Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
ANNUAL REPORT PURSUANT TO SECTIONS 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2009
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission File Number: 000-29101
SEQUENOM, INC.
(Exact name of Registrant as specified in its charter)
| DELAWARE | 77-0365889 | |
| (State or other jurisdiction or incorporation or organization) |
(I.R.S. Employer Identification No.) | |
| 3595 John Hopkins Court San Diego, California |
92121 | |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code: (858) 202-9000
Securities registered pursuant to Section 12(b) of the Act:
Common Stock, $.001 par value
(Title of class)
The NASDAQ Stock Market, LLC
(Name of Each Exchange on Which Registered)
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ¨ | Accelerated filer x | Non-accelerated filer ¨ | Smaller reporting company filer ¨ | |||
| (Do not check if a smaller reporting company) |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No x
The aggregate market value of the voting stock held by non-affiliates of the registrant, based upon the closing sale price of the Common Stock on June 30, 2009 as reported on The NASDAQ Global Market, was approximately $236.7 million. Shares of Common Stock held by each executive officer and director and by each person who owns 10% or more of the outstanding Common Stock have been excluded in that such persons may be deemed to be affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
As of February 12, 2010, there were 62,096,371 shares of the registrant’s Common Stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Part III incorporates by reference information from the registrant’s definitive proxy statement to be filed with the Securities and Exchange Commission (the Commission) in connection with the solicitation of proxies for the registrant’s annual meeting of stockholders to be held on June 14, 2010. Such definitive proxy statement will be filed with the Commission no later than 120 days after December 31, 2009.
Table of Contents
SEQUENOM, Inc.
FORM 10-K
For the Fiscal Year Ended December 31, 2009
i
Table of Contents
All statements in this report that are not historical are forward-looking statements within the meaning of Section 21E of the Securities Exchange Act. These forward-looking statements can generally be identified as such because the context of the statement will include words such as “may,” “will,” “intend,” “plans,” “believes,” “anticipates,” “expects,” “estimates,” “predicts,” “potential,” “continue,” “opportunity,” “goals,” or “should,” the negative of these words or words of similar import. Similarly, statements that describe our future plans, strategies, intentions, expectations, objectives, goals or prospects are also forward-looking statements. These forward-looking statements are or will be, as applicable, based largely on our expectations and projections about future events and future trends affecting our business, and so are or will be, as applicable, subject to risks and uncertainties including but not limited to the risk factors discussed in this report, that could cause actual results to differ materially from those anticipated in the forward-looking statements. We caution investors that there can be no assurance that actual results or business conditions will not differ materially from those projected or suggested in such forward-looking statements. Our views and the events, conditions and circumstances on which these future forward-looking statements are based, may change. All forward statements are qualified in their entirety by this cautionary statement and we undertake no obligation to revise or update any such statements to reflect events or circumstances after the date hereof.
SEQUENOM®, SpectroCHIP®, iPLEX®, and MassARRAY® are registered trademarks and EpiTYPER™, SEQureDx™, MassCLEAVE™, iSEQ™, AttoSense™ and SensiGene™ are trademarks of Sequenom, Inc. This report may also refer to trade names and trademarks of other organizations.
Sequenom, Inc. was incorporated in 1994 under the laws of the State of Delaware. As used in this report, the words “we,” “us,” “our,” and “Sequenom” refer to Sequenom, Inc. and its wholly-owned subsidiaries on a consolidated basis, unless explicitly noted otherwise.
Overview
We are a molecular diagnostic testing and genetics analysis company committed to providing products, services, diagnostic testing, applications and genetic analysis products that translate the results of genomic science into solutions for biomedical research, translational research, molecular medicine applications, and agricultural, livestock, and other areas of research. Our development and commercialization efforts in various diagnostic areas include noninvasive women’s health related and prenatal diagnostics, age-related macular degeneration diagnostics, oncology, infectious diseases, and other disorders and diseases.
1
Table of Contents
Operating Segments
We operate our business on the basis of two reportable segments, Molecular Diagnostics and Genetic Analysis. A further description of the operations of these segments is below. The following table sets forth as of December 31, 2009, revenues, research and development expenses, sales and marketing expenses and operating (loss) income for our Molecular Diagnostic and Genetic Analysis segments:
| (In thousands) | ||||
| Revenues: |
||||
| Molecular Diagnostics |
$ | 94 | ||
| Genetic Analysis |
37,769 | |||
| $ | 37,863 | |||
| Research and development expenses: |
||||
| Molecular Diagnostics |
$ | 20,935 | ||
| Genetic Analysis |
5,587 | |||
| Unallocated |
10,932 | |||
| Total |
$ | 37,454 | ||
| Sales and marketing expenses: |
||||
| Molecular Diagnostics |
$ | 5,780 | ||
| Genetic Analysis |
13,644 | |||
| Unallocated |
7,421 | |||
| Total |
$ | 26,845 | ||
| Operating (loss) income: |
||||
| Molecular Diagnostics |
$ | (27,033 | ) | |
| Genetic Analysis |
4,379 | |||
| Unallocated |
(48,068 | ) | ||
| $ | (70,722 | ) | ||
| (1) | Management evaluates research and development expenses and sales and marketing expenses exclusive of share based compensation, indirect overhead expenses and allocated and absorbed costs and operating (loss) income is evaluated by management exclusive of general and administrative expenses, share based compensation, indirect overhead expenses and allocated and absorbed costs, as these costs are not allocated to our business segments for performance assessment by our chief operating decision maker. |
We do not discretely allocate assets to our operating segments, nor does our chief operating decision maker evaluate operating segments using discrete asset information.
Molecular Diagnostics and SEQureDx™ Technology
Molecular Diagnostics
We are committed to researching, developing and pursuing the commercialization of various noninvasive molecular diagnostic tests for prenatal genetic disorders and diseases, women’s health related disorders and diseases, age-related macular degeneration diagnostics, oncology, infectious diseases, and other diseases and disorders. We have branded our diagnostic technology for prenatal diagnostics under the trademark SEQureDx. Our efforts in molecular diagnostics are focused on noninvasive diagnostics currently using our proprietary MassARRAY system; however, we may in the future employ other instrumentation platforms with our diagnostic applications as may be more suitable on a case-by-case basis considering optimum test performance and commercialization factors.
Currently, we are primarily focused on developing and commercializing prenatal screening and diagnostic tests using our foundational, patent protected, noninvasive, circulating cell-free fetal (ccff) nucleic acid based assay technology. This technology uses a simple maternal blood draw (meaning noninvasive compared to invasive procedures such as amniocentesis, chorionic villus sampling, or surgery) for a prenatal diagnosis or risk
2
Table of Contents
assessment in order to provide reliable information about the status of the fetus early in pregnancy. In early 2010 we launched noninvasive Rhesus D genotyping and Fetalxy sex determination laboratory developed tests (LDTs) using this patented ccff technology which we in-license from Isis Innovation Limited (Isis). We also launched, in September 2009, a noninvasive molecular based cystic fibrosis carrier screening LDT. These tests have all been launched through our College of American Pathology (CAP) accredited and Clinical Laboratory Improvement Amendments (CLIA) certified laboratory, Sequenom Center for Molecular Medicine, LLC, (Sequenom CMM) located in Grand Rapids, Michigan. Most molecular genetic tests are LDTs. We have made substantial investments in our information technology infrastructure to enhance the capabilities of our laboratory to track samples and provide electronic ordering and reporting, and have put in place sample collection and transportation logistics that can be readily scaled. We have also expanded our molecular diagnostic sales force and our marketing efforts. We are entering into contracts with third party payors to establish pricing for our tests and provide reimbursement. We also plan to conduct the development, validation, and other activities necessary to file submissions with the Food and Drug Administration (FDA) seeking approval for selected diagnostic tests. Revenues from our cystic fibrosis test were not significant from September through the end of 2009.
We are in the process of researching and evaluating a potential LDT for Trisomy 21 (Down syndrome), and a potential LDT for age-related macular degeneration (a highly prevalent, late onset, genetically linked vision disorder that is a common cause of legal blindness in the elderly). The tests we are researching and evaluating are all expected to use simple blood draws from patients rather than more invasive procedures. In April 2009, we announced that the expected launch of our Trisomy 21 test had been delayed. We are no longer relying on our previously announced research and development test data and results as the basis for launching a Trisomy 21 test. Our current research and development efforts are focused on DNA based approaches using our MassARRAY platform and next-generation whole genome sequencing platforms, for use in connection with a potential Trisomy 21 test. Our goal is to design a test that has better specificity and sensitivity than currently available screening tests, that could be utilized during first and second trimesters of pregnancy, has maximum ethnic coverage of the global population and is a genetic test, not a surrogate marker. There is no guarantee that we will be able to achieve any or all of these goals. We are continuing to collect clinical samples for sponsored clinical studies through independent third parties, the results of which will be published in a peer-reviewed journal.
Supporting our initiatives in women’s health, oncology and infectious disease we completed our acquisition of the complete AttoSense portfolio of gene-based molecular tests and related assets from SensiGen, LLC in February 2009. The acquisition included highly-sensitive and specific tests for the detection and monitoring of human papillomavirus (HPV) (the primary cause of cervical and head and neck cancers), systemic lupus erythematosus (Lupus), chronic kidney disease (CKD), and other tests, all of which utilize our proprietary MassARRAY platform. These tests will require further development and have not been commercialized.
We are also in the process of researching and evaluating a potential LDT for age-related macular degeneration. Supporting this initiative, in February 2010, we entered into a license agreement with Optherion, Inc. (“Optherion”), under which we were granted an exclusive, worldwide, royalty-bearing license to know-how and a consolidated portfolio of patent rights that had been licensed to Optherion by a number of prominent academic institutions, for research and commercial use, including LDTs or in vitro diagnostic tests, in conjunction with various types of technology platforms. The licensed patent portfolio includes 17 issued or allowed United States and foreign patents, and 68 pending United States and foreign patent applications. The license agreement covers extensive intellectual property rights for significant age-related macular degeneration related genetic variants. Under the agreement we agreed to conduct developmental activities with respect to the licensed technology for use with therapeutic products being developed by Optherion, and we also agreed to use commercially reasonable efforts to develop and commercialize licensed products and to achieve certain commercial milestones.
Under the terms of the agreement, in the event that the first commercial sale of a licensed product in the United States has not occurred on or before January 31, 2011, we will pay Optherion a non-creditable license maintenance fee equal to $260,000 per year. The license maintenance fee will be pro-rated for any period less than a full year before the first commercial sale of a licensed product in the United States. Following the first commercial sale of a
3
Table of Contents
licensed product in the United States, we will no longer be required to pay the license maintenance fee, but instead we will pay Optherion a minimum royalty payment each year during the term of the agreement ranging between $260,000 and $270,000 per year and such minimum payment shall be creditable against any royalties due based upon licensed product sales. We have also agreed to make payments to Optherion upon the achievement of specified development, regulatory and commercial milestones, and during the life of the patent claims licensed under the agreement, royalties on the cumulative worldwide annual net sales of products successfully developed and commercialized covered by the patent claims and know-how licensed under the agreement. We also agreed, upon entry into the agreement, to reimburse Optherion for its prior patent related costs and expenses in the amount of approximately $1.1 million. The agreement will remain in force in each country until the expiration of our obligation to make royalty payments in such country, subject to earlier termination by either party upon uncured material breach or other specified circumstances. Optherion may terminate the agreement if we challenge the validity of any patent covered by the licensed technology, if we abandon or suspend our research, development, marketing or commercialization of the licensed products, or if we fail to comply with certain insurance requirements set forth in the agreement. We may terminate the agreement for any reason upon 90 days prior written notice, provided that if such notice of termination is delivered prior to the first anniversary of the effective date of the agreement, we shall be required to pay Optherion a non-creditable termination fee of $2,000,000. In the event that the agreement expires pursuant to its terms, we will retain the licenses and sublicenses granted under the agreement as fully paid and royalty free, subject to certain specified limitations.
Prenatal Diagnostics Licenses
We have exclusively in-licensed patent rights (U.S. Patent No. 6,250,540 and its foreign equivalents) to use cell-free fetal nucleic acids for diagnostic testing of serum and plasma samples obtained from pregnant women from Isis. Our exclusive license rights, which are platform independent and not limited to mass spectrometry, cover the general diagnostic use of cell-free fetal nucleic acids derived from maternal plasma or serum in territories including the United States, Europe, Australia, Canada, Hong Kong and Japan as well as non-exclusive rights in China.
In October 2005, and as amended thereafter, we entered into the agreement with Isis (the Isis Agreement), pursuant to which Isis granted us an exclusive royalty-bearing license in the United States, Canada, France, Germany, Great Britain and other countries in Europe, to develop, use and market products covered by the patent claims of U.S. Patent No. 6,250,540 and its foreign equivalents, licensed under the Isis Agreement (the Licensed Products), except for the field of Rhesus D blood typing by RT-PCR amplification platforms in Europe. The licensed technology, including improvements made by the inventors prior to the Isis Agreement, covers noninvasive prenatal genetic diagnostic testing on fetal nucleic acids.
In October 2006 we entered into an amendment to the Isis Agreement pursuant to which, in exchange for an upfront payment by us and entitlement to milestone and royalty payments, Isis granted us an expanded exclusive license including the field of prenatal gender determination for social or lifestyle purposes and an expanded territory for the field of gender determination for social or lifestyle purposes including Japan and Australia. In November 2007, we entered into a second amendment to the Isis Agreement pursuant to which, in exchange for an upfront payment by us, a right to a milestone fee upon completion of a specified event, and royalty payments on sales, Isis granted us an expanded licensed territory to include Japan, Australia, and Hong Kong, excluding in the case of Hong Kong the field of gender determination for social or lifestyle purposes. In November 2009, we entered into a third amendment to the Isis Agreement pursuant to which Isis agreed to a modification of certain time-based commercial launch milestones relating to aneuploidy and other Licensed Products. In exchange for this modification, we agreed to make an immediate one-time payment of $1,000,000, increase royalty payments under the agreement during the final 12 months of the patent term and increase the specified minimum royalty amounts.
We also have an exclusive option to negotiate a further license of any improvements made by Isis inventors. Subject to the license rights granted under the Isis Agreement, intellectual property rights created in connection with improvements made to the licensed technology will belong to the party developing the improvements. We also granted to Isis a perpetual royalty-free license to the University of Oxford to use and publish material
4
Table of Contents
relating to the licensed technology and any of our improvements solely for non-commercial use. The University of Oxford’s right to publish is subject to our right to delay publication of information to protect the licensed technology or our improvements.
We have agreed to make up-front payments to Isis and pay to Isis royalties on net sales of Licensed Products, including specified minimum royalty amounts, and milestone payments upon commercial events with respect to Licensed Products for particular indications.
The Isis Agreement will remain in force for the life of any patent issued in connection with the patent application covering the licensed technology, subject to earlier termination by either party upon uncured material breach or other specified circumstances. Isis may terminate the Isis Agreement if we file a petition to wind-up or dissolve or upon 30 days written notice if we were to challenge the validity of the patent rights covering the licensed technology or fail to make the up-front payments as provided in the Isis Agreement. After the third anniversary of the Isis Agreement, we may terminate the Isis Agreement for any reason with six months advance written notice. In the event we fail to achieve certain milestone requirements with respect to particular indications, Isis may convert the exclusive license into a non-exclusive license with respect to those indications.
We have also exclusively in-licensed numerous patent rights from the Chinese University of Hong Kong, and Xenomics Inc. (renamed TrovaGene, Inc.), covering the general use, on any technology platform, of fetal nucleic acids derived from maternal plasma, serum, urine, and in some cases whole blood, for noninvasive prenatal genetic diagnostic testing, including genetic, expression-based, sequencing-based and epigenetic-based assays and tests.
Our license agreement with Xenomics, Inc. provides us with exclusively licensed patent rights (including United States Patent Nos. 6,251,638; and RE 39,920) for the use of fetal nucleic acids obtained from maternal urine. The license provides us with the exclusive global right to use transrenal fetal DNA in maternal urine for noninvasive prenatal diagnostics and analysis on a technology-independent basis for all uses, excluding the limited field of fetal gender determination solely by the presence of Y chromosome. This intellectual property for urine-based tests provides us with additional options for test development and commercialization. We have collected and continue to collect urine samples for purposes of developing urine-based tests and we have initiated exploratory experiments. The licensed intellectual property includes issued patents in the United States and Europe and is part of our continuing strategy to expand and protect our SEQureDx franchise through the identification and licensing of new technologies and sampling methodologies. As described under Item 3 of this report, we are currently engaged in litigation with Xenomics regarding our rights under the license agreement.
We also hold exclusive rights to issued patents and pending patent applications providing fundamental patent rights for digital PCR technologies and methods through a licensing agreement with Genomic Nanosystems, LLC, a wholly-owned subsidiary of the Cytonix Corporation. The issued patents are United States Patent Nos. 6,143,496; 6,391,559; and 7,459,315 and will expire in 2017. The license provides us with the exclusive right to use patented and patent pending digital PCR methods on any platform for noninvasive prenatal diagnostics and analysis for any sample (for example, fetal cells, amniocentesis fluids, plasma, urine, etc.). We also secured the exclusive right to use digital PCR methods in conjunction with mass spectrometry for any commercial, diagnostic or research purpose, excluding second generation sequencing.
In January 2007, as part of our platform independent commercialization strategy, we announced our first commercial partnership with Lenetix Medical Screening Laboratory, Inc., on a non-exclusive basis, who has developed a CLIA validated test for Rhesus D blood incompatibility using real time polymerase chain reaction RT-PCR (the Lenetix Agreement). In December 2007, Lenetix received New York State approval of a noninvasive prenatal LDT performed on a real-time PCR (RT-PCR) platform to detect fetal Rhesus D status (including male sex determination as an internal control) in the second trimester of pregnancy, based on our technology licensed and the work performed under the Lenetix Agreement. Commercial sales of the test by Lenetix commenced in January 2008. We have not and do not expect to derive significant revenues from the Lenetix Agreement in the future.
5
Table of Contents
Molecular Diagnostics Market
The United States molecular diagnostics testing market represents one of the fastest growing areas of the $51.7 billion clinical laboratory industry in the United States. Within this market, the molecular diagnostics market segment is estimated to be $4 billion growing at a rate of approximately 17% per year.
The total available markets for our currently marketed and planned molecular diagnostics tests are as follows:
| • | Each year in the U.S. there are approximately 528,000 Rhesus D negative women who are pregnant and could benefit from an assessment of the RhD status of their fetus. We estimate the total dollar size of the U.S. market to be approximately $250 million per year. |
| • | Our Fetalxy test is a physician-ordered/patient pay test. Based on our market research we believe the market opportunity for our test to be approximately $50 million. |
| • | There are a number of tests available for cystic fibrosis carrier screening. In the U.S. about 1.1 million tests are performed annually and the average cost of these tests is between $200 and $400 per test. The total available market in the U.S. is estimated to be approximately $300 million. |
| • | We estimate the total available market for a noninvasive Trisomy 21 screening test to be approximately $1.5 billion in the U.S. down syndrome affects about 1 in every 800 pregnancies. |
| • | Age-related Macular Degeneration (AMD) affects 15-20 million people in the U.S., over 2.5 million people in Canada, and more than 50 million people worldwide. In North America there are 2 million people with vision loss and more than 600,000 people that are legally blind due to the disease. The worldwide incidence of the disease increases from 1 in 10 people over the age of 60 to more than 1 in 4 people over the age of 75. According to the AMD Alliance, macular degeneration is more common than Parkinson’s disease, Alzheimer’s disease, breast cancer and prostate cancer combined. |
Genetic Analysis
Our proprietary MassARRAY system, comprised of hardware, software applications, consumable chips and reagents, is a high performance (in speed, accuracy and cost efficiency) nucleic acid analysis platform that quantitatively and precisely measures genetic target material and variations. Our platform is widely accepted as a leading high-performance DNA analysis platform for the fine mapping genotyping market and continues to gain traction for newer applications, such as agricultural-biotechnology and clinical research. Our customers include premier clinical research laboratories, bio-agriculture, bio-technology and pharmaceutical companies, academic institutions, and various government agencies worldwide. To provide customer support for our expanding user base and in an effort to maximize market penetration, we have established direct sales and support personnel serving North America, Europe and Asia, in addition to distribution partners in several major countries throughout the world.
Our MassARRAY system provides reliable results for a wide range of DNA/RNA analysis applications including single nucleotide polymorphism (SNP) genotyping detection of mutations, analysis of copy number variants and other structural genome variations. In addition, the system provides quantitative gene expression analysis, quantitative DNA methylation analysis, comparative sequence analysis of haploid organisms, SNP discovery, and oligonucleotide quality control. These applications are provided through proprietary application software that operates on the MassARRAY platform and through the purchase of consumable chips and reagent sets. While the MassARRAY system is versatile across many applications, it is a robust and cost-effective genotyping solution for fine mapping projects enabled through our iPLEX multiplexing assay, which permits multiplexed SNP analysis using approximately the same amount of reagents and chip surface area as is used for a single locus/SNP analysis.
Our research and development efforts in genetic analysis are committed to producing new and improved components and applications for the MassARRAY system that deliver greater system versatility and higher data quality at a competitive price per data point. These research and development activities and new applications also serve to facilitate and support our diagnostics initiatives.
6
Table of Contents
As a result of weaker demand for our MassARRAY systems resulting from the economic environment in early 2009, in April 2009 we formally approved and implemented a cost cutting initiative in our genetic analysis business, which resulted in approximately $8.0 million decrease in costs in 2009 and an overall annualized reduction in costs of approximately $12.1 million. This initiative included a decrease in the genetic analysis workforce that resulted in a cumulative charge of approximately $1.6 million for the year ended December 31, 2009 in connection with one-time termination benefits, office closures and other related costs.
Genetic Analysis Markets
Biomedical Research and Molecular Medicine
MassARRAY systems have been placed in academic, pharmaceutical, and clinical research institutions across the global biomedical research market to identify genetic markers with potential clinical utility. Whole-genome population studies are conducted for general research purposes to create SNP maps and to determine allele frequencies in different ethnicities or species. Whole genome association studies and linkage studies are conducted for genetic discovery purposes. In general, these studies are high throughput studies that analyze a small number of samples against a high number of SNPs. Candidate gene and candidate region association studies typically follow whole-genome population genetics studies, whole genome association studies, and linkage studies. Once target regions are identified and connections to disease are made, these institutions then typically perform fine mapping genotyping studies, which are conducted in an effort to apply genetics to diseases. Institutions conducting fine mapping genotyping studies use the MassARRAY system to perform candidate gene and candidate region association studies. Candidate gene association studies demonstrate that underlying genetic defects reside in specific biological pathways. From there, biomarker discovery efforts can potentially lead to clinical validation and use.
Oncology and Translational Research
Cancer is fundamentally a genetic disease and although the understanding of the genes, pathways, and signaling networks has increased exponentially over the past few decades, relatively little of this information has resulted in significant improvements in cancer mortality rates. The gap between the understanding of cellular and biological processes as they relate to tumor initiation and progression and improvements in patient survival may be due to an inability to comprehensively and systematically approach each cancer as an individual disease. The emerging field of translational medicine is directed at addressing this inability by integrating research inputs from the basic sciences and translating the results of clinical trials into changes in clinical practice. The molecular characterization of tumors is one of the areas of cancer research where science has made great strides in understanding the genetic changes associated with tumor initiation and progression and where it has lead to demonstrable improvements in patient care. We provide key research tools for translational medical research targeted at oncology. These tools allow evaluation of genomic alterations and mutations, which include base substitutions that inactivate tumor suppressor genes or cause constitutive activation of proto-oncogenes, large genomic deletions, large and small intragenic deletions, chromosomal translocations, as well as aberrant promoter methylation and other epigenetic events.
Clinical Research, Public Health Initiatives, Biodefense
Our iSEQ Comparative Sequencing Analysis application is directed to the clinical research market (with its focus on public health issues), healthcare industries, pharmaceutical sectors and homeland defense initiatives. In these areas nucleic acid based detection and identification of bacteria and viruses, especially pathogens of public health interest, have become reliable alternatives to classical detection methods. DNA based analyses are of increasing importance for pathogen typing and antibiotic resistance profiling. A large number of sequencing efforts in the past decade have provided reference sequences for massive parallel comparative sequencing of individuals to ascertain variations within populations and to identify informative genomic markers for routine DNA based microbial and viral typing and monitoring. This continuing effort requires accurate, reproducible, high-throughput technologies for large-scale comparative sequencing in extensive archives of microbes. The automation, throughput, accuracy, data portability and reproducibility of the MassARRAY iSEQ Comparative Sequence Analysis application serves these needs.
7
Table of Contents
Agricultural: Plant Crops and Livestock
There is market demand for genetic testing as it relates to trait selection and feedlot management. There is also demand for genetic analysis of crops, including maize, rice, and others for potentially growing agricultural products with enhanced traits, such as nutritional quality, disease resistance, and crop yields.
Our MassARRAY platform is widely accepted by livestock-focused service providers in the United States and Europe for genotyping, due to its suitability for routine testing of a large number of DNA samples with modest numbers of SNPs. Beginning with our first MassARRAY system placement with the U.S. Department of Agriculture in 1999, we have provided genotyping solutions for customers in the livestock industry. We serve the livestock market through product sales, panel development and optimization. Our competitive advantage in the livestock market is based upon the capability of the MassARRAY system to perform high-volume routine testing. While other genetic analysis platform companies have been successful in the whole genome mapping segment of the market, their platforms are not optimal for routine tests involving tens to hundreds of SNPs.
Strategic Direction
In our molecular diagnostics business we are focusing on developing and commercializing various noninvasive diagnostic tests. We plan to develop tests in prenatal care, women’s health and other disease areas including, oncology and infectious disease. In addition to our CLIA laboratory’s development of diagnostic tests for noninvasive prenatal diagnostics, we are pursuing partnering opportunities for the development and adaptation of the MassARRAY system for commercialization of molecular diagnostics in general. For example, in February 2010 we in-licensed age-related macular degeneration patent rights and plan to develop and commercialize a molecular diagnostic test for this eye disease.
Our genetic analysis business strategy leverages our technology, intellectual property and other assets to expand deeper into and beyond the fine mapping segment of the genetic analysis market, to more aggressively target pharmaceutical companies and other for-profit institutions, particularly in areas of translational research and molecular medicine and capitalizing on our potential in molecular diagnostics markets. In our core genetic analysis business, we are focusing on prioritizing key products that we believe will drive growth and create value.
Our strategy includes:
| • | Investing in our genetic analysis business by developing and commercializing new biomarker panels; |
| • | Launching and marketing a next-generation MassARRAY system, built under design control specifications for use in research and diagnostic applications; |
| • | Developing and commercializing noninvasive prenatal diagnostic assays and other proprietary tests for women’s health, age-related macular degeneration, oncology, infectious disease, and other areas; |
| • | Expanding our diagnostic offerings through in-licensing, partnering and acquisitions; |
| • | Investing in our CAP accredited and CLIA-certified laboratory, Sequenom CMM, enabling us to efficiently and effectively develop and market our proprietary molecular diagnostic tests. |
Intellectual Property
To establish and protect our proprietary technologies and products, we rely on a combination of patent, copyright, trademark and trade secret laws, as well as confidentiality provisions in our contracts.
We have implemented a diligent patent strategy, including in-licensing, designed to facilitate our research and development and commercialization of current and future products. Our patent portfolio, including in-licensed patent rights, includes approximately 487 issued or allowed patents and approximately 343 pending patent applications, in the United States and other major industrial nations throughout the world.
Our prenatal diagnostic patent portfolio includes numerous in-licensed issued patents and in-licensed pending patent applications. The issued patents include United States Patent Nos. 6,250,540, 6,927,028, and
8
Table of Contents
6,664,056, and foreign equivalents for portions of the portfolio that include Canada and Europe. These patents will expire between 2017 and 2022. Most of the patent applications that are in-licensed are in the early stages of patent prosecution and it is difficult to predict when patents will issue from those applications, if at all. These patents and patent applications cover methods of analyzing fetally-derived nucleic acids in maternal serum or plasma, methods of analyzing the methylation status of fetal nucleic acid to differentiate it from maternal nucleic acid, and various DNA and RNA markers which may be useful in detecting and diagnosing various fetal disorders, such as Down syndrome or maternal disorders, such as preeclampsia. We in-licensed United States Patent No. 6,250,540 and its foreign equivalents from ISIS in the United Kingdom. The European counterpart patent to U.S. Patent No. 6,250,540 is European Patent No. 994963. The 994963 Patent was the subject of an Opposition proceeding in the European Patent Office (the “EPO”), which was brought against ISIS by Ravgen, Inc. The Opposition concluded with the EPO’s decision to affirm the grant of the European 994963 Patent, however, with amended claims consistent with the issued claims of its counterpart United States Patent. Ravgen has appealed the EPO’s decision (Appeal No. T146/07-334) and the appeal remains currently pending before the EPO.
The majority of our issued United States patents pertaining to mass spectrometry-based nucleic acid analysis methods and technology will expire between 2013 and 2017. United States Patent Nos. 6,500,621, 6,300,076, 6,258,538, and 5,869,242 and European Patent No. EP 0815261 each claim nucleic acid analysis by mass spectrometry methods, including methods that may be performed using our MassARRAY system. Each of these patents expires in 2015.
Through our exclusive license agreement with Xenomics, Inc, we hold exclusive rights to patents for prenatal research and diagnostic uses and products using fetal nucleic acids found in maternal urine. The licensed patent rights include United States Patent Nos. 6,251,638; and RE 39,920, and foreign equivalents in Europe. These patents will expire between 2017 and 2018. The license provides us with exclusive rights to use transrenal fetal nucleic acids in maternal urine for noninvasive prenatal diagnostics and analysis on a platform and technology-independent basis for all uses, excluding fetal gender determination solely by the presence of Y chromosome. As described under Item 3 of this report, we are currently engaged in litigation with Xenomics regarding our rights under the license agreement.
Through our exclusive license agreement with Genomic Nanosystems, LLC, we hold exclusive rights to issued patents and pending patent applications providing fundamental rights for digital PCR technologies and methods. The issued patents are United States Patent Nos. 6,143,496; 6,391,559; and 7,459,315. These patents will expire in 2017. The license provides us with the exclusive right to use the technology on any platform for noninvasive prenatal diagnostics and analysis for any sample (for example, fetal cells, amniocentesis fluids, plasma, urine, etc.) and also in conjunction with mass spectrometry for any commercial, diagnostic or research purpose, excluding second generation sequencing.
Our success depends to a significant degree upon our ability to continue to develop proprietary products and technologies, to identify and validate useful genetic markers and to thoroughly understand their associations with disease, and to in-license desirable or necessary intellectual property as appropriate. We intend to continue to file patent applications as we develop new products and methods for nucleic acid analysis, and as we develop diagnostic and molecular medicine related technology and products. Patents provide some degree of protection for our intellectual property. However, the assertion of patent protection involves complex legal and factual determinations and is therefore uncertain. The laws governing patentability and the scope of patent coverage continue to evolve, particularly in the areas of genetics, molecular biology, and prenatal and molecular diagnostics that are of interest to us. There can be no assurance that patents will issue from any of our patent applications. The scope of any of our issued patents including U.S. Patent No. 6,250,540, may not be sufficiently broad to offer meaningful protection.
Our issued patents may be successfully challenged, invalidated, circumvented or declared unenforceable so that our patent rights would not create an effective competitive barrier. The laws of some foreign countries may not permit such assignments or may not protect our proprietary rights to the same extent, as do the laws of the United States. In view of these factors, our intellectual property positions bear some degree of uncertainty. We also rely in part on trade secret protection and confidentiality agreements for protection of our intellectual property. We attempt
9
Table of Contents
to protect our trade secrets and confidential information by entering into confidentiality agreements with outside parties and with our employees and consultants. Our employees also sign agreements requiring that they assign to us their intellectual property interests in work performed for us as a part of their employment. The laws of some foreign countries may not permit such assignments or may not protect our proprietary rights to the same extent, as do the laws of the United States. All employees sign an agreement not to compete unfairly with us during their employment and upon termination of their employment, through the misuse of confidential information, soliciting employees, soliciting customers, and the like. It is possible that these agreements may be breached or invalidated and if so, there may not be an adequate corrective remedy available. Parties may breach the confidentiality provisions in our contracts or infringe or misappropriate our patents, copyrights, trademarks, trade secrets, confidential information, and other proprietary rights. Outside parties may independently discover or invent competing technologies or reverse engineer our trade secrets or other technology. The measures we are taking to protect our proprietary rights may not be adequate due to factors beyond our control.
In the future, parties may file claims asserting that our technologies or products infringe on their intellectual property. We cannot predict whether parties will assert such claims against us, or whether those claims will harm our business. If we are forced to defend against such claims, we will face costly litigation and diversion of management’s attention and resources. As a result of such disputes, we may have to develop costly non-infringing technology or enter into licensing agreements. These agreements, if necessary, may be unavailable on terms acceptable to us, which could seriously harm our business and financial condition.
Competition
We face competition from various companies offering nucleic acid analysis systems and services, from various companies developing and commercializing diagnostic assays, and from various companies researching and developing prenatal diagnostic technology.
In the molecular diagnostic business, including the noninvasive prenatal diagnostic market, our tests are based on detection of circulating cell-free fetal nucleic acid in maternal plasma. Our exclusive license to the intellectual property surrounding the use of free fetal nucleic acids in maternal serum or plasma, combined with the precision and accuracy of our MassARRAY system provide us with a competitive advantage in this space. In addition to invasive techniques, our competition arises from alternative methods of noninvasive prenatal diagnostics such as fetal cell purification from maternal blood and trophoblast purification from cervical swabs, fetal cell approaches, and potentially from sequencing approaches. Competitors potentially include Ikonysis, Inc., Artemis Health, Inc., Celula Inc. and Fluidigm Corp. and others.
In the nucleic acid analysis marketplace, our MassARRAY system competes with alternative technology platforms that differ in cost per data point, throughput, sample amplification, analysis process, sample separation or method of DNA detection, turnaround time and quality of results. Most competitive technologies do not rely on direct detection methods such as mass spectrometry, but instead use indirect sample detection methods, such as hybridization or labeling. Competitive technologies are offered by Life Technologies, Corp. (formerly Applied Biosystems, Inc.), Beckman Coulter, Inc., Illumina Inc., Biotage AB, Fluidigm Corp., Ibis Biosciences, Inc. (now Abbott), Luminex and others.
Research and Development
We believe that investment in research and development is essential to establishing a long-term competitive position as a provider of genetic analysis tools and as a provider or an enabler of diagnostic tests. Our research and development expenses for the years ended December 31, 2009, 2008, and 2007, were $37.5 million, $27.5 million, and $14.4 million, respectively.
During 2009, we conducted most of our research and development activities at our facilities in the United States. Our research and development is augmented by advisory and collaborative relationships with others.
During 2009, we reviewed our research and development initiatives and determined to focus our research and development efforts on our key initiatives. Our efforts are primarily focused on our continuing efforts to
10
Table of Contents
develop a noninvasive ccff prenatal test for Trisomy 21, a new initiative to develop a noninvasive test for age-related macular degeneration, completion of the launch of a next-generation MassARRAY system with improved performance and reliability, expansion of the applications for our MassARRAY technology and the introduction of new panels for our research and translational medicine customers.
Government Regulation
Regulation by governmental authorities in the United States and other countries will be a significant factor in the development, testing, production and marketing of diagnostic products, including tests that may be developed by us or our corporate partners, collaborators or licensees. Certain diagnostic products developed by us or our collaborators may require regulatory approval by governmental agencies prior to commercialization. Products that we develop in the diagnostic markets, depending on their intended use, will be regulated as medical devices by the FDA and comparable agencies of other countries and require either premarket approval (PMA) or 510(k) clearance from the FDA prior to marketing. The 510(k) clearance pathway usually takes from three to six months from submission, but can take significantly longer. The premarket approval pathway is much more costly, lengthy, uncertain and generally takes from nine months to one year or longer from submission. The receipt and timing of regulatory clearances or approvals for the marketing of such products may have a significant effect on our future revenues. Human diagnostic products are subject to rigorous testing and other approval procedures by the FDA in the United States and similar health authorities in foreign countries. Various federal and state statutes and regulations also govern or influence the manufacturing, safety, labeling, storage, record keeping and marketing of diagnostic products.
Obtaining these approvals and the subsequent compliance with these regulations require the expenditure of substantial resources over a significant period of time, and there can be no assurance that any approvals will be granted. Any such delay in obtaining or failure to obtain such approvals could adversely affect our ability to earn sales revenues, royalties or other license-based fees. Current governmental regulations may change as a result of future legislation or administrative action and cannot be predicted.
As mentioned above, our strategy focuses on capitalizing on our potential in molecular diagnostics markets by commercializing various noninvasive diagnostic tests and laboratory platform systems. Our approach involves initial commercialization of tests as LDTs through our CLIA certified laboratory in Grand Rapids, Michigan. This approach involves transferring basic technology to the laboratory. The laboratory is solely responsible for the development, validation and commercialization of the assay. Such LDT testing is currently under the purview of CMS and State agencies that provide oversight of the safe and effective use of LDTs. To date, the FDA has exercised its regulatory discretion not to regulate LDTs, as LDTs are developed and used by a single laboratory. The FDA and the U.S. Department of Health and Human Services have been reviewing their approach to regulation in the area of genetic testing and LDTs, and the laws and regulations may undergo change in the near future. Although recent reforms and enforcement actions have focused on them, we have no current plans to utilize analyte specific reagents (ASRs) or In-Vitro Diagnostic Multivariate Index Assay (IVDMIAs) in our LDT strategy so the effect on us of any of these specific changes in FDA policy is currently considered remote to our business.
Sequenom CMM and any other CLIA certified laboratories that we may partner with are subject to CLIA regulations, which are designed to ensure the quality and reliability of clinical laboratories by mandating specific standards in the areas of personnel qualifications, administration and participation in proficiency testing, patient test management, quality control, quality assurance and inspections. The sanction for failure to comply with CLIA requirements may be suspension, revocation or limitation of a laboratory’s CLIA certificate, which is necessary to conduct business, as well as significant fines and/or criminal penalties. Sequenom CMM is also subject to regulation of laboratory operations under state clinical laboratory laws. State clinical laboratory laws may require that laboratories and/or laboratory personnel meet certain qualifications, specify certain quality controls or require maintenance of certain records. Certain states, including Florida, Maryland, New York, Pennsylvania and Rhode Island, each require that you obtain licenses to test specimens from patients residing in those states and additional states may require similar licenses in the future. Potential sanctions for violation of these statutes and regulations include significant fines and the suspension or loss of various licenses, certificates and authorizations.
11
Table of Contents
Our research and development activities involve the controlled use of hazardous materials and chemicals, however, the concentration and volumes of these chemicals are limited. We are subject to federal, state and local laws and regulations governing the use, storage, handling and disposal of such materials and chemicals, as well as certain waste products.
Employees
As of February 12, 2010, we employed 234 persons, of whom 46 hold Ph.D. or M.D. degrees and 44 hold other advanced degrees. Our success will depend in large part upon our ability to attract and retain employees. We face competition in this regard from other companies, research and academic institutions, government entities, and other organizations.
Executive Officers
Our executive officers, their positions with us, and their ages as of February 12, 2010 are as follows:
| Name |
Age | Position | ||
| Executive Officers |
||||
| Harry F. Hixson, Jr., Ph.D |
71 | Chief Executive Officer and Director | ||
| Charles R. Cantor, Ph.D. |
67 | Chief Scientific Officer and Director | ||
| Ronald M. Lindsay, Ph.D. |
62 | Interim Senior Vice President, Research and Development | ||
| Paul V. Maier, M.B.A. |
62 | Interim Chief Financial Officer | ||
| Allan Bombard, M.D., M.B.A. |
57 | Chief Medical Officer | ||
| Michael Monko, M.B.A. |
50 | Senior Vice President, Sales and Marketing | ||
| Larry Myres |
51 | Vice President, Operations | ||
| Clarke Neumann, J.D. |
46 | Vice President and General Counsel | ||
| Shawn Marcell |
48 | Vice President of Commercial Development, Prenatal Diagnostics | ||
| Karsten Schmidt, Ph.D. |
48 | Vice President, Business Development | ||
| Dereck Tatman, Ph.D., M.B.A. |
37 | Vice President, Business Development | ||
| Alisa Judge |
54 | Vice President, Human Resources | ||
| Gary Riordan |
51 | Vice President, Regulatory Affairs and Quality |
Harry F. Hixson, Jr., Ph.D. Dr. Hixson has served as our chief executive officer since September 28, 2009. Dr. Hixson has served as chairman of the board of directors since 2003. He also currently serves as a director of BrainCells, Inc., a biopharmaceutical company focused on central nervous system drug development, where he was chief executive officer from July 2004 until September 2005. Dr. Hixson served as chief executive officer of Elitra Pharmaceuticals, Inc., a biopharmaceutical company focused on anti-infective drug development, from February 1998 until May 2003. He served as president and chief operating officer of Amgen Inc., and as a member of its board of directors from 1988 to 1991. Prior to Amgen, Dr. Hixson held various management positions with Abbott Laboratories, including vice president, diagnostic products business group, and vice president, research and development, in the Diagnostics Division. Dr. Hixson also is a director of Arena Pharmaceuticals, Inc., Infinity Pharmaceuticals, Inc., and Novabay Pharmaceuticals. Dr. Hixson received his Ph.D. in Physical Biochemistry from Purdue University and an M.B.A. from the University of Chicago.
Charles R. Cantor, Ph.D. Dr. Cantor joined us as Chief Scientific Officer and Chairman of the Scientific Advisory Board in August 1998 and has served as a member of our board of directors since 1998. Dr. Cantor is also Chief Executive Officer of DiThera, Inc., a biotechnology company that he founded in 2007. Since 1992, Dr. Cantor has served as a professor in the Department of Biomedical Engineering and Co-Director of the Center for Advanced Biotechnology at Boston University. Prior to that time, Dr. Cantor held positions at Columbia University and the University of California, Berkeley. He was also Director of the Human Genome Center of the Department of Energy at Lawrence Berkeley Laboratory. Dr. Cantor published the first textbook on genomics, The Science and Technology of the Human Genome Project, and remains active in the Human Genome Project through his membership in a number of the project’s advisory committees and review boards. Dr. Cantor is a member of the National Academy of Sciences. He is also a scientific advisor to 12 biotechnology and life science companies and one venture capital firm. Dr. Cantor currently serves as a director of ExSAR, Inc., Human BioMolecular Research Institute, and Retrotrope, Inc. Dr. Cantor received his Ph.D. in Chemistry from the University of California, Berkeley.
12
Table of Contents
Ronald M. Lindsay, Ph.D. Dr. Lindsay has served as our interim senior vice president of research and development since September 28, 2009. Dr. Lindsay has served as a member of our board of directors since 2003. He currently operates Milestone Consulting, a biopharmaceutical consulting firm. Dr. Lindsay served as vice president, research and development, and chief science officer of diaDexus Inc., a biotechnology company, from 2000 to January 2004. From 1997 through 2000, Dr. Lindsay served in various senior management roles with Millennium Pharmaceuticals, Inc., a biopharmaceutical company. From 1989 to 1997, Dr. Lindsay served in various roles with Regeneron Pharmaceuticals Inc., of which he was a founding scientist. He is a director of Arqule Inc., and HistoRx Inc. Dr. Lindsay received his Ph.D. in Biochemistry from the University of Calgary.
Paul V. Maier, M.B.A. Mr. Maier has served as our interim chief financial officer since November 10, 2009. Mr. Maier served as senior vice president and chief financial officer of Ligand Pharmaceuticals Incorporated from 1992 until January 2007, where he helped build Ligand from a venture stage company to a commercial, integrated biopharmaceutical organization. Prior to Ligand, Mr. Maier spent six years in various management and finance positions at ICN Pharmaceuticals. Mr. Maier currently serves as a director of Hana Biosciences, Inc., Pure Bioscience, and International Stem Cell Corporation. Mr. Maier received his M.B.A. from Harvard University.
Allan Bombard, M.D., M.B.A. Dr. Bombard joined us as Chief Medical Officer in January 2009. From October 2008 to January 2009, Dr. Bombard was the Chief Executive Officer of Lenetix Medical Laboratory, which provides genetic screening and diagnostic testing for obstetricians, gynecologists, family practitioners, nurse midwives, laboratories, diagnostic facilities and other healthcare providers. From April 2005 to October 2008, Dr. Bombard was Chief Medical Officer of Sharp Mary Birch Hospital for Women. From 2002 to 2005, Dr. Bombard served as Senior Vice President, Chair, and Residency Program Director of the Department of Obstetrics and Gynecology at Lutheran Medical Center. Prior to Lutheran Medical Center, he served as the Western U.S. Medical Director for Women’s Health at Aetna. Since 1998, Dr. Bombard has been a clinical professor in the Department of Obstetrics and Gynecology & Women’s Health at the Albert Einstein College of Medicine and since 2004. Dr. Bombard received his M.D. from the George Washington University and his M.B.A. from the University of San Diego.
Michael Monko, M.B.A. Mr. Monko joined us as Senior Vice President, Sales and Marketing in August 2006. Mr. Monko served as Vice President of Sales for the organization that is now the diagnostics strategic business unit of Millipore, a bioscience research and biopharmaceutical manufacturing supplier, from 2005 to July 2006. Previously, he served 19 years in various sales roles at Invitrogen Corporation (now Life Technologies, Corp.), a biotechnology tools company. Mr. Monko received his M.B.A. from Babson College.
Larry Myres. Mr. Myres joined us as Vice President, Operations in November 2005. Mr. Myres was Vice President of Operations for DexCom, Inc., a medical device company, from 2000 to 2005 and Precision Vascular Systems, a medical device company, from 1997 to 2000. Mr. Myres received his Bachelor of Science degree from Westminster College of Salt Lake City.
Clarke Neumann, J.D. Mr. Neumann joined us in 1999 and has served as Vice President, General Counsel and Assistant Secretary since 2001. Prior to joining us, Mr. Neumann was an attorney at Lyon & Lyon, LLP, specializing in intellectual property litigation, strategic counseling, business litigation and transactional matters. Mr. Neumann holds a J.D. from Loyola Law School, Los Angeles.
Shawn Marcell. Mr Marcell joined us in 2008, through our acquisition of SensiGen, LLC, where he served as president and Chief Executive Officer. Prior to joining SensiGen Mr. Marcell was senior vice president of commercialization at the Science Center in Philadelphia where he was responsible for commercialization of a business incubator housing 36 companies. He also acted as Chief Executive Officer of the Science Center’s lead portfolio company, Neuro Diagnostic Devices, Inc., which he formed and for which he raised financing. Prior to joining the Science Center, Mr. Marcell was co-founder and Chief Executive Officer of Linguagen Corp. (now Redpoint Bio), a biotechnology company involved in the science of taste. Mr. Marcell was Global Head of Sales, Marketing and Business Development at Centocor, Inc., Diagnostics Division where he was responsible for businesses in the U.S., Europe and Japan. Prior to Centocor, Mr. Marcell held sales and marketing positions of
13
Table of Contents
increasing responsibility with Abbott Laboratories Diagnostic division. Mr. Marcell holds a B.A. in economics from the George Washington University, Washington, DC.
Karsten Schmidt, Ph.D. Dr. Schmidt joined us in January 1999 as Director, Business Development and has served as Vice President, Business Development, since December 2005. He has also served previously as Managing Director of our German subsidiary, Vice President, European Operations, and Vice President, Operations. Before joining us, Dr. Schmidt held a senior management position at Rhône-Poulenc Rorer where he was responsible for all drug regulatory affairs activities in the asthma and allergy area. Dr. Schmidt is a trained pharmacist. He received his Ph.D. in pharmaceutical biology from the University in Bonn.
Dereck Tatman, Ph.D., M.B.A. Dr. Tatman joined us in 2000 as a Business Development Analyst and has served as Vice President, Business Development since July 2004. Prior to joining us, Dr. Tatman was employed at Dow Agrosciences in the biotechnology business development group. Dr. Tatman holds a Ph.D. from Arizona State University and a M.S. in Management from Krannert School of Business at Purdue University.
Alisa Judge. Ms. Judge joined us in June 2007 as Vice President, Human Resources, and brings over 20 years of human resources experience having previously served as vice president of human resources at Claritas, a division of the Nielsen Company, the world’s leading provider of marketing information and audience measurement. Prior to Claritas, Ms. Judge held the same role for GKN Aerospace Chem-tronics, a leading global supplier to the world’s automotive and aerospace manufacturers. Prior to GKN, she was Director of Global Staffing and Retention at Invitrogen Corp. (now Life Technologies, Corp.), a global biotech manufacturer based in Carlsbad, Calif. She also held the title of Vice President, Human Resources, at Advanced Marketing Services, Inc., who provides wholesaling and distribution services to book retailers, where she spent 10 years. Ms. Judge holds a B.S. in Business from Humboldt State University.
Gary Riordan. Mr. Riordan joined us in September 2008 as Vice President, Regulatory Affairs and Quality. Prior to joining us, Mr. Riordan served as Director, Regulatory Affairs at Ventana Medical Systems, Inc., a diagnostic systems supplier, from November 2004 to September 2008, and at Roche Molecular Systems, Inc., from December 1997 to October 2004. Mr. Riordan worked at the U.S. Food and Drug Administration from June 1990 to December 1997 where he evaluated regulatory submissions for antibody- and nucleic acid-based HIV and Hepatitis diagnostic assays and conducted inspections of in vitro diagnostic manufacturers.
Available Information
We file our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, amendments to those reports, and other information with the SEC. We will supply a copy any document we file with the SEC, without charge. To request a copy, please contact Investor Relations, Sequenom, Inc., 3595 John Hopkins Court, San Diego, CA, 92121, USA. The public may also read and copy any document we file with the SEC at its public reference facilities at 100 F Street NE, Washington, D.C. 20549, or by calling the SEC at 1-800-SEC-0330, or by accessing the SEC’s website at www.sec.gov, where the SEC maintains reports, proxy and information statements and other information regarding us and other issuers that file electronically with the SEC. In addition, as soon as reasonably practicable after such materials are filed with or furnished to the SEC, we make copies available to the public free of charge through our website at www.sequenom.com. We also regularly post on our corporate website copies of our press releases as well as additional information about us. Interested persons can subscribe on our website to email alerts that are sent automatically when we issue press releases, file our reports with the SEC or certain other information is available.
Before deciding to invest in us or deciding to maintain or increase your investment, you should carefully consider the risks described below, in addition to the other information contained in this report and in our other filings with the SEC. The risks and uncertainties described below and in our other filings are not the only ones facing us. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also affect our business. If any of these known or unknown risks or uncertainties actually occurs, our business, financial condition and results of operations could be seriously harmed. In that event, the market price for our common stock could decline and you may lose your investment.
14
Table of Contents
If we fail to obtain the capital necessary to fund our operations, our financial results, financial condition and our ability to continue as a going concern will be adversely affected and we will have to delay or terminate some or all of our product development programs.
Our business will require additional financial investment that we have not yet secured. Our consolidated financial statements as of December 31, 2009 have been prepared assuming that we will continue as a going concern. As of December 31, 2009, we had an accumulated deficit of approximately $597.3 million. We expect to continue to incur losses for the foreseeable future and will have to raise substantial cash to fund our planned operations. As a result, there is substantial doubt about our ability to continue as a going concern unless we are able to successfully raise additional capital or we adopt measures to conserve our cash resources and prolong our ability to operate.
Our cash, cash equivalents and current marketable securities is $42.7 million as of December 31, 2009, which based on our current projections will not be sufficient to fund our obligations through the third quarter of 2010 if we continue our spending at our current levels. Our plans for research and development activities to expand our diagnostic test menu over the next 12 months can only be implemented if we are successful in raising significant funds during this period. In addition, there can be no assurances that our research and development activities will be successful. We need to collect a large number of patient samples in a timely manner in order to execute our molecular diagnostic research and development activities. If we do not make sufficient research and development progress, this could adversely impact our ability to raise significant additional funds, which could adversely impact our ability to continue as a going concern. The actual amount of funds that we will need and the timing of any such investment will be determined by many factors, some of which are beyond our control.
We intend to pursue raising additional capital during 2010. In addition to raising capital in 2010, we anticipate that we will need to raise additional funds in the future for the continued development and commercialization of our molecular diagnostic technology. We will need to sell equity or debt securities to raise significant additional funds. The sale of additional securities will likely result in dilution to our stockholders. Additional financing may not be available in amounts or on terms satisfactory to us or at all. We may be unable to raise additional financing due to a variety of factors, including our financial condition, the status of our research and development programs, the status of ongoing litigation and pending governmental investigations and the general condition of the financial markets. If we fail to raise significant additional financing, we will have to delay or terminate some or all of our research and development programs, our financial condition and operating results will be adversely affected and we may have to cease our operations.
If we obtain significant additional financing, we expect to continue to spend substantial amounts of capital on our operations for the foreseeable future. The amount of additional capital we will need depends on many factors, including:
| • | the size of our future operating losses; |
| • | our and our distributors’ success in selling our MassARRAY products and services; |
| • | our success in selling our cystic fibrosis carrier screening, Rhesus D genotyping, and Fetalxy sex determination tests, the level of reimbursement we receive and our collections for those tests; |
| • | the terms and conditions of sales contracts, including extended payment terms; |
| • | our ability to introduce and sell new products and services and successfully reduce inventory levels of earlier products; |
| • | the level of our selling, general and administrative expenses; |
| • | the extent of our investment in diagnostic technology, including prenatal genetic analysis technology, molecular diagnostics and noninvasive prenatal diagnostic technology, patient sample collection, development, commercialization, and regulatory approval; |
| • | our ability to validate our diagnostic tests and the level of sensitivity and specificity of our diagnostic tests; |
15
Table of Contents
| • | our success in, and the expenses associated with, researching, developing and commercializing diagnostic products, alone or in collaboration with our partners, and obtaining any required regulatory approval for those products; |
| • | the level of our success alone or in collaboration with our partners in launching and selling any diagnostic products and services; |
| • | the extent and success of our research and development pursuits, including our level of investment in MassARRAY product research and development, and diagnostic assay and other technology research and development; |
| • | the extent to which we enter into, maintain, and derive revenues from licensing agreements, including agreements to out-license our noninvasive prenatal analysis technology, research and other collaborations, joint ventures and other business arrangements; |
| • | the level of our legal expenses and insurance policy coverage, if any, including those expenses associated with the independent investigation, the investigations by the SEC, NASDAQ, the Office of the U.S. Attorney for the Southern District of California and the FBI, intellectual property protection, securities and stockholder derivative class actions and those expenses and any damages and settlement payments (as well as issuances of securities in connection with any settlements) associated with the securities, stockholder derivative, and Xenomics litigations and any future litigation that may arise; |
| • | the dilution from any issuance of securities in connection with the settlement of litigation; |
| • | the extent to which we acquire, and our success in integrating, technologies or companies; |
| • | the level of our expenses associated with the audit of our consolidated financial statements as well as compliance with other corporate governance and regulatory developments or initiatives; and |
| • | regulatory changes and technological developments in our markets. |
General market conditions, the market price of our common stock, uncertainty about our Trisomy 21 test, as well as other screening and diagnostic tests, the uncertainty regarding the results of ongoing litigation matters, the investigations by the SEC, NASDAQ, the Office of the U.S. Attorney for the Southern District of California and the FBI or other factors may not support capital raising transactions. In addition, our ability to raise additional capital may depend upon obtaining shareholder approval. There can be no assurance that we will be able to obtain shareholder approval if it is necessary. If we are unable to obtain sufficient additional funds on a timely basis or on terms favorable to us, we may be required to cease or reduce further commercialization of our products, to cease or reduce certain research and development projects, to sell, license or otherwise dispose of some or all of our technology or assets or business units, to merge all or a portion of our business with another entity or we may not be able to continue as a going concern. If we raise additional funds by selling shares of our capital stock (or otherwise issue shares of our capital stock or rights to acquire share of our capital stock), the ownership interest of our current stockholders will be diluted.
Uncertainty regarding our Trisomy 21 test and other planned tests could materially adversely affect our business, financial condition and results of operations.
We have announced that previously announced test data and results for our noninvasive prenatal test for Trisomy 21 cannot be relied upon. As a result, the launch of the test did not occur. While we are continuing our research and development program for the test, we are unable to provide guidance on the timetable for completing this program or for the potential commercialization of the test. In addition to mass-spectrometry-based approaches, one approach that we are exploring for a Trisomy 21 test is a nucleic acid sequencing-based approach. We have limited experience developing and no experience commercializing sequencing-based technology and would need to rely on collaborative partners and/or sequencing technology provided by others in order to commercialize a test utilizing sequencing. The launch of the test will require the completion of certain clinical development and commercialization activities, including the efforts of collaborative partners on which we rely, and the expenditure of additional cash resources. We can give no assurance that we will be able to successfully complete the clinical development of this test or that we will be able to maintain the collaborative
16
Table of Contents
relationships that are essential to our clinical development efforts. We also can give no assurance that we will be able to reduce our expenditures sufficiently or otherwise mitigate the risks associated with our business to raise enough capital to complete clinical development or commercialization for this test. Clinical development requires large numbers of patient samples and we may not be able to collect a sufficient number of samples in a timely manner to complete clinical development for a Trisomy 21 or any other planned molecular diagnostic test. Failure by us to collect a sufficient number of samples in a timely manner could prevent or significantly delay our ability to research, develop, complete clinical development and validation, and launch, any of our planned tests. Any failure to complete clinical development or commercialization of our Trisomy 21 test, as well as other planned screening and diagnostic tests could have a material adverse effect on our business, operating results or financial condition.
The launch of any of our diagnostic tests under development will require the completion of certain clinical development and commercialization activities and the expenditure of additional cash resources. We can give no assurance that we will be able to successfully complete the clinical development of any test under development or that we will have sufficient cash resources to do so. Other than mass-spectrometry-based and PCR-based approaches, we have limited experience working with alternative platforms and would need to rely on collaborative partners and/or technology provided by other parties in order to commercialize a test utilizing such alternative platforms. We can give no assurance that we will be able to reduce our expenditures sufficiently or raise enough capital to complete clinical development or commercialization for any test. Any failure to complete clinical development or commercialization of a test could have a material adverse effect on our business, operating results or financial condition.
We and certain of our former and current executive officers and directors have been named as defendants in litigation that could result in substantial costs, divert management’s attention and otherwise result in dilution to our stockholders.
We and certain of our current and former executive officers, have been sued for alleged violations of federal securities laws related to alleged false and misleading statements regarding our Trisomy 21 test under development. We have been engaged in a vigorous defense of such claims. On January 26, 2010, the U.S. District Court for the Southern District of California entered an order preliminarily approving a stipulation of settlement reached in our class action securities lawsuits related to alleged violations of federal securities laws consolidated under the caption In re Sequenom Inc. Securities Litigation. Even though a settlement has been preliminarily approved by the court, there is no guarantee that final approval of the settlement will be granted. If a final settlement of the claims is not approved and we are not successful in our defense of such claims, we could be forced to make significant payments to or otherwise enter into less favorable settlement arrangements with the class action plaintiffs and their lawyers in connection with such class action securities lawsuits. Even if we are able to settle the class action securities lawsuits, there are several other lawsuits and claims, including state and federal shareholder derivative actions that have not been settled. Although we intend to continue to vigorously defend such lawsuits and claims, there is no guarantee that we will be successful and we may be have to pay damages awards or otherwise may enter into settlement arrangements in connection with such other lawsuits and claims. Any such payments or settlement arrangements could have a material adverse effect on our business, operating results or financial condition. In connection with the pending settlement of class action lawsuits, if finally approved, we will be issuing shares of our common stock and may in the future be required to issue additional shares of our common stock or other securities convertible into or exchangeable for our common stock in connection with future settlements, which would result in dilution to our stockholders. Even if the pending claims are not successful, the litigation could result in substantial costs and significant adverse impact on our reputation and divert management’s attention and resources, which could have a material adverse effect on our business, operating results or financial condition.
We are the subject of an investigation by the SEC and have been contacted by representatives of NASDAQ, the Office of the U.S. Attorney for the Southern District of California and the FBI, each of which could adversely affect our reputation, business prospects, operating results, or financial condition.
In June 2009, we received written notification that the staff of the SEC has initiated an investigation relating to our April 29, 2009 announcement regarding our Trisomy 21 test under development. As part of this
17
Table of Contents
investigation, the SEC staff is also requiring us to produce information with respect to our announcements relating to our offer to acquire EXACT Sciences, Inc. in January 2009. We intend to continue to cooperate fully with the SEC on its investigation. Following our announcement on September 28, 2009 regarding the completion of the independent investigation by the special committee of our board of directors, representatives of NASDAQ, the Office of the U.S. Attorney for the Southern District of California and the FBI separately contacted us to inquire about our announcement. We intend to continue to cooperate fully with NASDAQ, the U.S. Attorney and the FBI. We cannot predict the duration, scope or outcome of the investigations by the SEC, NASDAQ, the U.S. Attorney and the FBI. If these matters continue for a prolonged period of time, it may have an adverse impact on our reputation, business prospects, operating results or financial condition regardless of the ultimate outcome of these matters. In the event that the investigations by the SEC, NASDAQ, the U.S. Attorney or the FBI leads to action against us, any current or former officer or director, our reputation, business prospects, operating results or financial condition may be adversely impacted. These matters may result in the incurrence of significant legal expenses and the diversion of management’s attention from our business, and are likely to have a negative effect on employee morale.
We have limited experience.
Our noninvasive prenatal and other molecular diagnostic tests are at an early stage of discovery and development or have just recently been launched. We continue to develop and commercialize new products and create new applications for our products. We are also researching, developing and pursuing the commercialization of additional noninvasive molecular diagnostic tests for prenatal genetic disorders and other diseases and disorders for use on our MassARRAY platform and potentially other platforms, and we have limited or no experience in these applications of our technology and operating and selling in these markets. Other than mass-spectrometry-based approaches, we have limited experience developing and no experience commercializing sequencing-based technology and would need to rely on collaborative partners and/or sequencing technology provided by others in order to commercialize a test utilizing sequencing. Among other risks, using an alternative platform provided by another party presents potential manufacturing supply and reliability, FDA regulatory compliance and design control, and intellectual property infringement risks. You should evaluate us in the context of the uncertainties and complexities affecting an early stage company developing products and applications for the life science industries and experiencing the challenges associated with entering into new markets that are highly competitive. Based on our limited experience, in developing new products and applications, we may not effectively execute on or focus our research and development efforts, properly model new opportunities to ensure appropriate resource allocation, create products that are appropriately developed to meet customer needs, perform adequate and timely validation testing of such products and applications, ensure appropriate communication between different departments responsible for commercialization activities, implement effective product launch or sales strategies, effectively design and manufacture products that achieve commercial success, or take other actions that ultimately lead to commercial success of any new products or applications that we develop. We may face setbacks in the development and commercialization of our noninvasive prenatal and other molecular diagnostic tests and technologies. As previously announced, we are no longer relying on prior studies related to our Trisomy 21 test. We need to make significant investments to ensure our genetic analysis products and applications and our diagnostics tests perform properly and are cost-effective, and we or our partners will likely need to apply for and obtain certain regulatory approvals to sell our products for diagnostic applications and it is uncertain whether such approvals will be granted. Even if we develop products for commercial use and obtain all necessary regulatory approvals, we may not be able to develop products that are accepted or satisfy customers in the genomic, diagnostic, noninvasive prenatal, clinical research, pharmaceutical, or other markets or the emerging field of molecular medicine and that can be marketed and sold successfully.
We may not be able to generate significant revenue from noninvasive prenatal diagnostic tests or any other tests we may develop.
Our business is substantially dependant on our ability to develop and launch our research-use-only, screening and diagnostic tests. We have committed significant research and development resources to the development of research-use-only and diagnostic tests, particularly noninvasive prenatal tests, for use on our
18
Table of Contents
MassARRAY system and other platforms. There is no guarantee that we will successfully generate significant revenues from any tests that we have launched or plan to launch in the future. In September 2009, we launched a carrier screening LDT for cystic fibrosis through our CAP/CLIA certified laboratory. In early 2010 we launched noninvasive prenatal LDTs for Rhesus D and fetal sex determination through our laboratory. We also plan to pursue the development and launch of a noninvasive prenatal screening LDT for Trisomy 21, a molecular LDT for assessment of risk for developing age-related macular degeneration, and additional tests in the future. However, there is no guarantee that we will be able to successfully launch these or other diagnostic tests on anticipated timelines or at all. We have limited experience in licensing, manufacturing, selling, marketing or distributing our SEQureDx technology, or diagnostic or other tests. If we, or our partners, are not able to successfully market or sell noninvasive prenatal research-use-only or diagnostic tests or other tests we may develop for any reason, including the failure to obtain any required regulatory approvals, we will not generate any revenue from the sale of such tests. Even if we are able to develop noninvasive prenatal research-use-only or diagnostic or other tests for sale in the marketplace, a number of factors could impact our ability to sell such tests or generate any significant revenue from the sale of such tests, including the following:
| • | the outcome of the pending lawsuits, SEC investigation and the investigations by NASDAQ, the Office of the U.S. Attorney for the Southern District of California and the FBI; |
| • | our ability to successfully implement the remedial measures recommended by the special committee following our independent investigation and the effectiveness of those measures and to continue to implement appropriate controls and risk management measures; |
| • | the availability of adequate study samples for validation studies for any diagnostic tests we develop; |
| • | reliance on Sequenom CMM and third-party CLIA certified laboratories, which are subject to routine governmental oversight and inspections for continued operation pursuant to CLIA, to process tests that we develop; |
| • | reliance on Sequenom CMM and third parties to manufacture any noninvasive prenatal research-use-only or diagnostic or other tests that we may develop; |
| • | our ability to establish and maintain adequate infrastructure to support the commercial launch and sale of our diagnostic tests through Sequenom CMM or a third-party CLIA certified laboratory, including establishing adequate laboratory space, information technology infrastructure, sample collection and tracking systems, electronic ordering and reporting systems and other infrastructure and hiring adequate laboratory and other personnel; |
| • | the success of the validation studies for our diagnostic tests under development and our ability to publish study results in peer-reviewed journals; |
| • | the availability of alternative and competing tests or products and technological innovations or other advances in medicine that cause our technologies to be less competitive; |
| • | compliance with federal, state and foreign regulations governing laboratory testing and the sale and marketing of research-use-only or diagnostic or other tests, including noninvasive prenatal tests; |
| • | the accuracy rates of such tests, including rates of false-negatives and/or false-positives; |
| • | concerns regarding the safety or effectiveness or clinical utility of noninvasive prenatal or other tests; |
| • | changes in the regulatory environment affecting health care and health care providers, including changes in laws regulating laboratory testing and/or device manufacturers and any laws regulating prenatal testing; |
| • | the extent and success of our sales and marketing efforts and ability to drive adoption of our diagnostic tests; |
| • | coverage and reimbursement levels by government payors and private insurers; |
| • | the level of physician and customer adoption of any diagnostic tests we develop; |
| • | pricing pressures and changes in third-party payor reimbursement policies; |
19
Table of Contents
| • | general changes or developments in the market for women’s and/or prenatal health diagnostics, or diagnostics in general; |
| • | ethical and legal issues concerning the appropriate use of the information resulting from noninvasive prenatal diagnostic tests or other tests; |
| • | the refusal by women to undergo such tests for moral, religious or other reasons, or based on perceptions about the safety or reliability of such tests; |
| • | our ability to provide effective customer support; |
| • | our ability to promote and protect our SEQureDx brand and technology and our other brands and technologies; and |
| • | intellectual property rights held by others or others infringing our intellectual property rights. |
Our operating results may fluctuate significantly.
Our revenues and results of operations may fluctuate significantly, depending on a variety of factors, including the following:
| • | our ability to manage costs and expenses and effectively implement our business strategy; |
| • | our ability to raise additional capital and continue as a going concern; |
| • | our and our distributors’ success in marketing and selling, and changes in the demand for, our products and services including our MassARRAY platform and iPLEX multiplex genotyping application and other applications and related consumables, and demand for products and services for genotyping, DNA methylation (epigenetic analysis) and QGE (gene expression analysis) applications; |
| • | our success in selling our cystic fibrosis carrier screening, Rhesus D genotyping, and Fetalxy sex determination tests, the level of reimbursement we receive and our collections for those tests; |
| • | our success in depleting or reducing current product inventories in view of new or upcoming product introductions and our success in transitioning to our next generation MassARRAY system and mass spectrometer; |
| • | the pricing of our products and services and those of our competitors; |
| • | our success in collecting payments from customers; |
| • | our success in responding to customer complaints effectively and managing relationships with our customers, variations in the timing of payments from customers and collaborative partners and the recognition of these payments as revenues; |
| • | the timing and cost of any new product or service offerings by us; |
| • | our ability to identify and develop in a cost-efficient manner new applications and products, such as noninvasive prenatal or other diagnostic assays and other diagnostic technologies, the success of such applications and products, and our ability to improve current products to increase demand for such products; |
| • | the potential need to acquire licenses to new technology, including genetic markers that may be useful in diagnostic applications, or to use our technology in new markets, which could require us to pay unanticipated license fees and royalties in connection with licenses we may need to acquire; |
| • | our research and development progress and how rapidly we are able to achieve technical milestones, including the milestone of sufficient fetal DNA enrichment and/or RNA based solutions with respect to our noninvasive prenatal technologies; |
| • | the cost, quality and availability of our next generation MassARRAY system and mass spectrometer, consumable chips, also known as SpectroCHIP bioarrays, oligonucleotides, DNA samples, tissue samples, reagents and related components and technologies; |
20
Table of Contents
| • | material developments in our customer and supplier relationships including our ability to successfully transition to new technologies to successfully maintain our relationships with our customers and suppliers; |
| • | our ability to clinically validate any potential noninvasive prenatal or other diagnostic related products and obtain regulatory approval of any potential diagnostic products; and |
| • | expenses and the payment of any damage awards or settlement amounts related to the pending lawsuits, the investigations by the SEC, NASDAQ, the Office of the U.S. Attorney for the Southern District of California and the FBI. |
Further, our revenues and operating results are difficult to predict because our diagnostic tests have only recently been launched and we do not have sufficient history to forecast revenues reliably for those tests, and also because our revenues and operating results depend on the number, timing, and type of MassARRAY system placements that we make during the year and the quantity and timing of consumables sales for the installed base of systems. Changes in the relative mix of our MassARRAY system and consumables sales, as well as service agreements can have a significant impact on our gross margin, as consumable sales and service agreements typically have margins significantly different than MassARRAY system sales. Our international revenues and operating results are also difficult to predict because they depend upon the activities of our distributors. We experienced reduced demand for our MassARRAY systems during 2009 as a result of the current economic environment and are uncertain whether demand for our products and services will further deteriorate in future periods as a result of the weak economy or other factors. The absence of or delay in generating revenues could cause significant variations in our operating results from year to year and could result in increased operating losses.
We believe that period-to-period comparisons of our financial results will not necessarily be meaningful. You should not rely on these comparisons as an indication of our future performance. If our operating results in any future period fall below the expectations of securities analysts and investors, our stock price will likely fall.
A reduction in revenues from sales of MassARRAY products would harm our business.
The demand for MassARRAY systems declined in 2009 compared to 2008 and any continued or further decline in demand will further reduce our total revenues. We expect that sales of MassARRAY systems and consumables will account for most of our total revenues throughout 2010 and perhaps thereafter, unless and until our noninvasive prenatal or other laboratory developed tests begin to generate significant revenues. The following factors, among others, would further reduce the demand for MassARRAY products and services:
| • | continued unstable, weak, or deteriorating economic conditions and fiscal policies or changes in fiscal policies that negatively impact customer buying decisions; |
| • | uncertainty about our ability to continue as a going concern and supply products and services to customers; |
| • | competition from other products and service providers or failure of our products or applications or services; and |
| • | negative publicity or evaluations, particularly with respect to product warranty and repair and troubleshooting services provided to existing customers, with respect to enabling or performing, or with respect to the independent investigation, the investigations by the SEC, NASDAQ, the Office of the U.S. Attorney for the Southern District of California and the FBI, ongoing litigation matters or developments or events in our prenatal diagnostic and other programs. |
Our revenues are subject to risks faced by our customers and potential customers.
We expect that our revenues throughout 2010 and perhaps thereafter, unless and until our noninvasive prenatal and/or other laboratory developed tests begin to generate significant revenues, will be derived primarily from MassARRAY system products provided to academic institutions and other research institutions.
21
Table of Contents
Our operating results could fluctuate substantially due to reductions and delays in research and development expenditures by these customers. These reductions and delays could result from factors such as:
| • | changes in economic conditions and possible country-based boycotts; |
| • | changes in government programs that provide funding; |
| • | changes in the regulatory environment affecting health care and health care providers, and, for example, recent draft FDA guidance which, if effected, may impose additional restrictions on CLIA certified laboratories performing laboratory diagnostic tests; |
| • | pricing pressures and reimbursement policies; |
| • | market-driven pressures on companies to consolidate and reduce costs; |
| • | other factors affecting research and development spending; and |
| • | uncertainty about our ability to continue as a going concern and fund operations and supply products and services to customers. |
None of these factors are within our control. We have broadened the markets to which we sell our products and applications and continue to develop new applications and products for use in new markets. We are targeting customers in clinical research and clinical marker validation, the emerging field of molecular medicine, genetic service laboratories, and animal testing laboratories and diagnostic testing markets. We have limited or no experience operating in certain of these potential markets and, as a result, may be unable to develop products and applications that allow us to penetrate these markets or successfully generate any revenue from sales in these markets. We will have limited ability to forecast future demand for our existing and any new products and applications in these markets.
We depend on sales of our consumable chips and other MassARRAY consumables for a significant portion of our revenues.
Sales of our consumable chips and other consumables for the MassARRAY system are an important source of revenue. Revenues from MassARRAY consumables totaled approximately 54% of our total revenues for the year ended December 31, 2009, compared to 41% of our total revenues for the year ended December 31, 2008. Factors which may limit the use of our consumable chips and other consumables or otherwise adversely affect our revenues from consumables include:
| • | the extent of our customers’ level of utilization of their MassARRAY systems; |
| • | our ability to provide timely repair services and our ability to secure replacement parts, such as lasers, for our MassARRAY systems and our ability to successfully transition to our next generation MassARRAY system and mass spectrometer; |
| • | the extent to which customers increase multiplexing levels using iPLEX applications; |
| • | the availability and adoption of new technologies and applications provided by our competitors; |
| • | failure to sell additional MassARRAY systems; |
| • | the termination of contracts with or adverse developments in our relations with suppliers of our consumables; |
| • | the training of customer personnel; |
| • | the acceptance of our technology by our customers; |
| • | any negative publicity with respect to ongoing litigation matters, our independent investigation, the SEC investigations, the investigations by NASDAQ, the Office of the U.S. Attorney for the Southern District of California and the FBI or developments or events in our prenatal diagnostic and other programs; |
| • | uncertainty about our ability to continue as a going concern and fund operations and supply products and services to customers; |
22
Table of Contents
| • | the ability to maintain necessary quality standards and specifications for our SpectroCHIP products; and |
| • | our inability to transition to new suppliers for components for our next generation MassARRAY system and mass spectrometer and our ability to maintain such relationships. |
We only recently acquired our CLIA certified laboratory and have limited experience operating a diagnostic laboratory. Our ability to successfully develop and commercialize diagnostic tests will depend on our ability to successfully operate our CLIA certified laboratory and obtain and maintain required regulatory approvals.
We have validated three LDT assays and commercialized them through Sequenom CMM, our CLIA certified laboratory located in Grand Rapids, Michigan. We acquired Sequenom CMM in 2008 and as a result have limited experience operating a CLIA certified laboratory. Because there is substantial distance between Sequenom CMM and us, we may have logistical and operational challenges in effectively managing and operating Sequenom CMM. For future tests, if we are unable to successfully transfer our diagnostic technology and tests to Sequenom CMM for validation or if Sequenom CMM is unable to successfully validate any LDT or other tests that we intend to commercialize through Sequenom CMM, we may not be able to successfully commercialize such tests on the anticipated timelines or at all. Although we have invested substantially in Sequenom CMM’s infrastructure, it is possible that we may not have adequate infrastructure in place to meet demand for our currently launched tests or for the commercial launch and sale of future diagnostic tests that we develop through Sequenom CMM. Our ability to successfully develop and commercialize diagnostic tests will depend on our ability to successfully operate Sequenom CMM and obtain and maintain required regulatory approvals.
Sequenom CMM as a clinical laboratory is subject to CLIA, which is designed to ensure the quality and reliability of clinical laboratories by mandating specific standards in the areas of personnel qualifications, administration and participation in proficiency testing, patient test management, quality control, quality assurance and inspections. The sanction for failure to comply with CLIA requirements may be suspension, revocation or limitation of a laboratory’s CLIA certificate, which is necessary to conduct business, as well as significant fines and/or criminal penalties. Sequenom CMM is also subject to regulation of laboratory operations under state clinical laboratory laws. State clinical laboratory laws may require that laboratories and/or laboratory personnel meet certain qualifications, specify certain quality controls or require maintenance of certain records. Certain states, including Florida, Maryland, New York, Pennsylvania and Rhode Island, each requires that laboratories obtain licenses to test specimens from patients residing in those states and additional states may require similar licenses in the future. If we are unable to obtain and maintain a license from New York, or are unable to maintain licenses from the other states, we will not be able to process any samples from patients located in those states. Potential sanctions for violation of these statutes and regulations include significant fines and the suspension or loss of various licenses, certificates and authorizations, which could adversely affect our business and results of operations.
We may not successfully obtain regulatory approval of any noninvasive prenatal or other diagnostic product or other product which we or our licensing or collaborative partners develop and we may not be able to successfully partner with CLIA certified laboratories with respect to diagnostic products.
Products that we or our collaborators develop in the molecular medicine, diagnostic, noninvasive prenatal diagnostic, or other markets, depending on their intended use, may be regulated as medical devices by the FDA and comparable agencies of other countries and require either PMA or 510(k) clearance from the FDA, prior to marketing. The 510(k) clearance process usually takes from three to six months from submission, but can take significantly longer. The PMA process is much more costly, lengthy, uncertain, and generally takes from nine months to one year or longer from submission. In addition, commercialization of any diagnostic or other product that our licensees or collaborators or we develop would depend upon successful completion of preclinical testing and clinical trials. Preclinical testing and clinical trials are long, expensive, and uncertain processes, and we do not know whether we, our licensees, or any of our collaborators, would be permitted or able to undertake clinical
23
Table of Contents
trials of any potential products. It may take us or our licensees or collaborators many years to complete any such testing, and failure could occur at any stage. Preliminary results of studies or trials do not necessarily predict final results, and acceptable results in early studies or trials may not be repeated in later studies or trials. A number of companies in the diagnostics industry, including biotechnology companies, have suffered significant setbacks in clinical trials, even after promising results in earlier trials. Delays or rejections of potential products may be encountered based on changes in regulatory policy for product approval during the period of product development and regulatory agency review. If our projects reach clinical trials, we or our licensees or collaborators could decide to discontinue development of any or all of these projects at any time for commercial, scientific, or other reasons.
We initially plan to validate assays and commercialize them in the form of LDTs through Sequenom CMM or a third-party CLIA certified laboratory. Although LDT testing is currently solely under the purview of CMS and state agencies who provide oversight of the safe and effective use of LDTs, the FDA and the U.S. Department of Health and Human Services have been reviewing their approach to regulation in the area of genetic testing and LDTs, and the laws and regulations may undergo change in the near future. Although we have no current plans to utilize in our LDT strategy ASRs or IVDMIAs, which have been the focus of recent reforms and enforcement actions by the FDA, we cannot predict the extent of the FDA’s future regulation and policies with respect to LDTs in general or our diagnostic tests in particular. We plan to conduct the development, validation, and other activities necessary to file submissions with the FDA seeking approval for selected diagnostic tests. If we are unable to successfully launch any diagnostic tests as LDTs or if we are otherwise required to obtain FDA premarket clearance or approval prior to commercializing any diagnostic tests, our ability to generate revenue from the sale of such tests may be delayed and we may never be able to generate significant revenues from sales of diagnostic products.
The results of preclinical studies and completed clinical trials are not necessarily predictive of future results, and our current diagnostic product candidates may not have favorable results in later studies or trials.
To date, long-term safety and efficacy have not yet been demonstrated in clinical trials for any of our diagnostic product candidates. Favorable results in our early studies or trials may not be repeated in later studies or trials that will be required to obtain either PMA or 510(k) clearance from the FDA prior to marketing any of our product candidates. Our product candidates, including any diagnostic or other tests we develop, may fail to demonstrate positive results in clinical trials despite having progressed through earlier-stage studies or trials. The limited results that we have obtained for our prenatal diagnostic tests may not predict results from studies in larger numbers of subjects drawn from more diverse populations over a longer period of time. Unfavorable results from ongoing preclinical studies or clinical trials could result in delays, modifications or abandonment of ongoing or future clinical trials, or abandonment of a product development program. Preclinical and clinical results or other study results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals or commercialization.
Because we exclusively licensed our noninvasive prenatal diagnostic and gender determination testing rights from Isis any dispute with Isis may adversely affect our ability to develop and commercialize diagnostic tests based on these licensed rights.
In October 2005, we entered into an exclusive license to noninvasive prenatal diagnostic rights (United States Patent No. 6,258,540 and foreign equivalents) with Isis, which we amended in October 2006 and in November 2007 to also include exclusive rights to intellectual property for noninvasive prenatal gender determination testing for social and lifestyle purposes. On November 3, 2009, we entered into a third amendment to modify certain time-based commercial launch milestones relating to aneuploidy and other products. In exchange for this modification, we agreed to make an immediate one-time payment of $1,000,000, increase royalty payments under the agreement during the final 12 months of the patent term and increase the specified minimum royalty amounts. We are using and intend to continue to use the rights that we acquired under the license to develop and commercialize noninvasive prenatal nucleic acid based tests, including gender
24
Table of Contents
determination tests. If there is any dispute between us and Isis regarding our rights under the license agreement, or we do not achieve the commercial launch milestones, as modified, in a timely manner, our ability to exclusively commercialize these diagnostic tests may be adversely affected and could delay or completely terminate our product development and commercialization efforts for these diagnostic tests.
We and our licensees and collaborators may not be successful in developing or commercializing diagnostic products, diagnostic assays including noninvasive prenatal diagnostic products, or other products using our products, services, or discoveries.
Development of diagnostic or other products by us, our licensees, or our collaborators including assays, are subject to risks of failure inherent in the development and commercial viability of any such product, such as demand for such product. These risks further include the possibility that such product would:
| • | be found to be ineffective, unreliable, or otherwise inadequate or otherwise fail to receive regulatory approval; |
| • | be difficult or impossible to manufacture on a commercial scale; |
| • | be uneconomical to market or otherwise not be effectively marketed; |
| • | fail to be successfully commercialized if adequate reimbursement from government health administration authorities, private health insurers, and other organizations for the costs of these products is unavailable; |
| • | be impossible to commercialize because they infringe on the proprietary rights of others or compete with products marketed by others that are superior; or |
| • | fail to be commercialized prior to the successful marketing of similar products by competitors. |
If a licensee discovers or develops diagnostic products or we or a collaborator discover or develop diagnostic or other products using our technology, products, services, or discoveries, we may rely on that licensee or collaborator (hereafter referred to as “partner”) for product development, regulatory approval, manufacturing, and marketing of those products before we can realize revenue and some or all of the milestone payments, royalties, or other payments we may be entitled to under the terms of the licensing or collaboration agreement. If we are unable to successfully achieve milestones or our partners fail to develop successful products, we will not earn the revenues contemplated and we may also lose exclusive (as in the case of our license agreement with Isis, under which we in-license our fundamental noninvasive prenatal diagnostic technology) or non-exclusive license rights to intellectual property that are required to commercialize such products. Our agreements may allow our partners significant discretion in electing whether to pursue any of these activities. We cannot control the amount and timing of resources our partners may devote to our programs or potential products. As a result, we cannot be certain that our partners will choose to develop or commercialize any products or will be successful in doing so. In addition, if a partner is involved in a business combination, such as a merger or acquisition, or changes its business focus, its performance under its agreement with us may suffer and, as a result, we may not generate any revenues or only limited revenues from the royalty, milestone, and similar payment provisions contained in our agreement with that partner.
Our ability to compete in the market may decline if we lose or do not obtain some of our intellectual property rights.
Our success will depend on our ability to obtain and protect patents on our technology, to protect our trade secrets, and to maintain our rights to licensed intellectual property or technologies. Our patent applications or those of our licensors may not result in the issue of patents in the United States or other countries. Our patents or those of our licensors may not afford meaningful protection for our technology and products. Others may challenge our patents or those of our licensors in litigation or by proceedings such as interference, oppositions and reexaminations, as is the case with the appeal pending before the European Patent Office with respect to the patent rights that we in-licensed from Isis for prenatal diagnostics (United States Patent No. 6,258,540 and European Patent No. 994963), and as a result, our patents or those of our licensors could be narrowed or
25
Table of Contents
invalidated or become unenforceable. Competitors may develop products similar to ours that do not conflict with our patents or patent rights. Others may develop noninvasive prenatal tests or other diagnostic tests or products, technologies or methods in violation of our patents or those of our licensors, or by operating around our patents or license agreements, which could reduce sales of our consumables or reduce or remove our noninvasive prenatal and other diagnostic commercialization opportunities. To protect or enforce our patent rights, we may initiate interference proceedings, oppositions, reexaminations or litigation against others. However, these activities are expensive, take significant time and divert management’s attention from other business concerns. We may not prevail in these activities. The patent position of biotechnology companies generally is highly uncertain and involves complex legal and factual questions that are often the subject of litigation. No consistent policy has emerged from the U.S. Patent and Trademark Office, the offices of foreign countries or the courts regarding the breadth of claims allowed or the degree of protection afforded under biotechnology patents. There is a substantial backlog of biotechnology patent applications at the U.S. Patent and Trademark Office and of the equivalent offices around the world and the approval or rejection of patent applications may take several years.
Claims by other companies that we infringe their intellectual property rights or that patents on which we rely are invalid could adversely affect our business.
From time to time, companies have asserted, and may again assert, patent, copyright and other intellectual proprietary rights against our products or products using our technologies. These claims have resulted and may in the future result in lawsuits being brought against us. We may not prevail in any lawsuits alleging patent infringement given the complex technical issues and inherent uncertainties in intellectual property litigation. If any of our products, technologies or activities, in particular our iPLEX products and our MassARRAY system and mass spectrometer (including our planned next-generation MassARRAY system and mass spectrometer), from which we derive a substantial portion of our revenues, were found to infringe on another company’s intellectual property rights, we could be subject to an injunction that would force the removal of our products from the market or we could be required to redesign our products, which could be costly. We could also be ordered to pay damages or other compensation, including punitive damages and attorneys’ fees to such other company. A negative outcome in any such litigation could also severely disrupt the sales of our marketed products to our customers or their customers, which in turn could harm our relationships with our customers, our market share and and/or product revenues. Even if we are ultimately successful in defending any intellectual property litigation, such litigation is expensive and time consuming to address, will divert our management’s attention from our business and may harm our reputation.
Other companies or entities also may commence actions seeking to establish the invalidity of our patents. In the event that one or more of our patents are challenged, a court may invalidate the patent(s) or determine that the patent(s) is not enforceable, which could harm our competitive position. If one or more of our patents are invalidated or found to be unenforceable, or if the scope of the claims in any of these patents is limited by a court decision, we could lose certain market exclusivity afforded by patents owned or in-licensed by us and potential competitors could more easily bring products to the market that directly compete with our own. Such adverse decisions may negatively impact our revenues.
The rights we rely upon to protect the intellectual property underlying our products may not be adequate, which could enable others to use our technology and reduce our ability to compete with them.
We require our employees, consultants, advisors, and collaborators to execute confidentiality agreements and in certain cases, assignment or license agreements. We cannot guarantee that these agreements will provide us with adequate intellectual property ownership or protection against improper or unauthorized use or disclosure of confidential information or inventions. In some situations, these agreements may conflict with or be subject to the rights of others with whom our employees, consultants, advisors, or collaborators have prior employment or consulting relationships. In some situations, as is the case with our employees in Germany, these types of agreements or relationships are subject to foreign law, which provides us with less favorable rights or treatment than under U.S. law. Others may gain access to our inventions, trade secrets or independently develop substantially equivalent proprietary materials, products, information, and techniques.
26
Table of Contents
We have a history of generating a large percentage of our revenue at the end of each quarterly accounting period.
Due to the manner in which many customers in our target markets allocate and spend their budgeted funds for acquisition of our products, a large percentage of our sales are booked at the end of each quarterly accounting period. Because of this timing of our sales, we may not be able to reliably predict order volumes and our quarterly revenues. A sales delay of only a few days may significantly impact our quarter-to-quarter comparisons. If our quarterly or year-end revenues fall below the expectations of securities analysts and investors, our stock price may decline. Similarly, if we are unable to ship our customer orders on time, or if extended payment terms are required, there could be a material adverse effect on revenues for a given quarter.
If our customers are unable to adequately prepare samples for our MassARRAY system, the overall market demand for our products may decline.
Before using the MassARRAY system, customers must prepare samples by following several steps that are subject to human error, including DNA isolation and DNA amplification. If DNA samples are not prepared appropriately, or the proposed assays are too complex, the MassARRAY system may not generate a reading or a correct reading. If our customers experience these difficulties, they might achieve lower throughput levels than specified for the system. If our customers are unable to generate expected levels of throughput, they might not continue to purchase our consumables, they could express their discontent with our products to others, or they could collaborate with others to jointly benefit from the use of our products. Any or all of these actions would reduce the overall market demand for our products. From time to time, we have experienced customer complaints regarding data quality and difficulty in processing more complex assays.
The sales cycles for our products are lengthy, and we may expend substantial funds and management effort with no assurance of successfully selling our products or services.
The sales cycles for our MassARRAY system products are typically lengthy. Our sales and licensing efforts require the effective demonstration of the benefits, value, and differentiation and validation of our products and services, and significant education and training of multiple personnel and departments within a customer organization. We may be required to negotiate agreements containing terms unique to each prospective customer or licensee which would lengthen the sales cycle. We may expend substantial funds and management effort with no assurance that we will sell our products or services. In addition, this lengthy sales cycle makes it more difficult for us to accurately forecast revenue in future periods and may cause revenues and operating results to vary significantly in such periods.
We may not be able to successfully adapt or maintain our products for commercial applications.
A number of potential applications of our MassARRAY technology, including research-use-only and diagnostic applications for noninvasive prenatal and other molecular testing, may require significant enhancements in our core technology or the in-licensing of intellectual property rights or technologies. In connection with developing new products and applications, we may not effectively deploy our research and development efforts in a cost-efficient manner or otherwise in a manner that leads to the successful commercialization and scale-up of such products and applications. If we are unable to complete the development, introduction, or scale-up of any product, or if any of our products or applications, such as gene expression analysis, epigenetic analysis or iPLEX multiplexing, do not achieve a significant level of market acceptance, our business, financial condition and results of operations could be seriously harmed. Achieving market acceptance will depend on many factors, including demonstrating to customers that our technology and products are cost competitive or superior to other technologies and products that are available now or that may become available in the future. We believe that our revenue growth and profitability will substantially depend on our ability to overcome significant technological and other challenges and successfully introduce our newly developed products, applications, and services into the marketplace.
27
Table of Contents
We have limited commercial production capability and experience and may encounter production problems or delays, which could result in lower revenue.
We partially assemble the MassARRAY system and partially manufacture our consumable chips and MassARRAY kits. During 2010 we plan to transition to a next-generation MassARRAY system and mass spectrometer which will require more outsourcing of component manufacturing and more internal assembly. To date, we have only produced our current products in moderate quantities. We may not be able to maintain acceptable quality standards as we transition to the next-generation MassARRAY system and mass spectrometer and ramp up production. To achieve anticipated customer demand levels, we will need to transition and scale-up our production capability and maintain adequate levels of inventory while manufacturing our products at a reasonable cost. We may not be able to produce sufficient quantities to meet market demand or manufacture our product at a reasonable cost. If we cannot achieve the required level and quality of production, we may need to abandon or reduce our internal efforts and fully outsource production or rely on licensing and other arrangements with third parties. This reliance could reduce our gross margins and expose us to the risks inherent in relying on others. We might not be able to successfully outsource our production or enter into licensing or other arrangements with these third parties, which would adversely affect our business. Also, from time to time we have experienced quality issues on some of our chips. We may not be able to maintain acceptable quality standards for production of our chips, which could harm our business and result in lower revenue.
We depend on third-party products and services and limited sources of supply to develop and manufacture our products.
We rely on outside vendors to supply certain products and the components and materials used in our products. Many of these products, components and materials are obtained from a single supplier or a limited group of suppliers and some have lead-times of several months. Our planned next-generation MassARRAY system and mass spectrometer is comprised of numerous components each provided to us from a single source and some of which have lead times of several months. Regarding other elements of our MassARRAY system, we also have sole suppliers for our chips, our pins for our nanodispenser and our liquid handling device.
Our consumables also include components provided by sole suppliers. In the event of any adverse developments with these vendors, our product supply may be interrupted and obtaining substitute components could be difficult and/or require us to re-design our products and assays which would have an adverse impact on our business. In the past, we have experienced quality problems with and delays in receiving components used to produce our consumable chips, quality issues with our chips, and also had technical difficulties with our pin-tool nanoliter dispenser device. We have also experienced software and operational difficulties with our MassARRAY system. Our reliance on outside vendors generally and a sole or a limited group of suppliers in particular involves several risks, including:
| • | the inability to obtain an adequate supply of properly functioning, required products, components, and materials due to capacity constraints, product defects, a discontinuance of a product by a supplier, or other supply constraints; |
| • | reduced control over quality and pricing of products, components, and materials; and |
| • | delays and long lead times in receiving products, components, or materials from vendors. |
If the validity of the consents from volunteers were to be challenged, we could be forced to stop using some of our resources, which would hinder our gene discovery outlicensing efforts and our diagnostic product development efforts.
We have attempted to ensure that all clinical data and genetic and other biological samples that we receive from our subsidiaries and our clinical collaborators have been collected from volunteers who have provided our collaborators or us with appropriate consents for the data and samples provided for purposes which extend to include commercial diagnostic product development activities. We have attempted to ensure that data and samples that have been collected by our clinical collaborators are provided to us on an anonymous basis. We have also attempted to ensure that the volunteers from whom our data and samples are collected do not retain or
28
Table of Contents
have conferred on them any proprietary or commercial rights to the data or any discoveries derived from them. Our clinical collaborators are based in a number of different countries, and, to a large extent, we rely upon our clinical collaborators for appropriate compliance with the voluntary consents provided and with local law and regulation. That our data and samples come from and are collected by entities based in different countries results in complex legal questions regarding the adequacy of consents and the status of genetic material under a large number of different legal systems. The consents obtained in any particular country could be challenged in the future, and those consents could prove invalid, unlawful or otherwise inadequate for our purposes. Any findings against us, or our clinical collaborators, could deny us access to or force us to stop using some of our clinical or genetic resources, which would hinder our diagnostic product development efforts. We could become involved in legal challenges, which could consume a substantial proportion of our management and financial resources.
If we cannot obtain licenses to patented SNPs and genes relevant to our diagnostic areas of interest, we could be prevented from obtaining significant revenue or becoming profitable.
The U.S. Patent and Trademark Office has issued and continues to issue patents claiming single SNP and gene discoveries and their related associations and functions. If certain SNPs and genes are patented, we will need to obtain rights to those SNPs and genes to develop, use, and sell related assays and other types of products or services utilizing such SNPs and genes. Required licenses may not be available on commercially acceptable terms. If we were to fail to obtain licenses to certain patented SNPs and genes, we might never achieve significant revenue from our diagnostic product development.
If the medical relevance of SNPs is not demonstrated or is not recognized by others, we may have less demand for our products and services and may have less opportunity to enter into diagnostic product development and commercialization collaborations with others.
Some of the products we hope to develop involve new and unproven approaches or involve applications in markets that we are only beginning to explore. They are based on the assumption that information about genes and SNPs may help scientists better understand conditions or complex disease processes. Scientists generally have a limited understanding of the role of genes and SNPs in diseases, and few products based on gene discoveries have been developed. We cannot be certain that genetic information will play a key role in the development of diagnostics or other products in the future, or that any genetic-based findings would be accepted by diagnostic, pharmaceutical, or biotechnology companies or by any other potential market or industry segment. If we or our customers or collaborators are unable to generate valuable information that can be used to develop diagnostics or other products, the demand for our products, applications, and services will be reduced and our business will be harmed.
We may not be able to form and maintain the collaborative relationships or the rights to third-party intellectual property and technologies that our business strategy requires and such relationships may lead to disputes over technology rights or product revenue, royalties, or other payments.
We form research collaborations and licensing arrangements with collaborators to operate our business successfully. To succeed, we will have to maintain our existing relationships and establish additional collaborations and licensing arrangements. Our current strategy includes pursuing partnering opportunities with larger companies interested in or involved in the development of pharmaceutical and diagnostic products. Our strategy also includes obtaining licenses to third-party intellectual property rights and technologies, such as our exclusive license to noninvasive prenatal analysis rights that we acquired from Isis (United States Patent No. 6,258,540 and foreign equivalents) and other rights we have acquired for the use of fetal nucleic acids obtained from maternal urine for noninvasive prenatal diagnostics and to pursue development of a molecular LDT for assessment of risk for developing age-related macular degeneration, to potentially expand our product portfolio and generate additional sources of revenue. If we do not achieve certain milestones in a timely manner, particularly with respect to our planned test for Trisomy 21, we risk losing our exclusive license rights from Isis and may also lose rights under our other licenses if we do not adequately pursue commercialization in the manner specified in those licenses. Disputes may also arise in connection with these collaborations and licensing arrangements, which may result in liability to us or may result in the loss of acquired technology that may
29
Table of Contents
adversely affect our business. For example, as described under Item 3 of this report, we are currently engaged in litigation with Xenomics regarding our rights under the license agreement and are being sued for substantial damages.
We cannot be sure that we will be able to establish any additional research collaborations, licensing arrangements, or other partnerships necessary to develop and commercialize products or that we can do so on terms favorable to us. If we are unable to establish these collaborations or licensing arrangements, we may not be able to successfully develop any diagnostic or other products or applications and generate any milestone, royalty, or other revenue from sales of these products or applications. If our collaborations or licensing arrangements are not successful or we are not able to manage multiple collaborations successfully, our programs will suffer and we may never generate any revenue from sales of products based on licensed rights or technologies or under these collaborative or licensing arrangements. If we increase the number of collaborations or licensing agreements, it will become more difficult to manage the various relationships successfully and the potential for conflicts among the collaborators and licensees or licensors will increase. Conflicts with our collaborators, licensees or licensors, or other factors may lead to disputes over technology or intellectual property rights or product revenue, royalties, or other payments, which may adversely affect our business.
In addition, our government grants provide the government certain license rights to inventions resulting from funded work. Our business could be harmed if the government exercises those rights.
If we do not succeed in obtaining development and marketing rights for products developed in collaboration with others, our revenue and profitability prospects could be substantially harmed.
Our business strategy includes, in part, the development of noninvasive prenatal diagnostic and other products in collaboration with others, or utilizing the technology of others, and we intend to obtain commercialization or royalty rights to those products or technologies. If we are unable to obtain such rights, or are unable to do so on favorable financial terms, our revenue and profitability prospects could be substantially harmed. To date, we have initiated limited activities towards commercializing products developed in collaboration with, or utilizing the technology of, others. Even if we obtain commercialization rights, commercialization of products may require resources that we do not currently possess and may not be able to develop or obtain, or commercialization may be financially unattractive based upon the revenue-sharing terms offered by potential licensors or provided for in the relevant agreement.
Ethical, privacy, or other concerns about the use of genetic information could reduce demand for our products and services.
Genetic testing, including gender determination and Trisomy 21 testing, has raised ethical issues regarding privacy and the appropriate uses of the resulting information. For these reasons, governmental authorities may limit or otherwise regulate the use of genetic testing or prohibit testing for genetic predisposition to certain conditions, particularly for those that have no known cure. Such concerns may lead individuals to refuse to use genetics tests even if permitted. Any of these scenarios could reduce the potential markets for our products and services, which would seriously harm our business, financial condition, and results of operations.
If we breach any of the terms of our license or supply agreements, or these agreements are otherwise terminated or modified, the termination or modification of such agreements could result in our loss of access to critical components and could delay or suspend our commercialization efforts.
We have sourced or licensed components of our technology from other parties. Our failure to maintain continued supply of such components, particularly in the case of sole suppliers, or the right to use these components would seriously harm our business, financial condition, and results of operations. As a result, in the event that demand for our products declines or does not meet our forecasts, we could have excess inventory or increased expenses or our margins could decrease which could have an adverse impact on our financial condition
30
Table of Contents
and business. In the event of any adverse developments with these vendors, our product supply may be interrupted, which would have an adverse impact on our business. Changes to or termination of our agreements or inability to renew our agreements with these parties or enter into new agreements with other suppliers could result in the loss of access to these aspects of our technology or other intellectual property rights or technologies that we may acquire from time to time and could impair, delay, or suspend our commercialization efforts. While we negotiate for agreement periods or notice of termination periods that provide us reasonable periods of time to secure alternative supplies, and require that such agreements may not be terminated without advance notice arbitrarily or without good reason, such as uncured breach or insolvency, these negotiations are often unsuccessful or such provisions may not provide us with adequate time to secure alternative supplies, provide us with access to alternative technologies on commercially acceptable terms, or otherwise provide us with adequate protection.
We may not successfully integrate acquired businesses and may not successfully complete the acquisition of businesses or technologies that we desire to acquire.
We may acquire additional businesses or technologies, or enter into other strategic transactions. For example, in November 2008, we completed the acquisition of the Center for Molecular Medicine, LLC, a CLIA certified laboratory facility and in February 2009 we completed the acquisition of substantially all of the assets of SensiGen, LLC.
Managing these and future acquisitions entails numerous operational and financial risks, including:
| • | the inability to retain key employees of any acquired businesses or hire enough qualified personnel to staff any new or expanded operations; |
| • | the impairment of relationships with key customers of acquired businesses due to changes in management and ownership of the acquired businesses; |
| • | the inability to sublease on financially acceptable terms excess leased space or terminate lease obligations of acquired businesses that are not necessary or useful for the operation of our business; |
| • | the exposure to federal, state, local and foreign tax liabilities in connection with any acquisition or the integration of any acquired businesses; |
| • | the exposure to unknown liabilities or disputes with the former stakeholders or management or employees of acquired businesses; |
| • | higher than expected acquisition and integration expenses that would cause our quarterly and annual operating results to fluctuate; |
| • | increased amortization expenses if an acquisition results in significant intangible assets; |
| • | combining the operations and personnel of acquired businesses with our own, which would be difficult and costly; |
| • | disputes over rights to acquired technologies or with licensors or licensees of those technologies; and |
| • | integrating or completing the development and application of any acquired technologies, which would disrupt our business and divert management’s time and attention. |
We may also attempt to acquire businesses or technologies or attempt to enter into strategic transactions that we are unable to complete. For example, in January 2009, we launched an exchange offer to acquire EXACT Sciences Corporation, but were not able to complete the transaction prior to EXACT Sciences selling and licensing a substantial portion of its assets and intellectual property to a third party. If we are unable to complete such transactions, we may expend substantial resources and ultimately not successfully complete the transaction. Such transactions may also distract management and result in other adverse effects on our business and operations. These transactions may also involve the issuance of shares of our capital stock, which may result in dilution to our stockholders.
31
Table of Contents
We may not be able to successfully compete in the biotechnology and diagnostic industries.
The biotechnology and diagnostic industries are highly competitive. We expect to compete with a broad range of companies in the United States and other countries that are engaged in the development and production of products, applications, services, and strategies to analyze genetic information and strategies to develop and commercialize diagnostic, noninvasive prenatal diagnostic, and other products for customers in the clinical research and clinical marker validation and molecular medicine fields as well as diagnostic service laboratories, animal testing and food safety labs, and customers in other markets. They include:
| • | biotechnology, pharmaceutical, diagnostic, chemical, and other companies; |
| • | academic and scientific institutions; |
| • | governmental agencies; and |
| • | public and private research organizations. |
Many of our competitors have much greater financial, technical, research, marketing, sales, distribution, service, and other resources than we do. Our competitors may offer broader product lines and services and have greater name recognition than we do. Several companies are currently making or developing products that compete with our products. Our competitors may develop or market technologies or products that are more effective or commercially attractive than our current or future products, or that may render our technologies or products obsolete. Our announcement in 2009 of the delay in the expected launch of our Trisomy 21 test, as well as the announcement of the independent investigation, the investigations by the SEC, NASDAQ, the Office of the U.S. Attorney for the Southern District of California and the FBI, and pending litigation may adversely affect our competitive position and the market acceptance of any tests that we may commercialize and may affect our ability to maintain and recruit key personnel.
We may potentially compete with our customers, which may adversely affect our business.
We have sold MassARRAY systems worldwide to pharmaceutical and biotechnology companies, academic research centers, and government laboratories. Some of our customers use our DNA analysis products to perform contract research services, or to perform genetics studies on their own disease populations for potential diagnostic and drug target identification in the same or similar manner as we have done. Although there are many potential contract research services opportunities and disease areas and diagnostic applications, our customers may seek service work or develop diagnostic assays or may target diseases areas that may overlap with those that we have chosen to pursue. In such cases we may potentially compete against our customers. Competition from our customers may adversely affect our services business or our ability to successfully commercialize diagnostic products.
If we cannot attract and retain highly-skilled personnel, our growth might not proceed as rapidly as we intend and our business may be adversely affected.
The success of our business will depend on our ability to identify, attract, hire, train, retain, maintain, and motivate highly skilled personnel, particularly sales, scientific, medical, and technical personnel, for our future success. Competition for highly skilled personnel is intense, and we might not succeed in attracting and retaining these employees. If we cannot attract and retain the personnel we require, we would not be able to expand our business as rapidly as we intend. Our announcement in 2009 of the delay in the expected launch of our Trisomy 21 test and subsequent announcements may have affected our ability to maintain and recruit key personnel. Following the completion of the investigation by the special committee of our board of directors in 2009, a number of our senior officers and members of the research and development program for our Trisomy 21 test left our company. We asked two of our directors, Harry F. Hixson, Jr. and Ronald M. Lindsay, to serve our company as chief executive officer and interim senior vice president of research and development, respectively, and appointed Paul V. Maier as our interim chief financial officer effective November 10, 2009. When we seek to hire personnel to fill these positions on a permanent basis, we can give no assurance that we will be able to hire
32
Table of Contents
qualified replacements for the positions that we need to fill, and there may be significant costs associated with the recruiting, hiring and retention of officers and employees for the open positions. The announcement of the results of the investigation and the departure of the officers and employees has had negative effect on employee morale. If we lose additional key employees, scientists, physician collaborators or if our management team is not able to effectively manage us through these events, our business, financial condition, and results of operations may be adversely affected. We do not carry “key person” insurance covering any of our officers or other employees.
If we do not effectively manage our business as it evolves, it could affect our ability to pursue opportunities and expand our business.
Evolution in our business, particularly our attempted transition to developing and commercializing molecular diagnostic tests, has placed and may continue to place a significant strain on our personnel, facilities, management systems, information technology infrastructure, disclosure controls, internal controls and resources. In 2009, we began implementing the remedial measures recommended by the special committee of our board of directors following its independent investigation, including:
| • | new disclosure controls and procedures, changes in our organizational and reporting structure, enhanced training in ethics and scientific processes for our employees; |
| • | new procedures for the conduct of research and development and clinical studies; |
| • | increased roles and responsibilities for independent third parties; |
| • | new procedures for the storage and management of samples for testing; and |
| • | creation of a science committee of our board of directors to oversee our research and development strategy and activities. |
In connection with entering into a stipulation of settlement with respect to the class action securities lawsuits related to alleged violations of federal securities laws, we also agreed to adopt or continue our implementation of changes and additions to certain corporate governance policies, protocols and practices. While we feel that the remedial measures recommended by the special committee and the other changes we have implemented have made our disclosure controls and procedures more effective, any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives and no evaluation of controls and procedures can provide absolute assurance that all control issues have been detected. We will need to continue to improve our operational and financial systems and managerial controls and procedures and train and manage our workforce and transition our business to execute on the commercialization of molecular diagnostic tests. If we fail to effectively manage the evolution of our business and the transition to also being a provider of diagnostic products, including the effective implementation these remedial measures and additional changes to our corporate governance policies, protocols and practices, or fail to take other necessary action to maintain close coordination among our various departments, our ability to execute on our business plan, rebuild credibility, pursue business opportunities, expand our business, and sell our products and applications in new markets may be adversely affected.
Certain of our molecular diagnostic tests may not be eligible for reimbursement by payors which may limit the demand for these tests by physicians and their patients. We may incur additional financial risk related to collections and reimbursement in connection with the commercialization of our molecular diagnostic tests.
In September 2009, we commercially launched our carrier screening LDT for cystic fibrosis, and in early 2010 we launched our noninvasive Rhesus D genotyping and Fetalxy sex determination LDTs, and we intend to continue launching additional molecular diagnostic tests in the future. Because these tests have only recently been launched, demand for and reimbursement by payors of these tests, in particular the cystic fibrosis carrier screening and Rhesus D genotyping tests, is uncertain. The Fetalxy sex determination test is paid for directly by the patient and is not eligible for reimbursement by payors because it is not a medically necessary test. Because
33
Table of Contents
certain of the molecular diagnostic tests we have launched or intend to launch may not be medically necessary or may otherwise not be subject to reimbursement by payors, it is difficult to know how much demand there will be for such tests by physicians and their patients. We generally bill third-party payors for our tests and pursue case-by-case reimbursement where policies are not in place for a particular test but we have very limited experience in billing and pursuing reimbursement and payment for molecular diagnostic tests. As a result of this lack of experience and uncertainty with respect to reimbursement, we may also face an increased risk in our collection efforts, including potential write-offs of doubtful accounts and long collection cycles for accounts receivable related to our diagnostic tests, which could adversely affect our business, results of operations and financial condition.
We must be in compliance with security and privacy regulations under the Health Insurance Portability and Accountability Act of 1996, or HIPAA, and other state regulations, which may increase our operational costs.
The HIPAA privacy and security regulations establish comprehensive federal standards with respect to the uses and disclosures of protected health information, or PHI, by health plans and healthcare providers, in addition to setting standards to protect the confidentiality, integrity and availability of electronic PHI. The regulations establish a complex regulatory framework on a variety of subjects, including:
| • | the circumstances under which uses and disclosures of PHI are permitted or required without a specific authorization by the patient, including but not limited to treatment purposes, to obtain payments for services and healthcare operations activities; |
| • | a patient’s rights to access, amend and receive an accounting of certain disclosures of PHI; |
| • | the content of notices of privacy practices for PHI; and |
| • | administrative, technical and physical safeguards required of entities that use or receive PHI electronically. |
In September 2009, we commercially launched our carrier screening test for cystic fibrosis, and intend to continue launching additional diagnostic tests in the future. As we launch additional commercial diagnostic tests, we must continue to implement policies and procedures related to compliance with the HIPAA privacy and security regulations, as required by law, which may increase our operational costs. Furthermore, the privacy and security regulations provide for significant fines and other penalties for wrongful use or disclosure of PHI, including potential civil and criminal fines and penalties. Although the HIPAA statute and regulations do not expressly provide for a private right of damages, we also could incur damages under state laws to private parties for the wrongful use or disclosure of confidential health information or other private personal information.
We are subject to risks associated with our foreign operations.
We expect that a significant portion of our sales will continue to be made outside the United States. Approximately 53% of our sales were made outside of the United States during the year ended December 31, 2009, compared to 50% for the year ended December 31, 2008. A successful international effort will require us to develop relationships with international customers and collaborators, including distributors. We may not be able to identify, attract, retain, or maintain suitable international customers or collaborators. Expansion into international markets will require us to establish and grow foreign operations, hire additional personnel to run these operations, and maintain good relations with our foreign customers and collaborators or distributors. International operations including many of the same risks to our business that affect our domestic operations, but also involve a number of risks not typically present in domestic operations, including:
| • | currency fluctuation risks; |
| • | changes in regulatory requirements; |
| • | costs and risks of deploying systems in foreign countries; |
34
Table of Contents
| • | licenses, tariffs, and other trade barriers; |
| • | political and economic instability and possible country-based boycotts; |
| • | difficulties in staffing and managing foreign operations; |
| • | potentially adverse tax consequences; |
| • | the burden of complying with a wide variety of complex foreign laws and treaties; and |
| • | different rules, regulations, and policies governing intellectual property protection and enforcement. |
Our international operations are also subject to the risks associated with the imposition of legislation and regulations relating to the import or export of high technology products. We cannot predict whether tariffs or restrictions upon the importation or exportation of our products will be implemented by the United States or other countries.
If our production and laboratory facilities are damaged, our business would be seriously harmed.
Our only production facility for genetic analysis products is located in San Diego, California, where we also have laboratories. We also have laboratory facilities in Grand Rapids, Michigan. Damage to our facilities due to war, fire, natural disaster, power loss, communications failure, terrorism, unauthorized entry, or other events could prevent us from conducting our business for an indefinite period, could result in a loss of important data or cause us to cease development and production of our products. We cannot be certain that our limited insurance to protect against business interruption would be adequate or would continue to be available to us on commercially reasonable terms, or at all.
Responding to claims relating to improper handling, storage or disposal of hazardous chemicals, and radioactive and biological materials which we use could be time consuming and costly.
We use controlled hazardous and radioactive materials in the conduct of our business, as well as biological materials that have the potential to transmit disease. The risk of accidental contamination or injury from these materials cannot be completely eliminated. If an accident with these substances occurs, we could be liable for any damages that result, which could seriously harm our business. Additionally, an accident could damage our research and manufacturing facilities and operations, resulting in delays and increased costs. Such damage and any expense resulting from delays, disruptions, or any claims may not be covered by our insurance policies.
We may not have adequate insurance if we become subject to product liability or other claims.
Our business exposes us to potential product liability and other types of claims and our exposure will increase as we and our partners and collaborators prepare to commercialize research-use-only or other types of molecular tests, including LDTs and diagnostics for prenatal and other applications. We have product and general liability insurance that covers us against specific product liability and other claims up to an annual aggregate limit of $20.0 million and $2.0 million, respectively. Any claim in excess of our insurance coverage would have to be paid out of our cash reserves, which would have a detrimental effect on our financial condition. It is difficult to determine whether we have obtained sufficient insurance to cover potential claims. Also, we cannot assure you that we can or will maintain our insurance policies on commercially acceptable terms, or at all.
The uncertainty of the current economic and political conditions could harm our revenues and operating results.
Current domestic and global economic conditions are uncertain and have continued to be volatile over the past year. The recent turmoil in the economic environment in many parts of the world may continue to put pressure on global economic conditions. Our revenues and operating results may be affected by uncertain or
35
Table of Contents
changing economic and market conditions, including the recent crisis in the credit markets and financial services industry and general conditions in the global capital markets. If global economic and market conditions, or economic conditions in the United States or other key markets, remain uncertain or persist, spread, or deteriorate further, we may experience material impacts on our business, operating results, and financial condition.
Our stock price has been and may continue to be volatile, and your investment could suffer a decline in value.
The trading price of our common stock has been volatile and could be subject to wide fluctuations in price in response to various factors, many of which are beyond our control, including but not limited to:
| • | our ability to raise additional capital and continue as a going concern; |
| • | actual or anticipated variations in quarterly and annual operating results; |
| • | announcements of technological innovations, research and development progress or setbacks by us or our competitors, and product launches; |
| • | our success in entering into, and the success in performing under, licensing and product development and commercialization agreements with others; |
| • | our success in and the expenses associated with researching, developing and commercializing diagnostic products, alone or in collaboration with our partners and obtaining any required regulatory approval for those products and services; |
| • | the status of litigation against us and certain of our former executive officers and directors; |
| • | the dilution from any issuance of securities in connection with the settlement of litigation; |
| • | our ability to successfully implement the remedial measures recommended by the special committee following our independent investigation and the effectiveness of those measures; |
| • | the status, duration, scope and outcome of the SEC investigation that has been initiated following our April 2009 announcement regarding our Trisomy 21 test and the investigations by NASDAQ, the Office of the U.S. Attorney for the Southern District of California and the FBI following our September 28, 2009 announcement regarding the completion of our independent investigation; |
| • | securities analysts’ earnings projections or securities analysts’ recommendations; and |
| • | general market conditions, including the recent crisis in global financial markets. |
The stock market in general, and The NASDAQ Global Market and the market for life sciences companies in particular, have experienced extreme price and volume fluctuations that may have been unrelated or disproportionate to the operating performance of the listed companies. There have been dramatic fluctuations in the market prices of securities of biotechnology companies. These price fluctuations may be rapid and severe and may leave investors little time to react. Broad market and industry factors may seriously harm the market price of our common stock, regardless of our operating performance. Sharp drops in the market price of our common stock expose us to securities class-action litigation. We have subsequently been named as defendants in various lawsuits alleging violations of federal securities laws related to alleged false and misleading statements regarding our Trisomy 21 test under development and are the subject of investigations by the SEC, NASDAQ, the Office of the U.S. Attorney for the Southern District of California and the FBI. Such litigation and investigations could result in substantial expenses and other liabilities, substantial dilution to our stockholders and a diversion of management’s attention and resources, which would seriously harm our business, financial condition, and results of operations.
Item 1B. UNRESOLVED STAFF COMMENTS
None.
36
Table of Contents
We are headquartered in San Diego, California, with wholly-owned subsidiaries located in Hamburg, Germany, Cambridge, England, Hong Kong, Grand Rapids, Michigan and Tokyo, Japan. We also have offices in Queensland, Australia and Beijing, China. Collectively, we lease approximately 135,000 square feet under leases that expire at various dates through September 2015, each of which contains laboratory, office, manufacturing, or storage facilities.
The San Diego site is our company headquarters and houses our selling, general, and administrative offices, research and development facilities and manufacturing operations. The site in Hamburg, Germany, is used to support sales and distribution in Europe. The site in Hong Kong is used for sales and support activities performed in Asia. The site in Cambridge, England is used for sales and support activities performed in Europe. The site in Grand Rapids, Michigan, houses our CLIA laboratory, Sequenom Center for Molecular Medicine, LLC. The site in Tokyo, Japan, is used for sales and support activities performed in Japan. We believe our facilities are adequate for our current needs.
In November 2001, we and certain of our current or former officers and directors were named as defendants in a class action shareholder complaint filed by Collegeware USA in the U.S. District Court for the Southern District of New York (now captioned In re Sequenom, Inc. IPO Securities Litigation) Case No. 01-CV-10831. In the complaint, the plaintiffs allege that our underwriters, certain of our officers and directors and we violated the federal securities laws because our registration statement and prospectus contained untrue statements of material fact or omitted material facts regarding the compensation to be received by and the stock allocation practices of the underwriters. The plaintiffs seek unspecified monetary damages and other relief. Similar complaints were filed in the same District Court against hundreds of other public companies that conducted initial public offerings of their common stock in the late 1990s and 2000 (the IPO Cases).
In October 2002, our officers and directors were dismissed without prejudice pursuant to a stipulated dismissal and tolling agreement with the plaintiffs. In February 2003, the District Court dismissed the claim against us brought under Section 10(b) of the Exchange Act, without giving the plaintiffs leave to amend the complaint with respect to that claim. The District Court declined to dismiss the claim against us brought under Section 11 of the Securities Act of 1933, as amended (the Securities Act).
In September 2003, pursuant to the authorization of a special litigation committee of our board of directors, we approved in principle a settlement offer by the plaintiffs. In September 2004, we entered into a settlement agreement with the plaintiffs. In February 2005, the District Court issued a decision certifying a class action for settlement purposes and granting preliminary approval of the settlement subject to modification of certain bar orders contemplated by the settlement. In August 2005, the District Court reaffirmed class certification and preliminary approval of the modified settlement. In December 2006, the U.S. Court of Appeals for the Second Circuit vacated the District Court’s decision certifying as class actions the six lawsuits designated as “focus cases.” Thereafter the District Court ordered a stay of all proceedings in all of the lawsuits pending the outcome of plaintiffs’ petition to the Second Circuit for rehearing en banc. In April 2007, the Second Circuit denied plaintiffs’ rehearing petition, but clarified that the plaintiffs may seek to certify a more limited class in the District Court. Accordingly, the settlement as originally negotiated was terminated pursuant to stipulation and will not receive final approval.
In February 2009, liaison counsel for plaintiffs informed the District Court that a new settlement of all IPO Cases had been agreed to in principle, subject to formal approval by the parties and preliminary and final approval by the District Court. In April 2009, the parties submitted a tentative settlement agreement to the District Court and moved for preliminary approval thereof. In June 2009, the District Court granted preliminary approval of the tentative settlement and ordered that notice of the settlement be published and mailed to class members. In September2009, the District Court held a final fairness hearing. On October 6, 2009, the District
37
Table of Contents
Court certified the settlement class in each IPO Case and granted final approval to the settlement. On or about October 23, 2009, three shareholders filed a Petition for Permission to Appeal Class Certification Order, asserting that the District Court’s certification of the settlement classes violates the Second Circuit’s earlier class certification decisions in the IPO Cases. Beginning on October 29, 2009, a number of shareholders also filed direct appeals, objecting to final approval of the settlement. Similar petitions and direct appeals may be filed by other shareholders. If the settlement is affirmed on appeal, the settlement will become effective and will result in the dismissal of all claims against us and our officers and directors with prejudice, and our pro rata share of the settlement fund will be fully funded by insurance.
In October 2008, we filed a patent infringement suit against Ibis Biosciences, Inc. (IBIS), formerly a subsidiary of Isis Pharmaceuticals, Inc. The complaint was served on the defendant in February 2009. IBIS has been acquired by Abbott Molecular. The lawsuit was filed in the U.S. District Court for the District of Delaware. The lawsuit alleged that the sale or offer for sale of the IBIS T5000 Biosensor System and related technology infringes three U.S. patents: 6,300,076, 6,500,621 and 7,419,787. Defendant has filed an answer and counterclaims against us seeking declaratory judgments that the patents are not infringed and are invalid and/or unenforceable. We sought a permanent injunction enjoining the defendant from further infringement and monetary damages, including enhanced damages pursuant to 35 U.S.C. § 284, costs, attorneys’ fees and other relief as the court deems just and proper. On October 22, 2009, we and IBIS entered into a non-exclusive license and settlement agreement and the court dismissed with prejudice all claims and counterclaims in the lawsuit. Pursuant to the terms of the agreement, we have agreed to grant IBIS and its affiliates a non-exclusive license under the three mass spectrometry-based patents involved in the lawsuit and certain pending mass-spectrometry-based applications and foreign counterparts to manufacture, use, practice, sell, offer to sell, and import products and methods protected by the licensed patents. As part of the agreement, we have also agreed to dismiss the litigation with prejudice and have granted IBIS, and its affiliates, immunity from suit for patent infringement for past, present or future damages related to the IBIS T5000 Biosensor System and T6000 System. Pursuant to the agreement, IBIS paid us $1.0 million.
In April 2009, we announced that the expected launch of our test for Trisomy 21 (Down syndrome) had been delayed and that we were no longer relying on our previously announced test data and results for that test. We also announced that our Board of Directors had formed a special committee of independent directors to oversee an independent investigation of activity related to the test data and results and that the committee had engaged independent counsel to assist the committee in the conduct of the investigation. In September 2009, we announced that the committee’s investigation had been completed. Based on the committee’s work and recommendations, the independent directors concluded that as a result of our attempted transition from researching potential molecular diagnostic tests to developing and commercializing those tests, we failed to put in place adequate protocols and controls for the conduct of studies in the Trisomy 21 program at our company. Certain of our employees also failed to provide adequate supervision. In the absence of such protocols, controls and supervision, the test data and results in our Trisomy 21 program included inadequately substantiated claims, inconsistencies and errors. Due to deficiencies in our disclosure controls and procedures, in a number of instances such test data and results were reported to the public in our press releases and other public statements. At the recommendation of the special committee, our Board of Directors began implementing a number of remedial measures including:
| • | new disclosure controls and procedures; |
| • | changes in our organizational and reporting structure; |
| • | enhanced training in ethics and scientific processes for our employees; |
| • | new procedures for the conduct of research and development and clinical studies, including increased roles and responsibilities for independent third parties; |
| • | new procedures for the storage and management of samples for testing; and |
| • | creation of a science committee of our board of directors to oversee our research and development strategy and activities. |
38
Table of Contents
We also terminated the employment of our president and chief executive officer, Harry Stylli, Ph.D., and our senior vice president of research and development, Elizabeth Dragon, Ph.D., effective immediately. In connection with the termination of Dr. Stylli’s employment, Dr. Stylli resigned as a director. We also obtained the resignation of our chief financial officer, Paul Hawran. We also terminated the employment of three other employees and obtained the resignation of one other officer. While each of these officers and employees denied wrongdoing, the committee’s investigation raised serious concerns, resulting in a loss of confidence by the independent directors in the personnel involved.
Our board of directors appointed our chairman of the board, Harry F. Hixson, Jr., Ph.D., to serve as our chief executive officer. Our board of directors appointed Ronald M. Lindsay, Ph.D., one of our directors, to serve as our interim senior vice president of research and development. Our board of directors appointed Paul V. Maier as our interim chief financial officer effective November 10, 2009. Our controller, Justin J. File, served as our principal financial and accounting officer until the effective date of Mr. Maier’s appointment as interim chief financial officer.
Following our April 2009 announcement, several complaints were filed in the U.S. District Court for the Southern District of California against us and certain of our current and former officers and directors on behalf of certain purchasers of our common stock. The complaints include claims asserted under Sections 10 and 20(a) of the Exchange Act and Sections 11 and 12(a)(2) of the Securities Act and have been brought as shareholder class actions. In general, the complaints allege that we and certain of our officers and directors violated federal securities laws by making materially false and misleading statements regarding our Trisomy 21 test under development, thereby artificially inflating the price of our common stock. The plaintiffs seek unspecified monetary damages and other relief. On September 1, 2009, the complaints were consolidated under the caption In re Sequenom, Inc. Securities Litigation, S.D. Cal. Case No. 09-CV-0921 LAB (WMc) and a lead plaintiff was appointed. On December 24, 2009, we entered into a stipulation of settlement with the lead plaintiff on behalf of the plaintiffs’ class which, if approved by the District Court, will resolve this action. Pursuant to the terms of the stipulation, we have agreed to pay $14 million, which will be funded by insurance proceeds. We have also agreed to issue to the plaintiffs’ class a number of shares of our common stock equal to 9.95% of our total shares outstanding at the time of determination, subject to certain limitations. We have also agreed to adopt or continue our implementation of changes and additions to certain corporate governance policies, protocols and practices. The court preliminarily approved the settlement on January 26, 2010. The court has scheduled a final settlement approval hearing on May 3, 2010.
In May 2009, a shareholder derivative complaint was filed in the Superior Court of California for the County of San Diego against certain of our current and former directors and officers. Thereafter, a number of similar actions, also styled as shareholder derivative suits, were filed in state court and have been consolidated in a single court. On July 1, 2009, the first of three shareholder derivative suits were filed in the U.S. District Court for the Southern District of California. The federal shareholder derivative actions have been consolidated before a single court under the caption In re Sequenom, Inc. Derivative Litigation, S.D. Cal. Case No. 09-CV-1341 LAB (WMc) and plaintiffs filed a single consolidated complaint. A separate federal derivative compliant, Ries, et al. v. Stylii, et al, case no. 09-CV-2517 LAB (WMc), was filed thereafter and it has been coordinated with the consolidated federal derivative action. The state and federal shareholder derivative actions are hereinafter collectively referred to as the “Derivative Actions.” The complaints in the Derivative Actions allege breaches of fiduciary duties by the defendants and other violations of law. In general, the complaints allege that our directors and certain of our officers caused or allowed for the dissemination of materially false and misleading statements regarding our Trisomy 21 test under development, thereby artificially inflating the price of our common stock. The plaintiffs are seeking unspecified monetary damages and other relief, including reforms and improvements to our corporate governance and internal procedures. We have not yet responded to the Derivative Actions, but will vigorously defend against the claims advanced.
In June 2009, we received written notification that the Enforcement staff of the SEC has initiated an investigation following our April 2009 announcement regarding our Trisomy 21 test under development. Following our September 2009 announcement, members of the special committee and its independent counsel
39
Table of Contents
met with the SEC staff in connection with its investigation. As part of this investigation, the SEC staff has also required us to produce information with respect to our announcement relating to our offer to acquire EXACT Sciences, Inc. in January 2009. We intend to continue to cooperate fully with the SEC in this matter.
In July 2009, an attorney who claims to represent certain stockholders who were issued an aggregate of 71,836 shares of our common stock when we acquired the assets of SensiGen in February 2009 sent us a letter claiming that we had breached our representations and warranties made in the asset purchase agreement and alleging that his clients had suffered approximately $1.3 million in damages as a result. On December 23, 2009, we entered into a stipulation of settlement with these stockholders. Pursuant to the terms of the settlement, in consideration of the stockholders’ release of claims, we issued an aggregate of 367,547 shares of our common stock to such stockholders.
Following our September 2009 announcement, representatives of the Office of the U.S. Attorney for the Southern District of California contacted us to inquire about the announcement. We have met with representatives of the U.S. Attorney and the Federal Bureau of Investigation (FBI) in connection with their investigations. We intend to continue to cooperate fully with the U.S. Attorney and the FBI in this matter.
Following our September 2009 announcement, representatives of NASDAQ also contacted us to inquire about the announcement. We have met with representatives from NASDAQ in connection with their investigation and we intend to continue to cooperate fully with NASDAQ in this matter in the event that NASDAQ has any further inquiries.
On October 28, 2009, plaintiff Xenomics, Inc. filed a complaint in the Supreme Court of the of the State of New York naming us as the defendant. In the complaint, the plaintiff alleges that due to materially false and misleading statements regarding our Trisomy 21 test under development, we have breached the license agreement entered into by the parties on October 29, 2008, which provides us with exclusively licensed patent rights for the use of fetal nucleic acids obtained from maternal urine, and that the plaintiff has suffered damages as a result. The plaintiff is seeking equitable relief and $300 million in damages. On December 15, 2009, we removed the case to the U.S. District Court for the Southern District of New York. On February 22, 2010, we filed a motion in the federal district court to, among other things, dismiss or stay the action in light of the fact that the License Agreement between the parties specifically provides that if Xenomics seeks to resolve a dispute arising under the agreement, it must do so by commencing an arbitration in San Diego. The district court has directed that the motion be fully briefed by March 26, 2010. Regardless of the forum in which the dispute is ultimately heard, we intend to vigorously defend against the claims advanced.
Legal expenses aggregating approximately $0.7 million relating to our insured legal expenses have been submitted directly to third party insurance carriers for reimbursement, but for which we are ultimately liable. Therefore, as of December 31, 2009, we have not accrued for these expenses in the accompanying consolidated financial statements. Should these expenses not be paid by our third party insurance carriers, we will be required to incur these expenses directly.
In addition, from time to time, we may be involved in litigation relating to claims arising out of our operations in the normal course of business.
Because of the uncertainties related to the incurrence, amount and range of loss on any pending litigation, investigation, inquiry or claim, management is currently unable to predict the ultimate outcome of any litigation, investigation, inquiry or claim, determine whether a liability has been incurred or make an estimate of the reasonably possible liability that could result from an unfavorable outcome. An adverse ruling or outcome in any lawsuit involving us could materially affect our business, liquidity, consolidated financial position or results of operations ability to sell one or more of our products or could result in additional competition. In view of the unpredictable nature of such matters, we cannot provide any assurances regarding the outcome of any litigation, investigation, inquiry or claim to which we are a party or the impact on us of an adverse ruling of such matters.
Item 4. (REMOVED AND RESERVED)
40
Table of Contents
| Item 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
(a) Our common stock is traded on The NASDAQ Global Market under the symbol “SQNM.” The following tables set forth the high and low sales prices for the Company’s common stock as reported on The NASDAQ Global Market for the periods indicated.
| High | Low | |||||
| Year Ended December 31, 2009: |
||||||
| Fourth Quarter |
$ | 4.49 | $ | 2.76 | ||
| Third Quarter |
6.61 | 3.23 | ||||
| Second Quarter |
16.27 | 2.93 | ||||
| First Quarter |
25.54 | 12.68 | ||||
| Year Ended December 31, 2008: |
||||||
| Fourth Quarter |
$ | 26.72 | $ | 12.71 | ||
| Third Quarter |
27.76 | 16.28 | ||||
| Second Quarter |
15.96 | 5.07 | ||||
| First Quarter |
9.40 | 5.06 | ||||
There were approximately 165 holders of record of our common stock as of February 18, 2010. We have not paid any cash dividends to date and do not anticipate any being paid in the foreseeable future.
41
Table of Contents
Performance Measurement Comparison*
The following graph compares the cumulative total stockholder return on our common stock between December 31, 2004 and December 31, 2009 with the cumulative total return of (i) the NASDAQ Composite Index (NASDAQ Index) and (ii) the NASDAQ Biotechnology Index (the NASDAQ Biotech Index), over the same period. This graph assumes the investment of $100.00 on December 31, 2004 in common stock, the NASDAQ Index and the NASDAQ Biotech Index, and assumes the reinvestment of any dividends.
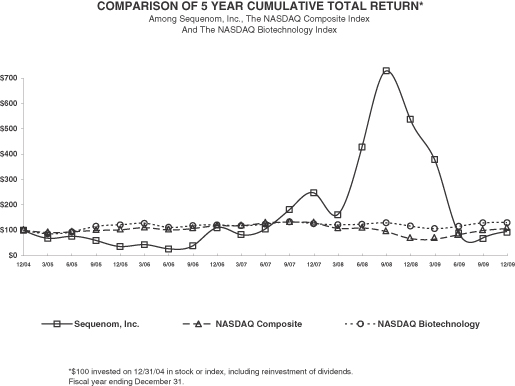
| * | This Section is not “soliciting material” is not deemed “filed” with the SEC and is not to be incorporated by reference in any of our filing under the Securities Act of 1933 or the Securities Exchange Act of 1934 whether made before or after the date hereof without regard to any general incorporation language in any such filing. |
42
Table of Contents
Item 6. SELECTED FINANCIAL DATA
The following selected consolidated financial data is derived from our audited consolidated financial statements and should be read in conjunction with the consolidated financial statements and the notes to such statements and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this report. Historical results are not necessarily indicative of the results to be expected in the future.
| Years ended December 31, | ||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||
| (In thousands, except per share data) | ||||||||||||||||||||
| Consolidated statements of operations data |
||||||||||||||||||||
| Revenues: |
||||||||||||||||||||
| Consumables, MassARRAY and other product related |
$ | 35,533 | $ | 42,259 | $ | 37,365 | $ | 27,051 | $ | 19,070 | ||||||||||
| Services |
2,209 | 4,817 | 3,524 | 1,023 | — | |||||||||||||||
| Diagnostic |
94 | — | — | — | — | |||||||||||||||
| Research and other |
27 | 73 | 113 | 422 | 351 | |||||||||||||||
| Total revenues |
37,863 | 47,149 | 41,002 | 28,496 | 19,421 | |||||||||||||||
| Costs and expenses: |
||||||||||||||||||||
| Cost of consumables, product, diagnostic and services revenue |
14,570 | 19,590 | 18,077 | 11,887 | 10,370 | |||||||||||||||
| Research and development |
37,454 | 27,455 | 14,352 | 11,939 | 11,930 | |||||||||||||||
| Selling and marketing, general and administrative |
54,972 | 42,735 | 31,148 | 22,425 | 22,382 | |||||||||||||||
| Restructuring and long-lived asset impairment charge |
1,589 | — | — | 10 | 593 | |||||||||||||||
| Amortization of acquired intangibles |
— | — | — | 1,511 | 2,014 | |||||||||||||||
| Total costs and expenses |
108,585 | 89,780 | 63,577 | 47,772 | 47,289 | |||||||||||||||
| Loss from operations |
(70,722 | ) | (42,631 | ) | (22,575 | ) | (19,276 | ) | (27,868 | ) | ||||||||||
| Other income (expense): |
||||||||||||||||||||
| Interest income |
442 | 1,592 | 1,781 | 906 | 633 | |||||||||||||||
| Interest expense |
(261 | ) | (139 | ) | (17 | ) | (20 | ) | (325 | ) | ||||||||||
| Loss on marketable securities |
(1,914 | ) | (2,584 | ) | (1,071 | ) | — | — | ||||||||||||
| Other income (expense), net |
1,560 | (181 | ) | (101 | ) | 191 | 94 | |||||||||||||
| Loss before income taxes |
(70,895 | ) | (43,943 | ) | (21,983 | ) | (18,199 | ) | (27,466 | ) | ||||||||||
| Income (expense) tax benefit |
(117 | ) | (211 | ) | — | 622 | 929 | |||||||||||||
| Net loss |
$ | (71,012 | ) | $ | (44,154 | ) | $ | (21,983 | ) | $ | (17,577 | ) | $ | (26,537 | ) | |||||
| Net loss per share, basic and diluted |
$ | (1.16 | ) | $ | (0.83 | ) | $ | (0.57 | ) | $ | (0.71 | ) | $ | (2.00 | ) | |||||
| Weighted average shares outstanding, basic and diluted |
61,171 | 53,129 | 38,865 | 24,842 | 13,276 | |||||||||||||||
| As of December 31, | |||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | |||||||||||
| (In thousands) | |||||||||||||||
| Consolidated balance sheet data |
|||||||||||||||
| Cash, cash equivalents, marketable securities and restricted cash |
$ | 44,100 | $ | 99,700 | $ | 52,150 | $ | 26,330 | $ | 8,678 | |||||
| Working capital |
45,473 | 103,246 | 52,690 | 23,651 | 5,403 | ||||||||||
| Total assets |
86,645 | 140,484 | 76,046 | 39,881 | 24,436 | ||||||||||
| Total long-term obligations |
5,226 | 4,779 | 5,744 | 3,525 | 1,363 | ||||||||||
| Total stockholders’ equity |
63,658 | 116,213 | 54,265 | 25,450 | 11,743 | ||||||||||
43
Table of Contents
| Item 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
Overview
We are a molecular diagnostic testing and genetics analysis company committed to providing products, services, diagnostic testing, applications and genetic analysis products that translate the results of genomic science into solutions for biomedical research, translational research, molecular medicine applications, and agricultural, livestock, and other areas of research. Our development and commercialization efforts in various diagnostic areas include noninvasive women’s health related and prenatal diagnostics, age-related macular degeneration diagnostics, oncology, infectious diseases, and other disorders and diseases.
Our proprietary MassARRAY system, comprised of hardware, software applications, consumable chips and reagents, is a high performance (in speed, accuracy and cost efficiency) nucleic acid analysis platform that quantitatively and precisely measures genetic target material and variations. Our platform is widely accepted as a leading high-performance DNA analysis platform for the fine mapping genotyping market and continues to gain traction for newer applications, such as agricultural-biotechnology and clinical research. Our customers include premier clinical research laboratories, bio-agriculture, bio-technology and pharmaceutical companies, academic institutions, and various government agencies worldwide. To provide customer support for our expanding user base and in an effort to maximize market penetration, we have established direct sales and support personnel serving North America, Europe and Asia, in addition to distribution partners in several major countries throughout the world.
We are researching, developing and pursuing the commercialization of various noninvasive molecular diagnostic tests for prenatal genetic disorders and diseases, women’s health related disorders and diseases, age-related macular degeneration diagnostics, oncology, infectious diseases, and other diseases and disorders. We have branded our diagnostic technology for prenatal diagnostics under the trademark SEQureDx. Our efforts in molecular diagnostics are focused on noninvasive diagnostics currently using our proprietary MassARRAY system; however, we may in the future employ other instrumentation platforms with our diagnostic applications as may be more suitable on a case-by-case basis considering optimum test performance and commercialization factors.
Currently, we are primarily focused on developing and commercializing prenatal screening and diagnostic tests using our foundational, patent protected, noninvasive, circulating cell-free fetal (ccff) nucleic acid based assay technology. This technology uses a simple maternal blood draw (meaning noninvasive compared to invasive procedures such as amniocentesis, chorionic villus sampling, or surgery) for a prenatal diagnosis or risk assessment in order to provide reliable information about the status of the fetus early in pregnancy. In early 2010 we launched noninvasive Rhesus D genotyping and Fetalxy sex determination laboratory developed tests (LDTs) using this patented ccff technology which we in-license from Isis Innovation Limited (Isis). We also launched, in September 2009, a noninvasive molecular based cystic fibrosis carrier screening LDT. These tests have all been launched through our College of American Pathology (CAP) accredited and Clinical Laboratory Improvement Amendments (CLIA) certified laboratory, Sequenom Center for Molecular Medicine, LLC, located in Grand Rapids, Michigan. Most molecular genetic tests are LDTs.
We have made substantial investments in our information technology infrastructure to enhance the capabilities of our laboratory to track samples and provide electronic ordering and reporting, and have put in place sample collection and transportation logistics that can be readily scaled. We are entering into contracts with third party payors to establish pricing for our tests and provide reimbursement. We also plan to conduct the development, validation, and other activities necessary to file submissions with the Food and Drug Administration (FDA) seeking approval for selected diagnostic tests. Revenues from our cystic fibrosis test were not significant from September through the end of 2009.
Our MassARRAY system provides reliable results for a wide range of DNA/RNA analysis applications including single nucleotide polymorphism (SNP) genotyping detection of mutations, analysis of copy number variants and other structural genome variations. In addition, the system provides quantitative gene expression
44
Table of Contents
analysis, quantitative DNA methylation analysis, comparative sequence analysis of haploid organisms, SNP discovery, and oligonucleotide quality control. These applications are provided through proprietary application software that operates on the MassARRAY platform and through the purchase of consumable chips and reagent sets. While the MassARRAY system is versatile across many applications, it is a robust and cost-effective genotyping solution for fine mapping projects enabled through our iPLEX multiplexing assay, which permits multiplexed SNP analysis using approximately the same amount of reagents and chip surface area as is used for a single locus/SNP analysis.
We have targeted customers conducting quality genotyping and performing fine mapping studies, candidate gene studies, comparative sequencing, gene expression analysis, and epigenetic analysis in the molecular medicine market. Epigenetic analysis is an important part of cancer and other research areas. DNA methylation analysis is the most frequently studied epigenetic change, and examines changes in the presence or absence of methyl groups in specific areas of the DNA.
We are targeting customers for our genetic analysis technology and products across four segments: biomedical research and molecular medicine; oncology and translational research; clinical research, public health initiatives, biodefense and agriculture. We believe the market and opportunities for growth for fine mapping genotyping are increasing as more researchers are completing their larger genomic studies such as whole genome scans. Epigenetic analysis is an emerging market that, along with gene expression analysis, is increasingly being utilized by researchers in conjunction with genotyping to attempt to fully understand genetic cause and effect.
As of December 31, 2009, our revenues consisted of sales of MassARRAY hardware, software, consumables, maintenance agreements, diagnostic testing and from services contracts through our genetic analysis contract research services business. The impact of our product offerings and contract research services business on future revenues, margins, expenses, and cash flows remains uncertain and depends on many factors as described in Item 1A of this report under the caption “Risk Factors.”
Expected revenues from our launched molecular tests (cystic fibrosis carrier screen, Rhesus D genotyping, and Fetalxy sex determination tests) are uncertain and difficult to predict. These tests have only recently been launched, demand for and acceptance of these tests by physicians and their patients is uncertain, and the level of reimbursement (applicable to the cystic fibrosis carrier screen and Rhesus D genotyping tests) is also uncertain. The Fetalxy sex determination test is paid for directly by the patient and is not subject to reimbursement because it is not a medically necessary test. As a result, our entitlement to, and the timing and amounts of, any revenues from molecular diagnostic products are uncertain and difficult to predict at this early point in time following their launch. Such revenues are uncertain and also depend on many factors as described in Item 1A of this report under the caption “Risk Factors.”
We have a history of recurring losses from operations and have an accumulated deficit of $597.3 million as of December 31, 2009 and management expects to incur further losses for the foreseeable future. Our capital requirements to sustain operations, including research and development projects, have been and will continue to be significant. As of December 31, 2009, we had available cash and cash equivalents and current marketable securities totaling $42.7 million and working capital of $45.5 million, which based on our current projections will not be sufficient to fund our obligations through the end of the third quarter of 2010 if we continue spending at our current levels. Therefore, we plan to pursue raising additional debt and/or equity financing through private or public offerings, but we cannot assure you that such financing or transaction will be available on acceptable terms, or at all. The uncertainty of this situation raises substantial doubt about our ability to continue as a going concern. The accompanying consolidated financial statements do not include any adjustments that might result from the failure to continue as a going concern.
Critical Accounting Policies
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions in certain circumstances that affect amounts reported in the accompanying consolidated financial statements and related notes. Certain of these
45
Table of Contents
accounting policies that we believe are the most critical to our investors’ understanding of our financial results and conditions are discussed below. Our significant accounting policies are more fully described in Note 2 to our Consolidated Financial Statements included elsewhere in this report. In preparing these financial statements, management uses its judgment to determine the appropriate assumptions to be used in the determination of certain estimates. Management considers many factors in selecting appropriate financial accounting policies and controls and in developing the estimates and assumptions that are used in the preparation of the consolidated financial statements. Management must apply significant judgment in this process. The estimation process often may yield a range of potentially reasonable estimates of the ultimate future outcomes and management must select an assessment that falls within the range of reasonable estimates. The application of these accounting policies involves the exercise of judgment and use of estimates and assumptions as to future uncertainties and, as a result, actual results could differ from these estimates.
Revenue Recognition
We recognize revenue for consumables, MassARRAY and other product related sales in accordance with current accounting rules, when persuasive evidence of an arrangement exists, delivery has occurred or services have been rendered, the price is fixed and determinable, and collectability is reasonably assured. Additionally, for MassARRAY system sales, the arrangement consideration is allocated among the separate units of accounting based on their relative fair values. The separate units of accounting are typically the system and software itself and maintenance contracts sold at the time of the system sale. Revenue is deferred for fees received before earned. Revenues from sales of consumables are recognized generally upon shipment and transfer of title to the customer. Revenue from sales of MassARRAY systems with general standard payment terms of 60 days or less are recognized upon shipment and transfer of title to the customer or when all revenue recognition criteria are met. Revenues from the sale or licensing of our proprietary software are recognized upon transfer of title to the customer or the duration of the software license. We recognize revenue on maintenance services for ongoing customer support over the maintenance period. Revenues from genetic services are recognized at the completion of key stages in the performance of the service, which is generally delivery of SNP assay information. Grant revenue is recorded as the research expenses relating to the grants are incurred, provided that the amounts received are not refundable if the research is not successful. Amounts received that are refundable if the research is not successful would be recorded as deferred revenue and recognized as revenue upon the grantor’s acceptance of the success of the research results.
Diagnostic revenues from our carrier screening test for cystic fibrosis, which was commercially launched in September 2009, have been recognized on a cash basis due to the limited number of contracts or agreements we have with third-party payors and our limited collections experience. We generally bill third-party payors upon generation and delivery of a report to the physician. As such, we take assignment of benefits and risk of collection with the third-party payor. We usually bill the patient directly for amounts not covered by their insurance carrier in the form of co-pays and deductibles, but only after multiple requests for full payment have been denied or only partially paid by the insurance carrier. Some payors may not cover our carrier screening test for cystic fibrosis, as ordered by the physician, under their reimbursement policies. Consequently, we pursue case-by-case reimbursement where policies are not in place.
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles requires management to make certain estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Significant estimates are as follows:
| • | Goodwill and impairment of long-lived assets. The purchase price allocation for acquisitions requires extensive use of accounting estimates and judgments to allocate the purchase price to the identifiable tangible and intangible assets acquired and liabilities assumed based on their respective fair values. |
46
Table of Contents
| Additionally, we must determine whether an acquired entity is considered to be a business or a set of net assets, because a portion of the purchase price can only be allocated to goodwill in a business combination. Goodwill and intangible assets deemed to have indefinite lives are not amortized, but are subject to annual impairment tests. The amounts and useful lives assigned to other intangible assets impact future amortization. Determining the fair values and useful lives of intangible assets requires the use of estimates and the exercise of judgment. These judgments can significantly affect our net operating results. |
| • | We periodically re-evaluate the original assumptions and rationale utilized in the establishment of the carrying value and estimated lives of our goodwill and long-lived assets. The criteria used for these evaluations include management’s estimate of the asset’s continuing ability to generate income from operations and positive cash flows in future periods as well as the strategic significance of any intangible assets in our business objectives. If assets are considered to be impaired, the impairment recognized is the amount by which the carrying value of the assets exceeds the fair value of the assets. No impairment of long-lived assets was recorded in 2009, 2008 or 2007. Intangible assets totaled $1.2 million, net of accumulated amortization, at December 31, 2009. |
| • | Allowance for Doubtful Accounts. We maintain an allowance for doubtful accounts for estimated losses resulting from the inability of our customers to make required payments. We evaluate the collectability of our accounts receivable balance based on a combination of factors. We regularly analyze customer accounts, review the length of time receivables are outstanding and review the historical loss rates. If the financial condition of our customers were to deteriorate, additional allowances could be required. |
| • | Reserves for obsolete and slow-moving inventory. We operate in an industry characterized by rapid improvements and changes to technology and products. The introduction of new products by us or our competitors can result in our inventory being rendered obsolete or requiring us to sell items at a discount to cost. We estimate the recoverability of our inventory by reference to our internal estimates of future demands and product life cycles. If we incorrectly forecast demand for our products or inadequately manage the introduction of new product lines, we could materially impact our financial statements by having excess inventory on hand. Our future estimates are subjective and could be incorrect. During 2009, slow-moving inventory reserves of $0.1 million were charged against cost of goods sold and the total reserve was $1.9 million at December 31, 2009. |
| • | Income taxes. Our provision for income taxes is computed using the asset and liability method, under which deferred tax assets and liabilities are recognized for the expected future tax consequences of temporary differences between the financial reporting and tax bases of assets and liabilities, and for the expected future tax benefit to be derived from tax loss and credit carryforwards. Deferred tax assets and liabilities are determined using the enacted tax rates in effect for the years in which those tax assets are expected to be realized. A valuation allowance is established when it is more likely than not the future realization of all or some of the deferred tax assets will not be achieved. The evaluation of the need for a valuation allowance is performed on a jurisdiction by jurisdiction basis, and includes a review of all available positive and negative evidence. As of December 31, 2009, we maintain a valuation allowance against our U.S. and foreign deferred tax assets that we concluded have not met the “more likely than not” threshold. |
We recognize excess tax benefits associated with share-based compensation to stockholders’ equity only when realized. When assessing whether excess tax benefits relating to share-based compensation have been realized, we follow the with-and-without approach, excluding any indirect effects of the excess tax deductions. Under this approach, excess tax benefits related to share-based compensation are not deemed to be realized until after the utilization of all other tax benefits available to us.
We recognize the impact of a tax position in our financial statements only if that position is more likely than not of being sustained upon examination by taxing authorities, based on the technical merits of the position. Any interest and penalties related to uncertain tax positions will be reflected in income tax expense.
47
Table of Contents
| • | Stock based compensation. Stock based compensation cost is estimated at the grant date based on the award’s fair-value as calculated by the Black-Scholes option pricing model (BSM) and is recognized as expense over the requisite service period. The BSM model requires various highly judgmental assumptions including volatility, forfeiture rates, and expected option life. If any of these assumptions used in the BSM model change significantly, stock-based compensation expense may differ materially in the future from that recorded in the current period. |
Recent Accounting Pronouncements
As of July 1, 2009, the Financial Accounting Standards Board (FASB) formally approved the FASB Accounting Standards Codification (Codification) as the single source of authoritative U.S. accounting and reporting standards, other than guidance issued by the SEC. At that time, the Codification superseded all then-existing non-SEC accounting and reporting standards. All other non-grandfathered, non-SEC accounting literature not included in the Codification became non-authoritative. The Codification is effective for interim and annual periods ending after September 15, 2009. We adopted the provisions of the Codification in the third quarter of 2009. The adoption did not have a material impact on our consolidated financial statements.
In May 2009, the FASB issued authoritative guidance that establishes general standards of accounting for and disclosure of events that occur after the balance sheet date but before financial statements are issued or are available to be issued. This guidance became effective for interim periods and fiscal years ending after June 15, 2009. We adopted the provisions of this guidance in the second quarter of 2009. The adoption of this guidance did not have a material impact on our consolidated financial statements.
In April 2009, the FASB issued authoritative guidance that requires publicly traded companies to include in their interim financial reports certain disclosures about the carrying value and fair value of financial instruments previously required only in annual financial statements and to disclose changes in significant assumptions used to calculate the fair value of financial instruments. This guidance became effective for all interim reporting periods ending after June 15, 2009, with early adoption permitted for interim reporting periods ending after March 15, 2009. We adopted the provisions of this guidance in the second quarter of 2009. The adoption of this guidance did not have a material impact on our consolidated financial statements. In December 2007, the FASB issued authoritative guidance that significantly changes the accounting and reporting requirements for business combination transactions, including capitalization of in-process research and development assets and expensing acquisition costs as incurred. This guidance became effective for business combination transactions occurring in fiscal years beginning after December 15, 2008. We adopted the provisions of this guidance in the first quarter of 2009. The adoption did not have a material impact on our 2009 consolidated financial statements.
In November 2008, the FASB issued authoritative guidance that clarifies how to account for acquired intangible assets subsequent to initial measurement in situations in which an entity does not intend to actively use the assets but intends to hold the asset to prevent others from obtaining access to the asset (a defensive intangible asset), except for intangible assets that are used in research and development activities. This guidance requires that a defensive intangible asset be accounted for as a separate unit of accounting and assigned a useful life that reflects the entity’s consumption of the expected benefits related to that asset. This guidance became effective for intangible assets acquired on or after December 15, 2008. We adopted the provisions of this guidance in the first quarter of 2009. The adoption of this guidance did not have a material impact on our consolidated financial statements.
In June 2008, the FASB issued authoritative guidance that clarifies the criteria for determining whether certain financial instruments should be classified as derivative instruments or equity instruments. The guidance became effective for years beginning after December 15, 2008. We adopted the provisions of the guidance in the first quarter of 2009. The adoption did not have a material impact on our consolidated financial statements.
In April 2008, the FASB issued authoritative guidance that amends the guidance for estimating the useful lives of recognized intangible assets and requires additional disclosure related to renewing or extending the useful lives of
48
Table of Contents
recognized intangible assets. This guidance became effective for fiscal years and interim periods beginning after December 15, 2008. We adopted the provisions of this guidance in the first quarter of 2009. The adoption of this guidance did not have a material impact on our consolidated financial statements.
In December 2007, the FASB issued authoritative guidance that defines collaborative arrangements and requires that transactions with third parties that do not participate in the arrangement be reported in the appropriate income statement line items pursuant to existing authoritative accounting literature. In accordance with this guidance, income statement classification of payments made between participants of a collaborative arrangement is to be based on other applicable authoritative accounting literature. If the payments are not within the scope or analogy of other authoritative accounting literature, a reasonable, rational and consistent accounting policy is to be elected. This guidance became effective for fiscal years beginning after December 15, 2008 and was to be applied as a change in accounting principle to all prior periods retrospectively for all collaborative arrangements existing as of the effective date. We adopted the provisions of this guidance in the first quarter of 2009. The adoption of this guidance did not have a material impact on our consolidated financial statements.
Results of Operations
Years ended December 31, 2009 and 2008
Revenues
Total revenues were $37.9 million and $47.1 million for the years ended December 31, 2009 and 2008, respectively. MassARRAY and other product related revenues are derived from the sale of MassARRAY systems, consumables, sales and licensing of our proprietary software, maintenance contracts, and license fees from end-users. Diagnostic revenues are from the sale of our carrier screening LDT for cystic fibrosis and consisted of cash collected from tests performed through December 31, 2009.
Consumable sales increased to $20.5 million in 2009 from $19.5 million in 2008. The increase in 2009 compared to 2008 was primarily due to increased consumables orders from our customers in the biomedical research and agricultural biology markets.
MassARRAY and other product related revenue decreased to $15.0 million in 2009 from $22.7 million in 2008. The decrease of $7.7 million was primarily due to a decrease in MassARRAY system hardware and software revenue to $10.7 million in 2009 from $19.5 million in 2008, which was attributable to fewer system placements during the year ended December 31, 2009. Revenue from other product sales, including MassARRAY system maintenance contracts, license fees and royalties for the years ended December 31, 2009 and 2008 was $4.3 million and $3.2 million, respectively. Maintenance revenue increased by approximately $1.1 million in 2009, as compared to 2008 due to higher service contracts in effect over our installed base.
We recorded genetic analysis service revenues of $2.2 million for the year ended December 31, 2009, compared to $4.8 million in service revenues for the year ended December 31, 2008. The decrease from 2008 is attributable to our cost cutting initiative that commenced in April 2009, which refocused our genetic analysis service business on fewer, higher margin studies and projects. We expect genetic analysis service revenues to be minimal going forward
We recognized diagnostic revenue of $94,000 and $0 for the years ended December 31, 2009 and 2008, respectively. Diagnostic revenue is currently generated only from our carrier screening test for cystic fibrosis, which we commercialized in September 2009. Diagnostic revenue is recognized upon cash collection as payments are received. Diagnostic revenue going forward is uncertain and difficult to predict due to the lack of historical sales trends associated with our recent commercial launch of this test.
Research and other revenue was $27,000 for the year ended December 31, 2009, compared to $73,000 for the year ended December 31, 2008. The timing of research revenues depends upon our expenditures on grant research and the receipt of the grant funding from the sponsoring agencies. We expect research revenue to be minimal going forward.
Domestic and non-U.S. revenues were $18.0 million and $19.8 million for the year ended December 31, 2009, respectively, and $23.8 million and $23.3 million for the year ended December 31, 2008, respectively.
49
Table of Contents
The following table presents revenue for each reportable segment for the year ended December 31, 2009. Prior to 2009, all revenue was derived from our Genetic Analysis operations.
| (In thousands) | |||
| Revenues: |
|||
| Molecular Diagnostics |
$ | 94 | |
| Genetic Analysis |
37,769 | ||
| $ | 37,863 | ||
Our revenues have historically fluctuated from period to period and likely will continue to fluctuate substantially in the future based upon the unpredictable sales cycle for the MassARRAY system, general economic conditions, revenue recognition criteria, the overall acceptance and demand for our new and existing commercial products and services, as well as the adoption rates of our cystic fibrosis carrier screening assay, Rhesus D genotyping assay, Fetalxy sex determination assay and future assays.
Costs of Consumables and Products, Services and Diagnostic Revenues and Gross Margins
Cost of consumables products revenues were $12.0 million and $15.1 million and gross margins were 66% and 64% for the years ended December 31, 2009 and 2008, respectively. The increase in gross margin for product revenues in 2009 compared to 2008 is attributable to increased consumable sales that generally have higher average gross margins compared to systems sales and is also due to fewer system placements, which historically sell at lower gross margins.
Cost of service revenues were $2.2 million and $4.5 million and gross margins were 1% and 7% for the years ended December 31, 2009 and 2008, respectively. Gross margins decreased compared to the prior year due to the completion of remaining unfavorable, low volume contracts. Gross margins on contract research services have been dependent on particular contract terms of the work undertaken, as well as the particular market in which the services are being performed, the size of the projects and the pricing terms.
Cost of diagnostic revenues are recognized at the completion of testing and were $0.4 million and $0 and gross margins were (338%) and 0% for the years ended December 31, 2009 and 2008, respectively. Gross margin on diagnostic tests are primarily affected by test volumes and overall reimbursement for the amount paid per test.
Our overall gross margins were 62% and 58% for the years ended December 31, 2009 and 2008, respectively. The increase in overall gross margin in 2009 is attributable to lower system and contract research revenues, which are sold at a lower margin than consumables. The increase in consumables revenue during 2009, as compared to 2008, also contributed to the improved margin.
We believe that gross margin in future periods will be affected by, among other things, the selling price for systems and consumables, consumable sales per MassARRAY system sold, the mix of product sales and the type of services, competitive conditions, sales volumes, discounts offered, sales through distributors, as well as the cost of goods sold, inventory reserves and obsolescence charges required and royalty payment obligations on in-licensed technologies. Our gross margin will also be affected by the adoption rates of our diagnostic LDTs we commercialize, the payor and other contracts we may enter into for laboratory developed or diagnostic tests and the volume of tests sold.
Research and Development Expenses
Research and development costs were $37.5 million and $27.5 million for the years ended December 31, 2009 and 2008, respectively. These expenses consist primarily of salaries and related personnel expenses, improvements to our existing products, clinical sample and related operations, validation of products under development and expenses relating to work performed under research contracts.
50
Table of Contents
The increase in research and development expenses of $10.0 million for 2009 compared to 2008 primarily resulted from increased headcount and related costs of $3.0 million associated with increased investment in our diagnostic development, an increase of $3.8 million for clinical trial costs associated with our prenatal diagnostic, molecular diagnostic and genetic analysis programs, headcount based overhead allocation expense related to our information technology and facilities departments of $1.8 million, share-based compensation expense of $2.2 million, as well as higher depreciation of $1.0 million associated with capital expenditures and our acquisition of SensiGen, LLC in February 2009, higher facilities operations expenses of $0.3 million and office expenses of $0.2 million related to higher freight and postage associated with sample collection activity with our clinical programs. These increases were offset by a decrease of $2.3 million in collaboration costs associated with various research and development projects and related licensing activities.
The following table presents a reconciliation of research and development expenses for each reportable segment for the year ended December 31, 2009.
Research and development expenses (in thousands):
| Molecular Diagnostics |
$ | 20,935 | |
| Genetic Analysis |
5,587 | ||
| Total segments |
26,522 | ||
| Share based compensation |
3,835 | ||
| Indirect overhead (1) |
3,898 | ||
| Allocated and absorbed costs (2) |
3,199 | ||
| Total research and development expenses |
$ | 37,454 | |
| (1) | Indirect overhead consists of expenses related to the following departments, which are unallocated to our operating segments for performance assessment by our chief operating decision maker: quality, regulatory, chief science officer and research and development collaborations (licensing costs). |
| (2) | Allocated costs consist of costs from the following departments, which are unallocated to our operating segments for performance assessment by our chief operating decision maker: human resources, information technology and facilities. Absorbed costs relate to costs incurred that are absorbed into cost of sales. |
We expect our research and development expenses to increase in 2010 compared to 2009, as we continue to expand our investment in the development of noninvasive prenatal nucleic acid based tests, including a potential Trisomy 21 LDT, and other noninvasive tests such as a potential LDT for age-related macular degeneration and as we continue to invest in new products and applications for our MassARRAY platform.
Sales and Marketing Expenses
Sales and marketing costs were $26.8 million and $24.3 million for the years ended December 31, 2009 and 2008, respectively. These expenses consist primarily of salaries and related expenses for sales and marketing, customer support, and business development personnel and their related department expenses.
The increase in selling and marketing expenses of $2.5 million for 2009 compared to 2008 primarily resulted from increased headcount and related costs of $1.1 million associated with building our marketing and contract sales force infrastructure for our noninvasive diagnostics business, $1.4 million for higher share-based compensation expense and $0.2 million related to increased travel, postage and freight charges and other general operating expenses as compared to 2008. These increases were offset by a decrease of $0.2 million in marketing expenses associated with our genetic analysis and diagnostic operations by reducing external consultant costs.
51
Table of Contents
The following table presents a reconciliation of sales and marketing expenses for each reportable segment for the year ended December 31, 2009.
Sales and marketing expenses (in thousands):
| Molecular Diagnostics |
$ | 5,780 | |
| Genetic Analysis |
13,644 | ||
| Total segments |
19,424 | ||
| Share based compensation |
3,590 | ||
| Indirect overhead (1) |
1,819 | ||
| Allocated and absorbed costs (2) |
2,012 | ||
| Total research and development expenses |
$ | 26,845 | |
| (1) | Indirect overhead consists of expenses related to the following departments, which are unallocated to our operating segments for performance assessment by our chief operating decision maker: business development and European sales administration. |
| (2) | Allocated costs consist of costs from the following departments, which are unallocated to our operating segments for performance assessment by our chief operating decision maker: human resources, information technology and facilities. Absorbed costs relate to costs incurred that are absorbed into cost of sales. |
We expect our sales and marketing expenses to remain higher in 2010 compared to 2009, due to our increased headcount and related expenditures as we strengthen our sales force and build our marketing and commercial development teams for our molecular diagnostic tests.
General and Administrative Expenses
General and administrative costs were $28.1 million and $18.4 million for the years ended December 31, 2009 and 2008, respectively. These expenses consist primarily of salaries and related expenses for legal, finance, and human resource personnel, and their related department expenses. General and administrative costs are not allocated to our business segments for performance assessment by our chief operating decision maker.
The increase in general and administrative expenses of $9.7 million for 2009 compared to 2008 primarily resulted from increased legal expense of $6.4 million associated with litigation, the independent investigation conducted by the Special Committee of our Board of Directors and the share-based payment related to the settlement of SensiGen, LLC’s claim against us, share-based compensation of $1.0 million, $0.9 million for increased headcount and related expenses, $1.0 million of higher rent, taxes and communications expenses associated with our San Diego facilities and $0.4 million in increased investor relations consulting, audit and tax related expenses.
We expect general and administrative costs to increase in 2010 compared to 2009, as we build our infrastructure in order to support our anticipated growth and due to expected continued legal costs related to ongoing investigations and litigation.
Restructuring and long-lived asset impairment charge
Restructuring and long-lived asset impairment charges were $1.6 million for the year ended December 31, 2009. These charges were associated with our April 2009 reduction in workforce, which included the closure of our leased facility in Boston, Massachusetts, the closure of our office located in New Delhi, India, as well as a decrease in our genetic analysis workforce primarily associated with our genetic analysis services business. These charges consist of one-time terminations benefits, office closure expenses and other related costs. There were no comparative charges for the year ended December 31, 2008.
52
Table of Contents
Interest Income
Interest income was $0.4 million and $1.6 million for the years ended December 31, 2009 and 2008, respectively. The decrease in 2009 compared to 2008 was attributable to a reduction of our cash, cash equivalents and marketable securities balances in the current period, as well as changes in our investment policy in the prior year that resulted in the overall reduction in the rates of return in our investment portfolio.
Loss on Marketable Securities
Loss on marketable securities was $1.9 million and $2.6 million for the years ended December 31, 2009 and 2008, respectively. The loss for the year ended December 31, 2009, was due to the sale of five auction rate security investments, which resulted in a realized loss of $0.8 million, as well as an other-than-temporary impairment in our remaining investments in auction rate securities of $1.1 million. Two of our four remaining investments in auction rate securities were subsequently sold in January 2010, with the realized loss of $0.7 million having been included within the $1.1 million recognized for the year ended December 31, 2009.
Interest Expense
Interest expense was $0.3 million and $0.1 million for the years ended December 31, 2009 and 2008, respectively. The increase in 2009 compared to 2008 is due to payments on our capital lease and debt obligations obtained in 2009, as well as continued payments on our asset-backed loans.
Other Income (Expense), net
Other income (expense), net was $1.6 million and ($181,000) for the years ended December 31, 2009 and 2008, respectively. The increase in 2009 primarily relates to a $1.0 million payment we received related to the settlement of our patent infringement lawsuit against Ibis Biosciences, Inc., favorable realized foreign currency translations and the receipt of a research and development tax credit from the U.S. Government of $0.3 million.
Income Tax Expense
Income tax expense was $117,000 and $211,000 for the year ended December 31, 2009 and 2008, respectively. Income tax expense in both periods was primarily due to statutory tax liabilities resulting from our foreign operations.
Years ended December 31, 2008 and 2007
Revenues
Total revenues were $47.1 million and $41.0 million for the years ended December 31, 2008 and 2007, respectively. MassARRAY and other product related revenues are derived from the sale of MassARRAY systems, consumables, sales and licensing of our proprietary software, maintenance contracts, and license fees from end-users.
Consumable sales increased to $19.5 million in 2008 from $16.5 million in 2007. The increase in 2008 compared to 2007 was a result of an increase in our installed base of MassARRAY Compact systems as well as increased demand for our iPLEX genotyping assay.
MassARRAY and other product related revenue increased to $22.7 million in 2008 from $20.8 million in 2007. The increase of $1.9 million was primarily due to an increase in MassARRAY system hardware and software revenue to $19.5 million in 2008 from $18.4 million in 2007, which was attributable to an increase in our selling price during 2008. Revenue from other product sales, including MassARRAY system maintenance contracts, license fees and royalties for the years ended December 31, 2008 and 2007 was $3.2 million and $2.5 million, respectively. Maintenance revenue increased by approximately $0.7 million from the comparative period due to higher service contracts in effect over our installed base.
53
Table of Contents
We recorded genetic analysis service revenues of $4.8 million for the year ended December 31, 2008, compared to $3.5 million in service revenues for the year ended December 31, 2007. The increase from 2007 is attributable to growth in our contract research service business primarily in the commercial, clinical analysis and the academic research markets.
Research and other revenue was $0.1 million in 2008 and $0.1 million 2007. The timing of research revenues depends upon our expenditures on grant research and the receipt of the grant funding from the sponsoring agencies.
Domestic and non-U.S. revenues were $23.8 million and $23.3 million, respectively, for the year ended December 31, 2008, and $22.2 million and $18.8 million, respectively, for the year ended December 31, 2007.
Costs of Consumables and Products, Services and Diagnostic Revenues and Gross Margins
Cost of product revenues were $15.1 million and $14.6 million and gross margins were 64% and 61% for the years ended December 31, 2008 and 2007, respectively. The increase in gross margin for product revenues in 2008 compared to 2007 is attributable to increased consumable sales that generally have higher average gross margins compared to systems sales, along with a favorable mix of new systems at higher selling prices with additional software at higher margins.
Cost of service revenues were $4.5 million and $3.5 million and gross margins were 7.0% and 1.2% for the years ended December 31, 2008 and 2007, respectively. Our genetic analysis contract research service business incurred higher expenses, primarily in salaries and related personnel expenses, as operations increased in scale to accommodate a higher volume of research contracts. Gross margins on contract research service revenues are dependent on the particular market the services are being performed, the size of the projects and the pricing terms.
Our overall gross margins were 58% and 56% for the years ended December 31, 2008 and 2007, respectively. The increase in overall gross margin in 2008 is attributable to increased consumables sales at a higher gross margin, a higher average selling price for new system sales and higher margins in contract research services due to a higher volume of activity.
Research and Development Expenses
Research and development costs were $27.5 million and $14.4 million for the years ended December 31, 2008 and 2007, respectively. These expenses consist primarily of salaries and related personnel expenses, improvements to our existing products, validation of products under development, and expenses relating to work performed under research contracts.
The increase in research and development expenses of $13.1 million for 2008 compared to 2007 primarily resulted from increased headcount and travel costs of $3.8 million, operating supplies of $2.8 million, clinical sample collection, consulting and collaboration costs of $2.9 million related to our noninvasive prenatal technology development, headcount based overhead allocation expense of $2.5 million, share-based compensation expense of $1.1 million, as well as depreciation and office expenses of $1.0 million. These increases were offset by $1.0 million in the absorption of research and development expenses to cost of service revenue.
Sales and Marketing Expenses
Sales and marketing costs were $24.3 million and $17.0 million for the years ended December 31, 2008 and 2007, respectively. These expenses consist primarily of salaries and related expenses for sales and marketing, customer support, and business development personnel and their related department expenses.
54
Table of Contents
The increase in selling and marketing expenses of $7.3 million for 2008 compared to 2007 primarily resulted from increased headcount and travel of $3.1 million, $1.6 million for higher share-based compensation expense, $0.9 million for higher headcount-based overhead allocation charges, $0.4 million for higher advertising, trade shows and public relations expenses, $0.3 million of consultant expenses for sales and marketing projects associated with diagnostic operations, $0.3 million in bad debt expense related to accounts receivable write-offs, $0.3 million for higher operating supplies, $0.3 million for higher office operating expenses and $0.1 million for higher sales bonus compensation.
General and Administrative Expenses
General and administrative costs were $18.4 million and $14.1 million for the years ended December 31, 2008 and 2007, respectively. These expenses consist primarily of salaries and related expenses for legal, finance, and human resource personnel, and their related department expenses.
The increase in general and administrative expenses of $4.3 million for 2008 compared to 2007 primarily resulted from increased legal expense of $2.9 million related to ongoing litigation, share-based compensation of $1.6 million, headcount and travel expense of $1.4 million, audit and tax related fees and expenses of $0.9 million, information technology expenses of $0.8 million for computers and software licenses, consultant expenses of $0.3 million and depreciation and other office expenses of $0.3 million. These increases were partially offset by reduced headcount-based overhead allocation of $3.3 million as well as higher absorption of overhead costs of $0.6 million.
Interest Income
Interest income was $1.6 million in 2008 compared to $1.8 million in 2007. The decrease in 2008 compared to 2007 was due to a change in our investment policy, which restricted our marketable securities investments exclusively to U.S. Government backed financial instruments that yield a lower overall return compared to our prior investment portfolio, despite higher cash balances following our public offering in July 2008.
Loss on Marketable Securities
Loss on marketable securities was $2.6 million in 2008 compared to $1.1 million in 2007. The recognized loss was due to an other-than-temporary impairment in our investments in auction rate securities. The increase in recognized losses in 2008 compared to 2007 is due to additional declines in the market value of these auction rate securities due to ongoing difficulties in global credit markets.
Interest Expense
Interest expense was $139,000 and $17,000 for 2008 and 2007, respectively. The increase in 2008 compared to 2007 is due to higher balances on our asset-backed loan commencing in September 2007.
Income Tax Expense
Income tax expense of $211,000 for the year ended December 31, 2008 was primarily due to statutory tax liabilities resulting from our foreign operations. There was no comparable income tax expense for the year ended December 31, 2007.
Liquidity and Capital Resources
As of December 31, 2009, cash, cash equivalents and current marketable securities totaled $42.7 million, compared to $98.3 million at December 31, 2008. Our cash equivalents and current marketable securities are held in U.S. Government securities with ratings of AAA and repurchase agreements collateralized by U.S. Government securities with ratings of AAA.
55
Table of Contents
As of December 31, 2009, we had $0.5 million of auction rate securities, which reflects a $3.8 million adjustment to the principal value of $4.3 million. Additional discussion with respect to the risks and uncertainties associated with our auction rate securities is included in “Quantitative and Qualitative Disclosures about Market Risk” in Item 7A of this report and in the notes to our consolidated financial statements included elsewhere in this report.
We have a history of recurring losses from operations and had an accumulated deficit of $597.3 million as of December 31, 2009. Our capital requirements to sustain operations, including research and development projects, have been and will continue to be significant. As of December 31, 2009 and 2008, we had working capital of $45.5 million and $103.2 million, respectively.
On July 1, 2008, we closed an underwritten public offering of our common stock totaling 5,500,000 shares of our common stock at $15.50 per share, with the underwriters exercising their option to purchase an additional 825,000 shares on July 8, 2008. Including the additional shares, the offering resulted in aggregate net proceeds of approximately $91.8 million after deducting underwriting discounts, commissions and estimated transaction expenses.
During 2007, we closed a $20.0 million registered direct offering of our common stock to several new and existing investors, as well as a $30.5 million private placement of our common stock. Under the terms of the registered direct offering we issued and sold 6,666,666 shares of our common stock at $3.00 per share, with aggregate net proceeds of approximately $18.3 million after deducting placement agents’ fees and transaction expenses. Under the terms of the private placement we issued and sold 3,383,335 shares of our common stock at $9.00 per share, with aggregate net proceeds of approximately $28.1 million after deducting placement agents’ fees and estimated transaction expenses.
We consider the material drivers of our cash flow to be sales volumes, working capital, inventory management and operating expenses. Our principal sources of liquidity are our cash, cash equivalents and current marketable securities. Cash used in operations for the year ended December 31, 2009 was $48.7 million compared to $34.6 million for the year ended December 31, 2008. Our use of cash was primarily a result of the net loss of $71.0 million for the year ended December 31, 2009, increased by $1.3 million from other current assets and prepaid expenses, $0.6 million in deferred rent, $1.7 million from lower accounts payable and accrued expense balances, $0.7 million of lower other longer-term liabilities and an adjustment to our bad debt provision resulting in a benefit of $0.1 million due to recoveries of previously reserved balances. Cash usages were primarily offset by non-cash adjustments for stock-based compensation of $11.8 million, depreciation and amortization of $5.2 million, losses on our auction rate securities of $1.9 million, restricted stock charges of $0.5 million, a loss on disposals of fixed assets of $0.2 million, a fair value adjustment to our contingent consideration payable related to our acquisition of SensiGen, LLC of $0.5 million, as well as a settlement payment made in our common stock of $1.5 million to certain shareholders of SensiGen, LLC. Additionally, cash usages were primarily offset by operating asset and liability changes of $1.9 million from lower accounts receivable and $2.9 million from lower inventory balances representing our efforts to maximize working capital and an increase in our deferred revenue balance of $0.3 million primarily associated with an increase in the sale of maintenance contracts from the prior year. At our current and anticipated level of operating loss, we expect to continue to incur an operating cash outflow for the next several years.
Investing activities, other than the net changes in our current marketable securities and restricted cash that provided $18.1 million, consisted of purchases for capital equipment, leasehold improvements and intangible assets that used $8.7 million in cash during the year ended December 31, 2009, compared to $4.9 million and $3.5 million for the same periods in 2008 and 2007, respectively. Additionally, we paid $2.0 million in cash related to our acquisition of Sensigen, LLC that closed in February 2009.
Net cash provided by financing activities was $0.2 million during the year ended December 31, 2009. Financing activities during the year ended December 31, 2009, included $1.9 million from the exercise of stock options and from employee contributions under our employee stock purchase plan, offset by approximately $1.6 million in payments on our long-term debt and $0.1 million on our capital lease obligation.
56
Table of Contents
The following table summarized our contractual obligations as of December 31, 2009 (in thousands):
| Contractual obligations |
Total | Less Than 1 Year |
1-3 Years | After 3 Years | ||||||||
| Open purchase orders |
$ | 5,002 | $ | 5,002 | $ | — | $ | — | ||||
| Long-term debt obligation |
3,157 | 1,320 | 1,522 | 315 | ||||||||
| Collaborations |
23,948 | 5,142 | 2,966 | 15,840 | ||||||||
| Operating leases |
30,431 | 7,041 | 15,505 | 7,885 | ||||||||
| Total contractual obligations |
$ | 62,538 | $ | 18,505 | $ | 19,993 | $ | 24,040 | ||||
Future operating lease commitments for leases have not been reduced by future minimum sublease rentals to be received through December 2010 aggregating $0.3 million. Open purchase orders are primarily for inventory items and research and development supplies. Collaborations primarily consist of agreements with institutions to conduct sponsored research and clinical study agreements.
In September 2005, we entered into an amendment to our lease for our corporate headquarters in San Diego. The lease amendment provides for the deferral of approximately $3.2 million of the monthly rent payments by reducing the monthly payments through September 30, 2007 and increasing the aggregate monthly payments by the deferred amount for the remaining term of the lease, from October 1, 2007 to September 30, 2015. The total obligation under the lease remains unchanged. The contractual obligation table above reflects the deferral of these rent payments.
Long-term debt obligations include the associated interest payable on these borrowings. Other commitments and contingencies that may result in contractual obligations to pay are described in the notes to our consolidated financial statements included elsewhere in this report.
Based on our current plans, we believe our cash, cash equivalents and current marketable securities will not be sufficient to fund our obligations through the third quarter of 2010 if we continue spending at our current levels. The actual amount of funds that we will need will be determined by many factors, some of which are beyond our control, and we may need funds sooner than currently anticipated. These factors include but are not limited to:
| • | the size of our future operating losses; |
| • | the level of our and our distributors’ success in selling our MassARRAY products and services and LDT services through Sequenom CMM; |
| • | the terms and conditions of sales contracts, including extended payment terms; |
| • | our ability to introduce and sell new products and services and successfully reduce inventory levels of earlier products; |
| • | the level of our selling, general and administrative expenses; |
| • | the extent of our investment in diagnostic technology, including prenatal genetic analysis technology, molecular diagnostics and noninvasive prenatal diagnostic technology, development, commercialization, and regulatory approval; |
| • | our success in, and the expenses associated with, researching, developing and commercializing diagnostic products, alone or in collaboration with our partners, and obtaining any required regulatory approval for those products; |
| • | the level of our success alone or in collaboration with our partners in launching and selling any diagnostic products and services; |
57
Table of Contents
| • | the extent of our research and development pursuits, including our level of investment in MassARRAY product research and development, and diagnostic assay and other technology research and development; |
| • | the extent to which we enter into, maintain, and derive revenues from licensing agreements, including agreements to out-license our noninvasive prenatal analysis technology, research and other collaborations, joint ventures and other business arrangements; |
| • | the level of our legal expenses, including those expenses associated with security class actions, intellectual property protection and those expenses and any damages or settlement payments associated with litigation or on-going investigations by government agencies; |
| • | the extent to which we acquire, and our success in integrating, technologies or companies; |
| • | the level of our expenses associated with the audit of our consolidated financial statements as well as compliance with other corporate governance and regulatory developments or initiatives; and |
| • | regulatory changes and technological developments in our markets. |
At December 31, 2009, we had outstanding stand-by letters of credit with financial institutions totaling $1.4 million related to our building, operating leases and customer guarantees. The letter of credit related to our Newton, Massachusetts building lease agreement will remain in place until its expiration in December 2010.
Item 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Marketable Securities
The primary objective of our investment activities is to preserve principal while at the same time maximizing the income we receive from our investments without significantly increasing risk. Some of the securities that we invest in may have market risk. This means that a change in prevailing interest rates may cause the fair value of the principal amount of the investment to fluctuate. For example, if we hold a security that was issued with a fixed interest rate at the then-prevailing rate and interest rates later rise, the fair value of the principal amount of our investment will probably decline. To minimize this risk in the future, we revised our investment policy in April 2008 to maintain our portfolio of cash equivalents and marketable securities in a variety of securities, including U.S. Government securities with ratings of AAA, and repurchase agreements collateralized by U.S. Government securities with ratings of AAA. Our investment policy includes a minimum quality rating for all new investments, as well as limits the amount of credit exposure to only issuances from the U.S. Government. If an investment we hold falls below this level, we research the reasons for the fall and determine if we should continue to hold the investment in order to minimize our exposure to market risk of the investment.
The appropriate classification of marketable securities is determined at the time of purchase and reevaluated as of each balance sheet date. Based on this determination, as of December 31, 2009 and 2008, all of our investments in marketable securities were classified as available for sale and were reported at fair value. We measure fair value based on the prices that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. Fair value is determined based on observable market quotes or valuation models using assessments of counterparty credit worthiness, credit default risk or underlying security and overall capital market liquidity. Declines in fair value that are considered other-than-temporary are charged to operations and those that are considered temporary are reported as a component of accumulated other comprehensive income in stockholders’ equity. We use the specific identification method of determining the cost basis in computing realized and unrealized gains and losses on the sale of our available-for-sale securities.
Consistent with our investment policy guidelines in effect when originally purchased, the auction rate security (ARS) investments held by us all had AAA/AA credit ratings at the time of purchase. At December 31,
58
Table of Contents
2009, $4.3 million of principal was invested in ARS. The ARS held are private placement securities with various long-term nominal maturities with interest rates reset through a Dutch auction each month. The monthly auctions historically have provided a liquid market for these securities. The investments in ARS represent interests in collateralized debt obligations supported by insurance securitizations and other structured credits, including corporate bonds and to a lesser degree, pools of residential and commercial mortgages. With the liquidity issues experienced in global credit and capital markets, the ARS held at December 31, 2009 had experienced multiple failed auctions as the amount of securities submitted for sale exceeded the amount of purchase orders.
Although the majority of our ARS continue to pay interest according to their stated terms, based on valuation models and an analysis of other-than-temporary impairment factors, we recognized a loss of approximately $1.1 million and $2.6 million for the years ending December 31, 2009 and 2008, respectively, which reflects the portion of ARS holdings that we have concluded have an other-than-temporary decline in value. During the fourth quarter of 2009, our management determined that it was no longer our intent to hold the remaining ARS to maturity and to actively pursue liquidation of our remaining ARS in the secondary market. As a result of this decision, during the fourth quarter of 2009 we sold ARS with an estimated fair value of $4.1 million, which resulted in a net realized loss of approximately $0.8 million for a total loss on our ARS of $1.9 million for the year ended December 31, 2009. As of December 31, 2009, our remaining ARS have a principal value of $4.3 million and an estimated fair market value of $0.5 million. Subsequent to year end, ARS with a principal value of $1.3 million and an estimated fair value of $0.5 million at December 31, 2009, were sold. The proceeds for these ARS were equal to their estimated fair value as of December 31, 2009. Our other remaining ARS with a principal value of $3.0 million have an estimated fair value of $0 as of December 31, 2009.
Since there is a lack of observable market quotes on our investment portfolio of marketable securities in ARS, we utilize valuation models including those that are based on expected cash flow streams and collateral values, including assessments of counterparty credit quality, default risk underlying the security, discount rates, overall capital market liquidity and our overall intent and ability to liquidate our ARS. The valuation of our investment portfolio is subject to uncertainties that are difficult to predict. Factors that may impact our valuation include changes to credit ratings of the securities as well as to the underlying assets supporting those securities, rates of default of the underlying assets, underlying collateral value, discount rates, counterparty risk and ongoing strength and quality of market credit and liquidity. In the event we need to access the ARS investments that are in an illiquid state, we will not be able to do so without the possible loss of principal, until a future auction for these investments is successful or they are redeemed by the issuer or they mature. The market value of these securities may decline.
Foreign currency rate fluctuations
We have foreign subsidiaries whose functional currencies are the Great British Pound (GBP), the Japanese Yen (Yen), the Indian Rupee (INR) and the Euro (EUR). The subsidiaries’ accounts are translated from the relevant functional currency to the U.S. dollar using the current exchange rate in effect at the balance sheet date, for balance sheet accounts, and using the average exchange rate during the period for revenues and expense accounts. The effects of translation are recorded as a separate component of stockholders’ equity. Our subsidiaries conduct their business with customers in local currencies. Additionally, we occasionally invoice Australian customers in their local currency. Exchange gains and losses arising from these transactions are recorded using the actual exchange differences on the date of the transaction. We have not taken any action to reduce our exposure to changes in foreign currency exchange rates, such as options or futures contracts, with respect to transactions with our subsidiaries or transactions with our customers where the invoicing currency is not the U.S. dollar.
59
Table of Contents
The table below sets forth our currency exposure (i.e., those transactional exposures that give rise to the net currency gains and losses recognized in the income and expenditure account) on our net monetary assets and liabilities. These exposures consist of our monetary assets and liabilities that are not denominated in the functional currency used by us or our subsidiary having the asset or liability.
| Functional currency of operations |
As of December 31,
2009 Net foreign monetary assets/(liabilities) | |||||
| AUS dollars | Euro | |||||
| ($ in millions) | ||||||
| USD |
$ | 0.9 | $ | 2.5 | ||
A movement of 10% in the U.S. dollar to Australian dollar exchange rate would create an unrealized gain or loss of approximately $85,000. A movement of 10% in the U.S. dollar to EUR exchange rate would create an unrealized gain or loss of approximately $252,000. We had no off balance sheet, or unrecognized, gains and losses in respect of financial instruments used as hedges at the beginning or end of the year ended December 31, 2009. We had no deferred gains or losses during the years ended December 31, 2009, 2008 or 2007.
Inflation
We do not believe that inflation has had a material adverse impact on our business or operating results during the periods presented.
Item 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Our consolidated financial statements and the reports of Ernst & Young LLP, our independent registered public accounting firm, are included in this report on Pages F-l through F-37.
| Item 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
Item 9A. CONTROLS AND PROCEDURES
On April 29, 2009, we announced that the expected launch of our noninvasive prenatal test for Trisomy 21 had been delayed and that we were no longer relying on our previously announced test data and results for that test. We also announced that our board of directors had formed a special committee of independent directors to oversee an independent investigation and that the committee had engaged independent counsel to assist the committee in the conduct of the investigation. The investigation was completed in September 2009. Based on the special committee’s work and recommendations, the independent members of our board of directors concluded that as a result of our attempted transition from researching potential molecular diagnostic tests to developing and commercializing those tests, we failed to put in place adequate protocols and controls for the conduct of studies in the Trisomy 21 program at our company. Certain employees also failed to provide adequate supervision. In the absence of such protocols, controls and supervision, the test data and results in our Trisomy 21 program included inadequately substantiated claims, inconsistencies and errors. Due to deficiencies in our disclosure controls and procedures, in a number of instances such test data and results were reported to the public in our press releases and other public statements.
At the recommendation of the special committee, in 2009 our board of directors began implementing, among other things, new disclosure controls and procedures. Our new enhanced disclosure controls and procedures have been designed to ensure that we record, process, summarize, and report information we are required to disclose in our periodic reports filed with the SEC in the manner and within the time periods specified in the SEC’s rules and forms. Our new enhanced disclosure controls and procedures are also designed to ensure
60
Table of Contents
that the information is accumulated and communicated to our management, including the principal executive officer and the principal financial officer, as appropriate to allow timely decisions regarding required disclosure.
As required by Rule 13a-15(b) under the Exchange Act, we carried out an evaluation, under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures as of the end of the year covered by this report. Based on the foregoing, our principal executive officer and principal financial officer concluded that our disclosure controls and procedures were effective as of December 31, 2009 to ensure that (a) the information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and (b) such information is accumulated and communicated to our management, including our principal executive officer and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure.
Our management does not expect that even our recently enhanced disclosure controls or our internal controls will prevent all error and all fraud. A control system, no matter how well conceived, implemented and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. The design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of a simple error or mistake. Controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the control. The design of any system of controls also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Over time, controls may become inadequate because of changes in conditions, or the degree of compliance with the policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
Management’s Report on Internal Control Over Financial Reporting
Internal control over financial reporting refers to the process designed by, or under the supervision of, our Chief Executive Officer and Interim Chief Financial Officer, and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that:
(1) Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets;
(2) Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorization of our management and directors; and
(3) Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
Internal control over financial reporting cannot provide absolute assurance of achieving financial reporting objectives because of its inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known
61
Table of Contents
features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk. Management is responsible for establishing and maintaining adequate internal control over financial reporting for the company, as defined in Exchange Act Rules 13a-15(f).
Management has used the framework set forth in the report entitled Internal Control-Integrated Framework published by the Committee of Sponsoring Organizations of the Treadway Commission, known as COSO, to evaluate the effectiveness of our internal control over financial reporting as of December 31, 2009. Based on our assessment, management, including our Chief Executive Officer and Interim Chief Financial Officer has concluded that our internal controls over financial reporting were effective as of December 31, 2009. The effectiveness of our internal control over financial reporting as of December 31, 2009 has been audited by Ernst & Young LLP, an independent registered public accounting firm, as stated in their report which is included herein.
Changes in Internal Control Over Financial Reporting
An evaluation was also performed under the supervision and with the participation of our management, including our Chief Executive Officer and Interim Chief Financial Officer, of any change in our internal control over financial reporting that occurred during our last fiscal quarter and that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting. That evaluation did not identify any change in our internal control over financial reporting that occurred during our latest fiscal quarter and that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
62
Table of Contents
Report of Independent Registered Public Accounting Firm
on Internal Control Over Financial Reporting
The Board of Directors and Stockholders of Sequenom, Inc.
We have audited Sequenom, Inc.’s internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Sequenom, Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Sequenom, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2009, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Sequenom, Inc. as of December 31, 2009 and 2008, and the related consolidated statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2009 of Sequenom, Inc. and our report dated March 15, 2010 expressed an unqualified opinion thereon.
/s/ ERNST & YOUNG LLP
San Diego, California
March 15, 2010
63
Table of Contents
On March 13, 2010, we entered into an employment agreement with Harry F. Hixson, Jr. Ph.D., formalizing the terms of our arrangement with Dr. Hixson for his service as our chief executive officer as previously disclosed by us in Current Reports filed with the SEC on September 28, 2009, October 7, 2009 and October 26, 2009.
Pursuant to the terms of the employment agreement, Dr. Hixson’s annual salary for service as our chief executive officer has been set at $475,000 and the target level for Dr. Hixson’s annual bonus is set at 50% of his base salary. For his service as our chief executive officer, on October 21, 2009 Dr. Hixson was granted pursuant to our 2006 Equity Incentive Plan two stock options, each with an exercise price per share equal to $3.39, which was the fair market value of our common stock on the date of grant. The first is an option to purchase 187,500 shares, vesting in 12 equal monthly installments from September 28, 2009 so long as Dr. Hixson provides service to us. The second is an option to purchase 187,500 shares, vesting in 12 equal monthly installments beginning September 28, 2010 so long as Dr. Hixson is serving as our chief executive officer. Dr. Hixson was also promised an additional stock option to be granted on or after September 28, 2010. This additional stock option will have an exercise price equal to the fair market value of our common stock on the date of grant and will be for a number of shares equal to the difference between 187,500 and the number determined by multiplying the fully diluted shares outstanding on the date of grant by the percentage that 187,500 shares represented of the fully diluted shares outstanding on October 21, 2009. Pursuant to the terms of the employment agreement, Dr. Hixson was also granted restricted stock units covering 225,000 shares, which will vest upon the achievement of specific performance-based milestones established by our board of directors, consistent with those established for all of our employees.
If Dr. Hixson’s employment is terminated by us, or by Dr. Hixson upon giving four weeks advanced notice, Dr. Hixson is entitled to continued health benefits for 90 days from the termination date and a proration of any target bonus determined to be appropriate after completion of the entire target bonus year.
The description of the employment agreement set forth above is qualified in its entirety by reference to the actual terms of the employment agreement, which is filed as an exhibit to this annual report on Form 10-K.
64
Table of Contents
Certain information required by Part III is omitted from this report because we will file with the Securities and Exchange Commission a definitive proxy statement within 120 days after the end of our fiscal year for our annual meeting of stockholder (Proxy Statement), and the information included in the Proxy Statement is incorporated herein by reference.
Item 10. DIRECTORS, AND EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this item is incorporated by reference to our Proxy Statement under the heading “Election of Directors.” Information regarding executive officers is set forth in Item 1 of Part I of this report and is incorporated herein by reference.
We have adopted a code of business conduct and ethics for directors, officers (including our principal executive, financial and accounting officers) and all employees, which we refer to as our Code of Business Conduct and Ethics. The Code of Business Conduct and Ethics is available on our website at http://www.sequenom.com. Stockholders may request a free copy of our Code of Business Conduct and Ethics from:
Sequenom, Inc.
Attention: Investor Relations
3595 John Hopkins Court
San Diego, CA 92121-1331
(858) 202-9000
If we make any substantive amendments to the code of business conduct and ethics or grant any waiver from a provision of the code to any executive officer or director, we will promptly disclose the nature of the amendment or waiver on our website. We will promptly disclose on our website (i) the nature of any amendment to the policy that applies to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions and (ii) the nature of any waiver, including an implicit waiver, from a provision of the policy that is granted to one of these specified individuals, the name of such person who is granted the waiver and the date of the waiver.
Section 16(a) Beneficial Ownership Reporting Compliance
Item 405 of Regulation S-K calls for disclosure of any known late filing or failure by an insider to file a report required by Section 16 of the Exchange Act. This disclosure is incorporated by reference from the information in the section entitled “Section 16(a) Beneficial Ownership Reporting Compliance” in the Proxy Statement.
Item 11. EXECUTIVE COMPENSATION
The information required by this item is incorporated herein by reference from the information in the section entitled “Executive Compensation” in the Proxy Statement.
| Item 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required by this item is incorporated herein by reference from the information in the sections entitled “Security Ownership of Certain Beneficial Owners and Management” and “Securities Authorized for Issuance under Equity Compensation Plans” in the Proxy Statement.
65
Table of Contents
| Item 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS AND DIRECTOR INDEPENDENCE |
The information required by this item is incorporated herein by reference from the information in the sections entitled “Certain Transactions” and “Independence of the Board of Directors” in the Proxy Statement.
| Item 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The information required by this item is incorporated herein by reference from the information in the section entitled “Principal Accountant Fees and Services” in the Proxy Statement.
66
Table of Contents
Item 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
| (a)(1) | Financial Statements |
The financial statements of Sequenom, Inc. are included herein as required under Item 8 of this report. See Index to Financial Statements on page F-l.
| (a)(2) | Financial Statement Schedules |
Schedule II—Valuation and Qualifying Accounts. The other financial statement schedules have been omitted because they are either not required, not applicable, or the information is otherwise included.
| (3) | Exhibits |
The exhibits listed below are required by Item 601 of Regulation S-K. Each management contract or compensatory plan or arrangement required to be filed as an exhibit to this report has been identified.
| Exhibit Number |
Description of Document | |
| 3.1(1) | Restated Certificate of Incorporation of the Registrant. | |
| 3.2(2) | Restated bylaws of Registrant, as amended. | |
| 3.3(3) | Registrant’s Certificate of Designation of Series A Junior Participating Preferred Stock. | |
| 4.1(1) | Specimen common stock certificate. | |
| 4.2(3) | Rights Agreement dated as of March 3, 2009, between the Registrant and American Stock Transfer and Trust Company, LLC. | |
| 4.3(3) | Form of Right Certificate. | |
| 10.1(4) | Form of Warrant Agreement between the Registrant and holders of the Series C Preferred Stock warrants. | |
| 10.2(1) | Form of Indemnification Agreement between the Registrant and each of its officers and directors. | |
| 10.3(4)# | 1994 Stock Plan. | |
| 10.4(4)# | 1994 Stock Plan Form of Non-Qualified Stock Option Grant. | |
| 10.5(4)# | 1994 Stock Plan Form of Incentive Stock Option Grant. | |
| 10.6(4)# | 1994 Stock Plan Form of Stock Restriction Agreement. | |
| 10.7(4)# | 1998 Stock Option/Stock Issuance Plan. | |
| 10.8(4)# | 1998 Stock Option/Stock Issuance Plan Form of Notice of Grant of Stock Option. | |
| 10.9(4)# | 1998 Stock Option/Stock Issuance Plan Form of Stock Option Agreement. | |
| 10.10(4)# | 1998 Stock Option/Stock Issuance Plan Form of Stock Purchase Agreement. | |
| 10.11(4)# | 1998 Stock Option/Stock Issuance Plan Form of Stock Issuance Agreement. | |
| 10.12(5)# | 1999 Stock Incentive Plan, as amended. | |
| 10.13(4)# | 1999 Stock Incentive Plan Form of Notice of Grant of Stock Option. | |
| 10.14(4)# | 1999 Stock Incentive Plan Form of Stock Option Agreement. | |
| 10.15(6)# | 1999 Employee Stock Purchase Plan, as amended. | |
| 10.16(7)# | 2006 Equity Incentive Plan, as amended. | |
| 10.17(1)# | 2006 Equity Incentive Plan Form of Stock Option Grant Notice. | |
| 10.18(1)# | 2006 Equity Incentive Plan Form of Stock Option Agreement. | |
67
Table of Contents
| Exhibit Number |
Description of Document | |
| 10.19(8)# | 2006 Equity Incentive Plan Form of Notice of Exercise. | |
| 10.20(9)# | 2006 Equity Incentive Plan Form of Restricted Stock Unit Award Grant Notice. | |
| 10.21(9)# | 2006 Equity Incentive Plan Form of Restricted Stock Unit Award Agreement. | |
| 10.22(10) | Business Loan Agreement, dated March 3, 2000, between the Registrant and Union Bank of California. | |
| 10.23(11) | Building Lease Agreement, dated March 29, 2000, between the Registrant and TPSC IV LLC. | |
| 10.24(12)# | Employment Agreement, dated September 15, 2005, by and between Registrant and Charles Cantor, Ph.D. | |
| 10.25(13)# | Form of Medical Expense Reimbursement Exec-U-Care Plan. | |
| 10.26(14)# | Employment Agreement, dated July 19, 2004, by and between the Registrant and Clarke Neumann. | |
| 10.27(15)* | Diagnostic Platform Benchmarking Study and Evaluation Agreement, dated October 25, 2004, by and between the Registrant and Siemens AG. | |
| 10.28(15)# | Form of Stock Issuance Agreement under 1999 Stock Incentive Plan. | |
| 10.29(16) | Amendment Number One to Lease dated March 29, 2000, by and between the Registrant and TPSC IV LLC dated September 9, 2005. | |
| 10.30(16) | Common Stock Warrant, dated September 9, 2005, issued to Kwacker, Ltd. | |
| 10.31(16)# | Employment Agreement Amendment, dated September 12, 2005, by and between the Registrant and Dr. Charles R. Cantor. | |
| 10.32(17)* | Exclusive License of Technology Agreement, dated October 14, 2005, by and between the Registrant and ISIS Innovation Limited. | |
| 10.33(18) | Amended and Restated Securities Purchase Agreement, dated March 30, 2006, by and among the Registrant, ComVest Investment Partners II LLC, LB I Group Inc., Pequot Private Equity Fund IV, L.P. and Siemens Venture Capital GmbH. | |
| 10.34(18) | Form of Warrant issued pursuant to the Amended and Restated Securities Purchase Agreement dated March 30, 2006. | |
| 10.35(19)# | Letter agreement dated April 6, 2006, by and between the Registrant and John E. Lucas. | |
| 10.36(1) | Registration Rights Agreement dated June 6, 2006 by and between the Registrant, ComVest Investment Partners II LLC, LB I Group Inc., Pequot Private Equity Fund IV, L.P. and Siemens Venture Capital GmbH. | |
| 10.37(20)* | Amendment to Exclusive License of Technology Agreement dated October 19, 2006, by and between the Registrant and ISIS Innovation Limited. | |
| 10.38(20)* | Supply Agreement dated November 3, 2006, by and between the Registrant and Bruker Daltonics Inc. | |
| 10.39(21)# | Form of Restricted Stock Bonus Grant Notice under 2006 Equity Incentive Plan. | |
| 10.40(21)# | Form of Restricted Stock Bonus Agreement under 2006 Equity Incentive Plan. | |
| 10.41(22)* | Collaboration and License Agreement dated January 24, 2007, between the Registrant and Lenetix Medical Screening Laboratory, Inc. | |
| 10.42(23) | Placement Agency Agreement dated April 25, 2007, between the Registrant and Lehman Brothers Inc. | |
| 10.43(24) | Letter agreement dated June 25, 2007, by and between the Registrant and Kathleen Wiltsey. | |
| 10.44(24) | Letter agreement dated July 2, 2007, by and between the Registrant and Richard Alan Lerner, M.D. | |
68
Table of Contents
| Exhibit Number |
Description of Document | |
| 10.45(25) | Form of Purchase Agreement, dated October 25, 2007, by and between the Registrant and the various purchasers of shares of the Registrant’s common stock. | |
| 10.46(26)* | Amendment to Exclusive License of Technology Agreement dated November 5, 2007, by and between the Registrant and ISIS Innovation, Limited. | |
| 10.47(27)# | Non-Employee Director Compensation Policy. | |
| 10.48(27)# | Amended and Restated Change in Control Severance Benefit Plan. | |
| 10.49(27)# | Deferred Compensation Plan, as amended. | |
| 10.50# | New-Hire Equity Incentive Plan. | |
| 10.51* | Amendment to Exclusive License of Technology Agreement dated November 3, 2009, by and between the Registrant and ISIS Innovation Limited. | |
| 10.52* | License Agreement dated February 4, 2010, by and between the Registrant and Optherion, Inc. | |
| 10.53# | Agreement dated March 13, 2010 by and between the Registrant and Harry F. Hixson, Jr., Ph.D. | |
| 10.54# | Letter agreement dated October 21, 2010 by and between the Registrant and Paul V. Maier. | |
| 21.1 | Subsidiaries of the Registrant. | |
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |
| 31.1 | Certification of Principal Executive Officer pursuant to Rule13a-14(a) and Rule 15d-14(a) of the Securities and Exchange Act, as amended. | |
| 31.2 | Certification of Principal Financial Officer pursuant to Rule13a-14(a) and Rule 15d-14(a) of the Securities and Exchange Act, as amended. | |
| 32.1 | Certification of Principal Executive Officer pursuant to 18 U.S.C. 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 32.2 | Certification of Principal Financial Officer pursuant to 18 U.S.C. 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| # | Management contract or compensatory plan. |
| * | Certain confidential portions of this Exhibit have been omitted pursuant to a request for confidential treatment. Omitted portions have been filed separately with the Securities and Exchange Commission. |
| (1) | Incorporated by reference to the Registrant’s Current Report on Form 8-K (No. 000-29101) filed June 6, 2006. |
| (2) | Incorporated by reference to the Registrant’s Current Report on Form 8-K (No. 000-29101) filed January 15, 2010. |
| (3) | Incorporated by reference to the Registrant’s Current Report on Form 8-K (No. 000-29101) filed March 4, 2009. |
| (4) | Incorporated by reference to the Registrant’s Registration Statement on Form S-1 (No. 333-91665), as amended. |
| (5) | Incorporated by reference to the Registrant’s Annual Report on Form 10-K (No. 000-29101) for the year ended December 31, 2006. |
| (6) | Incorporated by reference to the Registrant’s Current Report on Form 8-K (No. 000-29101) filed February 1, 2010. |
| (7) | Incorporated by reference to the Registrant’s Current Report on Form 8-K (No. 000-29101) filed June 2, 2008. |
| (8) | Incorporated by reference to the Registrant’s Current Report on Form 8-K (No. 000-29101) filed June 6, 2006. |
| (9) | Incorporated by reference to the Registrant’s Registration Statement on Form S-8 (No. 333-152230) filed July 10, 2008. |
69
Table of Contents
| (10) | Incorporated by reference to the Registrant’s Annual Report on Form 10-K (No. 000-29101) for the year ended December 31, 1999. |
| (11) | Incorporated by reference to the Registrant’s Quarterly Report on Form 10-Q (No. 000-29101) for the quarter ended March 31, 2000. |
| (12) | Incorporated by reference to the Registrant’s Registration Statement on Form S-1 (No. 333-91665), as amended, which exhibit is hereby supplemented with an additional Schedule A filed with the Registrant’s Annual Report on Form 10-K (No. 000-29101) for the year ended December 31, 2000. |
| (13) | Incorporated by reference to the Registrant’s Annual Report on Form 10-K (No. 000-29101) for the year ended December 31, 2003. |
| (14) | Incorporated by reference to the Registrant’s Annual Report on Form 10-K (No. 000-29101) for the year ended December 31, 2005. |
| (15) | Incorporated by reference to the Registrant’s Quarterly Report on Form 10-Q (No. 000-29101) for the quarter ended September 30, 2004. |
| (16) | Incorporated by reference to the Registrant’s Current Report on Form 8-K (No. 000-29101) filed September 14, 2005. |
| (17) | Incorporated by reference to the Registrant’s Quarterly Report on Form 10-Q (No. 000-29101) for the quarter ended September 30, 2005. |
| (18) | Incorporated by reference to the Registrant’s Current Report on Form 8-K (No. 000-29101) filed April 3, 2006. |
| (19) | Incorporated by reference to the Registrant’s Current Report on Form 8-K (No. 000-29101) filed April 10, 2006. |
| (20) | Incorporated by reference to the Registrant’s Quarterly Report on Form 10-Q (No. 000-29101) for the quarter ended September 30, 2006. |
| (21) | Incorporated by reference to the Registrant’s Current Report on Form 8-K (No. 000-29101) filed January 24, 2007. |
| (22) | Incorporated by reference to the Registrant’s Quarterly Report on Form 10-Q (No. 000-29101) for the quarter ended March 31, 2007. |
| (23) | Incorporated by reference to the Registrant’s Current Report on Form 8-K (No. 000-29101) filed April 25, 2007. |
| (24) | Incorporated by reference to the Registrant’s Quarterly Report on Form 10-Q (No. 000-29101) for the quarter ended June 30, 2007. |
| (25) | Incorporated by reference to the Registrant’s Current Report on Form 8-K (No. 000-29101) filed October 26, 2007. |
| (26) | Incorporated by reference to the Registrant’s Quarterly Report on Form 10-Q (No. 000-29101) for the quarter ended September 30, 2007. |
| (27) | Incorporated by reference to the Registrant’s Annual Report on Form 10-K (No. 000-29101) for the year ended December 31, 2008. |
70
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Date: March 15, 2010
| SEQUENOM, INC. | ||
| By: |
/S/ HARRY F. HIXSON, JR., PH.D. | |
| Harry F. Hixson, Jr., Ph.D. Chief Executive Officer | ||
POWER OF ATTORNEY
Know all men by these presents, that each person whose signature appears below constitutes and appoints Harry F. Hixson and Paul V. Maier, and each of them, as his attorneys-in-fact and agents, each with power of substitution in any and all capacities, to sign any amendments to this annual report on Form 10-K, and to file the same with exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that the attorney-in-fact or his substitute or substitutes may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ HARRY F. HIXSON, JR., PH.D. Harry F. Hixson, Jr., Ph.D. |
Chief Executive Officer and Chairman of the Board of Directors (Principal Executive Officer) |
March 15, 2010 | ||
| /s/ PAUL V. MAIER Paul V. Maier |
Interim Chief Financial Officer (Principal |
March 15, 2010 | ||
| /s/ CHARLES R. CANTOR, PH.D. Charles R. Cantor, Ph.D. |
Chief Scientific Officer and Director |
March 15, 2010 | ||
| /s/ ERNST-GUNTER AFTING, PH.D., M.D. Ernst-Gunter Afting, Ph.D., M.D. |
Director |
March 15, 2010 | ||
| /s/ KENNETH F. BUECHLER, PH.D. Kenneth F. Buechler, Ph.D. |
Director |
March 15, 2010 | ||
| /s/ JOHN A. FAZIO John A. Fazio |
Director |
March 15, 2010 | ||
| /s/ RICHARD A. LERNER, M.D. Richard A. Lerner, M.D. |
Director |
March 15, 2010 | ||
| /s/ RONALD M. LINDSAY, PH.D. Ronald M. Lindsay, Ph.D. |
Interim Senior Vice President of Research and Development and Director |
March 15, 2010 | ||
| /s/ DAVID PENDARVIS David Pendarvis |
Director |
March 15, 2010 | ||
| /s/ KATHLEEN M. WILTSEY Kathleen M. Wiltsey |
Director |
March 15, 2010 | ||
71
Table of Contents
SEQUENOM, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
F-1
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of
Sequenom, Inc.
We have audited the accompanying consolidated balance sheets of Sequenom, Inc. as of December 31, 2009 and 2008, and the related consolidated statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2009. Our audits also included the financial statement schedule listed in the Index at Item 15(a)(2). These financial statements and schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Sequenom, Inc. at December 31, 2009 and 2008, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2009, in conformity with U.S. generally accepted accounting principles.
The accompanying financial statements have been prepared assuming that Sequenom, Inc. will continue as a going concern. As more fully described in Note 2, the Company has incurred recurring operating losses and does not have sufficient working capital to fund operations through 2010. This condition raises substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to this matter is also described in Note 2. The 2009 financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities that may result from the outcome of this uncertainty.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Sequenom, Inc.’s internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 15, 2010 expressed an unqualified opinion thereon.
/s/ ERNST & YOUNG LLP
San Diego, California
March 15, 2010
F-2
Table of Contents
CONSOLIDATED BALANCE SHEETS
(In thousands, except share and per share information)
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 26,919 | $ | 68,338 | ||||
| Marketable securities |
15,762 | 29,991 | ||||||
| Restricted cash |
1,419 | 1,371 | ||||||
| Accounts receivable, net |
8,510 | 10,642 | ||||||
| Inventories, net |
7,722 | 10,631 | ||||||
| Other current assets and prepaid expenses |
2,598 | 1,311 | ||||||
| Total current assets |
62,930 | 122,284 | ||||||
| Equipment and leasehold improvements, net |
11,811 | 9,195 | ||||||
| Intangible assets, net |
1,172 | 114 | ||||||
| Goodwill |
10,007 | 2,398 | ||||||
| Marketable securities |
— | 5,748 | ||||||
| Other assets |
725 | 745 | ||||||
| Total assets |
$ | 86,645 | $ | 140,484 | ||||
| Liabilities and stockholders’ equity |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 6,064 | $ | 8,321 | ||||
| Accrued expenses |
8,202 | 8,626 | ||||||
| Deferred revenue |
1,871 | 1,444 | ||||||
| Current portion of debt and obligations |
1,320 | 647 | ||||||
| Total current liabilities |
17,457 | 19,038 | ||||||
| Deferred revenue, less current portion |
304 | 454 | ||||||
| Other long-term liabilities |
3,389 | 4,205 | ||||||
| Long-term portion of debt and obligations |
1,837 | 574 | ||||||
| Commitments and contingencies |
||||||||
| Stockholders’ equity: |
||||||||
| Convertible preferred stock, par value $0.001; authorized shares—5,000,000, no shares issued or outstanding at December 31, 2009 or 2008, respectively. |
— | — | ||||||
| Common stock, par value $0.001; authorized shares—185,000,000, issued and outstanding shares 61,988,473 and 60,943,469 at December 31, 2009 and 2008, respectively |
62 | 61 | ||||||
| Additional paid-in capital |
659,798 | 641,098 | ||||||
| Accumulated other comprehensive income |
1,084 | 1,328 | ||||||
| Accumulated deficit |
(597,286 | ) | (526,274 | ) | ||||
| Total stockholders’ equity |
63,658 | 116,213 | ||||||
| Total liabilities and stockholders’ equity |
$ | 86,645 | $ | 140,484 | ||||
See accompanying notes.
F-3
Table of Contents
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except per share information)
| Year ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Revenues: |
||||||||||||
| Consumables |
$ | 20,534 | $ | 19,535 | $ | 16,530 | ||||||
| MassARRAY and other product related |
14,999 | 22,724 | 20,835 | |||||||||
| Services |
2,209 | 4,817 | 3,524 | |||||||||
| Diagnostic |
94 | — | — | |||||||||
| Research and other |
27 | 73 | 113 | |||||||||
| Total revenues |
37,863 | 47,149 | 41,002 | |||||||||
| Costs and expenses: |
||||||||||||
| Cost of consumables and products revenue |
11,980 | 15,109 | 14,594 | |||||||||
| Cost of services revenue |
2,178 | 4,481 | 3,483 | |||||||||
| Cost of diagnostic revenue |
412 | — | — | |||||||||
| Research and development |
37,454 | 27,455 | 14,352 | |||||||||
| Selling and marketing |
26,845 | 24,299 | 17,015 | |||||||||
| General and administrative |
28,127 | 18,436 | 14,133 | |||||||||
| Restructuring |
1,589 | — | — | |||||||||
| Total costs and expenses |
108,585 | 89,780 | 63,577 | |||||||||
| Loss from operations |
(70,722 | ) | (42,631 | ) | (22,575 | ) | ||||||
| Interest income |
442 | 1,592 | 1,781 | |||||||||
| Loss on marketable securities |
(1,914 | ) | (2,584 | ) | (1,071 | ) | ||||||
| Interest expense |
(261 | ) | (139 | ) | (17 | ) | ||||||
| Other income (expense), net |
1,560 | (181 | ) | (101 | ) | |||||||
| Loss before income tax |
(70,895 | ) | (43,943 | ) | (21,983 | ) | ||||||
| Income tax expense |
(117 | ) | (211 | ) | — | |||||||
| Net loss |
$ | (71,012 | ) | $ | (44,154 | ) | $ | (21,983 | ) | |||
| Net loss per share, basic and diluted |
$ | (1.16 | ) | $ | (0.83 | ) | $ | (0.57 | ) | |||
| Weighted average shares outstanding, basic and diluted |
61,171 | 53,129 | 38,865 | |||||||||
See accompanying notes.
F-4
Table of Contents
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(In thousands, except share information)
| Common Stock | Additional Paid-In Capital |
Other Comprehensive Income |
Accumulated Deficit |
Total Stockholders’ Equity |
||||||||||||||||
| Shares | Amount | |||||||||||||||||||
| Balance at December 31, 2006 |
33,439,634 | $ | 33 | $ | 484,898 | $ | 656 | $ | (460,137 | ) | $ | 25,450 | ||||||||
| Net loss |
— | — | — | — | (21,983 | ) | (21,983 | ) | ||||||||||||
| Unrealized loss on available-for-sale securities |
— | — | — | (804 | ) | — | (804 | ) | ||||||||||||
| Translation adjustment |
— | — | — | 467 | — | 467 | ||||||||||||||
| Comprehensive loss |
— | — | — | — | — | (22,320 | ) | |||||||||||||
| Share-based compensation |
— | — | 3,058 | — | — | 3,058 | ||||||||||||||
| Exercise of stock options |
165,536 | — | 446 | — | — | 446 | ||||||||||||||
| Exercise of warrants |
1,197,012 | 1 | 1,255 | — | — | 1,256 | ||||||||||||||
| Purchases under Employee Stock Purchase Plan |
36,473 | — | 102 | — | — | 102 | ||||||||||||||
| Issuance of common stock, net of issuance costs |
10,050,001 | 10 | 46,263 | — | — | 46,273 | ||||||||||||||
| Balance at December 31, 2007 |
44,888,656 | $ | 44 | $ | 536,022 | $ | 319 | $ | (482,120 | ) | $ | 54,265 | ||||||||
| Net loss |
— | — | — | — | (44,154 | ) | (44,154 | ) | ||||||||||||
| Unrealized gain on available-for-sale securities |
— | — | — | 898 | — | 898 | ||||||||||||||
| Translation adjustment |
— | — | — | 111 | — | 111 | ||||||||||||||
| Comprehensive loss |
— | — | — | — | — | (43,145 | ) | |||||||||||||
| Share-based compensation |
— | — | 7,276 | — | — | 7,276 | ||||||||||||||
| Exercise of stock options and restricted stock |
340,936 | 1 | 1,718 | — | — | 1,719 | ||||||||||||||
| Exercise of warrants |
9,093,302 | 9 | 139 | — | — | 148 | ||||||||||||||
| Purchases under Employee Stock Purchase Plan |
107,781 | — | 568 | — | — | 568 | ||||||||||||||
| Issuance of common stock, net of issuance costs |
6,512,794 | 7 | 95,375 | — | — | 95,382 | ||||||||||||||
| Balance at December 31, 2008 |
60,943,469 | $ | 61 | $ | 641,098 | $ | 1,328 | $ | (526,274 | ) | $ | 116,213 | ||||||||
| Net loss |
— | — | — | — | (71,012 | ) | (71,012 | ) | ||||||||||||
| Unrealized gain on available-for-sale securities |
— | — | — | 2 | — | 2 | ||||||||||||||
| Translation adjustment |
— | — | — | (246 | ) | — | (246 | ) | ||||||||||||
| Comprehensive loss |
— | — | — | — | — | (71,256 | ) | |||||||||||||
| Share-based compensation |
— | — | 11,814 | — | — | 11,814 | ||||||||||||||
| Exercise of stock options and restricted stock |
503,377 | 1 | 2,664 | — | — | 2,665 | ||||||||||||||
| Purchases under Employee Stock Purchase Plan |
81,401 | — | 760 | — | — | 760 | ||||||||||||||
| Issuance of common stock, net of issuance costs |
460,226 | — | 3,462 | — | — | 3,462 | ||||||||||||||
| Balance at December 31, 2009 |
61,988,473 | $ | 62 | $ | 659,798 | $ | 1,084 | $ | (597,286 | ) | $ | 63,658 | ||||||||
F-5
Table of Contents
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| Year ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Operating activities |
||||||||||||
| Net loss |
$ | (71,012 | ) | $ | (44,154 | ) | $ | (21,983 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Stock-based compensation |
11,814 | 7,276 | 3,058 | |||||||||
| Depreciation and amortization |
5,201 | 2,893 | 1,940 | |||||||||
| Loss on marketable securities |
1,914 | 2,584 | 1,071 | |||||||||
| Loss on disposal of fixed assets |
193 | 232 | — | |||||||||
| Settlement of SensiGen, LLC claim |
1,522 | — | — | |||||||||
| Contingent consideration fair value adjustment |
514 | — | — | |||||||||
| Bad debt expense |
(139 | ) | 400 | 142 | ||||||||
| Restricted stock |
455 | 161 | — | |||||||||
| Deferred rent |
(569 | ) | (442 | ) | 1,631 | |||||||
| Other non-cash items |
7 | 41 | 462 | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Accounts receivable |
1,892 | (115 | ) | (6,044 | ) | |||||||
| Inventories |
2,923 | (6,492 | ) | (1,565 | ) | |||||||
| Other current assets and prepaid expenses |
(1,280 | ) | (212 | ) | (396 | ) | ||||||
| Other assets |
14 | (56 | ) | (107 | ) | |||||||
| Accounts payable and accrued expenses |
(1,733 | ) | 2,471 | 4,975 | ||||||||
| Deferred revenue |
267 | 686 | (554 | ) | ||||||||
| Other liabilities |
(691 | ) | 111 | (52 | ) | |||||||
| Net cash used in operating activities |
(48,708 | ) | (34,616 | ) | (17,422 | ) | ||||||
| Investing activities |
||||||||||||
| Purchase of equipment, leasehold improvements, and intangible assets |
(8,699 | ) | (4,878 | ) | (3,513 | ) | ||||||
| Restricted cash |
(49 | ) | (41 | ) | 75 | |||||||
| Acquisition of CMM, LLC |
— | (400 | ) | — | ||||||||
| Acquisition of SensiGen, LLC, net of cash acquired |
(2,017 | ) | — | — | ||||||||
| Purchases of marketable securities |
(30,297 | ) | (44,483 | ) | (70,781 | ) | ||||||
| Sales of marketable securities |
3,363 | 24,012 | (47,648 | ) | ||||||||
| Maturities of marketable securities |
45,000 | 21,683 | 5,621 | |||||||||
| Net cash provided by (used in) investing activities |
7,301 | (4,107 | ) | (20,950 | ) | |||||||
| Financing activities |
||||||||||||
| Repayment of long-term debt |
(1,576 | ) | (637 | ) | (70 | ) | ||||||
| Proceeds from long-term debt |
— | 610 | 1,318 | |||||||||
| Payments on capital lease obligations |
(80 | ) | — | — | ||||||||
| Proceeds from issuance of common stock, net of issuance costs |
— | 91,782 | 46,273 | |||||||||
| Proceeds from exercise of warrants, stock options and ESPP purchases |
1,905 | 2,011 | 1,803 | |||||||||
| Net cash provided by financing activities |
249 | 93,766 | 49,324 | |||||||||
| Net (decrease) increase in cash and cash equivalents |
(41,158 | ) | 55,043 | 10,952 | ||||||||
| Effect of exchange rate changes on cash and cash equivalents |
(261 | ) | 179 | 232 | ||||||||
| Cash and cash equivalents at beginning of year |
68,338 | 13,116 | 1,932 | |||||||||
| Cash and cash equivalents at end of year |
$ | 26,919 | $ | 68,338 | $ | 13,116 | ||||||
| Supplemental disclosure of cash flow information: |
||||||||||||
| Interest paid |
$ | 247 | $ | 134 | $ | 12 | ||||||
| Supplemental disclosure of non cash investing activities: |
||||||||||||
| Equipment purchased under capital lease obligation |
$ | 366 | $ | — | $ | — | ||||||
| Common stock issued for acquisition |
$ | — | $ | 3,600 | $ | — | ||||||
See accompanying notes.
F-6
Table of Contents
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2009
1. Nature of the Business
We are a molecular diagnostic testing and genetics analysis company committed to providing products, services, diagnostic testing, applications and genetic analysis products that translate the results of genomic science into solutions for biomedical research, translational research, molecular medicine applications, and agricultural, livestock, and other areas of research. Our development and commercialization efforts in various diagnostic areas include noninvasive women’s health related and prenatal diagnostics, age-related macular degeneration diagnostics, oncology, infectious diseases, and other disorders and diseases.
2. Summary of Significant Accounting Policies and Significant Accounts
Basis of Presentation and Consolidation
The accompanying consolidated financial statements have been prepared in conformity with U.S. generally accepted accounting principles (GAAP) and include the accounts of Sequenom, Inc. and our wholly-owned subsidiaries located in the United States, Germany, the United Kingdom, Japan, India and Hong Kong. All significant intercompany accounts and transactions are eliminated in consolidation. Our consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the settlement of liabilities and commitments in the normal course of business. Through December 31, 2009, we had accumulated losses of approximately $597.3 million. Management expects to incur further losses for the foreseeable future. We believe based on our current projections that our cash, cash equivalents and marketable securities at December 31, 2009 will not be sufficient to fund our obligations through the third quarter of 2010 and intend to pursue raising additional capital during 2010. Until we can generate sufficient levels of cash from our operations, we expect to continue to finance future cash needs primarily through proceeds from equity or debt financings, loans and/or collaborative agreements with partners in order to be able to sustain our operations until we can achieve profitability and positive cash flows, if ever.
We plan to seek additional debt and/or equity financing through private or public offerings, but we cannot assure you that such financing or transaction will be available on acceptable terms, or at all. The uncertainty of this situation raises substantial doubt about our ability to continue as a going concern. The accompanying consolidated financial statements do not include any adjustments relating to the recoverability and classification of assets or the amount and classification of liabilities that might result from the outcome of this uncertainty.
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles requires us to make certain estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Goodwill and Purchased Intangible Assets
Goodwill is recorded when the consideration paid for an acquisition exceeds the fair value of the identified net tangible and intangible assets acquired. The allocation of purchase price for acquisitions requires extensive use of accounting estimates and judgments to allocate the purchase price to the identifiable tangible and intangible assets acquired and liabilities assumed based on their respective fair values. Additionally, we must determine whether an acquired entity is considered to be a business or a set of net assets, because a portion of the purchase price can only be allocated to goodwill in a business combination. Goodwill and intangible assets
F-7
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
deemed to have indefinite lives are not amortized, but are subject to annual impairment tests. The amounts and useful lives assigned to other intangible assets, such as lab accreditations, patent rights and licenses, requires the use of estimates and the exercise of judgment. These judgments can significantly affect our net operating results. As of December 31, 2009 and 2008, we have goodwill recorded of $10.0 million and $2.4 million, respectively.
We annually evaluate our goodwill and purchased intangibles at the reporting unit level during the fourth quarter each fiscal year or more frequently if we believe indicators of impairment are present. Goodwill and certain intangible assets are assessed for impairment using fair value measurement techniques. Specifically, goodwill impairment is determined using a two-step process. The first step of the goodwill impairment test is used to identify potential impairment by comparing the fair value of a reporting unit with its carrying amount, including goodwill. Goodwill is allocated to reporting units based upon the type of products under development by the acquired company, which initially generated the goodwill. If the fair value of a reporting unit exceeds its carrying amount, goodwill of the reporting unit is considered not impaired and the second step of the impairment test is unnecessary. If the carrying amount of a reporting unit exceeds its fair value, the second step of the goodwill impairment test is performed to measure the amount of impairment loss. The second step of the goodwill impairment test compares the implied fair value of the reporting unit’s goodwill with the carrying amount of that goodwill. If the carrying amount of the reporting unit’s goodwill exceeds the implied fair value of that goodwill, an impairment loss is recognized in an amount equal to that excess. The implied fair value of goodwill is determined in the same manner as the amount of goodwill recognized in a business combination. That is, the fair value of the reporting unit is allocated to all of the assets and liabilities of that unit (including any unrecognized intangible assets) as if the reporting unit had been acquired in a business combination and the fair value of the reporting unit was the purchase price paid to acquire the reporting unit. The fair value is determined using a combination of the discounted cash flow analysis as well as market comparisons. The determination of fair values requires management to make significant judgment and estimates.
We periodically re-evaluate the original assumptions and rationale utilized in the establishment of the carrying value and estimated lives of our long-lived assets. The criteria used for these evaluations include management’s estimate of the asset’s continuing ability to generate income from operations and positive cash flows in future periods as well as the strategic significance of any intangible assets in our business objectives. If assets are considered to be impaired, the impairment recognized is the amount by which the carrying value of the assets exceeds the fair value of the assets.
Reserves for Obsolete and Slow-moving Inventory
We operate in an industry characterized by rapid improvements and changes to our technology and products. The introduction of new products by us or our competitors can result in our inventory being rendered obsolete or requiring us to sell items at a discount. We estimate the recoverability of our inventory by reference to our internal estimates of future demands and product life cycles. If we incorrectly forecast demand for our products or inadequately manage the introduction of new product lines, we could materially impact our consolidated financial statements by having excess inventory on hand. Our future estimates are subjective and could be incorrect. During 2009, slow-moving and obsolete inventory reserves of $0.1 million were charged against cost of goods sold and the total reserve was $1.9 million and $1.8 million at December 31, 2009 and 2008, respectively.
Shipping and Handling Costs
Shipping and handling costs are included within cost of product revenue on the statement of operations.
F-8
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
Cash and Cash Equivalents
Cash equivalents consist of short-term, highly liquid investments with original maturities of three months or less when purchased.
Marketable Securities
The classification of marketable securities is determined by management at the time of purchase and reevaluated as of each balance sheet date. As of December 31, 2009 and 2008, all of our investments in marketable securities were classified as available for sale and were reported at fair value. We measure fair value based on the prices that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. Fair value is determined based on observable market quotes or valuation models using assessments of counterparty credit worthiness, credit default risk or underlying security and overall capital market liquidity. Declines in fair value that are considered other-than-temporary are charged to operations and those that are considered temporary are reported as a component of accumulated other comprehensive income (loss) in stockholders’ equity. We use the specific identification method of determining the cost basis in computing realized and unrealized gains and losses on the sale of our available-for-sale securities.
Historically, we have invested in auction rate securities (ARS), commercial paper of prime quality, certificates of deposit, guaranteed bankers acceptance and U.S. Government instruments, and by policy, limit the amount of credit exposure to any one issuer. In April 2008, we revised our investment policy to maintain our portfolio of cash equivalents and marketable securities in a variety of securities, including U.S. Government securities with ratings of AAA, and repurchase agreements collateralized by U.S. Government securities with ratings of AAA at the time of acquisition. Our investment policy includes a minimum quality rating for all new investments, as well as limits the amount of credit exposure to only issuances from the U.S. Government. If an investment we hold falls below this level, we research the reasons for the fall and determine if we should continue to hold the investment in order to minimize our exposure to market risk of the investment. At December 31, 2009 and 2008, $4.3 million and $9.4 million of principal was invested in ARS, respectively. The ARS held are private placement securities with various long-term nominal maturities with interest rates reset through a Dutch auction each month, except for one ARS that resets every 92 days. The monthly auctions historically have provided a liquid market for these securities. The investments in ARS represent interests in collateralized debt obligations supported by insurance securitizations and other structured credits, including corporate bonds and to a lesser degree, pools of residential and commercial mortgages.
Consistent with our investment policy guidelines in affect when originally purchased, the ARS investments held by us all had AAA/AA credit ratings at the time of purchase. With the liquidity issues experienced in global credit and capital markets, the $4.3 million ARS held by us at December 31, 2009, had experienced multiple failed auctions as the amount of securities submitted for sale has exceeded the amount of purchase orders and we have been unable to liquidate these securities. Subsequent to year end, $1.3 million of our remaining ARS with a fair value of $0.5 million as of December 31, 2009, were sold. The proceeds for these ARS were equal to their estimated fair value as of December 31, 2009. See footnote 5 “Marketable Securities and Fair Value Measurements” for further discussion.
Restricted Cash
Restricted cash and investments of $1.4 million as of December 31, 2009 and 2008 are held in interest bearing cash accounts with restrictions on withdrawal, in support of certain borrowing agreements and stand-by letters of credit.
F-9
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
Concentration of Risks
We grant credit generally on an unsecured basis to customers throughout North America, Europe, and Asia. We establish an allowance for doubtful accounts based upon factors surrounding the credit risk of specific customers, historical trends, and other information. To reduce credit risk, certain sales are secured by letters of credit from commercial banks. The regional concentration of accounts receivables were as follows:
| Region |
December 31, 2009 |
Percent of receivable balance |
December 31, 2008 |
Percent of receivable balance |
||||||||
| (In thousands) | ||||||||||||
| Europe |
$ | 2,921 | 34 | % | $ | 3,624 | 34 | % | ||||
| Asia |
1,906 | 23 | % | 3,467 | 33 | % | ||||||
| North America |
3,683 | 43 | % | 3,551 | 33 | % | ||||||
| Total |
$ | 8,510 | 100 | % | $ | 10,642 | 100 | % | ||||
Our Asia-based major distributors represented $8.7 million and $10.1 million, or 23% and 24%, of our total product revenues during the year ended December 31, 2009 and 2008, respectively. At December 31, 2009, no customer had a year end accounts receivable balance greater than 10% of the total balance outstanding and no customer represented more than 10% of total world-wide revenue for the year ended December 31, 2009.
Our products incorporate components that are available from only one or a limited number of suppliers. Many of these components are manufactured with lead times, which can be significant. Shortages of various essential materials could occur due to interruption of supply. If we were unable to procure certain such components from suppliers or sub-contractors, it could affect our ability to meet demand for our products, which would have an adverse effect upon our results.
Inventories
Inventories are stated at the lower of cost (first-in, first-out) or market value (net realizable value). The components of inventories were as follows:
| December 31, | ||||||
| 2009 | 2008 | |||||
| (In thousands) | ||||||
| Raw materials |
$ | 4,657 | $ | 7,560 | ||
| Work in process |
11 | 282 | ||||
| Finished goods |
3,054 | 2,789 | ||||
| Total |
$ | 7,722 | $ | 10,631 | ||
Inventories are shown net of excess and obsolescence reserves of $1.9 million and $1.8 million at December 31, 2009 and 2008, respectively.
Equipment and Leasehold Improvements
Equipment is stated at cost and depreciated using the straight-line method over the estimated useful lives of the assets (generally 3 to 5 years). Leasehold improvements are amortized using the straight-line method over the
F-10
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
estimated useful life of the improvement or the remaining term of the lease, whichever is shorter. The maximum estimated useful life of any leasehold improvement is 15 years from the completion of the improvement. Maintenance and repairs are charged to operations as incurred. When assets are sold, or otherwise disposed of, the cost and related accumulated depreciation are removed from the accounts and any gain or loss is included in operating expense.
Equipment and leasehold improvements and related accumulated depreciation and amortization were as follows:
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| (In thousands) | ||||||||
| Laboratory equipment |
$ | 21,610 | $ | 18,793 | ||||
| Leasehold improvements |
5,092 | 4,537 | ||||||
| Office furniture and equipment |
10,980 | 7,670 | ||||||
| 37,682 | 31,000 | |||||||
| Less accumulated depreciation and amortization |
(25,871 | ) | (21,805 | ) | ||||
| $ | 11,811 | $ | 9,195 | |||||
Depreciation expense for the years ended December 31, 2009, 2008 and 2007 was $5.1 million, $2.8 million, and $1.7 million, respectively, and includes $92,000 of depreciation on equipment under capital lease.
Intangible Assets
Intangible assets consisted of the following:
| December 31, 2009 | December 31, 2008 | |||||||||||||||
| Weighted Average Life |
Gross Carrying Amount |
Accumulated Amortization |
Gross Carrying Amount |
Accumulated Amortization |
||||||||||||
| (In thousands) | ||||||||||||||||
| Clinical data collections |
5 | $ | 13,552 | $ | (13,552 | ) | $ | 13,552 | $ | (13,552 | ) | |||||
| Purchased patent rights and licenses |
3 | 5,621 | (4,539 | ) | 4,449 | (4,449 | ) | |||||||||
| Lab accreditation |
5 | 117 | (27 | ) | 117 | (3 | ) | |||||||||
| Total |
$ | 19,290 | $ | (18,118 | ) | $ | 18,118 | $ | (18,004 | ) | ||||||
Intangible assets are amortized using the straight-line method over their estimated useful lives. Amortization of intangible assets for the years ended December 31, 2009, 2008 and 2007 was $0.1 million, $0.1 million, and $0.3 million, respectively.
The following table is a schedule of future estimated amortization expense at December 31, 2009:
| (in thousands) | |||
| 2010 |
$ | 391 | |
| 2011 |
391 | ||
| 2012 |
336 | ||
| 2013 |
54 | ||
| Total |
$ | 1,172 | |
F-11
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
Warranty Cost and Reserves
We provide a warranty provision related to the sales of our MassARRAY equipment based on our experience of returns and repairs required under the warranty period.
We generally provide a one-year warranty on our MassARRAY system and related equipment. We establish an accrual for estimated warranty expenses associated with system sales based on historical amounts. This expense is recorded as a component of cost of product revenue.
Changes in our warranty liability during the three years ended December 31, 2009 are as follows (in thousands):
| Balance as of December 31, 2006 |
$ | 680 | ||
| Additions charged to cost of revenues |
314 | |||
| Repairs and replacements |
(468 | ) | ||
| Balance as of December 31, 2007 |
$ | 526 | ||
| Additions charged to cost of revenues |
385 | |||
| Repairs and replacements |
(305 | ) | ||
| Balance as of December 31, 2008 |
$ | 606 | ||
| Additions charged to cost of revenues |
199 | |||
| Repairs, replacements and reduction in liability requirements |
(630 | ) | ||
| Balance as of December 31, 2009 |
$ | 175 | ||
Fair Value of Financial Instruments
Financial instruments, including cash and cash equivalents, accounts receivable, accounts payable and accrued liabilities, are carried at cost, which management believes approximates fair value because of the short-term maturity of these instruments. The fair value of our asset-backed loan approximated carrying value because the terms are equivalent to borrowing rates currently available to us for debt with similar terms and maturities.
Accounts Receivable
Trade accounts receivable are recorded at net invoice values. We consider receivables past due based on the contractual payment terms. We review our exposure to amounts receivable and reserve specific amounts if collectability is no longer reasonably assured. We also reserve a percentage of our trade receivable balance based on collection history. We re-evaluate such reserves on a regular basis and adjust our reserves as needed. Amounts determined to be uncollectible are charged or written off against the reserve.
Revenue Recognition
Revenues are recognized when persuasive evidence of an arrangement exists, delivery has occurred or services have been rendered, the price is fixed and determinable and collectability is reasonably assured. In regard to MassARRAY system sales, the arrangement consideration is allocated among the separate units of accounting based on their relative fair values. The separate units of accounting are typically the system and software itself and maintenance contracts sold at the time of the system sale. Revenue is deferred for fees received before earned. Revenues from sales of consumables are recognized generally upon shipment and transfer of title to the customer. Revenue from sales of MassARRAY systems with standard payment terms of net
F-12
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
90 days or less are recognized upon shipment and transfer of title to the customer or when all revenue recognition criteria are met. Our contracts do not contain refund or cancellation clauses. Revenues from the sale or licensing of our proprietary software are recognized upon transfer of title to the customer. We recognize revenue on maintenance services for ongoing customer support over the maintenance period. Revenues from genetic services are recognized at the completion of key stages in the performance of the service, which is generally delivery of single nucleotide polymorphism (SNP) assay information. Grant revenue is recorded as the research expenses relating to the grants are incurred, provided that the amounts received are not refundable if the research is not successful. Amounts received that are refundable if the research is not successful would be recorded as deferred revenue and recognized as revenue upon the grantor’s acceptance of the success of the research results.
Diagnostic revenues from our carrier screening test for cystic fibrosis, which was commercially launched in September 2009, have been recognized on a cash basis due to the limited number of contract or agreements with third-party payors and limited collections experience. We generally bill third-party payors upon generation and delivery of a report to the physician. As such, we take assignment of benefits and risk of collection with the third-party payor. We usually bill the patient directly for amounts not covered by their insurance carrier in the form of co-pays and deductibles, but only after multiple requests for full payment have been denied or only partially paid by the insurance carrier. Some payors may not cover our carrier screening test for cystic fibrosis, as ordered by the physician, under their reimbursement policies. Consequently, we pursue case-by-case reimbursement where policies are not in place.
Research and Development Costs
Research and development costs are expensed as incurred. These costs include personnel expenses, fees paid to collaborators, laboratory supplies, facilities, miscellaneous expenses and allocation of corporate costs. These expenses are incurred during proprietary research and development activities, as well as providing services under collaborative research agreements.
Foreign Currency Translation and Transactions
The financial statements of our German, United Kingdom, India, and Japan subsidiaries are measured using, respectively, the Euro (EUR), Great British pound (GBP), the Indian Rupee (INR) and the Japanese Yen (JPY), as the functional currency. Assets and liabilities of these subsidiaries are translated at the rates of exchange at the balance sheet date. Income and expense items are translated at the average daily rate of exchange during the reporting period. Resulting remeasurement gains or losses are recognized as a component of other comprehensive income (loss). Transactions denominated in currencies other than the local currency are recorded based on exchange rates at the time such transactions arise. Subsequent changes in exchange rates result in transaction gains and losses, which are reflected in income as unrealized (based on period-end translations) or realized upon settlement of the transaction. Transaction gains or losses were not material for the years ended December 31, 2009, 2008 and 2007.
Income Taxes
Our provision for income taxes is computed using the asset and liability method, under which deferred tax assets and liabilities are recognized for the expected future tax consequences of temporary differences between the financial reporting and tax bases of assets and liabilities, and for the expected future tax benefit to be derived from tax loss and credit carryforwards. Deferred tax assets and liabilities are determined using the enacted tax rates in effect for the years in which those tax assets are expected to be realized. A valuation allowance is established when it is more likely than not the future realization of all or some of the deferred tax assets will not
F-13
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
be achieved. The evaluation of the need for a valuation allowance is performed on a jurisdiction by jurisdiction basis, and includes a review of all available positive and negative evidence. As of December 31, 2009 and 2008, we maintain a valuation allowance against U.S. and foreign deferred tax assets that we concluded have not met the “more likely than not” threshold. Changes in the valuation allowance when they are recognized in the provision for income taxes are included as a component of the estimated annual effective tax rate.
We recognize excess tax benefits associated with share-based compensation to stockholders’ equity only when realized. When assessing whether excess tax benefits relating to share-based compensation have been realized, we follow the with-and-without approach, excluding any indirect effects of the excess tax deductions. Under this approach, excess tax benefits related to share-based compensation are not deemed to be realized until after the utilization of all other tax benefits available to us.
We recognize the impact of a tax position in our financial statements only if that position is more likely than not of being sustained upon examination by taxing authorities, based on the technical merits of the position. Any interest and penalties related to uncertain tax positions will be reflected in income tax expense.
Stock-based Compensation
Share based compensation cost is measured at grant date based on the estimated fair value of the award using the Black-Scholes option pricing model and the portion that is ultimately expected to vest and is recognized as expense over the requisite service period for all share based awards granted, modified or cancelled. Our net loss for the years ended December 31, 2009, 2008 and 2007, included the following compensation expense related to our share based compensation awards:
| (In thousands) | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Research and development expense |
$ | 3,562 | $ | 1,306 | $ | 521 | |||
| Selling and marketing expense |
3,239 | 1,430 | 601 | ||||||
| General and administrative expense |
4,498 | 3,396 | 1,936 | ||||||
| $ | 11,299 | $ | 6,132 | $ | 3,058 | ||||
Cash flows resulting from tax deductions in excess of the cumulative compensation cost recognized for options exercised (excess tax benefits) are classified as cash inflows from financing activities and cash outflows from operating activities. Due to our net loss position, no tax benefits have been recognized in the consolidated statements of cash flows.
We have not recognized, and do not expect to recognize in the near future, any tax benefit related to stock-based compensation cost as a result of the full valuation allowance of our net deferred tax assets and our net operating loss carryforwards.
The fair value of options granted to non-employees is estimated at the measurement date using the Black-Scholes option pricing model. Total stock-based compensation for options granted to non-employees for the year ended December 31, 2009, 2008, and 2007, was $273,000, $731,000 and $128,000, respectively. Stock-based compensation for options granted to non-employees is included in general and administrative, research and development and selling and marketing expenses in the statement of operations for the year ended December 31, 2009, 2008 and 2007 totaling $1,000, $0 and $39,000; $59,000, $181,000 and $24,000; and $213,000, $550,000 and $65,000, respectively.
F-14
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
Comprehensive Income (Loss)
Comprehensive income (loss) and its components, which include net loss, unrealized gains and losses on our available-for-sale marketable securities and foreign currency translation gains and losses, are disclosed as a separate component of stockholders’ equity.
Net Loss Per Share
Basic net loss per share is computed by dividing the net loss for the period by the weighted average number of common shares outstanding during the period. Diluted net loss per share is computed by dividing the net loss for the period by the weighted average number of common and common equivalent shares outstanding during the period. Common stock equivalents consisting of exercisable stock options of 3,507,803, warrants of 59,035 and restricted stock of 59,001 were not included in the computation of diluted net loss per share as their effect was anti-dilutive for all periods presented.
Recent Accounting Pronouncements
As of July 1, 2009, the Financial Accounting Standards Board (FASB) formally approved the FASB Accounting Standards Codification (Codification) as the single source of authoritative U.S. accounting and reporting standards, other than guidance issued by the SEC. At that time, the Codification superseded all then-existing non-SEC accounting and reporting standards. All other non-grandfathered, non-SEC accounting literature not included in the Codification became non-authoritative. The Codification is effective for interim and annual periods ending after September 15, 2009. We adopted the provisions of the Codification in the third quarter of 2009. The adoption did not have a material impact on our consolidated financial statements.
In May 2009, the FASB issued authoritative guidance that establishes general standards of accounting for and disclosure of events that occur after the balance sheet date but before financial statements are issued or are available to be issued. This guidance became effective for interim periods and fiscal years ending after June 15, 2009. We adopted the provisions of this guidance in the second quarter of 2009. The adoption of this guidance did not have a material impact on our consolidated financial statements.
In April 2009, the FASB issued authoritative guidance that requires publicly traded companies to include in their interim financial reports certain disclosures about the carrying value and fair value of financial instruments previously required only in annual financial statements and to disclose changes in significant assumptions used to calculate the fair value of financial instruments. This guidance became effective for all interim reporting periods ending after June 15, 2009, with early adoption permitted for interim reporting periods ending after March 15, 2009. We adopted the provisions of this guidance in the second quarter of 2009. The adoption of this guidance did not have a material impact on our consolidated financial statements. In December 2007, the FASB issued authoritative guidance that significantly changes the accounting and reporting requirements for business combination transactions, including capitalization of in-process research and development assets and expensing acquisition costs as incurred. This guidance became effective for business combination transactions occurring in fiscal years beginning after December 15, 2008. We adopted the provisions of this guidance in the first quarter of 2009. The adoption did not have a material impact on our 2009 consolidated financial statements.
F-15
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
In November 2008, the FASB issued authoritative guidance that clarifies how to account for acquired intangible assets subsequent to initial measurement in situations in which an entity does not intend to actively use the assets but intends to hold the asset to prevent others from obtaining access to the asset (a defensive intangible asset), except for intangible assets that are used in research and development activities. This guidance requires that a defensive intangible asset be accounted for as a separate unit of accounting and assigned a useful life that reflects the entity’s consumption of the expected benefits related to that asset. This guidance became effective for intangible assets acquired on or after December 15, 2008. We adopted the provisions of this guidance in the first quarter of 2009. The adoption of this guidance did not have a material impact on our consolidated financial statements.
In June 2008, the FASB issued authoritative guidance that clarifies the criteria for determining whether certain financial instruments should be classified as derivative instruments or equity instruments. The guidance became effective for years beginning after December 15, 2008. We adopted the provisions of the guidance in the first quarter of 2009. The adoption did not have a material impact on our consolidated financial statements.
In April 2008, the FASB issued authoritative guidance that amends the guidance for estimating the useful lives of recognized intangible assets and requires additional disclosure related to renewing or extending the useful lives of recognized intangible assets. This guidance became effective for fiscal years and interim periods beginning after December 15, 2008. We adopted the provisions of this guidance in the first quarter of 2009. The adoption of this guidance did not have a material impact on our consolidated financial statements.
In December 2007, the FASB issued authoritative guidance that defines collaborative arrangements and requires that transactions with third parties that do not participate in the arrangement be reported in the appropriate income statement line items pursuant to existing authoritative accounting literature. In accordance with this guidance, income statement classification of payments made between participants of a collaborative arrangement is to be based on other applicable authoritative accounting literature. If the payments are not within the scope or analogy of other authoritative accounting literature, a reasonable, rational and consistent accounting policy is to be elected. This guidance became effective for fiscal years beginning after December 15, 2008 and was to be applied as a change in accounting principle to all prior periods retrospectively for all collaborative arrangements existing as of the effective date. We adopted the provisions of this guidance in the first quarter of 2009. The adoption of this guidance did not have a material impact on our consolidated financial statements.
3. Acquisitions
Center for Molecular Medicine, LLC
On November 14, 2008, we completed a tax free acquisition of certain assets of the Center for Molecular Medicine, LLC (CMM). The assets are now part of our wholly-owned subsidiary, the Sequenom Center for Molecular Medicine, LLC (Sequenom CMM). Under the terms of the asset purchase agreement, we paid to CMM $4.0 million for certain assets related to CMM’s business in advanced molecular pathology laboratory services relating to diagnostics, translational research and clinical trials, less all cash and cash equivalents. Ninety percent of the purchase price consisted of 187,794 shares of our common stock (valued at $3.6 million as of the acquisition closing date) and the remainder of the purchase price of $0.4 million was paid in cash, which was deposited into an escrow account. The escrow account will remain in place for 18 months following the closing of the transaction to satisfy potential indemnification claims. The acquisition of CMM provides us with a laboratory that has the required accreditations and certifications already in place to develop and market laboratory developed tests and have contributed to the purchase price for the acquisition of CMM, which resulted in the recognition of goodwill of approximately $3.0 million.
F-16
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
In connection with this acquisition, we completed a valuation study of the intangible assets acquired in order to allocate the purchase price. We have allocated the excess purchase price over the fair value of net tangible assets and identifiable intangible assets acquired to goodwill. We believe the fair values assigned to the CMM assets acquired were based on reasonable assumptions, as determined by management. The purchase price has been allocated as follows (in thousands):
| Net tangible assets |
$ | 893 | |
| Identifiable intangible asset |
117 | ||
| Goodwill |
2,990 | ||
| Total consideration |
$ | 4,000 | |
SensiGen, LLC
On February 27, 2009, we completed a taxable acquisition of certain assets and assumption of certain liabilities of SensiGen, LLC (SensiGen). The assets are now part of our wholly-owned subsidiary, Sequenom CMM. Under the terms of the asset purchase agreement (the Agreement), we acquired certain assets related to SensiGen’s business in gene-based molecular diagnostic tests relating to cervical cancer, head and neck cancer, chronic kidney disease and lupus. We paid SensiGen cash consideration of approximately $1.9 million, which included a loan advance of $340,000, and issued common stock valued at $1.9 million (utilizing the minimum floor price of $20.94 per share in accordance with the Agreement). An additional $1.3 million is contingently payable to SensiGen upon the completion of certain triggering events with either cash or shares of our common stock (priced at the average closing price of our common stock over the ten trading day period ending on the third trading day prior to the applicable triggering event for such payment). In June 2009 we satisfied one of the triggering events related to the Agreement with a cash payment of $130,000. This triggering event had previously been recorded at a fair value of $130,000 during our initial fair value measurement of contingent consideration associated with the allocation of purchase price. After the payment of this triggering event, our remaining fair value of contingent consideration in the allocation of purchase price at the date of closing is $27,000. In December 2009, we entered into stipulations with certain stockholders of SensiGen for their release of claims against us related to the acquisition in exchange for the issuance of an aggregate 367,547 shares of our common stock. The issuance of these additional shares was charged to operations for the year ended December 31, 2009, and was valued at approximately $1.5 million as of the date of share delivery.
The acquisition of the SensiGen assets provides us with intellectual property related to certain molecular diagnostics for women’s health and cancer and have contributed to the purchase price for the acquisition of SensiGen, which resulted in the recognition of goodwill of approximately $7.0 million.
The total purchase consideration, excluding potential contingent consideration fair values, consisted of (in thousands, except share and per share data):
| Cash paid to SensiGen |
$ | 1,887 | |
| Sequenom common stock issued on the closing date (92,679 shares at $20.94 per share) |
1,941 | ||
| Assumed liabilities |
3,242 | ||
| Write-off of preexisting receivables |
403 | ||
| Total purchase price |
$ | 7,473 | |
In connection with this acquisition we completed a valuation study of the intangible assets acquired in order to allocate the purchase price. We have allocated the excess purchase price over the fair value of net tangible
F-17
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
assets and liabilities assumed to goodwill, as well as provided an estimate of the fair value of the contingent consideration as of the closing date of the acquisition. We believe the fair values assigned to SensiGen’s assets acquired and contingent consideration were based on reasonable assumptions, as determined by management. The purchase price has been allocated as follows (in thousands):
| Net tangible assets |
$ | 613 | |
| Goodwill |
6,860 | ||
| 7,473 | |||
| Fair value of contingent consideration |
157 | ||
| Total consideration |
$ | 7,630 | |
At each reporting date, we re-measure the contingent consideration liability at fair value, until the contingency is resolved with the changes in fair value recognized as a charge to operations in the current period. This analysis, which includes a probability assessment regarding the likelihood of payment and a discounted present value factor, concluded that the fair value of the remaining contingently payable triggering events based on currently available information was $541,000 as of December 31, 2009, as compared to the $27,000 remaining accrued contingent liability discussed above. The increase in our contingent consideration liability of $514,000 was recognized as a component of research and development ($260,000) and general and administrative expenses ($254,000), respectively. Subsequent to year end, we paid $520,000 in cash to satisfy these triggering events.
4. Restructuring
In April 2009, we announced that we reduced our genetic analysis employee workforce in accordance with our initiative to reduce costs and restructure our workforce. Severance related charges are included in the restructuring charges in the accompanying consolidated statement of operations for the year ended December 31, 2009.
The beginning balance for facilities costs prior to the April 2009 reduction in workforce consisted of a $0.4 million in accrued facility exit costs associated with exiting our lease commitments for the Boston, Massachusetts facility. This facility was initially leased in conjunction with the acquisition of Gemini Genomics, plc in 2001.
The reduction in workforce included the closure of our leased facility near Boston, Massachusetts, with lease expiration on December 31, 2010, as well as the closure of our office located in New Delhi, India during the second quarter of 2009.
The following table summarizes the activity and balance of accrued restructuring charges included in accrued expenses in the consolidated balance sheet through December 31, 2009 associated with the April 2009 reduction in workforce (in thousands):
| Description |
Employee Related |
Facilities | Other | Total | ||||||||||||
| Balance at December 31, 2008 |
$ | — | $ | 484 | $ | — | $ | 484 | ||||||||
| Accrued restructuring charges |
605 | 553 | 460 | 1,618 | ||||||||||||
| Fixed asset retirements |
— | — | (96 | ) | (96 | ) | ||||||||||
| Cash payments |
(605 | ) | (502 | ) | (309 | ) | (1,416 | ) | ||||||||
| Balance at December 31, 2009 |
$ | — | $ | 535 | $ | 55 | $ | 590 | ||||||||
F-18
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
5. Marketable Securities and Fair Value Measurements
Marketable securities
The estimated fair market value of our ARS holdings at December 31, 2009, was $0.5 million and reflects a $3.8 million adjustment to the principal value of $4.3 million. The $3.8 million adjustment includes a $1.1 million other-than-temporary impairment loss charged to operations for the year ended December 31, 2009. The estimated fair market value of all ARS holdings at December 31, 2008, was approximately $5.7 million, which reflects a $3.7 million adjustment to the principal value of $9.4 million. Other-than-temporary impairment losses charged to operations related to our ARS during the years ended December 31, 2008 and 2007 were $2.6 million and $1.1 million, respectively. As of December 31, 2009, there are no accumulated unrealized losses in other comprehensive income related to these ARS. As of December 31, 2009, our ARS investments have contractual maturity dates ranging from 2011 to 2028.
Although the majority of our ARS continue to pay interest according to their stated terms, based on valuation models and an analysis of other-than-temporary impairment factors, we recognized a loss of approximately $1.1 million and $2.6 million for the years ending December 31, 2009 and 2008, respectively, which reflects the portion of ARS holdings that we have concluded have an other-than-temporary decline in value. During the fourth quarter of 2009, our management determined that it was no longer our intent to hold the remaining ARS to maturity and to actively pursue liquidation of our remaining ARS in the secondary market. As a result of this decision, during the fourth quarter of 2009 we sold ARS with an estimated fair value of $4.1 million, which resulted in a net realized loss of approximately $0.8 million for a total loss on our ARS of $1.9 million for the year ended December 31, 2009. As of December 31, 2009, our remaining ARS have a principal value of $4.3 million and an estimated fair market value of $0.5 million. Subsequent to year end, ARS with a principal value of $1.3 million and an estimated fair value of $0.5 million at December 31, 2009, were sold. The proceeds for these ARS were equal to their estimated fair value as of December 31, 2009. Our other remaining ARS with a principal value of $3.0 million have an estimated fair value of $0 as of December 31, 2009.
Approximately $1.0 million of the recorded loss for the year ended December 31, 2008, represented the reclassification of previously recorded unrealized losses in other comprehensive income and that we have now concluded have an other-than-temporary decline in value. At December 31, 2007, we had recorded an unrealized loss of approximately $0.8 million in accumulated other comprehensive income as a reduction in stockholders’ equity, reflecting adjustments to ARS holdings that we had concluded have a temporary decline in value. During 2008, the adjustments previously recorded to other comprehensive income at December 31, 2007, have been recognized as an other-than-temporary decline in value and are included within the $2.6 million of loss on marketable securities for the year ended December 31, 2008. During 2008, we reclassified our remaining ARS investments to non-current marketable securities available for sale. The $0.5 million of ARS sold subsequent to year end are presented as current marketable securities as of December 31, 2009.
Fair Value Measurements
SFAS 157 defines fair value as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants on the measurement date. SFAS 157 also establishes a fair value hierarchy that requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. The standard describes three levels of inputs that may be used to measure fair value:
Level 1—Quoted prices in active markets for identical assets or liabilities. We have determined that our investments in money market accounts, certificates of deposit and U.S. Government securities meet the criteria for definition within the Level 1 hierarchy.
F-19
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
Level 2—Inputs other than quoted prices included within Level 1 that are observable for the asset or liability, either directly or indirectly. These inputs include quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. We have determined that our investments in commercial paper and ARS that were sold subsequent to year end with moderate secondary market activity meet the criteria for definition within the Level 2 hierarchy.
Level 3—Unobservable inputs that are supported by little or no market activity and that with an estimated fair value of $0 at December 31, 2009 are significant to the fair value of the assets or liabilities. We have determined that our investments in ARS meet the criteria for definition within Level 3 hierarchy.
The fair values of our investments in ARS instruments of $0 are estimated utilizing a discounted cash flow analysis valuation model as of December 31, 2009. This analysis considers, among other items, the collateral underlying the security investments, the credit quality of the counterparty, the timing of expected future cash flows, the default risk underlying the security, discount rates, the expected time until a successful auction and the overall capital market liquidity. These securities were also compared, when possible, to other observable market data with similar characteristics to the securities. Management has also reviewed the valuation input criteria, which generally consists of the price of credit protection, information available on the trading of senior and subordinated securities and other debt in the market place for comparable types of maturities, the current credit rating of the trust sponsor and/or bond insurer, as well as the ultimate maturity and the underlying collateral of the securities and have deemed them to be reasonable assumptions in determining fair value. The valuation of our ARS investments is subject to uncertainties that are difficult to predict. Factors that may impact our valuation include changes to credit ratings of the securities, as well as to the underlying assets supporting those securities, rates of credit default of the underlying assets, underlying collateral value, discount rates, counterparty risk and the ongoing strength and quality of market credit and liquidity.
We endeavor to utilize the best available information in measuring fair value. Financial assets and liabilities are classified in their entirety based on the lowest level of input that is significant to the fair value measurement. All of the available for sale securities have a contractual maturity at December 31, 2009 of one year or less. The following table sets forth our financial assets and liabilities that were accounted for at fair value on a recurring basis as of December 31, 2009:
| Description |
Total | Level 1 | Level 2 | Level 3 | ||||||||
| (in thousands) | ||||||||||||
| Cash and cash equivalents |
$ | 14,941 | $ | 14,941 | $ | — | $ | — | ||||
| Restricted cash |
1,419 | 1,419 | — | — | ||||||||
| Marketable securities, current1 |
15,762 | 15,291 | 471 | — | ||||||||
| Total |
$ | 32,122 | $ | 31,651 | $ | 471 | $ | — | ||||
| 1 | Gains or losses considered to be temporary are recorded to other comprehensive income (loss) at each measurement date. Other than temporary losses are recorded to operations at each measurement date. |
F-20
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
The following table presents our assets measured at fair value on a recurring basis using significant unobservable inputs (Level 3) as defined in SFAS 157 at December 31, 2009:
| Level 3 Auction Rate Securities |
||||
| (in thousands) | ||||
| Balance at December 31, 2008 |
$ | 5,509 | ||
| Transfers from Level 3 |
(471 | ) | ||
| Total losses: |
||||
| Included in operations |
(1,993 | ) | ||
| Recognized loss previously included in other comprehensive income (loss) |
— | |||
| Included in other comprehensive income (loss) |
— | |||
| Purchases and settlements, net |
(3,045 | ) | ||
| Balance at December 31, 2009 |
$ | — | ||
| Losses included in operations (or changes in net assets) for the period relating to assets still held at December 31, 2009 |
— | |||
| Total net losses for the year ended December 31, 2009, included in operations |
$ | (1,914 | ) | |
| Total change in unrealized losses included in other comprehensive income (loss) |
$ | — | ||
6. Segment Reporting
At December 31, 2009, we reevaluated our segment reporting in light of changes to our management structure, internal performance reporting and incentive compensation plans that became effective in 2009. Prior to 2009, our business had been reported as a single segment with operating performance measured as a single unit and management incentive plans that were based on total life sciences segment performance. With changes during the current year, we are now reporting results for two life sciences segments, Molecular Diagnostics and Genetic Analysis.
Description of the types of products and services from which each reportable segment derives its revenues
We operate two primary business segments, Molecular Diagnostics and Genetic Analysis. Molecular Diagnostics researches, develops and commercializes noninvasive molecular diagnostic tests for prenatal genetic disorders and diseases, oncology, infectious diseases, and other diseases and disorders. Genetic Analysis designs, markets and provides maintenance services for our proprietary MassARRAY system, comprised of hardware, software applications, consumable chips and reagents which are marketed to premier clinical research laboratories, bio-agriculture, bio-technology and pharmaceutical companies.
Revenue for Molecular Diagnostics is generated from customers located within the United States. Revenue for Genetic Analysis is generated from customers and/or distributors located in North America, Europe and Asia.
Measurement of segment profit or loss and segment assets
We evaluate performance and allocate resources based on total segment revenue, operating expenses and operating profit or loss exclusive of general and administrative expenses, other indirect overhead costs and restructuring charges, which are not allocated to our segments for performance assessment by our chief operating decision maker. No evaluation of segment performance or allocation of resources is done in consideration of segment assets. The accounting policies of the reportable segments are the same as those described in the summary of significant accounting policies. Intersegment revenues and transfers are immaterial.
Unallocated expenses for research and development and sales and marketing expenses consist of share based compensation, indirect overhead expenses and allocated and absorbed costs. Unallocated operating loss consists of general and administrative expenses, share based compensation, indirect overhead expenses and unabsorbed costs.
F-21
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
Factors management used to identify our reportable segments
Our reportable segments are business units that offer different products and services and are each managed separately. Operating results for each segment are reported separately to senior management to make decisions as to the allocation of resources and to assess performance.
Operating segment financial data is as follows as of December 31, 2009:
| (In thousands) | ||||
| Revenues: |
||||
| Molecular Diagnostics |
$ | 94 | ||
| Genetic Analysis |
37,769 | |||
| $ | 37,863 | |||
| Research and development expenses: |
||||
| Molecular Diagnostics |
$ | 20,935 | ||
| Genetic Analysis |
5,587 | |||
| Share based compensation |
3,835 | |||
| Indirect overhead (1) |
3,898 | |||
| Allocated and absorbed costs (2) |
3,199 | |||
| Total |
$ | 37,454 | ||
| Sales and marketing expenses: |
||||
| Molecular Diagnostics |
$ | 5,780 | ||
| Genetic Analysis |
13,644 | |||
| Share based compensation |
3,590 | |||
| Indirect overhead (3) |
1,819 | |||
| Allocated and absorbed costs (4) |
2,012 | |||
| Total |
$ | 26,845 | ||
| Operating (loss) income: |
||||
| Molecular Diagnostics |
$ | (27,033 | ) | |
| Genetic Analysis |
4,379 | |||
| Unallocated (5) |
(48,068 | ) | ||
| $ | (70,722 | ) | ||
| (1) | Indirect overhead consists of expenses related to the following departments, which are unallocated to our operating segments for performance assessment by our chief operating decision maker: quality, regulatory, chief science officer and research and development collaborations (licensing costs). |
| (2) | Allocated costs consist of costs from the following departments, which are unallocated to our operating segments for performance assessment by our chief operating decision maker: human resources, information technology and facilities. Absorbed costs relate to costs incurred that are absorbed into cost of sales. |
| (3) | Indirect overhead consists of expenses related to the following departments, which are unallocated to our operating segments for performance assessment by our chief operating decision maker: business development and European sales administration. |
| (4) | Allocated costs consist of costs from the following departments, which are unallocated to our operating segments for performance assessment by our chief operating decision maker: human resources, information technology and facilities. Absorbed costs relate to costs incurred that are absorbed into cost of sales. |
| (5) | Unallocated costs consist of those reconciling items for research and development and sales and marketing expenses, as well as general and administrative expenses, which are unallocated to our operating segment for performance assessment by our chief operating decision maker. |
F-22
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
7. Debt and Obligations
Asset-backed Loan
On August 31, 2007, we signed an amendment to our existing asset-backed loan line that had previously expired. Under the terms of this amendment, we may elect to have individual minimum fundings of $100,000 up to an aggregate limit of $3.0 million through December 23, 2008. All borrowings are secured by the underlying financed equipment.
As of December 31, 2009, we had an aggregate $0.6 million outstanding on this asset-backed loan line relating to four fundings with interest rates from 10.6% to 9.73%. Payments are to be repaid in 36 monthly installments with final payments dates ranging from April 2010 to May 2011. Our rights to borrow funds under this facility expired in December 2008.
Debt
In connection with our acquisition of SensiGen in February 2009, we assumed two loans with the Michigan Economic Development Corporation with an aggregate balance at the closing date of approximately $3.2 million. The first loan of approximately $0.3 million has a stated interest rate of 1% with all payments deferred until March 2013. Commencing March 2013, principal payments of approximately $3,000 are due monthly through March 2018, at which time a final balloon payment of approximately $161,000 is due. The second loan of approximately $2.9 million has a stated interest rate of 7% with monthly principal payments of approximately $68,000 through September 2012. As of December 31, 2009, we had an aggregate of $2.3 million outstanding on these loans. Both loans are collateralized by all of Sequenom CMM’s tangible and intangible property and rights in which a security interest or lien may be taken.
The following is a schedule of future maturities on our asset-back loans and debt at December 31, 2009:
| Year Ending December 31, |
Payments | ||
| (In thousands) | |||
| 2010 |
$ | 1,205 | |
| 2011 |
744 | ||
| 2012 |
622 | ||
| 2013 |
25 | ||
| 2014 |
30 | ||
| Thereafter |
260 | ||
| $ | 2,886 | ||
Capital Lease
In the second quarter of 2009, we entered into a 36 month capital lease arrangement for new phone equipment, which was capitalized with office furniture and equipment at an aggregate balance of approximately $366,000.
F-23
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
The following is a schedule of minimum future rental payments on non-cancelable capital leases at December 31, 2009:
| Year Ending December 31, |
Payments | |||
| (In thousands) | ||||
| 2010 |
$ | 131 | ||
| 2011 |
131 | |||
| 2012 |
34 | |||
| Total minimum payments required |
296 | |||
| Less: amount representing interest |
(25 | ) | ||
| Present value of net minimum lease payments |
$ | 271 | ||
8. Commitments and Contingencies
Building Leases
We lease facilities in the United States, Germany, China, United Kingdom and Japan. In total, we lease space in 10 buildings under leases that expire at various dates through September 2015. Total rent expense under these leases was approximately $5.4 million, $5.0 million and $5.0 million in 2009, 2008 and 2007, respectively.
In September 2005, we entered into an amendment to our lease for our corporate headquarters in San Diego. The lease amendment provides for the deferral of approximately $3.2 million of the monthly rent payments by reducing the monthly payments during the period commencing October 1, 2005 and ending September 30, 2007 and increasing the aggregate monthly payments by the deferred amount for the remaining term of the lease, from October 1, 2007 to September 30, 2015. The total obligation under the lease remains unchanged. Rent expense is calculated on a straight-line basis. In connection with the lease amendment, we issued our landlord a warrant to purchase 50,000 shares of our common stock with an exercise price of $2.64 per share. The warrants are exercisable and have a ten year term. The fair value of the warrants, calculated using the Black-Scholes option pricing model, was recorded as prepaid rent and is being amortized as rent expense over the remaining life of the lease.
The following is a schedule of future minimum lease payments under non-cancelable operating lease commitments at December 31, 2009:
| Year Ending December 31, |
Operating Leases | ||
| (In thousands) | |||
| 2010 |
$ | 7,041 | |
| 2011 |
5,851 | ||
| 2012 |
5,341 | ||
| 2013 |
4,313 | ||
| 2014 |
4,456 | ||
| Thereafter |
3,429 | ||
| $ | 30,431 | ||
The above operating leases expire at various dates through 2015. Certain leases contain extension, return, or renewal provisions for two years at existing lease rates and/or purchase options. Future operating lease commitments for leases have not been reduced by future minimum sublease rentals aggregating $0.3 million.
F-24
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
Letters of Credit
At December 31, 2009, we had outstanding stand-by letters of credit with financial institutions totaling $1.4 million related to our building, operating leases and customer guarantees. The letter of credit related to our Newton, Massachusetts building lease agreement will remain in place until its expiration in December 2010.
Collaboration, Development and Licensing Agreements
In October 2005 we acquired exclusive rights in certain countries, including the United States, United Kingdom and other countries in Europe and elsewhere, to noninvasive prenatal diagnostic intellectual property from Isis Innovation Ltd. (ISIS), the technology transfer company of the University of Oxford. The intellectual property covers noninvasive prenatal genetic diagnostic testing on fetal nucleic acids derived from plasma or serum on any platform including mass spectrometry and real time polymerase chain reaction amplification platforms. In October 2006 and November 2007 we entered into additional related agreements with other entities, as well as amendments to the ISIS agreement that expanded the licensed applications and territory. Under the terms of this agreement and its amendments, we have paid up-front fees totaling $0.8 million and are required to pay up to approximately $0.5 million in aggregate milestone payments upon the achievement of initial sales or tests performed of various products or the issuance of a patent, as well as royalties on product sales.
In November 2009, we entered into a third amendment to the Isis Agreement pursuant to which Isis agreed to a modification of certain time-based commercial launch milestones relating to aneuploidy and other products. In exchange for this modification, we agreed to make an immediate one-time payment of $1,000,000, increase royalty payments under the agreement during the final 12 months of the patent term and increase the specified minimum royalty amounts.
We have entered into various license agreements since 1996 allowing us to utilize certain patents rights. If these patents are used in connection with a commercial product sale, we will pay royalties based on a percentage of the related product revenues. During the years ended December 31, 2009, 2008, and 2007, the amount of royalties incurred in connection primarily with product sales was $0.1 million, $0.1 million, and $0.1 million, respectively.
Litigation
In November 2001, we and certain of our current or former officers and directors were named as defendants in a class action shareholder complaint filed by Collegeware USA in the U.S. District Court for the Southern District of New York (now captioned In re Sequenom, Inc. IPO Securities Litigation) Case No. 01-CV-10831. In the complaint, the plaintiffs allege that our underwriters, certain of our officers and directors and we violated the federal securities laws because our registration statement and prospectus contained untrue statements of material fact or omitted material facts regarding the compensation to be received by and the stock allocation practices of the underwriters. The plaintiffs seek unspecified monetary damages and other relief. Similar complaints were filed in the same District Court against hundreds of other public companies that conducted initial public offerings of their common stock in the late 1990s and 2000 (the IPO Cases).
In October 2002, our officers and directors were dismissed without prejudice pursuant to a stipulated dismissal and tolling agreement with the plaintiffs. In February 2003, the District Court dismissed the claim against us brought under Section 10(b) of the Exchange Act, without giving the plaintiffs leave to amend the complaint with respect to that claim. The District Court declined to dismiss the claim against us brought under Section 11 of the Securities Act of 1933, as amended (the Securities Act).
F-25
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
In September 2003, pursuant to the authorization of a special litigation committee of our board of directors, we approved in principle a settlement offer by the plaintiffs. In September 2004, we entered into a settlement agreement with the plaintiffs. In February 2005, the District Court issued a decision certifying a class action for settlement purposes and granting preliminary approval of the settlement subject to modification of certain bar orders contemplated by the settlement. In August 2005, the District Court reaffirmed class certification and preliminary approval of the modified settlement. In December 2006, the U.S. Court of Appeals for the Second Circuit vacated the District Court’s decision certifying as class actions the six lawsuits designated as “focus cases.” Thereafter the District Court ordered a stay of all proceedings in all of the lawsuits pending the outcome of plaintiffs’ petition to the Second Circuit for rehearing en banc. In April 2007, the Second Circuit denied plaintiffs’ rehearing petition, but clarified that the plaintiffs may seek to certify a more limited class in the District Court. Accordingly, the settlement as originally negotiated was terminated pursuant to stipulation and will not receive final approval.
In February 2009, liaison counsel for plaintiffs informed the District Court that a new settlement of all IPO Cases had been agreed to in principle, subject to formal approval by the parties and preliminary and final approval by the District Court. In April 2009, the parties submitted a tentative settlement agreement to the District Court and moved for preliminary approval thereof. In June 2009, the District Court granted preliminary approval of the tentative settlement and ordered that notice of the settlement be published and mailed to class members. In September 2009, the District Court held a final fairness hearing. On October 6, 2009, the District Court certified the settlement class in each IPO Case and granted final approval to the settlement. On or about October 23, 2009, three shareholders filed a Petition for Permission To Appeal Class Certification Order, asserting that the District Court’s certification of the settlement classes violates the Second Circuit’s earlier class certification decisions in the IPO Cases. Beginning on October 29, 2009, a number of shareholders also filed direct appeals, objecting to final approval of the settlement. Similar petitions and direct appeals may be filed by other shareholders. If the settlement is affirmed on appeal, the settlement will become effective and will result in the dismissal of all claims against us and our officers and directors with prejudice, and our pro rata share of the settlement fund will be fully funded by insurance.
In October 2008, we filed a patent infringement suit against Ibis Biosciences, Inc. (IBIS), formerly a subsidiary of Isis Pharmaceuticals, Inc. The complaint was served on the defendant in February 2009. IBIS has been acquired by Abbott Molecular. The lawsuit was filed in the U.S. District Court for the District of Delaware. The lawsuit alleged that the sale or offer for sale of the IBIS T5000 Biosensor System and related technology infringes three U.S. patents: 6,300,076, 6,500,621 and 7,419,787. Defendant has filed an answer and counterclaims against us seeking declaratory judgments that the patents are not infringed and are invalid and/or unenforceable. We sought a permanent injunction enjoining the defendant from further infringement and monetary damages, including enhanced damages pursuant to 35 U.S.C. § 284, costs, attorneys’ fees and other relief as the court deems just and proper. On October 22, 2009, we and IBIS entered into a non-exclusive license and settlement agreement and the court dismissed with prejudice all claims and counterclaims in the lawsuit. Pursuant to the terms of the agreement, we have agreed to grant IBIS and its affiliates a non-exclusive license under the three mass spectrometry-based patents involved in the lawsuit and certain pending mass-spectrometry-based applications and foreign counterparts to manufacture, use, practice, sell, offer to sell, and import products and methods protected by the licensed patents. As part of the agreement, we have also agreed to dismiss the litigation with prejudice and have granted IBIS, and its affiliates, immunity from suit for patent infringement for past, present or future damages related to the IBIS T5000 Biosensor System and T6000 System. Pursuant to the agreement, IBIS paid us $1.0 million.
In April 2009, we announced that the expected launch of our test for Trisomy 21 (Down syndrome) had been delayed and that we were no longer relying on our previously announced test data and results for that test.
F-26
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
We also announced that our Board of Directors had formed a special committee of independent directors to oversee an independent investigation of activity related to the test data and results and that the committee had engaged independent counsel to assist the committee in the conduct of the investigation. In September 2009, we announced that the committee’s investigation had been completed. Based on the committee’s work and recommendations, the independent directors concluded that as a result of our attempted transition from researching potential molecular diagnostic tests to developing and commercializing those tests, we failed to put in place adequate protocols and controls for the conduct of studies in the Trisomy 21 program at our company. Certain of our employees also failed to provide adequate supervision. In the absence of such protocols, controls and supervision, the test data and results in our Trisomy 21 program included inadequately substantiated claims, inconsistencies and errors. Due to deficiencies in our disclosure controls and procedures, in a number of instances such test data and results were reported to the public in our press releases and other public statements. We also terminated the employment of our president and chief executive officer, Harry Stylli, Ph.D., and our senior vice president of research and development, Elizabeth Dragon, Ph.D., effective immediately. In connection with the termination of Dr. Stylli’s employment, Dr. Stylli resigned as a director. We also obtained the resignation of our chief financial officer, Paul Hawran. We also terminated the employment of three other employees and obtained the resignation of one other officer. While each of these officers and employees denied wrongdoing, the committee’s investigation raised serious concerns, resulting in a loss of confidence by the independent directors in the personnel involved.
Our board of directors appointed our chairman of the board, Harry F. Hixson, Jr., Ph.D., to serve as our chief executive officer. Our board of directors appointed Ronald M. Lindsay, Ph.D., one of our directors, to serve as our interim senior vice president of research and development. Our board of directors appointed Paul V. Maier as our interim chief financial officer effective November 10, 2009. Our controller, Justin J. File, served as our principal financial and accounting officer until Mr. Maier’s appointment as interim chief financial officer.
Following our April 2009 announcement, several complaints were filed in the U.S. District Court for the Southern District of California against us and certain of our current and former officers and directors on behalf of certain purchasers of our common stock. The complaints include claims asserted under Sections 10 and 20(a) of the Exchange Act and Sections 11 and 12(a)(2) of the Securities Act and have been brought as shareholder class actions. In general, the complaints allege that we and certain of our officers and directors violated federal securities laws by making materially false and misleading statements regarding our Trisomy 21 test under development, thereby artificially inflating the price of our common stock. The plaintiffs seek unspecified monetary damages and other relief. On September 1, 2009, the complaints were consolidated under the caption In re Sequenom, Inc. Securities Litigation, S.D. Cal. Case No. 09-CV-0921 LAB (WMc) and a lead plaintiff was appointed. On December 24, 2009, we entered into a stipulation of settlement with the lead plaintiff on behalf of the plaintiffs’ class, which if approved by the District Court, will resolve this action. Pursuant to the terms of the stipulation, we have agreed to pay $14 million, which will be funded by insurance proceeds. We have also agreed to issue to the plaintiffs’ class a number of shares of our common stock equal to 9.95% of our total shares outstanding at the time of determination, subject to certain limitations. We have also agreed to adopt or continue our implementation of changes and additions to certain corporate governance policies, protocols and practices. The court preliminarily approved the settlement on January 26, 2010. The court has scheduled a final settlement approval hearing on May 3, 2010.
In May 2009, a shareholder derivative complaint was filed in the Superior Court of California for the County of San Diego against certain of our current and former directors and officers. Thereafter, a number of similar actions, also styled as shareholder derivative suits, were filed in state court and have been consolidated in a single court. On July 1, 2009, the first of three shareholder derivative suits were filed in the U.S. District Court for the Southern District of California. The federal shareholder derivative actions have been consolidated before
F-27
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
a single court under the caption In re Sequenom, Inc. Derivative Litigation, S.D. Cal. Case No. 09-CV-1341 LAB (WMc) and plaintiffs filed a single consolidated complaint. A separate federal derivative compliant, Ries, et al. v. Stylii, et al, case no. 09-CV-2517 LAB (WMc), was filed thereafter and it has been coordinated with the consolidated federal derivative action. The state and federal shareholder derivative actions are hereinafter collectively referred to as the “Derivative Actions.” The complaints in the Derivative Actions allege breaches of fiduciary duties by the defendants and other violations of law. In general, the complaints allege that our directors and certain of our officers caused or allowed for the dissemination of materially false and misleading statements regarding our Trisomy 21 test under development, thereby artificially inflating the price of our common stock. The plaintiffs are seeking unspecified monetary damages and other relief, including reforms and improvements to our corporate governance and internal procedures. We have not yet responded to the Derivative Actions, but will vigorously defend against the claims advanced.
In June 2009, we received written notification that the Enforcement staff of the SEC has initiated an investigation following our April 2009 announcement regarding our Trisomy 21 test under development. Following our September 2009 announcement, members of the special committee and its independent counsel met with the SEC staff in connection with its investigation. As part of this investigation, the SEC staff has also required us to produce information with respect to our announcement relating to our offer to acquire EXACT Sciences, Inc. in January 2009. We intend to continue to cooperate fully with the SEC in this matter.
In July 2009, an attorney who claims to represent certain stockholders who were issued an aggregate of 71,836 shares of our common stock when we acquired the assets of SensiGen in February 2009 sent us a letter claiming that we had breached our representations and warranties made in the asset purchase agreement and alleging that his clients had suffered approximately $1.3 million in damages as a result. On December 23, 2009, we entered into a stipulation of settlement with these stockholders. Pursuant to the terms of the settlement, in consideration of the stockholders’ release of claims, we issued an aggregate of 367,547 shares of our common stock to such stockholders.
Following our September 2009 announcement, representatives of the Office of the U.S. Attorney for the Southern District of California contacted us to inquire about the announcement. We have met with representatives of the U.S. Attorney and the Federal Bureau of Investigation (FBI) in connection with their investigations. We intend to continue to cooperate fully with the U.S. Attorney and the FBI in this matter.
Following our September 2009 announcement, representatives of NASDAQ also contacted us to inquire about the announcement. We have met with representatives from NASDAQ in connection with their investigation and we intend to continue to cooperate fully with NASDAQ in this matter in the event that NASDAQ has any further inquiries.
On October 28, 2009, plaintiff Xenomics, Inc. filed a complaint in the Supreme Court of the of the State of New York naming us as the defendant. In the complaint, the plaintiff alleges that due to materially false and misleading statements regarding our Trisomy 21 test under development, we have breached the license agreement entered into by the parties on October 29, 2008, which provides us with exclusively licensed patent rights for the use of fetal nucleic acids obtained from maternal urine, and that the plaintiff has suffered damages as a result. The plaintiff is seeking equitable relief and $300 million in damages. On December 15, 2009, we removed the case to the U.S. District Court for the Southern District of New York. On February 22, 2010, we filed a motion in the federal district court to, among other things, dismiss or stay the action in light of the fact that the License Agreement between the parties specifically provides that if Xenomics seeks to resolve a dispute arising under the agreement, it must do so by commencing an arbitration in San Diego. The district court has directed that the motion be fully briefed by March 26, 2010. Regardless of the forum in which the dispute is ultimately heard, we intend to vigorously defend against the claims advanced.
F-28
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
Legal expenses aggregating approximately $0.7 million relating to our insured legal expenses have been submitted directly to third party insurance carriers for reimbursement, but for which we are ultimately liable. Therefore, as of December 31, 2009, we have not accrued for these expenses in the accompanying consolidated financial statements. Should these expenses not be paid by our third party insurance carriers, we will be required to incur these expenses directly.
In addition, from time to time, we may be involved in litigation relating to claims arising out of our operations in the normal course of business.
Because of the uncertainties related to the incurrence, amount and range of loss on any pending litigation, investigation, inquiry or claim, management is currently unable to predict the ultimate outcome of any litigation, investigation, inquiry or claim, determine whether a liability has been incurred or make an estimate of the reasonably possible liability that could result from an unfavorable outcome. An adverse ruling or outcome in any lawsuit involving us could materially affect our business, liquidity, consolidated financial position or results of operations ability to sell one or more of our products or could result in additional competition. In view of the unpredictable nature of such matters, we cannot provide any assurances regarding the outcome of any litigation, investigation, inquiry or claim to which we are a party or the impact on us of an adverse ruling of such matters.
9. Related Party Transactions
We had the following transactions with parties related to certain of our Board members:
| • | Boston University. Dr. Charles Cantor is our Chief Scientific Officer, a member of our Board and was previously the chair and professor of the department of biomedical engineering and biophysics, and Director of the Center for Advanced Biotechnology at Boston University. We recorded product revenue for MassARRAY hardware and consumables, totaling $0.1 million, $0.1 million, and $0.1 million, in the years ended December 31, 2009, 2008 and 2007, respectively. We have agreements with Boston University in which Dr. Cantor participates under which we paid $0.4 million, $0.9 million, and $0.4 million, in the years ended December 31, 2009, 2008 and 2007 respectively. |
| • | University of California, San Diego. Dr. Cantor is adjunct professor in the department of bioengineering at the University of California, San Diego (UCSD). We recorded product revenue for MassARRAY hardware and consumables, totaling $3,300, $24,000 and $2,000 in the years ended December 31, 2009, 2008 and 2007, respectively. We have agreements with UCSD under which we paid $56,600, $9,800 and $0, in the years ended December 31, 2009, 2008 and 2007, respectively. |
| • | Dr. Richard Lerner is a member of our Board of Directors and is President of The Scripps Research Institute (Scripps). For the years ended December 31, 2009, 2008, and 2007, we have recorded product revenue for MassARRAY hardware and consumables totaling approximately $35,200, $30,000 and $318,000, respectively. We have agreements with Scripps under which we paid $61,300, $14,700 and $0, in the years ended December 31, 2009, 2008 and 2007, respectively. |
At December 31, 2009, we had the following receivable and payable balances with the following related parties:
| Related party |
Receivables | Payables | ||||
| (in thousands) | ||||||
| Boston University |
$ | 47 | $ | 42 | ||
| Scripps Research Institute |
2 | 2 | ||||
| UCSD |
— | 26 | ||||
| Total |
$ | 49 | $ | 70 | ||
F-29
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
At December 31, 2008, we had the following receivable and payable balances with the following related parties:
| Related party |
Receivables | Payables | ||||
| (in thousands) | ||||||
| Boston University |
$ | 5 | $ | 90 | ||
| Scripps Research Institute |
13 | — | ||||
| UCSD |
— | — | ||||
| Total |
$ | 18 | $ | 90 | ||
10. Stockholders’ Equity
On July 1, 2008, we closed an underwritten public offering of our common stock totaling 5,500,000 shares of our common stock at $15.50 per share, with the underwriters exercising their option to purchase an additional 825,000 shares on July 8, 2008. Including the additional shares, the offering resulted in aggregate net proceeds of approximately $91.8 million after deducting underwriting discounts, commissions and estimated transaction expenses.
During 2007, we closed a $20.0 million registered direct offering of our common stock to several new and existing investors, as well as a $30.5 million private placement of our common stock. Under the terms of the registered direct offering we issued and sold 6,666,666 shares of our common stock at $3.00 per share, with aggregate net proceeds of approximately $18.3 million after deducting placement agents’ fees and transaction expenses. Under the terms of the private placement we issued and sold 3,383,335 shares of our common stock at $9.00 per share, with aggregate net proceeds of approximately $28.1 million after deducting placement agents’ fees and estimated transaction expenses.
Stock Compensation Plans
On May 31, 2006, the stockholders approved our 2006 equity incentive plan (the 2006 plan), as the successor to our 1999 stock option plan (the 1999 plan). In connection with the adoption of the 2006 plan, we terminated the automatic annual increase feature under the 1999 plan and resolved to cease to grant additional stock awards under the 1999 plan following the effectiveness of the 2006 plan. The aggregate number of shares of common stock that may be issued under the 2006 plan is 7,701,290, plus the number of shares subject to any stock awards under the 1999 plan that terminate or are forfeited or repurchased and would otherwise have been returned to the share reserve under the 1999 plan.
Stock Options
The estimated fair value of each stock option award granted was determined on the date of grant using the Black-Scholes option pricing model with the following weighted-average assumptions for stock option grants during the years ended December 31, 2009, 2008 and 2007:
| 2009 | 2008 | 2007 | ||||||||||
| Risk free interest rate |
2.81 | % | 3.17 | % | 4.51 | % | ||||||
| Volatility |
100 | % | 87 | % | 82 | % | ||||||
| Dividend yield |
0 | % | 0 | % | 0 | % | ||||||
| Expected option life (years) |
7.0 | 6.6 | 6.4 | |||||||||
| Weighted average fair value of stock option grants to employees |
$ | 11.17 | $ | 8.81 | $ | 4.09 | ||||||
F-30
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
The risk-free interest rate assumption is based upon observed interest rates appropriate for the expected term of our employee stock options. The expected volatility is based on the historical volatility of our stock. We have not paid any dividends on common stock since our inception and do not anticipate paying dividends on common stock in the foreseeable future. The computation of the expected option life assumption is based on a weighted-average calculation combining the average historical exercise activity with the estimated life of all unexercised stock options.
Forfeitures are required to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. Pre-vesting forfeitures were estimated to be 11.7% based on historical experience. Our determination of fair value is affected by our stock price as well as a number of assumptions that require judgment. Changes in forfeiture estimates impact compensation cost in the period in which the change in estimate occurs.
A summary of the status of our stock option plans as of December 31, 2009 and of changes in stock options outstanding under the plans during the years ended December 31, 2009, 2008 and 2007 is as follows:
| Outstanding |
Shares Subject to Options |
Weighted Average Exercise Price per Share |
Weighted Average Remaining Contractual Term (in years) |
Aggregate Intrinsic Value | |||||||
| Outstanding at December 31, 2006 |
3,285,783 | $ | 6.45 | ||||||||
| Granted |
2,232,976 | 5.38 | |||||||||
| Canceled |
(270,452 | ) | 5.20 | ||||||||
| Exercised |
(168,071 | ) | 2.98 | ||||||||
| Outstanding at December 31, 2007 |
5,080,236 | $ | 6.16 | ||||||||
| Granted |
1,705,652 | 11.63 | |||||||||
| Canceled |
(265,892 | ) | 6.73 | ||||||||
| Exercised |
(281,925 | ) | 4.55 | ||||||||
| Outstanding at December 31, 2008 |
6,238,071 | $ | 7.70 | ||||||||
| Granted |
2,263,091 | 14.09 | |||||||||
| Canceled |
(1,848,852 | ) | 10.73 | ||||||||
| Exercised |
(479,503 | ) | 2.38 | ||||||||
| Outstanding at December 31, 2009 |
6,172,807 | $ | 9.55 | 6.44 | $ | 3,656,718 | |||||
| Options vested and exercisable at December 31, 2009 |
3,507,803 | $ | 8.61 | 4.73 | $ | 2,910,320 | |||||
The aggregate intrinsic value of stock options exercised in 2009, 2008 and 2007 was $2.7 million, $4.9 million and $1.1 million, respectively. As of December 31, 2009, there was $22.9 million of unamortized compensation cost related to unvested stock option awards, which is expected to be recognized over a remaining weighted-average vesting period of 2.1 years. Cash received from stock option exercises for the years ended December 31, 2009 and 2008 was $1.1 million and $1.3 million, respectively. At December 31, 2009, 199,932 shares were available for future option grants.
F-31
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
Restricted Stock
On October 18, 2007, we granted 50,000 restricted stock units to an executive officer with a grant date fair value of $11.04. These units vest over 4 years, with 13/48th of the units vesting 13 months after the grant date, and the remaining units vesting in equal monthly installments, thereafter. During 2009, 23,929 of these restricted stock units vested and the remaining 26,071 have been cancelled.
On January 29, 2008, we granted 18,628 restricted stock awards and 20,974 restricted stock units to certain executive officers and employees with a weighted average grant date fair value of $8.16 per share. During 2008, restricted stock awards totaling 1,781 were cancelled. The remaining 16,847 restricted stock awards vested on January 29, 2009 and the 20,974 restricted stock units vested on February 28, 2009.
On July 17, 2008, we granted 55,555 restricted stock units to an executive officer with a grant date fair value of $20.00 per share. These units vest over 4 years, with 13/48th of the units vesting 13 months after the grant date, and the remaining units vesting in equal monthly installments, thereafter. During 2009, 16,159 of these restricted stock units vested and the remaining 39,396 have been cancelled.
On February 9, 2009, we granted 15,907 restricted stock units to certain executive officers with a grant date fair value of $17.60 per share, of which 7,234 vest after 12 months and 8,673 vest over 13 months, respectively. During 2009, 5,978 of these restricted stock units were cancelled and the remaining 9,929 restricted stock units are outstanding and unvested.
On February 17, 2009, we granted 5,552 restricted stock units to an executive officer with a grant date fair value of $16.38 per share, which vest after 13 months. During 2009, all of these restricted stock units were cancelled.
The cancellation of the restricted stock units referred to in the above paragraphs were related to the departure of certain former executive officers as disclosed in our current report on Form 8-K, filed with the SEC on September 28, 2009.
On January 2, 2009, we granted 5,754 restricted stock awards to certain of our board members with a grant date fair value of $20.58 per share, which vest quarterly over one year. During 2009, 5,069 of these restricted stock awards vested and 685 restricted stock awards were cancelled.
On October 20, 2009, we granted 26,000 restricted stock units to two employees with a grant date fair value of $3.38 per share. The restricted units are tied to the successful achievement of performance criteria to be met by June 30, 2010. As of December 31, 2009, all of these restricted units were unvested and outstanding.
Also on October 20, 2009, we granted 25,000 restricted stock units to a certain executive officer with a grant date value of $3.38 per shares and vest equally over four quarters from the grant date. As of December 31, 2009, all of these restricted units were unvested and outstanding.
On December 17, 2009, we granted 1,209,600 restricted units to our employees with a grant date fair value of $3.86 per share. The restricted units vest over four years and are tied to the successful achievement of various performance criteria. As of December 31, 2009, all of these restricted units were unvested and outstanding.
F-32
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
Employee Stock Purchase Plan
In 1999, we adopted the 1999 Employee Stock Purchase Plan (the 1999 ESPP). As of December 31, 2009, we had reserved 936,902 shares of common stock for issuance under the 1999 ESPP. Beginning in 2001, the amount of authorized shares available under the 1999 ESPP automatically increased each January 1st by an amount equal to 1% of the outstanding common stock on the last trading day of the prior year, subject to an annual increase limitation of 166,666 shares. Subsequent to yearend, the 1999 ESPP was amended to remove the automatic annual increase provision, as well as placing a per employee share limit of 10,000 shares per offering period. The 1999 ESPP provides for a series of concurrent offering periods, each with a maximum duration of 24 months. Shares are purchased semi-annually at 85% of the lower of the beginning or end of the period price.
In October 2006, our Board of Directors approved a change to all offerings under the 1999 ESPP that commence on or after February 1, 2007. New offerings are for a duration of six months and consist of one purchase interval, but do not impose either an individual or all-participant limitation on the number of shares purchasable on a purchase date, although the 1999 ESPP limits stock purchases to $25,000 per individual per calendar year. Participants had the option of: continuing under the current plan offering period until its expiration, or withdrawing from the current offering prior to its expiration and enrolling in the new offering commencing on February 1, 2007. Those employees not electing to enroll in the new offering period continued under the then current offering until the 24 month offering period expires. On July 31, 2008, the final 24 month offering period expired and all ESPP participants are now under six month offering periods. As of December 31, 2009, employees have contributed approximately $0.3 million to the current offering of the 1999 ESPP since the beginning of the offering period that commenced August 1, 2009. For the year ended December 31, 2009 and 2008, we have recognized approximately $243,000 and $360,000, respectively, as share-based compensation expense related to the 1999 ESPP Plan.
Warrants
In connection with the acquisition of Axiom Biotechnologies in 2002, we assumed an outstanding warrant to purchase 7,333 Axiom ordinary shares at an exercise price of $10.50, which was adjusted to become a warrant to purchase 1,535 shares of our common stock at an exercise price of $50.19 per share. As of December 31, 2009, this warrant has not been exercised and expires in December 2011.
In connection with an amendment to our lease for our corporate headquarters in San Diego, California in September 2005, we issued to the landlord a warrant to purchase 50,000 shares of our common stock with an exercise price of $2.64 per share. The warrant expires in October 2015. As of December 31, 2009, the warrant remains outstanding and exercisable.
In connection with the private placement financing completed in June 2006, we issued to the investors warrants to purchase an aggregate of 11,999,999 shares of our common stock at an exercise price of $2.10 per share. These warrants contained anti-dilution provisions that adjusted the exercise price and number of shares subject to the warrants upon reorganization, mergers, stock splits and combinations, reclassifications of our common stock, stock dividends, or other issuances of our common stock at purchase prices less than the warrants’ exercise price (other than certain exempt issuances, such as sales of common stock to our employees or conversions of convertible securities and options that were outstanding prior to the issuance of the warrants). During 2008, investors exercised all remaining warrants to purchase an aggregate of 11,301,499 shares of our common stock in a series of cashless exercises, which resulted in the purchase of 8,982,521 shares of our common stock. No warrants remain unexercised with the investor group from our private placement completed in June 2006.
F-33
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
Additionally in connection with the June 2006 private placement financing, we issued to our placement agent a warrant to purchase 866,666 shares of our common stock at an exercise price of $2.52 per share. This warrant contains anti-dilution provisions that adjust the exercise price and number of shares subject to the warrants upon reorganization, mergers, stock splits and combinations, reclassifications of our common stock, or stock dividends, but not for other issuances of our common stock. During 2007 the placement agent transferred portions of the warrant to certain of its employees. During 2008, the placement agent and its transferees had exercised warrants in both cash and cashless exercises to purchase an aggregate of 110,781 shares of our common stock. As of December 31, 2009, warrants to purchase an aggregate of 7,500 shares remained outstanding and exercisable and expire in June 2011.
11. Income Taxes
The Company recognizes the impact of an uncertain income tax position on our income tax return at the largest amount that is more-likely-than-not to be sustained upon audit by the relevant taxing authority. An uncertain income tax position will not be recognized if it has less than a 50% likelihood of being sustained.
There are no unrecognized tax benefits included in the consolidated balance sheets at December 31, 2009 and 2008.
Our practice is to recognize interest and/or penalties related to income tax matters in income tax expense. We had no accrual for interest and penalties on our balance sheets at December 31, 2009 and 2008 and have recognized no interest and/or penalties in the statement of operations for the year ended December 31, 2009.
We are subject to taxation in the U.S., foreign and various state jurisdictions. Our tax years for 1995 and forward are subject to examination by the Federal and California tax authorities due to the carryforward of unutilized net operating losses and research and development credits.
We completed a Section 382/383 analysis regarding the limitation of net operating loss and research and development credit carryforwards in April 2008. We are currently in the process of updating the Section 382/383 analysis and we are removing our federal and state net operating losses and research and development credits from the deferred table until this analysis is complete. As of December 31, 2009, we do not have any unrecognized tax benefits. Due to the existence of the valuation allowance, future changes in our unrecognized tax benefits will not impact our effective tax rate.
The reconciliation of income tax computed at the Federal statutory tax rate to the expense for income taxes is as follows:
| December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| (In thousands) | ||||||||||||
| Tax at statutory rate |
$ | (24,805 | ) | $ | (15,380 | ) | $ | (7,694 | ) | |||
| State taxes, net of federal benefit |
(2,910 | ) | (2,525 | ) | (1,263 | ) | ||||||
| Change in valuation allowance |
(30,458 | ) | 29,532 | (108,313 | ) | |||||||
| Change in state rate |
3,724 | — | — | |||||||||
| Credits and other |
54,566 | (11,416 | ) | 117,270 | ||||||||
| $ | 117 | $ | 211 | $ | — | |||||||
F-34
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
The 2009 and 2008 income tax expense of $117,000 and $211,000 are comprised of foreign current and deferred taxes.
Deferred income taxes reflect the net effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of our deferred tax assets and liabilities are shown below. A full valuation allowance has been recorded, as realization of such assets is uncertain.
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| (In thousands) | ||||||||
| Deferred tax assets: |
||||||||
| Net operating loss carryforwards |
$ | 3,161 | $ | 32,221 | ||||
| Capitalized research expenses |
8,260 | 11,387 | ||||||
| Depreciation |
1,605 | 683 | ||||||
| Stock options (FAS123r) |
3,243 | 1,384 | ||||||
| Accruals and reserves |
5,655 | 6,715 | ||||||
| Other, net |
490 | (98 | ) | |||||
| Total deferred tax assets |
22,414 | 52,292 | ||||||
| Valuation allowance |
(22,414 | ) | (52,292 | ) | ||||
| Net deferred tax assets (liabilities) |
$ | — | $ | — | ||||
At December 31, 2009, we have federal and state tax net operating loss carryforwards of approximately $132.8 million and $133.4 million, respectively. The federal tax loss carryforwards will begin to expire in 2025, unless previously utilized. The state tax loss carryforwards will begin to expire in 2010, unless previously utilized.
We also have German net operating loss carryforwards of approximately $10.6 million, which may be carried forward indefinitely. We have discontinued operations in the United Kingdom (U.K.) and therefore, removed our U.K. net operating loss carryforwards of $35.6 million from our deferred tax schedule as of December 31, 2007.
We also have federal and California research and development tax credit carryforwards of approximately $2.5 million and $9.7 million, respectively. The federal research and development credits have been reduced by the Section 383 limitation. The federal research and development tax credit carryforwards will begin to expire in 2026 unless previously utilized. The California research and development credit carryforward indefinitely.
In February 2009, the California legislature enacted 2009-2010 budget legislation containing various California tax law changes, including an election to apply a single sales factor apportionment formula for taxable years beginning on or after January 1, 2011. The Company anticipates not making the election, reflecting anticipated losses for the foreseeable future.
12. Savings and Pension Plans
We have a 401(k) savings plan covering most United States employees. In the United Kingdom we make contributions to defined contribution pension plans. Under these plans, individual employees may make contributions to the plan, which can be matched by us in an amount determined by the Board of Directors or as determined by local statutes. We made no matching contributions in 2009, 2008 and 2007.
F-35
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
13. Geographic Information
We have wholly-owned subsidiaries located in Germany, the United Kingdom, India, Hong Kong and Japan and have customer and vendor relationships worldwide. The following table presents information about us by geographic area. There were no material amounts of transfers between geographic areas. Included in the consolidated balance sheets and consolidated statements of operations are the following domestic and foreign components at December 31, 2009, 2008 and 2007:
| December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| (In thousands) | ||||||||||||
| Current assets: |
||||||||||||
| United States |
$ | 54,891 | $ | 111,717 | $ | 59,992 | ||||||
| Europe |
5,290 | 6,911 | 6,313 | |||||||||
| Asia |
2,749 | 3,656 | 2,087 | |||||||||
| $ | 62,930 | $ | 122,284 | $ | 68,392 | |||||||
| Equipment and leasehold improvements, net: |
||||||||||||
| United States |
$ | 11,306 | $ | 8,655 | $ | 5,559 | ||||||
| Europe |
262 | 433 | 276 | |||||||||
| Asia |
243 | 107 | 124 | |||||||||
| $ | 11,811 | $ | 9,195 | $ | 5,959 | |||||||
| Long-term assets: |
||||||||||||
| United States |
$ | 11,632 | $ | 8,928 | $ | 1,695 | ||||||
| Europe |
204 | 77 | — | |||||||||
| Asia |
68 | — | — | |||||||||
| $ | 11,904 | $ | 9,005 | $ | 1,695 | |||||||
| Total assets: |
||||||||||||
| United States |
$ | 77,829 | $ | 129,300 | $ | 67,245 | ||||||
| Europe |
5,756 | 7,421 | 6,590 | |||||||||
| Asia |
3,060 | 3,763 | 2,211 | |||||||||
| $ | 86,645 | $ | 140,484 | $ | 76,046 | |||||||
| Revenues: |
||||||||||||
| United States |
$ | 18,020 | $ | 23,806 | $ | 22,243 | ||||||
| Europe |
11,150 | 13,272 | 10,821 | |||||||||
| Asia |
8,693 | 10,071 | 7,938 | |||||||||
| $ | 37,863 | $ | 47,149 | $ | 41,002 | |||||||
| Net loss: |
||||||||||||
| United States |
$ | (52,644 | ) | $ | (21,256 | ) | $ | (12,690 | ) | |||
| Europe |
748 | (6,856 | ) | (2,527 | ) | |||||||
| Asia |
(19,116 | ) | (16,042 | ) | (6,766 | ) | ||||||
| $ | (71,012 | ) | $ | (44,154 | ) | $ | (21,983 | ) | ||||
F-36
Table of Contents
SEQUENOM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2009
14. Selected Quarterly Financial Data (unaudited)
| First Quarter |
Second Quarter |
Third Quarter |
Fourth Quarter |
Total Year |
||||||||||||||||
| (In thousands, except share information) | ||||||||||||||||||||
| 2009 |
||||||||||||||||||||
| Net sales |
$ | 8,688 | $ | 9,168 | $ | 9,220 | $ | 10,787 | $ | 37,863 | ||||||||||
| Gross profit |
5,264 | 6,042 | 6,542 | 5,445 | 23,293 | |||||||||||||||
| Net loss |
(17,489 | ) | (20,246 | ) | (14,875 | ) | (18,402 | ) | (71,012 | ) | ||||||||||
| Net loss per share, basic and fully diluted |
$ | (0.29 | ) | $ | (0.33 | ) | $ | (0.24 | ) | $ | (0.30 | ) | $ | (1.16 | ) | |||||
| Shares used in calculated per share amounts, historical, basic and fully diluted |
61,014 | 61,138 | 61,211 | 61,313 | 61,171 | |||||||||||||||
| 2008 |
||||||||||||||||||||
| Net sales |
$ | 10,574 | $ | 12,845 | $ | 11,570 | $ | 12,160 | $ | 47,149 | ||||||||||
| Gross profit |
5,903 | 7,383 | 7,042 | 7,231 | 27,559 | |||||||||||||||
| Net loss |
(8,626 | ) | (9,743 | ) | (10,371 | ) | (15,414 | ) | (44154 | ) | ||||||||||
| Net loss per share, basic and fully diluted |
$ | (0.19 | ) | $ | (0.21 | ) | $ | (0.18 | ) | $ | (0.25 | ) | $ | (0.83 | ) | |||||
| Shares used in calculated per share amounts, historical, basic and fully diluted |
45,330 | 47,147 | 59,115 | 60,775 | 53,129 | |||||||||||||||
15. Subsequent Events
In February 2010, our wholly-owned subsidiary Sequenom CMM signed a worldwide licensing agreement with Optherion, Inc., for the rights to develop and commercialize diagnostic tests to predict genetic predisposition to late stage age-related macular degeneration (AMD). Under the terms of the licensing agreement, in the event that the first commercial sale of a licensed product in the United States has not occurred on or before January 31, 2011, we will pay Optherion a non-creditable license maintenance fee equal to $260,000 per year. The license maintenance fee will be pro-rated for any period less than a full year before the first commercial sale of a licensed product in the United States. Following the first commercial sale of a licensed product in the United States, we will no longer be required to pay the license maintenance fee, but instead we will pay Optherion a minimum royalty payment each year during the term of the agreement ranging between $260,000 and $270,000 per year and such minimum payment shall be creditable against any royalties due based upon licensed product sales. We have also agreed to make payments to Optherion upon the achievement of specified development, regulatory and commercial milestones, and during the life of the patent claims licensed under the agreement, royalties on the cumulative worldwide annual net sales of products successfully developed and commercialized covered by the patent claims and know-how licensed under the agreement. We also agreed, upon entry into the agreement, to reimburse Optherion for its prior patent related costs and expenses in the amount of $1,071,651. We may terminate the agreement for any reason upon 90 days prior written notice, provided that if notice of termination is delivered prior to the first anniversary of the effective date of the agreement, we are required to pay Optherion a non-creditable termination fee of $2,000,000. In the event that the agreement expires pursuant to its terms, we will retain the licenses and sublicenses granted under the agreement as fully paid and royalty free, subject to certain specified limitations.
F-37
Table of Contents
Schedule II—SEQUENOM, INC.
Valuation and Qualifying Accounts
($ in thousands)
| Description |
Balance at Beginning of Period |
Charged to Costs and Expenses |
Deductions | Balance at End of Period | ||||||||||
| Year ended December 31, 2009: |
||||||||||||||
| Allowance for doubtful accounts |
$ | 405 | $ | (145 | ) | $ | 19 | $ | 241 | |||||
| Reserve for obsolete or excess inventory |
1,809 | 2,468 | 2,359 | (1) | 1,918 | |||||||||
| Year ended December 31, 2008: |
||||||||||||||
| Allowance for doubtful accounts |
$ | 186 | $ | 281 | $ | 62 | $ | 405 | ||||||
| Reserve for obsolete or excess inventory |
1,089 | 1,180 | 460 | (1) | 1,809 | |||||||||
| Year ended December 31, 2007: |
||||||||||||||
| Allowance for doubtful accounts |
$ | 117 | $ | 143 | $ | 74 | $ | 186 | ||||||
| Reserve for obsolete or excess inventory |
1,082 | 185 | 178 | (1) | 1,089 | |||||||||
| (1) | Write off of obsolete or excess inventory |
F-38