) on marketable securities
|
- | - | - | - | - | (19 | ) | - | (19 | ) | ||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (28,298 | ) | (28,298 | ) | ||||||||||||||||||||||
|
Total comprehensive loss
|
(28,317 | ) | ||||||||||||||||||||||||||||||
|
Balance at June 30, 2003
|
- | - | 16,230,101 | 2 | 275,829 | 144 | (186,565 | ) | 89,410 | |||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||
|
Issuance of common stock, net of issuance costs, for cash at an average price of $13.00 per share
|
- | - | 3,200,000 | - | 39,350 | - | - | 39,350 | ||||||||||||||||||||||||
|
Issuance of common stock upon exercise of stock options at an average price of $4.91 per share
|
- | - | 181,136 | - | 889 | - | - | 889 | ||||||||||||||||||||||||
|
Issuance of common stock upon exercise of purchase rights at an exercise price of $4.90 per share
|
- | - | 36,680 | - | 180 | - | - | 180 | ||||||||||||||||||||||||
|
Share-based compensation expense
|
- | - | - | - | 18 | - | - | 18 | ||||||||||||||||||||||||
|
Change in unrealized gain (loss) on marketable securities
|
- | - | - | - | - | (394 | ) | - | (394 | ) | ||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (29,165 | ) | (29,165 | ) | ||||||||||||||||||||||
|
Total comprehensive loss
|
(29,559 | ) | ||||||||||||||||||||||||||||||
|
Balance at June 30, 2004
|
- | - | 19,647,917 | 2 | 316,266 | (250 | ) | (215,730 | ) | 100,288 | ||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||
|
Issuance of common stock upon exercise of stock options at an average price of $4.46 per share
|
- | - | 61,014 | - | 272 | - | - | 272 | ||||||||||||||||||||||||
The accompanying notes are an integral part of these financial statements
PHARMACYCLICS, INC.
(a development stage enterprise)
STATEMENTS OF STOCKHOLDERS' EQUITY (DEFICIT) (Continued)
For the period from inception (April 19, 1991) through June 30, 2010
(in thousands, except share and per share amounts)
|
Deficit
|
||||||||||||||||||||||||||||||||
|
Accumulated
|
Accumulated
|
|||||||||||||||||||||||||||||||
|
Convertible
|
Additional
|
other
|
During
|
|||||||||||||||||||||||||||||
|
Preferred Stock
|
Common Stock
|
Paid-in
|
Comprehensive
|
Development
|
||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Income (Loss)
|
Stage
|
Total
|
|||||||||||||||||||||||||
|
Issuance of common stock upon exercise of purchase rights at an exercise price of $5.24 per share
|
- | - | 90,704 | - | 476 | - | - | 476 | ||||||||||||||||||||||||
|
Share-based compensation expense
|
- | - | - | - | 49 | - | - | 49 | ||||||||||||||||||||||||
|
Change in unrealized gain (loss) on marketable securities
|
- | - | - | - | - | (43 | ) | - | (43 | ) | ||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (31,048 | ) | (31,048 | ) | ||||||||||||||||||||||
|
Total comprehensive loss
|
(31,091 | ) | ||||||||||||||||||||||||||||||
|
Balance at June 30, 2005
|
- | - | 19,799,635 | 2 | 317,063 | (293 | ) | (246,778 | ) | 69,994 | ||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||
|
Issuance of common stock upon exercise of stock options and purchase rights at an average price of $3.80 per share
|
- | - | 147,059 | - | 559 | - | - | 559 | ||||||||||||||||||||||||
|
Issuance of common stock for purchase of Celera assets
|
- | - | 1,000,000 | - | 4,500 | - | - | 4,500 | ||||||||||||||||||||||||
|
Share-based compensation expense
|
- | - | - | - | 6,264 | - | - | 6,264 | ||||||||||||||||||||||||
|
Change in unrealized gain (loss) on marketable securities
|
- | - | - | - | - | 161 | - | 161 | ||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (42,158 | ) | (42,158 | ) | ||||||||||||||||||||||
|
Total comprehensive loss
|
(41,997 | ) | ||||||||||||||||||||||||||||||
|
Balance at June 30, 2006
|
- | - | 20,946,694 | 2 | 328,386 | (132 | ) | (288,936 | ) | 39,320 | ||||||||||||||||||||||
|
Issuance of common stock, net of issuance costs, for cash at $4.75 per share
|
- | - | 4,830,000 | 1 | 21,296 | - | - | 21,297 | ||||||||||||||||||||||||
|
Issuance of common stock upon exercise of stock options and purchase rights at an average price $4.16 per share
|
- | - | 191,495 | - | 796 | - | - | 796 | ||||||||||||||||||||||||
|
Share-based compensation expense
|
- | - | - | - | 3,082 | - | - | 3,082 | ||||||||||||||||||||||||
|
Change in unrealized gain (loss) on marketable securities
|
- | - | - | - | - | 123 | - | 123 | ||||||||||||||||||||||||
The accompanying notes are an integral part of these financial statements.
PHARMACYCLICS, INC.
(a development stage enterprise)
STATEMENTS OF STOCKHOLDERS' EQUITY (DEFICIT) (Continued)
For the period from inception (April 19, 1991) through June 30, 2010
(in thousands, except share and per share amounts)
|
Deficit
|
||||||||||||||||||||||||||||||||
|
Accumulated
|
Accumulated
|
|||||||||||||||||||||||||||||||
|
Convertible
|
Additional
|
other
|
During
|
|||||||||||||||||||||||||||||
|
Preferred Stock
|
Common Stock
|
Paid-in
|
Comprehensive
|
Development
|
||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Income (Loss)
|
Stage
|
Total
|
|||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (26,217 | ) | (26,217 | ) | ||||||||||||||||||||||
|
Total comprehensive loss
|
(26,094 | ) | ||||||||||||||||||||||||||||||
|
Balance at June 30, 2007
|
- | - | 25,968,189 | 3 | 353,560 | (9 | ) | (315,153 | ) | 38,401 | ||||||||||||||||||||||
|
Issuance of common stock upon exercise of stock options and purchase rights at an average price $1.34 per share
|
- | - | 47,200 | - | 63 | - | - | 63 | ||||||||||||||||||||||||
|
Share-based compensation expense
|
- | - | - | - | 2,260 | - | - | 2,260 | ||||||||||||||||||||||||
|
Change in unrealized gain (loss) on marketable securities
|
- | - | - | - | - | 19 | - | 19 | ||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (24,298 | ) | (24,298 | ) | ||||||||||||||||||||||
|
Total comprehensive loss
|
(24,279 | ) | ||||||||||||||||||||||||||||||
|
Balance at June 30, 2008
|
- | - | 26,015,389 | 3 | 355,883 | 10 | (339,451 | ) | 16,445 | |||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||
|
Issuance of common stock, net of issuance costs, for cash at $0.93 per share
|
- | - | 1,470,204 | - | 1,351 | - | - | 1,351 | ||||||||||||||||||||||||
|
Issuance of common stock in exchange for services provided
|
15,000 | - | 15 | - | - | 15 | ||||||||||||||||||||||||||
|
Issuance of common stock upon exercise of stock options and purchase rights at an average price $1.04 per share
|
- | - | 38,785 | - | 41 | - | - | 41 | ||||||||||||||||||||||||
|
Disount on note payable to related party
|
- | - | - | - | 570 | - | - | 570 | ||||||||||||||||||||||||
|
Share-based compensation expense
|
- | - | - | - | 3,293 | - | - | 3,293 | ||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||
|
Change in unrealized gain (loss) on marketable securities
|
- | - | - | - | - | (9 | ) | - | (9 | ) | ||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (23,447 | ) | (23,447 | ) | ||||||||||||||||||||||
|
Total comprehensive loss
|
(23,456 | ) | ||||||||||||||||||||||||||||||
|
Balance at June 30, 2009
|
- | - | 27,539,378 | 3 | 361,153 | 1 | (362,898 | ) | (1,741 | ) | ||||||||||||||||||||||
The accompanying notes are an integral part of these financial statements.
PHARMACYCLICS, INC.
(a development stage enterprise)
STATEMENTS OF STOCKHOLDERS' EQUITY (DEFICIT) (Continued)
For the period from inception (April 19, 1991) through June 30, 2010
(in thousands, except share and per share amounts)
|
Deficit
|
||||||||||||||||||||||||||||||||
|
Accumulated
|
Accumulated
|
|||||||||||||||||||||||||||||||
|
Convertible
|
Additional
|
other
|
During
|
|||||||||||||||||||||||||||||
|
Preferred Stock
|
Common Stock
|
Paid-in
|
Comprehensive
|
Development
|
||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Income (Loss)
|
Stage
|
Total
|
|||||||||||||||||||||||||
|
Issuance of common stock in a rights offering at $1.28 per share for cash and settlement of related party note in the amount of $6,100, net of issuance costs
|
- | - | 22,500,000 | 2 | 27,804 | - | - | 27,806 | ||||||||||||||||||||||||
|
Issuance of common stock in a registered direct offering for cash at $6.51 per share, net of issuance costs
|
- | - | 8,054,968 | 1 | 50,792 | - | - | 50,793 | ||||||||||||||||||||||||
|
Issuance of common stock upon exercise of stock options and purchase rights at an average price $1.58 per share
|
- | - | 1,105,060 | - | 1,744 | - | - | 1,744 | ||||||||||||||||||||||||
|
Share-based compensation expense
|
- | - | - | - | 3,190 | - | - | 3,190 | ||||||||||||||||||||||||
|
Change in unrealized gain (loss) on marketable securities
|
- | - | - | - | - | (7 | ) | - | (7 | ) | ||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | (15,024 | ) | (15,024 | ) | ||||||||||||||||||||||
|
Total comprehensive loss
|
- | (15,031 | ) | |||||||||||||||||||||||||||||
|
Balance at June 30, 2010
|
- | $ | - | 59,199,406 | $ | 6 | $ | 444,683 | $ | (6 | ) | $ | (377,922 | ) | $ | 66,761 | ||||||||||||||||
The accompanying notes are an integral part of these financial statements.
PHARMACYCLICS, INC.
(a development stage enterprise)
NOTES TO FINANCIAL STATEMENTS
Note 1 — The Company and Significant Accounting Policies:
Description of the Company
We are a clinical-stage biopharmaceutical company focused on developing and commercializing innovative small-molecule drugs for the treatment of cancer and immune mediated diseases. Our mission and goal is to build a viable biopharmaceutical company that designs, develops and commercializes novel therapies intended to improve quality of life, increase duration of life and resolve serious unmet medical healthcare needs; and to identify promising product candidates based on exceptional scientific development expertise, develop our products in a rapid, cost-efficient manner and pursue commercialization and/or development partners when and where appropriate. We exist to make a difference for the better and these are important times to do just that.
Presently, we have four product candidates in clinical development, a clinical development candidate in late-stage preclinical evaluation and several preclinical molecules in lead optimization. To date, substantially all of our resources have been dedicated to the research and development of our products, and we have not generated any commercial revenues from the sale of our products. We do not anticipate the generation of any product commercial revenues until we receive the necessary regulatory and marketing approvals to launch one of our products.
We continue to evolve into a company focused on licensing and co-development activities. In fiscal 2009, we signed a collaboration and licensing agreement for one of our key compounds allowing us to significantly expedite the development path outside of the U.S., while retaining U.S. rights and receiving upfront and potential future milestone payments. This partnership is indicative of our strategy going forward.
To date, substantially all of our resources have been dedicated to the research and development of our products, and we have not generated any commercial revenues from the sale of our products. We do not anticipate the generation of any product commercial revenues until we receive the necessary regulatory and marketing approvals to launch one of our products.
We have incurred significant operating losses since our inception in 1991, and as of June 30, 2010, had an accumulated deficit of approximately $377,922,000. Based upon the current status of our product development and plans, we believe that our existing cash, cash equivalents and marketable securities, will be adequate to satisfy our capital needs through at least the next twelve months. We expect research and development expenses, as a result of on-going and future clinical trials, to consume a large portion of our existing cash resources. Changes in our research and development plans or other changes affecting our operating expenses may affect actual future consumption of existing cash resources as well. In any event, we will need to raise substantial additional capital to fund our operations in the future. Our actual capital requirements will depend on many factors, including the following:
|
|
·
|
our ability to establish and the scope of any new partnership collaborations;
|
|
|
·
|
the progress and success of preclinical studies and clinical trials of our product candidates; and
|
|
|
·
|
the costs and timing of obtaining regulatory approvals.
|
Our forecast of the period of time through which our financial resources will be adequate to support our operations is a forward-looking statement that involves risks and uncertainties, and actual results could vary materially. The factors described above will impact our future capital requirements and the adequacy of our available funds. If we are required to raise additional funds, we cannot be certain that such additional funding will be available on terms attractive to us, or at all. Furthermore, any additional equity financing may be highly dilutive, or otherwise disadvantageous, to existing stockholders and debt financing, if available, may involve restrictive covenants. Collaborative arrangements, if necessary to raise additional funds, may require us to relinquish rights to certain of our technologies, products or marketing territories. Our failure to raise capital when needed and on acceptable terms, would require us to reduce our operating expenses and would limit our ability to respond to competitive pressures or unanticipated requirements, to develop our product candidates, and to continue operations, any of which would have a material adverse effect on our business, financial condition and results of operations.
Management's use of estimates and assumptions
The preparation of financial statements in accordance with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reported period. Actual results could differ from those estimates.
Basic and diluted net loss per share
Basic net loss per share is computed using the weighted average number of common shares outstanding during the period. Diluted net loss per share is computed using the weighted average number of common and potential common shares outstanding during the period. Potential common shares consist of shares issuable upon the exercise of stock options (using the treasury stock method). Options to purchase 8,395,394, 8,452,899 and 5,540,544 shares of common stock were outstanding at June 30, 2010, 2009 and 2008, respectively, but have been excluded from the computation of diluted net loss per share because their effect was anti-dilutive.
Cash and cash equivalents
All highly liquid investments purchased with an original maturity date of three months or less that are readily convertible into cash and have insignificant interest rate risk are considered to be cash equivalents.
Marketable securities and fair value measurements
Our marketable securities are classified as “available-for-sale”. We include these investments in current assets and carry them at fair value. Unrealized gains and losses on available-for-sale securities are included in accumulated other income (loss). The amortized cost of debt securities is adjusted for the amortization of premiums and accretion of discounts to maturity. Such amortization is included in interest income. Gains and losses on securities sold are recorded based on the specific identification method and are included in interest expense and other income (expense), net in the statement of operations.
Management assesses whether declines in the fair value of marketable securities are other than temporary. If the decline is judged to be other than temporary, the cost basis of the individual security is written down to fair value and the amount of the write down is included in the statement of operations. In determining whether a decline is other than temporary, management considers various factors including the length of time and the extent to which the market value has been less than cost, the financial condition and near-term prospects of the issuer and our intent and ability to retain our investment in the issuer for a period of time sufficient to allow for any anticipated recovery in market value. To date we have not recorded any impairment charges on marketable securities related to other-than-temporary declines in market value.
The fair value of our financial assets and liabilities is determined by using three levels of input which are defined as follows:
Level 1 - Quoted prices in active markets for identical assets or liabilities.
Level 2 - Observable inputs other than Level 1 prices such as quoted prices for similar assets or liabilities, quoted prices in markets that are not active, or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. Our short-term investments primarily utilize broker quotes in markets with infrequent transactions for valuation of these securities.
Level 3 - Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
We utilize the market approach to measure fair value for our financial assets and liabilities. The market approach uses prices and other relevant information generated by market transactions involving identical or comparable assets or liabilities.
Restricted investments
Under our lease agreement, we are required to maintain a $290,000 letter of credit as security for performance under the lease. The letter of credit is secured by a $290,000 certificate of deposit which is included in other assets at June 30, 2010 and 2009.
Concentration of credit risk and other risks and uncertainties
Financial instruments that potentially subject us to credit risk consist principally of cash, cash equivalents, marketable securities and accounts receivable. We place our cash and cash equivalents with high-credit quality financial institutions and invest in debt instruments of financial institutions, corporations and government entities with strong credit ratings. Our management believes it has established guidelines relative to credit quality, diversification and maturities that maintain safety and liquidity. Accounts receivable at June 30, 2010 and 2009 represent amounts due from Servier associated with reimbursement of certain costs and drug shipments, respectively.
Our products require approvals from the United States Food and Drug Administration (the “FDA”) and international regulatory agencies prior to commercial sales. There can be no assurance that our future products will receive required approvals. If we were denied such approvals or such approvals were delayed, it could have a materially adverse impact on us and the execution of our business strategy.
We have expended and will continue to expend substantial funds to complete the research, development and clinical testing of our products. We also will be required to expend additional funds to establish commercial-scale manufacturing arrangements and to provide for the marketing and distribution of our products. Specifically, we will require additional funds to commercialize our products. We are unable to entirely fund these efforts with our current financial resources.
Additional funds may not be available on acceptable terms, if at all. If adequate funds are unavailable on a timely basis from operations or additional sources of financing, we may have to delay, reduce the scope of or eliminate one or more of our research or development programs which would materially and adversely affect our business, financial condition and operations.
Property and equipment
Property and equipment are stated at cost. Depreciation is computed using the straight-line method over the shorter of the estimated useful lives of the assets, generally three to five years, or the lease term of the respective assets, if applicable. Amortization of leasehold improvements is computed using the straight-line method over the shorter of their estimated useful lives or lease terms.
Long-lived assets
Management reviews the carrying values of its long-lived assets for possible impairment whenever events or changes in business conditions indicate that the carrying amount of the assets may not be recoverable. Management evaluates impairment on the basis of undiscounted future cash flows from operations before interest relating to such assets for the remaining useful life of the assets. If present, impairment is measured based on the difference between fair value and the net book value of the related assets. No significant impairment losses have been recorded to date with respect to our long-lived assets, which consist primarily of property and equipment and leasehold improvements.
Revenue recognition
We recognize revenue when all four revenue recognition criteria have been met: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the fee is fixed or determinable; and collectability is reasonably assured. Revenue under our license and collaboration arrangements is recognized based on the performance requirements of the contract. Amounts received under such arrangements consist of up-front collaboration payments, periodic milestone payments and payments for research activities. Our collaborations with multiple elements are evaluated and are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value and whether there is verifiable objective and reliable evidence of the fair value of the undelivered items. The consideration we receive is combined and recognized as a single unit of accounting when criteria for separation are not met.
Up-front payments under agreements which include future performance requirements are recorded as deferred revenue and are recognized over the performance period. The performance period is estimated at the inception of the arrangement and is reevaluated at each reporting period. The reevaluation of the performance period may shorten or lengthen the period during which the deferred revenue is recognized. Revenues related to substantive, at-risk collaboration milestones are recognized upon achievement of the event specified in the underlying agreement. Revenues for research activities are recognized as the related research efforts are performed.
Research and development
Research and development expenses include personnel and facility-related expenses, outside contracted services including clinical trial costs, manufacturing and process development costs, research costs and other consulting services. Research and development costs are expensed as incurred.
Clinical development costs are a significant component of research and development expenses. We have a history of contracting with third parties that perform various clinical trial activities on our behalf in the ongoing development of our product candidates. The financial terms of these contracts are subject to negotiations and may vary from contract to contract and may result in uneven payment flow. We accrue and expense costs for clinical trial activities performed by third parties based upon estimates of the percentage of work completed over the life of the individual study in accordance with agreements established with contract research organizations and clinical trial sites. We determine our estimates through discussions with internal clinical personnel and outside service providers to the progress or stage of completion of trials or services and the agreed upon fee to be paid for such services.
We have purchased quantities of drug substances that are expected to be used in the future to support our clinical development. Until the commercial viability of such products has been demonstrated and the necessary regulatory approvals received, we will continue to charge all such amounts to research and development expense.
Income taxes
We provide for income taxes using the asset and liability method. This method requires that deferred tax assets and liabilities are recognized for the expected future tax consequences of temporary differences between the tax bases of assets and liabilities and their financial statement reported amounts. Valuation allowances are established when necessary to reduce deferred tax assets to the amounts expected to be realized.
Contingencies
We are not currently subject to any material legal proceedings. We may form time to time, however, become party to various legal proceedings arising in the ordinary course of business.
Fair value of financial instruments
The carrying value of our financial instruments including cash and cash equivalents, marketable securities, accounts payable and accrued liabilities, approximate fair value due to their short maturities. See Note 5, Related Party Notes Payable, for the fair value of notes payable to related party.
Accounting for share-based compensation
Share-based compensation cost for employee stock options is measured at the grant date based on the fair value of the award. The accounting grant date for employee stock options with performance obligations is the date on which the performance goals have been defined and a mutual understanding of the terms has been reached. Generally options with performance obligations vest over a four year period, with the goals set and agreed upon annually. Share-based compensation for non-employee stock options is re-estimated at each period-end through the vesting date.
The fair value of each stock option is estimated using the Black-Scholes option pricing model. Expected volatility is based on historical volatility data of our stock. The expected term of stock options granted is based on historical data and represents the period of time that stock options are expected to be outstanding. The expected term for each of our employee options, non-employee options and our Employee Stock Purchase Plan is calculated for and applied to one group of grants as we do not expect substantially different exercise or post-vesting termination behavior among our employee or non-employee population. The risk-free interest rate is based on a zero-coupon United States Treasury bond whose maturity period equals the expected term of the our options.
Options vest upon the passage of time or a combination of time and the achievement of certain performance obligations. The Compensation Committee of the Board of Directors will determine if the performance conditions have been met. Share-based compensation expense for the options with performance obligations is recorded when the company believes that the vesting of these options is probable.
Recent Accounting Pronouncements
In October 2009, the FASB issued two updates relating to revenue recognition. The first update eliminates the requirement that all undelivered elements in an arrangement with multiple deliverables have objective and reliable evidence of fair value before revenue can be recognized for items that have been delivered. The update also no longer allows use of the residual method when allocating consideration to deliverables. Instead, arrangement consideration is to be allocated to deliverables using the relative selling price method, applying a selling price hierarchy. Vendor specific objective evidence (VSOE) of selling price should be used if it exists. Otherwise, third party evidence (TPE) of selling price should be used. If neither VSOE nor TPE is available, the company’s best estimate of selling price should be used. The second update eliminates tangible products from the scope of software revenue recognition guidance when the tangible products contain software components and non-software components that function together to deliver the tangible products’ essential functionality. Both updates require expanded qualitative and quantitative disclosures and are effective for fiscal years beginning on or after June 15, 2010, with prospective application for new or materially modified arrangements or retrospective application permitted. Early adoption is permitted. The same transition method and period of adoption must be used for both updates. We are in the process of analyzing the impact of these updates.
In January 2010, the FASB issued ASU 2010-06, “Fair Value Measurements and Disclosures (ASC 820): Improving Disclosures about Fair Value Measurements.” This update will require (1) an entity to disclose separately the amounts of significant transfers in and out of Levels 1 and 2 fair value measurements and to describe the reasons for the transfers; and (2) information about purchases, sales, issuances and settlements to be presented separately (i.e., present the activity on a gross basis rather than net) in the reconciliation for fair value measurements using significant unobservable inputs (Level 3 inputs). This guidance clarifies existing disclosure requirements for the level of disaggregation used for classes of assets and liabilities measured at fair value and requires disclosures about the valuation techniques and inputs used to measure fair value for both recurring and nonrecurring fair value measurements using Level 2 and Level 3 inputs. The new disclosures and clarifications under this ASU are effective over a period of two fiscal years, for interim and annual reporting periods beginning after December 15, 2009 and after December 15, 2010. The updates for the first adoption date under ASU 2010-06 did not have a material impact on our financial statements. The adoption of the updates for the second date is not expected to have a material impact on our financial statements.
In March 2010, the FASB issued new guidance on the use of the milestone method of recognizing revenue for research and development arrangements under which consideration to be received by the vendor is contingent upon the achievement of certain milestones. The update provides guidance on the criteria that should be met for determining whether the milestone method of revenue recognition is appropriate. A vendor can recognize consideration in its entirety as revenue in the period in which the milestone is achieved only if the milestone meets all criteria to be considered substantive. Additional disclosures describing the consideration arrangement and the entity’s accounting policy for recognition of such milestone payments are also required. The new guidance is effective for fiscal years, and interim periods within such fiscal years, beginning on or after June 15, 2010, with early adoption permitted. The guidance may be applied prospectively to milestones achieved during the period of adoption or retrospectively for all prior periods. We do not believe the adoption of this guidance will have a significant impact on our financial statements.
Note 2 — Agreements:
Collaboration and License Agreement with Les Laboratoires Servier. In April 2009, we entered into a collaboration and license agreement with Les Laboratoires Servier ("Servier") to research, develop and commercialize PCI-24781, an orally active, novel, small molecule inhibitor of Pan HDAC enzymes. Under the terms of the agreement, Servier acquired the exclusive right to develop and commercialize the Pan HDAC inhibitor product worldwide except for the United States and will pay a royalty to Pharmacyclics on sales outside of the United States. Pharmacyclics will continue to own all rights within the United States. In May 2009, Pharmacyclics received an upfront payment of $11,000,000 from Servier, less applicable withholding taxes of $550,000, for a net receipt of $10,450,000.
Under the agreement Pharmacyclics will also receive $4,000,000 from Servier for research collaboration over a twenty-four month period, paid in equal increments every six months, of which $2,000,000 has been received through June 30, 2010. Servier is solely responsible for conducting and paying for all development activities outside the United States. In addition, Pharmacyclics could also receive from Servier up to approximately $24,500,000 upon the achievement of certain future milestones up to and including commercialization, as well as royalty payments.
Revenue associated with our collaboration and related agreements is recognized upon achieving the four general criteria for revenue recognition (i.e., evidence of arrangement, delivery, fixed or determinable amount and collectability). Our collaboration agreement with Servier is accounted for in accordance with accounting rules governing “Revenue Arrangements with Multiple Deliverables.” The non-refundable portion of upfront payments received under our existing agreements is deferred upon receipt and recognized on a straight-line basis over the period ending on the anticipated date of completion of the research activities associated with the Research Program, which we believe represents the conclusion of all significant obligations on our part. For our current agreement, this period was determined to be two years for reasons described further below.
Under the terms of the agreement, four company representatives are required to participate on a Joint Research and Development Committee ("JRDC"). The JRDC’s only responsibilities are to:
|
|
·
|
Meet at least twice a year during the agreement term,
|
|
|
·
|
Oversee the Research Program, Research Plan (as defined) and Development Plan (as defined),
|
|
|
·
|
Oversee the registration and commercialization of licensed products, and
|
|
|
·
|
Maintain a list of Option Compounds (as defined) existing prior to and identified during the Research Term.
|
We believe that our involvement in the JRDC over the term required to complete the research activities associated with the Research Program (currently expected to be the two year Research Term defined in the agreements) associated with the collaboration represents a substantive performance obligation or "deliverable.” However, following completion of such research activities, participation on the JRDC represents only a right and a governance role, rather than a substantive performance obligation.
Given that the deliverables under the collaboration do not meet criteria in the accounting rules for separation (e.g., no separately identifiable fair value), the arrangement is being treated as a single unit-of-accounting for purposes of revenue recognition. We recognize the combined unit of accounting over the estimated period required to complete the research activities under the collaboration (two years), which coincides with the delivery period for all substantive obligations or “deliverables” associated with the collaboration.
The collaboration and license agreement required us to enter into an agreement to supply drug product for Servier’s use in clinical trials. During the quarter ending December 31, 2009, the supply agreement, which is considered part of the arrangement, was completed and executed. Prior to the execution of the supply agreement, we did not meet the “evidence of an arrangement” criterion required for revenue recognition and therefore had deferred revenue recognition until the execution of the supply agreement. Accordingly upon the execution of the drug supply agreement with Servier, we began recognizing revenue from our collaboration and license agreement. Total revenue recognized in the year ended June 30, 2010 was $9,307,000, of which $1,211,000 represents the pro-rata portion of revenue attributable to the period from April 2009 (i.e., the signing of the collaboration agreement) to June 30, 2009, had the supply agreement been completed in April 2009.
Celera Corporation. In April 2006, we acquired multiple small molecule drug candidates for the treatment of cancer and other diseases from Celera Genomics, an Applera Corporation business (now Celera Corporation). Under the terms of the agreement, we acquired Celera technology and intellectual property relating to drugs that target histone deacetylase (HDAC) enzymes, selective HDAC enzymes, a Factor VIIa inhibitor targeting a tumor signaling pathway involved in angiogenesis, tumor growth and metastases, and B-cell associated tyrosine kinase inhibitors potentially useful for the treatment of lymphomas and autoimmune diseases. At the date of acquisition, the HDAC drug candidate was in a Phase I clinical trial and the other drug candidates were in pre-clinical development. Total consideration paid was $6,647,000 which consisted of 1,000,000 shares of our common stock, $2,000,000 of cash and $147,000 of transaction costs. We recorded an expense of $6,647,000 related to the consideration for the acquired drug candidates which had not yet reached technological feasibility and had no alternative future use due to the early stage of development and the significant regulatory requirements remaining.
In May 2008, we amended our agreement with Celera pertaining to the potential sublicensing of our HDAC compounds. Under the amendment, Celera may receive a portion of any upfront licensing payments we receive from sublicensing an HDAC product and the total future potential milestone payments due to Celera were reduced from $144,000,000 to $104,000,000. Celera will also be entitled to royalty payments in the mid- to high single digits based on annual sales of any drugs commercialized from these programs.
The agreement with Celera was amended for a second time in March 2009. Pursuant to this amendment, the total future milestone payments to Celera were reduced to approximately $98,000,000 of which approximately $97,000,000 is outstanding at June 30 2010. Approximately 90% of this amount will become due upon regulatory approval for the drug programs in different geographic markets and with the achievement of certain net sales levels of any drugs commercialized from the HDAC program.
The Celera agreement was again amended for the third time at the end of March 2009. That amendment changed the payment timeline of certain payments to Celera and also changed the obligations for us to pay royalties under certain conditions to Celera. In connection with this third amendment, we paid Celera $1,000,000 in April 2009. The amount was recorded as research and development expense in the quarter ended March 31, 2009, as the technology rights are being utilized in research and development and it is not clear that an alternative future use exists for such technology.
University of Texas License. We have entered into a license agreement with the University of Texas under which we received the exclusive worldwide rights to develop and commercialize porphyrins, expanded porphyrins (e.g. Motexafin Gadolinium) and other porphyrin-like substances covered by their patents. We have made payments, under the license, to the University of Texas of $50,000 in each of the years ended June 30, 2005 and 2004, respectively, and cumulative payments of $300,000 from the inception of the license. No payments are due after fiscal 2005.
Note 3 — Cash and Cash Equivalents and Marketable Securities
The following table sets forth our financial assets as of June 30, 2010 and June 30, 2009 (in thousands):
|
Estimated
|
||||||||||||||||
|
Fair Value as of
|
Basis of Fair Value Measurements
|
|||||||||||||||
|
June 30, 2010
|
Level 1
|
Level 2
|
Level 3
|
|||||||||||||
|
Money market funds
|
$ | 50,432 | $ | 50,432 | $ | - | $ | - | ||||||||
|
Corporate bonds
|
8,003 | - | 8,003 | - | ||||||||||||
|
Government agency securities
|
14,947 | - | 14,947 | - | ||||||||||||
|
Total cash equivalents and marketable securities
|
$ | 73,382 | $ | 50,432 | $ | 22,950 | $ | - | ||||||||
|
Estimated
|
||||||||||||||||
|
Fair Value as of
|
Basis of Fair Value Measurements
|
|||||||||||||||
|
June 30, 2009
|
Level 1
|
Level 2
|
Level 3
|
|||||||||||||
|
Money market funds
|
$ | 10,077 | $ | 10,077 | $ | - | $ | - | ||||||||
|
Corporate bonds
|
2,642 | - | 2,642 | - | ||||||||||||
|
Total cash equivalents and marketable securities
|
$ | 12,719 | $ | 10,077 | $ | 2,642 | $ | - | ||||||||
The following is a summary of our available-for-sale securities at June 30, 2010 and June 30, 2009 (in thousands):
|
Estimated
|
||||||||||||||||
|
Unrealized
|
Unrealized
|
Fair
|
||||||||||||||
|
As of June 30, 2010
|
Cost Basis
|
Gain
|
Loss
|
Value
|
||||||||||||
|
Money market funds
|
$ | 50,432 | $ | - | $ | - | $ | 50,432 | ||||||||
|
Corporate bonds
|
8,019 | - | (16 | ) | 8,003 | |||||||||||
|
Government agency securities
|
14,937 | 10 | - | 14,947 | ||||||||||||
| 73,388 | 10 | (16 | ) | 73,382 | ||||||||||||
|
Less cash equivalents
|
(50,432 | ) | - | - | (50,432 | ) | ||||||||||
|
Total marketable securities
|
$ | 22,956 | $ | 10 | $ | (16 | ) | $ | 22,950 | |||||||
|
Estimated
|
||||||||||||||||
|
Unrealized
|
Unrealized
|
Fair
|
||||||||||||||
|
As of June 30, 2009
|
Cost Basis
|
Gain
|
Loss
|
Value
|
||||||||||||
|
Money market funds
|
$ | 10,077 | $ | - | $ | - | $ | 10,077 | ||||||||
|
Corporate bonds
|
2,641 | 1 | - | 2,642 | ||||||||||||
| 12,718 | 1 | - | 12,719 | |||||||||||||
|
Less cash equivalents
|
(10,927 | ) | - | - | (10,927 | ) | ||||||||||
|
Total marketable securities
|
$ | 1,791 | $ | 1 | $ | - | $ | 1,792 | ||||||||
To date we have not recorded any impairment charges on marketable securities related to other-than-temporary declines in market value. In determining whether a decline is other than temporary, we consider various factors including the length of time and extent to which the market value has been less than cost, the financial condition and near-term prospects of the issuer and our intent and ability to retain our investment in the issuer for a period of time sufficient to allow for any anticipated recovery in market value.
Gross realized losses and gains on the sale of available-for-sale securities during the years ended June 30, 2010, 2009 and 2008, were not material.
At June 30, 2010, our marketable securities had the following contractual maturities (in thousands):
|
Amortized
|
Estimated
|
|||||||
|
Cost
|
Fair Value
|
|||||||
|
Less than one year
|
$ | 22,956 | $ | 22,950 | ||||
Note 4 — Balance Sheet Components:
Accounts receivable consist of the following (in thousands):
|
June 30,
|
||||||||
|
2010
|
2009
|
|||||||
|
Due from Servier
|
$ | 194 | $ | 632 | ||||
| $ | 194 | $ | 632 | |||||
Property and equipment consists of the following (in thousands):
|
June 30,
|
||||||||
|
2010
|
2009
|
|||||||
|
Equipment
|
$ | 6,462 | $ | 6,328 | ||||
|
Leasehold improvements
|
2,576 | 2,569 | ||||||
|
Furniture and fixtures
|
200 | 184 | ||||||
| 9,238 | 9,081 | |||||||
|
Less accumulated depreciation and amortization
|
(8,779 | ) | (8,611 | ) | ||||
| $ | 459 | $ | 470 | |||||
Accrued liabilities consist of the following (in thousands):
|
June 30,
|
||||||||
|
2010
|
2009
|
|||||||
|
Employee compensation
|
$ | 1,051 | $ | 732 | ||||
|
Accrued interest and other
|
3 | 69 | ||||||
| $ | 1,054 | $ | 801 | |||||
Note 5 – Related Party Notes Payable
In December 2008, we borrowed $5,000,000 and in March 2009, borrowed $1,400,000 from an affiliate of Robert W. Duggan, our Chairman of the Board and CEO. Under the terms of the unsecured loans, we repaid the principal sum of $6,400,000 and accrued interest in August 2009. The loans bore interest as follows: (i) 1.36% from December 30, 2008 until March 31, 2009, (ii) the rate of interest in effect for such day as publicly announced from time to time by Citibank N.A. as its “prime rate” from April 1, 2009 until the loan was repaid.
The principal amount of the loans were discounted to fair value for balance sheet presentation such that the stated interest rate together with the accretion of the discount reflected an estimate of the market interest rate during the term of the loan. Due to the lack of reliable and objective observable data available to estimate the fair value of the $5,000,000 loan, the value of the loan was determined using Level 3 inputs. These inputs included an estimate of the probable term of the loan. Due to already initiated corporate events and our then current cash situation, the probable term of the loan was estimated at 6.25 months.
With the term established, we used various methods to estimate the fair market interest rate of the $5,000,000 loan. The first and primary approach was a market risk approach, beginning with the risk-free rate on the effective date of the loan increased by the calculated estimates of the cost of each of the different premiums a lender would require for the various risks taken. These premiums included the non-marketability risk of the note, the risk of default, the cost of arrangement and the opportunity costs. Using this methodology, we estimated the fair market interest rate to be 23%. The reasonableness of this rate was then further supported by comparison to the outcome of the Black Scholes pricing model calculated using the following assumptions:
· Strike price: $5,000,000
· Expected term: 6.25 months
· Risk free interest rate: 0.28%
· Six month volatility: 139%
The calculation was performed using actual volatility for both the six-month and three-month periods prior to the loan effective date. Our fair market rate estimate was further supported by market data estimating the interest rate on high yield bonds that we believe would be of comparable quality to the loan.
The total discount related to the $5,000,000 loan was $484,000 at June 30, 2009.
As the terms of the $1,400,000 loan are the same as the $5,000,000 loan with the same expiration date, we applied the same methodology to determine the fair value of this loan. Due to the lack of reliable and objective observable data available to estimate the fair value of the $1,400,000 loan, the value of the loan was determined using Level 3 inputs. These inputs included first an estimate of the probable term of the loan. Due to already initiated corporate events and our cash situation, the probable term of the loan was estimated at 3.25 months.
With the term established, we used various methods to estimate the fair market interest rate of the $1,400,000 loan. The first and primary approach was a market risk approach, beginning with the risk-free rate on the effective date of the loan increased by the calculated estimates of the cost of each of the different premiums a lender would require for the various risks taken. These premiums included the non-marketability risk of the note, the risk of default, the cost of arrangement and the opportunity costs. Using this methodology, we estimated the fair market interest rate to be 23%. The reasonableness of this rate was then further supported by comparison to the outcome of the Black Scholes pricing model calculated using the following assumptions:
|
|
·
|
Strike price: $1,400,000
|
|
|
·
|
Expected term: 3.25 months
|
|
|
·
|
Risk free interest rate: 0.16%
|
|
|
·
|
Three month volatility: 115%
|
The calculation was performed using actual volatility for the three month period prior to the loan effective date. Our fair market rate estimate was further supported by market data estimating the interest rate on high yield bonds that we believe would be of comparable quality to the loan.
The total discount related to the $1,400,000 loan was $65,000 at June 30, 2009.
Total interest expense related to both loans was $43,000 for the year ended June 30, 2010 and $618,000 for the year ended June 30, 2009. The fair value of the loans was $6,379,000 at June 30, 2009. In accordance with the terms of the loans, both loans plus accrued interest were settled in August 2009 by the issuance of 4,754,870 shares of common stock in our rights offering and the payment of $404,000 in cash.
Note 6 — Stockholders' Equity (Deficit):
Common stock
Unregistered private placement
In February 2009, we sold approximately 1.5 million shares of unregistered common stock, at $0.93 per share for net proceeds of approximately $1,400,000. The purchasers of the shares were certain foreign and U.S. individuals and entities of which some were shareholders of Pacific Biopharma Group, Ltd. (“PBG”). Each investor acted individually and did not purchase shares for the account of PBG or any other affiliated company. Glenn Rice, our former President and Chief Operating Officer and current Director, is a principal of PBG but did not participate in the transaction.
Rights Offering
On July 17, 2009, we commenced a rights offering pursuant to which holders of our common stock were entitled to purchase additional shares of our common stock at a price of $1.28 per share (the “Rights Offering”).
In the Rights Offering, stockholders of record as of July 15, 2009, were issued, at no charge, one subscription right for each share of common stock then outstanding. Each right entitled the holder to purchase 0.6808 share of our common stock for $1.28 per share.
Fractional shares were not issued in the Rights Offering. The subscription rights issued pursuant to the Rights Offering expired on July 31, 2009. Stockholders who exercised their rights in full were also permitted an oversubscription right to purchase additional shares of common stock that remained unsubscribed at the expiration of the Rights Offering, subject to the availability of shares and a pro rata allocation of shares among persons exercising the oversubscription right.
As of the close of the Rights Offering on July 31, 2009, the Rights Offering was oversubscribed. The proration of available over-subscription shares was made in accordance with the Offering Prospectus. Approximately 22.5 million shares of our common stock were purchased in the Rights Offering for net proceeds (after offering costs of approximately $1,000,000 and the partial settlement of loans from an affiliate of Robert W. Duggan, our Chairman of the Board and Chief Executive Officer, of approximately $6,100,000) of approximately $21,700,000. Mr. Duggan participated in the Rights Offering for a total of $6,100,000.
Registered Direct Offering
In June 2010, we sold approximately 8.1 million shares to a group of institutional investors in a registered direct offering at $6.51 per share for net proceeds of approximately $50,800,000. Our Chairman and CEO, Robert W. Duggan, participated in the offering in the amount of $7,000,000.
Preferred stock
As amended, our Certificate of Incorporation authorizes 1,000,000 shares of preferred stock, par value $0.0001 per share. The Board of Directors is authorized to issue the preferred stock in one or more series and to fix the rights, preferences, privileges and restrictions thereof, including dividend rights, dividend rates, conversion rights, voting rights, terms of redemption, redemption prices, liquidation preferences and the number of shares constituting any series or the designation of such series, without further vote or action by the stockholders. No preferred stock was outstanding at June 30, 2010 or June 30, 2009.
The ability of our Board of Directors to issue shares of preferred stock without stockholder approval may have certain anti-takeover effects. We are also subject to provisions of the Delaware General Corporation Law, which may make certain business combinations more difficult.
Stock plans
2004 Equity Incentive Award Plan. In December 2004, stockholders approved the 2004 Equity Incentive Award Plan (the “2004 Plan”) as a replacement for both our 1995 Stock Option Plan (the “1995 Plan”) and the 1995 Non-Employee Directors Stock Option Plan (the “Directors Plan”). At June 30, 2009, we had reserved 4,600,000 shares of our common stock for issuance under the plan. In December 2009, the stockholders approved an increase of 2,000,000 shares available for issuance under the plan. The 2004 Plan provides for the issuance of various types of equity awards, such as incentive stock options, nonqualified stock options, restricted stock, stock appreciation rights and performance shares. The exercise price of all stock options granted under the 2004 Plan may not be less than the fair market value of our common stock on the date of grant and no stock option will be exercisable more than ten years after the date it is granted. Stock options for employees and consultants typically vest over four years. Non-employee Directors receive annual, automatic, non-discretionary grants of nonqualified stock options. Each new non-employee Director receives an option to purchase 10,000 shares as of the date he or she first becomes a Director. This option grant vests in equal annual installments over five years. In addition, on the date of each annual meeting, each individual re-elected as a non-employee Director will receive an automatic option grant to purchase an additional 7,500 shares of common stock, provided such individual has served as a Director for at least six months prior to the date of grant. This option grant vests in equal monthly installments over twelve months following the date of grant.
1995 Stock Option Plan. Our 1995 Plan was adopted by the Board of Directors in August 1995. Options issued under the 1995 Plan can, at the discretion of the plan administrator, be either incentive stock options or nonqualified stock options. In December 2003, the stockholders approved amendments to the 1995 Plan (i) such that the exercise price of all stock options must be at least equal to the fair value of Pharmacyclics’ common stock on the date of grant and (ii) that increased the total number of authorized shares under the plan to 5,345,724 shares of common stock. Generally, shares subject to options under the 1995 Plan vest over a four or five year period and are exercisable for a period of ten years. In December 2004, the remaining shares available for future grant under the 1995 Plan were transferred to the 2004 Plan. Additionally, if options granted under the 1995 Plan expire or otherwise terminate without being exercised, the shares of common stock reserved for such options again become available for future grant under the 2004 Plan.
1995 Non-Employee Directors Stock Option Plan. Our Directors Plan was adopted by the Board of Directors on August 2, 1995 and provides for issuance of common stock to non-employee Directors pursuant to a predetermined formula. The exercise price of options granted under the Directors Plan must be at least equal to the fair value of Pharmacyclics’ common stock on the date of grant. Each individual first elected or appointed as a non-employee Board member will automatically be granted, on the date of such election or appointment, a non-statutory option to purchase 10,000 shares of common stock vesting over five years. In addition, on the date of each annual stockholders’ meeting each individual who is to continue to serve as a non-employee Board member after that annual meeting and has been a member of the Board for at least six months will automatically be granted a non-statutory option to purchase 5,000 shares of common stock. A total of 271,667 shares of common stock have been reserved for issuance under the Directors Plan. In December 2004, the remaining shares available for future grant under the Directors Plan were transferred to the 2004 Plan. Additionally, if options granted under the Directors Plan expire or otherwise terminate without being exercised, the shares of common stock reserved for such options again become available for future grant under the 2004 Plan.
The following table summarizes our stock option activity (in thousands, except per share amounts):
|
Options Outstanding
|
||||||||
|
Weighted
|
||||||||
|
Average
|
||||||||
|
Number
|
Exercise
|
|||||||
|
of
|
Price Per
|
|||||||
|
Options
|
Share
|
|||||||
|
Authorized
|
- | $ | - | |||||
|
Granted
|
480 | 0.19 | ||||||
|
Balance at June 30, 1993
|
480 | 0.19 | ||||||
|
Exercised
|
(324 | ) | 0.12 | |||||
|
Granted
|
167 | 2.22 | ||||||
|
Forfeited or expired
|
(8 | ) | 0.11 | |||||
|
Balance at June 30, 1994
|
315 | 1.37 | ||||||
|
Exercised
|
(39 | ) | 0.24 | |||||
|
Granted
|
193 | 3.75 | ||||||
|
Forfeited or expired
|
(38 | ) | 1.82 | |||||
|
Balance as of June 30, 1995
|
431 | 2.50 | ||||||
|
Authorized
|
- | |||||||
|
Exercised
|
(92 | ) | 3.09 | |||||
|
Granted
|
492 | 10.03 | ||||||
|
Forfeited or expired
|
(11 | ) | 6.11 | |||||
|
Balance as of June 30, 1996
|
820 | 9.20 | ||||||
|
Authorized
|
- | |||||||
|
Exercised
|
(96 | ) | 2.74 | |||||
|
Granted
|
569 | 16.69 | ||||||
|
Forfeited or expired
|
(31 | ) | 12.21 | |||||
|
Balance as of June 30, 1997
|
1,262 | 11.58 | ||||||
|
Authorized
|
- | |||||||
|
Exercised
|
(89 | ) | 6.57 | |||||
|
Granted
|
577 | 25.33 | ||||||
|
Forfeited or expired
|
(158 | ) | 15.41 | |||||
|
Balance as of June 30, 1998
|
1,592 | 16.43 | ||||||
|
Authorized
|
- | |||||||
|
Exercised
|
(75 | ) | 5.10 | |||||
|
Granted
|
671 | 19.25 | ||||||
|
Forfeited or expired
|
(221 | ) | 20.37 | |||||
|
Balance as of June 30, 1999
|
1,967 | 17.38 | ||||||
|
Authorized
|
- | |||||||
|
Exercised
|
(103 | ) | 13.88 | |||||
|
Granted
|
723 | 56.97 | ||||||
|
Forfeited or expired
|
(53 | ) | 23.38 | |||||
|
Balance as of June 30, 2000
|
2,534 | 28.70 | ||||||
|
Authorized
|
- | |||||||
|
Exercised
|
(94 | ) | 16.17 | |||||
|
Granted
|
947 | 36.80 | ||||||
|
Forfeited or expired
|
(114 | ) | 45.70 | |||||
|
Balance as of June 30, 2001
|
3,273 | 29.78 | ||||||
|
Authorized
|
- | |||||||
|
Exercised
|
(13 | ) | 13.93 | |||||
|
Granted
|
1,634 | 8.76 | ||||||
|
Forfeited or expired
|
(625 | ) | 27.83 | |||||
|
Balance as of June 30, 2002
|
4,269 | 21.82 | ||||||
|
Options Outstanding
|
||||||||
|
Weighted
|
||||||||
|
Average
|
||||||||
|
Number
|
Exercise
|
|||||||
|
of
|
Price Per
|
|||||||
|
Options
|
Share
|
|||||||
|
Authorized
|
- | |||||||
|
Exercised
|
(3 | ) | 1.03 | |||||
|
Granted
|
749 | 4.35 | ||||||
|
Forfeited or expired
|
(837 | ) | 25.30 | |||||
|
Balance as of June 30, 2003
|
4,178 | 18.03 | ||||||
|
Authorized
|
- | |||||||
|
Exercised
|
(181 | ) | 4.91 | |||||
|
Granted
|
532 | 9.53 | ||||||
|
Forfeited or expired
|
(296 | ) | 28.55 | |||||
|
Balance as of June 30, 2004
|
4,233 | 16.78 | ||||||
|
Authorized
|
- | |||||||
|
Exercised
|
(61 | ) | 4.46 | |||||
|
Granted
|
814 | 8.08 | ||||||
|
Forfeited or expired
|
(200 | ) | 18.19 | |||||
|
Balance as of June 30, 2005
|
4,786 | 15.40 | ||||||
|
Authorized
|
- | |||||||
|
Exercised
|
(191 | ) | 7.02 | |||||
|
Granted
|
1,351 | 4.58 | ||||||
|
Forfeited or expired
|
(679 | ) | 12.85 | |||||
|
Balance as of June 30, 2006
|
5,267 | 13.26 | ||||||
|
Exercised
|
(133 | ) | 4.38 | |||||
|
Granted
|
1,310 | 3.01 | ||||||
|
Forfeited or expired
|
(855 | ) | 13.83 | |||||
|
Balance as of June 30, 2007
|
5,589 | 10.98 | ||||||
|
Exercised
|
- | - | ||||||
|
Granted
|
1,555 | 1.05 | ||||||
|
Forfeited or expired
|
(1,603 | ) | 11.31 | |||||
|
Balance as of June 30, 2008
|
5,541 | 8.10 | ||||||
|
Exercised
|
(4 | ) | 0.85 | |||||
|
Granted
|
2,186 | 1.11 | ||||||
|
Forfeited or expired
|
(668 | ) | 10.05 | |||||
|
Balance as of June 30, 2009
|
7,055 | 5.75 | ||||||
|
Exercised
|
(1,044 | ) | 1.58 | |||||
|
Granted
|
2,343 | 5.27 | ||||||
|
Forfeited or expired
|
(833 | ) | 15.98 | |||||
|
Balance as of June 30, 2010
|
7,521 | 5.05 | ||||||
The above table does not include 303,750 and 570,500 performance options granted in fiscal 2010 and 2009, respectively for which the performance criteria had not been established as of June 30, 2010.
The components of share-based compensation recognized in our statements of operations for the years ended June 30, 2010, 2009 and 2008 and since inception were as follows:
|
Period
|
||||||||||||||||
|
from
|
||||||||||||||||
|
Inception
|
||||||||||||||||
|
(April 19, 1991)
|
||||||||||||||||
|
through
|
||||||||||||||||
|
Year Ended June 30,
|
June 30,
|
|||||||||||||||
|
2010
|
2009
|
2008
|
2010
|
|||||||||||||
|
Research and development
|
$ | 1,998,000 | $ | 738,000 | $ | 961,000 | $ | 8,712,000 | ||||||||
|
General and administrative
|
1,192,000 | 2,555,000 | 1,299,000 | 10,294,000 | ||||||||||||
|
Total share-based compensation
|
$ | 3,190,000 | $ | 3,293,000 | $ | 2,260,000 | $ | 19,006,000 | ||||||||
There were no capitalized share-based compensation costs at June 30, 2010
The fair value of each stock option is estimated using the Black-Scholes option pricing model. Expected volatility is based on historical volatility data of our stock. The expected term of stock options granted is based on historical data and represents the period of time that stock options are expected to be outstanding. The expected term for each of the types is calculated for and applied to one group of stock options as we do not expect substantially different exercise or post-vesting termination behavior among our employee population. The risk-free interest rate is based on a zero-coupon United States Treasury bond whose maturity period equals the expected term of the our options.
|
Year Ended June 30,
|
|||||||
|
2010
|
2009
|
2008
|
|||||
|
Employee stock options:
|
|||||||
|
Expected dividend yield
|
-
|
%
|
-
|
%
|
-
|
%
|
|
|
Expected stock price volatility
|
98
|
%
|
90
|
%
|
74
|
%
|
|
|
Risk free interest rate
|
2.05
|
%
|
2.12
|
%
|
2.66
|
%
|
|
|
Expected life (years)
|
5.00
|
5.00
|
5.00
|
||||
|
Non-employee stock options:
|
|||||||
|
Expected dividend yield
|
-
|
%
|
-
|
%
|
*
|
|
|
|
Expected stock price volatility
|
88 - 90
|
%
|
87 - 91
|
%
|
*
|
|
|
|
Risk free interest rate
|
3.20 - 3.89
|
%
|
1.55 - 3.86
|
%
|
*
|
|
|
|
Expected life (years)
|
7.00 - 10.00
|
7.00 - 10.00
|
*
|
||||
|
* No non-employee options were granted during the year ended June 30, 2008.
|
|||||||
The weighted average estimated grant date fair value for employee options granted under our stock option plans during fiscal 2010, 2009 and 2008 was $4.55, $0.77 and $0.65 per share, respectively.
The total pre-tax intrinsic value of employee and employee stock options exercised during the years ended June 30, 2010, 2009 and 2008 was $3,990,000, $2,000 and $0, respectively. No income tax benefits were realized in the years ended June 30, 2010, 2009 and 2008.
Shares reserved for issuance and available for grant under the 2004 Plan were 1,664,000 shares as of June 30, 2010.
As of June 30, 2010, $6,669,000 of total unrecognized compensation costs related to non vested employee options were scheduled to be recognized over a weighted average period of 2.74 years. As of June 30, 2010, unrecognized compensation cost of $3,722,000 related to non vested non-employee stock options was scheduled to be recognized over a weighted average period of 3.71 years.
A summary of outstanding and vested stock options as of June 30, 2010 is as follows:
|
Options Outstanding
|
Options Vested
|
|||||||||||||||||||||||
|
Weighted
|
Weighted
|
Weighted
|
||||||||||||||||||||||
|
Average
|
Average
|
Average
|
||||||||||||||||||||||
|
Number
|
Remaining
|
Exercise
|
Aggregate
|
Number
|
Exercise
|
Aggregate
|
||||||||||||||||||
|
Range of
|
of
|
Contractual
|
Price
|
Intrinsic
|
of
|
Price
|
Intrinsic
|
|||||||||||||||||
|
Exercise Prices
|
Shares
|
Life
|
Per Share
|
Value
|
Shares
|
Per Share
|
Value
|
|||||||||||||||||
| $0.73 - $0.73 | 300,000 | 8.69 | $ | 0.73 | 75,000 | $ | 0.73 | |||||||||||||||||
| $0.75 - $0.75 | 845,481 | 8.66 | 0.75 | 214,858 | 0.75 | |||||||||||||||||||
| $0.78 - $0.86 | 946,582 | 6.32 | 0.85 | 659,314 | 0.84 | |||||||||||||||||||
| $0.91 - $1.94 | 863,092 | 6.80 | 1.29 | 393,281 | 1.27 | |||||||||||||||||||
| $2.00 - $2.76 | 972,756 | 4.63 | 2.59 | 770,163 | 2.64 | |||||||||||||||||||
| $2.90 - $4.16 | 891,035 | 4.42 | 3.97 | 795,432 | 4.05 | |||||||||||||||||||
| $4.25 - $6.56 | 983,397 | 5.84 | 5.33 | 481,814 | 4.61 | |||||||||||||||||||
| $6.63- $7.10 | 530,696 | 9.01 | 6.81 | 76,946 | 6.95 | |||||||||||||||||||
| $7.19 - $7.19 | 982,500 | 9.78 | 7.19 | 24,062 | 7.19 | |||||||||||||||||||
| $7.20 - $12.99 | 767,380 | 1.86 | 8.41 | 757,380 | 8.41 | |||||||||||||||||||
| $18.07 - $49.50 | 312,475 | 0.80 | 29.95 | 312,475 | 29.95 | |||||||||||||||||||
| 8,395,394 | 6.20 | $ | 4.83 | $24,588,000 | 4,560,725 | $ | 5.52 | $13,825,000 | ||||||||||||||||
We had outstanding exercisable options to purchase 6,754,206, 5,862,640, and 4,928,736 shares of common stock with a weighted average exercise price of $5.24, $6.73 and $8.86 at June 30, 2010, 2009 and 2008, respectively.
Employee Stock Purchase Plan. We adopted an Employee Stock Purchase Plan (the “Purchase Plan”) in August 1995. Qualified employees may elect to have a certain percentage of their salary withheld to purchase shares of our common stock under the Purchase Plan. The purchase price per share is equal to 85% of the fair market value of the stock on specified dates. Sales under the Purchase Plan in fiscal 2010, 2009 and 2008 were 61,026, 35,035 and 47,200 shares of common stock at an average price of $1.55, $1.06 and $1.34 per share, respectively. Shares available for future purchase under the Purchase Plan were 417,546 at June 30, 2010.
Compensation cost is estimated using the Black-Scholes valuation model using the assumptions noted in the following table.
|
Year Ended June 30,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Expected dividend yield
|
- | % | - | % | - | % | ||||||
|
Stock price volatility
|
105 | % | 144 | % | 71 | % | ||||||
|
Risk free interest rate
|
0.53 | % | 0.77 | % | 3.78 | % | ||||||
|
Expected life (years)
|
1.20 | 0.58 | 1.29 | |||||||||
The weighted average estimated grant date fair value of purchase awards under our employee stock purchase plan during fiscal 2010, 2009 and 2008 was $4.09, $0.73 and $0.97 per share, respectively.
During fiscal 2010 a modification to our Purchase Plan went into effect that increased both the maximum employee contribution and the limit on the number of shares that could be purchased. As a result, a number of our employees chose to increase their contribution percentage which was accounted for as a modification to the terms of the award and resulted in $306,000 of additional compensation cost during the fiscal year.
As of June 30, 2010, $1,518,000 of total unrecognized compensation costs related to purchase awards under our employee stock purchase plan were scheduled to be recognized over a weighted average period of 0.75 years.
Note 7 — Employee Benefit Plan:
We maintain a defined contribution plan covering substantially all employees under Section 401(k) of the Internal Revenue Code. Our matching contribution to the plan was $64,000, $58,000 and $103,000 for the years ended June 30, 2010, 2009 and 2008, respectively, and $1,071,000 for the period from inception (April 19, 1991) through June 30, 2010.
Note 8 — Income Taxes:
Deferred tax assets are summarized as follows (in thousands):
|
June 30,
|
||||||||
|
2010
|
2009
|
|||||||
|
Net operating loss carryforwards
|
$ | 53,005 | $ | 44,927 | ||||
|
Tax credit carryforwards
|
8,755 | 8,318 | ||||||
|
Capitalized start-up and R & D costs
|
3,553 | 3,997 | ||||||
|
Depreciation and amortization
|
2,670 | 2,887 | ||||||
|
Share-based compensation
|
3,107 | 3,235 | ||||||
|
Reserves and accruals
|
2,597 | 4,807 | ||||||
|
Gross deferred tax assets
|
73,687 | 68,171 | ||||||
|
Less valuation allowance
|
(73,687 | ) | (68,171 | ) | ||||
|
Net deferred tax assets
|
$ | - | $ | - | ||||
A full valuation allowance has been established for our deferred tax assets at June 30, 2010 and 2009 since realization of such assets through the generation of future taxable income is uncertain. The increase (decrease) in the valuation allowance was approximately $5,516,000, $(73,322,000) and $7,438,000 for the years ended June 30, 2010, 2009 and 2008, respectively.
The provision for income taxes differs from the amount determined by applying the United States statutory income tax rate to the loss before income taxes as summarized below (in thousands):
|
Year Ended June 30,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Tax benefit at statutory rate
|
$ | 6,204 | $ | 9,121 | $ | 9,561 | ||||||
|
Research and development credits
|
436 | 2,585 | 1,408 | |||||||||
|
Deferred tax assets not benefited
|
(6,136 | ) | (10,269 | ) | (10,058 | ) | ||||||
|
Share-based compensation
|
(488 | ) | (308 | ) | (432 | ) | ||||||
|
Other
|
(16 | ) | (1,129 | ) | (479 | ) | ||||||
|
Withholding tax
|
550 | (550 | ) | - | ||||||||
| $ | 550 | $ | (550 | ) | $ | - | ||||||
The $550,000 tax benefit for the year ended June 30, 2010 is the result of the reversal of the French withholding taxes related to our receipt of an $11,000,000 upfront payment from Servier in 2010. The withholding tax expense was no longer required as a result of a new treaty between the U.S. and France that was signed at the end of December 2009.
At June 30, 2010, we had federal and state net operating loss carryforwards of approximately $141,100,000 and $86,500,000, respectively. The federal and state net operating loss carryforwards will begin to expire in 2011. Federal and state tax credit carryforwards of $3,500,000 and $9,200,000, respectively, are available to offset future taxable income. The federal tax credits will begin to expire in 2025. State research and development credits can be carried forward indefinitely.
Under the Tax Reform Act of 1986, the amounts of and the benefit from net operating losses and tax credit carry-forwards that can be carried forward may be impaired or limited in certain circumstances. These circumstances include, but are not limited to, a cumulative stock ownership change of greater than 50%, as defined, over a three year period. Such an annual limitation may result in the expiration of net operating losses before utilization. We have determined that a cumulative stock ownership change happened and we have estimated that a significant portion of our net operating losses for federal and state tax purposes, as well as some amount of our federal research credits, will not be available for use in future periods due to these limitation rules. As a result we have revised our 2009 deferred tax footnote by reducing the amount of previously reported net operating losses and credits for the estimated impairment amounts. The reduction to the 2009 deferred tax assets had no effect on our statement of operations, earnings per share, balance sheet, statement of cash flows or statement of stockholders’ equity (deficit) for any period presented.
The following table summarizes the activity related to our gross unrecognized tax benefits (in thousands):
|
Year Ended June 30,
|
||||||||||
|
2010
|
2009
|
2008
|
||||||||
|
Beginning balance
|
$ | 1,285 | $ | 2,960 | $ | 2,890 | ||||
|
Additions based on tax
|
||||||||||
|
positions related to current year
|
- | - | - | |||||||
|
Additions (reduction) for
|
||||||||||
|
tax positions of prior years
|
- | (1,675 | ) | 70 | ||||||
|
Settlements
|
- | - | - | |||||||
|
Lapse of applicable statue of limitations
|
- | - | - | |||||||
|
Ending balance
|
$ | 1,285 | $ | 1,285 | $ | 2,960 | ||||
The revision to the 2009 deferred tax assets noted above had a corresponding impact on the amount of unrecognized tax benefit recorded at the beginning of 2009 that required a similar reduction to the amount of the previously reported unrecognized tax benefit. This reduction has no effect on our statement of operations, earnings per share, balance sheet, statement of cash flows or statement of stockholders’ equity (deficit) for any period presented.
We may from time to time be assessed interest or penalties by major tax jurisdictions, although there have been no such assessments historically. In the event we receive an assessment for interest and/or penalties, it would be classified in the financial statements as income tax expense. As of June 30, 2010, open tax years in major jurisdictions remain open due to the taxing authorities’ ability to adjust operating loss carry forwards. We do not expect any material changes to the unrecognized tax benefits reported above during the next twelve months.
Note 9 — Commitments:
We lease our facility under a non-cancelable operating lease that expires in fiscal 2012. Future minimum lease payments under the non-cancelable operating lease are as follows (in thousands):
|
Operating
|
||||
|
Lease
|
||||
|
Commitments
|
||||
|
2011
|
$ | 644 | ||
|
2012
|
330 | |||
|
Total minimum lease payments
|
$ | 974 | ||
Rent expense for the years ended June 30, 2010, 2009 and 2008 was $752,000, $905,000 and $952,000, respectively, and $17,764,000 for the period from inception (April 19, 1991) through June 30, 2010. Sublease income was $0 for each of the years ended June 30, 2010, 2009 and 2008, and $924,000 from the period from inception (April 19, 1991) through June 30, 2010. The terms of the facility lease provide for rental payments on a graduated scale. We recognize rent expense on a straight-line basis over the lease period and have accrued for rent expense incurred but not paid at June 30, 2010.
Note 10 — Quarterly Results (Unaudited)
The following table is in thousands, except per share amounts:
|
Quarter Ended
|
||||||||||||||||
|
September 30,
|
December 31,
|
March 31,
|
June 30,
|
|||||||||||||
|
Fiscal 2010
|
||||||||||||||||
|
Loss from operations
|
$ | (4,821 | ) | $ | (786 | ) | $ | (3,144 | ) | $ | (6,861 | ) | ||||
|
Net loss
|
(4,845 | ) | (218 | ) | (3,123 | ) | (6,838 | ) | ||||||||
|
Basic and diluted net loss per share(1)
|
$ | (0.12 | ) | $ | 0.00 | $ | (0.06 | ) | $ | (0.13 | ) | |||||
|
Shares used in computation of basic
and diluted net loss per share
|
40,993 | 50,076 | 50,536 | 51,771 | ||||||||||||
|
Quarter Ended
|
||||||||||||||||
|
September 30,
|
December 31,
|
March 31,
|
June 30,
|
|||||||||||||
|
Fiscal 2009
|
||||||||||||||||
|
Loss from operations
|
$ | (6,642 | ) | $ | (4,860 | ) | $ | (6,415 | ) | $ | (4,511 | ) | ||||
|
Net loss
|
(6,542 | ) | (4,834 | ) | (6,665 | ) | (5,406 | ) | ||||||||
|
Basic and diluted net loss per share(1)
|
$ | (0.25 | ) | $ | (0.19 | ) | $ | (0.25 | ) | $ | (0.20 | ) | ||||
|
Shares used in computation of basic
and diluted net loss per share |
26,015 | 26,034 | 26,696 | 27,533 | ||||||||||||
|
(1)
|
Basic and diluted net loss per share amounts are computed independently for each of the quarters presented. Therefore, the sum of quarterly basic and diluted net loss per share information may not equal annual basic and diluted net loss per share.
|
Note 11 – Separation Agreements
In September 2008, our President & CEO and our Vice President, Finance and Administration and CFO, resigned their positions and entered into separation agreements with us. Under the separation agreements, the two executives continued to provide services through September 30, 2008 and October 31, 2008, respectively. Under the agreements, we agreed to pay Dr. Miller and Mr. Lea one year of salary in severance payments, accelerate the vesting of all outstanding options, extend the exercise period of all outstanding options to three years after termination and provide healthcare benefits for twelve months following the termination of their employment. We recorded severance expense of $536,000 and share-based compensation expense of $1,394,000 associated with the separation agreements in the quarter ended September 30, 2008. We also recorded severance expense of $600,000 including approximately $200,000 relating to cash-based severance payments and share-based compensation expense of approximately $400,000 in the quarter ended December 31, 2008 associated with Mr. Lea’s separation agreement.
In October 2009, we entered into a separation agreement with Glenn Rice, our President and Chief Operating Officer. Under the agreement, Dr. Rice continued to provide services through February 2010 and vesting was accelerated on certain outstanding options. We recorded approximately $200,000 in additional share-based compensation expense related to this agreement in fiscal 2010.
Note 12 – Related Party Transaction
As discussed in Note 5 – Related Party Notes Payable, as of June 30, 2009, we had borrowed $6,400,000 from an affiliate of Robert W. Duggan, our Chairman of the Board and Chief Executive Officer, in the form of an unsecured loan. The related party notes and accrued interest were repaid in full during fiscal 2010 by the issuance of shares in our Rights Offering and a cash payment of $404,000.
As discussed in Note 6 - Robert W. Duggan, our Chairman of the Board and Chief Executive Officer, participated in our Rights Offering for a total of $7,000,000.
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure
Not Applicable.
Item9A. Controls and Procedures
(a) Evaluation of Disclosure Controls and Procedures:
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Vice President, Finance and Administration, as appropriate, to allow for timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and in reaching a reasonable level of assurance, management is required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
As of June 30, 2010, the end of the period covered by this report, we carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and our Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures. Based on the foregoing, our Chief Executive Officer and Vice President, Finance and Administration concluded that our disclosure controls and procedures were effective at the reasonable assurance level.
(b) Management’s Annual Report on Internal Control Over Financial Reporting:
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Under the supervision and with the participation of our management, including our Chief Executive Officer and Vice President, Finance and Administration, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of June 30, 2010 based on the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on that evaluation, our management concluded that our internal control over financial reporting was effective as of June 30, 2010.
Management’s assessment of the effectiveness of our internal control over financial reporting as of June 30, 2010 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report which is included on page 64 of this Annual Report on Form 10-K.
(c) Changes in Internal Control Over Financial Reporting:
There has been no change in our internal control over financial reporting during our most recent fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. Other Information
Not applicable.
PART III
Item 10. Directors, Executive Officers and Corporate Governance
Certain information required by this Item 10 is hereby incorporated by reference to the information under the captions, (i) “Election of Directors,” (ii) “Audit Committee,” (iii) “Code of Business Conduct and Ethics” and (iv) “Section 16(a) Beneficial Ownership Reporting Compliance,” contained in our Definitive Proxy Statement to be filed with the Securities and Exchange Commission no later than 120 days from the end of our last fiscal year.
Item 11. Executive Compensation
The information required by this Item 11 is incorporated by reference to the information under the caption “Executive Compensation and Other Information” in the Definitive Proxy Statement.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this Item 12 with respect to stock ownership of certain beneficial owners and management and securities authorized for issuance under equity compensation plans are incorporated by reference to the information under the captions “Stock Ownership of Management and Certain Beneficial Owners” and “Securities Authorized For Issuance Under Equity Compensation Plans” in the Definitive Proxy Statement.
Item 13. Certain Relationships and Related Transactions and Director Independence
The information required by this Item 13 is incorporated by reference to the information under the caption “Certain Relationships and Related Transactions” in the Definitive Proxy Statement.
Item 14. Principal Accountant Fees and Services
The information required by this Item 14 is incorporated by reference to the information in the Definitive Proxy Statement.
PART IV
Item 15. Exhibits and Financial Statement Schedules
(a) 1. Financial Statements
See Index to Financial Statements under Item 8 on page 63.
(a) 2. Financial Statement Schedules
All schedules are omitted because they are not applicable or are not required or the information required to be set forth therein is included in the Financial Statements or notes thereto.
(a) 3. Exhibits
See Index to Exhibits beginning on page 106.
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, (the “Exchange Act”) the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Dated: September 13, 2010
|
PHARMACYCLICS, INC.
|
||
|
By:
|
/s/ ROBERT W. DUGGAN
|
|
|
Robert W. Duggan
|
||
|
Chairman of the Board & Chief Executive Officer
|
||
POWER OF ATTORNEY
KNOW ALL MEN BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints jointly and severally, Robert W. Duggan and Rainer Erdtmann, or either of them as his or her true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution, for him or her and in his or her name, place and stead, in any and all capacities, to sign any and all amendments (including post-effective amendments) to this Report on Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or any of them, or their or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
IN WITNESS WHEREOF, each of the undersigned has executed this Power of Attorney as of the date indicated.
Pursuant to the requirements of the Exchange Act, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
|
Signature
|
Title
|
Date
|
||
|
/s/ ROBERT W. DUGGAN
|
Chairman of the Board and Chief Executive Officer
(Principal Executive Officer)
|
September 13, 2010
|
||
|
Robert W. Duggan
|
||||
|
/s/ RAINER M. ERDTMANN
|
Vice President, Finance and Administration and Secretary
(Principal Financial and Accounting Officer)
|
September 13, 2010
|
||
|
Rainer M. Erdtmann
|
||||
|
/s/ JASON T. ADELMAN
|
Director
|
September 13, 2010
|
||
|
Jason T. Adelman
|
||||
|
/s/ CYNTHIA C. BAMDAD, Ph.D.
|
Director
|
September 13, 2010
|
||
|
Cynthia C. Bamdad
|
||||
|
/s/ MINESH P. MEHTA, M.D.
|
Director
|
September 13, 2010
|
||
|
Minesh P. Mehta, M.D.
|
||||
|
/s/ GLENN C. RICE, Ph.D.
|
Director
|
September 13, 2010
|
||
|
Glenn C. Rice, Ph.D.
|
||||
|
/s/ DAVID D. SMITH, Ph.D.
|
Director
|
September 13, 2010
|
||
|
David D. Smith, Ph.D.
|
||||
|
/s/ RICHARD A. VAN DEN BROEK
|
Director
|
September 13, 2010
|
||
|
Richard A. van den Broek
|
EXHIBITS INDEX
|
Exhibit
Number
|
Description
|
|
|
3.1
|
Amended and Restated Certificate of Incorporation of the Company (incorporated by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on December 16, 2008).
|
|
|
3.2
|
Amended and Restated Bylaws of the Company (incorporated by reference to Exhibit of the same number to the Quarterly Report on Form 10-Q for the quarter ended December 31, 2001).
|
|
|
3.3
|
Amendment to the Amended and Restated Bylaws of the Company (incorporated by reference to Exhibit 3.1 to the Periodic Report on Form 8-K filed on August 9, 2006).
|
|
|
4.2
|
Specimen Certificate of the Company's Common Stock (incorporated by reference to Exhibit 4.2 to the Company's Registration Statement on Form S-1, Commission File No. 33-96048).
|
|
|
4.3
|
*
|
Stock Purchase Agreement By and Between Pharmacyclics, Inc. and Applera Corporation dated April 7, 2006 (incorporated by reference to Exhibit 4.3 to the Quarterly Report on Form 10-Q for the quarter ended March 31, 2006).
|
|
4.5
|
Form of Stock Purchase Agreement related to the February 19, 2009 sale of approximately 1.5 million shares of the Company’s common stock to certain foreign and U.S. individuals and entities of which some are shareholders of Pacific Biopharma Group, Ltd. (incorporated by reference to Exhibit 4.1 to the Quarterly Report on Form 10-Q for the quarter ended March 31, 2009).
|
|
|
10.6
|
*
|
Patent License Agreement entered into between the Company and The University of Texas, Austin entered into on or about July 1, 1991 (incorporated by reference to Exhibit 10.8 to the Company's Registration Statement on Form S-1, Commission File No. 33-96048).
|
|
10.9
|
Lease Agreement entered into between the Company and New England Mutual Life Insurance Company dated as of June 17, 1993, as amended on July 22, 1993, and as further amended on March 1, 1994 (incorporated by reference to Exhibit 10.11 to the Company's Registration Statement on Form S-1, Commission File No. 33-96048).
|
|
|
10.13
|
+
|
The Company's 1995 Stock Option Plan (incorporated by reference to Exhibit 99.1 to the Company's Registration Statement on Form S-8, Commission File No. 333-52881).
|
|
10.14
|
+
|
The Company's 1995 Non-Employee Directors' Stock Option Plan (incorporated by reference to Exhibit 99.7 to the Company's Registration Statement on Form S-8, Commission File No. 33-98514).
|
|
10.15
|
+
|
The Company's Employee Stock Purchase Plan as amended on October 9, 2008 (incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on December 16, 2008).
|
|
10.16
|
+
|
Employment Agreement entered into between the Company and Richard A. Miller, M.D. dated as of June 10, 1992 (incorporated by reference to Exhibit 10.19 to the Company's Registration Statement on Form S-1, Commission File No. 33-96048).
|
|
10.22
|
+
|
Form of Notice of Grant of Stock Option generally to be used under the 1995 Stock Option Plan (incorporated by reference to Exhibit 99.2 to the Company's Registration Statement on Form S-8, Commission File No. 33-98514).
|
|
10.23
|
+
|
Form of Stock Option Agreement (incorporated by reference to Exhibit 99.3 to the Company's Registration Statement on Form S-8, Commission File No. 333-52881).
|
|
10.25
|
+
|
Form of Addendum to Stock Option Agreement (Special Tax Election) (incorporated by reference to Exhibit 99.5 to the Company's Registration Statement on Form S-8, Commission File No. 33-98514).
|
|
10.26
|
+
|
Form of Addendum to Stock Option Agreement (Involuntary Termination following Change in Control) (incorporated by reference to Exhibit 99.6 to the Company's Registration Statement on Form S-8, Commission File No. 33-98514).
|
|
10.27
|
+
|
Form of Notice of Grant of Automatic Stock Option (Initial Grant) (incorporated by reference to Exhibit 99.8 to the Company's Registration Statement on Form S-8, Commission File No. 33-98514).
|
|
10.28
|
+
|
Form of Notice of Grant of Automatic Stock Option (Annual Grant) (incorporated by reference to Exhibit 99.9 to the Company's Registration Statement on Form S-8, Commission File No. 33-98514).
|
|
10.29
|
+
|
Form of Non-Employee Director Stock Option Agreement (incorporated by reference to Exhibit 99.10 to the Company's Registration Statement on Form S-8, Commission File No. 33-98514).
|
|
10.30
|
+
|
Form of Employee Stock Purchase Plan Enrollment/Change Form (incorporated by reference to Exhibit 99.12 to the Company's Registration Statement on Form S-8, Commission File No. 33-98514).
|
|
10.31
|
+
|
Form of Stock Purchase Agreement (incorporated by reference to Exhibit 99.13 to the Company's Registration Statement on Form S-8, Commission File No. 33-98514).
|
|
10.38
|
+
|
Employment Agreement, dated December 18, 1997, by and between the Company and Leiv Lea (incorporated by reference to Exhibit 10.38 to the Quarterly report on Form 10-Q for the quarter ended March 31, 1998).
|
|
10.49
|
Lease and Lease Termination Agreement dated June 14, 2000 by and between the Registrant and Metropolitan Life Insurance Company (incorporated by reference to Exhibit 10.49 to the Annual Report on Form 10-K for the year ended June 30, 2001).
|
|
|
10.50
|
First Amendment to New Lease dated April 10, 2001 by and between the Registrant and Metropolitan Life Insurance Company (incorporated by reference to Exhibit 10.50 to the Annual Report on Form 10-K for the year ended June 30, 2001).
|
|
|
10.51
|
Second Amendment to New Lease dated June 29, 2001 by and between the Registrant and Metropolitan Life Insurance Company (incorporated by reference to Exhibit 10.51 to the Annual Report on Form 10-K for the year ended June 30, 2001).
|
|
|
10.54
|
Third Amendment to New Lease dated February 5, 2003 by and between the Registrant and Metropolitan Life Insurance Company (incorporated by reference to Exhibit 10.1 to the Quarterly Report on Form 10-Q for the quarter ended March 31, 2003).
|
|
|
10.55
|
Form of Indemnification Agreement between the Company and its directors and executive officers (incorporated by reference to Exhibit 10.55 to the Annual Report on Form 10-K for the year ended June 30, 2004).
|
|
|
10.56
|
+
|
The Company's 2004 Equity Incentive Award Plan (the "2004 Plan") as amended on October 9, 2008 (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on December 16, 2008).
|
|
10.57
|
+
|
Form of Option Agreement for the 2004 Plan (incorporated by reference from Exhibit 99.1 to the Company's Current Report on Form 8-K filed with the Securities and Exchange Commission on December 22, 2004).
|
|
10.58
|
+
|
Form of Non-employee Director Option Agreement for the 2004 Plan (incorporated by reference from Exhibit 99.2 to the Company's Current Report on Form 8-K filed with the Securities Exchange Commission on December 22, 2004).
|
|
10.59
|
+
|
Form of Amendment to Form of Notice of Grant of Stock Option used under the Company's 1995 Stock Option Plan (the "1995 Plan") (incorporated by reference to Exhibit 10.5 to the quarterly Report on Form 10-Q for the quarter ended December 31, 2004).
|
|
10.60
|
+
|
Form of Non-Employee Directors Stock Option Election Option Agreement used under the Company's 1995 Plan (incorporated by reference to Exhibit 10.6 to the Quarterly Report on Form 10-Q for the quarter ended December 31, 2004).
|
|
10.61
|
First Amendment To Patent License Agreement entered into on or about July 1, 1991 by and between the Company and the University of Texas System, Austin (incorporated by reference to Exhibit 10.1 to the Quarterly Report on Form 10-Q for the quarter ended March 31, 2005).
|
|
|
10.64
|
*
|
Assignment Agreement By and Between Pharmacyclics, Inc. and Applera Corporation dated April 7, 2006 (incorporated by reference to Exhibit 10.64 to the Quarterly Report on Form 10-Q for the quarter ended March 31, 2006).
|
|
10.65
|
Fourth Amendment to New Lease dated August 14, 2006 by and between the Company and Metropolitan Life Insurance Company (incorporated by reference to Exhibit 10.5 to the Company's Current Report on Form 8-K filed with the Securities and Exchange Commission on August 17, 2006).
|
|
|
10.67
|
Fifth Amendment to New Lease dated July 11, 2008 by and between the Registrant and Metropolitan Life Insurance Company (incorporated by reference to Exhibit 10.6 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on July 15, 2008).
|
|
|
10.68
|
*
|
Amendment No. 1 to Assignment Agreement by and between Pharmacyclics, Inc. and Applera Corporation dated May 12, 2008 (incorporated by reference to Exhibit 10.68 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2008).
|
|
10.69
|
+
|
Form of Severance Agreement between the Company and certain executive officers (incorporated by reference to Exhibit 10.69 to the Company’s Annual Report on Form 10-K for the year ended June 30,2008).
|
|
10.71
|
+
|
Separation Agreement effective as of September 10, 2008 by and between Richard A. Miller, M.D. and Pharmacyclics, Inc. (incorporated by reference to Exhibit 10.1 to the Quarterly Report on Form 10-Q for the quarter ended September 30, 2008).
|
|
10.72
|
+
|
Separation Agreement effective as of September 10, 2008 by and between Leiv Lea and Pharmacyclics, Inc. (incorporated by reference to Exhibit 10.2 to the Quarterly Report on Form 10-Q for the quarter ended September 30, 2008).
|
|
10.73
|
+
|
Form of Restricted Stock Award Agreement for the 2004 Plan (incorporated by reference to Exhibit 10.3 to the Quarterly Report on Form 10-Q for the quarter ended September 30, 2008).
|
|
10.74
|
+
|
Offer letter dated April 13, 2006 by and between the Company and David J. Loury, Ph.D. (incorporated by reference to Exhibit 10.2 of the Registrant’s Current Report on Form 8-K filed with the Securities and Exchange Commission on November 13, 2008).
|
|
10.75
|
+
|
Severance benefit agreement dated November 5, 2008 by and between the Company and David J. Loury, Ph.D. (incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Securities and Exchange Commission on November 13, 2008).
|
|
10.76
|
Loan Agreement entered into between the Company and Robert W. Duggan & Associates dated as of December 30, 2008 (incorporated by reference to Exhibit 10.1 to the Quarterly Report on Form 10-Q for the quarter ended December 31, 2008).
|
|
|
10.77
|
*
|
Amendment No. 2 to Assignment Agreement by and between Pharmacyclics, Inc. and Celera Corporation dated March 2, 2009 (incorporated by reference to Exhibit 10.1 to the Quarterly Report on Form 10-Q for the quarter ended March 31, 2009).
|
|
10.78
|
*
|
Amendment No. 3 to Assignment Agreement by and between Pharmacyclics, Inc. and Celera Corporation dated March 30, 2009 (incorporated by reference to Exhibit 10.2 to the Quarterly Report on Form 10-Q for the quarter ended March 31, 2009).
|
|
10.79
|
Amendment No. 1 to Loan Agreement entered into between the Company and Robert W. Duggan & Associates dated as of March 30, 2009 (incorporated by reference to Exhibit 10.3 to the Quarterly Report on Form 10-Q for the quarter ended March 31,2009).
|
|
10.80
|
+
|
Offer letter dated February 2, 2009 by and between the Company and Glenn C. Rice, Ph.D. (incorporated by reference to Exhibit 10.4 to the Quarterly Report on Form 10-Q for the quarter ended March 31, 2009).
|
|
10.81
|
+
|
Offer letter dated February 5, 2009 by and between the Company and Rainer M. Erdtmann (incorporated by reference to Exhibit 10.5 to the Quarterly Report on Form 10-Q for the quarter ended March 31, 2009).
|
|
10.82
|
+
|
Offer letter dated February 26, 2009 by and between the Company and Ahmed Hamdy, M.B.B.Ch. (incorporated by reference to Exhibit 10.6 to the Quarterly Report on Form 10-Q for the quarter ended March 31, 2009).
|
|
10.83
|
*
|
Collaboration Agreement by and between Pharmacyclics, Inc. and Les Laboratoires Servier and Institut de Recherches Internationales Servier dated April 9, 2009 (incorporated by reference to Exhibit 10.83 to the Annual Report on Form 10-K for the year ended June 30, 2009).
|
|
10.84
|
Amendment No. 2 to Loan Agreement entered into between the Company and Robert W. Duggan and Blazon Corporation Profit Sharing Plan dated as of June 17, 2009 (incorporated by reference to Exhibit 10.84 to the Annual Report on Form 10-K for the year ended June 30, 2009).
|
|
|
10.85
|
*
|
Drug Supply Agreement by and between Pharmacyclics, Inc. and Les Laboratoires Servier dated December 18, 2009 (incorporated by reference to Exhibit 10.1 to the Quarterly Report on Form 10-Q for the quarter ended December 31, 2009).
|
|
10.86
|
Agreement and Release with Glenn Rice dated October 28, 2009 (incorporated by reference to Exhibit 10.2 to the Quarterly Report on Form 10-Q for the quarter ended December 31, 2009).
|
|
|
10.87
|
Form of Stock Purchase Agreement by and between Pharmacyclics, Inc. and various institutional investors dated June 16, 2010 (incorporated by reference to the Current Report on Form 8-K filed with the Securities and Exchange Commission on June 17, 2010).
|
|
|
23.1
|
Consent of Independent Registered Public Accounting Firm.
|
|
|
24.1
|
Power of Attorney (see page 72).
|
|
|
31.1
|
Section 302 Certification of Chief Executive Officer.
|
|
|
31.2
|
Section 302 Certification of Chief Financial Officer.
|
|
|
32.1
|
Section 906 Certification of Chief Executive Officer and Chief Financial Officer.
|
________________
* Confidential treatment has been granted as to certain portions of this agreement.
+ Indicates a management contract or compensatory plan or arrangement.
109