Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Fiscal Year Ended December 31, 2009
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Transition period from to .
Commission file number: 001-32836
MEDIVATION, INC.
(Exact name of Registrant as specified in its charter)
| Delaware | 13-3863260 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
201 Spear Street, 3rd Floor
San Francisco, California 94105
(Address of principal executive offices) (Zip Code)
Registrant’s telephone number, including area code: (415) 543-3470
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class |
Name of Each Exchange on Which Registered | |
| Common Stock, par value $0.01 per share |
The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. YES x NO ¨
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. YES ¨ NO x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. YES x NO ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ¨ Accelerated filer x
Non-accelerated filer (Do not check if a smaller reporting company) ¨ Smaller reporting company ¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act.). YES ¨ NO x
The aggregate market value of the voting stock held by non-affiliates of the registrant was approximately $599,169,357 as of June 30, 2009, based upon the closing sale price on The NASDAQ Global Market reported on June 30, 2009. Excludes an aggregate of 6,622,844 shares of the registrant’s common stock held by officers, directors and affiliated stockholders. For purposes of determining whether a stockholder was an affiliate of the registrant at June 30, 2009, the registrant assumed that a stockholder was an affiliate of the registrant at June 30, 2009 if such stockholder (i) beneficially owned 10% or more of the registrant’s common stock, as determined based on public filings, and/or (ii) was an executive officer or director or was affiliated with an executive officer or director of the registrant at June 30, 2009. Exclusion of such shares should not be construed to indicate that any such person possesses the power, direct or indirect, to direct or cause the direction of the management or policies of the registrant or that such person is controlled by or under common control with the registrant.
There were 34,004,318 shares of Registrant’s Common Stock, par value $0.01 per share, issued and outstanding as of March 8, 2010.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive Proxy Statement for the 2010 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission pursuant to Regulation 14A not later than 120 days after the end of the fiscal year covered by this Form 10-K are incorporated by reference in Part III, Items 10-14 of this Form 10-K.
Table of Contents
MEDIVATION, INC.
2009 ANNUAL REPORT ON FORM 10-K
| Page | ||||
| PART I |
||||
| Item 1. |
2 | |||
| Item 1A. |
20 | |||
| Item 1B. |
33 | |||
| Item 2. |
33 | |||
| Item 3. |
34 | |||
| Item 4. |
34 | |||
| PART II |
||||
| Item 5. |
35 | |||
| Item 6. |
38 | |||
| Item 7. |
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
39 | ||
| Item 7A. |
53 | |||
| Item 8. |
54 | |||
| Item 9. |
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
54 | ||
| Item 9A. |
54 | |||
| Item 9B. |
56 | |||
| PART III |
||||
| Item 10. |
57 | |||
| Item 11. |
57 | |||
| Item 12. |
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
57 | ||
| Item 13. |
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS , AND DIRECTOR INDEPENDENCE |
57 | ||
| Item 14. |
58 | |||
| PART IV |
||||
| Item 15. |
59 | |||
| 62 | ||||
| FINANCIAL STATEMENTS |
||||
| REPORT OF PRICEWATERHOUSECOOPERS LLP, INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM |
63 | |||
| 64 | ||||
| 65 | ||||
| 67 | ||||
| 66 | ||||
| 68 | ||||
i
Table of Contents
FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K includes forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, or the Exchange Act. All statements other than statements of historical facts contained in this Annual Report on Form 10-K, including statements regarding our anticipated future clinical and regulatory events, future financial position, business strategy and plans and objectives of management for future operations, are forward-looking statements. The words “believe,” “may,” “will,” “estimate,” “continue,” “anticipate,” “intend,” “expect” and similar expressions, as they relate to us, are intended to identify forward-looking statements. Such forward-looking statements include, without limitation, statements regarding the anticipated start dates, durations and completion dates of our ongoing and future clinical trials, statements regarding the anticipated designs of our future clinical trials, statements regarding anticipated future regulatory submissions and events, statements regarding our anticipated future cash position and statements regarding future events under our current and potential future collaborations. These forward-looking statements are subject to a number of risks, uncertainties and assumptions, including without limitation the risks described in “Risk Factors” in Part I, Item 1A of this Annual Report on Form 10-K. These risks are not exhaustive. Other sections of this Annual Report on Form 10-K include additional factors which could adversely impact our business and financial performance. Moreover, we operate in a very competitive and rapidly changing environment. New risk factors emerge from time to time and it is not possible for our management to predict all risk factors, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements. You should not rely upon forward-looking statements as predictions of future events. We cannot assure you that the events and circumstances reflected in the forward-looking statements will be achieved or occur and actual results could differ materially from those projected in the forward-looking statements. We assume no obligation to update or supplement forward-looking statements.
1
Table of Contents
PART I
| Item 1. | Description of Business. |
The Company
We are a biopharmaceutical company focused on the rapid development of novel small molecule drugs to treat serious diseases for which there are limited treatment options. Our product candidates in clinical development are dimebon (latrepirdine), which is in Phase 3 development for the treatment of Alzheimer’s disease and Huntington disease, and MDV3100, which is in Phase 3 development for the treatment of castration-resistant prostate cancer. Our dimebon program is partnered with Pfizer Inc, or Pfizer, and our MDV3100 program is partnered with Astellas Pharma Inc., or Astellas.
In September 2008, we announced a Collaboration Agreement with Pfizer, which became effective in October 2008. Due to the negative results in the CONNECTION study we reported in March 2010, Pfizer obtained the unilateral right to terminate our Collaboration Agreement at any time. We do not know whether or when Pfizer may choose to exercise this right. A more comprehensive discussion of the risks and uncertainties related to development of dimebon, including our dependence on Pfizer for the continued development of dimebon, can be found in the section entitled “Risk Factors—Risks Related to Our Product Development Candidates” under Item 1A of this Annual Report on Form 10-K. Under the terms of the agreement, we and Pfizer will develop and commercialize dimebon for the treatment of Alzheimer’s disease and Huntington disease. We and Pfizer will share the costs and expenses of developing and commercializing dimebon for the United States market on a 60% Pfizer/40% Medivation basis, and will share profits (or losses) resulting from commercialization of dimebon in the United States in the same proportions. Outside the United States, Pfizer will bear all development and commercialization costs, and will pay us tiered royalties on aggregate net sales of dimebon. In the fourth quarter of 2008, we received a non-refundable, up-front cash payment of $225.0 million pursuant to our Collaboration Agreement with Pfizer.
With Pfizer, we have been conducting a broad dimebon clinical development program, including five randomized, double-blind, placebo-controlled Phase 3 studies in Alzheimer’s and Huntington diseases. In March 2010, we reported negative results from the first of these studies—the CONNECTION trial, a six-month study of 598 patients with mild-to-moderate Alzheimer’s disease in the United States, Western Europe, Russia and Chile in which patients were randomized to receive dimebon 20 mg three times daily, dimebon 5 mg three times daily or placebo. Patients in the CONNECTION trial were not permitted to take any approved Alzheimer’s disease medicines for a period of 90 days before, or at any time during, the study. In the CONNECTION study, dimebon treated patients did no better than placebo patients on any of the primary and secondary efficacy endpoints at either dose tested. Dimebon was well tolerated in the CONNECTION study, as well as in a separate randomized, double-blind, placebo-controlled safety study of dimebon, also reported in March 2010, in which 742 patients in the United States and Canada were randomized to receive dimebon 20 mg three times daily or placebo and treated for three to six months. Approximately 85% of patients in the safety study were taking one or more approved Alzheimer’s disease medicines while on the trial. Further analysis of the results from both of these studies is currently underway.
Given the negative results in the CONNECTION trial, we are currently re-evaluating our approach to the clinical development of dimebon for Alzheimer’s disease and Huntington disease. This will be a complex analysis that will encompass scientific, clinical, regulatory and business issues. We do not know at this time which, if any, of our ongoing Phase 3 dimebon trials will be completed, or if Pfizer will elect to terminate our collaboration. If Pfizer elects to terminate our collaboration, we may be unable to fund the development costs on our own and may be unable to find a new collaborator, which could cause our dimebon program to fail. A more comprehensive discussion of the risks and uncertainties related to the development of dimebon, including our dependence on Pfizer for the continued development of dimebon, can be found in the section entitled “Risk Factors—Risks Related to our Product Development Candidates” under Item 1A of this Annual Report on Form 10-K.
In October 2009, we entered into a Collaboration Agreement with Astellas. Under the terms of the agreement, we and Astellas will develop and commercialize MDV3100 for the treatment of both late- and
2
Table of Contents
early-stage prostate cancer. We and Astellas will share equally the costs and expenses of developing and commercializing MDV3100 for the United States market, except that development costs for studies useful in both the United States market and either Europe or Japan, such as the ongoing Phase 3 AFFIRM study described below, will be shared two-thirds by Astellas and one-third by us. We and Astellas will share equally profits (or losses) resulting from commercialization of MDV3100 in the United States. Outside the United States, Astellas will bear all development and commercialization costs, and will pay us tiered royalties on aggregate net sales of MDV3100. In the fourth quarter of 2009, we received a non-refundable, up-front cash payment of $110.0 million pursuant to our Collaboration Agreement with Astellas, ten percent of which we paid to the academic institution from which we licensed our rights to MDV3100.
With Astellas, we are currently conducting a randomized, double-blind, placebo-controlled Phase 3 trial, known as the AFFIRM trial, of MDV3100 in men with a type of advanced prostate cancer known as castration-resistant prostate cancer, or CRPC.
We have funded our operations primarily through private and public offerings of our common stock, and from the up-front payment and cost-sharing payments from our Collaboration Agreements with Pfizer and Astellas. As of December 31, 2009, we had an accumulated deficit of $177.4 million and we expect to incur substantial additional losses in the future as we continue research and development activities designed to support potential approval of our present and potential future product candidates.
Our Pipeline
Dimebon (latrepirdine)
Alzheimer’s disease. Our product candidate dimebon (latrepirdine) is in Phase 3 development for Alzheimer’s disease, in collaboration with Pfizer. In March 2010, we reported top-line results from our CONNECTION trial, a randomized, double-blind, six-month, placebo-controlled Phase 3 trial in 598 patients with mild-to-moderate Alzheimer’s disease in the United States, Western Europe, Russia and Chile, and from a separate 742-patient safety study of dimebon in patients with mild-to-moderate Alhzeimer’s disease in the United States and Canada, approximately 85% of whom were also taking one or more approved Alzheimer’s disease medicines. In the CONNECTION trial, dimebon failed to show a statistically significant improvement over placebo on any of the primary or secondary efficacy endpoints, and thus did not meet any of the study endpoints. Dimebon was well tolerated in both the CONNECTION trial and in the 742-patient safety study. We designed the CONNECTION trial to confirm the results of our first clinical trial of dimebon in 183 patients with mild-to-moderate Alhzeimer’s disease in Russia, or the Russian Study, which was published in 2008 in the Lancet. In the Russian Study, dimebon showed a statistically significant improvement over placebo on all of the same primary and secondary efficacy endpoints used in the CONNECTION trial. Thus, the CONNECTION trial failed to replicate the efficacy results seen in the Russian Study. We and Pfizer currently are enrolling patients in three ongoing randomized, double-blind, placebo-controlled Phase 3 clinical trials in Alzheimer’s disease patients, ranging in duration from six to twelve months.
Huntington disease. Dimebon is also in Phase 3 clinical development for Huntington disease, also in collaboration with Pfizer. In July 2008, we announced top-line results of a Phase 2 study showing that dimebon was well tolerated and significantly improved cognitive function in Huntington disease patients compared to those treated with a placebo. The study, which was conducted in the U.S. and the United Kingdom, met its primary endpoint of safety and tolerability; in addition, dimebon showed statistically significant benefit versus placebo in cognition as measured by the Mini-Mental State Examination, or MMSE, a secondary endpoint in the study. However, dimebon failed to show a statistically significant benefit over placebo in this study on two other cognitive endpoints—the cognitive component of the Unified Huntington Disease Rating Scale, or UHDRS, and the ADAS-cog. Results of this study were published in March 2010 in Archives of Neurology. In July 2009, we enrolled the first patient in a randomized, double-blind, six-month, placebo-controlled Phase 3 study to treat Huntington disease, known as the HORIZON study.
3
Table of Contents
Impact of the negative CONNECTION trial results. Given the negative CONNECTION trial results, we and Pfizer are currently re-evaluating our approach to the clinical development of dimebon for Alzheimer’s disease and Huntington disease. This will be a complex analysis that will encompass scientific, clinical, regulatory and business issues. We do not know at this time which, if any, of our ongoing Phase 3 dimebon trials will be completed, or if Pfizer will elect to terminate our collaboration. The negative results in the CONNECTION trial had a material adverse effect on our stock price and market capitalization and resulted in Pfizer obtaining the right to terminate our collaboration at any time. Under our collaboration agreement, Pfizer is responsible for 60% of dimebon development costs for the United States market, and 100% of dimebon development costs for ex-US markets. If Pfizer were to terminate our collaboration, these costs would become our responsibility, following a six-month transition period after the date of termination, during which Pfizer would continue to bear its share of development costs. The negative CONNECTION results thus may have materially adverse effects on our business and financial position, including on our ability to enroll, successfully complete and fund our ongoing dimebon clinical trials, especially if Pfizer elects to terminate our collaboration. A more comprehensive discussion of the risks and uncertainties related to the development of dimebon, including our dependence on Pfizer for the continued development of dimebon, can be found in the section entitled “Risk Factors—Risks Related to Our Product Development Candidates” under Item 1A of this Annual Report on Form 10-K.
MDV3100
Our product candidate MDV3100 is in Phase 3 development for castration-resistant prostate cancer, in collaboration with Astellas. In June 2009, we presented data from our ongoing Phase 1-2 clinical trial of MDV3100 at the American Society of Clinical Oncology Annual Meeting. In the 140 men enrolled in this trial, including both men who had failed prior chemotherapy and men who were chemotherapy-naïve, MDV3100 consistently demonstrated anti-tumor activity across dose levels and endpoints, as evaluated by reductions in prostate-specific antigen, or PSA, levels, radiographic findings and circulating tumor cell, or CTC, counts. In this trial, MDV3100 was generally well tolerated at doses up to 240 mg per day. In September 2009, we enrolled the first patient in a randomized, double-blind, placebo-controlled Phase 3 study, known as the AFFIRM study, in patients with castration-resistant prostate cancer who have failed prior chemotherapy.
Our Corporate Structure
We have formed separate subsidiaries to hold the product candidates we are developing. Our subsidiary Medivation Neurology, Inc. holds our dimebon technology, and our subsidiary Medivation Prostate Therapeutics, Inc. holds our MDV3100 technology. Our subsidiary Medivation Technologies, Inc. holds our technologies that have not yet entered clinical development.
Our History
We are a corporation formed in Delaware in October 1995, under our former name Orion Acquisition Corp. II, to identify and consummate a business combination. Medivation Neurology, Inc. was formed in Delaware in September 2003 to acquire and develop dimebon. On December 17, 2004, Medivation Neurology, Inc. became our subsidiary pursuant to a merger. Medivation Prostate Therapeutics, Inc. was formed in Delaware as our subsidiary to acquire and develop our MDV300 series technology.
Our Dimebon Program
Dimebon (latrepirdine) is our investigational drug candidate in Phase 3 development for both Alzheimer’s disease and Huntington disease.
Alzheimer’s Disease
Alzheimer’s disease, the leading cause of dementia, is characterized by the progressive loss of memory, thinking and ability to perform activities of daily living (bathing, feeding, self-care, etc.), as well as significant
4
Table of Contents
behavioral disturbances (agitation, aggression, delusions, hallucinations, etc). There is currently no cure. According to the Alzheimer’s Association and the American Health Assistance Foundation:
| • | Alzheimer’s disease currently affects approximately 5.3 million people in the U.S., including as many as 13% of people aged 65 and older and approximately 50% of those aged 85 and older. |
| • | Worldwide, Alzheimer’s disease affects 26 million people, and that number is expected to reach 106 million by 2050. |
| • | There are approximately 454,000 new diagnoses of Alzheimer’s disease, and approximately 72,000 Alzheimer’s disease deaths, per year in the U.S. |
| • | Following initial diagnosis, patients live four to six years on average, but may live up to 20 years with the disease. |
| • | Total annual expenditures on Alzheimer’s disease in the U.S. exceed $208 billion annually, and the average lifetime cost per Alzheimer’s disease patient is $174,000. |
There are only four commonly-used drugs that the FDA has approved for the treatment of Alzheimer’s disease. Although the precise mechanism of action of these four drugs is unknown, three of them are believed to inhibit cholinesterase, and one is believed to inhibit the N-methyl-D-asparatate, or NMDA, receptor. These four drugs and their respective marketers, FDA approval dates (as listed in the FDA’s on-line edition of its Orange Book) and postulated mechanisms of action (as appearing in the package inserts for these drugs) are set forth in the following table.
| Drug (Trade Name/ Generic) |
Marketed by |
FDA Approval Date |
Postulated Mechanism | |||
| Aricept® (donepezil) |
Pfizer Inc./Eisai Co., Ltd. | November 25, 1996 | Cholinesterase inhibition | |||
| Exelon® (rivastigmine) |
Novartis AG | April 21, 2000 | Cholinesterase inhibition | |||
| Razadyne® (galantamine) |
Johnson & Johnson | February 28, 2001 | Cholinesterase inhibition | |||
| Namenda® (memantine) |
Forest Laboratories, Inc. | October 16, 2003 | NMDA-receptor antagonism | |||
According to IMS Health, the worldwide market for Alzheimer’s disease therapies currently exceeds $5 billion, with the largest selling cholinesterase inhibitor, Aricept, generating more than half of those sales, followed by Forest Laboratories, Inc.’s NMDA-receptor antagonist Namenda. In 2008, the first generic equivalent, galantamine, entered the market. Further generic entrants are expected as the remaining branded drugs lose patent protection in the United States and Europe between 2010 and 2015.
Huntington Disease
Huntington disease is a fatal neurological disorder characterized clinically by involuntary movements, loss of cognitive function and a wide spectrum of behavioral disorders. Common motor symptoms include chorea (involuntary writhing and spasming), clumsiness and progressive loss of the abilities to walk, speak and swallow. Cognitive symptoms include loss of intellectual speed, attention and short-term memory. Behavioral symptoms span the range of changes in personality, depression, irritability, emotional outbursts and apathy. Huntington disease is known to be caused by a specific genetic mutation, which results in degeneration of neurons in many different regions of the brain. This degeneration is particularly focused in neurons located in the basal ganglia, structures deep within the brain that control many important functions, including coordinating movement, and also in neurons on the outer surface of the brain or cortex, which controls thought, perception and memory.
There are no FDA-approved therapies to treat the cognitive impairment associated with Huntington disease. Everyone who carries at least one copy of the Huntington disease mutation and lives long enough will develop the disease. Symptoms generally begin between the ages of 30 and 45, but have been reported to appear as early as two years of age. The disease is invariably fatal and death usually occurs between 10 and 20 years after the onset of symptoms, making Huntington disease not only a devastating but also a protracted illness. According to the Hereditary Disease Foundation, in the United States alone approximately 30,000 patients currently suffer
5
Table of Contents
from Huntington disease, and an additional 150,000 are genetically at risk for developing it. The Huntington Disease Society of America estimates that the prevalence of Huntington disease in the U.S. population is approximately 1 in 10,000 persons.
Mechanism of Action
We believe that dimebon may operate through a novel mechanism of action involving enhancement of mitochondrial function. Mitochondria are intracellular structures that are responsible for generating energy within all cells and play important roles in mediating brain cell function and survival. Mitochondrial dysfunction has been linked in the published literature to both Alzheimer’s and Huntington diseases. In addition, autopsy studies of brains from patients with Alzheimer’s disease suggest that mitochondrial damage and synaptic dysfunction are early cellular events in Alzheimer’s disease development and progression.
In laboratory experiments, dimebon has been shown to improve mitochondrial function in the setting of cellular stress with very high potency. For example, dimebon treatment improved mitochondrial function and increased the number of surviving cells in a dose-dependent fashion after treatment with a cell toxin known as ionomycin, as well as with beta amyloid, a toxic substance often associated with Alzheimer’s disease and the loss of brain cells. Dimebon also has been shown in laboratory experiments to impact two aspects of brain cell function: promotion of neurite outgrowth and preservation of mitochondrial function after brain cells were challenged with beta amyloid. Results of the study showed that dimebon induced a statistically significant increase in neurite outgrowth from cortical, hippocampal and spinal cord neurons. Dimebon’s effect on neurite outgrowth was seen at low concentrations and was comparable to that achieved with maximally effective concentrations of a potent naturally occurring protein that is known to enhance brain cell function (Brain Derived Neurotrophic Factor).
We also believe, based on the results of laboratory experiments, that dimebon does not operate through the same mechanisms of action as the existing approved Alzheimer’s disease medicines—inhibition of cholinesterase or modulation of the NMDA receptor. These experiments have demonstrated that dimebon inhibits acetylcholinesterase much less potently (2,900 fold) than donepezil, an approved Alzheimer’s drug that acts by inhibiting this enzyme. Dimebon binds to the NMDA receptor 200 fold less potently than memantine, an approved Alzheimer’s drug that acts by inhibiting this receptor. Preclinical studies published on-line in the Journal of Pharmacology and Experimental Therapeutics in March 2010 suggest that dimebon’s cognition enhancing effect in animals is not mediated by acetylcholinesterase or the NMDA receptor.
Completed Alzheimer’s Disease Clinical Trials
The most important Alzheimer’s disease clinical studies we have completed to date are the following: (a) the Russian Study, a randomized, double-blind, placebo-controlled, twelve-month safety and efficacy study, in which we enrolled 183 patients with mild-to-moderate Alzheimer’s disease in Russia, the results of which were published in the Lancet in 2008; (b) the CONNECTION study, a randomized, double-blind, placebo-controlled, six-month safety and efficacy study designed to confirm the results of the Russian Study, in which we enrolled 598 patients with mild-to-moderate Alzheimer’s disease in the United States, Western Europe, Russia and Chile; and (c) the Safety Study, a randomized, double-blind, placebo-controlled three-to-six-month safety study, in which we enrolled 742 patients with mild-to-moderate Alzheimer’s disease in the United States and Canada.
Study Designs
The Russian and CONNECTION Studies. Both the Russian Study and the CONNECTION study enrolled patients with mild-to-moderate Alzheimer’s disease. The inclusion and exclusion criteria in both studies were substantially identical, and were designed to mimic those used in the pivotal registration trials of the currently approved medicines for mild-to-moderate Alzheimer’s disease. In the Russian Study, patients were randomized to two treatment groups—one of which received dimebon 20 mg three times per day and the other of which received placebo. In the CONNECTION study, patients were randomized to three treatment groups—dimebon 20 mg three times per day, dimebon 5 mg three times per day, and placebo. Patients were not permitted to take any other Alzheimer’s disease drugs during either trial.
6
Table of Contents
In both trials we used five widely-accepted clinical endpoints to assess dimebon’s potential effects on all of the primary aspects of Alzheimer’s disease—memory, thinking, activities of daily living (bathing, feeding, self-care, etc.), behavior (agitation, aggression, delusions, hallucinations, etc.) and overall clinical function. These endpoints were the ADAS-cog, the Clinician’s Interview-based Impression of Change-plus caregiver input, or CIBIC-plus, the Alzheimer’s Disease Cooperative Study-Activities of Daily Living, or ADCS-ADL, the Neuropsychiatric Inventory, or NPI, and the MMSE. The ADAS-cog and the CIBIC-plus are the two endpoints that have been accepted by the FDA to support approval of drugs for mild-to-moderate Alzheimer’s disease.
In both studies, patients were treated for six months. Patients who completed the initial six months of treatment in the Russian Study were offered the opportunity to continue treatment for an additional six months on a blinded basis in the same treatment group to which they originally were randomized. Patients who completed the blinded treatment periods, twelve months in the case of the Russian Study and six months in the case of the CONNECTION study, were offered the opportunity to receive dimebon 20 mg three times a day on an open label basis.
The Safety Study. Patients in the Safety Study were randomized to either dimebon 20 mg three times per day or placebo, and were treated for a period of either three or six months. Approximately 85% of patients enrolled in the Safety Study were taking one or more currently approved Alzheimer’s disease medicines while participating in the study.
Study Results
Efficacy. Dimebon met all of its primary and secondary efficacy endpoints in the Russian Study, but failed to meet any of its primary and secondary efficacy endpoints in the CONNECTION study.
In the Russian Study, dimebon caused statistically significant improvement over placebo on all five efficacy endpoints after six months and a full year of treatment. The mean drug-placebo difference on the ADAS-cog, which measures cognition, increased from 4.0 points at six months to 6.9 points at one year (p < 0.0001 at both six and twelve months). Compared to their starting scores at the beginning of the trial, dimebon-treated patients were significantly better on all five endpoints after six months of treatment, and remained stabilized on all five clinical endpoints after a full year of treatment. By contrast, scores of the placebo-treated patients declined significantly from their starting levels on all five endpoints after both six months and a full year of treatment.
By contrast, neither dose of dimebon tested in the CONNECTION trial demonstrated statistically significant improvement over placebo on any of the same five efficacy endpoints used in the Russian Study after six months of treatment, including on the co-primary endpoints—the ADAS-cog and the CIBIC-plus. The mean drug-placebo difference on the ADAS-cog at the 20 mg dimebon dose was 0.1 point, but statistical significance was not reached (p=0.86). Compared to their starting scores at the beginning of the trial, dimebon-treated patients at the 20 mg dose were significantly better on two of five endpoints (the NPI and the MMSE) after six months of treatment and not significantly changed from baseline on three of five endpoints (ADAS-cog, CIBIC-plus and ADCS-ADL), including both of the two co-primary endpoints. Scores of the placebo treated patients did not decline on any endpoint; they improved significantly from their starting baseline levels on one of five endpoints (the MMSE), and were not significantly changed from baseline on the other four endpoints (ADAS-cog, CIBIC-plus, ADCS-ADL and NPI). Results for the dimebon 5 mg dose were similar to the dimebon 20 mg dose and placebo, although they were numerically lower. Thus, in the CONNECTION study, dimebon-treated patients did no better than placebo patients on any endpoint at either dose tested.
Safety and Tolerability. Dimebon was well tolerated in all three of the Russian Study, the CONNECTION study and the Safety Study.
In the Russian Study, the number of patients with at least one adverse event was similar in the dimebon 20 mg and placebo groups after both six and twelve months of treatment (69% in the dimebon group vs. 66% in the placebo group after six months; 79% in the dimebon group vs. 75% in the placebo group after twelve months).
7
Table of Contents
Adverse events that occurred in five percent or more of dimebon patients and more frequently than in placebo patients after twelve months of treatment were dry mouth (18% vs. 1%) and depressed mood/depression (15% vs. 5%). Depressed mood/depression reflected reports from patients and their caregivers, not clinical diagnoses of depression. The reported depressed mood/depression was generally mild and did not cause any of the affected patients to discontinue participation in the trial.
In the CONNECTION study, the number of patients with at least one adverse event was similar in the dimebon 20 mg and placebo groups after six months of treatment (72% in the dimebon group vs. 74% in the placebo group). Adverse events that occurred in five percent or more of dimebon 20 mg patients and more frequently than in placebo patients after six months of treatment were somnolence (11% vs. 10%), dry mouth (9% vs. 7%), headache (10% vs. 6%), dizziness (8% vs. 5%), constipation (6% vs. 4%), cough (8% vs. 4%) and depression (6% vs. 4%).
In the Safety Study, adverse events that occurred in five percent or more of dimebon patients and more frequently than in placebo patients were somnolence (5% vs. 2%) and fatigue (5% vs. 2%).
Ongoing Phase 3 Alzheimer’s Disease Clinical Trials
We and Pfizer are enrolling patients in three ongoing Phase 3 clinical trials of dimebon in Alzheimer’s disease. These trials are known as the CONCERT trial, the CONTACT trial and the CONSTELLATION trial. Given the negative results in the CONNECTION study, we are currently re-evaluating our approach to the clinical development of dimebon for Alzheimer’s disease and Huntington disease. This will be a complex analysis that will encompass scientific, clinical, regulatory and business issues. We do not know at this time which, if any, of our ongoing Phase 3 dimebon trials will be completed, or if Pfizer will elect to terminate our collaboration. If Pfizer elects to terminate co-funding of dimebon development, we may be unable to fund the development costs on our own and may be unable to find a new collaborator, which could cause our dimebon program to fail. A more comprehensive discussion of the risks and uncertainties related to the development of dimebon, including our dependence on Pfizer for the continued development of dimebon, can be found in the section entitled “ Risk Factors—Risks Related to Our Product Development Candidates” under Item 1A of this Annual Report on Form 10-K.
The CONCERT Trial. The CONCERT trial is a twelve-month randomized, double-blind, placebo-controlled Phase 3 trial of dimebon in approximately 1,000 patients with mild-to-moderate Alzheimer’s disease who are also taking Aricept, the leading approved Alzheimer’s disease medication. Patients are being enrolled in the United States, Western Europe, Australia and New Zealand, and will be randomized to receive dimebon 20 mg three times daily, dimebon 5 mg three times daily or placebo in addition to their Aricept. The Safety Study demonstrated that dimebon is well tolerated when given in combination with Aricept. The CONCERT trial is designed to evaluate the potential benefits of dimebon over a one-year period when added to treatment with Aricept, as compared to treatment with Aricept alone. The primary endpoints are the ADAS-cog and the ADCS-ADL. We enrolled the first patient in the CONCERT trial in March 2009.
The CONTACT Trial. The CONTACT trial is a six-month randomized, double-blind, placebo-controlled Phase 3 trial of dimebon in approximately 600 patients with moderate-to-severe Alzheimer’s disease who are also taking Aricept. Patients are being enrolled in Europe and South America, and will be randomized to receive either dimebon 20 mg three times daily or placebo in addition to their Aricept. The CONTACT trial is designed to assess the potential benefits of adding dimebon to ongoing Aricept therapy on patients’ behavioral difficulties and ability to perform routine activities of daily living. The primary endpoints are the NPI and the Alzheimer’s Disease Cooperative Study-Activities of Daily Living (severe), or ADCS-ADLsev. We enrolled the first patient in the CONTACT trial in November 2009.
The CONSTELLATION Trial. The CONSTELLATION trial is a six-month randomized, double-blind, placebo-controlled Phase 3 trial of dimebon in approximately 570 patients with moderate-to-severe Alzheimer’s disease who are also taking Namenda. Patients are being enrolled in the United States, Canada and Europe, and
8
Table of Contents
will be randomized to receive either dimebon 20 mg three times daily or placebo in addition to their Namenda. The CONSTELLATION trial is designed to assess the potential benefits of adding dimebon to ongoing Namenda therapy on patients’ cognition, memory and ability to perform routine activities of daily living. The primary endpoints are the Severe Impairment Battery, or SIB, and the ADCS-ADLsev. We enrolled the first patient in the CONSTELLATION trial in October 2009.
Completed Huntington Disease Clinical Trial
In 2008, we announced top-line results of a randomized, double-blind, placebo-controlled Phase 2 clinical trial of dimebon in Huntington disease patients. The trial was conducted at 16 centers in the United States and the United Kingdom in collaboration with the Huntington Study Group, or HSG, a network of more than 250 experienced clinical trial investigators, coordinators and consultants from more than 60 academic and research institutions throughout the United States, Canada, Europe and Australia dedicated to clinical research of Huntington disease. The trial enrolled 90 patients with Huntington disease, with half randomized to dimebon 20 mg three times daily and the other half to placebo for a three-month dosing period. The primary endpoint of the trial was safety and tolerability. The secondary endpoint was efficacy, as measured by the MMSE, the cognition scale most widely used by clinicians to assess patients with neurodegenerative diseases, the UHDRS, a composite assessment tool that evaluates the impact of Huntington disease on cognition, motor function, behavior, overall function and level of independence, and the ADAS-cog, a cognition scale generally used in Alzheimer’s disease clinical trials.
In this study, dimebon was well tolerated and significantly improved cognitive function in Huntington disease patients compared to those treated with a placebo. The study met its primary endpoint of safety and tolerability; in addition, dimebon showed statistically significant benefit versus placebo in cognition as measured by the MMSE, a secondary endpoint in the study. However, dimebon failed to show a statistically significant benefit over placebo in this study on two other cognitive endpoints—the cognitive component of the UHDRS and the ADAS-cog. Results of this study were published in March 2010 in Archives of Neurology.
Dimebon was well tolerated in this trial. There were fewer patients reporting adverse events in the dimebon group than in the placebo group (70% vs. 80%). Huntington disease patients treated with dimebon had fewer falls (9%), a common problem in this patient population that often results in injury and associated health care costs, than did patients on placebo (16%). The most common adverse event in the dimebon group was headache, which occurred in 19% of treated patients compared to 7% of placebo patients. Headaches were generally mild in severity. Dry mouth and depressed mood were similar in both treated and placebo groups (4% and 7%, respectively).
Subgroup analysis data from the Phase 2 trial showed that dimebon’s positive effect on cognition, as measured by the MMSE, was 60% greater in the more cognitively impaired patients. The highest possible MMSE score is 30, which reflects the absence of any cognitive impairment. Because dimebon treatment resulted in improvement over baseline at the start of the trial and because patients with normal or near normal MMSE scores at baseline have little opportunity to improve, this analysis focused on the subgroup of patients with clear cognitive impairment (baseline MMSE scores £26) and found an almost 1.6 point improvement in the MMSE scores in the dimebon-treated group as compared to the placebo group (p=0.008).
Ongoing Phase 3 Huntington Disease Clinical Trial
We and Pfizer are enrolling patients in an ongoing Phase 3 clinical trial of dimebon in Huntington disease, known as the HORIZON trial. Given the negative results in the CONNECTION study, we are currently re-evaluating our approach to the clinical development of dimebon for Alzheimer’s disease and Huntington disease. This will be a complex analysis that will encompass scientific, clinical, regulatory and business issues. We do not know at this time which, if any, of our ongoing Phase 3 dimebon trials will be completed, or if Pfizer will elect to terminate our collaboration. If Pfizer elects to terminate our collaboration, we may be unable to fund the development costs on our own and may be unable to find a new collaborator, which could cause our dimebon
9
Table of Contents
program to fail. A more comprehensive discussion of the risks and uncertainties related to the development of dimebon, including our dependence on Pfizer for the continued development of dimebon, can be found in the section entitled “Risk Factors—Risks Related to our Product Development Candidates” under Item 1A of this Annual Report on Form 10-K.
The HORIZON Trial. The HORIZON trial is a six-month randomized, double-blind, placebo-controlled Phase 3 trial of dimebon in approximately 350 patients with Huntington disease. Patients are being enrolled in the United States, Europe and Australia, and will be randomized to receive either dimebon 20 mg three times daily or placebo. The HORIZON trial is designed to assess the potential benefits of dimebon on patients’ cognition and global function. The primary endpoints are the MMSE and the CIBIC-plus. We enrolled the first patient in the HORIZON trial in July 2009. Based on the subgroup analysis from our Phase 2 trial, we are enrolling only patients with MMSE scores of 26 or lower (i.e., those with clear cognitive impairment) in the HORIZON trial in an effort to enhance the chances of a positive outcome.
Orphan Drug Designation
The FDA has granted orphan drug designation to dimebon for the treatment of Huntington disease. Orphan drug designation is available to drugs intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States. Due to its receipt of orphan drug designation, if dimebon is approved for Huntington disease, it will be entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications to market dimebon for Huntington disease, except in very limited circumstances, for seven years. Orphan drug designation does not shorten the duration of the regulatory review or approval process.
Pfizer Inc. Collaboration Agreement
In September 2008, we announced a Collaboration Agreement with Pfizer. Due to the negative results in the CONNECTION study, Pfizer obtained the unilateral right to terminate our Collaboration Agreement at any time. We do not know whether or when Pfizer may choose to exercise this right. A more comprehensive discussion of the risks and uncertainties related to development of dimebon, including our dependence on Pfizer for the continued development of dimebon can be found in the section entitled “Risk Factors—Risks Related to Our Product Development Candidates” under Item 1A of this Annual Report on Form 10-K.
Under the Pfizer Collaboration Agreement, we and Pfizer will collaborate on development of dimebon for Alzheimer’s disease and Huntington disease for the United States market, including associated regulatory filings with the FDA. In addition, if approved by the FDA, following such approval and the launch of dimebon in the United States, we, at our option, and Pfizer have the right co-promote dimebon to specialty physicians in the United States, and Pfizer has the sole right to promote dimebon to primary care physicians in the United States. Pfizer will be responsible for development and seeking regulatory approval for, and commercialization of, dimebon outside the United States. Following a period of transition from our contract manufacturers to Pfizer, Pfizer has assumed responsibility for all manufacture of product for both clinical and commercial purposes. Both we and Pfizer have agreed not to commercialize for the treatment of specified indications any other products directed to the same primary molecular target as dimebon for a specified time period, subject to certain exceptions.
The agreement establishes several joint committees consisting of an equal number of representatives from both parties that operate by consensus to oversee the collaboration. In the event that a joint committee is unable to reach consensus on a particular issue, then, depending on the issue, a dispute may be decided at the joint committee level by the party with the final decision on the issue or escalated to senior management of the parties. If a dispute is escalated to senior management and no consensus is reached, then the dispute may be decided by the party to whom the contract grants final decision on such issue. Other issues can only be decided by consensus of the parties, and unless and until the parties’ representatives reach agreement on such issue, no decision on such issue will be made, and the status quo will be maintained.
10
Table of Contents
Under the Pfizer Collaboration Agreement, Pfizer paid us an up-front cash payment of $225 million in the fourth quarter of 2008. We are also eligible to receive payments of up to $500 million upon the attainment of development and regulatory milestones plus additional milestone payments upon the achievement of certain net sales levels for the product. We and Pfizer will share the costs and expenses of developing and commercializing dimebon for the United States market on a 60%/40% basis, with Pfizer assuming the larger share, and we and Pfizer will share profits (or losses) resulting from the commercialization of dimebon in the United States in such proportions. Outside the United States, Pfizer will bear all development and commercialization costs and will pay us tiered royalties on the aggregate net sales of dimebon.
If one of the parties merges with, or acquires or is acquired by, a third party and as a result such party must divest its interest in the dimebon collaboration due to a governmental requirement, then the other party has the first right to purchase the divesting party’s interest in the collaboration, on terms to be negotiated by the parties. In the event that the parties are unable to agree on the terms of this purchase after following the negotiation procedure outlined in the Collaboration Agreement, the divesting party will have a time-limited right to sell its interest in the collaboration to a third party. However, the terms of this sale must be more favorable than any terms offered by the non-divesting party and the third party will remain bound by the terms of the Collaboration Agreement. In the event the non-divesting party declines to purchase the divesting party’s interest, the divesting party may sell its interest in the collaboration to a third party on any terms but such third party will remain bound by the terms of the Collaboration Agreement.
We are permitted to terminate the Collaboration Agreement for an uncured material breach by Pfizer. Pfizer has a right to terminate the Collaboration Agreement unilaterally at any time. In the event of our uncured material breach of the Collaboration Agreement, Pfizer may elect either to terminate the Collaboration Agreement or to keep the Collaboration Agreement in place, but terminate our right to participate in development, commercialization (other than co-promoting dimebon) and other activities for dimebon, including the joint committees and decision making for dimebon. However, such termination would not affect our financial return or, unless we commit an uncured material breach of our co-promotion obligations, our co-promotion rights. Following any termination of the Collaboration Agreement, all rights to develop and commercialize dimebon will revert to us, and Pfizer will grant a license to us to enable us to continue such development and commercialization, remain responsible for its ongoing financial and other obligations under the Collaboration Agreement for a transition period of six months following termination, and is obligated to supply product to us for a reasonable period, not to exceed to eighteen months following termination, on terms to be negotiated between the parties in good faith.
Our MDV300 Series Prostate Cancer Program
We own an exclusive, worldwide commercial license to a series of novel small molecules, referred to as the MDV300 series compounds. Our lead development candidate from the MDV300 series is a molecule we refer to as MDV3100, which is in Phase 3 development for a type of advanced prostate cancer known as castration-resistant prostate cancer, or CRPC. We are conducting this program in collaboration with Astellas.
Prostate Cancer Statistics
According to the American Cancer Society, prostate cancer is the most commonly diagnosed cancer among men in the United States, other than skin cancer. The American Cancer Society estimates that approximately 192,000 new cases of prostate cancer were diagnosed, and approximately 27,000 men died of prostate cancer, in the United States alone during 2009. Prostate cancer is thus the second-leading cause of cancer death in men in the United States, after lung cancer.
Metastatic Prostate Cancer—The Hormone-Sensitive and Castration-Resistant States
Metastatic prostate cancer is cancer that has spread beyond the prostate and surrounding tissues into distant organs and tissues. The majority of men who die from prostate cancer die from the consequences of metastatic disease. According to the National Cancer Institute, the median survival of patients with prostate cancer that has
11
Table of Contents
metastasized to distant organs is usually one to three years, and most such patients will die of prostate cancer. Metastatic prostate cancer is generally divided into two states: the hormone-sensitive state and the castration-resistant state (also referred to as the hormone-refractory state).
The Hormone-Sensitive State. Testosterone and other male sex hormones, known collectively as androgens, can fuel the growth of prostate cancer cells. Androgens exert their effects on prostate cancer cells by binding to and activating the androgen receptor, which is expressed in prostate cancer cells. When they first metastasize to distant sites, most prostate cancers depend on androgens for growth. These prostate cancers are known as hormone-sensitive cancers.
Accordingly, the leading therapies currently used for the treatment of metastatic prostate cancer are focused on diminishing, or antagonizing, the effects of androgens on prostate cancer cells. This effect is achieved through two separate approaches. The first approach uses drugs known as anti-androgens, which directly block the interaction of androgens with the androgen receptor. Casodex® (bicalutamide), sold by AstraZeneca PLC, is the largest selling of these drugs, with global annual sales of more than $800 million in 2009 according to the public disclosures of AstraZeneca PLC. The second approach is to reduce the amount of androgens produced in the body, primarily in the testes. This can be achieved surgically by removal of both testicles (orchiectomy) or through use of drugs known as luteinizing hormone-releasing hormone, or LHRH, agonists, which lower the native production of testosterone in the testicles (sometimes called chemical castration). Anti-androgens and LHRH agonists often are given in combination therapy, an approach known as a combined androgen blockade. However, because these therapies operate by reducing the ability of androgens to fuel the growth of prostate cancer cells, they generally are effective only on prostate cancers that remain hormone-sensitive—i.e., those that still depend on androgens for growth.
The Castration-Resistant State. Most metastatic prostate cancer initially is hormone-sensitive and thus responds to hormonal therapies. However, according to a study published in the October 7, 2004 issue of The New England Journal of Medicine, virtually all hormone-sensitive metastatic prostate cancer undergoes changes that convert it to the castration-resistant state in a median of 18-24 months after initiation of hormonal therapy. Once in this state, prostate cancers generally continue to grow in an androgen-dependent manner despite the reduction of testosterone production to very low (i.e., post-castration) levels. Prostate cancer in this state is known as castration-resistant prostate cancer, or CRPC. The switch from the hormone-sensitive to the castration-resistant state following initiation of hormonal therapy is generally determined based on either rising levels of prostate-specific antigen, or PSA, or documented disease progression as evidenced by imaging tests or clinical symptoms. Metastatic prostate cancer that has become castration-resistant is extremely aggressive; these patients have a median survival of only 10 to 16 months.
A primary reason that CRPC is so deadly is that it no longer responds to hormonal therapies that are effective in the hormone-sensitive state. To further complicate the situation, due to biological changes in prostate cancer that has entered the castration-resistant state, drugs that initially block the androgen receptor and inhibit growth of hormone-sensitive prostate cancer may have precisely the opposite effect and start to fuel the growth of CRPC. Agents are clearly needed to improve the treatment options for patients with CRPC.
Switch from the Hormone-Sensitive to the Castration-Resistant State
One of the factors that historically hindered the development of drugs to treat CRPC was that the cause of the switch from the hormone-sensitive to the castration-resistant state was not known. This problem was addressed by Dr. Charles Sawyers and his colleagues, who discovered that one of the important mechanisms by which prostate cancer cells switch from the hormone-sensitive to the castration-resistant state appears to be through overexpression of the androgen receptor. In experiments comparing gene expression in hormone-sensitive and castration-resistant prostate cancer cells, the results of which were published in the January 1, 2004 issue of Nature Medicine, Dr. Sawyers and his colleagues reported that an increase in androgen receptor expression was the only gene change consistently associated with castration-resistant disease. In a series of experiments, scientists showed (as expected) that activation of the androgen receptor in hormone-sensitive
12
Table of Contents
human prostate cancer cell lines was inhibited by current androgen receptor blockers, including Casodex. However, when the prostate cancer cell lines were genetically engineered to overexpress the androgen receptor (converting them from the hormone-sensitive to the castration-resistant state), not only did Casodex fail effectively to inhibit the androgen receptor in these cells, but in some cases it became a stimulant of the androgen receptor. Androgen receptor activation is correlated with the growth of prostate cancer. This finding is consistent with the published human clinical experience with Casodex in CRPC.
The MDV300 Series Compounds
The MDV300 series compounds, a series of approximately 170 small molecules, were synthesized based on the discovery of a mechanism by which prostate cancer shifts from the hormone-sensitive to the castration- resistant state. These molecules bind the androgen receptor, the same target bound by Casodex and other marketed drugs for metastatic prostate cancer, but do so in a manner designed to render them effective in treating cancers that have become refractory to currently used drugs.
Mechanism of Action
While MDV3100 operates through the androgen receptor pathway, it does so in a manner that is distinct from that of currently approved drugs targeting the androgen receptor. Prostate cancer growth requires the male sex hormone testosterone, which binds to the androgen receptor. When testosterone binds to the androgen receptor, the receptor moves into the nucleus of the prostate cancer cell. This is a process known as nuclear translocation. Once inside the nucleus of the cell, the androgen receptor binds to the cell’s DNA and stimulates prostate cancer growth. This is a process known as DNA binding.
An article published in May 2009 in Science described the discovery and novel mechanism of action of MDV3100. In the Science article, researchers using various preclinical models of CRPC provided evidence that MDV3100 (a) potently blocks the androgen receptor with greater binding affinity than Casodex, (b) impairs nuclear translocation and blocks DNA binding of the androgen receptor, one of the key steps required for androgen-dependent prostate cancer growth and a step not blocked by Casodex, and (c) induces death of castration-resistant prostate tumor cells, an effect not seen with Casodex. These properties potentially explain why MDV3100 has demonstrated beneficial effects in patients whose tumors are no longer responding to the currently available treatments for prostate cancer, including Casodex.
Ongoing Clinical Trials
Phase 1-2 Clinical Trial
In December 2008, we completed enrollment in an open-label Phase 1-2 clinical trial of MDV3100 in patients with CRPC. We enrolled 140 patients in seven dose groups, ranging from 30 mg per day to 600 mg per day, at several clinical sites in the United States, including Memorial Sloan-Kettering Cancer Center in New York. Of the 140 patients, 65 had previously failed chemotherapy and 75 were chemotherapy-naïve. Patients are permitted to remain on study drug until their disease progresses (by biochemical, radiographic or clinical criteria) or until they cease tolerating the drug. Patients enrolled in the trial were heavily pretreated, with 77% having failed two or more lines of prior hormonal therapy and 43% having also failed prior chemotherapy. All patients had progressive disease upon enrollment into the trial. The study endpoints include safety, tolerability, pharmacokinetics, circulating tumor cell, or CTC, counts, serum prostate-specific antigen, or PSA, levels, radiographic change in soft tissue and bony metastases, and time on treatment.
In June 2009, we presented efficacy and safety data covering all 140 patients enrolled in the trial at the American Society of Clinical Oncology, or ASCO, 2009 Annual Meeting. The data presented at ASCO reported the study results as of April 1, 2009. These data confirmed earlier data from the trial, showing that in the 140 men enrolled in this trial, including both men who had failed prior chemotherapy and men who were chemotherapy-naïve, MDV3100 consistently demonstrated anti-tumor activity across dose levels and endpoints, as evaluated by reductions in PSA levels, radiographic findings and CTC counts.
13
Table of Contents
MDV3100 produced significant PSA declines (50% or more from baseline) and radiographic control (partial response or stable disease) in both chemotherapy naïve and post-chemotherapy patients, as follows (data as of April 1, 2009):
| PSA response > 50% | Radiographic control: soft tissue lesions (partial response or stable disease) |
Radiographic control: bony lesions (stable disease) |
|||||||
| Chemotherapy naïve |
62 | % | 80 | % | 63 | % | |||
| Post-chemotherapy |
51 | % | 65 | % | 51 | % |
The median time to PSA progression was 6.1 months for post-chemotherapy patients, and not yet reached for chemotherapy-naïve patients. In addition, the median time to radiographic progression was 6.6 months for post-chemotherapy patients, and not yet reached for chemotherapy-naïve patients.
Almost all patients with favorable CTC counts of four or less at the start of treatment maintained favorable counts while on MDV3100 treatment (91% of evaluable chemotherapy naïve patients and 91% of evaluable post-chemotherapy patients). Importantly, a significant number of patients with unfavorable CTC counts of five or higher at baseline converted to favorable counts of less than five following MDV3100 treatment (75% of chemotherapy naïve patients and 37% of post-chemotherapy patients). This CTC conversion rate is important in light of a study published in the October 2008 issue of Clinical Cancer Research, in which post-treatment conversion to a CTC count below five was associated with a 15-month survival benefit in CRPC patients.
MDV3100 has been generally well tolerated in this trial at doses of up to and including 240 mg/day. The most frequently reported adverse event was fatigue. Seizures were observed in two patients, one each at doses of 600 and 360 mg/day. Both patients were taking concomitant medications that can cause seizures. A possible but unwitnessed seizure was reported in a patient taking a dose of 480 mg/day.
The AFFIRM Trial
In September 2009, we enrolled the first patient in a randomized, double-blind, placebo-controlled Phase 3 study, known as the AFFIRM trial, of MDV3100 in approximately 1,200 patients with CRPC who have failed prior chemotherapy. Patients will be enrolled in the United States, Canada, Europe, South America, Australia and South Africa, and will be randomized to receive either MDV3100 160 mg per day or placebo. The primary endpoint is overall survival.
The Astellas Collaboration Agreement
Our global development and commercialization agreement with Astellas became effective in October 2009. Under the Astellas Collaboration Agreement, we and Astellas agreed to collaborate on the development of MDV3100 for prostate cancer for the United States market, including associated regulatory filings with the FDA. In addition, if approved by the FDA, following such approval and the launch of MDV3100 in the United States, we, at our option, and Astellas have the right to co-promote MDV3100 in the United States. Astellas is responsible for development of, seeking regulatory approval for, and commercialization of MDV3100 outside the United States. Astellas will be responsible for commercial manufacture of MDV3100 on a global basis. Both Medivation and Astellas have agreed not to commercialize certain other products having a similar mechanism of action as MDV3100 for the treatment of specified indications for a specified time period, subject to certain exceptions.
We and Astellas share the costs of developing and commercializing MDV3100 for the United States market on a 50%/50% basis, and we and Astellas will share profits (or losses) resulting from the commercialization of MDV3100 in the United States in such proportions. Costs of clinical trials supporting development in both the United States and in either Europe or Japan, including the ongoing AFFIRM trial, are borne two-thirds by Astellas and one-third by us. Outside the United States, Astellas will bear all development and commercialization costs and will pay us tiered, double-digit royalties on the aggregate net sales of MDV3100.
14
Table of Contents
Under the Astellas Collaboration Agreement, Astellas paid us a non-refundable, up-front cash payment of $110.0 million in November 2009, ten percent of which we paid to the academic institution from which we licensed our rights to MDV3100. We are also entitled to receive up to $335.0 million in development milestone payments, plus up to an additional $320.0 million in commercial milestone payments. We are required to share 10% of the up-front payment and any development milestone payments received under the Astellas Collaboration Agreement with the academic institution from which we licensed our rights to MDV3100. We paid 10% of the up-front payment, or $11.0 million, to the academic institution in the fourth quarter of 2009.
Each of Medivation and Astellas is permitted to terminate the Astellas Collaboration Agreement for an uncured material breach by the other party or for the insolvency of the other party. Astellas has a right to terminate the Astellas Collaboration Agreement unilaterally by advance written notice to us, but, except in certain specific circumstances, generally cannot exercise that termination right until the first anniversary of MDV3100’s first commercial sale. Following any termination of the Astellas Collaboration Agreement in its entirety, all rights to develop and commercialize MDV3100 will revert to us, and Astellas will grant a license to us to enable us to continue such development and commercialization. In addition, except in the case of a termination by Astellas for our uncured material breach, Astellas will supply MDV3100 to us during a specified transition period.
Intellectual Property
As of December 31, 2009, we owned issued patents in the United States and Europe claiming the use of dimebon and certain related compounds to treat neurodegenerative diseases, an issued patent in the United States claiming the use of dimebon to treat Alzheimer’s disease, and issued foreign counterpart patents in Canada and Hong Kong. The U.S. and European patents expire in October 2016. However, if we succeed in receiving regulatory approval to sell dimebon, under current laws our U.S. and European patent protection for dimebon for the first approved indication may be eligible for extension for up to five additional years. We also own multiple pending patent applications claiming, among other things, the use of dimebon to treat Huntington disease and other indications, and numerous novel dimebon-like molecules. We own all of the above dimebon intellectual property and have full control over prosecution and enforcement against potential infringers, subject to the terms of our Collaboration Agreement with Pfizer in the case of intellectual property we have sublicensed to Pfizer. In addition, we have an exclusive license to multiple pending patent applications covering the MDV300 series compounds, including our lead development candidate MDV3100, and their uses in the treatment and prevention of disease. We intend to prosecute our owned intellectual property, and request that our licensors prosecute our licensed intellectual property, in the United States, Europe and other jurisdictions that we deem appropriate.
We require our employees and consultants to execute non-disclosure and proprietary rights agreements at the beginning of employment or consulting arrangements with us. These agreements generally acknowledge our exclusive ownership of all inventions and intellectual property, including, but not limited to patents, developed by the individual during the course of his or her work with us and require that all proprietary information disclosed to the individual remain confidential. We intend to enforce vigorously our intellectual property rights if infringement or misappropriation occurs.
Government Regulation and Product Approvals
The clinical development, manufacturing and marketing of our products are subject to regulation by various authorities in the U.S., the E.U. and other countries, including, in the U.S., the FDA, and, in the E.U., the European Agency for the Evaluation of Medical Products, or EMEA. The Federal Food, Drug, and Cosmetic Act and the Public Health Service Act in the U.S., and numerous directives, regulations, local laws, and guidelines in the E.U. govern testing, manufacture, safety, efficacy, labeling, storage, record keeping, approval, advertising and promotion of our products. Product development and approval within these regulatory frameworks takes a number of years, and involves the expenditure of substantial resources.
Regulatory approval will be required in all markets in which we, or our partners, including Pfizer and Astellas, seek to test and market our drug candidates. At a minimum, approval requires evaluation of data
15
Table of Contents
relating to quality, safety and efficacy of a product for its proposed use. The specific types of data required and the regulations relating to these data differ depending on the territory, the drug involved, the proposed indication and the stage of development.
In the U.S., specific preclinical data, chemical data and a proposed clinical study protocol must be submitted to the FDA as part of an Investigational New Drug application, or IND, which, unless the FDA objects, will become effective 30 days following receipt by the FDA. Phase 1 trials may commence only after the IND application becomes effective. Prior regulatory approval for human healthy volunteer studies is also required in member states of the European Union, or E.U. Currently, in each member state of the E.U., following successful completion of Phase 1 trials, data are submitted in summarized format to the applicable regulatory authority in the member state in respect of applications for the conduct of later Phase 2 trials. The regulatory authorities in the E.U. typically have between one and three months in which to raise any objections to the proposed clinical trial, and they often have the right to extend this review period at their discretion. In the U.S., following completion of Phase 1 trials, further submissions to regulatory authorities are necessary in relation to Phase 2 and 3 trials. Authorities may require additional data before allowing the trials to commence and could demand discontinuation of studies at any time if there are significant safety issues.
In addition to regulatory review, a clinical trial involving human subjects has to be approved by an independent body. The exact composition and responsibilities of this body differ from country to country. In the U.S., for example, each clinical trial is conducted under the auspices of an Institutional Review Board at the institution at which the clinical trial is conducted. This board considers among other things, the design of the clinical trial, ethical factors, the safety of the human subjects and the possible liability risk for the institution. Equivalent rules apply in each member state of the E.U., where one or more independent ethics committees that typically operate similarly to an Institutional Review Board will review the ethics of conducting the proposed research. Other authorities elsewhere in the world have slightly differing requirements involving both execution of clinical trials and import or export of pharmaceutical products. It is our responsibility to ensure that we conduct our business in accordance with the regulations of each relevant territory.
Information generated in this process is susceptible to varying interpretations that could delay, limit, or prevent regulatory approval at any stage of the approval process. Failure to demonstrate adequately the quality, safety and efficacy of a therapeutic drug under development would delay or prevent regulatory approval of the product. There can be no assurance that if clinical trials are completed, either we or our collaborative partners will submit applications for required authorizations to manufacture or market potential products or that any such application will be reviewed and approved by appropriate regulatory authorities in a timely manner, if at all.
In order to gain marketing approval, we must submit an application to the relevant authority for review, which is known in the U.S. as an NDA and in the E.U. as a marketing authorization application or MAA. The format is usually specified by each authority, although in general it will include information on the quality, chemistry, manufacturing and pharmaceutical aspects of the product and non-clinical and clinical data. The FDA undertakes such reviews for the U.S. In the E.U., there is, for many products, a choice of two different authorization routes: centralized and decentralized. Under the centralized route, one marketing authorization is granted for the entire E.U., while under the decentralized route a series of national marketing authorizations are granted. In the centralized system, applications are reviewed by members of the Committee for Medicinal Products for Human Use, on behalf of the EMEA. The EMEA will, based upon the review of the Committee for Medicinal Products for Human Use, provide an opinion to the European Commission on the safety, quality and efficacy of the product. The decision to grant or refuse an authorization is made by the European Commission. In circumstances where use of the centralized route is not mandatory, we can choose to use the decentralized route, in which case the application will be reviewed by each member state’s regulatory agency. If the regulatory agency grants the authorization, other member states’ regulatory authorities are asked to “mutually recognize” the authorization granted by the first member state’s regulatory agency.
Approval by regulatory authorities can take several months to several years or be denied. The approval process can be affected by a number of factors. Additional studies or clinical trials may be requested during the
16
Table of Contents
review and may delay marketing approval and involve unbudgeted costs. Regulatory authorities may conduct inspections of relevant facilities and review manufacturing procedures, operating systems and personnel qualifications. In addition to obtaining approval for each product, in many cases each drug manufacturing facility must be approved. Further, inspections may occur over the life of the product. An inspection of the clinical investigation sites by a competent authority may be required as part of the regulatory approval procedure. As a condition of marketing approval, the regulatory agency may require post-marketing surveillance to monitor adverse effects, or other additional studies as deemed appropriate. After approval for the initial indication, further clinical studies are usually necessary to gain approval for additional indications. The terms of any approval, including labeling content, may be more restrictive than expected and could affect product marketability.
Competition
The biopharmaceutical industry is intensely competitive in general. Furthermore, our business strategy is to target large unmet medical needs, and those markets are even more highly competitive. For example, there are four branded drugs and one generic drug currently marketed to treat Alzheimer’s disease. These drugs are all dosed once or twice per day, while dimebon dosing is three times daily in all of our completed and ongoing Alzheimer’s disease and Huntington disease clinical trials. This difference in dosing regimen may make dimebon less competitive than alternative drugs if dimebon receives marketing approval based on a thrice per day dosing regimen. In addition, one of the four branded Alzheimer’s disease drugs has already lost patent protection, and the other three branded drugs are expected to lose patent protection between 2010 and 2015. Such loss of patent protection typically results in the entry of generic competition for, and significant reductions in the commercial pricing of, those approved drugs. A generic version of one Alzheimer’s disease drug received FDA marketing approval in 2008. This development would put significant competitive pressure on the prices we or our potential partners could charge for dimebon should it ever be approved. Companies marketing currently approved Alzheimer’s disease therapeutics include some of the world’s largest and most experienced pharmaceutical companies, such as Novartis AG and Johnson & Johnson. Pfizer also already markets a currently approved Alzheimer’s disease drug. In addition, in October 2009 Pfizer merged with Wyeth, Inc., which is co-developing an Alzheimer’s disease product candidate that, like dimebon, is currently in Phase 3 development. There are also dozens of additional small molecule and recombinant protein candidates in development targeting the clinical indications we are pursuing, particularly Alzheimer’s disease and CRPC, including compounds already in Phase 3 clinical trials. One or more such compounds may be approved in each of our target indications before any of our product candidates could potentially be approved, including an investigational chemotherapy drug from sanofi-aventis which demonstrated an overall survival benefit in a Phase 3 CRPC trial reported in March 2010, a prostate cancer vaccine from Dendreon which demonstrated an overall survival benefit in CRPC patients and has an application for approval pending with the FDA, and an investigational hormonal drug from Johnson & Johnson which, like MDV3100, operates through the androgen receptor pathway and is ahead of MDV3100 in clinical development. Most, if not all, of these competing drug development programs are being conducted by pharmaceutical and biotechnology companies with considerably greater financial resources, human resources and experience than ours. Any of our product candidates that receives regulatory approval will face significant competition from both approved drugs and from any of the drugs currently under development that may subsequently be approved. Bases upon which our product candidates would have to compete successfully include efficacy, safety, price and cost-effectiveness. Even if dimebon were ever to receive marketing approval, the negative data from the CONNECTION trial could be included in the product label, which could make dimebon less attractive to physicians and patients than other products that are currently, or that in the future may be, approved for Alzheimer’s disease, which could limit potential sales of dimebon. In addition, our product candidates would have to compete against these other drugs with several different categories of decision makers, including physicians, patients, government and private third- party payors, technology assessment groups and patient advocacy organizations. Even if one of our product candidates is approved, we cannot guarantee that we, Pfizer, Astellas or any of our potential future partners will be able to compete successfully on any of these bases. Any future product candidates that we may subsequently acquire will face similar competitive pressures. If we or our partners cannot compete successfully on any of the bases described above, our business will not succeed.
17
Table of Contents
Research and Development Expenses
A significant portion of our operating expenses is related to research and development, and we intend to maintain our strong commitment to research and development. For the years ended December 31, 2009, 2008 and 2007, we recorded $87.7 million, $54.9 million and $23.4 million, respectively, in research and development expenses. More information regarding our research and development expenses can be found in the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” under Item 7 of this Annual Report on Form 10-K.
Manufacturing
Our business strategy is to use cGMP-compliant contract manufacturers for manufacture of clinical supplies as well as for commercial supplies if required by our commercialization plans, and to transfer manufacturing responsibility to our corporate partners when possible.
The dimebon tablets and matching placebos we used in the Russian Study were produced by a Russian company that is licensed by the Russian government to manufacture dimebon tablets for human use in Russia and that engaged in such manufacture for several years. The dimebon tablets and matching placebos used in the CONNECTION study, the Safety Study, the Phase 2 Huntington disease study, and all of our other completed and ongoing clinical trials in both Alzheimer’s disease and Huntington disease were manufactured by cGMP compliant contract manufacturers in the U.S. and Western Europe or by our partner Pfizer. Pursuant to our Collaboration Agreement, Pfizer has assumed substantially all manufacturing responsibility for dimebon, including clinical and commercial manufacturing capacity. Should Pfizer elect to terminate our collaboration agreement, we would have the right to require Pfizer to continue to supply us with dimebon for a reasonable period of time not to exceed eighteen months following the date Pfizer terminates the collaboration, on terms to be negotiated in good faith.
The MDV3100 being used in our ongoing Phase 1-2 clinical trial in CRPC and in the AFFIRM trial was manufactured by cGMP-compliant contract manufacturers. Pursuant to our Collaboration Agreement, Astellas has agreed to assume manufacturing responsibility for MDV3100, including clinical and commercial manufacturing capacity, after we complete transfer of those responsibilities to Astellas. We have not yet manufactured MDV3100 at commercial scale. Based on currently available information, we believe that MDV3100 drug product can be manufactured at commercial scale on a cost-effective basis. However, we caution you that this is a forward-looking statement and that we cannot guarantee that we will be able to complete this work on a timely basis or at all.
Scientific and Clinical Advisory Board
We maintain a Scientific and Clinical Advisory Board comprised of scientists and physicians with experience relevant to our company and our product candidates. Members of our Scientific and Clinical Advisory Board have agreed to consult and advise us in their respective areas of expertise. We have placed special emphasis on identifying members of our Scientific and Clinical Advisory Board with expertise in the treatment of the clinical indications targeted by our programs. Our Scientific and Clinical Advisory Board consists of the following members:
Paul Aisen, M.D. Dr. Aisen is a Professor in the Department of Neurosciences at the University of California, San Diego, and the National Institute on Aging-appointed director of the Alzheimer’s Disease Cooperative Study. Dr. Aisen was one of the first Alzheimer’s disease clinical trialists in the United States, and was an investigator in the pivotal FDA registration studies for Namenda. Dr. Aisen received his M.D. from Columbia University, College of Physicians and Surgeons.
Sergey Bachurin, Ph.D., D.Sc., Prof. Dr. Bachurin is the Director of the Institute of Physiologically Active Compounds in Chernogolovka, Russia, and a member of the Russian Academy of Sciences. Dr. Bachurin has served as a visiting scholar at several U.S. academic research centers, including the University of California, San
18
Table of Contents
Francisco, Tufts University and St. Elizabeth’s Medical Center. Dr. Bachurin holds a Ph.D. in Chemical Catalysis and a D.Sc. in Biochemistry from Moscow State University. In addition, Dr. Bachurin holds a Professor degree in Bioorganic Chemistry from the Institute of Physiologically Active Compounds.
Rachelle Doody, M.D., Ph.D. Dr. Doody is a Professor of Neurology, the Effie Marie Cain Chair in Alzheimer’s Disease Research, and the Director of the Alzheimer’s Disease and Memory Disorder Center at Baylor College of Medicine. Dr. Doody participated in the development of CIBIC-plus, one of the primary cognitive assessment endpoints that the FDA has used for the currently approved Alzheimer’s disease drugs. Dr. Doody has worked on clinical studies for all of the FDA-approved cholinesterase inhibitors for Alzheimer’s disease. Dr. Doody received her M.D. from Baylor College of Medicine and holds a M.A. and Ph.D. in Cognitive Anthropology from Rice University.
Benjamin Lewin, Ph.D. Dr. Lewin is the founding editor of Cell, a leading international journal in the field of biology and, until 1999, also served as the Chief Executive Officer of Cell Press, the publisher of Cell. Dr. Lewin holds a M.Sc. from the University of London, and a M.A. and a Ph.D. from the University of Cambridge. Dr. Lewin also has authored multiple books and scientific publications in the field of genetics.
Marc A. Shuman, M.D. Dr. Shuman is Professor of Medicine and Urology at the University of California, San Francisco, and Co-Leader of the Prostate Cancer Program at the UCSF Helen Diller Family Comprehensive Cancer Center. Dr. Shuman also serves as Clinical Director of the California Institute for Quantitative Biosciences. Dr. Shuman is a member of the American Society of Clinical Investigation and the Association of American Physicians, and has published extensively in the field of prostate cancer. Dr. Shuman received his M.D. from the Thomas Jefferson Medical College and clinical training at the University of Pennsylvania and Washington University in St. Louis.
Roger Tung, Ph.D. Dr. Tung is the president and chief executive officer of Concert Pharmaceuticals. He has more than twenty years of experience in scientific and scientific management positions at the Squibb Institute for Medical Research, Merck Research Laboratories and Vertex Pharmaceuticals Incorporated. Prior to joining Concert, he served as vice president, drug discovery, of Vertex Pharmaceuticals Incorporated. He discovered two of Vertex’s currently marketed products, and is an inventor on multiple issued U.S. patents. Dr. Tung holds a Ph.D. in Pharmaceutical Chemistry from the University of Wisconsin-Madison.
Pablo Valenzuela, Ph.D. Dr. Valenzuela is a Professor of Biochemistry at the Universidad Andres Bello in Santiago, Chile. Dr. Valenzuela has founded several companies, including Chiron Corporation, a major biotechnology company that was acquired by Novartis AG, and Chilean biotechnology companies Bios-Chile Ingenieria Genetica SA, Austral Biologicals and Algisa SA. He also founded, and serves as the Director of, Fundacion Ciencia para la Vida, a private, non-profit foundation promoting the adoption and use of science-based innovation by Chilean companies. Dr. Valenzuela holds a Ph.D. in Biochemistry from Northwestern University.
Employees
As of December 31, 2009, we had 98 employees.
Available Information
Our website address is www.medivation.com; however, information found on, or that can be accessed through, our website is not incorporated by reference into this Annual Report on Form 10-K. We file or furnish electronically with the SEC our Annual Report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934. We make available free of charge on or through our website copies of these reports as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. The SEC maintains an internet site that contains reports, proxy and information statements and other information regarding
19
Table of Contents
our filings at www.sec.gov. You may also read and copy any of our materials filed with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. Information regarding the operation of the Public Reference Room can be obtained by calling the SEC at 1-800-SEC-0330.
| Item 1A. | Risk Factors. |
Our business faces significant risks, some of which are set forth below to enable readers to assess, and be appropriately apprised of, many of the risks and uncertainties applicable to the forward-looking statements made in this Annual Report on Form 10-K. You should carefully consider these risk factors as each of these risks could adversely affect our business, operating results and financial condition. If any of the events or circumstances described in the following risks actually occurs, our business may suffer, the trading price of our common stock could decline and our financial condition or results of operations could be harmed. Given these risks and uncertainties, you are cautioned not to place undue reliance on forward-looking statements. These risks should be read in conjunction with the other information set forth in this Annual Report on Form 10-K. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us, or that we currently believe to be immaterial, may also adversely affect our business.
Risks Related to Our Business
We have incurred net losses since inception, expect to incur additional losses in the future as we expand our development activities and may never achieve sustained revenues or profitability. Our only revenue to date has been collaboration revenue under our collaboration agreements with Pfizer and with Astellas. We have not completed development of any of our product candidates and do not expect that any of our present or future product candidates will be commercially available for a number of years, if at all. We have incurred losses since inception and expect to continue to incur substantial additional losses for the foreseeable future as we continue to finance clinical and preclinical studies of our existing and potential future product candidates and our corporate overhead costs. Our operating losses have had, and will continue to have, an adverse impact on our working capital, total assets and stockholders’ equity. We do not know when or if we will ever generate any additional revenue, including any milestone payments, profit sharing payments or royalty payments under our collaboration agreements with Pfizer and Astellas, or become profitable because of the significant uncertainties with respect to our ability to generate product revenue from, and obtain approval from the FDA for, any of our current or future product candidates.
Because we depend on financing from third parties for our operations, our business may fail if such financing becomes unavailable or is offered on commercially unreasonable terms. To date, we have financed our operations primarily through the sale of our debt and equity securities, and from up-front license payments and cost-sharing payments received pursuant to our collaborative arrangements with third parties. As of December 31, 2009 we had cash, cash equivalents and short-term investments of $278.2 million available to fund operations. Based upon our current expectations, we believe our capital resources at December 31, 2009 will be sufficient to fund our currently planned operations beyond the next twelve months. This estimate is based on a number of assumptions that may prove to be wrong and we could exhaust our available cash reserves. Our future capital requirements will depend on many factors, including without limitation:
| • | whether Pfizer terminates our collaboration agreement, which it obtained the right to do at any time as a result of the negative results in the CONNECTION study; |
| • | whether any changes are made to the scope of our ongoing clinical development of dimebon as a result of the negative results in the CONNECTION study or other factors; |
| • | the scope and results of our and our corporate partners’ preclinical and clinical trials; |
| • | whether we experience delays in our preclinical and clinical development programs, including potential delays in or inability to recruit patients into our ongoing dimebon trials as a result of the negative results in the CONNECTION study, or slower than anticipated product development; |
20
Table of Contents
| • | whether opportunities to acquire additional product candidates arise and the costs of acquiring and developing those product candidates; |
| • | whether we are able to enter into additional third-party collaborative partnerships to develop and/or commercialize any of our product candidates on terms that are acceptable to us; |
| • | the timing and requirements of, and the costs involved in, conducting studies required to obtain regulatory approvals for our product candidates from the FDA and comparable foreign regulatory agencies; |
| • | the availability of third parties to perform the key development tasks for our product candidates, including conducting preclinical and clinical studies and manufacturing our product candidates to be tested in those studies and the associated costs of those services; |
| • | expenses associated with the pending and potential additional related purported securities class action lawsuits, as well as any unforeseen litigation; and |
| • | the costs involved in preparing, filing, prosecuting, maintaining, defending the validity of and enforcing, patent claims and other costs related to patent rights and other intellectual property rights, including litigation costs and the results of such litigation. |
We may not be able to obtain additional financing when we need it on acceptable terms or at all. If we cannot raise funds on acceptable terms, we may not be able to continue developing our product candidates, acquire or develop additional product candidates or respond to competitive pressures or unanticipated requirements. For these reasons, any inability to raise additional capital when we require it would seriously harm our business.
Our business strategy depends on our ability to identify and acquire additional product candidates which we may never acquire or identify for reasons that may not be in our control, or are otherwise unforeseen or unforeseeable to us. A key component of our business strategy is to diversify our product development risk by identifying and acquiring new product opportunities for development. However, we may not be able to identify promising new technologies. In addition, the competition to acquire promising biomedical technologies is fierce, and many of our competitors are large, multinational pharmaceutical, biotechnology and medical device companies with considerably more financial, development and commercialization resources and experience than we have. Thus, even if we succeed in identifying promising technologies, we may not be able to acquire rights to them on acceptable terms or at all. If we are unable to identify and acquire new technologies, we will be unable to diversify our product risk. We believe that any such failure would have a significant negative impact on our prospects because the risk of failure of any particular development program in the pharmaceutical field, including our ongoing dimebon and MDV3100 development programs, is high.
Because we depend on our management to oversee the execution of development plans for our existing product candidates and to identify and acquire promising new product candidates, the loss of any of our executive officers would harm our business. Our future success depends upon the continued services of our executive officers. We are particularly dependent on the continued services of David Hung, M.D., our president and chief executive officer and a member of our board of directors. Dr. Hung identified all of our existing product candidates for acquisition and has primary responsibility for identifying and evaluating other potential product candidates. We believe that Dr. Hung’s services in this capacity would be difficult to replace. None of our executive officers is bound by an employment agreement for any specific term, and they may terminate their employment at any time. In addition, we do not have “key person” life insurance policies covering any of our executive officers. The loss of the services of any of our executive officers could delay the development of our existing product candidates and delay or preclude the identification and acquisition of new product candidates, either of which events could harm our business.
Our reliance on third parties for the operation of our business may result in material delays, cost overruns and/or quality deficiencies in our development programs. We rely on outside vendors to perform key product development tasks, such as conducting preclinical and clinical studies and manufacturing our product candidates at appropriate scale for preclinical and clinical trials and, in situations where we are unable to transfer those
21
Table of Contents
responsibilities to a corporate partner, for commercial use as well. In order to manage our business successfully, we will need to identify, engage and properly manage qualified external vendors that will perform these development activities. For example, we need to monitor the activities of our vendors closely to ensure that they are performing their tasks correctly, on time, on budget and in compliance with strictly enforced regulatory standards. Our ability to identify and retain key vendors with the requisite knowledge is critical to our business and the failure to do so could negatively impact our business. Because all of our key vendors perform services for other clients in addition to us, we also need to ensure that they are appropriately prioritizing our projects. If we fail to manage our key vendors well, we could incur material delays, cost overruns or quality deficiencies in our development programs, as well as other material disruptions to our business.
Risks Related to Our Product Development Candidates
We are currently reassessing our future approach to the clinical development of our product candidate dimebon, which is currently in Phase 3 development for both Alzheimer’s and Huntington diseases. Because dimebon failed to meet any of its primary and secondary efficacy endpoints in the CONNECTION study of dimebon for mild-to-moderate Alzheimer’s disease, we and Pfizer have begun a comprehensive analysis of the data from the CONNECTION study to determine how it may impact our ongoing dimebon development program. In addition, based on the negative results of the CONNECTION study, Pfizer obtained the unilateral right to terminate the dimebon Collaboration Agreement between our companies. If we and Pfizer, or either of us individually, determine that future clinical development of dimebon should be curtailed or abandoned, our potential future milestone payments and potential future revenues from the potential commercialization of dimebon would be reduced or eliminated.
Given the negative results in the CONNECTION trial, our existing dimebon clinical development plan, even if completed successfully, may be inadequate to obtain approval in mild-to-moderate Alzheimer’s disease. In February 2008, the FDA expressed to us its view that our Russian Study could be used as one of the two pivotal registration studies required to support the approval of dimebon to treat mild-to-moderate Alzheimer’s disease, so long as a significant proportion of the sites in the CONNECTION trial, which was designed to confirm the results of the Russian Study, were located in the United States. At the time of expressing that view, the FDA had not yet performed an in-depth review of the then-existing dimebon data, including the data from the Russian Study, so its views remained subject to change. While approximately 40% of the patients enrolled in the CONNECTION trial were from the United States, the trial failed in its objective of replicating the results of the Russian Study. Thus, even if we successfully complete other dimebon Alzheimer’s disease trials, the FDA may not accept the Russian Study as pivotal. Furthermore, even if the FDA does accept the Russian Study as pivotal, our dimebon development program includes only one other Phase 3 study in mild-to-moderate Alzheimer’s disease—the CONCERT study, which is evaluating dimebon in combination with Aricept, compared to Aricept alone, over a one-year treatment period. The FDA may conclude that those two studies together are inadequate to support the approval of dimebon to treat mild-to-moderate Alzheimer’s disease. Comparable regulatory agencies in other countries may reach similar conclusions. Were this to occur, we may never be able to obtain approval of dimebon to treat mild-to-moderate Alzheimer’s disease, which constitutes the largest segment of the total Alzheimer’s disease market.
Our product candidates require extensive, time-consuming and expensive preclinical and clinical testing to establish safety and efficacy. We may never attract additional partners for our technologies or receive marketing approval in any jurisdiction. The research and development of pharmaceuticals is an extremely risky industry. Only a small percentage of product candidates that enter the development process ever receive marketing approval. Except for dimebon’s approval in Russia as an antihistamine, which is not a commercially attractive opportunity for us, none of our product candidates is currently approved for sale anywhere in the world, and none of them may ever receive such approval. The process of conducting the preclinical and clinical testing required to establish safety and efficacy and obtain marketing approval is expensive and uncertain and takes many years. If we are unable to complete preclinical or clinical trials of any of our current or future product candidates, or if the results of these trials are not satisfactory to convince regulatory authorities or partners of their safety or efficacy,
22
Table of Contents
we will not be able to obtain marketing approval or attract additional partners for those product candidates. Furthermore, even if we or our partners are able to obtain marketing approvals for any of our product candidates, those approvals may be for indications that are not as broad as desired or may contain other limitations that would adversely affect our ability to generate revenue from sales of those products. If this occurs, our business will be materially harmed and our ability to generate revenue will be severely impaired.
Positive results in the Russian Study were not predictive of results in the CONNECTION study, which was designed as a confirmatory study, and positive results seen in any of our other clinical trials, including our Phase 2 clinical trial of dimebon in Huntington disease and our Phase 1-2 clinical trial of MDV3100 in CRPC, may not be predictive of future clinical trial results. The CONNECTION study was designed expressly to replicate the positive results seen in the Russian Study but failed to do so. As evidenced by this example, even where we achieve positive results in clinical trials, subsequent clinical trials may fail, even if those subsequent trials are designed very similarly to their predecessors. Accordingly, despite the positive results seen in our Phase 2 clinical trial of dimebon in Huntington disease, our ongoing Phase 3 HORIZON trial of dimebon in Huntington disease may fail, and despite the positive results seen to date in our Phase 1-2 trial of MDV3100 in CRPC, our ongoing Phase 3 AFFIRM trial of MDV3100 in CRPC may fail. In addition, despite the positive results seen in our Russian Study, any or all of our ongoing Phase 3 clinical trials in Alzheimer’s disease may fail. Product candidates in clinical trials, including Phase 3 clinical trials, often fail to show the desired safety and efficacy traits despite having progressed successfully through prior stages of preclinical and clinical testing. In addition, we do not know whether interim results from any of our ongoing clinical trials, including the results to date in our Phase 1-2 clinical trial of MDV3100 in CRPC, will be predictive of final results of any such trial.
We are dependent upon our collaborative relationships with Pfizer and Astellas to further develop, manufacture and commercialize dimebon and MDV3100, respectively. There may be circumstances that delay or prevent Pfizer’s or Astellas’ ability to develop, manufacture and commercialize dimebon or MDV3100, respectively, or give rise to Pfizer’s or Astellas’ ability to terminate our agreements with each of them. In September 2008, we announced that we had entered into a collaboration agreement with Pfizer for the development, manufacture and commercialization of dimebon to treat Alzheimer’s disease and Huntington disease. Under the agreement, Pfizer is responsible for development and seeking regulatory approval for, and commercialization of, dimebon outside the United States and is responsible globally for all manufacture of product for both clinical and commercial purposes. In the United States, we and Pfizer are responsible for jointly developing and commercializing dimebon, and we will share the costs, profits and losses on a 60%/40% basis, with Pfizer assuming the larger share. Under the terms of the agreement, Pfizer has the unilateral right to terminate the agreement at any time based on the negative results of the CONNECTION trial. In October 2009, we announced that we had entered into a collaboration agreement with Astellas for the development, manufacture and commercialization of MDV3100 to treat prostate cancer. Under the agreement, Astellas is responsible for developing, seeking regulatory approval for, and commercializing MDV3100 outside the United States and, following a transition period, is responsible globally for all manufacture of product for both clinical and commercial purposes. We and Astellas are jointly responsible for developing, seeking regulatory approval for, and commercializing MDV3100 in the United States. We and Astellas will share equally the costs, profits and losses arising from development and commercialization of MDV3100 in the United States. For clinical trials useful both in the United States and in Europe or Japan, including the ongoing Phase 3 AFFIRM trial, we will be responsible for one-third of the total costs and Astellas will be responsible for the remaining two-thirds.
We are subject to a number of risks associated with our dependence on our collaborative relationships with Pfizer and Astellas, including:
| • | the rights of Pfizer or Astellas to terminate the respective collaboration agreement with us on limited notice for convenience (subject to certain limitations in the case of Astellas), or for other reasons specified in the respective collaboration agreements; |
| • | the need for us to identify and secure on commercially reasonable terms the services of third parties to perform key activities currently performed by Pfizer and Astellas in the event that either or both of our |
23
Table of Contents
| partners were to terminate their collaborations with us, including clinical and commercial manufacturing, development activities outside of the United States and commercialization activities globally; |
| • | adverse decisions by Pfizer or Astellas regarding the amount and timing of resource expenditures for the development and commercialization of dimebon or MDV3100, respectively; |
| • | possible disagreements as to the timing, nature and extent of our development plans, including clinical trials or regulatory approval strategy; |
| • | changes in key management personnel that are members of each collaboration’s various committees; and |
| • | possible disagreements with Pfizer or Astellas, including those regarding the development and/or commercialization of products, interpretation of the collaboration agreement and ownership of proprietary rights. |
Due to these factors and other possible disagreements with Pfizer or Astellas, we may be delayed or prevented from further developing, manufacturing or commercializing dimebon or MDV3100, respectively, or we may become involved in litigation or arbitration, which would be time consuming and expensive.
If Pfizer or Astellas were to unilaterally terminate our collaborative relationship, we would need to undertake development, manufacturing and marketing activities for dimebon or MDV3100, respectively, solely at our own expense and/or seek another partner for some or all of these activities, worldwide. If we pursued these activities on our own, it would significantly increase our capital and infrastructure requirements, might limit the indications we are able to pursue and could prevent us from effectively developing and commercializing dimebon and MDV3100. If we sought to find another pharmaceutical company partner for some or all of these activities, we may not be successful in such efforts, or they may result in a collaboration that has us expending greater funds and efforts than our current relationships with Pfizer and Astellas.
We are dependent on the efforts of, and funding by, Pfizer and Astellas for the development of dimebon and MDV3100, respectively. Under the terms of both the Pfizer collaboration agreement and the Astellas collaboration agreement, we and each of Pfizer and Astellas must agree on any changes to the development plan for dimebon or MDV3100, respectively, that is set forth in each agreement. If we and Pfizer or we and Astellas cannot agree on any such changes, clinical trial progress could be significantly delayed or halted. Pfizer is free to terminate our collaboration agreement at its discretion on limited notice to us. If Pfizer terminates its co-funding of dimebon development, we may be unable to fund the development and commercialization costs on our own and may be unable to find a new collaborator, which could cause our business to fail. If Pfizer does not exercise its right to terminate our collaboration agreement and desires to change the development plan for dimebon, we may be asked to agree to a revised development plan that does not prioritize the development of dimebon for Alzheimer’s disease or Huntington disease in order to continue receiving funding from Pfizer for the development and commercialization of dimebon. Subject to certain limitations set forth in the Astellas Collaboration Agreement, Astellas is generally free to terminate the Astellas agreement at its discretion on limited notice to us. If Astellas terminates its co-funding of MDV3100 commercialization, we may be unable to fund the development and commercialization costs on our own and may be unable to find another partner, which could cause our business to fail. Similarly, in the event of an uncured material breach of the Astellas agreement by us, Astellas may elect to terminate the agreement, in which case all rights to develop and commercialize MDV3100 will revert to us. We may be unable to fund the continued development and commercialization of MDV3100 on our own and may be unable to find another partner, which could cause our business to fail. In the event of an uncured material breach of the Pfizer agreement by us, Pfizer may elect either to terminate the agreement or to keep the agreement in place, but terminate our right to participate in development, commercialization (other than co-promoting dimebon, which right Pfizer may terminate only if our uncured material breach pertains to our exercise of that right) and other activities for dimebon, including the joint committees and decision making for dimebon. If Pfizer terminates our right to participate in such activities, we
24
Table of Contents
would be entirely dependent on Pfizer’s actions with respect to the development and commercialization of dimebon. In addition, under the Pfizer agreement, Pfizer is solely responsible for the development and regulatory approval of dimebon outside the United States, so we are entirely dependent on Pfizer for the successful completion of those activities. Similarly, under the Astellas agreement, Astellas is solely responsible for the development and regulatory approval of MDV3100 outside the United States, so we are entirely dependent on Astellas for the successful completion of those activities.
The financial returns to us, if any, under our collaboration agreements with Pfizer and Astellas depend in large part on the achievement of development and commercialization milestones, plus a share of any profits from any product sales in the United States and royalties on any product sales outside of the United States. Therefore, our success, and any associated financial returns to us and our investors, will depend in large part on the performance of Pfizer and Astellas under each respective agreement. If Pfizer or Astellas fails to perform or satisfy its obligations to us, the development, regulatory approval or commercialization of dimebon or MDV3100, respectively, would be delayed or may not occur and our business and prospects could be materially and adversely affected for that reason.
We are dependent on the efforts of Pfizer and Astellas to market and promote dimebon and MDV3100, respectively. Under our collaboration with Pfizer, we and Pfizer have the right to co-promote dimebon to specialty physicians in the United States and Pfizer has the sole right to promote dimebon to primary care physicians in the United States. Outside the United States, Pfizer has the sole right to promote dimebon. We are thus solely dependent on Pfizer to successfully promote dimebon to primary care physicians in the United States and to all customers outside of the United States and are partially dependent on Pfizer to successfully promote dimebon to specialty physicians in the United States. Under our collaboration with Astellas, we and Astellas have the right to co-promote MDV3100 to all customers in the United States, and Astellas has the sole right to promote MDV3100 to all customers outside of the United States. We are thus partially dependent on Astellas to successfully promote MDV3100 in the United States and solely dependent on Astellas to successfully promote MDV3100 to all customers outside of the United States. We have limited ability to direct Pfizer or Astellas in its commercialization of dimebon or MDV3100, respectively, in any country, including the United States. If Pfizer or Astellas fail to adequately market and promote dimebon or MDV3100, respectively, whether inside or outside of the United States, we may be unable to obtain any remedy against Pfizer or Astellas. If this were to happen, sales of dimebon or MDV3100, respectively, may be harmed, which would negatively impact our business, results of operations, cash flows and liquidity.
We are dependent on Pfizer and Astellas to manufacture clinical and commercial requirements of dimebon and MDV3100, respectively, which could result in the delay of clinical trials or regulatory approval or lost sales. Under both of our agreements with each of Pfizer and Astellas, after a transition period, Pfizer and Astellas have the primary right and responsibility to supply dimebon and MDV3100, respectively, for clinical trials and all commercial requirements. We transitioned substantially all of the manufacturing obligations for dimebon to Pfizer in 2009, and are in the process of transitioning the manufacturing obligations for MDV3100 to Astellas. Consequently, we are, and expect to remain, dependent on Pfizer and Astellas to manufacture dimebon and MDV3100, respectively. In the event that Pfizer terminates the dimebon collaboration, at our request it will supply us with dimebon for clinical and commercial use for a reasonable period of time not to exceed eighteen months following its notice of termination, on terms to be negotiated by the parties in good faith. Pfizer or Astellas may encounter difficulties in production scale-up, including problems involving production yields, quality control and quality assurance, and shortage of qualified personnel. Pfizer or Astellas may not perform as agreed or may default in their obligations to supply clinical trial supplies and/or commercial product. Pfizer or Astellas may fail to deliver the required quantities of our products or product candidates on a timely basis. Any such failure by Pfizer or Astellas could delay our future clinical trials and our applications for regulatory approval, or could impair our ability to meet the market demand for dimebon or MDV3100, respectively, and therefore result in decreased sales. If Pfizer or Astellas does not adequately perform, we may be forced to incur additional expenses, delays, or both, to arrange for other third parties to manufacture products on our behalf, as we do not have any internal manufacturing capabilities.
25
Table of Contents
If Pfizer’s or Astellas’ business strategies change, any such changes may adversely affect our collaborative relationships with each party. Either Pfizer or Astellas may change its business strategy. Decisions by either Pfizer or Astellas to either reduce or eliminate its participation in the Alzheimer’s field or prostate cancer field, respectively, to emphasize other competitive agents currently in its portfolio at the expense of dimebon or MDV3100, respectively, or to add additional competitive agents to its portfolio, could reduce its financial incentives to continue to develop, seek regulatory approval for, or commercialize dimebon or MDV3100, respectively. For example, in October 2009 Pfizer completed its acquisition of Wyeth, which is co-developing an Alzheimer’s disease product candidate that, like dimebon, is currently in Phase 3 development. A change in Pfizer’s business strategy as a result of the Wyeth acquisition or for other reasons, including the negative results of the CONNECTION study, may adversely affect activities under our collaboration agreement with Pfizer, which could cause significant delays and funding shortfalls impacting the activities under the collaboration and seriously harming our business. In addition, Astellas has partnered with us based in part on Astellas’ desire to use MDV3100 as a component of building a global oncology franchise, which Astellas presently does not have. If Astellas’ strategic objective of building a global oncology franchise were to change, such change could negatively impact any commercial prospects of MDV3100.
Our industry is highly regulated by the FDA, and comparable foreign regulatory agencies. We must comply with extensive, strictly enforced regulatory requirements in order to develop and obtain marketing approval for any of our product candidates. Before we, Pfizer, Astellas or our potential future partners can obtain regulatory approval for the sale of our product candidates, our product candidates must be subjected to extensive preclinical and clinical testing to demonstrate their safety and efficacy for humans. The preclinical and clinical trials of any product candidates that we develop must comply with regulation by numerous federal, state and local government authorities in the United States, principally the FDA, and by similar agencies in other countries. We will be required to obtain and maintain an effective investigational new drug application to commence human clinical trials in the United States and must obtain and maintain additional regulatory approvals before proceeding to successive phases of our clinical trials. Securing FDA approval requires the submission of extensive preclinical and clinical data and supporting information for each therapeutic indication to establish the product candidate’s safety and efficacy for its intended use. It takes years to complete the testing of a new drug or medical device and development delays and/or failure can occur at any stage of testing. Any of our present and future clinical trials may be delayed or halted due to any of the following:
| • | any preclinical test or clinical trial may fail to produce safety and efficacy results satisfactory to the FDA or foreign regulatory authorities; |
| • | preclinical and clinical data can be interpreted in different ways, which could delay, limit or prevent regulatory approval; |
| • | negative or inconclusive results from a preclinical test or clinical trial, such as the negative results from the CONNECTION trial reported in March 2010, or adverse medical events during a clinical trial could cause a preclinical study or clinical trial to be repeated or a program to be terminated, even if other studies or trials relating to the program are ongoing or have been completed and were successful; |
| • | the FDA or foreign regulatory authorities can place a clinical hold on a trial if, among other reasons, it finds that patients enrolled in the trial are or would be exposed to an unreasonable and significant risk of illness or injury; |
| • | the FDA might not approve the clinical processes or facilities that we utilize, or the processes or facilities of our consultants, including without limitation the vendors who will be manufacturing drug substance and drug product for us or any potential collaborators; |
| • | any regulatory approval we, Pfizer, Astellas or any potential future collaborators ultimately obtain may be limited or subject to restrictions or post-approval commitments that render the product not commercially viable; and |
| • | we may encounter delays or rejections based on changes in FDA policies or the policies of foreign regulatory authorities during the period in which we develop a product candidate or the period required for review of any final regulatory approval before we are able to market our product candidates. |
26
Table of Contents
Enrollment and retention of patients in clinical trials is often an expensive and time-consuming process, could be made more difficult or rendered impossible by multiple factors outside our control, including the negative results from the CONNECTION trial reported in March 2010 and the potential availability of competing drugs for the treatment of CRPC, and could result in significant delays, cost overruns, or both, in our product development activities, or could result in the failure of such activities. We may encounter delays in enrolling a sufficient number of patients to complete our clinical trials, and the negative results of the CONNECTION study may make it more difficult for us to enroll patients in other dimebon clinical trials. Patient enrollment depends on many factors, including the size of the patient population, the nature of the trial protocol, the existing body of safety and efficacy data with respect to the study drug, the number and nature of ongoing clinical trials of competing drugs for the same indication, the proximity of patients to clinical sites and the eligibility criteria for the study. For example, to be successful in our ongoing Phase 3 AFFIRM study of MDV3100 in castration-resistant prostate cancer, MDV3100 must demonstrate a statistically significant difference in survival between MDV3100-treated and placebo-treated patients. If one or more other treatments for castration-resistant prostate cancer becomes available, either commercially or on a compassionate use basis, before completion of the AFFIRM trial, patients could elect to leave our trial and switch to an alternative treatment. The survival of these patients continues to be included in the analysis of the AFFIRM trial despite the fact that they have dropped out. Thus, use of other treatments by these patients could make it more difficult for the AFFIRM trial to succeed if such treatments prolong survival. Furthermore, because patients in the AFFIRM trial have a one-third chance of being randomized to placebo, the availability of competing treatments may make it more difficult, or impossible, to complete enrollment in our trial. Such potential competing treatments include an investigational chemotherapy drug from sanofi-aventis which demonstrated an overall survival benefit in a Phase 3 CRPC trial reported in March 2010 a prostate cancer vaccine from Dendreon which demonstrated an overall survival benefit in CRPC patients and has an application for approval pending with the FDA, and an investigational hormonal drug from Johnson & Johnson which, like MDV3100, operates through the androgen receptor pathway and is ahead of MDV3100 in clinical development. Delays or failures in planned patient enrollment and/or retention may result in increased costs, program delays or both, which could have a harmful effect on our ability to develop dimebon, MDV3100 or any other product candidates, or could render further development impossible.
If our product candidates cannot be manufactured in a cost-effective manner and in compliance with current good manufacturing practices and other applicable regulatory standards, they will not be commercially successful. All pharmaceutical and medical device products in the United States, Europe and other countries must be manufactured in strict compliance with current good manufacturing practices, or cGMP, and other applicable regulatory standards. Establishing a cGMP-compliant process to manufacture pharmaceutical and medical device products involves significant time, cost and uncertainty. Furthermore, in order to be commercially viable, any such process would have to yield product on a cost-effective basis, using raw materials that are commercially available on acceptable terms. We face the risk that our contract manufacturers may have interruptions in raw material supplies, be unable to comply with strictly enforced regulatory requirements, or for other reasons beyond their or our control, be unable to complete their manufacturing responsibilities on time, on budget, or at all. Under our collaboration agreements with Pfizer and Astellas, Pfizer and Astellas are responsible for all manufacture of dimebon and MDV3100, respectively, for commercial purposes, but we cannot guarantee that either Pfizer or Astellas will be able to manufacture dimebon or MDV3100, respectively, in a timely manner or at all. Furthermore, neither dimebon nor MDV3100 has been manufactured at commercial-scale under cGMP- compliant conditions. In the event that Pfizer elects to terminate our dimebon collaboration agreement, following a period of transition assistance from Pfizer, we will be responsible for manufacturing dimebon or finding a different third party to manufacture dimebon and neither we nor any other third party have experience manufacturing dimebon at commercial scale under cGMP-compliant conditions. We thus cannot guarantee that commercial-scale cGMP manufacture of dimebon and/or MDV3100 will be possible, on a cost-effective basis or at all, which would materially and adversely affect the value of these programs.
Any of our product development candidates that receive marketing approval will face significant competition from other approved products, including generic products and products with more convenient
27
Table of Contents
dosing regimens, and other products in development. The biopharmaceutical industry is intensely competitive in general. Furthermore, our business strategy is to target large unmet medical needs, and those markets are even more highly competitive. For example, there are four branded drugs and one generic drug currently marketed to treat Alzheimer’s disease. These drugs are all dosed once or twice per day, while dimebon dosing is three times daily in all of our completed and ongoing Alzheimer’s disease and Huntington disease clinical trials. This difference in dosing regimen may make dimebon less competitive than alternative drugs if dimebon receives marketing approval based on a thrice per day dosing regimen. In addition, one of the four branded Alzheimer’s disease drugs has already lost patent protection, and the other three branded drugs are expected to lose patent protection between 2010 and 2015. Such loss of patent protection typically results in the entry of generic competition for, and significant reductions in the commercial pricing of, those approved drugs. A generic version of one Alzheimer’s disease drug received FDA marketing approval in 2008. This development would put significant competitive pressure on the prices we or our potential partners could charge for dimebon should it ever be approved. Companies marketing currently approved Alzheimer’s disease therapeutics include some of the world’s largest and most experienced pharmaceutical companies, such as Novartis AG and Johnson & Johnson. Pfizer also already markets a currently approved Alzheimer’s disease drug. In addition, in October 2009 Pfizer merged with Wyeth, which is co-developing an Alzheimer’s disease product candidate that, like dimebon, is currently in Phase 3 development. There are also dozens of additional small molecule and recombinant protein candidates in development targeting the clinical indications we are pursuing, particularly Alzheimer’s disease and CRPC, including compounds already in Phase 3 clinical trials. One or more such compounds may be approved in each of our target indications before any of our product candidates could potentially be approved, including an investigational chemotherapy drug from sanofi-aventis which demonstrated an overall survival benefit in a Phase 3 CRPC trial reported in March 2010, a prostate cancer vaccine from Dendreon which demonstrated an overall survival benefit in CRPC patients and has an application for approval pending with the FDA, and an investigational hormonal drug from Johnson & Johnson which, like MDV3100, operates through the androgen receptor pathway and is ahead of MDV3100 in clinical development. Most, if not all, of these competing drug development programs are being conducted by pharmaceutical and biotechnology companies with considerably greater financial resources, human resources and experience than ours. Any of our product candidates that receives regulatory approval will face significant competition from both approved drugs and from any of the drugs currently under development that may subsequently be approved. Bases upon which our product candidates would have to compete successfully include efficacy, safety, price and cost-effectiveness. Even if dimebon were ever to receive marketing approval, the negative results from the CONNECTION trial could be included in the product label, which could negatively affect dimebon’s ability to compete. In addition, our product candidates would have to compete against these other drugs with several different categories of decision makers, including physicians, patients, government and private third-party payors, technology assessment groups and patient advocacy organizations. Even if one of our product candidates is approved, we cannot guarantee that we, Pfizer, Astellas or any of our potential future partners will be able to compete successfully on any of these bases. Any future product candidates that we may subsequently acquire will face similar competitive pressures. If we or our partners cannot compete successfully on any of the bases described above, our business will not succeed.
Any of our product candidates that is eventually approved for sale may not be commercially successful if not widely-covered and appropriately reimbursed by third-party payors. Third-party payors, including public insurers such as Medicare and Medicaid and private insurers, pay for a large share of health care products and services consumed in the United States. In Europe, Canada and other major international markets, third-party payors also pay for a significant portion of health care products and services and many of those countries have nationalized health care systems in which the government pays for all such products and services and must approve product pricing. Even if approved by the FDA and foreign regulatory agencies, our product candidates are unlikely to achieve commercial success unless they are covered widely by third-party payors and reimbursed at a rate which generates an acceptable commercial return for us and any collaborative partner. It is increasingly difficult to obtain coverage and acceptable reimbursement levels from third-party payors and we may be unable to achieve these objectives. Achieving coverage and acceptable reimbursement levels typically involves negotiating with individual payors and is a time-consuming and costly process. In addition, we would face competition in such negotiations from other approved drugs against which we compete, which may include other
28
Table of Contents
approved drugs marketed by Pfizer or Astellas, and the marketers of such other drugs are likely to be significantly larger than us and therefore enjoy significantly more negotiating leverage with respect to the individual payors than we may have. The competition for coverage and reimbursement level with individual payors will be particularly intense for dimebon, if approved to treat Alzheimer’s disease, because all four currently marketed Alzheimer’s disease drugs are expected to lose patent protection prior to, or shortly following, dimebon’s potential commercial launch, and thus are likely to be available at generic price levels that are more attractive to individual payors. One such drug already has lost patent protection and a generic version is on the market. Our commercial prospects would be further weakened if payors approved coverage for our product candidates only as second- or later-line treatments, or if they placed any of our product candidates in tiers requiring unacceptably high patient co-payments. Failure to achieve acceptable coverage and reimbursement levels could materially harm our or our partner’s ability to successfully market our product candidates.
We may be subject to product liability or other litigation, which could result in an inefficient allocation of our critical resources, delay the implementation of our business strategy and, if successful, materially and adversely harm our business and financial condition as a result of the costs of liabilities that may be imposed thereby. Our business exposes us to the risk of product liability claims that is inherent in the development of pharmaceutical products. If any of our product candidates harms people, or is alleged to be harmful, we may be subject to costly and damaging product liability claims brought against us by clinical trial participants, consumers, health care providers, corporate partners or others. We have product liability insurance covering our ongoing clinical trials, but do not have insurance for any of our other development activities. If we are unable to obtain insurance at an acceptable cost or otherwise protect against potential product liability claims, we may be exposed to significant litigation costs and liabilities, which may materially and adversely affect our business and financial position. If we are sued for injuries allegedly caused by any of our product candidates, our litigation costs and liability could exceed our total assets and our ability to pay. In addition, we may from time to time become involved in various lawsuits and legal proceedings which arise in the ordinary course of our business. Any litigation to which we are subject, including the purported securities class action lawsuit described in the section entitled “Legal Proceedings” under Item 3 of this Annual Report on Form 10-K, could require significant involvement of our senior management and may divert management’s attention from our business and operations. Litigation costs or an adverse result in any litigation that may arise from time to time may adversely impact our operating results or financial condition.
Risks Related to Intellectual Property
Intellectual property protection for our product candidates is crucial to our business, and is subject to a significant degree of legal risk—particularly in the life sciences industry. The success of our business will depend in part on our ability to obtain and maintain intellectual property protection—primarily patent protection—of our technologies and product candidates, as well as successfully defending these patents against third-party challenges. We and our collaborators will only be able to protect our technologies and product candidates from unauthorized use by third parties to the extent that valid and enforceable patents or trade secrets cover them. Furthermore, the degree of future protection of our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us or our potential future collaborators to gain or keep our competitive advantage.
The patent positions of life sciences companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. Further, changes in either the patent laws or in interpretations of patent laws in the United States or other countries may diminish the value of our intellectual property rights. Accordingly, we cannot predict the breadth of claims that may be granted or enforced for our patents or for third-party patents that we have licensed. For example:
| • | we or our licensors might not have been the first to make the inventions covered by each of our pending patent applications and issued patents; |
| • | we or our licensors might not have been the first to file patent applications for these inventions; |
29
Table of Contents
| • | others may independently develop similar or alternative technologies or duplicate any of our technologies; |
| • | it is possible that none of our pending patent applications or the pending patent applications of our licensors will result in issued patents; |
| • | our issued patents and future issued patents, or those of our licensors, may not provide a basis for protecting commercially viable products, may not provide us with any competitive advantages, or may be challenged by third parties and invalidated or rendered unenforceable; and |
| • | we may not develop additional proprietary technologies or product candidates that are patentable. |
Our existing and any future patent rights may not adequately protect any of our product candidates, which could prevent us from ever generating any revenues or profits. We cannot guarantee that any of our pending or future patent applications will mature into issued patents, or that any of our current or future issued patents will adequately protect our product candidates from competitors. For example, there is a large body of prior art, including multiple issued patents and published patent applications, disclosing molecules in the same chemical class as our MDV300 series compounds. Since our MDV300 series compounds include approximately 170 specific molecules, we expect that some members of this series may not be patentable in light of this prior art, or may infringe the claims of patents presently issued or issued in the future. Furthermore, we also cannot guarantee that any of our present or future issued patents will not be challenged by third parties, or that they will withstand any such challenge. If we are not able to obtain adequate protection for, or defend, the intellectual property position of our technologies and product candidates, then we may not be able to attract collaborators to acquire or partner our development programs. Further, even if we can obtain protection for and defend the intellectual property position of our technologies and product candidates, we or any of our potential future collaborators still may not be able to exclude competitors from developing or marketing competing drugs. Should this occur, we and our potential future collaborators may not generate any revenues or profits from our product candidates or our revenue or profits would be significantly decreased.
We could become subject to litigation or other challenges regarding intellectual property rights, which could divert management attention, cause us to incur significant costs, prevent us from selling or using the challenged technology and/or subject us to competition by lower priced generic products. In recent years, there has been significant litigation in the United States and elsewhere involving pharmaceutical patents and other intellectual property rights. In particular, generic pharmaceutical manufacturers have been very aggressive in challenging the validity of patents held by proprietary pharmaceutical companies, especially if these patents are commercially significant. If any of our present or future product candidates succeed, we may face similar challenges to our existing or future patents. For example, in the prosecution of our issued United States patents claiming the use of dimebon and certain related compounds to treat neurodegenerative diseases, including Alzheimer’s disease, the prior owners missed a filing deadline with the U.S. Patent & Trademark Office, or PTO, which resulted in the patent application being deemed abandoned. The prior owners petitioned the PTO to revive the patent application alleging that missing the deadline was unintentional and the PTO approved the petition and issued the patent. However, as with any other decision the PTO makes, this decision could be challenged in subsequent litigation in an attempt to invalidate this issued United States patent and any other United States patent that may issue based on the same patent application. If a generic pharmaceutical company or other third party were able to successfully invalidate any of our present or future patents, any of our product candidates that may ultimately receive marketing approval could face additional competition from lower priced generic products that would result in significant price and revenue erosion and have a significantly negative impact on the commercial viability of the affected product candidate(s).
In the future, we may be a party to litigation to protect our intellectual property or to defend our activities in response to alleged infringement of a third party’s intellectual property. These claims and any resulting lawsuit, if successful, could subject us to significant liability for damages and invalidation, or a narrowing of the scope, of our proprietary rights. These lawsuits, regardless of their success, would likely be time-consuming and expensive to litigate and resolve and would divert management time and attention. Any potential intellectual property litigation also could force us to do one or more of the following:
| • | discontinue our products that use or are covered by the challenged intellectual property; or |
30
Table of Contents
| • | obtain from the owner of the infringed intellectual property right a license to sell or use the relevant technology, which license may not be available on reasonable terms, or at all. |
If we are forced to take any of these actions, our business may be seriously harmed. Although we carry general liability insurance, our insurance does not cover potential claims of this type.
In addition, our patents and patent applications, or those of our licensors, could face other challenges, such as interference proceedings, opposition proceedings and re-examination proceedings. Any such challenge, if successful, could result in the invalidation of, or in a narrowing of the scope of, any of our patents and patent applications subject to the challenge. Any such challenges, regardless of their success, would likely be time-consuming and expensive to defend and resolve and would divert our management’s time and attention.
We may in the future initiate claims or litigation against third parties for infringement in order to protect our proprietary rights or to determine the scope and validity of our proprietary rights or the proprietary rights of competitors. These claims could result in costly litigation and the diversion of our technical and management personnel and we may not prevail in making these claims.
We may need to obtain licenses of third-party technology that may not be available to us or are available only on commercially unreasonable terms, and which may cause us to operate our business in a more costly or otherwise adverse manner that was not anticipated. From time to time we may be required to license technology from third parties to develop our existing and future product candidates. For example, in our industry there are a large number of issued patents and published patent applications with claims to treating diseases generically through use of any product that produces one or more biological activities—such as inhibiting a specific biological target. We are aware of several such issued patents relating to Alzheimer’s disease and expect to continue to encounter such patents relating to other diseases targeted by our present and future product candidates. We have not conducted experiments to analyze whether, and we have no evidence that, any of our product candidates produce the specific biological activities covered in any of the issued patents or published patent applications of which we are presently aware. We have not sought to acquire licenses to any such patents. In addition, the commercial scale manufacturing processes that we are developing for our product candidates may require licenses to third-party technology. Should we be required to obtain licenses to any third-party technology, including any such patents based on biological activities or required to manufacture our product candidates, such licenses may not be available to us on commercially reasonable terms, or at all. The inability to obtain any third-party license required to develop any of our product candidates could cause us to abandon any related development efforts, which could seriously harm our business and operations.
We may become involved in disputes with Pfizer, Astellas or potential future collaborators over intellectual property ownership, and publications by our research collaborators and scientific advisors could impair our ability to obtain patent protection or protect our proprietary information, which, in either case, could have a significant impact on our business. Inventions discovered under research, material transfer or other such collaborative agreements, including our collaboration agreements with Pfizer and Astellas, may become jointly owned by us and the other party to such agreements in some cases and the exclusive property of either party in other cases. Under some circumstances, it may be difficult to determine who owns a particular invention, or whether it is jointly owned, and disputes could arise regarding ownership of those inventions. These disputes could be costly and time consuming and an unfavorable outcome could have a significant adverse effect on our business if we were not able to protect or license rights to these inventions. In addition, our research collaborators and scientific advisors generally have contractual rights to publish our data and other proprietary information, subject to our prior review. Publications by our research collaborators and scientific advisors containing such information, either with our permission or in contravention of the terms of their agreements with us, may impair our ability to obtain patent protection or protect our proprietary information, which could significantly harm our business.
Trade secrets may not provide adequate protection for our business and technology. We also rely on trade secrets to protect our technology, especially where we believe patent protection is not appropriate or obtainable. However, trade secrets are difficult to protect. While we use reasonable efforts to protect our trade secrets, our or
31
Table of Contents
our potential collaborators’ employees, consultants, contractors or scientific and other advisors may unintentionally or willfully disclose our information to competitors. If we were to enforce a claim that a third party had illegally obtained and was using our trade secrets, our enforcement efforts would be expensive and time consuming, and the outcome would be unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets. Moreover, if our competitors independently develop equivalent knowledge, methods or know-how, it will be more difficult or impossible for us to enforce our rights and our business could be harmed.
Risks Related to Ownership of Our Common Stock
We have been named a defendant in a purported securities class action lawsuit. This, and potential similar or related litigation, could result in substantial damages and may direct management’s time and attention from our business and operations. On March 9, 2010 a purported securities class action lawsuit was commenced in the United States District Court for the Northern District of California, naming as defendants us and certain of our officers. The lawsuit alleges violations of the Securities Exchange Act of 1934 in connection with allegedly false and misleading statements made by us related to dimebon. The plaintiff alleges, among other things, that the defendants disseminated false and misleading statements about the effectiveness of dimebon for the treatment of Alzheimer’s disease making it impossible for stockholders to gain a realistic understanding of the drug’s progress toward FDA approval. The plaintiff seeks to represent a class of stockholders who purchased our common stock between July 17, 2008 and March 2, 2010. The plaintiff seeks damages, an award of its costs and injunctive and/or equitable relief. It is possible that additional suits will be filed, or allegations received from stockholders, with respect to these same matters and also naming us and/or our officers and directors as defendants.
Our management believes that we have meritorious defenses and intends to defend the lawsuit vigorously. This lawsuit and any other related lawsuits are subject to inherent uncertainties, and the actual cost will depend upon many unknown factors. The outcome of the litigation is necessarily uncertain, we could be forced to expend significant resources in the defense of these suits and we may not prevail. Monitoring and defending against legal actions is time-consuming for our management and detracts from our ability to fully focus our internal resources on our business activities. In addition, we may incur substantial legal fees and costs in connection with the litigation. We are not currently able to estimate the possible cost to us from this matter, as this lawsuit is currently at an early stage and we cannot be certain how long it may take to resolve this matter or the possible amount of any damages that we may be required to pay. We have not established any reserves for any potential liability relating to this lawsuit. It is possible that we could, in the future, incur judgments or enter into settlements of claims for monetary damages. A decision adverse to our interests on these actions could result in the payment of substantial damages, or possibly fines, and could have a material adverse effect on our cash flow, results of operations and financial position. In addition, the uncertainty of the currently pending litigation could lead to more volatility in our stock price.
Our stock price may be volatile, and our stockholders’ investment in our stock could decline in value. The market prices for our securities and those of other life sciences companies have been highly volatile and may continue to be highly volatile in the future. The following factors, in addition to other risk factors described in this Annual Report on Form 10-K, may have a significant impact on the market price of our common stock:
| • | the receipt or failure to receive the additional funding necessary to conduct our business; |
| • | the progress and success of preclinical studies and clinical trials of our product candidates conducted by us, Pfizer, Astellas or our future collaborative partners or licensees, if any; |
| • | selling by existing stockholders and short-sellers; |
| • | announcements of technological innovations or new commercial products by our competitors or us; |
| • | developments concerning proprietary rights, including patents; |
| • | developments concerning our collaboration with Pfizer, our collaboration with Astellas, or any future collaborations, including any potential decision by Pfizer to terminate our dimebon collaboration due to the negative results of the CONNECTION trial reported in March 2010; |
32
Table of Contents
| • | publicity regarding us, our product candidates or those of our competitors, including research reports published by securities analysts; |
| • | regulatory developments in the United States and foreign countries; |
| • | litigation, including the purported securities class action lawsuit commenced against us and certain of our officers and any other related lawsuits; |
| • | economic and other external factors or other disaster or crisis; and |
| • | period-to-period fluctuations in financial results. |
We do not intend to pay dividends on our common stock for the foreseeable future. We do not expect for the foreseeable future to pay dividends on our common stock. Any future determination to pay dividends on or repurchase shares of our common stock will be at the discretion of our board of directors and will depend upon, among other factors, our success in completing sales or partnerships of our programs, our results of operations, financial condition, capital requirements, contractual restrictions and applicable law.
Our principal stockholders exert substantial influence over us and may exercise their control in a manner adverse to your interests. Certain stockholders and their affiliates own a substantial amount of our outstanding common stock. These stockholders may have the power to direct our affairs and be able to determine the outcome of certain matters submitted to stockholders for approval. Because a limited number of persons controls us, transactions could be difficult or impossible to complete without the support of those persons. Subject to applicable law, it is possible that these persons will exercise control over us in a manner adverse to your interests.
Provisions of our charter documents, our stockholder rights plan and Delaware law could make it more difficult for a third party to acquire us, even if the offer may be considered beneficial by our stockholders. Provisions of the Delaware General Corporation Law could discourage potential acquisition proposals and could delay, deter or prevent a change in control. The anti-takeover provisions of the Delaware General Corporation Law impose various impediments to the ability of a third party to acquire control of us, even if a change in control would be beneficial to our existing stockholders. In addition, Section 203 of the Delaware General Corporation Law, unless its application has been waived, provides certain default anti-takeover protections in connection with transactions between the company and an “interested stockholder” of the company. Generally, Section 203 prohibits stockholders who, alone or together with their affiliates and associates, own more than 15% of the subject company from engaging in certain business combinations for a period of three years following the date that the stockholder became an interested stockholder of such subject company without approval of the board or the vote of two-thirds of the shares held by the independent stockholders. Our board of directors has also adopted a stockholder rights plan, or “poison pill,” which would significantly dilute the ownership of a hostile acquirer. Additionally, provisions of our amended and restated certificate of incorporation and bylaws could deter, delay or prevent a third party from acquiring us, even if doing so would benefit our stockholders, including without limitation, the authority of the board of directors to issue, without stockholder approval, preferred stock with such terms as the board of directors may determine.
| Item 1B. | Unresolved Staff Comments. |
None.
| Item 2. | Properties. |
For the conduct of our operations, we lease approximately 34,000 square feet of office space located at 201 Spear Street, San Francisco, California 94105 pursuant to leases that expire in July 2012 and May 2013. In November 2009 we signed a lease for approximately 64,000 square feet of office space located at 345 Spear Street, San Francisco, California 94105. Because of the negative CONNECTION trial results, we terminated the 345 Spear Street lease in March 2010, and paid a $1.5 million termination fee to the landlord. We also lease
33
Table of Contents
5,700 square feet of office space located at 55 Hawthorne Street, San Francisco, California 94105, our former office location. We have sub-leased the Hawthorne Street space to a third party through February 2011.
| Item 3. | Legal Proceedings. |
On March 9, 2010, a purported securities class action lawsuit was commenced in the United States District Court for the Northern District of California, naming as defendants us and certain of our officers. The lawsuit alleges violations of the Securities Exchanges Act of 1934 in connection with allegedly false and misleading statements made by us related to dimebon. The plaintiff alleges among other things that the defendants disseminated false and misleading statements about the effectiveness of dimebon for the treatment of Alzheimer’s disease making it impossible for stockholders to gain a realistic understanding of the drug’s progress toward FDA approval. The plaintiff seeks to represent a class of stockholders who purchased our common stock between July 17, 2008 and March 2, 2010. The plaintiff seeks damages, an award of its costs and injunctive and/or equitable relief. It is possible that additional suits will be filed, or allegations received from stockholders with respect to these same matters and also naming us and/or our officers and directors as defendants.
Our management believes that we have meritorious defenses and intends to defend the lawsuit vigorously. This lawsuit and any other related lawsuits are subject to inherent uncertainties, the actual cost may be significant, and we may not prevail.
| Item 4. | (Removed and Reserved). |
34
Table of Contents
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities. |
Market for Our Common Stock
Our common stock trades on The NASDAQ Global Market under the symbol “MDVN”. The following table sets forth on a per share basis the high and low intraday sales prices of our common stock as reported on The NASDAQ Global Market:
| High | Low | |||||
| 2009 |
||||||
| Quarter ended March 31, 2009 |
$ | 23.43 | $ | 13.36 | ||
| Quarter ended June 30, 2009 |
$ | 25.00 | $ | 17.12 | ||
| Quarter ended September 30, 2009 |
$ | 28.00 | $ | 21.18 | ||
| Quarter ended December 31, 2009 |
$ | 39.66 | $ | 24.82 | ||
| 2008 |
||||||
| Quarter ended March 31, 2008 |
$ | 18.85 | $ | 12.07 | ||
| Quarter ended June 30, 2008 |
$ | 17.47 | $ | 11.75 | ||
| Quarter ended September 30, 2008 |
$ | 34.40 | $ | 11.53 | ||
| Quarter ended December 31, 2008 |
$ | 27.34 | $ | 11.27 | ||
As of March 8, 2010, there were 29 stockholders of record of our common stock. On March 12, 2010, the last reported sales price per share of our common stock was $12.82 per share. We have never paid our stockholders cash dividends and we do not anticipate paying any cash dividends in the foreseeable future as we intend to retain any earnings for use in our business. Any future determination to pay dividends will be at the discretion of our board of directors.
35
Table of Contents
Performance Graph
The following graph shows the total stockholder return of an investment of $100 in cash on December 31, 2004 for: (i) the Company’s Common Stock; (ii) the Nasdaq Composite Index; and (iii) the Nasdaq Biotechnology Index. All values assume reinvestment of the full amount of all dividends and are calculated as of the last stock trading day of each year.
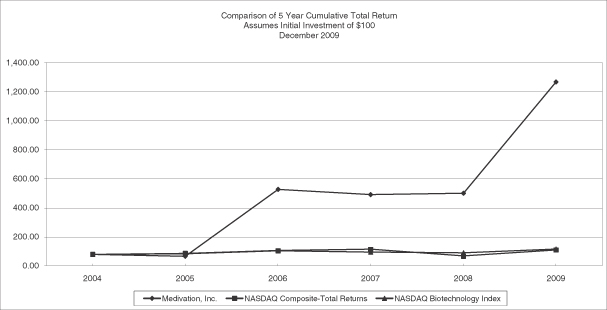
| December 31, 2004 |
December 30, 2005 |
December 29, 2006 |
December 31, 2007 |
December 31, 2008 |
December 31, 2009 | ||||||||
| Medivation, Inc. |
$ | 100 | 84.67 | 527.33 | 480.00 | 485.67 | 1255.00 | ||||||
| Nasdaq Composite Index |
$ | 100 | 102.12 | 112.73 | 124.74 | 74.87 | 108.83 | ||||||
| Nasdaq Biotechnology Index |
$ | 100 | 102.88 | 103.98 | 108.81 | 95.43 | 110.66 | ||||||
Source: Nasdaq.net. The information under “Performance Graph” is not deemed to be “soliciting material” or “filed” with the Securities and Exchange Commission or subject to Regulation 14A or 14C, or to the liabilities of Section 18 of the Securities Exchange Act of 1934, as amended, and is not to be incorporated by reference in any filing of Medivation under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date of this annual report on Form 10-K and irrespective of any general incorporation language in those filings.
Medivation, Inc. was formed in Delaware in October 1995, under our former name Orion Acquisition Corp. II, to identify and consummate a business combination. Medivation Neurology, Inc. was formed in Delaware in September 2003 to acquire and develop dimebon. On December 17, 2004, Medivation Neurology, Inc. became our subsidiary pursuant to a merger. Medivation Prostate Therapeutics, Inc. was formed in Delaware as our subsidiary to acquire and develop our MDV300 series technology.
36
Table of Contents
Recent Sales of Unregistered Securities, Issuer Purchases of Equity Securities
On October 26, 2009, we issued 1,263 shares of our common stock pursuant to the net exercise of a warrant held by one of our investors. The warrant was exercisable for 1,342 shares of common stock and had an exercise price of $1.55 per share. The number of shares issued upon the exercise of the warrant was reduced by 79 shares to effect the net exercise of the warrant in accordance with its terms.
On December 11, 2009, we issued 107,000 shares of our common stock pursuant to the exercise of a warrant held by one of our directors. The warrant, which was exercised for cash, had an exercise price of $1.55 per share resulting in aggregate consideration to us of $165,850.
For each of these transactions, we relied on the exemption provided by Section 4(2) of the Securities Act of 1933, as amended, and/or Regulation D promulgated thereunder as a transaction by an issuer not involving a public offering.
37
Table of Contents
| Item 6. | Selected Financial Data. |
The statement of operations data for the years ended December 31, 2009, 2008 and 2007, and the balance sheet data as of December 31, 2009 and 2008, are derived from our audited consolidated statements included in Item 8 of this Report. The statement of operations data for the years ended December 31, 2006 and 2005, and the balance sheet data as of December 31, 2007, 2006 and 2005, are derived from our financial statements not included in this Report. The information below is not necessarily indicative of results of future operations, and should be read in conjunction with Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” of this Annual Report on Form 10-K and the financial statements and related notes thereto, included in Item 8 of this Report to fully understand factors that may affect the comparability of the information presented below.
| Year Ended December 31, | ||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||||||
| Consolidated Statements of Operations Data |
||||||||||||||||||||
| Collaboration revenue |
$ | 69,254 | $ | 12,578 | $ | — | $ | — | $ | — | ||||||||||
| Operating expenses: |
||||||||||||||||||||
| Research and development |
87,728 | 54,895 | 23,399 | 11,825 | 5,272 | |||||||||||||||
| Selling, general and administrative |
28,983 | 21,865 | 10,364 | 4,321 | 3,143 | |||||||||||||||
| Total operating expenses |
116,711 | 76,760 | 33,763 | 16,146 | 8,415 | |||||||||||||||
| Loss from operations |
(47,457 | ) | (64,182 | ) | (33,763 | ) | (16,146 | ) | (8,415 | ) | ||||||||||
| Interest and other income and (expense), net |
976 | 1,712 | 2,022 | 785 | (1,401 | ) | ||||||||||||||
| Net loss before income tax expense |
(46,481 | ) | (62,470 | ) | (31,741 | ) | (15,361 | ) | (9,816 | ) | ||||||||||
| Income tax (benefit) expense |
8,272 | (10 | ) | 2 | 2 | 2 | ||||||||||||||
| Net loss |
(54,753 | ) | (62,460 | ) | (31,743 | ) | (15,363 | ) | (9,818 | ) | ||||||||||
| Basic and diluted net loss per common share |
$ | (1.71 | ) | $ | (2.12 | ) | $ | (1.14 | ) | $ | (0.63 | ) | $ | (0.70 | ) | |||||
| Shares used in computing basic and diluted net loss per share |
32,094 | 29,478 | 27,932 | 24,248 | 13,937 | |||||||||||||||
| December 31, | ||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Consolidated Balance Sheet Data: |
||||||||||||||||||||
| Cash and cash equivalents |
$ | 57,463 | $ | 71,454 | $ | 43,258 | $ | 4,649 | $ | 12,843 | ||||||||||
| Short-term investments |
220,781 | 149,968 | — | 42,534 | — | |||||||||||||||
| Working capital |
189,813 | 149,584 | 40,214 | 45,777 | 11,681 | |||||||||||||||
| Total assets |
296,690 | 229,272 | 45,596 | 47,612 | 13,281 | |||||||||||||||
| Long-term obligations, excluding current portion |
554 | 410 | 492 | — | — | |||||||||||||||
| Deferred revenue |
253,168 | 212,423 | — | — | — | |||||||||||||||
| Accumulated deficit |
(177,413 | ) | (122,660 | ) | (60,200 | ) | (28,457 | ) | (13,093 | ) | ||||||||||
| Total stockholders’ equity |
$ | 25,274 | $ | 3,408 | $ | 41,058 | $ | 45,873 | $ | 11,815 | ||||||||||
38
Table of Contents
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations. |
The following discussion should be read in conjunction with our audited consolidated financial statements and notes thereto for the fiscal year ended December 31, 2009, included elsewhere in this Report. The following Management’s Discussion and Analysis of Financial Condition and Results of Operations contains “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934. We intend that these forward-looking statements be subject to the safe harbors created by those provisions. Forward-looking statements are generally written in the future tense and/or are preceded by words such as “may,” “should,” “forecast,” “could,” “expect,” “suggest,” “believe,” “anticipate,” “intend,” “plan,” or other similar words. The forward-looking statements contained in this Report involve a number of risks and uncertainties, many of which are outside of our control. Factors that could cause actual results to differ materially from projected results include, but are not limited to, those discussed in “Risk Factors” elsewhere in this Report. Readers are expressly advised to review and consider those Risk Factors, which include risks associated with (1) the negative results we reported from our CONNECTION trial in March 2010 and the potential impact of those results on our ongoing clinical development of dimebon, including risks associated with Pfizer’s potential termination of our Collaboration Agreement, which Pfizer has the right to do at any time, (2) our ability to successfully conduct clinical and preclinical trials for our product candidates, (3) our ability to obtain required regulatory approvals to develop and market our product candidates, (4) our ability to raise additional capital on favorable terms, (5) our ability to execute our development plan on time and on budget, (6) our ability to obtain commercial partners, (7) our ability, whether alone or with commercial partners, to successfully commercialize any of our product candidates that may be approved for sale, and (8) our ability to identify and obtain additional product candidates. Although we believe that the assumptions underlying the forward-looking statements contained in this Report are reasonable, any of the assumptions could be inaccurate, and therefore there can be no assurance that such statements will be accurate. In light of the significant uncertainties inherent in the forward-looking statements included herein, the inclusion of such information should not be regarded as a representation by us or any other person that the results or conditions described in such statements or our objectives and plans will be achieved. Furthermore, past performance in operations and share price is not necessarily indicative of future performance. We disclaim any intention or obligation to update or revise any forward-looking statements, whether as a result of new information, future events or otherwise.
The Company
We are a biopharmaceutical company focused on the rapid development of novel small molecule drugs to treat serious diseases for which there are limited treatment options. Our product candidates in clinical development are dimebon (latrepirdine), which is in Phase 3 development for the treatment of Alzheimer’s disease and Huntington disease, and MDV3100, which is in Phase 3 development for the treatment of castration-resistant prostate cancer. Our dimebon program is partnered with Pfizer Inc, or Pfizer, and our MDV3100 program is partnered with Astellas Pharma Inc., or Astellas.
In September 2008, we announced a Collaboration Agreement with Pfizer, which became effective in October 2008. Under the terms of the agreement, we and Pfizer will develop and commercialize dimebon for the treatment of Alzheimer’s disease and Huntington disease. We and Pfizer will share the costs and expenses of developing and commercializing dimebon for the United States market on a 60% Pfizer/40% Medivation basis, and will share profits (or losses) resulting from commercialization of dimebon in the United States in the same proportions. Outside the United States, Pfizer will bear all development and commercialization costs, and will pay us tiered royalties on aggregate net sales of dimebon. In the fourth quarter of 2008, we received a non-refundable, up-front cash payment of $225.0 million pursuant to our Collaboration Agreement with Pfizer.
With Pfizer, we have been conducting a broad dimebon clinical development program, including five randomized, double-blind, placebo-controlled Phase 3 studies in Alzheimer’s and Huntington diseases. In March 2010, we reported negative results from the first of these studies—the CONNECTION trial, a six-month study of
39
Table of Contents
598 patients with mild-to-moderate Alzheimer’s disease in the United States, Western Europe, Russia and Chile in which patients were randomized to receive dimebon 20 mg three times daily, dimebon 5 mg three times daily or placebo. Patients in the CONNECTION trial were not permitted to take any approved Alzheimer’s disease medicines for a period of 90 days before, or at any time during, the study. In the CONNECTION study, dimebon treated patients did no better than placebo patients on any of the primary and secondary efficacy endpoints at either dose tested. Dimebon was well tolerated in the CONNECTION study, as well as in a separate randomized, double-blind, placebo-controlled safety study of dimebon, also reported in March 2010, in which 742 patients in the United States and Canada were randomized to receive dimebon 20 mg three times daily or placebo and treated for three to six months. Approximately 85% of patients in the safety study were taking one or more approved Alzheimer’s disease medicines while on the trial. Further analysis of the results from both of these studies is currently underway.
Given the negative results in the CONNECTION trial, we are currently re-evaluating our approach to the clinical development of dimebon for Alzheimer’s disease and Huntington disease. This will be a complex analysis that will encompass scientific, clinical, regulatory and business issues. We do not know at this time which, if any, of our ongoing Phase 3 dimebon trials will be completed, or if Pfizer will elect to terminate our collaboration. If Pfizer elects to terminate our collaboration, we may be unable to fund the development costs on our own and may be unable to find a new collaborator, which could cause our dimebon program to fail. A more comprehensive discussion of the risks and uncertainties related to the development of dimebon, including our dependence on Pfizer for the continued development of dimebon, can be found in the section entitled “Risk Factors—Risks Related to our Product Development Candidates” under Item 1A of this Annual Report on Form 10-K.
In October 2009, we entered into a Collaboration Agreement with Astellas. Under the terms of the agreement, we and Astellas will develop and commercialize MDV3100 for the treatment of both late- and early-stage prostate cancer. We and Astellas will share equally the costs and expenses of developing and commercializing MDV3100 for the United States market, except that development costs for studies useful in both the United States market and either Europe or Japan, such as the ongoing Phase 3 AFFIRM study, will be shared 2/3 by Astellas and 1/3 by us. We and Astellas will share equally profits (or losses) resulting from commercialization of MDV3100 in the United States. Outside the United States, Astellas will bear all development and commercialization costs, and will pay us tiered royalties on aggregate net sales of MDV3100. In the fourth quarter of 2009, we received a non-refundable, up-front cash payment of $110.0 million pursuant to our Collaboration Agreement with Astellas, ten percent of which we paid to the academic institution from which we licensed our rights to MDV3100.
With Astellas, we are currently conducting a randomized, double-blind, placebo-controlled Phase 3 trial, known as the AFFIRM trial, of MDV3100 in men with a type of advanced prostate cancer known as castration-resistant prostate cancer, or CRPC.
We have funded our operations primarily through private and public offerings of our common stock, and from the up-front payment and cost-sharing payments from our Collaboration Agreements with Pfizer and Astellas. As of December 31, 2009, we had an accumulated deficit of $177.4 million and we expect to incur substantial additional losses in the future as we continue research and development activities designed to support potential approval of our present and potential future product candidates.
Our Pipeline
Dimebon (latrepirdine)
Alzheimer’s disease. Our product candidate dimebon (latrepirdine) is in Phase 3 development for Alzheimer’s disease, in collaboration with Pfizer. In March 2010, we reported top-line results from our CONNECTION trial, a randomized, double-blind, six-month placebo-controlled Phase 3 trial in 598 patients
40
Table of Contents
with mild-to-moderate Alzheimer’s disease, and from a separate 742-patient randomized, double-blind, placebo-controlled three-to-six month safety study of dimebon in patients with mild-to-moderate Alhzeimer’s disease, or the Safety Study, approximately 85% of whom were also taking one or more approved Alzheimer’s disease medicines. In the CONNECTION trial, dimebon failed to show a statistically significant improvement over placebo on any of the primary or secondary efficacy endpoints, and thus did not meet any of the study’s efficacy endpoints. Dimebon was well tolerated in both the CONNECTION trial and in the Safety Study. We designed the CONNECTION trial to confirm the results of our first trial of dimebon in mild-to-moderate Alhzeimer’s disease, which was published in 2008 in the Lancet. In the first trial, in which we enrolled 183 patients in Russia with mild-to-moderate Alzheimer’s disease, dimebon showed a statistically significant improvement over placebo on all of the same primary and secondary efficacy endpoints used in the CONNECTION trial. Thus, the CONNECTION trial failed to replicate the efficacy results seen in our prior trial in Russia. We and Pfizer currently are enrolling patients in three ongoing randomized, double-blind, placebo-controlled Phase 3 clinical trials in Alzheimer’s disease patients, ranging in duration from six to twelve months.
Huntington disease. Dimebon is also in Phase 3 clinical development for Huntington disease, also in collaboration with Pfizer. In July 2008, we announced top-line results of a Phase 2 study showing that dimebon was well tolerated and significantly improved cognitive function in Huntington disease patients compared to those treated with a placebo. The study, which was conducted in the U.S. and the United Kingdom, met its primary endpoint of safety and tolerability; in addition, dimebon showed statistically significant benefit versus placebo in cognition as measured by the Mini-Mental State Examination, or MMSE, a secondary endpoint in the study. However, dimebon failed to show a statistically significant benefit over placebo in this study on two other cognitive endpoints—the cognitive component of the Unified Huntington Disease Rating Scale, or UHDRS, and the ADAS-cog. The study results were published in March 2010 in Archives of Neurology. In July 2009, we enrolled the first patient in a randomized, double-blind, six-month placebo-controlled Phase 3 study to treat Huntington disease, known as the HORIZON study.
Impact of the negative CONNECTION trial results. Given the negative results in the CONNECTION trial, we and Pfizer are currently re-evaluating our approach to the clinical development of dimebon for Alzheimer’s disease and Huntington disease. This will be a complex analysis that will encompass scientific, clinical, regulatory and business issues. We do not know at this time which, if any, of our ongoing Phase 3 dimebon trials will be completed, or if Pfizer will elect to terminate our collaboration. The negative results in the CONNECTION trial had a material adverse effect on our stock price and market capitalization and resulted in Pfizer obtaining the right to terminate our collaboration at any time. Under our collaboration agreement, Pfizer is responsible for 60% of dimebon development costs for the United States market, and 100% of dimebon development costs for ex-US markets. If Pfizer were to terminate our collaboration, these costs would become our responsibility, following a six-month transition period after the date of termination, during which Pfizer would continue to bear its share of development costs. The negative CONNECTION results thus may have materially adverse effects on our business and financial position, including on our ability to enroll, successfully complete and fund our ongoing dimebon clinical trials, especially if Pfizer elects to terminate our collaboration. A more comprehensive discussion of the risks and uncertainties related to the development of dimebon, including our dependence on Pfizer for the continued development of dimebon, can be found in the section entitled “Risk Factors—Risks Related to Our Product Development Candidates” under Item 1A of this Annual Report on Form 10-K.
MDV3100
Our product candidate MDV3100 is in Phase 3 development for castration-resistant prostate cancer, in collaboration with Astellas. In June 2009, we presented data from our ongoing Phase 1-2 clinical trial of MDV3100 at the American Society of Clinical Oncology Annual Meeting. In the 140 men enrolled in this trial, including both men who had failed prior chemotherapy and men who had were chemotherapy-naïve, MDV3100 consistently demonstrated anti-tumor activity across dose levels and endpoints, as evaluated by reductions in
41
Table of Contents
prostate-specific antigen, or PSA, levels, radiographic findings and circulating tumor cell, or CTC, counts. In this trial, MDV3100 was generally well tolerated at doses up to 240 mg per day. In September 2009, we enrolled the first patient in a randomized, double-blind, placebo-controlled Phase 3 study, known as the AFFIRM study, in patients with castration-resistant prostate cancer who have failed prior chemotherapy.
Critical Accounting Policies and the Use of Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the U.S., or U.S. GAAP. The preparation of our financial statements requires management to make estimates and assumptions that affect the amounts reported in our financial statements and accompanying notes. Actual results could differ materially from those estimates.
The items in our financial statements requiring significant estimates and judgments are as follows:
Revenue Recognition
Revenue recognized to date is attributable solely to the non-refundable up-front payments of $110.0 million and $225.0 million we received in the fourth quarter of 2009 and 2008, respectively, pursuant to our Collaboration Agreements with Astellas and Pfizer. For a description of the Collaboration Agreements, see Note 3 to our consolidated financial statements included elsewhere in this Report on Form 10-K.
We account for the various payment flows under our Collaboration Agreements in a consistent manner, as follows:
Upfront Payments
Both the Pfizer and Astellas Collaboration Agreements contain multiple elements and deliverables, and require evaluation pursuant to ASC 605-25 “Revenue Recognition—Multiple-Element Arrangements” (“ASC 605-25”). We evaluated the facts and circumstances of the Collaboration Agreements to determine whether we had obligations constituting deliverables under ASC 605-25. We concluded that we had multiple deliverables under both the Pfizer and Astellas Collaboration Agreements, including deliverables relating to grants of technology licenses, and performance of manufacturing, regulatory and clinical development activities in the U.S. In the case of the Pfizer Collaboration Agreement, our management estimated that the period in which we would perform our deliverables began in the fourth quarter of 2008 and will be completed in the first quarter of 2012. In the case of the Astellas Collaboration Agreement, our management estimated that the period in which we would perform our deliverables began in the fourth quarter of 2009 and will be completed in the fourth quarter of 2014. We also concluded that our deliverables under each Collaboration Agreement should be accounted for as a single unit of accounting under ASC 605-25. Accordingly, we will recognize the non-refundable, up-front payments from Pfizer and Astellas as revenue on a straight-line basis over the estimated performance period, using the proportional performance model.
Estimation of the performance period of our deliverables requires the use of our management’s judgment. Significant factors considered in management’s evaluation of the estimated performance period include, but are not limited to, our experience, along with Pfizer’s and Astellas’ experience, in conducting clinical development activities. Given the negative results in the CONNECTION study we reported in March 2010, we and Pfizer are currently re-evaluating which, if any, of our ongoing Phase 3 dimebon clinical trials should continue. This will be a complex analysis that will encompass scientific, clinical, regulatory and business issues. After this analysis is complete, we and Pfizer will be able to assess our respective future approaches to the clinical development of dimebon for Alzheimer’s and Huntington diseases. We do not know at this time which, if any, of our ongoing Phase 3 dimebon trials will be completed, or if Pfizer will elect to terminate our collaboration. If any changes to our ongoing dimebon development plan are made, or if Pfizer elects to terminate our collaboration, we may need
42
Table of Contents
to adjust our estimate of the performance period under our Pfizer collaboration. We will review the estimated duration of our performance periods under both collaborations on a quarterly basis and make any appropriate adjustments on a prospective basis. Future changes in estimates of either of our performance periods may materially impact the timing of future revenue recognized under the applicable Collaboration Agreement.
Milestone Payments
Under both the Pfizer and Astellas Collaboration Agreements, we are entitled to receive milestone payments based on achievement of specified development, regulatory and commercial events. Management evaluated the nature of the events triggering these contingent payments, and concluded that these events—except for (a) those relating to regulatory activities in Europe, development and regulatory activities in Japan, and commercial activities, all of which are areas in which we have no pertinent contractual responsibilities, and (b) the initiation of the second Phase 3 clinical trial under the Astellas Collaboration Agreement, an event which management deemed to be reasonably assured at the inception of the Astellas collaboration—constituted substantive milestones. This conclusion was based primarily on the facts that (i) each triggering event represents a specific outcome that can be achieved only through successful performance by us of one or more of our deliverables, (ii) achievement of each triggering event was subject to inherent risk and uncertainty and would result in additional payments becoming due to us, (iii) each of these milestones was substantive, based primarily on the facts that the payments they trigger are non-refundable, (iv) achievement of the milestone entails risk and was not reasonably assured at inception of the Collaboration Agreement, (v) substantial effort is required to complete each milestone, (vi) the amount of each milestone payment is reasonable in relation to the value created in achieving the milestone, (vii) a substantial amount of time is expected to pass between the up-front payment and the potential milestone payments, and (viii) the milestone payments relate solely to past performance. Based on the foregoing, we will recognize any revenue from these milestone payments under the substantive milestone method in the period in which the underlying triggering event occurs.
For the contingent payments triggered by events that do not constitute substantive milestones, management concluded that the appropriate revenue recognition treatment depends on whether the triggering event occurs during or after the performance period of the applicable Collaboration Agreement. Where the triggering event occurs during the applicable performance period, we will amortize any revenue from this event on a straight-line basis over the period beginning on the date the event occurs and ending at completion of the applicable performance period. Where the triggering event occurs after the applicable performance period, we will recognize the associated revenue in the period in which the event occurs.
Royalties and Profit Sharing Payments
Under both the Pfizer and Astellas Collaboration Agreements, we are entitled to receive profit sharing payments on sales of products in the U.S. and royalties on sales of products outside the U.S. We will recognize any revenue from these events based on the revenue recognition criteria set forth in ASC 605-10-25-1, “Revenue Recognition.” Based on those criteria, we consider these potential payments to be contingent revenues, and will recognize them as revenue in the period in which the applicable contingency is resolved.
Cost-Sharing True-Up Payments
Under both the Pfizer and Astellas Collaboration Agreements, we and our partners share certain development and commercialization costs in the U.S. The parties make quarterly true-up payments between themselves to ensure that each has borne its applicable percentage of the shared development and commercialization costs. Our policy is to account for cost-sharing true-up payments receivable by us as reductions in expense, and to account for cost-sharing true-up payments payable by us as increases in expense.
43
Table of Contents
Stock-based Compensation
We apply ASC 718, “Compensation—Stock Compensation” (ASC 718), which requires the measurement and recognition of non-cash compensation expense for all share-based payment awards made to employees and directors, including employee stock options and employee stock purchases related to our Restated 2004 Equity Incentive Award Plan, based on estimated fair values. We have applied the provisions of Staff Accounting Bulletin No. 107, or SAB 107, and Staff Accounting Bulletin No. 110, or SAB 110, in application of ASC 718.
Stock compensation arrangements with non-employee service providers are accounted for in accordance with ASC 505-50 Equity-Based Payments to Non-Employees using a fair value approach. The compensation costs of these arrangements are subject to remeasurement over the vesting terms as earned.
We recognized stock-based compensation expense of $10.7 million, $8.5 million and $5.9 million during the years ended December 31, 2009, 2008 and 2007, respectively, which increased our reported research and development expenses and selling, general and administrative expenses during those periods. These expenses break out as follows (in millions):
| Year Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Selling, general and administrative expense |
$ | 5.0 | $ | 4.3 | $ | 3.9 | |||
| Research and development expense |
$ | 5.7 | $ | 4.2 | $ | 2.0 | |||
We calculated these expenses based on the fair values of the stock-based compensation awards as estimated using the Black-Scholes model. Use of this model requires us to make assumptions about expected future volatility of our stock price and the expected term of the options that we grant. Calculating stock-based compensation expense under ASC 718 also requires us to make assumptions about expected future forfeiture rates for our option awards. As of December 31, 2009, total unrecognized compensation expense related to unvested share-based compensation arrangements previously granted was $42.3 million, which we expect to recognize over a weighted-average period of 2.8 years.
The Black-Scholes option valuation model requires the use of several subjective assumptions, including assumptions of expected stock price volatility, expected stock option term and forfeiture rates, and expected risk-free rates of return. If any of the assumptions used change significantly, stock-based compensation expense could differ materially in the future from that recorded in the current and past periods.
Research and Development Expenses and Accruals
Research and development expenses include personnel expenses, outside contracted services including clinical trial costs, manufacturing and process development costs, research costs and other consulting services. Research and development costs are expensed as incurred. In instances where we enter into agreements with third parties to provide research and development services to us, costs are expensed as services are performed. Amounts due under such arrangements may be either fixed fee or fee for service, and may include upfront payments, monthly payments, and payments upon the completion of milestones or receipt of deliverables.
Our cost accruals for clinical trials and other research and development activities are based on estimates of the services received and efforts expended pursuant to contracts with numerous clinical trial centers and contract research organizations. In the normal course of business we contract with third parties to perform various research and development activities in the on-going development of our product candidates, including without limitation, third party clinical trial centers and contract research organizations that perform and administer our clinical trials on our behalf. The financial terms of these agreements are subject to negotiation and vary from contract to contract and may result in uneven payment flows. Payments under these agreements depend on factors such as the achievement of certain events, the successful enrollment of patients, and the completion of
44
Table of Contents
portions of the clinical trial or similar conditions. The objective of our accrual policy is to match the recording of expenses in our financial statements to the actual services received and efforts expended. As such, expense accruals related to clinical trials and other research and development activities are recognized based on our estimate of the degree of completion of the event or events specified in the specific agreement.
Our estimates are dependent upon the timelines and accuracy of data provided by third parties regarding the status and cost of studies, and may not match the actual services performed by the organizations. This could result in adjustment to our research and development expense in future periods. To date, we have had no significant adjustments.
Operating Leases
On certain of the operating lease agreements, we may receive rent holidays and other incentives. We recognize operating lease costs on a straight-line basis without regard to deferred payment terms, such as rent holidays that defer the commencement date of required payments. In addition, lease incentives that we receive are treated as a reduction of rent expense over the term of the related agreements.
Accounting for Income Taxes
On January 1, 2007, we adopted ASC 740-10-25 (formerly FIN No. 48), Accounting for Uncertainty in Income Taxes, which clarifies the accounting for uncertainty in income taxes recognized in accordance with ASC 740-10 (formerly SFAS No. 109), Accounting for Income Taxes. Our policy is to recognize interest and/or penalties related to income tax matters in income tax expense. There were no accrued interest or penalties associated with uncertain tax positions as of December 31, 2009. We had $0.9 million of unrecognized tax benefits as of December 31, 2009 and we do not expect our unrecognized tax benefits to change significantly over the next twelve months.
We maintained a full valuation allowance on our net deferred tax assets as of December 31, 2009. The valuation allowance was determined in accordance with the ASC 740-10, Accounting for Income Taxes, which requires an assessment of both positive and negative evidence when determining whether it is more likely than not that deferred tax assets are recoverable; such assessment is required on a jurisdiction by jurisdiction basis. Cumulative historic losses represented sufficient negative evidence under ASC 740-10 and accordingly, a full valuation allowance was recorded against U.S. deferred tax assets. We intend to maintain a full valuation allowance on the U.S. deferred tax assets until sufficient positive evidence exists to support reversal of the valuation allowance.
Recent Accounting Pronouncements
Refer to Note 2(q), Recent Accounting Pronouncements to our consolidated financial statements included elsewhere in this Report on Form 10-K for a discussion of recent accounting pronouncements.
Results of Operations
Years Ended December 31, 2009, 2008 and 2007
| Year Ended December 31, | ||||||||||
| 2009 | 2008 | 2007 | ||||||||
| (In thousands) | ||||||||||
| Collaboration revenue |
$ | 69,254 | $ | 12,578 | $ | — | ||||
| Percentage increase |
451 | % | ||||||||
We recorded $69.3 million and $12.6 million of collaboration revenue in the year ended December 31, 2009 and 2008 respectively. The $56.7 million increase in collaboration revenue was driven by an additional $52.8 million of collaboration revenues related to our collaboration with Pfizer, which became effective in October 2008, and a $3.9 million increase of collaboration revenues related to our collaboration with Astellas, which became effective in October 2009.
45
Table of Contents
Under the Pfizer Collaboration Agreement, we received a non-refundable, up-front cash payment of $225.0 million in October 2008. This payment was recorded to deferred revenue, which we are amortizing on a straight-line basis over the expected performance period of our deliverables under the Collaboration Agreement. We currently expect this performance period to end in the first quarter of 2012. We recorded $65.4 million and $12.6 million of amortized collaboration revenue in the years ended December 31, 2009 and 2008, respectively. Further, we are eligible to receive payments of up to $500.0 million upon the attainment of certain development and regulatory milestones plus certain additional undisclosed commercial milestone payments. At present, we are unable to predict the timing or likelihood of such milestone payments. (See Note 3 to our consolidated financial statements included elsewhere in this Report on Form 10-K for further details on our collaboration with Pfizer).
The Astellas Collaboration Agreement became effective in October 2009 and we received a non-refundable, up-front cash payment of $110.0 million in November 2009, ten percent of which we paid to the academic institution from which we licensed our rights to MDV3100. This payment was recorded to deferred revenue, which we are amortizing on a straight-line basis over the expected performance period of our deliverables under the Collaboration Agreement. We currently expect this performance period to end in the last quarter of 2014. We recorded $3.9 million of amortized collaboration revenue in the year ended December 31, 2009. Further, we are eligible to receive up to $335.0 million in development milestone payments, plus up to an additional $320.0 million in commercial milestone payments. At present, we are unable to predict the timing or likelihood of such milestone payments. (See Note 3 to our consolidated financial statements included elsewhere in this Report on Form 10-K for further details on our collaboration with Astellas).
There were no revenues in 2007.
Research and Development Expense
| Year Ended December 31, | |||||||||||
| 2009 | 2008 | 2007 | |||||||||
| (In thousands) | |||||||||||
| Research and development expenses |
$ | 87,728 | $ | 54,895 | $ | 23,399 | |||||
| Percentage increase |
60 | % | 135 | % | |||||||
Research and development expenses increased by 60% or $32.8 million for the year ended December 31, 2009 as compared to the same period in 2008. The $32.8 million increase in research and development expenses was driven by a $29.1 million increase in clinical and preclinical study expenses, resulting from our initiation of six Phase 3 trials in 2009, an $11 million one-time payment to UCLA, representing 10% of our up-front payment from Astellas received in November 2009, a $7.2 million increase in payroll related expenses associated with increased headcount in research and development of 22 employees in 2009, a $5.5 million increase in occupancy and other operating costs principally in connection with our higher headcount and expanded research and development activities, partially offset by a $20.0 million increase in research and development cost true-up payments from Astellas and Pfizer.
Under the Pfizer Collaboration Agreement, specified costs incurred by Pfizer and us related to the development of dimebon for the United States market are shared by the parties on a 60% Pfizer, 40% Medivation basis. Costs related to the development of dimebon for ex-U.S. markets are borne 100% by Pfizer. The parties make quarterly true-up payments to ensure that each has borne its applicable percentage of the shared development costs incurred by both companies. We account for development cost true-up payments as additions to research and development expense when such payments are payable by us, and as reductions to research and development expense when such payments are payable to us. For the years ended December 31, 2009 and 2008, the net development cost true-up payments to us were $20.4 million and $3.2 million, respectively, from Pfizer.
Under the Astellas Collaboration Agreement, specified costs incurred by Astellas and us related to the development of MDV3100 for the United States market are shared equally by the parties, except for costs of clinical trials supporting development in both the United States and either Europe or Japan, including our ongoing Phase 3 AFFIRM trial, the costs of which are borne two-thirds by Astellas and one-third by us. Costs related to the development of MDV3100 for ex-U.S. markets are borne 100% by Astellas. The parties make quarterly true-up
46
Table of Contents
payments to ensure that each has borne its applicable percentage of the shared development costs incurred by both companies. We account for development cost true-up payments as additions to research and development expense when such payments are payable by us, and as reductions to research and development expense when such payments are payable to us. For the year ended December 31, 2009, total development cost true-up payments to us were $2.8 million, reflecting costs incurred following October 26, 2009, the effective date of the Collaboration Agreement. There were no such payments in prior period.
Research and development expenses increased by 135% or $31.5 million for the year ended December 31, 2008 as compared to the same period in 2007. The $31.5 million increase was driven by a $26.2 million increase in clinical and preclinical study expense primarily associated with significant expansions in our clinical development work for both dimebon and MDV3100, including initiation of the CONNECTION trial of dimebon in Alzheimer’s disease and continuation of our ongoing Phase 1-2 clinical trial of MDV3100 in castration-resistant prostate cancer, a $6.6 million increase in payroll related expenses associated with increased headcount in research and development of 17 employees in 2008, and a $1.8 million increase in occupancy and other operating costs principally in connection with our higher headcount and expanded research and development activities, partially offset by a $3.2 million increase in research and development cost true-up payments from Pfizer reflecting costs incurred following October 21, 2008, the effective date of the Pfizer Collaboration Agreement.
To date, we have been engaged in two major research and development programs: the development of dimebon for the treatment of Alzheimer’s disease and Huntington disease, and the development of MDV3100 for the treatment of castration-resistant prostate cancer. Other research and development programs consist of preclinical stage programs, primarily our dimebon analog program. Research and development costs are identified as either directly allocable to one of our research and development programs or as an indirect cost, with only direct costs being tracked by specific program. Direct costs consist primarily of clinical and preclinical study costs, cost of supplying drug substance and drug product for use in clinical and preclinical studies, contract research organization fees, and other contracted services pertaining to specific clinical and preclinical studies. Indirect costs consist of personnel costs (including both cash costs and non-cash stock-based compensation costs) corporate overhead costs, and other administrative and support costs. The following table summarizes the direct costs attributable to each program and the total indirect costs for each respective period.
| Years Ended December 31, | |||||||||||||||
| 12/31/2009 | 12/31/2008 | 12/31/2007 | 12/31/2006 | 12/31/2005 | |||||||||||
| (in thousands) | |||||||||||||||
| Direct Costs: |
|||||||||||||||
| Dimebon |
$ | 40,594 | $ | 27,910 | $ | 10,721 | $ | 5,186 | $ | 3,077 | |||||
| MDV 3100 |
12,054 | 8,845 | 2,619 | 3,021 | 261 | ||||||||||
| Other |
6,050 | 3,481 | 748 | 198 | — | ||||||||||
| Total direct costs |
58,698 | 40,236 | 14,088 | 8,405 | 3,338 | ||||||||||
| Indirect costs |
29,030 | 14,659 | 9,311 | 3,420 | 1,934 | ||||||||||
| Total research and development expenses |
$ | 87,728 | $ | 54,895 | $ | 23,399 | $ | 11,825 | $ | 5,272 | |||||
We did not track research and development expense by project prior to 2005.
Selling, General and Administrative Expense
| Year Ended December 31, | |||||||||||
| 2009 | 2008 | 2007 | |||||||||
| (In thousands) | |||||||||||
| Selling, general and administrative expense |
$ | 28,983 | $ | 21,865 | $ | 10,364 | |||||
| Percentage increase |
33 | % | 111 | % | |||||||
47
Table of Contents
Selling, general and administrative expenses consisted of primarily payroll and related costs associated with corporate administration and executive management, facilities, marketing and medical science personnel, professional services including legal and accounting services, medical education and other administrative costs.
The increase in selling, general and administrative expenses of 33% or $7.1 million in 2009 as compared to 2008 was due primarily to an increase of $4.9 million in payroll and related costs associated with increased selling, general and administrative headcount of 17 employees during 2009, a $1.0 million fee paid to our financial advisor in connection with our Collaboration Agreement with Astellas, and a $0.5 million increase in patent fees. These increases in selling, general and administrative costs were incurred primarily in support of our expanded research and development work, and our Collaboration Agreements with Pfizer and Astellas.
The increase in selling, general and administrative expenses of 111% or $11.5 million in 2008 as compared to 2007 was due primarily to an increase of $4.6 million in payroll and related costs associated with increased selling, general and administrative headcount of 14 employees during 2008, a $3.5 million fee paid to our financial advisor in connection with our Collaboration Agreement with Pfizer, and a $1.3 million increase in patent fees. These increases in selling, general and administrative costs were incurred primarily in support of our expanded research and development work, and our Collaboration Agreement with Pfizer.
Under our Collaboration Agreement with Pfizer, we share certain dimebon-related commercialization costs in the U.S., including pre-launch commercialization costs, on a 60% Pfizer, 40% Medivation basis. The parties make quarterly true-up payments between themselves to ensure that each has borne its applicable percentage of the shared commercialization costs. We account for commercialization cost true-up payments as additions to selling, general and administrative expense, when such payments are made by us, and as reductions to selling, general and administrative expense, when such payments are made to us. For the year ended December 31, 2009, total commercialization cost true-up payments to Pfizer were $0.7 million. For the year ended December 31, 2008, total commercialization cost true-up payments to us were $0.3 million, reflecting costs incurred following October 21, 2008, the effective date of the Pfizer Collaboration Agreement. There were no such collaboration payments in prior periods.
Under our Collaboration Agreement with Astellas, we share certain MDV 3100-related commercialization costs in the U.S., including pre-launch commercialization costs, on a 50%/50% basis. The parties make quarterly true-up payments between themselves to ensure that each has borne its applicable percentage of the shared commercialization costs. We account for commercialization cost true-up payments as additions to selling, general and administrative expense, when such payments are made by us, and as reductions to selling, general and administrative expense, when such payments are made to us. For the year ended December 31, 2009, total commercialization cost true-up payments to us were $0.1 million, reflecting costs incurred following October 26, 2009, the effective date of the Astellas Collaboration Agreement. There were no such collaboration payments in prior periods.
Due to the purported securities class action lawsuit recently filed against us, we expect that our legal expenses will increase in 2010 since we intend to vigorously defend the lawsuit.
Interest Income, net
| Year Ended December 31, | |||||||||||
| 2009 | 2008 | 2007 | |||||||||
| (In thousands) | |||||||||||
| Interest income, net |
$ | 1,128 | $ | 1,206 | $ | 2,022 | |||||
| Percentage change |
(6 | %) | (40 | %) | |||||||
The decrease in net interest income of 6% or $0.1 million in the year ended December 31, 2009 as compared to 2008 was primarily due to lower yields on cash and investment balances.
The decrease in net interest income of 40% or $0.8 million in the year ended December 31, 2008 as compared 2007 was primarily attributable to interest income on lower average cash balances and lower prevailing interest rates.
48
Table of Contents
Other Income (Expense), net
| Year Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| (In thousands) | |||||||||
| Other income (expense), net |
$ | (152) | $ | 506 | $ | — | |||
| Percentage change |
(130%) | ||||||||
The decrease in other income (expense), net of 130% or $0.7 million in 2009 as compared to 2008 was due to a payment of $0.6 million we received in 2008 for a securities law violation by one of our unaffiliated stockholders, one-time and realized losses on foreign exchange payables of $0.1 million in 2009.
Income Tax (Benefit) Expense
The following table presents our income tax expense (benefit), and effective tax rate for the periods presented:
| Year Ended December 31, | |||||||||||
| 2009 | 2008 | 2007 | |||||||||
| (In thousands) | |||||||||||
| Income tax (benefit) expense |
$ | 8,272 | $ | (10 | ) | $ | 2 | ||||
| Effective tax rate |
17.9 | % | — | — | |||||||
The income tax expense for 2009 was approximately $8.3 million, which mainly consisted of the federal and state income tax and represents an effective tax rate of 17.9%. The effective tax rate for 2009 has increased substantially from 0% in 2008 to 17.9% in 2009, was primarily attributed to the current California income tax payable due to the state’s suspension of net operating loss, or NOL, utilization. During the year ended December 31, 2009, we accelerated the recognition of revenue related to the Pfizer non-refundable, up-front payment received in 2008 for income tax purposes, while such revenue is deferred for financial statement purposes. Due to the suspension of California NOL utilization in 2009, we were not able to utilize the NOL carryforwards to offset the taxable income in 2009.
Of the $110.0 million up-front payment received from Astellas during the fourth quarter of 2009, we recognized $3.9 million for both financial statement and income tax purpose in the year ended December 31, 2009. The remaining balance of approximately $106.1 million will be recognized as revenue for income tax purposes in the year ended December 31, 2010 while for financial statement purposes, it will continue to be amortized on a straight-line basis over the remaining 60 months. Accordingly, we anticipate being profitable for income tax purposes for the year 2010 and may be subject to federal and state income taxes if there are not enough tax attributes (such as net operating loss and R&D credits) to offset the tax liability.
A reconciliation of the federal statutory income tax rate to our effective tax rate is set forth in Note 11 of our consolidated financial statements included elsewhere in this Annual Report on Form 10-K.
Liquidity and Capital Resources
Sources of Liquidity
We have incurred cumulative net losses of $177.4 million through December 31, 2009, and we expect to incur substantial additional losses in the future as we continue research and development activities designed to support potential approval of our present and potential future product candidates. We have not generated any revenue from product sales to date, and we and do not expect to generate product revenue for several years, if ever. In the year ended December 31, 2009, we generated $69.3 million in collaboration revenue from the Pfizer and Astellas Collaboration Agreements. All of our operations to date have been funded through the sale of our debt and equity securities, the $225.0 million up-front payment we received from Pfizer in October 2008, the
49
Table of Contents
$110.0 million up-front payment we received from Astellas in November 2009 and cost-sharing true-up payments from Pfizer and Astellas. As of December 31, 2009 we had cash, cash equivalents and short-term investments of $278.2 million available to fund operations. Based upon our current expectations, we believe our capital resources at December 31, 2009 will be sufficient to fund our currently planned operations beyond the next twelve months. This estimate is based on a number of assumptions that may prove to be wrong and we could exhaust our available cash reserves. Our future capital requirements will depend on many factors, including whether Pfizer terminates our collaboration agreement which it obtained the right to do any time as a result of the negative results in the CONNECTION study, as well as whether any changes are made to the scope of our ongoing clinical development of dimebon as a result of the negative results in the CONNECTION study.
Pfizer Collaboration Agreement
In September 2008, we announced a Collaboration Agreement with Pfizer. Due to the negative results in the CONNECTION study, Pfizer has the unilateral right to terminate our Collaboration Agreement at any time. We do not know whether or when Pfizer may choose to exercise this right. A more comprehensive discussion of the risks and uncertainties related to development of dimebon, including our dependence on Pfizer for the continued development of dimebon can be found in the section entitled “Risk Factors—Risks Related to Our Product Development Candidates” under Item 1A of this Annual Report on Form 10-K.
Under the Pfizer Collaboration Agreement, we and Pfizer will collaborate on development of dimebon for Alzheimer’s disease and Huntington disease for the United States market, including associated regulatory filings with the FDA. In addition, if approved by the FDA, following such approval and the launch of dimebon in the United States, we, at our option, and Pfizer have the right to co-promote dimebon to specialty physicians in the United States, and Pfizer has the sole right to promote dimebon to primary care physicians in the United States. Pfizer will be responsible for development and seeking regulatory approval for, and commercialization of, dimebon outside the United States. Following a period of transition from our contract manufacturers to Pfizer, Pfizer has assumed responsibility for all manufacture of product for both clinical and commercial purposes. Both we and Pfizer have agreed not to commercialize for the treatment of specified indications any other products directed to the same primary molecular target as dimebon for a specified time period, subject to certain exceptions.
The agreement establishes several joint committees consisting of an equal number of representatives from both parties that operate by consensus to oversee the collaboration. In the event that a joint committee is unable to reach consensus on a particular issue, then, depending on the issue, a dispute may be decided at the joint committee level by the party with the final decision on the issue or escalated to senior management of the parties. If a dispute is escalated to senior management and no consensus is reached, then the dispute may be decided by the party to whom the contract grants final decision on such issue. Other issues can only be decided by consensus of the parties, and unless and until the parties’ representatives reach agreement on such issue, no decision on such issue will be made, and the status quo will be maintained.
Under the Pfizer Collaboration Agreement, Pfizer paid us an up-front cash payment of $225 million in the fourth quarter of 2008. We are also eligible to receive payments of up to $500 million upon the attainment of development and regulatory milestones plus additional milestone payments upon the achievement of certain net sales levels for the product. We and Pfizer will share the costs and expenses of developing and commercializing dimebon for the United States market on a 60%/40% basis, with Pfizer assuming the larger share, and we and Pfizer will share profits (or losses) resulting from the commercialization of dimebon in the United States in such proportions. Outside the United States, Pfizer will bear all development and commercialization costs and will pay us tiered royalties on the aggregate net sales of dimebon.
If one of the parties merges with, or acquires or is acquired by, a third party and as a result such party must divest its interest in the dimebon collaboration due to a governmental requirement, then the other party has the first right to purchase the divesting party’s interest in the collaboration, on terms to be negotiated by the parties.
50
Table of Contents
In the event that the parties are unable to agree on the terms of this purchase after following the negotiation procedure outlined in the Collaboration Agreement, the divesting party will have a time-limited right to sell its interest in the collaboration to a third party. However, the terms of this sale must be more favorable than any terms offered by the non-divesting party and the third party will remain bound by the terms of the Collaboration Agreement. In the event the non-divesting party declines to purchase the divesting party’s interest, the divesting party may sell its interest in the collaboration to a third party on any terms but such third party will remain bound by the terms of the Collaboration Agreement.
We are permitted to terminate the Collaboration Agreement for an uncured material breach by Pfizer. Pfizer has a right to terminate the Collaboration Agreement unilaterally at any time. In the event of our uncured material breach of the Collaboration Agreement, Pfizer may elect either to terminate the Collaboration Agreement or to keep the Collaboration Agreement in place, but terminate our right to participate in development, commercialization (other than co-promoting dimebon) and other activities for dimebon, including the joint committees and decision making for dimebon. However, such termination would not affect our financial return or, unless we commit an uncured material breach of our co-promotion obligations, our co-promotion rights. Following any termination of the Collaboration Agreement, all rights to develop and commercialize dimebon will revert to us, and Pfizer will grant a license to us to enable us to continue such development and commercialization, remain responsible for its ongoing financial and other obligations under the Collaboration Agreement for a transition period of six months following termination, and is obligated to supply product to us for a reasonable period, not to exceed eighteen months following termination, on terms to be negotiated between the parties in good faith.
Astellas Collaboration Agreement
Our global development and commercialization agreement with Astellas became effective in October 2009. Under the Astellas Collaboration Agreement, we and Astellas agreed to collaborate on the development of MDV3100 for prostate cancer for the United States market, including associated regulatory filings with the FDA. In addition, if approved by the FDA, following such approval and the launch of MDV3100 in the United States, we, at our option, and Astellas have the right to co-promote MDV3100 in the United States. Astellas is responsible for development of, and seeking regulatory approval for, and commercialization of MDV3100 outside the United States. Astellas will be responsible for commercial manufacture of MDV3100 on a global basis. Both Medivation and Astellas have agreed not to commercialize certain other products having a similar mechanism of action as MDV3100 for the treatment of specified indications for a specified time period, subject to certain exceptions.
We and Astellas share the costs of developing and commercializing MDV3100 for the United States market on a 50%/50% basis, and we and Astellas will share profits (or losses) resulting from the commercialization of MDV3100 in the United States in such proportions. Costs of clinical trials supporting development in both the United States and in either Europe or Japan, including the ongoing AFFIRM trial, are borne two-thirds by Astellas and one-third by us. Outside the United States, Astellas will bear all development and commercialization costs and will pay to us tiered, double-digit royalties on the aggregate net sales of MDV3100.
Under the Astellas Collaboration Agreement, Astellas paid us a non-refundable, up-front cash payment of $110.0 million in November 2009, ten percent of which we paid to the academic institution from which we licensed our rights to MDV3100. We are also entitled to receive up to $335.0 million in development milestone payments, plus up to an additional $320.0 million in commercial milestone payments. We are required to share 10% of the up-front payment and any development milestone payments received under the Astellas Collaboration Agreement with the academic institution from which we licensed our rights to MDV3100. We paid 10% of the up-front payment, or $11.0 million, to the academic institution in the fourth quarter of 2009.
Each of Medivation and Astellas is permitted to terminate the Astellas Collaboration Agreement for an uncured material breach by the other party or for the insolvency of the other party. Astellas has a right to terminate the Astellas Collaboration Agreement unilaterally by advance written notice to us, but, except in
51
Table of Contents
certain specific circumstances, generally cannot exercise that termination right until the first anniversary of MDV3100’s first commercial sale. Following any termination of the Astellas Collaboration Agreement in its entirety, all rights to develop and commercialize MDV3100 will revert to us, and Astellas will grant a license to us to enable us to continue such development and commercialization. In addition, except in the case of a termination by Astellas for our uncured material breach, Astellas will supply MDV3100 to us during a specified transition period.
Cash Flow
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| (In thousands) | ||||||||||||
| Net cash provided by (used in): |
||||||||||||
| Operating activities |
$ | (7,585 | ) | $ | 162,172 | $ | (23,358 | ) | ||||
| Investing activities |
(72,568 | ) | (149,546 | ) | 40,931 | |||||||
| Financing activities |
66,162 | 15,570 | 21,036 | |||||||||
| Net change in cash and cash equivalents |
$ | (13,991 | ) | $ | 28,196 | $ | 38,609 | |||||
Cash, cash equivalents and short-term investments totaled $278.2 million at December 31, 2009, compared to $221.4 million at December 31, 2008 and $43.3 million at December 31, 2007. The net increase in cash, cash equivalents and short-term investments of $56.8 million in 2009 as compared to 2008 was due primarily to cash proceeds from an up-front license payment received from Astellas of $110.0 million, the issuance of $62.1 million of common stock, and cash proceeds of $3.2 million from exercise of stock options, partially offset by the cash used to fund operations in 2009. The net increase in cash, cash equivalents and short-term investments of $178.1 million in 2008 as compared to 2007 was due primarily to cash proceeds from an up-front license payment received from Pfizer of $225.0 million and the issuance of $15.0 million of common stock, partially offset by the cash used to fund operations in 2008.
Operating Activities
Net cash used in operations was $7.6 million in 2009. Cash used in operating activities during 2009 was primarily driven by our net loss of $54.8 million, increased prepaid expenses of $5.5 million and increased receivables from our corporate partners of $3.0 million, partially offset by increased deferred revenue of $40.8 million arising primarily from our $110 million up-front payment from Astellas, $10.7 million in non-cash stock-based compensation expense and increased accounts payable and accrued expenses of $4.0 million.
Net cash provided by operating activities totaled $162.2 million in 2008. Cash provided by operating activities during 2008 was primarily driven by the cash payment of $225.0 million from Pfizer, non-cash stock-based compensation expense of $8.5 million, and increases in accounts payable and accrued expenses of $5.4 million and $3.6 million, respectively, arising in the ordinary course of business as a result of increased research and development activities, partially offset by our net loss of $62.5 million and a cost-sharing receivable from Pfizer of $3.5 million.
Net cash used in operating activities totaled $23.4 million in 2007, which was primarily attributable to our net loss of $31.7 million, partially offset by non-cash stock-based compensation expense of $5.9 million and increased accrued expenses of $2.2 million.
Investing Activities
Net cash used in investing activities totaled $72.6 million and $149.5 million in 2009 and 2008, respectively. In both periods, cash used in investing activities primarily represented net investments from purchases and maturities of short-term investments.
52
Table of Contents
Net cash provided by investing activities totaled $40.9 million in 2007 and primarily represented net proceeds from purchases and maturities of short-term investments.
Financing Activities
Net cash provided by financing activities totaled $66.2 million in 2009. Cash provided by financing activities was primarily due to our sale of our common stock in a registered public offering, raising net proceeds of approximately $62.1 million, and $3.2 million in proceeds from the issuance of common stock related to the exercise of employee stock options.
Net cash provided by financing activities totaled $15.6 million in 2008. Cash provided by financing activities was primarily due to our sale of our common stock in a registered direct offering, raising net proceeds of approximately $14.9 million, and $0.7 million in proceeds from the issuance of common stock related to the exercise of employee stock options.
Net cash provided by financing activities totaled $21.0 million in 2007. Cash provided by financing activities was primarily due to our sale of common stock in a registered direct offering, raising net proceeds of $20.8 million, and $0.2 million in proceeds from the issuance of common stock related to the exercise of employee stock options.
Commitments and Contingencies
At December 31, 2009, we had minimum future payments under our operating leases as follows (in thousands):
| Payment due by Period | |||||||||||||||
| Total | Less than 1 Year |
1-3 Years | 3-5 Years | > 5 Years | |||||||||||
| Operating lease obligations(1) |
$ | 5,980 | $ | 2,909 | $ | 2,757 | $ | 314 | $ | — | |||||
| (1) | The lease agreements covering our present office facilities expire from July 2012 to May 2013. We are committed to pay a portion of the related operating expenses under these lease agreements. These operating expenses are not included in the table above. Certain of these leases have free or escalating rent payment provisions. We recognize rent expense under such leases on a straight-line basis over the term of the lease. Please refer to Note 13, “Commitments and Contingencies,” to our consolidated financial statements included elsewhere in this Report on Form 10-K for further discussion regarding our future operating lease commitments. |
Off-Balance Sheet Arrangements
We have no material off-balance sheet arrangements as defined in Regulation S-K 303(a)(4)(ii).
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk. |
Our exposure to market risk for changes in interest rates relates to our cash equivalents on deposit in highly liquid money market accounts and short-term investments in highly liquid U.S. Treasury securities. The primary objective of our cash investment activities is to preserve principal. We do not use derivative financial instruments in our investment portfolio. Our cash and investments policy emphasizes liquidity and preservation of principal over other portfolio considerations. Our investment portfolio is subject to interest rate risk and will fall in value if market interest rates rise.
53
Table of Contents
Interest Rate Risk
We have cash equivalents and short-term investments at December 31, 2009, which are exposed to the impact of interest rate changes and our interest income fluctuates as our interest rates change. Due to the short-term nature of our investments in money market funds and US Treasury bills, the carrying value of our cash equivalents and short-term investments approximate their fair value at December 31, 2009.
We maintain an investment portfolio in accordance with our investment policy. The primary objectives of our investment policy are to preserve principal, maintain proper liquidity to meet operating needs and maximize yields. Due to the nature of our short-term investments, we believe that we are not subject to any material market risk exposure.
Foreign Currency Exchange Risk
We do not have any material exposure to foreign currency rate fluctuations as we operate primarily in the U.S. Although we conduct some research and development work with vendors outside the U.S., most of our transactions are denominated in U.S. dollars. However, certain of our ex-U.S. clinical development activities are pursuant to contracts denominated in foreign currencies. For the year ended December 31, 2009, we recorded $0.1 million in foreign currency exchange losses. As of December 31, 2009, we have recorded the equivalent of approximately $0.7 million of foreign denominated vendor payables.
| Item 8. | Financial Statements and Supplementary Data. |
All information required by this item is included in Item 15 of Part IV of this Annual Report on Form 10-K and is incorporated into this item by reference.
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure. |
None.
| Item 9A. | Controls and Procedures. |
Evaluation of Disclosure Controls and Procedures
We maintain “disclosure controls and procedures”, as such term is defined in Rule 13a-15(e) under the Securities Exchange Act of 1934, or the Exchange Act, that are designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the Commission’s rules and forms and that such information is communicated to our management, including our principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. In designing and evaluating our disclosure controls and procedures, management recognizes that disclosure controls and procedures, no matter how well conceived and operated, can provide only reasonable, but not absolute, assurance that the objectives of the disclosure controls and procedures are met. Our disclosure controls and procedures have been designed to meet the reasonable assurance standards. Additionally, in designing disclosure controls and procedures, our management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible disclosure controls and procedures. The design of any disclosure controls and procedures is also based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions.
As required by Rule 13a-15(b) or Rule 15d-15(b) of the Exchange Act, we carried out an evaluation, under the supervision and with the participation of our management, including our principal executive officer and our principal financial officer, of the effectiveness of the design and operation of our disclosure controls and
54
Table of Contents
procedures as of December 31, 2009. Based on the foregoing, our principal executive officer and our principal financial officer concluded that as of December 31, 2009, our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15(d)-15(f). Management, with the participation of our principal executive offer and principal financial offer, has conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework set forth in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on our evaluation under the framework set forth in Internal Control—Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2009.
The effectiveness of our internal control over financial reporting as of December 31, 2009 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report which appears herein.
Changes in Internal Control Over Financial Reporting
There were no changes in internal control over financial reporting during the quarter ended December 31, 2009 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
55
Table of Contents
| Item 9B. | Other Information. |
None.
56
Table of Contents
PART III
The information required by Part III is omitted from this Annual Report on Form 10-K since we intend to file our definitive Proxy Statement for our 2010 Annual Meeting of Stockholders, pursuant to Regulation 14A of the Securities Exchange Act of 1934, as amended, not later than 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and certain information to be included in the Proxy Statement is incorporated herein by reference.
| Item 10. | Directors, Executive Officers and Corporate Governance. |
Information required by this item regarding directors and director nominees, executive officers, the board of directors and its committees, and certain corporate governance matters is incorporated by reference to the information set forth under the captions “Election of Directors,” “Information Regarding the Board of Directors and Corporate Governance” and “Executive Officers” in our Proxy Statement pursuant to Section 14(a) of the Securities Exchange Act of 1934 for the 2010 Annual Meeting of Stockholders. Information required by this item regarding compliance with Section 16(a) of the Securities Exchange Act of 1934, as amended, is incorporated by reference to the information set forth under the caption “Section 16(a) Beneficial Ownership Reporting Compliance” in our Proxy Statement for the 2010 Annual Meeting of Stockholders.
We have adopted a written code of business conduct and ethics that applies to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons serving similar functions. The code of business conduct and ethics is available on our corporate website at www.medivation.com. If we make any substantive amendments to our code of business conduct and ethics or grant to any of our directors or executive officers any waiver, including any implicit waiver, from a provision of our code of business conduct and ethics, we will disclose the nature of the waiver or amendment on our website or in a Current Report on Form 8-K.
| Item 11. | Executive Compensation. |
Information required by this item regarding executive compensation is incorporated by reference to the information set forth under the captions “Executive Compensation”, “Director Compensation,” “Compensation Committee Interlocks and Insider Participation” and “Compensation Committee Report” in our Proxy Statement for the 2010 Annual Meeting of Stockholders.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters. |
Information required by this item regarding security ownership of certain beneficial owners and management is incorporated by reference to the information set forth under the caption “Security Ownership of Certain Beneficial Owners and Management” in our Proxy Statement for the 2010 Annual Meeting of Stockholders. Information required by this item regarding securities authorized for issuance under our equity compensation plans is incorporated by reference to the information set forth under the caption “Equity Compensation Plan Information” in our Proxy Statement for the Annual Meeting of Stockholders to be filed with the Commission on or before April 30, 2010.
| Item 13. | Certain Relationships and Related Transactions, and Director Independence. |
Information required by this item regarding certain relationships and related transactions is incorporated by reference to the information set forth under the caption “Transactions with Related Persons” in our Proxy Statement for the 2010 Annual Meeting of Stockholders. Information required by this item regarding director independence is incorporated by reference to the information set forth under the caption “Information Regarding the Board of Directors and Corporate Governance” in our Proxy Statement for the 2010 Annual Meeting of Stockholders.
57
Table of Contents
| Item 14. | Principal Accountant Fees and Services. |
Information required by this item regarding principal accountant fees and services is incorporated by reference to the information set forth under the caption “Ratification of Selection of Independent Registered Public Accounting Firm” in our Proxy Statement for the 2010 Annual Meeting of Stockholders.
58
Table of Contents
PART IV
| Item 15. | Exhibits and Financial Statement Schedules. |
(a) The following documents are filed as part of this Annual Report:
1. Financial Statements. Our financial statements and the Report of Independent Registered Public Accounting Firm, are included herein on the pages indicated:
2. Financial Statement Schedules: None.
3. Exhibits:
Unless otherwise indicated below, the Commission file number to the exhibit is No. 001-32836 for filings made in or after March 2006 and No. 000-20837 for filings made prior to March 2006.
| Exhibit No. |
Exhibit Description | |
| 3.1(a) | Amended and Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.1(a) to the Quarterly Report on Form 10-QSB of Medivation, Inc. for the quarter ended June 30, 2005). | |
| 3.1(b) | Certificate of Amendment of Amended and Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.1(b) to the Quarterly Report on Form 10-QSB of Medivation, Inc. for the quarter ended June 30, 2005). | |
| 3.1(c) | Certificate of Amendment to the Amended and Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.1(c) to the Quarterly Report on Form 10-QSB of Medivation, Inc. for the quarter ended June 30, 2005). | |
| 3.1(d) | Certificate of Designations of the Series C Junior Participating Preferred Stock of Medivation, Inc. (incorporated by reference to Exhibit 3.1(d) to the Annual Report on Form 10-KSB of Medivation, Inc. for the fiscal year ended December 31, 2007). | |
| 3.2 | Amended and Restated Bylaws of Medivation, Inc. (incorporated by reference to Exhibit 3.2 to the Annual Report on Form 10-K for the year ended December 31, 2008). | |
| 4.1 | Common Stock Certificate (incorporated by reference to Exhibit 4.1 to the Registration Statement on Form SB-2 of Medivation, Inc. (formerly Orion Acquisition Corp. II) (No. 333-03252)). | |
| 4.2 | Rights Agreement, dated as of December 4, 2006, between Medivation, Inc. and American Stock Transfer & Trust Company, as Rights Agent, which includes the form of Certificate of Designations of the Series C Junior Participating Preferred Stock of Medivation, Inc. as Exhibit A, the form of Right Certificate as Exhibit B and the Summary of Rights to Purchase Preferred Shares as Exhibit C (incorporated by reference to Exhibit 4.1 to the Current Report on Form 8-K filed on December 4, 2006). | |
59
Table of Contents
| Exhibit No. |
Exhibit Description | |
| 10.1 | Registration Rights Agreement by and among Orion Acquisition Corp. II and Special Situations Fund III, L.P., Special Situations Cayman Fund, L.P. and Special Situations Private Equity Fund, L.P., dated as of December 17, 2004 (incorporated by reference to Exhibit 10.3(a) to the Registration Statement on Form SB-2 of Medivation, Inc. (formerly Orion Acquisition Corp. II) (No. 333-122431)). | |
| 10.2(a) | Warrant to purchase Common Stock of Medivation Neurology, Inc. assumed by Orion Acquisition Corp. II issued to Joseph J. Grano, Jr., dated as of June 8, 2004 (incorporated by reference to Exhibit 10.5(a) to the Registration Statement on Form SB-2 of Medivation, Inc. (formerly Orion Acquisition Corp. II) (No. 333-122431)). | |
| 10.2(b) | Warrant to purchase Common Stock of Medivation Neurology, Inc. assumed by Orion Acquisition Corp. II issued to Joseph J. Grano, Jr., dated as of August 1, 2004 (incorporated by reference to Exhibit 10.5(b) to the Registration Statement on Form SB-2 of Medivation, Inc. (formerly Orion Acquisition Corp. II) (No. 333-122431)). | |
| 10.2(c) | Warrant to purchase Common Stock of Medivation Neurology, Inc. assumed by Orion Acquisition Corp. II issued to Joseph J. Grano, Jr., dated as of September 1, 2004 (incorporated by reference to Exhibit 10.5(c) to the Registration Statement on Form SB-2 of Medivation, Inc. (formerly Orion Acquisition Corp. II) (No. 333-122431)). | |
| 10.2(d) | Amendment Agreement by and between Orion Acquisition Corp. II and Joseph J. Grano, Jr., dated as of December 17, 2004 (incorporated by reference to Exhibit 10.5(d) to the Registration Statement on Form SB-2 of Medivation, Inc. (formerly Orion Acquisition Corp. II) (No. 333-122431)). | |
| 10.3* | Warrant to purchase Common Stock of Medivation Neurology, Inc. assumed by Orion Acquisition Corp. II issued to David T. Hung, M.D., dated as of November 16, 2004 (incorporated by reference to Exhibit 10.6 to the Registration Statement on Form SB-2 of Medivation, Inc. (formerly Orion Acquisition Corp. II) (No. 333-122431)). | |
| 10.4(a)* | Amended and Restated 2004 Equity Incentive Award Plan (incorporated by reference to Exhibit 10.4(a) to the Annual Report on Form 10-KSB for the fiscal year ended December 31, 2007). | |
| 10.4(b)* | Form of Stock Option Agreement under the 2004 Equity Incentive Award Plan (incorporated by reference to Exhibit 10.7(b) to the Annual Report on Form 10-KSB of Medivation, Inc. (formerly Orion Acquisition Corp. II) for the year ended December 31, 2004). | |
| 10.4(c)* | Form of Stock Option Agreement for Early Excercisable Options under the 2004 Equity Incentive Award Plan (incorporated by reference to Exhibit 10.7(c) to the Annual Report on Form 10-KSB of Medivation, Inc. (formerly Orion Acquisition Corp. II) for the year ended December 31, 2004). | |
| 10.5 | Common Stock Purchase Agreement, dated as of September 7, 2007, by and between Medivation, Inc. and Azimuth Opportunity Ltd. (incorporated by reference to Exhibit 10.1 to Medivation, Inc.’s Current Report on Form 8-K filed on September 10, 2007). | |
| 10.6 | Form of Purchase Agreement, dated as of June 22, 2008, between Medivation, Inc. and the purchasers of its common stock (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed on June 23, 2008). | |
| 10.7** | Amended and Restated Collaboration Agreement, dated as of October 20, 2008, between Medivation, Inc. and Pfizer Inc. (incorporated by reference to Exhibit 10.8 to the Quarterly Report on Form 10-Q for the quarter ended September 30, 2008). | |
| 10.8* | Bonuses for Fiscal Year 2008 and Base Salaries for Fiscal Year 2009 for Certain Executive Officers (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed on November 5, 2008). | |
60
Table of Contents
| Exhibit No. |
Exhibit Description | |
| 10.9* | Medivation, Inc. 2009 Bonus Plan (incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K filed on November 5, 2008). | |
| 10.10* | Bonuses for Fiscal Year 2009 and Base Salaries for Fiscal Year 2010 for Named Executive Officers (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed on December 7, 2009). | |
| 10.11* | Medivation, Inc. 2010 Bonus Plan Summary (incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K filed on December 7, 2009). | |
| 10.12* | Change of Control Severance Benefits Agreement, dated as of February 2, 2009, between Medivation, Inc. and David Hung, M.D. (incorporated by reference to Exhibit 10.11 to the Annual Report on Form 10-K filed on March 16, 2009). | |
| 10.13* | Severance Benefits Agreement, dated as of February 9, 2009, between Medivation, Inc. and Rohan Palekar (incorporated by reference to Exhibit 10.12 to the Annual Report on Form 10-K filed on March 16, 2009). | |
| 10.14* | Form of Medivation, Inc. Change of Control Severance Benefits Agreement (incorporated by reference to Exhibit 10.13 to the Annual Report on Form 10-K filed on March 16, 2009). | |
| 10.15† | Collaboration Agreement, dated as of October 26, 2009, by and between Medivation, Inc. and Astellas US LLC. | |
| 10.16 | Office Lease Agreement, dated as of November 2, 2009, by and between Medivation, Inc. and PPF OFF 345 Spear Street, LP. | |
| 10.17* | Compensation Information for Non-Employee Directors. | |
| 21.1 | Subsidiaries of Medivation, Inc. | |
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |
| 24.1 | Power of attorney (contained on signature page). | |
| 31.1 | Certification pursuant to Rule 13a-14(a)/15d-14(a). | |
| 31.2 | Certification pursuant to Rule 13a-14(a)/15d-14(a). | |
| 32.1 | Certifications of Chief Executive Officer and Chief Financial Officer. | |
| * | Indicates management contract or compensatory plan or arrangement. |
| ** | Confidential treatment has been granted with respect to certain portions of this exhibit. |
| † | Confidential treatment requested as to certain portions, which portions were omitted and filed separately with the Securities and Exchange Commission. |
61
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| MEDIVATION, INC. | ||
| /s/ C. PATRICK MACHADO | ||
| C. Patrick Machado Chief Business Officer and Chief Financial Officer (Duly Authorized and Principal Financial and Accounting Officer) | ||
Dated: March 15, 2010
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints David T. Hung, M.D. and C. Patrick Machado, and each of them, as his true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution, for him and in his name, place, and stead, in any and all capacities, to sign any and all amendments to this Report, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming that all said attorneys-in-fact and agents, or any of them or their or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated:
| /s/ DAVID T. HUNG, M.D. David T. Hung, M.D. |
President, Chief Executive Officer and Director (Principal Executive Officer) |
March 15, 2010 | ||
| /s/ C. PATRICK MACHADO C. Patrick Machado |
Chief Business Officer and Chief Financial Officer (Principal Financial and Accounting Officer) | March 15, 2010 | ||
| /s/ DANIEL D. ADAMS Daniel D. Adams |
Director | March 15, 2010 | ||
| /s/ GREGORY H. BAILEY, M.D. Gregory H. Bailey |
Director | March 15, 2010 | ||
| /s/ W. ANTHONY VERNON W. Anthony Vernon |
Director | March 15, 2010 | ||
62
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To Board of Directors and Stockholders of
Medivation, Inc.
In our opinion, the accompanying consolidated balance sheets and the related consolidated statements of operations, of cash flows and of stockholders’ equity present fairly, in all material respects, the financial position of Medivation, Inc. and its subsidiaries (the “Company”) at December 31, 2009 and December 31, 2008 and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2009 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for these financial statements, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in Management’s Report on Internal Control over Financial Reporting appearing under Item 9A. Our responsibility is to express opinions on these financial statements and on the Company’s internal control over financial reporting based on our audits which were integrated audits in 2009 and 2008. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the Company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP
San Jose, California
March 15, 2010
63
Table of Contents
CONSOLIDATED BALANCE SHEETS
(In thousands, except share and per share data)
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| ASSETS |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 57,463 | $ | 71,454 | ||||
| Short-term investments |
220,781 | 149,968 | ||||||
| Receivable from collaboration partners (Note 3) |
6,490 | 3,522 | ||||||
| Prepaid expenses and other current assets |
9,343 | 1,957 | ||||||
| Total current assets |
294,077 | 226,901 | ||||||
| Property and equipment, net |
1,092 | 768 | ||||||
| Restricted cash |
843 | 843 | ||||||
| Other non-current assets |
678 | 760 | ||||||
| Total assets |
$ | 296,690 | $ | 229,272 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 4,840 | $ | 7,166 | ||||
| Accrued expenses |
12,054 | 5,772 | ||||||
| Deferred revenue |
86,570 | 64,286 | ||||||
| Other current liabilities |
800 | 93 | ||||||
| Total current liabilities |
104,264 | 77,317 | ||||||
| Deferred revenue, net of current |
166,598 | 148,137 | ||||||
| Other non-current liabilities |
554 | 410 | ||||||
| Total liabilities |
271,416 | 225,864 | ||||||
| Commitments and contingencies (Note 13) |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.01 par value per share; 1,000,000 shares authorized; no shares issued and outstanding |
— | — | ||||||
| Common stock, $0.01 par value per share; 50,000,000 shares authorized; issued and outstanding 33,823,062 shares at December 31, 2009 and 30,088,390 at December 31, 2008 |
338 | 301 | ||||||
| Additional paid-in capital |
202,361 | 125,074 | ||||||
| Accumulated other comprehensive gain (loss) |
(12 | ) | 693 | |||||
| Accumulated deficit |
(177,413 | ) | (122,660 | ) | ||||
| Total stockholders’ equity |
25,274 | 3,408 | ||||||
| Total liabilities and stockholders’ equity |
$ | 296,690 | $ | 229,272 | ||||
SEE ACCOMPANYING NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
64
Table of Contents
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except per share data)
| Years ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Collaboration revenue |
$ | 69,254 | $ | 12,578 | $ | — | ||||||
| Operating expenses: |
||||||||||||
| Research and development |
87,728 | 54,895 | 23,399 | |||||||||
| Selling, general and administrative |
28,983 | 21,865 | 10,364 | |||||||||
| Total operating expenses |
116,711 | 76,760 | 33,763 | |||||||||
| Loss from operations |
(47,457 | ) | (64,182 | ) | (33,763 | ) | ||||||
| Other income (expense): |
||||||||||||
| Interest income |
1,128 | 1,206 | 2,022 | |||||||||
| Other income (expense), net |
(152 | ) | 506 | — | ||||||||
| Total other income (expense) |
976 | 1,712 | 2,022 | |||||||||
| Net loss before income tax |
(46,481 | ) | (62,470 | ) | (31,741 | ) | ||||||
| Income tax (benefit) expense |
8,272 | (10 | ) | 2 | ||||||||
| Net loss |
$ | (54,753 | ) | $ | (62,460 | ) | $ | (31,743 | ) | |||
| Basic and diluted net loss per common share |
$ | (1.71 | ) | $ | (2.12 | ) | $ | (1.14 | ) | |||
| Weighted average common shares used in the calculation of basic and diluted net loss per share |
32,094 | 29,478 | 27,932 | |||||||||
SEE ACCOMPANYING NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
65
Table of Contents
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| Years ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Cash flows from operating activities: |
||||||||||||
| Net loss |
$ | (54,753 | ) | $ | (62,460 | ) | $ | (31,743 | ) | |||
| Adjustments to reconcile net loss to net cash flows from operating activities: |
||||||||||||
| Depreciation and amortization |
311 | 193 | 74 | |||||||||
| Loss on disposal of property and equipment |
— | — | 6 | |||||||||
| Accretion of discount on securities |
(1,081 | ) | (340 | ) | (70 | ) | ||||||
| Stock-based compensation |
10,726 | 8,547 | 5,893 | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Receivable from collaboration partners |
(2,968 | ) | (3,522 | ) | — | |||||||
| Prepaid expenses and other current assets |
(5,450 | ) | (966 | ) | (328 | ) | ||||||
| Other assets |
78 | (606 | ) | 12 | ||||||||
| Accounts payable |
(2,326 | ) | 5,419 | 55 | ||||||||
| Accrued expenses |
6,282 | 3,554 | 2,214 | |||||||||
| Other current liabilities |
707 | 23 | 37 | |||||||||
| Deferred revenue |
40,745 | 212,423 | — | |||||||||
| Other non-current liabilities |
144 | (93 | ) | 492 | ||||||||
| Net cash provided by (used in) operating activities |
(7,585 | ) | 162,172 | (23,358 | ) | |||||||
| Cash flows from investing activities: |
||||||||||||
| Purchase of short-term investments |
(342,437 | ) | (248,935 | ) | (24,692 | ) | ||||||
| Maturities of short-term investments |
272,000 | 100,000 | 66,956 | |||||||||
| Purchase of property and equipment |
(631 | ) | (268 | ) | (758 | ) | ||||||
| Purchase of restricted cash |
(1,500 | ) | (343 | ) | (575 | ) | ||||||
| Net cash provided by (used in) investing activities |
(72,568 | ) | (149,546 | ) | 40,931 | |||||||
| Cash flows from financing activities: |
||||||||||||
| Proceeds from issuance of common stock, net of issuance costs |
62,059 | 14,911 | 20,799 | |||||||||
| Warrant exercises |
167 | — | — | |||||||||
| Excess tax benefits from stock-based compensation |
714 | — | — | |||||||||
| Stock option exercises |
3,222 | 659 | 237 | |||||||||
| Net cash provided by financing activities |
66,162 | 15,570 | 21,036 | |||||||||
| Net increase (decrease) in cash and cash equivalents |
(13,991 | ) | 28,196 | 38,609 | ||||||||
| Cash and cash equivalents at beginning of year |
71,454 | 43,258 | 4,649 | |||||||||
| Cash and cash equivalents at end of year |
$ | 57,463 | $ | 71,454 | $ | 43,258 | ||||||
| Supplemental disclosure of cash flow information: |
||||||||||||
| Income taxes paid |
$ | 8,400 | $ | — | $ | — | ||||||
| Receivable from stock option exercises |
$ | 436 | $ | — | $ | — | ||||||
SEE ACCOMPANYING NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
66
Table of Contents
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(In thousands, except share data)
| COMMON STOCK | ADDITIONAL PAID-IN CAPITAL |
ACCUMULATED OTHER COMPREHENSIVE INCOME |
ACCUMULATED DEFICIT |
TOTAL STOCKHOLDERS’ EQUITY |
|||||||||||||||||
| SHARES | AMOUNT | ||||||||||||||||||||
| Balances at January 1, 2007 |
27,723,532 | $ | 277 | $ | 74,052 | $ | — | $ | (28,457 | ) | $ | 45,872 | |||||||||
| Common stock issued for: |
|||||||||||||||||||||
| Azimuth common stock purchase agreement |
1,023,548 | 10 | 21,240 | — | — | 21,250 | |||||||||||||||
| Cashless exercise of warrants |
24,020 | — | — | — | — | — | |||||||||||||||
| Cash upon exercise of stock options |
66,190 | 1 | 236 | — | — | 237 | |||||||||||||||
| Offering expenses |
— | — | (451 | ) | — | — | (451 | ) | |||||||||||||
| Stock-based compensation expense |
— | — | 5,893 | — | — | 5,893 | |||||||||||||||
| Net loss |
— | (31,743 | ) | (31,743 | ) | ||||||||||||||||
| Balances at December 31, 2007 |
28,837,290 | 288 | 100,970 | — | (60,200 | ) | 41,058 | ||||||||||||||
| Common stock issued for: |
|||||||||||||||||||||
| Cash in the June 2008 financing |
1,129,518 | 11 | 14,989 | — | — | 15,000 | |||||||||||||||
| Cashless exercise of warrants |
5,496 | 1 | (1 | ) | — | — | — | ||||||||||||||
| Cash upon exercise of stock options |
116,086 | 1 | 658 | — | — | 659 | |||||||||||||||
| Offering expenses |
— | — | (89 | ) | — | — | (89 | ) | |||||||||||||
| Stock-based compensation expense |
— | — | 8,547 | — | — | 8,547 | |||||||||||||||
| Comprehensive loss: |
|||||||||||||||||||||
| Net loss |
— | (62,460 | ) | (62,460 | ) | ||||||||||||||||
| Unrealized gain on securities available-for sale |
693 | — | 693 | ||||||||||||||||||
| Comprehensive loss |
(61,767 | ) | |||||||||||||||||||
| Balances at December 31, 2008 |
30,088,390 | 301 | 125,074 | 693 | (122,660 | ) | 3,408 | ||||||||||||||
| Common stock issued for: |
|||||||||||||||||||||
| Cash in the June 2009 financing |
3,162,500 | 32 | 62,396 | — | — | 62,428 | |||||||||||||||
| Cashless exercise of warrants |
101,576 | 1 | (1 | ) | — | — | — | ||||||||||||||
| Cash upon exercise of warrants |
107,000 | 1 | 166 | — | — | 167 | |||||||||||||||
| Cash upon exercise of stock options |
344,430 | 3 | 3,655 | — | — | 3,658 | |||||||||||||||
| Cash upon exercise of restricted stock award |
19,166 | — | — | — | |||||||||||||||||
| Offering expenses |
— | — | (369 | ) | — | — | (369 | ) | |||||||||||||
| Stock-based compensation expense |
— | — | 10,726 | — | — | 10,726 | |||||||||||||||
| Tax benefit from employee stock plan awards |
— | — | 714 | — | — | 714 | |||||||||||||||
| Comprehensive loss: |
|||||||||||||||||||||
| Net loss |
— | (54,753 | ) | (54,753 | ) | ||||||||||||||||
| Change in unrealized gain / (loss) on available-for-sale securities |
(705 | ) | — | (705 | ) | ||||||||||||||||
| Comprehensive loss |
(55,458 | ) | |||||||||||||||||||
| Balances at December 31, 2009 |
33,823,062 | $ | 338 | $ | 202,361 | $ | (12 | ) | $ | (177,413 | ) | $ | 25,274 | ||||||||
SEE ACCOMPANYING NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
67
Table of Contents
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2009
1. DESCRIPTION OF BUSINESS
The Company is a biopharmaceutical company focused on the rapid development of novel small molecule drugs to treat serious diseases for which there are limited treatment options. Its product candidates in clinical development are dimebon (latrepirdine), which is in Phase 3 development for the treatment of Alzheimer’s disease and Huntington disease, and MDV3100, which is in Phase 3 development for the treatment of castration-resistant prostate cancer. The Company’s dimebon program is partnered with Pfizer Inc, or Pfizer, and its MDV3100 program is partnered with Astellas Pharma Inc., or Astellas.
In September 2008, the Company announced a Collaboration Agreement with Pfizer, which became effective in October 2008. Under the terms of the agreement, the Company and Pfizer will develop and commercialize dimebon for the treatment of Alzheimer’s disease and Huntington disease. The Company and Pfizer will share the costs and expenses of developing and commercializing dimebon for the United States market on a 60% Pfizer/40% Medivation basis, and will share profits (or losses) resulting from commercialization of dimebon in the United States in the same proportions. Outside the United States, Pfizer will bear all development and commercialization costs, and will pay the Company tiered royalties on aggregate net sales of dimebon. In the fourth quarter of 2008, the Company received a non-refundable, up-front cash payment of $225.0 million pursuant to its Collaboration Agreement with Pfizer.
With Pfizer, the Company has been conducting a broad dimebon clinical development program, including five randomized, double-blind, placebo-controlled Phase 3 studies in Alzheimer’s and Huntington diseases. In March 2010, the Company reported negative results from the first of these studies—the CONNECTION trial, a six-month study of 598 patients with mild-to-moderate Alzheimer’s disease in the United States, Western Europe, Russia and Chile in which patients were randomized to receive dimebon 20 mg three times daily, dimebon 5 mg three times daily or placebo. Patients in the CONNECTION trial were not permitted to take any approved Alzheimer’s disease medicines for a period of 90 days before, or at any time during, the study. In the CONNECTION study, dimebon treated patients did no better than placebo patients on any of the primary and secondary efficacy endpoints at either dose tested. Dimebon was well tolerated in the CONNECTION study, as well as in a separate randomized, double-blind, placebo-controlled safety study of dimebon, also reported in March 2010, in which 742 patients in the United States and Canada were randomized to receive dimebon 20 mg three times daily or placebo and treated for three to six months. Approximately 85% of patients in the safety study were taking one or more approved Alzheimer’s disease medicines while on the trial. Further analysis of the results from both of these studies is currently underway.
Given the negative results in the CONNECTION trial, the Company is currently re-evaluating its approach to the clinical development of dimebon for Alzheimer’s disease and Huntington disease. This will be a complex analysis that will encompass scientific, clinical, regulatory and business issues. The Company does not know at this time which, if any, of the ongoing Phase 3 dimebon trials will be completed, or if Pfizer will elect to terminate the collaboration.
In October 2009, the Company entered into a Collaboration Agreement with Astellas. Under the terms of the agreement, the Company and Astellas will develop and commercialize MDV3100 for the treatment of both late-and early-stage prostate cancer. The Company and Astellas will share equally the costs and expenses of developing and commercializing MDV3100 for the United States market, except that development costs for studies useful in both the United States market and either Europe or Japan, such as the ongoing Phase 3 AFFIRM study, will be shared 2/3 by Astellas and 1/3 by the Company. The Company and Astellas will share equally profits (or losses) resulting from commercialization of MDV3100 in the United States. Outside the United States,
68
Table of Contents
Astellas will bear all development and commercialization costs, and will pay the Company tiered royalties on aggregate net sales of MDV3100. In the fourth quarter of 2009, the Company received a non-refundable, up-front cash payment of $110.0 million pursuant to its Collaboration Agreement with Astellas, ten percent of which the Company paid to the academic institution from which it licensed it rights to MDV3100.
With Astellas, the Company is currently conducting a randomized, double-blind, placebo-controlled Phase 3 trial, know as the AFFIRM trial, of MDV3100 in men with a type of advanced prostate cancer known as castration-resistant prostate cancer, or CRPC.
The Company has funded its operations primarily through private and public offerings of its common stock, and from the up-front payment and cost-sharing payments from its Collaboration Agreements with Pfizer and Astellas. As of December 31, 2009, the Company had an accumulated deficit of $177.4 million and expects to incur substantial additional losses in the future as it continues research and development activities designed to support potential approval of its present and potential future product candidates.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
(a) Basis of Consolidation; Business Segments
The consolidated financial statements incorporate the accounts of Medivation and its operating subsidiaries. All significant inter-company transactions have been eliminated in consolidation. The Company operates in only one business segment.
(b) Estimates
The preparation of financial statements in conformity with generally accepted accounting principles requires that management make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Estimates and assumptions principally relate to the performance periods of the Company’s deliverables under its Collaboration Agreements with Pfizer and Astellas, services performed by third parties but not yet invoiced, estimates of the fair value and forfeiture rates of stock options issued to employees and consultants, and estimates of the probability and potential magnitude of contingent liabilities. Actual results could differ from those estimates.
(c) Cash Equivalents
Cash and cash equivalents are stated at cost, which approximates fair market value. The Company considers all highly liquid investments with a remaining maturity of three months or less at the time of acquisition to be cash equivalents.
(d) Short-Term Investments
The Company considers all highly liquid investments with a remaining maturity at the time of acquisition of more than three months but no longer than twelve months to be short-term investments. The Company classifies its securities as available-for-sale, which are reported at fair value with related unrealized gains and losses included as a component of stockholders’ equity. The amortized cost of debt securities in this category is adjusted for amortization of premiums and accretion of discounts to maturity. Such amortization is included in interest income. Realized gains and losses and declines in value judged to be other-than-temporary, if any, on available-for-sale securities are included in other income or expense. The cost of securities sold is based on the specific identification method. Interest and dividends on securities classified as available-for-sale are included in interest income.
69
Table of Contents
(e) Restricted cash
Restricted cash represents certificates of deposit held in the Company’s name with a major financial institution to secure the Company’s contingent obligations under irrevocable letters of credit issued to the lessors of the Company’s office facilities. Please refer to Note 13 for additional information.
(f) Fair value of financial instruments
The fair value of the Company’s cash equivalents and marketable securities is based on quoted market prices. Other financial instruments, including accounts receivable, accounts payable and accrued expenses, are carried at cost, which the Company believes approximates fair value because of the short-term maturities of these instruments.
(g) Concentration of Credit Risk
Financial instruments that potentially subject the Company to credit risk consist of cash, cash equivalents, short-term investments and receivables from collaboration partners to the extent of the amounts recorded on the balance sheets. The Company’s current investment policy is to invest only in a) debt securities issued by, or backed by the full faith and credit of, the U.S. government, b) repurchase agreements that are fully collateralized by such debt securities, and c) money market funds invested exclusively in the types of securities described in a) and b) above. Given this investment policy, the Company does not believe its exposure to credit risk with respect to the issuers of the securities in which it invests is material, and accordingly has no formal policy for mitigating such risk. The Company’s cash and cash equivalents are primarily invested in deposits and money market accounts with one major bank in the United States. Deposits in this bank may exceed the amount of insurance provided on such deposits. The Company’s receivables from collaborative partners at December 31, 2009 (Note 3) were collected in full subsequent to December 31, 2009.
(h) Property and Equipment
Property and equipment purchases are recorded at cost. Repairs and maintenance costs are expensed in the period incurred. Property and equipment is depreciated on a straight-line basis over the estimated useful lives of the assets as follows:
| Description |
Estimated Useful Life | |
| Office equipment and furniture |
3 years | |
| Software and computer equipment |
3-5 years | |
| Laboratory equipment |
5 years | |
| Leasehold improvements and fixtures |
Lesser of estimated useful life or life of lease |
(i) Other Comprehensive Income (Loss)
Other comprehensive income (loss) is comprised of unrealized holding gains and losses on the Company’s available-for-sale securities that are excluded from net income (loss) and reported separately in stockholders’ equity. The reconciliation of the Company’s other comprehensive loss for the years ended December 31, 2009, 2008 and 2007 is as follows:
| Year ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| (In thousands) | ||||||||||||
| Net loss |
$ | (54,753 | ) | $ | (62,460 | ) | $ | (31,743 | ) | |||
| Change in unrealized gain / (loss) on available-for-sale securities |
(705 | ) | 693 | — | ||||||||
| Total comprehensive loss |
$ | (55,458 | ) | $ | (61,767 | ) | $ | (31,743 | ) | |||
70
Table of Contents
(j) Revenue Recognition
Revenue recognized to date is attributable solely to amortization of the non-refundable up-front payments of $110.0 million and $225.0 million the Company received in the fourth quarters of 2009 and 2008, respectively, pursuant to its Collaboration Agreements with Astellas and Pfizer, respectively. For a description of the Collaboration Agreements, see Note 3.
The Company accounts for the various payment flows under its Collaboration Agreements in a consistent manner, as follows:
Upfront Payments
Both the Pfizer and Astellas Collaboration Agreements contain multiple elements and deliverables, and require evaluation pursuant to ASC 605-25 “Revenue Recognition—Multiple-Element Arrangements” (“ASC 605-25”). The Company evaluated the facts and circumstances of the Collaboration Agreements to determine whether it had obligations constituting deliverables under ASC 605-25. The Company concluded that it had multiple deliverables under both the Pfizer and Astellas Collaboration Agreements, including deliverables relating to grants of technology licenses, and performance of manufacturing, regulatory and clinical development activities in the U.S. In the case of the Pfizer Collaboration Agreement, management estimated that the period in which the Company would perform its deliverables began in the fourth quarter of 2008 and will be completed in the first quarter of 2012. In the case of the Astellas Collaboration Agreement, management estimated that the period in which the Company would perform its deliverables began in the fourth quarter of 2009 and will be completed in the fourth quarter of 2014. The Company also concluded that its deliverables under each Collaboration Agreement should be accounted for as a single unit of accounting under ASC 605-25. Accordingly, the Company will recognize the non-refundable, up-front payments from Pfizer and Astellas as revenue on a straight-line basis over the estimated performance period, using the proportional performance model.
Estimation of the performance periods of the Company’s deliverables requires the use of management’s judgment. Significant factors considered in management’s evaluation of the estimated performance periods include, but are not limited to, the Company’s experience, along with Pfizer’s and Astellas’ experience, in conducting clinical development activities. Given the negative CONNECTION data the Company reported in March 2010, the Company and Pfizer are currently re-evaluating which, if any, of the ongoing Phase 3 dimebon clinical trials should continue. This will be a complex analysis that will encompass scientific, clinical, regulatory and business issues. After this analysis is complete, the Company and Pfizer will be able to assess their respective future approaches to the clinical development of dimebon for Alzheimer’s and Huntington diseases. The Company does not know at this time which, if any, of the ongoing Phase 3 dimebon trials will be completed, or if Pfizer will elect to terminate the collaboration. If any changes to the ongoing dimebon development plan are made, or if Pfizer elects to terminate the collaboration, the Company may need to adjust its estimate of the performance period under the Pfizer collaboration. The Company will review the estimated duration of its performance periods under both collaborations on a quarterly basis and make any appropriate adjustments on a prospective basis. Future changes in estimates of either of the Company’s performance periods may materially impact the timing of future revenue recognized under the applicable Collaboration Agreement.
Milestone Payments
Under both the Pfizer and Astellas Collaboration Agreements, the Company is entitled to receive milestone payments based on achievement of specified development, regulatory and commercial events. Management evaluated the nature of the events triggering these contingent payments, and concluded that these events—except for (a) those relating to regulatory activities in Europe, development and regulatory activities in Japan, and commercial activities, all of which are areas in which the Company has no pertinent contractual responsibilities, and (b) the initiation of the second Phase 3 clinical trial under the Astellas Collaboration Agreement, an event which management deemed to be reasonably assured at the inception of the Astellas collaboration—constituted substantive milestones. This conclusion was based primarily on the facts that (i) each triggering event represents
71
Table of Contents
a specific outcome that can be achieved only through successful performance by the Company of one or more of its deliverables, (ii) achievement of each triggering event was subject to inherent risk and uncertainty and would result in additional payments becoming due to the Company, (iii) each of these milestones was substantive, based primarily on the facts that the payments they trigger are non-refundable, (iv) achievement of the milestone entails risk and was not reasonably assured at inception of the Collaboration Agreement, (v) substantial effort is required to complete each milestone, (vi) the amount of each milestone payment is reasonable in relation to the value created in achieving the milestone, (vii) a substantial amount of time is expected to pass between the up-front payment and the potential milestone payments, and (viii) the milestone payments relate solely to past performance. Based on the foregoing, the Company will recognize any revenue from these milestone payments under the substantive milestone method in the period in which the underlying triggering event occurs.
For the contingent payments triggered by events that do not constitute substantive milestones, management concluded that the appropriate revenue recognition treatment depends on whether the triggering event occurs during or after the performance period of the applicable Collaboration Agreement. Where the triggering event occurs during the applicable performance period, the Company will amortize any revenue from this event on a straight-line basis over the period beginning on the date the event occurs and ending at completion of the applicable performance period. Where the triggering event occurs after the applicable performance period, the Company will recognize the associated revenue in the period in which the event occurs.
Royalties and Profit Sharing Payments
Under both the Pfizer and Astellas Collaboration Agreements, the Company is entitled to receive profit sharing payments on sales of products in the U.S. and royalties on sales of products outside the U.S. The Company will recognize any revenue from these events based on the revenue recognition criteria set forth in ASC 605-10-25-1, “Revenue Recognition.” Based on those criteria, the Company considers these potential payments to be contingent revenues, and will recognize them as revenue in the period in which the applicable contingency is resolved.
Cost Sharing True-Up Payments
Under both the Pfizer and Astellas Collaboration Agreements, the Company and its partners share certain development and commercialization costs in the U.S. The parties make quarterly true-up payments between themselves to ensure that each has borne its applicable percentage of the shared development and commercialization costs. The Company’s policy is to account for cost-sharing true-up payments receivable by it as reductions in expense, and to account for cost-sharing true-up payments payable by it as increases in expense.
(l) Research and Development
Research and development expenses include personnel and facility-related expenses, outside contracted services including clinical trial costs, manufacturing and process development costs, research costs and other consulting services. Research and development costs are expensed as incurred. In instances where the Company enters into agreements with third parties to provide research and development services to the Company, costs are expensed as services are performed. Amounts due under such arrangements may be either fixed fee or fee for service, and may include upfront payments, monthly payments, and payments upon the completion of milestones or receipt of deliverables.
The Company’s cost accruals for clinical trials and other research and development activities are based on estimates of the services received and efforts expended pursuant to contracts with numerous clinical trial centers and contract research organizations. In the normal course of business the Company contracts with third parties to perform various research and development activities in the on-going development of its product candidates, including without limitation, third party clinical trial centers and contract research organizations that perform and administer clinical trials on its behalf. The financial terms of these agreements are subject to negotiation and vary
72
Table of Contents
from contract to contract and may result in uneven payment flows. Payments under these agreements depend on factors such as the achievement of certain events, the successful enrollment of patients, and the completion of portions of the clinical trial or similar conditions. The objective of the Company’s accrual policy is to match the recording of expenses in the financial statements to the actual services received and efforts expended. As such, expense accruals related to clinical trials and other research and development activities are recognized based on the estimated degree of completion of the events specified in the specific agreement.
(l) Stock Based Compensation
The Company records compensation expense associated with stock options and other equity-based compensation in accordance with ASC 718. ASC 718 requires the measurement and recognition of non-cash compensation expense for all share-based payment awards made to employees and directors, including employee stock options granted under our Amended and Restated 2004 Equity Incentive Award Plan, based on estimated fair values. The Company has applied the provisions of Staff Accounting Bulletin No. 107, or SAB 107, and Staff Accounting Bulletin No. 110, or SAB 110, in its application of ASC 718.
Stock compensation arrangements with non-employee service providers are accounted for in accordance with ASC 718 and ASC 505-50 Equity-Based Payments to Non-Employees using a fair value approach. The compensation costs of these arrangements are subject to remeasurement over the vesting terms as earned.
The Company recognized stock-based compensation expense of $10.7 million, $8.5 million and $5.9 million in the years ended December 31, 2009, 2008 and 2007, respectively. Please refer to Note 9(f) “Stock-Based Compensation” for additional information.
(m) Promotional and Advertising Expense
Promotional and advertising costs are classified as selling, general and administrative expenses, and are expensed as incurred. Promotional and advertising expenses consist primarily of the costs of designing, producing and distributing materials promoting the Company or its product candidates, including its corporate website. Promotional and advertising expenses were insignificant in the years ended December 31, 2009, 2008 and 2007.
(n) Income Taxes
The Company accounts for income taxes using an asset and liability approach in ASC 740-10, Accounting for Income Taxes, which requires the recognition of taxes payable or refundable for the current year and deferred tax assets and liabilities for the future tax consequences of events that have been recognized in the Consolidated Financial Statements or tax returns. The measurement of current and deferred tax assets and liabilities is based on provisions of the enacted tax law; the effects of future changes in tax laws or rates are not anticipated. The measurement of deferred tax assets is reduced, if necessary, by the amount of any tax benefits that, based on available evidence, is not expected to be realized.
The Company records a valuation allowance to reduce its deferred tax assets to the amount that it believes is more likely than not to be realized. Due to the Company’s lack of earnings history, the Company intends to maintain a full valuation allowance on the U.S. deferred tax assets until sufficient positive evidence exists to support reversal of the valuation allowance.
On January 1, 2007, the Company adopted ASC 740-10-25 (formerly FIN No. 48), “Accounting for Uncertainty in Income Taxes—an interpretation of FASB Statement No.109”. The Company did not have any unrecognized tax benefits and there was no effect on its financial condition or results of operations as a result of adopting ASC 740-10-25 (formerly FIN No. 48). The Company’s practice is to recognize interest and/or penalties related to income tax matters in income tax expense as incurred.
73
Table of Contents
(o) Net Loss per Common Share
Basic net loss per share is computed by dividing the net loss by the weighted-average number of common shares outstanding during the period. Diluted net loss per common share is computed similarly to basic net loss per share, except that the denominator is increased to include all potential dilutive common shares, including outstanding options, warrants and common stock subject to repurchase. Potential dilutive common shares have been excluded from the diluted loss per common share computations in all periods presented because such shares have an anti-dilutive effect on loss per share due to the Company’s net losses. There are no reconciling items used to calculate the weighted average number of common shares outstanding for basic and diluted net loss per share data.
Potential common shares outstanding at December 31, 2009, 2008 and 2007 were as follows:
| At December 31, | ||||||
| 2009 | 2008 | 2007 | ||||
| (In thousands) | ||||||
| Outstanding options |
5,604 | 4,856 | 3,359 | |||
| Outstanding warrants |
100 | 316 | 322 | |||
| Outstanding restricted shares |
11 | 30 | — | |||
| Total |
5,715 | 5,202 | 3,681 | |||
(p) Reclassification
Certain amounts in prior years’ financial statements have been reclassified to conform to the current period presentation.
(q) Recently Issued Accounting Pronouncements
In April 2009, the Financial Accounting Standards Board (FASB) issued several pronouncements related to fair value measurement, recording and disclosure in financial reporting.
ASC 825-10 “Financial Instruments” and ASC 270-10, “Interim Reporting,” were issued to outline the required financial statement disclosures relating to fair value of financial instruments during interim reporting periods. ASC 820-10, “Fair Value Measurements and Disclosures,” was issued to provide additional guidance in evaluating the fair value of a financial instrument when the volume and level of activity for the asset or liability has significantly decreased. ASC 320-10, “Recognition Investments—Debt & Equity Securities,” was issued to provide additional guidance on presenting impairment losses on securities.
All of the fair value measurement pronouncements were effective for interim and annual reporting periods ending after June 15, 2009. The adoption of these new pronouncements did not have a material effect on the Company’s consolidated results of operations or financial condition.
In May 2009, the FASB issued ASC 855-10, “Subsequent Events.” ASC 855-10 is intended to establish general standards of accounting for and disclosure of events that occur after the balance sheet date but before financial statements are issued or are available to be issued. ASC 855-10 is effective for interim or annual financial periods ending after June 15, 2009. The adoption of ASC 855-10 did not have a material effect on the Company’s consolidated results of operations or financial condition.
In June 2009, the FASB issued ASC 810, “Consolidation” (ASC 810), which changes the approach in determining the primary beneficiary of a variable interest entity (VIE) and requires companies to more frequently assess whether they must consolidate VIEs. ASC 810 is effective for annual periods beginning after November 15, 2009. We are evaluating the impact, if any, the adoption of ASC 810 will have on our consolidated financial statements.
In October 2009, the FASB issued Accounting Standards Update (ASU) No. 2009-13, “Multiple-Deliverable Revenue Arrangements” (ASU 2009-13). This update removes the criterion that entities must use objective and reliable evidence of fair value in separately accounting for deliverables and provides entities with a
74
Table of Contents
hierarchy of evidence that must be considered when allocating arrangement consideration. The new guidance also requires entities to allocate arrangement consideration to the separate units of accounting based on the deliverables’ relative selling price. The provisions will be effective for revenue arrangements entered into or materially modified in the Company’s fiscal year 2011. The Company is currently evaluating the impact of the provisions of ASU 2009-13 but does not intend to early adopt.
3. COLLABORATION AGREEMENTS
(a) Collaboration Agreement with Pfizer
In September 2008, the Company announced a Collaboration Agreement with Pfizer. Under this agreement, the Company and Pfizer agreed to collaborate on development of dimebon for Alzheimer’s disease and Huntington disease for the United States market, including associated regulatory filings with the FDA. In addition, if approved by the FDA, following such approval and the launch of dimebon in the United States, the Company, at its option, and Pfizer will co-promote dimebon to specialty physicians in the United States, and Pfizer will promote dimebon to primary care physicians in the United States. Pfizer will be responsible for development and seeking regulatory approval for, and commercialization of, dimebon outside the United States. Pfizer is responsible for all manufacture of product for both clinical and commercial purposes. Both the Company and Pfizer have agreed not to commercialize for the treatment of specified indications any other products directed to the same primary molecular target as dimebon for a specified time period, subject to certain exceptions.
The agreement establishes several joint committees consisting of an equal number of representatives from both parties that will operate by consensus to oversee the collaboration. In the event that a joint committee is unable to reach consensus on a particular issue, then, depending on the issue, a dispute may be decided at the joint committee level by the party with the final decision on the issue or escalated to senior management of the parties. If a dispute is escalated to senior management and no consensus is reached, then the dispute may be decided by the party to whom the contract grants final decision on such issue. Other issues can only be decided by consensus of the parties, and unless and until the parties’ representatives reach agreement on such issue, no decision on such issue will be made, and the status quo will be maintained.
Under the Pfizer Collaboration Agreement, Pfizer paid the Company an up-front cash payment of $225 million in the fourth quarter of 2008. The Company is also eligible to receive payments of up to $500 million upon the attainment of development and regulatory milestones plus additional milestone payments upon the achievement of certain net sales levels for the product. The Company and Pfizer will share the costs and expenses of developing and commercializing dimebon for the United States market on a 60%/40% basis, with Pfizer assuming the larger share, and the Company and Pfizer will share profits (or losses) resulting from the commercialization of dimebon in the United States in such proportions. Outside the United States, Pfizer will bear all development and commercialization costs and will pay the Company tiered royalties on the aggregate net sales of dimebon.
The Company is permitted to terminate the Pfizer Collaboration Agreement for an uncured material breach by Pfizer. Pfizer has a right to terminate the Pfizer Collaboration Agreement unilaterally at any time. In the event of an uncured material breach of the Pfizer Collaboration Agreement by the Company, Pfizer may elect either to terminate the Pfizer Collaboration Agreement or to keep the Pfizer Collaboration Agreement in place, but terminate the Company’s right to participate in development, commercialization (other than co-promoting dimebon) and other activities for dimebon, including the joint committees and decision making for dimebon. However, such termination would not affect the Company’s financial return or, unless the Company commits an uncured material breach of its co-promotion obligations, the Company’s co-promotion rights. Following any termination of the Pfizer Collaboration Agreement, all rights to develop and commercialize dimebon will revert to the Company, and Pfizer will grant a license to the Company to enable the Company to continue such development and commercialization, remain responsible for its ongoing financial and other obligations under the Collaboration Agreement for a transition period of six months following termination, and is obligated to supply product to the Company for a reasonable period of time, not to exceed eighteen months following termination, on terms to be negotiated between the parties in good faith.
75
Table of Contents
For the years ended December 31, 2009 and 2008, collaboration revenue recognized pursuant to the Pfizer Collaboration Agreement was $65.4 million and $12.6 million, respectively, representing the amortized portion of the $225.0 million up-front payment received from Pfizer. The remaining $147.0 million of the up-front payment has been recorded as deferred revenue, and will be amortized on a straight-line basis over the expected performance period of the Company’s deliverables under the Collaboration Agreement. Management currently expects this performance period to end in the first quarter of 2012. Any milestone payments received by the Company pursuant to the Pfizer Collaboration Agreement will be recognized as revenue in the period in which the underlying milestone event is achieved, other than milestone payments triggered by events that occur during the performance period and do not qualify as substantive milestones, which will be amortized on a straight-line basis over the period beginning on the date such triggering event occurs and ending on completion of the performance period. Any profit sharing and royalty payments received by the Company pursuant to the Pfizer Collaboration Agreement will be recognized as revenue in the period in which the underlying product sales occur.
The Company’s policy is to present true-up payments of development and commercialization costs, whether to or from the Company, within the applicable expense line in the statement of operations. True-up payments by the Company to Pfizer are thus presented as an increase in expense in the Company’s statement of operations, while true-up payments by Pfizer to the Company are presented as a reduction in expense. For the year ended December 31, 2009, the Company recorded development cost true-up payments receivable from Pfizer of $20.4 million and commercialization cost true-up payments payable to Pfizer of $0.7 million, and a corresponding reduction in research and development expense and increase in selling, general and administrative expense. For the year ended December 31, 2008, the Company recorded development and commercialization cost true-up payments receivable from Pfizer of $3.2 million and $0.3 million, respectively, and corresponding reductions in research and development expense and selling, general and administrative expense.
(b) Collaboration Agreement with Astellas
In October 2009, the Company announced a collaboration Agreement with Astellas. Under the Astellas Collaboration Agreement, the Company and Astellas agreed to collaborate on the development of MDV3100 for prostate cancer for the United States market, including associated regulatory filings with the FDA. In addition, if approved by the FDA, following such approval and the launch of MDV3100 in the United States, the Company, at its option, and Astellas will co-promote MDV3100 in the United States. Astellas is responsible for development of, seeking regulatory approval for and commercialization of MDV3100 outside the United States. Astellas will be responsible for commercial manufacture of MDV3100 on a global basis. Both Medivation and Astellas have agreed not to commercialize certain other products having a similar mechanism of action as MDV3100 for the treatment of specified indications for a specified time period, subject to certain exceptions.
The agreement establishes several joint committees consisting of an equal number of representatives from both parties that will operate by consensus to oversee the collaboration. In the event that a joint committee is unable to reach consensus on a particular issue, then, depending on the issue, a dispute may be decided at the joint committee level by the party with the final decision on the issue or escalated to senior management of the parties. If a dispute is escalated to senior management and no consensus is reached, then the dispute may be decided by the party to whom the contract grants final decision on such issue. Other issues can only be decided by consensus of the parties, and unless and until the parties’ representatives reach agreement on such issue, no decision on such issue will be made, and the status quo will be maintained.
Under the Astellas Collaboration Agreement, Astellas paid the Company an up-front cash payment of $110.0 million in the fourth quarter of 2009. The Company is also entitled to receive up to $335.0 million in development milestone payments, plus up to an additional $320.0 million in commercial milestone payments. The Company is required to share 10% of the up-front payment and any development milestone payments received under the Astellas Collaboration Agreement with the academic institution from which it licensed its rights to MDV3100. The Company paid 10% of the up-front payment, or $11.0 million, to the academic institution in the fourth quarter of 2009. The Company and Astellas will share equally the costs and expenses of
76
Table of Contents
developing and commercializing MDV3100 for the United States market, except that development costs for studies useful in both the United States market and either Europe or Japan, such as the ongoing Phase 3 AFFIRM study, will be shared 2/3 by Astellas and 1/3 by the Company. The Company and Astellas will share profits (or losses) resulting from the commercialization of MDV3100 in the United States equally. Outside the United States, Astellas will bear all development and commercialization costs and will pay the Company tiered, double-digit royalties on the aggregate net sales of MDV3100.
The Company and Astellas each are permitted to terminate the Astellas Collaboration Agreement for an uncured material breach by, or the insolvency of, the other party. Astellas has a right to terminate the Astellas Collaboration Agreement unilaterally by advance written notice to the Company, but except in certain specific circumstances, generally cannot exercise that termination right until the first anniversary of MDV3100’s first commercial sale. Following any termination of the Astellas Collaboration Agreement in its entirety, all rights to develop and commercialize MDV3100 will revert to the Company, and Astellas will grant a license to the Company to enable the Company to continue such development and commercialization. In addition, except in the case of a termination by Astellas for an uncured material breach, Astellas will supply MDV3100 to the Company during a specified transition period.
For the year ended December 31, 2009, collaboration revenue recognized pursuant to the Astellas Collaboration Agreement was $3.9 million, representing the amortized portion of the $110.0 million up-front payment received from Astellas in the fourth quarter of 2009. The remaining $106.1 million of the up-front payment has been recorded as deferred revenue, and will be amortized on a straight-line basis over the expected performance period of the Company’s deliverables under the Astellas Collaboration Agreement. Management currently expects this performance period to end in the fourth quarter of 2014. Any milestone payments received by the Company pursuant to the Astellas Collaboration Agreement will be recognized as revenue in the period in which the underlying milestone event is achieved, other than milestone payments triggered by events that occur during the performance period and do not qualify as substantive milestones, which will be amortized on a straight-line basis over the period beginning on the date such triggering event occurs and ending on completion of the performance period. Any profit sharing and royalty payments received by the Company pursuant to the Astellas Collaboration Agreement will be recognized as revenue in the period in which the underlying product sales occur.
The Company’s policy is to present true-up payments of development and commercialization costs, whether to or from the Company, within the applicable expense line in the statement of operations. True-up payments by the Company to Astellas are thus presented as an increase in expense in the Company’s statement of operations, while true-up payments by Astellas to the Company are presented as a reduction in expense. For the year ended December 31, 2009, the Company recorded development and commercialization cost true-up payments receivable from Astellas of $2.8 and $0.1 million, respectively, and corresponding reductions in research and development expense and selling, general and administrative expense.
4. SHORT-TERM INVESTMENTS
As of December 31, 2009, the amortized cost, gross unrealized gain, and estimated fair value for available-for-sale securities, consisting solely of United States treasury notes maturing in February, July and August 2010, was $220.8 million, $0.0 million and $220.8 million, respectively. As of December 31, 2008, the amortized cost, gross unrealized gain, and estimated fair value for available-for-sale securities, consisting solely of United States treasury notes maturing in April and June 2009, was $149.3 million, $0.7 million and $150.0 million, respectively.
77
Table of Contents
5. PROPERTY AND EQUIPMENT
The components of the Company’s property and equipment and related accumulated depreciation and amortization at December 31, 2009 and 2008 were as follows (in thousands):
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Furniture and fixtures |
$ | 221 | $ | 72 | ||||
| Leasehold improvements |
630 | 594 | ||||||
| Computer equipment and software |
484 | 142 | ||||||
| Laboratory equipment |
311 | 163 | ||||||
| Construction in progress |
11 | 55 | ||||||
| 1,657 | 1,026 | |||||||
| Less: accumulated depreciation and amortization |
(565 | ) | (258 | ) | ||||
| $ | 1,092 | $ | 768 | |||||
Depreciation and amortization expense on property and equipment was $307,000, $189,000 and $70,000 for the years ended December 31, 2009, 2008 and 2007, respectively.
6. ACCRUED EXPENSES
Accrued expenses at December 31, 2009 and 2008 consisted of the following (in thousands):
| December 31, | ||||||
| 2009 | 2008 | |||||
| Payroll and payroll related |
$ | 759 | $ | 260 | ||
| Preclinical and clinical trials |
10,529 | 4,979 | ||||
| Other |
766 | 533 | ||||
| Total accrued expenses |
$ | 12,054 | $ | 5,772 | ||
7. DEFERRED REVENUE
Deferred revenue at December 31, 2009 and 2008 consisted of the following (in thousands):
| December 31, | ||||||
| 2009 | 2008 | |||||
| Current portion: |
||||||
| Deferred revenue related to Pfizer (see Note 3a) |
$ | 65,361 | $ | 64,286 | ||
| Deferred revenue related to Astellas (see Note 3b) |
21,209 | — | ||||
| Total |
$ | 86,570 | $ | 64,286 | ||
| Long-term portion: |
||||||
| Deferred revenue related to Pfizer (see Note 3a) |
$ | 81,701 | $ | 148,137 | ||
| Deferred revenue related to Astellas (see Note 3b) |
84,897 | — | ||||
| Total |
$ | 166,598 | $ | 148,137 | ||
78
Table of Contents
8. OTHER NON-CURRENT LIABILITIES
Other non-current liabilities at December 31, 2009 and 2008 consisted of the following (in thousands):
| December 31, | ||||||
| 2009 | 2008 | |||||
| Deferred rent and lease incentives |
$ | 262 | $ | 324 | ||
| Other non-current liabilities |
292 | 86 | ||||
| Total other non-current liabilities |
$ | 554 | $ | 410 | ||
9. STOCKHOLDERS’ EQUITY
(a) Common Stock
In June 2009 and 2008, the Company raised $62.4 million and $15.0 million respectively in public offerings of 3,162,500 and 1,129,518 shares of its Common Stock pursuant to an effective registration statement on Form S-3 previously filed with the Commission. Cash offering costs of $369,000 and $69,000 incurred in connection with these public offerings were charged against additional paid-in capital in the years ended December 31, 2009 and 2008, respectively.
(b) Stock Purchase Rights
All shares of the Company’s common stock, if issued prior to the termination by the Company of its rights agreement, dated as of December 4, 2006, include stock purchase rights. The rights are exercisable only if a person or group acquires twenty percent or more of the Company’s common stock or announces a tender or exchange offer which would result in ownership of twenty percent or more of the Company’s common stock. Following the acquisition of twenty percent or more of the Company’s common stock, the holders of the rights, other than the acquiring person or group, may purchase Medivation common stock at half of its fair market value. In the event of a merger or other acquisition of the Company, the holders of the rights, other than the acquiring person or group, may purchase shares of the acquiring entity at half of their fair market value. The rights were not exercisable at December 31, 2009.
(c) Medivation Equity Incentive Plan
The Medivation Amended and Restated 2004 Equity Incentive Award Plan, or the Medivation Equity Incentive Plan, which is stockholder-approved, provides for the issuance of options and other stock-based awards, including restricted stock and stock appreciation rights, covering up to 7,500,000 shares of Medivation’s common stock. Shares issued upon exercise of stock-based awards are new shares that have been reserved for issuance under the plan. The amendments to the Medivation Equity Incentive Plan were approved by the Board and by the stockholders in March and May 2007, respectively.
The Medivation Equity Incentive Plan is administered by the Board, or a committee appointed by the Board, which determines recipients and types of awards to be granted, including the number of shares subject to the awards, the exercise price and the vesting schedule. The term of stock options granted under the Medivation Equity Incentive Plan cannot exceed ten years. Options generally have an exercise price equal to the fair market value of the common stock on the grant date, and generally vest over a period of four years. The options may contain an early exercise feature, pursuant to which the optionee may exercise the option before it has vested. However, so long as an option remains unvested, all shares purchased upon early exercise remain subject to repurchase by Medivation at the option exercise price if the optionee’s service with Medivation terminates. For purposes of the following disclosures, early exercise options are not considered to have been exercised, or to be exercisable, until this repurchase right has lapsed. To date, the Company has not issued any shares upon early exercise of stock options. In addition, all outstanding awards under the Medivation Equity Incentive Plan will accelerate and become immediately exercisable upon a “change of control” of Medivation, as defined in the Medivation Equity Incentive Plan.
79
Table of Contents
The following table summarizes stock option activity under the Medivation Equity Incentive Plan for the years ended December 31, 2009, 2008 and 2007:
| Number of Shares |
Weighted Average Exercise price | |||||
| Options outstanding, December 31, 2006 |
2,211,750 | $ | 4.53 | |||
| Granted |
1,309,566 | $ | 19.83 | |||
| Exercised |
(66,190 | ) | $ | 3.57 | ||
| Forfeited |
(95,800 | ) | $ | 18.90 | ||
| Options outstanding, December 31, 2007 |
3,359,326 | $ | 10.10 | |||
| Granted |
1,629,448 | $ | 17.96 | |||
| Exercised |
(116,086 | ) | $ | 5.68 | ||
| Forfeited |
(16,783 | ) | $ | 20.83 | ||
| Options outstanding, December 31, 2008 |
4,855,905 | $ | 12.81 | |||
| Granted |
1,140,543 | $ | 31.53 | |||
| Exercised |
(344,430 | ) | $ | 10.62 | ||
| Forfeited |
(48,058 | ) | $ | 18.12 | ||
| Options outstanding, December 31, 2009 |
5,603,960 | $ | 16.71 | |||
The weighted-average grant-date fair value of options granted during the years ended December 31, 2009, 2008 and 2007 was $23.04 per share, $11.91 per share and $11.78 per share, respectively. Further information regarding the value of options vested and exercised during the years ended December 31, 2009, 2008 and 2007, is set forth below.
| Year Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| (in thousands) | |||||||||
| Fair value of options vested during period |
$ | 11,653 | $ | 7,717 | $ | 3,916 | |||
| Intrinsic value of options exercised during period |
$ | 6,208 | $ | 2,198 | $ | 915 | |||
Further information regarding the stock options outstanding and exercisable as of December 31, 2009, is as follows:
| Options Outstanding |
Options Exercisable | |||||||||||
| Range of Exercise Prices |
Number Outstanding |
Weighted Avg. Remaining Contractual Life (yrs) |
Weighted Avg. Exercise Price |
Number Exercisable |
Weighted Avg. Exercise Price | |||||||
| $ 0 .02 – 1.55 |
549,500 | 4.8 | $ | 0.96 | 549,500 | $ | 0.96 | |||||
| 2.60 – 4.30 |
572,970 | 5.4 | 3.50 | 571,413 | 3.51 | |||||||
| 4.45 – 11.76 |
718,900 | 6.7 | 7.87 | 577,786 | 7.81 | |||||||
| 13.31 – 20.00 |
1,898,207 | 8.3 | 17.55 | 727,963 | 17.53 | |||||||
| 20.83 – 26.03 |
1,864,383 | 9.1 | 27.96 | 394,396 | 21.57 | |||||||
| 5,603,960 | 7.7 | 16.71 | 2,821,058 | 10.04 | ||||||||
As of December 31, 2009, the aggregate intrinsic value of total options outstanding was $117.3 million, of which $77.9 million represented vested options.
As of December 31, 2009, the weighted average remaining contractual life of options outstanding and of options exercisable was 7.7 years and 6.6 years, respectively.
As of December 31, 2009, consultants held an aggregate of 86,435 unvested options at a weighted average exercise price of $27.30 per share.
80
Table of Contents
(d) Restricted Stock Awards:
The following table summarizes information about restricted stock award activity for the year ended December 31, 2009:
| Shares | Weighted Average Grant Date Fair Value |
Weighted Average Remaining Contractual Term (in years) |
Aggregate Intrinsic Value in $000s | |||||||
| Restricted Stock Awards: |
||||||||||
| Balance at December 31, 2007 |
— | — | ||||||||
| Granted |
30,000 | $ | 15.71 | |||||||
| Vested |
— | — | ||||||||
| Cancelled |
— | — | ||||||||
| Balance at December 31, 2008 |
30,000 | $ | 15.71 | 2.04 | $ | 437 | ||||
| Granted |
— | — | ||||||||
| Vested |
19,166 | 15.71 | ||||||||
| Cancelled |
— | — | ||||||||
| Balance at December 31, 2009 |
10,834 | $ | 15.71 | 1.04 | $ | 408 | ||||
The Company measures the fair value of restricted stock awards using the closing price of the Company’s stock on the grant date. The fair value of the restricted stock awards is being amortized on a straight-line basis over the requisite service period of the awards.
(e) Warrants
At December 31, 2009, warrants to purchase an aggregate of 100,323 shares of Medivation common stock at a weighted average exercise price of $2.78 per share were outstanding. These outstanding warrants expire between 2014 and 2017.
(f) Stock-Based Compensation
The Company applies Statement of Financial Accounting Standards ASC 718, Stock Compensation. ASC 718 requires the measurement and recognition of non-cash compensation expense for all share-based payment awards made to employees and directors, including employee stock options and employee stock purchases related to our Restated 2004 Equity Incentive Award Plan, based on estimated fair values. The Company has applied the provisions of Staff Accounting Bulletin No. 107, or SAB 107, and Staff Accounting Bulletin No. 110, or SAB 110, in our adoption of ASC 718.
Stock compensation arrangements with non-employee service providers are accounted for in accordance with ASC 505-50 Equity-Based Payments to Non-Employees using a fair value approach. The compensation costs of these arrangements are subject to remeasurement over the vesting terms as earned.
The Company estimates the fair value of stock options and warrants using the Black-Scholes option valuation model. Estimated volatility is based on the historical stock price volatility of the Company’s common stock, the historical stock price volatility of comparable companies’ common stock and the implied volatility of the Company’s common stock inherent in the market prices of publicly traded options in its common stock. Estimated dividend yield is 0%. The risk-free rate is estimated to equal U.S. Treasury security rates for the applicable terms. The Company does not have sufficient history of exercise behavior to develop estimates of expected term since as of December 31, 2009 only nine percent of the options it has issued since inception had been exercised. Accordingly, the Company uses the simplified method of estimating option term provided for in
81
Table of Contents
the Commission’s Staff Accounting Bulletins 107 and 110 for options granted to employees and directors, which results in an estimated option term of six years. For consultant options, the Company uses an estimated option term of four years, which is equal to the period required for the options to vest in full. As the Company gathers more history regarding its option exercise pattern, and as information regarding the option exercise pattern of comparable companies in its industry becomes publicly available, it will review these estimates and revise them as appropriate. Different estimates of volatility and expected term could materially change the value of an option and the resulting expense.
Stock-based awards granted to employees and directors are valued at their respective grant dates and expensed over the remaining vesting period of the award. The Black-Scholes assumptions used in the years ended December 31, 2009, 2008 and 2007 for employee and director options are as follows:
| Year Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Risk-free interest rate |
1.71–2.87 | % | 2.96–3.94 | % | 3.83–5.1 | % | |||
| Expected remaining term (in years) |
6 | 6 | 6 | ||||||
| Expected volatility |
72–88 | % | 62–84 | % | 58–60 | % | |||
| Expected dividends |
None | None | None | ||||||
Stock-based awards granted to consultants are valued at their respective measurement dates and recognized as expense based on the portion of the total consulting services provided during the applicable period. As further consulting services are provided in each period, the Company will revalue the associated awards and recognize additional expense based on their then-current fair values. The Black-Scholes assumptions used in the years ended December 31, 2009, 2008 and 2007 for consultant options are as follows:
| Year Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Risk-free interest rate |
0.37–1.70 | % | 0.37–3.36 | % | 3.23–4.94 | % | |||
| Expected remaining term (in years) |
0.3–4 | 0.2–4 | 1.3–4 | ||||||
| Expected volatility |
74–91 | % | 60–87 | % | 54–60 | % | |||
| Expected dividends |
None | None | None | ||||||
The Company recognized stock-based compensation expense of $10.7 million, $8.5 and $5.9 million in the years ended December 31, 2009, 2008 and 2007 respectively. The following table summarizes information related to stock-based compensation expense recognized in the income statement (in thousands):
| Year Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Stock compensation expense recognized in: |
|||||||||
| Research and development expense |
$ | 5,664 | $ | 4,216 | $ | 2,040 | |||
| General and administrative expense |
5,062 | 4,331 | 3,853 | ||||||
| Total stock-based compensation expense |
$ | 10,726 | $ | 8,547 | $ | 5,893 | |||
Stock-based compensation expense attributable to employee and director awards was $10.0 million, $7.1 million and $2.8 million in the years ended December 31, 2009, 2008 and 2007, respectively. Stock-based compensation expense attributable to consultant awards was $0.7 million, $1.5 million and $3.1 million in the years ended December 31, 2009, 2008 and 2007, respectively.
At December 31, 2009, the unrecognized compensation cost attributable to employee and director awards totaled $42.3 million, which is expected to be recognized as expense over a weighted-average remaining requisite service period of 2.8 years.
82
Table of Contents
10. RETIREMENT PLAN
The Company has a 401(k) defined contribution savings plan (401(k) Plan). The 401(k) Plan is for the benefit of all employees and permits voluntary contributions by employees up to 80% of their annual pretax compensation limited by the IRS-imposed maximum. Effective January 1, 2009, the Company matched 75% of each employee’s contributions up to a maximum 6% of the employee’s eligible earnings. Employer contributions to the plan were $0.4 million for the year ended December 31, 2009.
11. INCOME TAXES
The income tax expense (benefit) for the years ended December 31, 2009, 2008 and 2007 consisted of the following (in thousands):
| Year Ended December 31, | ||||||||||
| 2009 | 2008 | 2007 | ||||||||
| Current |
||||||||||
| Federal |
$ | 441 | $ | (12 | ) | $ | — | |||
| State |
7,831 | 2 | 2 | |||||||
| $ | 8,272 | $ | (10 | ) | $ | 2 | ||||
| Deferred |
||||||||||
| Federal |
$ | — | $ | — | $ | — | ||||
| State |
— | — | — | |||||||
| Total deferred |
$ | — | $ | — | $ | — | ||||
| Total income tax expense |
$ | 8,272 | $ | (10 | ) | $ | 2 | |||
A reconciliation of the statutory federal income tax to the Company’s effective tax rates for the periods ended is as follows:
| December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Federal tax provision at statutory rate |
35.00 | % | 35.00 | % | 34.00 | % | |||
| State taxes (net of federal benefit) |
6.91 | % | 5.73 | % | 5.83 | % | |||
| Stock-based compensation |
(1.26 | %) | (0.86 | %) | (0.25 | %) | |||
| Research and development credits |
6.54 | % | 1.53 | % | 1.31 | % | |||
| Other |
1.52 | % | 0.41 | % | (0.22 | %) | |||
| Valuation allowance |
(66.6 | %) | (41.81 | %) | (40.67 | %) | |||
| Provision for taxes |
(17.89 | %) | 0.00 | % | 0.00 | % | |||
83
Table of Contents
Deferred income taxes reflect the net tax effects of temporary difference between the carrying amounts of assets for financial reporting purposes and the amount used for income tax purposes. Significant components of the Company’s deferred tax assets for federal and state income taxes are follows (in thousands):
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Deferred tax assets |
||||||||
| Deferred Revenue |
$ | 59,922 | $ | — | ||||
| Net operating loss carry forward |
5,912 | 41,911 | ||||||
| Stock-based compensation |
7,552 | 4,924 | ||||||
| Research & development credit |
4,430 | 2,912 | ||||||
| Depreciation, amortization and other |
16 | 115 | ||||||
| Accruals and reserves |
3,091 | 256 | ||||||
| Total deferred tax assets |
80,923 | 50,118 | ||||||
| Valuation allowance |
(80,923 | ) | (50,118 | ) | ||||
| Net deferred tax assets |
$ | — | $ | — | ||||
During the year ended December 31, 2009, the Company accelerated the recognition of revenue related to the Pfizer non-refundable, up-front payment received in 2008 for income tax purposes, while such revenue is deferred for financial statement purposes. Due to the suspension of California Net Operating Loss utilization in 2009, the Company was not able to utilize the NOL carryforwards to offset the taxable income in 2009.
Due to the Company’s lack of earnings history, the net deferred tax assets have been fully offset by a valuation allowance. The valuation allowance increased by $30.8 million, $26.1 million and $12.9 million during the years ended December 31, 2009, 2008 and 2007 respectively.
On January 1, 2007, the Company adopted ASC 740-10-25, “Accounting for Uncertainty in Income Taxes” (ASC 740-10-25). The Company did not have any unrecognized tax benefits and there was no effect on our financial condition or results of operations as a result of adopting ASC 740-10-25. The Company’s practice is to recognize interest and/or penalties related to income tax matters in income tax expense as incurred.
At December 31, 2009 and 2008,, the Company has approximately $0.9 million and $0.4 million, respectively, in total unrecognized tax benefit. Approximately $0.2 million of the total gross unrecognized tax benefit at December 31, 2009, if recognized, would affect the effective tax rate. No interest and penalty has been recognized for the years ended December 31, 2009, 2008 and 2007. A reconciliation of the beginning and ending amount of unrecognized tax benefits is as follows (in thousands):
| December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Balance as of beginning of year |
$ | 358 | $ | — | $ | — | |||
| Additions based on tax positions related to the current year |
429 | 145 | — | ||||||
| Reductions based on tax positions related to prior years |
102 | 213 | — | ||||||
| Settlements |
— | — | — | ||||||
| Balance as of end of year |
$ | 889 | $ | 358 | $ | — | |||
The Company currently has no federal or state tax examinations in progress nor has it had any federal or state tax examinations since its inception. As a result of the Company’s net operating loss carryforwards, all of its tax years are subject to federal and state tax examination.
Federal and state tax laws impose substantial restrictions on the utilization of net operating loss (NOL) and credit carryforwards in the event of an “ownership change” for tax purposes, as defined in IRC Section 382. The Company completed Section 382 studies through December 31, 2009, and concluded that ownership changes
84
Table of Contents
occurred in 2004 and 2007. The ownership changes did not result in a reduction of its net operating loss or in its research and development credits expiring unused. During the year ended December 31, 2009, the Company was able to utilize only $104.7 million of its federal net operating loss due to the IRC Section 382 limitation. As of December 31, 2009, the Company has federal net operating loss carryforwards of approximately $1 million. As of December 31, 2008, the Company had federal and state net operating loss carryforwards of approximately $105.5 million, and $105.6 million, respectively. Approximately $0.5 million of the total net operating loss carryforwards is attributable to excess employee stock option deductions, the benefit from which will be credited to additional paid-in capital when subsequently utilized in future years.
In response to budgetary pressures, the State of California has temporarily suspended the use of net operating loss carryforwards for the year 2008 and 2009. The Company’s ability to utilize its California net operating loss carryforwards is limited to offset its current year taxable income. As of December 31, 2009, the Company has state net operating loss carryforwards of approximately $105.6 million, which will expire at various dates between the years 2014 and 2021, if not utilized.
In addition, the Company had federal research and development credit carryforwards of approximately $6.2 million and $2.6 million as of December 31, 2009 and December 31, 2008, respectively. The federal tax credit carryforwards expires in the year 2024 through 2029, if not used. At December 31, 2008, the Company had state research and development credit carryforwards of approximately $0.9 million. The Company utilized all of its California research and development credit carryforwards in 2009 and therefore, has no state research and development credit carryforwards at December 31, 2009.
12. FAIR VALUE MEASUREMENTS
The Company follows ASC 820-10, “Fair Value Measurements and Disclosures” (ASC 820-10), which among other things, defines fair value, establishes a consistent framework for measuring fair value and expands disclosure for each major asset and liability category measured at fair value on either a recurring or nonrecurring basis. Fair value is an exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is a market-based measurement that should be determined based on assumptions that market participants would use in pricing an asset or liability. As a basis for considering such assumptions, a three-tier fair value hierarchy has been established, which prioritizes the inputs used in measuring fair value as follows:
| • | Level 1—Inputs are unadjusted, quoted prices in active markets for identical assets or liabilities at the measurement date. |
Fair valued assets that are generally included in this category are cash equivalents comprised of money market funds, restricted cash and short-term investments.
| • | Level 2—Inputs (other than quoted prices included in Level 1) are either directly or indirectly observable for the asset or liability through correlation with market data at the measurement date and for the duration of the instrument’s anticipated life. |
At December 31, 2009 and 2008, the Company did not have any fair valued assets or liabilities classified as Level 2.
| • | Level 3—Inputs reflect management’s best estimate of what market participants would use in pricing the asset or liability at the measurement date. Consideration is given to the risk inherent in the valuation technique and the risk inherent in the inputs to the model. |
85
Table of Contents
At December 31, 2009 and 2008, the Company did not have any fair valued assets or liabilities classified as Level 3.
Assets measured at fair value as of December 31, 2009 and 2008 are classified below based on the three fair value hierarchy tiers described above (in thousands):
| Fair value measurements using | ||||||||||||
| Carrying Value | Level 1 | Level 2 | Level 3 | |||||||||
| December 31, 2009: |
||||||||||||
| Assets |
||||||||||||
| Cash equivalents |
$ | 41,761 | $ | 41,761 | $ | — | $ | — | ||||
| Short-term Investments |
220,781 | 220,781 | — | — | ||||||||
| Restricted cash |
2,343 | 2,343 | — | — | ||||||||
| Total assets |
$ | 264,885 | $ | 264,885 | $ | — | $ | — | ||||
| December 31, 2008: |
||||||||||||
| Assets |
||||||||||||
| Cash equivalents |
$ | 71,351 | $ | 71,351 | $ | — | $ | — | ||||
| Short-term Investments |
149,968 | 149,968 | — | — | ||||||||
| Restricted cash |
843 | 843 | — | — | ||||||||
| Total assets |
$ | 222,162 | $ | 222,162 | $ | — | $ | — | ||||
13. COMMITMENTS AND CONTINGENCIES
For the conduct of its operations, the Company leases approximately 34,000 square feet of office space located at 201 Spear Street, San Francisco, California 94105 pursuant to leases that expire in July 2012 and May 2013. In November 2009 the Company signed a lease for approximately 64,000 square feet of office space located at 345 Spear Street, San Francisco, California 94105. Because of the negative CONNECTION trial results, the Company terminated the 345 Spear Street lease in March 2010, and paid a $1.5 million termination fee to the landlord, half of which was recorded as expense in the fourth quarter of 2009 and the remaining half of which will be recorded as expense in the first quarter of 2010. This expense recognition represents amortization of the $1.5 million minimum lease payments over the term of the lease. The Company also leases 5,700 square feet of office space located at 55 Hawthorne Street, San Francisco, California 94105, its former office location. The Company has sub-leased the Hawthorne Street space to a third party through February 2011.
The Company is committed to pay a portion of the actual operating expenses under its office lease agreements. These operating expenses are not included in the table below. Certain of these arrangements have free or escalating rent payment provisions. The Company recognizes rent expense under such arrangements on a straight-line basis over the term of lease.
At December 31, 2009, future minimum payments under the Company’s noncancelable operating leases were as follows (in thousands):
| Facilities | Equipment | Total | |||||||
| 2010 |
$ | 2,885 | $ | 24 | $ | 2,909 | |||
| 2011 |
1,521 | 24 | 1,545 | ||||||
| 2012 |
1,192 | 20 | 1,212 | ||||||
| 2013 |
309 | 5 | 314 | ||||||
| Total minimum payments required |
$ | 5,907 | $ | 73 | $ | 5,980 | |||
86
Table of Contents
Rent expense, net of sublease income, for the years ended December 31, 2009, 2008, 2007 was $2.8 million, $0.6 million and $0.4 million, respectively. Sublease income was $0.2 million for the years ended December 31, 2009 and 2008, respectively, and $40,000 for the year ended December 31, 2007.
See also Note 14 with respect to the purported securities class action lawsuit was commenced on March 9, 2010 naming as defendants the Company and certain of its officers.
14. SUBSEQUENT EVENTS
With Pfizer, the Company has been conducting a broad dimebon clinical development program, including five randomized, double-blind, placebo-controlled Phase 3 studies in Alzheimer’s and Huntington diseases. In March 2010, the Company reported negative top-line results from the first of these studies—the CONNECTION trial, a six-month study of 598 patients with mild-to-moderate Alzheimer’s disease. In the CONNECTION study, dimebon treated patients did no better than placebo patients on any of the primary and secondary efficacy endpoints at either dose tested. Given the negative CONNECTION data, the Company and Pfizer are currently re-evaluating which, if any, of the ongoing Phase 3 Alzheimer’s disease clinical trials should continue. This will be a complex analysis that will encompass scientific, clinical, regulatory and business issues. After this analysis is complete, the Company and Pfizer will be able to assess their respective future approaches to the clinical development of dimebon for Alzheimer’s and Huntington diseases. The Company does not know at this time which, if any, of the ongoing Phase 3 dimebon trials will be completed, or if Pfizer will elect to terminate the collaboration. Should the ongoing dimebon development program be materially changed, or should Pfizer elect to terminate the collaboration agreement, management’s estimate of the performance period over which the Company is recognizing the $225.0 million up-front payment received from Pfizer in the fourth quarter of 2008 would change.
On March 9, 2010, a purported securities class action lawsuit was commenced in the United States District Court for the Northern District of California, naming as defendants the Company and certain of its officers. The lawsuit alleges violations of the Securities Exchange Act of 1934 in connection with allegedly false and misleading statements made by the Company related to dimebon. The plaintiff alleges, among other things, that the defendants disseminated false and misleading statements about the effectiveness of dimebon for the treatment of Alzheimer’s disease making it impossible for stockholders to gain a realistic understanding of the drug’s progress toward FDA approval. The plaintiff seeks to represent a class of stockholders who purchased the Company’s common stock between July 17, 2008 and March 2, 2010. The plaintiff seeks damages, an award of its costs and injunctive and/or equitable relief. It is possible that additional suits will be filed, or allegations received from stockholders, with respect to these same matters and also naming the Company and/or its officers and directors as defendants. This lawsuit and any other related lawsuits are subject to inherent uncertainties, and the actual cost will depend upon many unknown factors. The outcome of the litigation is necessarily uncertain, the Company could be forced to expend significant resources in the defense of those suits and we may not prevail. We are not currently able to estimate the possible cost to us from these matters as this lawsuit is currently at an early stage and the Company cannot ascertain how long it may take to resolve these matters. The Company has not established any reserve for any potential liability relating to this lawsuit. The Company’s management believes that the Company has meritorious defenses and intends to defend the lawsuit vigorously.
87
Table of Contents
15. SELECTED QUARTERLY FINANCIAL DATA (UNAUDITED)
The following table presents the unaudited quarterly results of operations of the Company for the years ended December 31, 2009 and 2008, respectively. The unaudited information is prepared on the same basis as the audited consolidated financial statements. The Company’s operating results for any quarter are not necessarily indicative of results for any future quarters or for a full year.
| Quarters Ended | ||||||||||||||||
| March 31, | June 30, | September 30, | December 31, | |||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| 2009 |
||||||||||||||||
| Collaboration revenue |
$ | 16,340 | $ | 16,340 | $ | 16,341 | $ | 20,233 | ||||||||
| Operating expenses |
(22,081 | ) | (24,156 | ) | (27,564 | ) | (42,910 | ) | ||||||||
| Loss from operations |
(5,741 | ) | (7,816 | ) | (11,223 | ) | (22,677 | ) | ||||||||
| Net loss |
(5,609 | ) | (8,923 | ) | (13,973 | ) | (26,248 | ) | ||||||||
| Basic and diluted net loss per common share |
$ | (0.19 | ) | $ | (0.29 | ) | $ | (0.42 | ) | $ | (0.78 | ) | ||||
| Weighted average common shares used in the calculation of basic and diluted net loss per share |
30,105 | 31,154 | 33,468 | 33,595 | ||||||||||||
| 2008 |
||||||||||||||||
| Collaboration revenue |
$ | — | $ | — | $ | — | $ | 12,578 | ||||||||
| Operating expenses |
(15,909 | ) | (18,715 | ) | (20,636 | ) | (21,500 | ) | ||||||||
| Loss from operations |
(15,909 | ) | (18,715 | ) | (20,636 | ) | (8,922 | ) | ||||||||
| Net loss |
(15,530 | ) | (18,543 | ) | (20,456 | ) | (7,931 | ) | ||||||||
| Basic and diluted net loss per common share |
$ | (0.54 | ) | $ | (0.64 | ) | $ | (0.68 | ) | $ | (0.26 | ) | ||||
| Weighted average common shares used in the calculation of basic and diluted net loss per share |
28,847 | 28,943 | 30,022 | 30,082 | ||||||||||||
88
Table of Contents
Exhibit Index
Unless otherwise indicated below, the Commission file number to the exhibit is No. 001-32836 for filings made in or after March 2006 and No. 000-20837 for filings made prior to March 2006.
| Exhibit No. |
Exhibit Description | |
| 3.1(a) | Amended and Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.1(a) to the Quarterly Report on Form 10-QSB of Medivation, Inc. for the quarter ended June 30, 2005). | |
| 3.1(b) | Certificate of Amendment of Amended and Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.1(b) to the Quarterly Report on Form 10-QSB of Medivation, Inc. for the quarter ended June 30, 2005). | |
| 3.1(c) | Certificate of Amendment to the Amended and Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.1(c) to the Quarterly Report on Form 10-QSB of Medivation, Inc. for the quarter ended June 30, 2005). | |
| 3.1(d) | Certificate of Designations of the Series C Junior Participating Preferred Stock of Medivation, Inc. (incorporated by reference to Exhibit 3.1(d) to the Annual Report on Form 10-KSB of Medivation, Inc. for the fiscal year ended December 31, 2007). | |
| 3.2 | Amended and Restated Bylaws of Medivation, Inc. (incorporated by reference to Exhibit 3.2 to the Annual Report on Form 10-K for the year ended December 31, 2008). | |
| 4.1 | Common Stock Certificate (incorporated by reference to Exhibit 4.1 to the Registration Statement on Form SB-2 of Medivation, Inc. (formerly Orion Acquisition Corp. II) (No. 333-03252)). | |
| 4.2 | Rights Agreement, dated as of December 4, 2006, between Medivation, Inc. and American Stock Transfer & Trust Company, as Rights Agent, which includes the form of Certificate of Designations of the Series C Junior Participating Preferred Stock of Medivation, Inc. as Exhibit A, the form of Right Certificate as Exhibit B and the Summary of Rights to Purchase Preferred Shares as Exhibit C (incorporated by reference to Exhibit 4.1 to the Current Report on Form 8-K filed on December 4, 2006). | |
| 10.1 | Registration Rights Agreement by and among Orion Acquisition Corp. II and Special Situations Fund III, L.P., Special Situations Cayman Fund, L.P. and Special Situations Private Equity Fund, L.P., dated as of December 17, 2004 (incorporated by reference to Exhibit 10.3(a) to the Registration Statement on Form SB-2 of Medivation, Inc. (formerly Orion Acquisition Corp. II) (No. 333-122431)). | |
| 10.2(a) | Warrant to purchase Common Stock of Medivation Neurology, Inc. assumed by Orion Acquisition Corp. II issued to Joseph J. Grano, Jr., dated as of June 8, 2004 (incorporated by reference to Exhibit 10.5(a) to the Registration Statement on Form SB-2 of Medivation, Inc. (formerly Orion Acquisition Corp. II) (No. 333-122431)). | |
| 10.2(b) | Warrant to purchase Common Stock of Medivation Neurology, Inc. assumed by Orion Acquisition Corp. II issued to Joseph J. Grano, Jr., dated as of August 1, 2004 (incorporated by reference to Exhibit 10.5(b) to the Registration Statement on Form SB-2 of Medivation, Inc. (formerly Orion Acquisition Corp. II) (No. 333-122431)). | |
| 10.2(c) | Warrant to purchase Common Stock of Medivation Neurology, Inc. assumed by Orion Acquisition Corp. II issued to Joseph J. Grano, Jr., dated as of September 1, 2004 (incorporated by reference to Exhibit 10.5(c) to the Registration Statement on Form SB-2 of Medivation, Inc. (formerly Orion Acquisition Corp. II) (No. 333-122431)). | |
| 10.2(d) | Amendment Agreement by and between Orion Acquisition Corp. II and Joseph J. Grano, Jr., dated as of December 17, 2004 (incorporated by reference to Exhibit 10.5(d) to the Registration Statement on Form SB-2 of Medivation, Inc. (formerly Orion Acquisition Corp. II) (No. 333-122431)). | |
Table of Contents
| Exhibit No. |
Exhibit Description | |
| 10.3* | Warrant to purchase Common Stock of Medivation Neurology, Inc. assumed by Orion Acquisition Corp. II issued to David T. Hung, M.D., dated as of November 16, 2004 (incorporated by reference to Exhibit 10.6 to the Registration Statement on Form SB-2 of Medivation, Inc. (formerly Orion Acquisition Corp. II) (No. 333-122431)). | |
| 10.4(a)* | Amended and Restated 2004 Equity Incentive Award Plan (incorporated by reference to Exhibit 10.4(a) to the Annual Report on Form 10-KSB for the fiscal year ended December 31, 2007). | |
| 10.4(b)* | Form of Stock Option Agreement under the 2004 Equity Incentive Award Plan (incorporated by reference to Exhibit 10.7(b) to the Annual Report on Form 10-KSB of Medivation, Inc. (formerly Orion Acquisition Corp. II) for the year ended December 31, 2004). | |
| 10.4(c)* | Form of Stock Option Agreement for Early Exercisable Options under the 2004 Equity Incentive Award Plan (incorporated by reference to Exhibit 10.7(c) to the Annual Report on Form 10-KSB of Medivation, Inc. (formerly Orion Acquisition Corp. II) for the year ended December 31, 2004). | |
| 10.5 | Common Stock Purchase Agreement, dated as of September 7, 2007, by and between Medivation, Inc. and Azimuth Opportunity Ltd. (incorporated by reference to Exhibit 10.1 to Medivation, Inc.’s Current Report on Form 8-K filed on September 10, 2007). | |
| 10.6 | Form of Purchase Agreement, dated as of June 22, 2008, between Medivation, Inc. and the purchasers of its common stock (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed on June 23, 2008). | |
| 10.7** | Amended and Restated Collaboration Agreement, dated as of October 20, 2008, between Medivation, Inc. and Pfizer Inc. (incorporated by reference to Exhibit 10.8 to the Quarterly Report on Form 10-Q for the quarter ended September 30, 2008). | |
| 10.8* | Bonuses for Fiscal Year 2008 and Base Salaries for Fiscal Year 2009 for Certain Executive Officers (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed on November 5, 2008). | |
| 10.9* | Medivation, Inc. 2009 Bonus Plan (incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K filed on November 5, 2008). | |
| 10.10* | Bonuses for Fiscal Year 2009 and Base Salaries for Fiscal Year 2010 for Named Executive Officers (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed on December 7, 2009). | |
| 10.11* | Medivation, Inc. 2010 Bonus Plan Summary (incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K filed on December 7, 2009). | |
| 10.12* | Change of Control Severance Benefits Agreement, dated as of February 2, 2009, between Medivation, Inc. and David Hung, M.D. (incorporated by reference to Exhibit 10.11 to the Annual Report on Form 10-K filed on March 16, 2009). | |
| 10.13* | Severance Benefits Agreement, dated as of February 9, 2009, between Medivation, Inc. and Rohan Palekar (incorporated by reference to Exhibit 10.12 to the Annual Report on Form 10-K filed on March 16, 2009). | |
| 10.14* | Form of Medivation, Inc. Change of Control Severance Benefits Agreement (incorporated by reference to Exhibit 10.13 to the Annual Report on Form 10-K filed on March 16, 2009). | |
| 10.15† | Collaboration Agreement, dated as of October 26, 2009, by and between Medivation, Inc. and Astellas US LLC. | |
| 10.16 | Office Lease Agreement, dated as of November 2, 2009, by and between Medivation, Inc. and PPF OFF 345 Spear Street, LP. | |
Table of Contents
| Exhibit No. |
Exhibit Description | |
| 10.17* | Compensation Information for Non-Employee Directors. | |
| 21.1 | Subsidiaries of Medivation, Inc. | |
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |
| 24.1 | Power of attorney (contained on signature page). | |
| 31.1 | Certification pursuant to Rule 13a-14(a)/15d-14(a). | |
| 31.2 | Certification pursuant to Rule 13a-14(a)/15d-14(a). | |
| 32.1 | Certifications of Chief Executive Officer and Chief Financial Officer. | |
| * | Indicates management contract or compensatory plan or arrangement. |
| ** | Confidential treatment has been granted with respect to certain portions of this exhibit. |
| † | Confidential treatment requested as to certain portions, which portions were omitted and filed separately with the Securities and Exchange Commission. |