Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
| x | Annual report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
for the fiscal year ended December 31, 2009
or
| ¨ | Transition report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
for the transition period from to
Commission file number 0-24517
ORTHOVITA, INC.
(Exact name of registrant as specified in its charter)
| Pennsylvania | 23-2694857 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |
| 77 Great Valley Parkway Malvern, Pennsylvania |
19355 | |
| (Address of principal executive offices) | (Zip Code) | |
Registrant’s telephone number, including area code: (610) 640-1775
Securities registered pursuant to Section 12(b) of the Act:
Common Stock, par value $.01 per share
(Title of class)
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in the definitive proxy statement incorporated by reference in Part III of this annual report on Form 10-K or any amendment to this annual report on Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of “accelerated filer and large accelerated filer” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer ¨ Accelerated filer x Non-accelerated filer ¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The approximate aggregate market value of Common Stock held by non-affiliates of the registrant was $343,615,757 as of June 30, 2009 (For purposes of determining this amount only, the registrant has defined affiliates as including (a) the executive officers of the registrant as of June 30, 2009, (b) all directors of the registrant as of June 30, 2009 and (c) each shareholder that informed the registrant that as of June 30, 2009 it was the beneficial owner of 10% or more of the outstanding common stock of the registrant).
As of March 8, 2010, there were 76,564,278 shares of the registrant’s Common Stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE:
Portions of Orthovita, Inc.’s Proxy Statement relating to the 2010 Annual Meeting of Shareholders (to be filed within 120 days after the end of the year covered by this annual report on Form 10-K) are incorporated into Part III of this annual report on Form 10-K by reference.
Table of Contents
Table of Contents
PART I
| ITEM 1. | BUSINESS |
In addition to historical facts or statements of current conditions, our disclosure and analysis in this Form 10-K contain some forward-looking statements. See the discussion of forward-looking statements in Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operation” included in this Form 10-K.
Unless the context indicates otherwise, the terms Orthovita, Company, we, us or our herein refer to Orthovita, Inc. and, where appropriate, one or more of our subsidiaries.
GENERAL DEVELOPMENT OF OUR BUSINESS
We are a specialty spine and orthopedic company with a portfolio of orthobiologic and biosurgery products. Our products are based on novel and unique proprietary biomaterials that have innovative mechanisms of action in the body.
Our orthobiologic platform includes products for the fusion, regeneration and fixation of human bone. Our orthobiologic products are based on our proprietary Vitoss™ Bone Graft Substitute technology and include the Imbibe™ Bone Marrow Aspiration System used with Vitoss. Vitoss is the market-leading synthetic bone graft in the United States. Several of our Vitoss products incorporate our proprietary bioactive ceramic glass technology which accelerates bone healing. Our orthobiologic products also include our Cortoss™ Bone Augmentation Material and the Aliquot™ Delivery System used with Cortoss, both of which were first available for sale in the U.S. in July 2009. Cortoss is an advanced synthetic biomaterial that hardens to mimic weight-bearing, cortical bone following injection into spinal vertebrae. Cortoss is the first clinically-proven, injectable alternative to polymethylmethacrylate (PMMA) bone cement cleared by the U.S. Food and Drug Administration (FDA) for the treatment of vertebral compression fractures, an often extremely painful condition that occurs in patients with osteoporosis and cancer. In addition to extensive clinical data showing statistically significant improvements in pain relief (at three months following implantation) and function (at 24 months following implantation) from the use of Cortoss in comparison to PMMA, we believe that Cortoss has significant ease-of-use advantages over current vertebral augmentation technology.
Our biosurgery products include Vitagel™ Surgical Hemostat, Vitasure™ Absorbable Hemostat, and the CellPaker™ Plasma Collection System used in conjunction with Vitagel, and other accessories and delivery products that complement our Vitagel product. These products incorporate advanced biosurgical materials to help control bleeding during surgeries.
We have expanded our product portfolio through internal product development efforts, co-development efforts with strategic partners and the in-licensing of products. We internally developed our Vitoss and Cortoss materials and maintain an ongoing internal research and development program. In addition, we worked with Kensey Nash Corporation (Kensey) to co-develop and commercialize certain Vitoss Foam products, and we have acquired rights from third parties to market Vitagel and Vitasure. We continue to pursue in-licensing, co-development and other opportunities to acquire complementary products and technologies to broaden our product offerings and further leverage our sales force.
Other Sales and Corporate Information
In the U.S., we market and sell our products through a field sales network of direct sales representatives and independent distributors. As of December 31, 2009, we had 90 direct sales representatives (DSRs), 13 associate sales representatives who support our DSRs, and arrangements with 42 independent distributors that we utilize to market and sell our products in the U.S. We continue to strengthen our field sales network through the addition of
1
Table of Contents
DSRs in those U.S. sales territories where either we do not have independent distributor coverage or the territories are underserved. Outside the U.S., we primarily utilize a network of independent stocking distributors to market our products, although we market these products through a direct sales force in the United Kingdom.
Information regarding our product sales by geographic markets for 2009, 2008 and 2007 is included in note 14 (Product Sales) to the consolidated financial statements included in this report.
We market our products for only the indication(s) or use(s) that have received regulatory approval or clearance.
We incorporated in Pennsylvania in 1992 and maintain a branch operation in Belgium, as well as a wholly-owned subsidiary in the United Kingdom, and a wholly-owned subsidiary incorporated in Delaware to hold certain intangible properties.
Our principal executive offices are located at 77 Great Valley Parkway, Malvern, Pennsylvania 19355, and our telephone number is (610) 640-1775. We maintain a website at www.orthovita.com and make available free of charge on this website our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, as soon as reasonably practicable after we electronically file these materials with, or furnish them to, the U.S. Securities and Exchange Commission. The reference to our website is intended to be an inactive textual reference only.
OUR PRODUCTS
As further discussed below under the caption “GOVERNMENT REGULATION,” our products and product candidates are subject to extensive regulation as medical devices by the FDA, regulatory authorities in Europe and regulatory authorities in other jurisdictions. Product approval or clearance applications for our products must be supported by valid scientific evidence, and may require clinical trial data, to demonstrate the safety and effectiveness of the products.
Our existing orthobiologic and biosurgery products are described below.
Orthobiologic Products: Vitoss and Cortoss and Related Delivery Systems and Accessories
Vitoss Bone Graft Substitute
| Vitoss Blocks and Morsels |
FDA 510(k) cleared December 2000 Approved in European Union July 2000 | |
| Vitoss Micro and Macro Morsels |
FDA 510(k) cleared December 2000 Approved in European Union July 2003 | |
| Vitoss Standard and Micro Canisters |
FDA 510(k) cleared November 2003 Approved in European Union July 2003 | |
| Vitoss Foam Product Platform, including: |
FDA 510(k) cleared December 2003 Approved in European Union May 2005 | |
| Vitoss Foam Strips and Cylinders Vitoss Foam Flow Vitoss Foam Shapes Vitoss Foam Pack |
||
| Vitoss Bioactive Foam Strips |
FDA 510(k) cleared September 2007 Approved in European Union October 2009 | |
| Vitoss Bioactive Foam Pack |
FDA 510(k) cleared June 2008 Approved in European Union October 2009 | |
2
Table of Contents
Vitoss is a high-porous resorbable beta-tricalcium phosphate bone graft substitute used to help the body guide the three-dimensional regeneration of the patient’s own bone. We launched Vitoss in the U.S. and the European Union (EU) in 2001. Vitoss’ high-porosity allows it to soak and hold its own volume in blood and bone marrow aspirate and has been shown to perform well in an array of applications in the spine, extremities and pelvis, such as spinal grafting and the treatment of bone defects due to trauma, degenerative disease and tumors, including posterolateral spine fusion procedures. Vitoss integrates well into existing bone and allows for bone in-growth and maturation. Vitoss, the Vitoss Foam products and the Vitoss Bioactive Foam products are covered by issued and pending U.S. and non-U.S. patents.
Since the initial U.S. regulatory clearance and subsequent Vitoss product launch in mid-2001, we have developed and are developing a number of new Vitoss-based products and related delivery devices that are designed to expand the market for our Vitoss products by broadening the range of surgical indications and by addressing additional surgeon preferences and clinical needs. In 2003, we entered into a development, manufacturing and supply agreement with Kensey, leading to the development and launch of the Vitoss Foam product platform. The Vitoss Foam products combine our base Vitoss technology with Kensey’s proprietary resorbable biomaterials to produce a wide array of pliant, flexible, flowable and compression resistant bone graft materials. The Vitoss Foam products have the ability to retain biological fluids such as blood and bone marrow aspirate in pliable and compression resistant forms. These forms can be designed into specific shapes and material characteristics to meet a surgeon’s need for handling and delivery in a variety of surgical approaches and applications. We launched our most recent commercialized iteration of this product line, Vitoss Bioactive Foam, in 2008. In addition to our proprietary Vitoss beta-tricalcium phosphate bone graft substitute and Kensey’s proprietary resorbable biomaterials, Vitoss Bioactive Foam products also contain our proprietary combeite bioactive glass to advance bone healing.
Bone Defect Grafting. Injury or trauma to the bone, degenerative conditions, disease and aging all affect the health and viability of the human skeleton. These conditions often result in the need for the repair of bone defects through a bone grafting procedure. We estimate that between 2 and 2.5 million bone grafting procedures on a worldwide basis are performed each year in the spine, extremities and pelvis. Bone grafting material is either (i) autograft material, which is often obtained or harvested from the iliac crest region of the patient’s own hip, (ii) allograft material, which is obtained from a cadaver, or (iii) synthetically derived materials that provide one or more components of either bone-like scaffold (such as Vitoss), cells or signals (such as bone morphogenic proteins). Vitoss has been used in bone grafting procedures as a bone graft substitute to provide a synthetic scaffold in a variety of applications, including those of the extremities, spine and pelvis. When used with the patient’s bone marrow, Vitoss provides all components of scaffold, cells and signals.
As a synthetic beta-tricalcium phosphate material, our Vitoss material has advantages over both autograft and allograft materials. For example, a harvest of autograft material involves an additional procedure that extends surgical time, adding to costs and increasing blood loss and patient risk of infection or adverse reaction from the additional time under anesthesia. In addition, harvesting bone for autograft sometimes causes protracted pain that may necessitate additional medical care after the surgical procedure. Disadvantages of allograft bone include the possibility of disease transmission, antigenic response and quality variability from donor to donor. Using Vitoss instead of autograft or allograft material avoids these potential complications.
Spinal Fusion and Grafting. Many patients affected by severe back pain due to degeneration of one or more discs are often treated with spinal surgical procedures. We estimate that each year approximately 530,000 spinal fusion procedures are performed in the United States. In cases where the patient has advanced disc degeneration or spinal instability, a fusion procedure can involve a surgical incision in the patient’s back or abdomen to access and remove the affected disc material. To provide initial stability and support of the surrounding vertebrae, the resulting defect is filled with a structural implant made of either titanium, shaped bone derived from a human cadaver, or a synthetic material known as polyetheretherketone (PEEK). Adjunctively, these procedures may require the use of bone grafting material to repair defects and facilitate the fusion of two bony elements. We believe the use of Vitoss provides a clinically proven alternative to patient- or cadaver-derived tissues and may be preferable for both patients and their surgeons for its efficacy and safety.
3
Table of Contents
Trauma. Physical trauma such as falls and accidents can result in bone fracture or damage. Fractures of broken bones are often realigned with hardware, such as plates, rods and screws. Once the hardware has been used to recreate the skeletal anatomy and to provide the stability of the bony structure, there are often defects or voids in the bone which remain. Those voids may require the use of bone graft material. The goal of bone grafting in trauma applications is to rapidly heal the damaged bone. Approximately 300,000 trauma-related bone graft repairs are performed annually on a worldwide basis. Autograft, cadaver allograft, as well as synthetic scaffolds like Vitoss, are used for trauma-related bone graft repairs.
Dental, Periodontal, Oral and Cranio-Maxillofacial. In August 2002, we entered into a supply agreement with BioMimetic Therapeutics, Inc. for our proprietary Vitomatrix™ particulate synthetic scaffold biomaterial, which we produce in the same process used to manufacture Vitoss. In January 2008, in connection with the sale of its dental business to Luitpold Pharmaceuticals, Inc., BioMimetic assigned its rights and obligations under the supply agreement to Luitpold with our consent. Under the agreement, we supply Vitomatrix to Luitpold for its clinical and commercial use in combination with rhPDGF-BB. Luitpold markets the combined product in the dental, periodontal, oral and cranio-maxillofacial bone grafting markets through its Osteohealth Company division. In March 2010, we entered into an agreement with Luitpold pursuant to which the Vitomatrix supply agreement will be terminated upon the following material conditions: (1) Luitpold will purchase approximately $1 million of Vitomatrix from us in March 2010; and (2) any remaining purchase obligations of Luitpold under the supply agreement will be eliminated.
Bone Marrow Aspiration System with Imbibe Needles, Syringes and Imbibe Disposable Delivery Instrumentation, used with Vitoss
| Bone Marrow Aspiration Needle |
FDA 510(k) cleared June 2005 | |
| Bone Marrow Aspiration Syringe |
FDA 510(k)s cleared September 2001 and March 2003 Approved in European Union September 2004 | |
| Disposable Delivery Funnel |
Class I exempt device (see GOVERNMENT REGULATION below) Approved in European Union September 2004 | |
| Disposable Delivery Tube |
Class I exempt device (see GOVERNMENT REGULATION below) Approved in European Union January 2005 | |
The disposable Imbibe devices provide spine and orthopedic surgeons with a simple method for harvesting a patient’s own bone marrow, mixing it with Vitoss, and delivering the mixture to the bone graft site. We believe Imbibe, when used together with Vitoss, provides greater flexibility and options for surgeons. Imbibe is covered by a U.S.-issued patent.
Cortoss Bone Augmentation Material
| Cortoss Bone Augmentation Material |
FDA 510(k) cleared June 2009 for vertebral augmentation Approved in European Union January 2003 for vertebral augmentation Approved in European Union October 2001 for screw augmentation |
4
Table of Contents
Cortoss is a polymer composite which mimics the structural characteristics of human bone that has been cleared by the FDA and approved in the EU under the CE mark process. For patients with poor bone healing, as seen in osteoporotic patients, Cortoss may be used in a number of surgical procedures to quickly provide structural stability and reinforcement of the bones after surgery. The surgeon’s goal is to repair the patient’s bone and enhance the patient’s mobility as quickly as possible since prolonged bed rest or inactivity may result in decreased overall health for older or osteoporotic patients. Cortoss’ simple mix-on-demand delivery system design allows for minimum waste and maximum ease of use and flexibility for the surgeon. Cortoss is an injectable material that is delivered through a pre-filled, unit dose, disposable cartridge. Delivery of Cortoss to the surgical site may be started and stopped for a prolonged period of time throughout the surgical procedure as polymerization is initiated only when Cortoss is expressed through its static mix-tip. The polymerization is a self-setting reaction that causes Cortoss to harden within minutes. Cortoss provides two stages of fixation: immediate mechanical interlock into porous bone, followed by intimate bone apposition over time along the contours of its surface. Cortoss is covered by several patents issued in the U.S. and other countries, and other U.S. and non-U.S. patent applications are pending.
Cortoss has regulatory clearance in the U.S. and EU for use in the treatment of vertebral compression fractures (VCFs) of the spine. Cortoss also has regulatory approval in the EU for use in screw augmentation. We began to sell Cortoss in the U.S. in July 2009 shortly after obtaining clearance from the FDA to market Cortoss for vertebral augmentation. We fully launched Cortoss in the U.S. after completing a limited, controlled launch of the product in September 2009. We have sold Cortoss in Europe since December 2001.
Vertebral Augmentation of VCFs. Vertebral augmentation of VCFs is a procedure for repairing fractured vertebrae that can be performed on a hospital inpatient or outpatient basis or in ambulatory surgery centers, clinics and physician offices. We estimate there are approximately 700,000 patients in the U.S. with VCFs caused by osteoporotic bone or bone cancer resulting in severe pain and immobility. Of these, we believe approximately 200,000 fractures are treated annually using vertebroplasty or kyphoplasty techniques. The traditional treatments, e.g., bed rest, bracing, narcotics or anesthetic injections, may not address the underlying fracture. Vertebral augmentation of VCFs has been reported to provide early pain relief in up to 90% of osteoporotic patients. Early relief of pain provided by vertebral augmentation of VCFs enables patients to maintain better functional capacity. Functional capacity, in turn, is believed to be directly related to the ability to live independently and unassisted. We are not aware of any product that has received FDA approval or a CE mark for use in this procedure on the basis of prospective, controlled clinical data. However, since 2004, several versions of PMMA bone cement have been 510(k) cleared by the FDA. Results from our pivotal clinical data comparing Cortoss to PMMA in vertebral augmentation showed statistically significant improvements for Cortoss patients in pain relief (at three months following implantation) and function (at 24 months following implantation). Results from this study further indicated that Cortoss patients treated at one vertebral level with no previous fracture had a lower incidence of subsequent adjacent level fracture. In addition, the material properties of Cortoss described below result in the following advantages over PMMA:
| • | Flow characteristics and fill patterns that enhance procedural safety and control; |
| • | The ability to be mixed on demand with no time constraints in the pre-injection phase and delivered on a start-stop basis, thereby enhancing ease of use; |
| • | Mechanical properties more closely resembling cortical bone; |
| • | A lower temperature setting that reduces the risk of tissue necrosis associated with PMMA; and |
| • | The absence of volatile, free unreacted methylmethacrylate monomer released into the patient’s body. Although documented complications of monomer released from PMMA may be rare, they are severe adverse events when they occur. |
For the foregoing reasons, we believe that Cortoss has meaningful clinical benefits over PMMA in the treatment of VCFs.
5
Table of Contents
Screw Augmentation. Screw augmentation is a procedure for securing the fixation of bone screws used in patients with weak bone caused by osteoporosis. Cortoss may be used in those procedures where the screws “strip” or fail to hold the integrity of the internal fixation construct due to poor bone quality, as is often the case with osteoporotic bone. Where screws fail to hold, current treatment options include: (i) replacing the screw with a screw of larger diameter, which may further weaken the bone and is not always possible because of the size of the screw holes and/or the bone, or (ii) augmenting the screws with PMMA bone cement, which is cumbersome and time consuming. PMMA must be manually mixed and transferred into a syringe for application. After mixing, there is only a small time window in which the PMMA can be used before it sets, making it difficult to augment more than one screw at a time. Additionally, PMMA bone cement is not approved in either Europe or the U.S. for pedicle, trauma or hip fracture screw augmentation. We believe the use of Cortoss to anchor the screw in a quick and efficient way enables the full function of the screw to be restored. We market Cortoss for use in screw augmentation outside of the U.S. only where we have obtained regulatory approval for this indication.
Aliquot Delivery System
Our Aliquot Delivery System facilitates delivery of materials to bony sites, including delivery of our Cortoss product directly to the surgical site. A coaxial system of catheter and syringe dispenser is designed to assure effective delivery of material in vertebral augmentation of VCFs and screw augmentation procedures. We launched the Aliquot system in the U.S. concurrently with the limited launch of Cortoss in July 2009. Aliquot is exempt from 510(k) clearance. During the second quarter of 2002, we received CE mark approval from our notified body for certain Aliquot kit configurations.
Biosugery Products: Vitagel and Vitasure and Related Components and Accessories
Vitagel Surgical Hemostat and Accessories
| Vitagel Surgical Hemostat |
PMA approved, distribution rights initially obtained June 2004 Approved in European Union July 2007 (See GOVERNMENT REGULATION below) | |
| CellPaker Plasma Collection System |
PMA approved, distribution rights initially obtained June 2004 Approved in European Union July 2007 (See GOVERNMENT REGULATION below) | |
| Malleable Extended Applicator |
PMA approved, distribution rights obtained June 2004 Approved in European Union July 2007 (See GOVERNMENT REGULATION below) | |
| Laparoscopic Extended Applicator |
PMA approved, U.S. distribution rights obtained June 2004 Approved in European Union July 2007 (See GOVERNMENT REGULATION below) | |
| Vitagel Spray Set |
FDA 510(k) cleared October 2005 Approved in European Union July 2007 | |
Vitagel is an FDA-approved, CE-marked composite liquid hemostat that combines the biomaterials bovine thrombin and bovine collagen with the patient’s autologous plasma. When applied to the surgical site, Vitagel creates a safe adherent matrix and an impermeable barrier to blood flow, and its unique microfibrillar collagen component facilitates healing. Vitagel has been marketed to general surgeons and is applicable in the spine, hip
6
Table of Contents
and knee replacement surgery markets, where bleeding is a significant complication for many routine surgeries. After obtaining the right to sell Vitagel and CellPaker from a third party, we commenced sales of the products in the U.S. in 2005 through our existing orthopedics and spine-based sales channel, utilizing the same marketing strategy that we use for all of our Vitoss-related products. This strategy provides our sales network additional functional biomaterial products to offer to customers. During the first half of 2006, we launched the Vitagel Spray Set in the U.S. We obtained the CE mark for Vitagel, CellPaker and related accessories in July 2007 and launched these products in the European Union in the first quarter of 2008. Vitagel and CellPaker are covered by U.S. and non-U.S. issued patents owned by the licensor of the products.
Vitasure Absorbable Hemostat
| Vitasure Absorbable Hemostat | PMA approved, distribution rights initially obtained April 2008 Approved in European Union September 2002 (See GOVERNMENT REGULATION below) |
Vitasure is an FDA-approved, CE-marked, plant-based hemostat product that can be deployed quickly throughout surgery. Through proprietary microporous polysaccharide hemosphere technology, Vitasure acts as a molecular sieve to quickly extract fluids from blood, creating an osmotic action that causes the Vitasure particles to swell and concentrate serum proteins, platelets, and other elements on the surface. The Vitasure particles and their coating of compacted cells then create a matrix for the formation of a tenacious fibrin clot. We originally obtained the right to distribute Vitasure in the U.S. and certain other territories for orthopedic and spine applications in 2008 from Medafor, Inc. We launched the Vitasure product in the U.S. in 2008 and in certain non-U.S. territories in January 2009. Vitasure is covered by U.S. and non-U.S. issued patents owned by Medafor and its licensor.
STRATEGIC PARTNERSHIPS
Kensey - Vitoss Foam
In March 2003, we entered into an agreement with Kensey to jointly develop and commercialize certain orthobiologic products based upon our Vitoss platform. The new products developed under this agreement are based on our internally developed, proprietary Vitoss bone graft substitute material in combination with proprietary resorbable Kensey biomaterials. To date, the products covered by the Kensey agreement for which we have received 510(k) clearance from the FDA are our Vitoss Foam and Vitoss Bioactive Foam products.
Kensey has the exclusive right to manufacture any approved or cleared products that were jointly developed under the agreement, and we market and sell these products worldwide. Following the regulatory approval or clearance of each new product, we have obligations under the agreement to pay Kensey for manufacturing the product and make royalty payments to Kensey based on the net sales of the product. These rights and obligations extend until at least February 2024 for our Vitoss Bioactive Foam product, and until February 2014 for our other products under the Vitoss Foam product platform.
Approximately 67% of our product sales during 2009 were from products based upon our Vitoss Foam platform covered by the Kensey agreement, including Vitoss Bioactive Foam.
Angiotech - Vitagel and CellPaker
We originally obtained rights to distribute Vitagel and CellPaker from Angiotech Pharmaceuticals (U.S.), Inc. (which we refer to collectively with its affiliates and parent company Angiotech Pharmaceuticals, Inc. as Angiotech) under a license agreement executed in June 2004 and launched these products in 2005. In December 2006, pursuant to a royalty sale agreement, we paid Angiotech $9.0 million in cash in order to eliminate our obligations to pay Angiotech royalties on future sales of Vitagel and CellPaker products under the license
7
Table of Contents
agreement. Concurrently with this agreement, we amended and restated our license agreement with Angiotech to eliminate our obligations to meet minimum sales requirements, extend the term of the license from December 31, 2014 through July 31, 2017, and eliminate certain termination rights in favor of Angiotech.
Under the amended and restated license agreement: (i) we have exclusive rights to manufacture, market and sell Vitagel products throughout the world for orthopedic indications, and non-exclusive rights to manufacture, market and sell CellPaker products throughout the world for all indications; and (ii) Angiotech has an option for co-exclusive rights outside the orthopedic field which, if exercised, would permit Angiotech to manufacture, market and sell an Angiotech-branded Vitagel product throughout the world. Until Angiotech elects to exercise its option for co-exclusive rights, we have exclusive rights to manufacture, market and sell Vitagel outside of the orthopedic field throughout the world. If Angiotech elects to exercise its option, we would then have co-exclusive rights to manufacture, market and sell Vitagel outside of the orthopedic field throughout the world.
We currently manufacture Vitagel at our facilities in Malvern, Pennsylvania and manufacture CellPaker at our subcontractor’s facility.
Medafor - Vitasure
After obtaining certain non-exclusive rights from Medafor, Inc. to distribute Vitasure Absorbable Hemostat pursuant to a distribution agreement, we launched the product in the third quarter of 2008. Our territory under the agreement currently consists of the United States, Australia, Belgium, Germany, Ireland, the Netherlands, South Africa and the United Kingdom. Pursuant to contractual obligations, we purchased an aggregate $1.5 million of Vitasure product inventory from Medafor in 2008 and 2009. The agreement with Medafor was amended in October 2009 to eliminate our remaining minimum purchase commitments for Vitasure and our unilateral right to renew the agreement in 2013 for another three-year term. The initial term was not amended and extends through December 2013.
SIGNIFICANT CONTRACTUAL OBLIGATIONS
See Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operation” of this report for a discussion of significant contractual arrangements relating to (i) our senior secured note facility, (ii) our leases, (iii) product development milestone and related payments and (iv) our joint product development and commercialization arrangement with Kensey.
RESEARCH & PRODUCT DEVELOPMENT
We employ the disciplines of composite engineering, polymer science, biologics processing, solution chemistry and nanoparticulate ceramic glass science to create our orthobiologic and biosurgery materials technology platforms. We then utilize technologies developed, co-developed or licensed by us to commercialize orthobiologic and biosurgery products for orthopedic and surgical applications. Patents have been issued and additional patent applications have been filed to protect our key developments. See “PATENTS AND PROPRIETARY INTELLECTUAL PROPERTY” below for additional information. We incurred approximately $6.8 million, $6.7 million, and $6.4 million in research and development expenses in 2009, 2008 and 2007, respectively.
We are continuing to develop new products under certain of our approved product platforms. In the near-term, we may seek to bring Vitoss, Cortoss, Vitagel and/or Imbibe product line extensions to market in the U.S. through the 510(k) or premarket approval (PMA) regulatory process (see “GOVERNMENT REGULATION” below).
In the long-term, we will continue to exploit our biomaterials expertise and manufacturing capacity to develop additional biomaterials for alternative indications related or unrelated to spine and orthopedic uses. Additionally we may utilize our knowledge of the spine and orthopedic markets to identify and develop products that may be unrelated to our biomaterials expertise.
8
Table of Contents
PATENTS AND PROPRIETARY INTELLECTUAL PROPERTY
Our strategy is to seek protection for our product technologies and manufacturing methods through the use of U.S. and non-U.S. patents. We have filed or intend to file applications as appropriate for patents covering our technologies, products and processes. As of March 1, 2010, certain of the products that we have developed or market are covered by one or more of our 24 issued U.S. patents, 19 pending U.S. patent applications and numerous counterparts of certain of these patents and pending patent applications outside the U.S., including Canada, Europe, Mexico and Japan. Our issued patents are set to expire between 2015 and 2025. Key patents covering Vitoss, Cortoss, Vitagel and Vitasure are set to expire in 2017, 2022, 2017 and 2019, respectively.
MANUFACTURING AND PRODUCT SUPPLY
The manufacture of our products is subject to regulation and periodic inspection by various regulatory bodies for compliance with current FDA Quality System Regulations, the EU Medical Devices Directive, International Organization for Standardization (ISO) 9000 Series standards, ISO 13485/European Norm (EN) 13485 and equivalent requirements.
In-House Manufacturing. Our 25,800-square foot Vitoss, Vitagel and Cortoss manufacturing facilities that produce our commercial products are leased through July 2017 and are certified as meeting the requirements of ISO 9000: 2000 and EN 13485 through July 1, 2012. These facilities are subject to inspection by the FDA for compliance with FDA device manufacture requirements. We believe our existing manufacturing facilities have the capacity to meet our commercial needs through at least 2012, assuming that, with respect to Vitagel only, we obtain the requisite regulatory approval for our Vitagel collagen processing facility as described in the following paragraph by the second half of 2010. In 2008, we completed renovations to our second production line for Vitagel and received FDA approval for these renovations in January 2009. During 2009, we further broadened the scope of our Vitagel manufacturing capabilities and received FDA approval for these changes in October 2009.
In October 2009, we completed renovations at our leased space to establish a 4,000-square foot facility to process collagen used as a raw material in the manufacture of Vitagel. This facility is leased through 2017. In December 2009, we submitted a PMA supplement to the FDA for this facility. We believe that we have sufficient inventory of processed collagen acquired from a third party to meet our commercial needs for Vitagel into the first half of 2011 (see—Raw Materials Supply below). We plan to fully transition the processing of collagen for Vitagel in-house by the second half of 2010. However, we cannot market commercial Vitagel product containing collagen processed at our facility until the requisite PMA supplement for this facility has been approved by the FDA (see “GOVERNMENT REGULATION” below), which we expect to occur by the second half of 2010. As part of the establishment of our collagen processing facility, in September 2008 we acquired collagen production equipment and a non-exclusive, perpetual, royalty-free, irrevocable license to certain collagen production process technology and know-how from Allergan, Inc. The license rights are limited to certain uses, including those related to the processing and production of collagen for surgical hemostats such as Vitagel.
In 2008, we completed renovations at our leased space to expand our capacity to manufacture Aliquot, Imbibe and CellPaker. We submitted an application to the FDA in the first quarter of 2010 for the manufacture of CellPaker at this renovated facility. We are not required to obtain regulatory approval to manufacture Aliquot or Imbibe at this facility.
Third Party Manufacturers. We are manufacturing CellPaker, Imbibe and Aliquot through outside third party contract manufacturers. Our Vitoss is converted to Vitoss Foam and Vitoss Bioactive Foam by Kensey, our development partner under a long-term supply agreement. We purchase Vitasure from Medafor, which uses third party manufacturers to make this product. Our and Medafor’s third party manufacturers are regulated by government agencies and are required to be ISO 9001/EN 13485 certified or otherwise meet our quality system requirements (See “GOVERNMENT REGULATION” below).
9
Table of Contents
Raw Materials Supply. Our ability to manufacture our products is dependent on a limited number of specialty suppliers of certain raw materials.
A key raw material for manufacturing Vitagel is processed collagen that is derived from bovine hides. In September 2008, we acquired a strategic amount of processed collagen raw material from Allergan for use in the manufacture of Vitagel after Allergan informed us of its intention to close its collagen processing plant and terminate a collagen supply agreement with us. We believe the strategic collagen reserve acquired from Allergan in 2008 will be sufficient to meet our commercial needs for Vitagel into the second half of 2011. While we believe that bovine hides are available from various sources, the type of collagen necessary for Vitagel in processed form is difficult to obtain. For this reason, we have established a facility to process collagen for Vitagel in-house. However, we must receive the requisite regulatory approvals for our collagen processing facility before we may sell Vitagel containing collagen processed in-house. We expect to receive this approval by the second half of 2010. We are also in the process of negotiating agreements for the supply of bovine hides.
We have various supply agreements with third parties for raw materials used in products we manufacture, and we have a supply agreement with Kensey for bovine collagen used in our Vitoss Foam and Vitoss Bioactive Foam product lines. However, we do not have supply agreements for all raw materials. The failure of any supplier to continue to provide us with materials at a price or quality acceptable to us, or at all, or our inability to find an alternative supplier with acceptable prices and quality, would have a material adverse effect on our ability to manufacture our products.
SALES AND MARKETING
In the U.S, we have a U.S. field sales network of direct sales representatives and independent non-stocking distributors who market our products. We continually seek to strengthen our field sales network through the addition of direct sales representatives in those U.S. sales territories where either we do not have independent distributor coverage or the territories are underserved. Outside the U.S., we primarily utilize a network of independent stocking distributors to market our products, although we sell these products through a direct sales force in the United Kingdom.
COMPETITION
Extensive research efforts and rapid technological change characterize the market for products in the orthopedic, spine and orthobiologic and biosurgery markets. We face intense competition from medical device and biomedical technology companies. Our products could be rendered noncompetitive or obsolete by competitors’ technological advances. We may be unable to respond to technological advances through the development and introduction of new products. Moreover, many of our existing and potential competitors have substantially greater financial, marketing, sales, distribution, manufacturing and technological resources than we do. These competitors may also be in the process of seeking FDA or other regulatory approvals, or patent protection, for new products. Our competitors could, therefore, commercialize new competing products in advance of our products. We cannot guarantee that we will be able to compete successfully against current or future competitors or that competition will not have a material adverse effect on our business, financial condition and results of operations.
We believe Vitoss faces competition from numerous synthetic products, bone morphogenic proteins (BMPs), stem cell-based products and cadaver-based products, such as demineralized bone matrix (DBM) products, currently on the market that may be used for the same indications as our products, as well as from other products and technologies that may enter the market in the future. Moreover, we believe that pressure generated by current economic conditions and the possibility of health care reform have resulted in hospitals becoming more proactive at controlling costs for medical devices. These efforts increase competition on a pricing level among bone graft products.
10
Table of Contents
Cortoss received regulatory clearance in the U.S. in 2009 for use in the vertebral augmentation of VCFs and received the CE mark several years ago for use in vertebral augmentation of VCFs and in screw augmentation. Cortoss faces competition from the several PMMA bone cement products that have been 510(k) cleared by the FDA since 2004 for this indication. Outside the U.S., Cortoss is facing competition from several other PMMA bone cement products that have received the CE mark since 2004 for this indication.
Vitagel is approved by the FDA as a surgical hemostat where bleeding may be a significant complication for many routine surgeries. As such, Vitagel can be marketed broadly to general surgeons and is applicable in the spine, hip and knee replacement surgery markets. Vitagel faces competition from several established surgical hemostat products marketed by other companies with substantially greater resources than us. However, we believe Vitagel offers unique advantages in comparison to these other products due to Vitagel’s ability to facilitate healing through its collagen component, as well as its use of the patient’s autologous plasma instead of pooled donated human plasma as is the case with the leading products in the market. In 2008, recombinant human thrombin was approved for sale in the U.S. We believe that recombinant human thrombin will be positioned as an improvement to bovine-derived thrombin, a raw material in our Vitagel product, which could adversely affect our sales of Vitagel. It is too early to estimate the effect of this new technology on our Vitagel product. If management believes this effect to be material and adverse to our Vitagel sales, it will consider alternative formulations of Vitagel and the regulatory approvals that may be required for any such alternative formulations.
Vitasure competes with several thrombin products and surgical hemostats on the basis of its safety profile and pricing.
GOVERNMENT REGULATION
In order to market our products, we must apply for, be granted and maintain all necessary regulatory approvals, clearances and certifications. In addition, we must comply with all regulatory requirements in each applicable jurisdiction. The section entitled “OUR PRODUCTS” above details the regulatory approvals, clearances and certifications currently in place for our products.
The following discussion summarizes the material regulatory requirements applicable to our products.
United States
The medical devices that we manufacture and market, or intend to market, are subject to extensive regulation by the FDA. Pursuant to the Federal Food, Drug and Cosmetic Act (FFD&C Act) and the regulations promulgated thereunder, the FDA regulates the clinical testing, manufacture, labeling, distribution and promotion of medical devices. Noncompliance with applicable requirements can result in, among other things, fines, injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, failure of the government to grant premarket clearance or premarket approval for devices, withdrawal of marketing approvals/clearances and criminal prosecution.
In the U.S., medical devices are classified into one of three classes (Class I, II or III) on the basis of the controls deemed necessary by the FDA to reasonably assure their safety and effectiveness. Under FDA regulations, Class I devices, the least regulated category, are subject to general controls and Class II devices are subject to general and special controls. Generally, Class III devices are those that must receive premarket approval by the FDA to ensure their safety and effectiveness.
Before we can introduce a new device into the market, we must generally obtain market clearance through a 510(k) notification or premarket approval through a PMA application. A 510(k) clearance will be granted if the submitted information establishes that the proposed device is “substantially equivalent” to a legally marketed Class I or II medical device, or to a Class III medical device for which the FDA has not required a PMA. The
11
Table of Contents
FDA may determine that a proposed device is not substantially equivalent to a legally marketed device or that additional information or data are needed before a substantial equivalence determination can be made. A request for additional data may require that clinical studies be performed to establish the device’s “substantial equivalence.”
Commercial distribution of a device for which a 510(k) notification is required can begin only after the FDA issues a letter finding the device to be “substantially equivalent” to a predicate device. Pursuant to the FFD&C Act, the FDA must make a determination with respect to a 510(k) submission within 90 days of its receipt. The FDA may, and often does, extend this time frame by requesting additional data or information.
A “not substantially equivalent” determination, or a request for additional information, could delay or prevent the market introduction of new products for which we file such notifications. For any of our products that are cleared through the 510(k) process, modifications or enhancements that could significantly affect the safety or efficacy of the device or that constitute a major change to the intended use of the device will require new 510(k) submissions. The FDA has implemented a policy under which certain device modifications may be submitted as a “Special 510(k),” which will require only a 30-day review. Special 510(k)s are limited to those device modifications that do not affect the intended use or alter the fundamental scientific technology of the device and for which substantial equivalence can be demonstrated through design controls.
We must file a PMA if our proposed device is not substantially equivalent to a legally marketed Class I or Class II device, or if it is a Class III pre-amendment device (on the market since prior to May 28, 1976) for which FDA has called for PMAs. A PMA must be supported by valid scientific evidence that typically includes extensive data, including pre-clinical and clinical trial data, to demonstrate the safety and effectiveness of the device, as well as extensive manufacturing information.
FDA review of a PMA generally takes one to two years from the date the PMA is accepted for filing, but may take significantly longer. The review time is often significantly extended should the FDA ask for more information or clarification of information already provided in the submission.
During the PMA review period, an advisory committee, typically a panel of clinicians, will likely be convened to review and evaluate the application and provide recommendations to the FDA as to whether the device should be approved. The FDA is not bound by the recommendations of the advisory panel. Toward the end of the PMA review process, the FDA generally will conduct an inspection of the manufacturer’s facilities to ensure that they comply with Quality System Regulation (QSR) requirements.
If the FDA’s evaluations of both the PMA and the manufacturing facilities are favorable, the FDA will either issue an approval letter or an “approvable letter,” which usually contains a number of conditions that must be met in order to secure final approval of the PMA. When, and if, those conditions have been fulfilled to the satisfaction of the FDA, the agency will issue an approval letter, authorizing commercial marketing of the device for certain indications. If the FDA’s evaluation of the PMA or manufacturing facilities is not favorable, the FDA will deny approval of the PMA or issue a “not approvable letter.” The FDA may also determine that additional clinical trials are necessary, in which case PMA approval may be delayed up to several years while additional clinical trials are conducted and submitted as an amendment to the PMA. The PMA process can be expensive, uncertain and lengthy, and a number of devices for which other companies have sought FDA approval have never been approved for marketing.
Modifications to a device that is the subject of an approved PMA (including modifications to its labeling, raw material components or manufacturing process) may require approval by the FDA in the form of a PMA supplement or new PMA. Supplements to a PMA often require the submission of the same type of information required for an initial PMA, except that the supplement is generally limited to that information needed to support the proposed change from the product covered by the original PMA.
12
Table of Contents
If clinical trials of a device are required in connection with either a 510(k) notification or a PMA and the device presents a “significant risk,” we will be required to file an investigational device exemption (IDE) application prior to commencing clinical trials. The IDE application must be supported by data, typically including the results of animal and laboratory testing. If the IDE application is reviewed and approved by the FDA and one or more appropriate Institutional Review Boards (IRBs), clinical trials may begin at a specific number of investigational sites with a specific number of patients, as approved by the FDA. If the device presents a “non-significant risk” to the patient, we may begin the clinical trials after obtaining approval for the study by one or more appropriate IRBs. For “significant risk” devices, we must submit an IDE supplement to the FDA and receive approval from the FDA before we, or our investigator, may make a change to the investigational plan that may affect its scientific soundness or the rights, safety or welfare of human subjects. IRB approval may be required for changes in the investigational plan for both non-significant risk and significant risk devices.
Any products manufactured or distributed by us pursuant to FDA clearances or approvals are subject to extensive regulation by the FDA, including reporting and record keeping requirements. Device manufacturers are required to register their establishments and list their devices with the FDA and certain state agencies, and are subject to periodic inspections by the FDA and certain state agencies. The FFD&C Act requires devices to be manufactured in accordance with QSR requirements that impose certain procedural and documentation requirements upon us with respect to manufacturing and quality assurance activities. Medical devices are also subject to post-market reporting requirements for deaths or serious injuries when the device may have caused or contributed to the death or serious injury, and for certain device malfunctions that would be likely to cause or contribute to a death or serious injury if the malfunction were to recur. If safety or efficacy problems occur after the product reaches the market, the FDA may impose severe limitations on the use of any approved or cleared product.
Our labeling and promotion activities are subject to scrutiny by the FDA and, in certain instances, by the Federal Trade Commission. The FDA actively enforces regulations prohibiting marketing of products for unapproved or uncleared uses. We, as well as our products, are also subject to a variety of state laws and regulations in those states or localities where our products are or will be marketed. Any applicable state or local regulations may hinder our ability to market our products in those states or localities. We are also subject to numerous federal, state and local laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control and disposal of hazardous or potentially hazardous substances. There can be no assurance that we will not be required to incur significant costs to comply with such laws and regulations now or in the future or that such laws or regulations will not have a material adverse effect upon our ability to do business. Noncompliance with applicable requirements can result in, among other things, fines, injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, failure of the government to grant the pre-market clearance or premarket approval for devices, withdrawal of marketing approvals, clearances and criminal prosecution.
European Union
In order to sell our products within the EU, we are required to achieve compliance with the requirements of the EU Medical Devices Directive, or MDD, and affix a CE mark on our products to attest such compliance. To achieve this, our products must meet the “essential requirements” defined under the MDD relating to safety and performance and we must successfully undergo a verification of our regulatory compliance by an independent notified body referred to as a conformity assessment. The nature of the conformity assessment will depend on the regulatory class of our products. Under European law, our products, other than Imbibe and Aliquot (which are Class I-sterile and Class II-A, respectively), are likely to be in Class III. In the case of Class III products, we must (as a result of the regulatory structure which we have elected to follow) establish and maintain a complete quality system for design and manufacture as described in Annex II of the MDD and demonstrated by compliance with EN 13485:2003. We are certified as meeting the requirements of ISO 9001: 2000 and EN 13485 through July 1, 2012. Our notified body has audited our quality system and determined that it meets the requirements of the MDD. In addition, the notified body must certify the specific design of each device in
13
Table of Contents
Class III, IIa and IIb. As part of the design certification process, the notified body must also verify that the products comply with the essential requirements of the MDD. In order to comply with these requirements, we must, among other things, complete a risk analysis and may be required to present clinical data. The clinical data presented by us must provide evidence that the products meet the performance specifications claimed by us, provide sufficient evidence of adequate assessment of unwanted side effects and demonstrate that the benefits to the patient outweigh the risks associated with the device. We are subject to continued surveillance by the notified body and are required to report any serious adverse incidents to the appropriate authorities of the European Union member states. The certification of the notified body is valid for a maximum of five years but may be extended by application for additional five year periods. We also are required to comply with additional national requirements that are outside of the scope of the MDD. There can be no assurance that we will not be required to incur significant costs to comply with such laws and regulations now or in the future or that such laws or regulations will not have a material adverse effect upon our ability to do business.
THIRD PARTY REIMBURSEMENT
Successful sales of our products in the U.S. and other markets will depend on the availability of adequate coverage and reimbursement from third party payers relative to our products. In the U.S., health care providers, such as hospitals and physicians that purchase or utilize our products for treatment of their patients, generally rely on independent third party payers, such as private insurance companies or HMOs, and governmental payers, such as Medicare and Medicaid, to reimburse all or part of the costs and fees associated with the provision of items and services using our products. Physicians, hospitals, and other health care providers may not purchase our products if they do not receive satisfactory reimbursement from these payers for the cost of procedures using our products.
Each payer has its own process and standards for determining whether it will cover and reimburse a procedure or particular product. Private payers often rely on the lead of the governmental payers in rendering coverage and reimbursement determinations. Therefore, achieving favorable Medicare coverage and reimbursement is usually a significant gating issue for successful introduction of a new product. Medicare is a federally funded program managed by the Centers for Medicare and Medicaid Services (CMS), through local contractors that administer coverage and reimbursement for certain health care items and services furnished to the elderly and disabled. Medicaid is an insurance program for the poor that is both federally and state funded and managed by each state. The federal government sets general guidelines for Medicaid and each state creates specific regulations that govern their individual program.
Medicare provides, among other things, health care benefits that cover, within prescribed limits, the major costs of most medically necessary care for such individuals, subject to certain deductibles and co-payments. The Medicare program has established, and continues to establish, standards and guidelines for the coverage and reimbursement of certain procedures, equipment, supplies and other items and services. Generally, in order to be covered and reimbursed by Medicare, a health care item or service furnished to a Medicare beneficiary must be included in a statutory benefit category, must not be excluded from coverage by statute, and must be reasonable and necessary for the diagnosis or treatment of an illness or injury or to improve the functioning of a malformed body part. Medicare’s coverage decisions can be made on a nationwide basis or at the local contractor level, and coverage of an item or service can be reconsidered at any time. Several of Medicare’s contractors have policies governing coverage of our products or procedures using our products, including vertebroplasty and kyphoplasty. In July 2008, CMS identified vertebroplasty and kyphoplasty, procedures for which our Cortoss product is cleared, as one of 20 potential national coverage determination topics; however, the agency has taken no further action since that time.
The methodology for determining (1) the coverage status of our products; and (2) the amount of Medicare reimbursement for our products varies based upon, among other factors, the setting in which a Medicare beneficiary received health care items and services. Acute care hospitals are generally reimbursed by Medicare for inpatient operating costs based upon prospectively determined rates. Under the inpatient Prospective Payment
14
Table of Contents
System (PPS), acute care hospitals receive a predetermined payment rate based upon the Medicare Severity Diagnosis-Related Group, or MS-DRG, into which each Medicare beneficiary stay is assigned, regardless of the actual cost of the services provided. These payment rates are adjusted for geographic variations in costs, medical education payments, and adjustments for hospitals that serve a disproportionate share of low-income patients. In some cases, hospitals also may receive adjustments for use of certain new technologies. Certain additional or “outlier” payments may be made to a hospital for cases involving unusually high costs. Accordingly, acute care hospitals generally do not receive direct Medicare reimbursement under the inpatient PPS for the distinct costs incurred in purchasing our products. Rather, reimbursement for these costs is deemed to be included within the MS-DRG-based payments made to hospitals for the services furnished to Medicare-eligible inpatients in which our products are utilized. Because inpatient PPS payments are based on predetermined rates and may be less than a hospital’s actual costs in furnishing care, acute care hospitals have incentives to lower their inpatient operating costs by utilizing equipment, devices and supplies, including those sold by us, that will reduce the length of inpatient stays, decrease labor or otherwise lower their costs. Our product revenue could be affected negatively if acute care hospitals discontinue product use due to insufficient reimbursement, or if other treatment options are perceived to be more cost-effective. Similarly, some states reimburse certain health care providers for inpatient services under their Medicaid programs by using prospective rates for diagnosis-related groups of illnesses.
Medicare also reimburses most hospital outpatient services on a prospective payment basis, subject to certain adjustments, exceptions, and limitations. Under the Medicare hospital outpatient PPS, services are grouped based on clinical and cost similarity into ambulatory payment classifications (APCs). All services in an APC have the same rate, intended to cover the equipment, supplies, and labor used to perform the service. Unlike the inpatient PPS, if a patient receives multiple services, the hospital will receive reimbursement through multiple APCs. Similar to the inpatient PPS, this system provides hospitals with an incentive to select equipment and supplies that reduce the costs of treating a patient. Therefore, health care providers may refuse to use our products if coverage or reimbursement is inadequate.
Any changes in federal legislation, regulations and policy affecting Medicare coverage and reimbursement relative to our products could have a material effect on our results of operations. Changes in the health care industry in the United States and elsewhere also could adversely affect the demand for our products as well as the way in which we conduct our business. Significantly, health reform legislation currently before Congress would require most individuals to have health insurance, establish new regulations on health plans, create insurance pooling mechanisms, and other expanded public health care measures. This legislation also would reduce Medicare spending on services provided by hospitals and other providers. The version of the legislation passed by the House of Representatives would impose a 2.5 percent tax on the first taxable sale of any medical device, and the version passed by the Senate includes a $2 billion annual fee or excise tax on the medical device manufacturing sector. Various health care reform proposals have also emerged at the state level. We cannot predict what health care initiatives, if any, will be implemented at the federal or state level, or the effect any future legislation or regulation will have on us. However, if the excise tax contained in the House or Senate health reform bills is enacted into law, our results of operations could be materially and adversely affected.
Inadequate coverage or reimbursement by private insurance companies and government programs could significantly reduce usage of our products. In addition, an increasing emphasis on managed care in the U.S. has placed, and we believe will continue to place, greater pressure on medical device pricing. Such pressures could have a material adverse effect on our ability to sell our products profitably. Failure by hospitals and other users of our products to obtain favorable coverage or reimbursement from third party payers or changes in governmental and private third party payers’ policies toward coverage or reimbursement for procedures employing our products could reduce demand for our products.
The European Union has not established a uniform process to govern the pricing and reimbursement of medical devices. Consequently, the individual EU member states operate various combinations of centrally financed health care systems and private health insurance systems. The relative importance of government and private systems varies from country to country. The choice of devices is subject to constraints imposed by the
15
Table of Contents
availability of funds within the purchasing institution and by hospital and authority priorities and procedures. Medical devices are most commonly sold to hospitals or health care facilities at a price set by negotiation between the buyer and the seller. A contract to purchase products may result from an individual initiative or as a result of a competitive bidding process. In either case, the purchaser pays the supplier, and payment terms vary widely throughout the EU. Failure to obtain favorable negotiated prices with hospitals or health care facilities could adversely affect sales of our products.
HEALTH CARE FRAUD AND ABUSE LAWS
We are subject to various federal and state laws pertaining to health care fraud and abuse, including anti-kickback laws and false claims laws. Similar regulations have been adopted by EU member states. Violations of these laws are punishable by criminal, civil and/or administrative sanctions, including, in some instances, fines, imprisonment, and exclusion from participation in federal, state and national health care programs, including Medicare, Medicaid and veterans’ health programs. Because of the far-reaching nature of these laws, there can be no assurance that the occurrence of one or more violations of these laws would not result in a material adverse effect on our business, financial condition and results of operations.
Anti-Kickback Laws. Our operations are subject to federal and state anti-kickback laws. The federal anti-kickback law prohibits entities such as us from knowingly and willfully offering, paying, soliciting or receiving any form of remuneration (including any kickback, bribe or rebate) in return for the referral of items or services for which payment may be made under a federal health care program, or in return for the recommendation, arrangement, purchase, lease or order of items or services for which payment may be made under a federal health care program. The definition of remuneration has been broadly interpreted to include anything of value, including for example gifts, discounts, the furnishing of supplies or equipment, payments of cash and waivers of payments. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals or otherwise generate business involving goods or services reimbursed in whole or in part under federal health care programs, the statute has been violated. Violation of the federal anti-kickback law is a felony, punishable by criminal fines and imprisonment for up to five years or both. In addition, the Department of Health and Human Services may impose civil penalties and exclude violators from participation in federal health care programs such as Medicare and Medicaid. Exclusion of a manufacturer would preclude any federal health care program from paying for its products. In addition, enforcement officials have argued (and some courts have agreed) that kickback arrangements can provide the basis for an action under the Federal False Claims Act, which is discussed below.
Government officials have focused recent enforcement efforts on, among other things, the sales and marketing activities of health care companies, and recently have brought cases against individuals or entities with personnel who allegedly offered unlawful inducements to potential or existing customers in an attempt to procure their business. Settlements of these cases by health care companies have involved significant fines and/or penalties and in some instances criminal pleas.
Many states have adopted similar prohibitions against payments intended to induce referrals of products or services paid by Medicaid or other third party payers.
False Claims. The federal False Claims Act imposes civil and criminal liability on individuals or entities who submit (or cause the submission of) false or fraudulent claims for payment to the government. Violations of the federal False Claims Act may result in penalties equal to three times the damages which the government sustained, an assessment of between $5,500 and $11,000 per claim, civil monetary penalties and exclusion from participation in the Medicare and Medicaid programs.
The federal False Claims Act also allows a private individual to bring a qui tam suit on behalf of the government against an individual or entity for violations of the False Claims Act. In a qui tam suit, the private plaintiff is responsible for initiating a lawsuit that may eventually lead to the government recovering money of
16
Table of Contents
which it was defrauded. After the private plaintiff has initiated the lawsuit, the government must decide whether to intervene in the lawsuit and become the primary litigant. In the event the government declines to join the lawsuit, the private plaintiff may choose to pursue the case alone, in which case the private plaintiff’s counsel will have primary control over the case (although the government must be kept apprised of the progress of the lawsuit). In return for bringing the suit on the government’s behalf, the statute provides that the private plaintiff is entitled to receive up to 30% of the recovered amount from the litigation proceeds if the litigation is successful plus reasonable expenses and attorneys fees. Recently, the number of qui tam suits brought against entities in the health care industry has increased dramatically. In addition, a number of states have enacted laws modeled after the False Claims Act that allow those states to recover money which was fraudulently obtained from the state.
Other Fraud and Abuse Laws. The Health Insurance Portability and Accountability Act of 1996 created, in part, two new federal crimes: Health Care Fraud and False Statements Relating to Health Care Matters. The Health Care Fraud statute prohibits the knowing and willful execution of a scheme or artifice to defraud any health care benefit program. A violation of the statute is a felony and may result in fines and/or imprisonment. The False Statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact by any trick, scheme or device or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for health care benefits, items or services. A violation of this statute is a felony and may result in fines and/or imprisonment.
The countries of the EU have individual and varying legislative provisions governing health care fraud and abuse, including provisions governing inducement to purchase. Violation of these provisions can lead to action both against the provider of the device and against the health care provider.
PRODUCT LIABILITY AND INSURANCE
We manufacture medical devices used on patients in surgery, and we may be subject to product liability lawsuits. We have experienced two product liability claims to date, which we have successfully resolved. There can be no assurance that other product liability claims will not be asserted against us in the future. Under certain of our agreements with our independent distributors and hospital customers, we provide indemnification from product liability claims. Any product liability claim brought against us, with or without merit, could result in the increase of our product liability insurance rates or the inability to secure coverage in the future. In addition, we would have to pay any amount awarded by a court in excess of policy limits. We maintain product liability insurance in the annual aggregate amount of up to $20 million, although our insurance policies have various exclusions. Thus, we may be subject to a product liability claim for which we have no insurance coverage, in which case we may have to pay the entire amount of any award.
EMPLOYEES
As of December 31, 2009, we had 281 full-time employees, with 144 employees at our Malvern, Pennsylvania headquarters, 121 employees in other parts of the U.S. and 16 employees in Europe. We consider our relations with our employees to be good.
| ITEM 1A. | RISK FACTORS |
We have organized the risk factors set forth below that may affect us or our securities into the following four categories: (i) risks related to our business and industry; (ii) risks related to our financial results and financing; (iii) risks related to our intellectual property and litigation; and (iv) risks related to investing in our stock and equity markets.
17
Table of Contents
RISKS RELATED TO OUR BUSINESS AND INDUSTRY
If our current products are not commercially successful, our operating results will be impaired and our viability will be jeopardized.
We are highly dependent on successfully selling our products that have received regulatory approval or clearance. To date, Vitoss, Imbibe, Cortoss, Vitagel, CellPaker and Vitasure are approved or cleared for specified uses in the United States. Vitoss, Cortoss, Aliquot, Vitagel, CellPaker and Vitasure are cleared or approved for specified uses in the European Union and countries adhering to the regulatory standards of the European Union. Vitoss, Cortoss and Aliquot are also cleared for specified uses in Australia. Revenues generated from sales of our approved or cleared products in their approved territories have not been and in the future may not be sufficient to fund operations. We received FDA clearance in June 2009 to market and sell Cortoss for use in vertebral augmentation and launched the product in the U.S. in July 2009. Cortoss represents a new technology which will generally need to be approved by technology review boards of target hospitals in order for the hospitals to carry the product. The amount of time to obtain approvals from the technology review boards can be difficult to predict and we cannot guarantee that we will be able to secure widespread hospital approvals for Cortoss. During 2009, we sold approximately $1.1 million of Cortoss and Aliquot in the U.S. We expect our U.S. Cortoss sales will increase as the product launch progresses, we obtain further hospital approvals for the product and our sales force trains more physicians on the product. There is no assurance that Cortoss or our other products will generate revenues sufficient to enable us to fund operations.
Certain factors that may also limit our ability to increase sales include:
| • | the ability of our direct sales representatives to develop and retain a customer base and compete effectively with larger sales forces offering more extensive product portfolios; |
| • | the introduction of new products based upon competing technologies into the market by competing orthopedic, biomedical and other companies; |
| • | increased efforts by hospitals to reduce costs of medical devices; |
| • | our dependence in certain geographical areas on the efforts of independent agents and distributors over which we have limited control to promote the use of our products; |
| • | our dependence on the continued publication of independent pre-clinical and clinical data to support the use of our products; |
| • | our need to educate and train a sufficient number of surgeons to create demand for our products; and |
| • | the need for payers to authorize insurance reimbursement for procedures using our products. |
Market acceptance of our products will largely depend on our ability to demonstrate their relative safety, efficacy, cost-effectiveness and ease of use. Surgeons will not use our products unless they determine, based on experience, clinical data and recommendations from prominent surgeons and mentors that our products are safe and effective. Our products are based on newer technologies and compete with more established treatments currently accepted as the standards of care. The attributes of some of our products may require some changes in surgical techniques that have become standard within the medical community, and there may be resistance to change. Therefore, for these products, we must be able to convince surgeons who currently favor existing techniques to switch to new procedures that would use our products. Many surgeons will not purchase our products until there is sufficient, long-term clinical evidence to convince them to alter their existing treatment methods. In addition, surgeons may be slow to change their medical treatment practices because of perceived liability risks arising from the use of new products and the uncertainty of third party reimbursement for our products. Any failure to gain market acceptance of our products could result in lower sales and our inability to become profitable.
18
Table of Contents
If we are unable to operate an effective sales and distribution network, our ability to generate sales and become profitable will be impaired.
We have assembled a field sales network of direct sales representatives and independent non-stocking distributors in the U.S. to market Vitoss, Imbibe, Cortoss, Aliquot, Vitagel, CellPaker and Vitasure. Outside of the U.S., we utilize a network of independent stocking distributors to market Vitoss, Cortoss, Aliquot, Vitagel, CellPaker and Vitasure. We are dependent upon our direct sales representatives and distributors for the sale of our products. Any failure to maintain and effectively manage our distribution network will impair our ability to generate additional sales and become profitable.
In an effort to further grow our sales, we continue to build our sales management team and add direct sales representatives to our organization in the U.S. for certain open or underserved territories as well as to support the launch of Cortoss. At the end of 2008, we added a direct sales force in the United Kingdom, which includes six direct sales representatives as of March 1, 2010. The addition of direct sales representatives will increase our operating expenses. Furthermore, there is no assurance that adding direct sales representatives will improve sales or that our direct sales representatives will be successful in generating sufficient sales to cover the cost of supporting their sales activities. There is no assurance that we will be able to attract and retain qualified personnel to market or sell our products or that we will successfully manage this type of sales and distribution method.
There can be no assurance that our distributors will perform effectively their obligations in their respective territories as expected, or that we will continue to derive any revenue from these arrangements. We cannot assure that our interests will continue to coincide with those of our distributors. In addition, we cannot assure that our distributors will not develop independently, or with other companies, competitive products.
Most of our product sales generated by independent U.S. distributors are from Vitoss. These distributors generally sell products for other orthopedic companies. A single distributor may sell Vitoss, as well as hardware manufactured by other orthopedic companies consisting of metal plates, screws and spinal cages, to end user hospitals. Our sales could be adversely affected if, for any reason, one or more of our successful distributors lost their hardware product line provided by other orthopedic companies since Vitoss is typically sold with such hardware products. Additionally, our independent distributors may be unwilling to carry or unable to effectively sell Vitoss as a result of the introduction of new products based upon other technologies that could compete with Vitoss. Our sales could be adversely affected if one or more of our successful distributors eliminated Vitoss from its product line for any other reason or terminated its arrangement with us. Our sales could also be adversely affected if our independent distributors become concerned that their arrangements with us could be eliminated as a result of another company’s offer or threat to acquire us. The complete product line represented by the distributors, including our products, is an important factor in the distributors’ ability to penetrate the markets for our products. Accordingly, our ability to penetrate the markets that we serve or intend to serve is highly dependent upon the quality and breadth of the other product lines carried by our distribution network, the components of which may change from time to time, and over which we have little or no control.
If we do not manage commercial scale manufacturing capability and capacity for our products in compliance with regulatory requirements and in a cost-effective manner, our product sales and operating results may suffer.
The manufacture of our products is subject to regulation and periodic inspection by various regulatory bodies for compliance with the FDA Quality System Regulation (the QSR), and EU Medical Devices Directive requirements, ISO 9000 Series standards, ISO/European Norm (EN) 13485 and equivalent requirements.
We manufacture our commercial Vitoss, Vitagel and Cortoss products at our 24,800-square foot FDA-registered manufacturing facilities located in Malvern, Pennsylvania. These facilities are leased through July 2017 and are certified as meeting the requirements of ISO 9000: 2000 and EN 13485 through July 1, 2012. We are currently required to demonstrate and maintain compliance with the QSR, which is complex regulatory
19
Table of Contents
scheme that covers the methods and documentation of the design, testing, control, manufacturing, labeling, quality assurance, packaging, storage and shipping of our products. The FDA enforces the QSR through periodic unannounced inspections. We have been, and anticipate in the future being, subject to such inspections. We assumed full responsibility for manufacturing Vitagel in 2006 and CellPaker in 2007 after receiving the requisite approvals from the FDA. There can be no assurance that we will be able to comply with FDA requirements to continue to manufacture Vitoss, Cortoss, Vitagel or CellPaker, which could result in delay or inability to manufacture and sell the products. We believe our existing manufacturing facilities have the capacity to meet our commercial needs through at least 2012 and plan to transition processing of collagen for Vitagel in-house after obtaining regulatory approval for such processing facility, which we expect will occur in the second half of 2010. If we do not receive regulatory approval for our collagen processing facility, we may not be able to manufacture Vitagel to adequately meet commercial demand, if at all.
Our third party manufacturers are also regulated by governmental agencies and are required to be ISO 9001/EN 13485-certified or otherwise meet our quality system requirements. We manufacture CellPaker, Imbibe and Aliquot through outside third party contract manufacturers. Our Vitoss is converted to Vitoss Foam and Vitoss Bioactive Foam by Kensey, and we purchase Vitasure finished product from Medafor, which uses third party manufacturers to make the product.
Our product sales depend upon, among other things, our ability to manufacture our products in commercial quantities, in compliance with regulatory requirements and in a cost-effective manner. There can be no assurance that regulatory authorities will not, during the course of an inspection of existing or new facilities, identify what they consider to be deficiencies in meeting the applicable quality standards and request or seek remedial action.
Failure to comply with such regulations or a delay in attaining compliance may result in:
| • | warning letters; |
| • | injunctions suspending the manufacture of products; |
| • | civil and criminal penalties; |
| • | refusal to grant premarket approvals or clearances, CE marks, CE mark renewals, or clearances to products that are subject to future or pending submissions; |
| • | product recalls or seizures of products; |
| • | total or partial suspensions of production; and |
| • | refusal to permit the import or export of our products. |
The imposition of any of these sanctions could cause our business and operating results to suffer. Furthermore, if we or any of our third-party manufacturers cannot produce our products, our product sales would suffer.
We are dependent on a limited number of specialty suppliers of certain raw materials, and our business and financial results could be harmed if a specialty supplier no longer provides materials to us.
Our ability to manufacture Vitoss, Cortoss and Vitagel and to have Vitoss Foam and Vitoss Bioactive Foam manufactured by and for us is dependent on a limited number of specialty suppliers of certain raw materials. We have several single-source suppliers for raw materials used in our products. The failure of a supplier to continue to provide us with these materials at a price or quality acceptable to us, or at all, would have a material adverse effect on our ability to manufacture these products. Although we believe that we maintain good relationships with our suppliers, we cannot assure that such supplies and services will continue to be available with respect to our current and future commercialized products.
20
Table of Contents
We are still in the process of ensuring that our collagen supply and manufacturing capabilities for Vitagel will be sufficient after Allergan, our former supplier of processed collagen for Vitagel, notified us in 2007 of its intention to shut down its collagen-processing facility and cease supplying us with this collagen raw material under our then-existing supply agreement. In November 2007, we entered into a new supply and license agreement under which Allergan agreed to sell us additional lots of processed collagen in 2008 before its facility closed. We used all of the strategic collagen reserve supply received from Allergan in 2008 to manufacture reserve Vitagel inventory and additional Vitagel intermediate goods, which we believe will be sufficient to meet our needs for Vitagel into the second half of 2011. In 2008, we renovated leased property to establish a facility to process collagen raw materials obtained after exhausting the strategic processed collagen reserve acquired from Allergan. We must obtain FDA approval of the new facility in order to sell Vitagel made with collagen processed at the facility, and this approval is pending. We are also in the process of negotiating supply agreements for collagen raw materials for Vitagel. The occurrence of any of the following could have a material adverse effect on our ability to manufacture and sell Vitagel and CellPaker, which is approved for use only in conjunction with Vitagel:
| • | delays caused by the need to secure regulatory approval of the collagen raw material processing facility that we recently built; or |
| • | our inability to secure an adequate supply of collagen raw materials meeting our pricing, quality and regulatory requirements. |
If we are unable to sell Vitagel and CellPaker for a period of time, our customers may seek alternative products and we may lose market share for Vitagel and CellPaker. Further, there is no assurance that we will regain any Vitagel and CellPaker market share that may be lost.
We also cannot assure that supply or manufacturing capability will be sufficient to meet our own needs and Angiotech’s needs if Angiotech elects to be supplied by us under our license agreement with them. Under our license agreement with Angiotech, in the future we may be obligated, under certain circumstances, to provide CoStasis (an Angiotech-branded Vitagel product) and certain CoStasis materials to Angiotech. Our collagen supply or manufacturing capabilities may be inadequate, and we may need to seek additional suppliers for CoStasis materials.
We are dependent on a limited number of specialized manufacturing suppliers for certain products, and our business and financial results could be harmed if a third-party manufacturer fails to supply us with these products.
We are dependent upon Kensey and other third-party manufacturers for the supply of certain products. Approximately 67% of our product sales during 2009 were from products based upon our Vitoss Foam platform co-developed with Kensey, including Vitoss Bioactive Foam. Our Vitoss is converted to Vitoss Foam and Vitoss Bioactive Foam by Kensey. We also manufacture certain delivery accessory products through third party specialty suppliers. There is no assurance that these manufacturers will continue to supply our requirements. If they fail to do so, we cannot guarantee that we will be able to engage another supplier for these products on a timely basis and on acceptable terms. A delay in our ability to obtain adequate supplies of these products may negatively impact our product sales.
If we fail to obtain and maintain the regulatory approvals or clearances necessary to make or sell our products, sales could be delayed or never realized.
The jurisdictions in which we seek to market our products will regulate these products as medical devices. In most circumstances, we and our distributors and agents must obtain regulatory clearances, approvals and certifications and otherwise comply with extensive regulations regarding safety, quality and efficacy standards. These regulations vary from country to country, and the regulatory review can be lengthy, expensive and uncertain. We may not obtain or maintain the regulatory clearances, approvals and certifications necessary to
21
Table of Contents
make or market our products in our targeted markets. Moreover, regulatory clearances, approvals and certifications that are obtained may involve significant restrictions on the anatomic sites and types of procedures for which our products can be used. In addition, we may be required to incur significant costs in obtaining or maintaining our regulatory clearances, approvals and certifications. If we do not obtain or maintain regulatory clearances, approvals and certifications to enable us to make or market our products in the U.S. or elsewhere, or if the clearances, approvals and certifications are subject to significant restrictions, we may never generate significant revenues. The regulatory requirements in some of the jurisdictions where we currently market or intend to market our products are summarized below.
United States
Regulation by FDA. The FDA regulates the clinical testing, manufacturing, labeling, distribution and promotion of medical devices. To date, we have received 510(k) clearance from the FDA to market our Vitoss, Vitoss Foam, Vitoss Bioactive Foam, Imbibe and Cortoss products. Aliquot is exempt from 510(k) clearance.
We market Vitoss, Imbibe, Cortoss, Aliquot, Vitagel, CellPaker and Vitasure in the U.S., and we market Vitoss, Cortoss, Aliquot, Vitagel, CellPaker and Vitasure outside the U.S. We directly manufacture Vitoss, Cortoss, Vitagel and CellPaker in the U.S., and we manufacture Imbibe and Aliquot in the U.S. through outside third party contract manufacturers. In addition, under our agreement with Kensey, Kensey has the exclusive right to manufacture any approved or cleared jointly developed products under the agreement. This right extends until February 2014 for the Vitoss Foam product platform and at least until 2024 for the Vitoss Bioactive Foam product platform. We purchase Vitasure from Medafor, which utilizes third party manufacturers to produce this product. Under our distribution agreement with Medafor, Medafor is responsible for maintaining regulatory approval and ensuring compliance with manufacturing requirements for Vitasure.
Vitoss, Cortoss, Vitagel and Vitasure, as well as any other products that we manufacture or distribute following their approval or clearance by the FDA, will be subject to extensive regulation by the FDA. If safety or efficacy problems occur after the product reaches the market, the FDA may impose severe limitations on the use of the product. Moreover, modifications to the approved or cleared product may require the submission of a new 510(k) notification or premarket approval application or premarket application supplement. We or our partners may not be successful in obtaining approval to market the modified product in a timely manner, if at all. Noncompliance with applicable requirements can result in, among other things, fines, injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, failure of the government to grant premarket clearance or premarket approval for devices, withdrawal of marketing approvals and criminal prosecution.
Medical devices may be marketed only for the indications for which they are approved or cleared. The FDA may not approve or clear indications that are necessary or desirable for successful commercialization. Indeed, the FDA may refuse our requests for 510(k) clearance or premarket approval of new products, new intended uses or modifications to existing products. Our clearances can be revoked if safety or effectiveness problems develop.
European Union and Other International Markets
General. International sales of medical devices are subject to the regulatory requirements of each country in which the products are sold. Accordingly, the introduction of our products in markets outside the United States will be subject to regulatory approvals or clearances in those jurisdictions. The regulatory review process varies from country to country. Many countries also impose product standards, packaging, labeling and language requirements and import restrictions on medical devices. In addition, the EU and each country outside of the EU have its own tariff regulations, duties and tax requirements. The approval or clearance by foreign government authorities is uncertain and can be expensive. Our ability to market our products could be substantially limited due to delays in receipt of, or failure to receive, the necessary approvals or clearances.
22
Table of Contents
Requirement of CE Mark Certification in the European Union. To market a product in the European Union, we must be entitled to affix a CE mark, an international symbol of adherence to quality assurance standards and compliance with applicable European medical device directives. A CE mark, preceded by the relevant certification process, enables us to market a product in all of the countries of the European Union, as well as in other countries, such as Switzerland and Turkey, that have mutual recognition agreements with the EU or have adopted the European Union’s regulatory standards. To date, we have received CE mark approval for the use of Vitoss as a bone graft substitute, for the use of Cortoss in screw augmentation and vertebral augmentation procedures, for Vitagel as a surgical hemostat and for certain delivery accessories for Vitoss, Cortoss and Vitagel. Vitasure has CE mark approval for use as a surgical hemostat. There can be no assurance that we will maintain CE mark approval for our products or that we will receive CE mark approval for other devices.
After clearance or approval of our products, we are subject to continuing regulation by the FDA. If we fail to comply with FDA regulations, our business could suffer.
Even after clearance or approval of a product, we are subject to continuing regulation by the FDA, including the requirements that our facility be registered and our devices listed with the agency. We are subject to Medical Device Reporting regulations, which require us to report to the FDA if our products may have caused or contributed to a death or serious injury or malfunction in a way that would likely cause or contribute to a death or serious injury if the malfunction were to recur. We must report corrections and removals to the FDA where the correction or removal was initiated to reduce a risk to health posed by the device or to remedy a violation of the Federal Food, Drug, and Cosmetic Act caused by the device that may present a risk to health, and maintain records of other corrections or removals. The FDA closely regulates promotion and advertising.
The FDA and state authorities have broad enforcement powers. Our failure to comply with applicable regulatory requirements could result in enforcement action by the FDA or state agencies, which may include any of the following sanctions:
| • | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
| • | repair, replacement, refunds, recall or seizure of our products; |
| • | operating restrictions or partial suspension or total shutdown of production; |
| • | refusing or delaying our requests for 510(k) clearance or premarket approval of new products or new intended uses; |
| • | withdrawing 510(k) clearance or premarket approvals that have already been granted; and |
| • | criminal prosecution. |
If any of these events were to occur, they could harm our business.
We have modified some of our products without FDA clearance. The FDA could retroactively determine that the modifications were improper and require us to stop marketing and recall the modified products until such clearance is obtained.
Any modification to an FDA-cleared device that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, requires a new 510(k) clearance; any change that affects the safety or effectiveness of a PMA approved device requires a premarket approval supplement. We have made modifications to some of our products in the past, and may make additional modifications in the future that we believe, based upon FDA’s regulations and guidance, do not or will not require additional clearances or approvals. If the FDA disagrees, we could be required to obtain new clearances or approvals for the modifications. We also may be required to recall and to stop marketing modified products until such clearance or approval is obtained, which could harm our operating results. We may be required to submit extensive pre-clinical and clinical data to support these supplementary submissions, depending on the nature of the changes. We may not be able to obtain additional 510(k) clearances or premarket approvals for modifications to,
23
Table of Contents
or additional indications for, our existing products in a timely fashion, or at all. Delays in obtaining future clearances or approvals would adversely affect our ability to introduce new or enhanced products in a timely manner, which in turn would harm our operating results.
If we cannot keep up with technological changes and marketing initiatives of competitors, sales of our products may be harmed.
Extensive research efforts and rapid technological change characterize the market for products in the orthopedic and biomaterials markets. We anticipate that we will face intense competition from medical device, medical products and pharmaceutical companies. There are many products that are competitive to our Vitoss platform products. For instance, the use of bone morphogenic proteins, or BMPs, in orthopedic surgery has increased significantly in the past five years and competitive stem cell products have recently entered the market, a trend we expect to continue. With respect to Cortoss, several PMMA bone cements have received 510(k) clearance since 2004 for vertebral augmentation of VCFs. Further, recombinant human thrombin was approved by the FDA in 2008 and may be positioned as a better alternative to bovine-derived collagen, a raw material in our Vitagel product. Our products could be rendered uncompetitive or obsolete by these and other competitors’ new technologies. We may be unable to respond to technological advances by others through the development and introduction of new products or the modification of existing products. Moreover, many of our existing and potential competitors have substantially greater financial, marketing, sales, distribution, manufacturing and technological resources than us. These competitors may be in the process of seeking FDA or other regulatory approvals or clearances, or patent protection, for competitive products. Our competitors could, therefore, commercialize competing products in advance of our products. They may also enjoy substantial advantages over us in terms of:
| • | research and development expertise; |
| • | experience in conducting clinical trials; |
| • | experience in regulatory matters; |
| • | manufacturing efficiency; |
| • | name recognition; |
| • | sales and marketing expertise; |
| • | established distribution channels; and |
| • | established relationships with health care providers and payers. |
These advantages may adversely affect our plans for market acceptance of our products.
If health care providers cannot obtain third party reimbursement for procedures using our products, or if such reimbursement is inadequate, we may never become profitable.
Successful sales of our products in the United States and other markets will depend on the availability of adequate reimbursement from third party payers. Each payer has its own process and standards for determining whether it will cover and reimburse a procedure or particular product. Private payers often rely on the lead of the governmental payers in rendering coverage and reimbursement determinations. Therefore, achieving favorable Medicare coverage and reimbursement is usually a significant issue for successful introduction of a new product. The Medicare program has established, and continues to establish, standards and guidelines for the coverage and reimbursement of certain procedures, equipment, supplies and other items and services. Medicare’s coverage decisions can be made on a nationwide basis or at the local contractor level, and coverage of an item or service can be reconsidered at any time. Several of Medicare’s contractors have policies governing coverage of our products or procedures using our products, including vertebroplasty and kyphoplasty. In July 2008, CMS identified vertebroplasty and kyphoplasty, procedures for which our Cortoss product is cleared, as one of twenty
24
Table of Contents
potential national coverage determination topics; however, the agency has taken no further action since that time. Decisions by CMS or its local contractors to limit coverage of our products could significantly reduce use of our products, particularly if other payers adopt similar policies.
Health care providers, such as hospitals and surgery centers that purchase medical devices for treatment of their patients, generally rely on third party payers to reimburse all or part of the costs and fees associated with the procedures performed with these devices. Both public and private insurance reimbursement plans are central to product acceptance and use as health care providers may refuse to use our products if reimbursement is inadequate. Inadequate reimbursement by private insurance companies and government programs could significantly reduce usage of our products.
Any changes in federal legislation, regulations and policy affecting Medicare coverage and reimbursement relative to our products could have a material effect on our results of operations. Changes in the health care industry in the United States and elsewhere also could adversely affect the demand for our products as well as the way in which we conduct our business. Significantly, health reform legislation currently before Congress would require most individuals to have health insurance, establish new regulations on health plans, create insurance pooling mechanisms, and other expanded public health care measures. This legislation also would reduce Medicare spending on services provided by hospitals and other providers. Various health care reform proposals have also emerged at the state level. We cannot predict what health care initiatives, if any, will be implemented at the federal or state level, or the effect any future legislation or regulation will have on us.
In addition, an increasing emphasis on managed care in the United States has placed, and we believe will continue to place, greater pressure on medical device pricing. Such pressures could have a material adverse effect on our ability to sell our products or to operate profitably.
If we do not successfully train a sufficient number of surgeons to use our products, demand for our products could be adversely affected.
It is critical to the commercial success of our products that our independent distributors and direct sales representatives succeed in training a sufficient number of surgeons and in providing them adequate instruction in the use of our products. This training requires a commitment of time and money by surgeons which they may be unwilling to give. Even if surgeons are willing, if they are not properly trained, they may misuse or ineffectively use our products. This may result in unsatisfactory patient outcomes, patient injury, negative publicity or lawsuits against us, any of which could damage our business, reduce product sales and subject us to claims and other liabilities.
We may incur significant liability if it is determined that we are promoting the “off-label” use of our medical devices. The unapproved or “off-label” use of our products could also adversely affect the reputation of our products and our company.
Under the Federal Food, Drug, and Cosmetic Act and other laws, we are prohibited from labeling, training, or promoting our products for off-label uses. This prohibition means that we cannot proactively discuss or provide information or training on the use of our products outside of their cleared or approved uses, except in certain limited scientific and other settings. In the practice of medicine, physicians may use devices for off-label uses. Because the interpretation of these requirements is subjective, the FDA could deem some of our activities as promotional even when we believe in good faith that such activities were not promotional. A company that is found to have improperly promoted off-label uses may be subject to significant liability, including civil and administrative remedies as well as criminal sanctions. Enforcement measures taken against us could harm our business and our reputation.
The medical devices that we manufacture and market, or intend to market, are subject to extensive regulation by the FDA, the European Union Medical Devices Directive and other worldwide regulatory agencies. In order to market our products, we must apply for, receive and maintain all necessary regulatory approvals or
25
Table of Contents
clearances in each applicable jurisdiction for specified uses of the products. Under FDA regulations and laws of other countries, we are only permitted to commercially distribute our products for approved indication(s) or use(s); failure to comply with these regulations may subject us to FDA and other enforcement action.
Cortoss received a CE Mark, an international symbol of adherence to quality assurance standards and compliance with applicable European medical device directives, in January 2003 for use in repairing vertebral compression fractures and in October 2001 for use in screw augmentation procedures. However, surgeons may have attempted to use Cortoss “off-label” in procedures to repair vertebral compression fractures performed prior to the European Union’s approval of Cortoss for this type of procedure. Furthermore, not all surgeons have been trained in the proper use of Cortoss to repair vertebral compression fractures. A surgeon who has not been properly trained to use Cortoss in a procedure to repair vertebral compression fractures could pose a risk to the reputation of our Cortoss product. In addition, the occurrence of an adverse event while using our product “off-label” in procedures other than the repair of vertebral compression fractures or screw augmentation could adversely affect the reputation of Cortoss or any of our other products as well as us and could subject us to liability.
We could be negatively impacted by future interpretation or implementation of federal and state fraud and abuse laws, including anti-kickback laws and false claims laws.
We are subject to various federal and state laws pertaining to health care fraud and abuse, including anti-kickback laws and false claims laws. Violations of these laws are punishable by criminal, civil, and/or administrative sanctions, including, in some instances, imprisonment and exclusion from participation in federal and state health care programs, including Medicare, Medicaid, and veterans’ health programs. Although we seek to structure our business practices to be in compliance with applicable requirements, these laws are broadly written and broadly interpreted, and it is often difficult to determine precisely how these laws will be applied in specific circumstances.
Health care fraud and abuse regulations are complex, and even minor, inadvertent irregularities can potentially give rise to claims that the law has been violated. Any violations of these laws could result in a material adverse effect on our business, financial condition and results of operations. If there is a change in law, regulation or administrative or judicial interpretations, we may have to change our business practices or our existing business practices could be challenged as unlawful, which could have a material adverse effect on our business, financial condition and results of operations. Any state or federal review of us, regardless of the outcome, could be costly and time consuming.
We could become subject to false claims litigation under federal or state statutes, which can lead to civil money penalties, criminal fines and imprisonment, and/or exclusion from participation in federal health care programs. These false claims statutes include the federal False Claims Act, which allows any person to bring suit alleging the false or fraudulent submission of claims for payment under federal programs or other violations of the statute and to share in any amounts paid by the entity to the government in fines or settlement. Such suits, known as qui tam actions, have increased significantly in recent years and have increased the risk that companies like us may have to defend a false claim action. We could also become subject to similar false claims litigation under state statutes. If we are unsuccessful in defending any such action, such action may have a material adverse effect on our business, financial condition and results of operations.
The difficulties of operating in international markets may harm sales of our products.
Our sales outside the U.S. totaled $5.1 million in 2009. The international nature of our business subjects us and our representatives, agents and distributors to the laws and regulations of the jurisdictions in which they operate, and in which our products are sold and used. The types of risks that we face in international operations include:
| • | the imposition of governmental controls; |
26
Table of Contents
| • | logistical difficulties in managing international operations; and |
| • | fluctuations in foreign currency exchange rates. |
Our international sales and operations may be limited or disrupted if we cannot successfully meet the challenges of operating internationally.
We may acquire technologies, products or businesses in the future, and these acquisitions could result in disruption of our business, increased costs and dilution to our shareholders.
An acquisition of assets, products or another business entails risks, uncertainties and potential disruptions to our business, and could divert management attention. We also could fail to assimilate the acquired business or company, which could lead to higher operating expenses. In addition, certain acquired technology could require significant additional development work and efforts to obtain regulatory clearance or approval, which could cause us to incur significant expense or to expend significant amounts of cash. Further, any acquired product may not provide the intended complementary fit with our existing product portfolio. Our shareholders could also be diluted if we issue shares of our stock to acquire another company, business or technology.
Our business could suffer if we cannot attract and retain the services of key employees.
We depend substantially upon the continued service and performance of our existing executive officers. We rely on key personnel in formulating and implementing our product research, development and commercialization strategies. Our success will depend in large part on our ability to attract and retain highly skilled employees. We compete for such personnel with other companies, academic institutions, government entities and other organizations. We may not be successful in hiring or retaining qualified personnel. If one or more of our key employees resigns, the loss of that employee could harm our business. If we lose any key personnel, we may not be able to prevent the unauthorized disclosure or use of our technical knowledge or other trade secrets by those former employees, or successfully prevent them from competing against us despite our use of confidentiality and non-compete agreements.
Negative economic conditions may adversely affect our sales, business or the profitability of our products.
The current economic conditions and recent financial crisis affecting the banking system and financial markets have resulted in a tightening in the credit markets, a low level of liquidity in many financial markets, and extreme volatility in fixed income, credit and equity markets. The impact of these conditions could result in a number of follow-on adverse effects on our business, liquidity and results of operations, including but not limited to:
| • | the inability of one or more of our key suppliers to obtain credit to finance their operations or the insolvency of one or more of our key suppliers, which could adversely affect our product supply and reduce our sales; |
| • | the inability of customers to obtain credit to finance purchases of our products and/or customer insolvencies, which could reduce our sales, profitability and collectability of our receivables; |
| • | an unwillingness or inability of our current or potential business partners to continue existing collaborations or enter into transactions with us; |
| • | a negative impact on our current investment portfolio; and |
| • | our inability to raise funds through equity or debt financings that may be necessary in the future for various purposes, including acquisitions of products, technologies or other companies. |
| • | In addition, U.S. unemployment levels have been increasing. If patients become unemployed and lose health insurance previously obtained through their employers’ plans, we could see a decrease in elective surgeries that use our products, which would adversely affect our sales and could result in excess inventory. |
27
Table of Contents
If Congress enacts health care reform legislation, our business and financial position could be adversely affected.
The recent focus by the Obama administration and Congress on health care reform could result in laws and regulations that could adversely affect our business. An expansion in government’s role in the U.S. health care industry may lower reimbursements for our products, reduce medical procedure volumes and adversely affect our business. With respect to our industry in particular, Congress has also proposed an excise tax on medical device companies. If enacted, this tax could delay our ability to become profitable or, if we become profitable, this tax could reduce our profitability.
RISKS RELATED TO OUR FINANCIAL RESULTS AND FINANCING
If our losses continue in the long term, they could limit our growth and jeopardize our viability.
We have experienced negative operating cash flows since our inception and have funded our operations primarily from proceeds received from sales of our securities and borrowings under a debt facility. In November and December 2006, we raised approximately $26.6 million in net proceeds from the sale of approximately 8.8 million shares of our common stock to institutional investors. In July 2007, we sold approximately 12.3 million shares of our common stock for net proceeds of approximately $32.2 million. In addition, in July 2007, we entered into a senior secured note purchase facility under which we have issued $35.0 million of our 10% senior secured notes due July 30, 2012. We believe our existing cash, cash equivalents, and short-term investments of approximately $23.1 million as of December 31, 2009 will be sufficient to meet our currently estimated operating and investing requirements for the foreseeable future. To date, we have not been profitable. We have incurred substantial operating losses since our inception and at December 31, 2009, had an accumulated deficit of $180.6 million. These losses have resulted principally from:
| • | the development and patenting of our technologies; |
| • | pre-clinical and clinical studies; |
| • | preparation of submissions to the FDA and foreign regulatory bodies; |
| • | the development of manufacturing, sales and marketing capabilities; and |
| • | the repurchase of a revenue interest from Royalty Securitization Trust I in July 2007. |
We may incur operating losses in the future unless or until we can consistently leverage our product portfolio, our direct sales force, our manufacturing capacity and our product development capability to generate product sales in amounts that exceed our cost of sales and operating expenses. We may never be able to achieve or maintain profitability in the future, and our products may never be commercially accepted or generate sufficient product sales to fund operations.
If we default under our senior secured note purchase facility, we may be required to repay amounts due under the facility, and we may incur additional interest and prepayment penalties. If we are unable to repay amounts due under the note facility, the lender could foreclose on certain assets that are essential to our operations.
We have $35 million in outstanding principal indebtedness under a senior secured note purchase facility. Notes issued under the facility are due July 30, 2012. We initially issued $25 million principal amount of our 10% senior secured notes under the facility and used most of the proceeds to repurchase a revenue interest obligation. On July 31, 2008 we issued an additional $10 million principal amount of notes under the facility primarily to fund our acquisition of the collagen raw material, equipment and technology license from Allergan. In connection with borrowings made under the facility, the note purchaser received five-year warrants, expiring in 2012, to purchase approximately 1.1 million shares of our common stock at an exercise price of $3.41 per share, which are currently exercisable and outstanding.
28
Table of Contents
Borrowings under the facility are guaranteed by us and one of our wholly-owned subsidiaries. The facility is secured by a first priority lien on substantially all of our assets (including intellectual property) other than those exclusively related to Cortoss and Aliquot. We are required to make quarterly interest only payments to the note holder. Outstanding principal amounts under the notes bear annual interest at 10%, provided that interest would accrue at 12% per year during the continuance of any event of default and would be payable on demand. Based on the amount outstanding under the facility as of December 31, 2009, we will incur quarterly interest expense of $875,000 during 2010. The unpaid principal amount under the notes and accrued interest and all other obligations would become due and payable immediately if we are insolvent, are in bankruptcy proceedings or have a custodian or receiver appointed for any substantial part of our property. If an event of default not described in the preceding sentence occurs and is continuing (including a change of control of the Company), then the holders of at least two-thirds in principal amount of notes then outstanding may declare the unpaid principal amount of the notes, accrued interest and all other obligations due and payable immediately. In addition, if the notes become due and payable, whether automatically or by declaration, by reason of any of the following events of default, then we must pay a prepayment premium of up to 16% of the outstanding principal balance of the notes then outstanding, depending on the timing of the prepayment:
| • | We fail to pay any principal on any note or prepayment premiums, if any, when due and payable; |
| • | We fail to pay any interest on any note or other amount (other than principal or prepayment premiums) for more than three business days after becoming due and payable; |
| • | We are insolvent, in bankruptcy proceedings or have a custodian or receiver appointed for any substantial part of our property; or |
| • | A change of control of our company occurs. |
Under the facility, we must comply with various financial and non-financial covenants. For example, we are required to maintain a minimum cash balance equal to at least 25% of the then outstanding principal amount under the notes in a separate account that is pledged as collateral for the loan. As of December 31, 2009, we must maintain a minimum cash balance of $8.75 million. In addition, if the balance in the separate account falls below an amount equal to 40% of the principal amount outstanding under the notes, we must obtain and maintain for the benefit of favor of the note holder a letter of credit in the amount of 25% of the principal amount outstanding under the notes. As of December 31, 2009, we must maintain a minimum balance of $14 million in cash, cash equivalents, and short-term investments in order to avoid obtaining a letter of credit. Currently we are not required to obtain a letter of credit. The primary non-financial covenants limit our ability to incur indebtedness or liens, sell assets, conduct mergers or acquisitions, make investments and pay dividends.
As of December 31, 2009, we had $35 million outstanding under the facility, had accrued $875,000 in interest thereon, and were in compliance with all covenants under the facility. Based on our current operating plans, we expect to be in compliance with the covenants under the facility within the next twelve months from the date of filing of this report. However, if we default under the facility and cannot repay amounts then due, the lender could foreclose on the pledged assets, we could be forced into bankruptcy and we could be otherwise harmed.
We may seek to obtain additional funds through equity and or debt financings, or strategic alliances with third parties either alone or in combination. This could adversely affect our financial condition and cause dilution to our existing shareholders.
There are factors that may cause our future capital requirements to be greater than anticipated or could accelerate our need for funds including, without limitation:
| • | inability to increase sales of our cleared or approved products; |
| • | unforeseen difficulties in operating an effective direct sales and distribution network; |
| • | delayed, insufficient or lack of market acceptance of our products; |
29
Table of Contents
| • | unanticipated expenditures in research and product development or manufacturing activities; |
| • | unforeseen changes in health care reimbursement for procedures using our products; |
| • | unforeseen developments during our pre-clinical and clinical trials; |
| • | unanticipated expenditures in the acquisition, enforcement and defense of intellectual property rights; |
| • | inability to train a sufficient number of surgeons to create demand for our products; |
| • | lack of financial resources to adequately support our operations; |
| • | difficulties in maintaining commercial scale manufacturing capacity and capability; |
| • | unforeseen problems with our third party manufacturers and service providers or with our specialty suppliers of certain raw materials; |
| • | delays in the timing of receipt of required regulatory approvals or clearances to make or sell products; |
| • | unanticipated difficulties in operating in international markets; |
| • | the need to respond to technological changes and increased competition; |
| • | unforeseen problems in attracting and retaining qualified personnel to market our products; |
| • | enactment of new legislation or administrative regulation or changes in the regulatory climate affecting our products or the markets in which we compete; |
| • | the application to our business of new court decisions and regulatory interpretations; |
| • | claims that might be brought in excess of our insurance coverage; |
| • | any imposition of penalties for failure to comply with regulatory guidelines; or |
| • | management’s perception that uncertainties relating to these factors may be increasing. |
In addition, we may seek to expand our operations and product portfolio through acquisitions or joint ventures. Any such acquisitions or joint ventures may increase our capital requirements.
We cannot be certain that funding will be available on terms acceptable to us, if at all, particularly in light of current economic conditions. Any public or private financings may involve issuances of debt or common stock or other classes of our equity, which could further dilute existing shareholders’ percentage ownership. If we are unable to obtain funding to support operations, we may be forced to curtail our operations or be unable to develop products.
We are required annually, or on an interim basis as needed, to review the carrying value of our long-lived assets for impairment. Our long-lived assets consist of property and equipment and intangible assets. If sales of our products decline because of, for example, competition or an inability to manufacture sufficient supply of product, the value of our long-lived assets could become impaired.
As of December 31, 2009, we had approximately $17.9 million of property and equipment, net of depreciation, and $11.4 million of license and technology intangible assets, net of accumulated amortization. Our property and equipment consist primarily of assets used to manufacture Vitoss, Vitagel and Cortoss. Our intangible assets consist primarily of (i) $6.5 million for our prepayment in December 2006 of the profit-sharing license royalties on Vitagel and CellPaker, (ii) $4.5 million recorded for the value of the collagen-processing technology license acquired from Allergan in the third quarter of 2008 and (iii) $0.4 million recorded for the value of a license acquired from a business partner related to the development of a new product. If a change in circumstances causes us to lower our future sales forecast for our products, we may be required to write off a portion or all of the net book value of the affected property and equipment or intangible assets associated with that product. Long-lived assets are reviewed for impairment whenever events or changes in circumstances
30
Table of Contents
indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by comparing the carrying amount of the asset against the estimated undiscounted future cash flows expected to be generated by this asset. If the sum of the expected future net cash flows is less than the carrying value of the asset being evaluated, an impairment charge would be recognized. The impairment charge would be calculated as the amount by which the carrying value of the asset exceeds its fair value. An impairment charge to our net intangible assets, depending on the size, could result in a material adverse effect on our business, financial condition and results of operations.
RISKS RELATED TO OUR INTELLECTUAL PROPERTY AND LITIGATION
Our business will be damaged if we are unable to protect our proprietary rights to our products, and we may be subject to intellectual property infringement claims by others.
We rely on patent protection, as well as a combination of copyright, trade secret and trademark laws, nondisclosure and confidentiality agreements and other contractual restrictions to protect our proprietary technology. However, these measures afford only limited protection and may not adequately protect our rights. For example, our patents may be challenged, invalidated or circumvented by third parties. As of March 1, 2010, we own or control 24 issued U.S. patents and 19 pending patent applications in the U.S., and counterparts of certain of these patents and pending patent applications worldwide, including Canada, Europe, Mexico and Japan. There can be no assurance that patents will issue from any of the pending patent applications. Moreover, we cannot be certain that we were the first creator of inventions covered by pending patent applications or we were the first to file patent applications for the relevant inventions for the following reasons:
| • | patent applications filed prior to December 2000 in the U.S. are maintained in secrecy until issued; |
| • | patent applications filed after November 2000 in the U.S. are maintained in secrecy until 18 months after the date of filing; and |
| • | the publication of discoveries in the scientific or patent literature tends to lag behind actual discoveries. |
If we do receive a patent, it may not be broad enough to protect our proprietary position in the technology or to be commercially useful to us. In addition, if we lose certain key personnel, we may not be able to prevent the unauthorized disclosure or use of our technical knowledge or other trade secrets by those former employees. Furthermore, the laws of foreign countries may not protect our intellectual property rights to the same extent as the laws of the United States. Even if our intellectual property rights are adequately protected, litigation or other proceedings may be necessary to enforce our intellectual property rights, which could result in substantial costs to us and divert management attention. If our intellectual property is not adequately protected, our competitors could use the intellectual property that we have developed to enhance their products and compete more directly with us, which could damage our business.
In addition to the risk of failing to adequately protect our proprietary rights, there is a risk that we may become subject to a claim that we infringe upon the proprietary rights of others. Although we do not believe that we are infringing the rights of others, third parties may claim that we are doing so. There is a substantial amount of litigation over patent and other intellectual property rights in the medical device industry generally, and in the spinal market segments particularly. If the holder of a proprietary right brought an infringement action against us, the cost of litigating the claim could be substantial and divert management attention. In addition, if a court determined that one of our products infringed a proprietary right, we could be prevented from selling that product unless we could obtain a license from the owner of the right. A license may not be available on terms acceptable to us, if at all. Modification of our products or development of new products to avoid infringement may require us to conduct additional clinical trials for these modified or new products and to revise our filings with the FDA, which is time consuming and expensive. If we are not successful in obtaining a license or redesigning our product, our business could suffer.
We have obtained certain of our intellectual property rights and our rights to develop and commercialize certain products, and made certain commitments regarding our intellectual property and these rights, through
31
Table of Contents
license and development agreements with third parties. These agreements generally may be terminated by our counterparty if we materially breach any of our obligations under the agreement. We may disagree with these third parties as to our and their obligations under the agreements or the interpretation of provisions in the agreements. Any such disagreements could cause these third parties to seek to terminate our rights under the particular agreement. If any of our agreements were terminated, we could lose the rights to the product, product candidate or intellectual property covered by the agreement, reducing our revenue, potential revenues and significantly harming our business. Whether or not such a termination were proper, if a dispute arises under any of our agreements, the need to resolve the dispute could consume significant resources and divert management’s time and attention.
We distribute Vitasure, which we purchase from Medafor and is manufactured by third parties on behalf of Medafor. Medafor and these third parties have patents, licenses, and proprietary technologies related to Vitasure. We cannot be certain that no one will challenge the validity or enforceability of any patent that they own. We cannot be certain that if anyone does make such a challenge, that these third parties will be able to successfully defend that challenge, with or without our assistance. In addition, others may infringe these third parties’ patents or use their proprietary rights inappropriately. If others infringe patents owned or licensed by these third parties and related to Vitasure, we may choose to assist in bringing an infringement action and may incur substantial costs in any efforts to uphold the validity and prevent infringement of a patent or to protect proprietary technologies and methods. Furthermore, we cannot be certain that competitors will not independently develop similar technologies, duplicate technologies, design around the patented aspects of such technologies, or attempt to duplicate their proprietary technologies that have no patent protection. In addition, we cannot be sure that these third parties’ technologies will not infringe patents or other rights owned by others.
If we are sued in a product liability action, we could be forced to incur substantial defense costs, pay substantial damages and the attention of our management team may be diverted from operating our business.
We manufacture medical devices that are used on patients in surgery, and we may be subject to product liability lawsuits. In particular, the market for spine products has a history of product liability litigation. Under certain of our agreements with our distributors and hospital customers, we indemnify the distributor or hospital from product liability claims. Any product liability claim brought against us, with or without merit, could result in the increase of our product liability insurance rates or the inability to secure coverage in the future. In addition, we could incur substantial costs to defend these claims and would have to pay any amount awarded by a court in excess of policy limits. We maintain product liability insurance in the annual aggregate amount of up to $20 million, although our insurance policies have various exclusions. Thus, we may be subject to a product liability claim for which we have no insurance coverage, in which case we may have to pay the entire amount of any defense costs, award or settlement. Even in the absence of a claim, our insurance rates may rise in the future to a point where we may decide to carry a lesser amount of this insurance or to not carry this type of insurance. A meritless or unsuccessful product liability claim would be time-consuming and expensive to defend and could divert management’s attention from our core business. A successful product liability claim or series of claims brought against us in excess of our coverage could have a material adverse effect on our business, financial condition and results of operations. Any product liability claim—even if unsuccessful—could adversely affect our reputation and otherwise adversely affect our business and the market for our common stock.
32
Table of Contents
RISKS RELATED TO INVESTING IN OUR STOCK AND EQUITY MARKETS
Our stock price may be volatile for many reasons, including fluctuations in our results of operations due to factors out of our control.
The market price of our common stock may fluctuate substantially, which could adversely affect the value of an investment in our common stock or shareholders’ ability to sell our shares. Our stock price may fluctuate due to fluctuations in our results of operations or for other reasons. Future levels of our product sales and the timing of regulatory clearances and product introductions are difficult to predict, and product sales to date may not be indicative of future sales levels. We currently sell Vitagel and CellPaker under a license agreement, the termination of which would have a material adverse effect on our sales revenues. Our results of operations may fluctuate significantly in the future as a result of a number of additional factors, many of which are outside of our control. These factors include, but are not limited to:
| • | the medical community’s acceptance of our products; |
| • | the public perception of our company and the reputation of our business practices and products; |
| • | issues in establishing adequate commercial-scale manufacturing capabilities; |
| • | disruptions with the manufacturing of our products, including with respect to our third party manufacturers and suppliers of raw materials; |
| • | difficulties in establishing and expanding our sales and distribution channels; |
| • | the mix of our products as profit margins differ between our products; |
| • | the timing of governmental approvals or clearances for our products and our competitors’ products; |
| • | the timing of product introductions by us or our competitors; |
| • | the timing in obtaining adequate third party reimbursement of our products; |
| • | unanticipated events associated with clinical and pre-clinical trials of our products; |
| • | product recalls; |
| • | the success of competitive products; |
| • | our ability to enter into strategic alliances with other companies; |
| • | expenses associated with development and protection of intellectual property matters; |
| • | events affecting logistics and elective surgery trends; |
| • | the timing of expenses related to commercialization of new products; |
| • | competitive disruptions to our sales and distribution channels; |
| • | fluctuations in sales levels, which may be caused by seasonality of elective surgeries, the timing of orders from hospitals or distributor stocking orders, and other factors; |
| • | news concerning economic or market conditions in the general economy or within our industry, or the movement of capital into or out of our industry; |
| • | changes in U.S. or foreign government regulations impacting our industry; |
| • | the adequate training of a sufficient number of surgeons in the use of our products; and |
| • | the timing of governmental approvals or clearances for us to manufacture the products we sell. |
The results of our operations may fluctuate significantly from quarter to quarter and may not meet expectations of securities analysts and investors. This may also cause our stock price to be volatile.
33
Table of Contents
In general, equity markets, including NASDAQ, have from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. These broad market fluctuations, including the following factors, may adversely affect the market price of our common stock:
| • | changes in the financial guidance we provide to the investment community; |
| • | changes in stock market analyst recommendations regarding our stock; |
| • | sales by existing holders of significant numbers of shares of our stock into the market, including sales by the three institutional investors that we believe held approximately one-quarter of our outstanding shares at or about December 31, 2009; |
| • | FDA and international regulatory actions regarding us or our competitors; |
| • | determinations by governments and insurance companies regarding reimbursement for medical procedures using our or our competitors’ products; |
| • | product sales growth rates; |
| • | public perception of our company and the reputation of our business practices and products; |
| • | developments with respect to patents or proprietary rights; |
| • | public concern as to the safety of products developed by us or by others; |
| • | changes in health care policy in the United States and internationally; |
| • | acquisitions or strategic alliances by us or our competitors; |
| • | business conditions affecting other medical device companies or the medical device industry generally; |
| • | general market conditions, particularly for companies with small market capitalizations; and |
| • | pending or threatened offers to acquire us. |
Provisions of Pennsylvania law or our Articles of Incorporation may deter a third party from seeking to obtain control of us or may affect your rights as a shareholder.
Certain provisions of Pennsylvania law could make it more difficult for a third party to acquire us, or could discourage a third party from attempting to acquire us. These provisions could limit the price that certain investors might be willing to pay in the future for shares of our common stock. In addition, our Articles of Incorporation enable our board of directors to issue up to 19,998,100 shares of preferred stock having rights, privileges and preferences as are determined by the board of directors. Accordingly, our board is empowered, without shareholder approval, to issue preferred stock with dividend, liquidation, conversion, voting or other rights superior to those of holders of our common stock. For example, an issuance of preferred stock could:
| • | adversely affect the voting power of the common shareholders; |
| • | make it more difficult for a third party to gain control of us; |
| • | discourage bids for our common stock at a premium; or |
| • | otherwise adversely affect the market price of our common stock. |
Future sales of our common stock in the public market could adversely affect our stock price.
We cannot predict the effect, if any, that future sales of our common stock or the availability for future sales of shares of our common stock or securities convertible into or exercisable for our common stock will have on the market price of our common stock prevailing from time to time. We have an effective registration statement on Form S-3 which allows us to sell up to approximately $67.5 million of securities in one or more public offerings. The registration statement provides us with the flexibility to determine the type of security we choose to sell, including common stock, preferred stock, warrants with debt securities, as well as the ability to time such sales when market conditions are favorable.
34
Table of Contents
As of December 31, 2009, we had outstanding options to purchase approximately 7.2 million shares of our common stock at a weighted average exercise price of $3.55 per share; restricted stock, restricted stock unit and performance-based equity awards granted to employees and/or directors under our equity compensation plan for an aggregate of 617,000 shares of our common stock; and warrants to purchase approximately 1.1 million shares of our common stock at an exercise price of $3.41 per share. In order to attract and retain key personnel, we may issue additional securities, including stock options, restricted stock grants and shares of common stock, in connection with our equity compensation plan. The sale, or the availability for sale, of substantial amounts of common stock by our existing shareholders pursuant to an effective registration statement or under Rule 144, through the exercise of registration rights or the issuance of shares of common stock upon the exercise of stock options or warrants, or the perception that such sales or issuances could occur, could adversely affect the prevailing market prices for our common stock.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None
| ITEM 2. | PROPERTIES |
Our headquarters are located at the Great Valley Corporate Center in Malvern, Pennsylvania, which is a suburb of Philadelphia. We lease several buildings within the Great Valley Corporate Center as follows:
| Purpose |
Approximate Square Feet |
Lease Expiration | ||
| Executive offices |
13,700 | July 2017 | ||
| Manufacturing |
25,800 | July 2017 | ||
| Processing facility for Vitagel collagen(*) |
4,000 | July 2017 | ||
| Warehouse, logistics and quality assurance |
36,500 | July 2017 | ||
| Offices and research and development laboratories |
7,900 | July 2017 |
| (*) | Operation of the facility is pending FDA approval. |
We believe our existing manufacturing facilities have the capacity to meet our commercial needs through at least 2012. We are also expanding our manufacturing capability for Vitagel at sites we currently lease (see—MANUFACTURING AND PRODUCT SUPPLY—Raw Materials Supply under Item 1. BUSINESS, above).
We conduct the majority of our international sales and marketing activities through administrative office space in Leuven, Belgium, which we have leased through May 2018.
| ITEM 3. | LEGAL PROCEEDINGS |
None.
35
Table of Contents
PART II
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Our common stock is quoted on the NASDAQ Global Market (NASDAQ) under the symbol “VITA.” The following table reflects the ranges of high and low sale prices for our common stock as reported on NASDAQ for the stated periods.
| High | Low | |||||
| Fiscal year 2009: |
||||||
| First quarter |
$ | 3.78 | $ | 2.47 | ||
| Second quarter |
6.10 | 2.56 | ||||
| Third quarter |
6.91 | 4.09 | ||||
| Fourth quarter |
4.46 | 3.33 | ||||
| Fiscal year 2008: |
||||||
| First quarter |
$ | 3.40 | $ | 2.21 | ||
| Second quarter |
2.60 | 1.99 | ||||
| Third quarter |
3.16 | 1.96 | ||||
| Fourth quarter |
3.61 | 2.12 | ||||
As of March 8, 2010 there were 201 holders of record for shares of our common stock. Since a portion of our common stock is held in “street” or nominee name, we are unable to determine the exact number of beneficial holders. On March 8, 2010, the last sale price of our common stock as reported by NASDAQ was $4.17 per share. We have never declared or paid cash dividends on shares of our common stock and do not intend to pay any cash dividends in the foreseeable future on shares of our common stock. See Item 12 “SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS” included in this report for information regarding our equity compensation plans approved by security holders, which information is incorporated herein by reference.
36
Table of Contents
| ITEM 6. | SELECTED CONSOLIDATED FINANCIAL DATA |
The following table presents selected historical consolidated financial data derived from the consolidated financial statements of Orthovita, Inc. and subsidiaries as of and for each of the five years in the period ended December 31, 2009. This data should be read in conjunction with our consolidated financial statements, including notes thereto, and “MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATION,” included in this report.
| Year ended December 31, | ||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||
| (In thousands except per share amounts) | ||||||||||||||||||||
| Statement of Operations Data: |
||||||||||||||||||||
| Product sales |
$ | 92,853 | $ | 76,915 | $ | 58,046 | $ | 46,828 | $ | 34,679 | ||||||||||
| Cost of sales |
29,914 | 25,929 | 20,544 | 17,783 | 12,652 | |||||||||||||||
| Gross profit |
62,939 | 50,986 | 37,502 | 29,045 | 22,028 | |||||||||||||||
| Operating expenses |
64,028 | 60,170 | 51,080 | 46,006 | 34,955 | |||||||||||||||
| Operating loss |
(1,089 | ) | (9,184 | ) | (13,578 | ) | (16,960 | ) | (12,927 | ) | ||||||||||
| Interest expense, net of interest income |
(2,830 | ) | (1,506 | ) | (339 | ) | (503 | ) | (436 | ) | ||||||||||
| (Loss) gain on sale of product line and related assets |
— | (72 | ) | 372 | — | — | ||||||||||||||
| Charge for repurchase of revenue interest obligation |
— | — | (16,605 | ) | — | — | ||||||||||||||
| Income tax benefit |
13 | 10 | 268 | — | — | |||||||||||||||
| Net loss |
$ | (3,906 | ) | $ | (10,752 | ) | $ | (29,882 | ) | $ | (17,464 | ) | $ | (13,363 | ) | |||||
| Net loss per share, basic and diluted |
$ | (0.05 | ) | $ | (0.14 | ) | $ | (0.44 | ) | $ | (0.33 | ) | $ | (0.28 | ) | |||||
| Shares used in computing basic and diluted net loss per share |
76,176 | 75,804 | 67,181 | 53,354 | 48,107 | |||||||||||||||
| As of December 31, | ||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Balance Sheet Data: |
||||||||||||||||||||
| Cash, cash equivalents and short-term investments |
$ | 23,106 | $ | 32,291 | $ | 48,408 | $ | 28,339 | $ | 24,665 | ||||||||||
| Accounts receivable, net |
12,324 | 10,881 | 8,438 | 8,755 | 6,224 | |||||||||||||||
| Inventories |
26,058 | 19,757 | 15,612 | 9,444 | 11,923 | |||||||||||||||
| Property and equipment, net |
17,940 | 14,438 | 8,252 | 5,295 | 5,032 | |||||||||||||||
| License and technology intangible assets, net |
11,376 | 12,354 | 8,150 | 9,000 | — | |||||||||||||||
| Long-term investments |
— | — | — | — | 3,007 | |||||||||||||||
| Total assets |
92,629 | 91,171 | 90,217 | 62,215 | 52,336 | |||||||||||||||
| Current liabilities |
13,367 | 11,410 | 11,282 | 8,164 | 9,123 | |||||||||||||||
| Notes payable, net of debt discount |
34,095 | 33,809 | 23,895 | 1,338 | 1,403 | |||||||||||||||
| Long-term liabilities |
408 | 310 | 511 | 9,451 | 7,434 | |||||||||||||||
| Total shareholders’ equity |
44,759 | 45,642 | 54,529 | 43,262 | 34,376 | |||||||||||||||
37
Table of Contents
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATION |
UNDERSTANDING OUR FINANCIAL INFORMATION
The following discussion and analysis provides information management believes to be relevant in understanding our financial condition and results of operation. In addition to reading management’s discussion and analysis, you should review our consolidated financial statements and accompanying notes thereto as of December 31, 2009 and 2008 and for each of the years in the three-year period ended December 31, 2009, which begin on page F-1 of this report.
This discussion and analysis contains forward-looking statements that provide our current expectations, forecasts of future events or goals. You can identify these statements as those that do not relate strictly to historical or current facts. These statements could include words such as “may,” “will,” “anticipate,” “estimate,” “expect,” “project,” “intend,” “plan,” “believe,” “seek” and other words and terms of similar meaning in connection with any discussion of future operating or financial performance. Any or all of our forward-looking statements in this Form 10-K may turn out to be incorrect. They can be affected by inaccurate assumptions we might make, or by known or unknown risks and uncertainties. Consequently, no forward-looking statement can be guaranteed. Actual future results may vary materially. There are many important factors that could cause actual events or results to differ materially from those expressed or implied by forward-looking statements including, without limitation, the development, demand, market acceptance and hospital approval of our products; third party reimbursement rates and health care reform; our ability to compete in increasingly price-sensitive markets; our ability to grow revenues by leveraging our existing infrastructure and sales force; our ability to manage our manufacturing facilities and requirements; the ability of our competitors to develop more technically advanced products; the regulatory approval of our products; the development of our sales network; sales product mix and related margins; capital expenditures; future liquidity; uses of cash; the achievement of product development goals and the amount and timing of related milestone and other payments; cost and availability of raw materials; inventory levels; development costs for existing and new products; equity compensation expense; foreign currency exchange rates; fluctuations in our stock price; and the other risk factors addressed in ITEM 1A. “RISK FACTORS” in this report.
We undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise. You are advised, however, to consult any further disclosures we make on related subjects in our filings with the U.S. Securities and Exchange Commission (the SEC). This discussion is provided as permitted by the Private Securities Litigation Reform Act of 1995.
EXECUTIVE LEVEL OVERVIEW
We are a specialty spine and orthopedic company with a portfolio of orthobiologic and biosurgery products. Our products are based on novel and unique proprietary biomaterials that have innovative mechanisms of action in the body.
Our orthobiologic platform includes products for the fusion, regeneration and fixation of human bone. Our orthobiologic products are based on our proprietary Vitoss™ Bone Graft Substitute technology and include the Imbibe™ Bone Marrow Aspiration System. Vitoss is the market-leading synthetic bone graft in the United States and has been used in more than 320,000 implantations worldwide. Several of our Vitoss products incorporate our proprietary ceramic glass technology which accelerates bone healing. Our orthobiologic products also include our Cortoss™ Bone Augmentation Material and the Aliquot™ Delivery System used with Cortoss, both of which were first available for sale in the U.S. in July 2009. Cortoss is an advanced synthetic biomaterial. Following injection into spinal vertebrae, Cortoss hardens to mimic weight-bearing, cortical bone. Cortoss is the first clinically-proven, injectable FDA-cleared alternative to polymethylmethacrylate (PMMA) bone cement for the treatment of vertebral compression fractures, an often extremely painful condition that occurs in patients with osteoporosis and cancer. In addition to extensive clinical data showing statistically significant improvements in
38
Table of Contents
pain relief (at three months following implantation) and function (at 24 months following implantation) from the use of Cortoss in comparison to PMMA, we believe that Cortoss has significant ease-of-use advantages over current vertebral augmentation technology.
Our biosurgery products include Vitagel™ Surgical Hemostat, Vitasure™ Absorbable Hemostat, and the CellPaker™ Plasma Collection System used in conjunction with Vitagel, and other accessories and delivery products that complement our Vitagel product. These products incorporate advanced biosurgical materials and product engineering to help control bleeding during surgeries.
We sell our products worldwide and focus on the US orthopedic and spine market. In the United States our products are primarily sold through our direct sales force although we also use third-party distributors in certain markets. Our sales force focuses its marketing efforts on orthopedic surgeons, interventional radiologists, neuroradiologists and hospital administrators. Our products can be used with hospital in-patient procedures, hospital out-patient procedures and with ancillary surgical center procedures. Outside the United States we use a direct sales force to sell our products in the United Kingdom and use distributors to sell our products throughout the rest of the world.
We manufacture the majority of our products at our headquarters in Malvern, Pennsylvania. We are highly dependent on Kensey as a sole source provider of certain raw materials used in the manufacture of Vitoss Foam products. We co-developed the Vitoss Foam platform with Kensey and our cost of goods sold includes royalty expense to Kensey. We also source other raw materials and a majority of the disposable components that accompany and are used with our products with a limited number of third party manufacturers. We expect to continue to use third party manufacturers for these elements in order to reduce costs and increase product gross margins.
We manage our orthobiologic and biosurgery platforms as a single business unit. These platforms are marketed and sold by a single sales force to the same customer base. Accordingly, we report our financial results under a single operating segment – the development, manufacture and sale of specialty spine and orthopedic products.
Our mission is to develop and market quality products backed by solid clinical research that will improve patient outcomes and meet the needs of our surgical customers with maximum effectiveness and safety. Our strategy to accomplish this is threefold: (i) maximize the growth of existing products by continuing to optimize our direct sales force, develop additional clinical data and manage the lifecycles of product portfolios; (ii) continue product pipeline development of our own internally developed products; and (iii) pursue in-licensing, joint venture and acquisition opportunities to augment our product portfolio.
We believe we can continue to grow revenues by leveraging our existing infrastructure, direct sales force and research and development capability. While we pay attention to all meaningful trends we identify in our financial results, we currently pay particular attention to revenue growth, gross margins, operating expenses, operating cash flow and cash.
We believe that the execution of our strategy will drive long-term shareholder value.
Summary Full Year 2009 Financial Results – For 2009, our net loss was $3.9 million compared to a net loss for 2008 of $10.8 million, a reduction of 64%.
Product sales for 2009 increased to $92.9 million from $76.9 million for 2008. This 21% increase is primarily attributable to higher sales of our Vitoss Bioactive Foam products, which we launched in 2008.
Gross profit for 2009 was $62.9 million, or 68% of product sales, compared to $51.0 million, or 66% of product sales, for 2008. This increase is primarily attributable to product mix and higher margins on the Vitoss Bioactive and Cortoss sales compared to our other Vitoss and biosurgery products.
39
Table of Contents
Operating expenses for 2009 were $64.0 million, or 69% of product sales, compared to $60.2 million, or 78% of product sales for 2008. The 6% increase in operating expenses resulted from higher commissions associated with increased sales, infrastructure expansion expenses, and increased marketing costs related to the launch of Cortoss, partially offset by more effective cost management of certain sales force discretionary spending.
You should read the additional detailed commentary of product sales, gross profit, operating expenses and other analyses of the year ended December 31, 2009 compared to 2008, as well as similar comparisons of the year ended December 31, 2008 to 2007, contained later in this discussion.
CRITICAL ACCOUNTING POLICIES
The preparation of the consolidated financial statements requires us to make assumptions, estimates and judgments that affect the reported amounts of assets and liabilities, the disclosures of contingent assets and liabilities as of the date of the consolidated financial statements, and the reported amounts of revenues and expenses during the reporting periods. On an ongoing basis, we evaluate our estimates, including, but not limited to, those related to accounts receivable, inventories and recoverability of long-lived assets. We use historical experience and other assumptions as the basis for making estimates. Illiquid credit markets, volatile equity, foreign currency and energy markets, and declines in consumer spending have combined to increase the uncertainty inherent in such estimates and assumptions. As future events and their effects cannot be determined with precision, actual results could differ significantly from these estimates. Changes in those estimates will be reflected in the financial statements in those future periods. The critical accounting policies addressed below have been reviewed with the Audit Committee of our Board of Directors and reflect our most significant judgments and estimates used in the preparation of our consolidated financial statements.
Revenue Recognition
Revenue is not recognized until persuasive evidence of an arrangement exists, performance or delivery has occurred, the sales price is fixed or determinable and collection is probable. In the U.S., product sales revenue is recognized upon either shipment of purchased product to the customer, or receipt of documentation from the customer indicating its consumption of product from consigned inventory. Beginning in 2008, our policy for transactions outside the U.S. was to recognize product sales revenue upon shipment of our product to our independent stocking distributors. Prior to 2008, we primarily recognized product sales outside the U.S. upon receipt of product by our independent stocking distributors. When we transitioned to a direct sales force in the U.K. in 2009, we began to recognize product sales revenue upon shipment of our product to our customers in the U.K. We do not allow product returns or exchanges. We have no obligations to our customers once title has been transferred. We generally require each of our U.S. and the U.K. customers to pay on a net 30-day basis. Outside of the U.S. and the U.K., independent stocking distributors are generally required to pay on a net 60-day basis.
Allowance for Doubtful Accounts
We maintain an accounts receivable allowance for an estimated amount of losses that may result from a customer’s failure to pay for product purchased. The allowance for doubtful accounts is reviewed quarterly and is estimated based on historical bad debts, customer concentrations, known trends with current customers, and changes in customer payment patterns. If the financial condition of our customers or the overall condition of the health care industry were to deteriorate and result in an impairment of our customers’ ability to make payments, additional allowances could be required.
Inventories
Inventories are stated at the lower of cost or market value using the first-in first-out basis, or FIFO and consist of raw materials, Kensey raw material conversion costs, labor and overhead. The majority of our
40
Table of Contents
inventories are subject to expiration dating. We continually evaluate the carrying value of our inventories, and when, in the opinion of management, factors indicate that impairment has occurred, either a reserve is established against the inventories’ carrying value to reduce inventory costs to their net realizable values or the inventories are completely written off. We base these decisions on the level of inventories on hand in relation to our estimated forecast of product demand, production requirements over the next twelve months and the expiration dates of raw materials and finished goods. The costs of producing inventory in the reporting periods prior to the receipt of regulatory approval or clearance are recorded as research and development expense.
Income Taxes
We account for income taxes in accordance with an asset and liability approach requiring the recognition of deferred tax assets and liabilities for the expected tax consequences of events that have been recognized in the financial statements or tax returns. Deferred tax assets and liabilities are recorded without consideration as to their ability to be realized. The deferred tax asset includes net operating loss and credit carryforwards, and the cumulative temporary differences related to certain research and patent costs, which have been charged to expense in our consolidated statements of operations but have been recorded as assets for tax return purposes. The portion of any deferred tax asset, for which it is more likely than not that a tax benefit will not be realized, must then be offset by recording a valuation allowance against the asset.
In assessing the realizability of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. Management considers the scheduled reversal of deferred tax liabilities, projected future taxable income and tax planning strategies in making this assessment. With the exception of the Texas margin tax credit described in note 15 to our consolidated financial statements included in this report, management believes it is more likely than not that we will not realize the deferred tax assets in excess of deferred tax liabilities. A valuation allowance is maintained against the net deferred tax assets in excess of the Texas margin tax credit.
While we believe that our tax positions are fully supportable, there is a risk that certain positions could be challenged successfully. In these instances, we look to establish reserves. If we determine that a tax position is more likely than not of being sustained upon audit, based solely on the technical merits of the position, we recognize the benefit. We measure the benefit by determining the amount that has likelihood greater than 50% of being realized upon settlement. We presume that all tax positions will be examined by a taxing authority with full knowledge of all relevant information. We regularly monitor our tax positions, tax assets and tax liabilities. We reevaluate the technical merits of our tax positions and recognize an uncertain tax benefit or derecognize a previously recorded tax benefit when (i) there is a completion of a tax audit, (ii) there is a change in applicable tax law including a tax case or legislative guidance, or (iii) there is an expiration of the statute of limitations. Significant judgment is required in accounting for tax reserves.
Accounting for Stock Options Issued to Employees
We measure all employee stock-based compensation awards using a fair value method and record such expense in our Consolidated Financial Statements. Such expense is amortized on a straight line basis over the requisite service period of the award.
We estimate the grant date fair value of stock options using the Black-Scholes option-pricing model which requires the input of highly subjective assumptions. These assumptions include estimating the expected term of the award and the estimated volatility of our stock price over the expected term. Changes in these assumptions and in the estimated forfeitures of stock option awards may materially affect the amount of stock-based compensation recognized in our consolidated statements of operations.
41
Table of Contents
Impairment of Long-Lived Assets
We test long-lived assets, including property and equipment and amortizable intangible assets, for recoverability whenever events or changes in circumstances indicate that we may not be able to recover an asset’s carrying amount. We evaluate the recoverability of an asset by comparing its carrying amount to the undiscounted cash flows expected to result from the use and eventual disposition of that asset. If the undiscounted cash flows are not sufficient to recover the carrying amount, we measure any impairment loss as the excess of the carrying amount of the asset over its fair value. Events which could trigger asset impairment include significant underperformance relative to historical or projected future operating results, significant changes in the manner or use of an asset or in our overall business strategy, significant negative industry or economic trends, shortening of product life-cycles, negative changes in third party reimbursement, or changes in technology. We currently believe the future cash flows to be received from all long-lived assets will exceed their carrying value, and accordingly we have not recognized any impairment losses for the years ended December 31, 2009, 2008 or 2007. Any unanticipated significant impairment in the future, however, could have a material adverse impact to our balance sheet and future operating results.
Recent Accounting Pronouncements
In April 2009, the Financial Accounting Standards Board (FASB) provided guidance as codified in Accounting Standards Codification (ASC) Subtopic 320-10 which amends the accounting for certain investments in debt and equity securities to modify the indicator of other-than-temporary impairment for debt securities. Additionally, this guidance changes the amount of an other-than-temporary impairment that is recognized in earnings when there are credit losses on a debt security that management does not intend to sell and it is more-likely-than-not that the entity will not have to sell prior to recovery of the noncredit impairment. In those situations, the portion of the total impairment that is attributable to the credit loss would be recognized in earnings, and the remaining difference between the debt security’s amortized cost basis and its fair value would be included in other comprehensive income. The guidance was effective for interim and annual reporting periods ending after June 15, 2009 and did not have an impact on our consolidated financial statements.
In April 2009, the FASB provided guidance as codified in ASC Topics 270 and 825 to require disclosures about the fair value of financial instruments in interim financial statements as well as in annual financial statements. In addition, the guidance requires disclosures of the methods and significant assumptions used to estimate the fair value of those financial instruments. We adopted this new guidance and have provided the necessary additional disclosures.
42
Table of Contents
RESULTS OF OPERATIONS
We have incurred annual operating losses since inception. We may continue to incur operating losses in the future unless or until we can consistently leverage our product portfolio, our direct sales force, our manufacturing capacity and our product development capability to generate product sales in amounts that exceed our cost of sales and operating expenses. Our ability to increase revenue and to attain profitability are affected by the risk factors addressed in ITEM 1A. “RISK FACTORS” in this report. The following table summarizes our results of operations for each of the years ended December 31, 2009, 2008 and 2007:
| (Dollars in thousands except per share data) |
Year Ended December 31, | |||||||||||||||||||||
| 2009 | 2008 | % change |
2008 | 2007 | % change |
|||||||||||||||||
| PRODUCT SALES: |
||||||||||||||||||||||
| Orthobiologics |
$ | 71,297 | $ | 58,170 | 23 | % | $ | 58,170 | $ | 46,138 | 26 | % | ||||||||||
| Biosurgery |
21,556 | 18,745 | 15 | % | 18,745 | 11,907 | 57 | % | ||||||||||||||
| TOTAL PRODUCT SALES |
92,853 | 76,915 | 21 | % | 76,915 | 58,046 | 33 | % | ||||||||||||||
| COST OF SALES |
29,914 | 25,929 | 15 | % | 25,929 | 20,544 | 26 | % | ||||||||||||||
| GROSS PROFIT |
62,939 | 50,986 | 23 | % | 50,986 | 37,502 | 36 | % | ||||||||||||||
| Gross profit percentage |
68 | % | 66 | % | 66 | % | 65 | % | ||||||||||||||
| OPERATING EXPENSES: |
||||||||||||||||||||||
| General and administrative |
12,172 | 10,753 | 13 | % | 10,753 | 10,700 | 0 | % | ||||||||||||||
| Selling and marketing |
45,070 | 42,706 | 6 | % | 42,706 | 33,945 | 26 | % | ||||||||||||||
| Research and development |
6,786 | 6,711 | 1 | % | 6,711 | 6,435 | 4 | % | ||||||||||||||
| TOTAL OPERATING EXPENSES |
64,028 | 60,170 | 6 | % | 60,170 | 51,080 | 18 | % | ||||||||||||||
| OPERATING LOSS |
(1,089 | ) | (9,184 | ) | -88 | % | (9,184 | ) | (13,578 | ) | -32 | % | ||||||||||
| OTHER INCOME (EXPENSE), NET |
(2,830 | ) | (1,578 | ) | 79 | % | (1,578 | ) | (16,572 | ) | -90 | % | ||||||||||
| LOSS BEFORE INCOME TAXES |
(3,919 | ) | (10,762 | ) | -64 | % | (10,762 | ) | (30,150 | ) | -64 | % | ||||||||||
| INCOME TAX BENEFIT |
13 | 10 | n.m | 10 | 268 | n.m | ||||||||||||||||
| NET LOSS |
$ | (3,906 | ) | $ | (10,752 | ) | -64 | % | $ | (10,752 | ) | $ | (29,882 | ) | -64 | % | ||||||
| NET LOSS PER SHARE, BASIC AND DILUTED |
$ | (0.05 | ) | $ | (0.14 | ) | -64 | % | $ | (0.14 | ) | $ | (0.44 | ) | -68 | % | ||||||
| SHARES USED IN COMPUTING BASIC AND DILUTED NET LOSS PER SHARE |
76,176 | 75,804 | 0 | % | 75,804 | 67,181 | 13 | % | ||||||||||||||
n.m. - not meaningful
43
Table of Contents
The following chart summarizes the three components of operating expenses as a percent of product sales:
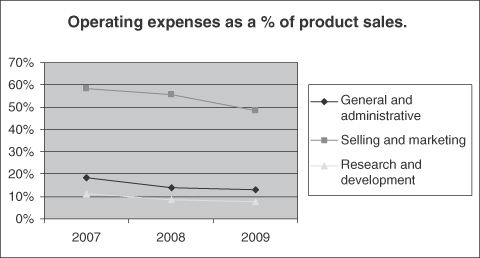
Comparison of the Year Ended December 31, 2009 to the Year Ended December 31, 2008
Product Sales. Product sales of $92.9 million in 2009 increased 21% from $76.9 million in 2008. Orthobiologic product sales and biosurgery product sales increased 23% and 15%, respectively. For both 2009 and 2008, 94% of product sales were in the United States and 6% were in Europe and the rest of the world. Approximately 67% of our product sales during 2009 were from products based upon our Vitoss Foam platform co-developed with Kensey, including Vitoss Bioactive Foam.
Orthbiologic product sales represented 77% of total product sales in 2009 compared to 76% in 2008. Orthobiologic product sales of $71.3 million in 2009 increased from $58.2 million in 2008 primarily due to higher sales of Vitoss Bioactive Foam products, which we launched in 2008. Sales of Cortoss in the U.S. also contributed to the higher orthobiologic product sales in 2009. We began to sell Cortoss in the U.S. in July 2009 as part of a limited, controlled launch shortly after obtaining clearance from the U.S. Food and Drug Administration to market Cortoss for the treatment of vertebral compression fractures. We fully launched Cortoss and Aliquot in the U.S. in September 2009. Sales of Cortoss and Aliquot in the United States were $1.1 million in 2009.
Biosurgery product sales represented 23% of total product sales in 2009 compared to 24% in 2008. Biosurgery product sales increased to $21.6 million in 2009 from $18.7 million in 2008, an increase of 15% primarily due to an increase in Vitagel sales.
Gross Profit. Gross profit for 2009 and 2008 was $62.9 million and $51.0 million, respectively. As a percentage of sales, gross profit was 68% for 2009, compared to 66% for 2008. Gross profit increases were seen in both the orthobiologic and biosurgery product families and primarily reflect more favorable product mix and successful efforts to reduce the cost of disposable and plastic components that are included in our product kits. Our gross profit may fluctuate from quarter to quarter based on the mix of products sold from period to period.
Operating expenses. While operating expenses increased 6% to $64.0 million in 2009 from $60.2 million in 2008, operating expenses as a percentage of product sales declined to 69% in 2009 from 78% of product sales in 2008. As illustrated in the line graph above, all components of operating expenses as a percentage of sales have been decreasing for the periods shown as we leverage our infrastructure to generate incremental sales. The increase in operating expenses for 2009 over 2008 was due to higher commissions associated with increased
44
Table of Contents
sales, infrastructure expansion expenses, and increased marketing costs related to the launch of Cortoss, partially offset by more effective cost management of certain sales force discretionary spending. We expect to continue to increase our staff, particularly for sales and marketing, for the next several years in response to anticipated product sales growth.
General and administrative expenses for 2009 were $12.2 million compared to $10.8 million for 2008, an increase of 13%. The increase is primarily due to additional finance, human resources and information technology staff required to provide sustainable infrastructure for future sales growth. Expenses for 2009 also included severance costs of $0.4 million associated with the departure of a senior executive officer. As a percentage of product sales, general and administrative expenses were 13% of product sales in 2009 compared to 14% of product sales in 2008. We expect general and administrative expenses to increase modestly to support the sales growth of our business.
Selling and marketing expenses for 2009 were $45.1 million, as compared to $42.7 million in 2008, an increase of 6%. Higher product sales commissions combined with costs associated with the launch of Cortoss were partially offset by reduced sales force discretionary spending and lower recruiting costs. Our field sales team increased to 111 employees at December 31, 2009, of which 90 were direct sales representatives, 13 were associate sales representatives and 8 were managers, from 96 employees at December 31, 2008, of which 76 were direct sales representatives, 11 were associate sales representatives and 9 were managers. As a percentage of product sales, selling and marketing expenses were 49% of product sales in 2009 compared to 56% of product sales in 2008. We expect selling and administrative costs to increase as we continue to expand our direct sales force; however, we also expect that these costs as a percentage of product sales will decrease as product sales increase and our sales force continues to become more efficient and effective.
Research and development expenses for 2009 were $6.8 million, compared to $6.7 million in 2008, an increase of 1%. The increase for 2009 was due to increased next generation and new product development costs offset by lower Cortoss development and clinical trial costs. As a percentage of product sales, research and development expenses were 7% of product sales in 2009 compared to 9% of product sales in 2008.
Other income (expense), net. Other income (expense), net, included interest income, interest expense and gain or loss on asset disposals. We recorded $2.8 million and $1.6 million of other income (expense), net for 2009 and 2008, respectively. Other income (expense), net for 2009 was higher than in 2008 primarily due to lower interest income resulting from lower interest rates and lower cash, cash equivalents and short-term investment balances in 2009, combined with higher interest expense in 2009 than in 2008. The higher 2009 interest expense was due to a higher level of borrowings outstanding for a longer period of time during 2009 as compared to 2008.
Comparison of the Year Ended December 31, 2008 to the Year Ended December 31, 2007
Product Sales. Product sales for 2008 increased 33% to $76.9 million as compared to $58.0 million for 2007. Sales growth in 2008 was primarily attributable to increased sales of Vitoss and Vitagel in the United States. Approximately 70% and 73% of our product sales during 2008 and 2007, respectively, were from products based upon our Vitoss platform. Vitagel contributed approximately 24% of our product sales during 2008, as compared to approximately 21% of our product sales during 2007.
For 2008 and 2007, 94% and 93% of product sales, respectively, were in the U.S., primarily from sales of Vitoss, Vitagel and Imbibe. The remaining sales during 2008 and 2007 were a result of Vitoss, Cortoss and Aliquot sales outside the U.S., primarily in Europe.
Gross Profit. Gross profit for 2008 and 2007 was $51.0 million and $37.5 million, respectively. As a percentage of sales, gross profit was 66% for 2008, as compared to 65% for 2007. The increase in the gross margin for 2008, as compared to 2007, primarily reflects more favorable product mix and lower Vitagel royalty expense as a percentage of product sales.
45
Table of Contents
Operating Expenses. Operating expenses for 2008 and 2007 were $60.2 million and $51.1 million, respectively, which represents an 18% increase in operating expenses as compared to a 33% increase in product sales and a 36% increase in gross profit. Operating expenses were 78% and 88% of product sales for 2008 and 2007, respectively.
General and administrative expenses for 2008 remained relatively constant at $10.8 million, as compared to $10.7 million for 2007. General and administrative expenses were 14% and 18% of product sales for 2008 and 2007, respectively.
Selling and marketing expenses were $42.7 million in 2008, a 26% increase from $33.9 million in 2007. Selling and marketing expenses increased in 2008 primarily due to higher expenses incurred in order to support the growth, and anticipated growth of U.S. product sales, and higher commissions paid in the U.S. as a result of increased product sales in 2008. Our field sales team increased to 96 employees at December 31, 2008, of which 76 were direct sales representatives, from 85 employees at December 31, 2007, of which 70 were direct sales representatives. Selling and marketing expenses were 56% and 58% of product sales for 2008 and 2007, respectively.
Research and development expenses increased 4% to $6.7 million in 2008 from $6.4 million for 2007. The increase for 2008 was primarily due to higher costs associated with our Cortoss product development and U.S. clinical trials. Research and development expenses were 9% and 11% of product sales for 2008 and 2007, respectively.
Other income (expense), net. Other income (expense), net included interest income, interest expense, gain or loss on asset disposals, a one-time charge for the repurchase of a revenue interest obligation, and revenue interest expense. We recorded $1.6 million and $16.3 million of other expense, net for 2008 and 2007, respectively. The decrease in other income (expense), net was primarily a result of the one-time charge of $16.6 million for the revenue interest obligation repurchase in 2007, partially offset by increased interest expense incurred under our debt facility (see note 10 to our consolidated financial statements included in this report) in 2008.
LIQUIDITY AND CAPITAL RESOURCES
We have experienced negative operating cash flows since our inception, and we have funded our operations primarily from the proceeds we received from sales of our common stock and other debt and equity securities. At December 31, 2009, we had cash, cash equivalents and short-term investments of $23.1 million, which represents 25% of total assets.
The Company’s cash flow activity was as follows:
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| (In thousands) | ||||||||||||
| Net cash used in operating activities |
$ | (5,064 | ) | $ | (14,210 | ) | $ | (14,174 | ) | |||
| Net cash provided by (used in) investing activities |
2,813 | 2,720 | (27,557 | ) | ||||||||
| Net cash provided by financing activities |
1,446 | 10,196 | 35,454 | |||||||||
| Effect of exchange rate changes on cash and cash equivalents |
44 | (374 | ) | 60 | ||||||||
| Net decrease in cash and cash equivalents |
$ | (761 | ) | $ | (1,668 | ) | $ | (6,217 | ) | |||
We may continue to use our existing cash, cash equivalents and short-term investments to fund our operations and fund our planned capital expenditures unless or until we are consistently profitable. Our operating cash flows are heavily dependent upon: (i) maintaining and growing product sales; (ii) the rates at which we add direct sales representatives; (iii) our product sales mix as relative increases in sales of our lower margin products result in lower gross profit; (iv) the amount of inventory, including raw materials and work-in-process, that we maintain to support product sales, anticipated product sales and anticipated product launches; and (v) the overall
46
Table of Contents
level of our research and development activities, which will depend on the development status and costs of products in our pipeline and any new products that we could pursue in the future. Accordingly, for the foreseeable future, our operating cash requirements may continue to be subject to quarterly volatility. If we are unable to improve cash flows from operations, we will need to consider and evaluate alternatives including reduction of capital expenditures, other cost reductions, and renegotiation of our outstanding debt, among others. Additionally, cash resources could be affected by in-licensing, joint ventures, acquisitions and other investment opportunities that we may pursue.
Discussion of Cash Flows
Cash Flows Used in Operating Activities
Net cash used in operating activities for 2009 was $5.1 million compared to $14.2 million used in operating activities for 2008, a decrease of $9.1 million. The decrease was primarily attributable to a $6.8 million decrease in our net loss (although non-cash charges increased by $1.3 million), and by increases in net working capital, primarily inventory to support higher sales levels and accounts receivable.
We expect to continue to focus our efforts on sales growth from our orthobiologic and biosurgery product platforms. We launched our Vitoss Bioactive Foam products in 2008 and our Cortoss product in the U.S. in 2009. After initiating a controlled launch of Cortoss in the U.S. in July 2009 using a subset of our sales force, we expanded the launch to our entire sales force at the end of the third quarter of 2009. We believe that the commercialization of Cortoss is benefiting and will continue to benefit from our infrastructure, core competencies and the call patterns of our sales force. Cortoss represents a new technology which will generally need to be approved by technology review boards of target hospitals in order for the hospitals to carry the product. The amount of time to obtain approvals from the technology review boards can be difficult to predict and we cannot guarantee that we will be able to secure widespread hospital approvals for Cortoss. We expect our U.S. Cortoss sales will increase as the product launch progresses, we obtain further hospital approvals for the product and our sales force trains more physicians on the product. We expect to continue to add direct sales representatives to our organization for those territories in the U.S. that are underserved. Also, we intend to fund studies to collect and publish data relating to the performance of our products to support our marketing and sales efforts. Our efforts to commercialize Cortoss could negatively impact our use of cash in the near term until sales staff hired in the fourth quarter of 2009 and in the first quarter of 2010 are fully trained and generating revenues in excess of their expenses.
Cash Flows Provided by (Used In) Investing Activities
Net cash provided by investing activities was $2.8 million for both 2009 and 2008. Net proceeds from the sale and maturity of investments decreased by $6.7 million in 2009 compared to 2008. Purchases of property and equipment totaled $4.9 million and $5.9 million in 2009 and 2008, respectively, and primarily related to the expansion of our Cortoss and Vitagel manufacturing facilities and we expect that this rate of capital expenditures will continue in 2010. Additionally, in 2008 we used $6.6 million to acquire collagen raw material, equipment and a technology license from Allergan Inc., which was recorded as an acquisition of a business (see note 3). There were no such cash outflows of this nature during 2009.
We invest our excess cash in highly liquid investment-grade marketable securities, including corporate debt securities and government-sponsored enterprise debt securities. Marketable securities having maturities greater than three months and less than one year are classified as short-term investments. Until we achieve sales at levels that consistently enable us to fund operations and investing activities, we expect to continue to use cash, cash equivalents and proceeds from sales of short-term investments to fund operating and investing activities.
Cash Flows Provided by Financing Activities
Net cash provided by financing activities for 2009 was $1.4 million, which was derived from the exercise of employee stock options and the sale of our common stock under our employee stock purchase plan. Net cash
47
Table of Contents
provided by financing activities for 2008 was $10.2 million, which was derived primarily from the issuance of an additional $10.0 million in debt under our debt facility (see note 10). The extent and timing of proceeds from future stock option and warrant exercises, if any, are primarily dependent upon future trading prices for our common stock and the expiration dates of these instruments.
CONTRACTUAL OBLIGATIONS AND COMMERCIAL COMMITMENTS
Senior Secured Note Purchase Facility. We have $35.0 million in outstanding principal indebtedness under a senior secured note purchase facility. Notes issued under the facility are due July 30, 2012. We issued $25.0 million in principal amount of notes under the facility on July 30, 2007 and used most of the proceeds to repurchase a revenue interest obligation. On July 31, 2008, we issued an additional $10.0 million principal amount of notes under the facility. We applied the proceeds of this note toward payment of (i) the purchase price of approximately $6.6 million for the collagen raw material, equipment and technology license acquired under our supply and license agreement with Allergan during the third quarter of 2008; and (ii) costs to expand our manufacturing capacity for Vitagel and ancillary products such as Aliquot, Imbibe and CellPaker.
Borrowings under the facility are guaranteed by us and one of our wholly-owned subsidiaries. The facility is secured by a first priority lien on substantially all of our assets (including intellectual property) other than those exclusively related to Cortoss and Aliquot. We are required to make quarterly interest-only payments to the note holder. Outstanding principal amounts under the notes bear annual interest at 10%, except that during the continuance of any event of defaults, interest would accrue at the rate of 12% per year and the notes would be payable on demand. We expect to incur quarterly interest payments of $875,000 under our debt facility during 2010 and each year thereafter until maturity on July 30, 2012.
Leases. We lease facilities under non-cancelable operating leases that extend through July 31, 2017. As we continue to expand, we expect to lease additional facilities.
Employment Agreements. Under the terms of employment agreements with two executive officers, extending through April 2011 and May 2011, subject to renewal, we are required to pay each individual a base salary for continuing employment with us. The agreements currently require aggregate payments of $688 and $255 in 2010 and 2011, respectively. The agreements also provide for other benefits, including certain obligations that may be triggered by a change in control, termination with cause or resignation for good reason.
Our contractual obligations under our notes, leases and employment agreements are set forth in the table below:
| (In Thousands) | Notes Payable Obligations |
Leases | Employment Agreements |
Total | ||||||||
| 2010 |
$ | — | $ | 862 | $ | 688 | $ | 1,550 | ||||
| 2011 |
— | 889 | 255 | 1,144 | ||||||||
| 2012 |
35,000 | 927 | — | 35,927 | ||||||||
| 2013 |
— | 964 | — | 964 | ||||||||
| 2014 and thereafter |
— | 3,689 | — | 3,689 | ||||||||
| $ | 35,000 | $ | 7,331 | $ | 943 | $ | 43,274 | |||||
In addition to the contractual obligations and commitments set forth above, we have the following contractual obligations:
Product Development Milestones and Related Payments. In connection with the development of new products with business partners, we may contractually agree to make milestone payments upon achievement of specified developmental goals. The timing and actual amount of these payments can be difficult to determine as they depend upon satisfactory achievement of product development and other milestones, which will be
48
Table of Contents
determined based on events that may occur in the future. During 2009, payments of $1.9 million were made for license fees and product development milestones pursuant to the contractual obligations that were incurred in 2009. If these products under development are brought to market, we may be obligated to make certain launch-related milestone payments of up to $1 million, as well as additional payments which may be made depending upon the potential achievements of clinical and sales milestones in the future.
Agreement with Kensey Nash Corporation. Approximately 67% of our product sales during 2009 were from products based upon our Vitoss Foam platform co-developed with Kensey, including Vitoss Bioactive Foam. As of December 31, 2009, we owed Kensey $2.4 million for manufactured product inventory and royalties, which amount is included in accounts payable and other accrued expenses on our consolidated balance sheet as of December 31, 2009 included in this report.
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
Foreign Currency Risk
The functional currency for our European branch operation is the Euro. The functional currency for our U.K. subsidiary operation is the British pound sterling. All assets and liabilities related to these operations are translated at the current exchange rates at the end of each period. Revenues and expenses are translated at average exchange rates in effect during the period. The resulting translation adjustments are accumulated in a separate component of shareholders’ equity (accumulated other comprehensive income (loss)). Foreign currency transaction gains and losses, if any, are included in our results of operations.
As of December 31, 2009, our total exposure to foreign currency risk in U.S. dollar terms was approximately $3.1 million, or 3.3%, of our total assets. The potential impact of a hypothetical 10% decline in the foreign exchange rates would result in a total decline in the fair value of our assets of approximately $0.3 million at December 31, 2009.
Market Risk
We may be exposed to market risk through changes in market interest rates that could affect the value of our short-term investments; however, we do not believe the fair value of our investment portfolio or related income would be significantly affected by changes in interest rates due mainly to the relatively short-term nature of the majority of our investment portfolio.
As of December 31, 2009, our short-term investments consisted of highly liquid investment-grade marketable securities. The impact on our future interest income and future changes in investment yields will depend on the gross amount of our investments and various external economic factors.
| ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
The consolidated financial statements of the Company and its subsidiaries and supplementary data required by this item are attached to this annual report on Form 10-K beginning on page F-1.
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
| ITEM 9A. | CONTROLS AND PROCEDURES |
Conclusion Regarding Effectiveness of Disclosure Controls and Procedures
A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to costs. Because of the inherent limitation in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within the Company have been detected. Because of the
49
Table of Contents
inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and may not be undetected. However, our disclosure controls and procedures are designed to provide reasonable assurance of achieving their objectives.
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2009. Based on that evaluation, the Chief Executive Officer and Chief Financial Officer concluded that, as of December 31, 2009 our disclosure controls and procedures are functioning effectively to provide reasonable assurance that the information required to be disclosed by us in reports filed under the Securities Exchange Act of 1934 is (i) recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms and (ii) accumulated and communicated to our management, including the Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding disclosure.
MANAGEMENT’S ANNUAL REPORT ON INTERNAL CONTROL OVER FINANCIAL REPORTING
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rule 13a-15(f) under the Securities Exchange Act of 1934. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our internal control over financial reporting includes those policies and procedures that pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets; provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, provide reasonable assurance that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management evaluated our internal control over financial reporting as of December 31, 2009. In making this assessment, management used the framework established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Management’s assessment included an evaluation of the design of our internal control over financial reporting and testing of the operational effectiveness of our internal control over financial reporting. Management reviewed the results of its assessment with the Audit Committee of our Board of Directors. Based on that assessment and based on the criteria in the COSO framework, management has concluded that, as of December 31, 2009, our internal control over financial reporting was effective.
Our independent registered public accounting firm, KPMG LLP, has audited the effectiveness of our internal control over financial reporting as of December 31, 2009. KPMG LLP’s report on the effectiveness of our internal control over financial reporting appears below.
| Antony Koblish | Nancy C. Broadbent | |
| President and Chief Executive Officer | Senior Vice President and Chief Financial Officer |
March 16, 2010
Changes in Internal Control Over Financial Reporting
There have been no changes in our internal control over financial reporting during the quarter ended December 31, 2009 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
50
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders
Orthovita, Inc.:
We have audited Orthovita, Inc. and subsidiaries’ (the Company) internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Orthovita, Inc. and subsidiaries’ management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Annual Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Orthovita, Inc. and subsidiaries maintained, in all material respects, effective internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control—Integrated Framework issued by the COSO.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Orthovita, Inc. and subsidiaries as of December 31, 2009 and 2008, and the related consolidated statements of operations, shareholders’ equity and comprehensive loss, and cash flows for each of the years in the three-year period ended December 31, 2009, and our report dated March 16, 2010 expressed an unqualified opinion on those consolidated financial statements.
/s/ KPMG LLP
Philadelphia, Pennsylvania
March 16, 2010
| ITEM 9B. | OTHER INFORMATION |
Not applicable.
51
Table of Contents
PART III
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
(a) Identification of Directors
Information with respect to the members of the Board of Directors of the Company is set forth under the caption “Election of Directors” in the Company’s definitive proxy statement, to be filed within 120 days after the end of the year covered by this annual report on Form 10-K, which information is incorporated herein by reference.
(b) Identification of Executive Officers
Information with respect to the executive officers of the Company is set forth under the caption “Executive Officers of Orthovita” in the Company’s definitive proxy statement, to be filed within 120 days after the end of the year covered by this annual report on Form 10-K, which information is incorporated herein by reference.
(c) Section 16(a) Beneficial Ownership Reporting Compliance
Information with respect to the Section 16(a) compliance of the directors and executive officers of the Company is set forth under the caption “Section 16(a) Beneficial Ownership Reporting Compliance” in the Company’s definitive proxy statement, to be filed within 120 days after the end of the year covered by this annual report on Form 10-K, which information is incorporated herein by reference.
(d) Code of Conduct and Ethics
Information with respect to the Company’s Code of Conduct is set forth under the caption “Governance of the Company-Code of Conduct” in the Company’s definitive proxy statement, to be filed within 120 days after the end of the year covered by this annual report on Form 10-K, which information is incorporated herein by reference.
(e) Audit Committee Financial Expert
Information with respect to the Company’s Audit Committee Financial Expert is set forth under the caption “Governance of the Company-Audit Committee” in the Company’s definitive proxy statement, to be filed within 120 days after the end of the year covered by this annual report on Form 10-K, which information is incorporated herein by reference.
| ITEM 11. | EXECUTIVE COMPENSATION |
Information required by this item is set forth under the caption “Compensation of Executive Officers and Directors” in the Company’s definitive proxy statement, to be filed within 120 days after the end of the year covered by this annual report on Form 10-K, which information is incorporated herein by reference.
52
Table of Contents
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
Equity Compensation Plan Information as of December 31, 2009
| Plan Category |
Number of securities to be issued upon exercise of outstanding options, warrants and rights (a) |
Weighted-average exercise price of outstanding options, warrants and rights (b) |
Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a) (c) | ||||
| Equity compensation plans approved by security holders |
7,200,000 | $ | 3.55 | 5,397,390 | |||
The information called for by this item not contained in this annual report on Form 10-K will be set forth under the captions “Stock Ownership-Securities Ownership of Certain Beneficial Owners and Management” in our definitive proxy statement, to be filed within 120 days after the end of the year covered by this annual report on Form 10-K, and is incorporated herein by reference.
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information called for by this item not contained in this annual report on Form 10-K will be set forth under the caption “Certain Relationships and Related Transactions, and Director Independence” in our definitive proxy statement, to be filed within 120 days after the end of the year covered by this annual report on Form 10-K, and is incorporated herein by reference.
| ITEM 14. | PRINCIPAL ACCOUNTING FEES AND SERVICES |
The information called for by this item not contained in this annual report on Form 10-K will be set forth under the caption “Ratification of Appointment of Independent Registered Public Accounting Firm” in our definitive proxy statement, to be filed within 120 days after the end of the year covered by this annual report on Form 10-K, and is incorporated herein by reference.
53
Table of Contents
PART IV
| ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES |
(a) Documents filed as part of this Report
1. Financial Statements. The following consolidated financial statements and notes thereto which are attached hereto beginning on page F-1 have been included by reference into Item 8 of this part of the annual report on Form 10-K:
2. Financial Statement Schedules. All schedules are omitted because they are inapplicable, or not required, or the information is shown in the Consolidated Financial Statements or notes thereto.
3. Exhibits. The information required by this item is set forth in the Exhibit Index hereto which is incorporated herein by reference.
54
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Date: March 16, 2010 | ORTHOVITA, INC. | |||
| By: | /s/ ANTONY KOBLISH | |||
| President and Chief Executive Officer | ||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| Signature |
Capacity |
Date | ||
| /s/ ANTONY KOBLISH |
President, Chief Executive Officer and Director (principal executive officer) |
March 16, 2010 | ||
| /s/ WILLIAM E. TIDMORE, JR. |
Chairman | March 16, 2010 | ||
| /s/ NANCY C. BROADBENT |
Senior Vice President and Chief Financial Officer (principal financial and accounting officer) |
March 16, 2010 | ||
| /s/ R. SCOTT BARRY |
Director | March 16, 2010 | ||
| /s/ MORRIS CHESTON, JR. |
Director | March 16, 2010 | ||
| /s/ MARY E. PAETZOLD |
Director | March 16, 2010 | ||
| /s/ PAUL G. THOMAS |
Director | March 16, 2010 | ||
| /s/ PAUL T. TOUHEY, JR. |
Director | March 16, 2010 | ||
55
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders
Orthovita, Inc.:
We have audited the accompanying consolidated balance sheets of Orthovita, Inc. and subsidiaries (the Company) as of December 31, 2009 and 2008, and the related consolidated statements of operations, shareholders’ equity and comprehensive loss, and cash flows for each of the years in the three-year period ended December 31, 2009. These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Orthovita, Inc. and subsidiaries as of December 31, 2009 and 2008, and the results of their operations and their cash flows for each of the years in the three-year period ended December 31, 2009, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Orthovita, Inc. and subsidiaries’ internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO), and our report dated March 16, 2010 expressed an unqualified opinion on the effectiveness of the Company’s internal control over financial reporting.
/s/ KPMG LLP
Philadelphia, Pennsylvania
March 16, 2010
F-1
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
CONSOLIDATED BALANCE SHEETS
(In thousands except per share data)
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| ASSETS | ||||||||
| CURRENT ASSETS: |
||||||||
| Cash and cash equivalents |
$ | 7,757 | $ | 8,518 | ||||
| Short-term investments |
15,349 | 23,773 | ||||||
| Accounts receivable, net of allowance for doubtful accounts of $217 and $252, respectively |
12,324 | 10,881 | ||||||
| Inventories |
26,058 | 19,757 | ||||||
| Other current assets |
784 | 693 | ||||||
| Total current assets |
62,272 | 63,622 | ||||||
| Property and equipment, net |
17,940 | 14,438 | ||||||
| License and technology intangible assets, net |
11,376 | 12,354 | ||||||
| Other assets |
1,041 | 757 | ||||||
| Total assets |
$ | 92,629 | $ | 91,171 | ||||
| LIABILITIES AND SHAREHOLDERS’ EQUITY | ||||||||
| CURRENT LIABILITIES: |
||||||||
| Accounts payable |
$ | 5,028 | $ | 4,032 | ||||
| Accrued compensation and related expenses |
2,805 | 2,329 | ||||||
| Other accrued expenses |
5,534 | 5,049 | ||||||
| Total current liabilities |
13,367 | 11,410 | ||||||
| LONG-TERM LIABILITIES: |
||||||||
| Notes payable, net of debt discount of $905 and $1,191, respectively |
34,095 | 33,809 | ||||||
| Other long-term liabilities |
408 | 310 | ||||||
| Total long-term liabilities |
34,503 | 34,119 | ||||||
| Total liabilities |
47,870 | 45,529 | ||||||
| COMMITMENTS (Note 16) |
||||||||
| SHAREHOLDERS’ EQUITY: |
||||||||
| Common stock, $.01 par value, 100,000 shares authorized, 76,542 and 75,930 shares issued and outstanding |
765 | 759 | ||||||
| Additional paid-in capital |
224,734 | 221,633 | ||||||
| Accumulated deficit |
(180,578 | ) | (176,672 | ) | ||||
| Accumulated other comprehensive loss |
(162 | ) | (78 | ) | ||||
| Total shareholders’ equity |
44,759 | 45,642 | ||||||
| Total liabilities and shareholders’ equity |
$ | 92,629 | $ | 91,171 | ||||
The accompanying notes are an integral part of these statements.
F-2
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands except per share data)
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| PRODUCT SALES |
$ | 92,853 | $ | 76,915 | $ | 58,046 | ||||||
| COST OF SALES |
29,914 | 25,929 | 20,544 | |||||||||
| GROSS PROFIT |
62,939 | 50,986 | 37,502 | |||||||||
| OPERATING EXPENSES: |
||||||||||||
| General and administrative |
12,172 | 10,753 | 10,700 | |||||||||
| Selling and marketing |
45,070 | 42,706 | 33,945 | |||||||||
| Research and development |
6,786 | 6,711 | 6,435 | |||||||||
| Total operating expenses |
64,028 | 60,170 | 51,080 | |||||||||
| OPERATING LOSS |
(1,089 | ) | (9,184 | ) | (13,578 | ) | ||||||
| OTHER INCOME (EXPENSE), NET: |
||||||||||||
| Interest expense |
(3,112 | ) | (2,777 | ) | (1,342 | ) | ||||||
| Interest income |
282 | 1,271 | 1,759 | |||||||||
| (Loss) gain on sale of product line and related assets |
— | (72 | ) | 372 | ||||||||
| Charge for repurchase of revenue interest obligation |
— | — | (16,605 | ) | ||||||||
| Revenue interest expense |
— | — | (756 | ) | ||||||||
| Total other income (expense), net |
(2,830 | ) | (1,578 | ) | (16,572 | ) | ||||||
| LOSS BEFORE INCOME TAXES |
(3,919 | ) | (10,762 | ) | (30,150 | ) | ||||||
| INCOME TAX BENEFIT |
13 | 10 | 268 | |||||||||
| NET LOSS |
$ | (3,906 | ) | $ | (10,752 | ) | $ | (29,882 | ) | |||
| NET LOSS PER SHARE, BASIC AND DILUTED |
$ | (0.05 | ) | $ | (0.14 | ) | $ | (0.44 | ) | |||
| SHARES USED IN COMPUTING BASIC AND DILUTED NET LOSS PER SHARE |
76,176 | 75,804 | 67,181 | |||||||||
The accompanying notes are an integral part of these statements.
F-3
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF SHAREHOLDERS’ EQUITY AND
COMPREHENSIVE LOSS
(In thousands)
| Common Stock | Additional Paid-in Capital |
Accumulated Deficit |
Accumulated Other Compre- hensive Income (Loss) |
Compre- hensive Loss |
Total | |||||||||||||||||||
| Number of shares |
Amount | |||||||||||||||||||||||
| BALANCE, JANUARY 1, 2007 |
61,313 | $ | 614 | $ | 178,555 | $ | (136,038 | ) | $ | 130 | $ | — | $ | 43,261 | ||||||||||
| Purchase of shares by employees, non-employees and exercise of warrants |
253 | 2 | 723 | — | — | — | 725 | |||||||||||||||||
| Issuance of shares under restricted stock award |
36 | — | 141 | — | — | — | 141 | |||||||||||||||||
| Issuance of shares for services |
4 | — | 11 | — | — | — | 11 | |||||||||||||||||
| Issuance of warrants in connection with debt facility |
— | — | 1,206 | — | — | — | 1,206 | |||||||||||||||||
| Sale of shares, net |
12,317 | 123 | 32,031 | — | — | — | 32,154 | |||||||||||||||||
| Issuance of shares as part of a revenue interest repurchase |
1,136 | 11 | 3,761 | — | — | — | 3,772 | |||||||||||||||||
| Issuance of shares in exchange for non-employee consultant stock options |
614 | 6 | 1,937 | — | — | — | 1,943 | |||||||||||||||||
| Stock-based compensation expense related to stock options and restricted stock units |
11 | — | 1,079 | — | — | — | 1,079 | |||||||||||||||||
| Comprehensive loss: |
||||||||||||||||||||||||
| Net loss |
— | — | — | (29,882 | ) | — | (29,882 | ) | (29,882 | ) | ||||||||||||||
| Other comprehensive loss: |
||||||||||||||||||||||||
| Unrealized gain on investments |
— | — | — | — | 41 | 41 | 41 | |||||||||||||||||
| Currency translation adjustment |
— | — | — | — | 77 | 77 | 77 | |||||||||||||||||
| Comprehensive loss |
$ | (29,764 | ) | |||||||||||||||||||||
| BALANCE, DECEMBER 31, 2007 |
75,684 | 756 | 219,444 | (165,920 | ) | 248 | 54,528 | |||||||||||||||||
| Purchase of shares by employees, non-employees and exercise of warrants |
80 | 1 | 195 | — | — | — | 196 | |||||||||||||||||
| Issuance of shares under restricted stock award |
28 | 1 | 40 | — | — | — | 41 | |||||||||||||||||
| Issuance of shares for services |
7 | — | 21 | — | — | — | 21 | |||||||||||||||||
| Issuance of warrants in connection with debt facility |
— | — | 286 | — | — | — | 286 | |||||||||||||||||
| Issuance of shares in exchange for non-employee consultant stock options |
131 | 1 | 415 | — | — | — | 416 | |||||||||||||||||
| Stock-based compensation expense related to stock options and restricted stock units |
— | 1,232 | — | — | — | 1,232 | ||||||||||||||||||
| Comprehensive loss: |
— | |||||||||||||||||||||||
| Net loss |
— | — | — | (10,752 | ) | — | (10,752 | ) | (10,752 | ) | ||||||||||||||
| Other comprehensive loss: |
||||||||||||||||||||||||
| Unrealized gain on investments |
— | — | — | — | 101 | 101 | 101 | |||||||||||||||||
| Currency translation adjustment |
— | — | — | — | (427 | ) | (427 | ) | (427 | ) | ||||||||||||||
| Comprehensive loss |
$ | (11,078 | ) | |||||||||||||||||||||
| BALANCE DECEMBER 31, 2008 |
75,930 | $ | 759 | $ | 221,633 | $ | (176,672 | ) | $ | (78 | ) | $ | 45,642 | |||||||||||
| Purchase of shares by employees, non-employees and exercise of warrants |
428 | 4 | 1,442 | — | — | — | 1,446 | |||||||||||||||||
| Issuance of shares under restricted stock award |
28 | — | 95 | — | — | — | 95 | |||||||||||||||||
| Stock-based compensation expense related to stock options and restricted stock units |
156 | 2 | 1,564 | — | — | — | 1,566 | |||||||||||||||||
| Comprehensive loss: |
||||||||||||||||||||||||
| Net loss |
— | — | — | (3,906 | ) | (3,906 | ) | (3,906 | ) | |||||||||||||||
| Other comprehensive loss: |
||||||||||||||||||||||||
| Unrealized loss on investments |
— | — | — | — | (150 | ) | (150 | ) | (150 | ) | ||||||||||||||
| Currency translation adjustment |
— | — | — | — | 66 | 66 | 66 | |||||||||||||||||
| Comprehensive loss |
$ | (3,990 | ) | |||||||||||||||||||||
| BALANCE DECEMBER 31, 2009 |
76,542 | $ | 765 | $ | 224,734 | $ | (180,578 | ) | $ | (162 | ) | $ | 44,759 | |||||||||||
The accompanying notes are an integral part of these statements.
F-4
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| OPERATING ACTIVITIES: |
||||||||||||
| Net loss |
$ | (3,906 | ) | $ | (10,752 | ) | $ | (29,882 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Charge for repurchase of revenue interest obligation |
— | — | 16,605 | |||||||||
| Amortization of discounts and premiums related to investments |
36 | (389 | ) | (832 | ) | |||||||
| Depreciation and amortization of property and equipment |
1,831 | 1,728 | 1,589 | |||||||||
| Amortization of license and technology intangible assets |
1,493 | 995 | 850 | |||||||||
| Amortization of debt discount |
286 | 199 | 102 | |||||||||
| Common stock issued for services rendered |
— | 21 | 11 | |||||||||
| Compensation related to restricted stock awards, restricted stock units and stock option accounting for employees and non-employees |
1,669 | 1,260 | 535 | |||||||||
| Charge related to exchange of non-employee stock options for common stock |
— | 170 | 1,161 | |||||||||
| Deferred tax expense (benefit) |
7 | 7 | (268 | ) | ||||||||
| Provision for doubtful accounts |
18 | 97 | 152 | |||||||||
| (Gain) on sale of product line and related assets |
— | — | (372 | ) | ||||||||
| Loss on disposal of property and equipment |
— | 72 | — | |||||||||
| (Increase) decrease in (net of business acquisition in 2008)— |
||||||||||||
| Accounts receivable |
(1,444 | ) | (2,578 | ) | 165 | |||||||
| Inventories |
(6,290 | ) | (3,187 | ) | (6,229 | ) | ||||||
| Other current assets |
(48 | ) | (71 | ) | 295 | |||||||
| Other assets |
(425 | ) | (212 | ) | (332 | ) | ||||||
| (Decrease) increase in — |
||||||||||||
| Accounts payable |
901 | (773 | ) | 2,393 | ||||||||
| Accrued compensation and related expenses |
357 | 202 | 331 | |||||||||
| Other accrued expenses |
375 | (1,056 | ) | (431 | ) | |||||||
| Other long-term liabilities |
76 | 57 | (17 | ) | ||||||||
| Net cash used in operating activities |
(5,064 | ) | (14,210 | ) | (14,174 | ) | ||||||
| INVESTING ACTIVITIES: |
||||||||||||
| Purchase of investments |
(29,936 | ) | (40,750 | ) | (61,050 | ) | ||||||
| Proceeds from sale and maturity of investments |
38,174 | 55,674 | 35,640 | |||||||||
| Purchases of property and equipment |
(4,910 | ) | (5,852 | ) | (2,606 | ) | ||||||
| Proceeds from sale of product line |
— | — | 459 | |||||||||
| Purchase of intangible asset |
(515 | ) | — | — | ||||||||
| Proceeds from sale of property and equipment |
— | 200 | — | |||||||||
| Acquisition of business |
— | (6,552 | ) | — | ||||||||
| Net cash provided by (used in) investing activities |
2,813 | 2,720 | (27,557 | ) | ||||||||
| FINANCING ACTIVITIES: |
||||||||||||
| Repayment of notes payable |
— | — | (2,033 | ) | ||||||||
| Repayments of capital lease obligations |
— | — | (392 | ) | ||||||||
| Repayment of revenue interest obligation |
— | — | (20,000 | ) | ||||||||
| Proceeds from notes payable |
— | 10,000 | 25,000 | |||||||||
| Proceeds from sales of common stock and warrants, net |
— | — | 32,154 | |||||||||
| Proceeds from issuance of common stock |
1,446 | 196 | 725 | |||||||||
| Net cash provided by financing activities |
1,446 | 10,196 | 35,454 | |||||||||
| EFFECT OF EXCHANGE RATE CHANGES ON CASH AND CASH EQUIVALENTS |
44 | (374 | ) | 60 | ||||||||
| NET DECREASE IN CASH AND CASH EQUIVALENTS |
(761 | ) | (1,668 | ) | (6,217 | ) | ||||||
| CASH AND CASH EQUIVALENTS, BEGINNING OF YEAR |
8,518 | 10,186 | 16,403 | |||||||||
| CASH AND CASH EQUIVALENTS, END OF YEAR |
$ | 7,757 | $ | 8,518 | $ | 10,186 | ||||||
The accompanying notes are an integral part of these statements.
F-5
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
1. The Company:
We are a specialty spine and orthopedic company with a portfolio of orthobiologic and biosurgery products. Our products are based on novel and unique proprietary biomaterials that have innovative mechanisms of action in the body. Our orthobiologic platform offers products for the fusion, regeneration and fixation of human bone. Our biosurgery platform offers products for controlling intra-operative bleeding, also known as hemostasis. In the United States, we primarily market our products through a direct sales force but also utilize non-stocking distributors and independent sales agents in certain geographical areas. Outside the United States, we market our products through a direct sales force in the United Kingdom and through independent stocking distributors in other countries.
Orthobiologics
Vitoss™ Bone Graft Substitute is our primary fusion and regeneration product, and Cortoss™ Bone Augmentation Material is our primary fixation product. Our Vitoss product line, which includes Vitoss Foam and Vitoss Bioactive Foam products, provides the non-structural bone graft market with synthetic, bioactive alternatives to patient- and cadaver-derived bone tissue. An injectable polymer composite which mimics the structural characteristics of human bone, Cortoss provides the vertebral compression fracture market with a synthetic, bioactive alternative to polymethylmethacrylate bone cement.
Biosurgery
The primary products in our hemostasis portfolio are Vitagel™ Surgical Hemostat, a proprietary, collagen-based matrix that controls bleeding and facilitates healing, and Vitasure™ Absorbable Hemostat, a proprietary, plant-based product that can be deployed quickly throughout surgery.
We also market accessories and delivery products which complement the orthobiologic and biosurgery platforms. In addition, we seek to expand our product portfolio through internal product development efforts, co-development efforts with strategic partners, and acquisition and in-licensing opportunities.
2. Summary of Significant Accounting Policies:
Basis of Presentation
The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries. All intercompany accounts and transactions have been eliminated in consolidation
The financial statements of our international operations are translated into U.S. dollars using the exchange rate at each balance sheet date for assets and liabilities, and an average exchange rate for each period of revenues, expenses, and gain and losses. The functional currency of our non-U.S. operations is the local currency. Adjustments resulting from the translation of financial statements are reflected in accumulated other comprehensive income. Transaction gains and losses are charged to operations.
Preparation of Financial Statements and Use of Estimates
The preparation of the consolidated financial statements requires us to make assumptions, estimates and judgments that affect the reported amounts of assets and liabilities, the disclosures of contingent assets and liabilities as of the date of the consolidated financial statements, and the reported amounts of revenues and expenses during the reporting periods. On an ongoing basis, we evaluate our estimates, including, but not limited to, those related to accounts receivable, inventories and recoverability of long-lived assets. We use historical
F-6
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
experience and other assumptions as the basis for making estimates. Illiquid credit markets, volatile equity, foreign currency and energy markets, and declines in consumer spending have combined to increase the uncertainty inherent in such estimates and assumptions. As future events and their effects cannot be determined with precision, actual results could differ significantly from these estimates. Changes in those estimates will be reflected in the financial statements in those future periods.
In June 2009, the Financial Accounting Standards Board’s (FASB) Accounting Standards Codification (referred to as the Codification or the ASC) officially became the single source of authoritative nongovernmental generally accepted accounting principles (GAAP), superseding existing accounting literature. Only one level of authoritative GAAP now exists and all other accounting literature is considered non-authoritative. The Codification reorganizes the thousands of GAAP pronouncements into roughly 90 accounting topics and displays them using a consistent structure. Also included in the Codification is relevant SEC guidance organized using the same topical structure in separate sections within the Codification. Disclosures in our consolidated financial statements to all references to authoritative accounting literature are now through the Codification.
Cash and Cash Equivalents and Short-Term Investments
We consider all highly liquid instruments with an original maturity of 90 days or less at the time of purchase to be cash equivalents. Cash equivalents consist of deposits that are readily convertible into cash (see note 4).
For financial reporting purposes, our entire short-term investment portfolio is classified as available-for-sale and is stated at fair value as determined by quoted market values. Accordingly, any unrealized holding gains and losses are included in accumulated other comprehensive income (loss), as a separate component of shareholders’ equity. Discounts and premiums are amortized over the term of the security and reported in interest income. The investments are reviewed on a periodic basis for other-than-temporary impairments (see notes 4 and 5).
Fair Value Measurements
ASC Topic 820, Fair Value Measurements, defines fair value, establishes a framework for measuring fair value and expands the related disclosure requirements. The guidance applies under other accounting pronouncements that require or permit fair value measurements and indicates, among other things, that a fair value measurement assumes that the transaction to sell an asset or transfer a liability occurs in the principal market for the asset or liability or, in the absence of a principal market, the most advantageous market for the asset or liability. ASC 820 defines fair value based upon an exit price model.
Our financial instruments are categorized into a three-level fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three broad levels. The fair value hierarchy gives the highest priority to quoted prices in active markets for identical assets or liabilities (Level 1) and the lowest priority to unobservable inputs (Level 3). If the inputs used to measure fair value fall within different levels of the hierarchy, the category level is based on the lowest priority level input that is significant to the fair value measurement of the instrument. Financial assets recorded at fair value on our consolidated balance sheets are categorized as follows:
| • | Level 1: Observable inputs such as quoted prices in active markets; |
| • | Level 2: Inputs, other than the quoted prices in active markets, that are observable either directly or indirectly; and |
| • | Level 3: Unobservable inputs in which there is little or no market data, which require the reporting entity to develop its own assumptions. |
F-7
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
Concentrations of Credit Risk
Our policy is to limit the amount of credit exposure to any one financial institution, and place investments with financial institutions evaluated as being creditworthy, or in short-term money market funds which are exposed to minimal interest rate and credit risk. We maintain our cash primarily in investment accounts within large financial institutions. Currently, the Federal Deposit Insurance Corporation insures these balances up to $250 per bank. We have not experienced any losses on our bank deposits and we believe these deposits do not expose us to any significant credit risk.
We grant credit, generally without collateral, to our customers, which are primarily in the health care market. Consequently, we are subject to potential credit risk related to changes in economic conditions within that market. However, we believe that our billing and collection policies are adequate to minimize the potential credit risk.
Concentrations of Supply Risk
We are highly dependent on Kensey Nash Corporation to convert our Vitoss to Vitoss Foam and Vitoss Bioactive Foam (see note 16). We also source other raw materials and a majority of the disposable components that accompany and are used with our products with a limited number of third party manufacturers and are highly dependent upon these manufacturers.
Inventories
Inventories are stated at the lower of cost or market value using the first-in first-out basis, or FIFO. A provision is recorded to reduce the cost of inventories to the estimated net realizable values, if required. The costs of producing inventory in the reporting periods prior to the receipt of regulatory approval or clearance are recorded as research and development expense (see note 6).
Property and Equipment
Property and equipment are recorded at cost. Depreciation is calculated on a straight-line basis over the estimated useful life of each asset, generally three to five years. The useful life for leasehold improvements is generally the term of the facility lease. Expenditures for major renewals and improvements are capitalized, and expenditures for maintenance and repairs are charged to operations as incurred. Property and equipment includes assets designated as construction in-progress and not placed in service. We begin to depreciate these assets when they are placed into service or, if the property and equipment requires approval from the U.S. Food and Drug Administration (FDA), when such approval is obtained and the asset is placed in service (see note 7).
Interest cost that is incurred on borrowed funds used to expand the manufacturing facilities is capitalized and is recorded as part of the asset to which it relates and is amortized over the asset’s estimated useful life.
Impairment of Long-Lived Assets
We evaluate the recoverability of long-lived assets whenever events or changes in circumstances indicate that an asset’s carrying amount may not be recoverable. Such circumstances could include, but are not limited to (1) a significant decrease in the market value of an asset, (2) a significant adverse change in the extent or manner in which an asset is used, or (3) an accumulation of costs significantly in excess of the amount originally expected for the acquisition of an asset. We measure the carrying amount of the asset against the estimated undiscounted future cash flows associated with it. If the sum of the expected future net cash flows is less than the
F-8
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
carrying value of the asset being evaluated, an impairment charge would be recognized. The impairment charge would be calculated as the amount by which the carrying value of the asset exceeds its fair value. The determination of fair value is based on quoted market prices, if available. If quoted market prices are not available, the estimate of fair value is based on various valuation techniques, including the discounted value of estimated future cash flows. At December 31, 2009 and 2008, the carrying amounts of our long-lived assets (property and equipment and intangible assets) were not impaired.
Revenue Recognition
Revenue is not recognized until persuasive evidence of an arrangement exists, performance or delivery has occurred, the sales price is fixed or determinable and collection is probable. In the U.S., we recognize product sales revenue upon either shipment of purchased product to the customer, or receipt of documentation from the customer indicating its consumption of product from consigned inventory. In 2008, for transactions outside the U.S., we modified shipping terms from FOB destination to FOB shipping point. Accordingly, outside the U.S., beginning in 2008, revenue from product sales was primarily recognized upon shipment of the product to the independent stocking distributors. Prior to 2008, we recognized revenue from product sales outside the U.S. upon receipt of product by the independent stocking distributors.
We do not allow product returns or exchanges. We have no obligations to our customers once title has been transferred. We generally require each of our U.S. and U.K. customers to pay on a net 30-day basis. Outside of the U.S. and the U.K., independent stocking distributors are generally required to pay on a net 60 day basis.
Net Loss per Common Share
Basic net loss per share excludes securities exercisable for or convertible into shares of common stock, and is computed by dividing net loss applicable to common shareholders by the weighted average number of shares of common stock outstanding for the period. Diluted net loss per common share data is computed assuming the conversion or exercise of all dilutive securities such as common stock options and warrants.
Equity awards and warrants exercisable to purchase 9,282 8,904 and 8,022 shares were excluded from the computation of diluted net loss per common share for 2009, 2008 and 2007, respectively, because the inclusion of the shares in the calculation would have been anti-dilutive due to the losses.
Income Taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to the differences between the financial statement carrying amounts of existing assets and liabilities and the respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date.
The asset and liability method requires that deferred tax assets and liabilities be recorded without consideration as to their realizability. The deferred tax asset primarily includes net operating loss and tax credit
F-9
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
carryforwards, accrued expenses not currently deductible and the cumulative temporary differences related to certain research and patent costs, which have been charged to expense in the accompanying statements of operations but have been recorded as assets for income tax purposes. The portion of any deferred tax asset for which it is more likely than not that a tax benefit will not be realized must then be offset by recording a valuation allowance. A valuation allowance has been established against all of the deferred tax assets, with the exception of the Texas margin tax credit, as it is more likely than not that these assets will not be realized given the history of operating losses.
While we believe that our tax positions are fully supportable, there is a risk that certain positions could be challenged successfully. In these instances, we look to establish reserves. If we determine that a tax position is more likely than not of being sustained upon audit, based solely on the technical merits of the position, we recognize the benefit. We measure the benefit by determining the amount that has a likelihood greater than 50% of being realized upon settlement. We presume that all tax positions will be examined by a taxing authority with full knowledge of all relevant information. We regularly monitor our tax positions, tax assets and tax liabilities. We reevaluate the technical merits of our tax positions and recognize an uncertain tax benefit or derecognize a previously recorded tax benefit when (i) there is a completion of a tax audit, (ii) there is a change in applicable tax law including a tax case or legislative guidance, or (iii) there is an expiration of the statute of limitations. Significant judgment is required in accounting for tax reserves (see note 15).
Stock-based Compensation
Stock-based compensation is accounted for at grant-date fair value and amortized on a straight-line basis over the vesting terms of the awards in the consolidated statements of operations based upon the portion of the awards that are expected to vest. We estimate pre-vesting forfeitures by analyzing historical data and revise those estimates in subsequent periods if actual forfeitures differ from those estimates (see note 13).
The following table sets forth the total stock-based compensation expense included in the consolidated statements of operations for the years ended December 31, 2009, 2008, and 2007.
| Year Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Cost of sales |
$ | 74 | $ | 42 | $ | 52 | |||
| General and administrative |
829 | 584 | 832 | ||||||
| Selling and marketing |
540 | 464 | 307 | ||||||
| Research and development |
218 | 183 | 29 | ||||||
| Total |
$ | 1,661 | $ | 1,273 | $ | 1,220 | |||
We estimated the fair value of each option grant using the Black-Scholes option-pricing model using the following weighted-average assumptions:
| Year Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Risk-free interest rate |
2.85 | % | 3.78 | % | 4.64 | % | |||
| Expected volatility |
55 | % | 58 | % | 54 | % | |||
| Dividend yield |
0 | % | 0 | % | 0 | % | |||
| Expected life |
6 years | 6 years | 5 years | ||||||
F-10
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
The weighted average fair value per share of options granted in 2009, 2008, and 2007 was $2.01, $1.70 and $1.61, respectively.
Expected volatility was calculated based upon the historical daily closing prices of our common stock as quoted on the NASDAQ Global Market (NASDAQ) over a prior period having a term equal to the expected life of the stock options.
Non-employee Stock Based Compensation
The cost of stock-based compensation awards issued to non-employees for services are recorded at the fair value of instruments issued in exchange for such services. Historically, the common stock options granted to non-employee consultants as compensation were fully vested on the date of the grant. In certain circumstances, these awards may have to be settled in cash through the expiration of the option or until it is exercised, whichever is earlier. Accordingly, the fair value of the fully-vested non-employee consultant stock options are classified as liabilities with changes in fair value between reporting periods recognized in our consolidated statements of operations.
The fair value of the derivative liability associated with non-employee consultant stock options was estimated using the Black-Scholes option pricing model. For the years ended December 31, 2009, 2008 and 2007, the income (expense) associated with the change in fair value of the derivative liability was $8, $(13) and $(675), respectively, and was recorded in selling and marketing expenses in our consolidated statements of operations.
Research & Development Costs
Research and development costs are expensed as incurred.
Segment Information
We are managed and operated as one business. The entire business is managed by a single management team that reports to the chief executive officer. We do not operate separate lines of business and do not prepare discrete financial information with respect to separate product areas or by location and therefore do not have separate reportable segments.
Comprehensive Income (Loss)
Comprehensive income (loss) is the change in equity of a business enterprise during a period from non-owner sources. Excluding net income (loss), our sources of other comprehensive income (loss) are from net unrealized appreciation (depreciation) on available-for-sale securities and foreign currency translation adjustments. Reclassification adjustments result from the recognition in net income (loss) of unrealized gains or losses that were included in comprehensive income (loss) in prior periods.
F-11
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
Supplemental Cash Flow Information
| Year Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Supplemental disclosure of non-cash transactions: |
|||||||||
| Warrants issued in connection with senior secured credit facility (note 10) |
$ | — | $ | 286 | $ | 1,206 | |||
| Exchange of stock options for common stock (note 13) |
— | 170 | 1,161 | ||||||
| Capital lease obligations |
— | — | 13 | ||||||
| Supplemental disclosure of cash flow information: |
|||||||||
| Cash paid for interest and revenue interest |
2,826 | 2,325 | 1,359 | ||||||
| Cash paid for taxes |
85 | 82 | 32 | ||||||
Recent Accounting Pronouncements
In April 2009, the FASB provided guidance as codified in ASC Subtopic 320-10 which amends the accounting for certain investments in debt and equity securities to modify the indicator of other-than-temporary impairment for debt securities. Additionally, this guidance changes the amount of an other-than-temporary impairment that is recognized in earnings when there are credit losses on a debt security that management does not intend to sell and it is more-likely-than-not that the entity will not have to sell prior to recovery of the noncredit impairment. In those situations, the portion of the total impairment that is attributable to the credit loss would be recognized in earnings, and the remaining difference between the debt security’s amortized cost basis and its fair value would be included in other comprehensive income. The guidance was effective for interim and annual reporting periods ending after June 15, 2009 and did not have an impact on our consolidated financial statements.
In April 2009, the FASB provided guidance as codified in ASC Topics 270 and 825 to require disclosures about the fair value of financial instruments in interim financial statements as well as in annual financial statements. In addition, the guidance requires disclosures of the methods and significant assumptions used to estimate the fair value of those financial instruments. We adopted this new guidance and have provided the necessary additional disclosures (see note 4).
3. Business Acquisition:
In September 2008, we purchased collagen raw materials, equipment and a technology license from Allergan, Inc. and its affiliate Allergan Sales, LLC under a supply and license agreement dated November 2007 for an aggregate purchase price of $6,552 in cash, which was funded through a senior secured note purchase facility (see note 10). We accounted for the purchase of the assets and the related business at fair value with the purchase price being allocated among the assets purchased. The raw materials, equipment and license acquired from Allergan relate primarily to the production of the Vitagel Surgical Hemostat product. The purchase price was allocated to the fair value of the assets purchased as follows: $980 of raw material inventory, $373 of equipment, and $5,199 of acquired technology. The purchase price allocation was based upon a third party independent valuation. The acquired technology is being amortized over approximately 9 years, which is the estimated remaining life of the relevant Vitagel patent. There were no liabilities assumed in the transaction.
F-12
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
4. Cash, Cash Equivalents and Short-Term Investments:
At December 31, 2009 and 2008 cash, cash equivalents and short-term investments consisted of the following:
| Gross Unrealized | |||||||||||||
| Amortized Cost | Gains | Losses | Fair Market Value | ||||||||||
| December 31, 2009: |
|||||||||||||
| Cash and cash equivalents |
$ | 7,757 | $ | — | $ | — | $ | 7,757 | |||||
| Short-Term Investments: |
|||||||||||||
| Corporate debt securities |
7,048 | — | (1 | ) | 7,047 | ||||||||
| Government-sponsored enterprise debt securities |
8,301 | 1 | — | 8,302 | |||||||||
| 15,349 | 1 | (1 | ) | 15,349 | |||||||||
| Total |
$ | 23,106 | $ | 1 | $ | (1 | ) | $ | 23,106 | ||||
| December 31, 2008: |
|||||||||||||
| Cash and cash equivalents |
$ | 8,518 | $ | — | $ | — | $ | 8,518 | |||||
| Short-Term Investments: |
|||||||||||||
| Corporate debt securities |
9,636 | 40 | (8 | ) | 9,668 | ||||||||
| Government-sponsored enterprise debt securities |
13,987 | 118 | — | 14,105 | |||||||||
| 23,623 | 158 | (8 | ) | 23,773 | |||||||||
| Total |
$ | 32,141 | $ | 158 | $ | (8 | ) | $ | 32,291 | ||||
Amortization of premiums and discounts related to investments resulted in expense of $36 and income of $389 and $832 for the years ended December 31, 2009, 2008 and 2007, respectively.
As of December 31, 2009 and 2008, all short-term investments mature within one year of the balance sheet date.
5. Fair Value Measurements:
The following tables provide the assets carried at fair value measured on a recurring basis as of December 31, 2009 and 2008:
| Carrying Value |
Fair Value Measurement at December 31, 2009 | |||||||||||
| Level 1 | Level 2 | Level 3 | ||||||||||
| Short-Term Investments: |
||||||||||||
| Corporate debt securities |
$ | 7,047 | $ | 7,047 | — | — | ||||||
| Government-sponsored enterprise debt securities |
8,302 | 8,302 | — | — | ||||||||
| $ | 15,349 | $ | 15,349 | $ | — | $ | — | |||||
F-13
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
| Carrying Value |
Fair Value Measurement at December 31, 2008 | |||||||||||
| Level 1 | Level 2 | Level 3 | ||||||||||
| Short-Term Investments: |
||||||||||||
| Corporate debt securities |
$ | 9,668 | $ | 9,668 | — | — | ||||||
| Government-sponsored enterprise debt securities |
14,105 | 14,105 | — | — | ||||||||
| $ | 23,773 | $ | 23,773 | $ | — | $ | — | |||||
6. Inventories:
As of December 31, 2009 and 2008, inventories consisted of the following:
| 2009 | 2008 | |||||
| Raw materials |
$ | 5,538 | $ | 3,883 | ||
| Work-in-process |
8,816 | 6,894 | ||||
| Finished goods |
11,704 | 8,980 | ||||
| $ | 26,058 | $ | 19,757 | |||
7. Property and Equipment:
As of December 31, 2009 and 2008, property and equipment consisted of the following:
| 2009 | 2008 | |||||||
| Construction in-progress |
$ | 9,898 | $ | 8,800 | ||||
| Machinery and equipment |
7,797 | 7,374 | ||||||
| Furniture and computer, sales, marketing and office equipment |
5,790 | 4,900 | ||||||
| Leasehold improvements |
9,083 | 6,283 | ||||||
| 32,568 | 27,357 | |||||||
| Less—Accumulated depreciation and amortization |
(14,628 | ) | (12,919 | ) | ||||
| $ | 17,940 | $ | 14,438 | |||||
Construction in-progress at December 31, 2009 consisted primarily of construction costs of our collagen manufacturing facility. We expect that this project will be approved by the FDA and placed in service by the second half of 2010. Construction in-progress at December 31, 2008 consisted of construction costs related to our collagen and aseptic manufacturing facilities.
Total interest cost capitalized was $674 and $265 for 2009 and 2008, respectively. Included in Construction in-progress at December 31, 2009 and 2008 is capitalized interest of $779 and $342, respectively.
Depreciation and amortization expense for the years ended December 31, 2009, 2008 and 2007 was $1,831, $1,728 and $1,589, respectively.
F-14
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
8. License and Technology Intangible Assets:
As of December 31, 2009 and 2008, license and technology intangible assets consisted of the following:
| Life |
2009 | 2008 | ||||||||
| Collagen-processing technology (note 17) |
11 years | $ | 9,000 | $ | 9,000 | |||||
| Acquired technology-Vitagel and CellPaker (note 3) |
9 years | 5,199 | 5,199 | |||||||
| Other-licensed technology (note 16) |
8 years | 515 | — | |||||||
| 14,714 | 14,199 | |||||||||
| Less — Accumulated amortization |
(3,338 | ) | (1,845 | ) | ||||||
| $ | 11,376 | $ | 12,354 | |||||||
Amortization expense was $1,492, $995 and $850 for 2009, 2008 and 2007, respectively. Amortization expense for the five years subsequent to the year ended December 31, 2009 is expected to be $1,492 per year.
9. Former Revenue Interest Obligation:
We had a prior obligation to pay a revenue interest on certain sales of our products under a $10,000 product development and equity financing completed in 2001. We repurchased the revenue interest obligation on July 30, 2007 and, as of that date, we were no longer obligated to pay royalties on our products that had been subject to the revenue interest obligation. The repurchase price was $23,773 which consisted of a payment of $20,000 in cash and the issuance of 1,136 shares of our common stock valued at $3,773, the market value of the common stock issued on the date of the transaction. We recorded a charge of $16,605 during 2007 to account for the repurchase of the revenue interest liability. We recorded revenue interest expense of $756 for 2007.
10. Senior Secured Note Purchase Facility:
We have $35,000 in outstanding principal indebtedness under a senior secured note purchase facility. Notes issued under the facility are due July 30, 2012. Outstanding principal amounts under the notes bear annual interest at 10%, except that during the continuance of any event of defaults, interest would accrue at the rate of 12% per year and the notes be payable on demand. In connection with entering into the facility, we issued to the note purchaser five-year warrants to purchase 1,466 shares of our common stock at an exercise price of $3.41 per share. Of these warrants, warrants to purchase 1,100 shares of our common stock have vested and, as of January 30, 2010, the remaining 366 warrants are no longer eligible to vest. All warrants expire in July 2012.
We issued a $25,000 principal amount of notes under the facility on July 30, 2007 and used most of the proceeds to repurchase a revenue interest obligation. In conjunction with the issuance of these notes, warrants to purchase 733 shares of common stock became exercisable. The fair value of these warrants of $1,206 was recorded in the consolidated balance sheet as a discount to the initial loan amount of $25,000 and is being amortized into interest expense over the remaining term of the facility.
On July 31, 2008, we issued an additional $10,000 principal amount of notes under the facility. We applied the proceeds of this note toward payment of (i) the $6,552 purchase price for the collagen raw material, equipment and technology license acquired under our supply and license agreement with Allergan (see note 3); and (ii) costs to expand our manufacturing capacity for Vitagel and ancillary products such as Aliquot, Imbibe and CellPaker. In conjunction with the issuance of the note, warrants to purchase 367 shares of common stock became exercisable. The fair value of these warrants of $286 has been recorded as a discount to the initial loan amount of $10,000 and is being amortized into interest expense over the remaining term of the facility.
F-15
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
Borrowings under the facility are guaranteed by us and one of our wholly-owned subsidiaries. The facility is secured by a first priority lien on substantially all of our assets (including intellectual property) other than those exclusively related to Cortoss and Aliquot. We are required to make quarterly interest-only payments to the note holder. Upon the occurrence of various events, including our receipt of (i) proceeds in excess of 5% of our total assets for certain asset dispositions; (ii) more than $1,000 in aggregate cash insurance proceeds for damaged or destroyed property that is not applied to the repair or replacement of the property within one year; or (iii) more than $7,500 in gross cash proceeds from judgment awards or settlements, the note holders are entitled to prepayment of the outstanding principal amount of the notes to the extent of the net cash proceeds that we receive. We may prepay at any time any part of the outstanding balance under the notes, in a minimum amount of $2,000 and in increments of at least $1,000 in excess of such minimum, together with interest accrued thereon. Both mandatory and optional prepayments are subject to a prepayment premium of up to 16% of the principal balance of the notes then outstanding, depending on the timing of the prepayment. The unpaid principal amount under the notes and accrued interest and all other obligations shall become due and payable immediately if we are insolvent, are in bankruptcy proceedings or have a custodian or receiver appointed for any substantial part of our property. If an event of default not described in the preceding sentence occurs and is continuing (including a change of control of the Company), then the holders of at least two-thirds in principal amount of notes then outstanding may declare the unpaid principal amount of the notes, accrued interest and all other obligations due and payable immediately. In addition, if the notes become due and payable, whether automatically or by declaration, by reason of any of the following events of default, then we must pay a prepayment premium of up to 16% of the principal balance of the notes then outstanding, depending on the timing of the prepayment:
| • | failure to pay any principal on any note or prepayment premiums, if any, when due and payable; |
| • | failure to pay any interest on any note or other amount (other than principal or prepayment premiums) for more than three business days after becoming due and payable; |
| • | we become insolvent, are in bankruptcy proceedings or have a custodian or receiver appointed for any substantial part of our property; or |
| • | the occurrence of a change of control of the Company as defined in the facility. |
Under the facility, we must comply with various financial and non-financial covenants. Under the financial covenant, we are required to maintain a minimum cash balance equal to at least 25% of the then-outstanding principal amount under the notes in a separate interest-bearing deposit or other similar demand investment account that is pledged as collateral for the loan. As of December 31, 2009, we must maintain a minimum cash, cash equivalent, and short-term investment balance of $8,750. If the balance in the separate account falls below an amount equal to 40% of the principal amount outstanding under the notes, we must obtain and maintain for the benefit of favor of the note holder a letter of credit in the amount of 25% of the principal amount outstanding under the notes. As of December 31, 2009, we must maintain a minimum balance of $14,000 in cash, cash equivalents, and short-term investments in order to avoid obtaining a letter of credit. Currently we are not required to obtain a letter of credit. The primary non-financial covenants limit our ability to incur indebtedness or liens, sell assets, conduct mergers or acquisitions as defined in the facility terms, make investments and pay dividends.
Borrowing obligation maturities as of December 31, 2009 are as follows:
| Notes Payable | |||
| 2010 |
$ | — | |
| 2011 |
— | ||
| 2012 |
35,000 | ||
| Total |
$ | 35,000 | |
F-16
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
Interest expense on notes payable was $3,112, $2,777 and $1,342 for the years ended December 31, 2009, 2008 and 2007, respectively.
11. Other Accrued Expenses:
As of December 31, 2009 and 2008, other accrued expenses consisted of the following:
| 2009 | 2008 | |||||
| Commissions payable |
$ | 2,423 | $ | 1,862 | ||
| Building improvements |
— | 496 | ||||
| Interest payable |
875 | 875 | ||||
| Other |
2,236 | 1,816 | ||||
| $ | 5,534 | $ | 5,049 | |||
12. Profit Sharing Plan:
We have a Section 401(k) plan for all qualified employees, as defined. Contributions are discretionary and determined annually and were $692, $667 and $480 for the years ended December 31, 2009, 2008 and 2007, respectively.
13. Shareholders’ Equity:
Common Stock
During 2007, we sold 12,317 shares of our common stock in a private placement transaction for proceeds of $32,199 and issued 1,136 shares of our common stock as partial consideration for the repurchase of a revenue interest obligation (see note 9). We also incurred costs of $45 in 2007 associated with prior sales of common stock.
During 2008 and 2007, we exchanged options to purchase 131 and 614 shares of common stock, respectively, for 131 and 614 common shares with the non-employee consultants. In connection with these exchanges, we incurred a non-cash charge of $170 in 2008 and $1,161 in 2007.
Equity Compensation Plan
Our equity compensation plan (the Plan) provides for incentive and nonqualified stock options, restricted stock awards, restricted stock units and other equity incentives to be granted to directors, employees, and consultants. Our shareholders have approved the Plan.
In June 2009, our shareholders approved an amendment to the plan to increase, by 4,000, the number of shares authorized for issuance under the Plan. At December 31, 2009, there were 17,850 shares authorized for issuance under the Plan with 5,397 underlying shares available for grant and 4,637 underlying shares currently outstanding.
Performance-based Equity Awards
In September 2008, we issued grants of performance-based equity awards under the Plan to certain executive officers whereby up to an aggregate 421 shares of our common stock, valued at $2.68 per share, could be issued. The number of shares of common stock payable under each award is dependent upon our achievement of pre-determined levels of U.S. sales of Cortoss Bone Augmentation Material and related delivery systems
F-17
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
during the twelve-month period commencing July 1, 2009 (the Twelve-Month Performance Period). As of December 31, 2009, we have determined that it is unlikely that U.S. Cortoss sales will achieve the predetermined level in the Twelve Month Performance Period. Accordingly, no related compensation expense has been recognized for the year ended December 31, 2009.
Common Stock
We did not issue common stock in 2009 as payment for consulting services. In 2008 and 2007, we issued 7 shares valued at $21 and 4 shares of common stock valued at $11, respectively, for consulting services rendered pursuant to consulting agreements.
Restricted Stock and Restricted Common Stock Units
We did not issue restricted common stock units during 2009 or 2008. During 2007, we issued an aggregate of 318 restricted common stock units valued at $1,041 to certain employees. In 2009, we issued 156 shares of our common stock upon the vesting of certain of the units granted in 2007. As of December 31, 2009, 113 restricted common stock units were outstanding and scheduled to vest in March 2011, subject to the continued employment of the holders of such unit.
During 2009, 2008 and 2007, we issued an aggregate of 28, 28 and 36 restricted common shares to non-employee directors in consideration of their services. These shares were valued at $138, $66 and $105, respectively. With respect to the shares granted in 2009 and 2008, the shares vest in equal one-third installments on each anniversary of the date of grant, or earlier upon either (i) a change of control of the Company, (ii) the recipient’s death or (iii) the recipient’s departure in good standing from the board of directors provided he or she served on the board for at least five years or the sum of his or her age and length of services is at least 65. The shares granted in 2007 vest on the five year anniversary of the date of grant, or earlier upon a change of control of the Company or when the recipient no longer serves on the board of directors. As of December 31, 2009, 83 restricted shares were outstanding.
We have $285 of unrecognized cost related to unvested restricted stock and restricted stock units as of December 31, 2009, which is expected to be recognized over a weighted average period of approximately 1.4 years. The compensation expense recorded for these awards during 2009, 2008 and 2007 was $266, $368 and $431, respectively, and is included in stock-based compensation expense (see note 2).
Common Stock Options
Options are granted with exercise prices equal to or greater than the fair market value of the common stock on the date of grant. Generally, incentive stock options become exercisable in equal installments over a four-year period and nonqualified stock options to non-employee consultants are fully vested at the date of grant. The options generally remain exercisable for a maximum period of ten years. Upon exercise of options new shares of common stock are issued.
In March 2007, we issued options to purchase 25 shares of common stock to a non-employee director in connection with his appointment to the board of directors. These options have an exercise price equal to $2.83 per share, were fully vested upon grant and expire 10 years after the date of grant.
In June 2007, we issued to non-employee directors options to purchase 70 shares of common stock in the aggregate. These options have an exercise price of $2.90 per share, vested as to 50% of the underlying shares on the date on grant and vested as to 25% of the underlying shares on each of the two successive anniversaries of the date of grant.
F-18
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
In July 2007, we issued options to purchase an aggregate 50 shares of common stock to two new non-employee directors appointed to our board of directors. These options have an exercise price equal to $3.32 per share, were fully vested upon grant and expire 10 years after the date of grant.
In July 2008, we issued to non-employee directors options to purchase 84 shares of common stock in the aggregate. These options have an exercise price of $2.35 per share and vest as to 25% of the underlying shares on each of the four successive anniversaries of the date of grant.
In July 2009, we issued to non-employee directors options to purchase 84 shares of common stock in the aggregate. These options have an exercise price of $4.89 per share and vest as to 25% of the underlying shares on each of the four successive anniversaries of the date of grant.
As of December 31, 2009, we had $3,860 of unrecognized compensation cost related to unvested stock options which we expect to recognized over a weighted average period of approximately 2.5 years.
For all options outstanding as of December 31, 2009, the weighted average exercise price per share was $3.55 with a weighted average remaining contractual life of approximately 6 years and an aggregate intrinsic value of $2,186.
At December 31, 2009, there were 4,613 options exercisable. These options have weighted average exercise price per share was $3.67 with a weighted average remaining contractual life of approximately 5.4 years and an aggregate intrinsic value of $983.
Summary stock option information is as follows:
| Aggregate Number |
Aggregate Exercise Price |
Exercise Price Range |
Weighted Average Exercise Price | |||||||||
| Outstanding, December 31, 2006 |
8,013 | 30,770 | $ | 1.65 – 11.25 | $ | 3.84 | ||||||
| Granted |
477 | 1,522 | 2.59 – 3.67 | 3.19 | ||||||||
| Exercised |
(201 | ) | (525 | ) | 1.85 – 3.36 | 2.49 | ||||||
| Cancelled |
(2,680 | ) | (9,953 | ) | 1.65 – 7.25 | 3.73 | ||||||
| Expired |
(197 | ) | (837 | ) | 4.25 – 4.25 | 4.25 | ||||||
| Outstanding, December 31, 2007 |
5,412 | 20,977 | $ | 1.65 – 11.25 | 3.74 | |||||||
| Granted |
1,787 | 4,861 | 2.01 – 3.40 | 2.72 | ||||||||
| Exercised |
(5 | ) | (12 | ) | 2.32 – 3.50 | 2.32 | ||||||
| Cancelled |
(464 | ) | (1,494 | ) | 1.69 – 6.60 | 3.22 | ||||||
| Expired |
(54 | ) | (273 | ) | 4.25 – 11.25 | 5.05 | ||||||
| Outstanding, December 31, 2008 |
6,676 | 24,059 | 1.69 – 11.25 | 3.55 | ||||||||
| Granted |
1,310 | 4,757 | 2.60 – 6.73 | 3.63 | ||||||||
| Exercised |
(360 | ) | (1,206 | ) | 2.48 – 4.48 | 3.35 | ||||||
| Cancelled |
(290 | ) | (1,014 | ) | 2.01 – 7.80 | 3.49 | ||||||
| Expired |
(137 | ) | (703 | ) | 4.75 – 6.60 | 5.17 | ||||||
| Outstanding, December 31, 2009 |
7,199 | 25,893 | $ | 1.65 – 6.75 | $ | 3.55 | ||||||
The intrinsic value of stock options exercised in 2009, 2008 and 2007 was $423, $2 and $6, respectively.
F-19
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
As of December 31, 2009, non-employee consultants held fully-vested stock options to purchase 62 shares of common stock, at an average exercise price of $3.20 per share and an average remaining term of approximately three years and are classified as a non-current liability (included in other long-term liabilities) as management does not believe that it will need to be satisfied using current assets within the next twelve months.
Employee Stock Purchase Plan
We have an Employee Stock Purchase Plan (ESPP) under which eligible employees may purchase shares of our common stock at a per share price equal to 95% of the closing price of our common stock as reported on NASDAQ on the last trading day of each calendar quarter. Under the terms of the ESPP, eligible employees may have up to 10% of eligible compensation deducted from their pay to purchase common stock. The term of the ESPP expires in 2018.
Employees purchased 68, 75 and 42 shares under the ESPP during 2009, 2008 and 2007, respectively, for cash proceeds of $240, $184 and $125, respectively. In June 2009, our shareholders approved an amendment to the ESPP to increase, by 200, the number of shares available for issuance under the ESPP. As of December 31, 2009, there were 700 shares of common stock authorized for issuance under the ESPP, of which 350 shares remained available for purchase and issue.
Common Stock Purchase Warrants
As of December 31, 2009, we had outstanding warrants to purchase 1,466 shares of common stock at an exercise price of $3.41 per share. All of these warrants were granted pursuant to the Senior Secured Note Purchase Facility (see note 10). Of the outstanding warrants, warrants to purchase 1,100 shares were exercisable as of December 31, 2009 and will expire in July 2012 if not exercised before that date. As of January 30, 2010, 366 warrants were no longer eligible to vest.
During 2007, warrants to purchase 10 shares of common stock were exercised for gross proceeds of $17.
14. Product Sales:
For 2009, 2008 and 2007, product sales by geographic market were as follows:
| Year Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| United States |
$ | 87,719 | $ | 71,997 | $ | 53,968 | |||
| Outside the United States |
5,134 | 4,918 | 4,078 | ||||||
| Total product sales |
$ | 92,853 | $ | 76,915 | $ | 58,046 | |||
Approximately 67%, 63% and 61% of product sales during 2009, 2008 and 2007, respectively, were from products based upon the VITOSS FOAM platform co-developed with Kensey (see note 16). Vitagel contributed approximately 22%, 24% and 21% of the product sales for 2009, 2008 and 2007, respectively.
F-20
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
15. Income Taxes:
The components of income taxes are as follows:
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Current |
$ | 20 | $ | 17 | $ | (35 | ) | |||||
| Deferred |
1,263 | 3,317 | 11,177 | |||||||||
| 1,283 | 3,334 | 11,142 | ||||||||||
| Valuation allowance |
(1,270 | ) | (3,324 | ) | (10,874 | ) | ||||||
| Income tax benefit |
$ | 13 | $ | 10 | $ | 268 | ||||||
Reconciliation between the provision (benefit) for income taxes, computed by applying the statutory federal income tax rate of 34% to income before income taxes, and the actual provision (benefit) for income taxes follows:
| December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Federal income tax provision at statutory rate |
(34 | )% | (34 | )% | (34 | )% | |||
| State taxes (benefit)—Texas Gross Margin Tax |
1 | — | (1 | ) | |||||
| State income taxes, net of federal income tax provision |
(2 | ) | (4 | ) | (4 | ) | |||
| Change in valuation allowance |
32 | 31 | 36 | ||||||
| Net operating losses (foreign) |
(11 | ) | |||||||
| Non-deductible expenses |
17 | 6 | 2 | ||||||
| Other |
(3 | ) | 1 | — | |||||
| Actual income tax provision (benefit) effective tax rate |
— | % | — | % | (1 | )% | |||
Components of deferred tax asset as of December 31, 2009 and 2008 were as follows:
| December 31 | ||||||||
| 2009 | 2008 | |||||||
| Deferred tax assets: |
||||||||
| Net operating loss carryforwards |
$ | 50,458 | $ | 50,332 | ||||
| Accrued expenses not currently deductible and other |
4,164 | 3,329 | ||||||
| Research and patent costs capitalized for tax purposes |
12,395 | 12,345 | ||||||
| Research and development credits |
2,042 | 1,783 | ||||||
| Texas margin tax temporary credit |
289 | 296 | ||||||
| 69,348 | 68,085 | |||||||
| Valuation allowance |
(69,059 | ) | (67,789 | ) | ||||
| Net deferred tax asset |
$ | 289 | $ | 296 | ||||
In October 2008, the United States Congress enacted the Housing Assistance Act of 2008 (Public Law 100-289), which enabled us to make a federal tax election to claim accelerated research tax credits in lieu of bonus depreciation. As a result, we recognized a current income tax benefit of $64 and $55 in the years ended December 31, 2009 and 2008, respectively.
F-21
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
In 2006, Texas enacted a margin tax which restructured the state business tax by replacing the taxable capital components of the current franchise tax with a new “taxable margin” component. The Texas margin tax was effective for taxable years ended on or after January 1, 2007. As a result of the Texas margin tax, during 2009, 2008, and 2007, we recognized current and deferred state income tax expense of $51, $45 and $35, respectively, and a deferred tax asset of $289, $296 and $303, respectively, for the Texas temporary credit on taxable margin. Under the Texas margin tax, we have the right to claim a temporary credit against our Texas margin tax liability over a 20 year period. As a result, we have recorded a deferred tax asset of $289 and $296 as of December 31, 2009 and 2008, respectively, which is included in Other Assets on our consolidated balance sheet. We have recognized a deferred tax asset related to the Texas tax credit as we believe it will more likely than not to be realized based on projections of future Texas margin tax liabilities.
As of December 31, 2009 we had approximately $118,735 of federal net operating loss carryforwards, $108,610 of state net operating loss carryforwards and $12,736 of foreign net operating loss carryforwards. Federal net operating loss carryforwards began to expire in 2009 and will fully expire in 2028 while the state net operating carryforwards began to expire prior to 2009 and will fully expire in 2028. The federal net operating losses have a carryforward period of 20 years. The state net operating losses have carryforward periods ranging from five to 20 years, with the Pennsylvania net operating loss having a carryforward period of 20 years. The foreign net losses have an unlimited carryforward period. The amount of U.S. federal net operating loss and credit carryforwards which can be utilized in any one period may be limited by federal and state income tax regulations, since a change in ownership as defined in Section 382 and 383 of the Internal Revenue Code may have occurred in prior years. The amount of net operating loss carryforwards which can be utilized in any one period on the Pennsylvania corporate income tax return is limited to the greater of $3,000 or 15% of apportioned net taxable income for tax years beginning after December 31, 2008. Federal and Pennsylvania research and development tax credits of $1,772 and $268 begin to expire in 2010 and 2014, respectively. The Texas margin tax temporary credit of $289 has a carryforward period of 20 years.
As of December 31, 2009, we determined that we had no liability for uncertain income taxes. Our policy is to recognize potential accrued interest as a component of interest expense and penalties as a component of other expense related to the liability for uncertain income taxes, if applicable. The Federal and State tax returns for 2006 through 2009 are subject to review by the appropriate taxing authority.
16. Commitments:
Operating Leases
For the years ended December 31, 2009, 2008 and 2007, lease expense was $897, $783 and $690, respectively. At December 31, 2009, future minimum rental payments under operating leases are as follows:
| 2010 |
$ | 862 | |
| 2011 |
889 | ||
| 2012 |
927 | ||
| 2013 |
964 | ||
| 2014 and thereafter |
3,689 | ||
| $ | 7,331 | ||
F-22
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
Agreement with Kensey Nash Corporation
We have an agreement with Kensey Nash Corporation (Kensey) to jointly develop and commercialize certain biomaterials-based products based upon the Vitoss platform. The products developed under this agreement are based on our internally developed proprietary Vitoss bone void filler material in combination with proprietary resorbable Kensey biomaterials. Kensey has the exclusive right to manufacture any approved or cleared jointly developed product under the agreement, and we will market and sell the product worldwide. Under the agreement, we are obligated to pay Kensey both a transfer price for manufacturing products and royalties based on the net sales of such products. These rights and obligations extend until February 2014 for the Vitoss Foam product platform and until February 2024 for the Vitoss Bioactive Foam product platform.
During 2009, 2008 and 2007, we purchased $8,497, $7,887 and $8,194, respectively, of product inventory manufactured by Kensey. As of December 31, 2009 and 2008, we owed Kensey $2,420 and $3,155, respectively, for manufactured product inventory and royalties, which are included in accounts payable and other accrued expenses in the consolidated balance sheets. All product royalty expense payable to Kensey is included in cost of sales on the consolidated statements of operations as we recognize product sales revenue from ours customers.
Separate from the royalty payment obligation described above, we pay additional royalties to Kensey pursuant to a contractual royalty obligation that Kensey purchased from a co-inventor of Vitoss technology. Under this arrangement, we are obligated to pay no more than $5,000 in aggregate royalties on Vitoss product sales. For the years ended December 31, 2009, 2008 and 2007, we recognized $645, $593 and $551, respectively, as royalty expense under this contractual arrangement. From inception of the royalty arrangement through December 31, 2009, we have made aggregate royalty payments of $3,259.
Product Development Milestones
In 2009, we contractually agreed to make milestone payments upon achievement of specified goals in connection with the development of new products with one or more business partners. The timing and actual amount of these payments can be difficult to determine as these payments depend upon satisfactory achievement of product development and other milestones, which will be determined based on events that may occur in the future. During 2009, we made payments of $1,915 related to this product development program: $500 has been recorded as a long term prepaid royalty, $515 has been recorded as an intangible asset for the value of a license acquired and is being amortized over the life of the licensed patents and the remaining amount of $900 has been recorded as research and development expense.
If these products under development are brought to market, we may be obligated to make certain launch-related milestone payments of up to $1,000, as well as additional payments which may be made depending upon the potential achievements of clinical and sales milestones in the future.
Employment Agreements
Under the terms of employment agreements with two executive officers, extending through April 2011 and May 2011, subject to renewal, we are required to pay each individual a base salary for continuing employment with us. The agreements currently require aggregate payments of $688 and $255 in 2010 and 2011, respectively. The agreements also provide for other benefits, including certain obligations that may be triggered by a change in control, termination with cause or resignation for good reason.
17. Agreements with Angiotech Pharmaceuticals, Inc.:
In December 2006, we paid Angiotech Pharmaceuticals, Inc. (Angiotech) $9,000 in cash in order to eliminate our obligations to pay Angiotech royalties on future sales of Vitagel and CellPaker products under a
F-23
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
license agreement. Concurrently with this purchase, we amended and restated our license agreement with Angiotech to eliminate our obligations to meet minimum sales requirements, extend the term of the license from December 31, 2014 through July 31, 2017, and eliminate certain termination rights in favor of Angiotech.
Under the amended and restated license agreement: (i) we have exclusive rights to manufacture, market and sell Vitagel products throughout the world for orthopedic indications, and non-exclusive rights to manufacture, market and sell CellPaker products throughout the world for all indications; and (ii) Angiotech has an option for co-exclusive rights outside the orthopedic field which, if exercised, would permit Angiotech to manufacture, market and sell an Angiotech-branded Vitagel product throughout the world. Until Angiotech elects to exercise its option for co-exclusive rights, we have exclusive rights to manufacture, market and sell Vitagel outside of the orthopedic field throughout the world. If Angiotech elects to exercise its option, we would then have co-exclusive rights to manufacture, market and sell Vitagel outside of the orthopedic field throughout the world.
The $9,000 payment described above has been recorded as a license right intangible in the accompanying consolidated balance sheets. This amount is being amortized based upon the greater of (a) straight-line amortization through July 31, 2017, or (b) actual units sold in a given period in relation to the total estimated units to be sold over the expected life of the applicable patent, which is July 31, 2017. Amortization of the license right intangible amounting to $850 was recorded during each year in the three year period ended December 31, 2009, and is included in cost of sales on the accompanying consolidated statements of operations.
18. Quarterly Financial Data (Unaudited):
| Three Months Ended | ||||||||||||||||||||
| March 31 | June 30 | September 30 | December 31 | Total | ||||||||||||||||
| 2009 |
||||||||||||||||||||
| Product sales |
$ | 21,690 | $ | 24,491 | $ | 22,295 | $ | 24,377 | $ | 92,853 | ||||||||||
| Gross profit |
14,680 | 16,825 | 15,145 | 16,289 | 62,939 | |||||||||||||||
| Total operating expenses |
15,053 | 16,922 | 16,149 | 15,904 | 64,028 | |||||||||||||||
| Net interest expense |
(789 | ) | (587 | ) | (722 | ) | (732 | ) | (2,830 | ) | ||||||||||
| Net loss |
(1,176 | ) | (698 | ) | (1,741 | ) | (291 | ) | (3,906 | ) | ||||||||||
| Net loss per share, basic and diluted |
$ | (0.02 | ) | $ | (0.01 | ) | $ | (0.02 | ) | $ | (0.00 | ) | $ | (0.05 | ) | |||||
| Three Months Ended | ||||||||||||||||||||
| March 31 | June 30 | September 30 | December 31 | Total | ||||||||||||||||
| 2008 |
||||||||||||||||||||
| Product sales |
$ | 16,164 | $ | 19,341 | $ | 20,563 | $ | 20,847 | $ | 76,915 | ||||||||||
| Gross profit |
10,352 | 12,985 | 13,881 | 13,768 | 50,986 | |||||||||||||||
| Total operating expenses |
14,370 | 16,276 | 15,085 | 14,439 | 60,170 | |||||||||||||||
| Net interest expense |
(180 | ) | (171 | ) | (549 | ) | (606 | ) | (1,506 | ) | ||||||||||
| (Loss) Gain on sale of assets |
— | (146 | ) | 74 | — | (72 | ) | |||||||||||||
| Net loss |
(4,212 | ) | (3,623 | ) | (1,694 | ) | (1,223 | ) | (10,752 | ) | ||||||||||
| Net loss per share, basic and diluted |
$ | (0.06 | ) | $ | (0.05 | ) | $ | (0.02 | ) | $ | (0.02 | ) | $ | (0.14 | ) | |||||
F-24
Table of Contents
ORTHOVITA, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(All amounts in thousands except per share data)
19. Valuation and Qualifying Accounts:
| Description |
Balance, Beginning of Period |
Cost and Expenses |
Deductions(1) | Balance, End of Period | ||||||||
| For the year ended December 31, 2009: |
||||||||||||
| Reserve for doubtful accounts |
$ | 252 | $ | 18 | $ | 53 | $ | 217 | ||||
| For the year ended December 31, 2008: |
||||||||||||
| Reserve for doubtful accounts |
$ | 218 | $ | 97 | $ | 63 | $ | 252 | ||||
| For the year ended December 31, 2007: |
||||||||||||
| Reserve for doubtful accounts |
$ | 202 | $ | 152 | $ | 136 | $ | 218 | ||||
| (1) | Represents write-offs of specific accounts receivable. |
20. Gain on Sale of Assets:
On February 15, 2007, we sold the assets associated with the Endoskeleton TA Vertebral Body Replacement structural device product line for $458, which was recorded as a gain of $372 on the sale of assets during 2007. As a result of the sale, we no longer manufacture or sell Endoskeleton products. We did not record any revenue in 2009, 2008 or 2007 for sales of Endoskeleton products.
F-25
Table of Contents
EXHIBIT INDEX
| Exhibit |
Description | |
| 3.1 | Composite Amended and Restated Articles of Incorporation of the Company, as amended(14) | |
| 3.2 | Amended and Restated Bylaws of the Company, as amended(14) | |
| 4.1 | Specimen of Common Stock Certificate of the Company(2) | |
| *10.1 | Amended and Restated Employment Agreement, as amended as of March 9, 2010, by and between the Company and Antony Koblish(1) | |
| *10.2 | Amended and Restated Employment Agreement, dated as of March 9, 2010, by and between the Company and Nancy C. Broadbent(1) | |
| *10.3 | Employment Letter, dated as of March 12, 1999, by and between the Company and Dr. Maarten Persenaire(3) | |
| *10.4 | Confidentiality and Non-Disclosure Agreement by and between the Company and Dr. Maarten Persenaire, dated as of July 3, 2007(5) | |
| *10.5 | Retention Agreement, dated as of May 15, 2009, between the Company and Albert J. Pavucek, Jr.(16) | |
| *10.6 | Form of Change of Control Agreement between Orthovita and certain officers(6) | |
| *10.7 | Form of Indemnification Agreement between the Company and directors(7) | |
| *10.8 | 2007 Omnibus Equity Compensation Plan, as amended on June 23, 2009(17) | |
| *10.9 | Form of Incentive Stock Option Grant(6) | |
| *10.10 | Form of Nonqualified Stock Option Grant(6) | |
| *10.11 | Form of Restricted Stock Unit Agreement(12) | |
| *10.12 | Form of Performance Share Agreement(15) | |
| *10.13 | Amended and Restated Employee Stock Purchase Plan(9) | |
| 10.14 | Securities Purchase Agreement, dated as of July 30, 2007, among the Company and the Buyers listed on the Schedule of Buyers attached thereto(13) | |
| 10.15 | Purchase and Sale Agreement, dated as of July 30 2007, between Royalty Securitization Trust I and the Company(13) | |
| 10.16 | Assignment Agreement, dated as of July 30, 2007, between Royalty Securitization Trust I, Royalty Financial Company LLC, and Paul Royalty Fund, L.P., as Assignors, and the Company, as Assignee(13) | |
| 10.17 | Consent and Release, dated as of July 30, 2007, by Royalty Securitization Trust I, Royalty Financial Company LLC, Paul Royalty Fund, L.P., Vita Special Purpose Corp., Vita Licensing, Inc. and the Company(13) | |
| 10.18 | Securities Disposition Agreement, dated as of July 30, 2007, between the Company and Paul Royalty Fund, L.P.(13) | |
| 10.19 | Senior Secured Note and Warrant Purchase Agreement, dated as of July 30, 2007, among the Purchasers named therein, the Company, and LB I Group Inc., as Collateral Agent, including form of 10% Senior Secured Promissory Note due July 30, 2012(13) | |
| 10.20 | Form of Warrant to Purchase Common Stock of the Company(13) | |
| 10.21 | Registration Rights Agreement, dated as of July 30, 2007, between the Company and the Purchasers named therein(13) | |
Table of Contents
| Exhibit |
Description | |
| 10.22 | Security Agreement, dated as of July 30, 2007, among the Company, the other entities listed on Schedule A thereto, as Pledgors, and LB I Group Inc., as Collateral Agent(13) | |
| 10.23 | Company Stock Pledge Agreement, dated as of July 30, 2007, by the Company in favor of LB I Group Inc. for the benefit of holders of those certain 10% Senior Secured Promissory Notes described in the Note Purchase Agreement(13) | |
| 10.24 | Company Intellectual Property Security Agreement, dated as of July 30, 2007, by the Company in favor of LB I Group Inc. for the benefit of holders of those certain 10% Senior Secured Promissory Notes described in the Note Purchase Agreement(13) | |
| 10.25 | Subsidiary Guaranty Agreement, dated as of July 30, 2007, among Vita Licensing, Inc., Orthovita International Services, Inc., Partisyn Corp. and Vita Special Purpose Corp., as Guarantors, and LB I Group Inc., as Collateral Agent(13) | |
| 10.26 | Subsidiary Stock Pledge Agreement, dated as of July 30, 2007, by Vita Licensing, Inc. in favor of LB I Group Inc. for the benefit of holders of those certain 10% Senior Secured Promissory Notes described in the Note Purchase Agreement(13) | |
| 10.27 | Subsidiary Intellectual Property Security Agreement, dated as of July 30, 2007, by each of Vita Licensing, Inc., Orthovita International Services, Inc., Partisyn Corp. and Vita Special Purpose Corp. in favor of LB I Group Inc. for the benefit of holders of those certain 10% Senior Secured Promissory Notes described in the Note Purchase Agreement(13) | |
| 10.28 | Development, Manufacturing and Supply Agreement dated as of March 25, 2003 between the Company and Kensey Nash Corporation (portions of this document have been omitted pursuant to the Company’s confidential treatment request under Exchange Act Rule 24b-2)(10) | |
| 10.29 | Amendment to Development, Manufacturing and Supply Agreement dated as of February 25, 2005 between the Company and Kensey Nash Corporation (portions of this document have been omitted and were filed separately with the SEC pursuant to a confidential treatment request under Exchange Act Rule 24b-2)(8) | |
| 10.30 | Fifth Amendment to the Development, Manufacturing and Supply Agreement between the Company and Kensey Nash Corporation dated as of February 21, 2007 (portions of this document have been omitted and were filed separately with the SEC pursuant to a confidential treatment request under Exchange Act Rule 24b-2)(5) | |
| 10.31 | Amended and Restated License Agreement dated as of December 29, 2006 between the Company and Angiotech Pharmaceuticals (U.S.), Inc. (portions of this document have been omitted and were filed separately with the SEC pursuant to a confidential treatment request under Exchange Act Rule 24b-2)(4) | |
| 10.32 | Royalty Sale Agreement dated as of December 5, 2006 between the Company and Angiotech Pharmaceuticals (U.S.), Inc.(11) | |
| 10.33 | Supply and License Agreement, dated as of November 5, 2007, by and between Allergan, Inc. and Allergan Sales, LLC and Orthovita, Inc. (portions of this document have been omitted and were filed separately with the SEC pursuant to a confidential treatment request under Exchange Act Rule 24b-2)(5) | |
| 21.1 | Subsidiaries(1) | |
| 23.1 | Consent of KPMG LLP(1) | |
| 31.1 | Certification of Chief Executive Officer pursuant to Rule 13a-14(a) and Rule 15d-14(a) of the Securities Exchange Act, as amended(1) | |
Table of Contents
| Exhibit |
Description | |
| 31.2 | Certification of Chief Financial Officer pursuant to Rule 13a-14(a) and Rule 15d-14(a) of the Securities Exchange Act, as amended(1) | |
| 32.1 | Certification of Chief Executive Officer pursuant to 18 U.S.C. 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002(1) | |
| 32.2 | Certification of Chief Financial Officer pursuant to 18 U.S.C. 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002(1) | |
| * | Constitutes management contract or compensatory plan or arrangement required to be filed as an exhibit to this form. |
| (1) | Filed herewith. |
| (2) | Filed as an Exhibit to the Company’s Annual Report on Form 10-K for the year ended December 31, 2000 and incorporated herein by reference. |
| (3) | Filed as an Exhibit to the Company’s Annual Report on Form 10-K for the year ended December 31, 2002 and incorporated herein by reference. |
| (4) | Filed as an Exhibit to the Company’s Annual Report on Form 10-K for the year ended December 31, 2006 and incorporated herein by reference. |
| (5) | Filed as an Exhibit to the Company’s Annual Report on Form 10-K for the year ended December 31, 2007 and incorporated herein by reference. |
| (6) | Filed as an Exhibit to the Company’s Annual Report on Form 10-K for the year ended December 31, 2008 and incorporated herein by reference. |
| (7) | Filed as an Exhibit to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2004 and incorporated herein by reference. |
| (8) | Filed as an Exhibit to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2005 and incorporated herein by reference. |
| (9) | Filed as an Exhibit to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2008 and incorporated herein by reference. |
| (10) | Filed as an Exhibit to the Company’s Current Report on Form 8-K filed on June 30, 2004 and incorporated herein by reference. |
| (11) | Filed as an Exhibit to the Company’s Current Report on Form 8-K filed on December 7, 2006 and incorporated herein by reference. |
| (12) | Filed as an Exhibit to the Company’s Current Report on Form 8-K filed on March 12, 2007 and incorporated herein by reference. |
| (13) | Filed as an Exhibit to the Company’s Current Report on Form 8-K filed on July 31, 2007 and incorporated herein by reference. |
| (14) | Filed as an Exhibit to the Company’s Current Report on Form 8-K filed on December 27, 2007 and incorporated herein by reference. |
| (15) | Filed as an Exhibit to the Company’s Current Report on Form 8-K filed on September 18, 2008 and incorporated herein by reference. |
| (16) | Filed as an Exhibit to the Company’s Current Report on Form 8-K filed on May 19, 2009 and incorporated herein by reference. |
| (17) | Filed as an Exhibit to the Company’s Current Report on Form 8-K/A filed on June 25, 2009 and incorporated herein by reference. |