Attached files
| file | filename |
|---|---|
| EX-32.2 - EXHIBIT 32.2 - PONIARD PHARMACEUTICALS, INC. | a2197312zex-32_2.htm |
| EX-32.1 - EXHIBIT 32.1 - PONIARD PHARMACEUTICALS, INC. | a2197312zex-32_1.htm |
| EX-23.2 - EXHIBIT 23.2 - PONIARD PHARMACEUTICALS, INC. | a2197312zex-23_2.htm |
| EX-31.1 - EXHIBIT 31.1 - PONIARD PHARMACEUTICALS, INC. | a2197312zex-31_1.htm |
| EX-23.1 - EXHIBIT 23.1 - PONIARD PHARMACEUTICALS, INC. | a2197312zex-23_1.htm |
| EX-10.21 - EXHIBIT 10.21 - PONIARD PHARMACEUTICALS, INC. | a2197312zex-10_21.htm |
| EX-31.2 - EXHIBIT 31.2 - PONIARD PHARMACEUTICALS, INC. | a2197312zex-31_2.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| (Mark One) | ||
ý |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the fiscal year ended December 31, 2009 |
||
or |
||
o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the transition period from to
Commission File No. 0-16614
PONIARD PHARMACEUTICALS, INC.
(Exact name of Registrant as specified in its charter)
| Washington (State or other jurisdiction of incorporation or organization) |
91-1261311 (IRS Employer Identification No.) |
7000 Shoreline Court, Suite 270, South San Francisco, CA 94080
(Address of principal executive offices)
Registrant's telephone number, including area code: (650) 583-3774
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Name of each exchange on which registered | |
|---|---|---|
| Common Stock, $0.02 par value | The Nasdaq Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
$2.4375 Convertible Exchangeable Preferred Stock, Series 1, $0.02 par value
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicated by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.45 of the chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ý
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer o | Accelerated filer ý | Non-accelerated filer o (Do not check if a smaller reporting company) |
Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No ý
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant was approximately $137.6 million as of June 30, 2009, based on a per share closing price of $5.97 on the Nasdaq Global Market on that date.
As of March 9, 2010, 42,844,262 shares of the registrant's common stock, $0.02 par value per share, were outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Specified portions of the registrant's Definitive Proxy Statement for the 2010 Annual Meeting of Shareholders, to be filed within 120 days of the end of the fiscal year covered by this Annual Report on Form 10-K are incorporated by reference into Part III of this Annual Report on Form 10-K. Except with respect to information specifically incorporated by reference into this Annual Report on Form 10-K, the Proxy Statement for the 2010 Annual Meeting of Shareholders is not deemed part hereof.
IMPORTANT INFORMATION REGARDING FORWARD-LOOKING STATEMENTS
This Form 10-K contains forward-looking statements within the meaning of the Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, and in reliance upon the safe harbor provisions of the Private Securities Litigation Reform Act of 1995. Forward-looking statements are those that predict or describe future events or trends and that do not relate solely to historical matters. In some cases, you can identify forward-looking statements by terminology such as "may," "will," "could," "should," "expect," "plan," "intend," "anticipate," "believe," "estimate," "predict," "project," "potential," "propose," "continue," "assume" or other similar expressions, or the negatives of those expressions. All statements contained in this Form 10-K or incorporated in this Form 10-K by reference regarding our corporate objectives and strategies, future operations, potential partners and other strategic relationships, projected financial position, planned clinical and regulatory activities, proposed products, future regulatory approvals, proposed product commercialization, estimated future revenue, projected costs, potential sources of capital, future prospects, the future of our industry, and results that might be obtained by pursuing management's current plans and objectives are forward-looking statements.
You should not place undue reliance on our forward-looking statements because these statements reflect our current views with respect to future events and are based on assumptions and are subject to risks and uncertainties that are difficult to predict. Our forward-looking statements are based on the information currently available to us and speak only as of the date of this report or, in the case of forward-looking statements incorporated herein by reference, the date of the filing that includes the statement. Over time, our actual results, performance or achievements may differ from those expressed or implied by our forward-looking statements, and such difference might be significant and materially adverse to our security holders. Except as required by law, we undertake no obligation to update publicly any forward-looking statements to reflect new information, events or circumstances after the date of this report, or to reflect the occurrence of unanticipated events.
We have identified some of the important factors that could cause future events to differ from our current expectations and they are described under the headings "Risk Factors" in Item 1A below and "Management's Discussion and Analysis of Financial Condition and Results of Operations" in Item 7 below. Please consider our forward-looking statements in light of these risks as you read this report and any information incorporated by reference in this report.
Unless otherwise indicated, all common stock-related amounts in this report have been adjusted to reflect our one-for-six reverse stock split effective September 22, 2006.
The Company
Poniard is a biopharmaceutical company focused on the development and commercialization of cancer therapeutics. Our lead product candidate is picoplatin, a new generation platinum-based cancer therapy that has the potential to become a platform product for use in different formulations, as a single agent or in combination with other anti-cancer agents, to treat multiple cancer indications, including small cell lung, colorectal, prostate and ovarian cancers. Picoplatin is an intravenous platinum-based chemotherapeutic designed to treat solid tumors that are resistant to existing platinum-based cancer therapies. Clinical studies to date suggest that picoplatin has an improved safety profile relative to existing platinum-based cancer therapies. We have completed enrollment and initial statistical analysis of a pivotal Phase 3 SPEAR (Study of Picoplatin Efficacy After Relapse) trial of picoplatin in the second-line treatment of patients with small cell lung cancer. This trial did not meet its primary endpoint of overall survival and we have initiated a process with the U.S. Food and Drug
2
Administration, or the FDA, to identify a potential regulatory path forward for picoplatin in this indication. We are also conducting two separate Phase 2 trials evaluating picoplatin as a first-line treatment of metastatic colorectal cancer and castration-resistant (hormone-refractory) prostate cancer. Additionally, we have completed a Phase 1 cardiac safety trial of picoplatin and a Phase 1 study evaluating an oral formulation of picoplatin in solid tumors. Our corporate strategy currently is focused on identifying potential regulatory pathways forward for picoplatin and exploring potential partnering and other relationships to support the continued development of picoplatin in multiple indications and two formulations.
We have financed our operations to date primarily through the sale of equity securities, technology licensing, collaborative agreements and borrowings under debt instruments. Entities affiliated with MPM Capital Management, or MPM, beneficially owned an aggregate of approximately 18.6% of our common stock outstanding on December 31, 2009. Entities affiliated with Bay City Capital Management IV LLC, or Bay City Management, beneficially owned an aggregate of approximately 13.0% of our common stock outstanding on December 31, 2009. Nicholas J. Simon, a representative of MPM, and Fred B. Craves and Carl S. Goldfischer, managing directors of Bay City Capital LLC, an affiliate of Bay City Management, serve on our board of directors.
During 2009, we sold an aggregate of approximately 7.0 million shares of our common stock to Azimuth Opportunity Ltd., or Azimuth, pursuant to two draw downs under an equity line of credit facility with Azimuth dated August 19, 2009, as amended. In the first draw down on November 23, 2009, we sold approximately 3.5 million common shares to Azimuth at a purchase price of approximately $2.15 per share. We sold Azimuth approximately 3.5 million common shares for approximately $1.87 per share in the second draw down on December 22, 2009. The equity financing facility terminated by its terms on December 22, 2009. We received aggregate net proceeds from the draw downs of approximately $13.7 million. In September 2008, we borrowed approximately $20.0 million of additional net cash proceeds under an amended and restated loan facility with GE Business Financial Services, Inc. and Silicon Valley Bank. On April 30, 2007, we completed a public offering of 11.8 million common shares at an offering price of $6.33 per share. Net proceeds of the offering were approximately $70.0 million. Additionally, in April 2006, we received $62.0 million in net proceeds from an equity financing, pursuant to which we issued 15.5 million common shares at a purchase price of $4.20 per share. Investors in the 2006 financing also received warrants to purchase an aggregate of 4.6 million common shares at a purchase price of $4.62 per share. The proceeds of these transactions were used to support our research and clinical development activities and commercialization efforts over the past three years, as well as for general corporate purposes, including working capital.
In 2010, we plan to refocus our resources on regulatory and partnering strategies to support the continued development of picoplatin. On February 5, 2010, we implemented a restructuring plan to conserve capital resources, which reduced our workforce by approximately 57%, to 22 employees. We believe that our current workforce is sufficient to support our regulatory and partnering strategies focused on the continued development of picoplatin. On March 15, 2010, we completed a draw down and sale of 4,229,000 shares of our common stock, at a price of approximately $1.49 per share, to Commerce Court Small Cap Value Fund, Ltd., or Commerce Court, under our equity line of credit facility with Commerce Court, dated February 23, 2010. Net proceeds of approximately $6,154,000 were received, after deducting estimated offering costs of approximately $166,000. A description of this equity line of credit facility is set out in the section entitled "Management's Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources" in Item 7 below.
Since our inception in 1984, we have dedicated substantially all of our resources to research and development. We have not generated significant revenue from any product sales and have operated at a loss in each year of our existence. We had a net loss of $45.7 million for the year ended December 31, 2009, a net loss of $48.6 million for the year ended December 31, 2008, and a net loss of $32.8 million
3
for the year ended December 31, 2007. We do not anticipate that our picoplatin product candidate will be commercially available before 2011. We expect to incur additional operating losses in the future. Clinical studies are inherently uncertain, and current and future trials of picoplatin may not confirm the results achieved in earlier clinical and preclinical studies. If picoplatin is not shown to be safe and effective, we will not receive the required regulatory approvals for commercial sale of such product. We are exploring potential partnering and other relationships to support the continued development of picoplatin. We may not be able to secure corporate partners or enter into strategic relationships on a timely basis or on terms that ultimately prove favorable to us.
Our consolidated financial statements have been prepared assuming that we will continue as a going concern, which contemplates realization of assets and the satisfaction of liabilities in the normal course of business for a reasonable period following the date of these financial statements. As of December 31, 2009, we had net working capital of $27.4 million, an accumulated deficit of $408.2 million and total shareholders' equity of $23.6 million. Cash, cash equivalents and investment securities, net of restricted cash of $0.3 million, totaled $43.4 million at December 31, 2009.
Our current loan facility with GE Business Financial Services, Inc. and Silicon Valley Bank, the terms of which are described below under the heading, "Management's Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources," requires us to maintain a minimum amount of unrestricted cash during the term of the loan equal to the lesser of (i) $17.9 million or (ii) the outstanding aggregate principal balance of the term loans plus $4.0 million. Taking into account the minimum unrestricted cash requirement under the loan facility and our projected operating results, we believe that our current cash, cash equivalent and investment securities balances, including the net proceeds received from our March 15, 2010 sale of common stock to Commerce Court under our equity line of credit facility, will provide adequate resources to fund operations at least through the end of 2010. However, given the uncertainties of outcomes of our regulatory and partnering strategies to support the continued development of picoplatin, there is no assurance that we can achieve our projected operating results. Thereafter, unless we raise additional funds, we will be in default of the loan facility. The occurrence of an event of default would increase the applicable rate of interest by 5% and could result in the acceleration of our payment obligations under the loan agreement. We have no assurance, especially in light of the current difficult economic environment, that the lenders will be willing to waive or renegotiate the terms of the loan agreement to address or avoid financial or other defaults.
If an event of default were to occur, we might not have sufficient funds to repay the loan or to fund our continuing operations. In such case, we would need to delay, scale back or curtail some or all of our current picoplatin clinical and regulatory efforts, further reduce our workforce, license picoplatin for development and commercialization by third parties, or attempt to sell the company. Provisions of the loan agreement would limit our ability to dispose of certain assets, engage in certain mergers, incur certain indebtedness, make certain distributions and engage in certain investment activities without the prior consent of the lenders.
Picoplatin Development Program
Overview of Cancer and its Treatment
Cancer is a disease characterized by the uncontrolled growth and spread of abnormal cells. Cancer cells often originate from one tissue site and invade, spread and damage other tissues and organs, leading to death. Cancer is the second highest cause of death in the United States, exceeded only by heart disease. In the United States, cancer accounts for one of every four deaths. In 2009, approximately 562,340 Americans were expected to die of cancer, more than 1,500 people a day. The National Cancer Institute estimated that 1,479,350 new cancer cases would be diagnosed in 2009 (American Cancer Society: Cancer Facts & Figures 2009).
4
In recent years, the diagnosis and treatment of human cancers have greatly improved. However, there is still considerable need for new cancer therapies, as well as treatments that improve upon existing therapies. Current treatments for cancer include surgery, external-beam radiation, chemotherapy, hormone therapy, cytokines, interferons and antibodies. It is anticipated that chemotherapeutics and targeted anti-cancer agents will be used both as single agents and in combination to provide benefit to cancer patients. Often patients are treated with multiple agents in combination and in varying sequences depending on the particular cancer type and severity of disease. In this regard, chemotherapeutics have continued to have significant impact on cancer treatment, especially when combined with other agents that have anti-cancer properties. We believe that new treatment combinations that incorporate approved targeted agents with chemotherapeutics that exhibit improved efficacy and safety features and provide benefit to risk ratios for specific patient populations, will be supported by physicians and their patients. In recent years, many new classes of agents providing modest increases in patient survival have been approved for use. We anticipate that the use of multiple agents, either in combination or in sequence, will continue to provide benefits to cancer patients. In addition, we believe that individualized therapies will become more prominent as enhanced tumor diagnostics and agents with different mechanisms of anti-cancer effect are approved and become available to the practicing oncologist. We also expect that early diagnosis and cancer prevention will provide for interventions that will allow patients to live longer and have a better quality of life.
Picoplatin and Platinum-Based Chemotherapeutics
Over the past three decades, platinum-based drugs have become a critical part of cancer treatment, administered primarily in combination with other chemotherapeutics, and more recently with approved targeted anti-cancer agents. Platinum-based agents, such as cisplatin, carboplatin and oxaliplatin, are currently used to treat a variety of tumors, including testicular, ovarian, colorectal and lung cancers. The mechanism that underlies the use of platinum-based agents relies upon the targeting of tumor DNA where the platinum compound binds. Cells that undergo active cell division are prevented from completing the cell cycle by the presence of the platinum drug that is chemically bound to the DNA. The inability to proceed through normal cell division ultimately causes cell death. In some cases, treatment of cancer patients with platinum compounds leads to reduction in tumor mass due to a higher rate of tumor cell death compared with tumor cell replication.
All platinum-based chemotherapeutic agents exhibit toxicity to the blood forming cells in the bone marrow, or myelosuppression, as a major adverse effect. The degree and characteristics of myelosuppression vary by platinum compound, dose and regimen. In addition, some current platinum agents show different degrees of other adverse side effects, including kidney damage (nephrotoxicity), hearing loss (ototoxicity), nausea, vomiting and nerve damage (neurotoxicity). As in the case of myelosuppression, these side effects vary with platinum agent, dose, combination therapy and regimen.
For most cancers that are treated with platinum-containing regimens, patients whose cancer initially responds to platinum-containing chemotherapy, subsequently experience progression of their disease due to acquired resistance to the chemotherapy. We believe that patients would benefit from a rationally designed platinum-based agent that is active in patients who have become resistant after receiving prior platinum-containing treatment and is potentially synergistic in combination with other agents.
In April 2004, we acquired the rights to develop, manufacture and commercialize picoplatin. In September 2006, we renegotiated the financial terms of our April 2004 license agreement and obtained exclusive worldwide rights to picoplatin. Picoplatin is a new-generation platinum-based chemotherapeutic agent designed to overcome platinum resistance associated with chemotherapy in solid tumors. We believe that picoplatin has the potential to become a platform product for use in different formulations, as a single agent or in combination with other anti-cancer agents, and to treat multiple cancer indications, including small cell lung, colorectal, prostate and ovarian cancers. Study
5
data to date suggest that picoplatin has an improved safety profile relative to existing platinum-based cancer therapies and can be safely administered in combination with multiple approved oncology products. Over 1,100 patients have received picoplatin in clinical trials to date. Results obtained suggest that decreased production of blood cells, or myelosuppression, is common but manageable. Kidney damage, or nephrotoxicity, and nerve damage, or neurotoxicity, have been less frequent and less severe than is commonly observed with other currently-marketed platinum chemotherapy drugs. Picoplatin has shown evidence of anti-tumor activity in a variety of solid tumors, including tumors that have been pre-treated with existing platinum-based therapeutics.
Picoplatin Clinical Studies
We have completed enrollment and initial statistical analysis of a pivotal Phase 3 SPEAR trial of picoplatin in the second-line treatment of patients with small cell lung cancer. We have completed enrollment of two separate Phase 2 trials evaluating picoplatin as a first-line treatment of metastatic colorectal cancer and castration-resistant (hormone-refractory) prostate cancer. Additionally, we have completed a Phase 1 cardiac safety trial of picoplatin and a Phase 1 study evaluating an oral formulation of picoplatin in solid tumors. These programs are described below and in the section of this report entitled "Management's Discussion and Analysis of Financial Condition and Results of Operations—Research and Development." It is important to keep in mind that clinical studies are inherently uncertain, and later trials may not confirm the results achieved in earlier clinical and preclinical studies and may not be supported by the results obtained in subsequent trials. You should refer to the section of this report entitled "Risk Factors" for a discussion of some of the factors that could materially affect our picoplatin development program.
Small Cell Lung Cancer
Small Cell Lung Cancer and its Treatment. There were an estimated 219,440 new cases of lung cancer in the United States in 2009. Lung cancer accounts for the most cancer related deaths in both men and women. An estimated 159,390 deaths, accounting for about 28% of all cancer deaths in the United States, were expected in 2009 (American Cancer Society: Cancer Facts and Figures, 2009). Small cell lung cancer accounts for approximately 10% to 15% of all lung cancer cases and is the most aggressive type of lung cancer. According to IntrinsiQ, the leading provider of United States oncology market data, 52,619 small cell lung cancer patients were treated in the United States in 2008. Small cell lung cancer metastasizes rapidly and is most often discovered after it has spread. At the time of diagnosis, approximately two-thirds of small cell lung cancer patients have metastases beyond the chest region. Few patients can be cured. Surgery is seldom an option for these patients because of the extent of the disease at diagnosis. Most patients receive chemotherapy, and patients whose disease is limited to one side of the chest may also receive radiation therapy.
Platinum-based combination therapy is used in the first-line treatment of small cell lung cancer. According to IntrinsiQ, more than 80% of patients with small cell lung cancer in the United States were treated with either carboplatin or cisplatin plus etoposide as first-line chemotherapy in 2008. Despite a response rate of 40% to 90% to first-line therapy, long-term survival is rare because patients develop resistance to chemotherapy and the cancer progresses or the disease relapses.
The prognosis for patients who relapse is poor, and the expected mean survival after relapse is two to four months without any treatment. There are no FDA-approved drugs for small cell lung cancer patients who do not respond to initial platinum-based therapy. Hycamtin® is the only FDA-approved therapy for the treatment of relapsing small cell lung cancer patients who initially responded to the first-line chemotherapy treatment; however, survival is still only approximately six months. Effective second-line treatment is a major unmet medical need.
6
Based on clinical and preclinical data to date, we believe that picoplatin has potential activity in the second-line treatment of small cell lung cancer patients who have failed first-line platinum-containing therapy. A Phase 2 study conducted by a prior licensee during 2001 and 2002 to assess the activity and tolerability of picoplatin when given intravenously as a second-line therapy to patients with small cell lung cancer demonstrated that median survival of 13 patients who were resistant to initial platinum-based chemotherapy was approximately 27 weeks.
Phase 2 Clinical Trial. In October 2004, we filed an investigational new drug application, or IND, with the FDA to conduct a Phase 2 clinical trial of intravenous picoplatin as a second-line therapy for small cell lung cancer patients whose disease failed to respond to, or relapsed or progressed after completion of, first-line platinum-containing therapy. The clinical endpoints of the study included safety, objective tumor response rate (tumor shrinkage), time to tumor progression and overall survival.
We completed enrollment of the Phase 2 study in August 2006. In November 2006, we announced positive interim overall survival results from the study, indicating a median overall survival of 27 weeks in 71 evaluable patients. This data served as the basis for our decision to initiate our pivotal Phase 3 SPEAR trial. In June and September 2007, we announced additional data from our Phase 2 trial, including longer follow-up on more patients, which confirmed the interim results, with median overall survival of 27 weeks in 77 evaluable patients.
Phase 3 Clinical Trial. We initiated our pivotal Phase 3 SPEAR trial and enrolled the first patient in April 2007. The Phase 3 trial was undertaken pursuant to a Special Protocol Assessment, or SPA, with the FDA. An SPA is a written agreement between a sponsor and the FDA regarding the objectives, design and endpoints of a study to be used as a basis of filing a New Drug Application, or NDA, and the data analysis plan necessary to support full regulatory approval. The Phase 3 trial is an international, multi-center, open-label, controlled study to compare the efficacy and safety of picoplatin plus best supportive care with best supportive care alone as a second-line therapy for small cell lung cancer. We were blinded to any analysis of the aggregate data until the database was locked after the occurrence of 320 evaluable events (patient deaths). The study was designed to enroll approximately 400 patients with small cell lung cancer whose disease is non-responsive (refractory) to first-line platinum-containing (cisplatin or carboplatin) chemotherapy or whose disease responded initially to first-line platinum-containing therapy but then progressed within six months after treatment was completed. Patients were randomized in a 2:1 ratio to receive picoplatin plus best supportive care or best supportive care alone. Best supportive care includes all medical, radiation and surgical interventions that small cell lung cancer patients should receive to relieve the symptoms and treat the complications caused by small cell lung cancer, but excludes treatment with other chemotherapy. We conducted the study at clinical sites in Eastern Europe, India and South America, where we believed the greater availability of patients could enable us to more rapidly complete patient enrollment. We completed patient enrollment in March 2009.
The primary endpoint of our Phase 3 SPEAR study is overall survival, as measured in time from randomization to death. Secondary endpoints include overall response rates, disease control and progression-free survival. In September 2009, we announced that 320 evaluable events (patient deaths) had occurred in our Phase 3 SPEAR trial, allowing us to begin analysis of trial data. On November 16, 2009, based on 321 patient deaths, we announced that our pivotal Phase 3 SPEAR trial did not meet its primary endpoint of overall survival in the intent-to-treat population. The analysis showed a hazard ratio of 0.82 with a p value of 0.089 (n=321). An imbalance in the use of post-study chemotherapy was observed in favor of patients who received best supportive care alone compared to patients who received picoplatin plus supportive care. Safety data was consistent with previous Phase 2 studies of picoplatin in small cell lung cancer. Additional data analysis and our SPEAR study are ongoing. We have initiated a process with the FDA to identify a potential regulatory path forward to enable a registrational filing seeking approval of picoplatin as a second-line treatment for patients with refractory disease. We have no assurance regarding the outcome of our process with the FDA.
7
The FDA has designated picoplatin as an orphan drug for the treatment of small cell lung cancer under the provisions of the Orphan Drug Act, as amended. To qualify for orphan drug status, a proposed drug must be intended for use in the treatment of a condition that affects fewer than 200,000 people in the United States. Orphan drug status entitles us to exclusive marketing rights for picoplatin in the United States for seven years following marketing approval, if any, and qualifies us for research grants to support clinical studies, tax credits for certain research expenses and an exemption from certain application user fees.
In August 2007, the FDA also granted picoplatin Fast Track designation for the second-line treatment of small cell lung cancer. The FDA's Fast Track programs are designed to facilitate the development and expedite the review of drugs that are intended to treat a serious or life-threatening condition and that demonstrate the potential to address unmet medical needs. Fast Track designation provides for priority interactions with the FDA to improve the efficiency of clinical development and support the expeditious review of promising drug candidates.
The European Commission, in 2007, designated picoplatin as an orphan medicinal product for the treatment of small cell lung cancer in the European Union. To qualify for this designation, a proposed drug must be intended for the treatment of life-threatening or serious conditions that are rare and affect not more than five in 10,000 persons in the European Union. Orphan medicinal product designation entitles us to certain incentives, such as regulatory assistance with protocol design and possible exemptions or reductions of certain regulatory fees during development or at the time of application for marketing approval in the European Union. If such approval is received, picoplatin would qualify for ten years of marketing exclusivity in the European Union.
Metastatic Colorectal Cancer
Colorectal Cancer and its Treatment. According to the American Cancer Society, cancers of the colon and rectum are the third most common cancer among American men and women. An estimated 146,970 new cases of colon and rectal cancer were diagnosed in 2009, with an estimated 49,920 deaths in 2009, accounting for almost 9% of all cancer deaths in the United States (American Cancer Society: Cancer Facts and Figures 2009). A FOLFOX-based regimen is the standard of care in the United States for treatment of advanced colorectal cancer, or CRC, and adjuvant (post surgical) treatment of colon cancer in patients who have their primary tumors surgically removed. FOLFOX is a combination chemotherapy containing 5-fluorouracil and leucovorin and oxaliplatin (Eloxatin®) administered every two weeks. According to IntrinsiQ, 41.8% of colorectal cancer patients in the United States received oxaliplatin-containing treatment regimes in 2008. However, approximately 82% of the patients previously untreated for advanced CRC who receive this treatment develop neuropathy, and approximately 19% of patients develop severe neuropathy, according to the oxaliplatin package insert. Neuropathy is a peripheral nerve function problem that can result in numbness, tingling and pricking sensations, sensitivity to touch, pain, and muscle weakness or wasting. The National Comprehensive Cancer Network Guidelines for Physicians recommends discontinuation of oxaliplatin after three months of therapy, or sooner if severe neuropathy develops, with the other two drugs maintained until time of tumor progression. In contrast to the nerve damaging effects of oxaliplatin, picoplatin has been generally well-tolerated when given as a single agent, with approximately 13% of patients developing mild or moderate neuropathy and < 1% of the patients developing severe neuropathy in 334 patients evaluated. There presently is no approved test to predict which patients will experience neuropathy and, if so, the extent thereof.
Phase 1-2 Clinical Trial. In May 2006, we treated the first patient in our ongoing Phase 1-2 study of intravenous picoplatin in the first-line treatment of patients with metastatic CRC. The trial is being conducted in Russia, and enrollment was completed in May 2008. The Phase 1 component of the trial was designed to determine an appropriate dose of picoplatin, either once every two weeks or once every four weeks, in combination with the chemotherapy agents 5-fluorouracil and leucovorin for
8
further testing in the Phase 2 component of the trial. This combination is called FOLPI. Based on final Phase 1 data, which was presented at the American Association of Cancer Research Annual Meeting in April 2009, both dosing regimens were generally well-tolerated. Twenty-two percent of the patients treated developed neuropathy. In the majority of patients the neuropathy was mild. Four percent of patients experienced moderate neuropathy. No severe neuropathy was observed. The most frequent dose limiting toxicity was hematological, or to the blood cells. The maximum tolerated dose was established in the every-four-week schedule at 150 mg/m2. The maximum tolerated dose for the every-two-week regimen was 85 mg/m2.
We initiated a Phase 2 trial in November 2007 to generate proof-of-concept data to demonstrate that picoplatin can be used as a first-line chemotherapeutic agent as a neuropathy-sparing alternative to oxaliplatin in patients with metastatic CRC who had not received prior chemotherapy. Enrollment of 101 patients in the randomized, controlled Phase 2 trial was completed in May 2008. The trial's primary objective was to measure the relative incidence and severity of neuropathy in the FOLPI regimen compared to the FOLFOX regimen. In addition, the study measured comparative safety and efficacy (assessed by disease control, progression-free survival, and overall survival); however, the study was not powered to assess the statistical significance of these efficacy endpoints. The final Phase 2 data indicate that we achieved the primary objective of the study in demonstrating that picoplatin is a neuropathy-sparing alternative to oxaliplatin and is active in metastatic CRC.
The Phase 2 data presented at the American Society of Clinical Oncology (ASCO) Gastrointestinal Cancers Symposium in January 2010 indicate that:
- •
- FOLPI is associated with a statistically significant reduction in neurotoxicity compared to FOLFOX (HR <0.30; p
<0.004). Neuropathy is less frequent and less severe with FOLPI. Neuropathy was 26% in FOLPI-treated patients and 64% in FOLFOX-treated patients. No severe neuropathy was observed in
patients who received the FOLPI regimen.
- •
- FOLPI had similar efficacy to FOLFOX as measured by:
- •
- Disease control rate of 75% and 76% for FOLPI and FOLFOX, respectively; (Relative risk 1.02 (95% Confidence Interval (CI)
0.79-1.32), p=0.9)
- •
- Progression-free survival of 6.8 months and 7.0 months for FOLPI and FOLFOX, respectively; HR
0.95 (95% CI 0.63-1.45), p=0.82
- •
- Overall survival of 13.6 months and 15.6 months for FOLPI and FOLFOX, respectively; HR 1.17 (95% CI
0.72-1.91), p=0.53.
- •
- Six-month and one-year survival rates were 80% and 52% for FOLPI and 83% and 55% for FOLFOX,
respectively.
- •
- More patients who discontinued FOLFOX had associated neuropathy; neurotoxicity was not dose-limiting for
FOLPI. More patients who discontinued FOLPI had associated hematological events than with FOLFOX, but the hematological events were manageable.
- •
- FOLPI had more frequent and severe, but manageable, thrombocytopenia and neutropenia; complications were rare, with only 1
patient (2%) having febrile neutropenia and 2 patients (4%) having minor, transient bleeding.
- •
- No hypersensitivity, cardiac toxicity or nephrotoxicity was observed with FOLPI or FOLFOX.
- •
- Most other toxicities, including gastrointestinal toxicity, were similar for both regimens except for alopecia (hair loss), which was more frequent with FOLPI.
9
Castration-Resistant Prostate Cancer
Castration-Resistant Prostate Cancer and its Treatment. Prostate cancer has the highest number of new cases among men in the United States and is the second leading cause of death in American men. The American Cancer Society estimated that, in 2009, there would be approximately 192,280 new cases of prostate cancer in the United States and that approximately 27,360 men would die from this disease. Ten to twenty percent of men with prostate cancer present with metastatic disease, and all patients with metastatic prostate cancer become resistant to hormone treatment.
Many patients diagnosed with prostate cancer initially receive surgery or radiation therapy, and some of these patients are cured. For many, however, the disease recurs. At this point, the recurrent disease is treated with hormone therapy, and most patients initially respond well. The average duration of response is only 10 to 12 months, however, and the tumor cells eventually become resistant to the hormones, or hormone-refractory, and the tumor again progresses. Hormone-refractory prostate cancer is also known as "castration-resistant prostate cancer," or CRPC. Increasingly, chemotherapy is being used as a first-line treatment for CRPC, but few effective drugs have been identified. Docetaxel in combination with prednisone was approved by the FDA in 2004 for the treatment of patients with metastatic (stage IV) CRPC. According to IntrinsiQ, 88.5% of U.S. patients received a docetaxel-containing regimen for first-line treatment of stage IV CRPC in 2008. Docetaxel or mitoxantrone, each as a single agent, were the two most commonly prescribed second-line treatment therapies for CRPC in the United States in 2008. We believe that the combination of picoplatin and docetaxel has the potential to be more effective as a first-line treatment of CRPC than either docetaxel or picoplatin alone.
Phase 1-2 Clinical Trial. In May 2006, we treated the first patient in our Phase 1-2 study of intravenous picoplatin in the treatment of patients with CRPC that had not previously been treated with chemotherapy. The trial was conducted in Russia, and enrollment was completed in December 2007. The Phase 1 component of the trial was designed to evaluate increasing doses of picoplatin in combination with 60 or 75 mg/m2 of the chemotherapy agent docetaxel (Taxotere®) administered every three weeks with 5 mg prednisone twice daily, to establish a dose of picoplatin for further testing in the Phase 2 component of the trial. Interim Phase 1 safety data, which was presented at the ASCO Gastrourinary Satellite Symposium in February 2008, showed that the picoplatin and docetaxel combination was generally well-tolerated, with only mild neuropathy in three of 33 patients (9%), with a prostate specific antigen, or PSA, response rate of 65% (20 of 31 evaluable patients). Myelosuppression was the dose limiting toxicity. We initiated the Phase 2 component of the trial in July 2007 and completed patient enrollment in December 2007.
The Phase 2 trial evaluated the efficacy and safety of intravenous picoplatin (120 mg/m2) administered every three weeks in combination with full doses of docetaxel (75 mg/m2) with daily prednisone (5 mg) as a first-line treatment in patients with metastatic CRPC who have not received prior chemotherapy. PSA response is the primary endpoint. Secondary endpoints include duration of PSA response, time to progression, radiologic response, survival and safety. Thirty-two patients were enrolled, and 29 patients received picoplatin in combination with docetaxel and prednisone.
The Phase 2 data presented at the ASCO Genitourinary Cancers Symposium in March 2010 indicate that:
- •
- PSA response was achieved in 78% of patients with sufficient data to evaluate response (n=27). In contrast, data from the
published literature report a PSA response of 45% in patients who received docetaxel 75 mg/m2 and prednisone 5 mg. (Source: Tannock et al, NEJM 2004;351:1502-12; docetaxel
package insert).
- •
- The median progression-free survival in 29 patients who received picoplatin in combination with docetaxel and prednisone was 7.4 months.
10
- •
- The median overall survival in 29 patients who received picoplatin in combination with docetaxel and prednisone was
21.4 months. In comparison, the published data showed the median overall survival for patients who received docetaxel and prednisone was 18.9 months. (Source: Tannock et al, NEJM
2004;351:1502-12; docetaxel package insert).
- •
- Picoplatin can be safely administered with full-dose docetaxel and prednisone. No neurotoxicity was observed
in this study. In contrast, data from the published literature report evidence of neuropathy in 30% of patients receiving docetaxel and prednisone, including severe neuropathy in almost 2% of
patients.
- •
- Neutropenia was the main hematologic toxicity. The data suggest that a combination of a taxane-type product (such as docataxel) and picoplatin may have a platelet-sparing effect: thrombocytopenia was less severe and less frequent with a taxane-picoplatin regimen than with picoplatin monotherapy. In addition, when comparing the magnitude of reduction in platelets or thrombocytopenia in prior studies employing picoplatin monotherapy, thrombocytopenia was less severe and less frequent with picoplatin administered in combination with docetaxel and prednisone.
Although the Phase 2 trial is a small single-arm study, we believe that the safety and efficacy results support further development of picoplatin in combination with docetaxel and prednisone for the first-line treatment of CRPC. Further, we believe that picoplatin could play a role in the treatment of other tumor types where platinum and taxane therapies are currently used.
Ovarian Cancer
Ovarian Cancer and its Treatment. An estimated 21,550 new cases of ovarian cancer were expected in the United States in 2009. Ovarian cancer accounts for about 3% of all cancers among women and ranks second among gynecologic cancers. An estimated 14,600 deaths were expected in 2009. Ovarian cancer causes more deaths than any other cancer of the female reproductive system. (American Cancer Society: Cancer Facts and Figures 2009). The treatment of ovarian cancer is based on the stage of the disease, which is a reflection of the extent or spread of the cancer to other parts of the body. The initial treatment for advanced ovarian cancer is to remove as much of the tumor as possible, and subsequently to administer chemotherapy using drugs such as cisplatin, carboplatin and paclitaxel. A carboplatin-based regimen is the standard of care for first-line treatment of advanced ovarian cancer in the United States. According to IntrinsiQ, 83% of ovarian cancer patients received carboplatin-based therapy for first-line treatment in the United States in 2008. Although most ovarian cancers respond to initial chemotherapy, the majority of ovarian cancer patients, including those who achieve a complete response to first-line chemotherapy, will relapse and eventually die. About 75% of women with ovarian cancer in the United States survive at least one year after diagnosis. Less than half (46%) of women with ovarian cancer are still alive at least five years after diagnosis. (American Cancer Society: Cancer Facts and Figures 2009).
Phase 2 Clinical Trial. In 2002, a prior licensee reported results of a Phase 2 open-label, non-comparative, multicenter study of picoplatin monotherapy in the second-line treatment of women whose ovarian cancer had relapsed or progressed after completion of prior platinum-containing treatment. The study, which assessed tumor response, time to progression, time to death and safety (adverse effects), was conducted in multiple study locations in Europe and Australia. A total of 94 patients were enrolled. The dosing schedule was 120 to 150 mg/m2 picoplatin as a one-hour intravenous infusion, once every three weeks until disease progression, with a median number of three doses per patient. An objective response of 41% was achieved in 82 evaluable patients, including eight patients with complete responses. Picoplatin appeared to be well tolerated, with manageable myelosuppression. No clinically significant ototoxicity, nephrotoxicity or neurotoxicity was observed. The results of this trial suggest that picoplatin has a manageable toxicity profile and encouraging activity in advanced ovarian cancer.
11
Phase 1 Combination Clinical Trial. In June 2008, we announced safety and efficacy results from a previously unpublished Phase 1 clinical trial of picoplatin and pegylated liposomal doxorubicin, or PLD, in patients with advanced solid tumor malignancies, including ovarian cancer. PLD is a chemotherapeutic agent approved for treatment of advanced ovarian cancer in women who failed both platinum and paclitaxel-based chemotherapy. The trial enrolled 16 patients who had received up to three prior regimens for metastatic disease. Patients were administered picoplatin followed by PLD on day one of a 28-day cycle. A total of 62 courses of treatment were delivered to 16 patients over four dose levels, with a median number of four cycles per patient. A total of 12 patients were evaluable for response. One patient with primary peritoneal cancer experienced a complete response and four patients experienced a partial response, including three of the five patients who had ovarian cancer. Hematologic and non-hemotologic toxicities were mild. We believe that this study suggests that picoplatin and PLD may be an active combination that can be given at standard recommended dose levels with minimal increase in toxicity.
Oral Picoplatin
Phase 1 Clinical Trial. We have completed a Phase 1 randomized, open-label, dose-ranging study of the safety (adverse effects), tolerability, pharmacokinetics (how the body processes the drug) and clinical pharmacology (how the drug works in the body) of picoplatin administered orally compared with picoplatin administered intravenously in patients with advanced solid tumor malignancies. This trial was conducted at clinical sites in the United States. We believe that oral picoplatin has significant potential for use in combination with radiation therapies, oral chemotherapies and targeted therapies, including in a refractory setting following relapse from first-line therapies. In preclinical studies, picoplatin has been shown to have up to 40% oral bioavailability. Bioavailability refers to the fraction of an administered dose of an unchanged drug that reaches systemic circulation. The final Phase 1 data were presented at the American Association for Cancer Research Annual Conference in April 2009. Results showed that the bioavailability of oral picoplatin is nearly 100% at doses of 50 mg and 100 mg, indicating sufficient bioavailability to support further clinical development.
Picoplatin Source of Supply
We have entered into separate agreements with W.C. Heraeus GmbH, or Heraeus, for the manufacture of picoplatin active pharmaceutical ingredient, or API, for use in our clinical studies and for commercial purposes. We similarly have entered into separate agreements with Baxter Oncology GmbH, or Baxter, for the bulk production and distribution of finished picoplatin drug product for clinical and commercial use.
Clinical Supply. Heraeus is our sole supplier of API and Baxter is our sole supplier of finished drug product for our clinical trials. Manufacturing services are provided on a purchase order, fixed-fee basis. The API clinical supply agreement continues in effect until it is terminated by one or both of the parties in accordance with its terms. The finished drug product clinical supply agreement had an initial term ending December 31, 2009, and in December 2009, we exercised our first renewal option, extending the term to December 31, 2010. This agreement remains subject to renewal, at our option, for an additional one-year term. The agreements generally provide that they may be terminated:
- •
- by mutual agreement of the parties;
- •
- by either party, if there is a material breach by the other party that remains uncured;
- •
- by either party, in the event of solvency or bankruptcy of the other party;
- •
- in the case of the API clinical supply agreement:
- •
- by either party, if the other party or any its personnel performing services is debarred;
12
- •
- by us, if there is a change of control of Heraeus; and
- •
- in the case of the finished drug product clinical supply agreement:
- •
- by us at any time with one year's advance notice; and
- •
- by Baxter, with 24 months prior written notice if we enter into a partnership or transfer rights to picoplatin involving a direct competitor of Baxter.
Commercial Supply. We entered into a picoplatin API commercial supply agreement with Heraeus in March 2008 and a finished drug product commercial supply agreement with Baxter in November 2008. Under these agreements, Heraeus and Baxter will produce picoplatin API and finished drug product, respectively, for commercial use. Manufacturing services are provided on a purchase order, fixed-fee basis, subject to certain purchase price adjustments and minimum quantity requirements. The API commercial supply agreement continues for an initial term ending December 31, 2013, and the finished drug product commercial supply agreement continues for an initial term ending November 22, 2013, in each case subject to extension. The agreements generally may be terminated upon the same terms and conditions as the clinical supply agreements described above.
We have no assurance that our current suppliers will be able to continue to manufacture sufficient picoplatin API and/or finished drug product on a timely or cost-effective basis at all times in the future. We believe that there are other contract manufacturers with the capacity to manufacture picoplatin API and finished drug product. If we are required to seek out alternative manufacturers, we may incur significant additional costs and suffer delays in, or be prevented from, subject to FDA approval, initiating commercial sales of picoplatin.
Patents and Proprietary Rights
Our policy is to aggressively protect our proprietary technologies. We have filed applications for United States and foreign patents on many aspects of our technologies.
We hold an exclusive worldwide license granted from Genzyme Corporation (successor to AnorMED, Inc.) for the development and commercial sale of picoplatin. Under the license agreement, as amended, Genzyme retains the right to prosecute its patent applications and maintain all licensed patents, with us reimbursing such expenses. We have the right to sue any third party infringers of the picoplatin patents. If we do not file suit, Genzyme, in its sole discretion, has the right to sue the infringer at its expense.
The parties executed the license agreement in April 2004, at which time we paid a one-time upfront milestone payment of $1.0 million in common stock and $1.0 million in cash. The original license agreement excluded Japan from the licensed territory and provided for $13.0 million in development and commercialization milestones, payable in cash or a combination of cash and common stock, and a royalty rate of up to 15% of product net sales after regulatory approval. The parties amended the license agreement on September 18, 2006, modifying several key financial terms and expanded the licensed territory to include Japan, thereby providing us worldwide rights. In consideration of the amendment, we paid Genzyme $5.0 million in cash on October 12, 2006 and paid Genzyme an additional $5.0 million in cash on March 30, 2007. The amendment eliminated all development milestone payments to Genzyme. We remain obligated to pay a total of $5.0 million in commercialization milestones upon the attainment of certain levels of annual net sales of picoplatin after regulatory approval. The amendment also reduced the royalty payable to Genzyme to a maximum of 9% of annual net product sales and eliminated sharing of sublicense revenues with Genzyme. The license agreement may be terminated by either party for breach, or if the other party files a petition in bankruptcy or insolvency or for reorganization or is dissolved, liquidated or makes assignment for the benefit of creditors. We can terminate the license at any time upon prior written notice to Genzyme. If not earlier terminated, the license agreement will continue in effect, in each country in the territory in
13
which the licensed product is sold or manufactured, until the earlier of (i) expiration of the last valid claim of a pending or issued patent covering the licensed product in that country or (ii) a specified number of years after first commercial sale of the licensed product in that country.
Our picoplatin portfolio includes United States and foreign patents and applications licensed from Genzyme, which cover the picoplatin product. With respect to picoplatin, we expect to rely primarily on U.S. Patent No. 5,665,771 (expiring February 7, 2016), which is licensed to us by Genzyme, and additional licensed patents expiring in 2016 covering picoplatin in the European Union and other countries. The FDA designated picoplatin as an orphan drug for the treatment of small cell lung cancer under the provisions of the Orphan Drug Act, which entitles us to exclusive marketing rights for picoplatin in the United States for seven years following market approval. In addition, the European Commission has designated picoplatin as an orphan medicinal product for the treatment of small cell lung cancer in the European Union, which entitles us to exclusive marketing rights for picoplatin in the European Union for ten years following market approval.
We and our licensors are continually assessing and seeking to strengthen the intellectual property estate for picoplatin. On May 8, 2009, patent owners, Genzyme Corporation and The Institute of Cancer Research, together with our company, filed with the United States Patent & Trademark Office, or USPTO, an application to reissue U.S. Patent No. 5,665,771 ('771 patent) to the picoplatin compound. Like many composition of matter patents, the claims of the '771 patent include many compounds in addition to picoplatin. The reissue seeks to narrow certain claims by removing a number of compounds other than picoplatin from these claims. In the reissue proceeding, the USPTO will review the patent on its merits and an initial rejection is not unusual. We will have the opportunity to respond to any rejections and, if necessary, pursue appeals with the USPTO and the United States federal courts. During this process the '771 patent stays in force and can be asserted in an infringement action. We believe that filing the reissue to amend these claims and strengthen the intellectual property estate is a prudent course of action. Picoplatin is currently covered by additional issued process patents and other pending applications in the United States and abroad.
A number of additional potential avenues exist which may further extend our picoplatin patent protection and exclusivity. In the United States, these include The Drug Price and Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Act, which, among other things, generally provides for patent term extension for up to five years for an issued patent covering a drug product which has undergone regulatory review before marketing. In addition, since picoplatin has not been previously approved for marketing in the United States, picoplatin may qualify for new chemical entity data exclusivity, under which the FDA bans for a period of time submissions of applications from competitors based on published data or Abbreviated New Drug Applications for a drug containing the same active agent. Certain patent term restoration procedures and marketing exclusivity rights also may be available for qualifying drug products in the European Union or individual foreign countries. We intend to evaluate the availability of these mechanisms for extending the patent term and marketing exclusivity for picoplatin on an individual regional or country basis. We cannot be certain that we will be successful in any efforts to extend the term of any patent relating to picoplatin or that picoplatin will be granted additional marketing exclusivity rights in the United States or abroad.
Risks associated with the protection of our patents and other proprietary technologies are described under the heading "Risk Factors" in Item 1A below. Pending or future patent applications by us or our collaborators will not necessarily result in issued patents. Moreover, the current patents that we own or license may not provide substantial protection or commercial benefit. In addition to patent protection, we rely upon trade secrets, unpatented proprietary know-how and continuing technological innovation to develop and maintain our competitive position. Third parties could acquire or independently develop the same or similar technology or our issued patents or those licensed by us could be circumvented, invalidated or rendered obsolete by new technology. Third parties also could
14
gain access to or disclose our proprietary technology, and we may be unable to meaningfully protect our rights in such unpatented proprietary technology.
Under United States law, although a patent has a statutory presumption of validity, the issuance of a patent is not conclusive as to its validity or as to the enforceable scope of its claims. Accordingly, the patents owned or licensed by us could be invalidated, infringed or designed around by third parties. Also, third parties could obtain patents that we would need to license or design around.
Corporate Strategy and Goals
We are focused on optimizing picoplatin's value proposition as a preferred platinum agent that is complementary to the products of potential partners. We intend to utilize the promising safety and efficacy data generated from the approximately 1,100 patients who have received picoplatin in clinical trials to date to explore potential partnering and other relationships to support the continued development of picoplatin in multiple indications and two formulations. As part of this strategy, we plan to work with our clinical advisors and the FDA to identify potential regulatory paths forward for picoplatin in small cell lung, prostate, colorectal and ovarian cancers.
Competition
The competition for development of cancer therapies is substantial. There is intense competition from biotechnology and pharmaceutical companies, as well as academic research institutions, clinical reference laboratories and government agencies that are pursuing research and development activities similar to ours in the United States and abroad. Our initial focus for picoplatin has been small cell lung cancer, the most aggressive and deadly form of lung cancer. Although platinum therapies are the preferred treatment, there are no FDA-approved drugs for small cell lung cancer patients who do not respond to initial platinum-based therapy, and only one FDA-approved drug for the treatment of small cell lung cancer patients who relapse after initially responding to first-line treatment. If approved, picoplatin will be competing with existing treatment regimens, as well as emerging therapies for small cell lung cancer, and other platinum-based therapeutics. Large pharmaceutical/biotechnology companies, including Abbott, Amgen, AstraZeneca, Baxter Healthcare, Bristol-Myers Squibb Company, Celgene Corporation, Dainippon Sumitomo Pharma Co. Ltd., Eli Lilly and Company, Genentech, Inc., GlaxoSmithKline PLC, Merck & Co., Nippon Kayaku Co. Ltd., Novartis AG, Pfizer Inc., OSI Pharmaceuticals, Sanofi-Aventis Group, Shionogi & Co. Ltd. and SK Pharma, are marketing and/or developing therapeutics in late-stage clinical trials for the treatment of small cell lung cancer or platinum agents for the treatment of cancer. Multiple biotechnology companies are engaged in clinical trials for the treatment of small cell lung cancer and other platinum-based therapeutics, including Abraxis BioScience Inc., Access Pharmaceuticals Inc, Ascenta Therapeutics, Gemin X, ImmunoGen, Inc., Ipsen Group, Keryx Biopharmaceuticals Inc., Meabco A/S, MolMed S.p.A., Onyx Pharmaceuticals Inc., PharmaMar (Zeltia Group), Proacta, Inc., Regulon, Inc., Simcere Pharmaceuticals, Sunesis Pharmaceuticals Inc., Theradex, Transave Inc., Vertex Pharmaceuticals and Vion Pharmaceuticals Inc. As we seek to expand the use of picoplatin into other oncology indications, such as prostate, colorectal and ovarian cancers, we will be facing additional competition from major pharmaceutical companies, biotechnology companies, research institutions and government agencies. We cannot assure you that we will be able to effectively compete with these or future third party product development programs.
Many of our existing or potential competitors have, or have access to, substantially greater financial, research and development, marketing and production resources than we do, and are better equipped than we are, to develop, manufacture and market competing products. Further, our competitors may have, or may develop and introduce, new products that would render our picoplatin or any other proposed product candidates less competitive, uneconomical or obsolete.
15
Our ability to commercialize picoplatin and to compete effectively will depend in large part on:
- •
- our ability to meet all necessary regulatory requirements and to advance picoplatin through the FDA approval process;
- •
- the perception by physicians and other members of the health care community of the safety, efficacy and benefits of
picoplatin compared to those of competing products or therapies;
- •
- our ability to acquire picoplatin API and finished drug product on a commercial scale;
- •
- timing of market introduction;
- •
- the effectiveness of our sales and marketing efforts;
- •
- the willingness of physicians to adopt new or modified treatment regimens using picoplatin;
- •
- our ability to secure reimbursement for picoplatin;
- •
- the price of picoplatin relative to competing products; and
- •
- our ability to develop a commercial scale infrastructure, either on our own or with a collaborator, which would include the development of a distribution network and other operational and financial systems necessary to support our increased scale.
We believe that competition among products approved for sale will be based, among other things, on product safety, efficacy, reliability, availability, third party reimbursement, price and patent position. Our competitiveness also will depend on our ability to advance our product candidates, license additional technology, maintain a proprietary position in our technologies and product candidates, obtain required government and other approvals on a timely basis, attract and retain key personnel, and enter into corporate partnering or other relationships that enable us and our collaborators to develop effective products that can be manufactured cost-effectively and marketed successfully.
Government Regulation and Product Testing
The FDA and comparable regulatory health authorities in state and local jurisdictions and in foreign countries impose substantial requirements upon the clinical development, manufacture, marketing and distribution of drugs. These health authorities and other federal, state, local and foreign entities regulate research and development activities and the testing, manufacture, quality control, safety, storage, record-keeping, approval, advertising and promotion of picoplatin and any other future drug candidates. Product development and approval within these regulatory frameworks take a number of years to accomplish, if at all, and involve the expenditure of substantial resources.
U.S. Government Regulation
In the United States, drugs and biologics are subject to regulation by the FDA under the Federal Food, Drug and Cosmetic Act of 1976, as amended, and implementing regulations. The process required by the FDA before picoplatin and any other future drug candidates may be marketed in the United States generally involves the following:
- •
- completion of extensive preclinical laboratory tests, in vivo preclinical
studies and formulation studies;
- •
- submission to the FDA of an Investigational New Drug Application, or IND, which must become effective before clinical
trials can commence;
- •
- performance of properly designed and well-controlled clinical trials to establish the safety and efficacy of
the product candidate for each proposed indication;
- •
- submission of a New Drug Application, or an NDA, to the FDA; and
16
- •
- FDA review and approval of the NDA prior to any commercial sale or shipment of the drug.
In addition to obtaining FDA approval for each product, each domestic drug manufacturing establishment must be registered with and inspected by the FDA. Domestic manufacturing establishments are subject to biennial inspections by the FDA and must comply with current Good Manufacturing Practice, or cGMP, regulations, which are enforced by the FDA through its facilities inspection program for biologics, drugs and devices. To supply products for use in the United States, foreign manufacturing establishments must also comply with cGMP regulations and are subject to periodic inspection by the FDA or by corresponding regulatory health authorities in such countries under reciprocal agreements with the FDA.
Nonclinical studies include laboratory evaluation of product chemistry and formulation, as well as animal studies, to assess the potential safety and efficacy of the proposed product. Laboratories that comply with the FDA regulations regarding Good Laboratory Practice must conduct preclinical safety tests. The results of the preclinical studies are submitted to the FDA as part of an IND and are reviewed by the FDA prior to commencement of clinical trials. Unless the FDA provides comments to an IND, the IND will become effective 30 days following its receipt by the FDA. Submission of an IND does not assure FDA authorization to commence clinical trials or to allow clinical studies to continue once initiated.
Clinical trials involve the administration of the investigational new drug to healthy volunteers or to patients under the supervision of a qualified principal investigator. Clinical trials are conducted in accordance with the FDA's Protection of Human Subjects regulations and Good Clinical Practices under protocols that detail the objectives of the study, the parameters to be used to monitor safety, and the efficacy criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND. Further, each clinical study must be conducted under the auspices of an independent Institutional Review Board, or IRB, at the institution where the study will be conducted. The IRB will consider, among other things, ethical factors, the safety of human subjects and the possible liability of the institution.
Clinical trials are typically conducted in three sequential phases, but the phases may overlap. In Phase 1, the drug is tested for:
- •
- safety (adverse effects);
- •
- dosage tolerance;
- •
- pharmacokinetics (how the body processes the drug) and
- •
- clinical pharmacology (how the drug works in the body).
In Phase 2, a limited patient population is studied to:
- •
- determine the efficacy of the drug for specific, targeted indications;
- •
- determine the dosage tolerance and optimal dosage; and
- •
- identify possible adverse effects and safety risks.
If a compound is found to have potential activity in a disease or condition and to have an acceptable safety profile in Phase 2 clinical trials, Phase 3 clinical trials are undertaken to further evaluate clinical activity and to further test for safety within an expanded patient population at geographically dispersed clinical study sites. Often, Phase 4 (post-marketing) studies are required by the FDA in order to gain more data on safety and efficacy of a drug after it has transitioned into general medical practice. With respect to picoplatin or any proposed products subject to clinical trials, there can be no assurance that Phase 1, Phase 2 or Phase 3 studies will be completed successfully within any specific time period, if at all. Clinical studies are inherently uncertain, and our current picoplatin and
17
any future clinical trials may not confirm the results achieved in earlier clinical or preclinical trials. If picoplatin is not shown to be safe and effective, we will not be able to obtain the required regulatory approvals for commercial sale of that product. Furthermore, we or the FDA may suspend clinical trials at any time if it is determined that the subjects or patients are being exposed to an unacceptable health risk.
The results of the pharmaceutical development, preclinical studies and clinical trials are submitted to the FDA in the form of an NDA for approval of the marketing and commercial shipment of the drug. The testing and approval processes are likely to require substantial cost, time and effort, and there is no assurance that approval will be granted on a timely basis, or at all. The FDA may deny an NDA if applicable regulatory criteria are not satisfied, may require additional testing or information, or may require post-market testing and surveillance to monitor the safety of the product. If regulatory approval is granted, such approval may entail limitations on the indicated uses for which the product may be marketed. The FDA may withdraw product approvals if compliance with regulatory standards is not maintained or if problems occur following initial marketing. Among the conditions for NDA approval is the requirement that the prospective manufacturers' quality control and manufacturing procedures conform to cGMP regulations. In complying with standards set forth in these regulations, manufacturers must continue to expend time, money and effort in the areas of production and quality control to ensure full technical compliance.
Foreign Regulation
In addition to regulation in the United States, we are subject to a variety of foreign regulations governing clinical trials and will be subject to foreign regulations with respect to commercial sales and distribution of picoplatin and any proposed future products. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by comparable regulatory health authorities of foreign countries before we can commence clinical trials or marketing of the product in those counties. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement also vary greatly from country to country.
Under the European Union regulatory systems, marketing authorizations may be submitted either under a centralized or mutual recognition procedure. For oncology products, a centralized procedure is required. It provides for the grant of a single marketing authorization that is valid for all European Union member states. The mutual recognition procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marketing authorization may submit an application to the remaining member states. Within 90 days of receiving the application and assessment report, each member state must decide whether to recognize approval.
Our Employees
On February 5, 2010, we implemented a restructuring plan to conserve capital resources, which reduced the Company's workforce by approximately 57%, to 22 employees. We implemented an earlier restructuring plan, effective March 31, 2009, which resulted in the discontinuation of our preclinical research operations and reduced our workforce by approximately eight employees.
As of March 9, 2010, we had 20 full-time employees and two part-time employees. Of these full-time employees, five hold PhD degrees, one holds an M.D. degree, one holds a D.V.M. degree, one holds a J.D. degree and two hold an M.B.A. degree. Of the total full-time employees, ten employees are engaged in regulatory and clinical activities and ten are in general administration. We consider our relations with employees to be good. None of our employees is covered by a collective bargaining agreement. We believe that our current workforce is sufficient to support our regulatory and partnering strategies focused on the continued development of picoplatin.
18
Our Executive Officers
Information with respect to the Company's executive officers is set forth below:
Name
|
Age | Position with the Company | |||
|---|---|---|---|---|---|
| Ronald A. Martell | 48 | Chief Executive Officer | |||
| Michael S. Perry, DVM, PhD | 50 | President and Chief Medical Officer | |||
| Gregory L. Weaver | 53 | Chief Financial Officer and Senior Vice President, Finance | |||
Business Experience
Ronald A. Martell was appointed Chief Executive Officer in February 2010. He served as President and Chief Operating Officer of the Company from May 2007 to February 2010. Mr. Martell joined the Company's board of directors in June 2006. Mr. Martell served as Senior Vice President, Commercial Operation of ImClone Systems Incorporated from January 2004 to August 2006. While at ImClone, Mr. Martell was responsible for overseeing the company's sales, marketing, and project and alliance management. Mr. Martell joined ImClone in November 1998 as Vice President, Marketing. From 1988 to 1998, he served in a variety of positions at Genentech, Inc., most recently as Group Manager, Oncology Products.
Michael S. Perry was appointed President and Chief Medical Officer in February 2010. Dr. Perry has been a consultant to the Company since September 2009 and is a Venture Partner with Bay City Capital LLC (since November 2005). He was Chief Development Officer at VIA Pharmaceuticals, Inc., a publicly held drug development company, from April 2005 until May 2009. Prior thereto, he served as Chairman and Chief Executive Officer of Extropy Pharmaceuticals, Inc., a privately held pediatric specialty pharmaceutical company, from June 2003 to April 2005. From 2002 to 2003, Dr. Perry served as President and Chief Executive Officer of Pharsight Corporation, a publicly held software and consulting services firm. From 2000 to 2002, Dr. Perry served as Global Head of Research and Development for Baxter BioScience. From 1997 to 2000, Dr. Perry was President and Chief Executive Officer of both SyStemix Inc. and Genetic Therapy Inc., two wholly owned subsidiaries of Novartis Corp., and from 1994 to 1997, he was Vice President of Regulatory Affairs for Novartis Pharma (previously Sandoz Pharmaceuticals). Prior to 1994, Dr. Perry held various management positions with Syntex Corporation, Schering-Plough Corporation and BioResearch Laboratories, Inc. Dr. Perry holds a Doctor of Veterinary Medicine, a Ph.D. in Biomedical Pharmacology and a B.Sc. in Physics from the University of Guelph, Ontario, Canada. He is also a graduate of the International Management Program at Harvard Business School.
Gregory L. Weaver was appointed Chief Financial Officer and Senior Vice President, Finance in February 2009. Prior to joining the Company, Mr. Weaver served as Chief Financial Officer of Talyst Inc., a privately-held pharmacy automation information technology company, from April 2007 to December 2008. Prior to that, he served as Senior Vice President and Chief Financial Officer of Sirna Therapeutics, a public RNAI therapeutics company, from February 2006 until sale of the company to Merck, Inc. in December 2006. From April 2002 to September 2005, Mr. Weaver was Chief Financial Officer of Nastech Pharmaceuticals, a public drug delivery company. From April 1999 to April 2002, Mr. Weaver was Chief Financial Officer of Ilex Oncology, Inc., a public cancer drug development company, and from 1996 to 1998, he was Chief Financial Officer of Prism Technologies, a private medical device manufacturer. In addition, Mr. Weaver held increasingly senior positions with Fidelity Capital in Boston and Arthur Andersen LLP. Mr. Weaver has served as a director and the chairman of the audit committee of Celsion Corp., a public oncology drug development company, since 2005, and as a director and the chairman of the audit committee of SCOLR Pharmaceuticals, a public drug delivery company, since 2007. Mr. Weaver is a certified public accountant and received his M.B.A. in finance from Boston College and his B.S. in accounting from Trinity University.
19
Corporate Background
We are a Washington corporation that was originally incorporated as NeoRx Corporation in 1984. We changed our name to Poniard Pharmaceuticals, Inc. in June 2006 and relocated our corporate headquarters from Seattle, WA to South San Francisco, CA in September 2006. Our principal executive office and mailing address is 7000 Shoreline Court, Suite 270, South San Francisco, California 94080, and our telephone number is (650) 583-3774.
WHERE YOU CAN FIND MORE INFORMATION
We file annual, quarterly and current reports, as well as registration and proxy statements and other information, with the U.S. Securities and Exchange Commission, or SEC. These documents may be read and copied at the SEC's public reference rooms in Washington, DC, New York, NY and Chicago, IL. Please call the SEC at 1-800-SEC-0330 for further information on the public reference rooms. Our SEC filings also are available to the public at the Internet web site maintained by the SEC at www.sec.gov. Our reports filed with the SEC after January 1, 2003, also are available on our web site, www.poniard.com. The information contained in our web site does not constitute part of, nor is it incorporated by reference into, this report. We will provide paper copies of our SEC filings free of charge upon request.
Investing in our common stock or other securities involves a high degree of risk. You should carefully read the risks and uncertainties described below and all information contained in this report before you decide to purchase our common stock. If any of the possible adverse events described below actually occurs, we may be unable to conduct our business as currently planned and our financial and operating results could be harmed. In addition, the trading price of our common stock could decline due to the occurrence of any of these risks and you may lose all or part of your investment. Please see "Important Information Regarding Forward-Looking Statements" at the beginning of this report.
Risks Related to Our Business
We have a history of operating losses, we expect to continue to incur losses, and we may never become profitable.
We have not been profitable since our formation in 1984. As of December 31, 2009, we had an accumulated deficit of $408.2 million. Our net loss for the year ended December 31, 2009 was $45.7 million. We had net losses of $48.6 million for the year ended December 31, 2008 and $32.8 million for the year ended December 31, 2007. These losses resulted principally from costs incurred in our research and development programs and from our general and administrative activities. To date, we have dedicated substantially all of our resources to research and development activities and have not generated significant revenue from any product sales. We do not anticipate that our picoplatin product candidate will be commercially available before 2011, if ever. We expect to incur additional operating losses in the future. These losses may increase significantly as we seek to identify potential regulatory pathways forward for picoplatin and explore potential partnering and other relationships to support the continued development of picoplatin in multiple indications and two formulations.
Our ability to achieve long-term profitability is dependent upon achieving successful results in clinical trials and obtaining regulatory approvals for our picoplatin product candidate and successfully commercializing our product alone or with third parties.
20
Our Phase 3 trial of picoplatin in small cell lung cancer failed to meet the primary endpoint of overall survival and there can be no assurance that picoplatin will be approved in this indication.
On November 16, 2009, based on 321 patient deaths, we announced that our pivotal Phase 3 SPEAR trial did not meet its primary endpoint of overall survival in the intent-to-treat population. The analysis showed a hazard ratio of 0.82 with a p value of 0.089 (n=321). An imbalance in the use of post-study chemotherapy was observed in favor of patients who received best supportive care alone compared to patients who received picoplatin plus supportive care. Additional data analysis and our SPEAR study are ongoing. We have initiated a process with the FDA to identify a potential regulatory path forward to enable a registrational filing seeking approval of picoplatin as a second-line treatment for patients with refractory disease. We have no assurance regarding the outcome of our process with the FDA, including whether the FDA will agree to moving forward based on our current clinical data, require one or more additional trials, or discourage further efforts to obtain marketing approval of picoplatin in this indication.
We will need to raise substantial additional capital to pursue our regulatory activities and explore potential partnering and other relationships to support the development of picoplatin and to fund operations, and our future access to capital is uncertain and additional financing may have dilutive or adverse effects on our shareholders.
Taking into account the minimum unrestricted cash requirement under our current loan facility and our projected operating results, we believe that our current cash, cash equivalent and investment securities balances, including the net proceeds received from our March 15, 2010 sale of common stock to Commerce Court Small Cap Fund, Ltd., or Commerce Court, under our equity line of credit facility, will provide adequate resources to fund operations at least through the end of 2010. However, given the uncertainties of outcomes of our regulatory and partnering strategies to support the continued development of picoplatin, there is no assurance that we can achieve our projected operating results. Thereafter, unless we raise additional funds, we will be in default of the loan agreement with GE Business Financial Services and Silicon Valley Bank, as described in the risk factor below. Our consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States, assuming that we will continue as a going concern.
It is expensive to develop cancer therapy products and conduct clinical trials for these products. We have not generated revenue from the commercialization of our picoplatin product candidate, and we expect to continue to incur substantial net operating losses and negative cash flows from operations for the foreseeable future. We will require substantial additional funding to pursue our regulatory and partnering strategies to support the continued development of picoplatin and to fund our future operations.
Management is continuously exploring financing alternatives, including:
- •
- raising additional capital through the public or private sale of equity or debt securities or through the establishment of
credit or other funding facilities; and
- •
- entering into strategic collaborations, which may include joint ventures or partnerships for product development and commercialization, merger, sale of assets or other similar transactions.
We may not be able to obtain the required additional capital or secure corporate partners or enter into other strategic relationships on a timely basis, on terms that ultimately prove favorable to us, or at all. Conditions in the capital markets in general, and in the life science capital market specifically, may affect our potential financing sources and opportunities for strategic partnering. Uncertainty about current global conditions and the current financial uncertainties affecting capital and credit markets may make it particularly difficult for us to obtain capital market financing or credit on favorable terms, if at all, or to attract potential partners or enter into other strategic relationships.
21
If we raise additional funds by issuing common stock or securities convertible into or exercisable for common stock, our shareholders may experience substantial dilution, and new investors could have rights superior to current security holders. If we are unable to obtain sufficient additional cash when needed, we may be forced to reduce expenses through the delay, reduction or curtailment of some or all of our current picoplatin trials and regulatory activities or through other cost-savings measures, including the further reduction of our workforce, the sale of our company or licensing or sale of our assets.
The amount of additional financing we will require in the future will depend on a number of factors, including:
- •
- the costs of performing our obligations under our loan facility with GE Business Financial Services and Silicon Valley
Bank, including the cost of interest and other payment obligations and penalties and the cost of complying with the covenants and restrictions under the loan agreement;
- •
- actions taken by the FDA and other regulatory authorities, specifically including the results of our ongoing process with
the FDA to identify a potential regulatory path forward to enable a registrational filing seeking approval of picoplatin for the second-line treatment of small cell lung cancer patients
with refractory disease and future FDA guidance regarding potential registrational strategies for picoplatin in prostate, colorectal and ovarian cancers;
- •
- the scope, timing and success of our current and any future picoplatin clinical studies and regulatory and partnering
activities to support the continued development of picoplatin in multiple indications and in both intravenous and oral formulations;
- •
- our access to clinical supplies of picoplatin API and finished drug product in a timely and cost effective manner;
- •
- the timing and amount of any milestone or other payments we might receive from or be obligated to pay to potential
strategic partners;
- •
- our degree of success in commercializing picoplatin;
- •
- the emergence of competing technologies and products, and other adverse market developments;
- •
- the acquisition or in-licensing of other products or intellectual property;
- •
- the costs of any strategic partnerships or other collaborations established; and
- •
- the costs of preparing, filing, prosecuting, maintaining and enforcing patent claims and other intellectual property rights.
Restrictions imposed under the terms of our current loan facility may limit our ability to utilize capital for operations and may limit our ability to raise capital through the sale of assets or a merger not approved by the lenders.
On September 2, 2008, we entered into an amended and restated loan and security agreement, with GE Business Financial Services and Silicon Valley Bank in the principal amount of $27.6 million. The loan agreement, the terms of which are described in detail under the heading "Management's Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources," contains restrictions on our ability, without the prior consent of the lenders, to:
- •
- dispose of certain assets,
- •
- engage in certain mergers and acquisition transactions,
- •
- incur indebtedness,
22
- •
- create liens on assets,
- •
- make investments,
- •
- pay dividends, and
- •
- repurchase stock.
The loan agreement also contains covenants requiring us to maintain a minimum amount of unrestricted cash during the term of the loan equal to the lesser of (i) $17.9 million or (ii) the outstanding aggregate principal balance of the term loans plus $4.0 million. This minimum unrestricted cash requirement may limit our ability to utilize a portion of our cash in 2010 to pay for operating costs and to pursue our regulatory and partnering strategies focused on the continued development of picoplatin.
The loan agreement contains events of default that include:
- •
- nonpayment of principal, interest or fees,
- •
- breaches of covenants,
- •
- material adverse changes,
- •
- bankruptcy and insolvency events,
- •
- cross defaults to any other indebtedness,
- •
- material judgments,
- •
- inaccuracy of representations and warranties, and
- •
- events constituting a change of control.
We have no assurance that, especially in light of the distressed economic environment, that the lenders will be willing to waive or renegotiate the terms of the loan agreement to address or avoid financial or other defaults.
Presently, taking into account the minimum unrestricted cash requirement under the loan agreement and our projected operating results, we believe that our current cash, cash equivalent and investment securities balances, including the net proceeds received from our March 15, 2010 sale of common stock to Commerce Court under our equity line of credit facility, will provide adequate resources to fund operations at least through the end of 2010. However, given the uncertainties of outcomes of our regulatory and partnering strategies to support the continued development of picoplatin, there is no assurance that we can achieve our projected operating results. Thereafter, unless we raise additional funds, we will be in default of the loan agreement. The occurrence of an event of default would increase the applicable rate of interest by 5% and could result in the acceleration of our payment obligations under the loan agreement. If an event of default were to occur, we might not have sufficient funds to repay the loan or to fund our continuing operations. In such case, we would need to delay, scale back or curtail some or all of our current picoplatin trials and regulatory efforts; further reduce our workforce, license picoplatin for development and commercialization by third parties, or attempt to sell the company. We have no assurance that we can obtain financing or otherwise raise additional funds, if at all, on terms acceptable to us or to our lenders.
Our potential products must undergo rigorous clinical testing and regulatory approvals, which are costly and time consuming, and may subject us to unanticipated delays or prevent us from marketing any products.
The development, manufacture and marketing of picoplatin and any other potential product candidates are subject to regulation for safety, efficacy and quality by the FDA in the United States and by comparable regulatory authorities in foreign countries.
23
The process of obtaining FDA and other required regulatory approvals, including any foreign approvals, is expensive, often takes many years and can vary substantially depending on the type, complexity and novelty of the products involved. We will not be able to commercialize picoplatin unless and until we obtain regulatory approvals, and consequently any delay in obtaining, or our inability to obtain, regulatory approvals could materially adversely affect our business. We have had only limited experience in filing and pursuing applications necessary to gain regulatory approvals. This may impede our ability to obtain timely approvals from the FDA or foreign regulatory agencies.
If we violate regulatory requirements at any stage, whether before or after marketing approval is obtained, we may be fined, forced to remove a product from the market or experience other adverse consequences, including delay of the approval of our marketing applications, which would materially harm our business and financial results. Additionally, we may not be able to obtain the labeling claims necessary or desirable for product promotion and could be required to conduct post-marketing studies on the safety or effectiveness of our products. If we or other parties identify serious side effects after any of our products are on the market, or if manufacturing or regulatory problems occur, regulatory approval may be withdrawn and reformulation of our products, additional clinical trials, changes in labeling of our products, and/or additional marketing applications may be required.
The requirements governing the conduct of clinical trials and manufacturing and marketing of our proposed products outside the United States vary widely from country to country. Foreign approvals may take longer to obtain than FDA approvals and can involve additional testing. Foreign regulatory approval processes include all of the risks associated with the FDA approval processes. Also, approval of a product by the FDA does not ensure approval of the same product by the health authorities of other countries.
We may take longer to complete our clinical trials than we project, or we may be unable to complete them at all.
We have completed enrollment and initial statistical analysis of a pivotal Phase 3 SPEAR trial of picoplatin in the second-line treatment of patients with small cell lung cancer. This trial did not meet its primary endpoint of overall survival and we have initiated a process with the FDA to identify a potential regulatory path forward for picoplatin in this indication. We are also conducting two separate Phase 2 trials evaluating picoplatin as a first-line treatment of metastatic colorectal cancer and castration-resistant (hormone-refractory) prostate cancer. Additionally, we have completed a Phase 1 cardiac safety trial of picoplatin and a Phase 1 study evaluating an oral formulation of picoplatin in solid tumors. These programs are described in the sections of this report entitled "Business" and "Management's Discussion and Analysis of Financial Condition and Results of Operations—Research and Development."
The actual times for completion of our current picoplatin trials or the initiation of any future picoplatin development activities depend upon numerous factors, including:
- •
- our ability to obtain adequate additional funding or enter into strategic partnerships or other relationships;
- •
- approvals and other actions by the FDA and other regulatory agencies and the timing thereof;
- •
- our ability to open clinical sites;
- •
- our ability to recruit and enroll qualified patients into our studies;
- •
- our ability to obtain sufficient, reliable and affordable supplies of the picoplatin API and finished drug product;
- •
- the extent of competing trials at the clinical institutions where we conduct our trials;
24
- •
- reaching agreement on acceptable terms with prospective clinical research organizations, or CROs, and trial sites, the
terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites;
- •
- obtaining institutional review board, or IRB, approval to conduct a clinical trial at a prospective site;
- •
- our ability to assure that clinical trials are conducted in accordance with regulatory requirements or our clinical
protocols;
- •
- results of inspections of the clinical trial operations or trial sites by the FDA or other regulatory authorities,
including the risk of the imposition of a clinical hold;
- •
- unforeseen safety and efficacy issues;
- •
- the extent of scheduling conflicts with participating clinicians and clinical institutions; and
- •
- the identified endpoints of the studies, the extent of patient disease and patient performance status.
We may not complete our current picoplatin clinical studies as projected or achieve successful results.
We rely on academic institutions and CROs to conduct, supervise or monitor some or all aspects of clinical trials involving picoplatin. Further, to the extent that we now or in the future participate in partnering or other collaborative arrangements in connection with the development and commercialization of picoplatin or any other proposed products, we will have less control over the timing, planning and other aspects of our clinical trials. If we fail to initiate, advance or complete, or experience delays in or are forced to curtail our current or any future clinical trials, our stock price and our ability to conduct our business could be materially negatively affected.
If testing of a particular product does not yield positive results, we will be unable to commercialize that product.
Our clinical program is designed to test the safety and efficacy of our proposed products in humans. We may experience numerous unforeseen events during, or as a result of, the testing process that could delay or prevent commercialization of picoplatin, including the following:
- •
- the safety and efficacy results obtained in early human clinical trials may not be indicative of results obtained in later
clinical trials;
- •
- the results of preclinical studies may be inconclusive or they may not be indicative of results that will be obtained in
human clinical trials;
- •
- after reviewing test results, we or any potential collaborators may abandon projects that we previously believed were
promising;
- •
- we, our potential collaborators or regulators may suspend or terminate clinical trials if the participating subjects or
patients are being exposed to unacceptable health risks; and
- •
- the effects of our potential products may not be the desired effects or may include undesirable side effects or other characteristics that preclude regulatory approval or limit their commercial use if approved.
Clinical testing is very expensive, can take many years, and the outcome is uncertain. The data that we may collect from our picoplatin clinical trials may not be sufficient to support regulatory approval of our proposed picoplatin product. The clinical trials of picoplatin may not be initiated or completed as planned and the FDA or foreign regulatory agencies may not ultimately approve picoplatin for
25
commercial sale. Our failure to adequately demonstrate the safety and efficacy of picoplatin would delay or prevent regulatory approvals, which would prevent us from marketing the product.
Success in early clinical trials may not be indicative of results obtained in later trials.
Results of early preclinical and clinical trials are based on a limited number of patients and may, upon review, be revised or negated by authorities or by later stage clinical results. Historically, the results from preclinical testing and early clinical trials often have not been predictive of results obtained in later clinical trials. A number of new drugs and therapeutics have shown promising results in initial clinical trials, but subsequently failed to establish sufficient safety and effectiveness data to obtain necessary regulatory approvals. Data obtained from preclinical and clinical activities are subject to varying interpretations, which may delay, limit or prevent regulatory approval.
If we cannot successfully maintain and protect our current license for the development and commercial sale of picoplatin, we would be unable to move forward with our picoplatin studies and our current business and prospects will be materially negatively affected.
We have entered into an exclusive worldwide license, as amended, with Genzyme Corporation (successor to AnorMED, Inc.) for the development and commercial sale of picoplatin. Under that license, we are solely responsible for the development and commercialization of picoplatin. Genzyme retains the right, at our cost, to prosecute its patent applications and maintain all licensed patents. The parties executed the license agreement in April 2004, at which time we paid a one-time upfront payment of $1.0 million in common stock and $1.0 million in cash. The original agreement excluded Japan from the licensed territory and provided for $13.0 million in development and commercialization milestones, payable in cash or a combination of cash and common stock, and a royalty rate of up to 15% on product net sales after regulatory approval. The parties amended the license agreement on September 18, 2006, modifying several key financial terms and expanding the licensed territory to include Japan, thereby providing us worldwide rights. In consideration of the amendment, we paid Genzyme $5.0 million in cash on October 12, 2006 and an additional $5.0 million in cash on March 30, 2007. The amendment eliminated all development milestone payments to Genzyme. Genzyme remains entitled to receive up to $5.0 million in commercialization milestones upon the attainment of certain levels of annual net sales of picoplatin after regulatory approval. The amendment also reduced the royalty payable to Genzyme to a maximum of 9% of annual net product sales and eliminated sharing of sublicense revenues with Genzyme. We cannot currently predict the actual timing for completion of our current trials, the length of time to regulatory approval, if any, or the extent of annual sales, if any, of picoplatin and, therefore, cannot predict when or if the milestone and royalty payments under our license agreement with Genzyme may be triggered.
The license agreement may be terminated by either party for breach, or if the other party files a petition in bankruptcy or insolvency or for reorganization or is dissolved, liquidated or makes assignment for the benefit of creditors. We can terminate the license at any time upon prior written notice to Genzyme. If not earlier terminated, the license agreement will continue in effect, in each country in the territory in which the licensed product is sold or manufactured, until the earlier of (i) expiration of the last valid claim of a pending or issued patent covering the licensed product in that country or (ii) a specified number of years after first commercial sale of the licensed product in that country. If Genzyme were to breach its obligations under the license, or if the license expires or is terminated and we cannot renew, replace, extend or preserve our rights under the license agreement, we would be unable to move forward with our picoplatin clinical studies and commercialization efforts and our current business and prospects would be materially harmed.
26
The successful growth of our business may depend, in part, on our ability to find third party collaborators to assist or share in the costs of product development.
Our current strategy for the development and commercialization of picoplatin includes the formation of strategic partnerships and other collaborative arrangements with third parties. Potential third parties include pharmaceutical and biotechnology companies and other entities. Third party collaborators may assist us in:
- •
- funding or performing research, preclinical development, clinical trials and manufacturing;
- •
- seeking and obtaining regulatory approvals; and
- •
- successfully commercializing existing and future product candidates.
If we are unable to establish collaboration arrangements, we may be required to undertake product development and commercialization at our own expense. Such an undertaking may limit the number of indications in which we evaluate picoplatin, significantly increase our capital requirements and place additional strain on our internal resources. Our failure to enter into strategic partnerships or other collaborations could materially harm our business, financial condition, results of operations, cash flow or future prospects.
Strategic partnerships and other collaborative arrangements may give rise to disputes over commercial terms, contract interpretations and ownership of our intellectual property and may adversely affect the commercial success of our potential products.
Strategic partnerships and other collaborative relationships are generally complex and may give rise to disputes regarding the relative rights, obligations and revenues of the parties, including the ownership of intellectual property and associated rights and obligations. Such disputes can delay collaborative research, development or commercialization of potential products and can lead to lengthy, expensive litigation or arbitration. The terms of strategic partnerships and other collaborative arrangements may also include or preclude us from developing products or technologies developed pursuant to such collaborations. Additionally, the collaborators under these arrangements might breach the terms of their respective agreements or fail to prevent infringement of the licensed patents by third parties. Moreover, negotiated strategic partnerships or other collaboration arrangements often take considerably longer to conclude than parties initially anticipate, which could cause us to enter into less favorable agreement terms that delay or defer recovery of our development costs and reduce funding available to support key programs.
We may not be able to enter into strategic partnerships or other collaboration agreements on acceptable terms, which would harm our ability to develop and commercialize picoplatin and any other potential future products. Further, if we do enter into strategic partnerships or other collaboration arrangements, it is possible that our collaborative partners would choose not to develop and commercialize our picoplatin product. Other factors relating to strategic partnerships or other collaborations that may adversely affect commercial success of picoplatin or any proposed future products include:
- •
- any parallel development by a collaborative partner of competitive technologies or products;
- •
- arrangements with collaborative partners that limit or preclude us from developing products or technologies;
- •
- premature termination of a collaborative agreement; or
- •
- failure by a collaborative partner to devote sufficient resources to the development and commercialization of our proposed products.
27
A strategic partnership or other collaborative arrangement would not necessarily restrict our collaborative partners from competing with us or restrict their ability to market or sell competitive products. Our potential collaborative partners may pursue existing or other development-stage products or alternative technologies in preference to those being developed in collaboration with us. Our potential collaborative partners may also terminate their partnership or other collaborative relationships with us or otherwise decide not to proceed with development and commercialization of our products.
We are dependent on third party suppliers for the timely delivery of materials and services and may experience future interruptions in supply.
For picoplatin to be successful, we need sufficient, reliable and affordable supplies of the picoplatin API and finished drug product. Sources of these supplies may be limited, and third party suppliers may be unable to manufacture picoplatin API and finished drug product in amounts and at prices necessary for successful commercialization. Moreover, third party manufacturers must continuously adhere to current Good Manufacturing Practice, or cGMP, regulations enforced by the FDA through its facilities inspection program. If the facilities of these manufacturers cannot pass a Pre-Approval Inspection, the FDA will not approve the NDA for our proposed products. In complying with cGMP and foreign regulatory requirements, any of our third party manufacturers will be obligated to expend time, money and effort in production, record-keeping and quality control to assure that our products meet applicable specifications and other requirements. If any of our third party manufacturers or suppliers fails to comply with these requirements, we may be subject to regulatory action.
We have limited experience in drug formulation or manufacturing, and we lack the resources and capability to manufacture picoplatin on a clinical or commercial scale. As a result, we rely on third parties to manufacture picoplatin API and finished drug product for our clinical trials and for our proposed commercialization activities. The finished drug product has been demonstrated to be stable for up to 30 months from the date of manufacture.
We currently have separate agreements with one supplier each of picoplatin API and finished drug product for clinical and commercial use. Manufacturing services are provided on a purchase order, fixed-fee basis pursuant to separate clinical and commercial API and finished drug product supply agreements. Our API clinical supply agreement continues in effect until it is terminated by mutual agreement of the parties or by either party in accordance with its terms. Our finished drug product clinical supply agreement had an initial term ending December 31, 2009, and, in December 2009, we exercised our first renewal option, extending the term of the agreement until December 31, 2010. The agreement remains subject to renewal, at our option, for an additional one-year term. Our commercial API and finished drug supply agreements have initial terms ending in late 2013. Additional information about these agreements, including the termination rights of the parties, can be found in the section entitled "Picoplatin Source of Supply" in Section 1 above.
We have no assurance that our current suppliers will be able to continue to manufacture sufficient picoplatin API and finished drug product on a timely or cost-effective basis at all times in the future. The recent tightening of global credit may increase the risk of disruptions or delays of performance by our third party manufacturers and other contractors. We believe that there are other contract manufacturers with the capacity to manufacture picoplatin API and finished drug product. However, if we are required to seek out alternative manufacturers, we may incur significant additional costs and suffer delays in, or be prevented from, subject to FDA approval, initiating commercial sales of picoplatin.
We also rely on third party contractors to perform for us, or assist us with, the set-up, conduct, support and management of our clinical studies. Because these contractors provide specialized services, their activities and quality of performance may be outside our direct control. If these contractors do not perform their contractual duties or obligations, do not meet expected deadlines, or need to be
28
replaced, or if the quality or accuracy of the clinical data they obtain is compromised due to their failure to adhere to our clinical trial protocols or for any other reasons, we may need to enter into new arrangements with alternative third parties. If any of these circumstances were to occur, our clinical trials may be extended, delayed or terminated or may need to be repeated, we may not be able to obtain regulatory approval for picoplatin, and we may be subject to regulatory action.
We currently have no sales and marketing staff or distribution organization. If we are unable to develop sales, marketing and distribution capabilities on our own or through collaborations with corporate partners, we may not be successful in commercializing any future products.
We have limited experience in selling, marketing or distributing therapeutic drug products. To the extent we are successful in obtaining approval for the commercial sale of picoplatin, we will need to secure one or more corporate partners to conduct these activities. We may not be able to enter into partnering arrangements in a timely manner, on terms that ultimately prove favorable to us, or at all. To the extent that we enter into co-promotion or other licensing arrangements, our net product revenues are likely to be lower than if we directly marketed and sold our products, and any revenues we receive would depend upon the efforts of third parties, which efforts may not be successful. If we are not able to secure adequate partnering arrangements, we would have to hire additional employees or consultants with expertise in sales, marketing and distribution. Employees with relevant skills may not be available to us. Additionally, any increase in the number of employees would increase our expenses and could have a material adverse effect on our financial position. If we are not successful in commercializing picoplatin, either on our own or through collaborations with one or more parties, we will incur significant additional losses.
We face substantial competition in the development of cancer therapies and may not be able to compete successfully, and our potential products may be rendered obsolete by rapid technological change.
The competition for development of cancer therapies is substantial. There is intense competition from biotechnology and pharmaceutical companies, as well as academic research institutions, clinical reference laboratories and government agencies that are pursuing research and development activities similar to ours in the United States and abroad. Our initial focus for picoplatin has been small cell lung cancer, the most aggressive and deadly form of lung cancer. Although platinum therapies are the preferred treatment, there are no FDA-approved drugs for small cell cancer patients who do not respond to initial platinum-based therapy and only one FDA-approved treatment for small cell lung cancer patients who relapse after initially responding for first-line treatment. If approved, picoplatin will be competing with existing treatment regimens, as well as emerging therapies for small cell lung cancer and other platinum-based therapeutics. Large pharmaceutical/biotechnology companies, including Abbott, Amgen, AstraZeneca, Baxter Healthcare, Bristol-Myers Squibb Company, Celgene Corporation, Dainippon Sumitomo Pharma Co. Ltd., Eli Lilly and Company, Genentech, Inc., GlaxoSmithKline PLC, Merck & Co., Nippon Kayaku Co. Ltd., Novartis AG, Pfizer Inc., OSI Pharmaceuticals, Sanofi-Aventis Group, Shionogi & Co. Ltd. and SK Pharma, are marketing and/or developing therapeutics in late-stage clinical trials for the treatment of small cell lung cancer or platinum agents for the treatment of cancer. Multiple biotechnology companies are engaged in clinical trials for the treatment of small cell lung cancer and other platinum-based therapeutics, including Abraxis BioScience Inc., Access Pharmaceuticals Inc., Antigenics, Inc., Ascenta Therapeutics, Gemin X, GPC Biotech AG (whose merger with Agennix was announced in February 2009), ImmunoGen, Inc., Ipsen Group, Keryx Biopharmaceuticals Inc., Meabco A/S, MolMed S.p.A., Onyx Pharmaceuticals Inc., PharmaMar (Zeltia Group), Proacta, Inc., Regulon, Inc., Schering-Plough, Simcere Pharmaceuticals, Sunesis Pharmaceuticals Inc., Theradex, Transave Inc., Vertex Pharmaceuticals and Vion Pharmaceuticals Inc. As we seek to expand picoplatin into other oncology indications such as prostate, colon and ovarian cancers, we will be facing additional competition from major pharmaceutical companies, biotechnology companies, research institutions and government
29
agencies. We cannot assure you that we will be able to effectively compete with these or future third party product development programs.
Many of our existing or potential competitors have, or have access to, substantially greater financial, research and development, marketing and production resources than we do and are better equipped than we are to develop, manufacture and market competing products. Further, our competitors may have, or may develop and introduce, new products that would render our picoplatin product candidates less competitive, uneconomical or obsolete.
Even if any of our drug candidates receives regulatory approval, our drug candidates will still be subject to extensive post-marketing regulation.
If we or our collaborators receive regulatory approval for our drug candidates, we will also be subject to ongoing FDA obligations and continued regulatory review, such as cGMP regulations and continued adverse event reporting requirements. We may also be subject to additional FDA post-marketing obligations, all of which may result in significant expense and limit our ability to commercialize such drugs.
If our picoplatin drug candidate receives U.S. regulatory approval, the FDA may still impose significant restrictions on the indicated uses for which such drug may be marketed or impose ongoing requirements for potentially costly post-approval studies. In addition, regulatory agencies subject a drug, its manufacturer and the manufacturer's facilities to continual review and inspections. The subsequent discovery of previously unknown problems with a drug, including adverse events of unanticipated severity or frequency, or problems with the facility where the drug is manufactured, may result in restrictions on the marketing of that drug, up to and including withdrawal of the drug from the market. Failure to comply with applicable regulatory requirements may result in:
- •
- issuance of warning letters by the FDA;
- •
- imposition of fines and other civil penalties;
- •
- criminal prosecution;
- •
- injunction, suspension or revocation of marketing approvals;
- •
- suspension of any ongoing clinical trials;
- •
- suspension of manufacturing;
- •
- delays in commercialization;
- •
- refusal by the FDA to approve pending applications or supplements to approved applications filed by us or our
collaborators;
- •
- bans on the import or export of the drugs to or from the United States;
- •
- restrictions on operations, including costly new manufacturing requirements; and
- •
- product recalls or seizures.
The FDA's policies may change and additional government regulations may be enacted that could prevent or delay regulatory approval of picoplatin or further restrict or regulate post-approval activities. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are not able to maintain regulatory compliance, we might not be permitted to market picoplatin and our business could suffer.
30
If we are unable to protect our proprietary rights, we may not be able to compete effectively or operate profitably.
Our success is dependent in part on obtaining, maintaining and enforcing our patents and other proprietary rights and our ability to avoid infringing the proprietary rights of others. The United States Patent and Trademark Office, or the USPTO, may not issue patents from the patent applications owned by or licensed to us. If issued, the patents may not give us an advantage over competitors with similar technologies. The issuance of a patent is not conclusive as to its validity or enforceability and it is uncertain how much protection, if any, will be given to our patents if we attempt to enforce them and they are challenged in court or in other proceedings, such as oppositions, which may be brought in foreign jurisdictions to challenge the validity of a patent. A third party may challenge the validity or enforceability of a patent after its issuance by the USPTO. It is possible that a competitor may successfully challenge our patents or that a challenge will result in limiting their coverage. Moreover, the cost of litigation to uphold the validity of patents and to prevent infringement can be substantial. If the outcome of litigation is adverse to us, third parties may be able to use our patented invention without payment to us. Moreover, it is possible that competitors may infringe our patents or successfully avoid them through design innovation. We may need to file lawsuits to stop these activities. These lawsuits can be expensive and would consume time and other resources, even if we were successful in stopping the violation of our patent rights. In addition, there is a risk that a court would decide that our patents are not valid and that we do not have the right to stop the other party from using the inventions. There is also the risk that, even if the validity of our patents was upheld, a court would refuse to stop the other party on the ground that its activities do not infringe our patents. The protection afforded by issued patents is limited in duration. With respect to picoplatin, in the United States we expect to rely primarily on U.S. Patent No. 5,665,771 (expiring February 7, 2016), which is licensed to us by Genzyme, and additional licensed patents expiring in 2016 covering picoplatin in Europe and other countries. The FDA has designated picoplatin as an orphan drug for the treatment of small cell lung cancer under the provisions of the Orphan Drug Act, which entitles us to exclusive marketing rights for picoplatin in the United States for seven years following market approval. If approved, we may also be able to extend the term of a U.S. patent covering picoplatin under the Hatch-Waxman Act, which Act permits the extension of the term of a U.S. patent on a new drug for up to a maximum of five years. In addition, the European Commission has designated picoplatin as an orphan medicinal product for the treatment of small cell lung cancer in the European Union, which entitles us to exclusive marketing rights for picoplatin in the European Union for ten years following market approval in the European Union. Additional potential avenues exist which may supplement patent protection and exclusivity for picoplatin in Europe.
On May 8, 2009, patent owners, Genzyme Corporation and The Institute of Cancer Research, together with our company, filed with the USPTO an application to reissue U.S. Patent No. 5,665,771 ('771 patent) to the picoplatin compound. Like many composition of matter patents, the claims of the '771 patent include many compounds in addition to picoplatin. The reissue seeks to narrow certain claims by removing a number of compounds other than picoplatin from these claims. In the reissue proceeding, the USPTO will review the patent on its merits and an initial rejection is not unusual. We will have the opportunity to respond to any rejections and, if necessary, pursue appeals with the USPTO and the United States federal courts. During this process the '771 patent stays in force and can be asserted in an infringement action. We believe that filing the reissue to amend these claims and strengthen the intellectual property estate is a prudent course of action. Picoplatin is currently covered by additional issued process patents and other pending applications in the United States and abroad.
Under our license agreement with Genzyme, Genzyme retains the right to prosecute its patent applications and maintain all licensed patents, with us reimbursing such expenses. We have the right to sue any third party infringers of the picoplatin patents. If we do not file suit, Genzyme, in its sole discretion, has the right to sue the infringer at its expense. U.S. Patent No. 5,665,771 is co-owned by
31
Genzyme and a third party, which has exclusively licensed its rights to the patent to Genzyme (as successor to AnorMED, Inc.).
In addition to the intellectual property rights described above, we rely on unpatented technology, trade secrets and confidential information. Therefore, others may independently develop substantially equivalent information and techniques or otherwise gain access to or disclose our technology. We may not be able to effectively protect our rights in unpatented technology, trade secrets and confidential information. We require each of our employees, consultants and advisors to execute a confidentiality agreement at the commencement of an employment or consulting relationship with us. However, these agreements may not provide effective protection of our information or, in the event of unauthorized use or disclosure, may not provide adequate remedies.
The use of our technologies could potentially conflict with the rights of others.
Our competitors or others may have or may acquire patent rights that they could enforce against us. In such case, we may be required to alter our products, pay licensing fees or cease activities. If our products conflict with patent rights of others, third parties could bring legal actions against us claiming damages and seeking to enjoin manufacturing and marketing of the affected products. If these legal actions are successful, in addition to any potential liability for damages, we could be required to obtain a license in order to continue to manufacture or market the affected products. We may not prevail in any legal action and a required license under the patent may not be available on acceptable terms.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights.
The cost to us of any litigation or other proceedings relating to intellectual property rights, even if resolved in our favor, could be substantial. Some of our competitors may be better able to sustain the costs of complex patent litigation because they have substantially greater resources. If there is litigation against us, we may not be able to continue our operations. If third parties file patent applications, or are issued patents claiming technology also claimed by us in pending applications, we may be required to participate in interference proceedings in the USPTO to determine priority of invention. We may be required to participate in interference proceedings involving our issued patents and pending applications. We may be required to cease using the technology or license rights from prevailing third parties as a result of an unfavorable outcome in an interference proceeding. A prevailing party in that case may not offer us a license on commercially acceptable terms.
Product liability claims in excess of the amount of our insurance would adversely affect our financial condition.
The testing, manufacture, marketing and sale of picoplatin and any other proposed cancer therapy products, including past clinical and manufacturing activities in connection with our terminated skeletal targeted radiotherapy, or STR, development program, may subject us to product liability claims. We are insured against such risks up to a $10.0 million annual aggregate limit in connection with clinical trials of our products under development and intend to obtain product liability coverage in the future. However, insurance coverage may not be available to us at an acceptable cost. We may not be able to obtain insurance coverage that will be adequate to satisfy any liability that may arise. Regardless of merit or eventual outcome, product liability claims may result in decreased demand for a product, injury to our reputation, withdrawal of clinical trial volunteers and loss of revenues. As a result, regardless of whether we are insured, a product liability claim or product recall may result in losses that could be material.
32
Our past use of radioactive and other hazardous materials exposes us to the risk of material environmental liabilities, and we may incur significant additional costs to comply with environmental laws in the future.
Our past research and development and manufacturing processes, as well as the manufacturing processes that may have been used by our collaborators, involved the controlled use of hazardous and radioactive materials. As a result, we are subject to foreign, federal, state and local laws, rules, regulations and policies governing the use, generation, manufacture, storage, air emission, effluent discharge, handling and disposal of certain materials and wastes in connection with our use of these materials. Although we believe that our safety procedures for handling and disposing of such materials complied with the standards prescribed by such laws and regulations, we may be required to incur significant costs to comply with environmental and health and safety regulations in the future. We terminated our STR manufacturing operations in Denton, Texas in May 2005. We recorded costs associated with the closure of the Denton facility of $0.5 million in 2005 and $0.3 million in 2006. We completed the sale of the Denton facility on October 1, 2007. Our current insurance does not cover liability for the clean-up of hazardous waste materials or other environmental risks.
Changes in health care reimbursement could adversely affect our ability to effectively price our products or obtain adequate reimbursement for sales of our products.
Potential sales of our products may be affected by the availability of reimbursement from governments or other third parties, such as insurance companies. It is difficult to predict the reimbursement status of newly approved, novel medical products. A government or third party payer decision not to approve pricing for, or provide adequate coverage and reimbursements of, our drugs, if any, could limit market acceptance of such drugs. In addition, third party payers are increasingly challenging the prices charged for medical products and services. If we succeed in bringing picoplatin to market, we cannot be certain that it will be considered cost-effective and that reimbursement to the consumer will be available or will be sufficient to allow us to competitively or profitably sell this product.
The levels of revenues and profitability of biotechnology companies may be affected by the continuing efforts of government and third party payers to contain or reduce the costs of health care through various means. For example, in certain foreign markets, pricing or profitability of prescription pharmaceuticals is subject to governmental control. In the United States, there have been, and we expect that there will continue to be, a number of federal and state proposals to implement similar governmental controls. It is uncertain what legislative proposals will be adopted or what actions federal, state or private payers for health care goods and services may take in response to any health care reform proposals or legislation. Even in the absence of statutory change, market forces are changing the health care sector. We cannot predict the effect health care reforms may have on the development, testing, commercialization and marketability of our picoplatin product. To the extent that such proposals or reforms have a material adverse effect on the business and potential revenues of other companies that are potential strategic partners or collaborators, our ability to commercialize opportunities may be adversely affected and the amount a potential collaborator may be willing to pay to license picoplatin in the future may be reduced.
The loss of key employees could adversely affect our operations.
On February 5, 2010, we implemented a restructuring plan to conserve capital resources, which reduced our workforce by approximately 57%, to 22 employees. We implemented an earlier restructuring plan, effective March 31, 2009, which resulted in the discontinuation of our preclinical research operations and reduced our workforce by approximately eight employees.
As part of that restructuring, Gerald McMahon, PhD stepped down as our chief executive officer and Robert De Jager, M.D., stepped down as our chief medical officer. Dr. McMahon will continue to
33
serve as non-executive chairman of our board of directors. Dr. De Jager will continue as a consultant to our company until December 31, 2010, subject to earlier termination by either party. We did not experience any material disruptions as a consequence of the management changes or the reductions in force. Ronald A. Martell, our former president and chief operating officer, was appointed as our new chief executive officer, and Michael S. Perry, DVM, PhD, was appointed as our new president and chief medical officer.
Caroline M. Loewy resigned as our chief financial officer effective November 28, 2008. We did not experience any material disruptions as a consequence of Ms. Loewy's resignation. Gregory L. Weaver was appointed as our chief financial officer effective February 18, 2009.
On November 21, 2008, we terminated David A. Karlin, M.D., our senior vice president of clinical development and regulatory affairs. The termination was based on our determination that we no longer required the services of Dr. Karlin, and we did not experience any material disruptions as a consequence of Dr. Karlin's termination.
As of March 9, 2010, we had 20 full-time employees and two part-time employees. Of these full-time employees, five hold PhD degrees, one holds an M.D. degree, one holds a D.V.M. degree, one holds a J.D. degree and two hold an M.B.A. degree. Of the total full-time employees, ten employees are engaged in regulatory and clinical activities and ten are in general administration. We consider our relations with employees to be good. None of our employees is covered by a collective bargaining agreement. We believe that our current workforce is sufficient to support our regulatory and partnering strategies focused on the continued development of picoplatin.
Our success depends, to a significant extent, on our principal management and clinical and regulatory personnel continuing to contribute to and participate in our regulatory and partnering efforts to enable the continued development of picoplatin. We have limited or no redundancy of personnel in key development areas, including finance, legal, clinical operations, regulatory affairs, strategic planning, quality control and assurance. The loss of the services of one or more of our employees could adversely affect our regulatory and partnering efforts. We do not maintain key-person life insurance on any of our officers, employees or consultants.
Competition for qualified employees among companies in the biotechnology and biopharmaceutical industry is intense. Our future success depends upon our ability to attract, retain and motivate highly skilled employees and consultants. In order to successfully develop and commercialize picoplatin, we may in the future be required to substantially expand our workforce.
We have change of control agreements and severance agreements with all of our officers and consulting agreements with several of our scientific advisors. Our agreements with our officers provide for "at will" employment, which means that each officer may terminate his or her service with us at any time. In addition, our scientific advisors may terminate their services to us at any time.
Our operations might be interrupted by the occurrence of a natural disaster or other catastrophic event.
Our principal executive offices are in South San Francisco, California, and we maintain clinical and regulatory activities in Seattle, Washington. We depend on our facilities and on our collaborators, contractors and vendors for the continued operation of our business. Natural disasters or other catastrophic events, including terrorist attacks, power interruptions, wildfires and other fires, actions of animal rights activists, earthquakes and wars could disrupt our operations or those of our collaborators, contractors and vendors. Even though we believe we carry commercially reasonable business interruption and liability insurance, and our contractors may carry liability insurance that protect us in certain events, we might suffer losses as a result of business interruptions that exceed the coverage available under our and our contractors' insurance policies or for which we or our contractors do not have coverage. Any natural disaster or catastrophic event could have a significant negative impact on
34
our operations and financial results. Moreover, any such event could delay our regulatory and partnering goals for the continued development of picoplatin.
Risks Relating to Our Securities
Our common stock may be delisted from The Nasdaq Global Market if we are unable to maintain compliance with Nasdaq Global Market continued listing requirements.
Our common stock listing has been listed on the The Nasdaq Global Market since October 1, 2007. Prior to that time, our common stock was listed on the Nasdaq Capital Market. In order to continue to be included in the Nasdaq Global Market, we must meet the Nasdaq Global Market continued listing standards, including maintaining a closing bid price of $1.00 per share (the Minimum Bid Price Requirement). Our common stock has in the past, and may in the future, fall below the Minimum Bid Price Requirement, or we may in the future fail to meet other requirements for continued listing on the Nasdaq Global Market. If we are unable to cure any events of noncompliance in a timely or effective manner, our common stock could be delisted from The Nasdaq Global Market.
If our common stock were threatened with delisting from The Nasdaq Global Market, we may, depending on the circumstances, seek to extend the period for regaining compliance with Nasdaq listing requirements by moving our common stock to the Nasdaq Capital Market. Failing that, we may seek quotation on a regional stock exchange, if available. Any such change in listing could reduce the market liquidity for our common stock. If our common stock is not eligible for quotation on another market or exchange, trading of our common stock could be conducted in the over-the-counter market on an electronic bulletin board established for unlisted securities such as the Pink Sheets or the OTC Bulletin Board. As a result, an investor would find it more difficult to dispose of, or obtain accurate quotations for the price of, our common stock.
If our common stock were to be delisted from The Nasdaq Stock Market, and our trading price remained below $5.00 per share, trading in our common stock might also become subject to the requirements of certain rules promulgated under the Securities Exchange Act of 1934, as amended, or the Exchange Act, which require additional disclosure by broker-dealers in connection with any trade involving a stock defined as a "penny stock" (generally, any equity security not listed on a national securities exchange or quoted on Nasdaq that has a market price of less than $5.00 per share, subject to certain exceptions). Many brokerage firms are reluctant to recommend low-priced stocks to their clients. Moreover, various regulations and policies restrict the ability of shareholders to borrow against or "margin" low-priced stocks, and declines in the stock price below certain levels may trigger unexpected margin calls. Additionally, because brokers' commissions on low-priced stocks generally represent a higher percentage of the stock price than commissions on higher priced stocks, the current price of the common stock can result in an individual shareholder paying transaction costs that represent a higher percentage of total share value than would be the case if our share price were higher. This factor may also limit the willingness of institutions to purchase our common stock. Finally, the additional burdens imposed upon broker-dealers by these requirements could discourage broker-dealers from facilitating trades in our common stock, which could severely limit the market liquidity of the stock and the ability of investors to trade our common stock.
Our stock price is volatile and, as a result, you could lose some or all of your investment.
There has been a history of significant volatility in the market prices of securities of biotechnology companies, including our common stock. In 2009, the reported high and low closing sale prices of our common stock were $8.63 and $1.64. During 2008, the reported high and low closing sale prices of our common stock were $6.18 and $1.40. The reported high and low closing sale prices during 2007 were $8.89 and $4.09, respectively. Our stock price has been and may continue to be affected by this type of
35
market volatility, as well as our own performance. Our business and the relative price of our common stock may be influenced by a large variety of factors, including:
- •
- the progress and results of our clinical trials;
- •
- the results of our discussions with the FDA and other regulatory agencies and our success in identifying potential
regulatory paths for the continued development of picoplatin;
- •
- our available cash or other sources of funding;
- •
- future sales of significant amounts of our common stock by us or our shareholders;
- •
- the expense and time associated with, and the extent of our ultimate success in, securing regulatory approvals;
- •
- developments concerning potential agreements with strategic partners and collaborators;
- •
- announcements by us or our competitors concerning acquisitions, strategic alliances, technological innovations, new
commercial products or changes in product development strategies; and
- •
- the availability of critical materials used in developing our proposed picoplatin product.
In addition, potential public concern about the safety and efficacy of picoplatin, comments by securities analysts, our ability to maintain the listing of our common stock on the Nasdaq Stock Market, and conditions in the capital markets in general and in the life science capital market specifically, may have a significant effect on the market price of our common stock. The realization of any of the risks described in this report, as well as other factors, could have a material adverse impact on the market price of our common stock and may result in a loss of some or all of your investment in our securities.
In the past, securities class action litigation often has been brought against companies following periods of volatility in their stock prices. We may in the future be the target of similar litigation. Securities litigation could result in substantial costs and divert our management's time and resources, which could cause our business to suffer.
Certain investors beneficially own significant blocks of our common stock; these large shareholders may take actions that are contrary to your interests, including selling their stock.
A small number of our shareholders hold a significant amount of our outstanding stock. As of December 31, 2009, entities affiliated with Bay City Management beneficially owned an aggregate of approximately 13.0% of our outstanding common stock. Two of our directors, Fred B. Craves and Carl S. Goldfischer, are managing directors of Bay City Capital LLC, an affiliate of Bay City Management, and possess capital and carried interests in the Bay City Management entities holding our shares. Entities affiliated with MPM beneficially owned an aggregate of approximately 18.6% of our outstanding common stock as of December 31, 2009. Nicholas J. Simon III, a director of our company, is a general partner of certain of the MPM entities that hold those shares. As a result, these shareholders will collectively be able to significantly influence all matters requiring approval of our shareholders, including the election of directors and approval of significant corporate transactions. These shareholders may support competing transactions and have interests that are different from yours.
36
Sales of a large number of shares of our stock by one or more of these large shareholders or other shareholders within a short period of time, or the expectation that such sales may occur, could significantly reduce the market price of our common stock. For example, certain of our officers and directors and their affiliates have established, or may in the future establish, selling plans under Rule 10b5-1 of the Exchange Act for the purpose of effecting specified sales of our common stock over a specified period of time. If any of these events cause a large number of our shares to be sold in the public market, the sales could reduce the trading price of our common stock and impede our ability to raise future capital.
Any future equity or debt issuances by us may have dilutive or adverse effects on our existing shareholders.
We have financed our operations, and we expect to continue to finance our operations, primarily by issuing and selling our common stock or securities convertible into or exercisable for shares of our common stock. In light of our need for additional financing, we may issue additional shares of common stock or convertible securities that could dilute your ownership in our company and may include terms that give new investors rights that are superior to yours. Moreover, any issuances by us of equity securities may be at or below the prevailing market price of our common stock and in any event may have a dilutive impact on your ownership interest, which could cause the market price of our common stock to decline. For example, in February 2010, we entered into an equity line of credit facility with Commerce Court, pursuant to which we may sell shares of our common stock to Commerce Court at a discount to the prevailing market price. Specifically, the per share purchase price for common shares purchased under the facility will equal the daily volume weighted average price of our common stock during the draw down period over which such shares are purchased, less a discount of 3.125% to 5.0%, based on the trading price of our common stock, excluding an additional placement agent fee of 1.0% payable by us on the gross proceeds.
We may also raise additional funds through the incurrence of debt, and the holders of any debt we may issue would have rights superior to your rights in the event we are not successful and are forced to seek the protection of bankruptcy laws.
We have never paid cash dividends on our common stock, and we do not anticipate paying any cash dividends in the foreseeable future; however, holders of our outstanding Series 1 preferred stock do receive payments of dividends and have certain other rights and preferences superior to those our common shareholders.
We have paid no cash dividends on our common stock to date, and we currently intend to retain our future earnings, if any, to fund our business operations. We do not anticipate paying any cash dividends on our common stock in the foreseeable future. Furthermore, our loan facility with GE Business Financial Services and Silicon Valley Bank restricts our ability to pay dividends on our common stock. As a result, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future.
We had 78,768 shares of $2.4375 convertible exchangeable preferred stock, or Series 1 preferred shares, outstanding as of March 9, 2010. These shares were originally issued in 1989. Pursuant to the terms of the designation of the Series 1 preferred shares, holders of Series 1 preferred shares are entitled to receive annual dividends of $2.4375 per Series 1 preferred share outstanding. Dividends on the Series 1 preferred shares are cumulative, which means that if they are not paid, the amount of the dividends accrue and, unless full cumulative dividends on the Series 1 preferred shares have been paid, no dividends may be paid on any stock ranking junior to the Series 1 preferred shares, including the common stock. In addition, holders of our Series 1 preferred shares have certain redemption rights, liquidation preferences and voting rights that are greater than or superior to the rights of our common shareholders. These rights may decrease the amount of earnings and assets available for distribution to our common shareholders.
37
Certain provisions in our articles of incorporation and Washington state law could discourage a change of control and may adversely affect the rights and interests of our common shareholders.
Our articles of incorporation authorize our board of directors to issue up to 200,000,000 shares of common stock and up to 2,998,425 shares of preferred stock. Up to 1,120,000 preferred shares have been designated Series 1 preferred shares, 78,768 of which currently are outstanding. The remaining authorized but unissued preferred shares are presently undesignated. We currently have no plans to issue any additional shares of preferred stock.
Under our articles of incorporation, our board of directors is authorized generally, without shareholder approval, to issue shares of preferred stock in one or more series and, in connection with the creation of each such series, to fix the number of shares of such series and designate the powers, preferences and rights of such series, including dividend rights, redemption rights, liquidation preferences, sinking fund provisions, conversion rights and voting rights, any or all of which may be greater than or superior to the rights of the common stock.
Washington law imposes restrictions on certain transactions between a corporation and significant shareholders. Chapter 23B.19 of the Washington Business Corporation Act prohibits a target corporation, with some exceptions, from engaging in particular significant business transactions with an acquiring person, which is defined as a person or group of persons that beneficially owns 10% or more of the voting securities of the target corporation, for a period of five years after the date the acquiring person first became a 10% beneficial owner of voting securities of the target corporation, unless (i) the business transaction or the acquisition of shares is approved by a majority of the members of the target corporation's board of directors prior to the time the acquiring person first became a 10% beneficial owner of the target corporation's voting securities or (ii) at or after the acquiring first person became a 10% beneficial owner of the target corporation, the business transaction is approved by a majority of the members of the target corporation's board of directors and at least 2/3 of the outstanding voting shares of the target corporation (excluding shares held by the acquiring person). Prohibited business transactions include, among other things:
- •
- a merger or consolidation with, disposition of assets to, or issuance or redemption of stock to or from the acquiring
person;
- •
- termination of 5% or more of the employees of the target corporation; or
- •
- receipt by the acquiring person of any disproportionate benefit as a shareholder.
After the five-year period, a significant business transaction may occur if it complies with "fair price" provisions specified in the statute. A corporation may not opt out of this statute. This provision may have an anti-takeover effect with respect to transactions that our board does not approve in advance.
The foregoing provisions of Washington law, together with the provisions of our articles of incorporation authorizing the board, without further vote or action by the shareholders, to issue shares of preferred stock with powers, preferences and privileges fixed by the board, may have the effect of delaying, deterring or preventing a change of control of our company, even if this change would be beneficial to our shareholders. These provisions also may discourage bids for our common stock at a premium over market price and may adversely affect the market price of, and the voting and other rights of the holders of, our common stock. In addition, these provisions could make it more difficult to replace or remove our current directors and management in the event our shareholders believe this would be in the best interests of the corporation and our shareholders.
38
Item 1B. UNRESOLVED STAFF COMMENTS
Not Applicable.
Our corporate headquarters currently is located at 7000 Shoreline Court in South San Francisco, CA, where we lease 17,045 square feet of office space (the "Premises") under a lease agreement that expires on July 10, 2011. On February 12, 2010, we executed a sublease agreement, whereby we leased to Veracyte, Inc. approximately 11,000 square feet of the Premises, effective March 1, 2010. On September 1, 2010, the sublease will expand to encompass the entire 17,045 square feet of the Premises. The sublease will expire on July 10, 2011, at which time Veracyte, Inc. will lease the Premises directly from the landlord. We currently are evaluating our facility space requirements and plan to explore alternative sites in the San Francisco area for our executive offices.
We also currently occupy approximately 21,000 square feet of office space located at 300 Elliott Avenue West in Seattle, WA, under an amended lease that expires December 31, 2010. The lease may be renewed for one five-year term, effective upon notice by us of our intent to renew six months prior to the expiration of the current term.
We believe that the South San Francisco and Seattle facilities are in good condition and are adequate for their present uses.
Not Applicable.
Item 4. SUBMISSION OF MATTERS TO A VOTE OF SECURITY HOLDERS
Not Applicable.
39
Item 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common stock has been listed on The Nasdaq Global Market since October 1, 2007. The following table sets forth, for the periods indicated, the high and low sales prices for our common stock as reported on The Nasdaq Global Market.
| |
High | Low | |||||
|---|---|---|---|---|---|---|---|
2009 |
|||||||
First Quarter |
$ | 3.73 | $ | 1.50 | |||
Second Quarter |
6.19 | 1.97 | |||||
Third Quarter |
9.14 | 6.50 | |||||
Fourth Quarter |
8.55 | 1.60 | |||||
2008 |
|||||||
First Quarter |
$ | 6.39 | $ | 3.27 | |||
Second Quarter |
5.29 | 3.31 | |||||
Third Quarter |
4.91 | 3.50 | |||||
Fourth Quarter |
4.38 | 1.05 | |||||
The closing sale price of our common stock on The Nasdaq Global Market was $1.56 on March 9, 2010.
There were approximately 783 shareholders of record on March 9, 2010. This figure does not include the number of shareholders whose shares are held on record by a broker or clearing agency, but includes such a brokerage house or clearing agency as one holder of record.
We have not declared or paid any cash dividends with respect to our common stock to date, and we currently intend to retain our earnings, if any, to fund our business operations. We do not anticipate paying any cash dividends on our common stock in the foreseeable future. Furthermore, our secured loan facility with GE Business Financial Services and Silicon Valley Bank restricts our ability to pay dividends on our common stock. See "Management's Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources" for a more detailed discussion of the terms of such loan facility.
See Part III, Item 12, for information regarding securities authorized for issuance under our incentive compensation plans.
40
Stock Price Performance Graph
The graph below compares the cumulative total shareholder return on our common stock with the cumulative shareholder return of the Nasdaq Stock Market Index (US) and the Nasdaq Pharmaceuticals Stocks Index. Stock price performance shown below is historical and not necessarily indicative of future price performance.
Comparison of Five-Year Cumulative Total Return Among Poniard Pharmaceuticals, Inc.,
Nasdaq Stock Market (US) and Nasdaq Pharmaceuticals Stocks Index(1)
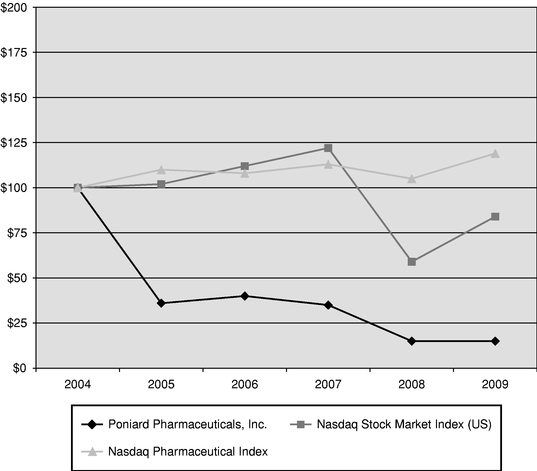
| |
2004 | 2005 | 2006 | 2007 | 2008 | 2009 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Poniard Pharmaceuticals, Inc. |
$ | 100 | $ | 36 | $ | 40 | $ | 35 | $ | 15 | $ | 15 | |||||||
Nasdaq Stock Market Index (US) |
100 | 102 | 112 | 122 | 59 | 84 | |||||||||||||
Nasdaq Pharmaceutical Index |
100 | 110 | 108 | 113 | 105 | 119 | |||||||||||||
- (1)
- Assumes $100 invested on December 31, 2004, in our common stock, the Nasdaq Stock Market Index and the Nasdaq Pharmaceutical Stocks Index, an index of approximately 214 companies with common stock quoted on the Nasdaq National Market. The Primary Standard Industrial Classification Code Number (SIC) of these companies is #2835—Pharmaceutical Companies. Total return performance for the Nasdaq Stock Market Index and the Nasdaq Pharmaceutical Stocks Index is weighted based on the market capitalization of the firms included in each index and assumes that dividends are reinvested. The Nasdaq Stock Market Index and the Nasdaq Pharmaceutical Stocks Index are produced and published by the Center for Research in Securities Pricing at the University of Chicago.
41
Item 6. SELECTED FINANCIAL DATA
The following table shows selected financial data. It is important to read this selected financial data along with the "Financial Statements and Supplementary Data," as well as the "Management's Discussion and Analysis of Financial Condition and Results of Operations" in Item 7 below.
| |
Years Ended December 31, | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | 2006 | 2005 | |||||||||||
| |
(in thousands, except per share data) |
|||||||||||||||
Consolidated Statement of Operations Data: |
||||||||||||||||
Revenues |
$ | — | $ | — | $ | — | $ | — | $ | 15 | ||||||
Operating expenses |
42,978 | 49,157 | 35,353 | 21,234 | 21,075 | |||||||||||
Loss from operations |
(42,978 | ) | (49,157 | ) | (35,353 | ) | (21,234 | ) | (21,060 | ) | ||||||
Net loss |
(45,715 | ) | (48,565 | ) | (32,782 | ) | (23,294 | ) | (20,997 | ) | ||||||
Net loss applicable to common shareholders |
(46,215 | ) | (49,065 | ) | (33,282 | ) | (23,794 | ) | (21,497 | ) | ||||||
Net loss per common share-basic and diluted |
$ | (1.31 | ) | $ | (1.41 | ) | $ | (1.08 | ) | $ | (1.37 | ) | $ | (3.83 | ) | |
Weighted average common shares outstanding—basic and diluted |
35,272 | 34,686 | 30,762 | 17,376 | 5,611 | |||||||||||
Consolidated Balance Sheet Data: |
||||||||||||||||
Cash, cash equivalents and restricted cash |
$ | 16,219 | $ | 44,425 | $ | 29,616 | $ | 44,284 | $ | 4,523 | ||||||
Investment securities |
27,451 | 28,611 | 63,286 | 9,562 | — | |||||||||||
Working capital (deficit) |
27,369 | 54,873 | 84,383 | 42,299 | (1,880 | ) | ||||||||||
Total assets |
52,442 | 84,232 | 105,140 | 69,067 | 10,114 | |||||||||||
Note payable and capital lease obligations, net of current portion and debt discounts |
11,671 | 17,445 | 6,561 | 9,975 | — | |||||||||||
Shareholders' equity |
$ | 23,644 | $ | 47,647 | $ | 89,105 | $ | 46,891 | $ | 3,173 | ||||||
Item 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Introduction
The following discussion of results of operations and liquidity and capital resources contains forward-looking statements that involve risks and uncertainties. As described under the heading "Important Information Regarding Forward-Looking Statements" at the beginning of this report, our actual results may differ materially from the results discussed in these forward-looking statements. Factors that might cause or contribute to such differences include those discussed below and in the section above entitled "Risk Factors."
Unless otherwise indicated, all common stock related amounts have been adjusted to reflect our one-for-six reverse stock split effective September 22, 2006.
Critical Accounting Policies
Impairment of Long-Lived and Intangible Assets: As of December 31, 2009, we had net facilities and equipment of approximately $0.2 million and a net intangible asset of approximately $7.6 million, which represents capitalized payments for our picoplatin license. In accounting for these long-lived and intangible assets, we estimate the expected useful lives of the assets, the expected residual values of the assets, and the potential for impairment based on events or circumstances, such as changes in our business strategy and plans, a significant decrease in market value, a significant change in asset condition or a significant adverse change in regulatory climate. Specifically, the value of the picoplatin intangible asset could be impaired as a result of negative results of clinical trials or as a result of adverse decisions or rulings of regulatory bodies, such as the FDA. Application of the test for
42
impairment requires significant judgment, taking into account potentially unfavorable factors, such as those mentioned above, that could adversely affect the carrying value of the asset.
In March 2009, we recognized an asset impairment loss of $0.6 million on certain facilities and equipment resulting from the discontinuation of the Company's preclinical research operations. The loss on these assets was determined based on estimates of potential sales values of used equipment and other selling costs. Additionally, at December 31, 2009, we recognized an impairment charge of approximately $1.5 million for our dedicated manufacturing equipment asset. The impairment charge was determined based on the delay in our plans for the commercialization of picoplatin, which we do not anticipate will occur before 2011.
Long-Term Debt: On September 2, 2008, we entered into an Amended and Restated Loan and Security Agreement (loan agreement), with GE Business Financial Services (formerly known as Merrill Lynch Capital) and Silicon Valley Bank that provided for a senior secured term loan facility of $27.6 million. The loan agreement contains covenants requiring the Company to maintain unrestricted cash in an amount equal to the lesser of (i) $17.9 million or (ii) the outstanding aggregate principal balance of the term loans plus $4.0 million. We use judgment to determine our compliance with the minimum cash covenant and other covenants under the loan agreement. We classify the portion of the loan that is due for payment in 2010 as a current liability and the portion due thereafter as a long-term payable.
Stock Compensation: We account for share-based compensation arrangements in accordance with the Financial Accounting Standards Board, or FASB, accounting standards for equity instruments exchanged for services, which require the measurement and recognition of compensation expense for all share-based payment awards based on estimated fair values.
- •
- Stock Options We use the Black-Scholes option valuation
model to estimate the fair value of our stock options at the date of grant. The Black-Scholes option valuation model was developed for use in estimating the fair value of traded options which have no
vesting restrictions and are fully transferable. Our employee stock options, however, have characteristics significantly different from those of traded options. For example, employee stock options are
generally subject to vesting restrictions and are generally not transferable. In addition, option valuation models require the input of highly subjective assumptions, including the expected stock
price volatility, the expected life of an option and the number of awards ultimately expected to vest. Changes in subjective input assumptions can materially affect the fair value estimates of an
option. Furthermore, the estimated fair value of an option does not necessarily represent the value that will ultimately be realized by an employee. We use historical data, and other related
information, as appropriate, to estimate the expected price volatility, the expected option life and the expected forfeiture rate. The risk-free rate is based on the U.S. Treasury yield
curve in effect at the time of a grant. If actual results are not consistent with our assumptions and judgments used in estimating the key assumptions, we may be required to increase or decrease
compensation expense, which could be material to our results of operations.
- •
- Restricted Stock Units ("RSUs") We award RSUs, which are exchangeable for our common shares upon vesting. We use the closing market price of our common stock on the award date to estimate the fair value of awarded RSUs. For RSUs that contain performance-based vesting or vesting based on the achievement of defined milestones, we use judgment to determine the probability of achievement of a milestone to determine whether compensation should be recognized. For RSUs with milestones probable of achievement, we estimate the probable date of achievement and recognize compensation over the resulting implied service period.
43
Results of Operations
Years Ended December 31, 2009, 2008 and 2007
Research and Development
Research and development expenses decreased 24% to $25.7 million in 2009 and increased 51% to $33.7 million in 2008. Our research and development expenses are summarized as follows:
| |
($ in thousands) | Annual Percentage Change |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | 2009-2008 | 2008-2007 | ||||||||||||
Research |
$ | 764 | $ | 3,551 | $ | 3,401 | -78 | % | 4 | % | |||||||
Contract manufacturing |
5,638 | 4,248 | 4,149 | 33 | % | 2 | % | ||||||||||
Clinical |
16,999 | 24,352 | 13,511 | -30 | % | 80 | % | ||||||||||
Share-based compensation |
2,338 | 1,581 | 1,320 | 48 | % | 20 | % | ||||||||||
Total |
$ | 25,739 | $ | 33,732 | $ | 22,381 | -24 | % | 51 | % | |||||||
Research expenses include, among other things, personnel, occupancy and external laboratory expenses associated with the discovery and identification of new therapeutic agents for the treatment of cancer. Research expenses also include research activities associated with our product candidate, picoplatin, including formulation and in vitro and in vivo studies. Research expenses decreased 78% to $0.8 million in 2009 due to the discontinuation of our research operations effective March 31, 2009. Research expenses increased 4% to $3.6 million in 2008, largely as a result of higher personnel and laboratory supply costs, offset by reduced outside laboratory costs.
Contract manufacturing expenses include personnel and occupancy expenses and external contract manufacturing costs for the scale up and manufacturing of drug product for use in our clinical trials, in addition to drug product stability and toxicology studies. Contract manufacturing costs increased 33% to $5.6 million in 2009, primarily due to higher drug production and scale-up costs. Contract manufacturing costs increased 2% to $4.2 million in 2008, primarily due to slightly higher personnel costs.
Clinical expenses include personnel expenses, travel, occupancy costs and external clinical trial costs, including clinical research organization charges, principal investigator fees, clinical site expenses and regulatory activities associated with conducting human clinical trials. Clinical expenses also include quality control and assurance activities, such as storage and shipment services for our picoplatin drug product. Clinical costs decreased 30% to $17.0 million in 2009, primarily due to reduced external clinical trial costs from the winding down of our trials, partially offset by costs from the increased use of consultants and contract labor. Clinical costs increased 80% to $24.4 million in 2008, principally due to expanded external clinical trial costs associated with our picoplatin trials and increased personnel costs. Prior to 2009, we included external legal costs associated with patent filings, maintenance and litigation in clinical expenses. We now report patent related external legal costs in our general and administrative. For the years ended 2009, 2008 and 2007, these costs totaled approximately $1.0 million in each year. The table above has been adjusted for this change in presentation.
Share-based compensation expenses reflect the non-cash charge recognized in accordance with the accounting rules for share-based compensation under which the fair value of all employee and non-employee share-based payments is charged to expense over the vesting period of the share-based awards. Share-based compensation expense increased 48% to $2.3 million in 2009. This increase was primarily a result of recognition of expense for restricted stock unit awards, partially offset by the departure of an officer at the end of 2008 for whom there was no expense in 2009, the expense of vesting of certain options held by certain officers in 2008 for which there was no similar acceleration in 2009, and a lower number and value of stock options outstanding in 2009 reflecting lower staffing
44
levels. Share-based compensation expense increased 20% to $1.6 million in 2008, primarily due to a higher number and value of outstanding stock options reflecting higher staffing levels.
Our major research and development program during the years ended December 31, 2009, 2008 and 2007 was picoplatin. Picoplatin is an intravenous platinum-based chemotherapeutic, designed to treat solid tumors that are resistant to existing platinum-based cancer therapies. We have completed enrollment and initial statistical analysis of a pivotal Phase 3 SPEAR trial of picoplatin in the second-line treatment of patients with small cell lung cancer. This trial did not meet its primary endpoint of overall survival and we have initiated a process with the FDA to identify a potential regulatory path forward for picoplatin in this indication. We are also conducting two separate Phase 2 trials evaluating picoplatin as a first-line treatment of metastatic colorectal cancer and castration-resistant (hormone-refractory) prostate cancer. Additionally, we have completed a Phase 1 cardiac safety trial of picoplatin and a Phase 1 study evaluating an oral formulation of picoplatin in solid tumors.
As of December 31, 2009, we have incurred external costs of approximately $73.4 million in connection with our entire picoplatin clinical program. Total estimated future costs of our picoplatin Phase 3 trial in small cell lung cancer is in the range of $1.5 million to $2.0 million through 2010. Total estimated future costs of both our picoplatin Phase 2 trial in colorectal cancer and our Phase 2 trial in castration-resistant prostate cancer are in the range of $1.0 million to $1.5 million through 2010. These future costs reflect activities to complete our trials through data analysis and data lock and could be substantially higher if we have to repeat, revise or expand the scope of any of our trials. Material cash inflows relating to the commercialization of picoplatin will not commence unless and until we complete required clinical studies and obtain FDA marketing approvals, and then only if picoplatin finds acceptance in the marketplace. To date, we have not received any revenues from sales of picoplatin.
Recap of Development and Clinical Program Costs. Our research and development administrative overhead costs, consisting of rent, utilities, consulting fees and other various overhead costs, are included in total research and development expense for each period, but are not allocated among our various projects. Our total research and development costs include the costs of various research efforts directed toward the identification and evaluation of future product candidates. These other research projects are preclinical and not considered major projects. We implemented a restructuring on March 31, 2009, which resulted in the discontinuation of our preclinical research operations. Our total research and development costs are summarized below:
| |
($ in thousands) | Annual Percentage Change |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | 2009-2008 | 2008-2007 | ||||||||||||
Picoplatin |
$ | 20,737 | $ | 24,161 | $ | 14,399 | -14 | % | 68 | % | |||||||
Other unallocated costs and overhead |
2,664 | 7,990 | 6,662 | -67 | % | 20 | % | ||||||||||
Share-based compensation |
2,338 | 1,581 | 1,320 | 48 | % | 20 | % | ||||||||||
Total research and development costs |
$ | 25,739 | $ | 33,732 | $ | 22,381 | -24 | % | 51 | % | |||||||
Our external costs for picoplatin in 2009, 2008 and 2007 reflect costs associated with our various picoplatin clinical studies and the manufacture of drug product to support our clinical trials. We expect our external costs for picoplatin to decrease in 2010, reflecting lower costs for our fully enrolled small cell lung, colorectal and prostate cancer trials and our completed oral picoplatin study, partially offset by clinical, consulting and other development costs associated with our regulatory and partnering activities focused on the continued development of picoplatin in multiple indications and two formulations.
45
The risks and uncertainties associated with completing the development of picoplatin on schedule, or at all, include the following, as well as the other risks and uncertainties described in this report:
- •
- we may not have adequate funds to complete the development of picoplatin;
- •
- picoplatin may not be shown to be safe and efficacious in clinical trials; and
- •
- we may be unable to obtain regulatory approvals of the drug or may be unable to obtain such approvals on a timely basis.
If we fail to obtain marketing approvals for picoplatin, are unable to secure adequate commercial supplies of picoplatin active pharmaceutical ingredient and finished drug product, or do not complete development and obtain United States and foreign regulatory approvals on a timely basis, our operations, financial position and liquidity could be severely impaired, including as follows:
- •
- we would not earn any sales revenue from picoplatin, which would increase the likelihood that we would need to obtain
additional financing for our ongoing product development efforts and continued operations; and
- •
- our reputation among investors might be harmed, which could make it more difficult for us to obtain equity capital on attractive terms, or at all.
Because of the many risks and uncertainties relating to completion of clinical trials, receipt of marketing approvals and acceptance in the marketplace, we cannot predict the period in which material cash inflows from our picoplatin program will commence, if ever.
General and Administrative
| |
($ in thousands) | Annual Percentage Change |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | 2009-2008 | 2008-2007 | ||||||||||||
General and administrative |
$ | 9,743 | $ | 9,883 | $ | 8,928 | -1 | % | 11 | % | |||||||
Share-based compensation |
4,955 | 5,542 | 4,149 | -11 | % | 34 | % | ||||||||||
Total |
$ | 14,698 | $ | 15,425 | $ | 13,077 | -5 | % | 18 | % | |||||||
General and administrative expenses decreased 5% to approximately $14.7 million in 2009 and increased 18% to approximately $15.4 million in 2008. General and administrative expenses, excluding share-based compensation expense, decreased 1% to approximately $9.7 million in 2009 and increased 11% to approximately $9.9 million in 2008. In 2009, the decrease was due primarily to lower personnel and travel costs, partially offset by higher investor relations costs. The increase in 2008 was primarily due to higher personnel costs, resulting from increased headcount, and consulting costs. Share-based compensation expense in 2009, 2008 and 2007 reflects non-cash charges recognized in accordance with the accounting rules for share-based compensation, under which the fair value of all employee and non-employee share-based payments is charged to expense over the vesting period of the share-based award. Share-based compensation expense decreased 11% to approximately $5.0 million in 2009 and increased 34% to approximately $5.5 million in 2008. In 2009, the decrease was primarily due to the departure of an officer at the end of 2008 for whom there was no expense in 2009, the acceleration of vesting of certain stock options held by certain officers in 2008 for which there was no similar expense in 2009, and a lower number and value of stock options outstanding in 2009 reflecting lower staffing levels, partially offset by expense recognized on restricted stock unit awards. The increase in 2008 was primarily due to a higher number and value of outstanding stock options reflecting higher staffing levels.
46
Restructuring and Asset Impairments
Effective March 31, 2009, we implemented a strategic restructuring plan to refocus our cash resources on clinical and commercial development of picoplatin, which resulted in the discontinuation of our preclinical research operations and reduced our workforce by approximately 12%, or eight employees. This restructuring resulted in charges of $0.5 million in the first quarter of 2009 consisting of $0.3 million in severance charges and $0.2 million in other expenses related to the closure of our lab facilities in South San Francisco, California.
In conjunction with the decision to discontinue our preclinical research operations, we recognized an asset impairment loss of $0.6 million on certain facilities and equipment related to the lab in South San Francisco, California. The loss on the assets was determined based on estimates of potential sales values of used equipment. These impairment charges established new cost bases for the impaired assets, which are included in assets held for sale and reported in prepaid expenses and other assets on our consolidated balance sheet for 2009. Additionally, at December 31, 2009, we recognized an impairment charge of approximately $1.5 million for our dedicated manufacturing equipment asset. The impairment charge was determined based on the delay in our plans for the commercialization of picoplatin, which we do not anticipate will occur before 2011.
Other Income and Expense
| |
($ in thousands) | Annual Percentage Change |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | 2009-2008 | 2008-2007 | |||||||||||
Interest expense |
$ | (3,143 | ) | $ | (1,897 | ) | $ | (1,727 | ) | 66 | % | 10 | % | |||
Interest income and other, net |
406 | 2,489 | 4,298 | -84 | % | -42 | % | |||||||||
|
$ | (2,737 | ) | $ | 592 | $ | 2,571 | -562 | % | -77 | % | |||||
Interest expense increased 66% to approximately $3.1 million in 2009 and increased 10% to approximately $1.9 million in 2008. The increases in interest expense in 2009 and 2008 were primarily due to increased interest costs resulting from additional borrowings that were effected in September 2008 under our bank loan. Interest income and other, net, decreased 84% to $0.4 million in 2009 and decreased 42% to $2.5 million in 2008. The decreases were primarily due to lower average yields from and decreasing balances in our investment securities portfolio.
Liquidity and Capital Resources
| |
December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | |||||||
| |
($ in thousands) |
|||||||||
Cash, cash equivalents and investment securities |
$ | 43,389 | $ | 72,755 | $ | 92,621 | ||||
Working capital |
27,369 | 54,873 | 84,383 | |||||||
Shareholders' equity |
23,644 | 47,647 | 89,105 | |||||||
| |
Years Ended December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | ||||||||
| |
($ in thousands) |
||||||||||
Cash provided by (used in): |
|||||||||||
Operating activities |
$ | (35,491 | ) | $ | (33,487 | ) | $ | (24,653 | ) | ||
Investing activities |
1,160 | 34,282 | (55,542 | ) | |||||||
Financing activities |
6,125 | 14,014 | 65,382 | ||||||||
We have historically experienced recurring operating losses and negative cash flows from operations. Cash, cash equivalents and investment securities, net of restricted cash of $0.3 million,
47
totaled $43.4 million at December 31, 2009. As of December 31, 2009, we had net working capital of $27.4 million, an accumulated deficit of $408.2 million and total shareholders' equity of $23.6 million.
We have historically maintained our financial position through strategic management of our resources, including, the sale of equity securities, borrowings under debt instruments, technology licensing and collaborative agreements. We invest excess cash in investment securities that will be used to fund future operating costs. Cash used for operating activities for the year ended December 31, 2009 totaled $35.5 million.
Recent Developments
During 2010, we plan to refocus our resources on regulatory and partnering strategies to support the continued development of picoplatin. On February 5, 2010, we implemented a restructuring plan to conserve capital resources, which reduced our workforce by approximately 57%, to 22 employees. We currently estimate that we will incur total restructuring charges of approximately $1.2 million, consisting mainly of one-time employee termination benefits, in connection with the restructuring.
On February 12, 2010, we entered into a sublease agreement with Veracyte, Inc., pursuant to which Veracyte is subleasing, effective March 1, 2010, approximately 11,000 square feet of our 17,045 square feet of executive office space, or the Premises, located at 7000 Shoreline Court, South San Francisco, California. Base sublease rental income for this space is $17,600 per month. On September 1, 2010, the subleased space will expand to encompass the entire 17,045 square feet of the Premises. After delivery of this expanded sublease space, base sublease rental income will be $28,124 per month until expiration of the sublease on July 10, 2011, at which time Veracyte will lease the Premises directly from the landlord. Additional rent under the sublease will be payable monthly to us by Veracyte, based on Veracyte's share of operating expenses attributable to the Premises. Our sublease with Veracyte does not modify or limit the terms and conditions of our original lease with the landlord, or waive any rights or remedies of the landlord, except that the landlord releases us from obligations under the original lease to remove alterations or repair or restore the Premises upon expiration of the original lease or following a casualty occurring during the original lease term. We currently are evaluating our facility space requirements and plan to explore alternative sites in the San Francisco area for executive office space. We expect to save approximately $700,000 in future rental and operating expenses as a consequence of the sublease.
On February 23, 2010, we entered into an equity line of credit facility with Commerce Court, pursuant to a Common Stock Purchase Agreement, or the Agreement, which provides that, upon the terms and subject to the conditions set forth in the Agreement, Commerce Court is committed to purchase up to $20.0 million of registered shares of our common stock over the approximately 18-month term of the Agreement. From time to time over the term of the Agreement, we may, at our sole discretion, present Commerce Court with draw down notices to purchase our common stock over eight consecutive trading days or such other period mutually agreed upon by us and Commerce Court. Each draw down is subject to limitations based on the price of the Company's common stock and a limit of 2.5% of our market capitalization at the time of the draw down (which limitations may be modified or waived by mutual agreement of the parties). In no event may we sell Commerce Court more than 8,423,431 shares of our common stock under the Agreement, which is the amount equal to one share less than 20% of our outstanding shares of common stock on the closing date of the Agreement, minus the number of share of common stock issued to Commerce Court in payment of its commitment fee. In addition, Commerce Court may not at any time acquire shares under the Agreement if, after giving effect to such purchase, Commerce Court would beneficially own 9.9% or more of our outstanding common stock. The Agreement does not require Commerce Court to purchase shares at prices below $1.00 per share.
Once presented with a draw down notice, Commerce Court is required to purchase a pro rata portion of the shares on each trading day during the trading period on which the daily volume
48
weighted average price for our common stock exceeds a threshold price determined by us for such draw down. The per share purchase price for these shares will equal the daily volume weighted average price of our common stock on each date during the draw down period on which shares are purchased, less a discount ranging from 3.125% to 5.0%, based on the trading price of our common stock. In consideration of Commerce Court's execution and delivery of the Agreement, we issued 121,183 shares of our common stock on February 25, 2010. All shares issued pursuant to the Agreement are covered by a registration statement on Form S-3 filed with the Securities and Exchange Commission.
On March 15, 2010, we completed a draw down and sale of 4,229,000 shares of our common stock, at a price of approximately $1.49 per share, to Commerce Court under the equity line of credit facility. Net proceeds of approximately $6,154,000 were received, after deducting estimated offering expenses of approximately $166,000. Proceeds of this draw down will be used to fund our efforts to enable a registrational filing to seek regulatory approval of picoplatin as a second-line treatment for small cell lung cancer patients with refractory disease and to explore strategic partnering and other relationships to support the continued development of picoplatin in other indications.
Capital Resources
Equity Financing. During 2009, we sold an aggregate of approximately 7.0 million shares of our common stock to Azimuth, pursuant to two draw downs under our equity line of credit facility with Azimuth. In the first draw down on November 23, 2009, we sold approximately 3.5 million common shares to Azimuth at a purchase price of approximately $2.15 per share. We sold Azimuth approximately 3.5 million common shares for $1.87 per share in the second draw down on December 22, 2009. The equity facility terminated by its terms on December 22, 2009. We received aggregate net proceeds from the draw downs of approximately $13.7 million. See Note 7 to the Notes to consolidated financial statements for additional information. The proceeds of the draw downs under the Azimuth facility were used for general corporate purposes, including working capital, following our November 16, 2009 announcement that our Phase 3 SPEAR trial failed to meet its primary endpoint of overall survival.
On April 30, 2007, we completed a public offering of 11.8 million shares of our common stock at a price of $6.33 per share. Net proceeds of the offering were $70.0 million. Additionally, in April 2006, we received $62.0 million in net proceeds from an equity financing, pursuant to which we issued 15.5 million shares of common stock at a purchase price of $4.20 per share. Investors in the 2006 financing also received warrants to purchase an aggregate of 4.6 million shares of common stock at a purchase price of $4.62 per share. The proceeds of these transactions were used to support our research and clinical development activities and commercialization efforts, as well as for general corporate purposes, including working capital.
Secured Loan Facility. On September 2, 2008, we entered into an amended and restated loan and security agreement with GE Business Financial Services, Inc. and Silicon Valley Bank, establishing a $27.6 million senior secured loan facility. The loan agreement amends and restates our earlier loan and security agreement with Silicon Valley Bank and Merrill Lynch Capital dated as of October 25, 2006, pursuant to which we obtained a $15.0 million capital loan that was to mature on April 1, 2010. Funds under the loan facility were made available as follows: (i) an initial term loan advance in the amount of $17.6 million, which was comprised of (a) the outstanding principal balance of $7.6 million remaining on the original loan and (b) an additional cash advance of approximately $10.0 million, which was fully funded on September 2, 2008; and (ii) a second term loan advance in the amount of $10.0 million, which was fully funded on September 30, 2008. The advances under the loan facility are repayable over 42 months, commencing on October 1, 2008. Interest on the advances is fixed at 7.80% per annum. Final payments in the amounts of $1.1 million and $0.9 million are due upon maturity or earlier repayment of the initial term loan advance and the second term loan advance, respectively. Additionally, as a condition to the amendment and restatement of the original loan, we agreed to
49
modification of the final payment obligations under the original loan, pursuant to which we paid $0.6 million to Silicon Valley Bank on September 2, 2008, the effective date of the loan facility, and will pay $0.7 million to GE Business Financial Services on March 31, 2010. The loan facility is secured by a first lien on all of our non-intellectual property assets. In connection with the loan agreement, we issued to the lenders ten-year warrants to purchase an aggregate of 219,920 shares of common stock at an exercise price of $4.297 per share. In October 2009, Silicon Valley Bank exercised its warrants. In November 2009, we amended the warrants held by GE Business Financial Services to change the exercise price to $2.00 per share. At December 31, 2009, the outstanding principal amount under the loan facility was $18.7 million.
The loan agreement contains restrictions on our ability to, among other things, dispose of certain assets, engage in certain mergers and acquisition transactions, incur indebtedness, create liens on assets, make investments, pay dividends and repurchase stock. The loan agreement also contains covenants requiring us to maintain a minimum amount of unrestricted cash during the term of the loan equal to the lesser of (i) $17.9 million or (ii) the outstanding aggregate principal balance of the term loans plus $4.0 million. The loan agreement contains events of default that include nonpayment of principal, interest or fees, breaches of covenants, material adverse changes, bankruptcy and insolvency events, cross defaults to any other indebtedness, material judgments, inaccuracy of representations and warranties and events constituting a change of control. The occurrence of an event of default would increase the applicable rate of interest by 5% and could result in the acceleration of our payment obligations under the loan agreement. We were in compliance with all loan covenants as of December 31, 2009.
Taking into account the minimum unrestricted cash requirement under the loan agreement and our projected operating results, we believe that our current cash, cash equivalent and investment securities balances, including the net proceeds from our March 15, 2010 sale of common stock to Commerce Court under our equity line of credit facility, will provide adequate resources to fund operations at least through the end of 2010. However, given the uncertainties of outcomes of our regulatory and partnering strategies to support the continued development of picoplatin, there is no assurance that we can achieve our projected operating results. Thereafter, unless we raise additional funds, we will be in default of the loan agreement. We have no assurance that, especially in light of the current difficult economic environment, the lenders will be willing to waive or renegotiate the terms of the loan agreement to address or avoid financial or other defaults.
If an event of default were to occur, we might not have sufficient funds to repay the loan or to fund our continuing operations. In such case, we would need to delay, scale back or curtail some or all of our current picoplatin clinical and regulatory efforts, further reduce our workforce, license picoplatin for development and commercialization by third parties, or attempt to sell the company. Provisions of the loan agreement would limit our ability to dispose of certain assets, engage in certain mergers, incur certain indebtedness, make certain distributions and engage in certain investment activities without the prior consent of the lenders. We have no assurance that we can obtain financing or otherwise raise additional funds, if at all, on terms acceptable to us or to our lenders.
Operating Agreements. We have entered into clinical supply agreements with Heraeus and Baxter, pursuant to which they produce picoplatin API and finished drug product, respectively, for our clinical trials. Manufacturing services under these clinical supply agreements are provided on a purchase order, fixed-fee basis. Our API clinical supply agreement continues in effect until it is terminated by mutual agreement of the parties or by either party in accordance with its terms. Our finished drug product clinical supply agreement had an initial term ending December 31, 2009, and in December 2009, we exercised our first renewal option, extending the term to December 31, 2010. This agreement remains subject to renewal, at our option, for an additional one-year term. The total aggregate cost of clinical supplies of picoplatin API and finished drug product purchased during the year ended December 31, 2009 was $4.1 million. We do not have any purchase commitments under these agreements.
50
We also have entered into a picoplatin API commercial supply agreement with Heraeus in March 2008 and a finished drug product commercial supply agreement with Baxter in November 2008. Under these agreements, Heraeus and Baxter will produce picoplatin API and finished drug product, respectively, for commercial use. Manufacturing services are provided on a purchase order, fixed-fee basis, subject to certain purchase price adjustments and minimum quantity requirements. The API commercial supply agreement continues for an initial term ending December 31, 2013, and the finished drug product commercial supply agreement continues for an initial term ending November 22, 2013, in each case subject to extension. The costs to Heraeus for the purchase and set-up of dedicated manufacturing equipment costing approximately $1.5 million will be repaid by us in the form of a surcharge on an agreed upon amount of the picoplatin API ordered and delivered on or before December 31, 2013. If we order and take delivery of less than the agreed upon amount of picoplatin API through December 31, 2013, we will be obligated to pay the balance of the equipment cost as of that date. Heraeus completed construction of the equipment as of December 31, 2009. We determined that the equipment should be accounted for as a capital lease and accordingly recognized an asset and long term obligation for the equipment of $1.5 million, respectively. We will reflect the surcharge payments as reductions in the capital lease balance outstanding and will accrete a finance charge to interest expense as specified under the agreement. Due to the delay in our plans for the commercialization of picoplatin, which we do not anticipate will occur before 2011, we determined that our capital lease asset for equipment under the Heraeus agreement was impaired as of December 31, 2009 and therefore recognized an impairment charge of $1.5 million. We do not have any purchase commitments under these agreements.
During the year ended December 31, 2009, we paid total rent (base rent and additional rent based on our share of facility common operating expenses) of $1.4 million under the operating leases for our South San Francisco headquarters facility and our Seattle facility. Of this amount, $1.2 million represents total aggregate minimum lease payments under these leases. As discussed under the heading "Recent Events" above, we have entered into a sublease of most, and as of September 1, 2010, all of our headquarter facilities in South San Francisco.
Potential Milestone and Royalty Obligations. If we are successful in our efforts to commercialize picoplatin, we would, under our amended license agreement with Genzyme, be required to pay Genzyme up to $5.0 million in commercialization milestones upon the attainment of certain levels of annual net sales of picoplatin. Genzyme also would be entitled to royalty payments of up to 9% of annual net sales of picoplatin related products.
We will require substantial additional capital to pursue our regulatory and partnering strategies to support the continued development of picoplatin and to fund our future operations. Management is continuously exploring financing alternatives, including:
- •
- raising additional capital through the public or private sale of equity or debt securities or through the establishment of
credit or other funding facilities; and
- •
- entering into strategic collaborations, which may include joint ventures or partnerships for product development and commercialization, merger, sale of assets or other similar transactions.
The amount of additional financing we will require in the future will depend on a number of factors, including:
- •
- the costs of performing our obligations under our loan facility with GE Business Financial Services and Silicon Valley
Bank, including the cost of interest and other payment obligations and penalties and the cost of complying with the covenants and restrictions under the loan agreement;
- •
- actions taken by the FDA and other regulatory authorities, specifically including the results of our ongoing process with the FDA to identify a potential regulatory path forward to enable a
51
- •
- the scope, timing and success of our current and any future picoplatin clinical trials and regulatory and partnering
activities to support the continued development of picoplatin in multiple indications and in both intravenous and oral formulations;
- •
- our access to clinical supplies of picoplatin API and finished drug product in a timely and cost effective manner;
- •
- the timing and amount of any milestone or other payments we might receive from or be obligated to pay to potential
strategic partners;
- •
- our degree of success in commercializing picoplatin;
- •
- the emergence of competing technologies and products, and other adverse market developments;
- •
- the acquisition or in-licensing of other products or intellectual property;
- •
- the costs of any strategic partnerships or other collaborations established; and
- •
- the costs of preparing, filing, prosecuting, maintaining and enforcing patent claims and other intellectual property rights.
registrational filing seeking approval of picoplatin for the second-line treatment of small cell lung cancer patients with refractory disease and future FDA guidance regarding potential regulatory strategies for picoplatin in prostate, colorectal and ovarian cancers;
We may not be able to obtain required additional capital or enter into relationships with potential corporate partners on a timely basis, on terms that ultimately prove favorable to us, or at all. Conditions in the capital markets in general, and in the life science capital market specifically, may affect our potential financing sources and opportunities for strategic partnering. Our consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States, assuming that we will continue as a going concern and do not include any adjustments that might result from the outcome of this uncertainty.
Contractual Obligations and Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenue or expenses, results of operations, liquidity, capital expenditures or capital resources that is material to investors.
At December 31, 2009, we had the following contractual obligations (in thousands):
| |
Payments due by period | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Total | Less than 1 year |
1 - 3 years | 3 - 5 years | More than 5 years |
|||||||||||||
Contractual Obligations |
||||||||||||||||||
Long-term debt obligations: |
||||||||||||||||||
Note payable(1)(2) |
$ | 22,025 | $ | 9,678 | $ | 12,347 | $ | — | $ | — | ||||||||
Operating lease obligations: |
||||||||||||||||||
Seattle premises |
548 | 548 | — | — | — | |||||||||||||
South San Francisco premises |
1,058 | 664 | 394 | — | — | |||||||||||||
|
1,606 | 1,212 | 394 | — | — | |||||||||||||
Capital Lease obligations(3) |
1,884 |
40 |
1,015 |
829 |
— |
|||||||||||||
Total |
$ | 25,515 | $ | 10,930 | $ | 13,756 | $ | 829 | $ | — | ||||||||
- (1)
- Amounts include interest payments.
52
- (2)
- Amount
in "Total" column includes total principal payment of $18,747 as reflected on the Consolidated Balance Sheet for the year ended December 31,
2009.
- (3)
- Amount in "Total" column includes total principal payment of $1,485 as reflected on the Consolidated Balance Sheet for the year ended December 31, 2009.
New Accounting Pronouncements
See Item 8, Note 1 to our consolidated financial statements, "Business Overview and Summary of Significant Accounting Policies," for a discussion of new accounting standards.
Item 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Interest Rate Risk
Our exposure to market rate risk for changes in interest rates relates primarily to the debt securities included in our investment portfolio. We do not invest in any derivative financial instruments. We invest in money market funds, debt instruments of the U.S. Government and its agencies and high-quality corporate issuers. Investments in both fixed rate and floating rate interest earning instruments carry a degree of interest rate risk. The fair market value of fixed rate securities may be adversely impacted due to a rise in interest rates, while floating rate securities may produce less income than expected if interest rates decrease. Due in part to these factors, our future investment income may fall short of expectations due to changes in interest rates or we may experience losses in principal if forced to sell securities that have declined in market value due to changes in interest rates. At December 31, 2009, we owned government debt instruments totaling $9.3 million and corporate debt securities totaling $18.2 million. Our exposure to losses as a result of interest rate changes is managed through investing primarily in securities with relatively short maturities of two years or less and in securities with variable interest rates. We owned corporate debt securities totaling $4.3 million at December 31, 2009 with maturities greater than one year.
Our only material outstanding debt is our loan obligation to GE Business Financial Services and Silicon Valley Bank. The outstanding balance of this loan was $18.7 million on December 31, 2009. The loan, which matures on March 1, 2012, bears interest at a fixed rate of 7.80%. The occurrence of an event of default under the loan, as described above, would increase the applicable rate of interest by 5% during the continuance of the event of default and could result in acceleration of our payment obligations under the loan agreement. As described elsewhere in this report, unless we raise additional capital, obtain a waiver or renegotiate the loan agreement, there is a likelihood that we will be in default of the minimum unrestricted cash requirement and potentially other requirements under the loan.
53
Item 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
All financial schedules are omitted since the required information is not applicable or has been presented in the consolidated financial statements and the notes thereto.
54
Report of Ernst & Young LLP, Independent Registered Public Accounting Firm
The Board of Directors and Shareholders of Poniard Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheet of Poniard Pharmaceuticals, Inc. as of December 31, 2009, and the related consolidated statements of operations, shareholders' equity, and cash flows for the year ended December 31, 2009. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Poniard Pharmaceuticals, Inc. at December 31, 2009, and the consolidated results of its operations and its cash flows for year ended December 31, 2009, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Poniard Pharmaceuticals, Inc.'s internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 16, 2010 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Palo
Alto, California
March 16, 2010
55
Report of KPMG, LLP, Independent Registered Public Accounting Firm
The
Board of Directors and Shareholders
Poniard Pharmaceuticals, Inc.:
We have audited the accompanying consolidated balance sheet of Poniard Pharmaceuticals, Inc. and subsidiary as of December 31, 2008, and the related consolidated statements of operations, shareholders' equity, and cash flows for the years ended December 31, 2008 and 2007. These consolidated financial statements are the responsibility of Poniard Pharmaceuticals, Inc.'s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Poniard Pharmaceuticals, Inc. and subsidiary as of December 31, 2008, and the results of their operations and their cash flows for the years ended December 31, 2008 and 2007, in conformity with U.S generally accepted accounting principles.
The accompanying consolidated financial statements have been prepared assuming the Company will continue as a going concern. As discussed in note 1 to the consolidated financial statements, the Company has suffered recurring losses from operations and negative cash flows. Furthermore, the Company's long-term debt agreement contains certain covenants that require the Company to maintain a certain level of unrestricted cash and cash equivalents, and contains certain subjective acceleration clauses related to material adverse changes which raise substantial doubt about its ability to continue as a going concern. Management's plans in regard to these matters are also described in note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ KPMG LLP
Seattle,
Washington
March 16, 2009
56
PONIARD PHARMACEUTICALS, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except share data)
| |
As of December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | |||||||||
ASSETS |
|||||||||||
Current assets: |
|||||||||||
Cash and cash equivalents |
$ | 15,938 | $ | 44,144 | |||||||
Cash—restricted |
281 | 281 | |||||||||
Investment securities |
27,451 | 28,611 | |||||||||
Prepaid expenses and other current assets |
826 | 977 | |||||||||
Total current assets |
44,496 | 74,013 | |||||||||
Facilities and equipment, net of depreciation of $1,199 and $1,319 at December 31, 2009 and 2008, respectively |
219 | 1,123 | |||||||||
Other assets |
135 | 289 | |||||||||
Licensed products, net |
7,592 | 8,807 | |||||||||
Total assets |
$ | 52,442 | $ | 84,232 | |||||||
LIABILITIES AND SHAREHOLDERS' EQUITY |
|||||||||||
Current liabilities: |
|||||||||||
Accounts payable |
$ | 849 | $ | 604 | |||||||
Accrued liabilities |
7,679 | 10,618 | |||||||||
Current portion of note payable and capital lease obligations |
8,599 | 7,918 | |||||||||
Total current liabilities |
17,127 | 19,140 | |||||||||
Long-term liabilities: |
|||||||||||
Note payable, noncurrent portion, net of debt discounts |
10,186 | 17,407 | |||||||||
Capital lease obligations |
1,485 | 38 | |||||||||
Total long-term liabilities |
11,671 | 17,445 | |||||||||
Commitments and contingencies |
|||||||||||
Shareholders' equity: |
|||||||||||
Preferred stock, $0.02 par value, 2,998,425 shares authorized: |
|||||||||||
Convertible preferred stock, Series 1, 205,340 shares issued and outstanding (entitled in liquidation to $5,175) |
4 | 4 | |||||||||
Common stock, $0.02 par value, 200,000,000 shares authorized: |
|||||||||||
42,079,468 and 34,687,724 shares issued and outstanding as of December 31, 2009 and 2008, respectively |
842 | 694 | |||||||||
Additional paid-in capital |
430,971 | 409,244 | |||||||||
Other comprehensive loss |
(17 | ) | (354 | ) | |||||||
Accumulated deficit |
(408,156 | ) | (361,941 | ) | |||||||
Total shareholders' equity |
23,644 | 47,647 | |||||||||
Total liabilities and shareholders' equity |
$ | 52,442 | $ | 84,232 | |||||||
See notes to the consolidated financial statements.
57
PONIARD PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except per share amounts)
| |
Years Ended December 31, | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | ||||||||||
Operating expenses: |
|||||||||||||
Research and development |
$ | 25,739 | $ | 33,732 | $ | 22,381 | |||||||
General and administrative |
14,698 | 15,425 | 13,077 | ||||||||||
Gain on sale of real estate and equipment |
— | — | (105 | ) | |||||||||
Restructuring |
468 | — | — | ||||||||||
Asset impairment loss |
2,073 | — | — | ||||||||||
Total operating expenses |
42,978 | 49,157 | 35,353 | ||||||||||
Loss from operations |
(42,978 | ) | (49,157 | ) | (35,353 | ) | |||||||
Other (expense) income: |
|||||||||||||
Interest expense |
(3,143 | ) | (1,897 | ) | (1,727 | ) | |||||||
Interest income and other, net |
406 | 2,489 | 4,298 | ||||||||||
Total other (expense) income, net |
(2,737 | ) | 592 | 2,571 | |||||||||
Net loss |
(45,715 | ) | (48,565 | ) | (32,782 | ) | |||||||
Preferred stock dividends |
(500 | ) | (500 | ) | (500 | ) | |||||||
Net loss applicable to common shareholders |
$ | (46,215 | ) | $ | (49,065 | ) | $ | (33,282 | ) | ||||
Net loss per share applicable to common shareholders—basic and diluted |
$ |
(1.31 |
) |
$ |
(1.41 |
) |
$ |
(1.08 |
) |
||||
Weighted average common shares outstanding—basic and diluted |
35,272 | 34,686 | 30,762 | ||||||||||
See notes to the consolidated financial statements.
58
PONIARD PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF SHAREHOLDERS' EQUITY
(In thousands)
| |
Preferred Stock, Series 1 |
Preferred Stock, Series B |
Common Stock |
|
|
|
|
|||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
|
|
Accumulated Other Comprehensive (Loss)/Income |
|
||||||||||||||||||||||||||||
| |
Shares | Par Value |
Shares | Par Value |
Shares | Par Value |
Additional Paid-In Capital |
Accumulated Deficit |
Shareholders' Equity |
|||||||||||||||||||||||
Balance, December 31, 2006 |
205 | $ | 4 | — | $ | — | 22,808 | $ | 456 | $ | 326,025 | $ | (279,594 | ) | $ | — | $ | 46,891 | ||||||||||||||
Exercise of stock options and warrants |
— | — | — | — | 6 | — | 22 | — | — | 22 | ||||||||||||||||||||||
Common stock issued, net of offering costs of $5,054 |
— | — | — | — | 11,849 | 237 | 69,709 | — | — | 69,946 | ||||||||||||||||||||||
Share-based employee compensation expense |
— | — | — | — | — | — | 5,414 | — | — | 5,414 | ||||||||||||||||||||||
Stock options and warrants issued for services |
— | — | — | — | — | — | 55 | — | — | 55 | ||||||||||||||||||||||
Comprehensive loss: |
||||||||||||||||||||||||||||||||
Net loss |
— | — | — | — | — | — | — | (32,782 | ) | — | (32,782 | ) | ||||||||||||||||||||
Unrealized gain on investment securities |
— | — | — | — | — | — | — | — | 59 | 59 | ||||||||||||||||||||||
Total comprehensive loss |
— | — | — | — | — | — | — | — | — | (32,723 | ) | |||||||||||||||||||||
Preferred stock dividends |
— | — | — | — | — | — | — | (500 | ) | — | (500 | ) | ||||||||||||||||||||
Balance, December 31, 2007 |
205 | 4 | — | — | 34,663 | 693 | 401,225 | (312,876 | ) | 59 | 89,105 | |||||||||||||||||||||
Exercise of stock options and warrants |
— | — | — | — | 25 | 1 | 90 | — | — | 91 | ||||||||||||||||||||||
Warrants issued in connection with issuance of debt |
— | — | — | — | — | — | 806 | — | — | 806 | ||||||||||||||||||||||
Share-based employee compensation expense |
— | — | — | — | — | — | 7,004 | — | — | 7,004 | ||||||||||||||||||||||
Stock options and warrants issued for services |
— | — | — | — | — | — | 119 | — | — | 119 | ||||||||||||||||||||||
Comprehensive loss: |
||||||||||||||||||||||||||||||||
Net loss |
— | — | — | — | — | — | — | (48,565 | ) | — | (48,565 | ) | ||||||||||||||||||||
Unrealized loss on investment securities |
— | — | — | — | — | — | — | — | (413 | ) | (413 | ) | ||||||||||||||||||||
Total comprehensive loss |
— | — | — | — | — | — | — | — | — | (48,978 | ) | |||||||||||||||||||||
Preferred stock dividends |
— | — | — | — | — | — | — | (500 | ) | — | (500 | ) | ||||||||||||||||||||
Balance, December 31, 2008 |
205 | 4 | — | — | 34,688 | 694 | 409,244 | (361,941 | ) | (354 | ) | 47,647 | ||||||||||||||||||||
Exercise of stock options and warrants |
— | — | — | — | 347 | 7 | 813 | — | — | 820 | ||||||||||||||||||||||
Common stock issued, net of offering costs of $253 |
— | — | — | — | 6,956 | 139 | 13,584 | — | — | 13,723 | ||||||||||||||||||||||
Common stock issued for fully vested restricted |
||||||||||||||||||||||||||||||||
stock units |
— | — | — | — | 88 | 2 | 277 | — | — | 279 | ||||||||||||||||||||||
Share-based employee compensation expense |
— | — | — | — | — | — | 6,795 | — | — | 6,795 | ||||||||||||||||||||||
Stock options and warrants issued for services |
— | — | — | — | — | — | 225 | — | — | 225 | ||||||||||||||||||||||
Amendment of warrant exercise price |
— | — | — | — | — | — | 33 | — | — | 33 | ||||||||||||||||||||||
Comprehensive loss: |
||||||||||||||||||||||||||||||||
Net loss |
— | — | — | — | — | — | — | (45,715 | ) | — | (45,715 | ) | ||||||||||||||||||||
Unrealized gain on investment securities |
— | — | — | — | — | — | — | — | 337 | 337 | ||||||||||||||||||||||
Total comprehensive loss |
— | — | — | — | — | — | — | — | — | (45,378 | ) | |||||||||||||||||||||
Preferred stock dividends |
— | — | — | — | — | — | — | (500 | ) | — | (500 | ) | ||||||||||||||||||||
Balance, December 31, 2009 |
205 | $ | 4 | — | $ | — | 42,079 | $ | 842 | $ | 430,971 | $ | (408,156 | ) | $ | (17 | ) | $ | 23,644 | |||||||||||||
See notes to the consolidated financial statements.
59
PONIARD PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| |
Years Ended December 31, | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | ||||||||||
Cash flows from operating activities: |
|||||||||||||
Net loss |
$ | (45,715 | ) | $ | (48,565 | ) | $ | (32,782 | ) | ||||
Adjustments to reconcile net loss to net cash used in operating activities: |
|||||||||||||
Depreciation and amortization |
1,470 | 1,603 | 1,526 | ||||||||||
Amortization of discount on notes payable |
1,417 | 790 | 775 | ||||||||||
Accretion (amortization) of premium (discount) on investment securities |
428 | (411 | ) | (1,167 | ) | ||||||||
Gain on disposal of real estate and equipment |
— | — | (105 | ) | |||||||||
Asset impairment loss |
2,073 | — | — | ||||||||||
Restructuring |
18 | — | — | ||||||||||
Amendment of exercise price for certain warrants |
33 | — | — | ||||||||||
Stock-based compensation issued for services |
219 | 119 | 55 | ||||||||||
Stock-based employee compensation |
7,074 | 7,004 | 5,414 | ||||||||||
Change in operating assets and liabilities: |
|||||||||||||
Prepaid expenses and other assets |
186 | (22 | ) | (301 | ) | ||||||||
Accounts payable |
245 | (73 | ) | (98 | ) | ||||||||
Accrued liabilities |
(2,939 | ) | 6,068 | 2,030 | |||||||||
Net cash used in operating activities |
(35,491 | ) | (33,487 | ) | (24,653 | ) | |||||||
Cash flows from investing activities: |
|||||||||||||
Proceeds from maturities of investment securities |
53,676 | 80,950 | 51,560 | ||||||||||
Purchases of investment securities |
(52,607 | ) | (46,277 | ) | (104,058 | ) | |||||||
Facilities and equipment purchases |
(21 | ) | (391 | ) | (773 | ) | |||||||
Proceeds from disposals of equipment and facilities |
112 | — | 2,729 | ||||||||||
Purchase of licensed product |
— | — | (5,000 | ) | |||||||||
Net cash provided by (used in) investing activities |
1,160 | 34,282 | (55,542 | ) | |||||||||
Cash flows from financing activities: |
|||||||||||||
Net proceeds from bank note payable |
— | 19,997 | — | ||||||||||
Repayment of principal on note payable |
(7,886 | ) | (5,345 | ) | (3,905 | ) | |||||||
Payment of debt issuance costs |
— | (200 | ) | — | |||||||||
Proceeds from stock options and warrants exercised |
820 | 91 | 22 | ||||||||||
Repayment of capital lease obligation |
(32 | ) | (29 | ) | (36 | ) | |||||||
Net proceeds from issuance of common stock and warrants |
13,723 | — | 69,946 | ||||||||||
Increase in restricted cash |
— | — | (145 | ) | |||||||||
Payment of preferred dividends |
(500 | ) | (500 | ) | (500 | ) | |||||||
Net cash provided by financing activities |
6,125 | 14,014 | 65,382 | ||||||||||
Net (decrease) increase in cash and cash equivalents |
(28,206 | ) | 14,809 | (14,813 | ) | ||||||||
Cash and cash equivalents: |
|||||||||||||
Beginning of period |
44,144 | 29,335 | 44,148 | ||||||||||
End of period |
$ | 15,938 | $ | 44,144 | $ | 29,335 | |||||||
Supplemental disclosure of non-cash financing activities: |
|||||||||||||
Accrual of preferred dividend |
$ | 500 | $ | 500 | $ | 500 | |||||||
Increase in capital leases |
1,485 | — | 134 | ||||||||||
Debt discount capitalized in shareholders' equity |
— | 806 | — | ||||||||||
Supplemental disclosure of cash paid during the period for: |
|||||||||||||
Interest |
$ | 1,746 | $ | 1,001 | $ | 974 | |||||||
See notes to the consolidated financial statements.
60
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note 1. Business Overview and Summary of Significant Accounting Policies
Overview
Poniard Pharmaceuticals, Inc. is a biopharmaceutical company focused on the development and commercialization of cancer therapeutics. The accompanying consolidated financial statements include the accounts of Poniard Pharmaceuticals, Inc. and its wholly owned subsidiary, NeoRx Manufacturing Group, Inc. (the "Company"). All inter-company balances and transactions have been eliminated.
Reclassifications
Certain balances and results from prior years have been reclassified to conform to the Company's current year presentation. In particular, certain legal expenses related to intellectual property and patents have been reclassified from research and development expense to general and administration expense in the consolidated statements of operations to conform with the current year's presentation. The amounts reclassified for these patent-related legal expenses for 2009, 2008 and 2007 are $995,000, $982,000 and $992,000, respectively. The Company's reclassifications had no effect on total operating expenses, net loss or shareholders' equity.
Significant Risks and Uncertainties
These consolidated financial statements have been prepared assuming that the Company will continue as a going concern, which contemplates realization of assets and the satisfaction of liabilities in the normal course of business for a reasonable period following the date of these financial statements. The Company has historically suffered recurring operating losses and negative cash flows from operations. As of December 31, 2009, the Company had net working capital of $27,369,000, an accumulated deficit of $408,156,000 and total shareholders' equity of $23,644,000. The Company's total cash, cash equivalent and investment securities balances, net of restricted cash of $281,000, was $43,389,000 at December 31, 2009. The Company has financed its operations to date primarily through the sale of equity securities, borrowings under debt instruments, technology licensing and collaborative agreements. The Company invests excess cash in investment securities that will be used to fund future operating costs. Cash used for operating activities for the year ended December 31, 2009 totaled $35,491,000. The Company's primary activities consist of research and development to further its primary product candidate, picoplatin. As a result, the Company does not have a predictable source of revenue or other cash flows and does not expect to generate cash from operations for the foreseeable future. Thus, the Company is dependent upon its ability to sell equity instruments, borrow, or enter into licensing agreements in amounts sufficient to sustain its operations. The Company may not be able to obtain required additional capital or enter into relationships with corporate partners on a timely basis, on terms ultimately favorable to the Company, or at all. Conditions in the capital markets in general, and in the life science capital markets specifically, may affect the Company's potential financing sources and opportunities for strategic partnering.
The Company's loan facility with GE Business Financial Services and Silicon Valley Bank, described in Note 6 below, requires that the Company maintain a minimum amount of cash, cash equivalents and investments ("unrestricted cash") during the term of the loan equal to the lesser of (i) $17,940,000 or (ii) the outstanding aggregate principal balance of the term loans plus $4,000,000. Taking into account the minimum unrestricted cash requirement under the Company's loan agreement and its projected operating results, the Company believes that its current cash, cash equivalent and investment securities balances, including the net proceeds from the Company's March 15, 2010 sale of
61
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 1. Business Overview and Summary of Significant Accounting Policies (Continued)
common stock to Commerce Court Small Cap Value Fund, Ltd. ("Commerce Court") under its equity line of credit facility (see Note 15 for discussion of subsequent events), will provide adequate resources to fund operations at least through the end of 2010. However, given the uncertainties of outcomes of the Company's regulatory and partnering strategies to support the continued development of picoplatin, there is no assurance that the Company can achieve its projected operating results. Thereafter, unless the Company raises additional funds, it will be in default of the minimum unrestricted cash requirement and potentially other provisions of the loan agreements. The Company has no assurance that, especially in light of the current distressed economic environment, the lenders will be willing to waive or renegotiate the terms of the loan agreement to address or avoid financial or other defaults. The occurrence of an event of default would increase the applicable rate of interest by 5% and could result in the acceleration of the Company's payment obligations under the loan agreement. If an event of default were to occur, the Company might not have sufficient funds to repay the loan or to fund its continuing operations. In such case, it would need to delay, scale back or curtail some or all of its current picoplatin trials and regulatory efforts, further reduce its workforce, license picoplatin for development and commercialization by third parties or attempt to sell the Company. Provisions of the loan agreement would limit the Company's ability to dispose of certain assets, engage in certain mergers, incur certain indebtedness, make certain distributions, and engage in certain investment activities without the prior consent of the lenders.
Significant Accounting Policies
Use of Estimates: The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Research and Development Expenses: Research and development costs are expensed as incurred. Research and development expenses include, but are not limited to, payroll and personnel expense, lab supplies, clinical studies, external contract manufacturing costs for clinical trial drug product supplies, consulting, travel, and related overhead.
Cash Equivalents and Investment Securities: All highly liquid investments with an original maturity of three months or less when purchased are considered to be cash equivalents. Cash equivalents represent cash invested primarily in money market funds, federal government and agency securities, and corporate debt securities. The Company considers all investment securities to be available-for-sale. All securities are carried at fair value. The Company does not invest in derivative financial instruments. Unrealized losses and gains on investment securities are reported as a component of comprehensive income or loss and classified as other comprehensive income/(loss) in shareholders' equity. The Company monitors investment securities for other than temporary declines in fair value and charges impairment losses to income when an other than temporary decline in estimated value occurs. Investment in cash equivalents and investment securities in both fixed and floating rate interest earning instruments carry a degree of credit risk. The Company's exposure to losses as a result of credit risk is managed through investing primarily in securities with relatively short maturities of two years or less and in securities with variable interest rates. See Note 3 below for further discussion of investment securities.
62
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 1. Business Overview and Summary of Significant Accounting Policies (Continued)
Facilities and Equipment: Facilities and equipment are stated at acquired cost, less any charges for impairment, and are depreciated using the straight-line method over estimated useful lives of the assets as follows:
| |
Years | |
|---|---|---|
Lab equipment |
3 - 7 | |
Office furniture and fixtures |
5 - 7 | |
Computer equipment and software |
3 |
Leasehold improvements are depreciated using the straight-line method over the shorter of the assets' estimated useful lives or the remaining lease term.
When assets are retired or otherwise disposed of the cost of the assets and related accumulated depreciation or amortization are removed from the accounts and any resulting gains or losses are reflected in the consolidated statement of operations at the time of disposition. Expenditures for additions and improvements to the Company's facilities are capitalized and expenditures for maintenance and repairs are charged to expense as incurred.
Impairment of Long-Lived and Intangible Assets: Long-lived assets, including facilities and equipment and intangible assets, including capitalized license payments for the Company's picoplatin product candidate, are reviewed for possible impairment whenever significant events or changes in circumstances, including changes in the Company's business strategy and plans, a significant decrease in market value, a significant change in asset condition, or a significant adverse change in regulatory climate, indicate an impairment may have occurred. An impairment is indicated when the sum of the expected future undiscounted net cash flows identifiable to that asset or asset group is less than its carrying value. Impairment losses are determined from actual or estimated fair values, which are based on market values, net realizable values or projections of discounted net cash flows, as appropriate. The Company reviews long-lived and intangible assets on an as-needed basis to determine if there have been any adverse events or circumstances that would indicate an impairment exists. In particular, the value of the picoplatin intangible asset was reviewed as a result of negative results of clinical trials during 2009, with no impairment charge recorded. See Note 11 for additional details related to the Company's picoplatin intangible asset. As discussed in Note 13, the Company recorded impairment charges on certain facilities and equipment related to restructuring activities during 2009, and as discussed in Note 8, during 2009 the Company recorded impairment charges on certain equipment related to commercial manufacturing arrangements.
Debt Issuance Costs: Costs incurred in connection with the securing of long-term bank loans and other long-term debt are deferred and amortized as interest expense over the term of the related debt using a method that approximates the effective interest method.
Licensed Products: Licensed products represent an exclusive license to develop, manufacture and commercialize picoplatin, a platinum-based anti-cancer agent. Licensed products are amortized using the straight-line method over their estimated useful life of twelve years. The Company evaluates the recoverability of licensed products periodically and takes into account events or circumstances that might indicate that an impairment exists as discussed above under "Impairment of Long-Lived and Intangible Assets." No impairment of licensed products was identified during 2009, 2008 or 2007. See Note 11 below for additional information.
63
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 1. Business Overview and Summary of Significant Accounting Policies (Continued)
Income Taxes: The Company computes income taxes using the asset and liability method, under which deferred income taxes are provided for the temporary differences between the financial reporting basis and the tax basis of the Company's assets and liabilities and for operating loss and tax credit carryforwards. A valuation allowance is established when necessary to reduce deferred tax assets to the amount, if any, which is expected more likely than not to be realized.
The Company accounts for uncertain tax positions in accordance with Financial Accounting Standards Board ("FASB") accounting standards which prescribe a recognition threshold and measurement attribute for financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return and also provide guidance on various related matters such as derecognition, interest and penalties, and disclosure. The Company has been in a net operating loss position since its inception and, by providing a full valuation allowance, has not recognized any tax benefits for any of its income taxes. The Company has adopted a policy whereby amounts related to interest and penalties associated with tax matters are classified as additional income tax expense when incurred. Historically, the Company has not incurred any interest or penalties associated with tax matters and no interest or penalties were recognized during the years ended December 31, 2009, 2008 or 2007.
Net Loss Per Common Share: Basic and diluted loss per share are based on net loss applicable to common shares, which is comprised of net loss and preferred stock dividends in all periods presented. Shares used to calculate basic loss per share are based on the weighted average number of common shares outstanding during the period. Shares used to calculate diluted loss per share are based on the potential dilution that would occur upon the exercise or conversion of securities into common stock using the treasury stock method to the extent such common stock equivalents are not anti-dilutive. The computation of diluted net loss per share excludes the following options and warrants to acquire shares of common stock for the years indicated because their effect would not be dilutive.
| |
2009 | 2008 | 2007 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Common stock options |
5,606,000 | 5,893,000 | 4,650,000 | |||||||
Restricted stock units |
561,000 | — | — | |||||||
Common stock warrants |
5,085,000 | 6,060,000 | 5,947,000 | |||||||
Additionally, aggregate common shares of 39,015, issuable as of December 31, 2009 upon conversion of the Company's Series 1 convertible exchangeable preferred stock are not included in the calculation of diluted loss per share for 2009, 2008, and 2007 because the share increments would not be dilutive.
Share-Based Compensation: The Company has an incentive plan that rewards employees, directors and non-employee consultants with stock options and restricted stock units. Share-based payments are accounted for in accordance with FASB accounting standards for equity instruments exchanged for services. Under the provisions of these standards, share-based compensation cost is measured at the grant date, based on the fair value of the award, and is recognized as an expense over the requisite service period (generally the vesting period of the equity grant). See Note 10 below for further details on share-based compensation.
Concentration in the Available Sources of Supply of Materials: The Company relies on third parties to manufacture picoplatin active pharmaceutical ingredient ("API") and finished drug product for its
64
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 1. Business Overview and Summary of Significant Accounting Policies (Continued)
clinical trials and for its future commercialization activities. The Company currently has separate agreements with one supplier each of picoplatin API and finished drug product for clinical and commercial use. The Company's API clinical supply agreement continues in effect until it is terminated by mutual agreement of the parties or by either party in accordance with its terms. The Company's finished drug product clinical supply agreement had an initial term ending December 31, 2009, and in December 2009, the Company exercised its first renewal option, extending the term to December 31, 2010. This agreement remains subject to renewal, at the Company's option, for an additional one-year term. The Company's commercial API and finished drug supply agreements have initial terms ending in late 2013. The Company has no assurance that its current suppliers will be able to manufacture sufficient picoplatin API and/or finished drug product on a timely or cost-effective basis at all times in the future. The Company believes that there are other contract manufacturers with the capacity to manufacture picoplatin API and finished drug product.
Fair Value Measurements: In September 2006, the FASB issued accounting standards associated with fair value measurements. This guidance defined fair value, established a framework for measuring fair value, and expanded disclosures about fair value measurements. In February 2008, the FASB delayed the effective date of the guidance for all non-financial assets and non-financial liabilities, except those that are measured at fair value on a recurring basis. Accordingly, the Company adopted this guidance for assets and liabilities recognized at fair value on a recurring basis effective January 1, 2008 and adopted the guidance for non-financial assets and liabilities measured on a non-recurring basis effective January 1, 2009. The application of the fair value framework did not have a material impact on the Company's consolidated financial position, results of operations or cash flows.
Subsequent Events: In May 2009, the FASB issued standards related to the accounting for and disclosure of events that occur after the balance sheet date, but before financial statements are issued or are available to be issued. The Company adopted the provisions of these standards, which became effective for interim and annual reporting periods ending after June 15, 2009. The Company has reviewed and evaluated material subsequent events through the date that the financial statements were available for issuance. See Note 15 for additional details.
Segment Reporting: The Company has one operating business segment, cancer therapeutics development.
Recent Accounting Standards
In October 2009, the FASB issued new standards for revenue recognition with multiple deliverables. These new standards impact the determination of when the individual deliverables included in a multiple-element arrangement may be treated as separate units of accounting. Additionally, these new standards modify the manner in which the transaction consideration is allocated across the separately identified deliverables by no longer permitting the residual method of allocating arrangement consideration. These new standards are required to be adopted in the first quarter of 2011; however, early adoption is permitted. The Company believes that these new standards would have an impact on its consolidated financial statements in the future were it to enter into an arrangement with multiple deliverables.
In January 2010, the FASB issued amended standards that require additional fair value disclosures. These amended standards require disclosures about inputs and valuation techniques used to measure
65
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 1. Business Overview and Summary of Significant Accounting Policies (Continued)
fair value as well as disclosures about significant transfers, beginning in the first quarter of 2010. Additionally, these amended standards require presentation of disaggregated activity within the reconciliation for fair value measurements using significant unobservable inputs (Level 3), beginning in the first quarter of 2011. The Company does not believe that these new standards will have a material effect on its consolidated financial statements.
Note 2. Fair Value Measurements
The Company categorizes assets and liabilities recorded at fair value in its consolidated balance sheets based upon the level of judgment associated with inputs used to measure their value. Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. The Company uses valuation techniques that maximize the use of observable inputs and minimize the use of unobservable inputs when determining fair value and then ranks the estimated values based on the reliability of the inputs used following the fair value hierarchy set forth by the FASB. The three levels of the FASB fair value hierarchy are as follows:
- •
- Level 1—Quoted prices in active markets for identical
assets or liabilities.
- •
- Level 2—Observable inputs other than Level 1
prices such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for
substantially the full term of the assets or liabilities.
- •
- Level 3—Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
The determination of a financial instrument's level within the fair value hierarchy is based on an assessment of the lowest level of any input that is significant to the fair value measurement. The Company utilizes the market approach to measure fair value for its financial assets and liabilities. The market approach uses prices and other relevant information generated by market transactions involving identical or comparable assets or liabilities.
The following tables present a summary of the Company's assets that are measured at fair value on a recurring basis as of December 31, 2009 and 2008 (in thousands):
| |
Fair Value Measurements as of December 31, 2009 |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Total | Level 1 | Level 2 | Level 3 | |||||||||
Cash and cash equivalents |
$ | 15,938 | $ | 15,938 | $ | — | $ | — | |||||
Investment securities |
27,451 | — | 27,451 | — | |||||||||
|
$ | 43,389 | $ | 15,938 | $ | 27,451 | $ | — | |||||
66
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 2. Fair Value Measurements (Continued)
| |
Fair Value Measurements as of December 31, 2008 |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Total | Level 1 | Level 2 | Level 3 | |||||||||
Cash and cash equivalents |
$ | 44,144 | $ | 44,144 | $ | — | $ | — | |||||
Investment securities |
28,611 | — | 28,611 | — | |||||||||
|
$ | 72,755 | $ | 44,144 | $ | 28,611 | $ | — | |||||
As of December 31, 2009 and 2008, the Company's cash, cash equivalents and investment securities are recorded at fair value as determined through market prices and other observable and corroborated sources. At December 31, 2009 the cash and cash equivalents balance consists of $491,000 in cash and $15,447,000 in money market funds. Investment securities are comprised of corporate debt securities and federal government and agency securities. See Note 3 below for further details on investment securities.
When the estimated fair value of a security is below its carrying value, the Company evaluates whether it is more likely than not that it will be required to sell the security before its anticipated recovery in market value and whether evidence indicating that the cost of the investment is recoverable within a reasonable period of time outweighs evidence to the contrary. The Company also evaluates whether or not it intends to sell the investment. If the impairment is considered to be other-than-temporary, the security is written down to its estimated fair value. In addition, the Company considers whether credit losses exist for any securities. A credit loss exists if the present value of cash flows expected to be collected is less than the amortized cost basis of the security. Other-than-temporary declines in estimated fair value and credit losses are charged to investment income. The Company has not deemed it necessary to record any charges related to impairments or other-than-temporary declines in the estimated fair values of its marketable debt securities or credit losses as of December 31, 2009.
Note 3. Investment Securities
The Company's investment securities, consisting of debt securities, are classified as available-for-sale. Unrealized holding gains or losses on these securities are included in other comprehensive loss on the consolidated balance sheets. Realized gains and losses and declines in value judged to be other-than-temporary (of which there have been none to date) on available-for-sale securities are included in interest income and other, net, in the consolidated statements of operations.
67
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 3. Investment Securities (Continued)
Investment securities consisted of the following at December 31, 2009 (in thousands):
| |
|
Gross Unrealized | |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Amortized Cost |
Estimated Fair Value |
|||||||||||||
| |
Gains | (Losses) | |||||||||||||
Type of security: |
|||||||||||||||
Corporate debt securities, with unrealized gains |
$ | 12,608 | $ | 8 | $ | — | $ | 12,616 | |||||||
Corporate debt securities, with unrealized losses |
5,565 | — | (13 | ) | 5,552 | ||||||||||
Federal government and agency securities, with unrealized gains |
1,509 | 1 | — | 1,510 | |||||||||||
Federal government and agency securities, with unrealized losses |
7,786 | — | (13 | ) | 7,773 | ||||||||||
|
$ | 27,468 | $ | 9 | $ | (26 | ) | $ | 27,451 | ||||||
Net unrealized loss |
$ | (17 | ) | ||||||||||||
Maturity: |
|||||||||||||||
Less than one year |
$ | 23,199 | $ | 23,198 | |||||||||||
Due in 1 - 2 years |
4,269 | 4,253 | |||||||||||||
|
$ | 27,468 | $ | 27,451 | |||||||||||
Investment securities consisted of the following at December 31, 2008 (in thousands):
| |
|
Gross Unrealized | |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Amortized Cost |
Estimated Fair Value |
|||||||||||||
| |
Gains | (Losses) | |||||||||||||
Type of security: |
|||||||||||||||
Corporate debt securities, with unrealized gains |
$ | 10,706 | $ | 26 | $ | — | $ | 10,732 | |||||||
Corporate debt securities, with unrealized losses |
13,261 | — | (382 | ) | 12,879 | ||||||||||
Federal government and agency securities, with unrealized gains |
4,998 | 2 | — | 5,000 | |||||||||||
|
$ | 28,965 | $ | 28 | $ | (382 | ) | $ | 28,611 | ||||||
Net unrealized loss |
$ | (354 | ) | ||||||||||||
Maturity: |
|||||||||||||||
Less than one year |
$ | 27,912 | $ | 27,561 | |||||||||||
Due in 1 - 2 years |
1,053 | 1,050 | |||||||||||||
|
$ | 28,965 | $ | 28,611 | |||||||||||
68
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 4. Facilities and Equipment
Facilities and equipment consisted of the following (in thousands):
| |
December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | ||||||
Lab equipment |
$ | — | $ | 574 | ||||
Office furniture and fixtures |
681 | 681 | ||||||
Computer equipment and software |
659 | 691 | ||||||
Leasehold improvements |
78 | 496 | ||||||
|
1,418 | 2,442 | ||||||
Less: accumulated depreciation |
(1,199 | ) | (1,319 | ) | ||||
Total |
$ | 219 | $ | 1,123 | ||||
Certain facilities and equipment balances for the Company's lab in South San Francisco, CA, were deemed to be impaired during 2009. Additionally, the Company recognized impairment on dedicated manufacturing equipment in 2009. Refer to Note 13 below for details on the impaired assets. Depreciation expense on facilities and equipment totaled $255,000, $388,000 and $311,000 for the years ended December 31, 2009, 2008 and 2007, respectively.
Note 5. Accrued Liabilities
Accrued liabilities consist of the following (in thousands):
| |
December 31, 2009 |
December 31, 2008 |
|||||
|---|---|---|---|---|---|---|---|
Clinical trials |
$ | 6,550 | $ | 8,266 | |||
Accrued expenses |
803 | 903 | |||||
Compensation |
326 | 1,164 | |||||
Severance |
— | 285 | |||||
|
$ | 7,679 | $ | 10,618 | |||
Note 6. Note Payable
On September 2, 2008, the Company entered into an Amended and Restated Loan and Security Agreement ("loan agreement"), with GE Business Financial Services Inc. (formerly known as Merrill Lynch Capital) and Silicon Valley Bank. The loan agreement amends and restates in its entirety the earlier Loan and Security Agreement dated as of October 25, 2006 ("original loan"), with the lenders, pursuant to which the Company obtained a $15,000,000 capital loan that was to mature on April 1, 2010.
The loan agreement provides for a $27,600,000 senior secured term loan facility ("loan facility") to be made available as follows: (i) an initial term loan advance in the amount of $17,600,000, which is comprised of (a) the outstanding principal balance of $7,600,000 remaining on the original loan and (b) an additional cash advance in the amount of $10,000,000 ("cash portion"), which was fully funded on September 2, 2008; and (ii) a second term loan advance in the amount of $10,000,000, which was fully funded on September 30, 2008. The cash portion of the initial term loan advance and the
69
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 6. Note Payable (Continued)
proceeds of the second term loan advance are being used to fund the Company's clinical trials and for general corporate purposes. The advances under the loan facility are repayable over 42 months, commencing on October 1, 2008. Interest on the advances is fixed at 7.8% per annum. Final loan payments in the amounts of $1,070,000 and $900,000 are due upon maturity or earlier repayment of the initial term loan advance and the second term loan advance, respectively. Additionally, as a condition to the amendment and restatement of the original loan, the Company agreed to modification of the final payment obligations under the original loan, pursuant to which the Company paid $600,000 to Silicon Valley Bank on September 2, 2008, the effective date of the loan facility, and will pay $675,000 to GE Business Financial Services on March 31, 2010. All final payment amounts are being accreted to the note payable balance over the term of the loan facility using the effective interest rate method and reflected as additional interest expense. All interest payable under the loan agreement and the full amount of the final payments must be paid upon any prepayment of a term loan advance. The loan facility is secured by a first lien on all of the non-intellectual property assets of the Company. At December 31, 2009, the outstanding principal balance under the loan facility was $18,747,000, net of discount of $1,641,000.
In connection with the loan agreement, the Company issued the lenders ten-year warrants to purchase an aggregate 219,920 shares of the Company's common stock at an exercise price of $4.297 per share. The fair value of the warrants using the Black-Scholes option-pricing model was approximately $928,000 based upon assumptions of expected volatility of 90%, a contractual term of ten years, an expected dividend rate of zero and a risk-free interest rate of 3.74%. The portion of the loan proceeds allocable to the warrants is approximately $806,000 based on the relative fair value of the warrants, which the Company recorded as discount to notes payable. The total of the final loan payments and the proceeds allocated to the warrants of approximately $4,051,000 are being amortized to interest expense using an effective interest rate of 13.8%. The warrants became exercisable upon issuance and are exercisable anytime during their term. In October 2009, Silicon Valley Bank exercised its warrants to purchase an aggregate of 197,169 common shares, net of equivalent shares at market to cover the total exercise price (see Note 9 for further details on this exercise). In November 2009, the Company amended the warrants held by GE Business Financial Services in connection with the 2006 and 2008 loan agreements to purchase an aggregate of 197,169 common shares to change the exercise price to $2.00 per share resulting in an increase in shareholders' equity and a charge to interest expense of $33,000.
The loan agreement contains restrictions on the Company's ability to, among other things, dispose of certain assets, engage in certain mergers and acquisition transactions, incur indebtedness, create liens on assets, make investments, pay dividends and repurchase stock. The loan agreement also contains covenants requiring the Company to maintain unrestricted cash in an amount equal to the lesser of (i) $17,940,000 or (ii) the outstanding aggregate principal balance of the term loans plus $4,000,000. The loan agreement contains events of default that include nonpayment of principal, interest or fees, breaches of covenants, material adverse changes, bankruptcy and insolvency events, cross defaults to any other indebtedness, material judgments, inaccuracy of representations and warranties, and events constituting a change of control. The occurrence of an event of default would increase the applicable rate of interest by 5% and could result in the acceleration of the Company's payment obligations under the loan agreement. The Company was in compliance with all loan covenants as of December 31, 2009.
70
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 6. Note Payable (Continued)
Maturities of the note payable and capital leases as of December 31, 2009 are as follows (see Note 8 for additional capital lease details) (in thousands):
Year
|
Capital Leases |
Note Payable |
Total | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
2010 |
$ | 38 | $ | 8,561 | $ | 8,599 | ||||
2011 |
— | 7,886 | 7,886 | |||||||
2012 |
— | 3,941 | 3,941 | |||||||
Thereafter |
1,485 | — | 1,485 | |||||||
|
1,523 | 20,388 | 21,911 | |||||||
Less: discount |
— | (1,641 | ) | (1,641 | ) | |||||
|
$ | 1,523 | $ | 18,747 | $ | 20,270 | ||||
Note 7. Common Stock Purchase Agreement
On August, 19, 2009, the Company entered into an equity line of credit arrangement with Azimuth Opportunity Ltd. ("Azimuth") pursuant to a Common Stock Purchase Agreement, as amended by Amendment 1 dated November 20, 2009, ("Purchase Agreement"), which provided that, upon the terms and subject to the conditions set forth therein, Azimuth was committed to purchase up to $60 million shares of the Company's common stock over the 18-month term of the Purchase Agreement. Under the Purchase Agreement, the Company could sell to Azimuth up to 6,955,606 shares of its common stock (the "Trading Market Limit").
On November 23, 2009, the Company completed a draw down and sale to Azimuth under the Purchase Agreement of 3,465,878 common shares, for net proceeds of $7,284,000. On December 22, 2009, the Company completed a second draw down and sale of 3,489,728 common shares for net proceeds of $6,439,000. With the completion of the second draw, the Company sold to Azimuth the maximum aggregate number of shares that could be sold under the Trading Market Limit. As a consequence of reaching the Trading Market Limit, the Purchase Agreement automatically terminated by its terms on December 22, 2009. The common stock sold to Azimuth has been registered with the Securities and Exchange Commission ("SEC").
Note 8. Commitments and Contingencies
The Company entered into a picoplatin active pharmaceutical ingredient ("API") commercial supply agreement with W.C. Heraeus ("Heraeus") in March 2008. Under this agreement Heraeus will produce picoplatin API to be used for preparing picoplatin finished drug product for commercial use. Manufacturing services are provided on a purchase order, fixed-fee basis, subject to certain purchase price adjustments and minimum quantity requirements. The costs to Heraeus for the purchase and set-up of dedicated manufacturing equipment ("equipment") as required under the commercial supply agreement will be repaid by the Company in the form of a surcharge on an agreed upon amount of the picoplatin API ordered and delivered on or before December 31, 2013. If the Company orders and takes delivery of less than the agreed upon amount of picoplatin API through December 31, 2013, it will be obligated to pay the balance of the equipment cost as of that date. Heraeus completed construction of the equipment as of December 31, 2009, at a total cost of approximately $1,485,000. The Company determined that the equipment should be accounted for as a capital lease and
71
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 8. Commitments and Contingencies (Continued)
accordingly recognized an asset and long term obligation for the equipment of $1,485,000 on its consolidated balance sheet as of December 31, 2009. The Company will reflect the surcharge payments as reductions to the capital lease balance outstanding, and will accrete a finance charge to interest expense as specified under the agreement. The Company does not anticipate beginning production of commercial supplies of picoplatin API, thereby utilizing the dedicated equipment, before 2011 and has, therefore, classified the obligation as long-term. Due to the delay in the Company's plans for the commercialization of picoplatin, which it does not anticipate will occur before 2011, the Company determined that its capital lease asset for equipment under the Heraeus agreement was impaired as of December 31, 2009 and therefore recognized an impairment charge of $1,485,000 in the consolidated statement of operations for 2009.
The Company leases the office space for its principal locations under various leasing arrangements. The Company's headquarters are located in South San Francisco, California, where it leases approximately 17,000 square feet of office space under a lease agreement that expires on July 10, 2011. Base rental payments under this lease are subject to annual adjustment based on the Consumer Price Index in the San Francisco metropolitan market (CPI-SFMM) and a one-time adjustment for reimbursement for tenant improvements. Initial monthly base rent of $45,200 was increased by $1,400, $1,400 and $1,500 following the adjustments for the 2007, 2008, and 2009 CPI-SFMM, respectively, for a current base rate of $49,500 per month. In December 2007, the Company accessed approximately $251,000 for tenant improvements that resulted in a further $5,400 increase for total base rent of $54,900 per month as of December 31, 2009. Additional rental payments under this lease are paid based on the Company's share of operating expenses of the project in which the leased facilities are located.
On February 12, 2010, the Company entered into a sublease agreement with Veracyte, Inc., pursuant to which Veracyte will sublease, effective March 1, 2010, approximately 11,000 square feet of the Company's 17,000 square feet of office space, located in South San Francisco, California. Base rent for this subleased space is $17,600 per month. On September 1, 2010, the subleased space will expand to encompass the entire 17,000 square feet of office space. After delivery of this expanded sublease space, base rent will be $28,124 per month until expiration of the sublease on July 10, 2011, at which time Veracyte will lease the office space directly from the landlord. Additional rent under the sublease will be payable monthly to the Company by Veracyte, based on Veracyte's share of operating expenses attributable to the subleased office space. The Company's sublease with Veracyte does not modify or limit the terms and conditions of the Company's original lease with the landlord, or waive any rights or remedies of the landlord, except that the landlord releases the Company from obligations under the original lease to remove alterations or repair or restore the office space upon expiration of the original lease or following a casualty occurring during the term of the original lease. The Company currently is evaluating its facility space requirements and plans to explore alternative sites in the San Francisco area for executive office space.
The Company also leases approximately 21,000 square feet of office space in Seattle, Washington, under an amended lease that expires December 31, 2010. The lease may be renewed for one five-year term, effective upon notice by the Company of its intent to renew six months prior to the expiration of the current term. Monthly base rent on this property is $45,000 and additional rent payments under this lease are paid based on the Company's share of operating expenses of the facility.
72
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 8. Commitments and Contingencies (Continued)
Total rent expense under non-cancelable operating leases was approximately $1,399,000, $1,485,000 and $1,355,000 for the years 2009, 2008 and 2007, respectively. The Company recognizes rent expense on a straight-line basis over the term of each lease, including any periods of free rent. Minimum lease payments under non-cancelable operating leases as of December 31, 2009 were as follows (in thousands):
Year
|
|
|||
|---|---|---|---|---|
2010 |
$ | 1,212 | ||
2011 |
390 | |||
2012 |
4 | |||
Thereafter |
— | |||
Total minimum lease payments |
$ | 1,606 | ||
At December 31, 2009 and 2008, the Company had restricted cash of $281,000 in the form of certificates of deposit. The certificates of deposit serve as collateral for standby letters of credit issued by Silicon Valley Bank on behalf of the Company.
Note 9. Shareholders' Equity
Common Stock Transactions: In 2009, the Company issued 6,955,606 shares of common stock under an equity line of credit arrangement with Azimuth. See Note 7 above for further details on the Purchase Agreement and 2009 stock transactions under this arrangement.
In connection with the Company's 2007 public offering, the Company issued approximately 11,849,000 shares of common stock at a purchase price of $6.33 per share. Net proceeds of the public offering, after payment of underwriters' discounts and commissions and offering expenses, were approximately $69,946,000.
During November 2009, the Company issued approximately 63,000 shares of common stock upon the exercise of warrants acquired by it in conjunction with 2006 financing activities. The exercise was for approximately 214,000 underlying shares of common stock, net of equivalent shares at market to cover the total exercise price. The Company also issued approximately 82,000 shares of common stock to Silicon Valley Bank upon the exercise of warrants it acquired in connection with the Company's 2006 and 2008 loan facilities as discussed in Note 6 above. The exercise was for approximately 197,000 underlying shares of common stock, net of equivalent shares at market to cover the total exercise price.
The Company received net proceeds from the issuance of shares of common stock related to the exercise of employee stock options in each of the years ended December 31, 2009, 2008 and 2007. Refer to Note 10 for further details on option exercises and share-based compensation.
In 2009, the Company issued approximately 88,000 shares of common stock upon the vesting and release of restricted stock units awarded to employees as part of its 2008 Incentive Plan. Each unit converted to one share of common stock. See Note 10 for further details on share-based compensation.
Preferred Stock Transactions: The Company had approximately 205,000 shares of Series 1 Convertible Exchangeable Preferred Stock ("Series 1 preferred stock") outstanding at December 31, 2009. Holders of the Series 1 preferred stock are entitled to receive an annual cash dividend of $2.4375
73
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 9. Shareholders' Equity (Continued)
per share if declared by the Board, payable semi-annually on June 1 and December 1. Dividends are cumulative. Each share of Series 1 preferred stock is convertible into 0.19 share of common stock, subject to adjustment in certain events. The Series 1 preferred stock is redeemable at the option of the Company at $25.00 per share. Holders of Series 1 preferred stock have no voting rights, except in limited circumstances. Dividends of $500,000 were paid in each of the years 2009, 2008 and 2007, respectively.
The Company's board of directors may, without further action by the shareholders, issue preferred stock in one or more series and fix the rights and preferences thereof, including dividend rights, dividend rates, conversion rates, voting rights, terms of redemption, redemption price or prices, liquidation preferences and the number of shares constituting any series or the designations of such series.
Warrants: The Company had outstanding warrants to purchase an aggregate of 5,085,000 and 6,060,000 shares of the Company's common stock as of December 31, 2009 and 2008, respectively. The weighted average exercise price of warrants outstanding was $4.83 and $5.51 per share for 2009 and 2008, respectively.
The detail of the warrants outstanding as of December 31, 2009 and 2008 is as follows (in thousands, except exercise price):
| |
|
|
|
|
Warrants Outstanding at December 31 |
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Issuance Date |
Expiration Date |
Exercise Price |
Exercise Dates |
||||||||||||||||||
| |
2008 | Exercised | Expired | 2009 | ||||||||||||||||||
Bank loan |
||||||||||||||||||||||
Lenders(1) |
9/2008 | 9/2018 | 2.00 | 10/2009 | 220 | 110 | — | 110 | (2) | |||||||||||||
Lenders(1) |
10/2006 | 10/2016 | 2.00 | 10/2009 | 174 | 87 | — | 87 | (2) | |||||||||||||
2006 financing |
||||||||||||||||||||||
Investors |
4/2006 | 4/2011 | 4.62 | 11/2009 | 4,231 | 195 | — | 4,036 | (3) | |||||||||||||
Investors (bridge notes) |
2/2006 | 2/2011 | 4.62 | 11/2009 | 412 | 19 | 393 | (3) | ||||||||||||||
Placement Agent |
4/2006 | 4/2011 | 4.62 | — | 139 | — | — | 139 | (3) | |||||||||||||
2005 financing |
||||||||||||||||||||||
Investors |
3/2005 | 9/2010 | 9.54 | — | 278 | — | — | 278 | ||||||||||||||
Placement Agent |
3/2005 | 9/2010 | 9.54 | — | 42 | — | — | 42 | ||||||||||||||
2004 financing |
||||||||||||||||||||||
Investors |
2/2004 | 2/2009 | 11.58 | — | 558 | — | 558 | — | ||||||||||||||
Placement Agent |
2/2004 | 2/2009 | 33.24 | — | 6 | — | 6 | — | ||||||||||||||
|
6,060 | 411 | 564 | 5,085 | ||||||||||||||||||
- (1)
- The
exercises in 2009 were at the original exercise price of $4.30; the remaining warrants were amended in November 2009 to change the price to $2.00 per
share.
- (2)
- See
Note 6 for additional details.
- (3)
- Issued in connection with the bridge notes that were issued as part of the 2006 financing.
74
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 10. Share-based Compensation
As of December 31, 2009, the Company's Amended and Restated 2004 Incentive Compensation Plan (the "2004 Plan") was the only equity compensation plan under which awards were available for grant. The Company's 1991 Stock Option Plan for Non-Employee Directors (the "Directors Plan") terminated on March 31, 2005, and no further options can be granted under that plan. The Company's Restated 1994 Stock Option Plan (the "1994 Plan") terminated on February 17, 2004, and no further options can be granted under that plan. Although no Company securities are available for issuance under the Directors Plan or the 1994 Plan, options granted prior to termination of those plans continue in effect in accordance with their terms.
The 2004 Plan, as amended and restated on June 14, 2007, authorizes the Company's board of directors or a committee appointed by the board of directors to grant share-based awards for an aggregate 7,634,000 shares of common stock. The 2004 Plan contains an evergreen provision pursuant to which the number of shares available under the plan automatically increase each year, beginning in 2008, according to certain limits set forth in the plan. The aggregate of 7,634,000 shares reflects an increase of 1,734,000 shares on January 1, 2009, and an increase of 1,733,000 shares on January 1, 2008, pursuant to the operation of the evergreen provision. The 2004 Plan allows for the issuance of incentive stock options, nonqualified stock options, restricted stock and RSUs to employees, officers, directors, agents, consultants, advisors and independent contractors of the Company, subject to certain restrictions. All option grants expire ten years from the date of grant, except in the event of earlier termination of employment or service. Option grants to employees with less than one year of service generally become exercisable at a rate of 25% after one year from the grant date and then in monthly increments at a rate of 1/36th of the remaining balance over the following three years. Option grants to employees with at least one year of service and employees receiving promotions become exercisable at a rate of 1/48th per month over four years from the grant date.
Share-based compensation cost is measured at the grant date, based on the fair value of the award, and is recognized as an expense over the employee's requisite service period, generally the vesting period of the equity grant. The Company utilizes the Black-Scholes pricing model to estimate the fair value of each option award granted. The Company recorded expense for share-based compensation, not including expense for awards granted to non-employee consultants, for the periods presented are as follows (in thousands):
| |
Years Ended December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | ||||||||
Research and development expense |
$ | 2,253 | $ | 1,462 | $ | 1,265 | |||||
General and administrative expense |
4,821 | 5,542 | 4,149 | ||||||||
Total |
$ | 7,074 | $ | 7,004 | $ | 5,414 | |||||
The share-based compensation expense for the twelve months ended December 31, 2009 includes the grant of stock options during the first quarter of 2009 to a Company officer to purchase an aggregate 200,000 shares of common stock over a four year vesting period and a grant in the fourth quarter of 2009 to a Company officer for 100,000 RSUs that vest annually over four years, subject to accelerated vesting based upon the achievement of specific performance goals.
Certain options that were granted to Company officers during 2006 and 2007 vest 50% in equal monthly installments over four years from the date of grant and vest another 50% on the seven-year
75
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 10. Share-based Compensation (Continued)
anniversary of the date of grant, subject to accelerated vesting of up to 25% of such portion of the options, based on the Company's achievement of annual performance goals established under its annual incentive plan, at the discretion of the equity awards subcommittee of the Company's board of directors. Based on the overall achievement of corporate goals in 2007 and 2008, the equity awards subcommittee accelerated vesting with respect to 20% of the shares subject to the seven-year vesting schedule in the second quarter of 2007 and both the first and fourth quarters of 2008. There was no acceleration in 2009, resulting in cumulative accelerated vesting of 60% of the shares subject to the seven-year vesting schedule as of December 31, 2009.
During 2008, the Company awarded 91,974 RSUs under the 2004 Plan to non-officer employees as an incentive for future performance (the "2008 Incentive Plan"). An additional 4,000 RSUs were awarded in 2009 under this incentive program. Upon vesting, each RSU is payable with one share of the Company's common stock. The average fair value of the RSUs was $3.13 per unit, or approximately $299,000 in total, based upon the closing market price of the Company's common stock on the award dates. The RSUs vest based on the achievement of certain performance milestones estimated to be achieved during 2009 and 2010. As of December 31, 2009, all three performance milestones had been achieved and 100% of the then outstanding RSUs under the 2008 Incentive Plan, approximately 88,000, vested and were released. Total share-based compensation expense recognized in 2009 for the 2008 Incentive Plan was $279,000.
On July 23, 2009 the Company awarded an additional 290,000 RSUs under the 2004 Plan to non-officer employees as an incentive for future performance (the "2009 Incentive and Retention Plan"). The fair value of the RSUs was $7.34 per unit, or approximately $2,132,000 in total, based upon the closing market price of the Company's common stock on the award date. The RSUs vest based on the achievement of certain performance milestones expected to be achieved during 2010, subject to acceleration under specific conditions defined in the award agreement. Based on events occurring in the first quarter of 2010 (refer to Note 15 below for details of subsequent events), vesting of approximately 157,000 RSUs awarded under the 2009 Incentive and Retention Plan was accelerated. The Company determined the expense for these awards on a straight-line basis from grant date through the accelerated vesting date and recognized share-based compensation expense of $1,582,000 for the 2009 portion.
On July 11, 2009, a Company director was awarded 170,000 RSUs as compensation for consulting services. The RSUs vest 50% on each of the first two anniversaries of the grant and are subject to accelerated vesting upon the achievement of a specific performance milestone defined in the agreement. The fair value of the award was approximately $311,000 at December 31, 2009, and will be re-measured at each reporting date as it is accounted for as a non-employee award. As of December 31, 2009, the Company determined that the performance milestone is probable of being achieved in 2010 and is thus recognizing share-based compensation expense for the fair value of the award on a pro-rata basis through the estimated date of achievement. Total share-based compensation expense recognized in 2009 for this award was $134,000.
On October 6, 2009, a Company officer was awarded 100,000 RSUs as incentive compensation. The RSUs vest 25% on each of the first four anniversaries of the grant, subject to continuous employment, and are subject to accelerated vesting under specific conditions defined in the award agreement. The fair value of the award was $727,000 based upon the closing market price of the Company's common stock on the award date. As of December 31, 2009, the Company determined that
76
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 10. Share-based Compensation (Continued)
it is probable that the award would be accelerated per the terms of the agreement and is thus recognizing share-based compensation expense for the award on a pro-rata basis through the estimated date of vesting. Total share-based compensation expense recognized in 2009 for this award was $157,000.
As of December 31, 2009, there were approximately 1,292,000 shares of common stock available for issuance as new awards under the 2004 Plan. As of January 1, 2010, 2,104,000 additional shares of common stock became available under the 2004 Plan due to the automatic annual increase under the evergreen provision. Accordingly, as of January 1, 2010, an aggregate of 3,396,000 shares of common stock were available for issuance as new awards under the 2004 Plan.
During 2008, the Company modified certain stock options which had been granted to an officer of the Company upon his conversion to consultant status effective with his termination of employment such that any vested options would remain exercisable until the earliest of (a) thirty days after the Company receives approval from the FDA of its NDA for picoplatin, (b) twenty-four months after termination of service, and (c) the option expiration date (as defined in the 2004 Plan) for the options. As a result of this modification, the Company incurred expense of $50,000 for the outstanding stock options during the fourth quarter of 2008.
The Company records compensation expense for employee stock options based on the estimated fair value of the options on the date of grant using the Black-Scholes option-pricing model. This fair value is amortized on a straight-line basis over the requisite service periods for the grants, which is generally the vesting period. The remaining unrecognized compensation cost related to unvested awards at December 31, 2009, was approximately $8,312,000 and the weighted-average period of time over which this cost will be recognized is 2.1 years. The Company uses historical data, and other related information as appropriate, to estimate the expected price volatility, the expected option life and the expected forfeiture rate. The risk-free rate is based on the U.S. Treasury yield curve in effect at the time of the grant. The weighted-average fair value per share of the Company's stock options granted to employees was estimated to be $2.68, $2.20, and $5.83 for the years ended December 31, 2009, 2008 and 2007, respectively, using the Black-Scholes model with the following weighted-average assumptions:
| |
Years Ended December 31, |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | |||||||
Expected term (in years) |
5.9 | 4.6 | 6.4 | |||||||
Risk-free interest rate |
2.32 | % | 2.26 | % | 5.03 | % | ||||
Expected stock price volatility |
95 | % | 90 | % | 101 | % | ||||
Expected dividend rate |
0 | % | 0 | % | 0 | % | ||||
77
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 10. Share-based Compensation (Continued)
The Company issues previously authorized but unissued shares of common stock upon exercise of stock options. A summary of option activity as of December 31, 2009 and changes during the three years then ended are as follows (shares and intrinsic value in thousands):
| |
Number of Shares |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Term in Years |
Aggregate Intrinsic Value |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Outstanding at December 31, 2006 |
1,660 | ||||||||||||
Granted |
3,283 | $ | 5.93 | ||||||||||
Exercised |
(6 | ) | 3.66 | ||||||||||
Forefeited/cancelled/expired |
(287 | ) | 13.14 | ||||||||||
Outstanding at December 31, 2007 |
4,650 | 7.12 | 8.6 | $ | 1,773 | ||||||||
Exercisable at December 31, 2007 |
1,251 | 9.66 | 7.2 | $ | 633 | ||||||||
Granted |
1,599 | 3.27 | |||||||||||
Exercised |
(25 | ) | 3.61 | ||||||||||
Forefeited/cancelled/expired |
(331 | ) | 6.97 | ||||||||||
Outstanding at December 31, 2008 |
5,893 | 6.10 | 7.9 | $ | 103 | ||||||||
Exercisable at December 31, 2008 |
2,583 | 7.69 | 6.8 | $ | — | ||||||||
Granted |
395 | 3.53 | |||||||||||
Exercised |
(202 | ) | 4.06 | ||||||||||
Forefeited/cancelled/expired |
(480 | ) | 5.74 | ||||||||||
Outstanding at December 31, 2009 |
5,606 | 6.02 | 7.0 | $ | 33 | ||||||||
Exercisable at December 31, 2009 |
3,363 | $ | 7.06 | 6.4 | $ | 7 | |||||||
Information relating to stock options outstanding and exercisable at December 31, 2009 is as follows (shares in thousands):
| |
Options Outstanding | Options Exercisable | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Range of Exercise Prices
|
Number of Shares |
Weighted Average Remaining Life in Years |
Weighted Average Exercise Price |
Number of Shares |
Weighted Average Exercise Price |
|||||||||||
$1.72 - $3.66 |
1,488 | 7.6 | $ | 2.63 | 613 | $ | 3.18 | |||||||||
3.72 - 4.70 |
955 | 7.6 | 4.30 | 436 | 4.21 | |||||||||||
4.80 - 6.48 |
1,080 | 6.9 | 6.01 | 763 | 6.01 | |||||||||||
6.68 - 7.17 |
1,032 | 7.4 | 6.89 | 686 | 6.91 | |||||||||||
7.27 - 15.00 |
958 | 6.0 | 9.07 | 772 | 9.31 | |||||||||||
15.42 - 109.50 |
93 | 1.7 | 36.88 | 93 | 36.88 | |||||||||||
|
5,606 | 7.0 | 6.02 | 3,363 | 7.06 | |||||||||||
78
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 10. Share-based Compensation (Continued)
No income tax benefit has been recorded for share-based compensation expense as the Company has recorded a full valuation allowance and management has concluded it is more likely than not that the Company's net deferred tax assets will not be realized.
Cash proceeds and intrinsic value related to total stock options exercised during the years ended December 31, 2009, 2008 and 2007 are provided in the following table (in thousands):
| |
Years Ended December 31, |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | |||||||
Proceeds from stock options exercised |
$ | 820 | $ | 91 | $ | 22 | ||||
Intrinsic value of stock options exercised |
$ | 702 | $ | 38 | $ | 6 | ||||
In connection with various consulting and service contracts, the Company has granted stock options to non-employees. The fair value of these options is re-measured quarterly using the Black-Scholes option-pricing model and the total value of the stock options is recognized as expense over the service period. Stock options to purchase 1,000, 40,000 and 44,998 shares of common stock were granted to non-employees during 2009, 2008 and 2007, respectively. The Company recorded compensation expense of $88,000, $119,000 and $55,000 during 2009, 2008 and 2007, respectively, for non-employee options.
Note 11. Picoplatin License and Amendment
The Company has entered into an exclusive worldwide license, as amended, with Genzyme Corporation (successor to AnorMED, Inc.) for the development and commercial sale of picoplatin. Under that license, the Company is solely responsible for the development and commercialization of picoplatin. Genzyme retains the right, at the Company's cost, to prosecute its patent applications and maintain all licensed patents. The parties executed the license agreement in April 2004, at which time the Company paid a one-time up-front payment of $1,000,000 in common stock and $1,000,000 in cash. The original agreement excluded Japan from the licensed territory and provided for $13,000,000 in development and commercialization milestones, payable in cash or a combination of cash and common stock, and a royalty rate of up to 15% on product net sales after regulatory approval. The parties amended the license agreement on September 18, 2006, modifying several key financial terms and expanding the licensed territory to include Japan, thereby providing the Company worldwide rights. In consideration of the amendment, the Company paid Genzyme $5,000,000 in cash on October 12, 2006 and an additional $5,000,000 in cash on March 30, 2007. The amendment eliminated all development milestone payments to Genzyme. Genzyme remains entitled to receive up to $5,000,000 in commercialization milestones upon the attainment of certain levels of annual net sales of picoplatin after regulatory approval. The amendment also reduced the royalty payable to Genzyme to a maximum of 9% of annual net product sales and eliminated the sharing of sublicense revenues with Genzyme.
79
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 11. Picoplatin License and Amendment (Continued)
The Company accounted for all payments made in consideration of the picoplatin license, as amended, by capitalizing them as an intangible asset. The Company's capitalization of the total $12,000,000 of picoplatin license payments is based on the Company's reasonable expectation at the time of acquisition and through the date of the amendment that the intravenous formulation of picoplatin, as it existed at the time of the acquisition of the picoplatin license and the license amendment, would be used in research and development ("R&D") projects and therefore had alternative future uses in the treatment of different cancer indications. At the time of acquisition, the Company planned to use intravenous picoplatin in a Phase 2 clinical trial in patients with small cell lung cancer and reasonably expected that the intravenous formulation could be used in additional, then identifiable R&D projects in the form of clinical trials for other solid tumor cancer indications, such as prostate and colorectal cancers.
The Company, at the time of acquisition of the picoplatin license, reasonably anticipated using intravenous picoplatin in clinical trials that could be conducted during the remaining term of the primary patent, which is active through 2016. The Company concluded that the twelve years remaining for the primary patent term was the appropriate useful life for the picoplatin intangible asset and is amortizing the initial $2,000,000 license payment over this twelve-year useful life beginning in April 2004. The Company concluded that no change in the twelve-year useful life of the picoplatin intangible asset occurred as a result of the 2006 license amendment and is, therefore, continuing to amortize the initial $2,000,000 license payment over the twelve-year useful life and is amortizing the license amendment payment of $10,000,000 over the remainder of the twelve-year useful life of the picoplatin intangible asset.
The Company reviews its long-lived assets for possible impairment whenever significant events indicate such impairment may have occurred. In November 2009, the Company announced that its pivotal Phase 3 SPEAR trial of picoplatin in the second-line treatment of patients with small cell lung cancer did not meet its primary endpoint of overall survival. The Company considers this event to be a trigger for testing its picoplatin intangible asset for possible impairment; however, upon review of the expected future undiscounted net cash flows identifiable to the picoplatin license, the Company determined that the picoplatin intangible is recoverable and that no impairment occurred.
Licensed products consists of the picoplatin amortizable intangible asset with a gross amount of $12,000,000 less accumulated amortization of $4,408,000 and $3,193,000 at December 31, 2009 and 2008, respectively. The Company recognized amortization expense of $1,215,000 in each of the years ended December 31, 2009, 2008 and 2007. The estimated annual amortization expense for licensed products is approximately $1,215,000 for each of the years 2010 through 2014.
80
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 12. Income Taxes
Temporary differences and carryforwards giving rise to deferred tax assets (liabilities) were as follows (in thousands):
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | |||||
Net operating loss carryforwards |
$ | 45,777 | $ | 38,104 | |||
Research and experimentation credit carryforwards |
2,932 | 2,030 | |||||
Capitalized research and development |
22,771 | 19,768 | |||||
Stock compensation |
4,569 | 3,362 | |||||
Property and equipment |
191 | 6 | |||||
Other |
3,287 | 1,768 | |||||
Net deferred tax assets |
79,527 | 65,038 | |||||
Deferred tax assets valuation allowance |
(79,527 | ) | (65,038 | ) | |||
Net deferred income taxes |
$ | — | $ | — | |||
The Company has established a valuation allowance equal to the amount of its net deferred tax assets because the Company has not had taxable income since its inception and significant uncertainty exists regarding the ultimate realization of its deferred tax assets. Accordingly, no tax benefits have been recorded in the accompanying statements of operations. The valuation allowance increased $14,489,000 in 2009 and increased $17,809,000 in 2008.
The Company's total tax provision of zero differs from the expected tax benefit calculated as a product of the federal statutory rate of 34% and the book loss by approximately $15,543,000 primarily due to permanent differences such as meals and entertainment expense, the research and experimentation credit, and stock option adjustments, as well as the change in the Company's valuation allowance.
In April 2006, the Company experienced a significant change to its capital structure which resulted in an ownership change, as defined under Section 382 of the Internal Revenue Code ("IRC"). Consequently, the amount of net operating loss carryforwards and research and experimentation credit carryforwards available to be used in future years are limited under IRC Sections 382 and 383, respectively ("Section 382/383 limitation"). The preliminary calculation of this limitation, as disclosed in the Company's consolidated financial statements for the year ended December 31, 2008, resulted in the loss of approximately $93,300,000 (approximately $31,700,000 in tax benefits) of the Company's net operating loss carryforwards. During 2009, the Company performed an additional analysis of the Section 382 limitation and revised the amount of net operating loss carryforwards that would be lost to approximately $96,900,000 (approximately $32,900,000 in tax benefits). Accordingly, the deferred tax asset and related valuation allowance associated with these carryforwards were reduced in 2006 initially by approximately $40,800,000 and by a revised amount of approximately $41,700,000 as of December 31, 2009.
The preliminary calculation of the Section 383 limitation, which was not revised, resulted in the loss of approximately $9,100,000 of the Company's research and development credit carryforwards.
At December 31, 2009, the Company has total net operating loss carryforwards of approximately $134,638,000 for federal taxes (net of the impact of the above referenced change in ownership under IRC Section 382) and approximately $21,543,000 for state taxes, which expire from 2010 through 2029
81
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 12. Income Taxes (Continued)
and from 2015 through 2029, respectively. Research and experimentation credits expire from 2010 to 2029. Future changes in the Company's ownership could result in additional limitations on the Company's ability to utilize its remaining net operating loss carryforwards and research and experimentation credit carryforwards.
Approximately $21,343,000 of the Company's federal net operating loss carryforwards at December 31, 2009, result from deductions associated with the exercise of non-qualified employee stock options, the realization of which would result in a credit to shareholders' equity.
The Company accounts for uncertain tax provisions as required under financial accounting rules issued by the FASB. Due to the Company's full valuation allowance against its deferred tax assets, coupled with the Section 382 limitation on prior years' net operating loss carryforwards (as discussed above), there are no material unrecognized tax benefits as of December 31, 2009 or December 31, 2008. Furthermore, the Company does not anticipate any significant changes in its unrecognized tax benefits over the next twelve months.
Historically, the Company has not incurred any material interest or penalties associated with tax matters and no material interest or penalties were recognized during the years ended December 31, 2009, 2008 or 2007. The Company has adopted a policy whereby amounts related to interest and penalties associated with tax matters are classified as income tax expense when incurred. The Company is subject to income taxes in the U.S. federal and various states jurisdictions. Tax regulations within each jurisdiction are subject to the interpretation of the related tax laws and regulations and require significant judgment to apply. The Company is no longer subject to tax examinations for years before 2006, except to the extent that it utilizes net operating losses or tax credit carryforwards that originated before 2006.
Note 13. Restructuring and Asset Impairment
Effective March 31, 2009, the Company implemented a strategic restructuring plan to refocus its cash resources on clinical and commercial development of picoplatin, resulting in the discontinuation of the Company's preclinical research operations and a reduction of its workforce by approximately 12%, or eight employees. The Company incurred severance charges totaling $296,000 related to the reduction in staff. All severance charges related to the restructuring have been paid as of December 31, 2009. The Company incurred additional charges totaling approximately $172,000 related to the closure of its lab facilities in South San Francisco, California. Of this amount, $6,000 was incurred as share-based compensation expense, $12,000 was a write-off of prepaid expenses, and $154,000 was incurred for contract and other termination costs. All outstanding liabilities for contract and termination costs have been paid as of December 31, 2009.
82
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 13. Restructuring and Asset Impairment (Continued)
The following table summarizes the impact of the restructuring charges through December 31, 2009 (in thousands):
Description
|
Initial Restructuring Charge March 31, 2009 |
Payment of Restructuring Obligations |
Accrued Restructuring Charge as of December 31, 2009 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Employee termination benefits |
$ | 296 | $ | (296 | ) | $ | — | ||||
Contract termination costs |
125 | (125 | ) | — | |||||||
Other termination costs |
47 | (47 | ) | — | |||||||
Subtotal |
172 | (172 | ) | — | |||||||
Total |
$ | 468 | $ | (468 | ) | $ | — | ||||
In conjunction with the decision to discontinue the Company's preclinical research operations, the Company recognized an asset impairment loss of $588,000 on certain facilities and equipment related to the lab in South San Francisco, California. The loss on the assets was determined based on estimates of potential sales values of used equipment. These impairment charges established new cost bases for the impaired assets, which are included in assets held for sale and reported in prepaid expenses and other current assets on the accompanying 2009 consolidated balance sheet. The remaining impaired assets held for sale were sold in January 2010.
The following table summarizes information related to the impairment charges (in thousands):
| |
So. San Francisco Lab Equipment & Leasehold Improvements |
|||
|---|---|---|---|---|
Impairment Loss |
$ | 588 | ||
Impaired Carrying Value Upon Restructuring March 31, 2009 |
$ | 57 | ||
Disposals of Assets |
(52 | ) | ||
Post Impairment Carrying Value as of December 31, 2009 |
$ | 5 | ||
Additionally, at December 31, 2009, the Company recognized an impairment charge of $1,485,000 for its dedicated manufacturing equipment capital lease asset. This impairment reduced the carrying value of the capital lease asset to zero as of December 31, 2009. See Note 8 for additional details.
83
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 14. Employee Benefit Plan
The Company sponsors a 401(k) plan that covers substantially all employees. In its sole discretion, the Company may make contributions to the plan on a percentage of participants' contributions. The Company made contributions of approximately $22,000, $19,000, and $15,000 for the years ended December 31, 2009, 2008 and 2007, respectively. The Company has no other post-employment or post-retirement benefit plans.
Note 15. Subsequent Events
On January 29, 2010, the Company approved a restructuring plan to conserve its capital resources, resulting in a reduction in the Company's workforce by approximately 57% to 22 employees, effective February 5, 2010. The Company currently estimates that it will incur total restructuring charges of approximately $1,200,000, primarily consisting of one-time employee termination benefits. All of the restructuring charges, except for related $174,000 share-based compensation expense, will result in future cash expenditures. The Company also recognized a $62,000 charge for impaired assets in connection with the restructuring. As a consequence of the restructuring, vesting was accelerated for approximately 130,000 RSUs issued under the 2009 Incentive and Retention Plan for the terminated employees in accordance with the original terms of the underlying RSU agreements. These RSUs were converted to common stock on a one-for-one basis in February 2010.
On February 23, 2010, the Company entered into an equity line of credit facility with Commerce Court. The facility provides that, upon the terms and subject to the conditions therein, Commerce Court is committed to purchase up to $20,000,000 worth of shares of the Company's registered common stock over approximately 18 months; provided, however, that in no event may the Company issue more than 8,423,431 shares of common stock, which is equal to one share less than 20% of the Company's outstanding common shares on the closing date of the facility, less 121,183 shares issued to Commerce Court as a commitment fee. The purchase price of the shares will be equal to the daily volume weighted average price over eight consecutive trading days or such other draw down period mutually agreed upon by the parties, less a discount ranging from 3.125% to 5.0%, based on the trading price of the Company's common stock. In addition, Commerce Court may not at any time acquire shares if, after giving effect to such purchase, Commerce Court would beneficially own 9.9% or more of the Company's outstanding common stock. Commerce Court is not obligated to purchase shares at prices below $1.00 per share. On March 15, 2010, the Company completed a draw down and sale of 4,229,000 shares of its common stock, at a price of approximately $1.49 per share, to Commerce Court under the equity line of credit facility. Net proceeds of approximately $6,154,000 were received, after deducting estimated offering expenses of approximately $166,000.
84
PONIARD PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 16. Condensed Quarterly Financial Data (Unaudited)
The following table presents summarized unaudited quarterly financial data (in thousands, except per share data):
| |
First Quarter |
Second Quarter |
Third Quarter |
Fourth Quarter |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
2009 |
||||||||||||||
Operating expenses |
$ | 12,244 | $ | 9,088 | $ | 9,198 | $ | 12,448 | ||||||
Net loss |
(12,950 | ) | (9,720 | ) | (9,877 | ) | (13,168 | ) | ||||||
Net loss applicable to common shares |
(13,075 | ) | (9,845 | ) | (10,002 | ) | (13,293 | ) | ||||||
Net loss per common share: |
||||||||||||||
Basic |
(0.38 | ) | (0.28 | ) | (0.29 | ) | (0.36 | ) | ||||||
Diluted |
(0.38 | ) | (0.28 | ) | (0.29 | ) | (0.36 | ) | ||||||
2008 |
||||||||||||||
Operating expenses |
$ | 10,519 | $ | 12,874 | $ | 12,407 | $ | 13,357 | ||||||
Net loss |
(9,855 | ) | (12,531 | ) | (12,237 | ) | (13,942 | ) | ||||||
Net loss applicable to common shares |
(9,980 | ) | (12,656 | ) | (12,362 | ) | (14,067 | ) | ||||||
Net loss per common share: |
||||||||||||||
Basic |
(0.29 | ) | (0.36 | ) | (0.36 | ) | (0.41 | ) | ||||||
Diluted |
(0.29 | ) | (0.36 | ) | (0.36 | ) | (0.41 | ) | ||||||
Note: Net loss per common share, basic and diluted, may not add to net loss per common share for the year due to rounding.
85
Item 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
On May 22, 2009, the Board of Directors of the Company, based on its Audit Committee's recommendation, dismissed KPMG LLP ("KPMG") as the Company's independent registered public accountants and approved the engagement of Ernst & Young LLP ("E&Y") to serve as the Company's independent registered public accountants for the fiscal year 2009.
The audit reports of KPMG on the consolidated financial statements of the Company for the years ended December 31, 2008 and 2007 did not contain an adverse opinion or disclaimer of opinion and were not qualified or modified as to uncertainty, audit scope or accounting principle, except for KPMG's report dated March 16, 2009, which contained an explanatory paragraph that cited certain conditions that raised substantial doubt about the Company's ability to continue as a going concern. The audit reports of KPMG on the effectiveness of internal control over financial reporting as of December 31, 2008 and 2007 did not contain any adverse opinion or disclaimer of opinion nor were they qualified or modified as to uncertainty, audit scope, or accounting principles.
During the Company's fiscal years ended December 31, 2008 and 2007 and the subsequent interim period through May 22, 2009, there were no disagreements with KPMG on any matter of accounting principles or practices, financial statement disclosure or auditing scope or procedure, which disagreements, if not resolved to KPMG's satisfaction, would have caused KPMG to make reference to the subject matter of such disagreements in connection with its reports on the Company's consolidated financial statements for such years.
There were no "reportable events," as defined in Item 304(a)(1)(v) of Regulation S-K, during the Company's fiscal years ended December 31, 2008 and 2007 and the subsequent interim period through May 22, 2009.
The Company provided KPMG with a copy of the above disclosures prior to its filing with the Securities and Exchange Commission ("SEC") and requested KPMG to furnish the Company with a letter addressed to the SEC stating whether or not KPMG agrees with the above statements. A copy of KPMG's letter dated May 26, 2009 was furnished to the SEC as an exhibit to our 8-K filed on filed on May 26, 2009.
During the Company's fiscal years ended December 31, 2008 and 2007 and the subsequent interim period through May 22, 2009, neither the Company nor anyone on its behalf consulted E&Y regarding either (i) the application of accounting principles to a specified transaction, either completed or proposed, or the type of audit opinion that might be rendered on the Company's financial statements, and no written report or oral advice of E&Y was provided to the Company that E&Y concluded was an important factor considered by the Company in reaching a decision as to an accounting, auditing or financial reporting issue; or (ii) any matter that was the subject of a disagreement or reportable event as defined in Regulation S-K, Item 304(a)(1)(iv) and Item 304(a)(1)(v), respectively.
Item 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
The Company maintains disclosure controls and procedures (as defined under Rules 13a-15(e) and 15d-15(e) of the Securities Exchange Act of 1934, as amended) that are designed to ensure that material information relating to the Company is made known to the officers who certify the Company's financial reports and to other members of senior management and the Board of Directors and, further, to ensure that information required to be disclosed in the Company's reports that are filed or submitted under the Exchange Act, are recorded, processed, summarized and reported within the time periods specified in the SEC's rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by the
86
Company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the Company's management, including the Company's principal executive and principal financial officers, or persons performing similar functions, as appropriate, to allow timely decisions regarding required disclosure.
Under the supervision and with the participation of management, including the Chief Executive Officer and the Chief Financial Officer, the Company has evaluated the effectiveness and design of its disclosure controls and procedures pursuant to Exchange Act Rule 13a-15(b) as of December 31, 2009. Based on that evaluation, the Chief Executive Officer and the Chief Financial Officer have concluded that these disclosure controls and procedures were effective as of December 31, 2009.
Management's Annual Report on Internal Control Over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting for the Company and for the assessment of the effectiveness of internal control over financial reporting. The Company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America. The Company's internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the Company's assets; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with accounting principles generally accepted in the United States of America, and that the Company's receipts and expenditures are being made only in accordance with authorizations of the Company's management and directors; and (iii) provide reasonable assurance regarding the prevention or timely detection of unauthorized acquisition, use or disposition of the Company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies and procedures may deteriorate.
Management conducted an evaluation of the effectiveness of the system of internal control over financial reporting based on the framework in Internal Control Integrated Framework issued by the Committee Sponsoring Organizations of the Treadway Commission. Based on this evaluation, management concluded that the Company's internal control over financial reporting was effective as of December 31, 2009. The Company's internal control over financial reporting as of December 31, 2009 has been audited by Ernst & Young LLP, a registered independent public accounting firm, as stated in its report set forth herein.
Changes in Internal Control Over Financial Reporting
There have been no changes in the Company's internal control over financial reporting that occurred during the Company's fourth fiscal quarter that have materially affected, or are reasonably likely to materially affect, the Company's internal control over financial reporting.
87
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders of Poniard Pharmaceuticals, Inc.
We have audited Poniard Pharmaceuticals, Inc.'s internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Poniard Pharmaceuticals, Inc.'s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management's Annual Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Poniard Pharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2009, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheet of Poniard Pharmaceuticals, Inc. as of December 31, 2009, and the related consolidated statements of operations, shareholders' equity, and cash flows for the year ended December 31, 2009 and our report dated March 16, 2010 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Palo
Alto, California
March 16, 2010
88
Not Applicable.
Item 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
(a) Directors. The information required by this item is incorporated herein by reference to the sections captioned "Election of Directors" and "Board of Directors and Corporate Governance" in the Company's definitive Proxy Statement for the 2010 Annual Meeting of Shareholders, to be filed with the Commission not later than 120 days after December 31, 2009.
(b) Executive Officers. The information concerning our executive officers is set forth in Item 1 of this report under the heading "Our Executive Officers."
(c) Compliance with Section 16(a) of the Exchange Act. The information required by this item is incorporated herein by reference to the section captioned "Section 16(a) Beneficial Ownership Reporting Compliance" in the Company's definitive Proxy Statement for the 2010 Annual Meeting of Shareholders, to be filed with the Commission not later than 120 days after December 31, 2009.
(d) Code of Ethics. The information required by this item is incorporated herein by reference to the section captioned "Board of Directors and Corporate Governance" in the Company's definitive Proxy Statement for the 2010 Annual Meeting of Shareholders, to be filed with the Commission not later than 120 days after December 31, 2009.
(e) Audit Committee. The information required by this item is incorporated herein by reference to the section captioned "Board of Directors and Corporate Governance" in the Company's definitive Proxy Statement for the 2010 Annual Meeting of Shareholders, to be filed with the Commission not later than 120 days after December 31, 2009.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this item is incorporated herein by reference to the sections captioned "Executive Compensation" and "Board of Directors and Corporate Governance—Compensation Committee Interlocks and Insider Participation" in the Company's definitive Proxy Statement for the 2010 Annual Meeting of Shareholders, to be filed with the Commission not later than 120 days after December 31, 2009.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
Equity Compensation Plan Information
The following table presents information as of December 31, 2009 with respect to the Company's compensation plans, including individual compensation arrangements, under which equity securities of
89
the Company are authorized for issuance to employees and non-employees of the Company, such as directors, lenders, consultants, and advisors:
Plan Category
|
(a) Number of Securities to be Issued Upon Exercise of Outstanding Options, Warrants and Rights(3) |
(b) Weighted- Average Exercise Price of Outstanding Options, Warrants and Rights(4) |
(c) Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans (excluding securities reflected in column (a))(5) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Equity Compensation Plans |
|||||||||||
Approved by Security Holders(1) |
6,167,207 | $ | 6.02 | 1,291,633 | |||||||
Equity Compensation Plans |
|||||||||||
Not Approved by Security Holders(2) |
5,085,196 | $ | 4.83 | — | |||||||
Total |
11,252,403 | $ | 5.45 | 1,291,633 | |||||||
- (1)
- Includes
the Company's 1991 Stock Option Plan for Non-Employee Directors (Directors Plan), the Restated 1994 Stock Option Plan (1994 Plan) and
the Amended and Restated 2004 Incentive Compensation Plan (2004 Plan). The Directors Plan was terminated on March 31, 2005 and the 1994 Plan was terminated on February 17, 2004.
Accordingly, no further awards can be issued under the Directors and 1994 Plans. For a description of the 2004 Plan, see Note 10 to the notes to the consolidated financial statements in
Item 8 of this report.
- (2)
- Reflects
a warrant issued for placement agent services in connection with our 2006 equity financing and warrants issued to financial institutions
participating in our term loan facility. For a description of these warrants, see Note 9 to the notes to the consolidated financial statements in Item 8 of this report.
- (3)
- Includes
561,044 shares subject to outstanding restricted stock unit awards granted under the 2004 Plan.
- (4)
- The
weighted-average exercise price does not include the common shares subject to outstanding restricted stock unit awards which have no exercise price. If
the restricted stock unit awards were included, the weighted-average exercise price would be $5.47 per share and the total weighted-average exercise price would be $5.18 per share.
- (5)
- All common shares remaining available for issuance under equity compensation plans are issuable under our 2004 Plan. The 2004 Plan contains an evergreen provision, pursuant to which the number of common shares available under the plan will automatically increase on the first day of each of the Company's fiscal years beginning in 2008. The number of additional common shares made available each year is equal to the lesser of (i) 3,000,000 common shares, (ii) 5% of the outstanding shares of common stock as of the end of the Company's immediately preceding fiscal year, (iii) any lesser number of common shares determined by the Company's board of directors, or (iv) a number of common shares that, when added to the sum of (x) the number of common shares subject to outstanding awards under the 2004 Plan as of the end of the Company's immediately preceding fiscal year (other than awards not subject to vesting or forfeiture conditions) and (y) the number of common shares that could be made subject to outstanding awards as of the end of the Company's immediately preceding fiscal year, does not exceed 20% of the outstanding shares of common stock on a fully diluted basis as of the end of the Company's immediately preceding fiscal year. Any additional common shares made available under the evergreen provision shall continue to be available for issuance under the 2004 Plan for subsequent years. Giving effect to the evergreen provision of the 2004 Plan, as of January 1, 2010, the aggregate number of common shares available for issuance as new awards was 3,395,606 shares.
90
Other information required by this item is incorporated herein by reference to the section captioned "Security Ownership of Certain Beneficial Owners and Management" in the Company's definitive Proxy Statement for the 2010 Annual Meeting of Shareholders, to be filed with the Commission not later than 120 days after December 31, 2009.
Item 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this item is incorporated herein by reference to the sections captioned "Certain Relationships and Related Transactions with Management" and "Board of Directors and Corporate Governance" in the Company's definitive Proxy Statement for the 2010 Annual Meeting of Shareholders, to be filed with the Commission not later than 120 days after December 31, 2009.
Item 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this item is incorporated herein by reference to the sections captioned "Independent Registered Public Accounting Firm" and "Ratification of Appointment of Independent Registered Public Accounting Firm" in the Company's definitive Proxy Statement for the 2010 Annual Meeting of Shareholders, to be filed with the Commission not later than 120 days after December 31, 2009.
91
Item 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
- (a)
- (1)
Financial Statements—See Index to Financial Statements.
- (2)
- Financial
Statement Schedules—Financial statement schedules have been omitted since they are either not required, not applicable, or the
information is otherwise included.
- (3)
- Exhibits—See
Exhibit Index of this Annual Report on Form 10-K contained herein.
- (b)
- Exhibits—See Exhibit Index of this Annual Report on Form 10-K contained herein.
92
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| PONIARD PHARMACEUTICALS, INC. (Registrant) |
||
/s/ GREGORY L. WEAVER Gregory L. Weaver Chief Financial Officer and Senior Vice President, Finance |
Date: March 16, 2010
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and as of the dates indicated:
| /s/ RONALD A. MARTELL Ronald A. Martell |
Director and Chief Executive Officer | March 16, 2010 | ||
/s/ GERALD MCMAHON Gerald McMahon |
Director (Chairman of the Board) |
March 16, 2010 |
||
/s/ ROBERT S. BASSO Robert S. Basso |
Director |
March 16, 2010 |
||
/s/ FRED B. CRAVES Fred B. Craves |
Director |
March 16, 2010 |
||
/s/ E. ROLLAND DICKSON E. Rolland Dickson |
Director |
March 16, 2010 |
||
/s/ CARL S. GOLDFISCHER Carl S. Goldfischer |
Director |
March 16, 2010 |
||
/s/ ROBERT M. LITTAUER Robert M. Littauer |
Director |
March 16, 2010 |
93
| /s/ GARY A. LYONS Gary A. Lyons |
Director | March 16, 2010 | ||
/s/ DAVID R. STEVENS David R. Stevens |
Director |
March 16, 2010 |
||
/s/ NICHOLAS J. SIMON III Nicholas J. Simon III |
Director |
March 16, 2010 |
||
/s/ GREGORY L. WEAVER Gregory L. Weaver |
Chief Financial Officer and Senior Vice President, Finance |
March 16, 2010 |
||
/s/ MICHAEL K. JACKSON Michael K. Jackson |
Principal Accounting Officer |
March 16, 2010 |
94
| Exhibit | Description | |
|||
|---|---|---|---|---|---|
| 3.1 | Amended and Restated Articles of Incorporation, as amended February 7, 2007 | (N) | |||
3.2 |
Restated Bylaws, as amended June 23, 2009 |
(C) |
|||
10.1 |
1991 Stock Option Plan for Non-Employee Directors, as amended(‡) |
(E) |
|||
10.2 |
Restated 1994 Stock Option Plan(‡) |
(F) |
|||
10.3 |
Stock Option Grant Program for Nonemployee Directors under the NeoRx Corporation 1994 Restated Stock Option Plan(‡) |
(M) |
|||
10.4 |
2004 Incentive Compensation Plan, as amended and restated June 14, 2007(‡) |
(B) |
|||
10.5 |
Stock Option Grant Program for Nonemployee Directors under the 2004 Incentive Compensation Plan, as amended June 14, 2007(‡) |
(X) |
|||
10.6 |
Form of Non-Qualified Stock Option Agreement under 2004 Incentive Compensation Plan, as amended June 14, 2007(‡) |
(D) |
|||
10.7 |
Form of Incentive Stock Option Agreement under 2004 Incentive Compensation Plan(‡) |
(O) |
|||
10.8 |
Non-Qualified Stock Option Agreement, dated December 19, 2000, between NeoRx Corporation and Carl S. Goldfischer(‡) |
(I) |
|||
10.9 |
Non-Qualified Stock Option Agreement, dated January 17, 2001, between NeoRx Corporation and Carl S. Goldfischer(‡) |
(I) |
|||
10.10 |
License Agreement dated as of April 2, 2004, between the Company and AnorMED, Inc. Certain portions of the agreement have been omitted pursuant to a request for confidential treatment |
(Q) |
|||
10.11 |
Amendment No. 1 to License Agreement effective as of September 18, 2006, between the Company and AnorMED, Inc. Certain portions of the agreement have been omitted pursuant to a request for confidential treatment |
(Y) |
|||
10.12 |
Facilities Lease dated February 15, 2002, between NeoRx Corporation and Selig Real Estate Holdings Six |
(A) |
|||
10.13 |
Amendment to Lease dated November 21, 2008, between the Company and Selig Holdings Company, LLC |
(S) |
|||
10.14 |
Change of Control Agreement dated as of February 5, 2010, between the Company and Michael S. Perry(‡) |
(L) |
|||
10.15 |
Key Executive Severance Agreement dated as of February 5, 2010, between the Company and Michael S. Perry(‡) |
(L) |
|||
10.16 |
Amended and Restated Key Executive Severance Agreement dated as of February 24, 2009, between the Company and Gerald McMahon(‡) |
(V) |
|||
10.17 |
Amended and Restated Key Executive Severance Agreement dated as of February 24, 2009, between the Company and Ronald A. Martell(‡) |
(V) |
|||
10.18 |
Amended and Restated Change of Control Agreement dated as of February 24, 2009, between the Company and Ronald A. Martell(‡) |
(V) |
|||
95
| Exhibit | Description | |
|||
|---|---|---|---|---|---|
| 10.19 | Amendment No. 1 dated as of February 5, 2010, to Amended and Restated Key Executive Severance Agreement dated as of February 24, 2009, between the Company and Ronald A. Martell(‡) | (L) | |||
10.20 |
Amendment No. 1 dated as of February 5, 2010, to Amended and Restated Change of Control Agreement dated as of February 24, 2009, between the Company and Ronald A. Martell(‡) |
(L) |
|||
10.21 |
Restricted Stock Unit Award Notice and Restricted Stock Award Agreement, dated October 6, 2009,with Ronald A Martell(‡) |
(Z) |
|||
10.22 |
Amended and Restated Key Executive Severance Agreement dated as of February 24, 2009, between the Company and Robert De Jager(‡) |
(V) |
|||
10.23 |
Consulting Agreement dated as of February 6, 2010, between the Company and Robert De Jager, M.D.(‡) |
(L) |
|||
10.24 |
Amended and Restated Key Executive Severance Agreement dated as of February 18, 2009, between the Company and Gregory L. Weaver(‡) |
(V) |
|||
10.25 |
Amended and Restated Change of Control Agreement dated as of February 18, 2009, between the Company and Gregory L. Weaver(‡) |
(V) |
|||
10.26 |
Letter Agreement dated as of February 3, 2009, between the Company and Gregory L. Weaver(‡) |
(V) |
|||
10.27 |
Amended and Restated Key Executive Severance Agreement dated as of February 24, 2009, between the Company and Cheni Kwok(‡) |
(V) |
|||
10.28 |
Amended and Restated Change of Control Agreement dated as of February 24, 2009, between the Company and Cheni Kwok(‡) |
(V) |
|||
10.29 |
Amended and Restated Key Executive Severance Agreement dated as of February 24, 2009, between the Company and Anna Wight(‡) |
(V) |
|||
10.30 |
Amended and Restated Change of Control Agreement dated as of February 24, 2009, between the Company and Anna Wight(‡) |
(V) |
|||
10.31 |
Amended and Restated Key Executive Severance Agreement dated as of February 24, 2009, between the Company and Janet Rea(‡) |
(V) |
|||
10.32 |
Amended and Restated Change of Control Agreement dated as of February 24, 2009, between the Company and Janet Rea(‡) |
(V) |
|||
10.33 |
Amended and Restated Key Executive Severance Agreement dated as of February 24, 2009, between the Company and Michael K. Jackson(‡) |
(V) |
|||
10.34 |
Form of Directors' Indemnification Agreements(‡) |
(K) |
|||
10.35 |
Lease Agreement dated as of July 10, 2006, between the Company and ARE San Francisco No. 17 LLC |
(W) |
|||
10.36 |
Sublease Agreement dated as of February 10, 2010, between the Company and Veracyte, Inc., including Consent to Sublease dated as of February 10, 2010, by ARE-San Francisco No. 17 LLC, the Company and Veracyte, Inc. |
(R) |
|||
10.37 |
Amended and Restated Loan and Security Agreement dated as of September 2, 2008, by and among the Company, GE Business Financial Services Inc. and Silicon Valley Bank |
(J) |
|||
96
| Exhibit | Description | |
|||
|---|---|---|---|---|---|
| 10.38 | Commercial Picoplatin Active Pharmaceutical Ingredient Manufacturing Agreement between the Company and W.C. Heraeus GmbH, dated as of March 24, 2008. Certain portions of the agreement have been omitted pursuant to a request for confidential treatment. | (J) | |||
10.39 |
Separation/Consulting Agreement and General Release, dated as of November 21, 2008, by and between David A. Karlin and the Company(‡) |
(S) |
|||
10.40 |
Commercial Supply Agreement between the Company and Baxter Oncology GmbH, dated as of November 22, 2008. Certain portions of the agreement have been omitted pursuant to a request for confidential treatment. |
(V) |
|||
10.41 |
Consulting Agreement dated as of April 1, 2009, between the Company and Gary A. Lyons, as amended by Amendment One to Consulting Agreement effective July 11, 2009(‡) |
(G) |
|||
10.42 |
Restricted Stock Unit Award Notice and Restricted Stock Award Agreement, dated July 1, 2009, with Gary A. Lyons(‡) |
(G) |
|||
10.43 |
Poniard Pharmaceuticals, Inc. Management Incentive Plan, as amended February 17, 2010(‡) |
(U) |
|||
10.44 |
Common Stock Purchase Agreement dated as of February 23, 2010, by and between the Company and Commerce Court Small Cap Value Fund, Ltd. |
(P) |
|||
23.1 |
Consent of Ernst & Young LLP, Independent Registered Public Accounting Firm |
(Z) |
|||
23.2 |
Consent of KPMG LLP, Independent Registered Public Accounting Firm |
(Z) |
|||
31.1 |
Rule 13a-14(a)/15d-14(a) Certification of Chief Executive Officer |
(Z) |
|||
31.2 |
Rule 13a-14(a)/15d-14(a) Certification of Chief Financial Officer |
(Z) |
|||
32.1 |
Section 1350 Certification of Chief Executive Officer |
(Z) |
|||
32.2 |
Section 1350 Certification of Chief Financial Officer |
(Z) |
|||
- (‡)
- Management
contract or compensatory plan
- (A)
- Filed
as an exhibit to the Company's Form 10-K for the fiscal year ended December 31, 2001, and incorporated herein by reference.
- (B)
- Incorporated
by reference to Annex A of the Company's definitive proxy statement on Schedule 14A filed May 8, 2007.
- (C)
- Filed
as an exhibit to the Company's Current Report on Form 8-K filed on June 25, 2009, and incorporated herein by reference.
- (D)
- Filed
as an exhibit to the Company's Form 10-Q for the quarterly period ended June 30, 2007, and incorporated herein by reference.
- (E)
- Incorporated
by reference to Exhibit A to the Company's definitive proxy statement on Schedule 14A filed April 10, 1996.
- (F)
- Filed
as an exhibit to the Company's Form 10-K for the fiscal year ended December 31, 1995, and incorporated herein by reference.
- (G)
- Filed as an exhibit to the Company's Current Report on Form 8-K filed on July 13, 2009, and incorporated herein by reference.
97
- (H)
- Reserved.
- (I)
- Filed
as an exhibit to the Company's Form 10-K for the fiscal year ended December 31, 2000, and incorporated herein by reference.
- (J)
- Filed
as an exhibit to the Company's Current Report on Form 8-K filed on September 6, 2008, and incorporated herein by reference.
- (K)
- Filed
as an exhibit to the Company's Current Reports on Form 8-K filed on April 28, 2006, June 27, 2006. May 9, 2007
and July, 13, 2009, and incorporated herein by reference.
- (L)
- Filed
as an exhibit to the Company's Current Report on Form 8-K filed on February 11 2010, and incorporated herein by reference.
- (M)
- Filed
as an exhibit to the Company's Form 10-Q for the quarterly period ended June 30, 2002, and incorporated herein by reference.
- (N)
- Filed
as an exhibit to the Company's Current Report on Form 8-K filed on February 8, 2007, and incorporated herein by reference.
- (O)
- Filed
as an exhibit to the Company's Form 10-Q for the quarterly period ended September 30, 2004, and incorporated herein by
reference.
- (P)
- Filed
as an exhibit to the Company's Report on Form 8-K filed on February 23, 2010, and incorporated herein by reference.
- (Q)
- Filed
as an exhibit to the Company's Form 10-Q for the quarterly period ended March 31, 2004, and incorporated herein by
reference.
- (R)
- Filed
as an exhibit to the Company's Current Report on Form 8-K filed February 18, 2010, and incorporated herein by reference.
- (S)
- Filed
as an exhibit to the Company's Current Report on Form 8-K filed November 26, 2008, and incorporated herein by reference.
- (T)
- Reserved.
- (U)
- Filed
as an exhibit to the Company's Report on Form 8-K filed on February 19, 2010, and incorporated herein by reference.
- (V)
- Filed
as an exhibit to the Company's Form 10-K for the fiscal year ended December 31, 2009, and incorporated herein by reference.
- (W)
- Filed
as an exhibit to the Company's Current Report Form 8-K filed on July 13, 2006 and incorporated herein by reference.
- (X)
- Filed
as an exhibit to the Company's Form 10-Q for the quarterly period ended September 30, 2007 and incorporated herein by
reference.
- (Y)
- Filed
as an exhibit to the Company's Form 10-Q for the quarterly period ended September 30, 2006 and incorporated herein by
reference.
- (Z)
- Filed
herewith.
- **
- In reviewing the agreements included as exhibits to this Annual Report on Form 10-K, please remember that they are included to provide you with information regarding their terms and are not intended to provide any other factual or disclosure information about the Company or the other parties to the agreements. The agreements contain representations and warranties of each of the parties to the applicable agreement. These representations and warranties have been made solely for the benefit of the other parties to the applicable agreements and
98
- •
- should not be treated as categorical statements of fact, but rather as a way of allocating risk to
one of the parties if those statements prove to be inaccurate;
- •
- have been qualified by disclosures that were made to the other party in connection with the
negotiation of the applicable agreement, which disclosures are not necessarily reflected in the agreement;
- •
- may apply standards of materiality in a way that is different from what may be viewed as material
to you and other investors; and
- •
- were made only as of the date of the applicable agreement or such other date or dates as may be specified in the agreement and are subject to more recent developments.
Accordingly, these representations and warranties may not describe the actual state of affairs of as of the date they were made or any other time. Additional information about the Company may be found elsewhere in this Form 10-K and the Company's other public filings which are available without charge through the SEC's website at http://www.sec.gov. See "Where You Can Find Other Information."
99