Attached files
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark one)
|
x
|
Annual Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934
|
for the Fiscal Year Ended March 31, 2010
OR
|
o
|
Transition Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934
|
Commission File Number 000-26372
ADAMIS PHARMACEUTICALS CORPORATION
(Exact name of registrant as specified in its charter)
|
Delaware
|
82-0429727
|
|
|
(State or other jurisdiction of
|
(I.R.S. Employer
|
|
|
incorporation or organization)
|
Identification No.)
|
|
2658 Del Mar Heights Rd., #555, Del Mar, CA 19512
|
|
(Address of Principal Executive Offices) (zip code)
|
Registrant’s telephone number, including area code: (858) 401-3984
Securities registered pursuant to Section 12(b) of the Act:
|
None
|
None
|
|
|
(Title of each class)
|
(Name of each exchange on which registered)
|
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, $0.0001 par value
(Title of class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
|
YES ¨
|
NO x
|
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
|
YES o
|
NO x
|
Note - Checking the box above will not relieve any registrant required to file reports pursuant to Section 13 or 15(d) of the Exchange Act from their obligations under those sections.
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
|
YES x
|
NO o
|
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of “accelerated filer and large accelerated filer” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large accelerated filer o
|
Accelerated filer o
|
Non-accelerated filer o
|
Smaller reporting company x
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act).
|
YES o
|
NO x
|
The aggregate market value of the voting stock held by non-affiliates of the Registrant as of September 30, 2009 was $ 8,059,665.
At June 30, 2010, the Company had 51,447,953 shares outstanding.
Documents Incorporated by Reference: None
ADAMIS PHARMACEUTICALS CORPORATION ANNUAL REPORT ON FORM 10-K FOR THE FISCAL YEAR ENDED MARCH 31, 2010
|
Part I
|
||
|
Item 1.
|
BUSINESS
|
6
|
|
Item 1A.
|
RISK FACTORS
|
33
|
|
Item 1B.
|
UNRESOLVED STAFF COMMENTS
|
47
|
|
Item 2.
|
PROPERTIES
|
47
|
|
Item 3.
|
LEGAL PROCEEDINGS
|
48
|
|
Item 4.
|
(REMOVED AND RESERVED)
|
48
|
|
Part II
|
||
|
Item 5.
|
MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS, AND ISSUER PURCHASES OF EQUITY SECURITIES
|
49
|
|
Item 7.
|
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
|
50
|
|
Item 8.
|
FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
|
61
|
|
Item 9.
|
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
|
61
|
|
Item 9A(T).
|
CONTROLS AND PROCEDURES
|
61
|
|
Item 9B.
|
OTHER INFORMATION
|
62
|
|
Part III
|
||
|
Item 10.
|
DIRECTORS AND EXECUTIVE OFFICERS OF THE REGISTRANT
|
63
|
|
Item 11.
|
EXECUTIVE COMPENSATION
|
63
|
|
Item 12.
|
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
|
63
|
|
Item 13.
|
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS
|
63
|
|
Item 14.
|
PRINCIPAL ACCOUNTANT FEES AND SERVICES
|
63
|
|
Part IV
|
||
|
Item 15.
|
EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
|
64
|
Information Relating to Forward-Looking Statements
This Annual Report on Form 10-K includes “forward-looking” statements within the meaning of the Private Securities Litigation Reform Act of 1995 which provides a “safe harbor” for these types of statements. These forward-looking statements are not historical facts, but are based on current expectations, estimates and projections about our industry, our beliefs and our assumptions. These forward-looking statements include statements about our strategies, objectives and our future achievement. To the extent statements in this Annual Report involve, without limitation, our expectations for growth, estimates of future revenue, our sources and uses of cash, our liquidity needs, our future products, expense, profits, cash flow balance sheet items or any other guidance on future periods, these statements are forward-looking statements. These statements are often, but not always, made through the use of word or phrases such as “believe,” “will,” “expect,” “anticipate,” “estimate,” “intend,” “plan,” and “would.” These forward-looking statements are not guarantees of future performance and concern matters that could subsequently differ materially from those described in the forward-looking statements. Actual events or results may differ materially from those discussed in this Annual Report on Form 10-K. We undertake no obligation to release publicly the results of any revisions to these forward-looking statements or to reflect events or circumstances arising after the date of this Report. Important factors that could cause actual results to differ materially from those in these forward-looking statements are disclosed in this Annual Report on Form 10-K, including, without limitation, those discussion under “Item 1A. Risk Factors,” in “Item 1. Business” and in “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations,” as well as other risks identified from time to time in our filing’s with the Securities and Exchange Commission, press releases and other communications.
Unless the context otherwise requires, the terms “we,” “our,” and “the Company” refer to Adamis Pharmaceuticals Corporation, a Delaware corporation (formerly Cellegy Pharmaceuticals, Inc.), and its subsidiaries. Savvy®, Aerokid®, AeroOtic®, and Prelone® are our trademarks. We also refer to trademarks of other corporations and organizations in this document.
PART I
In the discussion below, all statements concerning market sizes, annual U.S. sales of products, U.S. prescriptions and rates of prescriptions, the incidence of diseases or conditions in the general population, and similar statistical or market information are based on data published by the following sources: IMS Health Sales Perspectives, Retail and Non-Retail Combined Report, referred to as the IMS Report; National Data Corporation’s Epinephrine Prescription and Dollar Data, referred to as the NDC Report; Commercial and Pipeline Insight: Allergic Rhinitis, published by DataMonitor, referred to as the DataMonitor Report; AAAAI — American Academy of Allergy, Asthma and Immunology Allergy Statistics for the U.S., referred to as the AAAAI Statistics; American Cancer Society, Cancer, Facts & Figures 2009, referred to as ACS Statistics; and SEER Cancer Statistics Review, 1975-2007, National Cancer Institute, referred to as the NCI Statistics.
Company Overview
Adamis Pharmaceuticals Corporation (“Adamis”) was founded in June 2006 as a Delaware corporation. Adamis has three wholly-owned subsidiaries: Cellegy Holdings, Inc.; Adamis Corporation; and Biosyn, Inc. Adamis Corporation has two wholly-owned subsidiaries: Adamis Viral Therapies, Inc. (biotechnology), or Adamis Viral; and Adamis Laboratories, Inc. (specialty pharmaceuticals), or Adamis Labs.
Adamis Labs is a specialty pharmaceutical company that Adamis acquired in April 2007. Adamis has a line of prescription products in the anti-inflammatory, allergy and respiratory field that are currently commercialized by the Company. These products generated net revenues to Adamis of approximately $290,000 and $660,000 for Adamis’ fiscal years ended March 31, 2010 and 2009, respectively. Adamis’ Epinephrine Injection USP 1:1000 (0.3mg Pre-Filled Single Dose Syringe) product, or the single dose PFS Syringe product, a pre-filled epinephrine syringe product for use in the emergency treatment of extreme acute allergic reactions, or anaphylactic shock, was launched in July 2009; however, full commercial launch has been slowed by insufficient funding. Based on Adamis’ knowledge of a previously marketed pre-filled syringe indicated for anaphylaxis, the anticipated lower price of the PFS Syringe product relative to the leading syringe products currently marketed, and the ease of use of its product, Adamis believes that the PFS Syringe product has the potential to compete successfully after full commercial introduction of the product, although there can be no assurance that this will be the case. To date, Adamis’ ability to fully execute its plan for the full commercial launch of the PFS Syringe product has been hampered because of limited funding to support the full launch.
Additional product candidates in its product pipeline include a generic inhaled nasal steroid for the treatment of seasonal and perennial allergic rhinitis, and two other respiratory products (a generic HFA albuterol inhaler and a generic HFA beclomethsone inhaler). Adamis’ goal is to commence initial commercial sales of the nasal steroid product in the third quarter of 2012 and two other respiratory products in 2013, assuming adequate funding and no unexpected delays.
Adamis recently acquired and entered into agreements to acquire exclusive license agreements covering three small molecule compounds, named CPC-100, CPC-200 and CPC-300, that Adamis believes are promising small molecule anti-inflammatory drug candidates for the potential treatment of human prostate cancer (PCa). The intellectual property covered by the agreements was licensed from the Wisconsin Alumni Research Foundation, or WARF. The company has acquired the license agreement relating to CPC-300 and will, upon completion of an equity offering of more than $2 million, acquire the agreements relating to CPC-100 and CPC-200. In 2006 and 2007, CPC-100 and CPC-200, respectively, received the National Cancer Institute's multi-year, multi-million dollar RAPID (Rapid Access to Preventative Intervention Development) Award. Each year, this award is given by the NCI Division of Cancer Prevention, under the RAPID Program, to promising new preventative/ therapeutic anti-cancer drugs. Adamis’ objective is to file an Investigational New Drug application, or IND, with the U.S. Food and
6
Drug Administration, or FDA, by the end of calendar year 2010, and to subsequently commence a Phase 1/2a prostate cancer clinical study relating to the CPC-100 product and for men who have failed-Androgen Deprivation Therapy, or ADT, assuming adequate funding and no unexpected delays.
Adamis is also focused on developing patented preventative and therapeutic vaccines. The vaccine technology is applicable for certain viral-induced diseases such as Influenza, Hepatitis B and C, known to be involved in hepatocellular carcinomas or Human Papillomavirus, known to be involved in head and neck squamous cell carcinomas, as well as prostate cancer. However, Adamis currently intends to focus initially on the development of one or more of the recently licensed prostate cancer product candidates, and as a result the timing of initiation of trials relating to viral vaccine products is subject to uncertainty and the availability of sufficient funding, and there are no assurances concerning whether such a product will be developed or launched.
Future potential disease targets might include therapeutic vaccines for Influenza, Hepatitis B and C Virus, Human Papilloma Virus, and prostate cancer.
Adamis’ general business strategy is to increase revenue through a full commercial launch of the Single Dose Epinepherine Pre-Filled Syringe and its existing and proposed allergy and respiratory products in order to generate cash flow to help support the cancer and vaccine product development efforts of Adamis Viral. Adamis believes that the potential for increased revenues of specialty pharmaceuticals will be driven by two products.
|
•
|
Commercial sales of the Single Dose-Epinephrine PFS Syringe product commenced in July 2009, although commercial launch efforts have been materially slowed by insufficient funding. The product competes in a well-established U.S. market estimated to be over $150 million in annual sales, based on industry data published in the NDC Report.
|
|
|
•
|
Adamis Labs intends to introduce an aerosolized inhaled nasal steroid that is designed to take a small share of the U.S. market for nasal steroid products, estimated by Adamis to be approximately $3 billion in annual sales, based on the NDC Report. Adamis currently believes that this product could be introduced as early as the third calendar quarter of 2012, although the actual date of introduction will depend on a number of factors and the actual launch date could be later than that date. Factors that could affect the actual launch date include the outcome of discussions with the FDA concerning the number and kind of clinical trials that the FDA will require before the FDA will consider regulatory approval of the product, any unexpected difficulties in licensing or sublicensing intellectual property rights for other components of the product such as the inhaler, any unexpected difficulties in the ability of suppliers to timely supply quantities for commercial launch of the product, any unexpected delays or difficulties in assembling and deploying an adequate sales force to market the product, and the receipt of adequate funding to support sales and marketing efforts.
|
To achieve these goals, as well as to support the overall strategy, Adamis will need to raise a substantial amount of funding and make substantial investments in equipment, new product development and working capital. Adamis estimates that approximately $1.5 million to $2 million will be required to support the continued commercial launch of the PFS Syringe product, and that approximately an additional $4 million or more must be invested in the Adamis Labs operations to support development and commercial introduction of the aerosolized nasal steroid product candidate. The capital that is expected to be provided from expected sales of these products will be important to help fund expansion of those businesses and the research and development of the cancer and vaccine technologies. If adequate funding is obtained, clinical trials proceed successfully, regulatory approvals are obtained and sales are consistent with Adamis’ current expectations, following a period of initial commercial introduction, Adamis believes that revenues generated by Adamis Viral’s cancer drug or vaccine products could exceed revenues from Adamis Labs operations.
Effective April 1, 2009, Adamis completed a business combination transaction with Cellegy Pharmaceuticals, Inc., or Cellegy. The stockholders of Cellegy and the stockholders of former Adamis Pharmaceuticals Corporation, or Old Adamis, approved a merger transaction and related matters at an annual meeting of Cellegy’s stockholders and at a special meeting of Old Adamis’ stockholders, each held on March 23, 2009. On April 1, 2009, Cellegy completed the merger transaction with Old Adamis. Before the merger, Cellegy was a public company and Old Adamis was a private company.
7
In connection with the consummation of the merger and pursuant to the terms of the definitive merger agreement relating to the transaction, Old Cellegy changed its name from Cellegy Pharmaceuticals, Inc. to Adamis Pharmaceuticals Corporation, and Old Adamis changed its corporate name to Adamis Corporation.
Pursuant to the terms of the merger agreement, Old Cellegy effected a reverse stock split of its common stock immediately before the consummation of the merger. Pursuant to this reverse stock split, each approximately 10 shares of common stock of Old Cellegy that were issued and outstanding immediately before the effective time of the merger were converted into one share of Old Cellegy common stock and any remaining fractional shares held by a stockholder (after aggregating the fractional shares) were rounded up to the nearest whole share.
As a result, the total number of shares of Old Cellegy that were outstanding immediately before the effective time of the merger were converted into approximately 3,000,000 shares of post-reverse split shares of common stock of Old Cellegy. Pursuant to the terms of the merger agreement, at the effective time of the merger each share of Old Adamis common stock that was issued and outstanding immediately before the effective time of the merger ceased to be outstanding and was converted into the right to receive one share of Adamis common stock. As a result, approximately 44,000,00 shares of Adamis common stock were issued and/or are issuable to the holders of the outstanding shares of common stock of Old Adamis before the effective time of the merger. Old Adamis, renamed Adamis Corporation, was the surviving entity as a wholly-owned subsidiary of Adamis.
Allergy and Respiratory Specialty Pharmaceutical Drug Products
On April 23, 2007, Adamis completed the acquisition of a specialty pharmaceutical drug company named Healthcare Ventures Group, Inc., or HVG. HVG had previously acquired a group of allergy and respiratory products and certain related assets from a third party company. The third party also transferred to HVG members of its sales force and management team. Adamis created the Adamis Laboratories subsidiary, which then acquired HVG in a stock-for-stock exchange. Adamis issued approximately 12.6 million new shares of Adamis common stock to the shareholders of HVG. Under the terms of the transaction agreements, approximately 6.7 million of these shares are subject to restrictions on transfer as well as repurchase by Adamis if certain performance targets based on revenue over a period of three years are not achieved by Adamis Labs and if the holders do not remain employed by Adamis during that period.
Net revenues to Adamis from sales of Adamis Labs’ allergy and respiratory products from April 23, 2007, the date on which Adamis acquired Adamis Labs, through Adamis’ fiscal year ended March 31, 2010, were approximately $1,572,000. During Adamis’ fiscal year ended March 31, 2010, two customers, McKesson and Cardinal Health, accounted for approximately 44% and 31%, respectively, of Adamis’ revenues. The products have not been heavily promoted in the past due to funding limitations and the competitive market for antihistamine/decongestant products. Adamis believes there is limited growth potential for these products, due in part to the widespread substitution of generic products at the dispensing pharmacy level for the conditions indicated for the Adamis Labs products.
Specialty Pharma Drug Product Pipeline
Adamis Labs’ product pipeline includes the Single Dose-Epinephrine PFS Syringe product and an inhaled nasal steroid product candidate. The first product, the PFS Syringe product, was commercially launched in July 2009, although full commercial launch efforts have been materially slowed by insufficient funding. The second product, an aerosolized inhaled nasal steroid product for the treatment of seasonal and perennial allergic rhinitis, is targeted for commercial availability in 2012, assuming adequate funding to support product development and launch and no unanticipated delays in obtaining regulatory approvals. Adamis Labs has an agreement with Catalent Pharma Solutions, Inc. for sterile manufacturing product supply for the PFS Syringe product and is in discussions with an aerosol inhaler supplier for the aerosolized nasal steroid product candidate.
Single Dose Epinephrine Pre-Filled Syringe Product
There is a well-defined, growing market in the United States for patient-administered emergency epinephrine injectors used in the treatment of anaphylaxis. Based on information in the AAAAI Statistics, in the U.S., an estimated 5% of the population suffers from insect sting anaphylaxis, up to 6% are latex sensitive and up to 1.5% of adults and 5% of children under three years of age experience food related anaphylaxis. Adamis believes that anaphylaxis may be under-diagnosed. In January 2001, a published study by AAAAI revealed that up to 40 million Americans (15% of the total population) may be at risk for anaphylaxis, a significantly higher number than the
8
historically estimated at-risk population. According to information in the AAAAI Statistics, approximately 3,000 people in the U.S. die each year from anaphylaxis.
The number of prescriptions for epinephrine products has grown annually, as the risk of anaphylaxis has become more widely understood. According to the IMS Report, total prescriptions for EpiPen products more than doubled in the five year period from 2001 to 2005. Based on information in the IMS Report and more recently from NDC data, the U.S. epinephrine injector market was approximately $220 million in sales in 2008 and has historically grown at a rate of approximately 15% per year. Adamis estimates that the growth rate of annual prescriptions will decline to a growth rate of approximately 4-5% per year by 2010 consistent with IMS reports, although there are no assurances that this will be the case.
EpiPen® was originally developed by Meridian Medical Technologies, Inc. as an auto-injection system for use by military personnel. It was designed for self-administration as an antidote for chemical warfare agents and morphine. Meridian Medical Systems, which is the manufacturer of the EpiPen and EpiPen Jr., continues to focus on products for the military, and its major customer is the United States Department of Defense. The EpiPen® products were introduced to the market in 1982, and were the only epinephrine injectors for allergic emergencies that were available until 2005. In August 2005, another company introduced a competing product, Twinject ® Dual Pack, (and now Adrenaclick®) 0.3mg epinephrine auto injectors, which, Adamis believes due to pricing and ease of use issues, has enjoyed only a small market share in the United States. Twinject is currently owned by Sciele Pharma, Inc.
Adamis believes that there are barriers to market entry for new competitors based on epinephrine’s susceptibility to contamination, sensitivity to heat and light and a short shelf-life, as well as the need for a competitor to possess the expertise to overcome the packaging and delivery challenges of introducing a competing product to the market. Adamis also believes that the size of the market is too small to be a major focus of the large pharmaceutical companies, although there can be no guarantees that this will be the case.
Adamis believes that the primary opportunity lies in the 0.3 mg segment, which constitutes approximately 72% of the total market (measured as a percent of U.S. sales), based on EpiPen unit sales history and the NDC Report. When sales of dual packs of EpiPen and TwinJect/Adrenaclick are converted to single units, the total target market in the U.S. is about 2.5 million single units per year and growing.
Adamis believes that there is an opportunity for a simple, low-cost, intuitive and user-friendly pre-filled syringe to compete in this largest segment of the market. Adamis believes that its new product has the potential to compete effectively against EpiPen ® based on the following factors, among others:
|
•
|
Market Knowledge. Mr. Richard Aloi, President of Adamis Labs, is a U.S. leading authority on the commercialization of epinephrine injectors. He had responsibility for the worldwide introduction of EpiPen® and EpiPen® Jr. in 1982 and contributed to the subsequent growth of sales of the product line.
|
|
•
|
Lower Price. Adamis believes that a lower-priced option would be particularly attractive to individuals potentially susceptible to anaphylaxis as well as managed healthcare drug reimbursement plans providing patient prescription reimbursement. Adamis expects to introduce the Epi Syringe at a price point reflecting a discount to the price of the market leader, EpiPen, in part to make the product more attractive to customers. At this price Adamis believes it can still obtain significant gross margins, although there are no assurances that this will be the case.
|
|
•
|
Ease of Use. The EpiPen®, EpiPen® Jr., Twinject® and Adrenaclick® are powerful spring-loaded auto-injector devices. If not administered properly, they can misfire or be misused. Adamis’ Epi pre-filled 0.3mg syringe will allow patients to self-administer (self-inject) a pre-measured epinephrine dose quickly with a device that does not have moving parts that the user cannot control, which Adamis believes may increase product safety and sales, although there are no assurances that this will be the case.
|
There are four key supply components used in the manufacture of the PFS Syringe Product: the pre-filled single does syringe containing the epinephrine; the formulation solution; a specially designed plunger rod that expels only the appropriate emergency amount of 0.3mg of epinephrine; and the plastic carrying case.
9
Adamis believes that the market for emergency epinephrine injectors will grow, driven by increasing awareness, lower cost alternatives, and traditional or online promotions by new market entrants. Adamis expects that the total market unit growth rate will continue to grow as additional lower priced epinephrine products are introduced, but total dollar market will plateau as the market matures with multiple lower priced products. Adamis believes that the PFS Syringe product may acquire a share of the market in a manner somewhat similar to the pattern established by generic drugs, in that the price differential between the expected price of the PFS Syringe product and the price at which the market-leading product is currently sold will motivate purchasers and reimbursing payors to choose the lower cost alternative. Adamis also believes, however, that if its product competes successfully, at least one of the current competitors may introduce a competing, low-priced, pre-filled single dose syringe while maintaining the price points of its existing product lines. Adamis believes that such a competing product might have a comparable or lower price than the Adamis product. Adamis believes that the PFS Syringe product has the potential to compete successfully after full commercial activities are initiated, although there can be no assurance that this will be the case. To date, Adamis’ ability to fully execute its plan for the commercial launch of the PFS Syringe product has been materially hampered because of limited funding to support the full commercial launch.
Inhaled Nasal Steroid Product
Adamis Labs is developing an aerosolized inhaled nasal steroid for the treatment of seasonal and perennial allergic rhinitis. The active ingredient is beclomethasone diproprionate, a synthetic steroid that demonstrates potent glucocorticoid activity. Glucocorticosteroids are hormones produced by the adrenal cortex. Corticosteroids inhibit inflammation in allergic reactions by interfering with the synthesis of prostaglandins and leukotrienes, chemicals that are normally synthesized as part of the inflammatory process. Adamis refers to the product as Beclomethasone Aerosolized Nasal Steroid, or BANS.
The market for inhaled nasal steroids, or INS, as estimated by Adamis based on the DataMonitor Report, is about $3 billion annually in the U.S. and growing steadily. Although the market is dominated by two multi-national pharmaceutical companies, Adamis believes there is a niche that can be exploited, and that an Adamis product candidate can achieve a small but significant percentage share of this large market.
INS products are sold under prescription for seasonal allergic rhinitis. In addition to inhaled nasal steroids, many different types of products treat the symptoms of allergic rhinitis: oral antihistamines and decongestants are among the most popular for self-medication/patient treatment. All physician specialties report that the majority of their allergic rhinitis patients receive intranasal steroids, either alone or in combination with oral antihistamines. In general, physicians view intranasal steroids as safe and effective.
There are four major physician specialties that treat patients with allergic rhinitis: Allergists, Otolaryngologists, or ENTs; Primary Care Physicians, or PCPs; and Pediatricians. Allergists, along with ENTs, tend to be the most aggressive in terms of pharmacological treatment of allergic rhinitis. On an individual basis, the allergist is the largest prescriber of products within the INS category. ENT physicians contribute half as many prescriptions as allergists, but that is still about five times the volume of the average primary care physician.
The INS market is highly seasonal with most of the sales occurring in two periods: a spring season from April through May or June; and a fall season occurring in September and October. Based on information in the DataMonitor Report, Adamis estimates that the INS market grew at an annual rate of over 5% from 31.7 million prescriptions in 2002 to an estimated 38.7 million prescriptions in 2006. In the same period, total U.S. market sales grew from $1.89 billion in 2002 to an estimated $3 billion for 2006. This average growth rate is about 10% per year, and resulted primarily from steady price increases. Adamis expects that the growth rate in average price increases will likely decline and possibly reach zero by 2011, due to increasing competition from generic products.
Currently, the INS market is dominated by aqueous solution formulations delivered by a pump. These aqueous pump spray formulations have replaced CFC propellant INS products, which once dominated the INS market. The propellant inhaled nasal steroids that were previously available have been discontinued due to CFC concerns for the environment. Based on information in the IMS Report concerning 2005 sales, the two leading products account for over 70% of total product sales in this market.
Adamis believes that, in general, prescribing physicians view all INS products as being generally similar in terms of efficacy and safety. As a result, the INS market is sensitive to promotion, and companies spend a great deal of effort and money each year in the attempt to differentiate these products from one another. Adamis believes that
10
large amounts are spent on direct-to-consumer advertising for the two largest holders of market share, Nasonex®, marketed by Schering, and Veramyst® (fluticasone) marketed by GlaxoSmithKline. In addition to direct-to-consumer advertisement, GSK and Schering also spend large amounts of dollars in personal promotion detailing physicians and distributing samples as well as journal advertisement.
Adamis does not anticipate competing directly against the two leading companies in this market by attempting to out-spend or out-promote them in the marketplace. Adamis believes that its market opportunity lies in taking a small portion of the market with a new aerosolized HFA version of a well-established product but at a substantial discount to the current prices of the leading branded products.
Adamis expects BANS to be considered a “new” drug by the FDA, and accordingly Adamis believes that it will be required to submit data for an application for approval to market BANS pursuant to Section 505(b)(2) of the Food Drug and Cosmetics Act, although there are no assurances that this will be the case. Total time to develop the BANS product is expected to be approximately 25 months from inception of full product development efforts, assuming sufficient funding and no unexpected delays. The table below shows the estimated development timeline for the BANS product based on the number of months from inception of full product development efforts.
Projected Developmental Timeline for BANS
(beclomethasone diproprionate)
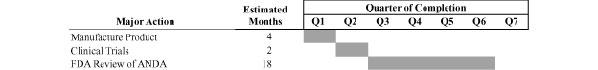
Adamis estimates that approximately $4 million or more must be invested from the date of this Annual Report on Form 10-K in the Adamis Labs operations to support development and commercial introduction of the aerosolized nasal steroid product candidate. The capital that is expected to be provided from expected sales of these products will be important to help fund expansion of those businesses and the research and development of the anti-cancer small molecule therapeutic drugs as well as the therapeutic vaccine technology. Currently, neither manufacturing nor clinical trials have begun for that product candidate. Adamis estimates that approximately a total of $6-$9 million is required to support the development and commercial introduction of the inhaled nasal steroid product candidate and its two other respiratory products, although there are no assurances that funds for such an investment will be available. Factors that could affect the actual launch date for the nasal steroid product candidate include the outcome of discussions with the FDA concerning the number and kind of clinical trials that the FDA will require before the FDA will consider regulatory approval of the product, any unexpected difficulties in licensing or sublicensing intellectual property rights for other components of the product such as the inhaler, any unexpected difficulties in the ability of our suppliers to timely supply quantities for commercial launch of the product, any unexpected delays or difficulties in assembling and deploying an adequate sales force to market the product, and adequate funding to support sales and marketing efforts. Significant delays in obtaining funding to support the development and introduction of the steroid product could reduce revenues and income to Adamis, require additional funding from other sources, and potentially have an adverse effect on the ability to fund Adamis’ research and development efforts for tumor indications, as well as vaccine product candidates by Adamis Viral.
Factors that could affect the actual launch date for the BANS product candidate include the outcome of discussions with the FDA concerning the number and kind of clinical trials that the FDA will require before the FDA will consider regulatory approval of the product, any unexpected difficulties in licensing or sublicensing intellectual property rights for other components of the product such as the inhaler, any unexpected difficulties in the ability of suppliers to timely supply quantities for commercial launch of the product, any unexpected delays or difficulties in assembling and deploying an adequate sales force to market the product, and adequate funding to support sales and marketing efforts.
Cancer and Vaccine Product Candidates
The company’s Adamis Viral subsidiary is focused on the development of Adamis’ therapeutic vaccine product candidates and prostate cancer drugs for prostate cancer patients with unmet medical needs in the multi-billion dollar global prostate market. Adamis Viral has previously focused on vaccine technologies only, with initial emphasis on developing a novel avian influenza vaccine. However, the recent acquisition by Adamis of license agreements relating to prostate cancer therapeutic anti-inflammatory small molecule drugs that the company believes are proprietary and promising; as well as recent published information regarding the revised incidence estimates of avian influenza, which are lower than previous estimates, and the resulting decrease in the likelihood of an imminent avian influenza pandemic, have all led the company to focus their efforts on both the small molecule cancer therapeutic drugs and on therapeutic vaccine opportunities.
Prostate Cancer Technologies
On February 24, 2010, Adamis entered into a definitive agreement to acquire exclusive license agreements covering three small molecule compounds, named CPC-100, CPC-200 and CPC-300. Adamis believes these compounds to be promising drug candidates for the potential treatment of human prostate cancer, or PCa. The intellectual property covered by the agreements was licensed from the Wisconsin Alumni Research Foundation, or WARF. The company has acquired the license agreement relating to CPC-300 and will, upon completion of an equity offering of more than $2 million, acquire the license agreements and obligations relating to CPC-100 and CPC-200. In 2006 and 2007, CPC-100 and CPC-200, respectively, each received the National Cancer Institute's multi-year, multi-million dollar Rapid Access to Preventative Intervention Development, or RAPID, Award. Each year, this award is given by the NCI Division of Cancer Prevention, under the RAPID Program, to promising new
11
preventative/ therapeutic anti-cancer drugs. Collectively, more than $16 million has been spent through government and private foundation grants and private investor funding for the development of these three new small molecule drug candidates. Adamis’ objective is to file an Investigational New Drug application, or IND, with the U.S. Food and Drug Administration, or FDA, by the end of calendar year 2010, and to subsequently commence a Phase 1/2a prostate cancer clinical study relating to the CPC-100 product candidate, assuming adequate funding and no unexpected delays.
The Human Prostate and Prostate Cancer; Disease and Market Background
In the discussion below concerning prostate cancer, all statistics, data and information concerning incidence of disease or other conditions in the general population, market sizes, annual U.S. sales of products, U.S. prescriptions and rates of prescriptions, and similar statistical or market information are based on data published sources: MedTrack and IMMS data reports, American Cancer Society, or ACS, Statistics and National Cancer Institute, or NCI, Statistics.
The prostate is a walnut-sized gland located in front of the rectum and underneath the urinary bladder. It is found only in men. The prostate starts to develop before birth and continues to grow until a man reaches adulthood. This growth is fueled by male hormones, the so-called androgens. The main androgen produced by men is the hormone testosterone. Testosterone can be converted by the body into dihydrotestosterone (DHT), which in turn signals the prostate to grow. The prostate stays at adult size in adult males as long as the male hormone is present at physiological levels. In older men, the inner part of the prostate around the urethra very often keeps growing, leading to a common urological condition called benign prostatic hyperplasia (BPH). In BPH, the prostate tissue can press on the urethra, leading to flow problems in passing urine. BPH is a serious medical problem, however BPH is not prostate cancer.
A prostate cancer develops when cells in the prostate begin to grow out of control, and a cancerous tumor can form. Several types of cells are found in the prostate, but over 99% of prostate cancers develop from gland cells within the prostate. The medical term for a cancer that starts in gland cells is an “Adenocarcinoma”. As the tumor grows, it can spread to the interior of the prostate, to tissues near the prostate, to the sac-like structures attached to the prostate known as the seminal vesicles, and to distant parts of the body, such as the bones, liver lobes or lungs. Prostate cancer (PCa) is one of the most invasive malignancies and a leading cause of cancer related deaths in many countries. According to the American Cancer Society and the National Cancer Institute, prostate cancer is the second-most common cancer in American men, and the second leading cause of cancer death in American men. The latest ACS estimates for prostate cancer in the United States for 2009 indicate that about 192,280 new cases of prostate cancer will be diagnosed and 27,360 men will die of prostate cancer. The NCI has estimated that approximately 20% of patients present with locally advanced or metastatic prostate cancer at the time of diagnosis. Metastatic prostate cancer is advanced prostate cancer that has spread beyond the prostate and surrounding tissues into distant organs and tissues. The majority of men who die from prostate cancer die from the consequences of metastatic disease. According to the National Cancer Institute, the five-year survival rate of patients with prostate cancer that has metastasized to distant organs is only about 30.6%. Metastatic prostate cancer is generally divided into two states: the Androgen hormone-sensitive, Androgen-dependent or castrate sensitive PCa state (CS-PCa) and the castrate-resistant PCa state (CR-PCa), also referred to as the Androgen hormone-refractory, Androgen-independent or the Androgen Deprivation Therapy (ADT) resistant state.
Testosterone and other male sex hormones, known collectively as androgens, can fuel the growth of prostate cancer cells. Androgens exert their effects on prostate cancer cells by binding to and activating the Androgen Receptor, which is expressed in prostate cancer and other cells. When they first metastasize to distant sites, most prostate cancers depend on androgen hormone for tumor growth. These prostate cancers are CS-PCa prostate cancers. The CS-PCa tumors treated with Androgen Deprivation Therapy (ADT) are often already inflamed or can also become chronically inflamed and invariably become CR-PCa tumors.
For patients with advanced, metastatic CS-PCa prostate cancer, the standard of care is treatment with hormonal ablation therapy, also known as androgen deprivation therapy or ADT. ADT is used to suppress production or block the action of androgens. Accordingly, the leading therapies currently used for the treatment of
12
prostate cancer, after it recurs following radiation or surgery, are focused on diminishing the production of androgens, or antagonizing the effects of androgens by blocking the Androgen Ligand Binding Domain on the Androgen Receptor inside prostate cancer cells with drugs known as anti-androgens. Thus, theses two different effects are achieved through two separate therapeutical approaches. The first approach is often to reduce the amount of androgens produced in the body, primarily in the testes. This can be achieved by surgical castration by removal of both testicles, referred to as an orchiectomy, or alternatively through use of one and /or two different kinds of ADT drugs, called chemical castration.
One chemical castrating therapeutic drug is known as a luteinizing hormone-releasing hormone (LHRH) agonist drug. This type of drug is exemplified by compounds like Zolodex (AstraZeneca PLC) that lower the native production of testosterone from the adrenal gland. A second chemical castrating therapeutic approach uses a drug known as anti-androgen, which directly block the interaction of androgens from binding to the ligand binding domain of the Androgen Receptor (AR-LBD). For example, Bicalutamide (Casodex®), is an anti-androgen drug that binds to the AR-LBD and displaces or blocks androgen binding to the AR-LBD and thus inhibits normal AR function. Bicalutamide is now a generic. Additional generic anti-androgens include Flutamide (also known as Nilutamide). Bicalutamide is still one of the largest selling of the anti-androgen CS-PCa therapeutic drugs, with global annual sales of about $1B and more than $800 million in 2009 from AstraZeneca PLC, according to their own public disclosures of sales. Anti-androgens and LHRH agonists often are given in combination therapy, an approach known as a Combined Androgen Blockade (CAB). However, because these ADT therapies operate by reducing the ability of androgen hormone to bind and activate the AR to fuel the growth of prostate cancer cells, they generally are effective only on prostate cancers that remain hormone-sensitive, that is, those men with CS-PCa tumors that still depend on androgen and the AR-LBD for PCa cell growth. Adamis, collaborators, and many others now commonly recognize that androgen deprivation therapy causes prostate cancer cell programmed cell death (apoptosis) and can also contribute to pathophysiological chronic inflammation in men with CS-PCa. There is significant published data supporting the important role of chronic inflammation in the change from CS-PCa to CR-PCa.
Most animal and human prostate cancer initially is hormone-sensitive and thus initially responds to ADT. However, according to a study published in the October 7, 2004 issue of The New England Journal of Medicine, and other studies, virtually all hormone-sensitive metastatic prostate cancer (CS-PCa) are commonly believed to undergo changes that convert CS-PCa to the castration-resistant (CR-PCa) state within a median of 18-24 months after initiation of ADT. Once in this ADT resistant CR-PCa state, CR-PCa generally continues to grow even when there is a significant reduction of testosterone production. The change to the castration-resistant state is generally determined based on monitoring either rising levels of prostate-specific antigen, or PSA, in prostate patients’ blood serum, or by documented disease progression as evidenced by radiographic imaging tests (via patient MRI or bone scans) or the CR-PCa patients’ presentation of significant clinical symptoms, including pain with or without chronic fatigue. Metastatic prostate cancer that has become castration-resistant most often becomes more highly advanced, resistant to all forms of therapy, and extremely aggressive; These patients have a median survival of often only 10 to 16 months because, at present, there is no successful medium- or long-term chemotherapy or immunotherapy treatment for advanced metastatic CR-PCa. Treatment of patients with CR-PCa remains a clinical challenge.
In summary, the standard treatment for localized advanced, recurrent, and metastatic prostate cancer is ADT, which blocks the growth promoting effects of androgens and activates apoptosis. After an initial favorable response, progression to androgen-independence or castration resistance is the usual outcome, for which there are currently no curative treatment options. Some brief survival extensions can sometimes be achieved using current Taxol-based chemotherapy protocols.
Adamis believes that the recently in-licensed prostate cancer therapeutic anti-inflammatory drugs, CPC-100, -200 and -300, may offer significant new treatments for prostate cancer and inflammation. In animal studies conducted to date, all three of these compounds are found to be safe and well tolerated, and are active not only against castrate sensitive but also against castrate resistant prostate tumors.
13
Drug Product Candidates in Development
CPC-100. CPC-100 is the most advanced of the three small molecule anti-inflammatory drug candidates. In animal studies conducted to date, CPC-100 demonstrated potent anti-androgenic and anti-inflammatory activities against prostate tumors growing in animal models and showed a strong safety profile in preclinical safety studies.
To date, CPC-100 has demonstrated desirable pharmacological characteristics as an oral or injectable anti-inflammatory and anti-androgenic drug candidate with multiple mechanisms of action. CPC-100 significantly decreases secretion of human PSA (Prostate Specific Antigen) by human prostate cancer cells growing in mice and also significantly increases the time-to-tumor progression and survival of PCa mice with CS-PCa and CR-PCa tumors. In animal studies conducted to date, at the maximum tolerated oral dose, or MTD, of CPC-100 (equal to 3 milligrams of CPC-100 per kilogram of mouse body weight), approximately 90% of mice developing metastatic prostate cancer achieved a highly statistically significant and highly repeatable therapeutic benefit. This is compared with approximately 55% of mice achieving a therapeutic benefit for the leading ADT drug Casodex, also known as Biculatamide (used at the oral MTD of 5 milligrams per kilogram of mouse body weight). CPC-100 is also significantly more effective than Flutamide, another generic anti-androgen sold world-wide and, like Bicalutamide, only effective with CS-PCa and not with CR-PCa patients.
Based on studies to date, Adamis believes that the CPC-100 drug candidate may offer important advantages over existing anti-androgen standard of care drugs that are used in hormonal therapies in prostate cancer patients. CPC-100 has the potential to be used for both castrate-sensitive and castrate-resistant prostate cancer patients. Currently, there is no drug specifically approved as a second-line hormonal agent for the treatment of prostate cancer. Rather, the standard of care for second-line hormonal therapies includes using existing drugs, such as steroids (hydrocortisone, dexamethasone), hormones (estrogen, aminoglutethimide) and anti-fungal agents (ketoconazole) in “off-label” drug use settings. Each of these drugs has characteristics limiting its usefulness as a treatment for prostate cancer. We believe that CPC-100 may have potential advantages over such existing treatments, most notably due to its being anti-inflammatory, anti-androgenic and multi-targeted, as well as safe and well tolerated in animal testing.
A variety of serious side effects have been associated with the use of existing second-line hormonal treatments, which are limiting their uses. To date, however, no serious side effects appear to be associated with the use of CPC-100. Should CPC-100 continue to demonstrate a continued lack of serious side effects, we believe it would be favorably positioned against other therapeutic PCa agents. Finally, agents used as second-line hormonal PCa agents for castration resistant prostate cancer must be taken multiple times during the day. In pre-clinical testing to date, CPC-100 has shown the potential to be administered once per day as an oral drug. Such a convenient oral dosing schedule may result in better patient at home compliance, when compared to other agents that are used as second-line hormonal treatments.
In 2006, CPC-100 was awarded the National Cancer Institute (NCI) Rapid Award. The award is given for promising new drugs for the treatment of cancer and resulted in significant funding for research and development of CPC-100. The development of CPC-100 has been funded by Michael Milken's Prostate Cancer Foundation (PCF, formerly CapCure), the Department of Defense's Congressionally Directed Medical Research Programs' (CDMRP) Prostate Cancer Research Program (PCRP), as well as grants and contracts from the U.S. Public Health Service and the National Cancer Institute (NCI).
All IND enabling efficacy and toxicology studies, as well as the proposed Phase 1/2a human clinical trial protocol for both castrate sensitive (CS-PCa) and castrate-resistant prostate cancer (CR-PCa), have been completed for CPC-100, and Adamis intends to discuss and submit an Investigational New Drug Application with the FDA during the calendar year 2010, assuming adequate funding and no unexpected delays.
CPC-200. The small molecule acetyl polyamine oxidase enzyme inhibitor, or CPC-200, is a drug candidate for both castrate-sensitive and castrate resistant prostate cancer. CPC-200 is an irreversible inhibitor of the acetyl polyamine oxidase enzyme and blocks androgen-induced hydrogen peroxide production and inflammation and inhibits mouse PCa. Whereas acute inflammation is important for host defenses, for example against acute bacterial and viral infections in the prostate, chronic inflammation can contribute significantly to prostate tumor initiation, growth, progression and metastic PCa. In animal studies conducted to date, CPC-200 was an excellent inhibitor of chronic inflammation, also completely inhibiting oxidase mediated high rates of hydrogen peroxide
14
production in vivo, and significantly delaying prostate cancer progression and death in the standard mouse prostate cancer model (TRAMP - transgenic adenocarcinoma of the mouse prostate – mouse model). TRAMP mice have spontaneously developing prostate cancer, where all animals usually die from metastatic PCa at 22 weeks of age. In the TRAMP animal studies conducted to date, CPC-200 repeatedly demonstrated a statistically significant therapeutic efficacy and a strong safety profile with highly desirable pharmacological therapeutic characteristics and with the capacity to be administered as either an oral or injectable drug.
CPC-200 is being developed as an oral, injectable or implantable drug, specifically in appropriate formulations for patients with PCa for whom Androgen Deprivation Therapy, or ADT, is currently not approved or appropriate with standard-of-care therapeutics, for example prior to surgery or radiation of the primary prostate cancer. Additionally, CPC-200 may fulfill another unmet medical need for which there is no approved drug on the market, in that it might be given after surgery or radiation but before or with ADT, since it has been shown to be a potent anti-inflammatory drug in the animal studies conducted to date. CPC-200 effectively inhibits the androgen-induced oxidase-mediated increased production of hydrogen peroxide in prostate tissues and inhibits inflammation which has been recognized to be an important factor in the induction and progression of prostate cancer. In the TRAMP mouse PCa model, CPC-200 increased survival and time to tumor progression, and demonstrated inhibition of PSA secretion by human tumors and low toxicity with no pro-estrogenic or other negative side-effects. In 2007, CPC-200 was awarded the NCI Rapid Award, which is adequate for funding of all IND-enabling data including the ongoing large animal GLP toxicology measurements and with extra funds for formulation studies in development.
Approximately two-thirds of all IND enabling studies have been completed for CPC-200 to date, and Adamis anticipates being able to meet with the FDA by the end of calendar year 2010 and to subsequently file and open the Adamis-sponsored IND relating to the clinical investigation of oral CPC-200 in PCa patients pre-ADT. These studies are anticipated to be initiated sometime during the first half of calendar year 2011, assuming adequate funding and no unexpected delays.
CPC-300. CPC-300 is a multi-targeted small molecule therapeutic drug that the company believes has the potential to demonstrate anti-inflammatory, pro-apoptotic anti-cancer activities for prostate cancer patients, including men with advanced metastatic CR-PCa. In pre-clinical in vivo studies conducted to date, CPC-300 repeatedly demonstrated a significant ability to inhibit human tumor growth and kill both castrate-sensitive and castrate-resistant human prostate cancer tumors. It also materially decreased human tumor volumes and suppressed local metastasis in human xenograft models, where malignant human prostate or human melanoma tumor tissue was grafted onto athymic immunosuppressed experimental mice.
CPC-300 inhibited human androgen receptor protein production in these studies. It also inhibited PSA secretion by human PCa cells, which is a serum marker for human prostate cancer. Based on the pre-clinical studies conducted to date, CPC-300 clearly targets microtubule assembly and regulation, inhibits inflammation and is a potent pro-apoptotic therapeutic oral drug with potential for human prostate cancer patients. Based on pre-clinical studies conducted to date, CPC-300 also (i) inhibits prostate growth with simultaneous effects on the level of alpha-tubulin and beta-tubulin (the microtubule structural proteins), Stathmin (a micotubule regulating protein) and Survivin (a microtubule-regulatory down stream target/pro-survival protein), (ii) induces Fas receptor-mediated apoptotic signaling, (iii) decreases the level of the anti-apoptotic protein cFLIP, (iv) decreases transcriptional activation of Survivin and cFLIP, and (v) has a strong safety profile and desirable pharmacological characteristics with the capacity to be administered as either an oral or injectable drug or as a nutraceutical. Because of its multiple mechanisms of action, Adamis believes that CPC-300 may have potential applications in the treatment of other tumor types in which microtubule inhibitors have already been shown to be effective, including melanoma, as well as in prostate cancer.
Adamis' strategy regarding CPC-100, CPC-200 and CPC-300 is to retain United States territorial rights, while out-licensing certain rights, including those to other geographic territories.
Vaccine Technologies
Adamis Viral is also focused on developing patented vaccine technology that has the potential to provide protection against a number of different viral infectious agents. This novel vaccination strategy, which employs
15
DNA plasmids, appears, based on preclinical studies conducted to date, to have the ability to “train” a person’s immune system to recognize and mount a defense against particular aspects of a virus’ structure. If successful, Adamis believes this technology will give physicians a new tool in generating immunity against a number of viral infections that have been difficult to target in the past.
The first target indication for this technology has yet to be determined, but will be based on market, technology, and patent position considerations. Disease targets might include therapeutic vaccines for Influenza, Hepatitis B and C, Human Papillomavirus, and prostate cancer.
The technology that provides the basis of Adamis Viral’s research and development was developed by Dr. Maurizio Zanetti, M.D., a professor at the Department of Medicine at the University of California, San Diego. Dr. Zanetti has developed and patented a method of DNA vaccination by somatic transgene immunization, or STI. Adamis has entered into a world-wide exclusive license with Dr. Zanetti, through a company of which he is the sole owner, Nevagen, LLC, to utilize the technology within the field of viral infectious agents. Adamis believes that the technology has broad applications and is targeting viral disease indications for its initial proof of concept.
STI (also sometimes called TLI) has already been tested in Phase I studies in humans for other vaccine applications. An immune response was elicited in the study, and the results suggested that the procedure was safe. Testing, for instance for influenza, is currently at the preclinical stage. If successful, STI may provide a vaccine for immunity to all forms of influenza, including avian flu, although there are no guarantees that any of the trials will be successful or that a commercial product will be developed or marketed.
Many current vaccines act by giving the immune system a preview of certain protein antigens expected to be found on the target structure; Pathogens, such as influenza, however, demonstrate the limitations of this approach: the influenza virus changes its coat, often by recombination with swine or human viruses or other variation processes approximately every flu season. The changes make each year’s new version of the flu unrecognizable to the immune system, and therefore immunity to influenza viral variants must be usually reestablished with a new vaccine every fall. The following summarizes the method proposed by Adamis to develop long lasting and cross-reactive immunity against, e.g. influenza, but also against other therapeutic vaccine targets using STI:
|
•
|
Draw a small amount of blood from patient
|
|
|
•
|
Separate the white blood cells
|
|
|
•
|
Add plasmid (DNA) to the white blood cells
|
|
|
•
|
Incubate overnight to allow the plasmid to enter the white blood cells (ex vivo transgenesis)
|
|
|
•
|
Inject white blood cells back to the individual to induce immunity to the target of choice, i.e., influenza, hepatitis, HPV, and prostate cancer.).
|
There are a number of factors, including those identified in the Risk Factors section of this Annual Report of Form 10-K that could cause actual events to differ from Adamis’ expectations concerning the timeline for product development and the regulatory approval process. Adamis believes that it will be able to obtain sufficient funding for its clinical trials and product launches as discussed, but there can be no assurance that this will be the case. Similarly, there are no assurances that the clinical trials will be successful or that Adamis will be able to submit an application for, or obtain approval from the FDA for, any vaccine or therapeutic drug product.
Experiments conducted by third parties for Adamis utilizing the STI technology in mice have shown that T-cell immunity can be induced in vivo by a single intravenous inoculation of naïve B lymphocytes genetically programmed by ex vivo transgenesis. This is accomplished by administering a plasmid DNA under control of a B cell specific promoter. The process is entirely spontaneous and mimics the process of viral infection, which is intracellular replication. Results show the induction of systemic effector CD4 and CD8 T-cell responses within 14 days after administration of the transgenic B cells. Durable immunologic memory is also induced. It has been demonstrated that a single injection of 5 x 103 transgenic B lymphocyte induces complete protection from a lethal virus challenge. The following outlines the protocol used in the mouse trial:
|
•
|
A small amount of blood was drawn from mice
|
16
|
•
|
B cells were separated from the blood and transfected with DNA from flu virus
|
|
|
•
|
Transfected lymphocyctes, or priming B cells, were re-infused into the mice
|
|
|
•
|
A lethal challenge of virus was administered via aerosol 14-21 days after re-infusion
|
|
|
•
|
For controls, mice were injected with priming B cells transfected with DNA not specific for the flu
|
A single injection of transgenic B lymphocytes in this trial was sufficient to generate specific CD8 T-cell memory responses, which protected mice from a lethal viral challenge. The immune response that was induced was a reaction against the common components of the influenza virus, and was cross-reactive, meaning that it reacted against various types of flu virus (avian or any other). Thus, this type of vaccine may be utilized to protect individuals from various strains of influenza that may occur.
License Agreements
License Agreements Relating to Cancer Technologies
On February 24, 2010, Adamis entered into an agreement with Colby Pharmaceutical Company, a private corporation, to acquire three separate exclusive license agreements, covering three small molecule anti-inflammatory compounds, named CPC-100, CPC-200 and CPC-300, for the potential treatment of human prostate cancer, or PCa, in exchange for shares of Adamis common stock. Colby licensed the patents, patent applications and related intellectual property relating to the compounds pursuant to license agreements with the Wisconsin Alumni Research Foundation (WARF).
The completion of all the transactions contemplated by the agreement is subject to certain customary closing conditions. An initial closing was held on February 25, 2010, pursuant to which Colby assigned and transferred to Adamis the license agreement relating to the CPC-300 compound, in consideration of the issuance to Colby of 1,000,000 shares of Adamis common stock. The transfer of the license agreements relating to CPC-100 and CPC-200 will occur at a subsequent closing, upon satisfaction of closing conditions, which include the receipt by Adamis of equity funding after the date of the agreement in excess of $2 million. The consideration for the transfer of these additional agreements will be 7,500,000 shares of Adamis common stock to Colby. Upon the completion of an equity offering of at least $2 million, Adamis will acquire the license agreements relating to the CPC-100 and CPC-200 compounds.
The discussion below will assume that Adamis has been assigned all rights under all three license agreements, and that Adamis is the licensee under the license agreements.
The CPC-100 and CPC-200 license agreements are dated January 26, 2007. The CPC-300 license agreement is dated January 2, 2008. The licensor under the agreements is Wisconsin Alumni Research Foundation, or WARF. Under each separate agreement, the licensor grants to licensee) an exclusive right and license, with rights of sublicense, Adamis under the patents and patent applications identified in the agreement, for the fields of human nutraceuticals, preventatives, therapeutics and diagnostics and for all territories worldwide that are covered by any of the licensed patents.
If the licensee grants a sublicense, the licensee remains responsible to pay to the licensor an amount equal to what licensee would have been required to pay to licensor had licensee sold the amount of products covered by the license agreement that were sold by the licensee. In addition, if the licensee receives any fees or other payments in consideration for any rights granted under a sublicense, and the fees or payments are not based directly on the amount or value of products sold by the sublicensee or provided as reimbursement for research and development costs incurred by licensee, then licensee is obligated to pay to licensor a percentage of such payments, ranging from 10% to 40% depending on what the stage of regulatory approval and clinical trial development at the time the payments are received.
In each agreement, the licensee agrees to use reasonable efforts to diligently develop, manufacture, market and sell products in each licensed field and licensed territory. The agreements include a development plan relating to covered products, and the licensee is obligated to provide periodic updates to the licensor concerning the development plan.
17
The license agreements include milestones that licensee agrees to meet by certain dates, relating to obtaining cumulative funding by certain dates, the filing of an IND relating to a covered product, enrollment of a first patient under a Phase II clinical trial by certain dates, and filing of an NDA with the FDA relating to a covered product by certain dates. The licensor has the right to terminate the license agreement with advance notice if the licensee fails to meet any of the funding milestones or commercialization milestones.
Each agreement provides that the licensee will pay the licensor minimum royalties of $25,000 per year, commencing in years 2020 under the CPC-100 and CPC-200 agreements and 2021 under the CPC-300 agreement. Under all of the agreements, the licensee agrees to pay product royalties to licensor based on net sales of covered products, at a rate of 5% of net sales. The agreements include customary stacking provisions providing for a reduction in royalties if the licensee is obligated to pay royalties to other third parties on sales of covered products, but in all events the rate will be not less than 2.5% of net sales.
Each agreement provides that the licensee will pay to licensor milestone payments as follows: (i) $25,000 upon the filing of the first IND or comparable regulatory filing for a human therapeutic product covered by the agreement; (ii) $150,000 upon the enrollment of the first patient under a full Phase II clinical trial for the covered first human therapeutic product; (iii) $200,000 upon the enrollment of the first patient under a full Phase III clinical trial for the first covered human therapeutic product; and $250,000 for the first NDA or comparable regulatory approval for a covered human therapeutic product. Licensee is obligated to make only one milestone payment under each of the milestones and will not be obligated to make a second payment for any subsequent occurrence of the same milestone.
Each agreement provides that the licensee will reimburse licensor for legal fees and other costs incurred in filing, prosecuting and maintaining the licensed patents during the term of the agreement. These amounts will accrue for a period of four years after the date of the agreement, after which time the accrued amounts will be paid in four annual installments.
Each agreement contains customary representations and warranties of licensor and licensee, record keeping obligations of licensee, inspection and audit rights of licensor, and penalties for underpayment of royalties. The licensee agrees to indemnify the licensor against any loss or expense arising out of the production, manufacture, sale, use, lease, consumption or advertisement of any covered product under the agreement.
The term of each agreement continues until the date that none of the licensed patents under the agreement remains an enforceable patent. License may terminate the agreement at any time with 90 days prior notice to the licensor. Licensor may terminate the agreement if the date of first commercial sale of a covered product does not occur by December 31, 2020 under the CPC-100 and CPC-200 agreements and December 31, 2021 under the CPC-300 agreement. Licensor may also terminate the agreement following licensee’s failure to meet a funding or commercialization milestone, fails to pay amounts when due or deliver a development report or commits a material breach of the agreement, and fails to cure the default within 90 days.
Each agreement provides that the licensee may not assign the agreement without the licensor’s consent, which shall not be unreasonably withheld, except that licensee may assign the agreement to a person or entity that acquires all or substantially all of licensee business or assets to which the agreements relate provided that the assignee agrees to be bound by the provisions of the agreement.
License Agreement Relating to Vaccine Technologies
On July 28, 2006, Adamis entered into a worldwide exclusive license agreement with Dr. Zanetti, through a company of which he is the sole owner, Nevagen, to utilize the technology within the field of viral infectious agents. The intellectual property, or IP, licensed by Adamis includes the use of the technology known as “Transgenic Lymphocyte Technology,” or TLI, covered by patent applications titled “Somatic Transgene Immunization and related methods” including but not limited to “ex vivo treatment of an individual’s lymphocytes. The vaccine is constituted of the individual’s lymphocytes harboring plasmid DNA, for example, DNA coding for selected epitopes of influenza virus. The IP includes rights under two issued U.S. patents, three U.S. patent applications and related patent applications filed in European Union, Japan and Canada. The U.S. patent was issued on October 9, 2007 and will expire on April 27, 2019, 20 years from the filing date of the earliest U.S. non-provisional application upon which the patent claims priority.
18
The field for this exclusive license is the prevention and treatment and detection of viral infectious diseases. The geographic area covered by the exclusive license is worldwide. The license will terminate with the expiration of the U.S. patent for the IP.
As part of the initial license fee Adamis granted Dr. Zanetti the right to purchase one million shares of Adamis common stock at a price of $0.001 per share, and he subsequently exercised that right. In addition, Adamis paid the licensor an initial license fee of $55,000. For the first product, Adamis will make payments upon reaching specified milestones in clinical development and submission of an application regulatory approval, potentially aggregating $900,000 if all milestone payments are made. As of the date of this Annual Report, no milestones have been achieved and no milestone payments have been made. The agreement also provides that Adamis will pay the licensor royalties, in the low single digits, payable on net sales received by Adamis of products covered by the IP. If additional technologies are required to be licensed to produce a functional product, the royalty rate will be reduced by the amount of the royalty paid to the other licensor, but not more than one-half the specified royalty rate. Royalties and incremental payments with respect to influenza will continue until reaching a cumulative total of $10 million.
Adamis and the licensor have the right to sublicense with written permission of the other party. In the event that the licensor sublicenses or sells the improved technology to a third party, then a portion of the total payments, to be decided by mutual agreement, will be due to Adamis. If Adamis sublicenses the IP for use in influenza to a third party, the licensor will be paid a fixed percentage of all license fees, royalties, and milestone payments, in addition to royalties due and payable based on net sales.
If the IP is sublicensed by Adamis to another company for any indication in the field covered by the license agreement other than with respect to influenza, the licensor will be paid a portion of all license fees, royalties and milestone payments, with the percentage declining over time based on the year in which the sublicense is granted. Certain incremental non-flu virus related sublicensing payments described in the license agreement are specifically excluded from the royalty cap.
All improvements of the IP conceived of, or reduced to practice by Adamis, or made jointly by Adamis and the licensor will be owned solely by Adamis. Adamis granted Nevagen a royalty-free nonexclusive license to use any improvements made on the existing technology for research purposes only but not for any commercial purposes of any kind. Adamis has agreed to grant to Nevagen a royalty-free license for any improvement needed for the commercialization of the IP for Nevagen’s use outside the field licensed to Adamis. If Nevagen sublicenses or sells the improved technology to a third party, then a portion of the total payments, to be decided by mutual agreement, will be due to Adamis.
Adamis will have the right of first offer to license the following additional technology from the licensor, if and when it becomes available:
|
•
|
Technology for the application of related intellectual property as a prophylactic or therapeutic cancer vaccine; and
|
|
|
•
|
Any additional technology developed by the licensor related to the IP.
|
Adamis has the right to terminate the agreement if it is determined that no viable product can come from the licensed technology. Upon such termination, Adamis would be required to transfer and assign to the licensor all filings, rights and other information in its control if termination occurs. Adamis would retain the same royalty rights for license, or sublicense, agreements if the technology is later developed into a product. Either party may terminate the license agreement in the event of a material breach of the agreement by the other party that has not been cured or corrected within 90 days of notice of the breach.
Drug Development Process
The statements below, and elsewhere in this Annual Report regarding anticipated future events concerning the development process of Adamis’ vaccine product candidates, the clinical trial process, and the regulatory approval process including the actions of the FDA, are subject to several uncertainties and contingencies that could cause actual results to differ in material respects from the results and timelines anticipated in the discussion below. Some of these uncertainties and contingencies are described herein under the heading “Risks Factors.” There is no guarantee that Adamis will be able to complete clinical development and obtain approval from the FDA for any vaccine product candidate.
19
Vaccine Development. Adamis may file for an Investigational New Drug Application, or IND, based on previously published data on this technology. Adamis believes that having this data could shorten the process of preclinical development and preparation of the IND, although there can be no assurance that this will be the case. Adamis believes that clinical trials could start within 60-90 days after acceptance of the IND by the FDA. The total time to complete an IND application is expected to be about one year following receipt of sufficient funding.
Phase 1/2a CPC-100 Prostate Cancer Trial. The Phase 1/2 clinical trial that would be specified in the IND would probably require about 18 months in total. Adamis estimates the total cost of the clinical trial to be about $2,100,000. After completion of the anticipated Phase 1/2a CPC-100 trial, Adamis expects that it would meet with the FDA to review the trial results and determine extension of the Phase 2a to Phase 2b.
All of the therapeutic product candidates will require extensive testing. Both pre-clinical and clinical testing will be necessary before we will be able to apply for regulatory approval. The entire process is expensive and time consuming and requires assistance from outside consultants. The clinical testing process is normally conducted at major cancer centers throughout the U.S., and possibly in specific clinical centers in Europe.
All of the pre-clinical testing is and will be conducted in accordance with Good Laboratory Practices (GLP). Likewise, the clinical testing will be done in accordance with Good Clinical Practice Standards (GCP). GCP is the standard that is required by the FDA and European authorities.
Other Product Candidates
Adamis’ Biosyn subsidiary has intellectual property relating to a microbicide contraceptive product candidate named Savvy®. Savvy underwent Phase III clinical trials in Ghana and Nigeria for reduction in the transmission of Human Immunodeficiency Virus/Acquired Immunodeficiency Disease, or HIV/AIDS, both of which were suspended in 2005 and 2006 and were terminated before completion. Savvy is the subject of a Phase III contraception trial in the United States. The trial has been completed and the analysis of the results is being conducted by the National Institute of Child Health and Human Development (NICHD). The analysis is expected to be completed by mid-2010. Biosyn is not directly involved with the conduct and funding thereof, and significant doubt exists concerning whether Savvy will be commercialized or that Biosyn will ever realize revenues therefrom.
Sources and Availability of Raw Materials
Adamis purchases, in the ordinary course of business, necessary raw materials, components and supplies essential to its operations from several suppliers in the U.S. and overseas. Adamis Labs has entered into a contract with a contract manufacturing organization for the development and production of its PFS Syringe product. Adamis intends to monitor these situations and to seek to provide a continued supply of both raw materials and components.
Sales and Marketing
Adamis Labs’ field force includes sales management, customer service representatives, trade relations/reimbursement specialists and executive management. Adamis’ expansion plan, depending upon securing adequate funding, includes hiring and training approximately 15-30 additional sales representatives to be strategically deployed in the most valuable prescribing U.S. markets to support the ongoing launch of the PFS Syringe product. For future field force expansion and before the launch of Adamis’ aerosolized inhaled nasal steroid, Adamis has identified the top prescribing markets in the U.S. by utilizing physician data aligned with zip code alignment data. Adamis expects to expand to approximately 50 specialty field force sales representatives before introducing the aerosolized nasal steroid product. Physician calls by Adamis’ sales force are expected to be to the highest prescribers of emergency epinephrine injectors in each market and then modified, if required, when the aerosolized nasal steroid product introduction occurs.
Governmental Regulation
The production and marketing of Adamis’ products and potential products and its ongoing research and development, preclinical testing and clinical trial activities are currently subject to extensive regulation and review by numerous governmental authorities in the United States and will face similar regulation and review for overseas approval and sales from governmental authorities outside of the United States. Many of the products Adamis is currently developing must undergo rigorous preclinical and clinical testing and an extensive regulatory approval
20
process before they can be marketed. This process makes it longer, more difficult and more costly to bring Adamis’ potential products to market, and Adamis cannot guarantee that any of its potential products will be approved. The pre-marketing approval process can be particularly expensive, uncertain and lengthy, and a number of products for which FDA approval has been sought by other companies have never been approved for marketing. In addition to testing and approval procedures, extensive regulations also govern marketing, manufacturing, distribution, labeling, and record-keeping procedures. If Adamis or its collaboration partners do not comply with applicable regulatory requirements, such violations could result in non-approval, suspensions of regulatory approvals, civil penalties and criminal fines, product seizures and recalls, operating restrictions, injunctions, and criminal prosecution.
Withdrawal or rejection of FDA or other government entity approval of Adamis’ potential products may also adversely affect Adamis’ business. Such rejection may be encountered due to, among other reasons, lack of efficacy during clinical trials, unforeseen safety issues, inability to follow patients after treatment in clinical trials, inconsistencies between early clinical trial results and results obtained in later clinical trials, varying interpretations of data generated by clinical trials, or changes in regulatory policy during the period of product development in the United States and abroad. In the United States, there is stringent FDA oversight in product clearance and enforcement activities, causing medical product development to experience longer approval cycles, greater risk and uncertainty, and higher expenses. Internationally, there is a risk that Adamis may not be successful in meeting the quality standards or other certification requirements. Even if regulatory approval of a product is granted, this approval may entail limitations on uses for which the product may be labeled and promoted, or may prevent Adamis from broadening the uses of Adamis’ current or potential products for different applications. In addition, Adamis may not receive FDA approval to export Adamis’ potential products in the future, and countries to which potential products are to be exported may not approve them for import.
Manufacturing facilities for Adamis’ products will also be subject to continual governmental review and inspection. The FDA has stated publicly that compliance with manufacturing regulations will continue to be strictly scrutinized. To the extent Adamis decides to manufacture its own products, a governmental authority may challenge Adamis’ compliance with applicable federal, state and foreign regulations. In addition, any discovery of previously unknown problems with one of Adamis’ potential products or facilities may result in restrictions on the potential product or the facility. If Adamis decides to outsource the commercial production of its products, any challenge by a regulatory authority of the compliance of the manufacturer could hinder Adamis’ ability to bring its products to market.
To the extent that Adamis is able to successfully advance a product candidate through clinical trials, it will be required to obtain regulatory approval prior to marketing and selling such product. Adamis is subject to extensive government regulation that increases the cost and uncertainty associated with its efforts to gain regulatory approval of its product candidates. Preclinical development, clinical trials, manufacturing, and commercialization of its product candidates are all subject to extensive regulation by U.S. and foreign governmental authorities. It takes many years and significant expenditures to obtain the required regulatory approvals for biological products. Satisfaction of regulatory requirements depends upon the type, complexity and novelty of the product candidate and requires substantial resources. Adamis cannot be certain that any of its product candidates will be shown to be safe and effective, or that it will ultimately receive approval from the FDA or foreign regulatory authorities to market these products. In addition, even if granted, product approvals and designations such as “fast-track” may be withdrawn or limited at a later time.
The process of obtaining FDA and other required regulatory approvals is expensive. The time required for FDA and other approvals is uncertain and typically takes a number of years, depending on the complexity or novelty of the product. The process of obtaining FDA and other required regulatory approvals for many of Adamis’ products under development is further complicated because some of these products use non-traditional or novel materials in non-traditional or novel ways. For example:
21
|
•
|
the FDA has not established guidelines concerning the scope of clinical trials required for gene-based therapeutic and vaccine products;
|
|
|
•
|
the FDA has provided only limited guidance on how many subjects it will require to be enrolled in clinical trials to establish the safety and efficacy of gene-based products; and
|
|
|
•
|
current regulations and guidance are subject to substantial review by various governmental agencies.
|
Therefore, U.S. or foreign regulations could prevent or delay regulatory approval of Adamis’ products or limit its ability to develop and commercialize products. These delays could:
|
•
|
impose costly procedures;
|
|
|
•
|
diminish any competitive advantages; or
|
|
|
•
|
negatively affect results of operations and cash flows.
|
Adamis believes that the FDA and comparable foreign regulatory bodies will separately regulate each product containing a particular gene depending on its intended use. Presently, to commercialize any product Adamis must sponsor and file a regulatory application for each proposed use. Adamis must conduct clinical studies to demonstrate the safety and efficacy of the product necessary to obtain FDA approval. The results obtained so far in clinical trials may not be replicated in future trials. This may prevent any of the potential products from receiving FDA approval.
Adamis will utilize recombinant DNA molecules in its product candidates, and therefore must comply with guidelines instituted by the NIH and its Office of Biotechnology Activities. The NIH could restrict or delay the development of its product candidates. In March 2004, the NIH Office of Biotechnology Activities and the FDA Center for Biologics Evaluation and Research launched the jointly developed Genetic Modification Clinical Research Information System, or GeMCRIS, an Internet-based database of human gene transfer trials. In its current form, GeMCRIS enables individuals to easily view information on particular characteristics of clinical gene transfer trials, and includes special security features designed to protect patient privacy and confidential commercial information. These security features may be inadequate in design or enforcement, potentially resulting in disclosure of confidential commercial information.
The FDA and the NIH are considering rules and regulations that would require public disclosure of additional commercial development data that is presently confidential. In addition, the NIH, in collaboration with the FDA, has developed an Internet site, ClinicalTrials.gov, which provides public access to information on clinical trials for a wide range of diseases and conditions. Such disclosures of confidential commercial information, whether by implementation of new rules or regulations, by inadequacy of GeMCRIS security features, or by intentional posting on the Internet, may result in loss of advantage of competitive secrets.
A rule published in 2002 by the FDA, known commonly as the “Animal Rule,” established requirements for demonstrating effectiveness of drugs and biological products in settings where human clinical trials for efficacy are not feasible or ethical. The rule requires as conditions for market approval the demonstration of safety and biological activity in humans, and the demonstration of effectiveness under rigorous test conditions in up to two appropriate species of animal. Adamis believes, that with appropriate guidance from the FDA, it may seek and win market approval under the Animal Rule for certain DNA-based products for which human clinical efficacy trials are not feasible or ethical. At the moment, however, it cannot determine whether the Animal Rule would be applied to any products of Adamis, or if applied, that its application would result in expedited development time or regulatory review.
Any regulatory approval to market a product may be subject to limitations on the indicated uses for which Adamis may market the product. These limitations may restrict the size of the market for the product and affect reimbursement by third-party payers. In addition, regulatory agencies may not grant approvals on a timely basis or may revoke or significantly modify previously granted approvals.
Adamis, or its collaborative partners, are subject to numerous foreign regulatory requirements governing the manufacturing and marketing of Adamis’ potential future products outside of the United States. The approval procedure varies among countries, additional testing may be required in some jurisdictions, and the time required to obtain foreign approvals often differs from that required to obtain FDA approvals. Moreover, approval by the FDA does not ensure approval by regulatory authorities in other countries, and vice versa.
22
In addition to regulations imposed by the FDA, Adamis may also be subject to regulation under the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act, the Research Conservation and Recovery Act, as well as regulations administered by the Nuclear Regulatory Commission, national restrictions on technology transfer, import, export and customs regulations and certain other local, state or federal regulations. From time to time, other federal agencies and congressional committees have indicated an interest in implementing further regulation of biotechnology applications. Adamis cannot predict whether any such regulations will be adopted or whether, if adopted, such regulations will apply to its business, or whether Adamis would be able to comply with any applicable regulations.
Even if Adamis’ products are approved by regulatory authorities, if it fails to comply with ongoing regulatory requirements, or if there are unanticipated problems with the products, these products could be subject to restrictions or withdrawal from the market. Even if regulatory approval of a product is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. Later discovery of previously unknown problems with the products, including unanticipated adverse events or adverse events of unanticipated severity or frequency, or failure to comply with regulatory requirements, may result in restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary or mandatory recall, fines, suspension of regulatory approvals, product seizures, injunctions or the imposition of civil or criminal penalties.
As a result of these factors, Adamis may not successfully begin or complete clinical trials in the time periods estimated, if at all. Moreover, if Adamis incurs costs and delays in development programs or fails to successfully develop and commercialize products based upon its technologies, Adamis may not become profitable, and its stock price could decline.
FDA Approval Process
General
In the United States, the FDA regulates drug products under the Federal Food, Drug, and Cosmetic Act, or FFDCA, and its implementing regulations, and regulates biological drug products under both the Public Health Service Act, or PHS Act, and its implementing regulations, as well as the FFDCA. Adamis’ product candidates include both biological drug products and drug products. The process required by the FDA before Adamis’ drug and biological drug product candidates may be marketed in the United States generally involves the following:
|
•
|
completion of extensive preclinical laboratory tests, preclinical animal studies and formulation studies all performed in accordance with the FDA’s current Good Laboratory Practice, or cGLP, regulations;
|
|
|
•
|
submission to the FDA of an IND, which must become effective before human clinical trials may begin;
|
|
|
•
|
performance of adequate and well controlled human clinical trials to establish the safety and efficacy of the product candidate for each proposed indication;
|
|
|
•
|
submission to the FDA of a new drug application, or NDA, for drug products, or a Biologic License Application, or BLA, for biological drug products;
|
|
|
•
|
satisfactory completion of an FDA pre-approval inspection of the manufacturing facilities at which the product is produced to assess compliance with cGMP regulations; and
|
|
|
•
|
FDA review and approval of the NDA or BLA prior to any commercial marketing, sale or shipment of the drug or biological drug.
|
Preclinical tests include laboratory evaluation of product chemistry, formulation and stability, as well as studies to evaluate toxicity in animals. The results of preclinical tests, together with manufacturing information and analytical data, are submitted as part of an IND to the FDA. The IND becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about the conduct of the clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development, and the FDA must grant permission before each clinical trial can begin. Further, an independent
23
institutional review board, or IRB, for each medical center proposing to conduct the clinical trial must review and approve the plan for any clinical trial before it commences at that center and it must monitor the study until completed. The FDA, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive good clinical practices, or GCPs, regulations and regulations for informed consent.
Clinical Trials
For purposes of an NDA or BLA submission and approval, human clinical trials are typically conducted in the following three sequential phases, which may overlap:
|
•
|
Phase 1 Clinical Trials. Studies are initially conducted in a limited population to test the product candidate primarily for safety, dose tolerance, pharmacokinetics and, for vaccine products, immunogenicity, is n healthy humans or in patients. In some cases, a sponsor may decide to conduct what is referred to as a “Phase 1b” evaluation, which is a second, safety-focused Phase 1clinical trial typically designed to evaluate the impact of the drug candidate in combination with currently approved drugs;
|
|
|
•
|
Phase 2 Clinical Trials. Studies are generally conducted in a limited patient population to identify possible adverse effects and safety risks, to determine the efficacy of the product for specific targeted indications and to determine dose tolerance and optimal dosage. Multiple Phase 2clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more extensive Phase 3 clinical trials. In some cases, a sponsor may decide to run what is referred to as a “Phase 2b” evaluation, which is a second, confirmatory Phase 2 clinical trial;
|
|
|
•
|
Phase 3 Clinical Trials. These are commonly referred to as pivotal studies. When Phase 2 evaluations demonstrate that a dose range of the product is effective and has an acceptable safety profile, Phase 3 clinical trials are undertaken in large patient populations to further evaluate dosage, to provide substantial evidence of clinical efficacy and to further test for safety in an expanded and diverse patient population at multiple, geographically-dispersed clinical trial sites; and
|
|
|
•
|
Phase 4 Clinical Trials. In some cases, the FDA may condition approval of an NDA or BLA for a product candidate on the sponsor’s agreement to conduct additional clinical trials to further assess the drug’s safety and effectiveness after NDA or BLA approval. Such post-approval trials are typically referred to as Phase 4 studies.
|
There can be no assurance that Phase I, Phase II trials or Phase III will be completed successfully within any specific time period, if at all, with respect to any of Adamis’ potential products subject to such testing.
After completion of the required clinical testing, an NDA or BLA is prepared and submitted to the FDA. FDA approval of the NDA is required before marketing of the product may begin in the United States. The NDA and BLA must include the results of all preclinical, clinical and other testing and a compilation of data relating to the product’s pharmacology, chemistry, manufacture and controls. The cost of preparing and submitting an NDA or BLA is substantial, and there can be no assurance that any approval will be granted on a timely basis, if at all. Under federal law, the submission of most NDAs and BLAs are additionally subject to a substantial application user fee, and the manufacturer and/or sponsor under an approved new drug application is also subject to annual product and establishment user fees. These fees are typically increased annually.
The FDA has 60 days from its receipt of an NDA or BLA to determine whether the application will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA has agreed to certain performance goals in the review of new drug applications. Most such applications for non-priority drug products are reviewed within ten months. The review process may be extended by the FDA for three additional months to consider certain information or clarification regarding information already provided in the submission. The FDA may also refer applications for novel drug products or drug products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when
24
making decisions. The FDA may deny approval of an NDA or BLA if the applicable regulatory criteria are not satisfied, or it may require additional information including clinical or CMC data. Even if such data are submitted, the FDA may ultimately decide that the NDA or BLA does not satisfy the criteria for approval. Data from clinical trials are not always conclusive and the FDA may interpret data differently than Adamis or its collaborators interpret data. Once issued, the FDA may withdraw product approval if ongoing regulatory requirements are not met or if safety problems occur after the product reaches the market.
Before approving an NDA or BLA, the FDA will typically inspect one or more clinical sites to assure compliance with Good Clinical Practices, or GCP. Additionally, the FDA will inspect the facility or the facilities at which the drug is manufactured. The FDA will not approve the product unless compliance with current good manufacturing practices, or cGMP, is satisfactory and the NDA or BLA contains data that provides substantial evidence that the drug is safe and effective in the indication studied. Failure to comply with GMP or other applicable regulatory requirements may result in withdrawal of marketing approval, criminal prosecution, civil penalties, recall or seizure of products, warning letters, total or partial suspension of production, suspension of clinical trials, FDA refusal to review pending marketing approval applications or supplements to approved applications, or injunctions, as well as other legal or regulatory action against Adamis or its corporate partners.
After the FDA evaluates the NDA or BLA and the manufacturing facilities, it issues an approval letter, an approvable letter or a not-approvable letter. Both approvable and not-approvable letters generally outline the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If and when those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA or BLA, the FDA will issue an approval letter.
An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. As a condition of NDA or BLA approval, the FDA may require substantial post-approval testing and surveillance to monitor the drug’s safety or efficacy and may impose other conditions, including labeling or distribution restrictions or other risk-management mechanisms which can materially affect the potential market and profitability of the drug. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing.
Adamis Labs Products
Several of Adamis Labs’ products, including AeroHist Caplets, AeroHist Plus Caplets, AeroKid Oral Liquid and AeroOtic HC Ear Drops, and the PFS Syringe product, were not the subject of a new drug application or abbreviated new drug application and have not been specifically approved by the FDA for marketing by Adamis. These products have been marketed for many years and, Adamis believes, are similarly situated to products marketed by many companies that are marketed without an approved new drug application or abbreviated new drug application. The products are drug listed with the FDA in the National Drug Code Directory but such listing does not constitute FDA approval of the products. In June 2006, the FDA issued a Compliance Policy Guide for Marketed Unapproved Drugs, which addressed some of the considerations utilized by the FDA in exercising its discretion with respect to products marketed without FDA approval. The guide does not establish legally enforceable responsibilities on the FDA and generally only represents the agency’s current thinking on a topic. The guide emphasizes that any product that is being marketed without required FDA approvals is subject to FDA enforcement action at any time. If the FDA were to issue a Federal Register Notice outlining revised conditions for marketing, which could include calling for the submission of an application for products such as Adamis’ cough/cold products, then Adamis would take appropriate action so as to be in compliance with any such policies. The FDA might also require clinical trials in support of any such applications, and Adamis would need to evaluate its alternatives in light of the costs required to conduct such trials, which could be substantial, compared to the economic benefit to Adamis from such products. In addition, independently of such actions, at any time the FDA could also exercise its discretion to proceed against Adamis and require immediate withdrawal of the PFS Syringe product or other products from the market, or prohibit Adamis from marketing the PFS Syringe product or one or more of such other products without first conducting required trials and obtaining approvals, or impose other penalties on Adamis. Some of Adamis Labs’ unapproved products include extended release formulations, which may subject Adamis to a higher risk of FDA enforcement action. Such actions could have a material adverse effect on Adamis’ business, financial condition and results of operations.
The Prelone product is the subject of an ANDA approval from the FDA. As Adamis believes is common with many drug products, the Prelone product has been manufactured by a third party manufacturer which holds the
25
ANDA approval relating to the product. Adamis owns the trademark and intellectual property rights relating to the product and distributes the product pursuant to those rights.
These products have not been heavily promoted recently due to funding limitations and the competitive market for antihistamine/decongestant products and liquid steroids (Prelone). Adamis has discontinued efforts related to these products, due in part to the widespread substitution of generic products at the dispensing pharmacy level for the conditions indicated for the Adamis Labs products, limited funding, the temporary suspension of its field force, and manufacturing and regulatory challenges facing this category of pharmaceutical products.
The Hatch-Waxman Act
Abbreviated New Drug Applications
In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent with claims that cover the applicant’s product. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential competitors in support of approval of an ANDA. An ANDA provides for marketing of a drug product that has the same active ingredients in the same strengths and dosage form as the listed drug and has been shown through bioequivalence testing to be therapeutically equivalent to the listed drug. ANDA applicants are not required to conduct or submit results of pre-clinical or clinical tests to prove the safety or effectiveness of their drug product, other than the requirement for bioequivalence testing. Drugs approved in this way are commonly referred to as “generic equivalents” to the listed drug, and can often be substituted by pharmacists under prescriptions written for the original listed drug.
The ANDA applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA’s Orange Book. Specifically, the applicant must certify that: (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (iv) the listed patent is invalid or will not be infringed by the new product. A certification that the new product will not infringe the already approved product’s listed patents or that such patents are invalid is called a Paragraph IV certification. If the applicant does not challenge the listed patents, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired.
If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit or a decision in the infringement case that is favorable to the ANDA applicant.
The ANDA application also will not be approved until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired. Federal law provides a period of five years following approval of a drug containing no previously approved active ingredients, during which ANDAs for generic versions of those drugs cannot be submitted unless the submission contains a Paragraph IV challenge to a listed patent, in which case the submission may be made four years following the original product approval. Federal law provides for a period of three years of exclusivity following approval of a listed drug that contains previously approved active ingredients but is approved in a new dosage form, route of administration or combination, or for a new use, the approval of which was required to be supported by new clinical trials conducted by or for the sponsor, during which FDA cannot grant effective approval of an ANDA based on that listed drug.
Section 505(b)(2) New Drug Applications
Most drug products obtain FDA marketing approval pursuant to an NDA or an ANDA. A third alternative is a special type of NDA, commonly referred to as a Section 505(b)(2) NDA, which enables the applicant to rely, in part, on the FDA’s findings of safety and efficacy of an existing product, or published literature, in support of its application. Section 505(b)(2) NDAs often provide an alternate path to FDA approval for new or improved
26
formulations or new uses of previously approved products. Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. The applicant may rely upon the FDA’s findings with respect to certain pre-clinical or clinical studies conducted for an approved product. The FDA may also require companies to perform additional studies or measurements to support the change from the approved product. The FDA may then approve the new product candidate for all or some of the label indications for which the referenced product has been approved, as well as for any new indication sought by the Section 505(b)(2) applicant.
To the extent that the Section 505(b)(2) applicant is relying on studies conducted for an already approved product, the applicant is subject to existing exclusivity for the reference product and is required to certify to the FDA concerning any patents listed for the approved product in the Orange Book to the same extent that an ANDA applicant would. Thus approval of a Section 505(b)(2) NDA can be stalled until all the listed patents claiming the referenced product have expired, until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired, and, in the case of a Paragraph IV certification and subsequent patent infringement suit, until the earlier of 30 months, settlement of the lawsuit or a decision in the infringement case that is favorable to the Section 505(b)(2) applicant.
Fast Track Designation
The FDA’s fast track program is intended to facilitate the development and to expedite the review of drugs and biological drugs that are intended for the treatment of a serious or life-threatening condition for which there is no effective treatment and which demonstrate the potential to address unmet medical needs for the condition. Under the fast track program, the sponsor of a new drug or biological drug candidate may request the FDA to designate the drug candidate for a specific indication as a fast track drug concurrent with or any time after the filing of the IND for the drug or biological drug candidate. The FDA must determine if the candidate qualifies for fast track designation within 60 days of receipt of the sponsor’s request.
If fast track designation is obtained, the FDA may initiate review of sections of an NDA or BLA before the application is complete. This rolling review is available if the applicant provides and the FDA approves a schedule for the submission of the remaining information and the applicant pays applicable user fees. However, the time period specified in PDUFA, which governs the time period goals the FDA has committed to reviewing an application, does not begin until the complete application is submitted. Additionally, the fast track designation may be withdrawn by the FDA if the FDA believes that the designation is no longer supported by data emerging in the clinical trial process.
In some cases, a fast track designated candidate may also qualify for one or more of the following programs:
|
•
|
Priority Review. Under FDA policies, a drug or biological drug candidate is eligible for priority review, or review within a six-month time frame from the time a complete NDA or BLA is accepted for filing, if the candidate provides a significant improvement compared to marketed drugs or biological drugs in the treatment, diagnosis or prevention of a disease. A fast track designated drug or biological drug candidate would ordinarily meet the FDA’s criteria for priority review, however, fast track designation is not required to be eligible for priority review.
|
|
|
•
|
Accelerated Approval. Under the FDA’s accelerated approval regulations, the FDA is authorized to approve drug and biological drug candidates that have been studied for their safety and efficacy in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit to patients over existing treatments based upon either an endpoint that is reasonably likely to predict clinical benefit or on the basis of an effect on a clinical endpoint other than patient survival. In clinical trials, surrogate endpoints are alternative measurements of the symptoms of a disease or condition that are substituted for measurements of observable clinical symptoms. A candidate approved on the basis of a surrogate endpoint is subject to rigorous post-marketing compliance requirements, including the completion of Phase IV or post-approval clinical trials to validate the surrogate endpoint or confirm the effect of the drug candidate on the clinical endpoint. Failure to conduct required post-approval studies, or to validate a surrogate endpoint or confirm a clinical benefit during post-marketing studies, will result in the FDA withdrawing the drug or biological drug from the market on an expedited basis. All promotional materials for drug and biological drug candidates approved under accelerated regulations are subject to prior review by the FDA.
|
27
When appropriate, Adamis intends to seek fast track designation, accelerated approval or priority review for its biological drug candidates.
Satisfaction of FDA regulations and approval requirements or similar requirements of foreign regulatory agencies typically takes several years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease. Typically, if a drug or biological drug candidate is intended to treat a chronic disease, safety and efficacy data must be gathered over an extended period of time. Government regulation may delay or prevent marketing of drug or biological drug candidates for a considerable period of time and impose costly procedures upon our activities. The FDA or any other regulatory agency may not grant approvals for new indications for our drug and biological drug candidates on a timely basis, or at all. Even if a drug or biological drug candidate receives regulatory approval, the approval may be significantly limited to specific disease states, patient populations and dosages. Further, even after regulatory approval is obtained, later discovery of previously unknown problems with a drug or biological drug may result in restrictions on the product or even complete withdrawal of the drug or biological drug from the market. Delays in obtaining, or failures to obtain, regulatory approvals for any of Adamis’ drug or biological drug candidates would harm Adamis’ business. In addition, Adamis cannot predict what adverse governmental regulations may arise from future U.S. or foreign governmental action.
Citizen Petitions
FDA regulations set forth procedures under which parties can petition the FDA to take or refrain from taking certain actions. NDA applicants occasionally submit such citizen petitions requesting that the FDA deny or delay approval of an ANDA, or impose specific additional requirements for approval on ANDAs for a particular drug product. Many such petitions are eventually denied by the FDA, but the submission of such petitions, especially when submitted near the end of an ANDA review, has often delayed the approval of an ANDA while the FDA considers and responds to the issues presented. Congress included provisions to address this practice in the recently enacted FDA Amendments Act of 2007, or FDAAA. The FDAAA prohibits the FDA from delaying approval of an ANDA due to the submission of a citizen petition unless the delay is necessary to protect the public health, and requires that the FDA take final action on any such petition within 180 days of its submission. In addition, the FDAAA requires that petitioners certify, among other things, the date upon which the petitioner first became aware of the information that forms the basis of the request and the name of the person or entity funding the petition.
Post-Approval Requirements
Once an NDA is approved, a product will be subject to certain post-approval requirements. For instance, the FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet.
Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved labeling. Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or BLA or NDA or BLA supplement before the change can be implemented. An NDA or BLA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA or BLA supplements as it does in reviewing NDAs or BLAs.
Adverse event reporting and submission of periodic reports is required following FDA approval of an NDA or BLA. The FDA also may require post-marketing testing, known as Phase IV testing, risk minimization action plans and surveillance to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product. In addition, quality control as well as drug manufacture, packaging, and labeling procedures must continue to conform to cGMP after approval. Drug manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA during which the agency inspects manufacturing facilities to assess compliance with cGMP. Accordingly, manufacturers must continue to expend time, money and effort in the areas
28
of production and quality control to maintain compliance with cGMP. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with regulatory standards, if it encounters problems following initial marketing, or if previously unrecognized problems are subsequently discovered.
The distribution of prescription pharmaceutical products is also subject to the Prescription Drug Marketing Act, or PDMA, which regulates the distribution of drugs and drug samples at the federal level, and sets minimum standards for the registration and regulation of drug distributors by the states. Both the PDMA and state laws limit the distribution of prescription pharmaceutical product samples and impose requirements to ensure accountability in distribution.
Anti-Kickback, False Claims Laws & The Prescription Drug Marketing Act
In addition to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal laws have been applied to restrict certain marketing practices in the pharmaceutical industry in recent years. These laws include anti-kickback statutes and false claims statutes. The federal healthcare program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Violations of the anti-kickback statute are punishable by imprisonment, criminal fines, civil monetary penalties and exclusion from participation in federal healthcare programs. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor.
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to have a false claim paid. In addition, certain marketing practices, including off-label promotion, may also violate false claims laws. The majority of states also have statutes or regulations similar to the federal anti-kickback law and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
New Legislation
On September 27, 2007, the President of the United States signed into law the Food and Drug Administration Amendments Act of 2007, or FDAAA. The legislation grants significant new powers to the FDA, many of which are aimed at improving drug safety and assuring the safety of drug products after approval. In particular, the new law authorizes the FDA to, among other things, require post-approval studies and clinical trials, mandate changes to drug labeling to reflect new safety information, and require risk evaluation and mitigation strategies for certain drugs, including certain currently approved drugs. In addition, it significantly expands the federal government’s clinical trial registry and results databank and creates new restrictions on the advertising and promotion of drug products. Under the FDAAA, companies that violate these and other provisions of the new law are subject to substantial civil monetary penalties.
While the above provisions of the FDAAA, among others, will undoubtedly have a significant effect on the pharmaceutical industry, the extent of that effect is not yet known. As regulations, guidance and interpretations are issued by the FDA relating to the new legislation, its impact on the industry, as well as our business, will become clearer. The changes and new requirements it imposes on the drug review and approval process and post-approval activities could make it more difficult, and certainly more costly, to obtain approval for new pharmaceutical products, or to produce, market, and distribute existing products.
Approval Outside the United States
In order to market any product outside of the United States, Adamis must comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy and governing, among other things, clinical trials and commercial sales and distribution of our products. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from and be longer than that required to obtain FDA approval. Regulatory
29
approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others.
To date, Adamis has not initiated any discussions with the European Medicines Agency, or EMEA, or any other foreign regulatory authorities with respect to seeking regulatory approval for any indication in Europe or in any other country outside the United States. As in the United States, the regulatory approval process in Europe and in other countries is a lengthy and challenging process. If Adamis fails to comply with applicable foreign regulatory requirements, it may be subject to fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Product Liability Insurance
The manufacture and sale of human therapeutic products involve an inherent risk of product liability claims and associated adverse publicity. Adamis currently has only limited product liability insurance, and there can be no assurance that Adamis will be able to maintain existing or obtain additional product liability insurance on acceptable terms or with adequate coverage against potential liabilities. Such insurance is expensive, difficult to obtain and may not be available in the future on acceptable terms, or at all. An inability to obtain sufficient insurance coverage on reasonable terms or to otherwise protect against potential product liability claims could inhibit Adamis’ business. A product liability claim brought against Adamis in excess of its insurance coverage, if any, could have a material adverse effect upon its business, financial condition and results of operations.
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. Many of Adamis’ competitors, including biotechnology and pharmaceutical companies, academic institutions and other research organizations, are actively engaged in the discovery, research and development of products that could compete directly or indirectly with Adamis’ products under development.
Adamis Labs Allergy Products. Adamis Labs’ current line of allergy and respiratory products compete with numerous prescription and non-prescription over-the-counter products targeting similar conditions, including, in the seasonal or perennial rhinitis areas, cough and cold, as well as prescription generic products. In addition, a number of companies, including GSK, Merck, and AstraZeneca, produce pharmaceutical products, such as antihistamines, corticosteroids and anti-leukotriene agents, which manage allergy symptoms.
PFS Syringe Product. Adamis’ PFS Syringe product competes against other self-administered epinephrine products, including EpiPen, EpiPen Jr. and Twinject.
BANS. Adamis’ inhaled nasal steroid BANS product, if developed, launched and marketed, is expected to compete with several inhaled nasal steroid products that are currently marketed, including Flonase, marketed by GlaxoSmithKline, Nasonex, marketed by Schering, Nasacort AQ, marketed by Aventis and Rhinocort, marketed by AstraZeneca.
Vaccine Technology. If Adamis successfully develops a vaccine product for viral infections based on the STI technology, that product is expected to compete with traditional and emerging vaccines from companies currently marketing these products, including GSK, Novartis, Sanofi-Pasteur, Medimmune/AstraZeneca and CSL. In addition, Adamis is aware of several companies developing potentially competing vaccines, including Acambis, VaxInnate, Merck, Vical and Dynavax Technologies Corporation, and other companies of which Adamis is not aware are also likely developing products intended to address indications targeted by Adamis. For instance, the prevalence during 2009 of the H1N1 influenza virus (“swine-flu”) and of illnesses and deaths throughout the world resulting from the H1N1 virus may cause additional companies to seek to develop vaccines or other therapies for influenza.
Prostate Cancer. The development and commercialization of new drugs for cancer is highly competitive. Many of the organizations that are in the cancer area have far greater financial and technical capabilities than we do. Additionally, many of the larger pharmaceutical companies have extensive experience in clinical and regulatory development, manufacturing and commercialization. The competition has been and will continue to be intense. In the area of prostate cancer, there are a number of companies actively involved. These include, but are not limited to: Astra-Zeneca, Sanofi-Aventis, Abbott Laboratories, Bristol-Myers Squibb, Eli Lilly, Takeda, J&J, Merck, Pfizer, Dendreon and Medivation.
30
Savvy. Biosyn’s Savvy contraceptive product candidate, if developed, launched and marketed, would be subject to competition from other microbicides that are currently undergoing clinical trials and which may be sold by prescription or over-the-counter, as well as non-microbicidal products such as condoms. There is also a number of existing contraception products currently on the market, which could greatly limit the marketability of the Savvy contraception product candidate. As a result, there can be no assurance that Biosyn’s Savvy product candidate, even if developed, would be able to compete successfully with existing products or other innovative products under development.
Many of the entities developing and marketing these competing products have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing than Adamis. Smaller or early-stage companies may also prove to be significant competitors, particularly for collaborative agreements with large, established companies and access to capital. These entities may also compete with Adamis in recruiting and retaining qualified scientific and management personnel, as well as in acquiring technologies complementary to, or necessary for, Adamis’ programs.
The pharmaceutical industry is characterized by extensive research efforts and rapid and significant technological change and intense competition. Adamis is much smaller in terms of size and resources than many of its competitors in the United States and abroad, which include, among others, major pharmaceutical, chemical, consumer product, and biotechnology companies, specialized firms, universities and other research institutions. Adamis’ competitors may succeed in developing technologies and products that are safer, more effective or less costly than any developed by Adamis, thus rendering its technology and potential products obsolete and noncompetitive. Many of these competitors have substantially greater financial and technical resources, clinical production and marketing capabilities and regulatory experience than Adamis.
Patents and Proprietary Technologies
Patents and other proprietary rights are important to Adamis’ business. Adamis’ policy is to file patent applications and protect inventions and improvements to inventions that are commercially important to the development of its business. Adamis also relies on trade secrets, know-how, confidentiality agreements, continuing technology innovations and licensing opportunities to protect its technology and develop and maintain its competitive position.
It is Adamis’ policy to require its employees to execute an invention assignment and confidentiality agreement upon employment. Each agreement provides that all confidential information developed or made known to the employee during the course of employment will be kept confidential and not disclosed to third parties except in specific circumstances. The invention assignment generally provides that all inventions conceived by the employee will be the exclusive property of Adamis. In addition, it is Adamis’ policy to require collaborators and potential collaborators to enter into confidentiality agreements. There can be no assurance, however, that these agreements will provide meaningful protection of Adamis’ trade secrets.
Adamis is the exclusive licensee, under the license agreement with Nevagen, of rights under two issued U.S. patents, three U.S. patent applications and related patent applications filed in the European Union, Japan and Canada, relating to the TLI technology, in the field of prevention and treatment and detection of viral infectious diseases.
The licensed intellectual property, or IP, includes the use of the technology known as “Transgenic Lymphocyte Technologym,” or TLI, covered by patent applications entitled “Somatic Transgene Immunization and Related Methods” and related know how. TLI includes, but is not limited to, creating a vaccine by exposing an individual’s lymphocytes to plasmid DNA encoding certain epitopes and re-administering the treated lymphocytes to the individual. The vaccines are made up of the individual’s lymphocytes harboring plasmid DNA encoding epitopes, e.g., selected epitopes of influenza virus. Virtually all of Adamis’ current vaccine product candidate plan are based on technology covered by these patents and applications.
In addition, Adamis recently acquired and entered into agreements to acquire exclusive license agreements covering three small molecule anti-inflammatory compounds, named CPC-100, CPC-200 and CPC-300, that Adamis believes are promising drug candidates for the potential treatment of human prostate cancer (PCa). The intellectual property covered by the agreements was licensed from the Wisconsin Alumni Research Foundation, or
31
WARF. The company has acquired the license agreement relating to CPC-300 and will, upon completion of an equity offering of at least $2 million, acquire the agreements relating to CPC-100 and CPC-200.
The IP includes rights under the following patents, including all divisionals, continuations, continuations-in-part, reexaminations and reissues:
|
•
|
US Patent #7,279,462 entitled SOMATIC TRANSGENE IMMUNIZATION AND RELATED METHODS, filed April 27, 1999, granted October 9, 2007. The expiration date for this patent is 2019.
|
|
|
•
|
European Patent Application 009301284.7 entitled SOMATIC TRANSGENE IMMUNIZATION AND RELATED METHODS, international filing date April 27, 2000.
|
|
|
•
|
Canadian Patent Application #2,369, 616 entitled SOMATIC TRANSGENE IMMUNIZATION AND RELATED METHODS, international filing date April 27, 2000.
|
|
|
•
|
Japan Patent Application #2000-613478 entitled SOMATIC TRANSGENE IMMUNIZATION AND RELATED METHODS, international filing date April 27, 2000.
|
|
|
•
|
US Patent Application No. 11,640,778, entitled SOMATIC TRANSGENE IMMUNIZATION AND RELATED METHODS, filed December 16, 2006.
|
|
|
•
|
US Patent Application No. 11,713,477 entitled SOMATIC TRANSGENE IMMUNIZATION AND RELATED METHODS, filed March 2, 2007.
|
|
|
•
|
Japan Patent Application #2008-328814 entitled SOMATIC TRANSGENE IMMUNIZATION AND RELATED METHODS, international filing date December 24, 2008.
|
|
|
•
|
US Patent Application No. 10/789385 entitled CHROMAN-DERIVED ANTI-ANDROGENS FOR TREATMENT OF ANDROGEN-MEDIATED DISORDERS.
|
|
|
•
|
Hong Kong Patent Application No. 06105362.3 entitled CHROMAN-DERIVED ANTI-ANDROGENS FOR TREATMENT OF ANDROGEN-MEDIATED DISORDERS.
|
|
|
•
|
European Patent Application No. 04785845.1 entitled CHROMAN-DERIVED ANTI-ANDROGENS FOR TREATMENT OF ANDROGEN-MEDIATED DISORDERS.
|
|
|
•
|
Canadian Patent Application No. 2517390 entitled CHROMAN-DERIVED ANTI-ANDROGENS FOR TREATMENT OF ANDROGEN-MEDIATED DISORDERS.
|
|
|
•
|
Australian Patent Application No. 2004260631 entitled CHROMAN-DERIVED ANTI-ANDROGENS FOR TREATMENT OF ANDROGEN-MEDIATED DISORDERS.
|
|
|
•
|
US Patent Application No. 60/797142 entitled DEVELOPMENT OF N1, N4-BIS (BUTA-1, 3-DIENYL) BUTANE-1, 4-DIAMINE, A PROSTATE TARGET ANTI-OXIDANT FOR PROSTATE CANCER PREVENTION.
|
|
|
•
|
US Patent Application No. 10/906691 entitled LUPEOL ANTI-TUMOR AGENT AND USES THEREOF.
|
BioSyn currently holds five patents worldwide relating to Savvy gel for contraception and the reduction in transmission of HIV infection. These patents expire at various dates between 2017 and 2021. Adamis is not aware of any organization that currently has legally blocking proprietary rights relating to the Savvy product candidate. However, it is impossible to anticipate the breadth or degree of protection that any such patents will afford, or whether we can meaningfully protect our rights to our unpatented trade secrets. No assurance can be given that competitors will not independently develop substantially equivalent proprietary information and techniques, or otherwise gain access to our trade secrets or disclose such technology.
32
Adamis’ failure to obtain patent protection or otherwise protect its proprietary technology or proposed products may have a material adverse effect on Adamis’ competitive position and business prospects. The patent application process takes several years and entails considerable expense. There is no assurance that additional patents will issue from these applications or, if patents do issue, that the claims allowed will be sufficient to protect Adamis’ technology.
The patent positions of pharmaceutical and biotechnology firms are often uncertain and involve complex legal and factual questions. Furthermore, the breadth of claims allowed in biotechnology patents is unpredictable. Adamis cannot be certain that others have not filed patent applications for technology covered by the pending STI applications or that the licensor of the TLI technology was the first to invent the technology that is the subject of such patent applications. Competitors may have filed applications for, or may have received patents and may obtain additional patents and proprietary rights relating to compounds, products or processes that block or compete with those of Adamis. Adamis is aware of patent applications filed and patents issued to third parties relating to HFA propellant technology and aerosolized inhalers, and there can be no assurance that any patent applications or patents will not have a material adverse effect on potential products Adamis is developing or may seek to develop in the future.
Patent litigation is widespread in the biotechnology industry. Litigation may be necessary to defend against or assert claims of infringement, to enforce patents issued to Adamis, to protect trade secrets or know-how owned or licensed by Adamis, or to determine the scope and validity of the proprietary rights of third parties. Except as described in "Item 3. Legal Proceedings” below, no third party has asserted that Adamis is infringing such third party’s patent rights or other intellectual property, there can be no assurance that litigation asserting such claims will not be initiated, that Adamis would prevail in any such litigation or that Adamis would be able to obtain any necessary licenses on reasonable terms, if at all. Any such claims against Adamis, with or without merit, as well as claims initiated by Adamis against third parties, can be time-consuming and expensive to defend or prosecute and to resolve. If other companies prepare and file patent applications in the United States that claim technology also claimed by Adamis, it may have to participate in interference proceedings to determine priority of invention which could result in substantial cost to Adamis even if the outcome is favorable to Adamis. There can be no assurance that third parties will not independently develop equivalent proprietary information or techniques, will not gain access to Adamis’ trade secrets or disclose such technology to the public or that Adamis can maintain and protect unpatented proprietary technology. Adamis typically requires its employees, consultants, collaborators, advisors and corporate partners to execute confidentiality agreements upon commencement of employment or other relationships with Adamis. There can be no assurance, however, that these agreements will provide meaningful protection or adequate remedies for Adamis’ technology in the event of unauthorized use or disclosure of such information, that the parties to such agreements will not breach such agreements or that Adamis’ trade secrets will not otherwise become known or be discovered independently by its competitors.
ITEM 1A: RISK FACTORS
Risks Related to Our Business and Industry
We have notes outstanding in the aggregate principal amount of $2,309,000, which become due in 2010 and which we may be unable to repay at maturity.
We have a convertible note outstanding to The G-Max Trust in the principal amount of $500,000 which is due and payable by Adamis on December 31, 2010. We also have several Senior Notes outstanding to a small number of institutional investors in the principal amount of $1,500,000 which become due and payable by Adamis on October 11, 2010. In addition, we have several notes outstanding to Dennis J. Carlo, Ph.D, our chief executive officer and a director, in the principal amount of $309,565 which become due on various dates in 2010. The notes accrue interest at the rate of 10%. We may not have the funds to repay the holders of these notes at maturity or to continue to pay the interest on the notes in the ordinary course which may result in our defaulting under the notes. If this occurs, the holders of the notes would have rights senior to those of our common stockholders, and holders of the Senior Notes would have rights with respect to all of our assets pursuant to the security agreement relating to the Senior Notes.
Additionally, the obligations of the Senior Notes are guaranteed by our principal subsidiaries, including Adamis Corporation, Adamis Laboratories, Inc. and Adamis Viral, Inc., and are secured by a security interest in all of our assets and those of our subsidiaries, which is described in the security agreement relating to the Senior Notes. The obligations of the
33
notes issued to Dennis J. Carlo, Ph.D are secured by a subordinated security interest in the assets of Adamis Laboratories, Inc. In the event of a default in any of Adams’ obligations under the Senior Notes, the holders may foreclose on our assets and those of our subsidiaries.
Adamis’ limited operating history may make it difficult to evaluate its business to date and Adamis’ future viability.
Adamis is in the early stage of operations and development, and has only a limited operating history on which to base an evaluation of its business and prospects, having just commenced operations in 2006. Moreover, Adamis acquired Adamis Labs, formerly Healthcare Ventures Group, during calendar year 2007. Adamis is subject to the risks inherent in the ownership and operation of a company with a limited operating history, such as regulatory setbacks and delays, fluctuations in expenses, competition, the general strength of regional and national economies, and governmental regulation. Any failure to successfully address these risks and uncertainties could seriously harm Adamis’ business and prospects. Adamis may not succeed given the technological, marketing, strategic and competitive challenges it will face. The likelihood of Adamis’ success must be considered in light of the expenses, difficulties, complications, problems and delays frequently encountered in connection with the growth of a new business, the continuing development of new drug development technology, and the competitive and regulatory environment in which Adamis operates or may choose to operate in the future.
Adamis will require additional financing.
At March 31, 2010, Adamis and its subsidiaries together had cash and cash equivalents of approximately $290,000 and accounts receivable of approximately $6,000. As described in greater detail below under the heading “Adamis’ Management’s Discussion and Analysis of Financial Condition and Results of Operations — Liquidity and Capital Resources — Recent Convertible Note Transactions,” in December 2009 and January 2010, Adamis raised gross proceeds of approximately $2,000,000 through the issuance of convertible promissory notes and shares of Adamis common stock. These notes become due and payable during calendar year 2010. Adamis will require additional cash resources to continue operations during 2010. Adamis will have capital needs at various times during calendar year 2010, and such capital needs will depend in part on the level of product revenues, the amount of additional funds raised in equity or debt transactions, the amounts spent on product development efforts, and whether outstanding convertible notes convert with common stock before their maturity dates in 2010. Adamis’ capital needs could include $4 million or more for ongoing sales, general and administrative activities and expenses and $9 million or more on product development. However, lower revenues or other factors could result in operations, sales and product development activities, expenses and capital needs significantly below these levels. Additional funding will be used to satisfy existing obligations and liabilities and future working capital needs, to build working capital reserves and to fund a number of projects, which may include the following:
|
•
|
market the Adamis Labs PFS Syringe product and the generic nasal steroid product candidate;
|
|
|
•
|
pursue the development of other product candidates;
|
|
|
•
|
fund clinical trials and seek regulatory approvals;
|
|
|
•
|
expand research and development activities;
|
|
|
•
|
access manufacturing and commercialization capabilities;
|
|
|
•
|
implement additional internal systems and infrastructure;
|
|
|
•
|
maintain, defend and expand the scope of Adamis’ intellectual property portfolio; and
|
|
|
•
|
hire additional management, sales, research, development and clinical personnel.
|
Statements in this Annual Report, including concerning Adamis’ anticipated or hoped-for target dates for introduction of its nasal steroid, tumor and vaccine product candidates, and for the commencement of clinical trials relating to the steroid, tumor and vaccine product candidates, assume that Adamis will have sufficient funding to support the timely introduction of products and the conduct of clinical trials. Failure to have sufficient funding could require Adamis to delay product launches or clinical trials, which would have an adverse effect on its business and results of operations and which could increase the need for additional financing in the future.
34
Until Adamis can generate a sufficient amount of revenue to finance its cash requirements, which Adamis may never do, Adamis expects to finance future cash needs primarily through public or private equity offerings, debt financings, or licensing revenues from strategic collaborations. Sales of additional equity securities will dilute current stockholders’ ownership percentage in Adamis. Adamis does not know whether additional financing will be available on acceptable terms, or at all. If Adamis is not able to secure additional equity or debt financing when needed on acceptable terms, Adamis may have to sell some of its assets or enter into a strategic collaboration for one or more of Adamis’ product candidate programs at an earlier stage of development than would otherwise be desired. This could lower the economic value of these collaborations to Adamis. In addition, Adamis may have to delay, reduce the scope of, or eliminate one or more of its clinical trials or research and development programs, or ultimately, cease operations.
Some of Adamis’ potential products and technologies are in early stages of development.
The development of new pharmaceutical products is a highly risky undertaking, and there can be no assurance that any future research and development efforts Adamis might undertake will be successful. Adamis’ potential products in oncology and viral fields will require extensive additional research and development before any commercial introduction, and development work on the generic nasal steroid product must still be completed. There can be no assurance that any future research, development or clinical trial efforts will result in viable products or meet efficacy standards. Future clinical or preclinical results may be negative or insufficient to allow Adamis to successfully market its product candidates. Obtaining needed data and results may take longer than planned or may not be obtained at all. Any such delays or setbacks could have an adverse effect on the ability of Adamis to achieve its financial goals.
Adamis’ corporate compliance programs cannot guarantee that Adamis is now or will be in compliance with all potentially applicable regulations.
The development, manufacturing, pricing, sales, and reimbursement of pharmaceutical products, together with Adamis’ general operations, are subject to extensive regulation by federal, state and other authorities within the United States and numerous entities outside of the United States. Adamis is a small company and it relies on third parties to conduct certain important functions. Adamis relies on a third party clinical regulatory organization, Health Decisions, Inc., pursuant to an agreement between Adamis and the National Institute of Child Health and Human Development, to conduct its Phase 3 Savvy clinical trial, and will rely on third parties to assist in evaluation of the results of that trial. In addition, Adamis also has significantly fewer employees than many other companies that have the same or fewer product candidates in clinical development. If Adamis fails to comply with any of these regulations, Adamis could be subject to a range of regulatory actions, including suspension or termination of clinical trials, restrictions on its products or manufacturing processes, or other sanctions or litigation. In addition, as a publicly-traded company, Adamis is subject to significant regulations, including the Sarbanes-Oxley Act of 2002. While Adamis has developed and instituted a corporate compliance program and continues to update the program in response to newly implemented or changing regulatory requirements, Adamis cannot assure you that it is now or will be in compliance with all such applicable laws and regulations. Failure to comply with potentially applicable laws and regulations could also lead to the imposition of fines, cause the value of Adamis’ common stock to decline and impede Adamis’ ability to raise capital or lead to the failure of Adamis’ common stock to continue to be traded on the OTC Bulletin Board.
Adamis is subject to substantial government regulation, which could materially adversely affect Adamis’ business.
The production and marketing of Adamis’ products and potential products and its ongoing research and development, pre-clinical testing and clinical trial activities are currently subject to extensive regulation and review by numerous governmental authorities in the United States and will face similar regulation and review for overseas approval and sales from governmental authorities outside of the United States. Some of the product candidates that Adamis is currently developing must undergo rigorous pre-clinical and clinical testing and an extensive regulatory approval process before they can be marketed. This process makes it longer, more difficult and more costly to bring Adamis’ potential products to market, and Adamis cannot guarantee that any of its potential products will be approved. The pre-marketing approval process can be particularly expensive, uncertain and lengthy, and a number of products for which FDA approval has been sought by other companies have never been approved for marketing. In addition to testing and approval procedures, extensive regulations also govern marketing, manufacturing, distribution, labeling, and record-keeping procedures. If Adamis or its collaboration partners do not comply with applicable regulatory requirements, such violations could result in non-approval, suspensions of regulatory
35
approvals, civil penalties and criminal fines, product seizures and recalls, operating restrictions, injunctions, and criminal prosecution.
Withdrawal or rejection of FDA or other government entity approval of Adamis’ potential products may also adversely affect Adamis’ business. Such rejection may be encountered due to, among other reasons, lack of efficacy during clinical trials, unforeseen safety issues, inability to follow patients after treatment in clinical trials, inconsistencies between early clinical trial results and results obtained in later clinical trials, varying interpretations of data generated by clinical trials, or changes in regulatory policy during the period of product development in the United States and abroad. In the United States, there is stringent FDA oversight in product clearance and enforcement activities, causing medical product development to experience longer approval cycles, greater risk and uncertainty, and higher expenses. Internationally, there is a risk that Adamis may not be successful in meeting the quality standards or other certification requirements. Even if regulatory approval of a product is granted, this approval may entail limitations on uses for which the product may be labeled and promoted, or may prevent Adamis from broadening the uses of Adamis’ current or potential products for different applications. In addition, Adamis may not receive FDA approval to export Adamis’ potential products in the future, and countries to which potential products are to be exported may not approve them for import.
Manufacturing facilities for Adamis’ products will also be subject to continual governmental review and inspection. The FDA has stated publicly that compliance with manufacturing regulations will continue to be strictly scrutinized. To the extent Adamis decides to manufacture its own products, a governmental authority may challenge Adamis’ compliance with applicable federal, state and foreign regulations. In addition, any discovery of previously unknown problems with one of Adamis’ potential products or facilities may result in restrictions on the potential product or the facility. If Adamis decides to outsource the commercial production of its products, any challenge by a regulatory authority of the compliance of the manufacturer could hinder Adamis’ ability to bring its products to market.
Some of Adamis Labs’ products that have been drug listed with the FDA are marketed without an approved new drug application or abbreviated new drug application. The FDA could at some future date seek to prevent marketing of these products, require that such products be marketed only after submission and approval of drug applications, or take other regulatory action against Adamis with respect to these products, which could have an adverse effect on Adamis.
Several of Adamis Labs’ products, including AeroHist Caplets, AeroHist Plus Caplets, AeroKid Oral Liquid and AeroOtic HC Ear Drops, and the Epi Syringe, were not the subject of a new drug application or abbreviated new drug application, or ANDA, and have not been specifically approved by the FDA for marketing by Adamis. These products have been marketed for many years and, Adamis believes, are similarly situated to products marketed by many companies that are marketed without an approved new drug application or abbreviated new drug application. The products are drug listed with the FDA in the National Drug Code Directory but such listing does not constitute FDA approval of the products. In June 2006, the FDA issued a Compliance Policy Guide for Marketed Unapproved Drugs, which addressed some of the considerations utilized by the FDA in exercising its discretion with respect to products marketed without FDA approval. The guide does not establish legally enforceable responsibilities on the FDA and generally only represents the agency’s current thinking on a topic. The guide emphasizes that any product that is being marketed without required FDA approvals is subject to FDA enforcement action at any time. If the FDA were to issue a Federal Register Notice outlining revised conditions for marketing, which could include calling for the submission of an application for products such as Adamis’ cough/cold products, then Adamis would take appropriate action so as to be in compliance with any such policies. The FDA might also require clinical trials in support of any such applications, and Adamis would need to evaluate its alternatives in light of the costs required to conduct such trials, which could be substantial, compared to the economic benefit to Adamis from such products. In addition, independently of such actions, at anytime the FDA could also exercise its discretion to proceed against Adamis and require immediate withdrawal of the PFS Syringe product or other products from the market, or prohibit Adamis from marketing the PFS Syringe product or one or more of such other products without first conducting required trials and obtaining approvals, or to impose other penalties on Adamis. Some of Adamis Labs’ unapproved products include extended release formulations, which may subject Adamis to a higher risk of FDA enforcement action. Such actions could have a material adverse effect on Adamis’ business, financial condition and results of operations.
Adamis relies on third parties to conduct its clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, Adamis may be unable to obtain, or may experience delays in
36
obtaining, regulatory approval, or may not be successful in commercializing Adamis’ planned and future products.
Like many companies its size, Adamis does not have the ability to conduct preclinical or clinical studies for its product candidates without the assistance of third parties who conduct the studies on its behalf. These third parties are usually toxicology facilities and clinical research organizations, or CROs, that have significant resources and experience in the conduct of pre-clinical and clinical studies. The toxicology facilities conduct the pre-clinical safety studies as well as all associated tasks connected with these studies. The CROs typically perform patient recruitment, project management, data management, statistical analysis, and other reporting functions. Adamis intends to rely on third parties to conduct clinical trials of its product candidates and to use different toxicology facilities and CROs for its pre-clinical and clinical studies.
Adamis’ reliance on these third parties for development activities will reduce its control over these activities. If these third parties do not successfully carry out their contractual duties or obligations or meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to Adamis’ clinical protocols or for other reasons, Adamis’ clinical trials may be extended, delayed or terminated. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, Adamis may be required to replace them. Although Adamis believes there are a number of third-party contractors it could engage to continue these activities, replacing a third-party contractor may result in a delay of the affected trial. Accordingly, Adamis may not be able to obtain regulatory approval for or successfully commercialize its product candidates.
Delays in the commencement or completion of clinical testing of Adamis’ product candidates could result in increased costs to Adamis and delay its ability to generate significant revenues.
Delays in the commencement or completion of clinical testing could significantly impact Adamis’ product development costs. Adamis does not know whether current or planned clinical trials will begin on time or be completed on schedule, if at all. The commencement of clinical trials can be delayed for a variety of reasons, including delays in:
|
•
|
obtaining regulatory approval to commence a clinical trial;
|
|
|
•
|
reaching agreement on acceptable terms with prospective contract research organizations and clinical trial sites;
|
|
|
•
|
obtaining sufficient quantities of clinical trial materials for any or all product candidates;
|
|
|
•
|
obtaining institutional review board approval to conduct a clinical trial at a prospective site; and
|
|
|
•
|
recruiting participants for a clinical trial.
|
In addition, once a clinical trial has begun, it may be suspended or terminated by Adamis or the FDA or other regulatory authorities due to a number of factors, including:
|
•
|
failure to conduct the clinical trial in accordance with regulatory requirements;
|
|
|
•
|
inspection of the clinical trial operations or clinical trial site by the FDA or other regulatory authorities resulting in the imposition of a clinical hold;
|
|
|
•
|
failure to achieve certain efficacy and/or safety standards; or
|
|
|
•
|
lack of adequate funding to continue the clinical trial.
|
Clinical trials require sufficient participant enrollment, which is a function of many factors, including the size of the target population, the nature of the trial protocol, the proximity of participants to clinical trial sites, the availability of effective treatments for the relevant disease, the eligibility criteria for Adamis’ clinical trials and competing trials. Delays in enrollment can result in increased costs and longer development times. Adamis’ failure to enroll participants in its clinical trials could delay the completion of the clinical trials beyond current expectations. In addition, the FDA could require Adamis to conduct clinical trials with a larger number of participants than it may project for any of its product candidates. As a result of these factors, Adamis may not be able to enroll a sufficient number of participants in a timely or cost-effective manner.
37
Furthermore, enrolled participants may drop out of clinical trials, which could impair the validity or statistical significance of the clinical trials. A number of factors can influence the discontinuation rate, including, but not limited to: the inclusion of a placebo in a trial; possible lack of effect of the product candidate being tested at one or more of the dose levels being tested; adverse side effects experienced, whether or not related to the product candidate; and the availability of numerous alternative treatment options that may induce participants to discontinue from the trial.
Adamis, the FDA or other applicable regulatory authorities may suspend clinical trials of a product candidate at any time if Adamis or they believe the participants in such clinical trials, or in independent third-party clinical trials for drugs based on similar technologies, are being exposed to unacceptable health risks or for other reasons.
We are subject to certain legal proceedings that may adversely affect our results of operations, financial condition and liquidity.
Adamis and its current officers and directors have been named defendants in a lawsuit alleging, among other things, that Adamis made material misrepresentations in private placement memoranda used to offer Adamis’ common stock to the plaintiffs, and that Adamis’ officers and directors have breached their fiduciary duties in connection with certain transactions. In addition, a lawsuit has been filed against Adamis for declaratory relief seeking a declaration that certain patent licenses held by Adamis are invalid. Although we believe the lawsuits are without merit and that we have substantial defenses to these lawsuits, there can be no assurance as to the outcome of these matters, and a loss in any of these cases could adversely affect our results of operations, our financial condition and liquidity. In addition, a loss of the license for certain patents could lead to a significant loss of sales and could materially affect future results of operations.
Adamis is subject to the risk of clinical trial and product liability lawsuits.
The testing of human health care product candidates entails an inherent risk of allegations of clinical trial liability, while the marketing and sale of approved products entails an inherent risk of allegations of product liability. Adamis currently maintains liability insurance coverage of $5,000,000. However, as Adamis conducts additional clinical trials and introduces products into the United States market, the risk of adverse events increases and Adamis’ requirements for liability insurance coverage are likely to increase. Adamis is subject to the risk that substantial liability claims from the testing or marketing of pharmaceutical products could be asserted against it in the future. There can be no assurance that Adamis will be able to obtain or maintain insurance on acceptable terms, particularly in overseas locations, for clinical and commercial activities or that any insurance obtained will provide adequate protection against potential liabilities. Moreover, Adamis’ current and future coverages may not be adequate to protect Adamis from all of the liabilities that it may incur. If losses from liability claims exceed Adamis’ insurance coverage, Adamis may incur substantial liabilities that exceed its financial resources. In addition, a product or clinical trial liability action against Adamis would be expensive and time-consuming to defend, even if Adamis ultimately prevailed. If Adamis is required to pay a claim, Adamis may not have sufficient financial resources and its business and results of operations may be harmed.
Adamis does not have commercial-scale manufacturing capability, and it lacks commercial manufacturing experience. Adamis will likely rely on third parties to manufacture and supply its product candidates.
Adamis does not own or operate manufacturing facilities for clinical or commercial production of product candidates. Adamis does not have any experience in drug formulation or manufacturing, and it lacks the resources and the capability to manufacture any of its product candidates on a clinical or commercial scale. Accordingly, Adamis expects to depend on third-party contract manufacturers for the foreseeable future. Any performance failure on the part of Adamis’ contract manufacturers could delay clinical development, regulatory approval or commercialization of Adamis’ current or future product candidates, depriving Adamis of potential product revenue and resulting in additional losses.
The manufacture of pharmaceutical products requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of pharmaceutical products often encounter difficulties in production, particularly in scaling up initial production.
38
These problems include difficulties with production costs and yields, quality control (including stability of the product candidate and quality assurance testing), shortages of qualified personnel, as well as compliance with strictly enforced federal, state and foreign regulations. If Adamis’ third-party contract manufacturers were to encounter any of these difficulties or otherwise fail to comply with their obligations or under applicable regulations, Adamis’ ability to provide product candidates to patients in its clinical trials or commercially would be jeopardized. As an example, Adamis’ PFS Syringe product is currently manufactured by Catalent Pharma Solutions (an FDA licensed and approved cGMP facility) in Brussels, Belgium and, therefore, is subject to regulation by the Belgian Ministry of Health as well as the FDA. Any delay or interruption in the supply of product could delay the completion of Adamis’ commercial launch or clinical trials, increase the costs associated with maintaining Adamis’ commercial production or clinical trial programs and, depending upon the period of delay, require Adamis to commence new trials or qualify new manufacturers at significant additional expense, possibly causing commercial delays or termination of the trials.
Adamis’ products can only be manufactured in a facility that has undergone a satisfactory inspection by the FDA and other relevant regulatory authorities. For these reasons, Adamis may not be able to replace manufacturing capacity for its products quickly if it or its contract manufacturer(s) were unable to use manufacturing facilities as a result of a fire, natural disaster (including an earthquake), equipment failure, or other difficulty, or if such facilities were deemed not in compliance with the regulatory requirements and such non-compliance could not be rapidly rectified. An inability or reduced capacity to manufacture Adamis products would have a material adverse effect on Adamis’ business, financial condition, and results of operations.
If Adamis fails to obtain acceptable prices or appropriate reimbursement for its products, its ability to successfully commercialize its products will be impaired.
Government and insurance reimbursements for healthcare expenditures play an important role for all healthcare providers, including physicians and pharmaceutical companies such as Adamis that plan to offer various products in the United States and other countries in the future. Adamis’ ability to earn sufficient returns on its products and potential products will depend in part on the extent to which reimbursement for the costs of such products will be available from government health administration authorities, private health coverage insurers, managed care organizations, and other organizations. In the United States, Adamis’ ability to have its products eligible for Medicare, Medicaid or private insurance reimbursement will be an important factor in determining the ultimate success of its products. If, for any reason, Medicare, Medicaid or the insurance companies decline to provide reimbursement for Adamis’ products, its ability to commercialize its products would be adversely affected. There can be no assurance that Adamis’ potential drug products will be eligible for reimbursement.
There has been a trend toward declining government and private insurance expenditures for many healthcare items and this trend may accelerate with proposed healthcare reform legislation. Third-party payors are increasingly challenging the price of medical and pharmaceutical products.
If purchasers or users of Adamis’ products and related treatments are not able to obtain appropriate reimbursement for the cost of using such products, they may forego or reduce such use. Even if Adamis’ products are approved for reimbursement by Medicare, Medicaid and private insurers, of which there can be no assurance, the amount of reimbursement may be reduced at times or even eliminated. This would have a material adverse effect on Adamis’ business, financial condition and results of operations.
Significant uncertainty exists as to the reimbursement status of newly approved pharmaceutical products, and there can be no assurance that adequate third-party coverage will be available.
Legislative or regulatory reform of the healthcare system may affect Adamis’ ability to sell its products profitably.
In both the United States and certain foreign jurisdictions, there have been and are expected to be a number of legislative and regulatory changes to the healthcare system in ways that could impact Adamis’ ability to sell its products profitably, including the Affordable Care Act signed into law in the United States on March 22, 2010. In recent years, new legislation has been enacted in the United States at the federal and state levels that effects major changes in the healthcare system, either nationally or at the state level. These new laws include a prescription drug benefit plan for Medicare beneficiaries and certain changes in Medicare reimbursement. Given the recent enactment of these laws, it is still too early to determine their impact on the biotechnology and pharmaceutical industries and Adamis’ business. Further, the U.S. Congress is considering a significant healthcare overhaul proposal that may affect Adamis’ ability to market and sell products on favorable terms, which would affect Adamis’ results of
39
operations as well as its ability to raise capital, obtain additional collaborators or profitably market its products. Such proposals may reduce Adamis’ revenues, increase its expenses or limit the markets for its products. In particular, Adamis expects to experience pricing pressures in connection with the sale of its products due to the trend toward managed health care, the increasing influence of health maintenance organizations and additional legislative proposals.
Adamis has limited sales, marketing and distribution experience.
Adamis has limited experience in the sales, marketing, and distribution of pharmaceutical products. There can be no assurance that Adamis will be able to establish sales, marketing, and distribution capabilities or make arrangements with its current collaborators or others to perform such activities or that such efforts will be successful. If Adamis decides to market any of its new products directly, it must either acquire or internally develop a marketing and sales force with technical expertise and with supporting distribution capabilities. The acquisition or development of a sales, marketing and distribution infrastructure would require substantial resources, which may not be available to Adamis or, even if available, divert the attention of its management and key personnel, and have a negative impact on further product development efforts.
Adamis may seek to enter into arrangements to develop and commercialize its products. These collaborations, if secured, may not be successful.
Adamis has entered into arrangements with third parties regarding development and commercialization of some of its products and may in the future seek to enter into collaborative arrangements to develop and commercialize some of its potential products both in North America and international markets. There can be no assurance that Adamis will be able to negotiate collaborative arrangements on favorable terms or at all or that its current or future collaborative arrangements will be successful.
Adamis’ strategy for the future research, development, and commercialization of its products is expected to be based in part on entering into various arrangements with corporate collaborators, licensors, licensees, health care institutions and principal investigators and others, and its commercial success is dependent upon these outside parties performing their respective contractual obligations responsibly and with integrity. The amount and timing of resources such third parties will devote to these activities may not be within Adamis’ control. There can be no assurance that such parties will perform their obligations as expected. There can be no assurance that Adamis’ collaborators will devote adequate resources to its products.
If Adamis is not successful in acquiring or licensing additional product candidates on acceptable terms, if at all, Adamis’ business may be adversely affected.
As part of its strategy, Adamis may acquire or license additional product candidates that it believes have growth potential. There are no assurances that Adamis will be able to identify promising product candidates. Even if Adamis is successful in identifying promising product candidates, Adamis may not be able to reach an agreement for the acquisition or license of the product candidates with their owners on acceptable terms or at all.
Adamis may not be able to successfully identify any other commercial products or product candidates to in-license, acquire or internally develop. Moreover, negotiating and implementing an economically viable in-licensing arrangement or acquisition is a lengthy and complex process. Other companies, including those with substantially greater resources, may compete with Adamis for the in-licensing or acquisition of product candidates and approved products. Adamis may not be able to acquire or in-license the rights to additional product candidates and approved products on terms that it finds acceptable, or at all. If it is unable to in-license or acquire additional commercial products or product candidates, Adamis’ ability to grow its business or increase its profits could be severely limited.
If Adamis’ competitors develop and market products that are more effective than Adamis’ product candidates or obtain regulatory and marketing approval for similar products before Adamis does, Adamis’ commercial opportunity may be reduced or eliminated.
The development and commercialization of new pharmaceutical products that target certain cancers and viral conditions, and allergy and other respiratory conditions addressed by the current and future products of Adamis Labs, is competitive, and Adamis faces competition from numerous sources, including major biotechnology and pharmaceutical companies worldwide. Many of Adamis’ competitors have substantially greater financial and technical resources, and development, production and marketing capabilities than Adamis does. In addition, many of these companies have more experience than Adamis in pre-clinical testing, clinical trials and manufacturing of
40
compounds, as well as in obtaining FDA and foreign regulatory approvals. Adamis also competes with academic institutions, governmental agencies and private organizations that are conducting research in the same fields.
Competition among these entities to recruit and retain highly qualified scientific, technical and professional personnel and consultants is also intense. As a result, there is a risk that one of Adamis’ competitors will develop a more effective product for the same indications for which Adamis is developing a product or, alternatively, bring a similar product to market before Adamis can do so. Failure of Adamis to successfully compete will adversely impact the ability to raise additional capital and ultimately achieve profitable operations.
If Adamis suffers negative publicity concerning the safety of its products in development, its sales may be harmed and Adamis may be forced to withdraw such products.
If concerns should arise about the safety of any of Adamis’ products that are marketed, regardless of whether or not such concerns have a basis in generally accepted science or peer-reviewed scientific research, such concerns could adversely affect the market for these products. Similarly, negative publicity could result in an increased number of product liability claims, whether or not these claims are supported by applicable law.
Adamis’ failure to adequately protect or to enforce its intellectual property rights or secure rights to third party patents could materially harm its proprietary position in the marketplace or prevent the commercialization of its products.
Adamis’ success depends in part on its ability to obtain and maintain protection in the United States and other countries for the intellectual property covering or incorporated into its technologies and products. The patents and patent applications in Adamis’ existing patent portfolio are either owned by Adamis or licensed to Adamis. Adamis’ ability to protect its product candidates from unauthorized use or infringement by third parties depends substantially on its ability to obtain and maintain valid and enforceable patents. Due to evolving legal standards relating to the patentability, validity and enforceability of patents covering pharmaceutical inventions and the scope of claims made under these patents, Adamis’ ability to obtain and enforce patents is uncertain and involves complex legal and factual questions for which important legal principles are unresolved.
There is a substantial backlog of patent applications at the United States Patent and Trademark Office, or USPTO. Patents in the United States are issued to the party that is first to invent the claimed invention. There can be no assurance that any patent applications relating to Adamis’ products or methods will be issued as patents, or, if issued, that the patents will not be challenged, invalidated or circumvented or that the rights granted thereunder will provide a competitive advantage. Adamis may not be able to obtain patent rights on products, treatment methods or manufacturing processes that it may develop or to which Adamis may obtain license or other rights. Even if Adamis does obtain patents, rights under any issued patents may not provide it with sufficient protection for its product candidates or provide sufficient protection to afford Adamis a commercial advantage against its competitors or their competitive products or processes. It is possible that no patents will be issued from any pending or future patent applications owned by Adamis or licensed to Adamis. Others may challenge, seek to invalidate, infringe or circumvent any patents Adamis owns or licenses. Alternatively, Adamis may in the future be required to initiate litigation against third parties to enforce its intellectual property rights. The defense and prosecution of patent and intellectual property claims are both costly and time consuming, even if the outcome is favorable to Adamis. Any adverse outcome could subject Adamis to significant liabilities, require Adamis to license disputed rights from others, or require Adamis to cease selling its future products.
In addition, many other organizations are engaged in research and product development efforts that may overlap with Adamis’ products. For example, Adamis’ PFS Syringe product competes against other self-administered epinephrine products, including EpiPen, EpiPen Jr. and Twinject; Adamis Labs’ line of allergy and respiratory products compete with numerous prescription and non-prescription over-the-counter products targeting similar conditions; and with regard to the Savvy product candidate, Ortho Pharmaceuticals and many other companies offer contraceptive vaginal gel products. Such organizations may currently have, or may obtain in the future, legally blocking proprietary rights, including patent rights, in one or more products or methods under development or consideration by Adamis. These rights may prevent Adamis from commercializing technology, or may require Adamis to obtain a license from the organizations to use the technology. Adamis may not be able to obtain any such licenses that may be required on reasonable financial terms, if at all, and cannot be sure that the patents underlying any such licenses will be valid or enforceable. As with other companies in the pharmaceutical industry, Adamis is subject to the risk that persons located in other countries will engage in development, marketing
41
or sales activities of products that would infringe Adamis’ patent rights if such activities were conducted in the United States.
Adamis’ patents also may not afford protection against competitors with similar technology. Adamis may not have identified all patents, published applications or published literature that affect its business either by blocking Adamis’ ability to commercialize its product candidates, by preventing the patentability of its products or by covering the same or similar technologies that may affect Adamis’ ability to market or license its product candidates. For example, patent applications filed with the USPTO are maintained in confidence for up to 18 months after their filing. In some cases, however, patent applications filed with the USPTO remain confidential for the entire time before issuance as a U.S. patent. Patent applications filed in countries outside the United States are not typically published until at least 18 months from their first filing date. Similarly, publication of discoveries in the scientific or patent literature often lags behind actual discoveries. Therefore, Adamis or its licensors might not have been the first to invent, or the first to file, patent applications on Adamis’ product candidates or for their use. The laws of some foreign jurisdictions do not protect intellectual property rights to the same extent as in the United States, and many companies have encountered significant difficulties in protecting and defending these rights in foreign jurisdictions. If Adamis encounters such difficulties or is otherwise precluded from effectively protecting its intellectual property rights in either the United States or foreign jurisdictions, Adamis’ business prospects could be substantially harmed.
If adequate funding is not obtained, the board of directors of Adamis will be required to explore alternatives for Adamis’ business and assets. These alternatives might include seeking the dissolution and liquidation of Adamis, seeking to merge or combine with another company, selling or licensing some of Adamis’ intellectual property, or initiating bankruptcy proceedings. There can be no assurance that any third party will be interested in merging with Adamis or would agree to a price and other terms that Adamis would deem adequate. If Adamis filed for bankruptcy, it would most likely not be able to raise any type of funding from any source. In that event, the creditors of Adamis would have first claim on the value of the assets of Adamis which, other than remaining cash, would most likely be liquidated in a bankruptcy sale. Adamis can give no assurance as to the magnitude of the net proceeds of such sale and whether such proceeds would be sufficient to satisfy Adamis’ obligations to its creditors, let alone to permit any distribution to its equity holders.
Adamis’ management will be required to devote substantial time to comply with public company regulations.
The Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, as well as rules subsequently implemented by the SEC, impose various requirements on public companies, including with respect to corporate governance practices. Adamis’ management and other personnel will need to devote a substantial amount of time to these requirements.
In addition, the Sarbanes-Oxley Act requires, among other things, that Adamis maintain effective internal controls for financial reporting and disclosure controls and procedures. In particular, Adamis must perform system and process evaluation and testing of its internal controls over financial reporting to allow management to report on the effectiveness of its internal controls over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act. Adamis’ compliance with Section 404 will require that it incur substantial accounting and related expense and expend significant management efforts. Adamis may need to hire additional accounting and financial staff to satisfy the ongoing requirements of Section 404. Moreover, if Adamis is not able to comply with the requirements of Section 404, or if Adamis or its independent registered public accounting firm identifies deficiencies in its internal controls over financial reporting that are deemed to be material weaknesses, the market price of Adamis’ stock could decline and Adamis could be subject to sanctions or investigations by the SEC or other regulatory authorities.
Adamis has incurred losses since its inception and anticipates that Adamis will continue to incur losses. Adamis may never achieve or sustain profitability.
Adamis incurred net losses of approximately $18,740,000 since inception and net losses of approximately $6,160,000 for its fiscal year ended March 31, 2010.These losses may increase as Adamis continues its research and development activities, seeks regulatory approvals for its product candidates and commercializes any approved products. These losses may cause, among other things, Adamis’ stockholders’ equity and working capital to decrease. The future earnings and cash flow from operations of Adamis’ business are dependent, in part, on its ability to further develop its products and on revenues and profitability from sales of products and product candidates of its Adamis Labs operations.
42
There can be no assurance that Adamis will grow and be profitable. There can be no assurance that Adamis will be able to generate sufficient product revenue to become profitable at all or on a sustained basis. Adamis expects to have quarter-to-quarter fluctuations in revenues and expenses, some of which could be significant, due to expanded manufacturing, marketing, research, development, and clinical trial activities. If Adamis’ product candidates fail in clinical trials or do not gain regulatory approval, or if Adamis’ products do not achieve market acceptance, Adamis may never become profitable. Adamis will need to increase product marketing and brand awareness and conduct significant research, development, testing and regulatory compliance activities that, together with projected general and administrative expenses, are expected to result in substantial operating losses for the foreseeable future. Even if Adamis does achieve profitability, it may not be able to sustain or increase profitability on a quarterly or annual basis.
Adamis has received a “going concern” opinion from its independent registered public accounting firm, which may negatively impact Adamis’ business. Adamis’ audit opinions from its independent registered public accounting firm regarding the consolidated financial statements for the years ended March 31, 2010 and 2009 include an explanatory paragraph indicating that Adamis has incurred recurring losses from operations and has limited working capital to pursue its business alternatives, and that these factors raise substantial doubt about its ability to continue as a going concern. Without additional funds from debt or equity financing, sales of assets, intellectual property or technologies, or from a business combination or a similar transaction, Adamis will exhaust its resources and will be unable to continue operations. These factors raise substantial doubt about Adamis’ ability to continue as a going concern.
Adamis may be required to suspend, repeat or terminate its clinical trials if the trials are not well designed, do not meet regulatory requirements or the results are negative or inconclusive, which may result in significant negative repercussions on Adamis’ business and financial condition.
Before regulatory approval for any potential product can be obtained, Adamis must undertake extensive clinical testing on humans to demonstrate the tolerability and efficacy of the product, both on its own terms, and as compared to the other principal drugs on the market that have the same therapeutic indication. Adamis cannot assure you that it will obtain authorization to permit product candidates that are already in the preclinical development phase to enter the human clinical testing phase. In addition, Adamis cannot assure you that any authorized preclinical or clinical testing will be completed successfully within any specified time period by Adamis, or without significant additional resources or expertise to those originally expected to be necessary. Adamis cannot assure you that such testing will show potential products to be safe and efficacious or that any such product will be approved for a specific indication. Further, the results from preclinical studies and early clinical trials may not be indicative of the results that will be obtained in later-stage clinical trials. In addition, Adamis or regulatory authorities may suspend clinical trials at any time on the basis that the participants are being exposed to unacceptable health risks.
Completion of clinical tests depends on, among other things, the number of patients available for testing, which is a function of many factors, including the number of patients with the relevant conditions, the nature of the clinical testing, the proximity of patients to clinical testing centers, the eligibility criteria for tests as well as competition with other clinical testing programs involving the same patient profile but different treatments. Adamis will rely on third parties, such as contract research organizations and/or co-operative groups, to assist it in overseeing and monitoring clinical trials as well as to process the clinical results and manage test requests, which may result in delays or failure to complete trials, if the third parties fail to perform or to meet the applicable standards. A failure by Adamis or such third parties to keep to the terms of a product program development for any particular product candidate or to complete the clinical trials for a product candidate in the envisaged time frame could have significant negative repercussions on Adamis’ business and financial condition.
Even if Adamis receives regulatory approval to market its product candidates, such products may not gain the market acceptance among physicians, patients, healthcare payors and the medical community.
Any products that Adamis may develop may not gain market acceptance among physicians, patients, healthcare payors and the medical community even if they ultimately receive regulatory approval. If these products do not achieve an adequate level of acceptance, Adamis, or future collaborators, may not be able to generate material product revenues and Adamis may not become profitable. The degree of market acceptance of any of Adamis’ product candidates, if approved for commercial sale, will depend on a number of factors, including:
|
•
|
demonstration of efficacy and safety in clinical trials;
|
43
|
•
|
the prevalence and severity of any unexpected side effects;
|
|
|
•
|
the introduction and availability of generic substitutes for any of Adamis’ products, potentially at lower prices (which, in turn, will depend on the strength of Adamis’ intellectual property protection for such products);
|
|
|
•
|
potential or perceived advantages over alternative treatments;
|
|
|
•
|
the timing of market entry relative to competitive treatments;
|
|
|
•
|
the ability to offer Adamis’ product candidates for sale at competitive prices;
|
|
|
•
|
relative convenience and ease of administration;
|
|
|
•
|
the strength of marketing and distribution support;
|
|
|
•
|
sufficient third party coverage or reimbursement; and
|
|
|
•
|
the product labeling or product insert (including any warnings) required by the FDA or regulatory authorities in other countries.
|
Adamis may not complete its clinical trials in the time expected, which could delay or prevent the commercialization of its products, which may adversely affect Adamis’ future revenues and financial condition.
Although for planning purposes Adamis will forecast the commencement and completion of clinical trials, the actual timing of these events can vary dramatically due to factors such as delays, scheduling conflicts with participating clinicians and clinical institutions and the rate of patient enrollment. Clinical trials involving Adamis’ product candidates may not commence or be completed as forecast. In certain circumstances, Adamis will rely on academic institutions or clinical research organizations to conduct, supervise or monitor some or all aspects of clinical trials involving Adamis’ products. Adamis will have less control over the timing and other aspects of these clinical trials than if it conducted them entirely on its own. These trials may not commence or be completed as Adamis expects and may not be conducted successfully. Failure to commence or complete, or delays in, any of Adamis’ planned clinical trials could delay or prevent the commercialization of Adamis’ products and harm its business and may adversely affect Adamis’ future revenues and financial condition.
If Adamis fails to keep pace with rapid technological change in the biotechnology and pharmaceutical industries, its products could become obsolete, which may adversely affect Adamis’ future revenues and financial condition.
Biotechnology and related pharmaceutical technology have undergone and are subject to rapid and significant change. Adamis expects that the technologies associated with biotechnology research and development will continue to develop rapidly. Adamis’ future will depend in large part on its ability to maintain a competitive position with respect to these technologies. Any compounds, products or processes that Adamis develops may become obsolete before Adamis recovers any expenses incurred in connection with developing these products, which may adversely affect Adamis’ future revenues and financial condition.
If Adamis is unable to retain its management, research, development, and clinical teams and scientific advisors or to attract additional qualified personnel, Adamis’ product operations and development efforts may be seriously jeopardized.
Adamis’ success will be dependent upon the efforts of a small management team and staff, including Dennis J. Carlo, Ph.D. The employment of Dr. Carlo may be terminated at any time by either Adamis or Dr. Carlo. Adamis currently does not, and Adamis will not, have key man life insurance policies covering any of its executive officers or key employees. If key individuals leave Adamis, Adamis could be adversely affected if suitable replacement personnel are not quickly recruited. There is competition for qualified personnel in all functional areas, which makes it difficult to attract and retain the qualified personnel necessary for the operation of Adamis’ business.
The loss of the services of any principal member of Adamis’ management and research, development and clinical teams could significantly delay or prevent the achievement of Adamis’ scientific and business objectives. Competition among biotechnology and pharmaceutical companies for qualified employees is intense, and the ability to retain and attract qualified individuals is critical to Adamis’ success. Adamis may be unable to attract and retain key personnel on acceptable terms, if at all.
44
Adamis has relationships with consultants and scientific advisors who will continue to assist Adamis in formulating and executing its research, development, regulatory and clinical strategies. These consultants and scientific advisors are not Adamis employees and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to Adamis. Adamis will have only limited control over the activities of these consultants and scientific advisors and can generally expect these individuals to devote only limited time to Adamis’ activities. Adamis also relies on these consultants to evaluate potential compounds and products, which may be important in developing a long-term product pipeline for Adamis. Consultants also assist Adamis in preparing and submitting regulatory filings. Adamis’ scientific advisors provide scientific and technical guidance on the company’s drug discovery and development. Failure of any of these persons to devote sufficient time and resources to Adamis’ programs could harm its business. In addition, these advisors may have arrangements with other companies to assist those companies in developing technologies that may compete with Adamis’ products.
Risks Related to Our Common Stock
Provisions of our charter documents could discourage an acquisition of our company that would benefit its stockholders and may have the effect of entrenching, and making it difficult to remove, management.
Provisions of Adamis’ certificate of incorporation and bylaws may make it more difficult for a third party to acquire control of Adamis, even if a change of control would benefit Adamis’ stockholders. In particular, shares of Adamis preferred stock may be issued in the future without further stockholder approval, and upon such terms and conditions, and have such rights, privileges and preferences, as Adamis’ board of directors may determine, including, for example, rights to convert into Adamis common stock. The rights of the holders of Adamis common stock will be subject to, and may be adversely affected by, the rights of the holders of any of Adamis’ preferred stock that may be issued in the future. The issuance of Adamis preferred stock, while providing desirable flexibility in connection with possible acquisitions and other corporate purposes, could have the effect of making it more difficult for a third party to acquire control of Adamis. This could limit the price that certain investors might be willing to pay in the future for shares of Adamis common stock and discourage those investors from acquiring a majority of Adamis common stock. Further, the existence of these corporate governance provisions could have the effect of entrenching management and making it more difficult to change Adamis’ management.
We may be required to pay liquidated damages to certain investors if we do not maintain an effective registration statement relating to common stock issuable upon conversion of our Senior Notes.
We have several senior secured notes outstanding, referred to as the Senior Notes, to a small number of institutional investors in the principal amount of $1,500,000 which become due and payable by Adamis on October 11, 2010. The definitive agreements relating to the Senior Notes require, among other things, that Adamis maintain an effective registration statement for common stock issuable upon conversion of the Senior Notes if they are not freely tradable under Rule 144 on or after six months after the date the Senior Notes were issued. If such shares are not freely tradable, then failure to maintain an effective registration statement relating to the common stock issuable upon conversion of the Senior Notes may result in liquidated damages payable to the Senior Note holders.
We have issued and outstanding Senior Notes with rights and preferences superior to those of our common stock.
The issued and outstanding shares of the Senior Notes grants the holders of such Senior Notes anti-dilution and liquidations rights that are superior to those held by the holders of our common stock. Should Adamis issue additional common stock or other securities for a price below $0.20 per share, the conversion price of the Senior Notes shall be lowered to the price at which such securities are issued, which will have the effect of immediately diluting the holders of our common stock.
Adamis’ common stock price is expected to be volatile.
The market price of Adamis’ common stock could be subject to significant fluctuations following this offering. Market prices for securities of early-stage pharmaceutical, biotechnology and other life sciences companies have historically been particularly volatile. Some of the factors that may cause the market price of Adamis’ common stock to fluctuate include:
|
•
|
the results of Adamis’ current and any future clinical trials of its product candidates;
|
|
|
•
|
the timing and results of ongoing preclinical studies and planned clinical trials of Adamis’ preclinical product candidates;
|
45
|
•
|
the entry into, or termination of, key agreements, including, among others, key collaboration and license agreements;
|
|
|
•
|
the results and timing of regulatory reviews relating to the approval of Adamis’ product candidates;
|
|
|
•
|
the initiation of, material developments in, or conclusion of, litigation to enforce or defend any of Adamis’ intellectual property rights;
|
|
|
•
|
failure of any of Adamis’ product candidates, if approved, to achieve commercial success;
|
|
|
•
|
general and industry-specific economic conditions that may affect Adamis’ research and development expenditures;
|
|
|
•
|
the results of clinical trials conducted by others on drugs that would compete with Adamis’ product candidates;
|
|
|
•
|
issues in manufacturing Adamis’ product candidates or any approved products;
|
|
|
•
|
the loss of key employees;
|
|
|
•
|
the introduction of technological innovations or new commercial products by competitors of Adamis;
|
|
|
•
|
changes in estimates or recommendations by securities analysts, if any, who cover Adamis’ common stock;
|
|
|
•
|
future sales of Adamis’ common stock;
|
|
|
•
|
period-to-period fluctuations in Adamis’ financial results;
|
|
|
•
|
publicity or announcements regarding regulatory developments relating to Adamis’ products;
|
|
|
•
|
clinical trial results, particularly the outcome of more advanced studies, or negative responses from both domestic and foreign regulatory authorities with regard to the approvability of Adamis’ products;
|
|
|
•
|
period-to-period fluctuations in Adamis’ financial results, including Adamis’ cash and cash equivalents balance, operating expenses, cash burn rate or revenue levels;
|
|
|
•
|
common stock sales in the public market by one or more of Adamis’ larger stockholders, officers or directors;
|
|
|
•
|
Adamis’ filing for protection under federal bankruptcy laws;
|
|
|
•
|
a negative outcome in any litigation or potential legal proceedings; or
|
|
|
•
|
other potentially negative financial announcements including: a review of any of Adamis’ filings by the SEC, changes in accounting treatment or restatement of previously reported financial results or delays in Adamis’ filings with the SEC.
|
The stock markets in general have experienced substantial volatility that has often been unrelated to the operating performance of individual companies. These broad market fluctuations may also adversely affect the trading price of Adamis’ common stock.
In the past, following periods of volatility in the market price of a company’s securities, stockholders have often instituted class action securities litigation against those companies. Such litigation, if instituted, could result in substantial costs and diversion of management attention and resources, which could significantly harm Adamis’ profitability and reputation.
Adamis’ common stock is expected to be traded on the OTC Bulletin Board and be subject to additional trading restrictions as a “penny stock,” which could adversely affect the liquidity and price of such stock.
Adamis’ common stock trades on the OTC Bulletin Board, or OTCBB. The OTCBB and Pink Sheets are viewed by most investors as a less desirable, and less liquid, marketplace. As a result, an investor may find it more difficult to purchase, dispose of or obtain accurate quotations as to the value of our common stock.
46
Because Adamis’ common stock is not listed on any national securities exchange, such shares will also be subject to the regulations regarding trading in “penny stocks,” which are those securities trading for less than $5.00 per share. The following is a list of the general restrictions on the sale of penny stocks:
|
•
|
Before the sale of penny stock by a broker-dealer to a new purchaser, the broker-dealer must determine whether the purchaser is suitable to invest in penny stocks. To make that determination, a broker-dealer must obtain, from a prospective investor, information regarding the purchaser’s financial condition and investment experience and objectives. Subsequently, the broker-dealer must deliver to the purchaser a written statement setting forth the basis of the suitability finding and obtain the purchaser’s signature on such statement.
|
|
|
•
|
A broker-dealer must obtain from the purchaser an agreement to purchase the securities. This agreement must be obtained for every purchase until the purchaser becomes an “established customer.” A broker-dealer may not effect a purchase of a penny stock less than two business days after a broker-dealer sends such agreement to the purchaser.
|
|
|
•
|
The Securities Exchange Act of 1934, or the Exchange Act, requires that before effecting any transaction in any penny stock, a broker-dealer must provide the purchaser with a “risk disclosure document” that contains, among other things, a description of the penny stock market and how it functions and the risks associated with such investment. These disclosure rules are applicable to both purchases and sales by investors.
|
|
|
•
|
A dealer that sells penny stock must send to the purchaser, within ten days after the end of each calendar month, a written account statement including prescribed information relating to the security.
|
These requirements can severely limit the liquidity of securities in the secondary market because few brokers or dealers are likely to be willing to undertake these compliance activities. As a result of Adamis’ common stock not being listed on a national securities exchange and the rules and restrictions regarding penny stock transactions, an investor’s ability to sell to a third party and Adamis’ ability to raise additional capital may be limited. Adamis makes no guarantee that its market-makers will continue to make a market in its common stock, or that any market for its common stock will continue.
Adamis’ principal stockholders have significant influence over Adamis, and your interests as a stockholder may conflict with the interests of those persons.
Based on the number of outstanding shares of Adamis common stock held by Adamis stockholders as of March 31, 2010, Adamis’ ten largest stockholders beneficially own approximately 49.6% of the outstanding Adamis common stock. As a result, those stockholders will be able to exert a significant degree of influence or actual control over Adamis’ management and affairs after the merger and over matters requiring stockholder approval, including the election of directors, any merger, consolidation or sale of all or substantially all of Adamis’ assets, and any other significant corporate transaction. The interests of these persons may not always coincide with the interests of Adamis or its other stockholders. For example, such persons could delay or prevent a change of control of Adamis even if such a change of control would benefit Adamis’ other stockholders. The significant concentration of stock ownership may adversely affect the trading price of Adamis’ common stock due to investors’ perception that conflicts of interest may exist or arise.
ITEM 1B: UNRESOLVED STAFF COMMENTS
None.
Adamis currently leases approximately 1,800 square feet of office/warehouse space in Coconut Creek, Florida, relating to its Adamis Labs operations. The Company is currently evaluating its space requirements and expects to either extend its current leases or move into new facilities, assuming the company secures adequate funding, that will better accommodate its needs.
47
In addition to the matters described below, we may become involved in or subject to, routine litigation, claims, disputes, proceedings and investigations in the ordinary course of business, which in our opinion will not have a material adverse effect on our financial condition, cash flows or results of operations.
Cosmo Bioscience, Inc. et. al. v. Adamis Pharmaceuticals Corp. and Maurizio Zanetti
Cosmo Bioscience, Inc. et. al. v. Adamis Pharmaceuticals Corp. and Maurizio Zanetti, Case No. 37-2010-00088584, was filed in San Diego Superior Court in May 2010. Plaintiffs are affiliated Cosmo Bioscience entities who claim to have sublicensed certain patented technology from Eurogen BV, an entity wholly owned and controlled by Maurizio Zanetti. Plaintiffs claim that Zanetti wrongfully terminated their license, and further that Zanetti improperly licensed the same technology to Adamis in violation of plaintiffs’ exclusive license agreement. Plaintiffs assert a single claim for declaratory relief seeking a declaration that the Cosmo sublicense is in full force and effect, and that the Adamis license is invalid. In a previous effort to assert claims with respect to the technology, one of the principals of Cosmo had previously claimed to be a co-inventor of the patents involved in the lawsuit – a claim which was rejected by a U.S. federal district court.
Adamis believes that the plaintiffs’ complaint is without merit and intends to vigorously assert and defend its rights in the technology that are the subject of the lawsuit. Zanetti intends to file a motion to compel arbitration and Adamis intends to file a demurrer and a motion to stay pending resolution of the arbitration. In addition, Adamis, through its counsel, has notified the Cosmo entities that it has reason to believe that Cosmo is engaging in activities that violate or interfere with Adamis’ rights to the technologies licensed to Adamis, and that any use of the technologies by Cosmo may be an unlawful infringement on the patents exclusively licensed to Adamis.
Curtis Leahy, et. al. v. Dennis J. Carlo, et al.
In May 2010, Curtis Leahy, et. al. v. Dennis J. Carlo, et al., Case Number 37-2010-00092524-CU-FR-CTL, was filed in San Diego Superior Court, and plaintiffs subsequently filed an amended complaint on June 18, 2010. The plaintiffs — Antaeus Capital Partners, Curtis Leahy, and David Amron – are Adamis shareholders. The defendants named in the Complaint are the Company, Dennis Carlo, David Marguglio, Robert Hopkins, and Richard Aloi, who are officers and/or directors of the company. Plaintiffs allege that defendants misrepresented and omitted material information in private placement memoranda distributed by Adamis in 2006 and 2008 regarding, among other things, Adamis’ license rights with respect to certain patented anti-viral technology; this claim appears to be based in part on the allegations of the Cosmo plaintiffs in the Cosmo lawsuit described above. Based on these purported misrepresentations and omissions, plaintiffs assert claims for violations of Sections 25401, 25501 and 25504 of the California Corporations Code, and claims for common law fraud and negligent misrepresentation on behalf of a putative class of shareholders who purchased stock pursuant to either or both of the Company’s 2006 and 2008 Private Placement Memoranda. Plaintiffs seek damages amounting to the difference between the purchase price of their stock and the current share price, or the price at which they previously sold their stock.
Plaintiffs also allege that defendants breached their fiduciary duties as directors and officers of Adamis with respect to certain corporate transactions, including the HVG transaction in 2007, the Cellegy merger in 2008, and the Gemini and G-Max financing transactions in 2010. Plaintiffs allege that these transactions were not in the best interest of the Company and did not achieve their stated objectives. Plaintiffs further allege that the director defendants collected excessive compensation in fiscal years 2008 and 2009, and assert that the Company should have exercised its right to repurchase certain shares issued to defendants and other senior managers pursuant to the Stock Repurchase Agreements in 2008 rather than amend those agreements to extend the dates for meeting the applicable performance criteria. Based on these allegations, plaintiffs assert claims for breach of fiduciary duty, unjust enrichment and constructive trust, declaratory relief, and injunctive relief.
Adamis believes that the complaint is without merit, and Adamis intends to vigorously defend the claim and may assert any available counterclaims. Adamis intends to file a demurrer and motion to strike relating to the complaint by the end of July 2010.
The litigation described in this section could divert management time and attention from the Company, could involve significant amounts of legal fees and other fees and expenses. An adverse outcome in any such litigation could have a material adverse effect on Adamis.
48
PART II
ITEM 5: MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS, AND ISSUER PURCHASES OF EQUITY SECURITIES
Adamis’ common stock has traded on the OTC Bulletin Board, or OTCBB, under the trading symbol ADMP.OB. The following table sets forth the range of high and low sales prices for the common stock as reported on the OTCBB for the periods indicated below. The quotations below reflect inter-dealer prices, without retail mark-up, markdown or commission, and may not represent actual transactions.
|
High
|
Low
|
|||||||
|
Fiscal 2009
|
||||||||
|
First Quarter (April 2008 - June 2008)
|
$ | 0.10 | $ | 0.04 | ||||
|
Second Quarter (July 2008 - September 2008)
|
$ | 0.09 | $ | 0.04 | ||||
|
Third Quarter (October 2008 - December 2008)
|
$ | 0.06 | $ | 0.02 | ||||
|
Fourth Quarter (January 2009 - March 2009)
|
$ | 0.07 | $ | 0.02 | ||||
| Fiscal 2010 | ||||||||
|
First Quarter (April 2009 - June 2009)
|
$ | 1.15 | $ | 0.04 | ||||
|
Second Quarter (July 2009 - September 2009)
|
$ | 0.40 | $ | 0.15 | ||||
|
Third Quarter (October 2009 - December 2009)
|
$ | 0.32 | $ | 0.19 | ||||
|
Fourth Quarter (January 2010 - March 2010)
|
$ | 0.56 | $ | 0.18 | ||||
As of July 9, 2010, there were approximately 129 holders of record common stock, excluding beneficial holders of stock held in street name.
Dividend Policy
We have have never declared or paid any cash dividends on our common stock, and we do not intend to do so in the foreseeable future. Accordingly, the stockholders of the Company will not receive a return on their investment unless the value of the Company’s shares increases, which may or may not occur. Any future determination to pay cash dividends will be at the discretion of the Company’s board of directors and will depend upon its financial condition, operating results, capital requirements, any applicable contractual restrictions and such other factors as the Company’s board of directors deems relevant.
Equity Compensation Plan Information
The following table sets forth, as of July 12, 2010, information with respect to our equity compensation plans, including our 1995 Equity Incentive Plan, the 1995 Directors’ Stock Option Plan, the 2005 Equity Incentive Plan and the 2009 Equity Incentive Plan, and with respect to certain other options and warrants.
49
|
Number of securities
to be issued upon exercise
of outstanding options,
warrants and rights
(a)
|
Weighted average exercise
price of outstanding options,
warrants and rights
(b)
|
Number of securities
remaining available for
future issuance under
equity compensation
plans (excluding securities
reflected in column (a))
(c)
|
||||||||||
|
Equity compensation plans approved by security holders
|
463,008 | (1) | $ | 11.21 | 8,998,234 | |||||||
|
Equity compensation plans not approved by security holders
|
431 | (2) | 2.90 | — | ||||||||
| — | ||||||||||||
|
Total
|
463,439 | $ | 11.22 | 8,998,234 | ||||||||
|
(1)
|
Represents shares subject to outstanding options with exercise prices ranging from $173.92 to $1.99 per share and expire between the years 2010 and 2015.
|
|
|
(2)
|
Represents shares subject to outstanding options and are fully vested with an exercise price $2.90 expire in 2010.
|
Recent Sales of Unregistered Securities
In May 2010, the Company issued 250,000 shares of common stock to a consultant for public relations efforts with relevant investors and public capital markets. The issuance was exempt from registration pursuant to Section 4(2) of the Securities Act promulgated thereunder, as transactions not involving a public offering.
ITEM 7: MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis of financial condition and results of operations should be read together with the consolidated financial statements and accompanying notes of the Company appearing elsewhere in this Report. This discussion of Adamis' financial condition and results of operations contains certain statements that are not strictly historical and are “forward-looking” statements within the meaning of the Private Securities Litigation Reform Act of 1995 and involve a high degree of risk and uncertainty. Actual results may differ materially from those projected in the forward-looking statements due to other risks and uncertainties that exist in Adamis' operations, development efforts and business environment, including those set forth in this Item 7, and in the sections entitled “1A. Risk Factors” and “1. Business” in this Report and uncertainties described elsewhere in this Report. All forward-looking statements included in this Report are based on information available to the Company as of the date hereof, and except as may be required under the Securities Exchange Act of 1934 and the rules and regulations promulgated thereunder, the Company assumes no obligation to update any such forward-looking statement.
General
Adamis Pharmaceuticals Corporation (“Adamis”) was founded in June 2006, as a Delaware corporation. Adamis has three wholly-owned subsidiaries: Cellegy Holdings, Inc., Adamis Corporation; and Biosyn, Inc. Adamis Corporation has two wholly-owned subsidiaries: Adamis Viral Therapies, Inc. (biotechnology), or Adamis Viral; and Adamis Laboratories, Inc. (specialty pharmaceuticals), or Adamis Labs.
Adamis Labs is a specialty pharmaceutical company. Adamis Labs currently has a line of prescription products that it markets for a variety of allergy, respiratory disease and pediatric conditions. Adamis acquired these products in April 2007 by acquiring all of the outstanding shares of Healthcare Ventures Group, a private company that had previously acquired the products and related intellectual property, assets and personnel from another corporation in February 2007, and subsequently renaming Healthcare Ventures Group as Adamis Labs. Adamis’ PFS Syringe product, a pre-filled epinephrine syringe product for use in the emergency treatment of extreme acute allergic reactions, or anaphylactic shock, was launched in July 2009; however, commercial launch has been materially slowed by insufficient funding. An additional product candidate in its specialty pharmaceutical drug product pipeline is a generic inhaled nasal steroid for the treatment of seasonal and perennial allergic rhinitis. Adamis’ goal is to commence commercial sales of the nasal steroid product in the third quarter of 2012, assuming adequate funding and no unexpected delays.
50
Adamis estimates that approximately $4 million or more must be invested from the date of this Annual Report on Form 10-K in the Adamis Labs operations to support development and commercial introduction of the aerosolized nasal steroid product candidate. The capital that is expected to be provided from expected sales of these products may be important to help fund expansion of those businesses and the research and development of the anti-cancer small molecule therapeutic drugs as well as the therapeutic vaccine technology. Currently, neither manufacturing nor clinical trials have begun for that product candidate. Adamis estimates that approximately a total of $6-$9 million is required to support the development and commercial introduction of the inhaled nasal steroid product candidate and its two other respiratory products, although there are no assurances that funds for such an investment will be available. Factors that could affect the actual launch date for the nasal steroid product candidate include the outcome of discussions with the FDA concerning the number and kind of clinical trials that the FDA will require before the FDA will consider regulatory approval of the product, any unexpected difficulties in licensing or sublicensing intellectual property rights for other components of the product such as the inhaler, any unexpected difficulties in the ability of our suppliers to timely supply quantities for commercial launch of the product, any unexpected delays or difficulties in assembling and deploying an adequate sales force to market the product, and adequate funding to support sales and marketing efforts. Significant delays in obtaining funding to support the development and introduction of the steroid product could reduce revenues and income to Adamis, require additional funding from other sources, and potentially have an adverse effect on the ability to fund Adamis’ research and development efforts for tumor indications, as well as vaccine product candidates by Adamis Viral.
Adamis recently acquired and entered into agreements to acquire exclusive license agreements covering three small molecule anti-inflammatory compounds, named CPC-100, CPC-200 and CPC-300, that Adamis believes are promising drug candidates for the potential treatment of human prostate cancer (PCa). The intellectual property covered by the agreements was licensed from the Wisconsin Alumni Research Foundation, or WARF. The company has acquired the license agreement relating to CPC-300 and will, upon completion of an equity offering of at least $2 million, acquire the agreements relating to CPC-100 and CPC-200. In 2006 and 2007, CPC-100 and CPC-200, respectively, received the National Cancer Institute's multi-year, multi-million dollar RAPID (Rapid Access to Preventative Intervention Development) Award. Each year, this award is given by the NCI Division of Cancer Prevention, under the RAPID Program, to promising new preventative/therapeutic anti-cancer drugs. Adamis’ objective is to file an Investigational New Drug application, or IND, with the U.S. Food and Drug Administration, or FDA, by the end of calendar year 2010, assuming adequate funding and no unexpected delays and subsequently to commence a Phase 1/2a prostate cancer clinical study relating to the CPC-100 product candidate, although there are no assurances that this will be the case.
To date, Adamis’ development efforts have been focused on development of its drug and vaccine technologies, and Adamis formed Adamis Viral to focus on developing patented preventative and therapeutic vaccines for a variety of viral diseases such as chronic hepatitis, human papillomavirus infection, influenza, as well as possibly prostate cancer. The first target indication for this technology has yet to be determined, but will be based on market, technology, and patent position considerations. However, Adamis currently intends to focus initially on development of one or more of the recently in-licensed small molecule prostate cancer product candidates, and as a result the timing of initiation of trials relating to vaccines is subject to uncertainty and the availability of sufficient funding, and there are no assurances concerning whether such a product will be developed or launched. Future potential disease targets might include therapeutic vaccines for Hepatitis B and C, Human Papillomavirus and possibly prostate cancer. As the prostate cancer and other product candidates are at an early stage of development, Adamis cannot estimate with any precision the amount that will be required to support these product developments, clinical trials and commercial introduction of the candidate products, although the amounts are most likely to be larger than those required to support the nasal steroid product candidate. Factors that could affect the costs of developing such product candidates include, but are not limited to, those described above for the steroid product candidate. Adamis’ lead small molecule prostate cancer pharmaceutical drug and vaccine product candidates are in the late-preclinical stage and have not generated any revenues, other than grants, to date. From June 6, 2006 (date of inception) through March 31, 2010, Adamis has spent a total of approximately $440,000 to in-license and develop the Adamis Viral vaccine technology. Research and development efforts for cancer small molecule therapeutic drug candidate and therapeutic vaccine candidates will be required. This will also require conducting and managing pre-clinical and/or clinical studies and significant additional funding, and even if development, partnering, and marketing efforts are successful, substantial time may pass before significant revenues will be realized. Accordingly, even if Adamis Labs generates the projected revenues and net income, during this period Adamis will still most likely require additional funds for its Adamis Viral operations, the availability of which cannot be assured. Consequently, Adamis is subject to many of the risks associated with early stage companies, including the need for additional financings; the uncertainty of research and development efforts resulting in successful commercial products, as well as the marketing and customer acceptance of such products; competition from larger organizations; reliance on the proprietary technology of others; dependence on key personnel; uncertain patent protection and competition; and dependence on corporate partners and collaborators.
51
To achieve successful operations of both its Adamis Viral and Adamis Labs subsidiaries, Adamis will require additional capital to continue research and development and marketing efforts and to make capital investments in its operations. No assurance can be given as to the timing or ultimate success of obtaining future funding, and there are no assurances that Adamis will be successful, with the limited experience and resources Adamis has available at the present time, in developing and commercializing its initial inhaled nasal steroid product and tumor treatment product.
The process of developing new therapeutic products is inherently complex, time-consuming, expensive and uncertain. Adamis must make long-term investments and commit significant resources before knowing whether its development programs will result in products that will receive regulatory approval and achieve market acceptance. Product candidates that may appear to be promising at all stages of development may not reach the market for a number of reasons. Product candidates may be found ineffective or may cause harmful side effects during clinical trials, may take longer to progress through clinical trials than had been anticipated, may not be able to achieve the pre-defined clinical endpoint due to statistical anomalies even though clinical benefit may have been achieved, may fail to receive necessary regulatory approvals, may prove impracticable to manufacture in commercial quantities at reasonable cost and with acceptable quality, or may fail to achieve market acceptance. For these reasons, as well as other reasons identified under the heading “Risk Factors” elsewhere in this Annual Report, Adamis is unable to predict the period in which material net cash inflows from product candidates incorporating the therapeutic vaccine and the small molecule therapeutic anti-inflammatory drug technologies will commence.
Merger of Cellegy and Adamis; Change of Corporate Name
Effective April 1, 2009, Adamis completed a business combination transaction with Old Cellegy Pharmaceuticals, Inc., or Cellegy. The stockholders of Cellegy and the former Adamis Pharmaceuticals Corporation, or Old Adamis, approved a merger transaction and related matters at an annual meeting of Old Cellegy’s stockholders and at a special meeting of Old Adamis’ stockholders each held on March 23, 2009. On April 1, 2009, Old Cellegy completed the merger transaction with Old Adamis. Before the merger, Old Cellegy was a public company and Old Adamis was a private company.
In connection with the consummation of the merger and pursuant to the terms of the definitive merger agreement relating to the transaction, Old Cellegy changed its name from Cellegy Pharmaceuticals, Inc. to Adamis Pharmaceuticals Corporation, and Old Adamis changed its corporate name to Adamis Corporation.
Pursuant to the terms of the merger agreement, immediately before the consummation of the merger Cellegy effected a reverse stock split of its common stock. Pursuant to this reverse stock split, each approximately 10 shares of common stock of Cellegy that were issued and outstanding immediately before the effective time of the merger were converted into one share of common stock and any remaining fractional shares held by a stockholder (after the aggregating fractional shares) were rounded up to the nearest whole share.
As a result, the total number of shares of Old Cellegy that were outstanding immediately before the effective time of the merger were converted into approximately 3,000,000 shares of post-reverse split shares of common stock of Adamis. Pursuant to the terms of the merger agreement, at the effective time of the merger each share of Old Adamis common stock that was issued and outstanding immediately before the effective time of the merger ceased to be outstanding and was converted into the right to receive one share of common stock of Adamis.
52
As a result, approximately 44,000,000 shares of Adamis were issued and/or are issuable to the holders of the outstanding shares of common stock of Old Adamis before the effective time of the merger. Old Adamis, renamed Adamis Corporation, was the surviving entity as a wholly-owned subsidiary of Adamis.
LaJolla Pharmaceuticals
On December 4, 2009, Adamis entered into an Agreement and Plan of Reorganization with La Jolla Pharmaceutical Company, or La Jolla, a public pharmaceuticals company, providing for the acquisition of La Jolla by Adamis. On March 3, 2010, Adamis and La Jolla agreed to terminate the Agreement and Plan of Reorganization. The termination followed the announcement by La Jolla that its common stock would be suspended and delisted from the NASDAQ stock market and that holders of only 13% of La Jolla’s outstanding common stock returned their proxy cards or otherwise indicated their votes with respect to proposals related to the proposed merger transaction and that as a result the La Jolla stockholder meeting and the solicitation of further votes had been cancelled.
Critical Accounting Policies and Estimates
Adamis has identified below some of its more significant accounting policies. For further discussion of Adamis’ accounting policies, see Note 1 in the Notes to the Adamis Consolidated Financial Statements.
Accounting Standards Codification. In July 2009, the Financial Accounting Standards Board (“FASB”) issued Statement of Financial Accounting Standards Number 168, “The FASB Accounting Standards Codification and the Hierarchy of Generally Accepted Accounting Principles”, which identified the FASB’s Accounting Standards Codification (“ASC”) as the single source of authoritative nongovernmental U.S. generally accepted accounting principles (“GAAP”). The ASC reorganized the thousands of U.S. GAAP pronouncements into roughly 90 accounting topics and displays all topics using a consistent structure. It also includes relevant Securities and Exchange Commission (“SEC”) guidance that follows the same topical structure in separate sections in the ASC. All previously existing accounting standards documents were superseded by the ASC, which was effective for interim and annual periods ending after September 15, 2009. All other accounting literature not included in the ASC is nonauthoritative. We believe that our adoption of this standard on its effective date (July 1, 2009) did not have a material effect on our consolidated financial statements.
Principles of Consolidation. The accompanying consolidated financial statements include Adamis Pharmaceuticals and its wholly owned subsidiaries. All significant intercompany balances and transactions have been eliminated in consolidation.
Use of Estimates. The preparation of consolidated financial statements, in conformity with accounting principles generally accepted in the United States, requires management to make estimates, judgments and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from those estimates.
Cash and Cash Equivalents. For purposes of the consolidated statements of cash flows, Adamis considers all highly liquid investments with original maturities at the date of purchase of three months or less to be cash equivalents.
Accounts Receivable, Allowance for Doubtful Accounts and Sales Returns. Trade accounts receivable are stated net of an allowance for doubtful accounts and sales returns. Adamis estimates an allowance based on its historical experience of the relationship between actual bad debts and net credit sales. At March 31, 2010 and 2009, no allowance for doubtful accounts was recorded. Adamis has established an allowance for sales returns based on management’s best estimate of probable loss inherent in the accounts receivable balance. Management determines the allowance based on current credit conditions, historical experience, and other currently available information. The allowance for sales returns was $75,899 and $19,501 at March 31, 2010 and 2009, respectively.
Adamis has established an allowance for sales returns based on management’s best estimate of probable loss inherent in the accounts receivable balance. Management determines the allowance based on current credit conditions, historical experience, and other currently available information.
53
Registration Payment Arrangements. Generally Accepted Accounting Principles, or GAAP, for registration payment arrangements specifies that the contingent obligation to make future payments under a registration payment arrangement should be separately recognized and measured.
Fair Value Measurements. The carrying amounts of cash and cash equivalents, accounts receivable, accounts payable and accrued liabilities approximate fair value due to the short maturity of the instruments. The notes payable to stockholders approximates fair value, based on current rates available to the Company for loans with similar maturities.
Effective October 1, 2008, we adopted the provisions of the Fair Values Measurements and Disclosures topic of the ASC, with respect to our financial assets and liabilities. Under the ASC accounting standards, fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value should maximize the use of observable inputs and minimize the use of unobservable inputs. The accounting standards describe a fair value hierarchy based on three levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value:
Level 1 — Quoted prices in active markets for identical assets or liabilities.
Level 2 — Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
Level 3 — Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
Inventory. Inventory, consisting of allergy and respiratory products, is recorded at the lower of cost or market, using the weighted average method.
Property and Equipment. Property and equipment are recorded at cost less accumulated depreciation. Depreciation is computed using the straight-line method over the estimated useful lives of the related assets. The cost of leasehold improvements are amortized over the lesser of the lease term or the life of the improvement, if shorter.
Useful lives used to depreciate property and equipment are as follows:
|
Life in
Years
|
||||
|
Office Furniture and Equipment
|
7 | |||
|
Computer Equipment and Software
|
3 | |||
|
Vehicles
|
3 | |||
Deferred Acquisition Costs. Adamis incurred certain professional fees associated with specific potential acquisition targets. These costs were capitalized as part of the purchase price paid for the acquisition.
Revenue Recognition. Our primary customers are pharmaceutical wholesalers. In accordance with our revenue recognition policy, revenue is recognized when title and risk of loss are transferred to the customer, the sale price to the customer is fixed and determinable, and collectability of the sale price is reasonably assured. Reported revenue is net of estimated customer returns and other wholesaler fees. Our policy regarding sales to customers is that we do not recognize revenue from, or the cost of, such sales, where we believe the customer has more than a demonstrably reasonable level of inventory. We make this assessment based on historical demand, historical customer ordering patterns for purchases, business considerations for customer purchases and estimated inventory levels. If our actual experience proves to be different than our assumptions, we would then adjust such allowances accordingly.
We estimate allowances for revenue dilution items using a combination of information received from third parties, including market data, inventory reports from our major U.S. wholesaler customers, when available, historical information and analysis that we perform. The key assumptions used to arrive at our best estimate of revenue dilution reserves are estimated customer inventory levels and purchase forecasts provided. Our estimates of
54
inventory at wholesaler customers and in the distribution channels are subject to the inherent limitations of estimates that rely on third-party data, as certain third-party information may itself rely on estimates, and reflect other limitations. We believe that such provisions are reasonably ascertainable due to the limited number of assumptions involved and the consistency of historical experience.
Sales returns and discounts for the period ended March 31, 2009 were approximately $12,700. The table below reconciles the “Sales Returns Reserve Adjustment” for the same period ended March 31, 2009:
|
Beginning Balance Sales Returns & Discounts as of March 31, 2008
|
$ | 21,022 | ||
|
|
||||
|
Less Actual Sales Returns & Discounts during Fiscal 2009
|
(12,725 | ) | ||
|
Reserve needed to replenish correct reserve
|
11,204 | |||
|
Sub total
|
(1,521 | ) | ||
|
Ending Sales Returns & Discounts as of March 31, 2009
|
$ | 19,501 |
The $(1,521) adjustment to “Sales Returns Reserve Adjustment” is the difference between the actual sales returns & discounts $(12,725) and the amount needed to replenish the reserve, $11,204.
Sales returns and discounts for the period ended March 31, 2010 were approximately $54,530. The table below reconciles the “Sales Returns Reserve Adjustment” for the same period ended March 31, 2010:
|
Beginning Balance Sales Returns & Discounts as of March 31, 2009
|
$ | 19,501 | ||
|
Less Actual Sales Returns & Discounts during Fiscal 2010
|
(54,530 | ) | ||
|
Reserve needed to replenish correct reserve
|
110,928 | |||
|
Sub total
|
56,398 | |||
|
Ending Sales Returns & Discounts as of March 31, 2010
|
$ | 75,899 |
The $56,398 adjustment to “Sales Returns Reserve Adjustment” is the difference between the actual sales returns & discounts $(54,530) and the amount needed to replenish the reserve, $110,928.
Revenues under license and royalty agreements are recognized in the period the earnings process is completed based on the terms of the specific agreement. Advanced payments received under these agreements are recorded as deferred revenue at the time the payment is received and are subsequently recognized as revenue on a straight-line basis over the longer of the life of the agreement or the life of the underlying patent. Royalties payable to Adamis under license agreements are recognized as earned when the royalties are no longer refundable under the terms defined in the agreement. To date no royalties have been paid.
Goodwill and Intangible Assets. Intangible assets include intellectual property and other patent rights acquired. Consideration paid in connection with acquisitions is required to be allocated to the acquired assets, including identifiable intangible assets, and liabilities acquired. Acquired assets and liabilities are recorded based on Adamis’ estimate of fair value, which requires significant judgment with respect to future cash flows and discount rates. For intangible assets other than goodwill, Adamis is required to estimate the useful life of the asset and recognize its cost as an expense over the useful life. Adamis uses the straight-line method to expense long-lived assets (including identifiable intangibles). In accordance with GAAP, goodwill and other intangible assets with indefinite lives are no longer systematically amortized, but rather Adamis performs an annual assessment for impairment by applying a fair-value based test. This test is generally performed each year in the fourth fiscal quarter. Additionally, goodwill and intangible assets are reviewed for impairment whenever events or circumstances indicate that the carrying amount of the asset may not be recoverable. An impairment loss would be recognized based on the difference between the carrying value of the asset and its estimated fair value, which would be determined based on either discounted future cash flows or other appropriate fair value methods. The evaluation of goodwill and other intangibles for impairment requires management to use significant judgments and estimates including, but not limited to, projected future revenue, operating results and cash flows. An impairment would require Adamis to charge to earnings the write-down in value of such assets.
55
Long Lived Assets. Adamis periodically assesses whether there has been permanent impairment of its long-lived assets held and used in accordance with GAAP, which requires Adamis to review long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by comparison of the carrying amount of the asset to future net undiscounted cash flows expected to be generated from the use and eventual disposition of the asset.
Research and Development Expenses. Adamis’ research and development costs are expensed as incurred. Non-refundable advance payments for goods and services to be used in future research and development activities are recorded as an asset and are expensed when the research and development activities are performed. Research and development costs were approximately $585,000 and $740,000 for the fiscal years ended March 31, 2010 and 2009, respectively, which were expensed. For fiscal 2009, approximately $728,000 of the costs related to the PFS Syringe product, and approximately $12,000 of the costs related to the vaccine product development efforts. For fiscal 2010, approximately $145,000 of the costs were related to the PFS Syringe product, and approximately $40,000 of the costs were related to the vaccine product development efforts.
Shipping and Handling Costs. Shipping and handling costs are included in selling, general and administrative expenses. Shipping and handling costs were $13,722 and $13,651 for the years ended March 31, 2010 and 2009, respectively.
Advertising Costs. Advertising costs are expensed as incurred. The Company incurred $1,853 and $2,848 of advertising expense for the years ended March 31, 2010 and 2009, respectively.
Net Loss per Share. Adamis computes net loss per share by dividing the income attributable to holders of common stock for the period by the weighted average number of shares of common stock outstanding during the period. Since the effect of common stock equivalents was anti-dilutive, all such equivalents were excluded from the calculation of weighted average shares outstanding. There were 1,922,139 and 1,000,000 outstanding warrants at March 31, 2010 and 2009.
Income Taxes. Adamis accounts for income taxes using an asset and liability approach for financial accounting and reporting for income taxes. Under the asset and liability approach, deferred taxes are provided for the net tax effect of temporary differences between the carrying amount of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Valuation allowances are established where management determines that it is more likely than not that some portion or all of a deferred tax asset will not be realized.
Effective April 1, 2009, Financial Accounting Standards Board (“FASB”) issued guidance that addresses accounting for uncertainty in income taxes which requires that a position taken or expected to be taken in a tax return be recognized in the consolidated financial statements when it is more likely than not (i.e., a likelihood of more than fifty percent) that the position would be sustained upon examination by tax authorities. A recognized tax position is then measured at the largest amount of benefit that is greater than fifty percent likely of being realized upon ultimate settlement. The adoption of this guidance did not have a significant impact on the Company’s consolidated financial statements.
Discontinued Operations. The results of operations for the year ended March 31, 2009, and the assets and liabilities at March 31, 2009 related to International Laboratories, Inc. (“INL”), a formerly wholly owned subsidiary of Adamis, have been accounted for as discontinued operations in accordance with GAAP.
Results of Operations
Adamis’ consolidated results of operations are presented for the fiscal year ending March 31, 2010 and for the fiscal year ending March 31, 2009.
Year Ended March 31, 2010 and Year Ended March 31, 2009
Revenues and Cost of Sales. Adamis had revenues of $290,288 and $659,538 for the year ending March 31, 2010 and March 31, 2009 respectively. The $369,250 decrease in revenues compared to the year ended 2009 was primarily the result of decreased sales of Aerohist, Aerohist plus, and Aerokid, offset in part by an increase of prefilled PFS syringe product sales.
56
Research and Development Expense. Adamis incurred research and development expenses of $585,758 in fiscal 2010 and $740,000 in fiscal 2009. The decrease was primarily due to the majority of development costs expended in fiscal 2009 for the PFS Syringe product.
Selling, General and Administrative Expenses. Selling, general and administrative expenses for fiscal 2010 and 2009 were $3,274,505 and $4,852,966, respectively. Selling, general and administrative expenses consist primarily of legal fees, accounting and audit fees, professional fees and employee salaries. The decrease in expenses for fiscal 2010 was primarily due to expenses associated with the merger with Cellegy Pharmaceuticals in 2009. The decrease in legal, accounting, salaries, professional and consulting, with increases in insurance, printing and telephone expenses accounted for approximately $1,598,000 of the variance.
Other Income (Expenses). Interest and other income (expense) for fiscal 2010 and 2009 were $(2,726,049) and $(451,038), respectively. Interest and other income (expense) consist primarily of interest expense paid in connection with various notes payable. The increase in interest expense for fiscal 2010 compared to fiscal 2009 was primarily due to interest expense charged from the notes with Gemini and G-Max during fiscal 2010.
Liquidity and Capital Resources
Fiscal 2010 and 2009
Since its inception, June 6, 2006, through March 31, 2010, Adamis has financed its operations principally through debt financing and through private issuances of common stock. Since inception, Adamis has raised a total of approximately $12 million in debt and equity financing transactions, consisting of approximately $6.3 million in debt financing and approximately $5.7 million in equity financing transactions. Adamis expects to finance future cash needs primarily through proceeds from equity or debt financings, loans, and/or collaborative agreements with corporate partners. Adamis has used the net proceeds from debt and equity financings for general corporate purposes, which have included funding for research and development, selling, general and administrative expenses, working capital, reducing indebtedness, pursuing and completing acquisitions or investments in other businesses, products or technologies, and for capital expenditures.
Adamis’ cash was $290,299 and $17,697 as of March 31, 2010 and March 31, 2009, respectively. The increase in cash was primarily the result of funds received from financing transactions.
Net cash used in operating activities from continuing operations for fiscal 2010 and 2009 were approximately $2.0 million and $4.2 million, respectively, due primarily to Adamis’ reduction of sales operations and the conclusion of the merger with Cellegy. Adamis expects net cash used in operating activities to increase going forward as it continues product development, launches new products, engages in additional product research and development activities and pursues additional expansion of its sales base and other business activities. The increase in accounts payable/accrued expenses of approximately $1,218,000 from fiscal 2009 relates primarily to the reduction in sales/cash collections. The reduced cash sales and collections have created a greater build up of payables/accrued expenses.
Net cash provided by investing activities from continuing operations was approximately $60,000 for fiscal 2010, compared to net cash provided by investing activities from continuing operations for fiscal 2009 of $6.6 million. The increase of net cash from investing activities in 2009 was provided by the sale of INL. Net cash provided by (used in) financing activities from continuing operations was approximately $2.2 million in fiscal 2010 and approximately $(2.6 million) in fiscal 2009, primarily due to the receipt of proceeds from debt financing in 2010 and the repayment of the debt financing in 2009.
As of March 31, 2010, Adamis had outstanding a total of fifteen secured promissory notes to Dennis J. Carlo, President and Chief Executive Officer of Adamis, in the aggregate outstanding principal amount of $309,565, reflecting loans made by Dr. Carlo to Adamis. Each of these notes bears interest at an annual rate of 10% and the total outstanding balance remain under these loan agreements.
Recent Convertible Note Transactions
Unsecured Convertible Note. On December 29, 2009, Adamis issued to The G-Max Trust, or G-Max, an unsecured convertible promissory note, referred to as the G-Max Note, in the principal amount of $500,000 and also issued 500,000 shares of common stock of Adamis, in a private placement transaction for aggregate gross proceeds of $500. Interest on the outstanding principal balance of the G-Max Note accrues at a rate of 10% per annum
57
compounded monthly and is payable monthly commencing February 1, 2010. All unpaid principal and interest on the G-Max Note is due and payable on December 31, 2010, sometimes referred to as the maturity date.
At any time on or before the maturity date, G-Max may convert some or all of the unpaid principal and interest into shares of Adamis common stock at a conversion price equal to $0.20 per share (subject to adjustment for stock dividends, stock splits, reverse stock splits, reclassifications or other similar events affecting the number of outstanding shares of Adamis common stock). Events of default under the G-Max Note include payment defaults and uncured material breaches of the Note. Upon an uncured event of default, G-Max may declare the entire unpaid amount owed under the G-Max Note immediately due, subject to the subordination provisions set forth in the G-Max Note, and may, at its option, charge default interest at a rate of 18% per annum.
The G-Max Note includes piggyback registration rights providing that at any time after one year after the date of the G-Max Note, if the shares that Adamis issued to G-Max and the shares of Adamis common stock that are issuable upon conversion of the G-Max Note, together referred to as the Transaction Shares, cannot be sold without restriction pursuant to SEC Rule 144, then if Adamis files a registration statement pursuant to the Securities Act of 1933, as amended, or the Act, at any time on or before December 29, 2010, relating to an offering for the account of others under the Act of any of its equity securities, other than registration statements on Form S-4 or Form S-8, Adamis will include in such registration any Transaction Shares specified by G-Max. The G-Max Note also includes subordination provisions providing that payment of principal, interest and any other amounts that may become due pursuant to the G-Max Note and any other obligation that Adamis may have to G-Max is subordinated to the payment in full of all secured debt or other senior indebtedness of Adamis.
Senior Secured Convertible Notes. In January 2010, Adamis completed a private placement financing transaction with a small number of institutional investors led by Gemini Master Fund, Ltd., pursuant to a Securities Purchase Agreement, referred to as the SPA. Adamis issued 10% Senior Secured Convertible Notes, referred to as the Secured Notes, in the aggregate principal amount of $1.5 million and 1,500,000 shares of Adamis common stock, and received gross proceeds of $1.5 million, excluding transaction costs and expenses.
Interest on the Secured Notes is payable at a rate of 10% per annum and is payable monthly on the first business day of each month. Principal and any accrued and unpaid interest is due and payable October 11, 2010. The Secured Notes are convertible into shares of Adamis common stock at any time at the discretion of the investor at an initial conversion price per share of $0.20, subject to adjustment for stock splits, stock dividends and other similar transactions and subject to the terms of the Notes. The conversion price is also subject to price anti-dilution adjustments providing that if Adamis issues equity securities or securities convertible into equity securities at an effective price per share below the conversion price (subject to certain exceptions), the conversion price will be adjusted downward to equal the price of the new securities.
Adamis’ obligations under the Secured Notes and the other transaction agreements are guaranteed by Adamis’ principal subsidiaries, including Adamis Corporation, Adamis Laboratories and Adamis Viral, and are secured by a security interest in all of the assets of Adamis and those subsidiaries, pursuant to a security agreement. The transaction agreements include restrictions on Adamis’ ability to engage in certain kinds of transactions while the Secured Notes are outstanding without the consent of two-thirds in interest of the holders of the Secured Notes, including incurring or paying certain kinds of indebtedness, entering into certain kinds of financing transactions at prices below $0.20 per share, or encumbering Adamis’ assets. In addition to the rights under the security agreement to foreclose on the collateral in the event of a default, the transaction documents include a variety of liquidated damages, penalties and default provisions upon events of default by Adamis, including without limitation an increase in the principal amount and interest rate and a potential decrease in the conversion price of the Secured Notes, and in connection with certain other breaches of covenants of Adamis. If the shares underlying the Secured Notes are not freely tradable under SEC Rule 144 after six months from the closing of the Secured Note transaction, Adamis intends to file a registration statement covering the resale of such shares. In connection with the transaction, Adamis’ officers entered into lockup agreements restricting sales of Adamis securities owned by them for as long as any Notes are outstanding, subject to certain limited exceptions.
Off Balance Sheet Arrangements
At March 31, 2010, the Company did not have any off balance sheet arrangements.
58
Recent Accounting Pronouncements
The Fair Value Measurements and Disclosures topic of the ASC includes certain concepts first set forth in September 2006, which define the use of fair value measures in financial statements, establish a framework for measuring fair value and expand disclosure related to the use of fair value measures. In February 2008, the FASB provides a one-year deferral of the effective date of those concepts for non-financial assets and non-financial liabilities, except those that are recognized or disclosed in the financial statements at fair value at least annually. The application of these concepts was effective for our fiscal year beginning April 1, 2009, excluding the effect of the one-year deferral noted above. See “Fair Value Measurements” above. We are currently evaluating the impact of adopting these concepts with respect to non-financial assets and non-financial liabilities on our consolidated financial statements, which will be effective beginning April 1, 2010.
The Financial Instruments topic of the ASC includes certain concepts first set forth in February 2007, under which we may elect to report most financial instruments and certain other items at fair value on an instrument-by-instrument basis with changes in fair value reported in earnings. After the initial adoption, the election is made at the acquisition of an eligible financial asset, financial liability, or firm commitment or when certain specified reconsideration events occur. The fair value election may not be revoked once an election is made. The application of these concepts was effective for our fiscal year beginning April 1, 2008 — however, we have elected not to measure eligible financial assets and liabilities at fair value. Accordingly, the adoption of these concepts did not have a significant impact on our consolidated financial statements.
The Business Combinations topic of the ASC establishes principles and requirements for how an acquirer recognizes and measures in its financial statements the identifiable assets acquired, the liabilities assumed, and any noncontrolling interest in an acquiree, including the recognition and measurement of goodwill acquired in a business combination. The provisions of this guidance became effective for our fiscal year beginning April 1, 2009. Under these provisions, we would have recorded the $147,747 of deferred acquisition costs included in other non-current assets on our March 31, 2009 balance sheet as expense during the year then ended. This amount was recorded as a reduction of paid-in capital on April 1, 2009.
The Subsequent Events topic of the ASC establishes general standards of accounting for and disclosure of events that occur after the balance sheet date but before financial statements are issued or are available to be issued. Certain of these requirements, which were first effective for interim or annual financial periods ending after June 15, 2009, relate to the concept of financial statements being “available to be issued” and require the disclosure of the date through which an entity has evaluated subsequent events and the basis for that date (i.e., whether that date represents the date the financial statements were issued or were available to be issued). Other than providing this disclosure, our adoption of these requirements as of and for the period ended June 30, 2009 did not have a significant impact on our interim condensed consolidated financial statements.
SFAS No. 157, Fair Value Measurements
SFAS No. 157, “Fair Value Measurements” (“SFAS 157”), has been issued by the Financial Accounting Standards Board (the “FASB”). This new standard provides guidance for using fair value to measure assets and liabilities. SFAS 157 applies whenever other standards require (or permit) assets or liabilities to be measured at fair value but does not expand the use of fair value in any new circumstances. Currently, over 40 accounting standards within GAAP require (or permit) entities to measure assets and liabilities at fair value. The standard clarifies that for items that are not actively traded, such as certain kinds of derivatives, fair value should reflect the price in a transaction with a market participant, including an adjustment for risk, not just the Company’s mark-to-model value. SFAS 157 also requires expanded disclosure of the effect on earnings for items measured using unobservable data. Under SFAS 157, fair value refers to the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants in the market in which the reporting entity transacts. In this standard, FASB clarified the principle that fair value should be based on the assumptions market participants would use when pricing the asset or liability. In support of this principle, SFAS 157 establishes a fair value hierarchy that prioritizes the information used to develop those assumptions. The fair value hierarchy gives the highest priority to quoted prices in active markets and the lowest priority to unobservable data, for example, the reporting entity’s own data. Under the standard, fair value measurements would be separately disclosed by level within the fair value hierarchy.
59
In February 2008, the FASB issued FSP SFAS No. 157-1, "Application of FASB Statement No. 157 to FASB Statement No. 13 and Its Related Interpretive Accounting Pronouncements That Address Leasing Transactions," and FSP SFAS No. 157-2, "Effective Date of FASB Statement No. 157." FSP SFAS No. 157-1 removes leasing from the scope of SFAS No. 157. FSP SFAS No. 157-2 delays the effective date of SFAS No. 157 for the Company from its fiscal 2009 to its fiscal 2010 year for all nonfinancial assets and nonfinancial liabilities, except those that are recognized or disclosed at fair value in the financial statements on a recurring basis (at least annually). The Company does not expect its adoption of the provisions of FSP SFAS No. 157-1 and FSP SFAS No. 157-2 will have a material effect on its financial condition, results of operations or cash flows.
In February 2007, FASB issued SFAS No. 159, “The Fair Value Option for Financial Assets and Financial Liabilities.” Under SFAS No. 159, a company may elect to measure at fair value various eligible items that are not currently required to be so measured. Eligible items include, but are not limited to, accounts receivable, available-for-sale securities, equity method investments, accounts payable and firm commitments. SFAS No. 159 is effective in fiscal years beginning after November 15, 2007 and is required to be adopted by the Company in the first quarter of its fiscal 2009 year.
SFAS No. 141 (Revised 2007), Business Combinations
On December 4, 2007, the FASB issued SFAS No. 141 (Revised 2007), Business Combinations (“SFAS 141R”). Under SFAS 141R, an acquiring entity will be required to recognize all the assets acquired and liabilities assumed in a transaction at the acquisition date fair value with limited exceptions. SFAS 141R will change the accounting treatment for certain specific items, including:
|
•
|
acquisition costs will be generally expensed as incurred;
|
|
|
•
|
non-controlling interests will be valued at fair value at the acquisition date;
|
|
|
•
|
acquired contingent liabilities will be recorded at fair value at the acquisition date;
|
|
|
•
|
in-process research and development will be recorded at fair value as an indefinite-lived intangible asset at the acquisition date until the completion or abandonment of the associated research and development efforts;
|
|
|
•
|
restructuring costs associated with a business combination will be generally expensed subsequent to the acquisition date; and
|
|
|
•
|
changes in deferred tax asset valuation allowances and income tax uncertainties after the acquisition date generally will affect income tax expense.
|
SFAS 141R also includes a substantial number of new disclosure requirements. SFAS 141R applies prospectively to business combinations for which the acquisition date is on or after the beginning of the first annual reporting period beginning on or after December 15, 2008. Earlier adoption is prohibited.
SFAS No. 160, “Non-controlling Interests in Consolidated Financial Statements — An Amendment of ARB No. 51”
On December 4, 2007, the FASB issued SFAS No. 160, “Non-controlling Interests in Consolidated Financial Statements — An Amendment of ARB No. 51” (“SFAS 160”). SFAS 160 establishes new accounting and reporting standards for the non-controlling interest in a subsidiary and for the deconsolidation of a subsidiary. Specifically, this statement requires the recognition of a non-controlling interest (minority interest) as equity in the consolidated financial statements and separate from the parent’s equity. The amount of net income attributable to the non-controlling interest will be included in consolidated net income on the face of the income statement. SFAS 160 clarifies that changes in a parent’s ownership interest in a subsidiary that do not result in deconsolidation are equity transactions if the parent retains its controlling financial interest. In addition, this statement requires that a parent recognize a gain or loss in net income when a subsidiary is deconsolidated. Such gain or loss will be
60
measured using the fair value of the non-controlling equity investment on the deconsolidation date. SFAS 160 also includes expanded disclosure requirements regarding the interests of the parent and its non-controlling interest. SFAS 160 is effective for fiscal years, and interim periods within those fiscal years, beginning on or after December 15, 2008. Earlier adoption is prohibited.
In June 2007, the FASB ratified EITF Issue No. 07-3, “Accounting for Nonrefundable Advance Payments for Goods or Services to Be Used in Future Research and Development Activities.” EITF 07-3 requires non-refundable advance payments for goods and services to be used in future research and development activities to be recorded as an asset and expensing the payments when the research and development activities are performed. EITF 07-3 applies prospectively for new contractual arrangements entered into in fiscal years beginning after December 15, 2007. The Company currently recognizes these non-refundable advanced payments as an asset upon payment, and expenses costs as goods are used and services are provided. There was no effect on the Company’s financial statement from the adoption of this pronouncement.
In July 2006, the FASB issued FASB Interpretation No. 48, “Accounting for Uncertainty in Income Taxes, an Interpretation of SFAS No. 109, Accounting for Income Taxes,” (“FIN 48”) which clarifies the accounting for income taxes by prescribing the minimum recognition threshold a tax position is required to meet and the measurement attribute for financial statement disclosure of tax positions taken or expected to be taken on a tax return. The Company adopted FIN 48 effective on January 1, 2007 , and there was no material effect on its results of operations or financial position.
ITEM 8: FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The financial statements and financial information required by Item 8 are set forth below on pages F-1 through F-33 of this report.
|
Index to Financial Statements
|
F-1
|
|
|
Report of Independent Registered Public Accounting Firm
|
F-2
|
|
|
Consolidated Balance Sheets
|
F-3
|
|
|
Consolidated Statements of Operations
|
F-4
|
|
|
Consolidated Statements of Stockholders’ Equity (Deficit) and Comprehensive Income
|
F-5
|
|
|
Consolidated Statements of Cash Flows
|
F-6
|
|
|
Notes to Consolidated Financial Statements
|
F-8
|
ITEM 9: CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURES
ITEM 9A(T): CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of our disclosure controls and procedures, as such term is defined under Rule 13a-15(e) and 15d-15(e) promulgated under the Securities Exchange Act of 1934, as amended (the “Exchange Act”) as of the end of the period covered by this Form 10-K. Based on their evaluation, our principal executive officer and principal accounting officer concluded that our disclosure controls and procedures were effective.
61
Internal Control over Financial Reporting
Management’s report on Adamis’ internal control over financial reporting (as such term is defined in Rules 13a-15(f) and 15d-15(f) in the Exchange Act), is included in this Annual Report on Form 10-K, under the headings, “Management’s Annual Report on Internal Control Over Financial Reporting” and is incorporated herein by reference. This report shall not be deemed to be filed for purposes of Section 18 of the Exchange Act or otherwise subject to the liabilities of that section, unless Adamis specifically states that the report is to be considered “filed” under the Exchange Act or incorporates it by reference into a filing under the Securities Act of the Exchange Act.
Management’s Annual Report on Internal Control over Financial Reporting
The management of the Company is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is a process designed by, or under the supervision of, our Chief Executive Officer and Chief Financial Officer and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to risk that controls may become inadequate due to changes in conditions, or that the degree of compliance with policies and procedures may deteriorate.
With the participation of the Chief Executive Officer and the Chief Financial Officer, our management conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework and criteria established in Internal Control—Integrated Framework, issued by the Committee of Sponsoring Organizations (“COSO”) of the Treadway Commission. Based on this evaluation, our management has concluded that our internal control over financial reporting was effective as of March 31, 2010.
This Annual Report does not include an attestation report of our registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to the attestation by our registered public accounting firm pursuant to the rules of the SEC that permit us to provide only management’s report in this Annual Report.
ITEM 9B: OTHER INFORMATION
None.
62
PART III
Part III will be filed by means of an amendment to the March 31, 2010 10K.
ITEM 10 — DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by Item 10 of Part III is incorporated by reference to the registrant’s proxy statement, to be filed within 120 days of the registrant’s fiscal year end, or will be included in an amendment to this Form 10-K.
ITEM 11 — EXECUTIVE COMPENSATION
The information required by Item 11 of Part III is incorporated by reference to the registrant’s proxy statement, to be filed within 120 days of the registrant’s fiscal year end, or will be included in an amendment to this Form 10-K.
ITEM 12 — SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by Item 12 of Part III is incorporated by reference to the registrant’s proxy statement, to be filed within 120 days of the registrant’s fiscal year end, or will be included in an amendment to this Form 10-K.
ITEM 13 — CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by Item 13 of Part III is incorporated by reference to the registrant’s proxy statement, to be filed within 120 days of the registrant’s fiscal year end, or will be included in an amendment to this Form 10-K.
ITEM 14 — PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required by Item 14 of Part III is incorporated by reference to the registrant’s proxy statement, to be filed within 120 days of the registrant’s fiscal year end, or will be included in an amendment to this Form 10-K.
63
PART IV
ITEM 15: EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
Exhibits
The following exhibits are attached hereto or incorporated herein by reference.
|
Incorporated by
Reference
|
||||||||
|
Exhibit
Number
|
Exhibit Description
|
Filed
Herewith |
Form/File
No. |
Date
|
||||
|
2.1
|
Agreement and Plan of Reorganization dated as of February 12, 2008, by and among Cellegy, Cellegy Holdings, Inc. and Adamis Pharmaceuticals Corporation (the “Merger Agreement”).
|
8-K
|
2/13/08
|
|||||
|
2.2
|
Agreement dated November 11, 2008, between the Company and Adamis amending the Merger Agreement.
|
8-K
|
11/13/08
|
|||||
|
2.3
|
Agreement dated January 8, 2009, between the Company and Adamis amending the Merger Agreement.
|
8-K
|
1/8/09
|
|||||
|
3.1
|
Certificate of Amendment to Amended and Restated Certificate of Incorporation.
|
8-K
|
4/3/09
|
|||||
|
3.2
|
Amended and Restated Certificate of Incorporation of the Registrant
|
8-K
|
4/3/09
|
|||||
|
3.3
|
Amended and Restated Bylaws of the Company
|
S-4/A
333-155322
|
1/12/09
|
|||||
|
4.1
|
Specimen stock certificate for common stock.
|
8-K
|
4/3/09
|
|||||
|
10.1
|
2005 Equity Incentive Plan
|
10-K
|
3/31/06
|
|||||
|
10.2
|
Form of Option Agreement under the 2005 Equity Incentive Plan.
|
10-K
|
3/31/06
|
|||||
|
*10.3
|
1995 Equity Incentive Plan
|
10-Q
|
8/13/02
|
|||||
|
10.4
|
Exclusive License Agreement dated as of December 31, 2002, by and between Cellegy and PDI, Inc.
|
10-K
|
3/21/03
|
|||||
|
*10.5
|
Retention and Severance Plan and Form of Agreement of Plan Participation under Retention and Severance Plan.
|
10-Q
|
5/8/03
|
|||||
|
*10.6
|
Letter agreement dated November 5, 2003, between Cellegy and Richard C. Williams.
|
10-K
|
4/6/04
|
|||||
|
*10.7
|
Stock option agreement dated November 5, 2003, between Cellegy and Richard C. Williams.
|
10-K
|
4/6/04
|
|||||
|
*10.8
|
Form of Indemnity Agreement between Cellegy and its directors and executive officers.
|
Proxy Statement
|
4/28/04
|
|||||
|
10.9
|
Agreement dated as of October 8, 1996 by and among Biosyn, Inc., Edwin B. Michaels and E.B. Michaels Research Associates, Inc. (Confidential treatment has been requested with respect to portions of this agreement)
|
10-K
|
3/31/05
|
|||||
|
10.10
|
Patent License Agreement by and among Biosyn, Inc., and certain agencies of the United States Public Health Service.
|
10-K
|
3/31/05
|
|||||
|
10.11
|
License Agreement dated as of May 22, 2001, by and between Crompton Corporation and Biosyn, Inc. (Confidential treatment has been requested for portions of this agreement.)
|
10-K
|
3/31/05
|
|||||
|
10.12
|
License Agreement dated January 30, 2006, by and between CONRAD, Eastern Virginia Medical School, and Biosyn, Inc. (Confidential treatment has been requested for portions of this agreement.)
|
10-K
|
4/02/07
|
|||||
64
|
Incorporated by
Reference
|
||||||||
|
Exhibit
Number
|
Exhibit Description
|
Filed
Herewith |
Form/File
No. |
Date
|
||||
|
*10.13
|
2009 Equity Incentive Plan.
|
8-K
|
4/3/2009
|
|||||
|
*10.14
|
Form of Option Agreement under 2009 Equity Incentive Plan.
|
8-K
|
4/3/2009
|
|||||
|
10.15
|
Amendment to License Agreement dated as of March 15, 2006, by and between Crompton Corporation and Biosyn, Inc.
|
S-4/A
333-155322
|
1/12/09
|
|||||
|
10.16
|
Funding Agreement dated October 12, 1992, by and between Ben Franklin Technology Center of Southeaster Pennsylvania and Biosyn, Inc.
|
S-4/A
333-155322 |
1/12/09
|
|||||
|
10.17
|
License Agreement dated July 28, 2006, by and between Nevagen, LLC and Adamis Pharmaceuticals Corporation.
|
S-4/A
333-155322 |
1/12/09
|
|||||
|
10.18
|
Amendment to License Agreement dated December 29, 2008, by and between Nevagen, LLC and Adamis Pharmaceuticals Corporation.
|
S-4/A
333-155322 |
1/12/09
|
|||||
|
*10.19
|
Stock Repurchase Agreement dated November 3, 2008, by and between Richard Aloi and Adamis Pharmaceuticals Corporation.
|
S-4/A
333-155322 |
1/12/09
|
|||||
|
*10.20
|
Stock Repurchase Agreement dated November 3, 2008, by and between Dennis J. Carlo and Adamis Pharmaceuticals Corporation
|
S-4/A
333-155322 |
1/12/09
|
|||||
|
*10.21
|
Stock Repurchase Agreement dated November 3, 2008, by and between Robert Hopkins and Adamis Pharmaceuticals Corporation
|
S-4/A
333-155322 |
1/12/09
|
|||||
|
*10.22
|
Stock Repurchase Agreement dated November 3, 2008, by and between David J. Marguglio and Adamis Pharmaceuticals Corporation.
|
S-4/A
333-155322 |
1/12/09
|
|||||
|
10.23
|
Amendment to License Agreement dated October 18, 2007, by and between CONRAD, Eastern Virginia Medical School, and Biosyn, Inc.
|
S-4/A
333-155322 |
1/12/09
|
|||||
|
10.24
|
Lease Agreement dated January 1, 2007, by and between HRM II Ltd and Healthcare Ventures Group.
|
S-4/A
333-155322 |
1/12/09
|
|||||
|
10.25
|
Amendment to Lease Agreement dated October 30, 2007, by and between HRM II Ltd and Healthcare Ventures Group.
|
S-4/A
333-155322 |
1/12/09
|
|||||
|
10.26
|
Clinical Trial Agreement between Biosyn, Inc. and the National Institute of Child Health and Human Development.
|
S-4/A
333-155322 |
1/12/09
|
|||||
|
10.27
|
Convertible Promissory Note dated December 29, 2009 between the Registrant and The G-Max Trust.
|
8-K
|
1/04/10
|
|||||
|
10.28
|
Securities Purchase Agreement dated January 11, 2010 between the Registrant and the investors listed therein.
|
8-K
|
1/14/10
|
|||||
|
10.29
|
Form of 10% Senior Secured Convertible Note dated January 11, 2010.
|
8-K
|
1/14/10
|
|||||
|
10.30
|
Form of Security Agreement dated January 11, 2010.
|
8-K
|
1/14/10
|
|||||
|
10.31
|
Assignment, Assumption and Stock Acquisition Agreement dated February 24, 2010 between the Registrant and Colby Pharmaceutical Company.
|
X
|
||||||
|
21.1
|
Subsidiaries of the Registrant
|
X
|
||||||
|
23.1
|
Consent of Mayer Hoffman McCann PC, Independent Registered Public Accounting Firm.
|
X
|
||||||
|
24.1
|
Power of Attorney (See signature page)
|
X
|
||||||
* Represents a compensatory plan or arrangement.
65
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Del Mar, State of California.
|
ADAMIS PHARAMCEUTICALS CORPORATION
|
|||
|
|
|||
|
By:
|
/s/ DENNIS J. CARLO
|
||
|
Dennis J. Carlo
Chief Executive Officer
|
|||
|
Dated: July 14, 2010
|
|||
Power of Attorney
Each person whose signature appears below constitutes and appoints each of Dennis J. Carlo and Robert O. Hopkins, true and lawful attorney-in-fact, with the power of substitution, for him in any and all capacities, to sign amendments to this Annual Report on Form 10-K, and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that said attorneys-in-fact, or his substitute or substitutes, may do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this report has been signed by the following persons in the capacities and on the dates indicated
|
Name
|
Title
|
Date
|
||
|
Principal Executive Officer:
|
||||
|
/s/ DENNIS J. CARLO
|
Chief Executive Officer and Director
|
July 14, 2010
|
||
|
Dennis J. Carlo
|
|
|||
|
Principal Financial Officer
|
||||
|
and Principal Accounting Officer:
|
||||
|
/s/ ROBERT O. HOPKINS
|
Vice President, Finance, Chief Financial Officer and Secretary
|
July 14, 2010
|
||
|
Robert O. Hopkins
|
|
|||
|
Directors:
|
||||
|
/s/ DAVID J. MARGUGLIO
|
Director
|
July 14, 2010
|
||
|
David J. Marguglio
|
||||
|
/s/ RICHARD L. ALOI
|
Director
|
July 14, 2010
|
||
|
Richard L. Aloi
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
TABLE OF CONTENTS
MARCH 31, 2010 AND 2009
|
PAGE
|
|
|
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM MAYER HOFFMAN MCCANN P.C.
|
F1
|
|
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM GOLDSTEIN LEWIN & CO.
|
F2
|
|
FINANCIAL STATEMENTS:
|
|
|
Consolidated Balance Sheets
|
F3
|
|
Consolidated Statements of Operations
|
F4
|
|
Consolidated Statements of Changes in Stockholders’ Equity (Deficit)
|
F5
|
|
Consolidated Statements of Cash Flows
|
F6 - F7
|
|
Notes to the Consolidated Financial Statements
|
F8 - F33
|
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Adamis Pharmaceuticals Corporation and Subsidiaries
Adamis Pharmaceuticals Corporation and Subsidiaries
Del Mar, California
We have audited the accompanying consolidated balance sheet of Adamis Pharmaceuticals Corporation and Subsidiaries as of March 31, 2010 and the related consolidated statements of operations, stockholders’ equity(deficit) and cash flows for the year then ended. These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the consolidated financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall consolidated financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Adamis Pharmaceuticals Corporation and Subsidiaries as of March 31, 2010, and the results of their operations and their cash flows for the year then ended in conformity with U.S. generally accepted accounting principles.
The accompanying consolidated financial statements have been prepared assuming the Company will continue as a going concern. As discussed in Note 14 to the consolidated financial statements, the Company has incurred recurring losses from operations and has limited working capital to pursue its business alternatives. These factors raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans with regard to these matters are also described in Note 14. The 2010 consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ Mayer Hoffman McCann P.C.
MAYER HOFFMAN MCCANN P.C.
Certified Public Accountants
Boca Raton, Florida
July 14, 2010
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Adamis Corporation and Subsidiaries
Adamis Corporation and Subsidiaries
Del Mar, California
We have audited the accompanying consolidated balance sheet of Adamis Pharmaceuticals Corporation and Subsidiaries as of March 31, 2009 and the related consolidated statements of operations, stockholders’ equity (deficit) and cash flows for the year then ended. These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall consolidated financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Adamis Pharmaceuticals Corporation and Subsidiaries as of March 31, 2009, and the results of their operations and their cash flows for the year then ended in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming the Company will continue as a going concern. As discussed in Note 14 to the consolidated financial statements, the Company has incurred recurring losses from operations and has limited working capital to pursue its business alternatives. These factors raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans with regard to these matters are also described in Note 14. The 2009 consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ Goldstein Lewin & Co.
GOLDSTEIN LEWIN & CO.
Certified Public Accountants
Boca Raton, Florida
July 30, 2009
F-2
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
|||
|
CONSOLIDATED BALANCE SHEETS
|
|
March 31,
|
||||||||
|
ASSETS
|
2010
|
2009
|
||||||
|
CURRENT ASSETS
|
||||||||
|
Cash
|
$ | 290,299 | $ | 17,697 | ||||
|
Accounts Receivable
|
5,555 | 136,283 | ||||||
|
Inventory, Net
|
2,709 | 195,167 | ||||||
|
Prepaid Expenses and Other Current Assets
|
13,004 | 4,087 | ||||||
|
Assets from Discontinued Operations
|
350,000 | 350,000 | ||||||
|
Total Current Assets
|
661,567 | 703,234 | ||||||
|
PROPERTY AND EQUIPMENT, Net
|
14,667 | 31,726 | ||||||
|
DEFERRED ACQUISITION COSTS
|
- | 147,747 | ||||||
|
Total Assets
|
$ | 676,234 | $ | 882,707 | ||||
|
LIABILITIES AND STOCKHOLDERS' EQUITY (DEFICIT)
|
||||||||
|
CURRENT LIABILITIES
|
||||||||
|
Accounts Payable
|
$ | 1,560,312 | $ | 972,522 | ||||
|
Accrued Other Expenses
|
205,981 | 98,778 | ||||||
|
Accrued Bonuses
|
1,401,821 | 625,118 | ||||||
|
Notes Payable
|
1,472,631 | - | ||||||
|
Notes Payable to Related Parties
|
309,565 | 599,765 | ||||||
|
Total Current Liabilities
|
4,950,310 | 2,296,183 | ||||||
|
COMMITMENTS AND CONTINGENCIES
|
||||||||
|
STOCKHOLDERS' EQUITY (DEFICIT)
|
||||||||
|
Preferred Stock – Par Value $.0001; 10,000,000 Shares Authorized; Issued and Outstanding-None
|
- | - | ||||||
|
Common Stock – Par Value $.0001; 175,000,000 Shares Authorized; 50,149,639 and 37,306,704 Issued, 49,047,953 and 36,990,704 Outstanding, Respectively
|
5,015 | 3,731 | ||||||
|
Additional Paid-in Capital
|
14,461,488 | 10,762,963 | ||||||
|
Accumulated Deficit
|
(18,739,477 | ) | (12,179,854 | ) | ||||
|
Treasury Stock - 1,101,686 and 316,000 Shares, Respectively
|
(1,102 | ) | (316 | ) | ||||
|
Total Stockholders' Equity (Deficit)
|
(4,274,076 | ) | (1,413,476 | ) | ||||
| $ | 676,234 | $ | 882,707 | |||||
F-3
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
|
CONSOLIDATED STATEMENTS OF OPERATIONS
|
|
Year Ended March 31,
|
|||||||||
|
2010
|
2009
|
||||||||
|
REVENUE
|
$ | 290,288 | $ | 659,538 | |||||
|
COST OF GOODS SOLD
|
264,599 | 262,008 | |||||||
|
Gross Margin
|
25,689 | 397,530 | |||||||
|
SELLING, GENERAL AND ADMINISTRATIVE EXPENSES
|
3,274,505 | 4,852,966 | |||||||
|
RESEARCH AND DEVELOPMENT
|
584,758 | 740,437 | |||||||
|
Loss from Operations
|
(3,833,574 | ) | (5,195,873 | ) | |||||
|
OTHER INCOME (EXPENSE)
|
|||||||||
|
Interest Expense
|
(2,726,049 | ) | (434,933 | ) | |||||
|
Gain on Fixed Asset Disposal
|
- | 5,766 | |||||||
|
Loss on Deposit
|
- | (21,871 | ) | ||||||
|
Total Other Income (Expense)
|
(2,726,049 | ) | (451,038 | ) | |||||
|
(Loss) from Continuing Operations
|
(6,559,623 | ) | (5,646,911 | ) | |||||
|
Income (Loss) from Discontinued Operations
|
- | 3,751,482 | |||||||
|
Net (Loss)
|
$ | (6,559,623 | ) | $ | (1,895,429 | ) | |||
|
Basic and Diluted (Loss) Income Per Share:
|
|||||||||
|
Continuing Operations
|
$ | (0.23 | ) | $ | (0.23 | ) | |||
|
Discontinued Operations
|
- | 0.16 | |||||||
|
Basic and Diluted (Loss) Per Share
|
$ | (0.23 | ) | $ | (0.07 | ) | |||
|
Basic and Diluted Weighted Average Shares Outstanding
|
28,837,700 | 24,886,573 | |||||||
F-4
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
|||||||||||||||
|
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY (DEFICIT)
|
|
Common Stock
|
Additional
|
Treasury Stock
|
Accumulated
|
|||||||||||||||||||||||||
|
Shares
|
Amount
|
Paid-In Capital
|
Shares
|
Amount
|
Deficit
|
Total
|
||||||||||||||||||||||
|
Balance March 31, 2008
|
35,390,129 | $ | 3,539 | $ | 8,788,417 | $ | - | $ | (10,284,425 | ) | $ | (1,492,469 | ) | |||||||||||||||
|
Issuance of Common Stock for Cash - $0.75 per share
|
1,339,651 | 134 | 1,004,604 | - | - | - | 1,004,738 | |||||||||||||||||||||
|
Unexcercised Beneficial Conversion Feature
|
- | - | (80,000 | ) | - | - | - | (80,000 | ) | |||||||||||||||||||
|
Issuance of Common Stock for Cash - $0.65 per share
|
76,924 | 8 | 49,992 | - | - | - | 50,000 | |||||||||||||||||||||
|
Issuance of Common Stock for Services
|
500,000 | 50 | 999,950 | - | - | - | 1,000,000 | |||||||||||||||||||||
|
Purchase of Treasury Stock
|
- | - | - | (316,000 | ) | (316 | ) | - | (316 | ) | ||||||||||||||||||
|
Net (Loss)
|
- | - | - | - | - | (1,895,429 | ) | (1,895,429 | ) | |||||||||||||||||||
|
Balance March 31, 2009
|
37,306,704 | 3,731 | 10,762,963 | (316,000 | ) | (316 | ) | (12,179,854 | ) | (1,413,476 | ) | |||||||||||||||||
|
Issuance of Common Stock for Merger with Cellegy Pharmaceutials
|
3,000,000 | 300 | (634,046 | ) | - | - | - | (633,746 | ) | |||||||||||||||||||
|
|
||||||||||||||||||||||||||||
|
Note Payable Converted to Equity
|
- | - | 777,902 | - | - | - | 777,902 | |||||||||||||||||||||
|
Issuance of Options to Employees
|
- | - | 29,918 | - | - | - | 29,918 | |||||||||||||||||||||
|
Release of Shares from Escrow
|
6,732,285 | 673 | (673 | ) | - | - | - | |||||||||||||||||||||
|
Purchase of Treasury Stock
|
- | - | - | (785,686 | ) | (786 | ) | (786 | ) | |||||||||||||||||||
|
Issuance of Common Stock for Employees and for Services
|
60,650 | 6 | 16,176 | - | - | - | 16,182 | |||||||||||||||||||||
|
Issuance of Common Stock for Payment of Payables
|
50,000 | 5 | 9,995 | - | - | - | 10,000 | |||||||||||||||||||||
|
Issuance of Common Stock for Cash at $1
|
2,000,000 | 200 | 1,800 | - | - | - | 2,000 | |||||||||||||||||||||
|
Discount on Notes Payable
|
- | - | 738,000 | - | - | - | 738,000 | |||||||||||||||||||||
|
Beneficial Conversion Features
|
- | - | 2,438,000 | - | - | - | 2,438,000 | |||||||||||||||||||||
|
Issuance of Warrants for Services
|
- | - | 69,300 | - | - | - | 69,300 | |||||||||||||||||||||
|
Issuance of Common Stock for Licensing Agreement
|
1,000,000 | 100 | 399,900 | - | - | - | 400,000 | |||||||||||||||||||||
|
Net (Loss)
|
- | - | - | - | - | (6,559,623 | ) | (6,559,623 | ) | |||||||||||||||||||
|
Balance March 31, 2010
|
50,149,639 | $ | 5,015 | $ | 14,609,235 | (1,101,686 | ) | $ | (1,102 | ) | $ | (18,739,477 | ) | $ | (4,274,076 | ) | ||||||||||||
F-5
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
||||||
|
CONSOLIDATED STATEMENTS OF CASH FLOWS
|
|
Year Ended March 31,
|
||||||||
|
CASH FLOWS FROM OPERATING ACTIVITIES
|
2010
|
2009
|
||||||
|
Net (Loss) from Continuing Operations
|
$ | (6,559,623 | ) | $ | (5,646,911 | ) | ||
|
Adjustments to Reconcile Net (Loss) from Continuing Operations to Net
|
||||||||
|
Cash (Used in) Operating Activities:
|
||||||||
|
Deferred Acquisition Cost Amortization
|
- | 320,000 | ||||||
|
Depreciation Expense
|
21,948 | 19,519 | ||||||
|
Amortization of Discounts
|
210,631 | - | ||||||
|
Gain on Fixed Asset Disposal
|
- | (5,766 | ) | |||||
|
Beneficial Conversion Feature
|
2,438,000 | (72,000 | ) | |||||
|
Inventory Reserve Adjustment
|
162,565 | (42,714 | ) | |||||
|
Issuance of Stock for Services
|
46,100 | 1,000,000 | ||||||
|
Issuance of Warrants for Services
|
63,900 | - | ||||||
| Issuance of Common Stock for licensing agreement | 400,000 | - | ||||||
|
Loss on Deposit
|
- | 21,871 | ||||||
|
Sales Returns Reserve Adjustment
|
56,398 | (120,712 | ) | |||||
|
Change in Assets and Liabilities:
|
||||||||
|
(Increase) Decrease in:
|
||||||||
|
Accounts Receivable
|
130,728 | (60,013 | ) | |||||
|
Inventory
|
29,893 | (128,190 | ) | |||||
|
Prepaid Expenses and Other Current Assets
|
17,446 | 140,134 | ||||||
|
Deferred Acquisition Costs
|
- | (46,500 | ) | |||||
|
Increase (Decrease) in:
|
370,666 | (18,623 | ) | |||||
|
Accounts Payable
|
(169,392 | ) | (160,491 | ) | ||||
| Accrued Other Expenses | 776,703 | 625,118 | ||||||
|
Accrued Bonuses
|
||||||||
|
Net Cash (Used in) Operating Activities from Continuing Operations
|
(1,998,637 | ) | (4,175,278 | ) | ||||
|
Net Cash (Used in) Operating Activities from Discontinued Operations
|
(811,960 | ) | ||||||
|
Net Cash (Used in) Operating Activities
|
(1,998,637 | ) | (4,987,238 | ) | ||||
|
CASH FLOWS FROM INVESTING ACTIVITIES
|
||||||||
|
Cash Acquired in Cellergy Pharmaceuticals Acquisition
|
65,114 | - | ||||||
|
Cash Received from Sale of International Laboratories, Inc.
|
- | 2,304,000 | ||||||
|
International Laboratories Corporation Obligation Repayments
|
- | 4,322,082 | ||||||
|
Sale of Property and Equipment
|
- | 8,501 | ||||||
|
Purchases of Property and Equipment
|
(4,889 | ) | - | |||||
|
|
||||||||
|
Net Cash Provided by Investing Activities from Continuing Operations
|
60,225 | 6,634,583 | ||||||
|
Net Cash (Used in) Investing Activities from Discontinued Operations
|
- | (862,122 | ) | |||||
|
Net Cash Provided by Investing Activities
|
60,225 | 5,772,461 | ||||||
|
CASH FLOWS FROM FINANCING ACTIVITIES
|
||||||||
|
Payments of Notes Payable to Related Parties
|
- | (1,752,316 | ) | |||||
|
Payments of Loans Payable
|
- | (2,000,000 | ) | |||||
|
Purchase of Treasury Stock
|
(786 | ) | - | |||||
|
Proceeds from Issuance of Common Stock
|
2,000 | 1,054,738 | ||||||
|
Proceeds from Issuance of Loans Payable
|
2,000,000 | - | ||||||
|
Proceeds from Issuance of Notes Payable to Related Parties
|
209,800 | 99,765 | ||||||
|
Net Cash Provided by (Used in) Financing Activities from Continuing Operations
|
2,211,014 | (2,597,813 | ) | |||||
|
Net Cash Provided by Financing Activities from Discontinued Operations
|
- | 1,829,746 | ||||||
|
Net Cash Provided by (Used in) Financing Activities
|
2,211,014 | (768,067 | ) | |||||
|
Increase in Cash
|
272,602 | 17,156 | ||||||
|
Cash:
|
||||||||
|
Beginning
|
17,697 | 541 | ||||||
|
Ending
|
$ | 290,299 | $ | 17,697 | ||||
F-6
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
||||||||
|
CONSOLIDATED STATEMENTS OF CASH FLOWS
|
||||||||
|
Year Ended
|
Year Ended
|
|||||||
|
March 31, 2010
|
March 31, 2009
|
|||||||
|
SUPPLEMENTAL DISCLOSURE OF CASH FLOW INFORMATION
|
||||||||
|
Cash Paid for Interest
|
$ | 50,232 | $ | 355,465 | ||||
|
SUPPLEMENTAL DISCLOSURE OF NON-CASH FINANCING AND INVESTING ACTIVITIES
|
||||||||
|
Accounts Payable Paid for in Common Stock
|
$ | 10,000 | $ | - | ||||
|
Issuance of Common Stock for Licensing Agreement
|
$ | 400,000 | $ | - | ||||
|
Release of shares of Common Stock from Escrow
|
$ | (673 | ) | $ | - | |||
|
Note Payable Converted to Equity
|
$ | 777,902 | $ | - | ||||
|
Warrants Issued for Services
|
$ | 69,300 | $ | - | ||||
|
Reduction of Capital from Unexercised Beneficial Conversion Feature
|
$ | - | $ | (80,000 | ) | |||
|
Increase in Capital from Beneficial Conversion Feature
|
$ | 2,438,000 | $ | - | ||||
|
Stock Issued as Discount on Note Payable
|
$ | 738,000 | $ | - | ||||
|
Stock Issued in Lieu of Services
|
$ | 46,100 | $ | 1,000,000 | ||||
F-7
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-8
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 1:
|
NATURE OF BUSINESS AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
|
Nature of Business
Adamis Pharmaceuticals Corporation and Subsidiaries (collectively “Adamis Pharmaceuticals”, the “Company”, “we”, “our”). is comprised of the following companies: Cellegy Holdings, Inc.; Adamis Corporation; and Biosyn, Inc. Adamis Corporation has two wholly-owned subsidiaries: Adamis Viral Therapies, Inc. (biotechnology), or Adamis Viral; and Adamis Laboratories, Inc. (specialty pharmaceuticals), or Adamis Labs. The Company’s strategic objective is to build a publicly-held company that combines the financial stability and sales force of a specialty pharmaceutical company with the near-term development of biopharmaceutical products.
Adamis Pharmaceuticals Corporation was established under the laws of the State of Delaware on June 6, 2006 and has devoted substantially all its efforts to establishing a new business. Adamis Pharmaceuticals merged with Cellegy Pharmaceuticals, Inc on April 1, 2009 (Note 2). Adamis Viral Therapies, Inc. was established under the laws of the State of Delaware on March 23, 2007, and was merged into Adamis Pharmaceuticals Corporation, the surviving entity, on March 30, 2007. The merged company changed its name to Adamis Viral Therapies, Inc. (“Viral”) on March 30, 2007. Viral had no activity during the years ended March 31, 2010 and 2009.
Adamis Holding Corporation was established under the laws of the State of Delaware on March 23, 2007. Adamis Holding Corporation changed its name to Adamis Pharmaceuticals Corporation on March 30, 2007. Viral transferred all of its authorized and outstanding shares of stock to Adamis Pharmaceuticals Corporation on March 30, 2007.
Adamis Laboratories, Inc. (formally known as HealthCare Ventures Group, Inc.) was established under the laws of the State of Delaware on September 2, 2005, and was acquired by the Company on April 23, 2007 (Note 2). On April 24, 2007, Healthcare Ventures Group, Inc. changed its name to Adamis Laboratories, Inc. (“Adamis Labs”). Adamis Labs is a distributor of respiratory products.
International Laboratories, Inc. (“INL”) was incorporated in the State of Florida in March 1981. INL’s operations consist of the packaging of prescription and non-prescription pharmaceutical and nutraceutical goods mainly for a major retailer (Notes 2 and 3).
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Accounting Standards Codification
In July 2009, the Financial Accounting Standards Board ("FASB") issued Statement of Financial Accounting Standards Number 168, "The FASB Accounting Standards Codification and the Hierarchy of Generally Accepted Accounting Principles", which identified the FASB's Accounting Standards Codification ("ASC") as the single source of authoritative nongovernmental U.S. generally accepted accounting principles ("GAAP"). The ASC reorganized the thousands of U.S. GAAP pronouncements into roughly 90 accounting topics and displays all topics using a consistent structure. It also includes relevant Securities and Exchange Commission ("SEC") guidance that follows the same topical structure in separate sections in the ASC. All other previously existing accounting standards documents were superseded by the ASC, which was effective for interim and annual periods ending after September 15, 2009.
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-9
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 1:
|
NATURE OF BUSINESS AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (CONTINUED)
|
Accounting Standards Codification (Continued)
All other accounting literature not included in the ASC is nonauthoritative. We believe that our adoption of this standard on its effective date (September 15, 2009) did not have a material effect on our consolidated financial statements.
Principles of Consolidation
The accompanying consolidated financial statements include Adamis Pharmaceuticals and its wholly-owned operating subsidiaries. All significant intra-entity balances and transactions have been eliminated in consolidation.
Accounting Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make certain estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements. Actual results could differ from those estimates, and the differences could be material.
Long-Lived Assets
The Company periodically assesses whether there has been permanent impairment of its long-lived assets held and used whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by comparison of the carrying amount of the asset to future net undiscounted cash flows expected to be generated from the use and eventual disposition of the asset.
Discontinued Operations
As discussed in Note 3, the results of operations for the year ended March 31, 2009, and the assets and liabilities at March 31, 2010 and 2009, related to INL have been accounted for as discontinued operations. There were no operations or related assets and liabilities of INL in the accompanying consolidated financial statements of prior periods.
Cash and Cash Equivalents
For purposes of the consolidated statements of cash flows, the Company considers all highly liquid investments with original maturities at the date of purchase of three months or less to be cash equivalents.
Income Taxes
The Company accounts for income taxes under the deferred income tax method. Under this method deferred income taxes are determined based on the estimated future tax effects of differences between the financial statement and tax basis of assets and liabilities given the provisions of enacted tax laws.
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-10
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 1:
|
NATURE OF BUSINESS AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (CONTINUED)
|
Income Taxes (Continued)
Deferred income tax provisions and benefits are based on changes to the assets of liabilities from year to year. In providing for deferred taxes, the Company considers tax regulations of the jurisdictions in which they operate, estimates of future taxable income, and available tax planning strategies. If tax regulations, operating results or the ability to implement tax planning strategies vary, adjustments to the carrying value of deferred tax assets and liabilities may be required. Valuation allowances are recorded related to deferred tax assets based on the "more likely than not" criteria.
The Company accounts for uncertain tax positions in accordance with accounting guidance which requires the Company to recognize the financial statement benefit of a tax position only after determining that the relevant tax authority would, more likely than not, sustain the position following an audit. For tax positions meeting the more likely than not threshold, the amount recognized in the financial statements is the largest benefit that has a greater than 50 percent likelihood of being realized upon ultimate settlement with the relevant tax authority. The Company at March 31, 2010 and 2009 did not recognize any additional liabilities for unrecognized tax benefits.
The Company is subject to income taxes in the United States Federal jurisdiction, California and Florida. The Company is potentially subject to the United States Federal California or Florida income examinations by tax authorities for all years. The Company recognizes interest and penalty accrued related to unrecognized tax benefits in its income tax expense, if any. No interest or penalties have been accrued for all presented periods
Revenue Recognition
Our primary customers are pharmaceutical wholesalers. In accordance with our revenue recognition policy, revenue is recognized when title and risk of loss are transferred to the customer, the sales price to the customer is fixed and determinable, and collectability of the sales price is reasonably assured. Reported revenue is net of estimated customer returns and other wholesaler fees. Our policy regarding sales to customers is that we do not recognize revenue from, or the cost of, such sales, where we believe the customer has more than a demonstrably reasonable level of inventory. We make this assessment based on historical demand, historical customer ordering patterns for purchases, business considerations for customer purchases and estimated inventory levels. If our actual experience proves to be different than our assumptions, we would then adjust such allowances accordingly.
We estimate allowances for revenue dilution items using a combination of information received from third parties, including market data, inventory reports from our major U.S. wholesaler customers, when available, historical information and analysis that we perform. The key assumptions used to arrive at our best estimate of revenue dilution reserves are estimated customer inventory levels and purchase forecasts provided. Our estimates of inventory at wholesaler customers and in the distribution channels are subject to the inherent limitations of estimates that rely on third-party data, as certain third-party information may itself rely on estimates, and reflect other limitations. We believe that such provisions are reasonably ascertainable due to the limited number of assumptions involved and the consistency of historical experience.
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-11
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 1:
|
NATURE OF BUSINESS AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (CONTINUED)
|
Fair Value of Financial Instruments
The carrying amounts of the Company's financial instruments, including cash, accounts receivable, accounts payable and accrued liabilities approximate their fair value due to their short-term nature. The Company’s notes payable approximate fair value based upon current rates available to the Company for loans with similar maturities.
Inventory
Inventory, consisting of allergy products, respiratory products, and epi inventory is recorded at the lower of cost or market, using the weighted average method.
Property and Equipment
Property and equipment are recorded at cost less accumulated depreciation. Depreciation is computed using the straight-line method over the estimated useful lives of the related assets.
Estimated useful lives used to depreciate property and equipment are as follows:
|
Estimated Useful Lives In Years
|
|
|
Office Furniture and Equipment
|
7
|
|
Computer Equipment and Software
|
3
|
|
Vehicles
|
3
|
Accounts Receivable, Allowance for Doubtful Accounts and Sales Returns
Trade accounts receivable are stated net of an allowance for doubtful accounts. Interest is not charged on outstanding balances. Accounts receivable are uncollateralized customer obligations due under normal trade terms requiring payment typically between 30 and 75 days from the invoice date. The Company estimates an allowance based on its historical experience of the relationship between actual bad debts and net credit sales. At March 31, 2010 and March 31, 2009, no allowance for doubtful accounts was recorded.
The Company has established an allowance for sales returns based on management’s best estimate of probable loss inherent in the accounts receivable balance. Management determines the allowance based on current credit conditions, historical experience, and other currently available information. The allowance for sales returns was $75,899 and $19,501 at March 31, 2010 and March 31, 2009, respectively, and is included in accrued expenses on the consolidated balance sheet.
Registration Payment Arrangements
Contingent obligations to make future payments under a registration payment arrangement are separately recognized and measured. At March 31, 2010 and March 31, 2009 the Company had no accrued estimated penalty. (Note 8)
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-12
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 1:
|
NATURE OF BUSINESS AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (CONTINUED)
|
Research and Development
Research and development costs are expensed as incurred. Non-refundable advance payments for goods and services to be used in future research and development activities are to be recorded as an asset and expensed when the research and development activities are performed.
Research and development expense, which primarily consist of salaries, contractor fees, facility and building costs, utilities, administrative expenses and other corporate costs, were $584,758 and $740,437 for the years ended March 31, 2010 and 2009, respectively. Expenses related to outside contractors for development of the Epinephrine Syringe (“epi”) during the years ended March 31, 2010 and 2009 were $144,858 and $135,634, respectively. Non-refundable advanced payments used and expensed during the years ended March 31, 2010 and 2009 were $0 and $225,459, respectively.
Expenses related to outside contractors for the development of influenza technology were $39,900 and $379,344 for the years ended March 31, 2010 and 2009, respectively.
Shipping and Handling
Shipping and handling costs are included in selling, general and administrative expenses. Shipping and handling costs were $13,722 and $13,651 for the years ended March 31, 2010 and 2009, respectively.
Advertising Expenses
Advertising costs are expensed as incurred. Advertising expenses were $1,853 and $2,848 during each of the years ended March 31, 2010 and 2009 respectively.
Goodwill and Intangible Assets
Intangible assets include intellectual property and other patent rights acquired. Consideration paid in connection with acquisitions is required to be allocated to the acquired assets, including identifiable intangible assets, and liabilities acquired. Acquired assets and liabilities are recorded based on Adamis’ estimate of fair value, which requires significant judgment with respect to future cash flows and discount rates. For intangible assets other than goodwill, Adamis is required to estimate the useful life of the asset and recognize its cost as an expense over the useful life. Adamis uses the straight-line method to expense long-lived assets (including identifiable intangibles). In accordance with GAAP, goodwill and other intangible assets with indefinite lives are assessed for impairment by applying a fair-value based test. This test is generally performed each year in the fourth fiscal quarter. Additionally, goodwill and intangible assets are reviewed for impairment whenever events or circumstances indicate that the carrying amount of the asset may not be recoverable. An impairment loss would be recognized based on the difference between the carrying value of the asset and its estimated fair value, which would be determined based on either discounted future cash flows or other appropriate fair value methods. The evaluation of goodwill and other intangibles for impairment requires management to use significant judgments and estimates including, but not limited to, projected future revenue, operating results and cash flows. An impairment would require Adamis to charge to earnings the write-down in value of such assets.
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-13
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 1:
|
NATURE OF BUSINESS AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (CONTINUED)
|
Net Loss Per Share
The Company computes basic loss per share by dividing the loss attributable to holders of common stock for the period by the weighted average number of shares of common stock outstanding during the period. Since the effect of common stock equivalents was anti-dilutive, all such equivalents were excluded from the calculation of weighted average shares outstanding. Outstanding warrants at March 31, 2010 and 2009 were 1,922,139 and 1,000,000. The outstanding options at March 31, 2010 and 2009 were 463,438 and 134,275, respectively.
In addition, the potential dilutive effects of the following convertible securities at March 31, 2010 have been excluded from the calculation of weighted average shares outstanding: (i) $2,000,000 of convertible notes which in the aggregate could potentially convert into up to 2,000,000 shares of common stock (ii) 1,000,000 restricted common shares for the purchase of intangible assets (iii) 6,732,295 of common shares released from escrow in conjunction with the Cellegy merger (Note 2) and (iv) 10,097,416 of additional common shares with various restrictions.
Recent Accounting Pronouncements
Fair Value Measurements and Disclosures require the use of fair value measures in financial statements, establish a framework for measuring fair value and expand disclosure related to the use of fair value measures. In February 2008, the FASB provided a one-year deferral of the effective date of those concepts for non-financial assets and non-financial liabilities, except those that are recognized or disclosed in the financial statements at fair value at least annually. The application of these concepts was effective for our fiscal year beginning April 1, 2009, excluding the effect of the one-year deferral noted above. See “Fair Value Measurements” above. We are currently evaluating the impact of adopting these concepts with respect to non-financial assets and non-financial liabilities on our consolidated financial statements, which will be effective beginning April 1, 2010.
We may elect to report most financial instruments and certain other items at fair value on an instrument-by-instrument basis with changes in fair value reported in earnings. After the initial adoption, the election is made at the acquisition of an eligible financial asset, financial liability, or firm commitment or when certain specified reconsideration events occur. The fair value election may not be revoked once an election is made. The application of these concepts was effective for our fiscal year beginning April 1, 2008 — however; we have elected not to measure eligible financial assets and liabilities at fair value. Accordingly, the adoption of these concepts did not have a significant impact on our consolidated financial statements.
Business Combinations require an acquirer to recognize and measure in its financial statements the identifiable assets acquired, the liabilities assumed, and any noncontrolling interest in an acquiree, including the recognition and measurement of goodwill acquired in a business combination. The provisions of this guidance became effective for our fiscal year beginning April 1, 2009. Under these provisions, we would have recorded the $147,747 of deferred acquisition costs included in other non-current assets on our March 31, 2009 balance sheet as expense during the year then ended. This amount was recorded as as a reduction of additional paid-in capital.
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-14
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 1:
|
NATURE OF BUSINESS AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (CONTINUED)
|
Recent Accounting Pronouncements (Continued)
In May 2009, the FASB established general standards of accounting for and disclosure of events that occur after the balance sheet date but before the financial statements are issued. It sets forth the period after the balance sheet date during which management of a reporting entity should evaluate events or transactions that occur for potential recognition or disclosure in the financial statements, the circumstances under which an entity should recognize events or transactions occurring after the balance sheet date in its financial statements and the disclosures that an entity should make about events or transactions that occurred after the balance sheet date and up to the date the financial statements are issued. ASC 855 was effective for financial statements issued for interim and annual periods ending after June 15, 2009 and did not have any impact on the Company’s financial statements.
|
NOTE 2:
|
MERGERS AND ACQUISITIONS
|
Cellegy Merger
The stockholders of Cellegy Pharmaceuticals, Inc. (“Old Cellegy”) and the former Adamis Pharmaceuticals Corporation (“Old Adamis”) approved a merger transaction and related matters at an annual meeting of Old Cellegy’s stockholders and at a special meeting of Old Adamis’ stockholders each eld on March 23, 2009. On April 1, 2009, Old Cellegy completed the merger transaction with Old Adamis. In connection with the closing of the merger transaction, a $500,000 promissory note issued by Old Cellegy to Old Adamis reflecting a loan made by Old Adamis to Old Cellegy in connection with the merger transaction was converted into shares of Old Adamis stock, and these shares were immediately cancelled.
In connection with the consummation of the merger and pursuant to the terms of the definitive merger agreement relating to the transaction (the “Cellegy Merger Agreement”), Old Cellegy changed its name from Cellegy Pharmaceuticals, Inc. to Adamis Pharmaceuticals Corporation (“Adamis” or the “Company”), and Old Adamis changed its corporate name to Adamis Corporation.
Pursuant to the terms of the Cellegy Merger Agreement, immediately before the consummation of the merger Old Cellegy effected a reverse stock split of its common stock. Pursuant to this reverse stock split, each 9.929060333 shares of common stock of Old Cellegy that were issued and outstanding immediately before the effective time of the merger were converted into one share of common stock and any remaining fractional shares held by a stockholder (after the aggregating fractional shares) were rounded up to the nearest whole share (the “Reverse Split”).
As a result, the total number of shares of Old Cellegy that were outstanding immediately before the effective time of the merger were converted into approximately 3,000,000 shares of post-Reverse Split shares of common stock of the Company. Pursuant to the terms of the Cellegy Merger Agreement, at the effective time of the merger, each share of Adamis common stock that was issued and outstanding immediately before the effective time of the merger ceased to be outstanding and was converted into the right to receive one share of common stock of the Company. As a result, the Company issued approximately 43,772,989 post-Reverse Split common stock, inclusive of 7,451,304 contingent shares held in escrow, which were issuable to the holders of the outstanding shares of common stock of Old
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-15
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 2:
|
MERGERS AND ACQUISITIONS (CONTINUED)
|
Cellegy Merger (Continued)
Adamis before the effective time of the merger. Old Adamis was the surviving entity and is a wholly-owned subsidiary of the Company.
Old Adamis security holders owned, immediately after the closing of the merger, approximately 93.5% of the combined company on a fully-diluted basis. Further, Old Adamis directors constitute a majority of the combined company’s board of directors and all members of executive management of the combined company were from old Adamis. Therefore, Old Adamis was deemed to be the acquiring company for accounting purposes and the merger transaction is accounted for as an asset acquisition recapitalization in accordance with accounting principles generally accepted in the United States. As a result, all of the assets and liabilities of Old Cellegy have been reflected in the financial statements at their respective fair market values and no goodwill or other intangibles were recorded as part of acquisition accounting and the cost of the merger is measured at the net liabilities acquired. Transaction costs amounting to $147,747 were considered as part of the assets acquired and included as a reduction of additional paid-in capital. The financial statements of the combined entity after the merger reflect the historical results of Old Adamis prior to the merger and do not include the historical financial results of Old Cellegy prior to the completion of the merger. Stockholders’ equity and earnings per share of the combined entity after the merger have been retroactively restated to include the number of shares received by Old Adamis security holders in the merger with the offset to additional paid-in capital.
In connection with the closing of the merger, the Company amended its certificate of incorporation to increase the authorized number of shares of common stock from 50,000,000 to 175,000,000 and the authorized number of shares of preferred stock from 5,000,000 to 10,000,000.
The assets acquired and liabilities assumed of Old Cellegy at April 1, 2009 are summarized as follows:
|
Current Assets
|
$ | 91,000 | ||
|
Current Liabilities
|
$ | 504,000 | ||
|
Notes Payable Long-Term
|
$ | 778,000 |
The operations of Old Cellegy prior to the merger is not considered significant to an understanding of the operations of the combined entities.
The cost of the acquisition to Adamis is equal to Cellegy’s stockholders’ deficit, which was approximately $1,191,000.
LaJolla Pharmaceutical Merger
On December 4, 2009, Adamis entered into an Agreement and Plan of Reorganization with La Jolla Pharmaceutical Company, or La Jolla, a public pharmaceuticals company, providing for the acquisition of La Jolla by Adamis. On March 3, 2010, Adamis and La Jolla agreed to terminate the Agreement and Plan of Reorganization. The termination followed the announcement by La Jolla that its common stock would be suspended and delisted from the NASDAQ stock market and that holders of only 13% of La Jolla’s outstanding common stock returned their proxy cards or otherwise indicated their votes with respect to proposals related to the proposed merger transaction and that as a result the La Jolla stockholder meeting and the solicitation of further votes had been cancelled.
Acquisition of HealthCare Ventures Group, Inc.
On April 23, 2007, the Company acquired all of the outstanding shares of HealthCare Ventures Group, Inc. in exchange for 5,159,807 shares of common stock valued at $0.50 per share, or approximately $2.6 million (the “HVG Acquisition”). The purchase agreement provides for an additional 7,451,304 restricted shares held in escrow with issuance conditional upon future earnings targets, 719,019 of which were subsequently cancelled. The acquired company’s name was changed to Adamis Labs.
The HVG Acquisition was accounted for as a business combination using the purchase method of accounting. Accordingly, the results of Adamis Labs’s operations have been included in the unaudited consolidated financial statements from the date of acquisition. The allocation of consideration for
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-16
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 2:
|
MERGERS AND ACQUISITIONS (CONTINUED)
|
Acquisition of HealthCare Ventures Group, Inc. (Continued
acquisitions requires extensive use of accounting estimates and management’s judgment to allocate the purchase price of tangible and identifiable intangible assets acquired and liabilities assumed based on their respective fair values. Management believes fair values assigned to the assets acquired and liabilities assumed are based on reasonable estimates and assumptions.
The purchase price for the HVG Acquisition was allocated to tangible and identifiable intangible assets acquired and liabilities assumed based on their estimated fair values at the acquisition date, with the excess being allocated to intangible assets, as follows:
|
Cash
|
$ | 12,611 | ||
|
Accounts Receivable
|
138,218 | |||
|
Inventory
|
37,373 | |||
|
Prepaid and Other Current Assets
|
71,915 | |||
|
Property
|
69,645 | |||
|
Other Assets
|
22,442 | |||
|
Intangible Assets
|
3,150,985 | |||
|
Accounts Payable
|
(148,657 | ) | ||
|
Accrued Liabilities
|
(191,611 | ) | ||
|
Interest Payable
|
(33,017 | ) | ||
|
Loan Payable
|
(550,000 | ) | ||
|
Net Assets Acquired
|
$ | 2,579,904 |
Acquisition of International Laboratories, Inc.
On December 31, 2007, the Company acquired all of the outstanding shares of INL in an all stock transaction for 2,000,000 shares of common stock valued at $0.50 per share, or $1.0 million (the “INL Acquisition”). The purchase agreement provided for an additional 8,000,000 restricted shares to be held in escrow with issuance conditional upon future earnings targets (Note 3).
The INL Acquisition was accounted for as a business combination using the purchase method of accounting. Accordingly, the results of International Laboratories, Inc.’s operations have been included
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-17
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 2:
|
MERGERS AND ACQUISITIONS (CONTINUED
|
Acquisition of International Laboratories, Inc. (Continued)
in the consolidated financial statements beginning December 31, 2007. The allocation of consideration for acquisitions requires extensive use of accounting estimates and management judgment to allocate the purchase price of tangible and identifiable intangible assets acquired and liabilities assumed based on their respective fair values. Management believes the fair values assigned to the assets acquired and liabilities assumed are based on reasonable estimates and assumptions.
The purchase price for the INL Acquisition was allocated to tangible and identifiable intangible assets acquired and liabilities assumed based on their estimated fair values at the acquisition date, with the excess being allocated to intangible assets, as Management deems a significant customer agreement with a finite term as the value purchased. The customer agreement was with a major retailer, and without such agreement Adamis would not have acquired INL. The length of the agreement at the time of INL’s purchase was in excess of two and one-half years.
The purchase price was allocated as follows:
|
Cash and Cash Equivalents
|
$ | 219,321 | ||
|
Accounts Receivable
|
707,101 | |||
|
Prepaid Expenses and Other Current Assets
|
29,155 | |||
|
Inventory
|
305,723 | |||
|
Property
|
434,935 | |||
|
Intangible Asset Acquired
|
6,328,704 | |||
|
Accounts Payable
|
(1,757,312 | ) | ||
|
Accrued Liabilities
|
(282,307 | ) | ||
|
Deferred Revenue
|
(114,437 | ) | ||
|
Current Portion of Long-Term Debt
|
(151,930 | ) | ||
|
Long-Term Debt
|
(4,718,953 | ) | ||
|
Net Assets Acquired
|
$ | 1,000,000 |
The customer agreement valued as the acquired intangible asset had a remaining term of approximately 2.5 years at the acquisition date. Amortization of the intangible asset recorded during the year ended March 31, 2009 was $759,444.
On July 18, 2008, INL was sold. Accordingly, the assets, liabilities, and results from operations are classified as discontinued operations in the consolidated financial statements (Note 3).
|
NOTE 3:
|
DISCONTINUED OPERATIONS
|
Effective July 18, 2008, the Company’s packaging division (INL) (Note 2) was sold for $2,654,000. On the closing date, $2,154,000 was paid to a lender to retire long-term debt. Additionally, $500,000 of the purchase price was held in escrow to secure any of the Company’s indemnification obligations. During 2010 the Company settled $150,000 of the amount held for indemnification obligations. In addition, INL repaid loans and accrued interest of $4,630,813 to Adamis Pharmaceuticals; however, the Company forgave $570,618 of outstanding loans to INL. The 8,000,000 shares of common stock held in escrow in connection with the Company’s purchase of INL were released and cancelled in conjunction with the sale agreement.
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-18
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 3:
|
DISCONTINUED OPERATIONS (CONTINUED)
|
The following table presents information regarding the calculation of the gain from the sale of INL:
|
Sale Price - Imperium note payment
|
$ | 2,154,000 | ||
|
Sale Price - Cash held in Escrow
|
350,000 | |||
|
Total Sale Price
|
2,504,000 | |||
|
INL Assets
|
9,615,763 | |||
|
INL Liabilities
|
(13,470,365 | ) | ||
|
Net Liabilities
|
(3,854,602 | ) | ||
|
Amount of inter-company loan not paid by Buyer
|
570,618 | |||
|
Total Basis
|
(3,283,984 | ) | ||
|
Gain on Sale
|
$ | 5,787,984 |
Operating loss from INL from the acquisition date through disposal, excluding the gain on sale, was $4,580,613.
Total loss from discontinued operations for the year ended March 31, 2009 was $3,751,482.
At March 31, 2010 and 2009, assets from discontinued operations consisted of $350,000 held in escrow.
|
NOTE 4:
|
CONCENTRATIONS OF CREDIT RISK
|
Financial instruments that potentially subject the Company to credit risk consist principally of cash, accounts receivable, purchases and accounts payable.
Cash
The Company at times may have cash in excess of the Federal Deposit Insurance Corporation (“FDIC”) limit. The Company maintains its cash with larger financial institutions. The Company has not experienced losses on these accounts and management believes that the Company is not exposed to significant risks on such accounts.
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-19
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 4:
|
CONCENTRATIONS OF CREDIT RISK (CONTINUED)
|
Sales and Accounts Receivable
The Company is dependent on a limited number of customers for a significant portion of its revenue. During the year ended March 31, 2010, the Company's two largest customers, Customers A and B accounted for approximately 44% and 31%, respectively, of the Company's net sales. During the year ended March 31, 2009, these two customers accounted for approximately 37% and 19% respectively, of the Company's net sales.
The Company grants credit to customers, substantially all of whom are pharmaceutical distribution and medical parties located throughout the United States. The Company typically does not require collateral from customers. The Company monitors exposure to credit losses and maintains allowance for anticipated losses considered necessary under the circumstances.
Accounts receivable from one Customer amounted to approximately 72% of total accounts receivable at March 31, 2009.
Trade accounts receivable were $5,555 and $124,755 at March 31, 2010 and 2009 respectively.
Trade accounts receivable do not include factor receivable of $0 and $11,528 at March 31, 2010 and 2009 respectively.
Purchases and Trade Accounts Payable
The Company had balances greater than 10% of trade accounts payable at March 31, 2010 with two vendors. Vendor A had a balance that accounted for 34% of total accounts payables and Vendor B had a balance of 20% at March 31, 2010. The company had no outstanding balance greater than 10% of trade accounts payable at March 31, 2009.
The Company is dependant on a limited number of vendors for a significant portion of its trade purchases. The Company had one vendor that comprised 47 % and 98% of the total trade purchases made during the years ended March 31, 2010 and 2009, respectively.
|
NOTE 5:
|
INVENTORY
|
Inventory consists of the following at March 31, 2010 and 2009, respectively:
|
2010
|
2009
|
|||||||
|
Respiratory and Allergy Products
|
$ | 226,710 | $ | 52,843 | ||||
|
Less: Obsolescence Reserve
|
(224,001 | ) | (37,175 | ) | ||||
|
Respiratory and Allergy Products, Net
|
2,709 | 15,668 | ||||||
|
Pre-Launch epi Inventory
|
- | 179,499 | ||||||
|
Inventory, Net
|
$ | 2,709 | $ | 195,167 | ||||
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-20
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 6:
|
PREPAID EXPENSES AND OTHER CURRENT ASSETS
|
Prepaid expenses and other current assets at March 31, 2010 and 2009:
|
2010
|
2009
|
|||||||
|
Prepaid Insurance
|
$ | 12,369 | $ | 3,452 | ||||
|
Prepaid Rent
|
635 | 635 | ||||||
| $ | 13,004 | $ | 4,087 | |||||
|
NOTE 7:
|
PROPERTY AND EQUIPMENT
|
Property and Equipment consists of the following at March 31, 2010 and 2009:
|
2010
|
2009
|
|||||||
|
Office Furniture and Equipment
|
$ | 128,883 | $ | 128,738 | ||||
|
Computer Equipment
|
22,707 | 22,707 | ||||||
|
Computer Software
|
64,382 | 59,639 | ||||||
| 215,972 | 211,084 | |||||||
|
Less: Accumulated Depreciation
|
(201,305 | ) | (179,358 | ) | ||||
| $ | 14,667 | $ | 31,726 | |||||
|
NOTE 8:
|
NOTES PAYABLE
|
Ben Franklin Note
Biosyn (a wholly owned subsidiary of Old Cellegy) issued a note payable to Ben Franklin Technology Center of Southeastern Pennsylvania (“Ben Franklin Note”) in October 1992, in connection with funding the development of Savvy, a compound to prevent the transmission of AIDS.
The Ben Franklin Note was recorded at its estimated fair value of $205,000 and was assumed by Old Cellegy as an obligation in connection with its acquisition of Biosyn in 2004. The repayment terms of the non-interest bearing obligation include the remittance of an annual fixed percentage of 3.0% applied to future revenues of Biosyn, if any, until the principal balance of $777,902 (face amount) is satisfied. Under the terms of the obligation, revenues are defined to exclude the value of unrestricted research and development funding received by Biosyn from nonprofit sources. Absent a material breach of contract or other event of default, there is no obligation to repay the amounts in the absence of future Biosyn revenues. Cellegy accreted the discount of $572,902 against earnings using the interest rate method (approximately 46%) over the discount period of five years, which was estimated in connection with the Ben Franklin Note’s valuation at the time of the acquisition.
Accounting principles generally accepted in the United States emphasize market-based measurement through the use of valuation techniques that maximize the use of observable or market-based inputs. The
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-21
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 8:
|
NOTES PAYABLE (CONTINUED)
|
Ben Franklin Note’s peculiar repayment terms outlined above affects its comparability with main stream market issues and also affects its transferability. The value of the Ben Franklin Note would also be impacted by the ability to estimate Biosyn’s expected future revenues which in turn hinge largely upon the outcome of its ongoing Savvy contraception trial, the results of which are currently under review and which are not known by the Company. Given the above factors and therefore the lack of market comparability, the Ben Franklin Note would be valued based on Level 3 inputs. As such, management has determined that the Ben Franklin Note will have no future cash flows, as we do not believe the product will create a revenue stream in the future. As a result, the Note had no fair market value at the time of the acquisition (Note 2).
G-Max Trust Note
On December 29, 2009, the Company issued a Convertible Promissory Note (the “G-Max Note”) to The G-Max Trust (the “Investor”) in connection with a private placement to the Investor for gross proceeds of $500,000, and 500,000 shares of common stock of the Company at par value for gross proceeds of $500 as an inducement to enter into the agreement. The market value of the common stock on the date issued was $0.25 per share, for a total value of $125,000. A discount on the note payable of $124,500 was recorded as a result, and is being amortized over the term of the G-Max Note. The stock is restricted for six months from the date issued. Subsequent to the six months, the investors can sell and have the restrictions removed under SEC Rule 144. Amortization of the discount, which is included in interest expense was $31,125 for the year ended March 31, 2010. As of March 31, 2010, the net carrying amount was $406,625 and the net unamortized discount was $93,375. The interest recognized in the contractual interest coupon was $12,917.
Interest on the outstanding principal balance of the G-Max Note accrues at a rate of 10% per annum compounded monthly and is payable monthly commencing February 1, 2010. All unpaid principal and interest on the G-Max Note is due and payable on December 31, 2010 (the “Maturity Date”).
At any time on or before the Maturity Date, the Investor has the right to convert part or all of the principal and interest owed under the G-Max Note into common stock at a conversion price equal to $0.20 per share (subject to adjustment for stock dividends, stock splits, reverse stock splits, reclassifications or other similar events affecting the number of outstanding shares of common stock). The conversion feature is considered beneficial to the Investor due to the purchase of the discounted shares. The estimated value of the beneficial conversion feature was $249,500. The entire amount was recorded as interest expense upon issuance as the G-Max Note is convertible at any time.
The effective annual interest rate of the G-Max Note is 84.8% after considering the discount and beneficial conversion feature.
Events of default under the Note include: (a) the Company fails to make payment of the principal amount of the G-Max Note when due and fails to cure the default within the permitted cure period; or (b) the Company fails in any material respect to comply with or to perform when due any other material term, obligation, covenant, or condition contained in the Note and fails to cure the default within the permitted cured period. In the event of a default by the Company, the Investor must provide the Company with written notice of default, and the Company will have five business days to cure the default. Upon an event of default that is not cured, the Investor may declare the entire unpaid amount owed under the G-Max Note immediately due, subject to the subordination provisions set forth in the G-Max Note. Upon the failure to pay the principal amount owed under the G-Max Note upon the final maturity date, the Investor, at its option, may charge default interest on the G-Max Note at a rate equal to the lesser of (i) 18% per annum and (ii) the maximum rate permitted under applicable usury or other laws.
The G-Max Note includes piggyback registration rights providing that at any time after one year after the date of the G-Max Note, if the Shares and the shares of common stock issuable upon conversion of the G-Max Note (together with the Shares, the “Transaction Shares”) cannot be sold without restriction pursuant to SEC Rule 144, then if the Company files a registration statement pursuant to the Securities Act of 1933, as amended (the “Act”) at any time on or before December 29, 2010, relating to an offering for the
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-22
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 8:
|
NOTES PAYABLE (CONTINUED)
|
G-Max Trust Note (Continued)
account of others under the Act of any of its equity securities (other than on Form S-4 or Form S-8 (each as promulgated under the Act) or their then equivalents), then the Company will promptly notify the Investor and will include in such registration and any related qualification under blue sky laws or other compliance, and in any underwriting involved therein, all Transaction Shares specified by the Investor. The Company will pay the registration fees relating to the inclusion of the Transaction Shares in the registration statement.
The G-Max Note includes subordination provisions providing that payment of principal, interest and any other amounts that may become due pursuant to the Note, and any other obligation that the Company may have to the Investor (“Subordinated Indebtedness”), is subordinated to the payment in full of all “Senior Indebtedness” of the Company, which is defined as any obligations of the Company outstanding on the date of the Note or created thereafter pursuant to any secured note of the Company and any agreements relating thereto, and that as between the Investor and any holder of Senior Indebtedness (a “Senior Lender”) the Senior Lender will hold a first priority lien in all collateral relating to the Senior Indebtedness. Until all of the Senior Indebtedness has been paid in full and the Senior Lender has released its lien in the collateral, the Investor may not, without the Senior Lender’s prior written consent, demand, receive or accept any payment, other than current interest payments, from the Company in respect of the Subordinated Indebtedness, or exercise any right of or permit any setoff in respect of the Subordinated Indebtedness. The Note includes other customary subordination provisions, including provisions subordinating the Subordinated Indebtedness to any Senior Indebtedness in the event of bankruptcy or similar proceedings or events. In addition, if an event of default occurs with respect to any Senior Indebtedness permitting the holder to accelerate the maturity thereof, then, unless the event of default has been cured or waived or has ceased to exist, or all Senior Indebtedness has been paid in full, no payment may be made in respect of the Note for a period of 180 days after the first occurrence of such event of default.
Gemini Master Fund, Ltd. Notes
The Company has completed the closing of a private placement financing transaction (the “January 2010 Financing”) with a small number of institutional investors led by Gemini Master Fund, Ltd., pursuant to a Securities Purchase Agreement. The Company issued 10% Senior Secured Convertible Notes (the “Notes”) in the aggregate principal amount of approximately $1.5 million and 1,500,000 shares of common stock (sold at par value) of the Company, and received gross proceeds of $1.5 million, excluding transaction costs and expenses. The fair market value of the Company's common stock on the date of the transaction was $ 0.41 per share. A discount of approximately $600,000 was calculated as a result, and is being amortized over the life of the Notes. The stock is restricted for six months from the date issued. Amortization of the discount, which is included in interest expense, was $179,506 for the year ended March 31, 2010. As of March 31, 2010, the net carrying amount was $1,066,006 and the net amortized discount was $433,994. Interest recognized on the contractual coupon was $32,500.
Interest on the Notes is payable at a rate of 10% per annum and is payable monthly on the first business day of each month. Principal and any accrued and unpaid interest is due and payable nine months after the date of the Notes. The Notes are convertible into shares of the Company’s common stock at any time at the discretion of the investor at an initial conversion price per share of $0.20, subject to adjustment for
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-23
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 8:
|
NOTES PAYABLE (CONTINUED)
|
Gemini Master Fund, Ltd. Notes (Continued)
stock splits, stock dividends and other similar transactions and subject to the terms of the Notes. The conversion price is also subject to price anti-dilution adjustments providing that if the Company issues equity securities or securities convertible into equity securities at an effective price per share below the conversion price of the Notes (subject to certain exceptions), the conversion price of the Notes will be adjusted downward to equal the price of the new securities. The conversion feature is considered beneficial to the investors due to the purchase of the discounted shares. The estimated value of the beneficial conversion feature is approximately $2.2 million. The entire amount was recorded as interest expense upon issuance since the Notes are convertible at any time.
The effective interest rate of the Notes is 210.4% after considering the discount and beneficial conversion feature.
The Company’s obligations under the Notes and the other transaction agreements are guaranteed by the Company’s principal subsidiaries, including Adamis Corporation, Adamis Laboratories, Inc. and Adamis Viral, Inc., and are collateralized by a security interest in all of the assets of the Company and those subsidiaries, pursuant to a Security Agreement.
The transaction agreements include restrictions on the Company’s ability to engage in certain kinds of transactions while the Notes are outstanding without the consent of two-thirds in interest of the Investors, including incurring or paying certain kinds of indebtedness, entering into certain kinds of financing transactions at prices below $.20 per share, or encumbering the Company’s assets. In addition to the rights under the Security Agreement to foreclose on the collateral in the event of a default, the transaction documents include a variety of liquidated damages, penalties and default provisions upon events of default by the Company, including without limitation an increase in the principal amount and interest rate and a potential decrease in the conversion price of the Notes, and in connection with certain other breaches of covenants of the Company. If the shares underlying the Notes are not freely tradable under SEC Rule 144, the Company intends to file a registration statement covering the resale of such shares.
In connection with the above, each officer and director of the Company was required to sign a lock up agreement covering their shares of Company common stock for the duration of the notes. The officers and directors agreed that during the restricted period, they will not (1) offer, pledge, sell, contract to sell, sell any option or contract to purchase, purchase any option or contract to sell, grant any option, right or warrant to purchase, or otherwise transfer or dispose of, directly or indirectly, any of their shares of Common Stock or any securities convertible into or exercisable or exchangeable for Common Stock or (2) enter into any swap or other agreement that transfers, in whole or in part, any of the economic consequences of ownership of the Common Stock. The lock up will not apply in connection with an offer made to all shareholders of the Company in connection with any merger, consolidation or similar transaction involving the Company or the purchase (but not the sale) of Common Stock upon the exercise of options or warrants.
Notes Payable to Related Parties
The Company had notes payable to related parties amounting to $309,565 and $599,765 at March 31, 2010 and 2009, respectively, which bear interest at 10%. Accrued interest, which is included in accrued expenses, in the consolidated balance sheet, related to the notes was $29,481 and 59,007 at March 31, 2010 and 2009, respectively.
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-24
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 8:
|
NOTES PAYABLE (CONTINUED)
|
Notes Payable to Related Parties (Continued)
On February 12, 2008, the Company issued a convertible promissory note to Old Cellegy for $500,000 bearing interest at 10% annum, with an original maturity date of June 12, 2009. As the merger with Old Cellegy became effective April 1, 2009 (Note 2) the note was converted into shares of Old Adamis and canceled immediately.
On various dates during the twelve months ended March 31, 2010 and included in the amount above, the Company issued promissory notes to shareholders for a total of $219,800, that bear interest at 10% with all principal and interest due on various maturity dates during May through June 2010, originally. The Company repaid $10,000 of the notes payable. Due to loan covenants under the Gemini Master Funds Ltd. Note Agreement, the Company is restricted from paying the outstanding loans until the Gemini notes are repaid or converted to commons stock. Interest continues to accrue on the unpaid balances.
|
NOTE 9:
|
LICENSING AGREEMENTS
|
On July 28, 2006, the Company entered into a nonexclusive, royalty free license agreement with an entity for the technology used to research and develop new viral therapies, and an exclusive royalty-bearing license requiring a small percentage of revenue received by the Company on future products developed and sold with a payment cap of $10,000,000. The Company paid the entity an initial license fee and granted one of the entity’s officers the right to purchase 1,000,000 Founder’s shares in the Company at price of $0.001 pursuant to a separate stock purchase agreement. The Company also granted the entity a royalty-free non-exclusive license to use any improvements made on the existing technology for research purposes only. The Company and the entity have the right to sublicense with written permission of each party. In the event that the entity sublicenses or sells the improved technology to a third party, then a portion of the total payments, to be decided by mutual agreement, will be due to the Company.
The Company is obligated to make the following milestone payments to the entity based on commencement of various clinical trials and submissions of an application to the FDA for regulatory approval:
|
Amount
|
Date due
|
|
|
$50,000
|
Within 30 days of commencement of Phase I/II clinical trial.
|
|
|
50,000
|
Within 30 days of commencement of a separate Phase II trial as required by the FDA.
|
|
|
300,000
|
Within 30 days of commencement of a Phase III trial.
|
|
|
500,000
|
Within 30 days of submission of a biological license application or a new drug application with the FDA.
|
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-25
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 9:
|
LICENSING AGREEMENTS
|
The total milestone payments are not to exceed $900,000 and can only be paid one time and will not repeat for subsequent products. At March 31, 2010, no milestones have been achieved.
The agreement will remain in effect as long as the patent rights remain in effect. Adamis has the right to terminate the agreement if it is determined that no viable product can come from the technology. Adamis would be required to transfer and assign all filings, rights and other information in its control if termination occurs. Adamis would retain the same royalty rights for license, or sublicense, agreements if the technology is later developed into a product. Either party may terminate the license agreement in the event of a material breach of the agreement by the other party that has not been cured or corrected within 90 days of notice of the breach.
On September 22, 2006, the Company entered into an agreement with an entity to manufacture an influenza vaccine for the Company. The agreement requires the Company to pay $70,000 upon commencement of the project, followed by monthly payments based upon services performed until the project is complete. No product has been manufactured and no payments have been made as of March 31, 2010. Once the project begins, the total payments will aggregate $283,420. The project has an open ended start time. Adamis may terminate the agreement upon notice to the other party, other than reimbursing the other party for non cancellable materials and supplies ordered, and work in progress, through the date of the termination.
On February 24, 2010, the Company entered into an agreement with Colby Pharmaceutical Company (the “Licensee”) to acquire three separate exclusive license agreements, covering three small molecule anti-inflammatory compounds, named CPC-100, CPC-200 and CPC-300, for the potential treatment of human prostate cancer, or PCa, in exchange for shares of the Company’s common stock. The Licensee licensed the patents, patent applications and related intellectual property relating to the compounds pursuant to license agreements with a third party (“WARF”). On February 25, 2010, the Company was assigned and transferred the license agreement relating to the CPC-300 compound in consideration of the issuance 1,000,000 shares of common stock to the Licensee valued at $400,000 (Note 11), which was expensed as research and development during the year ended March 31, 2010. The transfer of the license agreements relating to CPC-100 and CPC-200 will occur at a subsequent closing, upon satisfaction of closing conditions, which include the receipt by the Company of equity funding after the date of the agreement in excess of $2 million. The consideration for the transfer of these additional agreements will be 7,500,000 registered shares of the Company common stock to the Licensee.
With respect to sublicenses granted Licensee is to pay WARF according to the following schedule:
|
1.
|
Forty percent (40%) of amounts received under each agreement entered into before an Investigational New Drug ("IND") application is filed by Licensee with the Federal Drug Administration ("FDA") for a Product made a subject of the sublicense.
|
|
2.
|
Thirty percent (30%) of amounts received under each agreement entered into after the filing of an IND under item (1) above until completion of a Phase 1 clinical trial by Licensee for that Product.
|
|
3.
|
Twenty-five percent (25%) of amounts received under each agreement entered into after completion of item (2) above until completion of a Phase II clinical trial by Licensee for that Product.
|
|
4.
|
Twenty percent (20%) of amounts received under each agreement entered into after completion of item (3) above until a New Drug Application ("NDA") has been approved by the FDA for that Product.
|
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-26
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 9:
|
OTHER INTANGIBLES (CONTINUED)
|
|
5.
|
Ten percent (10%) of amounts received under each agreement entered into after the NDA has been approved by the FDA for that Product.
|
Milestone Payments are outlined below:
|
1.
|
$25,000 upon the filing of the first IND or comparable regulatory filing for a human therapeutic Product.
|
|
2.
|
$150,000 upon the enrollment of its first patient under a Phase II clinical trial for the first human therapeutic Product.
|
|
3.
|
$200,000 upon the enrollment of its first patient under a Phase III clinical trial for the first human therapeutic Product.
|
|
4.
|
$250,000 for the first NDA or comparable regulatory approval for a human therapeutic Product.
|
These milestone payments occur only once for each of the compounds
|
NOTE 10:
|
COMMITMENTS AND CONTINGENCIES
|
In addition to the matters described in Note 15, the Company may become involved in or subject to, routine litigation, claims, disputes, proceedings and investigations in the ordinary course of business, which in our opinion will not have a material adverse effect on our financial condition, cash flows or results of operations.
|
NOTE 11:
|
CAPITAL STRUCTURE
|
The Company is authorized to issue 175,000,000 shares of common stock and 10,000,000 shares of preferred stock with a par value of $0.0001 per share.
On various dates during the period ended March 31, 2009, the Company sold 1,339,651 shares of its common stock valued at $0.75, or $1,004,738, in a private placement.
On various dates during the period ended March 31, 2009, the Company sold 76,924 shares of its common stock valued at $0.65, or $50,000, in a private placement.
In December 2008, The Company issued 500,000 shares of its common stock as payment for past consulting services. According to the consulting agreement, the stock is guaranteed to have a value of $1,000,000 within ten business days of the agreement’s anniversary on March 20, 2010, and is non-refundable. As the 500,000, shares of common stock did not have a value of $1,000,000 in March 2010, effective May 1, 2010 the Company extended its consulting agreement to assist the Company in its public relations efforts and issued 1,500,000 shares of its common stock fulfilling the Company's obligation from its prior agreement.
The Company released the remaining 6,732,285 re-purchasable holdback shares related to the HVG Acquisition from escrow during the nine months ended December 31, 2009. The Company’s rights of repurchase related to such shares have not expired, are subject to certain restrictions and are still treated as contingent consideration related to the HVG Acquisition. As a result, no purchase price adjustment was recorded during the current period, and the shares were recorded at par value. The shares are considered anti-dilutive during the period due to the outstanding repurchase options. On August 14, 2009, the Company exercised its repurchase option and
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-27
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 11:
|
CAPITAL STRUCTURE (CONTINUED)
|
repurchased 785,686 shares of common stock for treasury at a total cost of $786 in connection with the resignation of the former Vice President of Operations.
On September 21, 2009, the Company issued 35,000 shares of its common stock in lieu of payment for consulting services with a value of $9,000.
On October 23, 2009, the board of directors of the Company authorized the Company to negotiate an amendment to a stock purchase agreement with the Company's Chief Financial Officer which would have the effect of waiving the Company's repurchase agreement with the Company’s Chief Financial Officer, which waived the repurchase option with respect to 580,500 shares of common stock, and as a result shares are considered vested and unrestricted. As of the date of this report on Form 10-K, such an agreement has not been executed.
During the three months ended December 31, 2009, the Company issued 75,650 shares of its common stock; 50,000 shares were issued in lieu of payment for consulting services with a value of $10,000 and 25,650 shares were issued to employees with a value of $7,182.
On December 29, 2009, the Company issued 500,000 shares of its common stock for par value as a discount to the note payable issued to the G-Max Trust (Note 8)
On January 12, 2010, the Company issued 1,500,000 shares of its common stock for par value as a discount to the note payable issued to the Gemini Master Fund, Ltd. (Note 8).
On February 24, 2010, the Company issued 1,000,000 share of its common stock for par value to acquire exclusive license agreements covering three small molecule compounds from Colby Pharmaceutical Company (Note 9).
|
NOTE 12:
|
STOCK OPTION PLANS, SHARES RESERVED AND WARRANTS
|
Old Cellegy’s stockholders approved a new 2009 Equity Incentive Plan (the “2009 Plan”), which became effective upon the closing of the merger with Old Cellegy. The 2009 Plan provides for the grant of incentive stock options, non-statutory stock options, restricted stock awards, restricted stock unit awards, stock appreciation rights, performance stock awards, and other forms of equity compensation (collectively “stock awards”). In addition, the 2009 Plan provides for the grant of performance cash awards. The aggregate number of shares of common stock that may be issued initially pursuant to stock awards under the 2009 Plan is 7,000,000 shares. The number of shares of common stock reserved for issuance will automatically increase on January 1 of each calendar year, from January 1, 2010 through and including January 1, 2019, by the lesser of (a) 5.0% of the total number of shares of common stock outstanding on December 31 of the preceding calendar year or (b) a lesser number of shares of common stock determined by the Company’s board of directors before the start of a calendar year for which an increase applies. On January 1, 2010, the number of shares reserved for this issuance increased by 2,327,398 to 9,327,398.
The Company granted options to purchase a total of 150,000 shares of common stock upon the closing of the merger to certain non-employee directors who were directors of Old Cellegy before the merger and who continued as directors of the company following the closing of the merger. The stock options have an exercise price of $0.60 per share, which is equal to the fair market value of the Company’s common stock on the date of the grant. The stock options vest over a period of three years from the date of the grant, and expire on the 10th anniversary of the grant date of the options. The Company estimated that the stock options have a fair market value of $0.30 per stock option using the Black-Scholes valuation model. Management’s assumptions included in the model assumed volatility of 35.4%, a risk-free interest rate of 2.7% based on the 10-year Treasury Rate at the
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-28
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 12:
|
STOCK OPTION PLANS, SHARES RESERVED AND WARRANTS (CONTINUED)
|
date of the grant and no dividends. The Company estimated a forfeiture rate of 5.5%. In June 2009, those directors resigned. Each director had 26,388 vested stock options for a total of 76,194. The remaining 73,806 stock options granted were cancelled during nine-months ended December 31, 2009. The total fair market value of only the vested stock options was recorded during the period.
In August 2009, the Company hired an employee, who was granted a stock option by the Company to purchase up to 250,000 shares of common stock. The stock options have an exercise price of $0.22 per share, which was equal to the fair market value of the Company’s common stock on the date of the grant. The stock options vest over a period of three years from the date of the grant, and expire on the 10th anniversary of the grant date of the option. The Company estimated that the stock option has a fair market value of $0.11 per share using the Black-Scholes valuation model. Management’s assumptions included in the model were volatility of 35.4%, a risk-free interest rate of 3.4% based on the 10-year Treasury Rate at the date of the grant and no dividends. The Company estimated a forfeiture rate of 5.5%. The Company recorded stock based compensation expense of $29,918 related to such stock options for the year-ended March 31, 2010.
In August 2009, the Company issued warrants to purchase up to 600,000 shares of common stock to consultants retained to assist the Company in fund-raising efforts. The warrants have an exercise price of $0.25 per share, which is equal to the fair market value of the Company’s common stock at the date of grant. The options had a five year term and expire on August 26, 2014. In January 2010, the Company terminated warrants to purchase up to 300,000 of these shares of common stock. The warrants had an exercise price of $0.25 per share. On the same date, the Company issued warrants to purchase up to 270,000 shares of common stock to the same consultants that have an exercise price of $0.20 per share. The fair market value of the Company's stock on the date of grant was $0.37 per share. The new, or modified, warrant had an intrinsic value of $45,900.
In October 2009 the Company issued warrants to purchase up to 200,000 shares of common stock to consultants retained to assist the Company in fund-raising efforts. The warrants have an exercise price of $0.29 per share, which is equal to the fair market value of the Company’s common stock at the date of grant. The options have a five year term and expire on October 26, 2014.
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-29
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 12:
|
STOCK OPTION PLANS, SHARES RESERVED AND WARRANTS (CONTINUED)
|
The following summarizes outstanding stock options at March 31, 2010:
|
|
Number of
Stock Options
|
Weighted
Average
Remaining
Contractual Life
|
Weighted
Average
Exercise
Price
|
Number of
Stock Options
Vested
|
|||||||||
|
1995 Equity Incentive Plan
|
20,641
|
4.18 Years
|
$
|
26.38
|
20,641
|
||||||||
|
2005 Equity Incentive Plan
|
4,834
|
5.50 Years
|
$
|
13.30
|
4,834
|
||||||||
|
Directors’ Stock Option Plan
|
7,654
|
3.41 Years
|
$
|
42.51
|
7,654
|
||||||||
|
Non-Plan Stock Options
|
100,714
|
3.61 Years
|
$
|
41.27
|
100,714
|
||||||||
|
Biosyn Stock Options
|
431
|
3.81 Years
|
$
|
2.90
|
431
|
||||||||
|
2009 Equity Incentive Plan
|
329,164
|
7.09 Years
|
$
|
0.31
|
120,831
|
||||||||
The Company has reserved shares of common stock for issuance upon exercise at March 31, 2010 as follows:
|
Biosyn Stock Options
|
431
|
|||
|
Director’s Plan
|
7,654
|
|||
|
Warrants
|
1,922,139
|
|||
|
Non-Plan Stock Options
|
100,714
|
|||
|
1995 Equity Incentive Plan
|
20,641
|
|||
|
2005 Equity Incentive Plan
|
4,834
|
|||
|
2009 Equity Incentive Plan
|
9,327,398
|
|||
|
Total Shares Reserved
|
11,383,811
|
The following summarizes warrants outstanding at March 31, 2010:
|
Warrant Shares
|
Exercise Price Per
Share
|
Date Issued
|
Expiration
Date
|
||||||||
|
Biosyn Warrants
|
8,245
|
$
|
57.97 - $173.92
|
October 22, 2004
|
2013 - 2014
|
||||||
|
May 2005 PIPE
|
|||||||||||
|
Series A
|
71,947
|
$
|
1.99
|
May 13, 2005
|
May 13, 2010
|
||||||
|
Series B
|
71,947
|
$
|
2.15
|
May 13, 2005
|
May 13, 2010
|
||||||
|
Old Adamis Warrants
|
1,000,000
|
$
|
0.50
|
November 15, 2007
|
November 15, 2012
|
||||||
|
Consultant Warrants
|
300,000
|
$
|
025
|
August 26, 2009
|
August 26, 2014
|
||||||
|
Consultant Warrants
|
270,000
|
$
|
0.20
|
|
January 29, 2010
|
January 25, 2015
|
|||||
|
Consultant Warrants
|
200,000
|
$
|
0.29
|
October 26, 2009
|
October 26, 2014
|
||||||
|
Total Warrants
|
1,922,139
|
||||||||||
Subsequent to year end, the May 2005 Series A and Series B warrants expired and no replacement warrants were issued.
|
NOTE 13:
|
INCOME TAXES
|
For the year ended March 31, 2009 and prior years, the Company did not file consolidated tax returns. Accordingly, the deferred tax assets for each of the consolidated companies can only be used to offset future tax expense of the respective company.
As a result of the merger with Old Cellegy (Note 2), the Company will file consolidated tax returns for the year ended March 31, 2010.
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-30
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 13:
|
INCOME TAXES (CONTINUED)
|
At March 31, 2009, the Company had net operating loss carryforwards of approximately $7 million for federal and state purposes. The federal and state net operating loss carryforwards expire through the year 2028.
At March 31, 2010, the Company had net operating loss carryforwards of approximately $105 million and $33 million for federal and state purposes, respectively. The federal and state net operating loss carryforwards expire through the year 2029. At March 31, 2010, the Company also had state research and development credit carryforwards of approximately $2.8 million and $200,000 for federal and state purposes, respectively. The federal credits expire through the year 2027 and the state credits expire through the year 2019. The Tax Reform Act of 1986 (the “Act) provides for a limitation on the annual use of net operating loss and research and development tax credit carryforwards following certain ownership changes that could limit the Company’s ability to utilize these carryforwards. The Company has experienced various ownership changes, as defined by the Act, as a result of past financings and the merger with Cellegy. Accordingly, the Company’s ability to utilize the aforementioned carryforwards may be limited. Additionally, U.S. tax laws limit the time during which these carryforwards may be applied against future taxes, therefore, the Company may not be able to take full advantage of these carryforwards for federal income tax purposes. The Company determined that the net operating loss carryforwards relating to Cellegy and Biosyn are limited due to the acquisitions, in 2009 and 2004 and has reflected the estimated amount of usuable net operating loss carryforwards in its deferred tax assets below.
The benefit for income taxes from continuing operations consists of the following for the years ended March 31, 2010 and 2009:
|
2010
|
2009
|
2009
|
||||||||||
|
Adamis Pharmaceuticals
|
Adamis Pharmaceuticals
|
Adamis Labs
|
||||||||||
|
Current
|
$ | - | $ | - | $ | - | ||||||
|
Deferred
|
(1,225,000 | ) | (188,000 | ) | (793,000 | ) | ||||||
|
Total
|
(1,225,000 | ) | (188,000 | ) | (793,000 | ) | ||||||
|
Change in Valuation Allowance
|
1,225,000 | 188,000 | 793,000 | |||||||||
|
Tax Benefit, net
|
$ | - | $ | - | $ | - | ||||||
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-31
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 13:
|
INCOME TAXES (CONTINUED)
|
At March 31, 2010 and 2009 the significant components of the deferred tax assets from continuing operations are summarized below:
|
2010
|
2009
|
2008
|
||||||||||
|
Adamis Pharmaceuticals
|
Adamis Pharmaceuticals
|
Adamis Labs
|
||||||||||
|
Net Operating Loss Carryforwards
|
$ | 39,322,000 | $ | 1,051,000 | $ | 1,559,000 | ||||||
|
Deferred Tax Assets
|
689,000 | 135,000 | 210,000 | |||||||||
|
Deferred Tax (Liabilities)
|
- | (181,000 | ) | - | ||||||||
|
Net Deferred Tax Assets
|
40,011,000 | 1,005,000 | 1,769,000 | |||||||||
|
Less Valuation Allowance
|
(40,011,000 | ) | (1,005,000 | ) | (1,769,000 | ) | ||||||
|
Net Deferred Tax Assets
|
$ | - | $ | - | $ | - | ||||||
We have determined at December 31, 2010 and 2009 that a full valuation allowance would be required against all of our operating loss carryforwards and deferred tax assets that we do not expect to be utilized by deferred tax liabilities.
The following table reconciles our losses from continuing operations before income taxes for the years ended March 31, 2010 and 2009.
|
2010
|
2009
|
2009
|
||||||||||||||
|
Adamis Pharmaceuticals
|
Adamis Pharmaceuticals*
|
Adamis Labs
|
||||||||||||||
|
Income (Loss) from
|
$ | (6,160,000 | ) | $ | 1,919,000 | $ | (1,778,000 | ) | ||||||||
|
Continuing Operations
|
||||||||||||||||
|
Permanent Differences:
|
||||||||||||||||
|
Non-Cash Interest
|
3,176,000 | 248,000 | -- | |||||||||||||
|
Non-Cash Services
|
125,000 | 1,000,000 | - | |||||||||||||
|
INL Gain
|
- | (4,064,000 | ) | - | ||||||||||||
|
Meals and Entertainment
|
1,000 | - | 3,000- | |||||||||||||
| $ | (2,858,000 | ) | $ | (897,700 | ) | $ | (1,175,000 | ) | ||||||||
|
Federal Statutory Rate
|
34.00 | % | $ | (2,230,000 | ) | $ | (652,000 | ) | (605,000 | ) | ||||||
|
State Income Tax, net of Federal Tax
|
3.63 | % | (238,000 | ) | 70,000 | (65,000 | ) | |||||||||
|
Intercompany Eliminations
|
37.63 | % | - | 353,000 | 124,000 | |||||||||||
|
Permanent Differences
|
37.63 | % | 1,243,000 | (1,263,000 | ) | 1,000 | ||||||||||
|
Change in Valuation Allowance
|
1,225,000 | 188,000 | 793,000 | |||||||||||||
|
Expected Tax Benefit
|
$ | - | $ | - | $ | - | ||||||||||
*Does not include operating loss from discontinued operations.
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-32
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 14:
|
GOING CONCERN
|
The Company's consolidated financial statements are prepared using the generally accepted accounting principles applicable to a going concern, which contemplates the realization of assets and liquidation of liabilities in the normal course of business. However, as shown in the accompanying consolidated financial statements, the Company has sustained substantial losses from continuing operations since inception and has not introduced new revenue producing products since inception. In addition, the Company has used, rather than provided, cash in its continuing operations. Without realization of additional capital, it would be unlikely for the Company to continue as a going concern. It is management's plan in this regard to obtain additional working capital through debt and equity financings.
The consolidated financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts and classification of liabilities that might be necessary should the Company be unable to continue in existence.
|
NOTE 15:
|
SUBSEQUENT EVENTS
|
The Company has evaluated subsequent events through July XX, 2010, which is the date the financial statements were available to be issued. Other than the events described below, no other subsequent events have been identified.
Termination of Consulting Agreement
On May 4, 2010 the Company and a consultant agreed to terminate the consulting services contract entered into on February 1, 2010. The Company agreed to pay the $70,000 owed under the agreement by issuing 350,000 share of the Company's common stock from the 2009 Equity Incentive Plan. Further, the Company will reduce 200,000 shares from the 1,000,000 consideration shares (Rule 144) issued to Colby Pharmaceuticals as part of the license agreement (Note 9).
EquiTrend Advisors
Effective May 1, 2010 the Company entered into a consulting agreement with EquiTrend Advisors LLC to assist the Company in its public relations efforts with relevant investors and public capital markets. As compensation, the Company issued 250,000 shares of its common stock and if Company does not terminate the engagement within 90 days it will be obligated to issue another 250,000 of its common shares.
Hansen Consulting
On April 6, 2010 the Company entered into an agreement with Christian Hansen to assist with the branding of the Company and its products. The Company issued 500,000 shares of its common stock for these services.
|
ADAMIS PHARMACEUTICALS CORPORATION AND SUBSIDIARIES
|
F-33
|
|
|
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
NOTE 15:
|
SUBSEQUENT EVENTS (CONTINUED)
|
Capital Group Communications
Effective May 1, 2010, the Company entered into a two year consulting agreement with Capital Group Communications for consulting services pertaining to public relations. The Company issued 1,500,000 shares of its common stock for these services.
Curtis Leahy, et. al. v. Dennis J. Carlo, et al.
In May 2010, Curtis Leahy, et. al. v. Dennis J. Carlo, et al., Case Number 37-2010-00092524-CU-FR-CTL, was filed in San Diego Superior Court, and plaintiffs subsequently filed an amended complaint on June 18, 2010. The plaintiffs — Antaeus Capital Partners, Curtis Leahy, and David Amron – are Adamis shareholders. The defendants named in the Complaint are the Company, Dennis Carlo, David Marguglio, Robert Hopkins, and Richard Aloi. Plaintiffs allege that defendants misrepresented and omitted material information in a private placement memoranda issued by Adamis in 2006 and 2008 regarding, among other things, Adamis’ license rights with respect to certain patented anti-viral technology. Based on these purported misrepresentations and omissions, plaintiffs assert claims for violations of Sections 25401, 25501 and 25504 of the California Corporations Code, and claims for common law fraud and negligent misrepresentation on behalf of a putative class of shareholders who purchased stock pursuant to either or both the Company’s 2006 and 2008 Private Placement Memoranda. Plaintiffs seek damages amounting to the difference between the purchase price of their stock and the current share price, or the price at which they previously sold their stock.
Plaintiffs also allege that defendants breached their fiduciary duties as directors and officers of Adamis with respect to certain corporate transactions, including the HVG transaction in 2007, the Cellegy merger in 2008, and the Gemini and G-Max financing transactions in 2009. Plaintiffs allege that these transactions were not in the best interest of the Company and did not achieve their stated objectives. Plaintiffs further allege that the director defendants collected excessive compensation in fiscal years 2008 and 2009, and assert that the Company should have exercised its right to repurchase certain shares issued to defendants and other senior managers pursuant to the Stock Repurchase Agreements in 2008 rather than amend those agreements to extend the dates for meeting the applicable performance criteria. Based on these allegations, plaintiffs assert claims for breach of fiduciary duty, unjust enrichment and constructive trust, declaratory relief, and injunctive relief.
Adamis believes that the complaint is without merit, and Adamis intends to vigorously defend the claim and may assert any available counterclaims. Adamis intends to file a demurrer and motion to strike the complaint by the end of July 2010.
Cosmo Bioscience, Inc. et. al. v. Adamis Pharmaceuticals Corp. and Maurizio Zanetti
Cosmo Bioscience, Inc. et. al. v. Adamis Pharmaceuticals Corp. and Maurizio Zanetti, Case No. 37-2010-00088584 was filed in San Diego Superior Court in May 2010. Plaintiffs are affiliated Cosmo Bioscience entities who claim to have sublicensed certain patented technology from Eurogen BV, an entity wholly owned and controlled by Maurizio Zanetti. Plaintiffs claim that Zanetti wrongfully terminated their license, and further that Zanetti improperly licensed the same technology to Adamis in violation of plaintiffs’ exclusive license agreement. Plaintiffs assert a single claim for declaratory relief seeking adeclaration that the Cosmo sublicense is in full force and effect, and that the Adamis license is invalid. Zanetti intends to file a motion to compel arbitration and Adamis intends to file a demurrer and a motion to stay pending resolution of the arbitration
The litigation described in this section could divert management time and attention from the company, could involve significant amounts of legal fees and other fees and expenses. An adverse outcome in any such litigation could have a material adverse effect on Adamis.