Attached files
| file | filename |
|---|---|
| EX-31.4 - SECTION 302 CFO CERTIFICATION - ZYMOGENETICS INC | dex314.htm |
| EX-31.3 - SECTION 302 CEO CERTIFICATION - ZYMOGENETICS INC | dex313.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 10-K/A
(Amendment No. 1)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15 (D) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2009
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(D) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File Number 0-33489
ZYMOGENETICS, INC.
(exact name of registrant as specified in its charter)
| Washington | 91-1144498 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) | |
|
1201 Eastlake Avenue East, Seattle, WA 98102 (Address of principal executive offices) | ||
Registrant’s telephone number, including area code (206) 442-6600
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class |
Name of Each Exchange on Which Registered | |
| Common Stock, no par value | The NASDAQ Stock Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark whether the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ¨. No x.
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes ¨. No x.
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months, and (2) has been subject to such filing requirements for the past 90 days.
Yes x. No ¨.
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes ¨ No ¨.
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendments to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ |
Accelerated filer x | Non-accelerated filer ¨ |
Smaller reporting company ¨ | |||||||
| (Do not check if a smaller reporting company) | ||||||||||
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Act).
Yes ¨ No x.
The aggregate market value of the common equity held by non-affiliates computed by reference to the price at which the common equity was last sold as of June 30, 2009 was: $173,248,521.
Common stock outstanding at February 19, 2010: 85,477,565 shares.
DOCUMENTS INCORPORATED BY REFERENCE
| (1) | Portions of the Company’s definitive Proxy Statement for the annual meeting of shareholders to be held on June 17, 2010 are incorporated by reference in Part III. |
Table of Contents
EXPLANATORY NOTE
This Amendment No. 1 on Form 10-K/A supplements our Annual Report on Form 10-K for the year ended December 31, 2009, which we filed with the Securities and Exchange Commission on March 1, 2010. We are filing this amendment to amend and restate Item 1 of Part I, Item 7A of Part II and Item 11 of Part III in response to comments we received from the staff of the Securities and Exchange Commission.
In addition, we have filed the following exhibits herewith:
| • | 31.3 Certifications of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
| • | 31.4 Certifications of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
Except as described above, no other amendments are being made to our Annual Report on Form 10-K filed on March 1, 2010.
Table of Contents
ZYMOGENETICS, INC.
AMENDMENT NO. 1 TO
ANNUAL REPORT ON FORM 10-K
For the Year Ended December 31, 2009
| Page No. | ||||
| PART I | ||||
| Item 1. |
2 | |||
| PART II | ||||
| Item 7A. |
25 | |||
| PART III | ||||
| Item 11. |
26 | |||
| PART IV | ||||
| 48 | ||||
1
Table of Contents
This Annual Report on Form 10-K contains, in addition to historical information, forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995 that involve risks and uncertainties. This Act provides a “safe harbor” for forward-looking statements to encourage companies to provide prospective information about themselves. All statements other than statements of historical fact, including statements regarding company and industry prospects and future results of operations, financial position and cash flows, made in this Annual Report on Form 10-K are forward-looking. We use words such as “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “future,” “intend,” “may,” “potential,” “seek,” “should,” “target” and similar expressions, including negatives, to identify forward-looking statements. Forward-looking statements reflect management’s current expectations, plans or projections and are inherently uncertain. Our actual results could differ significantly from the results discussed in the forward-looking statements. Readers are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date hereof. Factors that could cause or contribute to such differences include those discussed in “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” as well as those discussed elsewhere in this Annual Report on Form 10-K. We undertake no obligation to publicly release any revisions to these forward-looking statements that may be made to reflect events or circumstances after the date hereof or to reflect the occurrence of unanticipated events. Readers are urged, however, to review the information provided in reports that we file from time to time with the Securities and Exchange Commission or otherwise make public.
Overview
We are a biopharmaceutical company focused on the development and commercialization of therapeutic proteins for the treatment of human diseases. In 2009, through a series of strategic initiatives and workforce and cost reductions, we restructured our organization and are now focused on developing and commercializing a limited number of products, which we believe have substantial therapeutic and commercial potential and in which we retain a significant ownership position. Our current portfolio includes one commercial product, RECOTHROM® Thrombin, topical (Recombinant), and three immunology product candidates.
Commercial Product
RECOTHROM. We have developed and are marketing RECOTHROM in the United States for use as a topical hemostat to control moderate bleeding during surgical procedures. Our product, which is a recombinant version of the human blood-clotting protein thrombin, provides an effective and safe alternative to other thrombin products marketed in the United States in forms derived from bovine (cattle) plasma or human plasma. We hold worldwide rights to RECOTHROM, except for Canada, where Bayer Schering Pharma AG is responsible for commercializing the product.
Immunology Product Candidates
PEG-IFN-lambda. Pegylated Interferon-lambda (PEG-IFN-lambda) is being studied in collaboration with Bristol-Myers Squibb Company in a Phase 2 clinical trial for treatment of hepatitis C virus infection. In November 2009, we presented final results from an open-label Phase 1b study in patients with hepatitis C. We hold co-development rights to PEG-IFN-lambda in the United States and Europe and the option to co-promote and share profits on product sales in the United States. Bristol-Myers Squibb is responsible for commercializing the product outside the United States, for which we will receive milestones and royalties on sales.
IL-21. Interleukin-21 (IL-21) is currently being tested in an open-label Phase 2 clinical trial as a potential immunotherapy treatment for metastatic melanoma. In May 2009, we presented interim results from this study, with final results expected to be available in 2010. In addition, we presented results from an open-label Phase 2 study in renal cell carcinoma in May 2009. We hold worldwide rights to IL-21.
2
Table of Contents
IL-31 mAb. Interleukin-31 monoclonal antibody (IL-31 mAb) is currently in preclinical development as a potential treatment for atopic dermatitis. We hold worldwide rights to IL-31 mAb and expect to begin clinical testing for this candidate in 2011.
Our goal is to substantially increase the value of our company by advancing our products and candidates to key value inflection points associated with the achievement of development and/or commercial milestones. We intend to continue to build the market for RECOTHROM in the United States, which we expect will provide net cash flows that we may use to fund the further development of our immunology product candidates. Where appropriate, we intend to enter into strategic collaborations for the commercialization of our immunology product candidates, which we believe will enable us to maximize long-term value of these assets, while leveraging our internal resources and accessing complementary technologies, infrastructure and expertise. For example, we established the PEG-IFN-lambda collaboration with Bristol-Myers Squibb in January 2009, which provided substantial near-term cash and long-term commercial value retention.
We have out-licensed several product candidates previously identified through our discovery research efforts to third parties, including atacicept, FGF-18, IL-22RA mAb and IL-17RC soluble receptor to Merck Serono SA and IL-20 mAb and IL-21 mAb to Novo Nordisk A/S. These candidates are either outside our core area of interest or require levels of capital investment that we could not justify considering our available financial resources. We are not actively involved in the development of these product candidates. We are, however, eligible to receive milestone payments and royalties and, thus, we consider these product candidates to be important assets that could result in the generation of substantial value over the long term.
We intend to maintain strong patent protection for our asset portfolio. We file detailed patent applications with respect to our discoveries covering multiple patentable inventions, typically including composition of matter, method of making and method of use claims. We have issued patents or pending applications covering RECOTHROM and all of our internal product candidates. In total, we have more than 340 unexpired issued or allowed U.S. patents and over 180 U.S. patent applications pending. Outside of the United States, we have more than 790 issued or allowed foreign patents.
We were incorporated in the state of Washington in 1981. From 1988 to 2000, we were a wholly owned subsidiary of Novo Nordisk, one of the world’s largest producers of therapeutic proteins. In November 2000, as part of a restructuring by Novo Nordisk, we became an independent company. In February 2002, we completed our initial public offering. In addition to RECOTHROM, we have contributed to the discovery or development of seven recombinant protein products currently on the market. Our principal executive offices are located at 1201 Eastlake Avenue East, Seattle, Washington, 98102. Our telephone number is (206) 442-6600. Our website is www.zymogenetics.com. At the Investor Relations section of this website, we make available free of charge our annual report on Form 10-K, our annual proxy statement, our quarterly reports on Form 10-Q, any current reports on Form 8-K, and any amendments to these reports, as soon as reasonably practicable after we electronically file them with, or furnish them to, the Securities and Exchange Commission (SEC). The information on our website is not a part of, and is not incorporated into, this annual report on Form 10-K. In addition to our website, the SEC maintains an Internet site at www.sec.gov that contains reports, proxy and information statements, and other information regarding us and other issuers that file electronically with the SEC.
Commercial Product and Product Pipeline
Our current focus is the continued commercialization of our first product, RECOTHROM, and the development of three product candidates: PEG-IFN-lambda in partnership with Bristol-Myers Squibb, IL-21, and IL-31 mAb. We have out-licensed several product candidates that are outside of our core areas of interest or for which we could not justify the required capital investment. We are eligible to receive milestone payments and royalties related to these assets. The following table summarizes our commercial product and product candidates that are being internally developed or co-developed, as well as out-licensed product candidates.
3
Table of Contents
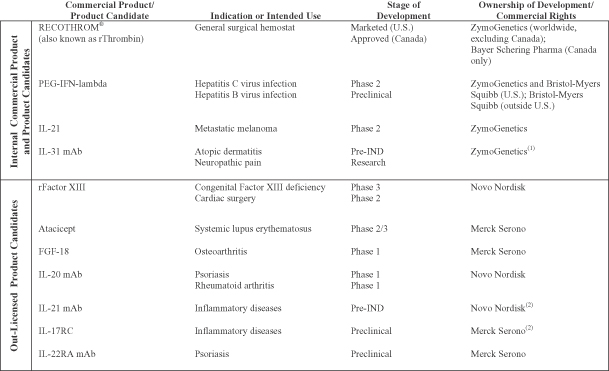
| (1) | Subject to certain opt-in rights granted to Merck Serono. |
| (2) | Subject to certain opt-in rights granted to ZymoGenetics. |
In the preceding table, “Research” refers to the stage in which we analyze the biology and therapeutic potential of proteins using a variety of laboratory methods. “Preclinical” refers to the stage in which safety, pharmacology and proof of efficacy in non-human animal models of specific human disease are evaluated. “Pre-IND” refers to the stage in which investigational new drug enabling preclinical toxicology studies are performed and materials in support of the investigational new drug (IND) and clinical studies are manufactured. “Phase 1” refers to clinical trials designed primarily to determine safety and pharmacokinetics in healthy volunteers or a limited patient population. “Phase 1b” refers to clinical trials designed to demonstrate biomarker or clinical outcome that could be considered for proof of concept in a limited patient population. “Phase 2” refers to clinical trials designed to evaluate preliminary efficacy, further characterize safety and optimize dosing in a limited patient population. “Phase 2/3” refers to large-scale clinical trials designed to establish safety and confirm efficacy in comparison to standard therapies or placebo in a patient population large enough to generate statistically significant results. “Phase 3” refers to clinical trials in a broad patient population with the intention of generating statistical evidence of efficacy and safety to support product approval.
Commercial Product
RECOTHROM®
RECOTHROM Thrombin, topical (Recombinant) was approved by the U.S. Food and Drug Administration (FDA) on January 17, 2008 for use as a general aid to control diffuse (non-arterial) bleeding during surgery. Net RECOTHROM sales in the United States totaled $8.8 million and $28.2 million during the years ended December 31, 2008 and December 31, 2009, respectively. RECOTHROM is available in a 5,000 international unit (IU) vial, a 20,000 IU vial and a 20,000 IU vial co-packaged with a spray applicator kit. Three wholesalers, AmerisourceBergen Corporation, Cardinal Health, Inc. and McKesson Corporation, accounted for approximately 90% of U.S. sales in 2009. If any of these wholesalers ceased distributing RECOTHROM, other wholesalers already distributing RECOTHROM would likely absorb the incremental sales volume with minimal interruption to the business or we would sell directly to hospitals.
4
Table of Contents
Thrombin is a specific blood-clotting enzyme that converts fibrinogen to fibrin, the primary protein contained in newly formed blood clots. Thrombin also promotes clot formation by activating Factor XIII, which cross-links the fibrin molecules and strengthens the newly forming clot. Topical thrombin is widely used to stop diffuse (non-arterial) bleeding occurring during surgical procedures, when control of bleeding by standard surgical techniques, such as direct pressure, ligation, or cautery, is ineffective or impractical. Minimizing bleeding during surgical procedures is important to maintain visibility in the operating field, limit the use of transfused blood products and reduce peri- and post-operative complications. Thrombin is generally sold as a lyophilized powder stored at room temperature, which is dissolved in saline and absorbed onto a surgical sponge, embedded onto a hemostatic pad or sprayed directly for topical application to wounds. Currently, there are three types of topical thrombin available in the United States: bovine (cattle) plasma-derived (Thrombin-JMI® marketed by King Pharmaceuticals), human-plasma derived (Evithrom® marketed by Ethicon, Inc. and the thrombin contained in GELFOAM Plus hemostasis kit marketed by Baxter Healthcare Corporation) and recombinant human thrombin (RECOTHROM marketed by ZymoGenetics). The thrombin market, based on the combined sales to hospitals of RECOTHROM, Thrombin-JMI, Evithrom and GELFOAM Plus, was estimated at $228 million in 2009, with full-year RECOTHROM sales representing approximately 13% market share.
We believe that there are several important advantages to recombinant human thrombin. Some patients may experience allergic reactions to plasma-derived products. Patients could also develop antibodies to bovine plasma-derived thrombin or to bovine Factor V or other protein impurities in the bovine plasma-derived product. In some cases, these antibodies can cross-react with analogous human proteins, creating a bleeding condition that can be difficult to manage and which has been fatal in patients who develop the most severe cases. Use of bovine plasma-derived thrombin in patients with pre-existing antibodies to bovine clotting factors may increase these risks and is, therefore, contraindicated. The package insert for bovine plasma-derived thrombin contains a black box warning, the most serious form of warning the FDA can require for approved products, describing these potential risks. In addition, all human plasma-derived products carry an FDA-mandated warning addressing a potential risk of transmitting infectious and other diseases, including HIV, hepatitis, parvovirus, Creutzfeldt-Jakob disease (CJD) and variant CJD. RECOTHROM, which is human thrombin produced using recombinant DNA technology, is inherently free from these potential risks and its package insert does not have a black box warning or any other warnings associated with the risk of transmitting blood-borne pathogens or infectious diseases.
In August 2009, we submitted a Citizen Petition to the FDA requesting that the FDA remove Thrombin-JMI Thrombin, topical (bovine origin) from the market in the interest of patient safety. The Citizen Petition was prompted by reports of serious or fatal bleeding-related adverse events in surgical patients exposed to bovine thrombin. In January 2010, we received an interim response from the FDA indicating that additional time was needed for the agency to reach a decision and provide a final response to the Citizen Petition. We can provide no assurance that the petition will be granted or that Thrombin-JMI will be removed from the market.
Prior to launch, we established the supply chain for RECOTHROM, from sourcing of critical raw materials and manufacturing to distribution to end customers, and built commercial inventories to satisfy expected market demand and provide what we believe are sufficient levels of safety stock. We have developed a patent-protected two-step process for the manufacture of recombinant thrombin. First, recombinant human prethrombin-1 is produced in mammalian cells. Then, using an enzyme activation step, prethrombin-1 is converted to recombinant human thrombin. The commercial-scale manufacturing process was developed in collaboration with Abbott Laboratories, our commercial manufacturer of the RECOTHROM bulk drug substance.
In December 2009, we announced a restructuring of our U.S. co-promotion agreement with Bayer HealthCare LLC and our license and collaboration agreement with Bayer Schering Pharma AG. Effective December 31, 2009 we ended the co-promotion with Bayer HealthCare such that Bayer HealthCare will no longer participate in the sales and marketing of RECOTHROM in the United States. We are currently in the process of increasing the size of our sales organization and intend to have the additional sales personnel fully trained and in the field by the end of first quarter of 2010. Outside the United States, we regained all rights to RECOTHROM, except for Canada, where the product was approved in December 2009. Bayer Schering Pharma
5
Table of Contents
will market and sell RECOTHROM in Canada and pay us royalties on net sales. Also, in December 2009, Bayer Schering Pharma withdrew the Marketing Authorization Application (MAA) to the European Medicines Agency (EMEA) for approval to market RECOTHROM in Europe. The application was withdrawn in response to indications from the European regulatory authorities that approval would require additional clinical trial data.
Clinical Trials. In September 2006, we completed a pivotal Phase 3 clinical study designed to evaluate the comparative efficacy of RECOTHROM and bovine thrombin, both administered with an absorbable gelatin sponge. The randomized, double-blind study was conducted at 34 sites in the United States and enrolled 411 patients in four surgical settings: spinal surgery, liver resection, peripheral artery bypass and arteriovenous graft construction. Both the primary and secondary endpoints of the study were met. RECOTHROM was shown to have comparable efficacy to bovine thrombin, as measured by the overall percentage of patients achieving hemostasis within 10 minutes. Both treatments were well tolerated and exhibited similar adverse event profiles. RECOTHROM also demonstrated a superior immunogenicity profile to bovine thrombin, based on a significantly lower incidence of post-treatment anti-product antibody development. The study was not powered to detect differences in clinical outcomes based on differences in antibody formation.
In 2007, we completed an open-label, non-comparative Phase 2 clinical study designed to evaluate safety and immunogenicity of RECOTHROM administered using a spray device in patients with burns undergoing autologous skin grafting. The study results demonstrated a safety and immunogenicity profile similar to that observed in the pivotal Phase 3 study.
In 2008, we completed an open-label Phase 3b study designed to evaluate the safety and immunogenicity of RECOTHROM in subjects at increased risk for having anti-bovine thrombin product antibodies as a result of prior surgical history. The study enrolled 205 subjects, 16% of whom had pre-existing antibodies to bovine thrombin. The study results demonstrated that no patients developed antibodies against RECOTHROM. Following a 29-day period after topical RECOTHROM application during a single spinal or vascular surgical procedure, the immunogenicity profile of RECOTHROM did not differ among subjects with or without pre-existing antibodies to bovine thrombin. RECOTHROM was well tolerated and observed adverse events were consistent with those commonly seen in post-surgical settings.
Adverse events observed in clinical trials with RECOTHROM were consistent with those commonly observed in surgical patients. In pooled results from completed clinical trials involving 583 patients exposed to RECOTHROM, the most common adverse events were incision site pain, procedural pain, and nausea. Of the 552 patients for whom complete immunogenicity observations were available, only 5 patients (0.9%) developed specific anti-product antibodies, and none of these antibodies were found to neutralize native human thrombin.
As part of our post-marketing approval commitments, we are conducting an open-label Phase 4 clinical study to evaluate the safety and immunogenicity of re-exposure to RECOTHROM in approximately 30 subjects previously treated with RECOTHROM. We expect to complete patient enrollment in the re-exposure Phase 4 study in 2011. In addition, pursuant to the Pediatric Research Equity Act, we are conducting an open-label Phase 4 clinical study to evaluate the safety of RECOTHROM as an aid to hemostasis in a pediatric population. We completed patient enrollment in the pediatric study in December 2009 and expect to report study results in 2010.
In November 2008, we borrowed $25 million under a $100 million financing arrangement with Deerfield Management. The draw entitles Deerfield to a royalty equal to 2% of RECOTHROM net sales in the United States until repayment, which is due by June 27, 2014. No further draws on the remaining $75 million available for borrowing under the facility were taken prior to expiration of the facility in February 2010.
We own issued U.S. and foreign patents directed to a genetically engineered thrombin precursor termed “prethrombin-1”, methods of producing recombinant human thrombin from prethrombin-1, formulations, and methods of activation and therapeutic use of the protein.
6
Table of Contents
Product Candidates
PEG-IFN-lambda
Interferon-lambda 1 (IFN-lambda 1, also known as IL-29) is a type III interferon that belongs to the 4-helical-bundle cytokine family. Native IFN-lambda 1 is generated in response to a viral infection and exhibits broad cellular anti-viral activity similar to type I interferons, such as interferon-alpha. However, IFN-lambda 1 signals through a receptor that is distinct from the type I interferon receptor and that has a more selective expression pattern compared to the widely expressed receptor for type I interferons. The difference in the receptor tissue distribution suggests that IFN-lambda 1 may serve as an alternative to interferon-alpha based therapy for viral infection by providing comparable antiviral activity with potentially fewer side effects.
In vitro studies have shown that IFN-lambda 1 has antiviral activity against human hepatitis C virus (HCV) in HCV preclinical models. Additionally, we have demonstrated that IFN-lambda 1 induces antiviral gene expression similar to interferon-alpha in primary human hepatocytes. Combined with the significant expression of the receptor for IFN-lambda 1 in liver samples from HCV positive individuals, these data provided the rationale for selecting HCV infection as our first clinical indication. Recent clinical studies have also demonstrated the importance of type III interferons in controlling HCV infection. Correlative data from these studies showed that patients with certain genotypes who had poor response to the current standard of care regimen of interferon-alpha plus ribavirin also produced lesser amounts of endogenous type III interferon, thus illustrating the importance of type III interferons in controlling the viral replication pathway.
Chronic infection with HCV is a leading cause of cirrhosis, liver failure, and hepatocellular carcinoma worldwide. It is estimated that there are over 170 million people worldwide infected with hepatitis C virus. In the United States, an estimated 4.0 million people have been exposed to HCV, and approximately 3.2 million have chronic HCV infection. HCV is associated with an estimated 8,000-10,000 deaths per year and is the main indication for liver transplantation in the United States. The current standard of care for chronic HCV infection involves treatment with the combination of pegylated interferon-alpha and ribavirin. Standard of care therapy has been associated with a number of significant side effects, including flu-like symptoms, anorexia, depression, hemolytic anemia and myelosuppression, which continue to be treatment-limiting factors. With a response rate to the current standard treatment for the most common form of HCV (genotype 1) in the United States of approximately 40%, there remains a need for better tolerated and more effective therapy for HCV infection. Our product candidate, PEG-IFN-lambda, is a pegylated version of the IFN-lambda 1 protein, produced using recombinant DNA technology. Pegylation extends the in vivo half-life of the protein, allowing for convenient dose scheduling, such as once per week.
In January 2009, we entered into an exclusive global collaboration with Bristol-Myers Squibb Company for PEG-IFN-lambda. Under the terms of the collaboration, we will co-develop PEG-IFN-lambda in the United States and Europe with Bristol-Myers Squibb and share development costs. We will have the option to co-promote PEG-IFN-lambda and to share profits on product sales in the United States, while receiving royalties on sales in the rest of the world. We may opt out of the co-development, co-promotion and profit sharing arrangement in the United States, in which case we would no longer be obligated to co-fund development or commercialization activities, and we would receive royalties on worldwide product sales.
In 2007, we completed a randomized, placebo-controlled, dose-escalation Phase 1a clinical trial in healthy volunteers to evaluate the safety, tolerability and pharmacokinetics of a single dose of PEG-IFN-lambda administered subcutaneously. The study enrolled 20 subjects who were randomized to four dose levels of PEG-IFN-lambda, ranging from 0.5 to 7.5 mcg/kg, or placebo. The results from this study demonstrated that administration of a single dose of PEG-IFN-lambda was associated with dose-related pharmacokinetic and pharmacodynamic effects, with evidence of biological activity, including up-regulation of interferon response markers, being observed at dose levels of 1.5 mcg/kg and above. No fever or hematologic effects, which are typically seen with interferon-alpha, were observed at all tested dose levels in this study.
7
Table of Contents
In 2009, we completed a Phase 1b study to evaluate the safety and antiviral effect of repeat dosing of PEG-IFN-lambda administered subcutaneously for four weeks as a single agent or in combination with ribavirin in genotype 1 HCV patients. Final study results were presented in November 2009 at the American Association for the Study of the Liver Diseases (AASLD) annual meeting. A total of 56 patients were enrolled in this three-part study. In Part 1, a total of 24 relapsed HCV patients were enrolled in 4 cohorts, consisting of 6 patients each. PEG-IFN-lambda was administered as a single agent weekly or bi-weekly at dose levels of 1.5 or 3.0 mcg/kg. In Part 2, a total of 25 relapsed HCV patients were treated in 4 cohorts, consisting of 6 or 7 patients each. PEG-IFN-lambda was administered weekly at dose levels of 0.5, 0.75, 1.5, or 2.25 mcg/kg in combination with ribavirin. In Part 3, a total of 7 previously untreated HCV patients were enrolled in a single cohort receiving 1.5 mcg/kg of PEG-IFN-lambda in combination with ribavirin. The results from this study demonstrated anti-viral activity of PEG-IFN-lambda at all dose levels tested in both relapsed and previously untreated HCV patients. A majority of patients across all treatment arms achieved a greater than 2 log reduction in HCV RNA measured by a test that identifies the presence of hepatitis C virus in patient’s blood. Minimal constitutional symptoms or hematologic effects were observed with PEG-IFN-lambda given as a single agent or in combination with ribavirin. The majority of adverse events and laboratory changes were mild or moderate. Dose-limiting elevations in liver enzymes, with or without an increase in bilirubin, were dose-dependent and reversible.
In October 2009, in collaboration with our partner Bristol-Myers Squibb, we initiated a two-part, randomized, controlled Phase 2 study of PEG-IFN-lambda administered subcutaneously for up to 48 weeks in combination with ribavirin in treatment-naïve patients with chronic genotype 1, 2, 3, or 4 HCV infection (the “EMERGE” study). The EMERGE study will evaluate the safety, tolerability and antiviral efficacy of PEG-IFN-lambda and ribavirin compared to PEGASYS® (PEG-IFN alfa-2a) and ribavirin. The primary efficacy endpoint of the study is the proportion of patients who achieve complete early virologic response (cEVR), defined as undetectable levels of HCV RNA after 12 weeks of treatment. A secondary efficacy endpoint is the proportion of patients who achieve sustained virologic response (SVR), defined as undetectable levels of HCV RNA 24 weeks after completed treatment. Part A of the EMERGE study is an open-label study that will explore four fixed doses of PEG-IFN-lambda compared to standard dose of PEGASYS in approximately 55 patients. Up to four doses of PEG-IFN-lambda will be selected from Part A for testing in the second part of the study. Part B of the study will be conducted as a randomized, blinded study and is designed to enroll up to 600 patients. We began enrolling patients in Part A of the study in October 2009 and expect to begin Part B in 2010.
We own issued patents for IFN-lambda 1 polypeptides, polynucleotides, expression vectors, cells, methods of treating a hepatitis infection, and a method of producing IFN-lambda 1, as well as patents on all known related molecules in the type III interferon family. We have filed patent applications for IFN-lambda 1 polypeptides, IFN-lambda 1 fusion proteins, antibodies, methods of expressing and purifying IFN-lambda 1, methods of using IFN-lambda 1 alone and in combination with other therapeutic agents to treat various viral diseases, cancers and autoimmune disorders. We will continue to file patent applications as new inventions are made. As part of our agreement with Bristol-Myers Squibb, we have assigned to Bristol-Myers Squibb a one-half ownership interest in each core patent relating to PEG-IFN-lambda filed outside the United States and a security interest in each core patent relating to PEG-IFN-lambda filed in the United States.
IL-21
IL-21 is a cytokine that activates several types of immune cells thought to be critical in eliminating cancerous or virally infected cells from the body. More specifically, IL-21 enhances the activity of mature natural killer (NK) cells; it has multiple effects on cytotoxic T lymphocyte cells (CTL), including increased activation and proliferation, extended longevity in circulation and improved ability to kill cancerous cells; and it enhances B-cell antibody production.
Preclinical studies have indicated that our recombinant version of IL-21 is an effective therapy in a number of animal models of cancer. In an animal model of metastatic melanoma, IL-21 was associated with significant
8
Table of Contents
anti-tumor activity. Animals in this model develop aggressive metastases to the lung, which can be readily measured. Treatment with IL-21 led to a significant reduction in the number of lung metastases relative to controls. IL-21 also was found to have potent inhibitory activity in other animal models of cancer. These models demonstrated that the in vivo effects of IL-21 were mediated through the activation of CTL and NK cells, which contribute to rejection of the tumors in the animal models. Moreover, this led to establishment of immunological memory, which protected animals from re-challenge with the parent tumor.
We believe that IL-21 could represent a potentially better tolerated and more efficacious immunotherapeutic agent than other cancer immunotherapies, such as interleukin-2 (IL-2) and interferon-alpha. In clinical practice, IL-2 produces durable responses in a very small percentage of patients with metastatic melanoma and metastatic renal cell carcinoma. Accompanying this relatively low level of efficacy are significant toxicities, including vascular leak syndrome and the release of pro-inflammatory cytokines, which profoundly limit the utility of IL-2 in treating disease. These side effects can be so severe that many patients are either hospitalized or stop the therapy before completion of the treatment program. Although somewhat better tolerated, interferon-alpha therapy is associated with significant chronic toxicities limiting its administration and produces a lower overall response rate with fewer complete responses compared to IL-2.
We own worldwide rights to our product candidate, recombinant human IL-21 protein. We had previously out-licensed rights to the IL-21 protein outside North America to Novo Nordisk and entered into a collaborative data sharing and cross-licensing agreement with them. In January 2009, subsequent to a strategic decision by Novo Nordisk to exit all of its oncology development programs, we reacquired rights to the IL-21 protein outside North America. Simultaneously, we and Novo Nordisk terminated a collaborative data sharing and cross-license agreement and a manufacturing agreement, under which Novo Nordisk was supplying clinical materials. As part of this termination, we acquired rights to all patent applications and data generated by Novo Nordisk as well as clinical product manufactured by Novo Nordisk. The reacquisition agreements did not require any upfront payment to Novo Nordisk. However, we will owe milestone payments and royalties to Novo Nordisk upon commercialization of IL-21 outside North America.
Metastatic Melanoma. We are pursuing metastatic melanoma as the lead indication for IL-21. There are an estimated 69,000 new cases of melanoma per year in the United States, with over 8,650 deaths per year attributed to this disease. Metastatic melanoma is essentially an incurable cancer with no established standard of care. Because of poor prognosis of overall survival for patients with advanced stages of metastatic melanoma, with a median of six to nine months, there is a significant unmet need for the development of therapies that can prolong overall survival. Several drugs have previously failed in late-stage clinical trials of metastatic melanoma. Most recently, Nexavar® (a product marketed by Bayer HealthCare AG and Onyx Pharmaceuticals, Inc.) failed to meet its endpoint of improved overall survival in a Phase 3 trial and a Phase 3 trial of elesclomol (Synta Pharmaceuticals Corp.) was suspended due to safety concerns. In October 2005, the FDA granted IL-21 orphan drug status for the treatment of melanoma patients with advanced or aggressive disease.
We are developing IL-21 as a single-agent treatment for metastatic melanoma. In 2007, we initiated an open-label Phase 2 clinical trial of IL-21 in previously untreated patients with metastatic or recurrent melanoma. The study, which is being conducted by the National Cancer Institute of Canada (NCIC), is designed to evaluate two dose levels of IL-21 at 30 and 50 mcg/kg. The interim results from 24 patients, presented at the World Congress on Melanoma annual meeting in May 2009, showed IL-21 to be biologically active, with 7 patients, or 29%, having a partial response and 8 patients, or 33%, having stable disease. Administered at a dose of 30 mcg/kg during three 5- day cycles, IL-21 was well tolerated. The most common adverse events were mild or moderate fatigue and rash. The 50 mcg/kg dose of IL-21 was poorly tolerated, with severe adverse events including neutropenia and skin rash. In August 2009, we completed study enrollment. A total of 30 patients received IL-21 at a dose of 30 mcg/kg and 10 patients received IL-21 at a dose of 50 mcg/kg. Final results from the study are expected to be available in 2010. In collaboration with the NCIC, we expect to initiate a larger randomized study versus DTIC (dacarbazine) in the first half of 2010. The study will evaluate safety and efficacy of IL-21 versus DTIC as first-line therapy in metastatic melanoma.
9
Table of Contents
Metastatic Renal Cell Carcinoma. In 2009, we completed an open-label Phase 2 clinical trial of IL-21 in combination with the tyrosine kinase inhibitor Nexavar in patients with advanced renal cell carcinoma. The study was designed to evaluate the safety, pharmacokinetics and anti-tumor activity of the combination therapy at the IL-21 maximum tolerated dose, previously established at 30 mcg/kg. The final study results were presented at the American Society of Clinical Oncology (ASCO) meeting in May 2009. The results indicated that the combination of IL-21 with Nexavar was well tolerated, with side effects that were manageable in an outpatient setting. The combination therapy was associated with anti-tumor activity both in terms of tumor response and duration of disease control, measured by progression-free survival (PFS). An independent data review completed for 33 patients showed an overall response rate of 21%, with 7 patients having a partial response. Median PFS was 5.7 months. No further investigation of IL-21 in renal cell carcinoma is planned at this time based on an evaluation of the competitive landscape and commercial opportunity.
B-cell Lymphoma. We have explored the use of IL-21 in combination with monoclonal antibodies, such as Rituxan® (a product marketed by Genentech, Inc. and Biogen Idec Inc.), an anti-CD20 antibody, that functions via antibody-dependent cellular cytotoxicity, a process enhanced by IL-21. In 2008, we completed an open-label Phase 1 clinical trial of IL-21 in combination with Rituxan in patients with relapsed low-grade B-cell lymphoma. The final study results, which were presented at the ASCO annual meeting in 2008, demonstrated that the combination of IL-21 at 100 mcg/kg with Rituxan was well tolerated and provided evidence of anti-tumor activity in this heavily pre-treated population, including one confirmed complete response, and three partial responses. No further investigation of IL-21 in B-cell lymphoma is planned.
We own issued patents for IL-21 polypeptides, polynucleotides and methods of using IL-21 to stimulate immune responses, particularly in tumor-bearing subjects as well as to the cell lines and methods of producing the recombinant IL-21 clinical product. We have filed patent applications for pharmaceutical compositions, IL-21 fusion proteins and other methods of using IL-21 for the treatment of disease. We have additional patent applications relating to IL-21 directed to methods for expressing and purifying recombinant IL-21; methods of treating specific cancers and viral diseases; combination therapies for IL-21 and monoclonal antibodies and IL-21 and tyrosine kinase inhibitors; and antagonist IL-21 ligands. We will continue to file patent applications as new inventions are made.
IL-31 mAb
Interleukin-31 (IL-31) is a cytokine derived from T cells. Analysis of IL-31 and IL-31 receptor levels in human and murine disease tissues suggests that IL-31 could play a role in atopic dermatitis (AD) and neuropathic pain. Transgenic animals over-expressing the IL-31 gene develop a severe type of dermatitis that resembles human AD, characterized by a destructive chronic scratching behavior in response to IL-31 mediated itch. Itch is a characteristic of human AD and the scratch response to itch is thought to be a major contributor to the severity of skin damage and disease. Treatment of animals in a murine model of spontaneous AD with a neutralizing antibody against IL-31 results in the reduction of scratching behavior. In addition, data shows that elevated levels of IL-31 mRNA and protein in skin correlate with both mouse and human AD, and that elevated circulating levels of IL-31 in serum correlate with severity of AD in patients. Analysis of peripheral blood T cells from human atopic dermatitis patients provides an association between IL-31 and skin-homing T cells, suggesting that skin diseases, such as AD, may be a promising therapeutic area for inhibition of IL-31.
Atopic dermatitis is a relapsing, chronic, inflammatory skin disorder that affects over 50 million people in the United States, major European countries and Japan. The disease has a high prevalence in children, with as many as 85% of cases developing AD before age 5. Some patients suffer from the disease into adulthood. AD typically resolves over time, but the disease becomes severe for those patients that do not go into remission, representing approximately 10% of the total affected population. Severe AD patients suffer from intense itching resulting in psychological problems, significant sleep loss and skin disfigurement. Current therapies on the market, such as topical corticosteroids, topical calcineurin inhibitors and antihistamines, are not effective for patients with severe disease, have safety concerns with long-term use, and do not target the disease mechanism. We believe that an inhibitor of IL-31 may be an effective therapy for treatment of these severely affected AD patients.
10
Table of Contents
Our product candidate is an IL-31 monoclonal antibody (IL-31 mAb) that has been shown to neutralize the activity of IL-31 in preclinical settings in a highly specific manner. IL-31 mAb is currently in the pre-IND development stage as a potential treatment for AD. We are in the process of manufacturing sufficient quantities of drug to supply toxicology studies, which are expected to begin in 2010, and Phase 1 clinical trials, which we intend to initiate in 2011. We own worldwide rights to IL-31, including protein products that target IL-31 such as monoclonal antibodies, subject to certain opt-in rights held by Merck Serono.
We have issued patents to IL-31 and IL-31 antibodies. We also have filed several patent applications relating to IL-31 and IL-31 antagonists, compositions and uses in disease on a worldwide basis, which cover protein products that target IL-31, including monoclonal antibodies, and therapeutic uses and will continue to file new patent applications as new inventions are made.
Out-licensed Product Candidates
Atacicept (formerly known as TACI-Ig)
Atacicept is a soluble form of the TACI receptor, a member of the tumor necrosis factor receptor family of proteins. Atacicept binds to and inhibits the activity of two ligands, BLyS and APRIL, which are implicated in B-cell survival, maturation and antibody production. We believe that atacicept could represent a more specific immunosuppressive agent for the treatment of autoimmune diseases.
Until August 2008, atacicept was developed jointly by us and Merck Serono SA pursuant to a collaborative development and marketing agreement established in 2001. In 2008, we converted this agreement to a worldwide royalty-bearing license, granting Merck Serono exclusive worldwide development and commercialization rights for atacicept.
Merck Serono is conducting a randomized, double-blind, placebo-controlled Phase 2/3 clinical trial of atacicept in patients with general systemic lupus erythematosus (SLE). In 2008, Merck Serono discontinued a Phase 2/3 study in patients with lupus nephritis. The study was discontinued due to the observation of the increased risk of severe infection, possibly resulting from significant underlying disease activity and the concomitant use of several immunosuppressive agents. In order to obtain regulatory approval in SLE, Merck Serono will need to complete additional studies of atacicept in patients with SLE.
Merck Serono is conducting three Phase 2 studies investigating atacicept in rheumatoid arthritis (RA): one in RA patients with inadequate response to TNF inhibitor therapy, another in RA patients who have not previously received TNF inhibitor therapy, and a third to evaluate the safety and efficacy of atacicept in combination with Rituxan. Preliminary results of the two single-agent atacicept Phase 2 studies confirmed the biological effect of atacicept on immunoglobulin and autoantibody production and no new safety signals were observed. However, the studies did not meet the pre-specified level of disease control activity to support moving directly into Phase 3 clinical testing. Following completion of further exploratory analysis, Merck Serono decided not to initiate further studies of atacicept in RA patients.
In September 2009, Merck Serono voluntarily discontinued two studies investigating atacicept in multiple sclerosis (MS) based upon a recommendation from an Independent Data Monitoring Committee (IDMC). In one of the MS studies, the IDMC observed an increase in MS disease activity in the atacicept treatment arms compared to the placebo arm. No comparable issues have been identified in the Phase 2/3 clinical trial in SLE or in the Phase 2 clinical trials in RA.
IL-21 monoclonal antibody (IL-21 mAb)
IL-21 mAb is a fully human monoclonal antibody we developed as an inhibitor of IL-21. IL-21 is a T-cell derived cytokine that exerts multiple effects on both T-cell and B-cell responses, which can be beneficial in fighting cancer or infections. In some situations, over expression of IL-21 can lead to autoimmune or
11
Table of Contents
inflammatory disease. In particular, IL-21 is a key regulator of two types of T cells: Th17 cells and T follicular helper (TFH) cells. Th17 cells are known to be involved in inflammation. By blocking IL-21 with IL-21 mAb, inflammation may be reduced in a number of diseases that share this pathway, such as psoriasis, Crohn’s disease and rheumatoid arthritis. TFH cells are specialized types of T cells that promote antibody responses from B cells. Blocking IL-21’s effect on B cells may have an impact on human diseases that are driven by antibody responses, such as systemic lupus erythematous (SLE). Murine models of psoriasis, Crohn’s disease (colitis), rheumatoid arthritis and SLE have demonstrated that inhibition of IL-21 leads to significant reductions in disease scores and pathology.
We and Novo Nordisk A/S have been parties to a license agreement for IL-21 since 2001, pursuant to which Novo Nordisk had certain rights to IL-21 outside North America, including monoclonal antibodies targeting IL-21. In December 2009, we and Novo Nordisk amended this license agreement, giving Novo Nordisk worldwide rights to IL-21 mAb and certain other embodiments of IL-21. Under the agreement, we have a right to co-promote the IL-21 mAb product in the United States and receive increased royalties on net sales in the United States if we contribute to Phase 3 clinical development costs. This license does not affect our IL-21 Phase 2 development candidate, which is recombinant human IL-21 protein and as to which we maintain worldwide rights.
IL-21 mAb is currently in the pre-IND stage. We expect to complete the transfer of IL-21 mAb technology and information to Novo Nordisk in 2010, which will enable them to begin clinical testing.
Other Out-licensed Product Candidates
rFactor XIII. rFactor XIII is a recombinant version of a human protein that is involved in blood clotting, and is being developed for the treatment of bleeding disorders. Novo Nordisk acquired rights to this protein from us in October 2004 after we completed several Phase 1 clinical trials in healthy volunteers and in patients with congenital Factor XIII deficiency. Novo Nordisk is conducting a Phase 3 study of rFactor XIII in patients with congenital Factor XIII deficiency and a Phase 2 study in patients undergoing cardiac surgery.
Fibroblast growth factor-18 (FGF-18). FGF-18 is a member of the fibroblast growth factor family of proteins. Our preclinical data suggest that FGF-18 may be useful for healing cartilage damaged by injury or disease. We out-licensed this protein to Merck Serono in October 2004 in conjunction with the strategic alliance. Merck Serono is conducting a Phase 1 clinical trial of FGF-18 for the treatment of osteoarthritis.
IL-17 receptor C (IL-17RC) soluble receptor. IL-17RC is a soluble receptor that binds to both IL-17A and IL-17F, the two most closely related cytokines in the IL-17 family. Both cytokines are highly expressed in a variety of inflammatory and autoimmune diseases, including rheumatoid arthritis, inflammatory bowel disease, multiple sclerosis and transplant rejection. We hypothesize that use of IL-17RC soluble receptor to neutralize the pro-inflammatory properties of IL-17A and IL-17F could have a beneficial therapeutic effect in any or all of these diseases. In August 2008, as part of the restructuring of our relationship, Merck Serono acquired an exclusive development and commercialization license to IL-17RC soluble receptor worldwide, subject to certain opt-in rights that we hold. The product candidate is currently in preclinical development.
IL-20 monoclonal antibody (IL-20 mAb). IL-20 is a member of the IL-10 cytokine family. In September 2001, Novo Nordisk licensed the rights to IL-20 outside North America pursuant to the option and license agreement. In March 2004, they licensed the rights to IL-20 in North America under a separate agreement. Our preclinical data suggest that IL-20 may play an important role in the regulation of skin inflammation and the pathology of psoriasis, as well as other inflammation diseases. Novo Nordisk is currently conducting Phase 1 clinical trials in psoriasis and rheumatoid arthritis patients.
IL-22 receptor subunit alpha monoclonal antibody (IL-22RA mAb). IL-22RA is a cytokine receptor that binds to both IL-20 and IL-22 cytokines and may be a potential target for the treatment of psoriasis. We
12
Table of Contents
out-licensed rights to IL-22RA monoclonal antibodies and to IL-22RA as a target to Merck Serono in October 2004 as part of the strategic alliance. The IL-22RA mAb product candidate is currently in preclinical development by Merck Serono.
Out-licensed Commercial Products
In addition to RECOTHROM, we have contributed to the discovery or development of seven recombinant protein products marketed by other companies.
Augment™ Bone Graft (formerly GEM-OS1TM)/Augment™ Injectable Bone Graft (formerly GEM-OS2 TM). Augment Bone Graft/Augment Injectable Bone Graft is a combination of platelet-derived growth factor (PDGF-BB) and a synthetic bone matrix. PDGF-BB is a growth factor that stimulates the growth of a variety of cell types, including bone forming cells. We cloned the gene that codes for platelet-derived growth factor, which we have out-licensed to BioMimetic Therapeutics, Inc. In November 2009, BioMimetic received approval to market Augment Bone Graft as an alternative to the use of autograft in foot and ankle fusion indications in Canada. In addition, BioMimetic completed submission of a modular Product Market Approval (PMA) to the U.S. FDA in February 2010.
Cleactor™ (tPA analog). Cleactor is a modified form of the protein tissue plasminogen activator, marketed in Japan by Eisai for the treatment of myocardial infarction, or heart attacks. In collaboration with Eisai, we developed this modified protein, which has enhanced properties that allow it to be given as a single injection.
GEM 21S® (platelet-derived growth factor). GEM 21S is a combination of a platelet-derived growth factor with a synthetic bone matrix, developed by BioMimetic Therapeutics, Inc. and marketed by Osteohealth Company, a division of Luitpold Pharmaceuticals, Inc. for the treatment of bone loss and gum tissue recession associated with advanced periodontal disease. We cloned the gene that codes for platelet-derived growth factor, the active agent in GEM 21S.
GlucaGen® (glucagon). GlucaGen is a protein therapeutic marketed by Novo Nordisk, Bedford Laboratories and Eisai Co., Ltd. (Eisai) for use as an aid for gastrointestinal motility inhibition and for the treatment of severe hypoglycemia in diabetic patients treated with insulin. In collaboration with Novo Nordisk, we developed a process for the production of this protein that is currently used by Novo Nordisk in the manufacture of GlucaGen.
Insulin and insulin analogs. Insulin and insulin analogs manufactured using recombinant DNA technology are marketed by Novo Nordisk worldwide for the treatment of diabetes. In collaboration with Novo Nordisk, we developed a process for the production of recombinant human insulin in yeast that is used by Novo Nordisk.
NovoSeven® (recombinant Factor VIIa). Factor VIIa is a protein involved in the generation of blood clots, and NovoSeven is marketed worldwide by Novo Nordisk for the treatment of patients with hemophilia and certain other coagulation deficiencies. We cloned the gene that codes for human Factor VII and developed the process for the production of activated recombinant human Factor VII, or recombinant Factor VIIa, which led to the establishment of the manufacturing process that Novo Nordisk currently uses to produce this protein.
Regranex® (platelet-derived growth factor). Regranex, until recently a product of Ethicon, Inc., a Johnson & Johnson Company, is a growth factor approved for the treatment of non-healing diabetic ulcers. In December 2008, One Equity Partners LLC announced the acquisition of Regranex from Ethicon Inc., which will be marketed and distributed by Systagenix Wound Management, a company created by One Equity Partners LLC. We cloned the gene that codes for platelet-derived growth factor and demonstrated the importance of this protein in stimulating wound healing.
13
Table of Contents
We have earned royalties on sales of some of these products. In the aggregate, from sales of these products and other technology licenses, we earned royalties of $1.3 million, $6.3 million, and $6.3 million for the years ended December 31, 2009, 2008, and 2007, respectively.
Commercialization
To commercialize RECOTHROM in the United States, we established our own dedicated commercial operations team with sales and sales operations, marketing, and supply chain and inventory management functions. We believe that the thrombin market, with its concentrated customer base, can be addressed with a relatively small sales force and that our recombinant technology gives us a competitive advantage in the current market.
In June 2007, we entered into a co-promotion agreement with Bayer HealthCare LLC, under which Bayer HealthCare provided sales people and medical liaisons to support RECOTHROM commercialization in the United States. During 2008 and 2009, the combined sales force worked to convert larger bovine thrombin accounts to RECOTHROM, by focusing on key surgeons, clinical pharmacists, operating room nurses and Pharmacy and Therapeutics (P&T) committee members within each account. Three wholesalers, AmerisourceBergen Corporation, Cardinal Health, Inc. and McKesson Corporation, accounted for approximately 90% of U.S. sales in 2009. If any of these wholesalers ceased distributing RECOTHROM, other wholesalers already distributing RECOTHROM would likely absorb the incremental sales volume with minimal interruption to the business or we would sell directly to hospitals.
In December 2009, we amended the U.S. co-promotion whereby Bayer HealthCare will no longer participate in the sales and marketing of RECOTHROM in the United States. We are currently in the process of increasing the size of our sales organization and intend to have the additional sales personnel fully trained and in the field by the end of first quarter 2010.
With our other product candidates, we intend, where appropriate, to enter into strategic collaborations for the commercialization of these candidates. We believe that this approach will enable us to maximize the long-term value of these assets.
Research and Development
In 2009, through a series of strategic initiatives and workforce and cost reductions, we restructured our organization to focus on developing and commercializing a smaller number of product candidates, which we believe have substantial therapeutic and commercial potential and in which we retain a significant ownership position. As part of these changes, we discontinued ongoing immunology and oncology discovery research programs, while retaining the research and development capabilities necessary to support the ongoing development programs for our product candidates.
Our research and development infrastructure draws upon a broad range of skills and technologies, including scientific computing, molecular and cellular biology, animal models of human disease, protein chemistry, antibody generation and engineering, pharmacology and toxicology, clinical development, medical and regulatory affairs, drug formulation, process development and protein manufacturing methods. We believe that this comprehensive approach enables us to effectively and efficiently develop our pipeline of therapeutic proteins.
We have a development organization with the skills and expertise to design and implement clinical trials for multiple product candidates and to file license applications with the FDA and other regulatory agencies. Our in-house development resources include a clinical development group responsible for designing, conducting and analyzing clinical trials. The group includes clinical research, clinical operations, biometrics, medical writing and drug safety. Our preclinical development group provides support in the areas of bioanalytical research and
14
Table of Contents
development, pharmacology (including pharmacogenomics), toxicology, pathology and pharmacokinetics. Our regulatory affairs group develops regulatory strategies and manages communications and submissions to regulatory agencies.
For additional details for research and development activities, refer to the Operating Expenses section under “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations Overview”.
Manufacturing
RECOTHROM®
We have established a RECOTHROM commercial supply chain, which relies on several single-source vendors. We have entered into a long-term manufacturing agreement with Abbott Laboratories for commercial-scale production of bulk recombinant human thrombin (rThrombin), the active drug substance in RECOTHROM. Under the agreement, Abbott manufactures rThrombin using the two-step process developed by ZymoGenetics, according to specifications developed and agreed upon by both companies. First, recombinant human prethrombin-1 is produced in mammalian cells. Then, using an enzyme activation step, prethrombin-1 is converted to rThrombin. Abbott has committed to annually supply up to a maximum amount, which we believe is sufficient to meet our projected market demand and provide adequate safety stock. We have agreed to purchase annual minimum amounts from Abbott. The agreement terminates in 2018. We have also entered into a manufacturing services agreement with Patheon Italia S.p.A. for fill and finish of rThrombin, which expires in December 2013, and are currently negotiating with an additional fill and finish supplier located in the United States. In addition, we have entered into agreements with two vendors for the final packaging of RECOTHROM and with Cardinal Health SPS, Inc. for third party logistics services. Furthermore, we have entered into agreements with several suppliers of critical raw materials, manufacturing aids, and components for RECOTHROM, some of which are located outside the United States.
Under the terms of the amended license and collaboration agreement, we agreed to supply vials of rThrombin approved for sale in the United States to Bayer Schering Pharma for sale in Canada throughout the term of the agreement.
PEG-IFN-lambda
We manufactured initial clinical supplies of PEG-IFN-lambda in our pilot-scale GMP manufacturing facility, using a high-yield internally developed E. coli process and believe we have adequate supply of product to support clinical development though Phase 2. Under the terms of our co-development and co-promotion agreement with Bristol-Myers Squibb, they will be responsible for all future manufacturing of PEG-IFN-lambda, including product for Phase 3 clinical trials and commercial sale.
IL-21
Our initial clinical supply of IL-21, which is made in E. coli, was manufactured by a third party using a process we developed. Subsequently, Novo Nordisk manufactured clinical materials for Phase 2 and initial Phase 3 development under a manufacturing agreement established in 2007. In January 2009, Novo Nordisk terminated the agreement and we acquired all rights to the manufacturing processes and obtained the existing supply of the product. We will need to identify and enter into an agreement with a third-party contract manufacturer for commercial supply of IL-21.
IL-31 mAb
Our initial supplies of IL-31 mAb product for toxicology studies and Phase 1 clinical trials are currently being manufactured by a third-party contractor.
15
Table of Contents
Manufacturing Changes
In 2009, we discontinued operation of our pilot-scale GMP manufacturing facility that was used to supply materials for toxicology studies and early-stage clinical trials. Going forward, we intend to rely on collaborative partners or third-party contractors for production of all preclinical, clinical and commercial supplies.
Collaborative Relationships
Bristol-Myers Squibb Co-Development and Co-Promotion Agreement for PEG-IFN-lambda
In January 2009, we entered into a co-development/co-promotion and license agreement with Bristol-Myers Squibb Company that covers all members of the type III interferon family, including interferon-lambda 1. Under the terms of the agreement, we are obligated to work exclusively with Bristol-Myers Squibb to develop biopharmaceutical products based on the type III interferon family. Currently, the companies intend to only develop PEG-IFN-lambda, a pegylated version of interferon-lambda 1, which is in development as a treatment for hepatitis C.
As part of the co-development/co-promotion and license agreement, Bristol-Myers Squibb receives an exclusive worldwide license to the core patents relating to the type III interferon family and a co-ownership interest in all core patents relating to the type III interferon family filed outside of the United States. In addition, Bristol-Myers Squibb receives a non-exclusive license to other intellectual property rights relating to the licensed products. We will be responsible for funding the first $100 million of development costs in the United States and Europe, which we expect to incur during Phase 1b and Phase 2 clinical testing, and 20% of all further development costs in the United States and Europe.
In return, during 2009 we have received:
| • | $105 million in license fees; and |
| • | $95 million in milestone payments related to initiation of Phase 2 activities. |
In addition, we may receive:
| • | Additional payments of up to $335 million based on pre-defined development and regulatory milestones for hepatitis C, up to $287 million in development and regulatory milestones for other potential indications, and up to $285 million based on pre-defined annual sales milestones; |
| • | 40% of the profits from the co-commercialization of any type III interferon family product within the United States. We will also be responsible for 40% of any loss from the co-commercialization of any product within the United States, provided that a portion of our share of losses incurred through the initial launch phase will be deferred, and deferred losses will subsequently be deducted from milestones, royalties and our share of profits; and |
| • | Royalties on product sales outside the United States. |
The research and development activities are governed by a steering committee made up of an equal number of representatives from each company. Bristol-Myers Squibb is responsible for all future manufacturing of PEG-IFN-lambda, including product for Phase 3 clinical trials and commercial sale.
We have the right to co-promote or co-fund PEG-IFN-lambda in the United States, and must exercise this right within 30 days after acceptance by the FDA of a Biologics License Application (BLA) filing, in which case we will share any profits or losses in the United States. In certain circumstances, we may opt out of co-promotion while retaining the option to co-fund and share product profits and losses. We have the right to discontinue co-promotion and co-funding in the United States, in which case we would be eligible to receive royalties on product sales in the United States. Under certain restricted circumstances, Bristol-Myers Squibb may terminate our right to co-promote in the United States, provided that, in certain of these circumstances, we will retain the
16
Table of Contents
option to co-fund and share product profits and losses. If Bristol-Myers Squibb terminates our co-promotion right and we do not have the option to co-fund or choose not to exercise that option, we would receive royalties on product sales instead of sharing profits and losses in the United States.
Royalties on sales vary based on annual sales volume and the degree of patent protection provided by the licensed intellectual property. Royalty payments may be reduced if Bristol-Myers Squibb is required to license additional intellectual property from one or more third parties in order to commercialize a product or, in certain circumstances, if product sales suffer from direct competition. Royalty obligations under the agreement continue on a country-by-country basis until the date on which no valid patent claims relating to a product exist or, if the product is not covered by a valid patent claim, eleven years after the date of first sale of the product in that country.
The term of the agreement began on February 26, 2009 and will continue for as long as a type III interferon product is the subject of an active development project or there is an obligation to pay royalties under the agreement.
Bayer License and Collaboration Agreement for rThrombin and U.S. Co-Promotion Agreement for RECOTHROM®
In June 2007, we executed a license and collaboration agreement with Bayer Schering Pharma AG and a U.S. co-promotion agreement with Bayer HealthCare LLC. Under the terms of the license and collaboration agreement, Bayer Schering Pharma was responsible for developing and commercializing rThrombin outside of the United States. Under the co-promotion agreement, Bayer HealthCare agreed to contribute sales people and medical science liaisons for the first three years following the launch of RECOTHROM in the United States. Through December 31, 2009, we received an initial milestone of $30.0 million upon signing of the agreements, an additional $40.0 million upon the U.S. approval of rThrombin, and $6.5 million upon the initial filings for approval in Canada, Europe and Asia.
In December 2009, we executed amendments to both agreements. Pursuant to the amended license and collaboration agreement, Bayer Schering Pharma will develop and commercialize the initial presentations of rThrombin in Canada, where it received marketing approval in December 2009, but will return all other rights to RECOTHROM outside the United States and Canada to us. As part of the agreement, we will also supply vials of rThrombin approved for sales in the United States to Bayer Schering Pharma for sale in Canada for the term of the license. Pursuant to the amended co-promotion agreement, Bayer HealthCare’s active role in promoting RECOTHROM in the United States ceased as of December 31, 2009, but they are entitled to commissions on sales in the United States for up to two years subject to an aggregate maximum amount of $12 million.
Merck Serono Development and Marketing Agreement for Atacicept
In August 2001, we entered into a collaborative development and marketing agreement with Ares Trading S.A., a wholly owned subsidiary of Serono S.A., focused on product candidates derived from two cellular receptors (designated TACI and BCMA) that are involved in the regulation of the human immune system. Following the acquisition of Serono by Merck KGaA in 2007, Serono’s rights under this agreement have been held by Merck Serono S.A. Pursuant to the collaborative development and marketing agreement, the parties had been co-developing atacicept in autoimmune diseases and cancer. In August 2008, we entered into an amended and restated development and marketing agreement, providing Merck Serono with exclusive worldwide rights to develop, market and sell products developed under the agreement, for which we will be entitled to receive milestone fees and royalties on worldwide net sales.
We granted Merck Serono an exclusive license to our intellectual property relating to TACI, BCMA and certain other related technologies to make, use, have made, sell, offer to sell and import products based on TACI and BCMA. Merck Serono is required to pay royalties on sales, which vary based on annual sales volume and the
17
Table of Contents
degree of patent protection provided by the licensed intellectual property. Royalty payments may be reduced if Merck Serono is required to license additional intellectual property from one or more third parties in order to commercialize a product or, in certain circumstances, if product sales suffer from direct competition. Royalty obligations under the agreement continue on a country-by-country basis until the date on which no valid patent claims relating to a product exist or, if the product is not covered by a valid patent claim, 15 years from the date of first sale of the product in that country.
The agreement will continue for as long as a TACI or BCMA product is the subject of an active development project or there is an obligation to pay royalties under the agreement. The agreement provides for an initial fee and milestone payments to be paid by Merck Serono in connection with the development and approval of products, up to an aggregate of $52.5 million of which $15.5 million has been received to date.
Novo Nordisk License Agreements for IL-21
As a result of a series of agreements, we now have worldwide development and commercialization rights for products based on our intellectual property to IL-21 protein and Novo Nordisk has development and commercialization rights for products based on our intellectual property to other embodiments of IL-21, including antibodies to IL-21.
In 2001, Novo Nordisk initially licensed the rights under our intellectual property to various embodiments of IL-21 in territories outside of North America. In 2005, we entered into a collaborative data sharing and cross-license agreement with Novo Nordisk to develop and execute a joint global clinical development plan for the IL-21 protein to achieve regulatory approval of a common product in the companies’ respective territories. In January 2007, the parties also entered into a manufacturing agreement whereby Novo Nordisk agreed to supply us with IL-21 protein for use in clinical trials. In January 2009, the parties restructured their relationship as it relates to IL-21. As part of the restructuring, the parties:
| • | Amended and restated the license agreement to no longer cover the IL-21 protein. However, Novo Nordisk continued to be responsible for developing other embodiments of IL-21, including antibodies to IL-21, outside North America. |
| • | Entered into a license and transfer agreement pursuant to which we received an exclusive license outside North America to the intellectual property rights that Novo Nordisk developed relating to the IL-21 protein, and are obligated to make milestone payments based on approval and sales and pay single-digit royalties on sales of any resulting products outside North America. In addition, we will pay Novo Nordisk a portion of any third-party license fees above a specified threshold. |
| • | Terminated the collaborative data sharing and cross-license agreement. However, our exclusive license in North America to the intellectual property rights that Novo Nordisk developed relating to the IL-21 protein survived termination. |
| • | Terminated the manufacturing agreements and Novo Nordisk transferred to us all manufacturing processes developed and its existing stock of IL-21 protein. |
In December 2009, the parties further amended and restated the license agreement to provide Novo Nordisk worldwide rights to embodiments of IL-21 (other than IL-21 protein), including antibodies to IL-21. Novo Nordisk paid an upfront license fee of $24.0 million and is obligated to make milestone payments based on the achievement of development milestones of up to an aggregate of $157.5 million (none of these milestone payments had been earned or paid as of December 31, 2009) and royalties on sales of any resulting products. Royalties on sales would vary from mid to high single digits based on annual sales volume and the degree of patent protection provided by the licensed intellectual property. In addition, we will have a right to co-promote the IL-21 mAb product in the United States if we contribute to Phase 3 clinical development costs directed achieving regulatory approval in the United States or European Union, in which case royalties on net sales in the United States would vary from high single digits to low double digits. We must make an election to contribute to Phase 3 clinical development costs within 90 days following receipt of data from the Phase 2b studies, in which case we will pay a one-time fee of $10 million and 15% of Phase 3 clinical development costs as they are incurred. Royalty obligations under the agreement continue on a country-by-country basis until the date on which no valid patent claims relating to a product exist or, if the product is not covered by a valid patent claim, 10 years from the date of first sale of the product in that country. Royalty payments may be reduced if Novo Nordisk is required to license additional intellectual property from one or more third parties in order to commercialize a product or, in certain circumstances, if product sales suffer from direct competition; provided that, in the aggregate, the relevant royalty rate may not be reduced by more than one half. The term of the agreement began on December 3, 2009 and will continue for as long as there is an obligation to pay royalties under the agreement. Novo Nordisk may terminate the agreement early for an uncured material breach, material safety concerns or, upon nine months notice, for its convenience.
18
Table of Contents
Merck Serono Strategic Alliance Agreement
In October 2004, we executed a strategic alliance agreement with Serono S.A. to research, develop and commercialize product candidates, including protein and antibody therapeutics, based on a specific portfolio of our proprietary genes. Following the acquisition of Serono in 2007 by Merck KGaA, Serono’s rights under this agreement have been held by Merck Serono SA, an affiliate of Merck KGaA. In August 2008, we amended the strategic alliance agreement, while retaining the original five-year term, which expired in October 2009.
At the time of the original agreement, we executed agreements granting Merck Serono exclusive worldwide licenses to two preclinical candidates, FGF-18 and IL-22RA mAb, and entered into a co-development agreement relating to IL-31 mAb. Subsequently, in June 2007, we entered into a co-development agreement with Merck Serono for the IL-17RC soluble receptor, under the strategic alliance agreement. In connection with the original agreement we received a $20.0 million upfront option fee and $11.25 million in license fees for FGF-18, IL-22RA mAb and IL-31 mAb. In addition, Merck Serono purchased approximately 3.2 million shares of our common stock for a total of $50.0 million, and entered into a related lockup agreement and a standstill agreement.
In connection with the 2008 amendment of the strategic alliance agreement, we amended and restated the co-development agreements for IL-31 mAb and IL-17RC soluble receptor to provide for exclusive licenses to, in the case of IL-31 mAb, ZymoGenetics and, in the case of IL-17RC soluble receptor, Merck Serono. Pursuant to these exclusive licenses we will receive (in the case of IL-17RC soluble receptor) and pay (in the case of IL-31 mAb):
| • | Potential milestone payments related to development progress, regulatory submissions and approvals for product candidates. |
| • | Royalties on worldwide sales of licensed products. The licensee is required to pay royalties on sales, which vary based on annual sales volume and the degree of patent protection provided by the licensed intellectual property. Royalty payments may be reduced if the licensee is required to license additional intellectual property from one or more third parties in order to commercialize a product or, in certain circumstances, if product sales suffer from direct competition. Royalty obligations under the agreements continue on a country-by-country basis until the date on which no valid patent claims relating to a product exist or, if the product is not covered by a valid patent claim, 15 years from the date of first sale of the product in that country. |
In addition, these exclusive licenses provide that if the licensee (whether us or Merck Serono) seeks a partner for the applicable product candidate, the licensor will have the right to opt in to co-develop and co-commercialize the product candidate on pre-negotiated terms, including retroactive and prospective cost sharing, royalties and milestone fees. In addition to having co-development and co-commercialization rights within the United States, Merck Serono would have an exclusive license outside of the United States whether Merck Serono opts in to develop a product candidate of ours or we opt in to develop a product candidate of Merck Serono.
Patents and Proprietary Rights
We seek appropriate patent protection for our proprietary technologies and product candidates by filing patent applications in the United States. We have more than 340 unexpired issued or allowed U.S. patents, and over 180 pending U.S. patent applications. When appropriate, we also seek foreign patent protection and to date have more than 790 issued or allowed foreign patents.
19
Table of Contents
Our patents and patent applications are primarily directed to therapeutic protein-based products. We commonly seek claims directed to compositions of matter for genes and proteins, including antibodies, methods of using and methods of making. When appropriate, we also seek claims to related technologies, such as reagents used in release assays and formulations. We maintain patents and prosecute applications, worldwide, for technologies that we have outlicensed. Similarly, for development projects that are partnered, we work closely with our development partners to coordinate patent efforts, including filings, prosecution, term extension, defense and enforcement. As our development product candidates advance through research and development, we seek to diligently identify and protect new inventions, such as combinations, improvements to methods of manufacturing or purification, and methods of treatment. We also work closely with our scientist personnel to identify and protect new inventions that could eventually add to our development pipeline.
Patents expire, on a country by country basis, at various times depending on various factors, including the filing date of the corresponding patent application(s), the availability of patent term extension and supplemental protection certificates and terminal disclaimers. For our commercial product and each of our product candidates, we have filed or expect to file multiple patent applications and expect to obtain multiple patents. These multiple patent applications and patents are filed and obtained over time and are directed to the various aspects of our commercial product and product candidates described in the preceding paragraph. The table below provides estimated expiration dates for our U.S. patents granted or expected to be granted from our first filed patent application covering our commercial product, RECOTHROM, and product candidates in our development pipeline. Each expiration date may be subject to patent term extension, where the length of term extension would not exceed five years under current law and depends on factors such as the amount of time taken by the FDA to review the first marketing approval application of a drug covered by the patent.
| Commercial Product/Product Candidate | Estimated Expiration Date for U.S.
Patent Arising from First Filed Application | |
| RECOTHROM | December 2012; expected to extend until July 2015 under patent term extension | |
| PEG-IFN-lambda | September 2021 | |
| IL-21 | March 2020 | |
| IL-31 mAb | January 2023 |
We require our scientific personnel to maintain laboratory notebooks and other research records in accordance with our policies, which are designed to strengthen and support our patent efforts. In addition to our patented intellectual property, we also develop and seek to protect unpatented technology, trade secrets and confidential information, including our genetic sequence database, bioinformatics algorithms, research, preclinical and clinical data, development and manufacturing strategies. Our policy is to require our employees, consultants and advisors to execute a confidentiality and proprietary information agreement before beginning their employment, consulting or advisory relationship with us. These agreements generally provide that the individual must keep confidential and not disclose to other parties any confidential information developed or learned by the individual during the course of their relationship with us except in limited circumstances. These agreements also generally provide that we shall own all inventions conceived by the individual in the course of rendering services to us. These agreements, however, may not provide effective protection of our technology, confidential information or, in the event of unauthorized use or disclosure, may not provide adequate remedies.
As part of our business strategy, we often work with third parties in our research and development activities. Accordingly, disputes may arise about inventorship, ownership and corresponding rights to know-how and inventions resulting from the joint creation or use of intellectual property by us and our corporate partners, licensors, licensees, scientific collaborators and consultants. In addition, other parties may circumvent any proprietary protection we do have. These parties may independently develop equivalent technologies or independently gain access to and disclose substantially equivalent information, and confidentially agreements and material transfer agreements we have entered into with them may not provide us with effective protection.
Refer to “Item 1A. Risk Factors” for additional information relating to our patents and proprietary rights.
20
Table of Contents
Government Regulation
Regulation by government authorities in the United States, Canada, Europe, Japan and other countries is a significant consideration in our ongoing research and product development activities and in the manufacture and marketing of our products and product candidates. Both before and after the approval of our products and product candidates, we, our products, our product candidates, our operations, our facilities, our suppliers, and our contract manufacturers, contract research organizations, and contract testing laboratories are subject to extensive regulation by government authorities in the United States and other countries. The FDA and comparable regulatory bodies in other countries currently regulate therapeutic proteins and related pharmaceutical products as biologics. Biologics cannot be lawfully marketed in the United States without FDA approval. Biologics are subject to extensive pre- and post-market regulation by the FDA, including regulations that govern the collection, testing, manufacture, safety, efficacy, potency, labeling, storage, record keeping, advertising, promotion, sale and distribution of the products. The time required for completing testing and obtaining approvals of our product candidates is uncertain and varies among product candidates for several reasons, but the process takes many years. In addition, we may encounter delays in product development or rejections of product applications due to changes in FDA or foreign regulatory laws or policies during the period of product development and testing. Failure to comply with regulatory requirements may subject us to, among other things, warning letters, civil penalties and criminal prosecution; fines and other monetary penalties; restrictions on product development and production; suspension, delay, rejection, or withdrawal of approvals; injunctions; and the seizure or recall of products. The lengthy process of obtaining regulatory approvals and ensuring compliance with appropriate statutes and regulations requires the expenditure of substantial resources. Any delay or failure, by us or our corporate partners, to obtain regulatory approvals could adversely affect our ability to commercialize product candidates, receive royalty payments and generate sales revenue.
The nature and extent of the governmental pre-market review process for our potential products will vary, depending on the regulatory categorization of particular products. The necessary steps before a new biological product may be marketed in the United States ordinarily include:
| • | nonclinical laboratory and animal tests; |
| • | submission to the FDA of an IND application, which must become effective before clinical trials may commence in the United States; |
| • | completion of adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug for its intended use; |
| • | submission to the FDA of a BLA; |
| • | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with current Good Manufacturing Practices (cGMP); and |
| • | FDA review and approval of the BLA. |
Nonclinical tests include laboratory evaluation of the product, as well as animal studies to assess the potential safety and efficacy of the product. The results of nonclinical tests, together with extensive manufacturing information, analytical data and proposed clinical trial protocols, are submitted to the FDA as part of an IND application, which must become effective before the initiation of clinical trials. The IND application will automatically become effective 30 days after receipt by the FDA unless before that time the FDA raises concerns or questions and imposes a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed. FDA may also impose a clinical hold on an ongoing clinical trial if, for example, safety concerns arise, in which case the trial cannot recommence without FDA authorization.
Clinical trials involve the administration of the product to human subjects under the supervision of qualified personnel. Clinical trials are conducted under protocols that detail the objectives of the trial, inclusion and exclusion criteria, and the parameters to be used to monitor safety and the efficacy criteria to be evaluated.
21
Table of Contents
Protocols for each phase of the clinical trials are submitted to the FDA as part of the original IND application or as an amendment to the IND application. Further, each clinical trial must be reviewed and approved by an independent institutional review board at each participating institution. The institutional review board will consider, among other things, ethical factors and the safety of human subjects, and may approve the protocol as submitted, require changes, or decline to approve it.
Clinical trials generally are conducted in three sequential phases that may overlap. In Phase 1, the potential product is administered to healthy human subjects or patients, or both, to assess safety, metabolism, pharmacokinetics and pharmacological actions, and side effects associated with increasing doses of the drug. Phase 2 usually involves one or more trials to evaluate the efficacy of the potential product for a particular indication or indications in patients with the disease or condition under study, to determine dosage tolerance and optimum dosage, and to further identify possible adverse reactions and safety risks. If a compound appears to be effective and to have an acceptable safety profile in Phase 2 evaluations, Phase 3 trials may be undertaken to evaluate further clinical efficacy, usually in comparison to standard therapies or placebo, generally within a broader patient population with the disease state or condition for which an indication for use will be sought, and at geographically dispersed clinical sites. Phase 1, Phase 2 or Phase 3 testing may not be completed successfully within any specific period of time, if at all, with respect to any of our potential products. Furthermore, we, an institutional review board, the FDA or other regulatory bodies may suspend a clinical trial at any time for any reason, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
The results of pharmaceutical development, nonclinical studies and clinical trials are submitted to the FDA in the form of a BLA for approval of the manufacture, marketing and commercial shipment of the biological product. A BLA contains extensive manufacturing information, and FDA usually inspects each manufacturing facility and quality system to assess compliance with cGMP before approving a BLA. The inspection and approval process require substantial time, effort and resources, and approvals may not be granted on a timely basis, if at all. If FDA determines that the BLA is not acceptable, it may issue a complete response letter outlining the deficiencies and stating what additional information or studies it requires for approval. Notwithstanding the submission of any requested additional testing or information, the FDA ultimately may decide that the application does not satisfy the criteria for approval, and deny the application. If a product is approved, the FDA may limit the indications for which the product may be marketed, require extensive warnings on the product label, impose restrictive distribution or other risk mitigation measures, and /or require post-approval clinical studies. After approval, certain changes to the approved product, such as adding new indications, manufacturing changes, or additional labeling claims, will require submittal of a new BLA or, in some instances, a BLA supplement, for further FDA review and approval.
Some of our product candidates may qualify as orphan drugs under the Orphan Drug provisions of the Food, Drug, and Cosmetic Act. Orphan drug designation must be requested before an application for marketing authorization is submitted. If granted, orphan drug designation applies to a drug for a specific indication and it is possible for more than one drug to receive orphan drug designation for the same indication. If a product which has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, which means the FDA may not approve any other application to market the same drug for the same indication for a period of seven years, except in limited circumstances, such as the showing of clinical superiority to the product with orphan exclusivity. The FDA granted IL-21 orphan drug status for the treatment of melanoma patients with advanced or aggressive disease. However, orphan drug designation for IL-21 may not have a positive effect on our revenues even if the product is approved, and it is possible that in the future none of our other product candidates will be designated as an orphan drug by the FDA.
In addition, the FDA regulates the advertising and promotion of biologic products and medical devices, emphasizing areas such as appropriate disclosure of risks, prohibition of off-label promotion, industry-sponsored scientific and educational activities, comparative advertising, and promotional activities involving the Internet.
22
Table of Contents
FDA marketing approval is only applicable in the United States. Marketing approval in foreign countries is subject to the regulations of those countries. The approval procedures vary among countries and can involve additional testing. The time required to obtain approval outside of the United States may differ from that required for FDA approval. There are centralized procedures for filings in the European Union (EU) countries, which allow submission of a single marketing authorization application to obtain approval in the approximately 25 countries of the EU. Outside of the EU, most countries generally have their own procedures and requirements, and compliance with these procedures and requirements may be expensive and time-consuming. Accordingly, there may be delays in obtaining required approvals from foreign regulatory authorities after the relevant applications are filed, if approvals are ultimately received at all.
We are also subject to various federal, state and local laws, regulations, industry guidelines and recommendations relating to employment practices; safe working conditions; laboratory and manufacturing practices; the experimental use of animals; the use and disposal of hazardous or potentially hazardous substances, including radioactive compounds and infectious disease agents; product liability; and unfair competition, including advertising and other promotional efforts. Government regulations that might result from future legislation or administrative action, including additions or changes to environmental laws, may materially affect our business operations and revenues.
Competition
We face competition from a broad range of biotechnology and pharmaceutical companies as well as academic and research institutions. We compete with these entities to develop and obtain proprietary rights to new therapeutic proteins and to commercialize the products we develop from these proteins. Some of our competitors have greater resources and experience than we have in discovering, developing, manufacturing and selling protein-based products. We expect that competition in our field will continue to be intense.
RECOTHROM, which was approved in January 2008 by the FDA for use as a topical hemostat in the United States, faces substantial competition in the topical hemostat market. In addition to RECOTHROM, there are two stand-alone thrombin products currently available in the United States: Thrombin-JMI, a bovine plasma-derived thrombin from King Pharmaceuticals, Inc., and Evithrom, a pooled human plasma-derived thrombin, from Ethicon, Inc., a Johnson & Johnson Company. Also, a number of other topical hemostatic agents are currently available on the market from Johnson & Johnson Wound Management, a division of Ethicon, Inc. and the BioSurgery business unit of Baxter BioScience, including GELFOAM Plus, a gelatin sponge product co-packaged with human-plasma derived thrombin. Furthermore, new products and technologies could be developed in the future to limit or control bleeding during surgeries.
We anticipate that our other product candidates currently in development will face intense competition in their respective therapeutic areas. PEG-IFN-lambda, which we are co-developing with Bristol-Myers Squibb as a potential treatment for hepatitis C virus (HCV), is targeting a market with an established standard of care, i.e., interferon-alpha based therapy plus ribavirin. We believe that PEG-IFN-lambda may be a better alternative to the current standard of care due to the potential for fewer side effects and equivalent or potentially better antiviral activity. However, we would have to compete with two well established interferon-alpha based drugs, Roche’s PEGASYS® and Schering-Plough’s PEG-Intron®. Furthermore, the introduction of a new class of drugs, such as HCV protease inhibitors or polymerase inhibitors or a combination thereof, may result in transition to a different treatment regimen and, if this regimen excludes an interferon component, may represent an additional competitive threat to our product. Two HCV protease inhibitors, Vertex’s telaprevir and Schering-Plough’s boceprevir, are currently in Phase 3 development.
Our other product candidates, such as IL-21 for metastatic melanoma and IL-31 mAb for severe atopic dermatitis, target diseases for which there is a significant unmet medical need; however, this may change with the introduction of new products currently in development by other companies. There are several product candidates currently in Phase 3 development for metastatic melanoma, including ipilimumab, an immunotherapy being developed by Bristol-Myers Squibb, and PLX4032, a targeted therapy being co-developed by Plexxikon Inc. and Roche.
23
Table of Contents
Although we believe that currently we are well positioned to compete effectively with respect to our existing and potential competitors, our ability to compete successfully in the future will depend on many factors, including our ability to:
| • | successfully maintain and expand as appropriate RECOTHROM commercial infrastructure, including the product supply and sales force, and establish commercial infrastructure for other product candidates as necessary; |
| • | develop products that are safer, more efficacious or more convenient to administer than other products in the marketplace; |
| • | leverage our established collaborations and enter into new collaborations to support the development of our products; |
| • | obtain timely regulatory approvals; |
| • | manufacture our products in a cost-effective manner in quantities sufficient to meet market demands; |
| • | obtain adequate reimbursement from government health administration authorities, private health insurers and health maintenance organizations; |
| • | obtain and enforce adequate patent protection for our product candidates and technologies; |
| • | maintain adequate capital levels; and |
| • | attract and retain key personnel. |
Employees
As part of our strategic refocus, we reduced headcount by approximately 33% in April 2009 and by an additional 15% in December 2009. As of December 31, 2009, we had 323 full-time employees, including 176 employees dedicated to research and development and 64 employees dedicated to sales and marketing. Each of our employees had signed confidentiality and intellectual property agreements, and no employees are covered by a collective bargaining agreement. We have never experienced employment-related work stoppages and consider our employee relations to be good.
Website Access to Our SEC Reports
Our Internet address is www.zymogenetics.com. We make our periodic SEC reports (Form 10-Q and Form 10-K), current reports (Form 8-K) and amendments to these reports available free of charge through our website as soon as reasonably practicable after they are filed electronically with the SEC. We may from time to time provide important disclosures to investors by posting them in the investor relations section of our website, as allowed by SEC rules.
24
Table of Contents
Item 7A. Qualitative and Quantitative Disclosures About Market Risk
Until recently, our exposure to market risk has been primarily limited to interest income sensitivity, which is affected by changes in the general level of United States interest rates, particularly because the majority of our investments are in short-term debt securities. The primary objective of our investment activities is to preserve principal while at the same time maximizing the income we receive without significantly increasing risk. To minimize risk, we maintain our portfolio of cash, cash equivalents and short-term investments in a variety of interest-bearing instruments, which have included United States government and agency securities, high-grade United States corporate bonds, asset-backed securities, commercial paper and money market funds.
In 2008 and continuing through 2009, due to deteriorating conditions in the debt markets, our exposure to market risk increased and impacted our investment portfolio. Overall liquidity for many debt issues has declined substantially, meaning that we may realize losses if we are required to liquidate securities upon short notice. Additionally, with respect to asset backed securities, overall economic conditions have generated concerns about the value of underlying assets held as collateral, and highlighted risks associated with insurance policies used to enhance the credit of the related debt issues. To date, we have not experienced defaults on any of our asset backed securities, but we have seen the time for expected repayment increase significantly. In 2009, we revised our investment guidelines to exclude future purchases of asset backed securities; however, we continue to hold several of these securities that were purchased prior to the revision of our guidelines. We continue to monitor these investments closely and, based on market conditions, recorded other-than-temporary impairment losses of $1.6 million and $400,000 in the fourth quarter of 2009 and the third quarter of 2008, respectively.
We have estimated the effect on our investment portfolio of a hypothetical increase in interest rates by one percent to be a reduction of $600,000 in the fair value of our investments as of December 31, 2009.
We have no material foreign currency exposure, nor do we hold derivative financial instruments.
25
Table of Contents
Item 11. Executive Compensation
The following is a discussion and analysis of our executive compensation program and describes the compensation earned in 2009 by each of the named executive officers identified in the Summary Compensation Table below.
Compensation Discussion and Analysis
Objectives and Components
The objectives of our executive compensation program are to:
| • | attract qualified, experienced executive personnel capable of achieving ZymoGenetics’ business objectives; |
| • | retain and motivate executives to achieve superior performance; |
| • | link individual compensation to individual and company performance; and |
| • | align executives’ financial interests with those of our shareholders. |
The components of our executive compensation program are:
| • | base salaries; |
| • | annual incentives in the form of cash bonuses; and |
| • | long-term incentives in the form of stock option grants. |
Total Compensation
Our executive compensation program has been designed to encourage and reward performance that we believe will increase our intrinsic value over the long term. Until the launch of RECOTHROM® recombinant thrombin in early 2008, we had no product sales and our commercial operations were limited. Accordingly, advancement of our product development programs toward approval by the United States Food and Drug Administration (“FDA”) and commercialization had represented the best way to build value for shareholders. With sales of RECOTHROM becoming important to our performance in 2008 and 2009, we have increased the emphasis on commercial accomplishments, such as achieving our sales budget.
We have included three components in our compensation structure, namely base salaries, cash bonuses and stock option grants, to compete with other companies in our industry. We would be unusual were we not to offer all three of these components, Not only are these components important for recruiting executives, they are also important for retaining them.
We have never viewed the total value of the three components as the appropriate amount for benchmarking with other companies. For companies like ours, without an established, profitable commercial business, stock options are highly speculative. Unless we are able to gain approval for additional products and ultimately generate significant sales and potential profits, these stock options are unlikely to have substantial value over the long term. We believe that stock options must be viewed and benchmarked independently from the two cash compensation components. Accordingly, we make no decisions regarding allocation of total compensation between cash and non-cash components; we benchmark each separately and set each at levels we believe will be competitive to our peer group of companies.
Determining Executive Compensation
The compensation committee of the Board of Directors is responsible for implementing our executive pay philosophy, evaluating compensation against the market and our pay philosophy, and approving the material
26
Table of Contents
terms of executive compensation arrangements. The compensation committee evaluates the performance of our Chief Executive Officer and determines his compensation based on this evaluation. Douglas E. Williams, Ph.D. has been our Chief Executive Officer since the retirement of Bruce L. A. Carter, Ph.D. in January 2009.
With respect to the other named executive officers, the compensation committee considers our Chief Executive Officer’s input as to performance evaluations and recommended compensation arrangements. The Chief Executive Officer’s recommendations on executive officer compensation are subject to final approval of the compensation committee.
Management and the compensation committee rely on outside advisors to ascertain competitive pay levels, evaluate pay program design, and assess emerging trends in executive and equity compensation. Generally, the compensation committee retains an independent consultant to perform such an assessment every two years. During 2007, the compensation committee engaged Radford Surveys + Consulting (“Radford”), as described below under the heading “Benchmarking.”
Benchmarking
In setting 2009 executive compensation levels, we relied on compensation statistics from various sources, including the 2007 edition of the Radford Global Life Sciences Survey.
In 2007, the compensation committee retained Radford to review our executive compensation program. In completing their 2007 review, Radford reviewed executive compensation data gathered from the most recent proxy statements of 22 U.S.-based biotechnology companies having the following characteristics:
| • | Either in late-stage clinical trials or the earlier stages of product launch (i.e., product revenues of less than $200 million); |
| • | Market capitalization between $500 million and $2 billion; and |
| • | Between 200 and 800 employees. |
The 22 companies in the peer group were:
| Alexion Pharmaceuticals Inc. | Exelixis Inc. | Progenics Pharmaceuticals Inc. | ||
| Arena Pharmaceuticals Inc. | GTx, Inc. | Regeneron Pharmaceuticals Inc. | ||
| Array Biopharma Inc. | Human Genome Sciences Inc. | Savient Pharmaceuticals Inc. | ||
| Auxilium Pharmaceuticals Inc. | InterMune Inc. | Theravance Inc. | ||
| BioMarin Pharmaceutical Inc. | MannKind Corporation | United Therapeutics Corporation | ||
| Cubist Pharmaceuticals, Inc. | Medarex Inc. | ViroPharma Incorporated | ||
| CV Therapeutics, Inc. | Myriad Genetics Inc. | Xenoport, Inc. | ||
| Onyx Pharmaceuticals Inc. | ||||
The compensation committee used the Radford’s findings as a component in establishing executive compensation levels in 2009. At the conclusion of their 2007 review, Radford made three principal recommendations in connection with executive compensation, namely, the target level for annual incentive awards, the target level for long-term incentive awards and the introduction of restricted stock grants as a part of our executive compensation program. The compensation committee adopted the first two recommendations of Radford but not the third.
27
Table of Contents
Base Salaries
We seek to establish base salaries that are competitive with salaries paid by other biotechnology companies of comparable size with similar business objectives. We believe that we compete with these companies for qualified executives.
We base a named executive officer’s salary on:
| • | an evaluation of salaries of individuals in similar positions in the biotechnology industry; and |
| • | an evaluation of the executive’s experience, scope of responsibilities and performance in achieving specific objectives. |
We conduct these evaluations sequentially. First, we establish a salary range for each named executive officer having a midpoint approximating the median base salary level for comparable positions within the biotechnology industry and a range from 37.5% below to 37.5% above the midpoint. The midpoint is determined annually using the benchmarking described above under the heading “Benchmarking.” Secondly, we fix each named executive officer’s actual base salary within the range based on his or her experience, scope of responsibilities and performance in achieving specific objectives. Each of these evaluations is performed annually for each named executive officer.
For 2009 salaries, annual base salary reviews for all our named executive officers other than our Chief Executive Officer, Dr. Williams, were conducted in conjunction with our company-wide performance management process. In light of the economic conditions prevailing at the time, the base salaries for executive officers were not increased for 2009 except for Dr. Williams in connection with his promotion to Chief Executive Officer in January 2009.
The base salary for each named executive officer identified in the Summary Compensation Table is subject to a minimum amount pursuant to an employment agreement. These minimum amounts are: for Dr. Williams, $550,000; for Mr. Zaruby $400,000; for Mr. Johnson, $369,200; for Ms. Franklin, $275,000; and for Dr. Ramos, $350,000.
Corporate and Individual Goals
We seek to structure incentive awards to encourage named executive officers to focus on achieving important near-term business objectives, which are comprised of both corporate and individual goals. Named executive officers are eligible for annual incentive awards, paid in the form of a cash bonus, and long-term incentive awards, paid in the form of stock options, based on the accomplishment of corporate goals, individual goals and important unanticipated contributions to our performance for the year. As part of the process of establishing our operating plan for the coming year, executive management identifies the goals most important to building our value and advancing our long-term business objectives. These corporate goals are then submitted to the Board of Directors for approval with regard to corporate strategy, and separately to the compensation committee for approval as the basis for incentive compensation. A named executive officer’s individual goals are supportive of the corporate goals and are developed in an iterative process of discussion between the named executive officer and our Chief Executive Officer.
For 2009, our corporate goals covered the following specific accomplishments and were given the following weights:
| • | RECOTHROM (35% weight) – executive plan to achieve sales budget; support Bayer in pursuing ex-U.S. regulatory approvals; and establish long-term surgical portfolio strategy. |
| • | Business development, alliance management and financial (30% weight) – establish effective structures to facilitate all necessary technology transfer, development and commercial plans and governance requirements for the PEG-interferon lambda collaboration; conduct IL-21 protein partnering process to reach term sheet stage; complete one or more partnership transactions related to preclinical programs; and end the year with more than two years of funding. |
| • | PEG-interferon lambda (25% weight) – complete Phase 1b study in Hepatitis C virus patients and supporting toxicology work for Phase 2 initiation; and initiate the first Phase 2 study to support registration program. |
| • | IL-21 (5% weight) – complete Phase 2 renal cell carcinoma study in combination with Nexavar® and complete enrollment of Phase 2 metastatic melanoma single-agent study. |
| • | Research (5% weight) – conduct toxicology studies for IL-21 mAb and manufacture IL-21 mAb clinical trial material to support Investigational New Drug application submission; and advance three designated molecules to lead development go/no go decision stage. |
Our compensation committee determined that we met or partially met all of our corporate goals in 2009. In addition, we made several major accomplishments that were not anticipated at the start of 2009, including successfully concluding a restructuring that eliminated $40 million from our expense base, redefining our strategic focus, renegotiating our relationship with Bayer Schering Pharma A.G., entering into an IL-21 mAb-related transaction with Novo Nordisk A/S and successfully positioning ourselves for an
28
Table of Contents
equity offering in the first week of 2010. Taking into consideration these unanticipated accomplishments and the prevailing economic conditions, the compensation committee concluded that our 2009 corporate performance was significantly above expectations and that the level of achievement of our corporate goals was 115%.
Within each named executive officer’s individual goals there is generally a range of difficulty such that there is a reasonable expectation that certain goals will be achieved while other goals will depend on superior or exceptional performance. Our Chief Executive Officer reports to the compensation committee regarding the performance of each named executive officer toward achievement of his or her individual goals. At the same time, the compensation committee assesses the achievement of corporate goals. Based on these evaluations, the compensation committee determines and approves the incentive amounts to be paid to each named executive officer. For 2009, the individual performance goals for our named executive officers covered the following specific accomplishments:
James A. Johnson
Mr. Johnson’s individual goals were:
| • | Supporting corporate restructuring efforts through reallocation of workloads, consolidation of space utilization and reductions to information technology overhead; |
| • | Managing operations to end the year with sufficient cash and cash equivalents to fund our operations for at least two years; |
| • | Ensuring effective collaboration with Bristol-Myers Squibb, including planning, budgeting, forecasting, time reporting and general management as one of our representatives on the joint steering committee; |
| • | Developing and implementing a revised strategic and tactical plan for corporate communications; |
| • | Completing an updated enterprise risk management assessment; |
| • | Coordinating improvements to our internal financial reporting process; and |
| • | Controlling actual expenses and capital costs within established budgets. |
Mr. Johnson’s level of achievement was determined to be 115%. In determining that Mr. Johnson had exceeded his individual goals the compensation committee considered his success in raising the profile of PEG-interferon lambda within the investment community and the high quality of the financial analyses that informed our strategic business review.
Stephen W. Zaruby
Mr. Zaruby’s individual goals were:
| • | Establishing a long-term surgical portfolio strategy; |
| • | Executing a plan to achieve established sales budget for Recothrom; |
| • | Managing our relationship with Bayer, including providing support for regulatory filings; |
| • | Maintaining positive investor interactions relating to Recothrom and other aspects of our business; and |
| • | Ensuring effective collaboration with Bristol-Myers Squibb, as one of our representatives on the joint steering committee. |
Mr. Zaruby’s level of achievement was determined to be 105%. In determining that Mr. Zaruby had exceeded his individual goals the compensation committee considered the measurable improvement in the performance of Recothrom that occurred shortly after Mr. Zaruby assumed responsibility for the brand.
Heather L. Franklin
Ms. Franklin’s individual goals were:
| • | Providing leadership during the strategic business review, including supporting the development of the long-term surgical portfolio strategy; |
| • | Integrating alliance and portfolio management and ensuring an effective interface with all parts of the organization; |
| • | Conducting an IL-21 partnering process to reach the term sheet stage: and |
| • | Establishing effective structures to facilitate all necessary technology transfer, development and commercial plans and governance requirements for the PEG-interferon lambda collaboration with Bristol-Myers Squibb. |
29
Table of Contents
Ms. Franklin’s level of achievement was determined to be 125%. In determining that Ms. Franklin had exceeded her individual goals the compensation committee considered the importance of the Bristol-Myers Squibb transaction consummated early in the year, the significant strategic and revenue generating transaction relating to IL-21 MAb, the successful restructuring of the Bayer relationship relating to Recothrom and that Ms. Franklin was a strong proponent of the structural and strategic changes we implemented during 2009 and, as such, made a significant contribution to the successful implementation of these changes.
Eleanor L. Ramos
Dr. Ramos’ individual goals were:
| • | With regard to PEG-interferon lambda facilitating the completion of the Phase lb study in Hepatitis C virus patients and the timely initiation of the Phase 2 study, and ensuring effective collaboration with Bristol-Myers Squibb, as one of our representatives on the joint steering committee; |
| • | With regard to IL-21, completing the Phase 2 renal cell carcinoma study and completing enrollment of the Phase 2a metastatic melanoma study; and |
| • | Maintaining positive investor interactions relating to our clinical development programs. |
Dr. Ramos’ level of achievement was determined to be 105%. In determining that Dr. Ramos had exceeded her individual goals the compensation committee considered the timely performance of our clinical organization and the resulting maintenance of credibility with our partners despite significant structural and personnel changes.
Annual Incentives
With regard to Dr. Williams, our Chief Executive Officer in 2009, 100% of his annual incentive award is based on actual company achievement of these corporate performance goals. With regard to each other named executive officer, 60% of his or her annual incentive award is based on actual company achievement of the corporate performance goals and the remaining 40% is based on actual individual achievement of their individual performance goals. This 60/40 (corporate/individual) formula reflects the compensation committee’s view that for annual incentive awards to be made at the target level both the company and the named executive officer must be performing well.
The compensation committee established the following payout ranges for 2009 annual incentive awards based on base salaries in effect as of the end of that year, as adjusted on February 1, 2009:
| Percentage of Base Salary | ||||||
| Name |
Target | Maximum | ||||
| Chief Executive Officer (Dr. Williams) |
50 | % | 75 | % | ||
| President (Mr. Zaruby) |
50 | % | 75 | % | ||
| Executive Vice President (Mr. Johnson) |
40 | % | 55 | % | ||
| Senior Vice President (Ms. Franklin, Dr. Onetto and Dr. Ramos) |
35 | % | 50 | % | ||
In determining the foregoing payout ranges, the compensation committee reviewed data from 22 similarly sized U.S. biotechnology companies (as reported in their proxy statements) and performed the other benchmarking described above under the heading “Benchmarking.” The survey data provided by Radford showed average annual incentive awards as a percentage of base salary as follows: Chief Executive Officer, 61%; President, 36%; Chief Scientific Officer, 49%; Chief Financial Officer, 36%; Chief Medical Officer, 40%; and Top Sales Executive, 47%.
Based on the compensation committee’s assessment of our company performance compared to corporate goals and, other than for our Chief Executive Officer, each named executive officer’s performance compared to individual goals, a percentage of overall target level is determined using the 60/40 formula described above. One hundred percent of the target level for corporate goals is awarded in cases where we met all of our goals and 100% of the target level for individual goals is awarded in cases where the executive has met all of his or her individual goals. Target levels may be exceeded to reward exceptional performance beyond expectations at the corporate or individual level. If goals are not achieved at the corporate or individual level, a percentage below 100% of target is awarded at the relevant level. A named executive officer might not receive any annual incentive award because there is no guaranteed minimum annual incentive award. Notwithstanding the forgoing, the compensation committee retains a general discretion to make an award of more or less than that determined by the 60/40 formula described above to take account of unforeseen or special circumstances.
30
Table of Contents
For 2009, annual incentive awards were paid to the named executive officers in early 2010 based on the percentages of target amount set forth in the table below.
| Name |
Percentage of Corporate Goals Achieved |
Percentage of Individual Goals Achieved |
Overall Percentage of Target Amount (1) | |||
| Douglas E. Williams, Ph.D. |
115% | Not Applicable | 115% | |||
| Bruce L.A. Carter, Ph.D.(2) |
Not Applicable | Not Applicable | Not Applicable | |||
| James A. Johnson |
115% | 115% | 115% | |||
| Stephen W. Zaruby |
115% | 105% | 111% | |||
| Heather L. Franklin |
115% | 125% | 119% | |||
| Nicole Onetto, M.D.(3) |
Not Applicable | Not Applicable | Not Applicable | |||
| Eleanor L. Ramos, M.D. |
115% | 105% | 111% |
| (1) | In determining the overall percentage of target amount, corporate goals bear a weight of, in the case of Dr. Williams, 100% and, in the case of the other named executive officers, 60%, and individual goals bear a weight of, in the case of Dr. Williams, 0% and, in the case of the other named executive officers, 40%. |
| (2) | Due to his retirement in January 2009, Dr. Carter was not awarded an annual incentive. |
| (3) | Due to her resignation in June 2009, Dr. Onetto was not awarded an annual incentive. |
The actual amount of annual incentive awards payable to each named executive officer for 2009 are reported in the Non-Equity Incentive Plan Compensation column of the Summary Compensation Table.
Long-Term Incentives
Long-term incentive awards, which consist of stock option grants, are a fundamental element in our executive compensation program. We believe that stock options are an effective way to emphasize long-term company performance, and reward named executive officers for the creation of value on the same basis as our shareholders.
A named executive officer typically receives a sizeable stock option grant when he or she joins us or receives a significant promotion. In determining the size of an initial option grant, level of responsibility and competitive factors are considered using the benchmarking described above under the heading “Benchmarking.”
Named executive officers also are eligible for annual stock option grants based on the achievement of the same individual and corporate goals that form the basis for the annual cash incentives. As with annual incentives, the long-term incentive award to our Chief Executive Officer is based entirely on actual company achievement of corporate performance goals. For the other named executive officers, 20% of the long-term incentive award is determined based on actual company achievement of corporate performance goals and the remaining 80% is based on actual achievement of individual performance goals related to his or her area of responsibility. This
31
Table of Contents
20/80 (corporate/individual) formula reflects the compensation committee’s view that individual performance is more significant in determining the size of a long-term incentive award because company performance, reflected through stock performance, is already factored into such an award by its nature. The compensation committee established the following ranges for annual stock option grants for 2009, which are set forth in the Grants of Plan-Based Awards table below:
| Number of Shares Granted | ||||
| Name |
Target | Maximum | ||
| Chief Executive Officer (Dr. Williams and Dr. Carter) |
150,000 | 225,000 | ||
| President (Mr. Zaruby) |
100,000 | 150,000 | ||
| Executive Vice President (Mr. Johnson) |
60,000 | 80,000 | ||
| Senior Vice President (Ms. Franklin, Dr. Onetto and Dr. Ramos) |
45,000 | 60,000 | ||
In determining the foregoing ranges, the compensation committee reviewed data from 22 similarly sized U.S. biotechnology companies (as reported in proxy statements) as described above under the heading “Benchmarking” and considered the foregoing ranges to be competitive.
A percentage of target level is determined for each named executive officer based on the compensation committee’s assessment of our overall performance and, other than for our Chief Executive Officer, each named executive officer’s individual performance compared to individual goals. One hundred percent of the target level for corporate goals is awarded in cases where we met all goals and 100% of the target level for individual goals is awarded in cases where the executive met all his or her individual goals. Target levels may be exceeded to reward exceptional performance beyond expectations at the corporate or individual level. If goals are not achieved at the corporate or individual level, a percentage below 100% of target is awarded at the relevant level. A named executive officer might not receive any long-term incentive award because there is no guaranteed minimum incentive award. Notwithstanding the forgoing, the compensation committee retains general discretion to make an award of more or less than that determined by the 20/80 formula described above to take account of unforeseen or special circumstances.
Stock option grants to the named executive officers for 2009 were awarded in January 2010 based on the percentages set forth in the table below.
| Name |
Percentage of Corporate Goals Achieved |
Percentage of Individual Goals Achieved |
Overall Percentage of Target Amount (1) |
Number of Shares Subject to Long-term Incentive Award | ||||
| Douglas E. Williams, Ph.D. (2) |
115% | Not Applicable | 115% | 250,000 | ||||
| Bruce L.A. Carter, Ph.D. (3) |
Not Applicable | Not Applicable | Not Applicable | — | ||||
| James A. Johnson |
115% | 115% | 115% | 69,000 | ||||
| Stephen W. Zaruby. |
115% | 105% | 107% | 107,000 | ||||
| Heather L. Franklin |
115% | 125% | 123% | 55,350 | ||||
| Nicole Onetto, M.D. (4) |
Not Applicable | Not Applicable | Not Applicable | — | ||||
| Eleanor L. Ramos, M.D. |
115% | 105% | 107% | 48,150 |
| (1) | In determining the overall percentage of target amount, corporate goals bear a weight of, in the case of Dr. Williams, 100% and, in the case of all other named executive officers, 20%, and individual goals bear a weight of, in the case of Dr. Williams, 0% and, in the case of all other named executive officers, 80%. |
| (2) | In the exercise of its discretion, the compensation committee increased Dr. Williams’ long-term incentive award by an additional 77,500 shares above that calculated using the formula. |
32
Table of Contents
| (3) | Due to his retirement in January 2009, Dr. Carter was not awarded a stock option as a long-term incentive. |
| (4) | Due to her resignation in June 2009, Dr. Onetto was not awarded a stock option as a long-term incentive. |
Annual stock option grants to the named executive officers and all other employees were awarded on the last business day of January. Stock options are granted under our 2001 Stock Incentive Plan (the “2001 Plan”), have an exercise price equal to the closing price of our common stock on the grant date and vest over a period of four years. We do not have a program, plan or practice to time stock option grants to executive officers in coordination with the release of material nonpublic information.
Stock Option Exchange Program
In early 2009, the compensation committee reviewed our use of equity incentives and noted that a large number of outstanding stock options were significantly out-of-the-money and therefore were no longer serving as effective incentive or retention tools, yet were being recorded as compensation expense by us and contributing to our potential employee equity overhang. In August 2009, the compensation committee recommended to the full board, and the board subsequently approved, a stock option exchange program, under which our employees, including executive officers, would be offered the opportunity to exchange eligible out-of-the-money stock options for new options having an exercise price per share equal to the closing price of our common stock on the day immediately following the expiration of the exchange offer. Outstanding stock options with an exercise price greater than $7.90 per share were eligible to participate in the stock option exchange program. Participants in the stock option exchange program received a new stock option to purchase a lesser number of shares for each eligible stock option surrendered in exchange at a lower exercise price than the options surrendered for cancellation. In addition, the new options vest over two or three years depending on whether the surrendered option was fully vested. The exchange ratio at which shares subject to eligible stock options were exchanged for shares subject to new stock options was set to result in the grant of replacement stock options for each grouping of surrendered options that, in the aggregate, had a fair value estimated to be approximately equal to the fair value of the cancelled stock options they replaced as of the date of the stock option exchange offer.
The compensation committee concluded that our executive officers should be eligible to participate in the stock option exchange program to provide them with improved incentives to increase shareholder value, increase the retention value of outstanding options and reduce the total number of potential shares directed toward employee incentive programs—all at virtually no additional compensation expense to the company.
The stock option exchange program was approved by our shareholders on November 10, 2009 and the exercise price of the replacement options was set at $6.35, the closing price of our common stock on December 16, 2009 (the day immediately following the expiration of the stock option exchange offer).
A total of 186 employees participated in the stock option exchange program, including three of our named executive officers. Information regarding the stock options surrendered and new options received by the named executive officers who participated in the stock option exchange program is set forth in the following table:
| Name |
Number of Shares Subject to Surrendered Options |
Exercise Price
of Surrendered Options ($) |
Number of Shares Subject to New Options |
Exercise Price of New Options ($) | ||||
| Douglas E. Williams, Ph.D. |
593,674 | 10.89 – 19.27 | 280,066 | 6.35 | ||||
| Bruce L.A. Carter, Ph.D. |
— | — | — | — | ||||
| James A. Johnson |
205,324 | 14.73 – 21.26 | 94,983 | 6.35 | ||||
| Stephen W. Zaruby. |
— | — | — | — | ||||
| Heather L. Franklin |
— | — | — | — | ||||
| Nicole Onetto, M.D. |
— | — | — | — | ||||
| Eleanor L. Ramos, M.D. |
50,000 | 15.11 | 28,570 | 6.35 |
33
Table of Contents
Perquisites
Although we prefer to limit perquisites, Dr. Carter received the use of an automobile, a health club membership and tax advice. These perquisites were negotiated prior to our separation from Novo Nordisk A/S in 2000 and were based on the perquisites Dr. Carter received during his employment by Novo Nordisk. Following his retirement as Chief Executive Officer in January 2009, Dr. Carter is no longer eligible to receive these benefits. Dr. Williams did not receive comparable perquisites upon his promotion to Chief Executive Officer.
Deferred Compensation Plan
Our named executive officers are eligible to participate in our Deferred Compensation Plan for Key Employees, which is intended to allow our executives to defer current income, without being limited by the Internal Revenue Code contribution limitations for 401(k) plans. Under the Deferred Compensation Plan, an executive may irrevocably elect, on an annual basis, to defer up to 50% of his or her salary and up to 100% of his or her bonus paid for services rendered in the relevant year. Each year, an account is established under the Deferred Compensation Plan to reflect the amount deferred by the executive for such year. The executive’s Deferred Compensation Plan accounts are adjusted for notional investment earnings. These earnings are based on the return of the investment tracking funds to which the executive has allocated his or her accounts under the Deferred Compensation Plan. The investment tracking funds are all publicly traded mutual funds available to participants in our 401(k) plan. Executives may change how their accounts are allocated among the tracking funds at any time. No named executive officers participated in the Deferred Compensation Plan in 2009, and the Board of Directors voted to freeze further deferrals under the Deferred Compensation Plan effective September 1, 2009.
Dr. Carter is the only named executive officers identified in the Summary Compensation Table who has participated in the Deferred Compensation Plan. Dr. Carter did not, however, participate in the Deferred Compensation Plan during 2009. Disclosures regarding Dr. Carter’s participation are provided in the Nonqualified Deferred Compensation table below and the narrative explanation that accompanies that table.
Stock Ownership Guidelines
Currently, there is no requirement for a named executive officer to own our stock. The compensation committee has discussed the possibility of introducing stock ownership guidelines for named executive officers in connection with the possible introduction of stock-based compensation vehicles other than stock options. There are, however, currently no plans to introduce stock ownership guidelines or stock-based compensation vehicles other than stock options for named executive officers.
Potential Post-Termination Payments
Based on the data from Radford described above under the heading “Benchmarking,” we believe that the payments described under the heading “Potential Payments Upon Termination or a Change in Control” below are customary in the industry and necessary to attract and retain qualified, experienced executive personnel.
Tax and Accounting
Section 162(m) of the Internal Revenue Code imposes a limitation on the deductibility of compensation payments in excess of $1 million to each of our Chief Executive Officer and our other named executive officers (other than our Chief Financial Officer). Certain performance-based compensation is not subject to the limitation on deductibility. For awards under our 2001 Plan to continue to be deductible under Section 162(m) as performance-based compensation, shareholders were asked at our 2006 annual meeting of shareholders to approve the material terms of the 2001 Plan. Shareholders approved the material terms of the 2001 Plan and, therefore, long-term awards granted under the 2001 Plan will continue to be designed to qualify for the performance-based exception to the $1 million limitation on deductibility of compensation payments. In 2009, compensation to our Chief Executive Officer and each of our other named executive officers did not exceed $1 million for purposes of Section 162(m), and we expect the same to be true for 2010. We may, however, in the future approve annual compensation that could exceed the $1 million limitation if we believe that it is in the best interests of our shareholders.
Summary Compensation Table for Fiscal Year 2009
The following table provides certain compensation information for the fiscal year ended December 31, 2009 for our Chief Executive Officer, Chief Financial Officer and our three most highly compensated executives other than our Chief Executive Officer and Chief Financial Officer. 2008 and 2007 compensation information is also provided for those executives who were included in the Summary Compensation Table for fiscal years 2008 and 2007.
| Name and Principal Position |
Year | Salary (1) (2) ($) |
Stock Awards (3) ($) |
Option Awards (4) ($) |
Non-Equity Incentive Plan Compensation (5) (6) ($) |
All Other Compensation (7) ($) |
Total ($) | |||||||
| Douglas E. Williams, Ph.D.* |
2009 | 549,745 | 443,188 | 316,250 | 9,116 | 1,318,299 | ||||||||
| Chief Executive Officer |
2008 2007 |
482,050 437,917 |
1,305 | 1,821,577 837,706 |
140,244 209,250 |
11,745 9,740 |
2,456,921 1,494,613 | |||||||
| Bruce L.A. Carter, Ph.D.* |
2009 | 4,839 | — | — | 222,718 | 227,557 | ||||||||
| Former Chief Executive Officer and Chairman of the Board |
2008 2007 |
636,681 612,551 |
1,305 | 789,615 1,301,100 |
185,231 276,373 |
43,116 41,225 |
1,655,948 2,231,249 | |||||||
| James A. Johnson |
2009 | 369,200 | 266,749 | 169,832 | 11,098 | 816,879 | ||||||||
| Executive Vice President, Chief Financial Officer, Treasurer and Secretary |
2008 2007 |
368,017 338,968 |
1,305 | 276,365 634,430 |
107,511 111,825 |
17,056 14,804 |
770,254 1,100,027 | |||||||
| Stephen W. Zaruby |
2009 | 398,485 | 740,703 | 222,000 | 242,605 | 1,603,793 | ||||||||
| President |
||||||||||||||
| Heather L. Franklin |
2009 | 283,021 | 246,077 | 125,991 | 15,260 | 670,349 | ||||||||
| Senior Vice President, Business Development |
||||||||||||||
| Nicole Onetto, M.D.† |
2009 | 219,319 | 91,612 | — | 417,486 | 728,417 | ||||||||
| Senior Vice President and Chief Medical Officer |
2008 2007 |
374,483 357,132 |
1,305 | 193,456 327,877 |
95,798 88,623 |
18,398 15,312 |
683,440 788,944 | |||||||
| Eleanor L. Ramos, M.D.† |
2009 | 335,261 | 299,298 | 135,975 | 12,661 | 783,195 | ||||||||
| Senior Vice President and Chief Medical Officer |
||||||||||||||
| * | Dr. Carter retired as our Chief Executive Officer in January 2009. Dr. Williams became our Chief Executive Officer effective as of Dr. Carter’s retirement. |
34
Table of Contents
| † | Dr. Onetto resigned as our Senior Vice President and Chief Medical Officer in May 2009. Dr. Ramos became our Senior Vice President and Chief Medical Officer in June 2009. |
| (1) | The amounts reported in the Salary column represent the dollar amounts actually earned by each executive in the year indicated, including adjustments for annual pay raises and pay raises in connection with promotions. |
| (2) | The amounts reported in the Salary column for Dr. Carter include amounts deferred under the Deferred Compensation Plan for Key Employees. Dr. Carter deferred $0 in 2009, $127,336 in 2008 and $122,510 in 2007. |
| (3) | The amounts reported in the Stock Awards column reflect the grant date fair value related to a grant of 100 unrestricted shares of our common stock to each of our employees, including the named executive officers, to mark the approval of RECOTHROM by the FDA. The grant date fair value for each named executive officer was equal to the product of 100 shares times $13.05 per share, the closing price of our common stock on the grant date of the shares. |
| (4) | The amounts reported in the Option Awards column reflect the aggregate grant date fair value of option awards, without adjustment for potential forfeiture. These amounts do not represent the actual amounts paid to or realized by the named executive officer for these awards during the relevant fiscal year. For fiscal 2009, these amounts also include the incremental fair value received by each named executive officer that participated in the stock option exchange program that was consummated in December 2009. See Note 14 of Notes to Consolidated Financial Statements in our Annual Report on Form 10-K for fiscal year 2009 for the assumptions used in determining the grant date fair value of stock options. |
| (5) | The amounts reported in the Non-Equity Incentive Plan Compensation column represent the amount of the annual incentive award earned for performance in the year indicated but paid out in the subsequent year. Annual incentive awards are based on the salaries in effect on the award date. |
| (6) | The amounts reported in the Non-Equity Incentive Plan Compensation column for Dr. Carter include amounts deferred under the Deferred Compensation Plan for Key Employees. Dr. Carter deferred $0 in 2009 (based on 2008 performance), $276,373 in 2008 (based on 2007 performance) and $297,415 in 2007 (based on 2006 performance). |
| (7) | For Dr. Carter, the amounts shown in the All Other Compensation column for 2009 include $15,035 for continuation of health insurance after his retirement and $206,909 paid in connection with Dr. Carter’s accrued unused vacation balance. For Mr. Zaruby, the amounts shown in the All Other Compensation column include a retention bonus that was grossed up for taxes resulting in a total cost to the company of $107,971 and payments in connection with his relocation totaling $132,874. For Dr. Onetto, the amounts shown in the All Other Compensation column for 2009 include severance payments of $375,975 and $24,155 paid in connection with Dr. Onetto’s accrued unused vacation balance. In addition, the amounts shown in the All Other Compensation column for 2009 include employer contributions to our 401(k) retirement plan of up to $9,800 for each named executive officer (other than Dr. Carter), the reimbursement of home telephone bills and premiums for supplemental term life insurance. Dr. Carter received $0 in employer matching contributions in 2009. |
35
Table of Contents
Grants of Plan-Based Awards for Fiscal Year 2009
The following table provides information regarding grants of plan-based awards to the named executive officers identified below under our 2001 Plan and under our annual incentive plan during the fiscal year ended December 31, 2009.
| Name |
Grant Date | Estimated Future Payouts under Non-Equity Incentive Plan Awards (1) |
Estimated Future Payouts under Equity Incentive Plan Awards (2) |
All Other Option Awards: Number of Securities Underlying Options (#) |
Exercise or Base Price of Option Awards (3) ($/Sh) |
Grant Date Fair Value of Stock and Option Awards (4) ($) | |||||||||||
| Target ($) |
Maximum ($) |
Target (#) |
Maximum (#) |
||||||||||||||
| Douglas E. Williams, Ph.D. |
01/30/2009 05/15/2009 12/16/2009 |
(5) (6) (7) |
87,000 100,000 280,066 |
4.25 4.37 6.35 |
202,188 241,000 — | ||||||||||||
| 275,000 | 412,500 | ||||||||||||||||
| 150,000 | 225,000 | ||||||||||||||||
| Bruce L.A. Carter, Ph.D. |
Not Applicable |
|
|||||||||||||||
| Not Applicable |
Not Applicable |
||||||||||||||||
| Not Applicable |
Not Applicable |
||||||||||||||||
| James A. Johnson |
01/30/2009 05/15/2009 12/16/2009 |
(5) (6) (7) |
52,560 60,000 94,983 |
4.25 4.37 6.35 |
122,149 144,600 — | ||||||||||||
| 147,680 | 203,060 | ||||||||||||||||
| 60,000 | 80,000 | ||||||||||||||||
| Stephen W. Zaruby |
01/02/2009 05/15/2009 06/10/2009 |
(8) (6) (6) |
300,000 50,000 50,000 |
3.00 4.37 4.61 |
489,900 120,500 130,303 | ||||||||||||
| 200,000 | 300,000 | ||||||||||||||||
| 100,000 | 150,000 | ||||||||||||||||
| Heather L. Franklin |
01/30/2009 05/15/2009 |
(5) (6) |
59,220 45,000 |
4.25 4.37 |
137,627 108,450 | ||||||||||||
| 96,250 | 137,500 | ||||||||||||||||
| 45,000 | 60,000 | ||||||||||||||||
| Nicole Onetto, M.D. |
01/30/2009 | (5) | 39,420 | 4.25 | 91,612 | ||||||||||||
| Not Applicable |
Not Applicable |
||||||||||||||||
| Not Applicable |
Not Applicable |
||||||||||||||||
| Eleanor L. Ramos, M.D. |
01/30/2009 05/15/2009 06/08/2009 12/16/2009 |
(5) (6) (9) (7) |
7,500 6,750 100,000 28,570 |
4.25 4.37 4.72 6.35 |
16,763 15,620 266,915 — | ||||||||||||
| 122,500 | 175,000 | ||||||||||||||||
| 45,000 | 60,000 | ||||||||||||||||
| (1) | The amounts shown in the Estimated Future Payouts under Non-Equity Incentive Plan Awards column are the payout levels for annual incentive awards described under the heading “Compensation Discussion and Analysis—Annual Incentives.” The amounts shown are based on salaries as of February 1, 2009. Because the lowest possible payment is zero, we have not indicated a threshold payout amount. Actual amounts earned for 2009 performance are reflected in the Non-Equity Incentive Plan Compensation column of the Summary Compensation Table. |
| (2) | The amounts shown in the Estimated Future Payouts under Equity Incentive Plan Awards column are the payout levels for long-term incentive awards described under the heading “Compensation Discussion and Analysis—Long-Term Incentives.” Actual stock option grants were approved in January 2010 and are |
36
Table of Contents
| discussed under the heading “Compensation Discussion and Analysis—Long-Term Incentives.” Due to his retirement in January 2009, Dr. Carter was not awarded a stock option as a long-term incentive. |
| (3) | The exercise price of the options is equal to the closing price of the common stock on the grant date as reported by the NASDAQ Global Market. |
| (4) | Represents grant date fair value of stock and stock options granted in 2009. See Note 14 of Notes to Consolidated Financial Statements in our Annual Report on Form 10-K for fiscal year 2009 for the assumptions used in determining the grant date fair value of stock options. The grant date fair value of the stock awards is equal to the fair market value of such awards on the grant date. The stock options granted pursuant to the stock option exchange program that was consummated in December 2009 did not have any incremental grant date fair value. |
| (5) | The option was granted as a long-term incentive award based on performance in 2008. |
| (6) | The option was granted as a retention bonus. |
| (7) | The options were granted pursuant to the stock option exchange program that was consummated in December 2009. Each option has a term of seven years from the grant date. Of the 280,066 options granted to Dr. Williams, (a) 134,842 vest over two years, with 50% of the shares subject to the option vesting on the first anniversary of the grant date, and 12.5% of the shares subject to the option vesting quarterly over the ensuing four quarters (the “Two-Year Vesting Schedule”) and (b) 145,224 vest over three years, with 33.3% of the shares subject to the option vesting on the first anniversary of the grant date, and 6.25% of the shares subject to the option vesting quarterly over the ensuing eight quarters (the “Three-Year Vesting Schedule”). Of the 94,983 options granted to Mr. Johnson, 50,347 vests according to the Two-Year Vesting Schedule and 44,636 vests according to the Three-Year Vesting Schedule. All of the 28,570 options granted to Dr. Ramos vest according to the Three-Year Vesting Schedule. |
| (8) | The option was granted in connection with the hiring of Mr. Zaruby as our President. |
| (9) | The option was granted in connection with Dr. Ramos’ promotion to Senior Vice President and Chief Medical Officer. |
The plans governing the awards included in the Summary Compensation Table and the Grants of Plan-Based Awards table are the annual incentive plan and the 2001 Plan. The annual incentive plan is described in Item 11 of this Annual Report on Form 10-K under the heading “Compensation Discussion and Analysis—Annual Incentives.” Each option shown in the Grants of Plan-Based Awards table was granted under the 2001 Plan, has a 10-year term, except as otherwise set forth in footnote (7) above, and is granted as an incentive stock option to the maximum extent permitted under applicable federal tax rules and, to the extent not so permitted, is granted as a nonqualified stock option. Except as otherwise set forth in footnote (7) above, each option shown in the Grants of Plan-Based Awards table vests as follows: 1/4th of the shares subject to the option vest upon the first anniversary of the grant date, and thereafter 1/12th of the shares subject to the option vest each quarter until the award is fully vested on the fourth anniversary of the grant date.
37
Table of Contents
Outstanding Equity Awards at 2009 Fiscal Year-End
The following table provides information about outstanding stock options held by the named executive officers as of December 31, 2009. The options granted in 2009 are also disclosed in the Grants of Plan-Based Awards table above.
| Name |
Option Awards | ||||||||
| Number of Securities Underlying Unexercised Options (#) |
Option Exercise Price ($) |
Option Expiration Date | |||||||
| Exercisable | Unexercisable | ||||||||
| Douglas E. Williams, Ph.D. |
— | 500,000 | (1) | 3.50 | 11/18/2018 | ||||
| — | 87,000 | (2) | 4.25 | 01/30/2019 | |||||
| — | 100,000 | (2) | 4.37 | 05/15/2019 | |||||
| — | 79,609 | (3) | 6.35 | 12/16/2016 | |||||
| — | 111,697 | (4) | 6.35 | 12/16/2016 | |||||
| — | 4,008 | (3) | 6.35 | 12/16/2016 | |||||
| — | 1,269 | (4) | 6.35 | 12/16/2016 | |||||
| — | 1,731 | (3) | 6.35 | 12/16/2016 | |||||
| — | 5,248 | (3) | 6.35 | 12/16/2016 | |||||
| — | 29,134 | (3) | 6.35 | 12/16/2016 | |||||
| — | 3,226 | (3) | 6.35 | 12/16/2016 | |||||
| — | 8,302 | (3) | 6.35 | 12/16/2016 | |||||
| — | 22,268 | (3) | 6.35 | 12/16/2016 | |||||
| — | 13,574 | (4) | 6.35 | 12/16/2016 | |||||
| Bruce L.A. Carter, Ph.D. |
426,000 | — | 2.78 | 08/23/2010 | |||||
| 288,000 | — | 2.78 | 03/08/2011 | ||||||
| 166,177 | — | 8.50 | 12/16/2012 | ||||||
| 119,761 | — | 17.15 | 01/07/2014 | ||||||
| 169,920 | — | 21.26 | 01/26/2015 | ||||||
| 50,000 | — | 20.61 | 02/03/2015 | ||||||
| 126,056 | 7,188 | (2) | 18.38 | 01/05/2016 | |||||
| 103,125 | 46,875 | (2) | 15.27 | 01/05/2017 | |||||
| 59,062 | 75,938 | (2) | 10.89 | 01/18/2018 | |||||
| — | 12,000 | (5) | 4.61 | 06/10/2019 | |||||
| — | 12,000 | (5) | 6.15 | 09/16/2019 | |||||
| James A. Johnson |
145,000 | — | 2.78 | 03/02/2011 | |||||
| 54,000 | — | 2.78 | 03/08/2011 | ||||||
| 35,000 | — | 9.80 | 03/21/2013 | ||||||
| 20,671 | 26,579 | (2) | 10.89 | 01/18/2018 | |||||
| — | 52,560 | (2) | 4.25 | 01/30/2019 | |||||
| — | 60,000 | (2) | 4.37 | 05/15/2019 | |||||
| — | 1,066 | (4) | 6.35 | 12/16/2016 | |||||
| — | 3,042 | (4) | 6.35 | 12/16/2016 | |||||
| — | 8,336 | (4) | 6.35 | 12/16/2016 | |||||
| — | 16,958 | (4) | 6.35 | 12/16/2016 | |||||
| — | 15,019 | (4) | 6.35 | 12/16/2016 | |||||
| — | 26,027 | (3) | 6.35 | 12/16/2016 | |||||
| — | 4,263 | (4) | 6.35 | 12/16/2016 | |||||
| — | 877 | (4) | 6.35 | 12/16/2016 | |||||
| — | 786 | (4) | 6.35 | 12/16/2016 | |||||
| — | 11,082 | (3) | 6.35 | 12/16/2016 | |||||
| — | 3,801 | (3) | 6.35 | 12/16/2016 | |||||
| — | 1,780 | (3) | 6.35 | 12/16/2016 | |||||
| — | 1,946 | (3) | 6.35 | 12/16/2016 | |||||
| Stephen W. Zaruby |
— | 300,000 | (2) | 3.00 | 01/02/2019 | ||||
| — | 50,000 | (2) | 4.37 | 05/15/2019 | |||||
| — | 50,000 | (2) | 4.61 | 06/10/2019 | |||||
38
Table of Contents
| Name |
Option Awards | ||||||||
| Number of Securities Underlying Unexercised Options (#) |
Option Exercise Price ($) |
Option Expiration Date | |||||||
| Exercisable | Unexercisable | ||||||||
| Heather L. Franklin |
2,579 | — | 9.80 | 03/21/2013 | |||||
| 7,800 | — | 15.45 | 09/12/2013 | ||||||
| 8,000 | — | 16.63 | 02/26/2014 | ||||||
| 15,000 | — | 31.26 | 01/26/2015 | ||||||
| 9,000 | — | 20.61 | 02/03/2015 | ||||||
| 8,437 | 563 | (2) | 21.25 | 02/08/2016 | |||||
| 5,156 | 2,344 | (2) | 15.91 | 02/05/2017 | |||||
| 3,500 | 4,500 | (2) | 9.10 | 02/15/2018 | |||||
| — | 5,000 | (6) | — | — | |||||
| 8,250 | 13,750 | (2) | 9.02 | 05/30/2018 | |||||
| — | 59,220 | (2) | 4.25 | 01/30/2019 | |||||
| — | 45,000 | (2) | 4.37 | 05/15/2019 | |||||
| Nicole Onetto, M.D. |
187,500 | — | 15.90 | 09/23/2015 | |||||
| 8,298 | — | 18.38 | 01/05/2016 | ||||||
| 23,625 | — | 15.27 | 01/05/2017 | ||||||
| 12,403 | — | 10.89 | 01/18/2018 | ||||||
| Eleanor L. Ramos, M.D. |
3,281 | 4,219 | (2) | 9.10 | 02/15/2018 | ||||
| — | 5,000 | (6) | — | — | |||||
| — | 7,500 | (2) | 4.25 | 01/30/2019 | |||||
| — | 6,750 | (2) | 4.37 | 05/15/2019 | |||||
| — | 100,000 | (2) | 4.72 | 06/08/2019 | |||||
| — | 13,130 | (3) | 6.35 | 12/16/2016 | |||||
| — | 15,440 | (3) | 6.35 | 12/16/2016 | |||||
| (1) | Vests 1/4th on the one-year anniversary of Dr. Williams promotion to Chief Executive Officer and then 1/12th quarterly thereafter such that the option is fully vested on the fourth anniversary of the grant date. |
| (2) | Vests 1/4th on the first anniversary of the grant date (which grant date is ten years prior to the expiration date of the relevant option set forth in the Option Expiration Date column), and then 1/12th quarterly thereafter such that the option is fully vested on the fourth anniversary of the grant date. |
| (3) | Vests 1/3rd on the first anniversary of the grant date (which grant date is ten years prior to the expiration date of the relevant option set forth in the Option Expiration Date column), and then 1/8th quarterly thereafter such that the option is fully vested on the third anniversary of the grant date. |
| (4) | Vests 1/2 on the first anniversary of the grant date (which grant date is ten years prior to the expiration date of the relevant option set forth in the Option Expiration Date column), and then 1/4th quarterly thereafter such that the option is fully vested on the second anniversary of the grant date. |
| (5) | Vests 100% on the first anniversary of the grant date (which grant date is ten years prior to the expiration date of the relevant option set forth in the Option Expiration Date column). |
| (6) | Represents the grant on February 22, 2008 of a Restricted Stock Unit (RSU) representing 7,500 shares of stock under the 2001 Plan, which RSU vests 1/3rd on each of February 22, 2009, February 22, 2010 and February 22, 2011. |
39
Table of Contents
Option Exercises and Stock Vested for Fiscal Year 2009
The following table shows for the fiscal year ended December 31, 2009, certain information regarding option exercises and stock vested during the last fiscal year with respect to the named executive officers:
| Option Awards | Stock Awards | |||||||
|
|
Number of Shares Acquired on Exercise (#) |
Value Realized on Exercise ($) (1) |
Number of Shares Acquired on Vesting (#) |
Value Realized on Vesting ($) (2) | ||||
| Douglas E. Williams, Ph.D. |
— | — | — | — | ||||
| Bruce L.A. Carter, Ph.D. |
— | — | — | — | ||||
| James A. Johnson |
10,000 10,000 |
32,223 42,223 |
— | — | ||||
| Stephen W. Zaruby |
— | — | — | — | ||||
| Heather L. Franklin |
— | — | 2,500 | 11,700 | ||||
| Nicole Onetto, M.D. |
— | — | — | — | ||||
| Eleanor L. Ramos, M.D. |
— | — | 2,500 | 11,700 | ||||
| (1) | The amounts shown in the Value Realized on Exercise column represent the difference between the stock option exercise price and the fair market value of our common stock on the date of exercise multiplied by the number of shares subject to the stock option that was exercised. |
| (2) | The amounts shown in the Value Realized on Vesting column represent the fair market value of our common stock on the date the award vested multiplied by the number of shares acquired on vesting. |
Nonqualified Deferred Compensation for Fiscal Year 2009
The following table provides information for each of our named executive officers regarding aggregate executive and company contributions and aggregate earnings for 2009 and year-end account balances under the Deferred Compensation Plan for Key Employees. All named executive officers identified in the Summary Compensation Table are eligible to participate in the Deferred Compensation Plan. To date, however, only Dr. Carter has elected to participate.
| Name |
Executive Contributions in Last Fiscal Year ($) |
Company Contributions in Last Fiscal Year (#) (1) |
Aggregate Earnings in Last Fiscal Year ($) (2) |
Aggregate Withdrawals/ Distributions ($) |
Aggregate Balance at Last Fiscal Year-End ($) (3) | |||||
| Douglas E. Williams, Ph.D. |
— | — | — | — | — | |||||
| Bruce L.A. Carter, Ph.D. |
— | — | 178,632 | 1,308,727 | 1,056,011 | |||||
| James A. Johnson |
— | — | — | — | — | |||||
| Stephen W. Zaruby |
— | — | — | — | — | |||||
| Heather L. Franklin |
— | — | — | — | — | |||||
| Nicole Onetto, M.D. |
— | — | — | — | — | |||||
| Eleanor L. Ramos, M.D. |
— | — | — | — | — |
| (1) | ZymoGenetics does not contribute to the Deferred Compensation Plan. |
| (2) | This amount represents the actual increase in the value of the investments selected by the executive. No related amount was included in the Summary Compensation Table for fiscal year 2009 because none of the amount shown was above-market or preferential. |
| (3) | $122,510 and $127,336 of the amount deferred by Dr. Carter was included in the Salary column of the Summary Compensation Table for fiscal years 2007 and 2008, respectively. $276,373 of the amount shown was included in the Non-Equity Incentive Compensation column of the Summary Compensation Table for fiscal year 2007. |
40
Table of Contents
The Deferred Compensation Plan is a nonqualified deferred compensation plan. As a result, it is subject to the requirements of Section 409A of the Internal Revenue Code, and we have administered it in good-faith compliance with those requirements since January 1, 2004, Section 409A’s effective date. During 2008, we amended the Deferred Compensation Plan to conform it to Section 409A’s requirements. Amounts deferred prior to 2005 (and any notional earnings attributable to those deferrals, as explained below), however, are not subject to Section 409A’s requirements and continue to be governed under the terms of the Deferred Compensation Plan as in effect on December 31, 2004. Under the Deferred Compensation Plan, an executive is fully vested in his or her deferrals (and any notional earnings attributable to those deferrals) at all times.
Under the Deferred Compensation Plan, a named executive officer may elect to defer up to 50% of his or her salary and up to 100% of his or her bonus for a year. Each year, an account is established under the Deferred Compensation Plan to reflect the amount deferred by the executive for such year. The executive’s Deferred Compensation Plan accounts are also adjusted for notional investment earnings, which are based on the return of the investment tracking funds to which the executive has allocated his or her accounts. The investment tracking funds are all publicly traded mutual funds available to participants in our 401(k) plan. Executives may change how their accounts are allocated among the tracking funds at any time. The Deferred Compensation Plan also permits us to make additional contributions on behalf of one or more participants. No such contributions have, however, ever been made. These additional contributions, if ever made, will be subject to the same vesting, distribution and notional investment rules as are executive deferrals.
The Board of Directors voted to freeze further deferrals under Deferred Compensation Plan effective September 1, 2009.
At the time the executive files his or her deferral election for a particular year, the executive must designate whether he or she wants the amount to be deferred for that year (and any notional earnings attributable to that amount) paid at termination of employment or on a different date. If an executive elects a date other than termination of employment, then that date may not be earlier than three years after the election. Similarly, at the time of the deferral election, the executive must irrevocably designate whether he or she wishes to have the amount deferred for the relevant year (and any notional earnings attributable to that amount) distributed in a lump sum or in substantially equal annual installments. Any annual installments can be paid over a period of up to ten years (as elected by the executive). Executives are not permitted to change their distribution elections. If an executive is a “specified employee” as defined in Section 409A of the Internal Revenue Code (generally, any company officer) on his or her termination date and the executive elected to have payment made or commence to be made upon termination of employment, then, as required by Section 409A, payment will be delayed until six months after the executive’s termination. As of December 31, 2009, Dr. Carter was a specified employee. If the executive dies before all of his or her deferrals (and related notional earnings) have been distributed, then any remaining amounts will be paid to his or her beneficiary in a lump sum.
There are two situations in which an executive can withdraw amounts from his or her Deferred Compensation Plan accounts early (that is, prior to the date(s) he or she has elected for distribution of those accounts). First, under certain circumstances, an executive may elect an early withdrawal from his or her accounts if he or she experiences an unforeseeable emergency. Second, for amounts that were deferred prior to 2005 (but not amounts that are deferred in 2005 or later), the executive may elect an early withdrawal from his or her accounts for any reason by paying a penalty equal to 10% of the amount withdrawn.
The Deferred Compensation Plan is considered to be an unfunded plan, which means its benefits are paid by us out of our general assets. To assist us in meeting our payment obligations under the Deferred Compensation Plan, we have established a trust to which we contribute amounts equal to the executives’ deferrals each year. The trust’s assets are considered to be part of our general assets, so that if we ever become bankrupt or insolvent, they will be available to help us satisfy our obligations to all of our general creditors, not just our obligations under the Deferred Compensation Plan. (The executives are considered to be general creditors with respect to their accounts under the Deferred Compensation Plan.) Benefits are paid from this trust, to the extent its assets are sufficient to pay them, and from our other general assets, to the extent the trust’s assets are not sufficient to pay them.
41
Table of Contents
Potential Payments Upon Termination or a Change in Control
The table below describes the potential payments to each of our named executive officers upon an involuntary termination of employment without cause by us or with good reason by the executive prior to, or in the event of, a change in control and a termination of employment by reason of death, or disability, resignation or retirement. The table reflects incremental compensation and does not show accrued liabilities, such as accrued salary, bonus or vacation, or benefits generally available to all employees. The amounts assume that each such termination was effective December 31, 2009 (the last business day of fiscal 2009). The amounts shown in the table are estimates only as the actual amounts that may be paid upon an executive’s termination of employment can only be determined at the actual time of such termination.
Dr. Carter retired as our Chief Executive Officer effective January 2, 2009. In connection with his retirement we paid Dr. Carter $206,909 in lieu of accrued vacation and agreed to pay his COBRA premiums for 24 months. The estimated cost of the COBRA premiums for Dr. Carter is $24,892. Dr. Carter did not receive any severance benefits as a result of his retirement.
Dr. Onetto resigned as our Senior Vice President and Chief Medical Officer effective June 5, 2009 and received a $375,975 severance payment pursuant to a separation agreement effective as of July 23, 2009. In addition, we paid Dr. Onetto $24,155 in lieu of accrued vacation.
| Name |
Benefit |
Involuntary (Without Cause or Good Reason) Termination Prior to Change in Control ($) |
Change in Control ($) (1) |
Death or Disability ($) | ||||||
| Douglas E. Williams, Ph.D. |
Base Salary |
550,000 | (5) | 1,100,000 | (6) | — | ||||
| Bonus |
— | 825,000 | — | |||||||
| Supplemental Life Insurance (2) |
— | — | 200,000 | |||||||
| Accelerated Stock Option Vesting (3) |
— | 1,844,383 | — | |||||||
| COBRA Premiums (4) |
18,322 | 18,322 | — | |||||||
| Total |
568,322 | 3,787,705 | 200,000 | |||||||
| James A. Johnson |
Base Salary |
369,200 | (5) | 553,800 | (6) | — | ||||
| Bonus |
— | 369,200 | — | |||||||
| Supplemental Life Insurance (2) |
— | — | 200,000 | |||||||
| Accelerated Stock Option Vesting (3) |
— | 237,478 | — | |||||||
| COBRA Premiums (4) |
18,322 | 18,322 | — | |||||||
| Total |
387,522 | 1,178,800 | 200,000 | |||||||
| Stephen W. Zaruby |
Base Salary |
400,000 | (5) | 800,000 | (6) | — | ||||
| Bonus |
— | 600,000 | — | |||||||
| Supplemental Life Insurance (2) |
— | — | 200,000 | |||||||
| Accelerated Stock Option Vesting (3) |
— | 1,207,000 | — | |||||||
| COBRA Premiums (4) |
10,854 | 10,854 | — | |||||||
| Total |
410,854 | 2,617,854 | 200,000 | |||||||
| Heather L. Franklin |
Base Salary |
302,500 | (5) | 302,500 | (6) | — | ||||
| Bonus |
— | 211,750 | — | |||||||
| Supplemental Life Insurance (2) |
— | — | 200,000 | |||||||
| Accelerated Stock Option Vesting (3) |
— | 217,631 | — | |||||||
| COBRA Premiums (4) |
12,446 | 12,446 | — | |||||||
| Total |
314,946 | 744,327 | 200,000 | |||||||
| Eleanor L. Ramos, M.D. |
Base Salary |
350,000 | (5) | 350,000 | (6) | — | ||||
| Bonus |
— | 245,000 | — | |||||||
| Supplemental Life Insurance (2) |
— | — | 200,000 | |||||||
| Accelerated Stock Option Vesting (3) |
— | 197,828 | — | |||||||
| COBRA Premiums (4) |
12,445 | 12,445 | — | |||||||
| Total |
362,445 | 805,273 | 200,000 | |||||||
| (1) | Except as described in footnote (4), all payments in this column occur only in the event the named executive officer is terminated without cause or the officer terminates his or her employment for good reason after a |
42
Table of Contents
| change in control, which termination must occur within two years after the change in control. Payments in this column have not been reduced in the event that such payments will result in golden parachute excise taxes under Section 280G of the Internal Revenue Code, which reduction in payments will occur if it results in a greater net, after-tax benefit to the named executive officers than full payment of all amounts otherwise due. |
| (2) | Benefits are paid under the supplemental term life insurance policy only in the event of death. |
| (3) | Pursuant to the terms of the 2001 Plan, an executive’s stock options will accelerate following a change in control, unless they are assumed or substituted by the successor company, even if employment is not terminated. |
| (4) | Represents estimated COBRA premiums to be paid by us for 12 months. |
| (5) | Payable in the course of our regularly scheduled payroll over 12 months. |
| (6) | Payable in a lump sum. |
2001 Stock Incentive Plan
If the employment of a participant, including a named executive officer, terminates by reason of death, each outstanding award under our 2001 Plan will automatically accelerate and become 100% vested and exercisable.
If a corporate transaction specified in our 2001 Plan occurs, each outstanding option under the 2001 Plan will automatically accelerate and become 100% vested and exercisable immediately before the corporate transaction, unless:
| • | the award is assumed, continued or replaced with a comparable award by the successor corporation or the parent of the successor corporation; or |
| • | individual agreements provide otherwise. |
Under the 2001 Plan, a corporate transaction is defined to include:
| • | a merger or consolidation of the company with or into another company, entity or person (other than a merger or consolidation in which the holders of voting securities of the company before the transaction continue to hold at least a majority of the successor company’s voting securities); or |
| • | a sale, lease, exchange or other transfer of all or substantially all of the company’s outstanding securities or assets (other than to a majority-owned subsidiary company). |
Any option or other equity award that a participant, including a named executive officer, holds that is assumed, continued or replaced with a comparable award in the corporate transaction, other than in specified related-party transactions, will accelerate and become 100% vested and exercisable if the participant’s employment is terminated by the successor corporation without cause within one year after the corporate transaction.
Deferred Compensation Plan
Our Deferred Compensation Plan for Key Employees is fully funded by the executives. Consequently, there would be no incremental cost to us in the event that an executive’s employment terminated and the deferred compensation was paid out.
Employment Agreements
We enter into employment agreements with each of our executive officers, the terms of which are summarized below for our named executive officers.
43
Table of Contents
Named Executive Officers other than Bruce L.A. Carter
Our employment agreements with Dr. Williams and Mr. Johnson were amended and restated in July 2008 to comply with Section 409A of the Internal Revenue Code and to make other changes recommended by Radford during 2008. Following Dr. Williams’ promotion to Chief Executive Officer in January 2009, his employment agreement, as amended and restated in July 2008, was further amended in March 2009 and continues to govern the terms and conditions of his employment with us. Mr. Zaruby entered into a Section 409A compliant employment agreement in December 2008, which was subsequently amended and restated in June 2009. Ms. Franklin and Dr. Ramos entered into a Section 409A compliant employment agreement in July 2008 and June 2009, respectively.
Each employment agreement has a two-year term, subject to automatic one-year renewal periods unless timely notice of non-renewal is given. Under the employment agreements, we may terminate an executive officer’s employment at any time, with or without cause. If we terminate an executive officer without cause or if an executive officer terminates employment for good reason, we must, however, pay him or her severance benefits. The type and amount of the severance benefits depend on the type of termination, such as whether it occurs within two years after a change in control.
Upon termination by us for cause or by the named executive officer for other than good reason, we must pay only benefits accrued but not yet paid as of the termination date, such as accrued salary, deferred compensation and vacation pay.
Upon termination by us for reasons other than cause or by the named executive officer for good reason, either prior to a change in control or more than two years after a change in control, we must pay severance benefits to the named executive officer, including (1) annual base salary for a period of one year after termination and (2) contributions toward the cost of COBRA health care continuation coverage under our group health plans until the earlier of one year after termination or until the executive is covered under a comparable group health plan or is no longer entitled to COBRA continuation coverage.
Upon termination within two years after a change in control by us (or a successor) for reasons other than cause or by the named executive officer for good reason, we must pay severance benefits to the named executive officer, including:
| • | a lump sum equal to the sum of the executive’s then current annual salary plus bonus at the target amount for the year of termination multiplied by, (a) in the case of Dr. Williams and Mr. Zaruby, two, (b) in the case of Mr. Johnson, one and one-half, and, (c) in the case of Ms. Franklin and Dr. Ramos, one; |
| • | a bonus at the target amount for the year of termination prorated through the date of the change in control (if the executive is not otherwise entitled to receive an annual bonus upon termination of employment); |
| • | contributions toward the cost of COBRA health care continuation coverage under our group health plans until the earlier of one year after termination or until the executive is covered under a comparable group health plan or is no longer entitled to COBRA continuation coverage; and |
| • | full vesting of all stock options. |
As a condition to payments and benefits under the employment agreements, each named executive officer must execute a general release and waiver of claims. The employment agreements also contain one-year noncompetition and nonsolicitation covenants. If payments due to a named executive officer under an employment agreement will result in golden parachute excise taxes imposed by Section 280G of the Internal Revenue Code, such payments will be reduced if and to the extent that doing so results in a greater net, after-tax benefit to the executive than full payment of all amounts otherwise due under the agreement.
44
Table of Contents
Under the employment agreement of Dr. Williams, “good reason” is defined to include:
| • | a demotion or other material reduction in the nature or status of Dr. Williams’ responsibilities; |
| • | a reduction in Dr. Williams’ annual base salary; |
| • | the requirement by a successor company that Dr. Williams relocate his principal place of employment to a location more than 50 miles from the principal place of employment where Dr. Williams was employed; |
| • | our failure to obtain an agreement from a successor company to assume and perform the obligations of Dr. Williams’ employment agreement; |
| • | following a change of control, Dr. Williams’ ceasing to hold the position of Chief Executive Officer; and |
| • | following a merger or similar transaction that does not constitute a change of control, someone other than Dr. Williams holding the position Dr. Williams held on the effective date of such individual’s employment agreement. |
Under the employment agreements of Mr. Johnson, Mr. Zaruby, Ms. Franklin and Dr. Ramos, “good reason” is defined to include:
| • | a material reduction in the executive’s base compensation; |
| • | a material reduction in the executive’s authority, duties or responsibilities; |
| • | a material reduction in the budget over which the executive retains authority; |
| • | the requirement by a successor company that the executive relocate his or her principal place of employment to a location more than 50 miles from the principal place of employment where the executive was employed; or |
| • | a material breach of the executive’s employment agreement by us or a successor company. |
Under each employment agreement, “cause” is defined to include:
| • | the executive’s willful misconduct or insubordination in the performance of his or her duties that results in a material adverse effect on us; |
| • | willful actions or intentional failures to act made in bad faith by the executive that materially impair our business, goodwill or reputation; |
| • | the executive’s use of controlled substances; deception, fraud, misrepresentation by the executive; or any incident materially compromising the executive’s reputation or ability to represent us with investors, customers or the public; |
| • | the executive’s conviction of a felony involving an act of dishonesty, moral turpitude or fraud; or |
| • | a material violation of the executive’s inventions agreement with us. |
Under each employment agreement, a “change in control” is deemed to happen upon the occurrence of the following events, provided such event or occurrence also constitutes a change in ownership or effective control of us, or in the ownership of a substantial portion of our assets, within the meaning of Section 409A of the Internal Revenue Code:
| • | certain mergers, reorganizations or sales or other dispositions of all or substantially all of our assets as a result of which: |
| • | our shareholders hold 50% or less of the outstanding common stock of the surviving entity; |
45
Table of Contents
| • | our shareholders hold 50% or less of the combined voting power of the outstanding voting securities of the surviving entity; |
| • | a single shareholder or group of affiliated shareholders holds more than 50% of the outstanding common stock of the surviving entity; |
| • | a single shareholder or group of affiliated shareholders holds more than 50% of the combined voting power of the outstanding voting securities of the surviving entity; or |
| • | less than a majority of the members of the board of directors of the surviving entity were members of our Board of Directors; or |
| • | approval by our shareholders of our complete liquidation or dissolution. |
Bruce L.A. Carter
Effective January 2, 2009, Bruce L.A. Carter, our Chief Executive Officer, retired and, under the terms of his employment agreement with us, he was not entitled to any benefits other than the right to receive accrued but unpaid benefits and the payment of health insurance premiums for him and his dependents for two years or until he receives similar benefits from another employer. Dr. Carter’s employment agreement includes a one-year noncompetition provision and a two-year nonsolicitation provision, which provisions survive his retirement. In addition, Dr. Carter’s employment agreement allows him a period of three years following his retirement to exercise stock options vested on the date of his retirement.
Director Compensation
All of our directors serving in 2009, with the exception of Dr. Williams, earned director compensation in the form of an annual retainer of $35,000. Additionally, our non-executive Chairman is paid an annual retainer of $35,000 and our lead independent director is paid an annual retainer of $15,000. Directors who serve on committees are paid the annual retainers described in the table set forth below. Employee directors are not paid separate compensation for service on the Board.
| Committee |
Chairman | Member | ||||
| Audit Committee |
$ | 20,000 | $ | 10,000 | ||
| Compensation Committee |
$ | 10,000 | $ | 6,000 | ||
| Nominating and Corporate Governance Committee |
$ | 6,000 | $ | 4,000 | ||
In addition, we reimburse our directors for their reasonable expenses incurred in attending meetings of the Board and its committees.
Immediately following each year’s annual meeting of shareholders, each of our continuing nonemployee directors automatically receives a nonqualified stock option grant of 12,000 shares under our Stock Option Grant Program for Nonemployee Directors, which is administered under the 2001 Plan. These options fully vest on the date of the next annual meeting of shareholders assuming continued service by the nonemployee director until that date. Upon the initial election of a new nonemployee director, the director receives a nonqualified stock option grant of 20,000 shares under our Stock Option Grant Program. This option vests 1/3 on each anniversary of the date of grant over a three-year period. If this initial option is granted within five months before an annual meeting, the first annual option grant of 12,000 shares will not be granted until immediately following the second annual meeting after the date of the initial grant. Options granted under the Stock Option Grant Program have exercise prices equal to the closing price of the common stock on the date of grant and expire ten years after the date of grant, or, if earlier, one year after the director’s termination of service with us. The options also become fully vested in the event of death and certain corporate transactions, such as a merger or sale of assets.
46
Table of Contents
Director Compensation for Fiscal Year 2009
The following table sets forth the compensation of our directors for 2009. As an employee director during 2009, Dr. Williams did not receive any separate compensation for his service on the Board. See the Summary Compensation Table for disclosure relating to Dr. Williams, who served as our Chief Executive Officer starting January 2, 2009.
| Name |
Fees Earned or Paid in Cash ($) |
Option Awards (1) (2) (3) ($) |
Total ($) | |||
| Bruce L. A. Carter, Ph.D. |
70,000 | 72,481 | 142,481 | |||
| James A. Harper |
51,000 | 31,273 | 82,273 | |||
| Judith A. Hemberger, Ph.D.* |
41,000 | 31,273 | 72,273 | |||
| David I. Hirsh, Ph.D. |
35,000 | 31,273 | 66,273 | |||
| Jonathan S. Leff |
45,000 | 31,273 | 76,273 | |||
| David H. MacCallum |
49,000 | 31,273 | 80,273 | |||
| Kurt Anker Nielsen |
61,000 | 31,273 | 92,273 | |||
| Edward E. Penhoet, Ph.D. |
60,000 | 31,273 | 91,273 | |||
| Lars Rebien Sørensen** (4) |
— | 31,273 | 31,273 |
| * | Dr. Hemberger resigned as a director in October 2009. |
| ** | Mr. Sørensen resigned as a director in March 2010. |
| (1) | Each of the directors identified in the table was granted a nonqualified stock option to purchase 12,000 shares of common stock under our Stock Option Grant Program for Nonemployee Directors with an exercise price of $4.61 (which was equal to the closing price of our common stock on June 10, 2009, the date of grant). Each option vests in full on the date of the 2010 annual meeting of shareholders and has a term of ten years. Dr. Carter received an additional award of 12,000 shares of common stock with an exercise price of $6.15 (which was equal to the closing price of our common stock on September 16, 2009, the date of grant). |
| (2) | The amounts reported in the Option Awards column reflect the aggregate grant date fair value of option awards. These amounts do not represent the actual amounts paid to or realized by the named executive officer for these awards during the relevant fiscal year. The value as of the grant date for stock options is recognized over the number of days of service required for the grant to become vested. See Note 14 of Notes to Consolidated Financial Statements in our Annual Report on Form 10-K for fiscal year 2009 for the assumptions used in determining the grant date fair value of stock options. |
| (3) | The following table sets forth the aggregate number of option awards held by each director as of December 31, 2009: |
| Name |
Aggregate Number of Option Awards | |
| Bruce L. A. Carter, Ph.D. |
1,662,102 | |
| James A. Harper |
71,000 | |
| Judith A. Hemberger, Ph.D.* |
44,000 | |
| David I. Hirsh, Ph.D. |
78,655 | |
| Jonathan S. Leff |
78,655 | |
| David H. MacCallum |
63,500 | |
| Kurt Anker Nielsen |
78,655 | |
| Edward E. Penhoet, Ph.D. |
222,655 | |
| Lars Rebien Sørensen** |
78,655 |
| (4) | At the request of Novo Nordisk A/S, no payments were made to Mr. Sørensen. |
Compensation Committee Interlocks and Insider Participation
None of the members of our compensation committee serves, or formerly has served, as an officer or employee of ZymoGenetics. None of our executive officers serves as a member of the compensation committee or board of directors of any entity that has an executive officer serving as a member of our compensation committee or Board of Directors.
47
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Company has duly caused this Amendment No. 1 to the Annual Report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized.
| ZYMOGENETICS, INC. | ||||
| Date: August 24, 2010 |
By: | /s/ Douglas E. Williams, Ph.D. | ||
| Douglas E. Williams, Ph.D. Chief Executive Officer | ||||
48
Table of Contents
Exhibit Index
| 31.3 | Certification of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | |
| 31.4 | Certification of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | |