Attached files
| file | filename |
|---|---|
| EX-31.1 - EX-31.1 - BIOSPHERE MEDICAL INC | a2197600zex-31_1.htm |
| EX-31.2 - EX-31.2 - BIOSPHERE MEDICAL INC | a2197600zex-31_2.htm |
| EX-32.2 - EX-32.2 - BIOSPHERE MEDICAL INC | a2197600zex-32_2.htm |
| EX-23.1 - EX-23.1 - BIOSPHERE MEDICAL INC | a2197600zex-23_1.htm |
| EX-32.1 - EX-32.1 - BIOSPHERE MEDICAL INC | a2197600zex-32_1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| ý | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
For the fiscal year ended: December 31, 2009 |
||
or |
||
o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the transition period from to |
||
Commission File Number: 0-23678
BioSphere Medical, Inc.
(Exact Name of Registrant as Specified in Its Charter)
| Delaware (State or Other Jurisdiction of Incorporation or Organization) |
04-3216867 (I.R.S. Employer Identification No.) |
|
1050 Hingham Street, Rockland, Massachusetts (Address of Principal Executive Offices) |
02370 (Zip Code) |
(781) 681-7900
(Registrant's telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of Class | Name of Each Exchange on Which Registered | |
|---|---|---|
| Common Stock, $.01 par value per share | NASDAQ Global Market |
Securities
registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically and posted to its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ý
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definition of "large accelerated filer," "accelerated filer," and "smaller reporting company" in rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer o | Accelerated filer o | Non-accelerated filer o (Do not check if a smaller reporting company) |
Smaller reporting company ý |
Indicate by check mark whether the registrant is a shell company (as defined in rule 12b-2 of the Act). Yes o No ý
The aggregate market value of voting common stock held by non-affiliates of the registrant on June 30, 2009, was $24,741,889 based on the closing price of the common stock as reported on the NASDAQ Global Market as of such date.
The Registrant had 18,419,895 shares of common stock outstanding as of March 1, 2010.
Documents incorporated by reference:
Portions of the information required by Part III of Form 10-K will appear in the registrant's definitive Proxy Statement on Schedule 14A for the 2010 Annual Meeting of Stockholders and are hereby incorporated by reference into this report.
2
This annual report on Form 10-K contains forward-looking statements that involve risks, uncertainties and assumptions that, if they never materialize or prove incorrect, could cause the results of BioSphere Medical, Inc. to differ materially from those expressed or implied by such forward-looking statements. All statements other than statements of historical fact are statements that could be deemed forward-looking statements, including any projections of revenue, expenses, earnings or losses from operations, or other financial items; any statements of the plans, strategies and objectives of management for future operations; any statements concerning product research, development, regulatory approval and commercialization timelines; any statements about our expectations regarding market acceptance and market penetration for our products and product liability challenges with respect to our products; any statements of expectation or belief; and any statements of assumptions underlying any of the foregoing. The risks, uncertainties and assumptions referred to above include risks that are described in "Risk Factors" and elsewhere in this annual report on Form 10-K and that are otherwise described from time to time in our Securities and Exchange Commission reports filed after this annual report on Form 10-K.
The forward-looking statements included in this annual report on Form 10-K represent our estimates as of the date of this annual report on Form 10-K. We specifically disclaim any obligation to update these forward-looking statements in the future. These forward-looking statements should not be relied upon as representing our estimates or views as of any date subsequent to the date of this annual report on Form 10-K. Unless the context otherwise requires, references in this annual report on Form 10-K to "BioSphere," "we," "us" and "our" refer to BioSphere Medical, Inc. and our subsidiaries.
We develop, manufacture and market products for medical procedures that use embolotherapy. Embolotherapy is the minimally invasive, image-guided therapeutic introduction of various biocompatible substances into a patient's circulatory system to occlude a blood vessel, either to arrest or prevent hemorrhaging, or to devitalize or destroy the structure by occluding its blood supply. Our core technologies consist of patented bioengineered polymers, which are chemical compounds created through the application of medical science, engineering principles and manufacturing methods. These core technologies are used to produce miniature spherical embolic particles, or microspheres, that are designed to have uniquely beneficial properties for a variety of medical applications.
We currently market and sell four microsphere products:
- •
- Embosphere Microspheres, which are marketed for symptomatic uterine
fibroids, hypervascularized tumors and arteriovenous malformations, in the United States, the European Union, the People's Republic of China and several other markets outside the United States;
- •
- EmboGold Microspheres, which are marketed for hypervascularized tumors and
arteriovenous malformations in the United States, the European Union and several other markets outside the United States;
- •
- HepaSphere Microspheres, which are marketed in the European Union, Brazil
and Russia for primary and metastatic liver cancer, and in the European Union and Russia for drug delivery in the treatment of primary and metastatic liver cancer; and
- •
- QuadraSphere Microspheres, which are marketed for the treatment of hypervascularized tumors and arteriovenous malformations in the United States.
Our Embosphere® Microspheres and EmboGold® Microspheres have a number of beneficial properties that we believe make them well suited for embolotherapy procedures. Because of their uniform spherical shape and soft, slippery surface, our particles are easy to inject through small
3
catheters, which enables an even distribution within the vessel network. Additionally, we offer these products to clinicians in calibrated size ranges so they can be selected to target the occlusion of specific-sized vessels. The use of appropriately size-ranged microspheres is designed to produce more predictable results and optimize therapeutic benefit.
Our expanding embolics, HepaSphere™ Microspheres and QuadraSphere® Microspheres, have different properties than Embosphere Microspheres and EmboGold Microspheres. Specifically, HepaSphere Microspheres and QuadraSphere Microspheres have an ability to absorb fluids and expand to four times their dry state in the body while maintaining their spherical form. HepaSphere Microspheres and QuadraSphere Microspheres occlude with a high degree of conformity to the vessel wall.
Our strategic priorities are to accelerate our revenue growth by expanding the market for the minimally invasive treatment of symptomatic uterine fibroids using a treatment called uterine fibroid embolization, or UFE, and by broadening treatment options for several medical conditions, including liver cancer.
Uterine fibroids are noncancerous, or benign, hypervascular tumors growing within or on the wall of the uterus. Such tumors are treatable with UFE. UFE is a minimally invasive procedure in which microspheres are injected through a microcatheter into the blood vessels that supply the uterus. Blood flow guides these particles into the network of vessels that preferentially flow toward the fibroids, thereby blocking the blood supply to the fibroids but not the surrounding healthy tissue. Most patients with uterine fibroids are not initially symptomatic and remain untreated until the patient experiences symptoms such as abnormal bleeding, increased urinary frequency, pain, pelvic discomfort or fertility difficulties. In each of the years ended December 31, 2009, 2008 and 2007, the majority of our revenue was derived from the sale of our Embosphere Microspheres for UFE, and we believe that UFE will remain the principal application for our microsphere products for the foreseeable future.
We believe that there are growth opportunities for other embolotherapy procedures, notably in the treatment of other hypervascularized tumors, such as primary liver cancer tumors. Our HepaSphere Microspheres are marketed and sold in Europe, Brazil and Russia for the treatment of primary and metastatic liver cancer. Our QuadraSphere Microspheres are identical in all respects to our HepaSphere Microspheres. However, FDA regulations require that we conduct clinical trials and submit a marketing application which includes positive data from the clinical trials to the United States Food and Drug Administration, or FDA, in order to obtain the approvals and clearances required to promote QuadraSphere Microspheres for the treatment of a specific disease or condition, including primary and metastatic liver cancer. European Union regulations do not require preclearance clinical trials for this class of medical device on an indication-by-indication basis. In October 2009, we submitted to the FDA an investigational device exemption, or IDE, application to commence a clinical trial to compare the effectiveness of our QuadraSphere Microspheres combined with the chemotherapeutic agent doxorubicin against conventional transarterial chemoembolization with doxorubicin in patients with primary liver cancer. The FDA has advised us that our study protocol will need to include survival as a primary endpoint for the trial, rather than overall tumor response rate at six months, which was the primary endpoint that we proposed in our initial IDE application. Our satisfactory resolution of the FDA's comments on the IDE is a condition to starting the clinical trial. We are currently evaluating the FDA's protocol requirements for the trial, including how the primary endpoint requirements will affect our plans regarding the size of the trial and the timeline and cost for completion. We have not reached a conclusion about whether or when to undertake the clinical trial.
On April 16, 2009, we entered into an international distribution agreement with Nippon Kayaku Co., Ltd., or Nippon Kayaku. The agreement grants Nippon Kayaku the exclusive right to distribute our HepaSphere Microspheres and Embosphere Microspheres in Japan upon regulatory approval. The agreement provides that Nippon Kayaku is responsible for filing, obtaining and
4
maintaining all regulatory approvals necessary for the sale, marketing, pricing and reimbursement of the products in Japan, including performing any clinical trials required as a result of seeking such regulatory approvals in Japan. We anticipate a clinical trial in Japan will be necessary to seek such regulatory approval. Assuming we successfully obtain product approval, we will provide HepaSphere Microspheres and Embosphere Microspheres to Nippon Kayaku for distribution and sale in Japan. Additionally, Nippon Kayaku has made a nonrefundable milestone payment of $1.00 million in 2009 and has agreed to make up to $3.00 million in additional milestone payments based upon specified objectives, including achievement of clinical, regulatory and sales goals.
We have a number of patent applications and issued U.S. patents related to the use of our microspheres for non-embolotherapy applications. Although our current focus is on embolotherapy markets, and, as such, we are not currently devoting significant resources to research relating to non-embolotherapy applications, we believe that these non-embolotherapy uses may provide us, at some point in the future, with development and commercialization opportunities through internal efforts or third-party licensing, collaboration or similar opportunities.
We were incorporated in Delaware in 1993. Our principal executive offices are located at 1050 Hingham Street, Rockland, Massachusetts 02370, and our telephone number is (781) 681-7900.
We maintain an Internet Web site with the address www.biospheremed.com. We are not including the information contained in our Web site as part of, or incorporating it by reference into, this annual report on Form 10-K. We make our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and all amendments to those reports available free of charge on our Web site as soon as reasonably practicable after we electronically file those materials with, or furnish those materials to, the United States Securities and Exchange Commission, or SEC. Our code of business conduct and ethics and the charters of the audit committee, compensation committee, and nominating and corporate governance committee of our board of directors are all available on the corporate governance section of our Web site. Stockholders may request a free copy of any of these documents by writing to Investor Relations, BioSphere Medical, Inc., 1050 Hingham Street, Rockland, Massachusetts, U.S.A. 02370 or submitting a request through the Web site.
BioSphere Medical®, Embosphere®, EmboGold®, EmboCath® Plus, Sequitor®, Segway®, HepaSphere™, QuadraSphere®, Tenor™, ask4UFE.com®, Ask4Tell4™ and PassThru® are trademarks of BioSphere Medical, Inc. Other trademarks appearing in this annual report on Form 10-K are the property of their respective holders.
INDUSTRY OVERVIEW
Embolotherapy Markets
Embolotherapy has been in use for more than 20 years by interventional radiologists to mechanically block the flow of blood to treat certain peripheral tumors and arteriovenous malformations and to control blood loss. Interventional radiologists around the world employ embolotherapy procedures, including UFE and embolization for the treatment of certain cancers, including primary liver cancer tumors. We believe that an increasing number of uterine fibroid and liver cancer patients are seeking treatments with embolotherapy due to their desire for less invasive treatment options than those presented by non-embolotherapy procedures.
Uterine Fibroids
Until recently, women suffering from uterine fibroids have had few treatment options. These treatment options include the following:
- •
- Hysterectomy. Hysterectomy is a surgical procedure to remove the uterus. A hysterectomy may be performed as an open surgery with or without robotic assistance or as a laparoscopic
5
- •
- Myomectomy. Myomectomy is the surgical removal of the uterine
fibroids without removal of the uterus. It is usually performed on women who wish to preserve their fertility. Only fibroids that can be easily accessed and excised are candidates for removal with
this technique. Because some fibroids are difficult to identify while others are difficult to remove, there is a relatively high recurrence rate, between 10% and 60%, after myomectomy.
- •
- Drug Therapy and "Watchful Waiting." Drug therapies include
nonsteroidal anti-inflammatory drugs, oral contraceptive pills, progestational agents and gonadotropin-releasing hormone agonists. Physicians may choose to monitor women with less severe
symptoms who elect against drug therapy and those seeking to conceive, and may determine to administer therapy only if the patient's condition worsens.
- •
- Other Treatments. Other treatments for uterine fibroids include high-intensity focused ultrasound and global endometrial ablation. High-intensity focused ultrasound is a method of delivering ultrasonic energy to a discrete point with resultant heat and tissue destruction, but without causing a significant temperature increase or cellular injury to tissue in the path of the ultrasound beam. Global endometrial ablation describes the minimally invasive application of energy to destroy the endometrial lining in women who are experiencing severe menstrual bleeding and who do not desire future pregnancy.
procedure. While hysterectomy has a relatively low complication rate, it requires a hospital stay of several days, a recovery period of up to six to eight weeks, and results in loss of fertility. Furthermore, hysterectomies have been tied to adverse psychological effects, sexual and urinary dysfunction, as well as the onset of early menopause. In addition, for many women who have their ovaries removed during hysterectomy, this treatment may result in the need for extended hormone replacement therapy.
Liver Cancer
Liver cancer is one of the most prevalent forms of cancer worldwide. There are several types of liver cancer. Primary liver cancer refers to cancer that begins within the liver itself. Chronic hepatitis B and chronic hepatitis C, inflammations of the liver associated with the hepatitis virus, are contributing factors to the development of primary liver cancer. Primary liver cancer is typically diagnosed at a stage that is too advanced to cure surgically. As a result, the majority of patients diagnosed with primary liver cancer are not surgical candidates. For these patients, existing treatment options are primarily designed to improve quality of life rather than cure the underlying disease. Metastatic liver cancer occurs when cancer begins in another part of the body, such as the colon or breast, and then migrates, or spreads, to the liver. Metastatic liver cancer is more prevalent than primary liver cancer. However, the rate of primary liver cancer is expected to increase due to the prevalence of hepatitis C, a key risk factor for primary liver cancer. Numerous studies and medical publications indicate that embolotherapy has been used for at least 20 years to treat liver cancer. For example, particle embolization is commonly used in Japan to manage liver cancer patients. Embolic particles are commonly injected with chemotherapeutic agents to control and target distribution of the chemotherapy agents, thereby increasing the therapeutic exposure at a specific area.
6
PRODUCTS
The following tables summarize information about our principal products.
| PRODUCT | CLEARED FOR THE FOLLOWING INTENDED USES |
PRINCIPAL GEOGRAPHIC APPROVALS | ||
|---|---|---|---|---|
Microsphere Products: |
||||
Embosphere Microspheres |
Uterine fibroids, hypervascularized tumors and other arteriovenous malformations | United States, Canada, European Union, Argentina, Brazil, Panama, Peru, Uruguay, Hong Kong, Taiwan, Israel, Australia, People's Republic of China, Russia, Switzerland, South Africa and New Zealand | ||
EmboGold Microspheres |
Hypervascularized tumors (other than uterine fibroids) and arteriovenous malformations |
United States, Canada, European Union, Brazil, Panama, Peru, Uruguay, Hong Kong, Taiwan, Israel, Australia, Russia, Switzerland, South Africa and New Zealand |
||
HepaSphere Microspheres |
Primary and metastatic liver cancer |
Canada, European Union, Argentina, Brazil, Hong Kong, Israel, Australia, Russia, Switzerland and South Africa |
||
HepaSphere Microspheres |
Transarterial chemoembolization, or TACE, of hepatocellular carcinoma in combination with doxorubicin |
European Union, Israel, Australia, Russia, Switzerland and South Africa |
||
QuadraSphere Microspheres |
Hypervascularized tumors and arteriovenous malformations |
United States |
||
Delivery System Products: |
||||
EmboCath Plus Infusion Microcatheter |
Infusion of various diagnostic, embolic and therapeutic agents and super-selective angiography within peripheral vasculature | United States, Canada, European Union, Switzerland, Israel, South Africa, Panama and Peru | ||
Sequitor Steerable Guidewire |
Various diagnostic and interventional procedures within peripheral vasculature |
United States, Canada, European Union, Switzerland, Israel, South Africa, Brazil, Panama, Peru and People's Republic of China |
||
Segway Guidewire |
Peripheral embolization procedures |
United States, Canada, Argentina, Brazil, Panama and People's Republic of China |
||
Tenor Steerable Guidewire |
Various diagnostic and interventional procedures within peripheral vasculature |
United States |
7
Embosphere Microspheres
Our Embosphere Microspheres are intended for use in embolotherapy to block or control the blood supply to certain tumors and other vascular malformations.
In November 2002, we received 510(k) clearance from the FDA to market our Embosphere Microspheres for UFE. We were the first company to gain regulatory clearance to market a product for UFE in the United States. Over the past three years, we have focused on growing our Embosphere Microsphere business through the development of physician-referral networks and patient-awareness programs. In each of the years ended December 31, 2009, 2008, and 2007, the majority of our revenue was derived from the sale of our Embosphere Microspheres for UFE. We believe that UFE will remain the principal application for our microsphere products for the foreseeable future.
Uterine fibroid embolization is a minimally invasive procedure, performed principally by interventional radiologists, in which microspheres are injected through a small catheter into the blood vessels that supply the uterus. Blood flow delivers these spheres into the network of vessels that preferentially supply the fibroids, thereby selectively blocking the blood supply to the fibroids and minimizing impact to the surrounding healthy uterine tissue. The goal of the uterine fibroid-embolization procedure is to eliminate the flow of blood to the uterine fibroids, thereby causing fibroid shrinkage and alleviating related symptoms, while attempting to preserve normal uterine and ovarian function.
We believe that embolotherapy is a significantly more attractive alternative for treatment of uterine fibroids when compared to the invasiveness of such surgical procedures as hysterectomy or myomectomy, or to drug therapy and "watchful waiting." Traditional therapies can have significant adverse side effects, including loss of fertility, lengthy recovery periods, high costs, discomfort and risk of recurrence of fibroids. Embolotherapy may not be an alternative for patients with certain types of fibroids, allergies to contrast medium, or other pre-existing medical conditions or patients who are pregnant. Third-party clinical data and publications support the safety, efficacy, cost-effectiveness and long-term durability of UFE. The January 2008 issue of Obstetrics & Gynecology concluded that over the long term, and in a broad range of practice settings, uterine artery embolization produces a high level of durable symptom control and results in a significant improvement in a woman's health-related quality of life. Three-year data from the largest, multicenter, prospective voluntary registry on any procedure for benign uterine fibroids showed that 90 percent of the women participating avoided a hysterectomy, and of these, 85 percent had a substantial improvement in symptoms and quality of life. In August 2008, the American College of Obstetricians and Gynecologists, or ACOG, recommended UFE as a Level A alternative to hysterectomy, which means it is based on "good and consistent scientific evidence." We believe the ACOG Level A recommendation for UFE will be a catalyst for increased acceptance of UFE as an effective alternative for patients who are on drug therapy or are considering undergoing surgery, such as hysterectomy or myomectomy, for treatment of their uterine fibroids. As such, we anticipate that the number of UFE procedures will continue to increase.
Most uterine fibroid embolization procedures can be performed in less than one hour, while the patient is sedated, but awake. The patient often stays overnight in the hospital to manage any discomfort and/or pain associated with the procedure and typically returns to everyday activities in several days. In contrast, hysterectomy patients undergo general anesthesia and typically stay in the hospital for two to three days and have a recovery period lasting up to six to eight weeks.
Embosphere Microspheres have a variety of characteristics that we believe make them preferable to other currently marketed particles. These include:
- •
- Uniform Spherical Shape/Calibrated Particle Size. We are able to synthesize embolic particles with uniform sizing and a spherical shape. When embolic materials are nonspherical or irregularly sized, as is the case with the polyvinyl alcohol, or PVA, particles that have been
8
- •
- Compliant and Resilient Properties. We have developed a soft,
elastic microsphere that has the capability to compress significantly, thus facilitating delivery through very small catheters known as microcatheters. Many clinicians prefer using microcatheters
during embolization, since these catheters minimize the frequency of artery or vessel spasm during the procedure. Vessel spasm can be of particular concern during uterine fibroid embolization as it
can disrupt the flow of blood, which clinicians rely on during embolization to direct the microspheres to the vessel targeted for occlusion. Immediately upon exiting the microcatheter, these resilient
microspheres resume their pre-compression dimensions thereby facilitating predictable vessel occlusion at the level of the pre-compressed particle size.
- •
- Hydrophilic Properties. As a result of the materials used to
manufacture microspheres, our products are hydrophilic, which means that they absorb moisture. This characteristic is important in that it minimizes the frequency of the microspheres from clumping in
the catheter or in the artery during the procedure.
- •
- Nonbiodegradability. Our microspheres are composed of a
synthetic three-component polymer that is designed to be compatible with the human body. This polymer is insoluble and nonbiodegradable. We believe, therefore, that our Embosphere Microspheres are an
appropriate agent for permanent vessel occlusion.
- •
- Cell Adhesion. Our Embosphere Microspheres feature a
cell-adhesion promoter composed of gelatin, which is designed to enhance a stable and complete occlusion of the vessel.
- •
- Charged Surface Property. Our microspheres are positively charged, enhancing attraction to the negatively charged blood vessel wall. This attachment to the vessel wall minimizes the potential for the microspheres to migrate to nontargeted vessels.
historically used in these applications, clinicians report that they find vessel targeting more difficult and may also experience an increased incidence in unwanted embolization of blood vessels away from the site of the tumor when compared to our embolic particles.
Embosphere Microspheres are currently available in size ranges, from 40 to 1,200 microns. They are designed for targeted and controlled occlusions. They can be used with our accessory catheter products or with other commercially available catheter and delivery systems.
EmboGold Microspheres
Our EmboGold Microsphere product contains a product enhancement that adds color to our Embosphere Microsphere product for improved visibility in the syringe during preparation and injection. Our EmboGold Microspheres received 510(k) clearance from the FDA in 2001 for the treatment of hypervascularized tumors and arteriovenous malformations. However, we do not have FDA clearance to market our EmboGold Microspheres for use in the treatment of uterine fibroids, and have determined not to seek such approval at this time.
HepaSphere Microspheres
In November 2004, we received CE Mark approval in the European Union to market our HepaSphere Microspheres for the treatment of primary and metastatic liver cancer and hepatic metastases, and in December 2007, we received CE Mark approval for use of HepaSphere Microspheres with the delivery of doxorubicin for the same treatments. CE Mark approval denotes conformity with European standards for quality and allows certified devices to be placed in the market in European Union countries.
In June 2007, we received approval to market our HepaSphere Microspheres in Brazil for the treatment of primary and metastatic liver cancer.
9
In February 2009, we received approval to market our HepaSphere Microspheres with or without delivery of doxorubicin in Russia for treatment of primary and metastatic liver cancer and hepatic metastases.
The product attributes of HepaSphere Microspheres are:
- •
- the ability to absorb, or "carry," a chemotherapeutic agent;
- •
- an ability to expand and absorb fluids, such as saline, contrast agents and human serum, that creates expansion to four
times its dry-state diameter in the body—64 times its initial volume—while maintaining its spherical form;
- •
- a high degree of conformity to vessel anatomy; and
- •
- a capability for complete occlusion of a vessel with, on average, just a single particle.
Like treatment of uterine fibroids, targeted liver embolotherapy is intended to starve the liver tumor without damaging the surrounding tissue or causing any adverse side effects to other parts of the body, as would be observed with alternative therapies such as chemotherapy and radiation.
In connection with the CE Mark approval of our HepaSphere Microspheres, we intend to conduct a post-market study in approximately ten European centers as part of an international multi-site clinical trial.
QuadraSphere Microspheres
In November 2006, the FDA granted marketing clearance for our QuadraSphere Microspheres in the United States for the treatment of hypervascularized tumors and peripheral arteriovenous malformations. The product attributes of QuadraSphere Microspheres are:
- •
- the ability to absorb, or "carry," a chemotherapeutic agent;
- •
- an ability to expand and absorb fluids, such as saline, contrast agents and human serum, that creates expansion to four
times its dry-state diameter in the body—64 times its initial volume—while maintaining its spherical form;
- •
- a high degree of conformity to vessel anatomy; and
- •
- a capability for complete occlusion of a vessel with, on average, just a single particle.
Our QuadraSphere Microsphere product is technically identical in all respects to our HepaSphere Microsphere product. However, FDA regulations require that we conduct clinical trials regarding the use of QuadraSphere Microspheres for the treatment of a specific disease or condition, including primary and metastatic liver cancer, while European Union regulations do not require trials for this class of medical device on an indication-by-indication basis. In October 2009, we submitted to the FDA an IDE application to commence a clinical trial to compare the effectiveness of our QuadraSphere Microspheres combined with the chemotherapeutic agent doxorubicin against conventional transarterial chemoembolization with doxorubicin in patients with primary liver cancer. The FDA has advised us that our study protocol will need to include survival as a primary endpoint for the trial, rather than overall tumor response rate at six months, which was the primary endpoint that we proposed in our initial IDE application. Our satisfactory resolution of the FDA's comments on the IDE is a condition to starting the clinical trial. We are currently evaluating the FDA's protocol requirements for the trial, including how the primary endpoint requirements will affect our plans regarding the size of the trial and the timeline and cost for completion. We have not reached a conclusion about whether or when to undertake the clinical trial.
10
Delivery Systems
Our EmboCath Plus Infusion Microcatheter, Sequitor Steerable Guidewire and Tenor Steerable Guidewire products are used to deliver embolization material into the target area. In developing these devices we sought to build on the advantages of our existing EmboCath Infusion Catheter and Segway Guidewire products by adding enhanced tracking, torque response (guidewire products), and coating technology to the product lines. These products were also specifically designed to be used together to optimize performance.
EmboCath Plus Infusion Microcatheter. The EmboCath Plus Infusion Microcatheter is a microcatheter that is designed to be used to deliver embolic, diagnostic and therapeutic agents into the peripheral vascular system for interventional procedures such as UFE and the embolization of other hypervascular tumors.
The product attributes of the EmboCath Plus Infusion Microcatheter are:
- •
- controlled delivery, featuring the largest internal lumen diameter in its class—0.028"—which
provides a greater flow rate;
- •
- a flexible, kink-resistant, durable design that offers optimal balance for agile tracking;
- •
- a clear, chemo-compatible hub designed for smooth, continuous injection of microspheres; and
- •
- enhanced fluoroscopic ability via an extra-bright tip.
Sequitor Steerable Guidewire. Our Sequitor Steerable Guidewire is designed specifically to complement our EmboCath Plus Infusion Microcatheter. Guidewires are used in most intravascular catheter procedures to establish a support structure for, and to facilitate placement of, the catheter. We designed our Sequitor Steerable Guidewire to address the needs of interventionalists. The product attributes of Sequitor Steerable Guidewire are:
- •
- a durable atraumatic polymer tip that is designed to reduce the risk of vascular spasm but retain its shape for selective
vessel access;
- •
- a highly visible distal segment, comprised of a radiopaque coil and polymer jacket, which provides visibility under live
imaging;
- •
- a specially tempered wire core designed to transmit one-to-one torque response without kinking;
and
- •
- PassThru® lubricious, hydrophilic coating that facilities wire trackability.
Segway Guidewire. Our Segway Guidewire is intended for placement of catheters for various peripheral vascular diagnostic and interventional procedures. It is a hydrophilic guidewire with composite construction providing a combination of support, softness and durability. The patented advanced composite construction combines a highly supporting PFTE coated stainless steel shaft with a kink-resistant nitinol distal segment. The radiopaque soft platinum tip allows for smooth, selective access and the ultra slippery surface enhances navigation through tortuous anatomy.
Tenor Steerable Guidewire. Our Tenor Steerable Guidewire is also designed specifically to complement our EmboCath Plus Infusion Microcatheter. We designed our Tenor Steerable Guidewire as a second generation to our Sequitor Steerable Guidewire. The product attributes of the Tenor Steerable Guidewire are:
- •
- exceptional pushability and torqueability for precise navigation;
- •
- a specially designed distal core with a highly shapeable tip to suit challenging anatomy;
11
- •
- a platinum coil supported by a radiopaque polymer jacket provides for accurate placement and increased durability;
- •
- hydrophilic coating for smooth tracking and superb handling; and
- •
- an environmentally friendly proximal polytetrafluoroethylene, or PTFE, coated core free of perfluorooctanic acid, or PFOA.
Other Products
We also sold barium delivery kits and other ancillary products in the European Union during 2008 and 2007. We purchased barium from a third party and resold it for use in gastrointestinal medical testing. While we generated 3% and 9% of our revenue in 2008 and 2007, respectively, from these nonstrategic products, we phased out this nonstrategic business during 2008 in order to focus on our core embotheraphy business.
MANUFACTURING AND SUPPLY
We currently produce and package all of our microsphere products at our facility in Roissy, France. Manufacturing of our microsphere products includes the synthesis and processing of raw materials and third-party manufactured compounds. The assembly and packaging of delivery systems, which include the EmboCath Plus Infusion Microcatheter, Sequitor Steerable Guidewire, Tenor Steerable Guidewire and Segway Guidewire are conducted by medical device contract manufacturers in the United States and Europe. We currently purchase key components and services with respect to our microspheres, catheters and guidewires from approximately ten third-party vendors, including third parties from whom we purchase guidewires for our catheters for our EmboCath Plus Infusion Microcatheter product and guidewires for our Sequitor Steerable Guidewire, Tenor Steerable Guidewire and Segway Guidewire products.
MARKETING AND SALES
In 2009, we marketed our embolotherapy and delivery systems products through a direct sales force covering 22 territories in the United States and two territories in France, and through distributors in Europe, Asia, Canada, the Middle East, South America and other parts of the world. Approximately 82% of our product revenue was generated through our direct sales force in 2009.
As part of our sales and marketing efforts, we attend major medical conventions throughout the world pertaining to our targeted markets and invest in market development, including physician training, practice building, referral network education and patient outreach. We work closely with major interventional radiology centers in the areas of training, therapy awareness programs, clinical studies and ongoing research. In 2009, one of our key initiatives was what we have called "Community Health Talks," or CHTs, an educational outreach to women likely to have symptomatic fibroids. These programs were executed in partnership with the hospital, with close collaboration between physicians in interventional radiology and gynecology. The goal of the CHT initiative is to educate women about fibroids and all their available treatment options, even though they many not seek immediate consult for UFE. We believe these women typically will seek a consult with their gynecologist or primary care or family practice physician who may then be referred back for a UFE sometime in the future.
No single customer accounted for more than 10% of our revenue in 2009. Our principal source of revenue in each of the last three fiscal years was from sales of our microsphere products. For the years ended December 31, 2009, 2008 and 2007, revenue from the sale of our microsphere products accounted for 94%, 92% and 85% respectively, of our total revenue.
On April 16, 2009, we entered into an international distribution agreement with Nippon Kayaku Co. Ltd., or Nippon Kayaku. The agreement grants Nippon Kayaku the exclusive right to
12
distribute our HepaSphere Microspheres and Embosphere Microspheres in Japan, upon regulatory approval. The agreement provides that Nippon Kayaku is responsible for filing, obtaining and maintaining all regulatory approvals necessary for the sale, marketing, pricing and reimbursement of the products in Japan, including performing any clinical trials required as a result of seeking such regulatory approvals in Japan. The distribution agreement establishes specified dates for the achievement of specified regulatory approval-related milestones by Nippon Kayaku and also provides that Nippon Kayaku's failure to achieve such milestones shall constitute a material breach of the distribution agreement, except in the circumstances of an excused delay. As further provided in the distribution agreement, Nippon Kayaku agrees to use diligent efforts to comply with its obligations thereunder.
Nippon Kayaku has not been granted manufacturing rights under the distribution agreement and we have the right to terminate the distribution agreement on 18 months' prior written notice if we determine to cease manufacturing the products; provided that we and Nippon Kayaku may elect to negotiate in good faith the terms of an exclusive royalty-bearing manufacturing license grant to Nippon Kayaku if it desires to continue distributing the products for the remainder of the term of the distribution agreement.
Assuming product approval, we will provide HepaSphere Microspheres and Embosphere Microspheres to Nippon Kayaku for distribution and sale in Japan in accordance with a predetermined formula that is indexed to the Japanese government reimbursement rate. Additionally, Nippon Kayaku made a non-refundable milestone payment of $1.00 million in 2009 and has agreed to pay up to $3.00 million in additional milestone payments based upon specified objectives, including achievement of clinical, regulatory and sales goals.
Assuming regulatory approvals have been obtained, thereafter Nippon Kayaku is subject to minimum purchase requirements. Failure to meet such requirements will constitute a breach of the distribution agreement, unless we elect to appoint Nippon Kayaku as a non-exclusive distributor in the territory. All intellectual property that arises out of Nippon Kayaku's performance under the distribution agreement will be jointly owned by the parties in Japan and solely by us outside of Japan, subject to the terms of the distribution agreement. In addition, Nippon Kayaku agrees not to compete with us during, or for a specified period of time after, the term of the distribution agreement, subject to limited exceptions.
The term of the agreement begins on April 16, 2009 and runs until April 16, 2022 unless earlier terminated (i) by either party as a result of a material breach or default under the agreement that remains uncured, (ii) as a result of bankruptcy, insolvency, reorganization, receivership or otherwise of either party, and (iii) on a product-by-product basis if the parties are unable to agree on a purchase price for a particular product or if Nippon Kayaku reasonably determines that a product infringes or is likely to infringe on the intellectual property of a third party.
RESEARCH AND DEVELOPMENT
Research and development expenses for the years ended December 31, 2009, 2008 and 2007 were $3.42 million, $3.31 million and $2.34 million, respectively, or 11%, 11% and 9%, respectively, of total revenue. Research and development expenses in these periods relate primarily to:
- •
- efforts to develop improved manufacturing processes for our currently marketed products;
- •
- research to identify and evaluate new and innovative embolotherapy products based on our platform microsphere technology,
including a smaller-sized microsphere for our HepaSphere Microsphere and QuadraSphere Microsphere product offerings;
- •
- research to identify and evaluate new applications for our products;
13
- •
- efforts to develop a new generation of steerable guidewire to augment and/or replace our current guidewire product
offerings; and
- •
- further preclinical testing and nonclinical trials to support initial and/or additional clinical indications and/or premarketing clearances for our Embosphere Microspheres, HepaSphere Microspheres, QuadraSphere Microspheres, Sequitor Steerable Guidewire and EmboCath Plus Infusion Microcatheter, all of which are currently cleared and marketed for specified indications and in specified geographic locations.
COMPETITION
We encounter, and expect to continue to encounter, competition in the sale of our current and future embolotherapy and delivery system products. Many of our current competitors have, and our future competitors are likely to have, greater financial, operational, sales and marketing resources and more experience in research and development than we have. We compete primarily on the basis of product performance, ease of use, degree of targeted embolization control, and quality of patient outcome.
Embolotherapy. The primary competitive embolotherapy product has been non-spherical polyvinyl alcohol, or PVA, particles, a product introduced into the market more than 20 years ago. Currently, the primary products with which our microspheres compete are spherical PVA, sold by Boston Scientific Corporation, Biocompatibles and Terumo Corporation; Embozene sold by CeloNova Biosciences, Inc.; gel foam, sold by Pfizer Inc.; and non-spherical (particle) PVA, sold by Boston Scientific and Cook Incorporated.
UFE. Our principal competitors in UFE are Biocompatibles, Boston Scientific, Cook, Cordis Corporation, a Johnson & Johnson company, Pfizer and Terumo, as well as companies selling or developing non-embolotherapy solutions for UFE.
Within the field of uterine artery embolization, we believe we are the market share leader and one of only three companies in the United States to have embolic products specifically indicated for use in UFE. Based on both research and clinical studies conducted on our product for UFE, we believe we offer physicians a high degree of consistent and predictable product performance, ease of use, targeted delivery, and durable vessel occlusion, and therefore satisfactory short- and long-term clinical outcomes validated by peer-reviewed publications, when compared to our competitors.
UFE competes with other treatments that are used to address symptoms related to fibroids. Endometrial ablation is a technique for addressing excessive uterine bleeding, or menorrhagia, in women. Most endometrial ablation systems are not indicated for use in treatment of fibroids; however, obstetricians and gynecologists may use this procedure to resolve symptoms secondary to fibroids. Robotic surgery is an additional option that offers a less invasive alternative to a full surgical hysterectomy or myomectomy. These procedures require purchase of an expensive piece of equipment and disposables required for the procedure. Although robotic surgery generally requires a smaller incision and therefore reduced time for healing, these procedures are still a surgical procedure requiring the surgical removal of either the tumor or entire uterus. Robotic surgery also requires extensive training and a specialized skill set.
Several companies have announced development programs intended to offer solutions that would enable gynecologists to treat uterine fibroids in their office using high-energy ultrasound, RF ablation, mechanical action or other energy-based techniques. No company is currently approved by the FDA to market a product in the gynecologist office market for this purpose.
14
Liver Cancer. We market and sell our HepaSphere Microspheres product for liver cancer indications in the European Union, Brazil and Russia. Our primary competitor in Europe for the treatment of liver cancer is Biocompatibles.
Delivery Systems. Our primary competitors in the field of delivery systems are Terumo Corporation, Cordis Corporation and Boston Scientific Corporation.
GOVERNMENT REGULATION
FDA Regulation. The FDA, and other federal, state, local, and foreign authorities regulate our products and manufacturing activities. Pursuant to the Federal Food, Drug, and Cosmetic Act and the regulations promulgated under that act, the FDA regulates the design, development, clinical trials, testing, manufacture, packaging, labeling, storage, distribution and promotion of medical devices. Before a new device that we develop can be introduced to the market, we must obtain market clearance through a 510(k) premarket notification or approval through a premarket approval application. Additionally, the new device may only be introduced to the market if the manufacturer quality system complies with the Quality System Regulation (21 CFR Part 820).
Changes in Cleared or Approved Devices. We must obtain new FDA 510(k) clearance or premarket approval when there is a major change or modification in the intended use or indications for use of a legally marketed device or a change or modification of the device, including product enhancements and product line extensions of a legally marketed device, as required by FDA regulations.
Current Good Manufacturing Practice / Quality System Regulation and Reporting. The Federal Food, Drug, and Cosmetic Act requires us to comply with the Quality System Regulation pertaining to all aspects of our product design and manufacturing process, including requirements for packaging, labeling and record keeping, complaint handling, corrective and preventive actions and internal auditing. The FDA enforces these requirements through periodic inspections of medical device manufacturers. In addition, the medical device reporting regulation requires us to inform the FDA whenever information reasonably suggests that one of our devices may have caused or contributed to a death or serious injury, or when one of our devices has malfunctioned, if the device would be likely to cause or contribute to a death or a serious injury in the event the malfunction were to recur. We believe that we, and all who manufacture our delivery systems, are in compliance with applicable Quality System Regulation and medical device reporting requirements.
Labeling and Advertising. Labeling and promotional activities are also subject to scrutiny by the FDA. Among other things, labeling violates the law if it is false or misleading in any respect or it fails to contain adequate directions for use. Moreover, product claims that are outside the labeling either approved or cleared by the FDA violate the Federal Food, Drug, and Cosmetic Act.
Our product promotion is also subject to regulation by the Federal Trade Commission under the Federal Trade Commission Act, which prohibits unfair methods of competition and unfair or deceptive acts or practices in or affecting commerce, as well as unfair or deceptive practices such as the dissemination of any false advertisement pertaining to medical devices.
Import Requirements. To import a device, the importer must file an entry notice and bond with the United States Customs Service pending an FDA decision on the product's admissibility. If the FDA refuses admission, Customs will issue a notice for redelivery. The failure to redeliver the product can result in monetary penalties. All devices are subject to FDA examination before release from Customs. Any article that appears to be in violation of the Federal Food, Drug, and Cosmetic Act may be refused admission and a notice of detention and hearing may be issued.
Export Requirements. Products for export from Europe and from the United States are subject to foreign countries' import requirements and the FDA's or European regulating bodies' exporting
15
requirements. In addition to the import requirements of foreign countries, we must also comply with the U.S. laws governing the export of products regulated by the FDA. Foreign countries often require, among other things, an FDA certificate for products for export, also called a Certificate for Foreign Government. To obtain this certificate from the FDA, the device manufacturer must apply to the FDA. The FDA certifies that the product has been granted clearance or approval in the United States and that the manufacturing facilities were in compliance with Quality Systems Regulation regulations at the time of the last FDA inspection.
Fines and Penalties for Noncompliance. Failure to comply with applicable FDA regulatory requirements could result in, among other things, withdrawal of market clearance or approval, injunctions, voluntary or mandatory patient/physician notifications, recalls, warning letters, product seizures, civil penalties, fines and criminal prosecutions. Federal Trade Commission enforcement can result in orders requiring, among other things, limits on advertising, corrective advertising, consumer redress, rescission of contracts and such other relief as may be deemed necessary.
Foreign Regulations. Medical device laws and regulations are also in effect in many countries outside of the United States. These range from comprehensive device approval requirements for some or all of our medical device products to more basic requests for product data or certification. The number and scope of these requirements are increasing. Sales of medical devices in the European Union are subject to compliance with the European Medical Device Directive. This directive contains requirements for quality system and essential requirements with which all manufacturers must comply. In February 2006, we obtained ISO 13485:2003 Quality Management Systems Requirements for Regulatory Purposes certification at our French facility and in April 2006 we obtained this certification at our facility in Rockland, Massachusetts.
Failure to Comply. Failure to materially comply with applicable federal, state and foreign medical device laws and regulations would likely have a material adverse effect on our business. In addition, federal, state and foreign regulations regarding the manufacture and sale of medical devices are subject to future changes.
Environmental Regulations. We are subject to various federal, state, local and foreign laws and regulations relating to the protection of the environment, as well as health and safety. In the course of our business, we are involved in the handling, storage and disposal of limited amounts of certain chemicals. The laws and regulations applicable to our operations include provisions that regulate the discharge of materials into the environment. Usually these environmental laws and regulations impose "strict liability," rendering a person liable without regard to negligence or fault on the part of such person. Such environmental laws and regulations may expose us to liability for the conduct of, or conditions caused by, others, or for acts that were in compliance with all applicable laws at the time the acts were performed. We have not been required to expend material amounts in connection with our efforts to comply with environmental requirements and do not believe that compliance with such requirements will have a material adverse effect upon our capital expenditures, results of operations or competitive position. Failure to comply with applicable environmental and related laws could have a material adverse effect on our business. In addition, because the requirements imposed by such laws and regulations are frequently changed, we are unable to predict the cost of compliance with such requirements in the future, or the effect of such laws on our capital expenditures, results of operations or competitive position.
Anti-Kickback Statutes. The Medicare and Medicaid Patient Protection Act of 1987, as amended, which is more commonly known as the federal health-care Anti-Kickback Statute, prohibits persons from, among other things, knowingly and willfully offering or paying remuneration, directly or indirectly, to a person to induce the purchase, order, lease, or recommendation of a good or service for which payment may be made in whole or part under a federal health-care program such as Medicare or Medicaid. The definition of remuneration has been broadly interpreted to include anything of value,
16
including, for example, gifts, discounts, the furnishing of supplies or equipment, credit arrangements, payments of cash and waivers of payments. Several courts have interpreted the statute's intended requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals or otherwise generate business involving goods or services reimbursed in whole or in part under federal health-care programs, the statute has been violated. The law contains several statutory exceptions, including payments to bona fide employees, certain discounts and certain payments to group purchasing organizations. Violations can result in significant penalties, imprisonment and exclusion from Medicare, Medicaid and other federal health-care programs. Exclusion of a manufacturer would preclude any federal health-care program from paying for its products. In addition, some courts have held that kickback arrangements can provide the basis for an action under the Federal False Claims Act, which is discussed in more detail below.
The Anti-Kickback Statute is broad and potentially prohibits many arrangements and practices that are lawful in businesses outside of the health-care industry. Recognizing that the Anti-Kickback Statute is broad and may technically prohibit many innocuous or beneficial arrangements, the Office of Inspector General of Health and Human Services, or OIG, issued a series of regulations, known as the safe harbors, beginning in July 1991. These safe harbors set forth provisions that, if all the applicable requirements are met, will assure health-care providers and other parties that they will not be prosecuted under the Anti-Kickback Statute. The failure of a transaction or arrangement to fit precisely within one or more safe harbors does not necessarily mean that it is illegal or that prosecution will be pursued. However, conduct and business arrangements that do not fully satisfy each applicable safe harbor may result in increased scrutiny by government enforcement authorities such as the OIG. Arrangements that implicate the Anti-Kickback Statute, and that do not fall within a safe harbor, are analyzed by the OIG on a case-by-case basis.
Government officials have focused recent enforcement efforts on, among other things, the sales and marketing activities of pharmaceutical, medical device, and other health-care companies, and recently have brought cases against individuals or entities with personnel who allegedly offered unlawful inducements to potential or existing customers in an attempt to procure their business. Settlements of these cases by health-care companies have involved significant fines and/or penalties and in some instances criminal pleas.
In addition to the Federal Anti-Kickback Statute, many states have their own anti-kickback laws. Often, these laws closely follow the language of the federal law, although they do not always have the same exceptions or safe harbors. In some states, these anti-kickback laws apply with respect to all payors, including commercial health insurance companies.
False Claims Laws. Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government or knowingly making, or causing to be made, a false statement to get a false claim paid. Manufacturers can be held liable under false claims laws, even if they do not submit claims to the government, if they are found to have caused submission of false claims. The Federal Civil False Claims Act also includes whistle blower provisions that allow private citizens to bring suit against an entity or individual on behalf of the United States and to recover a portion of any monetary recovery. Many of the recent highly publicized settlements in the health-care industry relating to sales and marketing practices have been cases brought under the False Claims Act. The majority of states also have statutes or regulations similar to the federal false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. Sanctions under these federal and state laws may include civil monetary penalties, exclusion of a manufacturer's products from reimbursement under government programs, criminal fines and imprisonment.
Privacy and Security. The Health Insurance Portability and Accountability Act of 1996, or HIPAA, and the rules promulgated thereunder require certain entities, referred to as covered entities, to comply
17
with established standards, including standards regarding the privacy and security of protected health information, or PHI. HIPAA further requires that covered entities enter into agreements meeting certain regulatory requirements with their business associates, as such term is defined by HIPAA, which, among other things, obligate the business associates to safeguard the covered entity's PHI against improper use and disclosure. While not directly regulated by HIPAA, a business associate may face significant contractual liability pursuant to such an agreement if the business associate breaches the agreement or causes the covered entity to fail to comply with HIPAA. In the course of our business operations, we have entered into several business associate agreements with certain of our customers that are covered entities. Pursuant to the terms of these business associate agreements, we have agreed, among other things, not to use or further disclose the covered entity's PHI except as permitted or required by the agreements or as required by law, to use reasonable safeguards to prevent prohibited disclosure of such PHI and to report to the covered entity any unauthorized uses or disclosures of such PHI. Accordingly, we incur compliance-related costs in meeting HIPAA-related obligations under business associates agreements to which we are a party. Moreover, if we fail to meet our contractual obligations under such agreements, we may incur significant liability.
In addition, HIPAA's criminal provisions potentially could be applied to a non-covered entity that aided and abetted the violation of, or conspired to violate, HIPAA, although we are unable at this time to determine conclusively whether our actions could be subject to prosecution in the event of an impermissible disclosure of health information to us. Also, many state laws regulate the use and disclosure of health information, and are not necessarily preempted by HIPAA, in particular those laws that afford greater protection to the individual than does HIPAA. Finally, in the event we change our business model and become a HIPAA-covered entity, we would be directly subject to HIPAA, its rules and its civil and criminal penalties.
PROPRIETARY TECHNOLOGY AND PATENT RIGHTS
We seek to establish and protect our proprietary technologies and products through a combination of patents, copyrights, trademarks and trade secrets, as well as by entering into licensing agreements and utilizing confidentiality agreements or provisions where appropriate. We have implemented a patent strategy designed to maximize our intellectual property rights. We are pursuing patent rights in the United States and foreign countries to protect the technology, inventions and improvements that we consider critical to the development of our products and business.
In 1998, we entered into an agreement with L'Assistance Publique-Hôpitaux de Paris, referred to as AP-HP, pursuant to which AP-HP has granted us the exclusive license to two United States patents and their foreign counterparts that we jointly own with AP-HP relating to Embosphere Microspheres. We are required to pay to AP-HP a royalty on the commercial sale of any products that incorporate technology covered by the patents. We may sublicense these exclusive rights under the agreement only with the prior written consent of AP-HP, which consent cannot be unreasonably withheld. Effective March 2, 2009, we and AP-HP amended our agreement such that our exclusive license was extended for the duration of both (i) the jointly owned U.S. and foreign counterpart patents which will expire in 2014 and 2012, respectively, and (ii) the products and specialties implementing the patents. The agreement can be terminated on three months' notice by either party if the other party does not perform one or more of its obligations under the agreement and fails to cure its nonperformance during the notice period.
We have a number of United States and foreign patents and pending applications related to our microsphere technologies and uses thereof. For example, we have at least six U.S. and twelve foreign patents, and four U.S. and four foreign counterpart pending applications related to microspheres and uses thereof for tissue bulking, tissue construction, dermal augmentation, and the treatment of gastroesophageal reflux disease, or GERD, and urinary incontinence that expire at various dates between 2019 and 2020. The U.S. and foreign counterpart patents expire at various dates between 2019
18
and 2020. We also have eleven issued foreign patents and at least four U.S. and fourteen foreign counterpart pending applications related to microspheres and uses thereof for drug delivery and gene therapy that expire at various dates between 2019 and 2020. Additionally, we have at least three patents in the U.S. and six foreign patents, as well as at least three U.S. and four foreign counterpart pending applications, related to PVA microspheres that expire in 2019. Other U.S. and foreign counterpart patent applications have also issued or are currently pending. The subjects of these patents and applications include new materials for embolization, new methods of using our materials for embolization and other applications, as well as new uses of our materials outside of embolization.
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the earliest date of filing a non-provisional patent application. In the United States, a patent's term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office in granting a patent, or may be shortened if a patent is terminally disclaimed over another patent.
The patent term of a patent that covers an FDA-approved product may also be eligible for patent term extension, which permits patent term restoration as compensation for the patent term lost during the FDA regulatory review process. The Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act, permits a patent term extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the drug is under regulatory review. Patent extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only one patent applicable to an approved product may be extended. Similar provisions are available in Europe and other foreign jurisdictions to extend the term of a patent that covers an approved product.
We currently own the following U.S. trademarks:
- •
- ask4UFE.com®
- •
- Ask4Tell4™
- •
- BioSphere Medical®
- •
- Embosphere®
- •
- EmboCath® Plus
- •
- EmboGold®
- •
- HepaSphere™
- •
- PassThru®
- •
- QuadraSphere®
- •
- Tenor™
- •
- Segway®
- •
- Sequitor®
19
Our success depends to a significant degree upon our ability to develop proprietary products and technologies and to obtain patent coverage for these products and technologies. We intend to continue to file patent applications covering any newly developed products and technologies. However, as discussed above, there can be no guarantee that any of our pending or future filed applications will issue as patents. There can be no guarantee that the United States Patent and Trademark Office or some third party will not initiate an interference proceeding involving any of our pending U.S. applications or U.S. patents, or that a third party will not oppose any granted patent in Europe. There can be no guarantee that a third party will not file an opposition or comparable proceeding against any of our foreign patents or pending patent applications. Finally, there can be no guarantee that our issued patents or future issued patents, if any, will provide adequate protection from competition.
Patents provide some degree of protection for our proprietary technology. However, the pursuit and assertion of patent rights, particularly in areas like medical device development, involve complex legal and factual determinations and, therefore, are characterized by significant uncertainty. Specifically, enforcement or defense of our patents against potential or actual third-party infringers may impose a significant burden on our financial and human resources, and we may be limited in our ability to protect all of our rights. If we enforce our patents against third parties, they may challenge the validity or enforceability of our patents. We cannot predict whether we will be successful in enforcing our patents or defending their validity or enforceability.
In addition, the laws governing patent issuance and the scope of patent coverage continue to evolve, particularly in the life sciences, and the patent rights we possess, or are pursuing, generally cover our technologies to varying degrees. As a result, we cannot ensure that patents will issue from any of our patent applications or from applications licensed to us, or that any of our issued patents will offer meaningful protection. In addition, our issued patents or patents licensed to us may be successfully challenged, invalidated, circumvented or rendered unenforceable so that our patent rights may not create an effective competitive barrier. Moreover, the laws of some foreign countries may not protect our proprietary rights to the same extent as do the laws of the United States. There can be no assurance that any patents issued to us will provide a legal basis for establishing an exclusive market for our products or provide us with any competitive advantages, or that the patents of others will not have an adverse effect on our ability to do business or to continue to use our technologies freely. In view of these factors, the value of our intellectual property position is uncertain.
We have a European Patent, EP 1128816, related to certain PVA microspheres useful for embolization and methods thereof. We have validated this European patent in Germany, Spain, France, United Kingdom and Italy. On January 13, 2005, we were notified of a Notice of Opposition filed in the European Patent Office, or EPO, by Biocompatibles UK Limited on December 23, 2004, challenging the patentability of the claims in this European patent. We filed a response to the Notice of Opposition in August 2005. Biocompatibles UK Limited subsequently filed a response in 2006. On December 10, 2007, the European Patent Office upheld the claims of our patent in amended form and rendered its formal written decision on December 27, 2007. We and Biocompatibles have appealed this decision. The EPO has not set a date, but we expect oral proceedings to be scheduled within the next twelve months. We intend to continue defending our European PVA patent in this appeal. While we are not able to predict the outcome of this proceeding, it will not impact our ability to sell our Embosphere Microsphere or HepaSphere Microsphere products in Europe.
We have a European Patent, EP 1267839, which relates to certain drug-loaded microspheres and their use in embolization. We have validated this European Patent in Austria, Belgium, Germany, Spain, France, United Kingdom, Greece, Italy, Luxembourg and the Netherlands. On July 1, 2008, Biocompatibles UK Limited filed a Notice of Opposition in the EPO challenging the patentability of the claims in this European Patent. We filed a reply to Notice of Opposition on March 19, 2009. Oral Proceedings are scheduled for September 23, 2010. We intend to continue to defend our European
20
patent and will file a timely response. We are not able to predict the outcome of this opposition proceeding.
We may be subject to third parties filing claims asserting that our technologies or products infringe on their intellectual property. We cannot predict whether third parties will assert such claims against us or our licensees or against the licensors of technology licensed to us, or whether those claims will harm our business. If we are forced to defend against such claims, regardless of their merit or whether they are resolved in favor of or against us, our licensees or our licensors, we may face costly litigation and diversion of management's attention and resources. As a result of such disputes, we may have to develop, at a substantial cost, non infringing technology, or enter into licensing agreements. These agreements, if necessary, may be unavailable on terms acceptable to us, or at all, which could seriously harm our business or financial condition.
We also rely in part on trade secret protection of our intellectual property. We attempt to protect our trade secrets by entering into confidentiality agreements with third parties, employees and consultants. Our employees also sign agreements assigning to us their interests in inventions and original expressions and any corresponding patents and copyrights arising from their work for us. However, it is possible that these agreements may be breached, invalidated or rendered unenforceable, and, if so, our trade secrets could be disclosed to others, including our competitors, and there may not be an adequate corrective remedy available. Despite the measures we have taken to protect our intellectual property, parties to our agreements may breach the confidentiality provisions in our contracts or infringe or misappropriate our patents, copyrights, trademarks, trade secrets and other proprietary rights. In addition, third parties may independently discover or invent competitive technologies, or reverse engineer our trade secrets or other technology. Therefore, the measures we are taking to protect our proprietary technology may not be adequate.
SEGMENT INFORMATION
We develop microspheres and other ancillary embolotherapy products for use in the treatment of hypervascularized tumors, including uterine fibroids and arteriovenous malformations. We operate exclusively in the embolotherapy product business, which we consider as one business segment. Further segment information can be found in Note 10 of the notes to our consolidated financial statements, included elsewhere in this annual report on Form 10-K.
EMPLOYEES
As of December 31, 2009, we had approximately 88 employees. Of these employees, 8 are primarily engaged in research, development and clinical activities, 27 are engaged in manufacturing, 42 are engaged in sales and marketing, and the remainder are engaged in finance and administration. Of these 88 employees, 54 are located in the United States and 34 are located in France.
Our employees in the United States are not covered by a collective bargaining agreement. In Europe, our employees are covered by the provisions of an agreement setting forth national guidelines and standards for labor relations within our industry. We consider our relations with our employees to be good.
21
Investing in our common stock involves a high degree of risk. The risks described below are not the only ones facing our company. Additional risks not presently known to us or that we deem immaterial may also impair our business operations. Any of the following risks could materially adversely affect our business, operating results and financial condition and could result in a complete loss of your investment.
Risks Relating to Our Future Profitability, Our Financial Results and Need For Financing
Because we have a history of losses and our future profitability is uncertain, our common stock is a speculative investment.
We have incurred operating losses since our inception and, as of December 31, 2009, had an accumulated deficit of approximately $93.38 million. We expect to spend substantial funds to continue research and product testing, to maintain sales, to perform clinical trials, for marketing, quality control, regulatory, manufacturing and administrative capabilities and for other general corporate purposes. We expect to continue to incur operating losses in 2010 as we seek to execute on our business plan, including continuing to establish sales and marketing capabilities and conducting research and development activities.
We may never become profitable. If we do become profitable, we may not remain profitable on a continuing basis. Our failure to become and remain profitable could depress the market price of our common stock and impair our ability to raise capital and expand, diversify or continue our operations.
We will continue to need additional funds, and if additional capital is not available, we may have to limit or scale back our operations.
We believe that our existing cash and other working capital, together with anticipated proceeds from sales of our products, will be sufficient to fund our currently planned operating and capital requirements through at least the next twelve months.
Our currently planned operating and capital requirements primarily include the need for working capital to:
- •
- produce and manufacture our products;
- •
- support our sales and marketing efforts for our Embosphere Microsphere products for UFE and other indications, as well as
our other products for sale;
- •
- support our QuadraSphere Microspheres clinical trial, if we commence such trial;
- •
- support our ongoing research and development activities; and
- •
- fund our general and administrative costs and expenses.
However, our cash requirements may vary materially from those now planned due to a number of factors, including, without limitation:
- •
- unanticipated changes in the amount of revenue we generate from sales of our products, in particular from sales of our
Embosphere Microspheres for UFE;
- •
- an adverse judgment in a product liability lawsuit which could materially adversely affect market acceptance of our
products, and, if not covered by our product liability insurance, could have a material adverse effect on our liquidity;
- •
- unplanned costs associated with our QuadraSphere Microspheres clinical trial, if we commence such trial;
22
- •
- costs resulting from changes in our research and development, regulatory and marketing strategies;
- •
- competitive advances that make it harder for us to market and sell our products;
- •
- the timing and cost of regulatory approvals and clearances; and
- •
- adverse global market and economic conditions.
We may also need additional funds for possible strategic acquisitions of synergistic businesses, products and/or technologies.
We will require substantial additional cash to fund our planned, and any unplanned, expenses. If adequate funds are not available, we could be required to reduce our capital expenditures, scale back or eliminate some or all of our research, development, sales and marketing initiatives, reduce our workforce, and license to others or divest products or technologies that we otherwise would seek to commercialize ourselves. We may seek additional funding through a combination of collaborative arrangements, debt financing or the sale of additional equity securities. We may not receive such additional funding on reasonable terms, or at all. Any sales of equity or debt securities are likely to dilute the ownership of our existing stockholders, and the new securities may have rights, preferences or privileges senior to those of existing holders of our capital stock. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through collaboration and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or products, or grant licenses on terms that are not favorable to us.
If our operating results fluctuate significantly from quarter to quarter, then our stock price may decline.
Our operating results could fluctuate significantly from quarter to quarter. These fluctuations may be due to a number of factors, including:
- •
- inflation and adverse global economic conditions, which may affect the rate of UFE procedures and the sales of our
products;
- •
- the timing and volume of customer orders for our products;
- •
- introduction or announcement of competitive products;
- •
- regulatory approvals or clearances;
- •
- product recalls;
- •
- product liability claims against our products, including any adverse judgments;
- •
- turnover in our direct sales force;
- •
- the timing and amount of expenses;
- •
- timing of orders by our distributors;
- •
- the effectiveness of new marketing and sales programs;
- •
- exchange rate fluctuations; and
- •
- liquidity constraints and losses in our invested cash due to the current adverse conditions in the global capital markets.
In addition, a large portion of our expenses, including expenses for facilities, equipment and personnel, are relatively fixed. Accordingly, if our revenue declines or does not grow as much as we anticipate, we might not be able to improve our operating margins. Failure to achieve anticipated levels
23
of revenue could significantly harm our operating results for a particular fiscal period. Due to these fluctuations, our operating results in some quarters may not meet the expectations of our investors and our stock price may decline as a result.
Unstable market and economic conditions may have serious adverse consequences on our business.
As widely reported, global credit and financial markets have been experiencing extreme disruptions in the past year, including severely diminished liquidity and credit availability, declines in consumer confidence, declines in economic growth, increases in unemployment rates, and uncertainty about economic stability. There can be no assurance that further deterioration in credit and financial markets and confidence in economic conditions will not occur. Our general business strategy may be adversely affected by the recent economic downturn and volatile business environment and continued unpredictable and unstable market conditions. The global economic crisis could adversely affect sales of our products. For example, worldwide product revenue from sales of our microspheres for use in interventional gynecology for UFE procedures declined in part due to current economic conditions, including the high rate of unemployment, which we believe reduced the demand for elective procedures and the use of our products. We may experience declines in revenue in 2010 and beyond as a result of these factors. Also, if the current equity and credit markets deteriorate further, or do not improve, it may make any necessary debt or equity financing more difficult, more costly, and more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance and stock price and could require us to delay or abandon clinical development plans. In addition, there is a risk that one or more of our current service providers, manufacturers and other partners may not survive these difficult economic times, which would directly affect our ability to attain our operating goals.
At December 31, 2009, we had $18.09 million of cash, cash equivalents and marketable securities consisting of corporate, bank, federal agency, mortgage-backed and government obligations. We are not aware of any downgrades, material losses, or other significant deterioration in the fair value of our cash equivalents or marketable securities since December 31, 2009, but no assurance can be given that further deterioration in conditions of the global credit and financial markets would not negatively impact our current portfolio of cash equivalents and marketable securities or our ability to meet our financing objectives. Further dislocations in the credit market may adversely impact the value and/or liquidity of marketable securities owned by us.
There is also a possibility that our stock price may decline because of the volatility of the stock market and the general economic downturn.
Compliance with changing regulation of corporate governance and public disclosure, as well as potential new accounting pronouncements, could impact our future financial position and results of operations.
Changing laws, regulations and standards relating to corporate governance and public disclosure, including SEC regulations and NASDAQ Global Market rules, could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. In addition, future changes in financial accounting standards may cause adverse, unexpected revenue fluctuations and affect our financial position or results of operations. New accounting pronouncements and varying interpretations of pronouncements have occurred with frequency in the past and may occur again in the future, and as a result we may be required to make changes in our accounting policies.
Our efforts to comply with evolving laws, regulations and standards have resulted in, and are likely to continue to result in, increased general and administrative expenses and management time related to compliance activities. We expect these efforts to require the continued commitment of significant resources. If our efforts to comply with new or changed laws, regulations and standards differ from the
24
activities intended by regulatory or governing bodies due to ambiguities related to practice, our reputation might be harmed and we might be subject to sanctions or investigation by regulatory authorities, such as the SEC. Any such action could adversely affect our financial results and tour stock price.
Failure to maintain effective internal controls in accordance with section 404 of the Sarbanes-Oxley Act could have a material adverse effect on our business and stock price.
Section 404 of the Sarbanes-Oxley Act of 2002 requires management's annual review and evaluation of our internal controls, and will require attestation of the effectiveness of our internal controls by our independent registered public accounting firm beginning with the fiscal year ended December 31, 2010. This process could require us to implement significant measures to improve our internal controls, may require us to hire additional personnel and outside advisory services, and will result in significant accounting and legal expenses. Any failure by us to maintain effective internal controls could have a material adverse effect on our business, operating results and stock price.
Changes to our performance in each jurisdiction in which we operate, resulting from either changes in our business or as a result of routine tax audits, could materially impact our deferred tax assets or could materially impact our future financial position or results of operations.
We use the asset and liability accounting method whereby deferred tax assets and liabilities are recognized based on temporary differences between the financial statement carrying amounts and tax bases of assets and liabilities using current statutory tax rates in each tax jurisdiction in which we operate. A valuation allowance against net deferred tax assets is recorded if, based on the available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized. Due to the size of our net operating loss carryforward in relation to our history of unprofitable operations, we have not recognized any of our net deferred tax assets. However, future improvements in operational performance in any tax jurisdiction in which we operate, while not guaranteed, could result in increased certainty of our ability to apply deferred tax assets against taxable income, which could, in turn, result in a significant impact on the value of our deferred tax assets and reported operating results.
Risks Relating to Our Industry, Business and Strategy
A significant portion of our revenue is derived from sales of our Embosphere Microspheres for UFE, and if we do not successfully commercialize and achieve widespread market acceptance of our Embosphere Microspheres for UFE, our business will be materially harmed and our stock price will decline.
The majority of our revenue for the year ended December 31, 2009 was derived from the sale of Embosphere Microspheres for use in UFE. Our principal business focus is to grow our embolotherapy business through increases in the utilization rate for UFE procedures versus other procedures to treat uterine fibroids and in the employment by medical providers of our Embosphere Microspheres in such procedures in lieu of competing products. We began marketing and selling Embosphere Microspheres for UFE in 2002, but to date we have not achieved widespread market acceptance of UFE as an alternative to other procedures, including hysterectomy. Our ability to grow our product revenue is substantially dependent upon growth in UFE procedures and our ability to achieve widespread acceptance of the use of Embosphere Microspheres for use in UFE procedures. If growth in UFE procedures does not occur; and if we do not achieve such market acceptance, our product revenue and our prospects for profitability and success will be materially adversely affected.
25
We face a number of significant risks relating to our ability to successfully commercialize Embosphere Microspheres for use in UFE, including risks relating to:
- •
- our ability to successfully market and sell Embosphere Microspheres for use in UFE with our limited sales force;
- •
- the success of our sales and marketing strategies for Embosphere Microspheres for use in UFE, including, but not limited
to, our ask4UFE and consumer health talk, or CHT, campaigns and other public relations campaigns, in which we are seeking to increase awareness among patients, referring physicians, interventional
radiologists and third-party payors of UFE as an alternative treatment for fibroids;
- •
- the unknown future impact that Health Care Reform and other measures may have on our ability to educate patients about
fibroids and treatment options on behalf of or in partnership with hospitals;
- •
- our ability to recruit and train our sales force and the effectiveness of our sales force, in influencing referral
behavior with gynecologists and other health-care providers;
- •
- reimbursement treatment from government and third-party insurers for our products;
- •
- longstanding use of established treatment options for uterine fibroids and/or the emergence of new treatment options;
- •
- our ability to gain market acceptance of UFE using Embosphere Microspheres as a safe, effective and medically necessary
treatment for uterine fibroids;
- •
- the availability of substantial amounts of cash to fund our commercialization plans;
- •
- competitive factors;
- •
- our ability to effectively develop adequate marketing, manufacturing, and distribution capabilities;
- •
- our ability to maintain the necessary patent protection and regulatory approvals required to market and sell Embosphere
Microspheres for UFE;
- •
- unemployment levels and adverse economic conditions, which may cause a decrease in UFE procedure rates and sales of our
products; and
- •
- the various other factors discussed in detail throughout this section titled "Risk Factors."
If the market concludes that our products are not safe or effective, we will not achieve widespread market acceptance of our microsphere products, and our business prospects will be seriously harmed.
In the United States, we began selling our first microsphere product in the first half of 2000. In November 2002, we received FDA clearance to market our Embosphere Microspheres in the United States for UFE procedures. We began to market and sell our HepaSphere Microspheres in the European Union in the fourth quarter of 2005 and received marketing clearance from the FDA for our QuadraSphere Microspheres in November 2006. Our success will depend upon increasing acceptance by the medical community, patients and third-party payors that our Embosphere Microspheres and other products are medically therapeutic and cost-effective. Our products may not gain widespread market acceptance for a variety of reasons, including:
- •
- Our microspheres are designed to occlude targeted blood vessels permanently. There is some risk that some or all of the microspheres used in a medical procedure may travel in the blood system to sites other than the intended treatment site and occlude, or block, other blood vessels,
26
- •
- To use our microspheres correctly for a particular medical procedure, trained physicians must correctly evaluate the
subject's vasculature, select and use the proper size and quantity of the product, and carry out appropriate placement of the product. Physician error potentially could have significant adverse health
effects on the patient, including death.
- •
- In UFE procedures, patients commonly experience a day or two of post-procedure abdominal pain or cramping.
Other infrequently occurring complications may include allergic reactions, rashes, early onset of menopause, infertility and infection that may, in some cases, require a hysterectomy. We are also
aware that a small number of the patient population, which we believe constituted approximately 2% of those receiving the UFE procedure using EmboGold Microspheres, reported a delayed onset of rash
and/or pain.
- •
- There are only limited data concerning the long-term health effects on persons receiving embolotherapy using
our microspheres. For example, the effect of UFE on continued fertility has not yet been specifically studied, and our FDA clearance for Embosphere Microspheres currently does not include women who
desire future pregnancy.
- •
- Product liability claims could create a perception that our products are unsafe. For example, we were named as a defendant
in two product liability lawsuits in which the plaintiffs claimed that they were harmed as a result of the use of our microspheres or the negligence of the health-care providers or both
factors combined.
- •
- Many health-care providers, including obstetricians and gynecologists, use other forms of treatment for
patients with uterine fibroids that do not require referral to an interventional radiologist.
- •
- We received approval to use our HepaSphere Microspheres to treat liver cancer using procedures such as targeted liver embolotherapy and transarterial chemoembolization in the European Union. Physicians may refrain from using our product for such procedures until further clinical data demonstrate its safety and efficacy as compared to other treatments. Physicians also may not elect to use our HepaSphere Microspheres to treat liver cancers for a number of other reasons, including, without limitation, unfavorable reimbursement from third-party payors, the effectiveness of our competitors in marketing their products, and our failure to convince physicians that our HepaSphere Microspheres have greater benefits than existing products or therapies.
resulting in the potential for significant adverse health effects on the patient or, in a worst case, even death.
In March 2006, we instituted a voluntary recall of our HepaSphere Microspheres in Europe to correct a packaging defect that we identified while conducting aging studies. HepaSphere Microspheres are contained in a prefilled vial that was in turn initially packaged inside a paper pouch. In the third quarter of 2006, we launched a new plastic packaging configuration for our HepaSphere MicroSphere product designed to correct this defect. Although we are not aware of any adverse events resulting from the defects in the paper packaging, our voluntary recall of this product, or any future recall, voluntary or mandatory, of any of our products, could result in reputational harm or a perception that the recalled product is not safe, either of which could adversely affect market acceptance of our products and result in decreased sales.
Other factors could also affect market acceptance of our products, including, without limitation, the introduction of competing products, safety concerns with similar products marketed by others, ineffective sales, marketing and distribution support and significant warranty claims.
27
If gynecologists, obstetricians, interventional radiologists and other health-care providers do not recommend and endorse our products, and if health-care providers do not make the necessary referrals to interventional radiologists who administer our embolotherapy products, our sales may decline or we may be unable to increase our sales and profits.
Our ability to establish and maintain favorable relationships with gynecologists, obstetricians, interventional radiologists and other health-care providers is critical to our continued growth. We believe that the success of these relationships is, and will be, based on, among other things, the quality of our products, such providers' perceptions concerning our commitment to embolotherapy treatments, our marketing efforts and our presence at medical society and trade association meetings. Any actual or perceived diminution in our reputation or the quality of our products, or our failure or inability to maintain these or other efforts, could damage our current relationships or prevent us from forming new relationships with health-care professionals and cause our growth to be limited and our business to be harmed.
In order for us to sell our products, health-care professionals must recommend and endorse them. For example, our embolotherapy techniques are administered by interventional radiologists. In the treatment of uterine fibroids, we believe that the UFE procedure utilizing our Embosphere Microspheres has not yet achieved widespread acceptance primarily because obstetrics and gynecology physicians may elect to offer and provide other forms of treatment to their patients with uterine fibroids that do not require a referral to another specialist, such as an interventional radiologist. The majority of our revenue is from the sale of our Embosphere Microspheres for UFE and, accordingly, our future success will depend upon obstetrics and gynecology physicians referring patients to interventional radiologists to receive treatment using our Embosphere Microspheres in lieu of, or in addition to, receiving other forms of treatment that the obstetrics and gynecology physicians can otherwise provide directly. We have not achieved widespread market acceptance for UFE as an alternative to other forms of treatment. Acceptance of UFE as a procedure, and our ability to obtain the necessary endorsements and referrals, depend on educating the medical community as to the distinctive characteristics, perceived benefits, safety, clinical efficacy and cost-effectiveness of our products compared to traditional methods of treatment and the products of our competitors, and on training health-care professionals in the proper application of our products. If we are not successful in obtaining the recommendations or endorsements of gynecologists, obstetricians, interventional radiologists and other health-care professionals for our products, our sales may decline or we may be unable to increase our sales and profits.
Product liability claims could create a perception that our products are unsafe. For example, we were named as a defendant in two product liability lawsuits in which the plaintiffs claimed that they were harmed as a result of the use of our microspheres or the negligence of the health-care providers or both factors combined.
If we experience delays, difficulties or unanticipated costs in establishing and growing the sales, distribution and marketing capabilities necessary to successfully commercialize our products, we will have difficulty maintaining and seeking to increase our sales.
We continue to develop sales, distribution and marketing capabilities primarily in the United States, the European Union, Asia and South America to promote UFE awareness and the benefits of our product for the treatment of uterine fibroids. It has been, and we expect it will continue to be, expensive and time-consuming for us to seek to develop a global sales and marketing force. At December 31, 2009, we had a sales and marketing force of 42 persons located principally in the United States. Competition for skilled salespersons in the medical device industry is intense, and we may not be able to provide adequate incentives to maintain our sales and marketing force or to attract new sales and marketing personnel to promote our products. We have only limited sales and marketing
28
experience in the United States and internationally and may not be successful in developing and implementing our strategy. Among other things, we need to:
- •
- provide or ensure that our distribution channels provide the technical and educational support customers need to use our
products successfully;
- •
- establish and implement successful sales and marketing and education programs that encourage our customers to purchase our
products;
- •
- manage geographically dispersed markets; and
- •
- modify our products and marketing and sales programs for foreign markets.
We currently have distribution agreements with a number of third-party distributors, and we may choose or find it necessary to enter into additional third-party agreements to sell, distribute or market our products in the future. Any third party with whom we have established a sales, distribution and/or marketing relationship may not devote sufficient time to the marketing and sales of our products, thereby adversely affecting our planned revenue and exposing us to potential expenses in terminating such distribution agreements. We and any of our third-party collaborators must also market our products in compliance with federal, state and local laws relating to the provision of incentives and inducements. Violation of these laws can result in substantial penalties. If we are unable to successfully motivate and expand our marketing and sales force and further develop our sales and marketing capabilities, or if our distributors fail to promote our products, we will have difficulty maintaining and increasing our sales, we may not achieve profitability and our stock price could decline.
We will be required to expend significant resources for research, development, testing and regulatory approval or clearance of our products under development, and these products may not be developed successfully.
We are developing and commercializing products for medical applications using embolotherapy techniques, including, without limitation, a smaller-sized version of our HepaSphere Microsphere and QuadraSphere Microsphere, which are still in preclinical development. Our products under development may not provide greater benefits than current treatments or products, or alternative treatments or products under development.
All of our products under development will require significant additional research, development, engineering and preclinical and/or clinical testing, as well as regulatory approval or clearance and a commitment of significant additional resources prior to their commercialization. For example, FDA regulations require that we conduct clinical trials and submit a marketing application which includes positive data from clinical trials to the FDA in order to obtain the approvals and clearances required to promote QuadraSphere Microspheres for the treatment of a specific disease or condition, including primary and metastatic liver cancer. In October 2009, we submitted to the FDA an IDE application to commence a clinical trial to compare the effectiveness of our QuadraSphere Microspheres combined with the chemotherapeutic agent doxorubicin against conventional transarterial chemoembolization with doxorubicin in patients with primary liver cancer. The FDA has advised us that our study protocol will need to include survival as a primary endpoint for the trial, rather than overall tumor response rate at six months, which was the primary endpoint that we proposed in our initial IDE application. Our satisfactory resolution of the FDA's comments on the IDE is a condition to starting the clinical trial. We are currently evaluating the FDA's protocol requirements for the trial, including how the primary endpoint requirements will affect our plans regarding the size of the trial and the timeline and cost for completion. We have not reached a conclusion about whether or when to undertake the clinical trial and if we determine to not undertake the trial, or if we undertake the trial and the results are not sufficient to obtain FDA approval, then we will not be able to promote our QuadraSphere Microspheres for liver cancer indications. Our potential products may not:
- •
- be developed successfully;
29
- •
- be proven safe and effective in clinical trials;
- •
- offer therapeutic or other improvements over current treatments and products;
- •
- meet applicable regulatory standards or receive regulatory approvals or clearances;
- •
- be capable of production in commercial quantities at acceptable costs and in compliance with regulatory requirements; or
- •
- be successfully marketed.
If we do not develop and introduce new products, our business may not grow and our future prospects may be adversely affected.
In order to grow our revenue in future periods we need to develop and introduce new applications for our embolotherapy technology and pursue opportunities for microsphere technology in other medical applications. Any such new application for our embolotherapy technology or microsphere technology will be subject to a number of risks inherent in the development and commercialization of a medical device product, including uncertainties with respect to the successful completion of clinical trials, our ability to achieve and maintain, and our willingness to seek, required regulatory approvals or clearances and our ability to successfully commercialize, market and sell these new applications, if FDA approval or clearance is achieved. If, as a result of these or other risks, we are not successful in developing new applications and products, our position in, and share of, the markets in which we participate, and our business, financial condition, results of operations and prospects may be adversely affected.
We have been subject to product liability claims in the past and may be subject to additional claims in the future, we may incur substantial costs and expenses in defending such claims, and if we are unable to obtain or maintain adequate product liability insurance, we may have to pay significant monetary damages in a successful product liability claim against us.
The development and sale of medical devices entails an inherent risk of product liability. For example, if physicians do not use our products properly, if patients experience adverse side effects in procedures in which our products are used, or if patients, health-care providers or other constituencies conclude that any of our products are not safe or effective for any reason, we may be exposed to product liability claims. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability, and a breach of warranties. Claims could also be asserted under state consumer protection acts. In 2005 and 2008, we were named as a defendant in two such product liability lawsuits in which the plaintiffs alleged, among other things, that they were harmed by the use of our microsphere products, the negligence of the health-care providers or both of these factors combined. Although these lawsuits settled within the limits of our product liability insurance, we are subject to the risk of additional product liability lawsuits, and our business, financial condition, results of operations and future prospects are subject to a number of significant risks relating to any such lawsuits, including the following:
- •
- Claims asserting medical product liability have in the past resulted in multiple-million-dollar damage awards for plaintiffs against various manufacturers of drugs and medical devices. Although we currently maintain product liability insurance coverage, if a plaintiff in a product liability lawsuit brought against us were to prevail in his or her claims against us and was awarded substantial damages, our insurance, which is subject to a $5.00 million cap on the maximum amount our insurer is required to pay for all claims within any one-year period and which is eroded by the costs of defense, may not provide us with adequate coverage for a judgment against us. If we are forced to satisfy a judgment in excess of our product liability
30
- •
- Although we maintain product liability insurance, any claim that may be brought against us could result in court judgments
or settlements that are not covered, in whole or in part, by our insurance. For example, our current product liability insurance policy contains an exclusion for punitive damages, which are typically
sought in product liability lawsuits. Our insurance policies also have various other exclusions, and we may be subject to a product liability claim for which we have no coverage. We will have to pay
any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and, in such case, we could be required to make a
substantial payment for which we may not have sufficient cash. There can be no assurance that, if required, we would be able to raise the additional funds required to make such payment on favorable
terms, or at all. In such case, we may be required to curtail our operations, which could have a material adverse effect on our financial condition, results of operations, the viability of our
business and our future prospects, and would likely cause our stock price to decline.
- •
- Any product liability claim brought against us, regardless of whether it has merit, could result in an increase in our
product liability insurance rates or our inability to secure additional insurance coverage in the future.
- •
- Defending litigation can be time consuming and can divert our management's attention from other priorities, including the
execution of business plans and strategies that are important to our ability to grow our business.
- •
- Our reputation with patients and health-care providers, including the referring gynecologists, oncologists and
interventional radiologists, who are key to our current sales and marketing strategies, could be harmed because we have been named as a defendant in product liability lawsuits in the past, and could
also be harmed if we are subject to any such claims in the future. Any reputational harm could adversely affect our ability to execute on our current sales and marketing strategies and, ultimately, on
the sales of our products, and our stock price would likely decline.
- •
- If, as a result of past or future product liability litigation, the market perceives that our products are unsafe, we
could be unsuccessful in our efforts to gain market acceptance of our products and experience a decline in our revenue, which could adversely affect our financial condition, results of operations and
the viability of our business, and cause our stock price to decline as a result.
- •
- In the prior lawsuits in which we were named as a party, the plaintiffs alleged that our microspheres were defective and that the manner in which the applicable product was marketed was insufficient. As is often the case when product liability lawsuits arise that assert claims of defects in the manufacturing and design of a medical device or of a failure to warn of risks inherent to such product, the FDA and comparable regulatory agencies outside of the U.S. could engage in a further review of the safety of these products and their approved conditions for use. On the basis of that review, the FDA or such other regulatory agency could decide to impose additional requirements regarding the manufacturing, marketing or promotion of these products, require changes to the labeling of these products, recall these products, or commence proceedings to withdraw its clearance of the products, any of which could harm our reputation, adversely affect market acceptance of these products or cause a decline in revenue from the sale
coverage, we may not have sufficient cash to pay such judgment. There can be no assurance that, if required, we would be able to raise the additional funds required to satisfy such judgment on favorable terms, or at all. In such case, we may be required to curtail our operations, which could have a material adverse effect on our financial condition, results of operations, the viability of our business and future prospects, and would likely cause our stock price to decline.
31
of the applicable product, which could adversely affect our financial condition, results of operations, the viability of our business and our future prospects, and cause our stock price to decline.
If we are not able to compete effectively, we may experience decreased demand for our products, which may result in price reductions.
The medical device market is characterized by extensive research and development, and rapid technological change. Our success depends upon our ability to develop and maintain a competitive position in both the embolotherapy and related delivery systems markets. We have many competitors in the United States and abroad, including medical device, biotechnology and other alternative therapeutic companies, universities and other private and public research institutions. Our key competitors in both the fields of embolotherapy and the delivery systems used in the UFE procedure are Biocompatibles Limited, Boston Scientific Corporation, Cook Incorporated, Cordis Corporation, a Johnson & Johnson company, Pfizer Inc., Terumo Corporation and CeloNova BioSciences, Inc.
Many of our competitors may have greater capabilities, experience and financial resources than we do. As a result, they may develop products more quickly or at less cost, that compete with our microsphere products and related delivery systems. For example, in recent years we have experienced increased competition from products that compete with Embosphere Microsphere products for UFE. Moreover, some of our competitors have provided free or reduced-price samples of competing forms of microspheres for use in medical procedures for which our Embosphere Microspheres are indicated. We believe the availability of these free or reduced-price samples may have adversely affected our revenue, and if this practice recurs our product revenue may continue to be adversely affected.
Currently, the primary products with which our microspheres compete for some of our applications are spherical PVA sold by Boston Scientific, Terumo and Biocompatibles, Embozene by CeloNova, gelfoam sold by Pfizer, and non-spherical (particle) PVA sold by Boston Scientific and Cook and drug-eluting beads manufactured by Biocompatibles.
In the treatment of symptomatic uterine fibroids, our customers compete with obstetrics and gynecology physicians who elect to offer and provide other forms of treatment to their patients with uterine fibroids that do not require referral to another specialist. These treatment options currently include hysterectomy, myomectomy, laparoscopic myomectomy, drug therapy and robotic-assisted hysterectomy.
Developments by other companies of new or improved products, processes or technologies, in particular in the market for treating uterine fibroids, may make our products or proposed products obsolete or less competitive and may negatively impact our revenue. As a result of these and other factors, we may not be able to improve our products or develop new products or technologies quickly enough to maintain a competitive position in our market and continue to develop our business commercially.
If we fail to maintain, or in some instances obtain, an adequate level of reimbursement for our products by third-party payors, there may be no commercially viable markets for our products.
The availability and levels of reimbursement by governmental and other third-party payors affects the market for any medical device. We may not be able to sell our products profitably if reimbursement is unavailable or limited in scope or amount. Some insurance companies do not fully reimburse for embolization procedures. These third-party payors may attempt to contain or reduce the costs of health care by lowering the rate at which providers are reimbursed for embolization procedures or challenging the prices that companies such as ours charge for medical products. For example, on January 1, 2007, the Centers for Medicare and Medicaid Services, or CMS, issued a rule providing for a single, all-inclusive reimbursement code for UFE. This code is inclusive of all services occurring on the day of
32
the procedure. This physician reimbursement rate is lower than the rate historically received by physicians. We believe that some physicians have shifted their procedural mix away from UFE in response to this change in reimbursement, which has, and may continue to, negatively affect our sales growth. Conversely, beginning January 1, 2008 the CMS assigned a new ambulatory procedure code, or APC, for UFE procedures performed in an outpatient setting. We believe that this new APC code has resulted in an increase in hospital reimbursement for UFE procedures. However, CMS could adversely change this APC code in the future or otherwise decrease the payment rate for such UFE procedures. Any change in reimbursement levels could have an adverse effect on utilization rates for UFE or liver embolization procedures.
In some foreign countries, particularly the countries of the European Union where our microsphere products are currently marketed and sold, the pricing of medical devices is subject to governmental control, and the prices charged for our products have in some instances been reduced as a result of these controls.
Initiatives to limit the growth of health-care costs, including price regulation, are underway in the United States and other major health-care markets. For example, payors may increase the complexity of patient precertification required prior to performing a UFE procedure. In addition, we may be affected by prescription drug benefit legislation recently enacted in the United States. It is unclear what, if any, impact on hospital and/or physician reimbursement levels for UFE may result from new health-care reform initiatives currently being debated in the U.S. Congress. While these initiatives have in many cases related to pharmaceutical pricing, implementation of more sweeping health-care reforms in significant markets may limit the price of, or the level at which reimbursement is provided for, our products and may influence a physician's selection of products used to treat patients.
If we do not recruit and retain senior management and other key employees, we may not be able to successfully implement our business strategy.
Our success is substantially dependent on our ability to recruit and retain members of our senior management, including Richard J. Faleschini, our president and chief executive officer; Martin J. Joyce, our executive vice president of finance and administration and chief financial officer; Melodie R. Domurad, Ph.D., our vice president of regulatory, medical affairs and quality systems; and Peter C. Sutcliffe, our vice president of manufacturing, and other key employees. Effective March 31, 2010, Willard W. Hennemann, Ph. D., our vice president of new products and business development, and Joel B. Weinstein, our vice president of global marketing and sales, will step down from their positions and cease to be employed by us. Disruptions in our business could result in the near term as a result of their departure or any other such departures. All of the agreements with our officers provide that their employment may be terminated either by the employee or by us at any time and without notice. The loss of the services of any of these persons might impede the achievement of our research, development and commercialization objectives. We do not carry key man life insurance on any of our executive officers or other personnel.
If we make any acquisitions, we will incur a variety of costs and may never successfully integrate the acquired businesses into ours.
We may attempt to acquire businesses, technologies, services or products that we believe are a strategic complement to our business model. We may encounter operating difficulties and expenditures relating to the integration of an acquired business, technology, service or product. These acquisitions may also absorb significant management attention that would otherwise be available for ongoing development of our business. Moreover, we may never realize the anticipated benefits of any acquisition. We may also make dilutive issuances of equity securities, incur debt or experience a decrease in the cash available for our operations, or incur contingent liabilities in connection with any future acquisitions.
33
Because key stockholders beneficially own a significant amount of our common stock, they may be able to exert control over us.
As of March 1, 2010, we believe that Sepracor Inc., an indirect wholly owned subsidiary of Dainippon Sumitomo Pharma Co., Ltd., or Sepracor, and funds affiliated with Cerberus Capital Management, L.P., or Cerberus, beneficially owned approximately 21% and 13% of our outstanding common stock, respectively, including shares of common stock issuable upon the exercise of series A preferred stock held by these stockholders. Moreover, we have granted board-observation rights to Sepracor and Cerberus. Accordingly, Sepracor and Cerberus may have significant influence over corporate actions requiring stockholder approval, such as the election of directors, amendment of our charter documents and the approval of merger or significant asset sale transactions. In addition, the shares of our series A preferred stock held by Sepracor and Cerberus entitle them to certain voting rights in accordance with the terms and conditions of the series A preferred stock. Specifically, we will need the consent of holders of at least 50% of the series A preferred stock initially purchased by Sepracor and Cerberus to undertake certain key corporate actions, including the following:
- •
- amending our charter or bylaws in a manner that adversely affects the holders of series A preferred stock;
- •
- authorizing or issuing any equity security that is senior to or pari passu with the series A preferred stock; and
- •
- declaring or paying any dividends on, or redeeming or repurchasing any shares of, our capital stock, subject to customary exceptions.
The ownership concentration of Sepracor and Cerberus could cause the market price of our common stock to decline. In addition, conflicts of interest between these key stockholders and us may arise, including with respect to competitive business activities and control of our management and our affairs.
The holders of shares of our series A preferred stock have rights that could adversely affect an investment in our common stock.
The holders of our series A preferred stock have the right to an adjustment in the conversion rate of the series A preferred stock if we issue securities at a price below the purchase price paid by these holders. These provisions could substantially dilute stockholders' interest in us in the event of future financing transactions. The holders of series A preferred stock also have the right to receive a 6% dividend per annum which, at our election, may be paid in cash or additional shares of series A preferred stock. To the extent such dividends are paid in stock, this dividend could also further dilute stockholders' ownership interest. In addition, the holders of our series A preferred stock have the right to participate in future capital-raising transactions by us. The existence of this right may reduce our ability to establish terms with respect to, or enter into, any financing with parties other than the holders of our series A preferred stock.
In the event that we enter into an acquisition or business combination in which we sell all or substantially all of our assets, or if there occurs a change of control of a majority of our common stock outstanding prior to such transaction, the holders of our series A preferred stock will have the right to receive, before any distributions or payments to the holders of our common stock, an amount in cash equal to their initial purchase price for the Series A preferred Stock, $8,000,000, plus an amount equal to any accrued but unpaid dividends, and will then participate with the holders of the common stock on a pro-rata basis with respect to the distribution of any remaining assets. The existence of this right may make it difficult for us to raise capital in financing transactions with third parties and will also result in holders of our common stock receiving smaller distributions or payments upon a change of control or
34
asset sale than they would be entitled to receive if no preferential payments were required to be made to holders of our series A preferred stock.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with FDA regulations, to provide accurate information to the FDA, to comply with manufacturing standards we have established, to comply with federal and state health-care fraud and abuse laws and regulations or the Foreign Corrupt Practices Act, to report financial information or data accurately or to disclose unauthorized activities to us. Employee misconduct could also involve the improper use of customer information or information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We have adopted a Code of Business Conduct and Ethics, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses.
The marketing claims the FDA specifically authorized us to make regarding our microspheres in the United States were set forth in the FDA clearance. Our EmboGold Microspheres have not been specifically cleared for use in UFE. Although our QuadraSphere Microspheres are identical to our HepaSphere Microspheres, which are currently marketed in the European Union for use in the embolization of hepatocellular carcinoma and hepatic metastasis, our QuadraSphere Microspheres are not indicated for use in hepatocellular carcinoma and hepatic metastasis. We will need to conduct a clinical trial and obtain approval of a marketing application from the FDA in order to claim the use of the QuadraSphere Microspheres for the treatment of a specific disease or condition, such as hepatocellular cancer or hepatic metastasis in the United States, while European Union regulations do not require such an application for this class of medical devices. In order for us to seek FDA approval or clearance to promote the use of QuadraSphere Microspheres for the embolization of hepatocellular carcinoma and hepatic metastasis, we will need to complete a clinical trial and submit positive clinical data to the FDA. In October 2009, we submitted to the FDA an IDE application to commence a clinical trial to compare the effectiveness of our QuadraSphere Microspheres combined with the chemotherapeutic agent doxorubicin against conventional transarterial chemoembolization with doxorubicin in patients with primary liver cancer. The FDA has advised us that our study protocol will need to include survival as a primary endpoint for the trial, rather than overall tumor response rate at six months, which was the primary endpoint that we proposed in our initial IDE application. Our satisfactory resolution of the FDA's comments on the IDE is a condition to starting the clinical trial. We are currently evaluating the FDA's protocol requirements for the trial, including how the primary endpoint requirements will affect our plans regarding the size of the trial and the timeline and cost for completion. We have not reached a conclusion about whether or when to undertake the clinical trial and if we determine to not undertake the trial, or if we undertake the trial and the results are not sufficient to obtain FDA approval, then we will not be able to promote our QuadraSphere Microspheres for liver cancer indications. Although we have not received approval or clearance from the FDA to market our QuadraSphere Microspheres for primary or metastatic liver cancer in the United States, we believe that some physicians are using QuadraSphere Microspheres off-label in the treatment of primary and metastatic liver cancer. If the FDA were to conclude that we have improperly promoted our products for unapproved indications, the FDA could allege that our promotional activities misbrand or adulterate our products.
35
In addition, during the course of our operations, our directors, executives and employees may have access to material, nonpublic information regarding our business, our results of operations or potential transactions we are considering. Despite our adoption of an Insider Trading Policy, we may not be able to prevent a director or employee from trading in our common stock on the basis of, or while having access to, material, nonpublic information. If a director or employee were to be investigated, or an action were to be brought against a director or employee for insider trading, it could have a negative impact on our reputation and our stock price. Such a claim, with or without merit, could also result in substantial expenditures of time and money, and divert attention of our management team from other tasks important to the success of our business.
Risks Relating to Regulatory Matters
If we do not obtain and maintain the regulatory approvals or clearances required to market and sell our products, then our business may be unsuccessful and the market price of our stock may decline.
We are subject to regulation by government agencies in the United States and abroad with respect to the design, manufacture, packaging, labeling, advertising, promotion, distribution and sale of our products. For example, our products are subject to approval or clearance by the FDA prior to commercial marketing in the United States. Similar regulations exist in most major foreign markets, including the European Union, Latin America and Asia. The process of obtaining necessary regulatory approvals and clearances is time-consuming and expensive for us. If we do not receive required regulatory approval or clearance to market our products, or if any approvals or clearances we have received are revoked or terminated, we may not be able to commercialize our products and become profitable, and the value of our common stock may decline.
We are also subject to numerous U.S. and foreign regulatory requirements governing the conduct of clinical trials, marketing authorization, pricing and third-party reimbursement. The foreign regulatory approval process includes all the risks associated with FDA approval or clearance described above, as well as risks attributable to the requirement to satisfy local regulations in foreign jurisdictions. Approval or clearance by the FDA does not ensure approval by regulatory authorities of some countries outside the United States. Many foreign regulatory authorities, including those in major markets such as Japan and the People's Republic of China, have different approval processes.
Clinical trials of new products or new indications for our products, if commenced, may not be successful, which may delay or prevent commercialization of such new products or new indications.
In order to obtain regulatory approval to market new products or new indications for our products, we may be required to complete clinical trials to demonstrate the safety and effectiveness of such new products or new indications. For example, FDA regulations require that we conduct clinical trials and submit a marketing application seeking to obtain the approvals and clearances required to promote our QuadraSphere Microspheres for primary liver cancer. In October 2009, we submitted to the FDA an IDE application to commence a clinical trial comparing the effectiveness of our QuadraSphere Microspheres combined with the chemotherapeutic agent doxorubicin against conventional transarterial chemoembolization with doxorubicin in patients with primary liver cancer. We are currently in discussions with the FDA about the clinical trial protocol and have not reached a conclusion about whether or when to undertake the clinical trial. Clinical testing is expensive, difficult to design and implement, can take multiple years to complete and is uncertain as to outcome. We may experience numerous unforeseen events during, or as a result of, any clinical trials that could delay or prevent our ability to receive the regulatory approval we are seeking. These unforeseen events may include:
- •
- regulatory authorities may not approve our application to commence such a trial, or we may be delayed in obtaining
approval of such application by the regulatory authority;
- •
- conditions imposed on us by the regulatory authority regarding the scope or design of such clinical trials;
36
- •
- difficulty obtaining or maintaining institutional review board approval of such clinical trials at one or more clinical
sites;
- •
- difficulty in complying with applicable regulations for conducting such clinical trials;
- •
- any clinical trials we may undertake may produce negative or inconclusive results, and we may decide, or regulators may
require us, to conduct additional clinical trials;
- •
- the number of patients required for such clinical trials may be larger than we anticipate, enrollment in such clinical
trials may be slower than we anticipate, or participants may drop out of such clinical trials at a higher rate than we anticipate, any of which would result in significant delays and increased costs;
- •
- we might have to suspend or terminate clinical trials if the participants are experiencing unacceptable health risks;
- •
- regulators may require that we hold, suspend or terminate clinical research for various reasons, including noncompliance
with regulatory requirements;
- •
- the cost of clinical trials may be greater than we anticipate; and
- •
- we may not achieve the desired favorable effects, the treatment may not compare favorably with other methods or procedures, may produce undesirable side effects or may have other unexpected characteristics.
If we are required to conduct additional clinical trials or other testing of the use of new products or products for new indications, if we are unable to successfully complete clinical trials or other testing, if the results of any such trials are not positive or are only modestly positive or if there are safety concerns, we may:
- •
- be delayed in obtaining marketing approval for the new products or new indications that are the subject of the clinical
trial;
- •
- not be able to obtain marketing approval; or
- •
- obtain approval for an indication that is not as broad as the indication that we sought.
The delay, suspension or discontinuation of clinical trials for any of the foregoing reasons could adversely affect our efforts to obtain regulatory approval for and to commercialize the new products or new indications that are the subject of the clinical trial, increase our operating expenses, and have a material adverse effect on our results of operations and financial condition.
If the FDA or another regulatory agency places restrictions on, or imposes additional approval or clearance requirements with respect to, products we are then marketing, we may incur substantial additional costs and experience delays or difficulties in continuing to market and sell these products.
Even if the FDA grants us approval or clearance with respect to marketing any product, such product will be subject to ongoing regulatory review and restrictions, including the review of clinical results which are reported after such product is made commercially available, and restrictions on the indications for which we can market the product. The FDA can propose to withdraw approval or clearance or impose additional restrictions if new clinical data on the use of a product indicates that a product may not be safe for use under the approved conditions of use. For example, we were named as a defendant in two product liability lawsuits in which the plaintiffs claimed that our microspheres were defective and not accompanied by proper warnings and instructions for use. Product liability claims like these can lead the FDA and comparable regulatory agencies outside of the U.S. to engage in a further review of the safety of these products and their approved conditions for use. On the basis of that review, the FDA or such other regulatory agency could determine to impose additional requirements regarding the manufacturing, marketing or promotion of these products, require changes to the labeling of these products, or commence proceedings to withdraw its clearance of these products, any of which
37
could harm our reputation, adversely affect market acceptance of these products or cause a decline in revenue from the sale of the applicable product, which could adversely affect our financial condition, results of operations, the viability of our business and our future prospects and cause our stock price to decline.
The marketing claims we are permitted to make in labeling or advertising regarding our microspheres in the United States are limited to those consistent with any FDA approval or clearance. For example, because our EmboGold Microspheres are not cleared for specific use in UFE, we may not promote them for this specific use. Although our QuadraSphere Microspheres are identical in all respects to our HepaSphere Microspheres, which are currently marketed in the European Union for use in the embolization of hepatocellular carcinoma and hepatic metastasis, our QuadraSphere Microspheres are not specifically indicated for use in hepatocellular carcinoma and hepatic metastasis. FDA regulations require that we conduct clinical trials prior to submitting a marketing application to claim the use of QuadraSphere Microspheres for the treatment of a specific disease or condition, such as hepatocellular cancer or hepatic metastasis, while European Union regulations do not require preclearance clinical trials for this class of medical device on an indication-by-indication basis. Accordingly, in order for us to seek FDA approval or clearance to promote the use of QuadraSphere Microspheres for the embolization of hepatocellular carcinoma and hepatic metastasis, we will be required to undertake clinical trials. In October 2009, we submitted to the FDA an IDE application to commence a clinical trial to compare the effectiveness of our QuadraSphere Microspheres combined with the chemotherapeutic agent doxorubicin against conventional transarterial chemoembolization with doxorubicin in patients with primary liver cancer. The FDA has advised us that our study protocol will need to include survival as a primary endpoint for the trial, rather than overall tumor response rate at six months, which was the primary endpoint that we proposed in our initial IDE application. Our satisfactory resolution of the FDA's comments on the IDE is a condition to starting the clinical trial. We are currently evaluating the FDA's protocol requirements for the trial, including how the primary endpoint requirements will affect our plans regarding the size of the trial and the timeline and cost for completion. We have not reached a conclusion about whether or when to undertake the clinical trial and if we determine to not undertake the trial, or if we undertake the trial and the results are not sufficient to obtain FDA approval, then we will not be able to promote our QuadraSphere Microspheres for liver cancer indications. If the FDA were to conclude that our advertisements, labeling or statements made by our sales representatives or other company officials, improperly promote our products for unapproved indications or otherwise violate the law, the FDA could allege that our promotional activities misbrand or adulterate our products. Specifically, the FDA could issue an untitled letter or warning letter, which may request, among other things, that we cease such promotional activities, including disseminating the advertisements and promotional labeling, and that we issue corrective labeling, including sending letters to health-care providers. The FDA also could take enforcement action, including seizure of product, injunction or criminal prosecution against us and our officers or employees, or seek civil penalties, disgorgement or restitution.
We may in the future make modifications to our microspheres or their labeling or the process through which they are manufactured, which we determine do not necessitate the filing of a new 510(k) notification or premarket approval application supplement, or PMA supplement. However, if the FDA does not agree with our determination, it may require us to make additional 510(k) filings for the modification, or to file a PMA supplement, and we may be prohibited from marketing the modified product or the new claims until we obtain FDA approval or clearance. The FDA may also institute an enforcement action against us in these circumstances. If we fail to comply with applicable federal, state or foreign laws or regulations, we could be subject to enforcement actions which could affect our ability to develop, market and sell our products and product candidates successfully and could harm our reputation and lead to decreased acceptance of our products by the market.
38
Even if we obtain the necessary FDA clearances or approvals, if we or our suppliers fail to comply with ongoing regulatory requirements, our products could be subject to corrections, removals or recalls from the market or other enforcement action.
We are subject to the Medical Device Reporting, or MDR, regulations that require us to report to the FDA if our products may have caused or contributed to a patient death or serious injury, or if our device malfunctioned and a recurrence of the malfunction would likely result in a death or serious injury. We must also file reports of device corrections and removals and adhere to the FDA's rules on labeling and promotion. We must also comply with the FDA's Good Manufacturing Practice requirements as set forth in the Quality System regulations. Our failure to comply with these or other applicable regulatory requirements could result in enforcement action by the FDA, which may include any of the following:
- •
- untitled letters, warning letters, fines, product seizures, injunctions and civil penalties;
- •
- administrative detention, which is the detention by the FDA of medical devices believed to be adulterated or misbranded;
- •
- customer notification of, or FDA orders for, repair, replacement or refund;
- •
- voluntary or mandatory recall of our products;
- •
- operating restrictions, partial suspension or total shutdown of production or a refusal to allow imported product into the
United States;
- •
- refusal to review premarket notification or premarket approval submissions;
- •
- rescission of a substantial equivalence order or suspension or withdrawal of a premarket approval; and
- •
- criminal prosecution.
If we are subject to an enforcement action, our ability to develop, market and sell our products successfully would be adversely affected, our reputation could be harmed, and we may experience decreased market acceptance of our products.
Existing or future legislation or regulations may make it more difficult and costly for us to obtain regulatory approval or clearance of our product candidates and to produce, market and distribute products after approval.
In September 2007, the President of the United States signed into law the Food and Drug Administration Amendments Act of 2007, or FDAAA. The FDAAA grants a variety of new powers to the FDA, many of which are aimed at improving the safety of drug products before and after approval. Under the FDAAA, companies that violate certain provisions of the new law are subject to substantial civil monetary penalties. The new requirements and other changes that the FDAAA imposes may make it more difficult, and likely more costly, to obtain approval or clearance of new medical device products and to produce, market and distribute products after approval. The FDA is currently reviewing its 510(k) regulatory program and may implement changes that will make it more difficult, time-consuming, or costly to obtain 510(k) clearance.
The FDAAA also establishes requirements for registering certain types of device clinical trials, and reporting the results of those trials, in a national, publicly accessible database. In addition, the FDA requires certification of compliance with this requirement to be submitted with applications or reports concerning such studies. The failure to comply with the clinical trial registry requirements is subject to civil monetary penalties.
Legislation or regulations enacted or modified in the future, including potential changes to the 510(k) premarket notification process, may also make it more difficult and/or costly to obtain clearance of new medical device products or to produce, market and distribute products after approval.
39
We may be subject, directly or indirectly, to federal and state health-care fraud and abuse laws and regulations and, if we are unable to fully comply with such laws, could face substantial penalties.
Our operations may be directly or indirectly affected by various broad state and federal health-care fraud and abuse laws, including the federal Anti-Kickback Statute, which prohibit any person from knowingly and willfully offering, paying, soliciting or receiving remuneration, directly or indirectly, to induce or reward either the referral of an individual, or the furnishing or arranging for an item or service, for which payment may be made under federal health-care programs, such as the Medicare and Medicaid programs.
If our past or present operations are found to be in violation of these laws, we and our officers may be subject to civil or criminal penalties, including large monetary penalties, damages, fines, imprisonment and exclusion from Medicare and Medicaid program participation. If enforcement action were to occur, our business and financial condition would be harmed.
Risks Relating to Our Intellectual Property
If we are unable to obtain patent protection for our products, their competitive value could decline.
We may not obtain meaningful protection for our technology and products with the patents and patent applications that we own or license relating to our microsphere technology or other ancillary products. In particular, the patent rights we possess or are pursuing generally cover our technologies to varying degrees, and these rights may not prevent others from designing products similar to or otherwise competitive with our Embosphere Microspheres and other products we commercialize. To the extent that our competitors are able to design products competitive with ours, we may experience less market penetration with our products and, consequently, we may have decreased revenue. The patent laws involving medical devices and life sciences technologies such as our microspheres are complex and vary from country to country. Thus, although we have a current policy of pursuing patent protection wherever possible for our new technologies, we cannot predict whether we will secure patent protection from any of our existing patent applications in the United States or abroad. We also cannot predict whether such coverage obtained in any of our United States or foreign patent applications will be meaningful.
We do not know whether competitors have similar U.S. patent applications on file, since U.S. patent applications filed before November 28, 2000, or for which no foreign patents will be sought, are secret until issued, and applications filed after November 28, 2000 are published approximately 18 months after their earliest priority date. Consequently, the United States Patent and Trademark Office could initiate interference proceedings involving our owned or licensed U.S. patent applications or issued patents. Further, there is a substantial backlog of patent applications at the United States Patent and Trademark Office, and the approval or rejection of patent applications may take several years.
We require our employees, consultants and advisors to execute confidentiality agreements. However, we cannot guarantee that these agreements will provide us with adequate protection against improper use or disclosure of confidential information. In addition, in some situations, these agreements may conflict with, or be subject to, the rights of third parties with whom our employees, consultants or advisors have prior employment or consulting relationships. Further, others may independently develop substantially equivalent proprietary information and techniques, or otherwise gain access to our trade secrets. Our failure to protect our proprietary information and techniques may inhibit or limit our ability to exclude certain competitors from the market.
40
If we become involved in expensive patent litigation or other proceedings to enforce or defend our patent rights, we could incur substantial costs and expenses or substantial liability for damages or be required to stop our product development and commercialization efforts.
On January 13, 2005, we were notified of a Notice of Opposition filed in the European Patent Office, or EPO, by Biocompatibles UK Limited on December 23, 2004, challenging the patentability of the claims in our granted European Patent, EP 1128816, which relates to certain PVA microspheres for use in embolization and methods thereof. On December 10, 2007, the EPO upheld the claims of our patent in amended form and rendered its formal written decision on December 27, 2007. We and Biocompatibles have appealed this decision. The EPO has not set a date, but we expect oral proceedings to be scheduled within the next twelve months. On July 1, 2008, Biocompatibles UK Limited filed a Notice of Opposition in the EPO challenging the patentability of the claims in our granted European Patent, EP 1267839, which relates to certain drug-loaded microspheres and their use in embolization. We filed a reply to Notice of Opposition on March 19, 2009. Oral Proceedings are scheduled for September 23, 2010. We intend to continue to defend our European patents. We are not able to predict the outcome of either of these opposition proceedings.
With the exception of the two European Opposition proceedings just described, we are not currently involved in any other litigation or actions with third parties to enforce or defend our patent rights. However, in order to protect or enforce our patent rights, we may have to initiate legal proceedings against third parties, such as infringement suits, opposition proceedings or interference proceedings or defend against such proceedings. By initiating legal proceedings to enforce our intellectual property rights, we may also provoke these third parties to assert claims against us. If we do not prevail in any such proceedings, our patents could be narrowed, invalidated or rendered unenforceable. Furthermore, we may be sued for infringing on the intellectual property rights of others. We may find it necessary, if threatened, to initiate a lawsuit seeking a declaration from a court regarding the proprietary rights of others. As we introduce new products into the market, we may be accused of infringing the patent rights of third parties. If we do not prevail in such a patent litigation brought against one of our products or its use, we may be required to pay damages, stop selling our product or obtain a royalty-bearing license if one is obtainable. Any required license might not be available to us on acceptable terms, or at all. In addition, some licenses may be nonexclusive, and therefore our competitors may have access to the same technology licensed to us. If we fail to obtain a required license or are unable to design around a patent, we may be prevented from selling some of our products, which could decrease our revenue. Intellectual property litigation is costly and, even if we prevail, could divert management attention and resources away from our business.
If our exclusive license to use the Embosphere Microsphere intellectual property that we jointly own with L'Assistance Publique-Hôpitaux de Paris is terminated, then our competitive position, financial condition, prospects and stock price could be adversely affected.
We have an agreement with L'Assistance Publique-Hôpitaux de Paris, or AP-HP, pursuant to which AP-HP has granted us exclusive rights to use two United States patents and their foreign counterparts that we jointly own with AP-HP relating to Embosphere Microspheres. This agreement can be terminated on short notice by AP-HP if we default on our obligations under the license and fail to cure such default after notice is provided. The license imposes commercialization, sublicensing, royalty, insurance and other obligations on us. Our failure, or any sublicensee's failure, to comply with the terms of the license could result in our loss of our exclusive rights to the jointly-owned intellectual property that is the subject of the license. If the AP-HP exclusive license is terminated, AP-HP will have the right to grant a non-exclusive license to our jointly-owned technology to a third party which would then have the freedom to seek regulatory approval of, and to market, products identical to our Embosphere Microspheres. This could have a material adverse effect on our competitive business position, financial condition and our business prospects and could cause our stock price to decline.
41
Risks Relating to the Production and Supply of Our Products
If we experience manufacturing delays or interruptions in production, then we may experience customer dissatisfaction and our reputation could suffer.
If we fail to produce sufficient products at our own manufacturing facility or at a third-party manufacturing facility, we may be unable to deliver products to our customers on a timely basis, which could lead to customer dissatisfaction and could harm our reputation and ability to compete. We currently produce and package all of our microsphere products in one manufacturing facility in France. We have contracted with two suppliers for our guidewire products. Either we or any third-party manufacturer would likely experience significant delays or cessation in producing our products if we or they experience difficulties, delays or failures in manufacturing processes, quality control processes, equipment calibration, process-critical equipment or in any other process necessary for the manufacture of our products, or if we or they experience a labor-based error or omission, or a labor strike, natural disaster, local or regional conflict or any disruption in supply. If we are unable to manufacture and package our products at our facility in France, we may be required to enter into arrangements with one or more alternative contract manufacturing companies.
Even if we are able to identify alternative facilities to manufacture our products, if necessary, we may experience disruption in the supply of our products until such facilities are available. Although we believe we possess adequate insurance for damage to our property and the disruption of our business from casualties, such insurance may not be sufficient to cover all of our potential losses and may not be available to us on acceptable terms or at all. Our failure to deliver products on a timely basis could lead to customer dissatisfaction and damage our reputation. In addition, if we are required to depend on third-party manufacturers, our profit margins may be lower, which will make it more difficult for us to achieve profitability.
Medical device manufacturers must adhere to current Good Manufacturing Practices and Quality System Regulations which are enforced by the FDA through its inspection program. We and other third-party manufacturers must comply with various quality system requirements pertaining to all aspects of our product design and manufacturing process, including requirements for packaging, labeling and record keeping, complaint handling, corrective and preventive actions, adoption of new manufacturing methods and internal auditing. In addition, medical device manufacturing laws are also in effect in many countries outside of the U.S. We or our third-party manufacturers may not be able to comply or maintain compliance. If we or any third-party manufacturers we engage fail to comply, such noncompliance could significantly delay our receipt of new product premarket approvals, result in FDA enforcement action, including an embargo on imported devices, or otherwise cause delays and disruptions in the manufacture and supply of our products, any of which would harm our reputation and could materially adversely affect our operating results.
Because we rely on a limited number of suppliers, we may experience difficulty in meeting our customers' demands for our products in a timely manner or within budget.
We currently purchase key components and services with respect to our microspheres, catheters and guidewires from approximately ten third-party vendors, including a third party from which we purchase guidewires for our Segway Guidewire product; a third party from which we purchase catheters for our EmboCath Plus Infusion Microcatheter product; and a third party from which we purchase guidewires for our Sequitor Steerable Guidewire and recently released Tenor Steerable Guidewire product. Our reliance on our suppliers exposes us to risks, including:
- •
- the possibility that one or more of our suppliers could terminate their services at any time without penalty;
- •
- the potential inability of our suppliers to obtain required components;
42
- •
- the potential delays and expenses of seeking alternative sources of supply;
- •
- reduced control over pricing, quality and timely delivery due to difficulties in switching to alternative suppliers; and
- •
- the possibility that one or more of our suppliers could fail to be compliant with Quality System Regulations, 21 CFR Part 820.
Consequently, in the event that our suppliers delay or interrupt the supply of components for any reason, our ability to produce and supply our products could be impaired, which could lead to customer dissatisfaction and harm our reputation.
Risks Relating to Our Foreign Operations
If we are unable to meet the operational, legal and financial challenges that we encounter in our international operations, we may not be able to grow our business.
Our worldwide manufacturing and European sales operations are currently conducted primarily through our French subsidiary. Furthermore, we currently derive a portion of our revenue from the sale of our microspheres and delivery system products outside the United States. For the years ended December 31, 2009, 2008 and 2007, approximately 22%, 20%, and 19%, respectively, of our revenue was derived from sales of our microspheres and delivery systems in geographic territories outside the United States. We are increasingly subject to a number of challenges that specifically relate to our international business activities. Our international operations may not be successful if we are unable to meet and overcome these challenges, which would limit the growth of our business. These challenges include:
- •
- failure of local laws to provide the same degree of protection against infringement of our intellectual property;
- •
- protectionist laws and business practices that favor local competitors, which could slow our growth in international
markets;
- •
- the requirement that we obtain regulatory approval or clearance in each country in which we choose to offer and sell our
products;
- •
- in some jurisdictions, strict government-regulated price controls;
- •
- complex reimbursement procedures;
- •
- potentially longer sales cycles to sell products, which could slow our revenue growth from international sales; and
- •
- potentially longer accounts receivable payment cycles and difficulties in collecting accounts receivables.
In 2008, we commercially launched our Embosphere Microsphere product in the People's Republic of China. Conducting business in China exposes us to a variety of risks and uncertainties that are unique to China. The economy of China has been transitioning from a planned economy to a market-oriented economy. Although in recent years the Chinese government has implemented measures emphasizing the utilization of market forces for economic reform, the reduction of state ownership of productive assets and the establishment of sound corporate governance in business enterprises, a substantial portion of productive assets in China is still owned by the Chinese government. In addition, the Chinese government continues to play a significant role in regulating industrial development. It also exercises significant control over China's economic growth through the allocation of resources, controlling payment of foreign currency-denominated obligations, setting monetary policy and providing preferential treatment to particular industries or companies. Efforts by the Chinese government to slow
43
the pace of growth of the Chinese economy could result in interruptions of our commercialization efforts in China. In addition, the Chinese legal system is a civil law system based on written statutes. Unlike common law systems, it is a system in which decided legal cases have little precedential value. In 1979, the Chinese government began to promulgate a comprehensive system of laws and regulations governing economic matters in general. Accordingly, we cannot predict the effect of future developments in the Chinese legal system, including the promulgation of new laws, changes to existing laws or the interpretation or enforcement thereof, or the preemption of local regulations by national laws. Our commercialization efforts in China could be materially harmed by any changes in the political, legal or economic climate in China or the inability to enforce applicable Chinese laws and regulations. If such commercialization efforts in China are materially harmed, we would not be able to grow sales of our Embosphere Microspheres in China and our operating results could be adversely affected.
Because we translate foreign currency from international sales into U.S. dollars and are required to make foreign currency payments, we may incur losses due to fluctuations in foreign currency exchange rates.
A significant portion of our business is conducted in the European Union euro. We recognize foreign currency gains or losses arising from our operations in the period incurred. As a result, currency fluctuations between the U.S. dollar and the currencies in which we do business will cause foreign currency translation gains and losses, which may cause fluctuations in our future operating results. We do not currently engage in foreign exchange hedging transactions to manage our foreign currency exposure.
Risk Relating to Our Stock Price
Because the market price of our stock is highly volatile, investments in our stock could rapidly lose their value and we may incur significant costs from class action litigation.
The market price of our stock is highly volatile. From January 1, 2008 through March 1, 2010, the price of our common stock has ranged from a low of $1.26 to a high of $6.00. As a result of this volatility, investments in our stock could rapidly lose their value.
Our stock price could fluctuate for many reasons, including, without limitation:
- •
- variations in our quarterly operating results or those of companies that are perceived to be similar to us;
- •
- third-party sales of large blocks of our common stock;
- •
- rumors relating to us or our competitors;
- •
- changes to our research and development plans and/or announcements regarding new technologies by us or our competitors;
- •
- our decision not to commence our planned QuadraSphere Microspheres clinical trial, or if commenced, suspension or
termination of such trial;
- •
- negative publicity or unfavorable media coverage;
- •
- filings of, results of or developments under lawsuits involving us or our competitors;
- •
- sales by us of equity or debt to fund our operations;
- •
- the loss of any of our key scientific or management personnel;
- •
- FDA or international regulatory actions or lawsuits concerning the safety of our products; and
- •
- market conditions, both in the medical device sector and generally.
44
In addition, the stock market often experiences extreme price and volume fluctuations, which affect the stock prices of many medical device companies and which are often unrelated to the operating performance of these companies.
When the market price of a stock has been as volatile as our stock price has been, holders of that stock may institute securities class action litigation against the company that issued the stock. If any of our stockholders were to bring a lawsuit of this type against us, even if the lawsuit is without merit, we could incur substantial costs in defending the lawsuit. The lawsuit could also divert the time and attention of our management.
Securities analysts may not initiate coverage for our common stock or may issue negative reports, and this may have a negative impact on the market price of our common stock.
Securities analysts may elect not to provide research coverage of our common stock. If securities analysts do not cover our common stock, the lack of research coverage may adversely affect the market price of our common stock. The trading market for our common stock may be affected in part by the research and reports that industry or financial analysts publish about us or our business. If one or more of the analysts who elect to cover us downgrades our stock, our stock price could decline. If one or more of these analysts ceases coverage of our company, we could lose visibility in the market, which in turn could cause our stock price to decline. It may be difficult for companies such as ours, with smaller market capitalizations, to attract independent financial analysts that will cover our common stock. This could have a negative effect on the market price of our stock.
Item 1B. UNRESOLVED STAFF COMMENTS
None.
We currently lease office and manufacturing facilities in Rockland, Massachusetts, and Roissy, France. Our Rockland, Massachusetts office includes approximately 13,000 square feet of corporate offices, laboratory and warehouse space pursuant to a lease expiring on February 28, 2011. Our Roissy, France facility, where we produce our Embosphere Microspheres, HepaSphere Microspheres and QuadraSphere Microspheres, includes approximately 18,000 square feet of office, laboratory and manufacturing space and is leased through May 2013.
We believe that the leased facilities in Rockland, Massachusetts and Roissy, France are suitable to meet our current requirements and that suitable additional or substitute space will be available to us on commercially reasonable terms, if needed in the future.
None.
Item 4. SUBMISSION OF MATTERS TO A VOTE OF SECURITY HOLDERS
No matters were submitted to a vote of our security holders, through solicitations of proxies or otherwise, during the quarter ended December 31, 2009.
45
EXECUTIVE OFFICERS
As of March 1, 2010, our executive officers, their respective ages and their positions are as follows:
Name
|
Age | Position | |||
|---|---|---|---|---|---|
Richard J. Faleschini |
63 | President and Chief Executive Officer |
|||
Martin J. Joyce |
56 | Executive Vice President and Chief Financial Officer |
|||
Melodie R. Domurad |
52 | Vice President of Regulatory, Medical Affairs, and Quality Systems |
|||
Willard W. Hennemann(1) |
55 | Vice President of New Product and Business Development |
|||
Peter C. Sutcliffe |
60 | Vice President of Manufacturing |
|||
Joel B. Weinstein(1) |
59 | Vice President of Global Marketing and Sales |
|||
- (1)
- Effective March 31, 2010 Mr. Hennemann and Mr. Weinstein will step down from their positions and cease to be employed by us.
Richard J. Faleschini has served as our President and Chief Executive Officer since November 2004 and as a director of BioSphere Medical since March 2005. From 2003 to 2004, Mr. Faleschini served as Vice President and General Manager of the gynecology division at American Medical Systems Holdings, Inc., a supplier of medical devices to physicians specializing in the treatment of urological and gynecological disorders. From 1999 to 2003, Mr. Faleschini was Vice President of Marketing and Sales for American Medical Systems Holdings, Inc. From 1995 to 1999, he held executive marketing and general management positions at Medtronic, Inc., a medical technology company, with responsibilities in several sectors of their cardiac rhythm management, cardiac surgery, and interventional vascular businesses. His previous experience also includes executive marketing and sales management responsibilities at Cordis Corporation, Biomagnetic Technologies, and ATL/ADR Ultrasound. Mr. Faleschini received his B.S. in biology and M.S. in physiology from Michigan Technological University.
Martin J. Joyce has served as our Executive Vice President and Chief Financial Officer since January 2006. He served as our Chief Financial Officer and Vice President from September 2004 to January 2006. From 2000 to 2004, Mr. Joyce served as Managing Partner of Stratex Group LLC, a provider of biopharmaceutical executive services to early-stage companies and venture investors. From 1996 to 2000, Mr. Joyce was North American Chief Financial Officer for Serono Inc. a biotechnology company. Prior to serving as North American Chief Financial Officer, Mr. Joyce held a variety of senior level positions within Serono in finance, sales, marketing and manufacturing. Mr. Joyce was previously employed at Millipore Corporation, a high technology bioscience company, and Bose Corporation, an audio equipment manufacturer, focusing on strategic planning, product rationalization and return on investment analysis. Mr. Joyce received a B.S. in finance from Northeastern University and a M.B.A. from Suffolk University, Boston, Massachusetts.
Melodie R. Domurad has served as our Vice President of Regulatory, Medical Affairs and Quality Systems since January 2008. From 1997 to 2007, Dr. Domurad served as Vice President of Clinical, Regulatory and Quality Affairs for Matritech, Inc., a developer of proteomics-based diagnostic products for the early detection of cancer. From 1994 to 1997, Dr. Domurad held the position of Director of Clinical Research and Clinical Research Manager at Ergo Science, Inc., where she focused on therapeutics for diabetes, obesity and cancer. Prior to joining Ergo Science, Inc., Dr. Domurad held leadership roles at the Center for the Study of Nutrition and Medicine at the New England Deaconess Hospital and at the Cambridge Center for Holistic Health. Dr. Domurad holds a B.A. from Cornell University and a Ph.D. from the University of Cincinnati.
46
Willard W. Hennemann has served as our Vice President of New Product and Business Development since February 2008. From 2006 to early 2008, Dr. Hennemann served as Vice President of Intravascular Systems/Marketing for Medeikon Corporation, an early-stage developer of proprietary disposable technologies to treat cardiovascular disease. From 2000 to 2006, Dr. Hennemann held the position of Vice President of Research and Development/Interventional Vascular at CryoCath Technologies, a leader in catheter-based products for the cryotherapeutic treatment of cardiovascular disease. From 1998 to 1999, Dr. Hennemann served as Director of Marketing and Product Development for InterVascular, Inc., a division of Datascope, which produces a broad line of vascular grafts. From 1996 to 1998, Dr. Hennemann served as Director of Marketing/International Clinical Studies of the Global Stent Business Unit for Medtronic, Inc., a medical technology company. Prior to Medtronic, Dr. Hennemann assumed roles of increasing responsibility over a 12-year period with Cordis Corporation, a pioneer in developing innovative diagnostic and therapeutic devices for interventional vascular medicine. Dr. Hennemann received his B.A. from the University of Maryland and a Ph.D. from the University of Florida.
Peter C. Sutcliffe has served as our Vice President of Manufacturing since October 2002. From 2001 to 2002, Mr. Sutcliffe served as the Vice President for North American Manufacturing for Whatman, Plc., a life science filtration company. From 1996 to 2001, he was the Chief Operating Officer for HemaSure Inc., a manufacturer and supplier of blood filters. From 1982 to 1996, Mr. Sutcliffe held the position of Vice President of Manufacturing for Corning Costar Company, a life science products company. Prior to Costar, he held manufacturing management positions with Millipore Corporation, a high technology bioscience company. Mr. Sutcliffe holds a B.S. in biology from the University of Richmond in Virginia and a M.B.A. from Sul Ross State University of Texas, Fort Bliss, Texas.
Joel B. Weinstein has served as our Vice President of Global Marketing and Sales since January 2008. Prior to joining BioSphere Medical, Mr. Weinstein founded and led several medical device companies, and founded his own firm, which provided strategic counsel to medical device companies and venture capital firms. From 1987 to 1998, Mr. Weinstein served as Vice President of Marketing and Business Development for Hologic, Inc., a medical device company focused on women's health. Prior to Hologic, Mr. Weinstein had progressively greater management responsibilities over a seven-year period with Advanced Technology Laboratories, a multi-modality diagnostic ultrasound company. He received his bachelor's degree in Electrical Engineering from City College of New York and a M.B.A. from Western New England College.
47
Item 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common stock trades on the NASDAQ Global Market under the symbol "BSMD." On March 1, 2010, the last reported sale price of our common stock on the NASDAQ Global Market was $3.09, and there were approximately 85 stockholders of record. This number does not include stockholders for whom shares are held in "nominee" or "street" name.
The following table shows the range of high and low sales prices per share of our common stock for the last two fiscal years as reported on the NASDAQ Global Market.
| |
2009 | ||||||
|---|---|---|---|---|---|---|---|
| |
High | Low | |||||
First Quarter |
$ | 2.98 | $ | 1.60 | |||
Second Quarter |
$ | 2.62 | $ | 1.26 | |||
Third Quarter |
$ | 4.19 | $ | 2.19 | |||
Fourth Quarter |
$ | 3.67 | $ | 2.51 | |||
| |
2008 | ||||||
|---|---|---|---|---|---|---|---|
| |
High | Low | |||||
First Quarter |
$ | 6.00 | $ | 3.30 | |||
Second Quarter |
$ | 5.12 | $ | 3.10 | |||
Third Quarter |
$ | 4.50 | $ | 3.15 | |||
Fourth Quarter |
$ | 3.59 | $ | 1.55 | |||
We have not paid any cash dividends on our common stock since our inception and do not intend to pay any cash dividends in the foreseeable future.
48
Comparative Stock Performance
The following graph compares the cumulative total stockholder return on our common stock for the last five fiscal years with the cumulative total return on (i) the Total Return Index for the NASDAQ Stock Market (U.S. Companies), which we refer to as the NASDAQ Composite Index (U.S.) and (ii) the NASDAQ Medical Equipment Index, which we refer to as the NASDAQ Medical Equipment Index. This graph assumes the investment of $100 on December 31, 2004 in our common stock and each of the indices listed above, and assumes dividends are reinvested. We have not paid any dividends on our common stock and no dividends are included in the representation of our performance. The stock price performance shown in the below graph is not necessarily indicative of future price performance. Measurement points are the last trading day of the fiscal years ended December 31, 2005, 2006, 2007, 2008 and 2009.
COMPARISON OF 5 YEAR CUMULATIVE TOTAL RETURN
AMONG BIOSPHERE MEDICAL, INC., THE NASDAQ COMPOSITE INDEX (U.S.)
AND THE NASDAQ MEDICAL EQUIPMENT INDEX
| |
12/05 | 12/06 | 12/07 | 12/08 | 12/09 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
BioSphere Medical, Inc. |
$ | 208.23 | $ | 171.72 | $ | 131.88 | $ | 49.61 | $ | 70.44 | ||||||
NASDAQ Composite Index (U.S.) |
$ | 101.33 | $ | 114.01 | $ | 123.71 | $ | 73.11 | $ | 105.61 | ||||||
NASDAQ Medical Equipment Index |
$ | 117.06 | $ | 122.50 | $ | 159.63 | $ | 88.67 | $ | 122.59 | ||||||
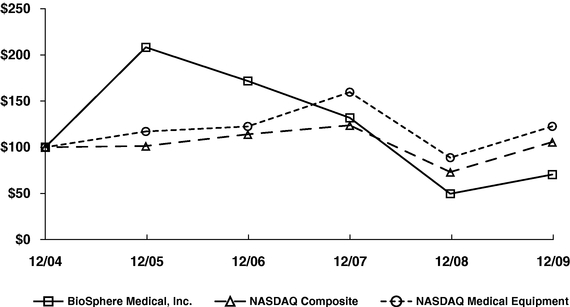
The graph and table above are not "soliciting material," are not deemed filed with the SEC and are not to be incorporated by reference in any filing of ours under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, which we refer to herein as the Exchange Act, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing.
49
Item 6. SELECTED FINANCIAL DATA
The following selected consolidated financial data should be read in conjunction with "Management's Discussion and Analysis of Financial Condition and Results of Operations" and our financial statements and related notes to those statements and other financial information included elsewhere in this annual report on Form 10-K. Historical results are not necessarily indicative of future results.
Year Ended December 31,
|
2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (in thousands, except per share amounts) |
|
|
|
|
|
||||||||||||||
Statement of Operations Data: |
|||||||||||||||||||
Revenue: |
|||||||||||||||||||
Product sales |
$ | 30,888 | $ | 28,842 | $ | 26,483 | $ | 22,787 | $ | 18,484 | |||||||||
Licensing and collaborative revenue |
555 | 416 | 417 | 104 | — | ||||||||||||||
Total revenue |
31,443 | 29,258 | 26,900 | 22,891 | 18,484 | ||||||||||||||
Costs and expenses: |
|||||||||||||||||||
Costs of product sales |
7,682 | 7,686 | 7,768 | 6,958 | 6,303 | ||||||||||||||
Research and development |
3,415 | 3,305 | 2,342 | 2,290 | 2,359 | ||||||||||||||
Sales |
10,636 | 10,374 | 7,671 | 7,550 | 5,792 | ||||||||||||||
Marketing |
5,243 | 6,514 | 5,290 | 3,699 | 2,473 | ||||||||||||||
General, administrative and patent |
7,839 | 7,204 | 6,439 | 5,561 | 4,219 | ||||||||||||||
Total costs and expenses |
34,815 | 35,083 | 29,510 | 26,058 | 21,146 | ||||||||||||||
Loss from operations |
(3,372 | ) | (5,825 | ) | (2,610 | ) | (3,167 | ) | (2,662 | ) | |||||||||
Other income (expense): |
|||||||||||||||||||
Interest income |
8 | 375 | 1,017 | 938 | 225 | ||||||||||||||
Interest expense |
(6 | ) | (9 | ) | (17 | ) | (15 | ) | (15 | ) | |||||||||
Other |
38 | 96 | (244 | ) | (80 | ) | (442 | ) | |||||||||||
Loss before income taxes |
(3,332 | ) | (5,363 | ) | (1,854 | ) | (2,324 | ) | (2,894 | ) | |||||||||
Income tax benefit (provision) |
658 | (129 | ) | — | — | 93 | |||||||||||||
Net loss |
(2,674 | ) | (5,492 | ) | (1,854 | ) | (2,324 | ) | (2,801 | ) | |||||||||
Preferred stock dividends |
(578 | ) | (578 | ) | (557 | ) | (525 | ) | (495 | ) | |||||||||
Net loss applicable to common stockholders |
$ | (3,252 | ) | $ | (6,070 | ) | $ | (2,411 | ) | $ | (2,849 | ) | $ | (3,296 | ) | ||||
Basic and diluted net loss per common share applicable to common stockholders |
$ | (0.18 | ) | $ | (0.34 | ) | $ | (0.14 | ) | $ | (0.17 | ) | $ | (0.22 | ) | ||||
Basic and diluted weighted average number of common shares outstanding |
18,044 | 17,983 | 17,647 | 17,027 | 14,653 | ||||||||||||||
As of December 31,
|
2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (in thousands) |
|
|
|
|
|
||||||||||||
Balance Sheet Data: |
|||||||||||||||||
Cash, cash equivalents and marketable securities |
$ | 18,088 | $ | 18,239 | $ | 23,579 | $ | 22,119 | $ | 8,774 | |||||||
Working capital |
21,254 | 21,919 | 26,555 | 24,719 | 10,832 | ||||||||||||
Total assets |
30,447 | 30,228 | 34,759 | 32,079 | 17,495 | ||||||||||||
Long-term debt and deferred revenue |
431 | 8 | 80 | 190 | 101 | ||||||||||||
Stockholders' equity |
23,647 | 24,746 | 29,109 | 26,965 | 13,088 | ||||||||||||
50
Item 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion of our financial condition and results of operations should be read in conjunction with our consolidated financial statements and the related notes included elsewhere in this report. Some of the information contained in this discussion and analysis and set forth elsewhere in this report, including information with respect to our plans, strategy and expectations for our business, financial condition and operations, includes forward-looking statements that involve risks and uncertainties. You should review the section titled "Item 1A—Risk Factors" for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
We develop, manufacture and market products for medical procedures that use embolotherapy. Embolotherapy is the minimally invasive, image-guided therapeutic introduction of various biocompatible substances into a patient's circulatory system to occlude a blood vessel, either to arrest or prevent hemorrhaging or to devitalize or destroy the structure by occluding its blood supply. Our core technologies consist of patented bioengineered polymers, which are chemical compounds created through the application of medical science, engineering principles and manufacturing methods. These core technologies are used to produce miniature spherical embolic particles, or microspheres, that are designed to have uniquely beneficial properties for a variety of medical applications. We currently market and sell four microsphere products:
- •
- Embosphere Microspheres, which are marketed for symptomatic uterine
fibroids, hypervascularized tumors and arteriovenous malformations, in the United States, the European Union, the People's Republic of China and several other foreign markets;
- •
- EmboGold Microspheres, which are marketed for hypervascularized tumors and
arteriovenous malformations in the United States, the European Union and several other foreign markets;
- •
- HepaSphere Microspheres, which are marketed in the European Union, Brazil
and Russia for primary and metastatic liver cancer, and in the European Union and Russia for drug delivery in the treatment of primary and metastatic liver cancer; and
- •
- QuadraSphere Microspheres, which are marketed for the treatment of hypervascularized tumors and arteriovenous malformations in the United States.
On April 16, 2009, we entered into an international distribution agreement with Nippon Kayaku Co., Ltd., or Nippon Kayaku. The agreement grants Nippon Kayaku the exclusive right to distribute our HepaSphere Microspheres and Embosphere Microspheres in Japan. The agreement provides that Nippon Kayaku is responsible for filing, obtaining and maintaining all regulatory approvals necessary for the sale, marketing, pricing and reimbursement of the products in Japan, including performing any clinical trials required as a result of seeking such regulatory approvals in Japan. Assuming product approval, we will provide HepaSphere Microspheres and Embosphere Microspheres to Nippon Kayaku for distribution and sale in Japan. Additionally, Nippon Kayaku has made a nonrefundable milestone payment of $1.00 million in 2009, which will be recognized as collaborative revenue ratably over the expected term of the research and development period, and has agreed to make up to $3.00 million in additional milestone payments based upon specified objectives, including achievement of clinical, regulatory and sales goals. For the year ended December 31, 2009, we recognized approximately $236,000 as collaborative revenue relating to the Nippon Kayaku agreement.
On September 15, 2009, we entered into a settlement agreement with the plaintiff in a product liability lawsuit filed against us and other previously dismissed defendants in the matter captioned Brett Pingel by next friend Dawn LaRose v. BioSphere Medical, Inc., Bruce Kirke Bieneman, M.D., St. Louis
51
University Hospital, John Stith, M.D. and St. Louis University. The parties agreed to settle the case without any admission of liability. We maintain product liability insurance, and our insurer agreed to pay the full amount of the settlement.
On March 24, 2010, we entered into a settlement agreement with the plaintiff in a product liability lawsuit filed against us and other defendants in the matter captioned Hamid Rashidi v. Franklin Moser, M.D., Cedars-Sinai Medical Center and Biosphere Medical, Inc. The parties agreed to settle the case without any admission of liability. We maintain product liability insurance, and our insurer has agreed to pay the full amount of the settlement.
Our QuadraSphere Microspheres are identical in all respects to our HepaSphere Microspheres. However, FDA regulations require that we conduct clinical trials and submit a marketing application which includes positive data from the clinical trials to the Unites States Food and Drug Administration, or FDA, in order to obtain the approvals and clearances required to promote QuadraSphere Microspheres for the treatment of a specific disease or condition, including primary liver cancer. European Union regulations do not require preclearance clinical trials for this class of medical device on an indication-by-indication basis. In October 2009, we submitted to the FDA an investigational device exemption, or IDE, application to commence a clinical trial to compare the effectiveness of our QuadraSphere Microspheres combined with the chemotherapeutic agent doxorubicin against conventional transarterial chemoembolization with doxorubicin in patients with primary liver cancer. The FDA has advised us that our study protocol will need to include survival as a primary endpoint for the trial, rather than overall tumor response rate at six months, which was the primary endpoint that we proposed in our initial IDE application. Our satisfactory resolution of the FDA's comments on the IDE is a condition to starting the clinical trial. We are currently evaluating the FDA's protocol requirements for the trial, including how the primary endpoint requirements will affect our plans regarding the size of the trial and the timeline and cost for completion. We have not reached a conclusion about whether or when to undertake the clinical trial.
For the years ended December 31, 2009 and 2008, we primarily generated revenue from product sales of our embolic products in North America and the European Union. We also recognized revenue from product sales in other geographic territories, including the Middle East, Africa, South America and Asia. Product revenue also includes the sale of accessory embolotherapy devices such as our EmboCath® Plus Infusion Microcatheter, Sequitor® Steerable Guidewire and Segway® Guidewire, as well our barium delivery kits and other ancillary medical devices sold during the first half of 2008 exclusively in Europe. We derive a majority of our revenue in the United States and the European Union from the sale of Embosphere Microspheres for use in the treatment of uterine fibroids, using a procedure called uterine fibroid embolization, or UFE. Although we have not received approval or clearance from the FDA to market our QuadraSphere Microspheres for primary or metastatic liver cancer, we believe that some physicians are using QuadraSphere Microspheres in the treatment of primary and metastatic liver cancer. For the years ended December 31, 2009 and 2008, we also derived a small portion of our revenue from our licensing of nonstrategic technology to a third party.
Our strategic priorities are to accelerate our revenue growth by expanding the market for the minimally invasive treatment of symptomatic uterine fibroids using UFE and broadening treatment options for other medical conditions for patients and physicians.
We have experienced operating losses in each period since our inception. As of December 31, 2009, we had $18.09 million in cash, cash equivalents and marketable securities, and an accumulated deficit of $93.38 million. Most of our expenditures to date have been for sales and marketing activities, general and administrative expenses and research and development activities. We expect to continue to incur operating losses in 2010 as we seek to execute on our business plan, including continuing to establish sales and marketing capabilities for our products and conducting research and development activities. Prior to 1999, we were engaged in chromatography programs that we divested in 1999. Of the
52
cumulative operating loss at the end of 2009, $31.54 million was related to chromatography and $61.84 million was related to embolotherapy platform development.
Research and Development
Research and development expense as a percentage of total revenue for the years ended December 31, 2009, 2008 and 2007 was 11%, 11% and 9%, respectively. Research and development expense in these periods relate primarily to:
- •
- efforts to develop improved manufacturing processes for our currently marketed products;
- •
- research to identify and evaluate new and innovative embolotherapy products based on our platform microsphere technology,
including a smaller-sized HepaSphere Microsphere and QuadraSphere Microsphere designed to allow for delivery into smaller vasculature, which is in preclinical development;
- •
- efforts to develop a new generation of steerable guidewire to augment and/or replace our current guidewire product
offerings; and
- •
- further preclinical testing and nonclinical trials to support initial and/or additional clinical indications and/or premarketing approvals for our Embosphere Microspheres, HepaSphere Microspheres, QuadraSphere Microspheres, Sequitor Steerable Guidewire and EmboCath Plus Infusion Microcatheter, all of which are currently approved and marketed for specified indications and in specified geographic locations.
In the first quarter of 2009, a third-party developer delivered a working prototype relating to a new process for the manufacture of Embosphere Microspheres. We evaluated the new process and at this time are not proceeding with plans to implement although we may choose to implement in the future.
Our research and development functions typically work on a number of projects concurrently. In addition, except for clinical expenses, a substantial amount of fixed research and development costs such as salary and salary-related benefits, facility costs, equipment depreciation and maintenance are shared among various programs. Accordingly, we have not historically tracked specific costs for each of our research and development projects.
We cannot reasonably estimate or know the nature, timing and estimated costs of the efforts necessary to complete the development of any of our product candidates that are currently in development, or the period in which material net cash inflows are expected to commence from any of our product candidates that are currently in development or from any of our currently marketed products for which we are seeking expanded marketing approvals or clearance in selected indications or geographic regions, due to the numerous risks and uncertainties associated with developing and commercializing medical devices, including uncertainties relating to:
- •
- the technical risks in new product research and development;
- •
- the timing, scope, rate of progress and cost of clinical trials and other research and development activities undertaken
by us;
- •
- future clinical trial results;
- •
- publicity with respect to our products or their indications;
- •
- the cost, timing and success of regulatory approvals or clearance;
- •
- the cost, timing and success of establishing sales, marketing and distribution capabilities;
53
- •
- the cost of establishing clinical and commercial supplies of our product candidates and any products that we may develop;
- •
- market acceptance of our approved products;
- •
- the effect of competing technological and market developments; and
- •
- the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights.
Any failure to complete the development of our product candidates in a timely manner, or at all, could have a material adverse effect on our operations, financial position and liquidity. A discussion of the risks and uncertainties associated with completing our projects on schedule, or at all, and some consequences of failing to do so, is set forth in "Part I, Item 1A—Risk Factors."
Critical Accounting Policies and Estimates
The discussion and analysis of our financial condition and results of operations are based upon our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States, or GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets and liabilities, revenue and expenses, and related disclosure at the date of our financial statements. We believe that the application of accounting policies relating to revenue recognition, stock-based compensation, accounts receivable, inventories, long-lived assets, income taxes and investments which are important to our financial position and results of operations, require significant judgments and estimates on the part of management. For a more detailed explanation of the judgments made in these areas, refer to Note 2 of the notes to our consolidated financial statements.
Revenue Recognition
We recognize revenue when products are shipped and the customer or distributor takes ownership and assumes risk of loss, collection of the relevant receivable is reasonably assured, persuasive evidence of an arrangement exists, for example, a valid purchase order from an approved customer, the sales price is fixed or determinable, payment is not contingent on resale and we do not have any continuing obligations to ensure resale. Revenue from licensing agreements generally is recognized ratably over the research and development period. We establish reserves for potential sales returns and evaluate the adequacy of those reserves based upon realized experience and expectations. Reductions to our reserves are offset against revenue. Any significant change in credit returns could have a material adverse impact on our revenue and operating results for the period or periods in which such returns materialize.
Stock Based Compensation
We measure the cost of employee services in exchange for equity awards based on the grant-date of an award and recognize the cost over the requisite service period. We recognize compensation expense on fixed awards with graded vesting on a straight-line basis over the awards' vesting period.
We estimate the fair value of each option on the date of grant using the Black-Scholes option-pricing model, which requires the consideration of several subjective assumptions, including the expected dividends on our common stock, the expected volatility of our common stock, the risk-free interest rate for the expected option term and the expected term of the option. Equity instrument valuation models, such as the Black-Scholes valuation model, are highly subjective. Any significant changes in any of our estimates and judgments, including those used to select the inputs for the Black-Scholes valuation model, could have a significant impact on the fair value of the equity instruments granted or sold and the associated compensation charge, if any, we record in our financial statements.
54
In 2009, 2008 and 2007, the weighted average assumptions used in the option pricing models were as follows:
| |
For the Years Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | |||||||
Dividend yield |
0 | % | 0 | % | 0 | % | ||||
Expected volatility |
68 | % | 65 | % | 68 | % | ||||
Risk-free interest rate |
2.33 | % | 2.79 | % | 4.41 | % | ||||
Expected term (years) |
6.16 | 5.67 | 5.79 | |||||||
Because share-based compensation expense recognized in our consolidated statements of operations is based on awards ultimately expected to vest, it has been reduced for estimated forfeitures. If factors change and we employ different assumptions in future periods, the compensation expense that we record may differ significantly from what we have recorded in the current year.
Allowance for Doubtful Accounts
We continuously monitor collections from our customers and maintain a provision for estimated credit losses based upon our historical payment experience and any specific customer collection issues that we have identified. While such credit losses have historically been within our expectations and the provisions established, we cannot guarantee that we will continue to experience the same credit loss rates that we have in the past. Substantially all of our receivables are due from hospitals, distributors, health care clinics, and managed care systems located throughout the United States, Canada, Europe, Asia and South America. A significant portion of products sold, both foreign and domestic, is ultimately funded through government reimbursement programs. As a consequence, changes in these programs can have an adverse impact on our operating results and cash flows.
Inventories
We value our inventory at the lower of the actual cost to purchase or manufacture the inventory or the net realizable market value for such inventory. We regularly review inventory quantities in process and on hand and record a provision for production loss and obsolete inventory based primarily on actual loss experience and on our estimated forecast of product demand. A significant decrease in demand could result in an increase in the amount of excess inventory quantities on hand. In the future, if our inventory is determined to be overvalued, we would be required to recognize such costs in our costs of product sales at the time of such determination. Although we make every effort to ensure the accuracy of our production process and forecasts of future product demand, any significant unanticipated changes in production yield or product demand could have a significant impact on the value of our inventory and our reported operating results.
Goodwill and Other Assets
Goodwill represents the difference between the purchase price and the fair value of the tangible and identifiable intangible assets acquired net of liabilities assumed when accounted for in accordance with the purchase method of accounting. Between February 1999 and November 2001, we recorded goodwill upon the step acquisition of BioSphere Medical SA, or BMSA.
We perform impairment reviews of our goodwill annually or whenever events or changes in circumstances indicate that the carrying value of the assets may not be recoverable. Goodwill was derived from the step acquisition of BMSA, our consolidated subsidiary that holds the license to the embolotherapy platform device that is the main focus of our business. In performing the review, we utilize the two-step approach. The first step requires a comparison of the carrying value of the reporting units, as defined, to the fair value of these units. If the carrying value of a reporting unit
55
exceeds its fair value, we will perform the second step of comparing the implied fair value of the reporting unit's goodwill to its carrying value. For purposes of performing the goodwill impairment review, management considers there to be one reporting unit. Based upon our review, we have not recorded any impairment charges.
Long-Lived Assets
Long-lived assets are recorded at cost and amortized over their estimated useful lives. We are required to evaluate our long-lived assets for potential impairment whenever events or changes in circumstances may indicate that the carrying amount of a recorded asset may not be recoverable. If the carrying amount of a long-lived asset exceeds its fair value, an impairment loss is recognized. We believe that the carrying value of our long-lived assets was realizable as of December 31, 2009.
Income Taxes
We use the asset and liability accounting method whereby deferred tax assets and liabilities are recognized based on temporary differences between the amount recorded in the financial statements and the tax bases of such assets and liabilities using current statutory tax rates. A valuation allowance against net deferred tax assets is recorded if, based on the available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized.
We are subject to income tax in numerous jurisdictions at various rates worldwide, and the use of estimates is required in determining the tax provision and valuation allowance against net deferred tax assets. We have substantial net operating loss carryforwards, or NOL, that have generated significant deferred tax assets in our tax jurisdictions. Due to the size of the net operating loss carryforward in relation to our history of unprofitable operations, we historically have not recognized any of our net deferred tax assets. However, future improvements in our operational performance in a tax jurisdiction or significant changes in any of our estimates and judgments, including the operating profitability of our French subsidiary, if any, could increase the certainty of our ability to apply our deferred tax assets against taxable income, which, if so applied, could have a significant impact on the valuation of our deferred tax assets and our reported operating results.
Marketable Securities
We review our financial investments in debt and equity securities for other-than-temporary impairments. If an impairment exists and we intend to sell the security or it is more likely than not that we will sell the debt or equity security before recovery, the impairment is considered other-than-temporary and the entire amount of the impairment shall be recognized in earnings. Additionally, if an impairment of a debt security exists and it is more likely than not that we will not sell the debt security before recovery of its cost basis, but it is probable that we will be unable to collect all amounts due according to the contractual terms of the security, the impairment is considered other-than-temporary. The amount of the impairment related to credit losses would be recognized in earnings and the amount of the impairment related to other factors, such as changes in interest rates, would be recognized in other comprehensive income. During the year ended December 31, 2009, we sold two asset-backed securities at a loss of approximately $52,000, of which we already recognized an approximate $21,000 other-than-temporary decline as of December 31, 2008.
56
Results of Operations
Years Ended December 31, 2009 and 2008
Revenue and Margin Overview
| |
For the Years Ended December 31, | |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Increase/ (Decrease) ($) |
Increase/ (Decrease) (%) |
|||||||||||
(in thousands)
|
2009 | 2008 | |||||||||||
Total revenue |
$ | 31,443 | $ | 29,258 | $ | 2,185 | 7 | % | |||||
Costs of product sales |
7,682 | 7,686 | (4 | ) | 0 | % | |||||||
Gross margin |
$ | 23,761 | $ | 21,572 | $ | 2,189 | 10 | % | |||||
Gross margin % |
76 | % | 74 | % | 2 | % | |||||||
Revenue. Total revenue increased for the year ended December 31, 2009 as compared to the year ended December 31, 2008, primarily due to an increase in sales of our microsphere products for interventional gynecology and interventional oncology procedures offset by decreases in the sales of gastric products. Specifically:
- •
- product revenue from sales of our microspheres for use in interventional gynecology for UFE increased
$1.00 million, or 5%, from the year ended December 31, 2008, primarily on higher sales of our Embosphere Microspheres in the United States. During the year ended December 31,
2009, sales of our microsphere products in the United States increased $824,000, or 5%, primarily resulting from a price increase. We believe that the high rate of unemployment in the United States
during 2009, which has reduced demand for elective procedures such as UFE, has had the most significant impact on our UFE sales, resulting in flat sales volumes as compared to the year ended
December 31, 2008. Sales outside the United States increased $176,000, or 5%, due to increased demand in the People's Republic of China and Brazil, offset by changes in foreign exchange rates,
which decreased sales by approximately $154,000.
- •
- product revenue from microsphere sales used in interventional oncology increased $1.73 million, or 32%, from the year ended December 31, 2008 due to increased sales in all of our geographic regions. During the year ended December 31, 2009, sales of our microsphere products for use in interventional oncology in the United States increased $923,000, or 25%. Sales of these products outside the United States increased $804,000, or 51%, principally due to increased sales from our HepaSphere Microspheres product in Europe totaling $475,000 and to increased demand in the People's Republic of China and Brazil totaling $322,000. We believe the increase in HepaSphere Microsphere sales in Europe included distributor stocking orders of approximately $300,000 resulting from the required pre-notification in our distributor agreements of a HepaSphere Microsphere price increase, which took effect in January 2010.
In addition, revenue increased during the year ended December 31, 2009, as compared to 2008, due to increased sales of our delivery systems. Product revenue from the sales of our delivery system products increased $139,000, or 11%, from the year ended December 31, 2008, on increased demand for our EmboCath Plus catheter in the United States and our Sequitor Guidewire outside the United States.
Also included in total revenue for the years ended December 31, 2009 and 2008 is $555,000 and $416,000, respectively, of revenue from our licensing of non-strategic technology to a third party and collaborative agreements. During 2009, we recognized $236,000 of revenue related to the $1.00 million nonrefundable payment we received upon signing of a distribution agreement with Nippon Kayaku for the exclusive distribution of our embolic products in Japan. In 2008, licensing revenue related to certain patent technologies licensed to a third party.
57
Offsetting the increase in revenue noted above is the effect of changes in foreign exchange rates. During the year ended December 31, 2009, as compared to the same period in 2008, revenue decreased $255,000 due to changes in foreign exchange rates as revenue from our French operations decreased due to the strengthening of the U.S. dollar versus the euro, which averaged 1.39 dollars to the euro during 2009, compared to 1.46 dollars to the euro during 2008.
Offsetting the increase in revenue noted above was the decrease in sales of nonstrategic gastric products, which totaled $822,000 in 2008. We phased out nonstrategic products in 2008.
Gross Margin. The gross margin improvement of 2% as a percentage of revenue for 2009 as compared to 2008 was primarily attributable to a favorable mix of product sales, resulting from the phase-out of certain non-strategic products during 2008. Sales from our embolic and delivery system products, which have higher gross margins than our non-strategic products, represented 98% of overall sales during the year ended December 31, 2009, an increase of 2% as compared to the year ended December 31, 2008.
We expect that future gross margin will be highly correlated with the following factors:
- •
- revenue growth;
- •
- mix of products sold and mix of geographic location of sales;
- •
- production levels;
- •
- foreign exchange rate movements;
- •
- terms and conditions of subcontracted manufacturer and supplier agreements; and
- •
- future inventory reserve requirements.
Expense Overview
| |
For the Years Ended December 31, | |
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Increase/ (Decrease) ($) |
Increase/ (Decrease) (%) |
||||||||||||
(in thousands)
|
2009 | 2008 | ||||||||||||
Research and development |
$ | 3,415 | $ | 3,305 | $ | 110 | 3 | % | ||||||
Sales |
10,636 | 10,374 | 262 | 3 | % | |||||||||
Marketing |
5,243 | 6,514 | (1,271 | ) | (20 | )% | ||||||||
General and administrative |
7,058 | 6,499 | 559 | 9 | % | |||||||||
Patent |
781 | 705 | 76 | 11 | % | |||||||||
Total operating expenses |
$ | 27,133 | $ | 27,397 | $ | (264 | ) | |||||||
Research and Development Expense. Total research and development expense in the year ended December 31, 2009 increased slightly over the year ended December 31, 2008, due primarily to the increase in development activities. In the fourth quarter of 2009, we submitted an IDE application to the FDA seeking to commence a multi-site clinical trial of our QuadraSphere Microspheres loaded with doxorubicin for treatment of primary liver cancer. During 2009, we incurred approximately $314,000 related to filing and preparation activities relating to this IDE application. In addition, we conducted a study comparing our Embosphere Microsphere product to a competitor's product and the cost of the study was approximately $225,000. The increases in 2009 were offset by a change in exchange rates, which resulted in a decrease in the U.S. dollar value of expenses at our facility in France. In 2008, we performed various small scale studies that totaled approximately $377,000.
Sales Expense. Sales expense for the year ended December 31, 2009 increased over the year ended December 31, 2008, primarily due to the addition of resources to manage our distributor relationships in our emerging markets. Our emerging markets territories consist of Asia, Central
58
America, South America, Pacific Rim and Canada. During 2009, we hired a full time sales person to manage our distributors in Asia and we utilized a part-time sales person to manage existing relationships and to support our expansion in Central and South America.
Marketing Expense. Marketing expense for the year ended December 31, 2009 decreased from the year ended December 31, 2008, primarily due to a reduction in national marketing events combined with a smaller presence at medical association conferences. In 2009, we did not continue our Ask4/Tell4 and associated national public relations campaigns, which resulted in a decrease of expenses of approximately $775,000. In addition, we decreased our conference participation in the United States and Europe as we had a smaller presence at the 2009 Cardiovascular and Interventional Radiological Society of Europe, or CIRSE, Global Embolization Symposium and Technologies, or GEST, and Society of Interventional Radiology, or SIR, conferences, which resulted in a $375,000 decrease in expenses compared to 2008.
General and Administrative Expense. General and administrative expense for 2009 increased 9% from the year ended December 31, 2008, primarily due to an increase in business development activities and to a higher estimated management incentive bonus on improved performance against financial and individual targets.
Patent Expense. Patent expense for 2009 increased 11% from the year ended December 31, 2008, primarily due to costs associated with a review of our patent portfolio performed during 2009, offset by a reduction in the initial registrations of our drug delivery intellectual property as compared to 2008.
Interest Income. Interest income decreased to $8,000 in the year ended December 31, 2009 from $375,000 in the year ended December 31, 2008. The decrease in 2009 as compared to 2008 was due primarily to lower interest rates on available investment-grade assets.
Foreign Exchange Gains (Losses). Foreign exchange gains and losses primarily resulted from euro-to-U.S. dollar currency fluctuations on euro-denominated short-term intercompany trade accounts. Such foreign exchange gains during the year ended December 31, 2009 totaled approximately $69,000, compared to the foreign exchange gains of approximately $113,000 in the comparable period of 2008.
Income tax benefit (provision). During 2009, we received approximately $658,000 in research and development tax credits related to research and development activities performed at our French facility from 2004 through 2009. In 2009, we received $411,000 in cash refunds from the French tax authorities as a portion of the credits were monetized under an economic stimulus program enacted by the French government. During the third quarter of 2009, we conducted a study of these research and development tax credits and determined the benefit of these credits could be recognized in our financial statements. As of December 31, 2009, $77,000 in outstanding research and development tax credits related to 2005 have been included in other current assets, as they were not received by the French tax authorities until 2010. An additional $170,000 in tax credits related to 2009 has been included in other long-term assets.
Years Ended December 31, 2008 and 2007
Revenue and Margin Overview
| |
For the Years Ended December 31, | |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Increase/ (Decrease) ($) |
Increase/ (Decrease) (%) |
|||||||||||
(in thousands)
|
2008 | 2007 | |||||||||||
Total revenue |
$ | 29,258 | $ | 26,900 | $ | 2,358 | 9 | % | |||||
Costs of product sales |
7,686 | 7,768 | (82 | ) | (1 | )% | |||||||
Gross margin |
$ | 21,572 | $ | 19,132 | $ | 2,440 | 13 | % | |||||
Gross margin % |
74 | % | 71 | % | 3 | % | |||||||
59
Revenue. Total revenue increased for the year ended December 31, 2008 as compared to the year ended December 31, 2007, primarily due to an increase in sales of our microsphere products for interventional gynecology and interventional oncology procedures offset by decreases in the sales of gastric products. Specifically:
- •
- product revenue from sales of our microspheres for use in UFE increased $3.20 million, or 18%, from the year ended
December 31, 2007, primarily on higher sales of our Embosphere Microspheres in the United States. During the year ended December 31, 2008, sales of our microsphere products in the United
States increased $2.40 million, or 16%. We believe the increase in product revenue from microsphere sales for use in interventional gynecology for UFE is due to increased acceptance of the UFE
procedure by obstetricians and gynecologists and to increased awareness of the UFE procedure among symptomatic women resulting from our additional selling and marketing activities by our expanded U.S.
sales organization. At the beginning of 2008, we increased the number of direct sales territories in the United States from 18 to 24. Sales outside the United States increased $802,000, or 27%, due to
increased demand in Europe, the commercial launch of Embosphere Microspheres in the People's Republic of China and changes in foreign exchange rates.
- •
- product revenue from microsphere sales used in interventional oncology increased $683,000, or 15%, from the year ended December 31, 2007 on higher unit sales of our Embosphere and QuadraSphere Microspheres in the United States and sales of our Embosphere Microspheres in the People's Republic of China. During the year ended December 31, 2008, sales of our microsphere products for use in interventional oncology in the United States increased $555,000, or 17%. Sales of these products outside the United States increased $128,000, or 9%, principally due to the commercial launch of our Embosphere Microsphere product in the People's Republic of China in January 2008.
Additional increases in revenue during the year ended December 31, 2008, as compared to 2007, were due to increased sales of our delivery systems and changes in foreign exchange rates. Product revenue from the sales of our delivery system products increased $129,000, or 12%, from the year ended December 31, 2007 on increased demand for our EmboCath Plus catheter in the United States.
Included in the increase in revenue noted above is the effect of changes in foreign exchange rates. During the year ended December 31, 2008, as compared to the same period in 2007, revenue increased $296,000 due to changes in foreign exchange rates as revenue from our French operations increased due to the weakening of the U.S. dollar versus the euro during the first three quarters of 2008.
Sales of nonstrategic gastric products in Europe, which include, barium and drainage kits, decreased $1.65 million, or 67%, from the year ended December 31, 2007 as we phased out nonstrategic products in 2008.
Also included in total revenue for the years ended December 31, 2008 and 2007 is $416,000 and $417,000, respectively, of revenue from our licensing of nonstrategic technology to a third party.
Gross Margin. The gross margin improvement of 3% as a percentage of revenue for 2008 as compared to 2007 was primarily attributable to a favorable mix of product sales resulting from the phaseout of certain nonstrategic products during 2008.
60
Expense Overview
| |
For the Years Ended December 31, | |
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Increase/ (Decrease) ($) |
Increase/ (Decrease) (%) |
||||||||||||
(in thousands)
|
2008 | 2007 | ||||||||||||
Research and development |
$ | 3,305 | $ | 2,342 | $ | 963 | 41 | % | ||||||
Sales |
10,374 | 7,671 | 2,703 | 35 | % | |||||||||
Marketing |
6,514 | 5,290 | 1,224 | 23 | % | |||||||||
General and administrative |
6,499 | 5,999 | 500 | 8 | % | |||||||||
Patent |
705 | 440 | 265 | 60 | % | |||||||||
Total operating expenses |
$ | 27,397 | $ | 21,742 | $ | 5,655 | ||||||||
Research and Development Expense. Total research and development expense in the fiscal year ended December 31, 2008 increased significantly over the fiscal year ended December 31, 2007, due primarily to higher employee costs due to an increased number of employees and higher third party development costs. During the first quarter of 2008, we hired a vice president of new product and business development and a vice president of regulatory, medical affairs and quality systems. In addition, in 2008 we engaged a third party to develop a new process for the manufacture of our Embosphere Microspheres. During the fourth quarter of 2008, we recognized $200,000 in additional expense associated with this new process development.
Sales Expense. Sales expense for the fiscal year ended December 31, 2008 increased over the fiscal year ended December 31, 2007, primarily due to an increase in salary, incentive compensation and other compensation-related expenses as a result of the expansion of our direct sales force in the United States and the addition of an employee dedicated to managing our distributor relationships in Asia and South America. At the beginning of 2008, we increased the number of U.S. sales territories from 18 to 24 and hired a fourth regional manager. During 2008, we also had three incremental quarters of employee-related costs associated with a sales manager we hired in the fourth quarter of 2007 to expand markets outside the United States and Europe.
Marketing Expense. Marketing expense for the fiscal year ended December 31, 2008 increased from the fiscal year ended December 31, 2007, primarily due to our increased marketing spending for UFE programs. During the third quarter of 2008, we launched our "Ask4Tell4" customer outreach program with supermodel actress and reality TV show celebrity, Beverly Johnson, who was featured on NBC's The Today Show, in an interview segment on NBC's iVillage Online, in a radio interview on the Tom Joiner Morning Show and in both online and newsletter segments on Womansday.com. In addition to the "Ask4Tell4" campaign, we increased local marketing activities during 2008, including increasing the number of comarketing programs run with hospitals.
General, Administrative and Patent Expense. General, administrative and patent expense for 2008 increased 12% from the fiscal year ended December 31, 2007, primarily due to higher intellectual property costs associated with our drug delivery platform and to legal costs related to our expansion into Asia.
Interest Income. Interest income decreased to $375,000 in the fiscal year ended December 31, 2008 from $1.02 million in the fiscal year ended December 31, 2007. The decrease in 2008 as compared to 2007 was due primarily to lower interest rates on available investment-grade assets.
Foreign Exchange Gains (Losses). The foreign exchange gains during the year ended December 31, 2008 totaled approximately $113,000, compared to the foreign exchange losses of approximately $239,000 in the comparable period of 2007. The increase was primarily the result of more favorable currency rates in 2008.
61
Liquidity and Capital Resources
As of December 31, 2009, we had $18.09 million of cash, cash equivalents and marketable securities, a decrease of $151,000 from $18.24 million at December 31, 2008. We have historically funded our operations from the net proceeds provided by public and private equity offerings, net revenue, bank financing and, to a lesser extent, the exercise of stock options.
Net cash provided by operating activities for the year ended December 31, 2009 was $452,000 and includes a net loss of $2.67 million, offset by $802,000 in working capital changes, non-cash charges primarily related to stock-based compensation and depreciation and amortization. Accounts receivable increased $435,000 from December 31, 2008 due to an increase in quarterly revenue during the fourth quarter of 2009 as compared to the fourth quarter of 2008. Days that sales were outstanding, or DSO, decreased to 56 days at December 31, 2009 from 59 days at December 31, 2008, primarily due to increased collection efforts in the United States. Accrued compensation increased $729,000 from December 31, 2008, due to an increase in the accrual for estimated management incentive bonuses based on higher year-to-date achievement of targets as compared to 2008. Other accrued expenses increased $605,000 from December 31, 2008, primarily due to deferred revenue relating to the Nippon Kayaku distribution agreement and an increase in accrued royalties on the sale of Embolic products.
The net cash used in operating activities in 2008 was $4.18 million and includes a net loss of $5.49 million and $1.14 million in working capital changes, offset by noncash charges primarily related to stock-based compensation and depreciation. Accounts receivable increased $722,000 as a result of a five-day increase in DSO, which increased to 59 days from 54 days at December 31, 2007. Accounts payable decreased $780,000, due primarily to the timing of payments on delivery system products, which are manufactured for us by third parties, production materials and capital equipment.
During 2009, we spent $235,000 to purchase new trade-show booths, computer hardware and software replacements, and to purchase equipment to increase the capacity of our research and development facility. In 2008, we spent $324,000 to purchase manufacturing equipment and information technology equipment to support the expansion of our direct sales force and our existing infrastructure. In August 2008, we acquired patent rights to enhance our intellectual property portfolio for a total cost of $345,000.
Net cash used in financing activities was $500,000 for the year ended December 31, 2009, which included $578,000 for the payment of a quarterly preferred stock dividend in cash, and scheduled principal payments on existing capital arrangements, offset by the proceeds from the issuance of common stock under our employee benefit and incentive plans.
We believe that the $18.09 million in cash, cash equivalents and marketable securities that we have as of December 31, 2009, together with anticipated proceeds from sales of our microspheres and delivery systems, will be sufficient to fund our operating and capital requirements as currently planned through at least the next twelve months. In the longer term, we expect to fund our operations and capital requirements through a combination of expected proceeds from product sales and capital equipment financings. If market conditions permit, we may seek additional funding through a combination of collaborative arrangements, debt financing, or the sale of additional equity securities.
Our currently planned operating and capital requirements primarily include the need for working capital to:
- •
- support our clinical trial for QuadraSphere Microspheres, if we commence such trial;
- •
- produce and manufacture our products;
- •
- support our United States sales force;
62
- •
- support our sales and marketing efforts directed at the use of our products in interventional gynecology, and other
indications, as well as our other products for sale;
- •
- support our ongoing research and development activities; and
- •
- fund our general and administrative costs and expenses.
However, our cash requirements may vary materially from those now planned due to a number of factors, including, without limitation:
- •
- unanticipated changes in the amount of revenue we generate from sales of our products, in particular from sales of our
Embosphere Microspheres for UFE;
- •
- changes in our UFE regulatory and marketing programs;
- •
- an adverse judgment in a product liability lawsuit which could materially adversely impact market acceptance of our
products and, if not adequately covered by our product liability insurance, could have a material adverse effect on our liquidity and our ability to continue all or a portion of our business
operations;
- •
- costs associated with our QuadraSphere Microspheres clinical trial, if we commence such trial;
- •
- costs resulting from changes in our research and development, regulatory and marketing strategies;
- •
- competitive advances that make it harder for us to market and sell our products;
- •
- the timing and cost of FDA regulatory review;
- •
- the market's acceptance of any approved products; and
- •
- adverse global market and economic conditions.
We also may need additional funds for possible strategic acquisitions of synergistic businesses, products and/or technologies.
We may require substantial additional cash to fund our planned, and any unplanned, expenses. If adequate funds are not available, we could be required to reduce our capital expenditures, scale back or eliminate some or all of our clinical research, development, sales and marketing initiatives, reduce our workforce, license to others, or divest products or technologies that we otherwise would seek to commercialize ourselves, or otherwise curtail our business operations. If market conditions permit, we may seek additional funding through a combination of collaborative arrangements, debt financing, or the sale of additional equity securities. We may not receive such additional funding on reasonable terms, or at all. Any sales of equity or debt securities are likely to dilute our existing stockholders, and the new securities may have rights, preferences or privileges senior to those of existing holders of our capital stock. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions such as incurring additional debt, or making capital expenditures. If we raise additional funds through collaboration and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or products, or grant licenses on terms that are not favorable to us.
Borrowing Arrangements
On June 30, 2009, our $3.00 million credit facility with a bank expired. There were no borrowings outstanding under this agreement as of June 30, 2009, and we did not borrow any amounts under this agreement during the prior five years. Previously, the credit facility acted as security for all outstanding letters of credit. As of December 31, 2009, we had $58,000 classified as restricted investments to be
63
held as collateral for our existing letters of credit relating to our facility leases. We do not anticipate seeking a new credit facility in the near term.
Other Contractual Obligations
As of December 31, 2009, we are party to two operating leases for our facilities in Rockland, Massachusetts, and Roissy, France. The Roissy, France, original operating lease was scheduled to expire in May 2010; however, in accordance with the lease agreement, it automatically renewed for a three-year period that ends in May 2013. In March 2010, we amended the lease for the office and laboratory facility that we currently occupy in Rockland, Massachusetts. Pursuant to that amendment, the term of the lease was extended from February 28, 2010 to February 28, 2011.
We are party to a noncancelable capital lease agreement with an equipment-financing company, related to the acquisition during 2005 of communication and computer equipment. The equipment lease has an initial term of 60 months, with an interest rate of 8.7%. Equipment leased under this arrangement serves as pledged capital with respect to the capital lease agreement.
Future cash payments, including interest, under contractual obligations in effect as of December 31, 2009, are as follows:
| |
Payments Due by Period | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in thousands)
|
Total | Less than 1 Year |
1 - 3 Years |
3 - 5 Years |
More than 5 Years |
||||||||||||
Capital lease |
$ | 8 | $ | 8 | $ | — | $ | — | $ | — | |||||||
Operating lease |
1,440 | 615 | 688 | 136 | 1 | ||||||||||||
Other contractual obligations |
1,289 | 1,226 | 48 | 15 | — | ||||||||||||
Total(1) |
$ | 2,737 | $ | 1,849 | $ | 736 | $ | 151 | $ | 1 | |||||||
- (1)
- This table excludes a liability for unrecognized tax benefits. Due to the uncertain nature of these tax matters, we are unable to make a reasonably reliable estimate as to if and when cash settlements with the appropriate taxing authorities will occur.
The forgoing table of contractual obligations does not include cash payments that we may make from time to time in satisfaction of the dividend payable on our outstanding shares of series A preferred stock. The holders of the series A preferred stock have the right to receive a 6% dividend per annum which, at our election, may be paid in cash or in additional shares of series A preferred stock. During 2009, we elected to pay this dividend in cash, in the aggregate annual amount of $578,000. We expect to make such payment in cash for the foreseeable future and estimate that such amount will equal approximately $578,000 per year in each of the next three years.
Related Party Transactions
We did not have any related party transactions during 2009.
Off-Balance Sheet Arrangements
We do not have any material off-balance sheet arrangements.
Inflation
We believe that the effects of inflation generally do not have a material adverse impact on our operations or financial condition.
64
New Accounting Pronouncements
Revenue Recognition (ASU 2009-13 Topic 605)—Multiple-Deliverable Revenue Arrangements
The amendments of ASU 2009-13 require an entity to allocate arrangement consideration at the inception of an arrangement to all of its deliverables based on their relative selling prices. When applying the relative-selling-price method, the determination of the selling price for each deliverable must be consistent with the objective of determining vendor-specific objective evidence of fair value, which is the price at which the entity does or would sell the element on a stand-alone basis. The amendments of ASU 2009-13 require both ongoing disclosures regarding an entity's multiple-element revenue arrangement as well as certain transitional disclosures during the periods after adoption. This guidance must be adopted no later than the beginning of the first fiscal year beginning on or after June 15, 2010. We do not believe the adoption of this standard will not have a material impact on our results of operations, financial position or cash flows.
Item 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Derivative Financial Instruments, Other Financial Instruments, and Derivative Commodity Instruments
As of December 31, 2009, we did not own any derivative financial instruments or other financial and commodity instruments. However, in the future, we may consider certain financing instruments, including foreign currency forward contracts or alternative instruments, which may be considered derivative in nature.
Primary Market Risk Exposures
Our primary market risk exposure is in the area of foreign currency exchange rate fluctuations. We are exposed to currency exchange rate fluctuations related to our operations in France. Operations in France are denominated in the euro, and as of December 31, 2009, approximately euro 3.06 million, or $4.39 million, remained outstanding within the intercompany trade accounts. We have not engaged in formal currency hedging activities to date, but we do have a limited natural hedge in that both our revenue and expenses in France are primarily denominated in the euro. We also attempt to minimize exchange rate risk by converting non-U.S. currency to U.S. dollars as often as practicable. We generally view our investment in foreign subsidiaries operating under a functional currency (the euro) other than our reporting currency (the U.S. dollar) as long term. Our investment in foreign subsidiaries is sensitive to fluctuations in foreign currency exchange rates. The effect of a change in foreign exchange rates on our net investment in foreign subsidiaries is reflected in the "Accumulated other comprehensive income" component of stockholders' equity. A hypothetical 100-basis-point increase or decrease in foreign exchange rates would not have a material impact on the fair value of our investment in foreign subsidiaries.
The primary objective of our cash, cash equivalent and marketable securities investment activities is to preserve principal while at the same time maximizing the income we receive from our investments without significantly increasing risk. We maintain our portfolio of cash equivalents and short-term investments in corporate, bank, federal agency and mortgage-backed obligations and, to a lesser extent, in treasuries. Due to the conservative nature of our investments, the relatively short duration of their maturities, our ability to convert some or all of our long-term investments to less interest rate-sensitive holdings and our general intent to hold most securities until maturity, we believe interest rate risk is not significant. A hypothetical 100-basis-point increase or decrease in interest rates would not have a material impact on the fair value of our short-term investments or their respective cash flows as of December 31, 2009. As of December 31, 2009, approximately 90% of the $14.72 million of investments classified as available-for-sale marketable securities and cash equivalents will mature within one year.
65
66
Item 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Shareholders of BioSphere Medical, Inc.
We have audited the accompanying consolidated balance sheets of BioSphere Medical, Inc. as of December 31, 2009 and 2008, and the related consolidated statements of operations, shareholders' equity, and cash flows for each of the three years in the period ended December 31, 2009. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company's internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of BioSphere Medical, Inc. at December 31, 2009 and 2008, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2009, in conformity with U.S. generally accepted accounting principles.
/s/ Ernst & Young LLP
Boston,
Massachusetts
March 26, 2010
67
BIOSPHERE MEDICAL, INC.
CONSOLIDATED BALANCE SHEETS
| |
December 31, | ||||||||
|---|---|---|---|---|---|---|---|---|---|
(in thousands except share data)
|
2009 | 2008 | |||||||
ASSETS |
|||||||||
Current Assets: |
|||||||||
Cash and cash equivalents |
$ | 8,573 | $ | 17,837 | |||||
Marketable securities |
9,515 | 402 | |||||||
Account receivable, net of allowance for doubtful accounts of $57 and $97 as of December 31, 2009 and 2008, respectively |
5,183 | 4,729 | |||||||
Inventories |
3,713 | 3,762 | |||||||
Prepaid and other current assets |
639 | 663 | |||||||
Total current assets |
27,623 | 27,393 | |||||||
Property and equipment, net |
829 | 989 | |||||||
Goodwill |
1,443 | 1,443 | |||||||
Other assets |
552 | 403 | |||||||
Total Assets |
$ | 30,447 | $ | 30,228 | |||||
LIABILITIES AND STOCKHOLDERS' EQUITY |
|||||||||
Current Liabilities: |
|||||||||
Accounts payable |
$ | 1,128 | $ | 1,175 | |||||
Accrued compensation |
2,796 | 2,051 | |||||||
Accrued preferred dividend |
144 | 144 | |||||||
Other accrued liabilities |
1,960 | 2,032 | |||||||
Current portion of capital lease obligations |
8 | 9 | |||||||
Current portion of deferred revenue |
333 | 63 | |||||||
Total current liabilities |
6,369 | 5,474 | |||||||
Long-term capital lease obligations |
— | 8 | |||||||
Long-term portion of deferred revenue |
431 | — | |||||||
Total Liabilities |
6,800 | 5,482 | |||||||
Commitments and contingencies (Note 8 and 15) Stockholders' equity: |
|||||||||
Preferred stock; $.01 par value; 1,000,000 shares authorized: |
|||||||||
6% series A convertible preferred stock, 12,000 authorized shares, 9,636 shares issued and outstanding, as of December 31, 2009 and 2008 |
8,521 | 8,521 | |||||||
Common stock; $.01 par value; 50,000,000 shares authorized; 18,419,895 and 18,347,022 shares issued and outstanding as of December 31, 2009 and 2008, respectively |
184 | 184 | |||||||
Additional paid-in capital |
107,742 | 105,729 | |||||||
Accumulated deficit |
(93,381 | ) | (90,129 | ) | |||||
Accumulated other comprehensive income |
581 | 441 | |||||||
Total stockholders' equity |
23,647 | 24,746 | |||||||
Total Liabilities and Stockholders' Equity |
$ | 30,447 | $ | 30,228 | |||||
The accompanying notes are an integral part of these consolidated financial statements.
68
BIOSPHERE MEDICAL, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
| |
For the Years Ended December 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in thousands except per share data)
|
2009 | 2008 | 2007 | |||||||||
Revenue: |
||||||||||||
Product sales |
$ | 30,888 | $ | 28,842 | $ | 26,483 | ||||||
Licensing and collaborative revenue |
555 | 416 | 417 | |||||||||
Total revenue |
31,443 | 29,258 | 26,900 | |||||||||
Costs and expenses: |
||||||||||||
Costs of product sales |
7,682 | 7,686 | 7,768 | |||||||||
Research and development |
3,415 | 3,305 | 2,342 | |||||||||
Sales |
10,636 | 10,374 | 7,671 | |||||||||
Marketing |
5,243 | 6,514 | 5,290 | |||||||||
General and administrative |
7,058 | 6,499 | 5,999 | |||||||||
Patent |
781 | 705 | 440 | |||||||||
Total costs and expenses |
34,815 | 35,083 | 29,510 | |||||||||
Loss from operations |
(3,372 | ) | (5,825 | ) | (2,610 | ) | ||||||
Interest income |
8 | 375 | 1,017 | |||||||||
Interest expense |
(6 | ) | (9 | ) | (17 | ) | ||||||
Foreign exchange gain (loss), net |
69 | 113 | (239 | ) | ||||||||
Other (expense) income, net |
(31 | ) | (17 | ) | (5 | ) | ||||||
Loss before income tax |
(3,332 | ) | (5,363 | ) | (1,854 | ) | ||||||
Income tax benefit (provision) |
658 | (129 | ) | — | ||||||||
Net loss |
(2,674 | ) | (5,492 | ) | (1,854 | ) | ||||||
Preferred stock dividends |
(578 | ) | (578 | ) | (557 | ) | ||||||
Net loss applicable to common stockholders |
$ | (3,252 | ) | $ | (6,070 | ) | $ | (2,411 | ) | |||
Net loss per common share applicable to common stockholders |
||||||||||||
Basic and diluted |
$ | (0.18 | ) | $ | (0.34 | ) | $ | (0.14 | ) | |||
Weighted average number of common shares outstanding |
||||||||||||
Basic and diluted |
18,044 | 17,983 | 17,647 | |||||||||
The accompanying notes are an integral part of these consolidated financial statements.
69
BIOSPHERE MEDICAL, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
AND COMPREHENSIVE LOSS
| |
Preferred Stock | Common Stock | |
|
Accumulated Other Comprehensive Income |
|
||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Additional Paid-in Capital |
Accumulated Deficit |
Total Stockholders' Equity |
|||||||||||||||||||||||
(in thousands)
|
Shares | Amount | Shares | Amount | ||||||||||||||||||||||
Balance at December 31, 2006 |
9,083 | $ | 7,970 | 17,958 | $ | 180 | $ | 100,275 | $ | (81,648 | ) | $ | 188 | $ | 26,965 | |||||||||||
Comprehensive loss: |
||||||||||||||||||||||||||
Net loss |
— | — | — | — | — | (1,854 | ) | — | (1,854 | ) | ||||||||||||||||
Unrealized gain on marketable securities |
— | — | — | — | — | — | 10 | 10 | ||||||||||||||||||
Translation adjustment |
— | — | — | — | — | — | 511 | 511 | ||||||||||||||||||
Total comprehensive loss |
(1,333 | ) | ||||||||||||||||||||||||
Dividends on convertible preferred stock |
553 | 557 | — | — | — | (557 | ) | — | — | |||||||||||||||||
Dividends paid in cash in lieu of partial shares |
— | (4 | ) | — | — | — | — | — | (4 | ) | ||||||||||||||||
Issuance of common stock under employee benefit and incentive plans |
— | — | 412 | 4 | 1,642 | — | — | 1,646 | ||||||||||||||||||
Issuance of restricted stock |
— | — | 17 | — | — | — | — | — | ||||||||||||||||||
Forfeiture of restricted stock |
— | — | (100 | ) | (1 | ) | — | — | — | (1 | ) | |||||||||||||||
Non-cash stock-based compensation |
— | — | — | — | 1,836 | — | — | 1,836 | ||||||||||||||||||
Balance at December 31, 2007 |
9,636 | 8,523 | 18,287 | 183 | 103,753 | (84,059 | ) | 709 | 29,109 | |||||||||||||||||
Net loss |
— | — | — | — | — | (5,492 | ) | — | (5,492 | ) | ||||||||||||||||
Unrealized loss on marketable securities |
— | — | — | — | — | — | (40 | ) | (40 | ) | ||||||||||||||||
Translation adjustment |
— | — | — | — | — | — | (228 | ) | (228 | ) | ||||||||||||||||
Total comprehensive loss |
(5,760 | ) | ||||||||||||||||||||||||
Dividends on convertible preferred stock |
— | — | — | — | — | (578 | ) | — | (578 | ) | ||||||||||||||||
Dividends paid in cash in lieu of partial shares |
— | (2 | ) | — | — | — | — | — | (2 | ) | ||||||||||||||||
Issuance of common stock under employee benefit and incentive plans |
— | — | 42 | 1 | 104 | — | — | 105 | ||||||||||||||||||
Issuance of restricted stock |
— | — | 18 | — | — | — | — | — | ||||||||||||||||||
Non-cash stock-based compensation |
— | — | — | — | 1,872 | — | — | 1,872 | ||||||||||||||||||
Balance at December 31, 2008 |
9,636 | 8,521 | 18,347 | 184 | 105,729 | (90,129 | ) | 441 | 24,746 | |||||||||||||||||
Comprehensive loss: |
||||||||||||||||||||||||||
Net loss |
— | — | — | — | — | (2,674 | ) | — | (2,674 | ) | ||||||||||||||||
Unrealized gain on marketable securities |
— | — | — | — | — | — | 33 | 33 | ||||||||||||||||||
Translation adjustment |
— | — | — | — | — | — | 107 | 107 | ||||||||||||||||||
Total comprehensive loss |
— | — | — | — | — | — | — | (2,534 | ) | |||||||||||||||||
Dividends on convertible preferred stock |
— | — | — | — | — | (578 | ) | — | (578 | ) | ||||||||||||||||
Issuance of common stock under employee benefit and incentive plans |
— | — | 55 | — | 87 | — | — | 87 | ||||||||||||||||||
Issuance of restricted stock |
— | — | 18 | — | — | — | — | — | ||||||||||||||||||
Non-cash stock-based compensation |
— | — | — | — | 1,926 | — | — | 1,926 | ||||||||||||||||||
Balance at December 31, 2009 |
9,636 | $ | 8,521 | 18,420 | $ | 184 | $ | 107,742 | $ | (93,381 | ) | $ | 581 | $ | 23,647 | |||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
70
BIOSPHERE MEDICAL, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
| |
For the Years Ended December 31, | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in thousands)
|
2009 | 2008 | 2007 | ||||||||||
Cash flows from operating activities: |
|||||||||||||
Net loss |
$ | (2,674 | ) | $ | (5,492 | ) | $ | (1,854 | ) | ||||
Adjustments to reconcile net loss to net cash (provided by) used in operating activities: |
|||||||||||||
Provision (recovery) of doubtful accounts |
3 | (8 | ) | (38 | ) | ||||||||
Provision for inventory obsolescence |
— | 257 | 328 | ||||||||||
Depreciation and amortization |
433 | 430 | 438 | ||||||||||
Non-cash stock compensation |
1,926 | 1,872 | 1,836 | ||||||||||
Foreign currency (gain) loss, net |
(69 | ) | (113 | ) | 239 | ||||||||
Realized loss on available-for-sale investments |
31 | 1 | 13 | ||||||||||
Unrealized loss on available-for-sale investments |
— | 20 | — | ||||||||||
Loss on disposal of property and equipment |
— | — | 1 | ||||||||||
Changes in operating assets and liabilities: |
|||||||||||||
Accounts receivable |
(435 | ) | (731 | ) | 240 | ||||||||
Inventories |
96 | (252 | ) | (1,128 | ) | ||||||||
Prepaid and other current assets |
(138 | ) | (24 | ) | 4 | ||||||||
Accounts payable |
(55 | ) | (780 | ) | 513 | ||||||||
Accrued compensation |
729 | 411 | (334 | ) | |||||||||
Deferred revenue |
701 | (83 | ) | (83 | ) | ||||||||
Other accrued expenses |
(96 | ) | 314 | 152 | |||||||||
Net cash provided by (used in) operating activities |
452 | (4,178 | ) | 327 | |||||||||
Cash flows from investing activities: |
|||||||||||||
Purchase of property and equipment |
(235 | ) | (324 | ) | (565 | ) | |||||||
Purchase of marketable securities |
(9,580 | ) | — | (11,162 | ) | ||||||||
Proceeds from the sale and maturity of marketable securities |
469 | 7,507 | 16,395 | ||||||||||
Acquisition of patent rights |
— | (345 | ) | — | |||||||||
Net cash (used in) provided by investing activities |
(9,346 | ) | 6,838 | 4,668 | |||||||||
Cash flows from financing activities: |
|||||||||||||
Proceeds from issuance of common stock under employee benefit and incentive plans |
87 | 105 | 1,646 | ||||||||||
Payment of cash dividends |
(578 | ) | (435 | ) | (4 | ) | |||||||
Principal payments under capital lease obligations |
(9 | ) | (27 | ) | (57 | ) | |||||||
Net cash (used in) provided by financing activities |
(500 | ) | (357 | ) | 1,585 | ||||||||
Effect of exchange rate changes on cash and cash equivalents |
130 | (74 | ) | 115 | |||||||||
Net change in cash and cash equivalents |
(9,264 | ) | 2,229 | 6,695 | |||||||||
Cash and cash equivalents at beginning of year |
17,837 | 15,608 | 8,913 | ||||||||||
Cash and cash equivalents at end of year |
$ | 8,573 | $ | 17,837 | $ | 15,608 | |||||||
The accompanying notes are an integral part of these consolidated financial statements.
71
BIOSPHERE MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Nature of the Business
BioSphere Medical, Inc. (the "Company") develops, manufactures and markets products for medical procedures that use embolotherapy. Embolotherapy is the minimally invasive, image-guided therapeutic introduction of various biocompatible substances into a patient's circulatory system to occlude a blood vessel, either to arrest or prevent hemorrhaging or to devitalize or destroy a structure by occluding its blood supply. The Company's core technologies consist of patented bioengineered polymers, which are chemical compounds that the Company creates through the application to medical science, engineering principles and manufacturing methods. These core technologies are used to produce miniature spherical embolic particles, or microspheres, that are designed to have uniquely beneficial properties for a variety of medical applications. The Company's principal focus is the application of its Embosphere® Microspheres for the treatment of symptomatic uterine fibroids using a procedure called uterine fibroid embolization ("UFE"). The Company's wholly-owned subsidiary, BioSphere Medical SA ("BMSA"), a French société anonyme, holds the license to the embolotherapy technology that is the main focus of the Company's business.
The Company believes that its existing working capital as of December 31, 2009, together with anticipated proceeds from sales of microspheres, delivery systems and other products will be sufficient to fund operating and capital requirements, as currently planned through at least the next twelve months. In the longer term, the Company expects to fund its operations and sustain its capital requirements through a combination of expected proceeds from product sales and capital equipment financing. However, cash requirements may vary materially from those now planned due to a number of factors, including the Company's failure to achieve expected revenue amounts, costs associated with changes in its UFE marketing programs, the outcome of product liability challenges, including the current product liability lawsuit described in Note 15, "Contingencies," for which an adverse judgment against the Company may not be adequately covered by product liability insurance, unanticipated research and development expenses, the scope and results of preclinical and clinical testing, changes in the focus and direction of research and development programs, competitive and technological advances, the timing and results of regulatory review at the United States Food and Drug Administration ("FDA") or comparable regulatory agencies in other countries, delays or failures in the market's acceptance of any approved products, including Embosphere Microspheres for UFE, HepaSphere™ Microspheres and QuadraSphere® Microspheres, and the need for additional funds for possible strategic acquisitions of synergistic businesses, products and/or technologies.
2. Summary of Significant Accounting Policies
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries BMSA, BioSphere Medical Japan, Inc. and BSMD Ventures, Inc. All intercompany balances and transactions have been eliminated in consolidation.
Translation of Foreign Currencies
The functional currency of each of the Company's foreign subsidiaries is its local currency. The assets and liabilities of the Company's foreign subsidiaries are translated into U.S. dollars using the exchange rates in effect as of each balance sheet date. Revenue and expense items are translated into U.S. dollars at average exchange rates prevailing during each reporting period. Resulting translation adjustments are recorded in the cumulative translation adjustment account in stockholders' equity.
72
Aggregate foreign exchange transaction gains and losses resulting from euro to U.S. dollar foreign currency fluctuations on euro-denominated intercompany trade accounts are included in the accompanying statement of operations.
Use of Estimates
The preparation of financial statements in conformity with United States generally accepted accounting principles requires management to make estimates and assumptions that affect the following: (1) the reported amounts of assets and liabilities, (2) the disclosure of contingent assets and liabilities at the date of the financial statements, and (3) the reported amounts of revenue and expenses during the reporting periods. Actual results could differ from those estimates.
Subsequent Events
In preparing these consolidated financial statements, the Company evaluated the events and transactions that occurred after December 31, 2009 and on or before the date these consolidated financial statements were issued.
Cash, Cash Equivalents and Marketable Securities
The Company considers all highly liquid investments with an original maturity of ninety days or less, as of the date of purchase, to be cash equivalents. In accordance with its investment policy, surplus cash is invested in investment grade corporate and U.S. government debt as well as certain asset-backed securities. The Company determines the appropriate classification of marketable securities at each balance sheet date. Available-for-sale marketable securities are carried at their fair value. Unrealized gains and unrealized losses that are not deemed to be other-than-temporary are included in accumulated other comprehensive income. The Company recognizes declines in fair value of its marketable securities that are deemed to be other-than-temporary impairments in the consolidated statement of operations. During the year ended December 31, 2009, the Company sold two asset-backed securities at a loss of approximately $52,000, of which it had already recognized a $21,000 other-than-temporary decline as of December 31, 2008.
As of December 31, 2009, the Company has restricted investments of $58,000, which consist of amounts the Company is required to keep as collateral for its existing letter of credit relating to its facility lease in Rockland, Massachusetts.
As of December 31, 2009 and 2008, $8.07 million and $17.23 million, respectively, of cash and cash equivalents held by financial institutions in the United States exceeded Federal Deposit Insurance Corporation-insured amounts.
Fair Value of Financial Instruments
The Company's financial instruments include cash and cash equivalents, marketable securities, accounts receivable and payable, capital lease obligations and accrued liabilities. The carrying amount of these instruments approximates fair value because of their short-term nature.
Concentration of Credit Risk and Off-Balance Sheet Risk
The Company has no material concentrations of credit risk, nor is it a party to any financial instruments with material off-balance sheet risk. Financial instruments that potentially subject the Company to concentration of credit risk consist primarily of cash equivalents, marketable securities and trade accounts receivable. The estimated fair value of the Company's financial instruments approximates their carrying value. Concentrations of credit risk with respect to trade accounts receivable are limited due to the large number of customers and their dispersion across many
73
geographic areas. No single customer accounted for greater than 10% of the outstanding receivables on December 31, 2009 or 2008, and no single customer accounted for greater than 10% of revenue in 2009, 2008 or 2007.
The Company places its cash, cash equivalents and marketable securities with major United States financial institutions. In accordance with the Company's investment policy, surplus cash is invested in investment-grade corporate and U.S. government debt as well as certain asset-backed securities. At December 31, 2009, all marketable securities were classified as current, since the Company had the intent and ability to use such securities to satisfy current liabilities as needed. Available-for-sale marketable securities are carried at their fair value. Unrealized gains and losses that are not deemed to be other-than-temporary are included in accumulated other comprehensive income (loss) in the accompanying balance sheet. The Company recognizes declines in fair value of its marketable securities that are deemed to be other-than-temporary impairments in the consolidated statement of operations.
Accounts Receivable and Allowance for Doubtful Accounts
Trade accounts receivable are recorded at the invoiced amount and do not bear interest. The allowance for doubtful accounts is the Company's best estimate of the amount of probable credit losses in its existing accounts receivable. The Company determines the allowance based on the creditworthiness of customers, age of receivables, historical write-off experience and future expectations by location. The Company reviews its allowance for doubtful accounts monthly. Account balances are charged-off against the allowance when the Company feels it is probable the receivable will not be recovered. The Company does not have any off-balance sheet credit exposure related to its customers. The Company does not require collateral from its customers.
Property and Equipment
Property and equipment are stated at cost. The Company provides for depreciation based upon expected useful lives using the straight-line method over the following estimated useful lives:
Office equipment |
3-5 years | |
Laboratory and manufacturing equipment |
3-5 years | |
Leasehold improvements |
Shorter of lease term or estimated useful life |
Maintenance and repairs are charged to expense as incurred. Upon retirement or sale, the cost of disposed assets and the related accumulated depreciation are removed from the accounts, and any resulting gain or loss is credited or charged to the statement of operations.
Goodwill and Other Assets
Goodwill represents the difference between the purchase price and the fair value of the tangible and identifiable intangible assets acquired net of liabilities assumed when accounted for in accordance with the purchase method of accounting. Between February 1999 and November 2001, the Company recorded goodwill upon the step acquisition of BMSA.
The Company performs impairment reviews of its goodwill annually or whenever events or changes in circumstances indicate that the carrying value of the assets may not be recoverable. Goodwill was derived from the step acquisition of BMSA, the consolidated subsidiary that holds the license to the embolotherapy platform device that is the main focus of the Company's business. In performing the review, the Company utilizes the two-step approach. The first step requires a comparison of the carrying value of the reporting units, as defined, to the fair value of these units. If the carrying value of a reporting unit exceeds its fair value, the Company will perform the second step of comparing the implied fair value of the reporting unit's goodwill to its carrying value. For purposes of performing the
74
goodwill impairment review, management considers the Company to be one reporting unit. Based upon the Company's review, the Company has not recorded any impairment charges.
Long-Lived Assets
Long-lived assets are recorded at cost and amortized over their estimated useful lives. The Company is required to evaluate its long-lived assets for potential impairment whenever events or changes in circumstances may indicate that the carrying amount of a recorded asset may not be recoverable. If the carrying amount of a long-lived asset exceeds its fair value, an impairment loss is recognized. The Company believes that the carrying value of its long-lived assets was realizable as of December 31, 2009.
Revenue Recognition
The Company recognizes revenue when products are shipped and the customer or distributor takes ownership and assumes risk of loss, collection of the relevant receivable is reasonably assured, persuasive evidence of an arrangement exists (such as a valid purchase order from an approved customer or distributor), the sales price is fixed or determinable, payment is not contingent on resale and the Company does not have any continuing obligations to ensure resale. The Company establishes reserves for potential sales returns and evaluates the adequacy of those reserves based upon realized experience and expectations. Any significant credit returns could have a material adverse impact on the Company's revenue and operating results for the period or periods in which such returns materialize. Shipping and handling costs are included in costs of product sales.
In September 2006, the Company entered into an agreement to license certain patent technologies to a third party in exchange for an upfront lump-sum payment of $250,000 and an additional 4% royalty on future net sales of the licensed products. Under the agreement, the third party paid a minimum royalty of $1.00 million over the first three years of the agreement. The Company is recognizing both the up-front payment and the minimum royalties over the estimated useful life of the patent. The Company recognized approximately $319,000 and $416,000 as licensing revenue relating to this agreement during the year ended December 31, 2009 and 2008, respectively.
On April 16, 2009, the Company entered into an international distribution agreement with Nippon Kayaku Co., Ltd. ("Nippon Kayaku"). The agreement grants Nippon Kayaku the exclusive right to distribute the Company's HepaSphere Microspheres and Embosphere Microspheres upon regulatory approval in Japan. The agreement provides that Nippon Kayaku is responsible for filing, obtaining and maintaining all regulatory approvals necessary for the sale, marketing, pricing and reimbursement of the products in Japan, including performing any clinical trials required as a result of seeking such regulatory approvals in Japan. Assuming product approval, the Company will provide HepaSphere Microspheres and Embosphere Microspheres to Nippon Kayaku for distribution and sale in Japan. Additionally, Nippon Kayaku made a nonrefundable milestone payment of $1.00 million in 2009 which is being recognized as collaborative revenue ratably over the expected term of the research and development period, and has agreed to make up to $3.00 million in additional milestone payments based upon specified objectives, including achievement of clinical, regulatory and sales goals. For the year ended December 31, 2009, the Company recognized approximately $236,000 as collaborative revenue relating to the Nippon Kayaku agreement, with the remaining $333,000 and $431,000 recorded as short- and long-term deferred revenue, respectively.
Research and Development
Research and development costs include payroll, facility costs, administrative expenses, and third-party costs related to developing new products, making technological improvements to existing products and production methods. Research and development costs are expensed in the period incurred.
75
Preclinical testing of product candidates and clinical trials and product validation costs associated with recently launched products are also included in research and development expenses. Once launched, these products no longer have costs included in research and development.
Income Taxes
The Company uses the asset and liability accounting method whereby deferred tax assets and liabilities are recognized based on temporary differences between the amount recorded in the financial statements and the tax bases of such assets and liabilities using current statutory tax rates. A valuation allowance against net deferred tax assets is recorded if, based on the available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized.
The Company is subject to income tax in numerous jurisdictions at various rates worldwide, and the use of estimates is required in determining the tax provision and valuation allowance against net deferred tax assets. The Company has substantial net operating loss carryforwards that have generated significant deferred tax assets in its tax jurisdictions. Due to the size of the net operating loss carryforward in relation to the Company's history of unprofitable operations, the Company historically has not recognized any of its net deferred tax assets. However, future improvements in the Company's operational performance in a tax jurisdiction or significant changes in any of the Company's estimates and judgments, including the operating profitability of its French subsidiary, if any, could increase the certainty of the Company's ability to apply its deferred tax assets against taxable income, which, if so applied, could have a significant impact on the valuation of the Company's deferred tax assets and reported operating results.
Comprehensive Income
Other comprehensive income includes certain changes in equity that are excluded from net loss; specifically, the effects of foreign currency translation adjustments and unrealized gains and losses on available-for-sale marketable securities, which are reflected separately in stockholders' equity in accumulated other comprehensive income. The components of accumulated other comprehensive income are as follows:
| |
December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
(in thousands)
|
2009 | 2008 | ||||||
Foreign exchange currency translation |
$ | 582 | $ | 475 | ||||
Unrealized losses on investments |
(1 | ) | (34 | ) | ||||
Total accumulated other comprehensive income |
$ | 581 | $ | 441 | ||||
Net Loss Per Share
The Company calculates net income (loss) per share in accordance with the two-class method. Basic net income (loss) per share is computed by dividing the net income (loss) available to common stockholders by the weighted-average number of common shares outstanding during the period. Should the Company become profitable diluted net income (loss) per share would be computed using the more dilutive of the (a) the two-class method, or (b) the if-converted method. Since the Company is in a net
76
loss position, shares used to compute dilutive net loss per share exclude the following common share equivalents as their inclusion would have an antidilutive effect.
| |
As of December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
(in thousands)
|
2009 | 2008 | 2007 | |||||||
Shares issuable upon exercise of stock options |
3,475 | 2,891 | 2,220 | |||||||
Shares issuable upon conversion of convertible securities |
2,409 | 2,409 | 2,409 | |||||||
Shares issuable upon exercise of outstanding warrants |
— | 400 | 400 | |||||||
Unvested restricted stock awards |
335 | 335 | 333 | |||||||
|
6,219 | 6,035 | 5,362 | |||||||
Stock Options
The Company measures the cost of employee services in exchange for an award of equity instruments based on the grant-date fair value of the award and recognizes the cost over the requisite service period. The Company will recognize compensation expense for (1) all share-based payments granted after the effective date and (2) all awards granted to employees prior to the effective date that remain unvested on the effective date. The Company recognizes compensation expense on fixed awards with graded vesting on a straight-line basis over the vesting period of such awards.
Reclassifications
Certain reclassifications have been made to prior year's consolidated financial statements to conform to the current-year presentation.
New Accounting Pronouncements
Revenue Recognition (ASU 2009-13 Topic 605)—Multiple-Deliverable Revenue Arrangements
The amendments of ASU 2009-13 require an entity to allocate arrangement consideration at the inception of an arrangement to all of its deliverables based on their relative selling prices. When applying the relative-selling-price method, the determination of the selling price for each deliverable must be consistent with the objective of determining vendor-specific objective evidence of fair value, which is the price at which the entity does or would sell the element on a stand-alone basis. The amendments of ASU 2009-13 require both ongoing disclosures regarding an entity's multiple-element revenue arrangement, as well as certain transitional disclosures during the periods after adoption. This guidance must be adopted no later than the beginning of the first fiscal year beginning on or after June 15, 2010. The Company does not believe the adoption of this standard will have a material impact on its results of operations, financial position or cash flows.
3. Marketable Securities and Cash Equivalents
All current fixed maturity securities are classified as "available-for-sale" and are reported at fair value. The Company has determined that all of its investment securities are available to support current operations and, accordingly, has classified such marketable securities as current assets without regard to contractual maturities. The unrealized gains or losses on these securities are included in accumulated other comprehensive income as a separate component of stockholders' equity unless the decline in value is deemed to be other-than-temporary, in which case securities are written-down to fair value and the loss is charged to income. The Company evaluates its investment securities for other-than-temporary declines based on quantitative and qualitative factors. During the year ended December 31, 2009, the Company sold two asset-backed securities, recognizing a loss of $31,000.
77
The Company's available-for-sale marketable securities and cash equivalents, including accrued interest receivable as of December 31, 2009, are as follows:
(in thousands)
|
Amortized Cost |
Unrealized Gains |
Unrealized Losses |
Estimated Fair Value |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Marketable securities: |
|||||||||||||||
Corporate obligations |
$ | 4,051 | $ | 1 | $ | — | $ | 4,052 | |||||||
Bank obligations |
2,648 | 2 | — | 2,650 | |||||||||||
Federal agency obligations |
2,794 | — | (4 | ) | 2,790 | ||||||||||
Mortgage-backed obligations |
23 | — | — | 23 | |||||||||||
Cash equivalents: |
|||||||||||||||
Treasury bills |
125 | — | — | 125 | |||||||||||
Bank obligations |
1,458 | — | — | 1,458 | |||||||||||
Federal agency obligations |
1,000 | — | — | 1,000 | |||||||||||
Government obligations |
2,623 | — | — | 2,623 | |||||||||||
Total marketable securities and cash equivalents |
$ | 14,722 | $ | 3 | $ | (4 | ) | $ | 14,721 | ||||||
The Company's available-for-sale marketable securities and cash equivalents, including accrued interest receivable as of December 31, 2008, are as follows:
(in thousands)
|
Amortized Cost |
Unrealized Gains |
Unrealized Losses |
Estimated Fair Value |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Marketable securities: |
|||||||||||||||
Asset-backed obligations |
$ | 436 | $ | — | $ | (34 | ) | $ | 402 | ||||||
Cash equivalents: |
|||||||||||||||
Treasury obligations |
17,000 | — | — | 17,000 | |||||||||||
Total marketable securities and cash equivalents |
$ | 17,436 | $ | — | $ | (34 | ) | $ | 17,402 | ||||||
The Company recognized $31,000 and $21,000 of net losses on its marketable securities during the years ended December 31, 2009 and December 31, 2008, respectively.
78
As of December 31, 2009, the contractual maturities of marketable securities are as follows:
(in thousands)
|
Estimated Fair Value |
|||||
|---|---|---|---|---|---|---|
Due within one year: |
||||||
Corporate obligations |
$ | 4,052 | ||||
Bank obligations |
2,650 | |||||
Federal agency obligations |
1,277 | |||||
Due between one and five years: |
||||||
Federal agency obligations |
1,513 | |||||
Due after ten years: |
||||||
Mortgage-backed obligations |
23 | |||||
Total marketable securities |
9,515 | |||||
Cash equivalents: |
||||||
Treasury bills |
125 | |||||
Bank obligations |
1,458 | |||||
Federal agency obligations |
1,000 | |||||
Government obligations |
2,623 | |||||
Total cash equivalents |
5,206 | |||||
Total marketable securities and cash equivalents |
$ | 14,721 | ||||
The following table presents information about the Company's assets that are measured at fair value on a recurring basis at each reporting period as of December 31, 2009, and indicates the fair value hierarchy of the valuation techniques utilized to determine such fair value. In general, fair values determined by Level 1 inputs utilize quoted prices (unadjusted) in active markets for identical assets or liabilities. Fair values determined by Level 2 inputs utilize data points that are observable, such as quoted prices, interest rates and yield curves. Fair values determined by Level 3 inputs are unobservable data points for the asset or liability, and include situations where there is little, if any, market activity for the asset or liability:
| |
|
Fair Value Measurements at Reporting Date Using |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in thousands)
|
Fair Value at December 31, 2009 |
Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
|||||||||||
Marketable securities: |
|||||||||||||||
Corporate obligations |
$ | 4,052 | $ | — | $ | 4,052 | — | ||||||||
Bank obligations |
2,650 | — | 2,650 | — | |||||||||||
Federal agency obligations |
2,790 | — | 2,790 | — | |||||||||||
Mortgage-backed obligations |
23 | 23 | |||||||||||||
Cash equivalents: |
|||||||||||||||
Treasury bills |
125 | 125 | — | — | |||||||||||
Bank obligations |
1,458 | — | 1,458 | — | |||||||||||
Federal agency obligations |
1,000 | — | 1,000 | — | |||||||||||
Government obligations |
2,623 | 2,623 | — | — | |||||||||||
Total marketable securities and cash equivalents |
$ | 14,721 | $ | 2,748 | $ | 11,973 | — | ||||||||
79
(in thousands)
|
Fair Value Measurements Using Significant Unobservable Inputs (Level 3) |
|||||
|---|---|---|---|---|---|---|
Beginning balance (at December 31, 2008): |
$ | 92 | ||||
Total realized losses: |
||||||
Included in earnings |
(31 | ) | ||||
Proceeds from sale |
(61 | ) | ||||
Ending balance (at December 31, 2009) |
$ | — | ||||
4. Inventories
Inventories are stated at the lower of cost (first-in, first-out) or market and consist of the following:
| |
December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
(in thousands)
|
2009 | 2008 | ||||||
Finished goods |
$ | 1,892 | $ | 2,395 | ||||
Work in progress |
1,394 | 1,039 | ||||||
Raw material |
427 | 328 | ||||||
Total inventory |
$ | 3,713 | $ | 3,762 | ||||
5. Property and Equipment
Property and equipment consists of the following:
| |
December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
(in thousands)
|
2009 | 2008 | ||||||
Office equipment |
$ | 1,303 | $ | 1,235 | ||||
Laboratory and manufacturing equipment |
3,314 | 3,185 | ||||||
Leasehold improvements |
224 | 222 | ||||||
Total property and equipment |
4,841 | 4,642 | ||||||
Less: accumulated depreciation |
(4,012 | ) | (3,653 | ) | ||||
Net property and equipment |
$ | 829 | $ | 989 | ||||
Property and equipment under capital lease agreements, net of accumulated depreciation, which are included in the table above, were $7,000 and $15,000, respectively, at December 31, 2009 and 2008.
Depreciation expense, including amortization on capital leases, was $410,000, $424,000 and $438,000 for the years ended December 31, 2009, 2008 and 2007, respectively.
6. Goodwill and Other Assets
Goodwill equaled $1.44 million as of December 31, 2009 and 2008, respectively, and was comprised of the unamortized purchase price paid in excess of the net BMSA assets acquired.
Other assets include an intangible asset related to a patent acquisition. The intangible asset has a carrying value of approximately $316,000 and is being amortized over its approximate useful life of
80
17 years on a straight-line basis. The Company believes amortization expense will be approximately $23,000 per year for the next five years.
| |
December 31, 2009 | December 31, 2008 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in thousands)
|
Gross Carrying Amount |
Accumulated Amortization |
Gross Carrying Amount |
Accumulated Amortization |
||||||||||
Amortizable intangible assets: |
||||||||||||||
Patent acquisition |
$ | 345 | $ | (29 | ) | $ | 345 | $ | (6 | ) | ||||
Unamortizable intangible assets: |
||||||||||||||
Goodwill |
$ | 1,443 | $ | — | $ | 1,443 | $ | — | ||||||
7. Other Accrued Expenses
Accrued expenses consist of the following:
| |
December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
(in thousands)
|
2009 | 2008 | ||||||
Accrued royalties |
$ | 1,423 | $ | 1,241 | ||||
Accrued other |
537 | 791 | ||||||
Total accrued expenses |
$ | 1,960 | $ | 2,032 | ||||
8. Contractual Obligations
Capital lease obligations consist of the following:
| |
December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
(in thousands)
|
2009 | 2008 | ||||||
Capital lease obligations |
$ | 8 | $ | 17 | ||||
Less: current portion |
(8 | ) | (9 | ) | ||||
Total long-term capital lease obligations |
$ | — | $ | 8 | ||||
On June 30, 2009, the Company's credit facility with a bank expired. There were no borrowings outstanding under this agreement as of June 30, 2009, and the Company did not borrow any amounts under this agreement during the prior five years. Previously, the credit facility acted as security for all outstanding letters of credit. As of December 31, 2009, the Company had $58,000 classified as restricted investments to be held as collateral for its existing letters of credit relating to its United States facility lease. The Company does not anticipate entering into a new credit facility in the near term.
Letters of credit issued in the ordinary course of business totaled $228,000 as of December 31, 2009, and were partially collateralized by the restricted investments noted above.
The Company leases approximately 13,000 square feet of office and laboratory space at its Rockland, Massachusetts, facility under an operating lease expiring in February 2011 for approximately $257,000 per year, exclusive of periodic operating and maintenance expenses. BMSA leases approximately 18,000 square feet of manufacturing and office space in Roissy, France, through May 2010 for approximately €230,000 per year (approximately $329,000 as of December 31, 2009). The Company also has several operating leases covering two vehicles located in France and certain pieces of office equipment through 2010.
81
The Company has a capital lease agreement in connection with the acquisition of certain computer and communication equipment. The lease has an initial term of 60 months with an interest rate of 8.7%. All equipment leased under this agreement serves as pledged capital.
Future minimum lease payments under noncancelable operating leases and the capital lease in effect as of December 31, 2009 are as follows:
(in thousands)
|
Operating | Capital | ||||||
|---|---|---|---|---|---|---|---|---|
2010 |
$ | 615 | $ | 8 | ||||
2011 |
374 | |||||||
2012 |
314 | |||||||
2013 |
134 | |||||||
Thereafter |
3 | |||||||
Total lease commitments |
$ | 1,440 | $ | 8 | ||||
Less amount representing interest |
— | |||||||
Present value of net minimum capital lease payments |
$ | 8 | ||||||
Total rent expense for the years ended December 31, 2009, 2008 and 2007 was approximately $590,000, $560,000 and $650,000, respectively.
The Company has committed to purchasing inventory and other contractual obligations. As of December 31, 2009, these commitments totaled $1.29 million, some of which may be cancelable.
9. Income Taxes
The components of the Company's pre-tax income (loss) by tax jurisdiction, net of any intercompany transactions, are as follows:
| |
For the years ended December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
(in thousands)
|
2009 | 2008 | 2007 | ||||||||
United States |
$ | (4,152 | ) | $ | (5,410 | ) | $ | (2,875 | ) | ||
France |
820 | 47 | 1,021 | ||||||||
Pretax loss |
$ | (3,332 | ) | $ | (5,363 | ) | $ | (1,854 | ) | ||
For the year ended December 31, 2009, the increase in the valuation allowance relating to losses not resulting in a current period tax benefit is the primary difference between the income tax provision (benefit) recorded by the Company and the amount of the income tax benefit that would have recorded at statutory income tax rates. In 2009, the Company recorded a tax benefit of $658,000 due to research and development tax credits related to research and development activities performed at the Company's French facility between 2004 through 2009. The Company received $411,000 in cash refunds from the French tax authorities as a portion of the credits were monetized under an economic stimulus program enacted by the French government. During 2009, the Company conducted a study of these research and development tax credits and determined the benefit of these credits could be recognized in its financial statements. As of December 31, 2009, $77,000 in outstanding research and development tax credits related to 2005 have been included in other current assets as they were not received from the French tax authorities until January 2010; and $170,000 in tax credits related to 2009 have been included in long-term assets.
In 2008, the Company recorded an income tax provision of $129,000 due to the reversal of a previously applied income tax credit that was disallowed by the French tax authorities during a tax audit of BMSA. For the year ended December 31, 2008, the increase in the valuation allowance
82
relating to losses not resulting in a current period tax benefit and the reversal of the income tax credit of BMSA are the primary differences between the income tax provision (benefit) recorded by the Company and the amount of the income tax benefit that would be at statutory income tax rates.
For the year ended December 31, 2007, the increase in valuation allowance relating to losses not resulting in a current period tax provision (benefit) is the primary difference between the income tax provision recorded by the Company and the amount of income tax benefit that the Company would have recorded at statutory income tax rates.
During the years presented, the income earned in France was fully offset by previous net operating loss ("NOL") carryforwards. The Company's practice is to classify interest and penalties related to income tax matters to income tax provision (benefit). For the years ended December 31, 2009, 2008 and 2007, there were no material income tax interest or penalties.
As of December 31, 2009, the Company had federal NOL carryforwards of approximately $76.95 million, which will expire through the year 2029, state NOL carryforwards of approximately $15.80 million, which will expire through the year 2019, and foreign NOL carryforwards of approximately $3.58 million, which do not expire. During the year ended December 31, 2009, approximately $5.49 million of state NOL carryforwards expired. The Company has $185,000 of federal research and development credit carryforwards to offset future income taxes, which will expire through the year 2018. The components of the Company's net deferred tax asset at December 31, 2009 and 2008 are as follows:
| |
December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
(in thousands)
|
2009 | 2008 | ||||||
Assets derived from the following: |
||||||||
NOL carryforwards |
$ | 27,335 | $ | 27,419 | ||||
Tax credit carryforwards |
185 | 185 | ||||||
Stock-based compensation |
1,137 | 807 | ||||||
Other |
997 | 234 | ||||||
Subtotal |
29,654 | 28,645 | ||||||
Valuation allowance |
(29,654 | ) | (28,645 | ) | ||||
Net deferred tax asset |
$ | — | $ | — | ||||
Utilization of the NOL carryforwards may be subject to a substantial annual limitation under Section 382 of the Internal Revenue Code of 1986 due to ownership change limitations that have occurred previously or that could occur in the future. These ownership changes may limit the amount of NOL credit carryforwards that can be utilized annually to offset future taxable income and tax, respectively. The Company has not completed a study to assess whether an ownership change has occurred, or whether there have been multiple ownership changes since its formation, due to the significant complexity and related cost associated with such study. There also could be additional ownership changes in the future which may result in additional limitations in the utilization of the carryforward NOLs and credits.
As discussed in Note 2, the Company accounts for share-based awards according to accounting standards update, ASU, Topic 718 Share-based Payment. Generally, tax return deductions are allowable on such arrangements but may arise in different amounts and periods from compensation costs recognized in the financial statements. If the tax return deduction for an award exceeds the cumulative compensation cost recognized in the financial statements, any excess tax benefit shall be recognized as additional paid-in-capital when the deduction reduces taxes payable. Prior to adoption, the Company recognized deferred tax assets, along with an offsetting valuation allowance, for net operating loss carryforwards that included deductions for excess tax benefits from stock-based compensation. Included
83
in the net operating loss carryforwards stated above is approximately $7.67 million of unrealized excess tax benefit. In addition, the Company also has $2.38 million of additional net operating losses resulting from excess tax benefits that were recognized after the adoption of ASU Topic 718.
The Company has established a full valuation allowance against its deferred tax assets as of December 31, 2009, as it considers the realizable value of any tax benefit against future taxable income to be uncertain. The change in the valuation allowance from December 31, 2008 to December 31, 2009 is a result of the increase in NOL carryforwards from the inclusion of the current period loss, offset by a decrease in state NOL carryforwards due to expiration and the utilization of foreign net operating loss carryforwards.
The Company adopted accounting guidance for uncertainty in income taxes which prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return and provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosure and transition. At the adoption date of January 1, 2007 and also at December 31, 2009, 2008 and 2007, the Company had no unrecognized tax benefits.
The statute of limitations for assessment by the Internal Revenue Service, or the IRS, and state tax authorities is closed for tax years prior to December 31, 2006, although carryforward attributes that were generated prior to tax year 2006 may still be adjusted upon examination by the IRS or state tax authorities if they either have been or will be used in a future period. The Company files income tax returns in the United States, various state jurisdictions and France. The Company has been audited in France through 2006. There is currently no federal audit in progress. The Company is currently under audit for the state of Massachusetts for the tax years 2006 through 2008.
10. Segment Information
The Company develops microspheres and other ancillary embolotherapy products for use in the treatment of hypervascularized tumors, including uterine fibroids and arteriovenous malformations. The Company operates exclusively in the embolotherapy product business, which the Company considers as one business segment. Financial information by geographic area, attributable to countries according to the location of customers and equipment, is as follows:
| |
For the years ended December 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in thousands)
|
2009 | 2008 | 2007 | |||||||||
Revenue: |
||||||||||||
United States |
$ | 24,234 | $ | 22,523 | $ | 19,395 | ||||||
France |
1,771 | 2,643 | 4,074 | |||||||||
Other European Union countries |
2,386 | 2,097 | 2,183 | |||||||||
Other foreign countries |
3,052 | 1,995 | 1,248 | |||||||||
Total revenue |
$ | 31,443 | $ | 29,258 | $ | 26,900 | ||||||
| |
As of December 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in thousands)
|
2009 | 2008 | 2007 | |||||||||
Property and equipment: |
||||||||||||
United States |
$ | 210 | $ | 291 | $ | 340 | ||||||
France |
619 | 698 | 784 | |||||||||
Total property and equipment, net |
$ | 829 | $ | 989 | $ | 1,124 | ||||||
84
11. Stockholders' Equity
Preferred Stock
Under the Certificate of Incorporation of the Company, the Board of Directors has the authority to issue up to 1,000,000 shares of $0.01 par value preferred stock from time to time in one or more series with such preferences terms and rights as the Board of Directors may determine without further action by the stockholders of the Company and as of December 31, 2009, a total of 9,636 shares had been issued. Accordingly, the Board of Directors has the power to establish the provisions, if any, relating to dividends, voting rights, redemption rates, liquidation preferences and conversion rights for any series of preferred stock issued in the future.
6% Series A Convertible Preferred Stock
In November 2004, the Company completed a private placement of $8.00 million of its series A convertible preferred stock ("series A preferred stock") and warrants to purchase common stock with Sepracor Inc., an indirect wholly owned subsidiary of Dainippon Sumitomo Pharma Co., Ltd., and affiliates of Cerberus Capital Management, L.P., two existing investors. These investors purchased a total of 8,000 shares of series A preferred stock, which are initially convertible into 2,000,000 shares of common stock based upon a conversion price of $4.00 per share. In addition, the Company has the right to convert the series A preferred stock into common stock, or redeem it, under specified circumstances. The series A preferred stock has a 6% dividend, which is payable quarterly in either cash or additional shares of series A preferred stock, at the Company's election. Additionally, the investors were issued warrants to purchase an aggregate of 400,000 shares of common stock. In November 2009, these warrants expired unissued, five years from the date of issuance. Through December 31, 2009, the Company issued 1,636 shares of series A preferred stock in payment of series A preferred stock dividends requirements.
12. Stock Plans
Stock Incentive Plans
As of December 31, 2009, the Company has granted options and/or restricted stock awards under the following stock-based compensation plans: (1) the 2006 Stock Incentive Plan (the "2006 Plan"), which was adopted by the Company's Board of Directors on March 9, 2006, approved by the Company's stockholders on May 10, 2006 and amended on May 14, 2009, and which authorizes the issuance of up to an aggregate of 3,000,000 shares of common stock to officers, directors, advisors, consultants and employees of the Company; (2) the 1997 Stock Option Plan (the "1997 Plan"), which expired March 2007, and, accordingly, has no shares available for future grant. The Company's 2006 Plan and 1997 Plan each provide for the grant of Incentive Stock Options ("ISOs") to officers and employees and Non-Statutory Stock Options ("NSOs") to officers, directors, advisors, consultants and employees of the Company. Options granted under such plans generally become exercisable in five equal annual installments beginning on the first anniversary of the date of the grant and have a maximum term of ten years from the date of grant. At December 31, 2009, there were 914,000 shares available for future grant under the 2006 Plan.
The 2006 Plan also provides for the grant of restricted stock awards to officers, directors, advisors, consultants and employees of the Company. Generally, the restricted stock awards are subject to a right of repurchase by the Company if service is terminated prior to specified dates and/or if specified performance conditions are not met, which right of repurchase lapses over time. Ownership of restricted stock cannot be transferred, except under specified circumstances, until the foregoing repurchase restrictions have lapsed. In connection with restricted stock grants, the Company records compensation expense based on the fair value of the shares at the time of grant, which is amortized on a straight-line basis over the vesting periods.
85
On June 1, 2006, the Board of Directors awarded an aggregate of 400,000 shares of restricted common stock to the Company's existing executive officers under the 2006 Plan. These shares of restricted common stock are subject to a right of repurchase by the Company, which lapses on June 1, 2010, subject to the achievement by the Company of specified gains in the market price of its common stock. If on June 1, 2010, the four-year cumulative total stockholder return on the Company's common stock is equal in dollar amount to the four-year cumulative total return for the NASDAQ Medical Equipment Index ("NASDAQ Index"), 25% of the restricted stock award will vest and no longer be subject to the repurchase option. An additional 1.6304% of the restricted stock award will vest and become free of the repurchase option for each one percentage that the four-year cumulative total stockholder return on the Company's common stock exceeds the four-year cumulative total return for the NASDAQ Index. The aggregate intrinsic value of the 400,000 shares of the Company's common stock at the date of grant underlying the restricted stock awards was $2.40 million, based on the closing price of the Company's common stock on the NASDAQ National Market on the date of grant. The Company utilized a Monte-Carlo simulation method to estimate a range of possible future stock prices over the four-year period for the Company's common stock and the NASDAQ Index to estimate the number of shares of restricted stock that may vest based upon such simulation. Using the Monte-Carlo simulation method, the Company calculated an aggregate compensation cost of $580,000 at the time of the grant. The Company is recognizing this compensation cost over the four-year service period whether or not the market condition is actually satisfied. However, in the event one or more of the participants voluntarily terminates before the end of the four-year period, some amounts of the charge will be reversed. In the event that a qualifying change in the control of the Company occurs prior to June 1, 2010, the Company's repurchase option will fully lapse, and the Company will then recognize a compensation charge equal to the full $2.40 million intrinsic value less any previously recognized compensation expense. In connection with the resignation of one of the Company's executive officers on July 27, 2007, the Company exercised its right to repurchase all 100,000 shares of the common stock issued to this executive at the price per share originally paid by the executive.
Pursuant to the Company's 2000 Employee Stock Purchase Plan (the "2000 ESPP"), the Company may issue and sell to its eligible employees up to an aggregate of 230,000 shares of common stock at a purchase price equal to 85% of the lower of the fair market value on the first or last day of each six-month offering period. Eligible employees may elect to have up to a maximum of 10% of their regular compensation withheld through payroll deductions to pay the purchase price of the shares at the end of the offering period, subject to limitations specified in the plan.
As discussed in Note 2, stock-based compensation expense relates to stock options, restricted stock and stock issued under the Company's employee stock purchase plan. The Company measures the cost of employee services in exchange for an award of equity instruments based on the grant-date fair value of the award and recognizes cost over the requisite service period. The Company recognizes compensation expense on fixed awards with graded vesting on a straight-line basis over the vesting period of such awards.
86
The fair value of stock options granted during the years ended December 31, 2009, 2008, and 2007 are estimated on the date of the grant using the Black-Scholes option-pricing model with the following weighted average assumptions:
| |
For the Years Ended December 31, |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | |||||||
Options granted (in thousands) |
654 | 763 | 441 | |||||||
Weighted average exercise price |
$ | 1.82 | $ | 4.13 | $ | 7.05 | ||||
Weighted average grant date fair value |
$ | 1.14 | $ | 2.45 | $ | 4.48 | ||||
Assumptions: |
||||||||||
Dividend yield |
0 | % | 0 | % | 0 | % | ||||
Expected volatility |
68 | % | 65 | % | 68 | % | ||||
Risk-free interest rate |
2.33 | % | 2.79 | % | 4.41 | % | ||||
Expected term (years) |
6.16 | 5.67 | 5.79 | |||||||
Historical Company information was the primary basis for the expected volatility and the expected term assumptions. The Company applies an estimated forfeiture rate to current-period expense to recognize stock-based compensation expense only for those awards expected to vest. The Company estimates forfeitures based upon historical data, adjusted for known trends, and will adjust its estimate of forfeitures where actual forfeitures differ, or are expected to differ from such estimates. Subsequent changes in estimated forfeitures are recognized through a cumulative adjustment in the period of change and will also impact the amount of stock-based compensation expense in future periods.
Changes in outstanding stock options for the year ended December 31, 2009 were as follows:
(in thousands, except exercise price and term)
|
Number of Stock Options |
Weighted-Average Exercise Price |
Weighted-Average Remaining Contractual Term (in years) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Outstanding at December 31, 2008 |
2,891 | $ | 4.98 | ||||||||
Granted |
654 | $ | 1.82 | ||||||||
Exercised |
(4 | ) | $ | 0.81 | |||||||
Forfeited and expired |
(66 | ) | $ | 4.72 | |||||||
Outstanding at December 31, 2009 |
3,475 | $ | 4.40 | 6.61 | |||||||
Exercisable at December 31, 2009 |
1,860 |
$ |
4.90 |
5.31 |
|||||||
Vested or expected to vest at December 31, 2009 |
3,288 | $ | 4.42 | 6.53 | |||||||
The aggregate intrinsic value of stock options outstanding at December 31, 2009 of $764,000 is calculated as the difference between the exercise price of the underlying stock options and the market price of the Company's common stock for the 1,249,000 shares of common stock underlying stock options that had exercise prices that were lower than the $2.74 closing market price of the Company's common stock at December 31, 2009. The aggregate intrinsic value of stock options vested or expected to vest at December 31, 2009 is $702,000. The aggregate intrinsic value of stock options exercisable at December 31, 2009 is $144,000. The total intrinsic value of stock options exercised was $4,000, $13,000 and $338,000 during the years ended December 31, 2009, 2008 and 2007, respectively, determined as of the date of exercise.
87
Changes in non-vested restricted stock awards for the year ended December 31, 2009 were as follows:
(in thousands, except fair value)
|
Number of Restricted Shares |
Weighted Average Grant Date Fair Value |
|||||
|---|---|---|---|---|---|---|---|
Non-vested at December 31, 2008 |
335 | $ | 1.85 | ||||
Awarded |
18 | $ | 1.80 | ||||
Vested |
(18 | ) | $ | 6.84 | |||
Forfeited |
— | — | |||||
Non-vested at December 31, 2009 |
335 | $ | 1.59 | ||||
The aggregate intrinsic value of restricted shares outstanding at December 31, 2009 is $913,000.
At December 31, 2009, there was $2.91 million and $80,000 of unrecognized compensation cost, net of estimated forfeitures, related to non-vested stock options and restricted stock awards, respectively, which the Company expects to recognize over weighted-average periods of 2.60 years and 0.67 years, respectively. However, the amount of stock compensation expense recognized in any future period cannot be predicted at this time because it will depend on levels of share-based payments granted in the future.
Employee Stock Purchase Plan
Under the 2000 ESPP, an aggregate of 230,000 shares of common stock may be purchased by employees at 85% of the fair market value on the first or last day of each six-month offering period, whichever is lower. During each offering period, the maximum number of shares that may be purchased by a participating employee is determined on the first day of the offering period and is equal to the number of shares of common stock determined by dividing $12,500 by the last reported sale price of the common stock on the NASDAQ Global Market on the first day of the offering. An eligible employee may elect to have up to a maximum of 10% deducted through payroll deductions from his or her regular salary. During 2009, 2008 and 2007, 51,373, 32,688 and 18,370 shares of the Company's common stock, respectively, were issued under the 2000 ESPP. During the years ended December 31, 2009, 2008 and 2007, the Company recognized $49,000, $18,000 and $17,000, respectively, of equity compensation related to the issuance of shares under the 2000 ESPP.
The following table presents the stock-based compensation expense relating to stock options, restricted stock and stock issued under the Company's employee stock purchase plan, for the years ended December 31, 2009, 2008 and 2007:
| |
For the Years Ended December 31, |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
(in thousands)
|
2009 | 2008 | 2007 | |||||||
Cost of product sales |
$ | 273 | $ | 230 | $ | 274 | ||||
Research and development |
$ | 176 | $ | 136 | $ | 93 | ||||
Sales |
$ | 233 | $ | 253 | $ | 237 | ||||
Marketing |
$ | 51 | $ | 68 | $ | 54 | ||||
General and administrative |
$ | 1,193 | $ | 1,185 | $ | 1,178 | ||||
13. Employee Savings Plan
The Company has a 401(k) savings plan for all domestic employees pursuant to which eligible employees may voluntarily contribute up to $16,500, subject to statutory limitations. In addition, the Company matches in cash 50% of the first $5,000 contributed by employees up to a $2,500 maximum
88
per employee per year. Employer cash-matching contributions amounted to approximately $102,000, $87,000, and $46,000 for the years ended December 31, 2009, 2008 and 2007, respectively.
The Company maintains a retirement indemnity plan for its employees located in France pursuant to which employees are eligible to receive a one-time payment calculated based on years of service. The indemnity is payable upon retirement and is forfeited upon discontinuation of service prior to retirement. As of December 31, 2009, the present value of the benefit under this plan is approximately $140,000, of which the Company has purchased an insurance contract covering approximately $34,000.
14. Allowance for Doubtful Accounts
The Company monitors the creditworthiness of its trade customers based upon historical payment experience. The allowance for doubtful accounts activity for the years ended December 31, 2009, 2008 and 2007 is as follows:
(in thousands)
|
Balance, Beginning of Period |
Recovery of Prior Costs and Expenses |
Deductions | Balance, End of Period |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Year ended December 31, 2009 |
$ | 97 | $ | 3 | $ | (43 | ) | $ | 57 | ||||
Year ended December 31, 2008 |
$ | 160 | $ | (8 | ) | $ | (55 | ) | $ | 97 | |||
Year ended December 31, 2007 |
$ | 218 | $ | (38 | ) | $ | (20 | ) | $ | 160 | |||
15. Contingencies
On September 15, 2009, the Company entered into a settlement agreement with the plaintiff in a product liability lawsuit filed against the Company and other previously dismissed defendants in the matter captioned Brett Pingel by next friend Dawn LaRose v. BioSphere Medical, Inc., Bruce Kirke Bieneman, M.D., St. Louis University Hospital, John Stith, M.D. and St. Louis University. The parties agreed to settle the case without any admission of liability. The Company maintains product liability insurance, and its insurer agreed to pay the full amount of the settlement.
On June 4, 2008, a lawsuit was filed in the Superior Court of California, County of Los Angeles, entitled Hamid Rashidi v. Franklin Moser, M.D., Cedars-Sinai Medical Center and BioSphere Medical, Inc. Plaintiff alleged, among other things, that he presented to an emergency room with a severe nosebleed and that he suffered permanent blindness in one eye following the treatment of his severe nosebleed with the Company's Embosphere Microspheres. Plaintiff claimed that the product was defective, that the manner in which it was marketed and sold was misleading, that the health-care providers were negligent, and that one or more of these factors combined to cause his injuries. Plaintiff sought compensatory and punitive damages.
On March 24, 2010, the Company entered into a settlement agreement with the plaintiff. The parties agreed to settle the case without any admission of liability. The Company maintains product liability insurance and expects its insurer will cover the full amount paid in the settlement.
89
16. Quarterly Financial Data (Unaudited)
The following is a summary of quarterly financial results:
(in thousands except per share amounts)
|
First Quarter |
Second Quarter |
Third Quarter |
Fourth Quarter |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Net revenue |
||||||||||||||
2009 |
$ | 7,283 | $ | 8,111 | $ | 7,686 | $ | 8,363 | ||||||
2008 |
$ | 7,214 | $ | 7,611 | $ | 7,198 | $ | 7,235 | ||||||
Gross profit |
||||||||||||||
2009 |
$ | 5,443 | $ | 6,197 | $ | 5,835 | $ | 6,286 | ||||||
2008 |
$ | 5,171 | $ | 5,621 | $ | 5,309 | $ | 5,471 | ||||||
Net Loss |
||||||||||||||
2009 |
$ | (1,640 | ) | $ | (522 | ) | $ | (216 | ) | $ | (296 | ) | ||
2008 |
$ | (1,401 | ) | $ | (1,159 | ) | $ | (1,398 | ) | $ | (1,534 | ) | ||
Net loss applicable to common stockholders |
||||||||||||||
2009 |
$ | (1,785 | ) | $ | (667 | ) | $ | (361 | ) | $ | (439 | ) | ||
2008 |
$ | (1,546 | ) | $ | (1,304 | ) | $ | (1,543 | ) | $ | (1,677 | ) | ||
Basic and diluted net loss per share |
||||||||||||||
2009 |
$ | (0.10 | ) | $ | (0.04 | ) | $ | (0.02 | ) | $ | (0.02 | ) | ||
2008 |
$ | (0.09 | ) | $ | (0.07 | ) | $ | (0.09 | ) | $ | (0.09 | ) | ||
17. Subsequent Events
On March 24, 2010, the Company entered into a settlement agreement in a product liability lawsuit filed against the Company and other defendants on behalf of Hamid Rashidi in Superior Court of California, Los Angeles. The settlement agreement has resolved all claims pending in the lawsuit in exchange for payment by the Company of an amount that the parties have agreed will be confidential. The parties agreed to settle the case without any admission of liability on the part of the Company. The Company maintains product liability insurance, and the Company's insurer has agreed to pay the full amount of the settlement.
90
Item 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
Item 9A. CONTROLS AND PROCEDURES
1. Disclosure Controls and Procedures
Our management, with the participation of our chief executive officer and chief financial officer, evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2009. The term "disclosure controls and procedures," as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC's rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company's management, including its principal executive and principal financial officers, as appropriate, to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2009, our chief executive officer and chief financial officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
2. Internal Control Over Financial Reporting
a) Management's Annual Report on Internal Control Over Financial Reporting
The management of the Company is responsible for establishing and maintaining adequate internal control over financial reporting for the Company. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the Exchange Act as a process designed by, or under the supervision of, the Company's principal executive and principal financial officers and effected by the Company's board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
- •
- pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and
dispositions of the assets of the Company;
- •
- provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in
accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the
Company; and
- •
- provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the
91
risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
The Company's management assessed the effectiveness of the Company's internal control over financial reporting as of December 31, 2009. In making this assessment, the Company's management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework.
Based on our assessment, management concluded that, as of as of December 31, 2009, the Company's internal control over financial reporting is effective based on those criteria.
b) Attestation Report of the Independent Registered Public Accounting Firm
This annual report on Form 10-K does not include an attestation report of our independent registered public accounting firm regarding internal control over financial reporting. Management's report was not subject to attestation by our registered independent public accounting firm pursuant to the rules of the SEC that permit us to provide only management's report in this annual report on Form 10-K.
c) Changes in Control Over Financial Reporting
No change in our internal control over financial reporting occurred during the fiscal quarter ended December 31, 2009 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
None.
92
Item 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Directors and Executive Officers
Information regarding our directors will be included in the definitive proxy statement for the 2010 Annual Meeting of Stockholders under "Nominees for Director" which we intend to file within 120 days of December 31, 2009 and is incorporated herein by reference.
Information regarding our executive officers is included in Part I, Item 4, under the heading "Executive Officers" and is incorporated herein by reference.
Audit Committee
We have a separately designated standing Audit Committee established in accordance with Section 3(a)(58)(A) of the Exchange Act. Additional information regarding the Audit Committee will be included in the definitive proxy statement for the 2010 Annual Meeting of Stockholders which we intend to file within 120 days of December 31, 2009 under "Board and Committee Meetings" and "Report of the Audit Committee" and is incorporated herein by reference.
Audit Committee Financial Expert
The Board of Directors has determined that John H. MacKinnon and William M. Cousins, Jr. are each an "audit committee financial expert" as defined by Item 401(h) of Regulation S-K of the Exchange Act and has determined that they are independent within the meaning of Item 7(d)(3)(iv) of Schedule 14A of the Exchange Act. In February 2010, Mr. William M. Cousins, Jr. notified the Company that he intends to resign and retire from the board of directors as of the 2010 Annual Shareholders Meeting.
Section 16(a) Beneficial Ownership Reporting Compliance
Information regarding Section 16(a) Beneficial Ownership Reporting Compliance will be included in the definitive proxy statement for the 2010 Annual Meeting of Stockholders which we intend to file within 120 days of December 31, 2009 under "Section 16(a) Beneficial Ownership Reporting Compliance" and is incorporated herein by reference.
Code of Ethics
We have adopted a code of business conduct and ethics that applies to our directors, officers (including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions) as well as our employees, a copy of which is listed as an exhibit to this annual report on Form 10-K. A copy of our code of ethics is also available on the Company's Web site at www.biospheremed.com.
Item 11. EXECUTIVE COMPENSATION
The response to this item will be included in the definitive proxy statement for the 2010 Annual Meeting of Stockholders which we intend to file within 120 days of December 31, 2009 under "Compensation of Executive Officers" and is incorporated herein by reference.
93
Item 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The response to this item will be included in the definitive proxy statement for the 2010 Annual Meeting of Stockholders which we intend to file within 120 days of December 31, 2009 under "Stock Ownership of Certain Beneficial Owners and Management" and "Equity Compensation Plan Information" and is incorporated herein by reference.
Item 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The response to this item will be included in the definitive proxy statement for the 2010 Annual Meeting of Stockholders which we intend to file within 120 days of December 31, 2009 under "Certain Relationships and Related Transactions" and "Nominees for Director" and is incorporated herein by reference.
Item 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The response to this item will be included in the definitive proxy statement for the 2010 Annual Meeting of Stockholders which we intend to file within 120 days of December 31, 2009 under "Report of the Audit Committee" and "Audit Fees, Audit-Related Fees, Tax Fees and All Other Fees" and is incorporated herein by reference.
Item 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
| (a) (1) | The following consolidated financial statements of BioSphere Medical, Inc. and subsidiaries are filed as part of this Form 10-K: | |
Statement |
||
| Report of Independent Registered Public Accounting Firm | ||
| Consolidated Balance Sheets—December 31, 2009 and 2008 | ||
| Consolidated Statements of Operations—Years ended December 31, 2009, 2008 and 2007 | ||
| Consolidated Statements of Stockholders' Equity and Comprehensive Loss—Years ended December 31, 2009, 2008 and 2007 | ||
| Consolidated Statements of Cash Flows—Years ended December 31, 2009, 2008 and 2007 | ||
| Notes to Consolidated Financial Statements | ||
(a) (2) |
All schedules are omitted because they are not applicable or the required information is shown in the Financial Statements or notes thereto. |
|
(a) (3) |
Exhibits included or incorporated herein: |
|
See Exhibit Index |
94
| |
|
Incorporated by Reference | |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Exhibit No.
|
Description | Form | SEC Filing Date | Exhibit No. | Filed with this 10-K | |||||||||||
| Articles of Incorporation and Bylaws | ||||||||||||||||
3.1 |
Certificate of Incorporation, as amended, of the Company |
S-8 |
07/23/1999 |
4.1 |
||||||||||||
3.2 |
Certificate of Amendment of Certificate of Incorporation of the Company |
10-K |
03/24/2007 |
4.4 |
||||||||||||
3.2 |
Bylaws of the Company |
S-8 |
06/10/1999 |
4.2 |
||||||||||||
3.3 |
Amendment to Bylaws of the Company |
8-K |
12/11/2007 |
3.1 |
||||||||||||
Instruments defining the rights of security holders |
||||||||||||||||
4.1 |
Specimen Certificate for shares of Common Stock, $.01 par value, of the Company |
10-K |
03/30/2000 |
4 |
||||||||||||
4.2 |
Certificate of Designations, Preferences and Rights of Series A Preferred Stock of the Company |
8-K |
11/15/2004 |
4.1 |
||||||||||||
4.3 |
Amendment No. 1 to Certificate of Designations, Preferences and Right of Series A Preferred Stock of the Company |
8-K |
05/23/2005 |
4.1 |
||||||||||||
Material Contracts—financing agreements |
||||||||||||||||
10.1 |
Share Purchase Agreement by and between Marie-Paule Leroy-Landercy and the Company, dated December 31, 1998 |
10-K |
03/30/2000 |
10.4 |
||||||||||||
10.2 |
Securities Purchase Agreement, dated as of November 10, 2004, by and among the Company and the investors named therein |
8-K |
11/15/2004 |
10.1 |
||||||||||||
10.3 |
Investor Rights Agreement, dated as of November 10, 2004, by and among the Company and the investors named therein |
8-K |
11/15/2004 |
10.2 |
||||||||||||
10.4 |
Restrictive Covenants Agreement, dated as of December 23, 2004, by and among the Company, Cerberus Partners, L.P. and Sepracor Inc. |
8-K |
12/30/2004 |
10.1 |
||||||||||||
95
| |
|
Incorporated by Reference | |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Exhibit No.
|
Description | Form | SEC Filing Date | Exhibit No. | Filed with this 10-K | |||||||||||
| 10.5 | Securities Purchase Agreement, dated as of February 17, 2006, by and among the Company and the investors named therein | 8-K | 2/21/2006 | 10.1 | ||||||||||||
10.6 |
Registration Rights Agreement, dated as of February 17, 2006, by and among the Company and the investors named therein |
8-K |
2/21/2006 |
10.2 |
||||||||||||
Material Contracts—leases |
||||||||||||||||
10.7 |
Lease Agreement dated January 7, 2000 by and between 1050 Hingham Street Realty Trust and the Company |
10-K |
03/30/2000 |
10.16 |
||||||||||||
10.8 |
First Amendment to Lease Agreement dated June 27, 2000 by and between 1050 Hingham Street Realty Trust and the Company |
10-K |
03/29/2001 |
10.15 |
||||||||||||
10.9 |
Second Amendment to Lease between Company and Thomas J. Teuten and John H. Spurr, Jr., Trustees of 1050 Hingham Street Realty Trust, dated January 24, 2005 |
8-K |
1/27/2005 |
10.15 |
||||||||||||
10.10 |
Third Amendment to Lease between the Company and Thomas J. Teuten and John H. Spurr, Jr., Trustees of 1050 Hingham Street Realty Trust, dated February 24, 2006 |
8-K |
2/28/2006 |
10.1 |
||||||||||||
10.11 |
Fourth Amendment to Lease between the Company and Thomas J. Teuten and John H. Spurr, Jr., Trustees of 1050 Hingham Street Realty Trust, dated January 5, 2009 |
8-K |
01/8/2009 |
10.1 |
||||||||||||
10.12 |
Fifth Amendment to Lease between the Company and Thomas J. Teuten and John H. Spurr, Jr., Trustees of 1050 Hingham Street Realty Trust, dated March 9, 2010 |
8-K |
3/12/2010 |
10.1 |
||||||||||||
10.13 |
Lease Agreement dated October 19, 2000 by and between the Company and Salamandre S.A. (translated from French) |
10-K |
03/29/2001 |
10.20 |
||||||||||||
96
| |
|
Incorporated by Reference | |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Exhibit No.
|
Description | Form | SEC Filing Date | Exhibit No. | Filed with this 10-K | |||||||||||
| Material Contracts—collaboration agreements and licenses | ||||||||||||||||
10.14+ |
Joint Ownership Contract between the Company and L'Assistance Publique Hôpitaux de Paris, dated January 5, 1998, together with amendment dated February 10, 2001 (translated from French) |
10-K |
03/30/2000 |
10.5 |
||||||||||||
10.15 |
Rider No. 2, dated June 20, 2000 to the Joint Ownership Contract between the Company and L'Assistance Publique Hôpitaux de Paris dated January 5, 1998 (translated from French) |
10-K |
04/01/2002 |
10.5 |
||||||||||||
10.16+ |
Rider No. 3, dated March 2, 2009 to the Joint Ownership Contract between the Company and L'Assistance Publique Hôpitaux de Paris, dated January 5, 1998 (translated from French) |
10-K |
03/20/2009 |
10.25 |
||||||||||||
10.17 |
International Distribution Agreement between the Company and Nippon Kayaku Co. Ltd., dated April 16, 2009 |
10-Q |
08/13/2009 |
10.1 |
||||||||||||
Material Contracts—management contracts and compensatory plans |
||||||||||||||||
10.18(1) |
1997 Stock Incentive Plan |
10-Q |
08/08/1997 |
10.2 |
||||||||||||
10.19(1) |
2006 Stock Incentive Plan |
8-K |
05/16/2006 |
10.1 |
||||||||||||
10.20(1) |
Amendment No. 1 to 2006 Stock Incentive Plan |
8-K |
08/09/2006 |
10.1 |
||||||||||||
10.21 |
Amendment No. 2 to 2006 Stock Incentive Plan |
8-K |
5/20/2008 |
10.1 |
||||||||||||
10.22 |
Amendment No. 3 to 2006 Stock Incentive Plan |
8-K |
5/20/2009 |
10.1 |
||||||||||||
10.23 |
2000 Employee Stock Purchase Plan |
8-K |
5/20/2008 |
10.2 |
||||||||||||
10.24 |
Amendment No. 1 to 2000 Employee Stock Purchase Plan |
8-K |
5/20/2008 |
10.2 |
||||||||||||
10.25 |
Amendment No. 2 to 2000 Employee Stock Purchase Plan |
8-K |
5/20/2008 |
10.2 |
||||||||||||
10.26 |
Amendment No. 3 to 2000 Employee Stock Purchase Plan |
8-K |
5/20/2009 |
10.2 |
||||||||||||
97
| |
|
Incorporated by Reference | |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Exhibit No.
|
Description | Form | SEC Filing Date | Exhibit No. | Filed with this 10-K | |||||||||||
| 10.27(1) | Form of Nonstatutory Stock Option Agreement granted under 1994 Stock Option Plan | 10-K | 03/29/2005 | 10.4 | ||||||||||||
10.28(1) |
Form of Incentive Stock Option Agreement granted under 1997 Stock Incentive Plan |
10-K |
03/29/2005 |
10.5 |
||||||||||||
10.29(1) |
Form of Nonstatutory Stock Option Agreement granted under 1997 Stock Incentive Plan |
10-K |
03/29/2005 |
10.6 |
||||||||||||
10.30(1) |
Form of Restricted Stock Agreement granted under 1997 Stock Incentive Plan |
10-K |
03/29/2005 |
10.7 |
||||||||||||
10.31(1) |
Form of Incentive Stock Option Agreement granted under 2006 Stock Incentive Plan |
8-K |
05/16/2006 |
10.2 |
||||||||||||
10.32(1) |
Form of Nonstatutory Stock Option Agreement granted under 2006 Stock Incentive Plan |
8-K |
05/16/2006 |
10.3 |
||||||||||||
10.33(1) |
Form of Restricted Stock Agreement granted under 2006 Stock Incentive Plan |
8-K |
05/16/2006 |
10.4 |
||||||||||||
10.34(1) |
Employment Agreement between the Company and Richard J. Faleschini, dated November 2, 2004 |
8-K |
11/08/2004 |
10.2 |
||||||||||||
10.35(1) |
Executive Retention Agreement between the Company and Richard J. Faleschini, dated November 2, 2004 |
8-K |
11/08/2004 |
10.3 |
||||||||||||
10.36(1) |
Second Acknowledgement and Amendment Agreement between the Company and Richard J. Faleschini, dated April 5, 2007 |
10-Q |
05/14/2007 |
10.2 |
||||||||||||
10.37(1) |
Third Acknowledgement and Amendment Agreement between the Company and Richard J. Faleschini, dated October 10, 2007 |
8-K |
10/12/2007 |
10.3 |
||||||||||||
10.38(1) |
Fourth Acknowledgement and Amendment Agreement between the Company and Richard J. Faleschini, dated December 23, 2008 |
8-K |
12/23/2008 |
10.6 |
||||||||||||
10.39(1) |
Letter Agreement between the Company and Martin J. Joyce, dated June 14, 2005 |
8-K |
06/17/2005 |
10.2 |
||||||||||||
98
| |
|
Incorporated by Reference | |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Exhibit No.
|
Description | Form | SEC Filing Date | Exhibit No. | Filed with this 10-K | |||||||||||
| 10.40(1) | Acknowledgement and Amendment Agreement between the Company and Martin J. Joyce, dated October 10, 2007 | 8-K | 10/12/2007 | 10.1 | ||||||||||||
10.41(1) |
Second Acknowledgement and Amendment Agreement between the Company and Martin J. Joyce, dated December 23, 2008 |
8-K |
12/23/2008 |
10.1 |
||||||||||||
10.42(1) |
Letter Agreement between the Company and Peter C. Sutcliffe, dated June 14, 2005 |
8-K |
06/17/2005 |
10.3 |
||||||||||||
10.43(1) |
Acknowledgement and Amendment Agreement between the Company and Peter C. Sutcliffe, dated October 10, 2007 |
8-K |
10/12/2007 |
10.2 |
||||||||||||
10.44(1) |
Second Acknowledgement and Amendment Agreement between the Company and Peter C. Sutcliffe, dated December 23, 2008 |
10-K |
12/23/2008 |
10.2 |
||||||||||||
10.45(1) |
Letter Agreement between the Company and Melodie R. Domurad, dated December 14, 2007 |
10-K |
03/20/2009 |
10.56 |
||||||||||||
10.46(1) |
Acknowledgement and Amendment Agreement between the Company and Melodie R. Domurad, dated December 23, 2008 |
8-K |
12/23/2008 |
10.3 |
||||||||||||
10.47(1) |
Letter Agreement between the Company and Joel B. Weinstein, dated January 3, 2008 |
10-K |
03/20/2009 |
10.58 |
||||||||||||
10.48(1) |
Acknowledgement and Amendment Agreement between the Company and Joel B. Weinstein, dated December 23, 2008 |
8-K |
12/23/2008 |
10.4 |
||||||||||||
10.49(1) |
Letter Agreement between the Company and Willard W. Hennemann, dated February 3, 2008 |
10-K |
03/20/2009 |
10.60 |
||||||||||||
10.50(1) |
Acknowledgement and Amendment Agreement between the Company and Willard W. Hennemann, dated December 23, 2008 |
8-K |
12/23/2008 |
10.5 |
||||||||||||
Additional Exhibits |
||||||||||||||||
14.1 |
Code of Business Conduct and Ethics of the Company |
10-K |
03/29/2005 |
14.1 |
||||||||||||
99
| |
|
Incorporated by Reference | |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Exhibit No.
|
Description | Form | SEC Filing Date | Exhibit No. | Filed with this 10-K | |||||||||||
| 21.1 | Subsidiaries of the Company | 10-K | 03/29/2001 | 21 | ||||||||||||
23.1 |
Consent of Ernst & Young LLP |
X |
||||||||||||||
31.1 |
Certification of the Chief Executive Officer pursuant to Rule 13a-14(a)/Rule 15d-14(a) of the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002, dated March 26, 2010 |
X |
||||||||||||||
31.2 |
Certification of the Chief Financial Officer pursuant to Rule 13a-14(a)/Rule 15d-14(a) of the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002, dated March 26, 2010 |
X |
||||||||||||||
32.1 |
Certification of the Chief Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, dated March 26, 2010 |
X |
||||||||||||||
32.2 |
Certification of the Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, dated March 26, 2010 |
X |
||||||||||||||
- (1)
- Management
contract or compensatory plan or arrangement filed as an exhibit to this form 10-K pursuant to Items 14(a) and 14(c) of
Form 10-K.
- +
- Confidential treatment requested as to certain portions.
100
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| BIOSPHERE MEDICAL, INC. | ||||
By: |
/s/ RICHARD J. FALESCHINI Richard J. Faleschini President and Chief Executive Officer |
|||
| Date: March 26, 2010 | ||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature
|
Title
|
Date
|
||
|---|---|---|---|---|
| /s/ RICHARD J. FALESCHINI Richard J. Faleschini |
Director, President and Chief Executive Officer (Principal Executive Officer) | March 26, 2010 | ||
/s/ MARTIN J. JOYCE Martin J. Joyce |
Executive Vice President and Chief Financial Officer (Principal Financial Officer) |
March 26, 2010 |
||
/s/ MICHAEL R. MEGNA Michael R. Megna, CPA |
Corporate Controller and Chief Accounting Officer (Principal Accounting Officer) |
March 26, 2010 |
||
/s/ TIMOTHY J. BARBERICH Timothy J. Barberich |
Director |
March 25, 2010 |
||
/s/ WILLIAM M. COUSINS, JR. William M. Cousins, Jr. |
Director |
March 26, 2010 |
||
/s/ MARIAN L. HEARD Marian L. Heard |
Director |
March 26, 2010 |
||
/s/ ALEXANDER M. KLIBANOV, PH.D. Alexander M. Klibanov, Ph.D. |
Director |
March 26, 2010 |
||
/s/ JOHN H. MACKINNON John H. MacKinnon, CPA |
Director |
March 26, 2010 |
||
/s/ RICCARDO PIGLIUCCI Riccardo Pigliucci |
Director |
March 26, 2010 |
||
/s/ DAVID P. SOUTHWELL David P. Southwell |
Director and Chairman of the Board |
March 26, 2010 |
101