Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
FOR ANNUAL & TRANSITION REPORTS PURSUANT TO
SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
(MARK ONE)
[ x ] Annual Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934
For the fiscal year ended December 31, 2009
or
[ ] Transition Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934
For the transition period from to
Commission File Number: 001-33221
A.P. PHARMA, INC.
(Exact name of registrant as specified in its charter)
| DELAWARE | 94-2875566 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification Number) | |
| 123 SAGINAW DRIVE, REDWOOD CITY, CALIFORNIA | 94063 | |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code:
(650) 366-2626
Securities registered pursuant to Section 12(b) of the Act:
| COMMON STOCK | THE NASDAQ CAPITAL MARKET |
Securities registered pursuant to Section 12(g) of the Act:
NONE
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Exchange Act. Yes [ ] No [ x ]
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes [ ] No [ x ]
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes [ x ] No [ ]
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes [ ] No [ ]
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405) is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. [ ]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check One)
Large accelerated filer [ ] Accelerated filer [ ] Non-accelerated filer [ ] Smaller reporting company [ x ]
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes [ ] No [ x ]
The aggregate market value of the voting and non-voting common equity of the registrant held by non-affiliates of the registrant as of June 30, 2009, was $14,349,934(1) based upon the closing sale price on the NASDAQ Global Market reported for such date.
As of February 28, 2010, 39,426,041 shares of registrant’s Common Stock, $.01 par value, were outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
| Document |
Form 10-K Part | |
| Definitive Proxy Statement to be used in connection with the 2010 Annual Meeting of Stockholders. |
III |
| (1) | Excludes 15,265,887 shares held by directors, officers and shareholders whose ownership exceeds 5% of the outstanding shares at June 30, 2009. Exclusion of such shares should not be construed as indicating that the holders thereof possess the power, directly or indirectly, to direct the management or policies of the registrant, or that such person is controlled by or under common control with the registrant. |
Table of Contents
2
Table of Contents
Introduction—Forward-Looking Statements
In this Annual Report on Form 10-K, the “Company,” “A.P. Pharma,” “we,” “us” and “our” refer to A.P. Pharma, Inc.
This Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended, and Section 27A of the Securities Act of 1933, as amended. All statements contained in this Form 10-K, other than statements of historical fact, are forward-looking statements. When used in this report or elsewhere by management from time to time, the words “believe,” “anticipate,” “intend,” “plan,” “estimate,” “expect,” “may,” “will,” “should,” “seeks” and similar expressions are forward-looking statements. Such forward-looking statements are based on current expectations, but the absence of these words does not necessarily mean that a statement is not forward-looking. Forward-looking statements made in this Form 10-K include, but are not limited to, statements about:
• the progress of our research, development and clinical programs and timing of regulatory approval and commercial introduction of APF530 and future product candidates;
• estimates of the dates by which we expect to report results of our clinical trials and the anticipated results of these trials;
• our ability to market, commercialize and achieve market acceptance for APF530 or other future product candidates;
• our ability to establish collaborations for our technology, APF530 and other future product candidates;
• uncertainties associated with obtaining and enforcing patents;
• our estimates for future performance; and
• our estimates regarding our capital requirements and our needs for additional financing.
Forward-looking statements are not guarantees of future performance and involve risks and uncertainties. Actual events or results may differ materially from those discussed in the forward-looking statements as a result of various factors. For a more detailed discussion of such forward-looking statements and the potential risks and uncertainties that may impact our actual results, see the “Risk Factors” section of this Form 10-K and the other risks and uncertainties described below under the headings: “Our Lead Product Candidate APF530,” “Development Pipeline,” “Our Technology Platform,” “Our Strategy,” “Patents and Trade Secrets,” “Competition,” and under “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” These forward-looking statements reflect our view only as of the date of this report. Except as required by law, we undertake no obligations to update any forward looking statements. Accordingly, you should also carefully consider the factors set forth in other reports or documents that we file from time to time with the Securities and Exchange Commission.
3
Table of Contents
Company Overview
A.P Pharma is a specialty pharmaceutical company developing pharmaceutical products using our proprietary Biochronomer™ polymer-based drug delivery technology. Our primary focus is on our lead product candidate, APF530, which is being developed for the prevention of chemotherapy-induced nausea and vomiting (CINV). In May 2009, we filed a New Drug Application (NDA) with the U.S. Federal Drug Administration (FDA) under Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act (FDCA) seeking approval for APF530. The FDA is currently reviewing the NDA, and based on the Prescription Drug User Fee Act (PDUFA), has issued an action date of March 18, 2010. We are seeking regulatory approval of APF530 for the prevention of acute CINV for patients undergoing both moderately and highly emetogenic chemotherapy, and for prevention of delayed CINV for patients undergoing moderately emetogenic chemotherapy. There are no guarantees that the FDA will provide its response by March 18, 2010, nor can there be any assurances that any such response will be favorable.
Our core Biochronomer technology, on which APF530 and our other products are based, consists of bioerodible polymers designed to release drugs over a defined period of time. We have completed over 100 in vivo and in vitro studies demonstrating that our Biochronomer technology is potentially applicable to a range of therapeutic areas, including prevention of nausea and vomiting, pain management, control of inflammation and treatment of ophthalmic diseases. We have also completed comprehensive animal and human toxicology studies that have established that our Biochronomer polymers are safe and well tolerated. Furthermore, our Biochronomer technology can be designed to deliver drugs over periods varying from days to several months.
In addition to our lead drug candidate, we have a pipeline of other product candidates that use our Biochronomer technology. Further development of our pipeline products has been temporarily deferred in order to focus both managerial and financial resources on the APF530 NDA and negotiations of a commercialization partnership for APF530. One of these pipeline products, APF112, incorporates the well-known local anesthetic, mepivacaine. It is designed to provide up to 36 hours of relief from post-surgical pain and to minimize the use of morphine-like drugs, or opiates, which are used extensively in the management of post-surgical pain. A second pipeline product, APF580, incorporates a presently unannounced opiate for extended relief of severe pain. An investigational new drug application (IND) for APF580 was successfully filed in the third quarter of 2008.
We were founded in February 1983 as a California corporation under the name AMCO Polymerics, Inc. (AMCO). AMCO changed its name to Advanced Polymer Systems, Inc. in 1984 and was reincorporated in the state of Delaware in 1987. We changed our name to A.P. Pharma, Inc. in May 2001 to reflect our new pharmaceutical focus. Our offices are located at 123 Saginaw Drive, Redwood City, California 94063. Our telephone number is (650) 366-2626. Our website is located at www.appharma.com. Information contained on, or that can be accessed through, our website is not part of this Annual Report on Form 10-K.
4
Table of Contents
Our Lead Product Candidate—APF530
CINV Background
Prevention and control of nausea and vomiting, or emesis, are paramount in the treatment of cancer patients. The majority of patients receiving chemotherapy will experience some degree of emesis if not prevented with an antiemetic. Chemotherapy treatments can be classified as moderately emetogenic, meaning that 30–90% of patients would experience CINV, or highly emetogenic, meaning that over 90% of patients would experience CINV, if they were not treated with an antiemetic prior to chemotherapy. Onset of CINV within the first 24 hours is described as “acute,” and CINV that occurs more than 24 hours after treatment is described as “delayed.” Delayed CINV may persist for several days. Prevention of CINV is significant because the distress caused by CINV can severely disrupt patient quality of life and can lead some patients to delay or discontinue chemotherapy.
Current Therapy
Chemotherapeutic agents activate or destroy cells in the lining of the gut, releasing a neurotransmitter called serotonin. When serotonin binds to 5-hydroxytryptamine type 3 (5-HT3) receptors, the patient experiences nausea and vomiting. Granisetron, like other 5-HT3 antagonists, inhibits the vomiting reflex by preventing serotonin from binding to 5-HT3 receptors. Physicians may combine 5-HT3 antagonists with other agents, such as corticosteroids or neurokinin-1 (NK1) antagonists, to better prevent CINV.
Current treatment options for preventing CINV include 5-HT3 antagonists such as palonosetron (Aloxi®), ondansetron (Zofran®), dolasetron (Anzemet®) and granisetron (Kytril®). Aprepitant (Emend®), an NK1 antagonist, is also used to prevent CINV and is always used in combination with a 5-HT3 antagonist. As shown in the table below, all of the 5-HT3 antagonists are approved for the prevention of acute CINV in patients receiving either moderately or highly emetogenic chemotherapy. Within the last several years, generic versions of granisetron and ondansetron have become available. Only Aloxi is approved for the prevention of delayed CINV in patients receiving moderately emetogenic chemotherapy. No 5-HT3 antagonist is approved for the prevention of delayed CINV in patients receiving highly emetogenic chemotherapy.
5
Table of Contents
According to IMS Health, Aloxi sales for the prevention of CINV were approximately $303 million in 2007, $391 million in 2008 and $437 million in 2009. We believe the total addressable U.S. market when calculated on a branded basis approaches $1 billion for use of 5-HT3 antagonists in the prevention of CINV.
| Chemotherapy Regimen |
Approved 5-HT3 Antagonists for Acute CINV |
Approved 5-HT3 Antagonists for Delayed CINV | ||
| Moderately Emetogenic |
Granisetron (Kytril) Ondansetron (Zofran) Dolasetron (Anzemet) Palonosetron (Aloxi) |
Palonosetron (Aloxi) | ||
| Highly Emetogenic |
Granisetron (Kytril) Ondansetron (Zofran) Dolasetron (Anzemet) Palonosetron (Aloxi) |
None |
Despite evidence that delayed CINV affects as many as 50–70% of patients, and that more patients experience delayed CINV than acute CINV, oncology nurses and physicians are likely to underestimate the magnitude of these problems in the patients for whom they care. This may occur in part since patients often do not report side effects they experience at home following chemotherapy treatments. Even though high percentages of chemotherapy patients experience such delayed nausea and emesis, presently Aloxi is the only 5-HT3 antagonist approved for dealing with this delayed CINV. We believe that APF530, if approved, could become the second long-acting product given in a single administration that is capable of dealing with this important medical need.
Our Solution—APF530
Our lead product candidate, APF530, is being developed for the prevention of acute CINV in patients receiving moderately or highly emetogenic chemotherapy and for the prevention of delayed CINV in patients receiving moderately emetogenic chemotherapy. APF530 is delivered by a single subcutaneous injection and contains the 5-HT3 antagonist granisetron. Granisetron, for infusion and oral tablets, is approved for the prevention of acute CINV, but not delayed CINV. We selected granisetron because it is a potent, well-tolerated drug and the applicable granisetron patent expired in the United States on December 29, 2007.
Granisetron and other 5-HT3 antagonists, as a class, have become the most common antiemetic agents used in chemotherapy. However, no 5-HT3 antagonist formulation is currently approved for the prevention of both acute and delayed CINV for both moderately and highly emetogenic chemotherapy. Results from our APF530 Phase 3 clinical trial demonstrated that we can prevent acute CINV for both moderately and highly emetogenic chemotherapy, and prevent delayed CINV in moderately emetogenic chemotherapy. The efficacy data involving delayed CINV in highly emetogenic chemotherapy showed results for the higher dose of APF530 that were numerically better than Aloxi and were statistically non-inferior. Because Aloxi is not approved for use in this setting, it would be necessary to demonstrate superiority in order to get approval for this use. However, APF530 did not achieve the statistically significant level of superiority necessary to support a claim for this use. If we
6
Table of Contents
obtain regulatory approval for all uses except the prevention of delayed CINV in highly emetogenic chemotherapy, we believe that we should have a product comparable to Aloxi, which despite the limitation of its claim for prevention of delayed CINV to only moderately emetogenic treatments, has achieved considerable commercial success.
Phase 2 Clinical Trial
In September 2005, we completed an open-label Phase 2 clinical trial for APF530. We evaluated the safety, tolerability and pharmacokinetics of APF530 in cancer patients. In addition, efficacy endpoints were evaluated relating to emetic events and the use of additional medication for treating CINV. The trial demonstrated that APF530 was well tolerated; there were no serious adverse events attributed to APF530 and less than 10% of participating patients had injection site reactions, all of which were mild.
Analysis of the efficacy data from our Phase 2 trial, in which patient groups received either moderately or highly emetogenic chemotherapy, was based on complete responders, patients who experienced no vomiting and no use of additional medication for CINV during the observation period. This efficacy data was compared to similar data for Aloxi, as reported from its Phase 3 trials. APF530 indications of efficacy compared favorably with the complete response results in the Aloxi trials data. Based on this comparison, we designed our Phase 3 clinical program to conclusively compare APF530 to Aloxi in a prospective randomized trial design.
Pivotal Phase 3 Clinical Trial Design
In December 2005, we held our end-of-Phase-2 meeting with the FDA, at which we discussed our regulatory approval strategy and our proposed design for the pivotal Phase 3 trial. Following this meeting, we finalized plans for our pivotal Phase 3 trial in accordance with FDA input. The trial’s primary objectives were to demonstrate:
• non-inferiority, or comparability, of APF530 to Aloxi for the prevention of acute CINV following the administration of either moderately emetogenic or highly emetogenic chemotherapy;
• non-inferiority of APF530 in comparison to Aloxi for the prevention of delayed CINV following administration of moderately emetogenic chemotherapy; and
• superiority of APF530 in comparison to Aloxi for the prevention of delayed CINV following administration of highly emetogenic chemotherapy.
7
Table of Contents
Our pivotal Phase 3 clinical trial was initiated in May 2006 as a multicenter, randomized, observer-blind, actively-controlled, double-dummy, parallel group study that compared the efficacy of APF530 with Aloxi. The trial stratified patients into two groups, one receiving moderately and the other receiving highly emetogenic chemotherapeutic agents in accordance with the Hesketh algorithm, which assigns emetogenic levels based on the chemotherapy agent, drug dosage and combinations employed. In each emetogenic group, patients were randomized during Cycle 1 to receive either APF530 high dose (10 mg), APF530 low dose (5 mg) or the currently approved dose of Aloxi. In subsequent treatment cycles up to three additional cycles (Cycles 2—4), the patients were re-randomized to either of the two APF530 doses. The diagram below provides further graphical representation of the patient stratification design and target enrollment for patient randomization in our clinical trial.
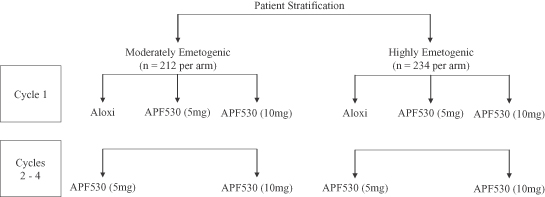
Pivotal Phase 3 Clinical Trial Results
During 2006 and the first half of 2007, all patient enrollments in the pivotal Phase 3 study of APF530 were located within the United States. Beginning in the second half of 2007, enrollments were broadened to include clinical trial sites in India and Poland. The study completed patient enrollment of 1,395 patients in June 2008, and we announced top-line results on September 30, 2008.
The goals of the trial were to demonstrate the safety and efficacy of APF530 in the treatment of CINV following the administration of highly or moderately emetogenic chemotherapy, and to establish an effective dose for APF530, creating a data package suitable for inclusion in the NDA. In the trial, 5 mg and 10 mg doses of granisetron were evaluated in patients, and based on the results, the 10 mg dose appears to provide greater efficacy with a side effect profile similar to the 5 mg dose. As such, we selected the APF530 10 mg dose for inclusion in the NDA.
The trial was structured to compare the two APF530 doses (5 mg and 10 mg) with Aloxi in four different primary efficacy endpoints:
• non-inferiority to Aloxi for the prevention of acute CINV in patients receiving moderately emetogenic chemotherapies;
• non-inferiority to Aloxi for the prevention of delayed CINV in patients receiving moderately emetogenic chemotherapies;
8
Table of Contents
• non-inferiority to Aloxi for the prevention of acute CINV in patients receiving highly emetogenic chemotherapies; and
• superiority to Aloxi for the prevention of delayed CINV in patients receiving highly emetogenic chemotherapies.
The 10 mg dose of APF530 achieved complete response (CR) rates that were numerically higher than Aloxi across all four assessments. These results demonstrated non-inferiority to Aloxi for all four assessments, but did not achieve the superiority endpoint for the delayed CINV assessment for highly emetogenic chemotherapies. Aloxi is not FDA approved for the prevention of delayed CINV in patients receiving highly emetogenic chemotherapies; therefore, APF530 needed to be deemed superior to Aloxi for this endpoint to obtain a corresponding label claim. CR was defined as the absence of emetic episodes or use of anti- emetic rescue medications during a specified period of time. The time periods studied for CINV onset were 0 to 24 hours after chemotherapy, which is known as acute CINV, and 24 to 120 hours after chemotherapy, which is known as delayed CINV.
The results summarized below are the primary endpoints from the study, with such data being drawn from the first cycle of treatment:
Complete Response by Treatment–Cycle 1
| Emetogenicity Level | Treatment Group | Statistics vs. Aloxi (Confidence Interval) | ||||||||
| APF530 (5 mg) |
APF530 (10 mg) |
Aloxi | 5 mg | 10 mg | ||||||
| Moderately emetogenic |
(n=214) | (n=212) | (n=208) | |||||||
| • Acute CINV |
74.8% | 76.9% | 75.0% | NI (-9.8, 9.3) | NI (-7.5, 11.4) | |||||
| • Delayed CINV |
51.4% | 59.0% | 57.7% | I (-17.1, 4.6) | NI (-9.5, 12.1) | |||||
| Highly emetogenic |
(n=229) | (n=240) | (n=238) | |||||||
| • Acute CINV |
77.7% | 81.3% | 80.7% | NI (-12.1, 6.1) | NI (-8.2, 9.3) | |||||
| • Delayed CINV |
64.6% | 68.3% | 66.4% | NS (-12.4, 8.8) | NS (-8.3, 12.2) | |||||
(NI) Non-inferior efficacy was established using a modified Bonferroni step down procedure. APF530 non-inferior to Aloxi (i.e. lower bound of adjusted 95% CI for APF530), Aloxi difference excludes less than or equal to negative 15%. The Confidence Intervals shown for the moderately emetogenic and highly emetogenic levels are 97.5% and 98.3%, respectively. (NS) = No significant difference. (I) = Inferior efficacy.
APF530 was generally well tolerated, with a side effect profile consistent with previous human use of granisetron and only one serious adverse event reported as possibly attributed to APF530. In Cycle 1, the data showed a low incidence of patients discontinuing therapy due to any adverse events (related or unrelated to study drugs): 0.5%, 0.9% and 0.9% in the moderately emetogenic patient group, and 2.0%, 3.5% and 1.2% in the highly emetogenic patient group for APF530 5 mg, APF530 10 mg and Aloxi, respectively. Further, of the patients completing the first cycle, 1,043 went on to receive a total of 2,374 additional doses of APF530 in Cycles 2 to 4. Of these patients, only 2 (or 0.2%) discontinued therapy due to treatment related adverse events.
9
Table of Contents
Additional data from the pivotal Phase 3 trial comparing APF530 with Aloxi was released on November 5, 2008, and is reported below. The additional data provided herein includes predetermined secondary efficacy endpoints and safety data that were not available at the time the top-line data were released. Review of the clinical data package demonstrates the robustness of the APF530 clinical response within and across chemotherapy cycles. Some of the additional key findings follow:
• Collectively, the Phase 3 efficacy and safety data support the conclusion that the APF530 10 mg dose is the most effective dose and therefore was the selected dose for the NDA.
• In patients receiving multiple cycles of APF530, CR rates were observed to generally increase over four cycles of chemotherapy. The data summarized below supports the continued benefits of APF530 over multiple cycles:
Complete Response(1) of APF530 10 mg Dose Over Four Chemotherapy Cycles
| Emetogenicity Level |
Cycle 1 | Cycle 2 | Cycle 3 | Cycle 4 | ||||
| Moderately Emetogenic |
(n=212) | (n=240) | (n=184) | (n=134) | ||||
| • Acute (0-24h) |
76.9% | 77.1% | 78.8% | 83.6% | ||||
| • Delayed (24-120h) |
59.0% | 62.1% | 61.4% | 66.4% | ||||
| • Overall (0-120h) |
54.2% | 58.8% | 60.3% | 63.4% | ||||
| Highly Emetogenic |
(n=240) | (n=263) | (n=202) | (n=148) | ||||
| • Acute (0-24h) |
81.3% | 84.8% | 89.6% | 87.8% | ||||
| • Delayed (24-120h) |
68.3% | 76.0% | 81.2% | 83.8% | ||||
| • Overall (0-120h) |
64.6% | 72.2% | 78.7% | 79.7% |
| (1) | No emetic episodes and no rescue medications |
• The evaluation of “time to first treatment failure,” defined as either time to first emetic episode or use of rescue medication, showed that a greater proportion of patients treated with APF530 10 mg dose (vs. Aloxi) remained “failure free” on days one through five following either moderate or highly emetogenic chemotherapy.
• The Phase 3 trial protocol predefined multiple primary and secondary endpoints, including complete response, complete control (no emesis, no rescue therapy and no-greater-than-mild nausea) and total response (no emesis, no rescue therapy and no nausea) measured over defined time intervals (acute, delayed and overall). Although there were no significant differences between the APF530 10 mg dose vs. Aloxi, the response rates for APF530 10 mg dose were higher than Aloxi in all nine analyses for moderately emetogenic chemotherapy and in five of nine analyses for highly emetogenic chemotherapy.
• The safety profile for APF530 was very similar to that for Aloxi; the most notable adverse event was constipation, observed in 15.4% and 13.4% of patients receiving APF530 10 mg and Aloxi, respectively. Headache was observed in 10.0% and 9.7% of patients receiving APF530 10 mg dose and Aloxi, respectively.
• Investigators were required to observe and record all reactions associated with the subcutaneous injection site on days one and five for each treatment cycle. Overall, greater than 90% of the recorded observations were mild in severity, the most common being redness and bruising. With each additional cycle of treatment, the frequency of injection site reactions decreased, indicating APF530 can safely be administered for multiple cycles.
10
Table of Contents
• During the trial, patients received more than 1,600 separate injections of APF530 10 mg dose. Assessment of any injection site pain was made on days one and five of treatment: on day one, less than 0.1% of injections produced any reports of pain; on day five approximately 4% of injections produced reported pain. All but four of these reports of pain were recorded as mild, with the four recorded as moderate.
Additional data from the pivotal Phase 3 trial was presented at the annual meeting of the American Society of Clinical Oncology on June 1, 2009, and is reported below. Some of the additional key findings follow:
• CR rates for APF530 10 mg dose were generally higher in patients that had received prior chemotherapy when compared to patients that had not received any previous chemotherapy. Additionally, in all instances, CR rates for APF530 in patients receiving prior chemotherapy were numerically higher than those observed for palonosetron. Based on previous clinical studies, many physicians believe that the risk of CINV increases with each additional cycle of chemotherapy. These new data may suggest potential utility for APF530 in treating patients who have received prior chemotherapy.
• Of the highly emetogenic chemotherapy regimens, those containing cisplatin are considered to be the most troublesome due to their ability to cause significant delayed CINV. The CR rates for patients receiving cisplatin based regimens were numerically higher for APF530 10 mg when compared to palonosetron in both acute and delayed CINV. Specifically, in acute CINV, APF530 had an 81.1% CR rate versus 75.5% for palonosetron, and 66.0% versus 60.4%, respectively, in delayed CINV.
• A pharmacokinetic analysis, conducted in a sub-group of patients, confirmed that a single APF530 10 mg dose successfully maintained blood levels of granisetron for the entire five-day period.
New Drug Application
In May 2009, we filed an NDA for APF530 with the FDA under Section 505(b)(2) of the FDCA. In July 2009 the FDA notified us that it had accepted the NDA and, under the PDUFA guidelines, issued an action date of March 18, 2010. There are no guarantees that the FDA will provide its response by March 18, 2010, nor can there be any assurances that any such response will be favorable. If the FDA does not take action on our NDA by the PDUFA date, does not approve our NDA or requests additional work or changes to the NDA, our continued ability to commercialize APF530 could be seriously impaired and our business would be adversely impacted.
Section 505(b)(2) of the FDCA permits the FDA, in its review of an NDA, to rely on previous FDA findings of safety and efficacy of the active ingredient in APF530, granisetron. Section 505(b)(2) applications may be submitted for drug products that represent a modification (e.g., a new indication or new dosage form) of an eligible approved drug and for which investigations other than bioavailability or bioequivalence studies are essential to the drug’s approval. The additional information in 505(b)(2) applications can be provided by literature or by reference to past FDA findings of safety and efficacy for approved drugs, or it can be based upon studies conducted by or for the applicant to which it has obtained a right of reference. The majority of 505(b)(2) applications are filed for new formulations of currently approved drugs, so there is an existing understanding—on the part of the FDA, as well as the medical community—of their safety and efficacy.
11
Table of Contents
Development Pipeline
In addition to our lead program, we have a pipeline of other product candidates using our Biochronomer technology. Further development of our pipeline products has been temporarily deferred in order to focus both managerial and financial resources on the development of APF530.
| Product Candidate |
Potential Application | Drug | Targeted Duration | Status | ||||
| APF112 |
Post-surgical pain relief | Mepivacaine | Up to 36 hours | Phase 2 | ||||
| APF580 |
Pain relief - human | Undisclosed Opiate | At least seven days | Preclinical | ||||
| APF580 |
Pain relief - veterinary | Undisclosed Opiate | At least seven days | Preclinical | ||||
APF112
APF112 utilizes our Biochronomer delivery technology to target post-surgical pain relief. The product is designed to provide up to 36 hours of localized pain relief by delivering mepivacaine directly to the surgical site. Mepivacaine is a well-known, short-acting, local anesthetic with an excellent safety profile. APF112 is designed to prolong the anesthetic effect of mepivacaine and thus minimize or eliminate the use of opiates. Opiates are currently used in the majority of surgical procedures as a means of managing post-operative pain, and while they are powerful and useful drugs, they may have side effects such as addiction, nausea, disorientation, sedation, constipation, vomiting, urinary retention and, in some situations, life-threatening respiratory depression. If efficacy in treating post-surgical pain can be demonstrated, we believe that there will be substantial potential for this product, as there are approximately 20 million surgical procedures performed annually in the United States for which the product could potentially be utilized.
During 2004, our Phase 2 clinical trial was conducted in surgeries for inguinal hernia repair, which is considered a moderately to severely painful procedure. The results indicated excellent safety and tolerability. The pharmacokinetics of APF112 showed sustained release of mepivacaine systemically over a period of three days (72 hours). No significant difference was shown between the two doses of APF112 and the standard of care (bupivacaine) in terms of pain scores and the amount of additional pain medication used. Mean Visual Analog Scale pain scores (VAS scores) in the standard of care group (bupivacaine) were significantly lower in this study when compared with other previously published studies in similar hernia trials. Based on published data, VAS scores for the standard of care in similar inguinal hernia studies ranged from 4.5 to 6.7, whereas in this study the mean score for the bupivacaine arm was 2.9 within the first 24 hours post-surgery. We believe that with a revised Phase 2 protocol we can demonstrate that APF112 is effective in controlling post-surgical pain. If we are successful in obtaining the required capital to support this program, we intend to reactivate and complete additional preclinical work on APF112 towards a revised protocol, followed by initiation of a Phase 2b clinical trial. Assuming successful completion of our Phase 2b clinical trial, we plan to explore corporate partnering opportunities to continue the development of APF112.
APF580
APF580 incorporates an opiate into our Biochronomer technology and is designed to provide analgesia lasting at least seven days from a single injection. It is targeted for situations where the intensity and duration of pain require use of an opiate rather than a local anesthetic. APF580 may find use in acute and chronic pain settings, improve patient compliance and reduce the
12
Table of Contents
risk of drug abuse. Our initial animal pharmacokinetic studies completed in 2006 present a promising profile, supporting future product development for post-surgical and chronic pain applications. In September of 2008 we filed an IND for APF580 with the FDA. If we are successful in obtaining the required capital to support this program, we plan to initiate a Phase 1 clinical trial of APF580. In addition, we announced in September 2009 that we entered into a licensing and development agreement with Merial Limited for a variant of APF580 for long-acting pain management in companion animals.
Our Technology Platform
We have made significant investments in the development of our bioerodible drug delivery technologies, which have produced tangible results. Specifically, we have developed a broad family of polymers with unique attributes, known collectively as poly (ortho esters), under the trade name Biochronomer. This technology has been specifically designed for use in drug delivery applications with a number of technical advantages, such as: ease of manufacturing, flexible delivery times, various physical forms and multiple potential applications due to a neutral pH environment for acid sensitive actives (nucleic acids, proteins, etc.).
Due to the inherent versatility of our Biochronomer technology, products can be designed to deliver drugs at a variety of implantation sites including: under the skin, at the site of a surgical procedure, in joints, in the eye or in muscle tissue. Our Biochronomer technology can provide sustained levels of drugs in systemic circulation for prolonged efficacy.
Reproducibility. Our Biochronomer technology is formed by the coupling of various monomers into a polymer chain. Our process knowledge underlying the commercial manufacture of our Biochronomers is based on extensive, well-documented, development studies. Commercial manufacturing campaigns to date have demonstrated that our Biochronomers may be produced in a highly reproducible manner.
Flexible Delivery Times. The Biochronomer “links,” or bonds, are stable at neutral pH conditions. Upon coming into contact with water-containing media, such as internal body fluids, the water reacts with these bonds. This reaction is known as hydrolysis. During the hydrolysis of the Biochronomer links, acidic elements are produced in a local micro-environment, in a controlled manner, without impacting the overall neutrality of the drug delivery technology. These elements assist in the continued, controlled erosion of the polymer with a simultaneous, controlled release of the active drug contained within the Biochronomer. By varying the amount of the acidic elements in the Biochronomer, different rates of hydrolysis may be effectively realized. In this manner, delivery times ranging from days to weeks to several months can be achieved.
Various Physical Forms. Our Biochronomers can be prepared in a variety of physical forms, ranging from hard, glassy materials to semi-solids that are injectable at room temperature, by proper selection of monomers. A significant advantage of our Biochronomer technology is that drugs can be incorporated by simple mixing procedures allowing the production of formulations in the form of injectable gels, microspheres, coatings and strands. All of these physical forms can be used in the controlled delivery of drugs without the undesirable incorporation of organic solvents in the final product.
Multiple Potential Applications. We have completed over 100 in vivo and in vitro studies demonstrating that our Biochronomer technology is potentially applicable to a range of therapeutic areas, including pain management, prevention of nausea, control of inflammation and treatment of ophthalmic diseases. We have also completed comprehensive animal and human toxicology
13
Table of Contents
studies that have established that our Biochronomer polymers are safe and well tolerated. All of our current development programs utilize the same semi-solid poly (ortho ester) delivery vehicle. The present forms of these products are stored under refrigeration. We are actively developing products that can be stored at room temperature.
Our Strategy
Our primary near-term objective is to obtain regulatory approval of the NDA for APF530 from the FDA. Longer-term, we intend to become a leading specialty pharmaceutical company focused on improving the effectiveness of existing pharmaceuticals using our proprietary drug delivery technologies. Implementation of the following activities will be subject to our success in obtaining additional capital. Key elements include:
• Maximize the value of our lead product, APF530. We believe that establishing a partnership with a pharmaceutical company for the commercialization of APF530 will maximize the value of APF530 for our shareholders. We intend to secure significant upfront license fees, followed by milestone payments and royalties. We also plan to evaluate either a single commercial partnership or multiple partnerships for the United States and the rest of world.
• Expand product pipeline. We plan to expand our product pipeline by leveraging our existing technology. We intend to develop new products based on our Biochronomer polymer-based drug delivery technology. Our research has indicated that our Biochronomer technology has potential applications across a range of therapeutic areas including: prevention of nausea, pain management, control of inflammation and treatment of ophthalmic diseases. With further work on our technology platforms, we may be able to develop products that deliver proteins, peptides, soluble RNA and RNA interference.
• Enter into strategic partnerships. We believe that selective partnering of our future product development programs can enhance the success of our product development and commercialization efforts, and enable diversification of our product portfolio by having partners fund the major portion of our late stage clinical trials. Additionally, such partnering will enable us to leverage the sales capabilities of our partners to commercialize our products.
• Minimize product development risk and time-to-market. We plan to apply our proprietary drug delivery technologies to improve the effectiveness of approved pharmaceutical products. By using our technologies to administer drugs for which clinical efficacy and safety data are available, we believe we will be able to reduce the cost and development risk inherent in traditional pharmaceutical product development.
Manufacturing and Supply
We do not currently own or operate manufacturing facilities for the production of clinical or commercial quantities of any of our product candidates. We rely on a small number of third-party manufacturers to produce our compounds and expect to continue to do so to meet the preclinical and clinical requirements of our potential products and for all of our commercial needs. We do not have long-term agreements with any of these third-parties. We require in our manufacturing and processing agreements that all third-party contract manufacturers and processors produce active pharmaceutical ingredients (APIs) and finished products in accordance with the FDA’s current Good Manufacturing Practices (cGMP) and all other applicable laws and regulations. We maintain confidentiality agreements with potential and existing manufacturers in order to protect our proprietary rights related to our drug candidates.
14
Table of Contents
With regard to our lead product candidate, APF530, we source the API, granisetron, from two suppliers. We use one supplier to source raw materials and prepare our proprietary polymer and another supplier to formulate the bulk drug product. We ship the bulk APF530 to a contract manufacturer for filling into cartridges. This supplier is one of a small number of companies with the ability to perform the syringe filling function with a highly viscous material like APF530. To date, APF530 has been manufactured in small quantities for preclinical studies and clinical trials. If APF530 is approved for commercial sale, we will need to manufacture such product in larger quantities. Significant scale-up of manufacturing may require additional process development and validation studies, which the FDA must review and approve. The commercial success of APF530, in the near-term, will be dependent upon the ability of our contract manufacturers to produce a product in commercial quantities at competitive costs of manufacture. If APF530 receives regulatory approval, we plan to scale-up manufacturing through our third-party manufacturers for APF530 in order to realize important economies of scale. These scale-up activities would take time to implement, require additional capital investment, process development and validation studies, and FDA approval. We cannot guarantee that we will be successful in achieving competitive manufacturing costs through such scale-up activities.
Sales and Marketing
A key part of our business strategy is to form collaborations with pharmaceutical partners. In the past, we have successfully partnered our development stage programs with leading pharmaceutical companies. In general, we grant limited marketing exclusivity in defined markets for defined periods to our partners. However, after development is completed and a partner commercializes a formulated product utilizing our delivery technologies, we can exert only limited influence over the manner and extent of our partner’s marketing efforts.
The status of our partnering arrangements is:
• In September 2009, we entered into a licensing and development agreement with Merial Limited for a long-acting pain management product for use with companion animals. The Company received an upfront payment and will receive on-going development funding and potential future milestones and royalties.
• In October 2009, we announced the termination of our license agreement with RHEI Pharmaceuticals, Inc. (RHEI) following its failure to make a milestone payment in connection with acceptance for filing of an NDA for APF530 by the FDA. We had granted an exclusive license in October 2006 to RHEI to seek regulatory approval and sell APF530 in China, Taiwan, Hong Kong and Macau.
• We have and will continue to engage in potential partnership discussions with domestic and international pharmaceutical companies.
Patents and Trade Secrets
Patents and other proprietary rights are important to our business. It is our policy to seek patent protection for our inventions, and to rely upon trade secrets, know-how, continuing technological innovations and licensing opportunities to develop and maintain our competitive position.
As part of our strategy to protect our current products and to provide a foundation for future products, we have filed a number of U.S. patent applications on inventions relating to the composition of a variety of polymers, specific products, product groups and processing technology. We have a total of 27 issued U.S. patents and an additional 49 issued (or registered) foreign pat -
15
Table of Contents
ents. The patents on our bioerodible technologies expire between January 2016 and November 2023. In addition, we have filed patent applications on further improvements to our polymer technology, which, if issued, would provide additional exclusivity beyond these dates.
Although we believe the bases for these patents and patent applications are sound, they are untested, and there is no assurance that they will not be successfully challenged. There can be no assurance that any patent previously issued will be of commercial value, that any patent applications will result in issued patents of commercial value, or that our technology will not be held to infringe patents held by others.
We also rely on unpatented trade secrets and know-how to protect certain aspects of our production technologies. Our employees, consultants, advisors and corporate partners have entered into confidentiality agreements with us. These agreements, however, may not necessarily provide meaningful protection for our trade secrets or proprietary know-how in the event of unauthorized use or disclosure. In addition, others may obtain access to, or independently develop, these trade secrets or know-how.
Competition
The pharmaceutical industry is highly competitive. Many of our competitors have substantially greater financial, research, development, manufacturing, sales, marketing, and distribution resources than we currently do. In addition, they may have significantly more experience in drug development, obtaining regulatory approval, and establishing strategic collaborations. We expect any future products we develop to compete on the basis of, among other things, product efficacy and safety, time to market, price, extent of adverse side effects experienced and convenience of administration and drug delivery. We also expect to face competition in our efforts to identity appropriate collaborators or partners to help commercialize our product candidates in our target commercial areas.
APF530 is expected to face significant competition for the prevention of delayed CINV, principally from Eisai/MGI Pharma’s Aloxi (palonosetron). In addition to Aloxi, APF530 will compete with entrenched products for the prevention of acute CINV, including Roche’s Kytril (granisetron), GlaxoSmithKline’s Zofran (ondansetron) and Aventis’ Anzemet (dolasetron). Generic versions of certain of these products are also marketed by other companies. We are also aware of several companies which have developed, or are developing, both generic and new formulations of granisetron, including additional transdermal formulations such as ProStrakan’s Sancuso® (granisetron transdermal patch) and Hana Biosciences’ Zensana™ (oral ondansetron), which has been licensed to Par Pharmaceuticals.
APF112 is expected to face competition from two injectable controlled release bupivicane products, Durect Corporation’s Posidur™ and Pacira Pharmaceutical’s Exparel™ DepoBupivacaine.
There are several companies that are developing new formulations of existing drugs using novel drug delivery technologies. The following are some of our major competitors among drug delivery system developers: Alkermes, Inc., Depomed, Inc., Durect Corporation and Pacira Pharmaceuticals, Inc.
Government Regulation and Product Approvals
The manufacturing and marketing of our potential products and our ongoing research and development activities are subject to extensive regulation by the FDA and comparable regulatory agencies in state and local jurisdictions and in foreign countries.
16
Table of Contents
United States Regulation
Before any of our products can be marketed in the United States, they must be approved by the FDA. To secure approval, any drug we develop must undergo rigorous preclinical testing and clinical trials that demonstrate the product candidate’s safety and effectiveness for each chosen indication for use. These extensive regulatory processes control, among other things: the development, testing, manufacture, safety, efficacy, record keeping, labeling, storage, approval, advertising, promotion, sale and distribution of biopharmaceutical products.
In general, the process required by the FDA before investigational drugs may be marketed in the United States involves the following steps:
• preclinical laboratory and animal tests;
• submission of an IND, which must become effective before human clinical trials may begin;
• adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug for its intended use;
• pre-approval inspection of manufacturing facilities and selected clinical investigators; and
• FDA approval of an NDA, or of an NDA supplement (for subsequent indications).
Preclinical Testing
In the United States, drug candidates are tested in animals until adequate proof-of-safety is established. These preclinical studies generally evaluate the mechanism of action of the product and assess the potential safety and efficacy of the product. Tested compounds must be produced according to applicable cGMP requirements and preclinical safety tests must be conducted in compliance with FDA and international regulations regarding good laboratory practices (GLP). The results of the preclinical tests, together with manufacturing information and analytical data, are generally submitted to the FDA as part of an IND, which must become effective before human clinical trials may commence. The IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA requests an extension or raises concerns about the conduct of the clinical trials as outlined in the application. If the FDA has any concerns, the sponsor of the application and the FDA must resolve the concerns before clinical trials can begin. Submission of an IND may not result in FDA authorization to commence a clinical trial. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development, and the FDA must grant permission for each clinical trial to start and continue. Regulatory authorities may require additional data before allowing the clinical studies to commence or proceed from one Phase to another, and could demand that the studies be discontinued or suspended at any time if there are significant safety issues. Furthermore, an independent institutional review board (IRB), for each medical center proposing to participate in the conduct of the clinical trial must review and approve the clinical protocol and patient informed consent before the center commences the study.
17
Table of Contents
Clinical Trials
Clinical trials for new drug candidates are typically conducted in three sequential phases that may overlap. In Phase 1, the initial introduction of the drug candidate into human volunteers, the emphasis is on testing for safety or adverse effects, dosage, tolerance, metabolism, distribution, excretion and clinical pharmacology. Phase 2 involves studies in a limited patient population to determine the initial efficacy of the drug candidate for specific targeted indications, to determine dosage tolerance and optimal dosage and to identify possible adverse side effects and safety risks. Once a compound shows evidence of effectiveness and is found to have an acceptable safety profile in Phase 2 evaluations, pivotal Phase 3 trials are undertaken to more fully evaluate clinical outcomes and to establish the overall risk/benefit profile of the drug, and to provide, if appropriate, an adequate basis for product labeling. During all clinical trials, physicians will monitor patients to determine effectiveness of the drug candidate and to observe and report any reactions or safety risks that may result from use of the drug candidate. The FDA, the IRB (or their foreign equivalents), or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk.
The data from the clinical trials, together with preclinical data and other supporting information that establishes a drug candidate’s safety, are submitted to the FDA in the form of an NDA, or NDA supplement (for approval of a new indication if the product candidate is already approved for another indication). Under applicable laws and FDA regulations, each NDA submitted for FDA approval is usually given an internal administrative review within 60 days following submission of the NDA. If deemed complete, the FDA will “file” the NDA, thereby triggering substantive review of the application.
The FDA can refuse to file any NDA that it deems incomplete or not properly reviewable. The FDA has established internal substantive review goals of six months for priority NDAs (for drugs addressing serious or life threatening conditions for which there is an unmet medical need) and ten months for regular NDAs. The FDA, however, is not legally required to complete its review within these periods, and these performance goals may change over time. Moreover, the outcome of the review, even if generally favorable, is not typically an actual approval, but a “complete response” that describes additional work that must be done before the NDA can be approved. The FDA’s review of an NDA may involve review and recommendations by an independent FDA advisory committee. The FDA may deny approval of an NDA, or NDA supplement, if the applicable regulatory criteria are not satisfied, or it may require additional clinical data and/or an additional pivotal Phase 3 clinical trial. Even if such data are submitted, the FDA may ultimately decide that the NDA or NDA supplement does not satisfy the criteria for approval.
Data Review and Approval
Satisfaction of FDA requirements or similar requirements of state, local, and foreign regulatory agencies typically takes several years and requires the expenditure of substantial financial resources. Information generated in this process is susceptible to varying interpretations that could delay, limit, or prevent regulatory approval at any stage of the process. Accordingly, the actual time and expense required to bring a product to market may vary substantially. We cannot assure you that we will submit applications for required authorizations to manufacture and/or market potential products or that any such application will be reviewed and approved by the appropriate regulatory authorities in a timely manner, if at all. Data obtained from clinical activities is not always conclusive and may be susceptible to varying interpretations, which could delay, limit, or prevent regulatory approval. Success in early stage clinical trials does not ensure success in later stage clinical trials. Even if a product candidate receives regulatory approval, the approval may be significantly limited to specific disease states, patient populations, and dosages, or have conditions placed on them that restrict the commercial applications, advertising, promotion, or distribution of these products.
18
Table of Contents
Once issued, the FDA may withdraw product approval if ongoing regulatory standards are not met or if safety problems occur after the product reaches the market. In addition, the FDA may require testing and surveillance programs to monitor the effect of approved products which have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs. The FDA may also request additional clinical trials after a product is approved. These so-called Phase 4 studies may be made a condition to be satisfied after a drug receives approval. The results of Phase 4 studies can confirm the effectiveness of a product candidate and can provide important safety information to augment the FDA’s voluntary adverse drug reaction reporting system. Any products manufactured or distributed by us pursuant to FDA approvals would be subject to continuing regulation by the FDA, including record-keeping requirements and reporting of adverse experiences with the drug. Drug manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMPs which impose certain procedural and documentation requirements upon us and our third-party manufacturers. We cannot be certain that we, or our present or future suppliers, will be able to comply with the cGMP regulations and other FDA regulatory requirements. If our present or future suppliers are not able to comply with these requirements, the FDA may halt our clinical trials, require us to recall a drug from distribution, or withdraw approval of the NDA for that drug. Furthermore, even after regulatory approval is obtained, later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market.
The FDA closely regulates the marketing and promotion of drugs. Approval may be subject to post-marketing surveillance and other record-keeping and reporting obligations, and involve ongoing requirements. Product approvals may be withdrawn if compliance with regulatory standards is not maintained or if problems occur following initial marketing. A company can make only those claims relating to safety and efficacy that are approved by the FDA. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties. Physicians may prescribe legally available drugs for uses that are not described in the product’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, restrict manufacturers’ communications on the subject of off-label use.
Section 505(b)(2) Applications
Some of our product candidates may be eligible for submission of applications for approval under the FDA’s Section 505(b)(2) approval process, which requires less information than the NDAs described above. Section 505(b)(2) was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Act. Section 505(b)(2) applications may be submitted for drug products that represent a modification (e.g., a new indication or new dosage form) of an eligible approved drug and for which investigations other than bioavailability or bioequivalence studies are essential to the drug’s approval. Section 505(b)(2) applications may rely on the FDA’s previous findings for the safety and effectiveness of the listed drug, scientific literature and information obtained by the 505(b)(2) applicant needed to support the modification of the listed drug. For this reason, preparing Section 505(b)(2) applications is generally less costly and time-consuming than preparing an NDA based entirely on new data and information from a full set of clinical trials. The law governing Section 505(b)(2) or FDA’s current policies may change in such a way as to adversely affect our applications for approval that seek to utilize the Section 505(b)(2) approach. Such changes could result in additional costs associated with additional studies or clinical trials and delays.
19
Table of Contents
The FDA provides that reviews and/or approvals of applications submitted under Section 505(b)(2) will be delayed in various circumstances. For example, the holder of the NDA for the listed drug may be entitled to a period of market exclusivity during which the FDA will not approve, and may not even review, a Section 505(b)(2) application from other sponsors. If the listed drug is claimed by a patent that the NDA holder has listed with the FDA, the Section 505(b)(2) applicant must submit a patent certification. If the 505(b)(2) applicant certifies that the patent is invalid, unenforceable or not infringed by the product that is the subject of the Section 505(b)(2) application, and within 45 days of its notice to the entity that holds the approval for the listed drug and the patent holder, the certification is challenged, the FDA will not approve the Section 505(b)(2) application until the earlier of a court decision favorable to the Section 505(b)(2) applicant or the expiration of 30 months. The regulations governing marketing exclusivity and patent protection are complex, and it is often unclear how they will be applied in particular circumstances.
In addition, both before and after approval is sought, we and our collaborators are required to comply with a number of FDA requirements. For example, we are required to report certain adverse reactions and production problems, if any, to the FDA, and to comply with certain limitations and other requirements concerning advertising and promotion for our products. Also, quality control and manufacturing procedures must continue to conform to cGMPs after approval, and the FDA periodically inspects manufacturing facilities to assess compliance with continuing cGMP. In addition, discovery of problems, such as safety problems, may result in changes in labeling or restrictions on a product manufacturer or NDA holder, including removal of the product from the market.
DEA Regulation
Our research and development processes involve the controlled use of hazardous materials, including chemicals. Some of these hazardous materials are considered to be controlled substances and subject to regulation by the U.S. Drug Enforcement Agency (DEA). Controlled substances are those drugs that appear on one of five schedules promulgated and administered by the DEA under the Controlled Substances Act (CSA). The CSA governs, among other things, the distribution, recordkeeping, handling, security and disposal of controlled substances. We must be registered by the DEA in order to engage in these activities, and are subject to periodic and ongoing inspections by the DEA and similar state drug enforcement authorities to assess ongoing compliance with the DEA’s regulations. Any failure to comply with these regulations could lead to a variety of sanctions, including the revocation, or a denial of renewal, of the DEA registration, injunctions or civil or criminal penalties.
Third-Party Payor Coverage and Reimbursement
Although none of our product candidates has been commercialized for any indication, if they are approved for marketing, commercial success of our product candidates will depend, in part, upon the availability of coverage and reimbursement from third-party payors at the federal, state and private levels. Government payor programs, including Medicare and Medicaid, private health care insurance companies and managed care plans have attempted to control costs by limiting coverage and the amount of reimbursement for particular procedures or drug treatments. The U.S. Congress and state legislatures from time to time propose and adopt initiatives aimed at cost containment. Ongoing federal and state government initiatives directed at lowering the total cost of health care will likely continue to focus on health care reform, the cost of prescription pharmaceuticals and on the reform of the Medicare and Medicaid payment systems. Examples of how limits on drug coverage and reimbursement in the United States may cause reduced payments for drugs in the future include:
• changing Medicare reimbursement methodologies;
20
Table of Contents
• fluctuating decisions on which drugs to include in formularies;
• revising drug rebate calculations under the Medicaid program; and
• reforming drug importation laws.
Some third-party payors also require pre-approval of coverage for new or innovative devices or drug therapies before they will reimburse health care providers that use such therapies. While we cannot predict whether any proposed cost-containment measures will be adopted or otherwise implemented in the future, the announcement or adoption of these proposals could have a material adverse effect on our ability to obtain adequate prices for our product candidates and operate profitably.
Foreign Approvals
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
We have not started the regulatory approval process in any jurisdiction other than the United States and we are unable to estimate when, if ever, we will commence the regulatory approval process in any foreign jurisdiction. Foreign approvals may not be granted on a timely basis, or at all. Regulatory approval of prices is required in most countries other than the United States. The prices approved may be too low to generate an acceptable return to us. If we fail to obtain approvals from foreign jurisdictions, the geographic market for our product candidates would be limited.
Employees
In May 2009, we implemented a reduction in force, releasing 11 members of our workforce.
As of February 28, 2010, we had 21 full-time employees, one of whom holds a Ph.D. degree and three full-time contract workers. There were 17 employees engaged in research and development and quality control, and four individuals working in finance, information technology, human resources and administration.
We consider our relations with our employees to be good. None of our employees are covered by a collective bargaining agreement.
21
Table of Contents
Our business is subject to various risks, including those described below. You should consider carefully these risk factors and all of the other information included in this Form 10-K. Any of these risk factors could materially adversely affect our business, operating results and financial condition. These risks are not the only ones we face. Additional risks not presently known to us or that we currently believe are immaterial may also significantly impair our business operations. In these circumstances, the market price of our common stock could decline, and you may lose all or part of your investment in our securities. Before you decide whether to purchase any of our common stock you should carefully consider the risk factors set forth below as may be updated from time to time by our future filings under the Securities Exchange Act of 1934, as amended, or the Exchange Act.
Risks Related To Our Business
We have a history of losses, we expect to generate losses in the near future, and we may never achieve or maintain profitability.
We have incurred recurring losses and had an accumulated deficit of $141.1 million as of December 31, 2009. We expect to continue to generate substantial losses over at least the next several years as we:
• expand drug product development efforts;
• conduct preclinical testing and clinical trials; and
• pursue additional applications for our existing delivery technologies.
To achieve and sustain profitability, we must, alone or with others, successfully develop, obtain regulatory approval for, manufacture, market and sell our products. We will incur substantial expenses in our efforts to develop and commercialize products and we may never generate sufficient revenue to become profitable or to sustain profitability.
We are substantially dependent upon the success of our APF530 product candidate. Clinical trial results and the NDA for this product may not lead to regulatory approval.
We have invested a significant portion of our time and financial resources in the development of our most advanced product candidate, APF530, which prevents chemotherapy-induced nausea and vomiting (CINV). Our near-term ability to generate revenues and our future success, in large part, depends on the development and commercialization of APF530.
We will not be able to commercialize our lead product candidate, APF530, until we obtain regulatory approval in the United States or foreign countries. To satisfy U.S. Food and Drug Administration (FDA) or foreign regulatory approval standards for the commercial sale of APF530, we must have demonstrated in adequate and controlled clinical trials that APF530 is safe and effective. APF530 is designed to provide at least five days prevention of CINV. In September and November 2008, we announced results of our pivotal Phase 3 human clinical trial of APF530 that achieved most of its primary and secondary endpoints. In May 2009 we submitted our new drug application (NDA) for approval of APF530 to the FDA. The NDA was accepted for review by the FDA in July 2009 and based on the Prescription Drug User Fee Act (PDUFA), the FDA has issued an action date of March 18, 2010. There are no guarantees that the FDA will provide its response by March 18, 2010, and we
22
Table of Contents
cannot assure you that any such response will be favorable. If the FDA does not take action on our NDA by the PDUFA date, does not approve our NDA or requests additional work or changes to the NDA, our continued ability to commercialize APF530 could be seriously impaired and our business would be adversely impacted. Obtaining regulatory approval of APF530 for the prevention of acute CINV for both moderately and highly emetogenic chemotherapy, and for the prevention of delayed CINV in moderately emetogenic chemotherapy, is subject to many variables. For example, the FDA’s review may not produce positive decisions as to:
• whether APF530 is safe and effective in its proposed use(s), and whether its benefits outweigh the risks;
• whether the proposed labeling (package insert) for APF530 is appropriate, and what it should contain; and
• whether the methods used in manufacturing APF530 and the controls used to maintain its quality are adequate to preserve its identity, strength, quality and purity.
Deficiencies on any of the above, or other factors, could prevent or delay obtaining regulatory approval of APF530, which would impair our reputation, increase our costs and prevent us from earning revenue.
We may not obtain regulatory approval for APF530 or any of our product candidates. Regulatory approval may also be delayed or cancelled or may entail limitations on the indicated uses of a proposed product.
The regulatory process, particularly for pharmaceutical product candidates like ours, is uncertain, can take many years and requires the expenditure of substantial resources. Any product that we or our collaborative partners develop must receive all relevant regulatory agency approvals or clearances, if any, before it may be marketed in the United States or other countries. In particular, human pharmaceutical therapeutic products are subject to rigorous preclinical and clinical testing and other requirements by the FDA in the United States and similar health authorities in foreign countries. We may not receive necessary regulatory approvals or clearances to market APF530 or any other product candidate.
Data obtained from preclinical and clinical activities is susceptible to varying interpretations that could delay, limit or prevent regulatory agency approvals or clearances. For example, the FDA may require additional clinical data to support approval, such as confirmatory studies, carcinogenicity studies and other data or studies to address questions or concerns that may arise during the FDA review process. Delays or rejections also may be encountered as a result of changes in regulatory agency policy during the period of product development and/or the period of review of any application for regulatory agency approval or clearance for a product. Delays in obtaining regulatory agency approvals or clearances could:
• significantly harm the marketing of any products that we or our collaborators develop;
• impose costly procedures upon our activities or the activities of our collaborators;
• diminish any competitive advantages that we or our collaborative partners may attain; or
• adversely affect our ability to receive royalties and generate revenue and profits.
23
Table of Contents
Even though we submitted our NDA for approval of APF530 to the FDA and intend to apply for approval of most of our other products in the United States under Section 505(b)(2) of the FDCA, which applies to reformulations of approved drugs and that may require smaller and shorter safety and efficacy testing than that for entirely new drugs, the approval process will still be costly, time-consuming and uncertain. We filed the NDA for APF530 under Section 505(b)(2) of the FDCA (which has a PDUFA action date of March 18, 2010), to rely on previous FDA findings of safety and efficacy of the active ingredient in APF530, granisetron. While we believe that Section 505(b)(2) is applicable to APF530, it is possible that the FDA may disagree and require us to submit a “stand-alone” or “full” Section 505(b)(1) NDA, which would require significantly more clinical studies and or other data collection or analysis. There are no guarantees that the FDA will provide its response by March 18, 2010, and we cannot assure you that any such response will be favorable.
We, or our collaborators, may not be able to obtain necessary regulatory approvals on a timely basis, if at all, for any of our potential products. Even if granted, regulatory approvals may include significant limitations on the uses for which products may be marketed. Failure to comply with applicable regulatory requirements can, among other things, result in warning letters, imposition of civil penalties or other monetary payments, delay in approving or refusal to approve a product candidate, suspension or withdrawal of regulatory approval, product recall or seizure, operating restrictions, interruption of clinical trials or manufacturing, injunctions and criminal prosecution.
In addition, the marketing and manufacturing of drugs and biological products are subject to continuing FDA review, and later discovery of previously unknown problems with a product, its manufacture or its marketing may result in the FDA requiring further clinical research or restrictions on the product or the manufacturer, including withdrawal of the product from the market.
Additional capital will be needed to enable us to implement our business plan, and we may be unable to raise capital when needed, which would force us to limit or cease our operations and related product development programs. Raising such capital may have to be accomplished on unfavorable terms, possibly causing dilution to our existing stockholders.
We believe that our existing cash, and cash equivalents on hand at December 31, 2009 will be sufficient to fund our operating expenses through the fourth quarter of 2010, based on our anticipated spending levels and other known sources of funding. However, our operating plan may change, and we may need additional funds sooner than anticipated to meet operational needs and capital requirements for product development and commercialization. If we are unable to complete a collaborative arrangement, equity offering, or otherwise obtain sufficient financing, we may be required to further reduce, defer, or discontinue our activities or may not be able to continue as a going concern entity. In addition, we may modify our operating plans to require additional funds in 2010. The amount of additional funding that we may require in 2010 depends on various factors, including the results of on-going regulatory review by the FDA of our APF530 NDA (which currently has a PDUFA date of March 18, 2010), our ability to establish a partnership with a pharmaceutical company for the commercialization of APF530, the time and costs related to manufacturing APF530, if approved, and competing technological and market developments. We do not currently have the financial resources to launch AP530. If AP530 is approved, we anticipate pursuing either a collaborative arrangement with a partner who will provide the necessary financial resources and expertise to launch AP530 or anticipate obtaining additional funding and resources that would be required to launch AP530 without a partner. There can be no assurance that AP530 will be approved and, if approved, that we will be successful in obtaining the additional necessary financial resources and expertise, with or without a partner, that will be required to launch AP530. Regardless of the FDA’s response with respect to the APF530 NDA, we believe we will require additional funding, from a collaborative arrangement, equity offer -
24
Table of Contents
ing, debt financing, licensing arrangement or otherwise, to support our operations in 2011. If we are not able to obtain adequate working capital and any necessary funding, we would have to curtail our development activities and adopt an alternative operating model to continue as a going concern.
The presently targeted source of additional capital is a corporate collaboration or partnership for APF530, but if efforts to secure such a collaboration or partnership fail to produce the necessary funds, we may need to seek alternate sources of capital.
In addition, the timing and degree of any longer-term capital requirements will depend on many factors, including:
• the number and characteristics of product development programs we pursue and the pace of each program;
• the scope, rate of progress, results and costs of preclinical testing and clinical trials;
• the time, cost and outcome involved in seeking regulatory approvals;
• scientific progress in our research and development programs;
• the magnitude and scope of our research and development programs;
• our ability to establish and maintain strategic collaborations or partnerships for research, development, clinical testing, manufacturing and marketing of our product candidates;
• the cost and timing of establishing sales, marketing and distribution capabilities for a specialty sales force if we commercialize any products independently;
• the cost of establishing clinical and commercial supplies of our product candidates and any products that we may develop; and
• general market conditions.
We do not anticipate that we will generate significant continuing revenues for at least several years, if ever. Until we can generate significant continuing revenues, we expect to satisfy our future cash needs through public or private equity offerings, debt financings, as well as strategic collaborations, in the form of license fees, research and development fees and milestone payments. If we issue additional equity securities or securities convertible into equity securities to raise funds, our stockholders will suffer dilution of their investment and it may adversely affect the market price of our common stock. Any debt financing we enter into may involve covenants that restrict our operations. These restrictive covenants may include, among other things, limitations on borrowing, specific restrictions on the use of our assets, as well as prohibitions on our ability to create liens, pay dividends, redeem capital stock or make investments. In the event that additional funds are obtained through arrangements with collaborative partners, these arrangements may require us to relinquish rights to some of our technologies, product candidates or products on terms that are not favorable to us or require us to enter into a collaboration arrangement that we would otherwise seek to develop and commercialize ourselves. If adequate funds are not available, we may again be required to delay, reduce the scope of, or eliminate one or more of our product development programs and reduce personnel-related and other costs, which will have a material adverse effect on our business.
25
Table of Contents
The general economic environment in which we operate is experiencing unprecedented weakness and volatility.
Our ability to secure the additional capital necessary for implementation of our longer-term business plans and general corporate continuity may be diminished by the recent and continuing negative business conditions and financial markets. For example, the difficulty in obtaining additional capital necessary to develop our other product candidates has led us to temporarily suspend such development programs.
We depend on our collaborators as a source of capital and to help us complete the process of developing and testing our products.
Our strategy for the development, clinical testing and commercialization of our products requires entering into collaborations with corporate partners, licensors, licensees and others. These collaborations are critical to funding our operations and our success in bringing our products and product candidates to the market and promoting such marketed products profitably. We are dependent upon the subsequent success of these other parties in performing their respective responsibilities and the cooperation of our partners. Our collaborators may not cooperate with us or perform their obligations under our agreements with them. We cannot control the amount and timing of our collaborators’ resources that will be devoted to our research activities related to our collaborative agreements with them. Our collaborators may choose to pursue existing or alternative technologies in preference to those being developed in collaboration with us. We may prioritize other programs ahead of collaboration activities such that funding from these other parties could be reduced or deferred. Failure to make or maintain these arrangements, or a delay in a collaborative partner’s performance, or factors that may affect our partner’s sales may materially adversely affect our business, results of operations and financial condition.
Under agreements with collaborators, we may rely significantly on them, among other activities, to:
• fund research and development activities with us;
• pay us fees upon the achievement of milestones; and
• market for or with us any commercial products that result from our collaborations.
Suspension of our pipeline development program will delay potential realization of value from new products.
Further development of our pipeline products has been temporarily deferred in order to focus, both managerial and financial resources on the approval of APF530. This action will delay the planned development of these products, reducing their potential commercial value.
Clinical trials are expensive and may not result in commercially viable products.
Conducting clinical trials is a lengthy, time-consuming and expensive process. For example, we have incurred significant expenses in developing APF530, and even if approved, it may not result in a commercially viable product. Before obtaining regulatory approvals for the commercial sale of any products, we, or our partners, must demonstrate through preclinical testing and clinical trials that our product candidates are safe and effective for use in humans. We have incurred and will continue to incur substantial expense and devote a significant amount of time to preclinical testing and clinical trials.
26
Table of Contents
Our business, results of operations and financial condition may be materially adversely affected by any delays in, or termination of, our clinical trials. Factors impacting our ability to generate commercially viable products through the conduct of clinical trials include:
• insufficient funds to continue necessary clinical trials;
• inability to find partners;
• failure of clinical trials to demonstrate the safety and efficacy of our products to the extent necessary to obtain regulatory approvals;
• failure of preclinical testing and early clinical trials to predict results of later clinical trials;
• delay in completion of clinical trials, resulting in increased costs; and
• inability to obtain regulatory approval of our products following completion of clinical trials, or delays in obtaining such approvals.
There can be no assurance that if our clinical trials are successfully initiated and completed, we will be able to obtain approval by the FDA in the United States or similar application to other regulatory authorities elsewhere in the world, in a timely manner, if at all. If we are unable to obtain approval by the FDA or other regulatory authorities elsewhere in the world, we will be unable to commercialize that product. These authorities can and do reject NDAs and require additional clinical trials, even when product candidates have performed well or have achieved favorable results in clinical trials. If we fail to commercialize one or more of our product candidates, we may be unable to generate sufficient revenues to attain profitability and our reputation in the industry and in the investment community would likely be damaged, each of which would cause our stock price to decrease.
Delays in clinical testing could increase our costs and delay our ability to obtain regulatory approval and commercialize our product candidates.
Before we, or our collaborators, can file for regulatory approval for the commercial sale of our potential products, the FDA requires extensive preclinical safety testing and clinical trials to demonstrate their safety and efficacy. Significant delays in preclinical and clinical testing could materially impact our product development costs and delay regulatory approval of our product candidates. For example, enrollment in the Phase 3 trial for APF530 was slower than we expected, resulting in delays in our development timeline and increased costs. Completing clinical trials in a timely manner depends on, among other factors:
• obtaining regulatory approval to commence a trial;
• obtaining clinical materials;
• reaching agreement on acceptable clinical study terms with prospective sites and clinical research organizations;
• obtaining institutional review board approval to conduct a study at a prospective site; and
• recruiting patients to participate in a study.
27
Table of Contents
Additionally, our current suspension of development work on our pipeline products will delay clinical testing of these products and their possible regulatory approvals, potentially reducing their eventual competitiveness and commercial values.
We rely on third parties to conduct our clinical trials, and their failure to perform their obligations in a timely and competent manner may delay development and commercialization of our product candidates.
We used clinical research organizations in the United States, India and Poland to oversee our clinical trial of APF530 and we expect to use the same or similar organizations for our future clinical trials. There are numerous alternative sources to provide these services; however, we may face delays outside of our control if these parties do not perform their obligations in a timely or competent fashion, or if we are forced to change service providers. Different cultural and operational issues in foreign countries could cause delays or unexpected problems with the patient enrollments or with the data obtained from those locations. If we experience significant delays in the progress of our clinical trials and in our plans to file NDAs or problems with the quality of data derived from various clinical sites, the prospects for approval our products in general and on a timely basis could decrease.
We have yet to demonstrate the full commercial viability of our delivery technology, and we cannot be certain that attainment of such a goal can be accomplished.
Our bioerodible drug delivery technology is at an early stage of development. We may not be able to substantiate the capability of our drug delivery technology for a variety of reasons:
• selection of inappropriate therapeutic compound for delivery;
• selection of inappropriate use or application for the particular product candidate;
• failure to receive regulatory approval on a timely basis or at all; or
• difficulties with manufacturing in commercial quantities at an acceptable cost.
Successful development of delivery technologies requires significant preclinical and clinical testing prior to regulatory approval, if any. Because of these scientific, regulatory and commercial hurdles, any program could be abandoned or otherwise fail, even after significant resources have been expended.
If any products that we or our collaborators may develop do not attain adequate market acceptance by healthcare professionals and patients, our business prospects and results of operations will suffer.
Even if a product candidate receives regulatory approval for commercial sale, the revenue received, or to be received, from the sale of the product may not be significant and will depend on many factors that are outside of our control. Factors that may affect revenue from our product candidates, if and when approved, include:
• perception of physicians and other members of the health care community of their safety and efficacy relative to that of competing products;
• cost-effectiveness;
28
Table of Contents
• patient and physician satisfaction with these products;
• ability to manufacture commercial products successfully and on a timely basis;
• cost and availability of raw materials;
• market size for these products;
• reimbursement policies of government and third-party payors;
• unfavorable publicity concerning these products or similar drugs;
• the introduction, availability and acceptance of competing treatments, including those of our collaborators;
• adverse event information relating to these products;
• product labeling or product insert required by the FDA or regulatory authorities in other countries;
• the regulatory developments related to the manufacture or continued use of these products;
• extent and effectiveness of sales and marketing and distribution support for the products; and
• our collaborators’ decisions as to the timing of product launches, pricing and discounting.
Our product revenue will be adversely affected if, due to these or other factors, the products we or our collaborators are able to commercialize do not gain significant market acceptance.
We depend on contract manufacturers and collaborators for manufacturing our products; if they do not perform as expected, our revenue and customer relations will suffer.
We do not own or operate manufacturing facilities for the production of clinical or commercial quantities of any product. Our ability to develop and commercialize any products we may develop will depend in part on our ability to manufacture, or arrange for collaborators or other parties to manufacture, our products at a competitive cost, in accordance with regulatory requirements, and in sufficient quantities for clinical testing and eventual commercialization. We do not intend to develop or acquire facilities to manufacture any of our product candidates for clinical trials or commercial purposes in the foreseeable future. We rely on a small number of third-party manufacturers to produce our compounds and expect to continue to do so to meet the preclinical and clinical requirements of our potential products and for all of our commercial needs, some of which are our sole source suppliers at present. We have no long-term agreements with any of these third parties. We may not be able to extend these agreements at satisfactory terms, or at all, and we may not be able to find a replacement contract manufacturer at satisfactory terms or on a timely basis. Additionally, difficult economic conditions may cause operational and financial problems for our third party suppliers, resulting in their failure and disruption to our operations.
Further, our contract manufacturers and our collaborators are required to comply with FDA requirements related to product testing, quality assurance, manufacturing and records and documentation. Our contract manufacturers, or our collaborators,
29
Table of Contents
may not be able to comply with the applicable FDA regulatory requirements. They may be required to pass an FDA pre-approval inspection for conformity with cGMPs before we can obtain approval to manufacture, and will be subject to ongoing, periodic, unannounced inspection by the FDA and corresponding state agencies to ensure strict compliance with cGMP, and other applicable government regulations and corresponding foreign standards. If we and our contract manufacturers fail to achieve and maintain high manufacturing standards in compliance with cGMP regulations, we may experience manufacturing errors resulting in patient injury or death, product recalls or withdrawals, delays or interruptions of production or failures in product testing or delivery, delay or prevention of filing or approval of marketing applications for our products, cost overruns or other problems that could seriously harm our business. We are aware that one particular contract manufacturer has previously received Form 483 notices from the FDA and has worked to address the deficiencies identified in these notices. The remediation efforts may not be adequate to address the deficiencies or the manufacturer may not stay in compliance with FDA requirements in the future. Not complying with FDA requirements could result in an enforcement or other action, prevent commercialization of our product candidates and impair our reputation and results of operations.
Any performance failure on the part of our contract manufacturers could delay clinical development or regulatory approval of product candidates or commercialization of our future products, depriving us of potential product revenue and resulting in additional losses. In addition, our dependence on a third party for manufacturing may adversely affect our future profit margins and limit our ability to commercialize products on a timely and competitive basis. Our ability to replace an existing manufacturer may be difficult because the number of potential manufacturers is limited, and the FDA must approve any replacement manufacturer before we can begin manufacturing APF530 or any of our other product candidates. Such approval would require new testing and compliance inspections. It may be difficult or impossible for us to identify and engage a replacement manufacturer on acceptable terms in a timely manner, or at all.
APF530 or any of our other product candidates may be in competition with other products for access to the facilities of third parties. Consequently, APF530 or any of our other product candidates may be subject to manufacturing delays if collaborators or outside contractors give other companies’ products greater priority than our products. For this and other reasons, our collaborators or third-party service providers may not be able to manufacture APF530 or any of our other product candidates in a cost-effective or timely manner. If not manufactured in a timely manner, the clinical development of any of our product candidates or their submission for regulatory approval could be delayed, and our ability to deliver products to market on a timely basis could be impaired or precluded.
To date, APF530 and our product candidates have been manufactured in small quantities for preclinical studies and clinical trials. If in the future APF530 or any of our product candidates are approved for commercial sale, we will need to manufacture our products in larger quantities. Significant scale-up of manufacturing may require additional process development and validation studies, which the FDA must review and approve. The commercial success of our products, including APF530 in the near-term, will be dependent upon the ability of our contract manufacturers to produce a product in commercial quantities at competitive costs of manufacture. The ability to do so cannot be presumed. Significant additional development work is required prior to any commercial launch of a product. In the case of APF530, the high viscosity of the product creates particularly challenging factors relative to attainable production rates and cost of manufacture. If APF530 receives regulatory approval, we plan to scale-up manufacturing for APF530 in order to realize important economies of scale. These scale-up activities would take time to implement, require additional capital investment, process development and validation studies, and FDA approval. We cannot guarantee that we will be successful in achieving competitive manufacturing costs through such scale-up activities.
30
Table of Contents
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to sell and market any products we may develop, we may be unable to generate product revenue.
We do not currently have a sales organization for the sales, marketing and distribution of pharmaceutical products. In order to commercialize any products, we must build our sales, marketing, distribution, managerial and other non-technical capabilities or make arrangements with third parties to perform these services. We have not yet determined whether we will attempt to establish internal sales and marketing capabilities or enter into agreements with third parties to sell and market any products we may develop. We have no experience in developing, training or managing a marketing and sales force. The establishment and development of our own sales force to market any products we may develop will be expensive and time consuming and could delay any product launch, and we cannot be certain that we would be able to successfully develop this capacity. If we are unable to establish our sales and marketing capability or any other non-technical capabilities necessary to commercialize any product we may develop, we will need to contract with third parties to market and sell any products we may develop. If we are unable to establish adequate sales, marketing and distribution capabilities, whether independently or with third parties, we may not be able to generate product revenue and may not become profitable.
If we fail to comply with continuing federal, state and foreign regulations, we could lose our approvals to market drugs and our business would be seriously harmed.
Following initial regulatory approval of any drugs we may develop, we will be subject to continuing regulatory review, including review of adverse drug experiences and clinical results that are reported after our drug products become commercially available. This would include results from any post-marketing tests or continued actions required as a condition of approval. The manufacturer and manufacturing facilities we use to make any of our drug candidates will be subject to periodic review and inspection by the FDA. If a previously unknown problem or problems with a product or a manufacturing and laboratory facility used by us is discovered, the FDA or foreign regulatory agency may impose restrictions on that product or on the manufacturing facility, including requiring us to withdraw the product from the market. Any changes to an approved product, including the way it is manufactured or promoted, often require FDA approval before the product, as modified, can be marketed. We and our contract manufacturers will be subject to ongoing FDA requirements for submission of safety and other post-market information. If we and our contract manufacturers fail to comply with applicable regulatory requirements, a regulatory agency may:
• issue warning letters;
• impose civil or criminal penalties;
• suspend or withdraw our regulatory approval;
• suspend or terminate any of our ongoing clinical trials;
• refuse to approve pending applications or supplements to approved applications filed by us;
• impose restrictions on our operations;
• close the facilities of our contract manufacturers; or
• seize or detain products or require a product recall.
31
Table of Contents
Additionally, such regulatory review covers a company’s activities in the promotion of its drugs, with significant potential penalties and restrictions for promotion of drugs for an unapproved use. Sales and marketing programs are under scrutiny for compliance with various mandated requirements, such as illegal promotions to healthcare professionals. We are also required to submit information on our open and completed clinical trials to public registries and databases; failure to comply with these requirements could expose us to negative publicity, fines and penalties that could harm our business.
If we are unable to recruit and retain skilled employees, we may not be able to achieve our objectives.
We depend on a small number of key management and technical personnel. Retaining our current employees and recruiting qualified scientific personnel to perform future research and development work will be critical to our success. While recent pharmaceutical and biotechnology industry layoffs have somewhat mitigated a usual shortage of skilled personnel in our industry, competition is always present for experienced scientists, and an inability to recruit or retain sufficient skilled personnel could result in delays to product development or approval, loss of sales and diversion of management resources. If we lose members of our senior management team, we may not be able to find suitable replacements and our business may be harmed as a result. Further proactive reductions in our workforce, such as occurred in November 2008 and May 2009, could create difficulties in our ability to perform the activities necessary to accomplish our operational and strategic goals.
We face intense competition from other companies.
APF530 is expected to face significant competition for the prevention of delayed CINV, principally from Eisai/MGI Pharma’s Aloxi (palonosetron). In addition to Aloxi, APF530 will compete with entrenched products for prevention of acute CINV including Roche’s Kytril (granisetron), GlaxoSmithKline’s Zofran (ondansetron) and Aventis’ Anzemet (dolasetron). Generic versions of certain of these products are also marketed by other companies. We are also aware of several companies which have developed or are developing both generic and new formulations of granisetron, including transdermal formulations such as ProStrakan’s Sancuso® (granisetron transdermal patch) and Hana Biosciences’ Zensana™ (oral ondansetron), which has been licensed to Par Pharmaceuticals. APF112 is expected to face competition from two injectable controlled release bupivicane products, Durect Corporation’s Posidur™ and Pacira Pharmaceutical’s Exparel™ DepoBupivacaine.
There are several companies that are developing new formulations of existing drugs using novel drug delivery technologies. Many of these companies have substantially greater financial, research and development, manufacturing, sales and marketing, distribution resources and experience than we do. The following are some of our major competitors among drug delivery system developers: Alkermes, Inc., Depomed, Inc., Durect Corporation, and Pacira Pharmaceuticals, Inc.
Smaller or early stage companies and research institutions may also prove to be significant competitors, particularly through collaborative arrangements with large and established pharmaceutical companies. We will also face competition from these parties in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, and acquiring and in-licensing technologies and products complementary to our programs or potentially advantageous to our business. If any of our competitors succeed in obtaining approval from the FDA or other regulatory authorities for their products sooner than we do or for products that are more effective or less costly than ours, our commercial opportunity could be significantly reduced.
32
Table of Contents
Major technological changes can happen quickly in the biotechnology and pharmaceutical industries, and the development of technologically improved or different products or drug delivery technologies may make our product candidates or platform technologies obsolete or noncompetitive.
Because we, or our collaborators must obtain regulatory approval to market our products in the United States and foreign jurisdictions, we cannot predict whether or when we will be permitted to commercialize our products.
Federal, state and local governments in the United States and governments in other countries have significant regulations in place that govern many of our activities. The preclinical testing and clinical trials of the products that we develop ourselves, or that our collaborators develop are subject to government regulation and may prevent us from creating commercially viable products from our discoveries. These regulations and their application may change making it more difficult or prohibitive to develop our products. In addition, the sale by us, or our collaborators, of any commercially viable product will be subject to government regulation from several standpoints, including the processes of:
• manufacturing;
• labeling;
• distributing;
• advertising and promoting; and
• selling and marketing.
If we cannot establish pricing of our product candidates acceptable to the United States or foreign governments, insurance companies, managed care organizations and other payors, or arrange for favorable reimbursement policies, any product sales will be severely hindered.
The continuing efforts of the United States and foreign governments, insurance companies, managed care organizations and other payors of health care costs to contain or reduce costs of health care may adversely affect our ability to generate adequate revenues and gross margins to make the products we develop commercially viable. Our ability to commercialize any product candidates successfully will depend in part on the extent to which governmental authorities, private health insurers and other organizations establish appropriate reimbursement levels for the cost of any products and related treatments.
In certain foreign markets, the pricing of prescription pharmaceuticals is subject to government control. In the United States, given recent federal and state government initiatives directed at lowering the total cost of health care, the U.S. Congress and state legislatures will likely continue to focus on health care reform, the cost of prescription pharmaceuticals and on the reform of the Medicare and Medicaid systems. The trend toward managed health care in the United States, which could significantly influence the purchase of health care services and products, as well as legislative proposals to reform health care, control pharmaceutical prices or reduce government insurance programs, may result in lower prices for our product candidates. While we cannot predict whether any legislative or regulatory proposals affecting our business will be adopted, the announcement or adoption of these proposals could have a material and adverse effect on our potential revenues and gross margins.
33
Table of Contents
Our business strategy includes the entry into additional collaborative agreements. We may not be able to enter into additional collaborative agreements or may not be able to negotiate commercially acceptable terms for these agreements.
Our current business strategy includes the entry into additional collaborative agreements for the development and commercialization of our delivery technologies. The negotiation and consummation of these types of agreements typically involve simultaneous discussions with multiple potential collaborators and require significant time and resources from our officers, business development, legal and research and development staff. In addition, in attracting the attention of pharmaceutical and biotechnology company collaborators, we compete with numerous other third parties with product opportunities as well the collaborators’ own internal product opportunities. We may not be able to consummate additional collaborative agreements, or we may not be able to negotiate commercially acceptable terms for these agreements. If we do not consummate additional collaborative agreements, we may have to consume money more rapidly on our product development efforts, continue to defer development activities or forego the exploitation of certain geographic territories, any of which could have a material adverse effect on our business.
If we or our collaborators cannot arrange for adequate third-party reimbursement for our products, our future revenue will suffer.
In both domestic and foreign markets, sales of our potential products will depend in substantial part on the availability of adequate reimbursement from third-party payors such as government health administration authorities, private health insurers and other organizations. Third-party payors often challenge the price and cost-effectiveness of medical products and services and such pressure may increase in the future. Significant uncertainty exists as to the adequate reimbursement status of newly approved health care products. Any products we are able to successfully develop may not be reimbursable by third-party payors. In addition, our products may not be considered cost-effective and adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize a profit. Legislation and regulations affecting the pricing of pharmaceuticals may change before our products are approved for marketing and any such changes could further limit reimbursement. Reimbursement policies utilized by our collaborators or ourselves may be challenged by regulatory entities, with resultant fines, negative publicity and the need to implement changes that reduce the utilization of our products. If any products we develop do not receive adequate reimbursement, our revenue will be severely limited.
Our inability to obtain specialized materials could slow down our product development process.
Some of the critical materials and components used in our products in development are sourced from a single supplier. An interruption in supply of a key material could significantly delay our research and development process or increase our expenses.
Specialized materials must often be manufactured for the first time for use in drug delivery technologies, or materials may be used in the technologies in a manner different from their customary commercial uses. The quality of materials can be critical to the performance of a drug delivery technology, so a reliable source of a consistent supply of materials is important. Materials or components needed for our drug delivery technologies may be difficult to obtain on commercially reasonable terms, particularly when relatively small quantities are required, or if the materials traditionally have not been used in pharmaceutical products.
34
Table of Contents
If we are unable to adequately protect or enforce our intellectual property rights or secure rights to third-party patents, we may lose valuable assets, experience reduced market share or incur costly litigation to protect our rights or our third-party collaborators may choose to terminate their agreements with us.
Our success will depend in part on our ability to obtain patents, maintain trade secret protection as well as successfully defending these patents against challenges, while operating without infringing the proprietary rights of others. We have filed a number of U.S. patent applications on inventions relating to the composition of a variety of polymers, specific products, product groups and processing technology. In addition to obtaining patents in a number of foreign countries, we have also filed U.S. and foreign patent applications on our polymer technology with the PCT, the European Patent Office, Australia, Canada, China, Hong Kong, Japan, South Korea, Singapore and Taiwan. We have a total of 21 issued U.S. patents and an additional 77 issued (or registered) foreign patents. The patents on the bioerodible technologies expire between January 2016 and November 2023. Our existing patents may not cover future products, additional patents may not be issued, and current patents, or patents issued in the future, may not provide meaningful protection or prove to be of commercial benefit.
The patent positions of pharmaceutical companies, including ours, are uncertain and involve complex legal and factual questions. In addition, the coverage claimed in a patent application can be significantly reduced before the patent is issued. Consequently, our patent applications, or those that are licensed to us, may not issue into patents, and any issued patents may not provide sufficient protection for our product candidates or provide sufficient protection to afford us a commercial advantage against competitive technologies or may be held invalid if challenged or circumvented. Patent applications in the United States are maintained in confidence for at least 18 months after their filing. Consequently, we cannot be certain that the patent applications we are pursuing will lead to the issuance of any patent or be free from infringement or other claims from other parties. Our competitors may also independently develop products similar to ours or design around or otherwise circumvent patents issued to us or licensed by us. In addition, the laws of some foreign countries may not protect our proprietary rights to the same extent as the U.S. law.
We are party to collaborative agreements. These agreements subject us to obligations which must be fulfilled and require us to manage complex relationships with third parties. If we are unable to meet our obligations or manage our relationships with our collaborators under these agreements, or enter into additional collaboration agreements, or if our existing collaborations are terminated or not extended on terms as beneficial as we anticipate, our revenue may decrease. Our third-party collaborators have entered into these agreements based on the exclusivity that our intellectual property rights confer on the products being developed. The loss or diminution of our intellectual property rights could result in a decision by our third-party collaborators to terminate their agreements with us. In addition, these agreements are generally complex and contain provisions that could give rise to legal disputes, including potential disputes concerning ownership of intellectual property and data under collaborations. Such disputes can lead to lengthy, expensive litigation or arbitration requiring us to devote management time and resources to such dispute which we would otherwise spend on our business.
We also rely upon trade secrets, technical know-how and continuing technological innovation to develop and maintain our competitive position. We require our employees, consultants, advisors and collaborators to execute appropriate confidentiality and assignment-of-inventions agreements with us. These agreements typically provide that all materials and confidential information developed or made known to the individual during the course of the individual’s relationship with us is to be kept confidential and not disclosed to third parties except in specific circumstances, and that all inventions arising out of the
individual’s relationship with us shall be our exclusive property. These agreements may be breached, and in some instances,
35
Table of Contents
we may not have an appropriate remedy available for breach of the agreements. Furthermore, our competitors may independently develop substantially equivalent proprietary information and techniques, reverse engineer our information and techniques, or otherwise gain access to our proprietary technology. We may be unable to meaningfully protect our rights in trade secrets, technical know-how and other non-patented technology. We may have to resort to litigation to protect our intellectual property rights, or to determine their scope, validity or enforceability. In addition, interference proceedings declared by the U.S. Patent and Trademark Office may be necessary to determine the priority of inventions with respect to our patent applications. Enforcing or defending our proprietary rights is expensive, could cause diversion of our resources and may not prove successful. In addition, courts outside the United States may be less willing to protect trade secrets. Costly and time-consuming litigation could be necessary to seek to enforce and determine the scope of our proprietary rights. Any failure to enforce or protect our rights could cause us to lose the ability to exclude others from using our technology to develop or sell competing products.
We may infringe on the intellectual property rights of others, and any litigation could force us to stop developing or selling potential products and could be costly, divert management attention and harm our business.
We must be able to develop products without infringing the proprietary rights of other parties. Because the markets in which we operate involve established competitors with significant patent portfolios, including patents relating to the composition of a variety of polymers, specific products, product groups and processing technology, it could be difficult for us to use our technologies or develop products without infringing the proprietary rights of others. We may not be able to design around the patented technologies or inventions of others and we may not be able to obtain licenses to use patented technologies on acceptable terms, or at all. If we cannot operate without infringing the proprietary rights of others, we will not be able to develop or commercialize some or all of our product candidates, and consequently will not be able to earn product revenue.
If we are required to defend ourselves in a lawsuit, we could incur substantial costs and the lawsuit could divert management attention, regardless of the lawsuit’s merit or outcome. These legal actions could seek damages and seek to enjoin testing, manufacturing and marketing of the accused product or process. In addition to potential liability for significant damages, we could be required to obtain a license to continue to manufacture or market the accused product or process and any license required under any such patent may not be made available to us on acceptable terms, if at all.
Periodically, we review publicly available information regarding the development efforts of others in order to determine whether these efforts may violate our proprietary rights. We may determine that litigation is necessary to enforce our proprietary rights against others. Such litigation could result in substantial expense, regardless of its outcome, and may not be resolved in our favor.
Furthermore, patents already issued to us or our pending patent applications may become subject to dispute, and any disputes could be resolved against us. In addition, because patent applications in the United States are currently maintained in secrecy for a period of time prior to issuance, and patent applications in certain other countries generally are not published until more than 18 months after they are first filed, and because publication of discoveries in scientific or patent literature often lags behind actual discoveries, we cannot be certain that we were the first creator of inventions covered by our pending patent applications or that we were the first to file patent applications on such inventions.
36
Table of Contents
We are exposed to risks and increased expenses as a result of laws requiring smaller reporting company filers to evaluate internal controls over financial reporting.
Section 404 of the Sarbanes-Oxley Act of 2002 (Section 404) requires our management to evaluate the effectiveness of our internal controls over financial reporting as of the end of each fiscal year, and to include a management report assessing the effectiveness of our internal control over financing reporting in our annual report on Form 10-K for each fiscal year. We expect that our independent auditors will be required to report on our internal control over financial reporting beginning with the year ending December 31, 2010. We and our independent registered public accounting firm may not be able to conclude on an ongoing basis that we have effective internal controls over financial reporting in accordance with Section 404. We have implemented an ongoing program to perform the system and process evaluation and testing we believe to be necessary to comply with these requirements. However, we cannot assure you that we will be successful in our efforts. We expect to incur increased expense and to devote additional management resources to Section 404 compliance. Any failure to implement required new or improved controls, or difficulties encountered in the implementation or operation of these controls, could harm our operating results and cause us to fail to meet our financial reporting obligations, which could adversely affect our business and reduce our stock price.
Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our internal controls over financial reporting will prevent all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. We cannot assure you that we or our independent registered public accounting firm will not identify a material weakness in our internal controls in the future, which would require management and our independent registered public accounting firm to evaluate our internal controls as ineffective. If our internal controls over financial reporting are not considered effective, we may experience a loss of public confidence, which could have an adverse effect on our business and on the price of our stock.
Legislative actions, potential new accounting pronouncements and higher insurance costs are likely to impact our future financial position or results of operations.
Future changes in financial accounting standards may cause adverse, unexpected fluctuations in the timing of the recognition of revenue or expenses and may affect our financial position or results of operations. New pronouncements and varying interpretations of pronouncements have occurred with frequency and may occur in the future and we may make changes in our accounting policies in the future. Compliance with changing regulation of corporate governance and public disclosure may result in additional expenses. Changing laws, regulations and standards relating to corporate governance and public disclosure, including the Sarbanes-Oxley Act of 2002, new Securities and Exchange Commission (SEC) regulations, the Public Company Accounting Oversight Board pronouncements and The NASDAQ Capital Market rules, are creating uncertainty for companies such as ours and insurance, accounting and auditing costs are increasing as a result of this uncertainty and other factors. We are committed to maintaining high standards of corporate governance and public disclosure. As a result, we intend to invest all reasonably necessary resources to comply with evolving standards, and this investment may result in increased general and administrative expenses and a diversion of management time and attention from revenue-generating activities to compliance activities.
37
Table of Contents
We could be exposed to significant product liability claims that could be time consuming and costly to defend, divert management attention and adversely impact our ability to obtain and maintain insurance coverage.
The testing, manufacture, marketing and sale of our products involve an inherent risk that product liability claims will be asserted against us. Although we are insured against such risks up to an annual aggregate limit in connection with clinical trials and commercial sales of our products, our present product liability insurance may be inadequate and may not fully cover the costs of any claim or any ultimate damages we might be required to pay. Product liability claims or other claims related to our products, regardless of their outcome, could require us to spend significant time and money in litigation or to pay significant damages. Any successful product liability claim may prevent us from obtaining adequate product liability insurance in the future on commercially desirable or reasonable terms. In addition, product liability coverage may cease to be available in sufficient amounts or at an acceptable cost. An inability to obtain sufficient insurance coverage at an acceptable cost or otherwise to protect against potential product liability claims could prevent or inhibit the commercialization of our products. A product liability claim could also significantly harm our reputation and delay market acceptance of our products.
Our use of hazardous materials could subject us to liabilities, fines and sanctions.
Our laboratory and clinical testing sometimes involve use of hazardous, radioactive or otherwise toxic materials. We are subject to federal, state and local laws and regulations governing how we use, manufacture, handle, store and dispose of these materials. Although we believe that our safety procedures for handling and disposing of such materials comply in all material respects with all federal, state and local regulations and standards, there is always the risk of accidental contamination or injury from these materials. In the event of an accident, we could be held liable for any damages that result and such liability could exceed our financial resources. If we fail to comply with these regulations and standards or with the conditions attached to our operating licenses, the licenses could be revoked, and we could be subjected to criminal sanctions and substantial financial liability or be required to suspend or modify our operations. Compliance with environmental and other laws may be expensive and current or future regulations may impair our development or commercialization efforts.
Earthquake damage to our facilities could delay our research and development efforts and adversely affect our business.
Our research and development facility in Redwood City, California, is located in a seismic zone, and there is the possibility of an earthquake, which could be disruptive to our operations and result in delays in our research and development efforts. In the event of an earthquake, if our facilities or the equipment in our facilities are significantly damaged or destroyed, we may not be able to rebuild or relocate our facility or replace any damaged equipment in a timely manner and our business, financial condition and results of operations could be materially and adversely affected.
Risks Related To Our Common Stock
Our common stock may be delisted from The NASDAQ Capital Market, which could negatively impact the price of our common stock and our ability to access the capital markets.
Our common stock is currently listed on The NASDAQ Capital Market. At the start of 2009, our common stock was listed on The NASDAQ Global Market, but we failed to meet the continued listing requirements for this market, so we applied for and were granted a move to The NASDAQ Capital Market. The listing standards of The NASDAQ Capital Market require that a
38
Table of Contents
company maintain stockholders’ equity of at least $2.5 million and a minimum bid price subject to specific requirements of $1.00 per share. During 2009 our minimum bid price fell below $1.00 per share and we received notice from NASDAQ Stock Market (NASDAQ) that we did not satisfy the $1.00 minimum bid price requirement. Our stock price subsequently closed above $1.00 for thirty consecutive days and on January 7, 2010, we received notice from NASDAQ confirming that we had regained compliance with the $1.00 minimum bid price requirement for continued listing on The NASDAQ Capital Market. Consequently, the NASDAQ Listing Qualifications Panel determined to continue the listing of our securities on NASDAQ.
Should we fail to comply with the minimum listing standards applicable to issuers listed on The NASDAQ Capital Market, our common stock may be delisted from The NASDAQ Capital Market. If our common stock is delisted, it could reduce the price of our common stock and the levels of liquidity available to our stockholders. In addition, the delisting of our common stock could materially adversely affect our access to the capital markets, and any limitation on liquidity or reduction in the price of our common stock could materially adversely affect our ability to raise capital on terms acceptable to us or at all. Delisting from The NASDAQ Capital Market could also result in other negative implications, including the potential loss of confidence by suppliers, customers and employees, the loss of institutional investor interest and fewer business development opportunities.
The price of our common stock has been and may continue to be volatile.
Our common stock has historically been volatile, with a trading price ranging from $0.30 to $10.92 over the past five years. The stock markets in general, and the markets for drug delivery and pharmaceutical stocks in particular, have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. These broad market fluctuations may adversely affect the trading price of our common stock. In addition, the limited trading volume of our stock may contribute to its volatility.
Price declines in our common stock could result from general market and economic conditions and a variety of other factors, including:
• delisting from The NASDAQ Capital Market;
• our ability to raise capital in the face of continuing cash depletion;
• non-approval of our product candidates, or delays in the FDA review process, particularly with respect to APF530, which has a PDUFA action date of March 18, 2010;
• adverse actions taken by regulatory agencies with respect to our product candidates, clinical trials, manufacturing processes or sales and marketing activities;
• continuing losses and failure to achieve or maintain profitability;
• adverse results, lack of success or delays in our clinical trials of our product candidates, including APF530;
• delays in preclinical and clinical testing;
• failure to substantiate the capability of our drug delivery technology;
39
Table of Contents
• failure to attain adequate market acceptance by healthcare professionals and patients;
• failure of our contract manufacturers and collaborators to perform their contractual obligations as expected;
• failure to comply with continuing federal, state and foreign regulations;
• market conditions relating to our segment of the industry or the securities markets in general;
• any lawsuit involving us or our product candidates;
• announcements concerning our competitors, or the biotechnology or pharmaceutical industries in general;
• changes in accounting principles; and
• loss of any of our key scientific or management personnel.
In the past, following periods of volatility in the market price of a particular company’s securities, litigation has often been brought against that company. If litigation of this type is brought against us, it could be extremely expensive and divert management’s attention and our company’s resources.
Our certificate of incorporation, our bylaws, Delaware law and our stockholder rights plan contain provisions that could discourage another company from acquiring us and may prevent attempts by our stockholders to replace or remove our current management.
Provisions of Delaware law, our certificate of incorporation, bylaws and stockholder rights plan may discourage, delay or prevent a merger or acquisition that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace or remove our board of directors. These provisions include:
• authorizing the issuance of “blank check” preferred stock without any need for action by stockholders; and
• providing for dilutive issuance of preferred stock, commonly referred to as a “poison pill”, which can be triggered after a person or a group acquires 20% or more of our common stock, except Tang Capital Partners and its affiliates, and Baker Brothers Investments and its affiliates, for which the potentially triggering level of ownership are 34% and 30%, respectively.
In addition, Section 203 of Delaware General Corporation Law may discourage, delay or prevent a change in control of our company by prohibiting stockholders owning in excess of 15% of our outstanding voting stock from merging or combining with us, unless certain approvals are obtained.
40
Table of Contents
Further concentration in shareholder ownership could influence strategic actions.
Our directors, executive officers, principal stockholders and affiliated entities currently beneficially own or control a majority of our outstanding securities. In October 2009, Tang Capital Partners, LP and its affiliates, including Tang Capital Management, LLC and its Managing Director, Kevin C. Tang, increased their beneficial ownership in our common stock to 31%, and Baker Brothers Investments and its affiliates increased their beneficial ownership in our common stock to 22%. Mr. Tang is also a member of our board. Baker Brothers Investments has the right to designate a representative to our Board of Directors. In February 2010, Stephen R. Davis was appointed to our Board of Directors as the Baker Brothers Investments designee. In the event Baker Brothers Investments exercises its right to purchase its designated additional shares under the terms of the Private Placement, Baker Brothers Investments shall have the right to designate an additional representative to our board of directors.
Such a concentration of common stock ownership could significantly influence corporate actions on various strategic matters, including, for example, receptivity to collaborations and merger or sale overtures.
Future sales of our common stock may cause our stock price to decline.
Our principal stockholders and affiliated entities hold a substantial number of shares of our common stock that they are able to sell in the public market. In addition, they currently own outstanding warrants exercisable through May 14, 2010 and January 7, 2015 for additional shares of our common stock. The exercise of these warrants or the sale by our current stockholders of a substantial number of shares, or the expectation that such exercises or sales may occur, could significantly reduce the market price of our common stock.
Future utilization of net operating loss carryforwards may be impaired due to recent changes in ownership.
As discussed in Note 11 to the Financial Statements, we believe our net operating losses and tax attributes may be subject to limitation under Section 382 of the Internal Revenue Code of 1986. As a result, our deferred tax assets, and related valuation allowance, have been reduced for the estimated impact of the net operating losses and credits that we currently estimate may expire unused. Utilization of our remaining net operating loss and research and development credit carryforwards may still be subject to substantial annual limitations due to ownership change limitations provided by the Internal Revenue Code and similar state provisions for ownership changes after December 31, 2009, including those that may come in conjunction with future equity financings or market trades by our shareholders.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
Available Information
We make available free of charge on or through our Internet website our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and all amendments to those reports as soon as reasonably practicable after they are electronically filed with, or furnished to, the SEC. Our Internet website address is “www.appharma.com.” The reference to our Internet website does not constitute incorporation by reference of the information contained on or hyperlinked from our Internet website. We file electronically with the SEC, our annual reports on Form 10-K, quarterly reports on Form 10-Q and current
41
Table of Contents
reports on Form 8-K pursuant to Section 13(a) or 15(d) of the Exchange Act. The SEC maintains an Internet site that contains reports, proxy information and information statements, and other information regarding issuers that file electronically with the SEC. The address of that website is http://www.sec.gov. The materials are also available at the SEC’s Public Reference Room, located at 100 F Street, Washington, D.C. 20549. The public may obtain information through the public reference room by calling the SEC at 1-800-SEC-0330.
We lease 26,067 square feet of laboratory, office and warehouse space in Redwood City, California under a lease expiring in 2011. The annual rent expense for the Redwood City facility is approximately $463,000.
We believe our facilities are adequate and suitable for current and anticipated needs. We plan to evaluate our options for leasing office and lab space in the proximity of our current offices and plans to enter a new lease following the end of the current lease. However, we can not be sure that we will be able to find suitable space at a reasonable lease rate.
While we are not currently a party to any material pending legal proceedings, from time to time we are named as a party to lawsuits in the normal course of its business. Litigation, in general, and intellectual property litigation in particular, can be expensive and disruptive to normal business operations. Moreover, the results of legal proceedings are difficult to predict.
42
Table of Contents
ITEM 5. MARKET FOR REGISTRANT’S COMMON STOCK RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Shares of our common stock trade on The NASDAQ Capital Market, under the symbol APPA. Prior to October 28, 2009 our stock traded on The NASDAQ Global Market. As of February 28, 2010, there were 108 holders of record of our common stock.
We have never paid cash dividends and do not anticipate paying cash dividends in the foreseeable future. The following table sets forth, for the fiscal periods indicated, the range of high and low sales prices for our common stock on the NASDAQ Global and NASDAQ Capital Markets.
| 2009 |
High | Low | 2008 |
High | Low | |||||||||
| First Quarter |
$ | 0.89 | $ | 0.41 | First Quarter | $ | 1.85 | $ | 1.08 | |||||
| Second Quarter |
1.21 | 0.38 | Second Quarter | 1.75 | 0.81 | |||||||||
| Third Quarter |
1.05 | 0.65 | Third Quarter | 1.67 | 0.63 | |||||||||
| Fourth Quarter |
1.39 | 0.77 | Fourth Quarter | 0.80 | 0.30 | |||||||||
On March 9, 2010, the closing sale price of our common stock was $1.87 per share.
In October 2009, in a private placement, we sold 7,954,543 shares of our common stock at $0.88 per share and warrants to purchase 3,977,270 shares of our common stock, exercisable through January 7, 2015, at $0.88 per share (Private Placement). The purchasers paid $0.125 per underlying share for the warrants. Additionally the purchasers have the right to purchase up to an additional 5,165,286 shares at $0.97 per share prior to May 14, 2010 and paid $0.125 per underlying share for the right to purchase such additional shares. Total proceeds were approximately $7.9 million, after deducting costs associated with the issuance. The sale of the securities is exempt from registration pursuant to the exemption for transactions by an issuer not involving any public offering under Section 4(2) the Securities Act of 1933, as amended, and Regulation D under the Securities Act of 1933, as amended. Pursuant to the terms of a registration rights agreement in connection with the Private Placement, we are required to prepare and file Form S-3 registration statements, as permissible under SEC rules and regulations, beginning within 30 days of October 22, 2009, with the SEC for the purpose of registering for resale of the securities sold in the Private Placement. We filed a Form S-3 covering 7,532,617 shares on November 6, 2009, which was declared effective by the SEC on November 17, 2009. However, if we fail to meet future filing or effectiveness deadlines for any additional registration statements required or fail to keep any registration statements continuously effective, we may be obligated to pay to the holders of the shares and warrants liquidated damages in the amount of 1% per month of the purchase price for the shares and warrants, up to a maximum cap of 8%. In addition, at the closing of the Private Placement, Baker Brothers Investments was given the right to designate a representative to our Board of Directors. In February 2010, Stephen R. Davis was appointed to our Board of Directors as the Baker Brothers Investments designee. In the event Baker Brothers Investments exercises its right to purchase its designated additional shares under the terms of the Private Placement, Baker Brothers Investments shall have the right to designate an additional representative to our board of directors.
43
Table of Contents
ITEM 6. SELECTED FINANCIAL DATA
The following selected financial data should be read in conjunction with the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the financial statements and notes thereto, included in Item 8 of this Annual Report on Form 10-K. The financial data does not purport to indicate results of operations as of any future date or for any future period.
| For the Years Ended December 31, (In thousands, except per share data) |
2009 | 2008 | 2007 | 2006 | 2005 | |||||||||||||||
| STATEMENTS OF OPERATIONS DATA |
||||||||||||||||||||
| Revenue: |
||||||||||||||||||||
| Royalties |
$ | — | $ | — | $ | — | $ | — | $ | 5,247 | ||||||||||
| Contract revenue |
1,261 | 369 | 412 | — | 144 | |||||||||||||||
| Total revenue |
1,261 | 369 | 412 | — | 5,391 | |||||||||||||||
| Operating expenses: |
||||||||||||||||||||
| Research and development |
7,796 | 19,507 | 19,364 | 15,236 | 10,299 | |||||||||||||||
| General and administrative |
3,707 | 4,307 | 4,681 | 3,628 | 3,565 | |||||||||||||||
| Operating loss |
(10,242 | ) | (23,445 | ) | (23,633 | ) | (18,864 | ) | (8,473 | ) | ||||||||||
| Gain on sale of interest in royalties |
— | — | 2,500 | 23,429 | — | |||||||||||||||
| Interest and other income, net |
24 | 520 | 1,333 | 952 | 290 | |||||||||||||||
| Income (loss) from continuing operations |
(10,218 | ) | (22,925 | ) | (19,800 | ) | 5,157 | (8,183 | ) | |||||||||||
| Gain (loss) from discontinued operations(1) |
68 | (200 | ) | (342 | ) | (188 | ) | (89 | ) | |||||||||||
| Gain on disposition of discontinued operations, net of taxes(2) |
— | — | 20 | 56 | 62 | |||||||||||||||
| Income (loss) before income taxes |
(10,150 | ) | (23,125 | ) | (20,122 | ) | 5,385 | (8,210 | ) | |||||||||||
| Benefit (provision) for income taxes |
122 | — | (41 | ) | (119 | ) | — | |||||||||||||
| Net income (loss) |
$ | (10,028 | ) | $ | (23,125 | ) | $ | (20,163 | ) | $ | 5,266 | $ | (8,210 | ) | ||||||
| Diluted income (loss) per common share: |
||||||||||||||||||||
| Income (loss) from continuing operations |
$ | (0.31 | ) | $ | (0.74 | ) | $ | (1.02 | ) | $ | 0.87 | $ | (1.30 | ) | ||||||
| Net income (loss) |
$ | (0.31 | ) | $ | (0.75 | ) | $ | (1.04 | ) | $ | 0.83 | $ | (1.31 | ) | ||||||
| Weighted-average common shares outstanding used to |
32,625 | 30,811 | 19,358 | 6,359 | 6,280 | |||||||||||||||
| (1) | Loss from discontinued operations represents the loss attributable to our Analytical Standards division that was sold to GFS Chemicals on February 13, 2003, and the loss attributable to our cosmeceutical and toiletries business that was sold to RP Scherer on July 25, 2000. See Note 9 to the Financial Statements. |
| (2) | See Note 9 to the Financial Statements. |
44
Table of Contents
| December 31, |
2009 | 2008 | 2007 | 2006 | 2005 | |||||||||||||||
| BALANCE SHEET DATA |
||||||||||||||||||||
| Cash, cash equivalents and marketable securities |
$ | 7,593 | $ | 10,538 | $ | 35,062 | $ | 15,522 | $ | 5,809 | ||||||||||
| Working capital |
6,426 | 7,629 | 29,589 | 12,014 | 4,882 | |||||||||||||||
| Total assets |
8,951 | 11,800 | 36,950 | 17,251 | 8,969 | |||||||||||||||
| Long-term liabilities |
268 | 1,015 | 1,269 | 1,000 | — | |||||||||||||||
| Accumulated deficit |
(141,079 | ) | (131,051 | ) | (107,926 | ) | (87,763 | ) | (93,029 | ) | ||||||||||
| Stockholders’ equity |
6,796 | 7,598 | 29,474 | 12,059 | 6,203 | |||||||||||||||
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
This Annual Report on Form 10-K contains “forward-looking statements” as defined by the Private Securities Litigation Reform Act of 1995. These forward-looking statements involve risks and uncertainties including uncertainties associated with anticipated availability of cash resources, corporate ability to continue operations, plans to secure additional capital, timely development, approval, launch and acceptance of new products, satisfactory completion of clinical studies, establishment of new corporate collaborations, progress in research and development programs and other risks and uncertainties identified below and in Item 1A “Risk Factors,” herein. We caution investors that forward-looking statements reflect our analysis only on their stated date. We do not intend to update them except as required by law.
Overview
A.P. Pharma is a specialty pharmaceutical company focused on developing pharmaceutical products using our proprietary Biochronomer polymer-based drug delivery technology. The Biochronomer technology consists of bioerodible polymers designed to release drugs over a defined period of time. Our primary focus is on our lead product candidate, APF530, which is being developed for the prevention of chemotherapy-induced nausea and vomiting (CINV). APF530 utilizes our Biochronomer technology and is a long-acting formulation of granisetron. In May 2009, we filed a New Drug Application (NDA) with the U.S. Federal Drug Administration (FDA) seeking approval for APF530. The FDA is currently reviewing the NDA, and based on the Prescription Drug User Fee Act (PDUFA), has issued an action date of March 18, 2010. During 2008, APF530 completed a pivotal Phase 3 clinical trial which was the basis for the application. In addition to APF530, we have a pipeline of other product candidates that use our Biochronomer technology. One product candidate, an undisclosed opiate for a long-acting pain management product, has been licensed on a world-wide basis to Merial Limited (Merial) for use with companion animals. Further development of our pipeline products has been temporarily deferred in order to focus both managerial and financial resources on the APF530 NDA and negotiations of a commercialization partnership for APF530.
Critical Accounting Policies and Estimates
Our accounting policies are more fully described in Note 2 of the Financial Statements. The preparation of financial statements in conformity with U.S. generally accepted accounting principles requires our management to make estimates and assumptions about future events that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ significantly from those estimates. We believe the following policies to be critical to understanding our financial condition,
45
Table of Contents
results of operations and expectations for 2010, because these policies require management to make significant estimates, assumptions and judgments about matters that are inherently uncertain.
Revenue Recognition
Our revenue arrangements with multiple deliverables are divided into separate units of accounting if certain criteria are met, including whether the delivered item has stand-alone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered items. The consideration we receive is allocated among the separate units based on their respective fair values, and the applicable revenue recognition criteria are considered separately for each of the separate units. Advance payments received in excess of amounts earned are classified as deferred revenue until earned. Milestones are recorded as revenue upon achievement of the milestone.
Royalties
Contractually required minimum royalties are recorded ratably throughout the contractual period. Royalties in excess of minimum royalties are recognized as earned when the related product is shipped to the end customer by our licensees based on information provided to us by our licensees.
Contract Revenue
We have licensing agreements that generally provide for a non-refundable license fee. The license agreements provide for us to earn future revenue through royalty payments. These non-refundable license fees are generally initially reported as deferred revenue and recognized as revenue over an appropriate period, depending on the license. Revenue recognized from deferred license fees is classified as Contract Revenue in the accompanying statements of operations.
Contract revenue also relates to research and development arrangements that generally provide for us to invoice research and development fees based on full-time equivalent hours for each project. Revenue from these arrangements is recognized as the related development services are rendered. This revenue approximates the costs incurred.
Clinical Trial Accruals
Our expenses related to clinical trials are based on estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and clinical research organizations that conduct and manage clinical trials on our behalf. Since the invoicing related to these services does not always coincide with our financial statement close process, we must estimate the level of services performed and fees incurred in determining the accrued clinical trial costs. The financial terms of these agreements are subject to negotiation and vary from contract to contract, which may result in uneven payment flows. Payments under the contracts depend on factors such as the successful enrollment of patients or achievement of certain events or the completion of portions of the clinical trial or similar conditions. The Phase 3 clinical trial of APF530 has had a significant effect on our research and development expenses. Expenses related to clinical trials generally are accrued based on the level of patient enrollment and services performed by the clinical research organization or related service provider according to the protocol. We monitor patient enrollment levels and related activity to the extent possible and adjust our estimates accordingly. Historically these estimates have been accurate and no material adjustments have had to be made.
46
Table of Contents
Income Taxes
We make certain estimates and judgments in determining income tax expense for financial statement purposes. These estimates and judgments occur in the calculation of certain tax assets and liabilities, which arise from differences in the timing of recognition of revenue and expense for tax and financial statement purposes. As part of the process of preparing our financial statements, we are required to estimate our income taxes in each of the jurisdictions in which we operate. This process involves us estimating our current tax exposure under the most recent tax laws and assessing temporary differences resulting from differing treatment of items for tax and financial statement purposes.
We assess the likelihood that we will be able to recover our deferred tax assets. We consider all available evidence, both positive and negative, including our historical levels of income and losses, expectations and risks associated with estimates of future taxable income and ongoing prudent and feasible tax planning strategies in assessing the need for a valuation allowance. If we do not consider it more likely than not that we will recover our deferred tax assets, we will record a valuation allowance against the deferred tax assets that we estimate will not ultimately be recoverable. At December 31, 2009, we believed that the amount of our deferred income taxes would not be ultimately recovered. Accordingly, we recorded a full valuation allowance for deferred tax assets.
Additionally, we believe that our deferred tax assets may have been limited in accordance with a provision of the Internal Revenue Code of 1986, whereby net operating loss and tax credit carryforwards available for use in a given period are limited upon the occurrence of certain events, including a significant change in ownership interests. As a result, our deferred tax assets and related valuation allowance were reduced for the estimated impact of the net operating losses and credits that may expire unused (see Note 11 to the Financial Statements).
Should there be a change in our ability to recover our deferred tax assets, we would recognize a benefit to our tax provision in the period in which we determine that it is more likely than not that we will recover our deferred tax assets.
Stock-Based Compensation
On January 1, 2006, we adopted the provisions of Accounting Standards Codification (ASC) 718-20, Stock Compensation – Awards Classified as Equity, (formerly Statement of Financial Accounting Standards No. 123(R), Share-Based Payment). ASC 718-20 requires companies to measure and recognize compensation expense for all employee stock-based payments at fair value over the service period underlying the arrangement. Accordingly, we are required to record the grant date fair value of stock options issued to employees and purchase date fair value of employee stock purchases. We adopted ASC 718-20 using the “modified prospective” method, whereby the fair value of all previously granted employee stock-based arrangements remaining unvested at January 1, 2006, based on the grant date value estimated in accordance with the pro forma provisions of ASC 718-20, and all grants made on or after January 1, 2006, based on fair value estimated in accordance with ASC 718-20, have been included in our determination of stock-based compensation expense in 2009, 2008 and 2007. We have not restated our operating results in prior periods to reflect charges for the fair value of stock-based arrangements.
47
Table of Contents
Results of Operations for the years ended December 31, 2009, 2008 and 2007
The following sets forth the statement of operations data and percentage changes as compared to the prior year (dollar amounts are presented in thousands):
| For the Years Ended December 31, |
Annual $ Change | Annual $ Change | |||||||||||||||||||||
| 2009 | 2008 | 2007 | 2009/2008 | 2008/2007 | 2009/2008 | 2008/2007 | |||||||||||||||||
| Total revenue |
$ | 1,261 | $ | 369 | $ | 412 | $ | 892 | $ | (43 | ) | 242 % | (10)% | ||||||||||
| Research and development |
7,796 | 19,507 | 19,364 | (11,711 | ) | 143 | (60)% | 1 % | |||||||||||||||
| General and administrative |
3,707 | 4,307 | 4,681 | (600 | ) | (374 | ) | (14)% | (8)% | ||||||||||||||
| Interest income |
29 | 587 | 1,326 | (558 | ) | (739 | ) | (95)% | (56)% | ||||||||||||||
| Gain on sale of royalty interests |
— | — | 2,500 | — | (2,500 | ) | — | (100)% | |||||||||||||||
| Gain (loss) from discontinued operations |
68 | (200 | ) | (342 | ) | 268 | 142 | 134 % | (42)% | ||||||||||||||
| Gain on disposition of discontinued operations, net of taxes |
— | — | 20 | — | (20 | ) | — | (100)% | |||||||||||||||
Revenue
Contract revenue increased in 2009 by $892,000, or 242%, compared to 2008 primarily from recognition of deferred revenue as a result of the termination of the RHEI license due to RHEI’s failure to make a required milestone payment. Revenue of $1.0 million, previously deferred in conjunction with our agreement with RHEI, was recognized in 2009 as a result of the termination and is included in contract revenue (see Note 12 to the Financial Statements).
Contract revenue decreased in 2008 by $43,000, or 10%, compared to 2007 as the proof-of-concept phase of the development program with Merial was completed early in the fourth quarter of 2008. Agreement on the next phase of development was reached with Merial in September 2009 (see Note 12 to the Financial Statements).
Research and Development
Research and development expense in 2009 decreased by $11,711,000, or 60%, compared to 2008 primarily due to decreased clinical and project related expenses as a result of the completion of clinical trials for APF530 and suspending other research and development projects to conserve resources. Additionally, research and development expense decreased as a result of headcount reductions.
Research and development expenses in 2008 increased by $143,000, or 1%, compared to 2007 primarily due to increased expenses for our pain programs, somewhat offset by decreases in the CINV program, as the APF530 Phase 3 clinical trial was completed in late 2008.
48
Table of Contents
The scope and magnitude of future research and development expense is difficult to predict given the number of studies that will need to be conducted for any of our potential products. In general, biopharmaceutical development involves a series of steps, beginning with identification of a potential target and includes proof-of-concept in animals and Phase 1, 2 and 3 clinical studies in humans. Each step of this process is typically more expensive than the previous one, so success in development results in increasing expenditures.
In addition to our lead program, APF530, we have a pipeline of other product candidates using our Biochronomer technology in various stages of development. Further development of our pipeline products has been temporarily deferred in order to focus both managerial and financial resources on the development of APF530. The following table sets forth the current opportunities for our own portfolio of product candidates, the compound selected, the delivery duration and the status:
| Product Candidate |
Potential Application |
Drug |
Targeted Duration |
Status | ||||
| APF112 |
Post-surgical pain relief |
Mepivacaine |
Up to 36 hours |
Phase 2 | ||||
| APF580 |
Pain relief - human |
Undisclosed Opiate |
At least seven days |
Preclinical | ||||
| APF580 |
Pain relief - veterinary |
Undisclosed Opiate |
At least seven days |
Preclinical | ||||
The major components of research and development expenses were as follows (in thousands):
| For the years December 31, |
2009 | 2008 | 2007 | ||||||
| Internal research and development costs |
$ | 4,783 | $ | 7,936 | $ | 6,264 | |||
| External development costs: |
|||||||||
| APF530 (CINV product) |
2,939 | 9,654 | 12,137 | ||||||
| APF112 and APF 580 (human pain products) |
3 | 1,258 | — | ||||||
| External general technology development costs |
71 | 659 | 963 | ||||||
| $ | 7,796 | $ | 19,507 | $ | 19,364 | ||||
Internal research and development costs consist of employee salaries and benefits, including stock-based compensation, laboratory supplies, depreciation and allocation of overhead. External general technology development costs include expenditures on polymer development and manufacturing, which are performed on our behalf by third parties.
General and Administrative
General and administrative expenses decreased in 2009 by $600,000, or 14%, compared to 2008 primarily as a result of lower headcount and associated expense reductions.
General and administrative expenses decreased in 2008 by $374,000, or 8%, compared to 2007 primarily related to severance costs recorded in 2007 associated with the departure of a former executive officer, as well as lower headcount, partially offset by increased consulting and other outside costs.
General and administrative expenses consist of salaries and related expenses, professional fees, directors’ fees, investor relations costs, insurance expense and related overhead cost allocation.
49
Table of Contents
Interest Income
Interest income consists primarily of income earned on our invested cash, cash equivalents and marketable securities. Interest income decreased by $558,000, or 95% in 2009 compared to 2008 and $739,000, or 56% in 2008 compared to 2007 due to lower average invested cash balances as a result of cash used in operations, as well as lower interest rate returns. We have currently invested our available cash in a money market fund containing U.S. Government-backed or collateralized overnight securities.
Discontinued Operations
On July 25, 2000, we completed the sale of certain technology rights for our topical pharmaceutical and cosmeceutical product lines and associated assets, referred to as our cosmeceutical and toiletry business, to RP Scherer Corporation (RP Scherer), a subsidiary of Cardinal Health, Inc. We received $25 million at closing of the transaction.
Under the terms of the agreement with RP Scherer, we guaranteed a minimum gross profit percentage on RP Scherer’s combined sales of products to Ortho Neutrogena (Ortho) and Dermik (Gross Profit Guaranty). The guaranty period initially commenced on July 1, 2000 and was to end on the earlier of July 1, 2010 or the end of two consecutive guaranty periods where the combined gross profit on sales to Ortho and Dermik equals or exceeds the guaranteed gross profit (the two period test). The Gross Profit Guaranty expense totaled $944,000 for the first seven guaranty years and in those years profits did not meet the two period test. Effective March 2007, in conjunction with a sale of assets by RP Scherer’s successor company to an Amcol International subsidiary (Amcol), we signed a new agreement with Amcol to provide continuity of product supply to Ortho and Dermik. This new agreement potentially extends the Gross Profit Guaranty period an additional three years to July 1, 2013, unless it is terminated earlier with the two period test. Amcol has indicated that its costs differ from those it charged historically to the RP Scherer successor company to produce the products. We have not paid any Gross Profit Guaranty amount asserted by Amcol, and have requested documentation of the actual costs, but have accrued the amount Amcol represents it is owed. As there is no minimum amount of Gross Profit Guaranty due, no accrual for the guaranty is estimable for future years. A liability of $553,000 related to the current amount due under the gross profit guarantees is included in accrued disposition costs as of December 31, 2009.
Gain (loss) from discontinued operations represents primarily the loss attributable to the Gross Profit Guaranty associated with the sale of our cosmeceutical and toiletry business.
On February 13, 2003, we completed the sale of certain assets of our Analytical Standards division to GFS Chemicals, Inc. (GFS), a privately held company based in Columbus, Ohio. In this transaction, we received $2.1 million at closing and were entitled to receive royalties on sales of Analytical Standards products for a period of five years following the sale at rates ranging from 5% to 15%. The gain on disposition of discontinued operations recorded in 2007 of $20,000 relates to the gain on the sale of our Analytical Standards division as measured by royalty receipts in excess of guaranteed minimum royalties.
50
Table of Contents
Liquidity and Capital Resources
We had cash, cash equivalents and marketable securities of $7,593,000 at December 31, 2009.
In October 2009, we sold 7,954,543 shares of our common stock in a private placement at $0.88 per share and warrants to purchase 3,977,270 shares of our common stock, exercisable through January 7, 2015, at $0.88 per share. The purchasers paid $0.125 per underlying share for the warrants. Additionally the purchasers have the right to purchase up to an additional 5,165,286 shares at $0.97 per share prior to May 14, 2010 and paid $0.125 per underlying share for the right to purchase such additional shares. Total proceeds were approximately $7.9 million, after deducting costs associated with the issuance. We are required to prepare and file Form S-3 registration statements, as permissible under SEC rules and regulations, with the SEC for the purpose of registering for resale the securities sold in this transaction.
In June 2007, we sold 24,393,939 shares of our common stock in a public offering at a price of $1.65 per share, for net proceeds of approximately $37.2 million after deducting underwriting fees and costs associated with the offering.
Net cash used in continuing operating activities for the year ended December 31, 2009 was $10,999,000 and related primarily to our net loss adjusted for changes in accrued expenses and deferred revenue, stock-based compensation and depreciation and amortization. The decrease in cash used in continuing operating activities for the year ended December 31, 2009, as compared to 2008, was primarily due to the change in net loss resulting from the completion of our Phase 3 trial for APF530 late in 2008, headcount reductions and suspending development of our other clinical product candidates.
Net cash used in continuing operating activities for the year ended December 31, 2008 was $24,294,000 and related primarily to our net loss, adjusted for changes in accrued expenses, accounts payable, stock-based compensation and depreciation and amortization. The increase in net cash used in continuing operating activities in 2008, as compared to 2007, was primarily due to changes in accrued expenses and accounts payable and the gain of $2.5 million in 2007 from the sale of our rights to royalties on sales of Retin-A Micro® and Carac®.
The cash provided by discontinued operations of $19,000 in 2008 relates primarily to the royalties received from GFS for sales of Analytical Standards products. The cash used in discontinued operations of $169,000 in 2007 related to Gross Profit Guaranty payments to RP Scherer, partially offset by the royalties received from GFS for sales of Analytical Standards products.
Net cash provided by investing activities was $612,000, $678,000 and $11,202,000 for the years ended December 31, 2009, 2008 and 2007, respectively. The decrease in net cash provided by investing activities in 2009 and 2008, compared with 2007, was primarily due to lower maturities and sales of marketable securities.
Our financing activities provided us with $8,013,000, $54,000 and $37,256,000 for the years ended December 31, 2009, 2008 and 2007, respectively. The net cash provided by financing activities in 2009 is primarily associated with a private equity financing in October 2009, as described in Note 7 to the Financial Statements. Total proceeds were approximately $7.9 million, after deducting expenses associated with the issuance. The net cash provided by financing activities in 2008 primarily related to proceeds from issuances of shares under the Employee Stock Purchase Plan. The net cash provided by financing activities in 2007 primarily related to the issuance of 24,393,939 shares of common stock at $1.65 per share for net proceeds of $37.2 million from our public offering in June 2007.
51
Table of Contents
Historically, we have financed our operations, including technology and product research and development, primarily through sales of our common stock, royalties received on sales of Retin-A Micro and Carac, the sale of our rights to royalties on sales of Retin-A Micro and Carac, income from collaborative research and development fees, proceeds received from the sales of our Analytical Standards division and our cosmeceutical and toiletry business and interest earned on short-term investments.
We believe our cash, cash equivalents and marketable securities as of December 31, 2009 will enable us to fund our operations through the fourth quarter of 2010, based on our anticipated spending levels and other known sources of funding. However, we may modify our operating plans to require additional funds in 2010. The amount of additional funding that we may require in 2010 depends on various factors, including the results of on-going regulatory review by the FDA of our APF530 NDA (which currently has a PDUFA date of March 18, 2010), our ability to establish a partnership with a pharmaceutical company for the commercialization of APF530, the time and costs related to manufacturing APF530, if approved, and competing technological and market developments.
Regardless of the FDA’s response with respect to the APF530 NDA, we believe we will require additional funding to support our operations in 2011. Our capital requirements going forward will depend on numerous factors including: the number and characteristics of product development programs we pursue and the pace of each program; the scope, rate of progress, results and costs of preclinical testing and clinical trials; the time, cost and outcome involved in seeking regulatory approvals; scientific progress in our research and development programs; the magnitude and scope of our research and development programs; our ability to establish and maintain strategic collaborations or partnerships for research, development, clinical testing; manufacturing and marketing of our product candidates; the cost and timing of establishing sales, marketing and distribution capabilities for a specialty sales force if we commercialize any products independently; the cost of establishing clinical and commercial supplies of our product candidates and any products that we may develop; and general market conditions.
We will be seeking additional financing to continue our activities, which may include a collaborative arrangement or an equity offering. If we are unable to complete a collaborative arrangement, equity offering, or otherwise obtain sufficient financing, we may be required to further reduce, defer, or discontinue our activities or may not be able to continue as a going concern entity.
We may not be able to raise sufficient additional capital when we need it or to raise capital on favorable terms. The sale of additional equity or convertible debt securities in the future may be dilutive to our stockholders, and debt financing arrangements may require us to pledge certain assets and enter into covenants that could restrict certain business activities or our ability to incur further indebtedness and may contain other terms that are not favorable to us or our stockholders. If we are unable to obtain adequate funds on reasonable terms, we may be required to curtail operations significantly or to obtain funds by entering into financing, supply or collaboration agreements on unattractive terms. We do not currently have the financial resources to launch AP530. If AP530 is approved, we anticipate pursuing either a collaborative arrangement with a partner who will provide the necessary financial resources and expertise to launch AP530 or anticipate obtaining additional funding and resources that would be required to launch AP530 without a partner. There can be no assurance that AP530 will be approved and, if approved, that we will be successful in obtaining the additional necessary financial resources and expertise, with or without a partner, that will be required to launch AP530.
52
Table of Contents
Contractual Obligations
Below is a summary of fixed payments related to certain contractual obligations (in thousands):
| Total | Less Than 1 year |
1-3 years |
3-5 years |
More Than 5 years | |||||||||||
| Operating Leases(1) |
$ | 719 | $ | 568 | $ | 147 | $ | 4 | $ | — | |||||
| Total |
$ | 719 | $ | 568 | $ | 147 | $ | 4 | $ | — | |||||
| (1) | See Note 6 to the Financial Statements. |
Under the terms of the agreement with RP Scherer, we guaranteed a minimum gross profit percentage on RP Scherer’s combined sales of products to Ortho Neutrogena and Dermik. See description of Gross Profit Guaranty in “Discontinued Operations” above.
Off-Balance-Sheet Arrangements
As of December 31, 2009, we did not have any off-balance-sheet arrangements, as defined in Item 303(a)(4)(ii) of Regulation S-K.
Recent Accounting Pronouncements
Recent accounting pronouncements are disclosed in Note 2 to the Financial Statements included in Item 8 of this Annual Report on Form 10-K.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURE ABOUT MARKET RISK
Our exposure to market rate risk for changes in interest rates relates primarily to our investment portfolio. We do not use derivative financial instruments. We manage our interest rate risk by maintaining an investment portfolio primarily consisting of debt instruments of high credit quality and relatively short average maturities. The interest rates as of December 31, 2009 and 2008 were 0.01% and 0.61%, respectively. Due to the recent financial crisis, we have invested our available cash, cash equivalents and marketable securities in a money market fund containing U.S. Government-backed or collateralized overnight securities. At December 31, 2009 and 2008, our available for sale, cash equivalents and marketable securities include corporate and other debt securities of $7,566,000 and $10,453,000, respectively, all of which are due in less than one year. Notwithstanding our efforts to manage interest rate risks, there can be no assurance that we will be adequately protected against risks associated with interest rate fluctuations.
53
Table of Contents
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
A.P. Pharma, Inc.
We have audited the accompanying balance sheets of A.P. Pharma, Inc. as of December 31, 2009 and 2008 and the related statements of operations, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2009. Our audits also included the financial data in the financial statement schedule listed in the Index at Item 15(a)2. These financial statements and schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements audited by us present fairly, in all material respects, the financial position of A.P. Pharma, Inc. at December 31, 2009 and 2008, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2009, in conformity with U.S. generally accepted accounting principles. Also in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly in all material respects the information set forth therein.
/s/ Odenberg, Ullakko, Muranishi & Co LLP
San Francisco, California
March 11, 2010
54
Table of Contents
A.P. PHARMA, INC.
BALANCE SHEETS
(in thousands except par value and shares)
| December 31, |
2009 | 2008 | ||||||
| ASSETS |
||||||||
| Current Assets: |
||||||||
| Cash and cash equivalents |
$ | 7,593 | $ | 9,967 | ||||
| Marketable securities |
— | 571 | ||||||
| Accounts receivable, net |
171 | 32 | ||||||
| Prepaid expenses and other current assets |
549 | 246 | ||||||
| Total current assets |
8,313 | 10,816 | ||||||
| Property and equipment, net |
510 | 881 | ||||||
| Other long-term assets |
128 | 103 | ||||||
| Total Assets |
$ | 8,951 | $ | 11,800 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY |
||||||||
| Current Liabilities: |
||||||||
| Accounts payable |
$ | 162 | $ | 344 | ||||
| Accrued expenses |
1,080 | 2,222 | ||||||
| Deferred revenue |
92 | — | ||||||
| Accrued disposition costs |
553 | 621 | ||||||
| Total current liabilities |
1,887 | 3,187 | ||||||
| Deferred revenue |
268 | 1,000 | ||||||
| Other long-term liabilities |
— | 15 | ||||||
| Total Liabilities |
2,155 | 4,202 | ||||||
| Commitments and Contingencies (Note 6) |
||||||||
| Stockholders’ Equity: |
||||||||
| Preferred stock, 2,500,000 shares authorized; none issued or outstanding at December 31, 2009 and 2008 |
— | — | ||||||
| Common stock, $0.01 par value, 100,000,000 shares authorized; 39,935,168 and 30,941,149 issued and outstanding at December 31, 2009 and 2008, respectively |
394 | 309 | ||||||
| Additional paid-in capital |
147,481 | 138,383 | ||||||
| Accumulated other comprehensive loss |
— | (43 | ) | |||||
| Accumulated deficit |
(141,079 | ) | (131,051 | ) | ||||
| Total Stockholders’ Equity |
6,796 | 7,598 | ||||||
| Total Liabilities and Stockholders’ Equity |
$ | 8,951 | $ | 11,800 | ||||
See accompanying Notes to Financial Statements.
55
Table of Contents
A.P. PHARMA, INC.
STATEMENTS OF OPERATIONS
(in thousands except per share amounts)
| For the Years Ended December 31, |
2009 | 2008 | 2007 | |||||||||
| REVENUE |
||||||||||||
| Contract revenue |
$ | 1,261 | $ | 369 | $ | 412 | ||||||
| OPERATING EXPENSES |
||||||||||||
| Research and development |
7,796 | 19,507 | 19,364 | |||||||||
| General and administrative |
3,707 | 4,307 | 4,681 | |||||||||
| Total operating expenses |
11,503 | 23,814 | 24,045 | |||||||||
| Operating loss |
(10,242 | ) | (23,445 | ) | (23,633 | ) | ||||||
| Interest income |
29 | 587 | 1,326 | |||||||||
| Gain on sale of royalty interest |
— | — | 2,500 | |||||||||
| Other income (loss), net |
(5 | ) | (67 | ) | 7 | |||||||
| Loss from continuing operations |
(10,218 | ) | (22,925 | ) | (19,800 | ) | ||||||
| Income (loss) from discontinued operations |
68 | (200 | ) | (342 | ) | |||||||
| Gain on disposition of discontinued operations |
— | — | 20 | |||||||||
| Loss before income taxes |
(10,150 | ) | (23,125 | ) | (20,122 | ) | ||||||
| Income tax benefit (provision) |
122 | — | (41 | ) | ||||||||
| Net loss |
$ | (10,028 | ) | $ | (23,125 | ) | $ | (20,163 | ) | |||
| Basic and diluted loss per share: |
||||||||||||
| Loss from continuing operations |
$ | (0.31 | ) | $ | (0.74 | ) | $ | (1.02 | ) | |||
| Net loss |
$ | (0.31 | ) | $ | (0.75 | ) | $ | (1.04 | ) | |||
| Weighted-average common shares outstanding—basic and diluted |
32,625 | 30,811 | 19,358 | |||||||||
See accompanying Notes to Financial Statements.
56
Table of Contents
A.P. PHARMA, INC.
STATEMENTS OF STOCKHOLDERS’ EQUITY
(in thousands)
| For the Years Ended December 31, 2009, 2008 and 2007 |
Common Stock |
Addi- tional Paid-in Capital |
Accu- mulated Deficit |
Accu- mulated Other Compre- hensive Income (Loss) |
Stock- holders’ Equity |
|||||||||||||||
| Shares | Amount | |||||||||||||||||||
| BALANCE, DECEMBER 31, 2006 |
6,360 | $ | 64 | $ | 99,771 | $ | (87,763 | ) | $ | (13 | ) | $ | 12,059 | |||||||
| Comprehensive loss |
||||||||||||||||||||
| Net loss |
— | — | — | (20,163 | ) | — | (20,163 | ) | ||||||||||||
| Net unrealized loss on marketable securities |
— | — | — | — | (25 | ) | (25 | ) | ||||||||||||
| Comprehensive loss |
(20,188 | ) | ||||||||||||||||||
| Common stock issued in public offering, net of issuance costs |
24,394 | 243 | 36,955 | — | — | 37,198 | ||||||||||||||
| Fair value of stock-based compensation issued to directors for restricted stock awards |
15 | — | 99 | — | — | 99 | ||||||||||||||
| Common stock issued to employees under the ESPP |
22 | — | 57 | — | — | 57 | ||||||||||||||
| Stock-based employee compensation related to stock options and ESPP |
— | — | 249 | — | — | 249 | ||||||||||||||
| BALANCE, DECEMBER 31, 2007 |
30,791 | 307 | 137,131 | (107,926 | ) | (38 | ) | 29,474 | ||||||||||||
| Comprehensive loss |
||||||||||||||||||||
| Net loss |
— | — | — | (23,125 | ) | — | (23,125 | ) | ||||||||||||
| Net unrealized loss on marketable securities |
— | — | — | — | (5 | ) | (5 | ) | ||||||||||||
| Comprehensive loss |
(23,130 | ) | ||||||||||||||||||
| Common stock issued upon exercise of stock options |
2 | — | 2 | — | — | 2 | ||||||||||||||
| Fair value of stock-based compensation issued to directors for restricted stock awards |
72 | 1 | 82 | — | — | 83 | ||||||||||||||
| Common stock issued to employees under the ESPP |
76 | 1 | 50 | — | — | 51 | ||||||||||||||
| Stock-based employee compensation related to stock options and ESPP |
— | — | 1,118 | — | — | 1,118 | ||||||||||||||
| BALANCE, DECEMBER 31, 2008 |
30,941 | 309 | 138,383 | (131,051 | ) | (43 | ) | 7,598 | ||||||||||||
| Comprehensive loss |
||||||||||||||||||||
| Net loss |
— | — | — | (10,028 | ) | — | (10,028 | ) | ||||||||||||
| Net unrealized gain on marketable securities |
— | — | — | — | 43 | 43 | ||||||||||||||
| Comprehensive loss |
(9,985 | ) | ||||||||||||||||||
| Common stock and warrants issued in private placement, net of issuance costs |
7,955 | 80 | 7,871 | — | — | 7,951 | ||||||||||||||
| Fair value of stock-based compensation issued to directors for restricted stock awards |
332 | 4 | 371 | — | — | 375 | ||||||||||||||
| Common stock issued upon exercise of stock options |
28 | 20 | — | — | 20 | |||||||||||||||
| Common stock issued to employees under the ESPP |
99 | 1 | 41 | — | — | 42 | ||||||||||||||
| Stock issued in lieu of bonus |
40 | — | 36 | — | — | 36 | ||||||||||||||
| Stock-based employee compensation related to stock options and ESPP |
— | — | 759 | — | — | 759 | ||||||||||||||
| BALANCE, DECEMBER 31, 2009 |
39,395 | $ | 394 | $ | 147,481 | $ | (141,079 | ) | $ | — | $ | 6,796 | ||||||||
See accompanying Notes to Financial Statements
57
Table of Contents
A.P. PHARMA, INC.
STATEMENTS OF CASH FLOWS
(in thousands)
| For the Years Ended December 31, |
2009 | 2008 | 2007 | |||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: |
||||||||||||
| Net loss |
$ | (10,028 | ) | $ | (23,125 | ) | $ | (20,163 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Loss (gain)from discontinued operations |
(68 | ) | 200 | 342 | ||||||||
| Gain on disposition of discontinued operations |
— | — | (20 | ) | ||||||||
| Loss on retirement of fixed assets |
17 | 85 | — | |||||||||
| Depreciation and amortization |
356 | 412 | 359 | |||||||||
| Stock-based compensation |
1,137 | 1,112 | 348 | |||||||||
| Stock issued in lieu of cash bonus |
36 | — | — | |||||||||
| Amortization of discount and accretion of premium on marketable securities |
— | — | (70 | ) | ||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Accounts receivable |
(139 | ) | 100 | (149 | ) | |||||||
| Prepaid expenses and other current assets |
(303 | ) | 336 | 27 | ||||||||
| Other long-term assets |
(25 | ) | (28 | ) | 18 | |||||||
| Accounts payable |
(182 | ) | (1,093 | ) | 665 | |||||||
| Accrued expenses |
(1,160 | ) | (2,293 | ) | 1,531 | |||||||
| Deferred revenue |
(640 | ) | — | — | ||||||||
| Net cash used in continuing operating activities |
(10,999 | ) | (24,294 | ) | (17,112 | ) | ||||||
| Cash provided by (used in) discontinued operations |
— | 19 | (169 | ) | ||||||||
| Net cash used in operating activities |
(10,999 | ) | (24,275 | ) | (17,281 | ) | ||||||
| CASH FLOWS FROM INVESTING ACTIVITIES: |
||||||||||||
| Purchases of property and equipment |
(2 | ) | (298 | ) | (481 | ) | ||||||
| Maturities of marketable securities |
614 | 976 | 4,875 | |||||||||
| Sales of marketable securities |
— | — | 6,808 | |||||||||
| Net cash provided by investing activities |
612 | 678 | 11,202 | |||||||||
| CASH FLOWS FROM FINANCING ACTIVITIES: |
||||||||||||
| Proceeds from the issuance of common stock, net of issuance costs |
7,951 | — | 37,198 | |||||||||
| Proceeds from the exercise of common stock options |
20 | 3 | — | |||||||||
| Proceeds from the issuance of shares under the Employee Stock Purchase Plan |
42 | 51 | 58 | |||||||||
| Net cash provided by financing activities |
8,013 | 54 | 37,256 | |||||||||
| Increase (decrease) in cash and cash equivalents |
(2,374 | ) | (23,543 | ) | 31,177 | |||||||
| Cash and cash equivalents at the beginning of the year |
9,967 | 33,510 | 2,333 | |||||||||
| Cash and cash equivalents at the end of the year |
$ | 7,593 | $ | 9,967 | $ | 33,510 | ||||||
| Supplemental Cash Flow Data: |
||||||||||||
| Cash paid for interest |
$ | 4 | $ | — | $ | — | ||||||
See accompanying Notes to Financial Statements.
58
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
NOTE 1 BUSINESS
A.P. Pharma is a specialty pharmaceutical company focused on developing pharmaceutical products using our proprietary Biochronomer polymer-based drug delivery technology. The Biochronomer technology consists of bioerodible polymers designed to release drugs over a defined period of time. Our primary focus is on our lead product candidate, APF530, which is being developed for the prevention of chemotherapy-induced nausea and vomiting (CINV). APF530 utilizes our Biochronomer technology and is a long-acting formulation of granisetron. In May 2009, we filed a New Drug Application (NDA) with the U.S. Federal Drug Administration (FDA) seeking approval for APF530. The FDA is currently reviewing the NDA, and based on the Prescription Drug User Fee Act (PDUFA), has issued an action date of March 18, 2010. During 2008, APF530 completed a pivotal Phase 3 clinical trial which was the basis for the application. In addition to APF530, we have a pipeline of other product candidates that use our Biochronomer technology. One product candidate, an undisclosed opiate for a long-acting pain management product, has been licensed on a world-wide basis to Merial Limited (Merial) for use with companion animals. Further development of our pipeline products has been temporarily deferred in order to focus both managerial and financial resources on the APF530 NDA and negotiations of a commercialization partnership for APF530.
NOTE 2 SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation and Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Estimates were made relating to useful lives of fixed assets, valuation allowances, impairment of assets, accruals for research and development expenses and stock-based costs. Actual results could differ materially from those estimates.
We have evaluated subsequent events through the date the audited financial statements were issued, and determined that there were no subsequent events that required disclosure in the financial statements other than those disclosed in Note 13.
Cash Equivalents and Marketable Securities
We consider all debt securities that have original maturities, from the date of purchase, of less than three months to be cash equivalents. Investments with maturities of three months and longer, from the date of purchase, are classified as marketable securities. Investments may consist primarily of money market funds containing U.S. Government-backed or collateralized overnight securities and high-grade corporate obligations, mortgage-backed securities, municipal bonds and corporate debt securities. We account for our marketable securities in accordance with Accounting Standard Codification (ASC) 320-10, Investments—Debt and Equity Securities (formerly Statement of Financial Accounting Standards (SFAS) No. 115, Accounting for Certain Investments in Debt and Equity Securities). We have classified all our investments in certain debt securities as “available-for-sale,” and, therefore, they are recorded at fair value with unrealized gains and losses reported as a separate component of stockholders’ equity. If the estimated fair value of a security is below its carrying value, we evaluate whether we intend to sell the security, or whether we would more likely than not be required to sell the security before the expected recovery of its amortized cost basis. If the impairment is considered to be other-than-temporary, the security is written down to its estimated fair value. Other-than-temporary declines in estimated fair value of all marketable securities are charged to “other income (loss), net.” The cost of all securities sold is based on the specific identification method.
59
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
Financial Instruments—Fair Value
The carrying values of our financial instruments, including marketable securities, accounts receivable and accrued liabilities, approximate their respective fair values due to their short maturities (see Note 3).
Property and Equipment
Property and equipment is stated at cost less accumulated depreciation. Depreciation is computed using the straight-line method over the estimated useful lives of the assets as follows: equipment and machinery, three to five years; furniture and fixtures, five years; and leasehold improvements, over the shorter of the respective lease terms or the respective useful lives of the leasehold improvements.
Long-Lived Assets
As circumstances dictate, we evaluate whether changes have occurred that would require us to consider whether long-lived assets have been impaired. Recoverability of assets to be held and used is determined by comparing the undiscounted net cash flows of long-lived assets to their respective carrying values. If such assets are considered to be impaired, the amount of impairment to be recognized is measured based on the projected discounted cash flows using an appropriate discount rate.
Stock-Based Compensation
On January 1, 2006, we adopted the provisions of ASC 718-20, Stock Compensation—Awards Classified as Equity (formerly SFAS No. 123(R), Share-Based Payment). ASC 718-20 requires companies to measure and recognize compensation expense for all employee stock-based payments at fair value over the service period underlying the arrangement. Accordingly, we are required to record the grant date fair value of stock options issued to employees and purchase date fair value of employee stock purchases. We adopted ASC 718-20 using the “modified prospective” method, whereby the fair value of all previously granted employee stock-based arrangements remaining unvested at January 1, 2006, based on the grant date value estimated in accordance with the pro forma provisions of ASC 718-20, and all grants made on or after January 1, 2006, based on fair value estimated in accordance with ASC 718-20, have been included in our determination of stock-based compensation expense in 2009, 2008 and 2007. We have not restated our operating results in prior periods to reflect charges for the fair value of stock-based arrangements.
In November 2005, the Financial Accounting Standards Board (FASB) issued ASC 718-10,Stock Compensation (formerly FASB Staff Position (FSP) No. SFAS 123(R)-3 Transition Election Related to Accounting for Tax Effects of Share-Based Payment Awards). We adopted the alternative transition method provided in ASC 718-10 for calculating the tax effects of stock-based compensation pursuant to ASC 718-20 in the fourth quarter of fiscal 2006. The alternative transition method includes simplified methods to establish the beginning balance of the additional paid-in capital pool (APIC pool) related to the tax effects for employee stock-based compensation, and to determine the subsequent impact on the APIC pool and Consolidated Statements of Cash Flows of the tax effects of employee stock-based compensation awards that are outstanding upon adoption of ASC 718-20.
60
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
Revenue Recognition
Our revenue arrangements with multiple deliverables are divided into separate units of accounting if certain criteria are met, including whether the delivered item has stand-alone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered items. The consideration we receive is allocated among the separate units based on their respective fair values, and the applicable revenue recognition criteria are considered separately for each of the separate units. Advance payments received in excess of amounts earned are classified as deferred revenue until earned. Milestones payments are recorded as revenue upon achievement of the milestone.
Royalties
Contractually required minimum royalties are recorded ratably throughout the contractual period. Royalties in excess of minimum royalties are recognized as earned when the related product is shipped to the end customer by our licensees based on information provided to us by our licensees.
Contract Revenue
We have licensing agreements that generally provide for a non-refundable license fee. The license agreements provide for us to earn future revenue through royalty payments. These non-refundable license fees are generally initially reported as deferred revenue and recognized as revenue over an appropriate period, depending on the license. Revenue recognized from deferred license fees is classified as Contract Revenue in the accompanying statements of operations.
Contract revenue relates to research and development arrangements that generally provide for us to invoice research and development fees based on full-time equivalent hours for each project. Revenue from these arrangements is recognized as the related development services are rendered. This revenue approximates the costs incurred.
Clinical Trial Accruals
Our expenses related to clinical trials are based on estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and clinical research organizations that conduct and manage clinical trials on our behalf. Since the invoicing related to these services does not always coincide with our financial statement close process, we must estimate the level of services performed and fees incurred in determining the accrued clinical trial costs. The financial terms of these agreements are subject to negotiation and vary from contract to contract, which may result in uneven payment flows. Payments under the contracts depend on factors such as the successful enrollment of patients or achievement of certain events or the completion of portions of the clinical trial or similar conditions. The Phase 3 clinical trials of APF530 have had a significant effect on our research and development expenses. Expenses related to clinical trials generally are accrued based on the level of patient enrollment and services performed by the clinical research organization or related service provider according to the protocol. We monitor patient enrollment levels and related activity to the extent possible and adjust our estimates accordingly. Historically these estimates have been accurate and no material adjustments have had to be made.
61
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
Research and Development
Research and development consists of costs incurred for Company-sponsored and collaborative research and development expenses. These costs consist primarily of employee salaries and other personnel-related expenses, facility-related expenses, laboratory consumables, polymer development manufacturing, clinical and pre-clinical related services performed by clinical research organizations, research institutions and other outside service providers.
Research and development expenses under collaborative agreements approximate the revenue recognized, excluding milestone and up-front payments received under such arrangements.
Net Income (Loss) Per Share
Basic income (loss) per share is estimated based on the weighted-average number of common shares outstanding. Diluted earnings per share are calculated using the weighted-average number of common shares outstanding and other dilutive securities. Dilutive securities are not included in the computation of diluted net loss per share if the inclusion of these potentially dilutive securities is anti-dilutive (see Note 8).
Concentrations of Credit Risk
Financial instruments that potentially subject us to concentrations of credit risk consist primarily of cash equivalents, short-term investments and trade accounts receivable. We invest excess cash in a variety of high-grade short-term, interest-bearing securities. This diversification of risk is consistent with our policy to ensure safety of principal and maintain liquidity.
Segment and Geographic Information
Our operations are confined to a single business segment, the design and commercialization of polymer technologies for pharmaceutical and other applications. Substantially all of our revenues are derived from customers within the United States.
Recent Accounting Pronouncements
Effective January 1, 2009, we adopted ASC 815-40, Derivatives and Hedging, Contracts in Entity’s Own Equity (formerly Emerging Issues Task Force (EITF) Issue No. 07-05, Determining Whether an Instrument (or an Embedded Feature) is indexed to an Entity’s Own Stock). ASC 815-40 provides that an entity should use a two step approach to evaluate whether an equity-linked financial instrument (or embedded feature) is indexed to its own stock, including evaluating the instrument’s contingent exercise and settlement provisions. ASC 815-40 is effective for financial statements issued for fiscal years and interim periods beginning after December 15, 2008. The adoption of ASC 815-40 did not have a material impact on our financial position or results of operations.
Effective January 1, 2009, we adopted ASC 808, Collaborative Arrangements (formerly EITF Issue No. 07-1, Accounting for Collaborative Arrangements). ASC 808 defines collaborative arrangements and establishes reporting requirements for transactions between participants in a collaborative arrangement. ASC 808 also establishes the appropriate income statement presentation and classification for joint operating activities and payments between participants, as well as the sufficiency of the
62
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
disclosure related to these arrangements. ASC 808 is effective for financial statements issued for fiscal years and interim periods beginning after December 15, 2008, and will be applied retrospectively to all prior periods presented for all collaborative arrangements existing as of the effective date. The adoption of ASC 808 did not have a material impact on our financial position or results of operations.
Effective April 1, 2009 we adopted ASC 855, Subsequent Events (formerly SFAS No. 165, Subsequent Events). ASC 855 provides guidance to establish general standards of accounting for and disclosures of events that occur after the balance sheet date, but before financial statements are issued or are available to be issued. ASC 855 also requires entities to disclose the date through which subsequent events were evaluated as well as the rationale for why that date was selected. ASC 855 is effective for financial statements issued for fiscal years and interim periods ending after June 15, 2009. The adoption of ASC 855 did not have a material impact on our financial position or results of operations.
Effective July 1, 2009, we adopted ASC 105, Generally Accepted Accounting Principles (formerly SFAS No. 168, The FASB Accounting Standards Codification and the Hierarchy of Generally Accepted Accounting Principles—a replacement of FASB Statement No. 162). ASC 105 establishes the FASB Accounting Standards Codification as the source of authoritative accounting principles recognized by the FASB. Following this statement, the FASB will issue new standards in the form of Accounting Standards Update’s (ASU). ASC 105 is effective for financial statements issued for fiscal years and interim periods ending after September 15, 2009. The issuance of ASC 105 does not change generally accepted accounting principles (GAAP) and therefore the adoption of ASC 105 only affects the specific references to GAAP literature in the notes to our consolidated financial statements. The adoption of ASC 105 did not have an impact on our financial position or results of operations.
In October 2009, the FASB issued ASU No. 2009-13, Multiple-Deliverable Revenue Arrangements (ASU 2009-13) (prior authoritative literature: EITF Issue No. 08-1, Revenue Arrangements with Multiple Deliverables). ASU 2009-13, amends existing revenue recognition accounting pronouncements that are currently within the scope of ASC 605-25, Multiple-Element Arrangements (formerly included within EITF Issue No. 00-21, Revenue Arrangements with Multiple Deliverables). ASU 2009-13 provides accounting principles and application guidance on whether multiple deliverables exist, how the arrangement should be separated, and the consideration allocated. This guidance eliminates the requirement to establish the fair value of undelivered products and services and instead provides for separate revenue recognition based upon management’s estimate of the selling price for an undelivered item when there is no other means to determine the fair value of that undelivered item. ASC 605-25 previously required that the fair value of the undelivered item be the price of the item either sold in a separate transaction between unrelated third parties or the price charged for each item when the item is sold separately by the vendor. Under ASC 605-25, if the fair value of all of the elements in the arrangement was not determinable, then revenue was deferred until all of the items were delivered or fair value was determined. ASU 2009-13 is effective prospectively for revenue arrangements entered into or materially modified in fiscal years and interim periods beginning on or after June 15, 2010. We are currently evaluating the potential impact of ASU 2009-13 on our financial position and results of operations.
NOTE 3 CASH EQUIVALENTS AND MARKETABLE SECURITIES
We consider our investments in marketable securities as available-for-sale and, accordingly, we have recorded these investments at fair value. Our available for sale securities as of December 31, 2009 consisted of money market funds primarily containing U.S. Government-backed or collateralized overnight securities with original maturities of ninety days or less.
63
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
At December 31, 2009 and 2008, the amortized cost and estimated fair value of investments in debt securities and cash equivalents are set forth in the tables below:
| December 31, 2009 (in thousands) |
Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Estimated Fair Value | ||||||||
| Available-for-sale: |
||||||||||||
| Money market fund (included in cash and cash equivalents) |
$ | 7,566 | — | — | $ | 7,566 | ||||||
| Total available-for-sale |
$ | 7,566 | $ | — | $ | — | $ | 7,566 | ||||
| December 31, 2008 (in thousands) |
Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Estimated Fair Value | |||||||||
| Available-for-sale: |
|||||||||||||
| Asset-backed securities (included in marketable securities) |
$ | 614 | $ | — | $ | (43 | ) | $ | 571 | ||||
| Money market fund (included in cash and cash equivalents) |
9,882 | — | — | 9,882 | |||||||||
| Total available-for-sale |
$ | 10,496 | $ | — | $ | (43 | ) | $ | 10,453 | ||||
Fair Value
Effective January 1, 2008, we adopted ASC 820-10, Fair Value Measurements and Disclosures (formerly SFAS No. 107, Disclosures About Fair Value of Financial Instruments), which defines fair value, establishes a framework for measuring fair value in generally accepted accounting principles and expands disclosures about fair value measurements. Broadly, the ASC 820-10 framework clarifies that fair value is an exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is a market-based measurement that should be determined based on assumptions that market participants would use in pricing an asset or liability.
As a basis for considering such assumptions, ASC 820-10 establishes a three tier value hierarchy, which prioritizes the inputs used in measuring fair value as follows: (Level 1) observable inputs such as quoted prices in active markets; (Level 2) inputs other than the quoted prices in active markets that are observable either directly or indirectly; and (Level 3) unobservable inputs in which there is little or no market data, which require us to develop our own assumptions. The hierarchy requires us to use observable market data, when available, and to minimize the use of unobservable inputs when determining fair value. On a recurring basis, we measure our available-for-sale securities at fair value.
The following methods and assumptions were used to determine the fair value of each class of assets recorded at fair value in the balance sheets:
Cash equivalents: Cash equivalents consist of highly rated money market funds with maturities of ninety days or less, and are purchased daily at par value with specified yield rates. Due to the high ratings and short-term nature of these funds, we consider all cash equivalents as Level 1 inputs.
64
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
Short-term available-for-sale investments at fair value: Fair values are based on quoted market prices, where available. These fair values are obtained from third party pricing services, which generally use Level 1 or Level 2 inputs for the determination of fair value in accordance with ASC 820-10. Third party pricing services normally derive the security prices through recently reported trades for identical or similar securities making adjustments through the reporting date based upon available market observable information. For securities not actively traded, the third party pricing services may use quoted market prices of comparable instruments or discounted cash flow analyses, incorporating inputs that are currently observable in the markets for similar securities. Inputs that are often used in valuation methodologies include, but are not limited to, benchmark yields, reported trades, broker/dealer quotes, issuer spreads, benchmark securities, bids, offers and reference data. We utilize third party pricing services to obtain fair value and we generally obtain one price for each individual security. We review the fair value hierarchy classification. Changes in the observability of valuation inputs may result in a reclassification of levels for certain securities within the fair value hierarchy.
The following tables summarize the basis used to measure certain assets at fair value on a recurring basis in our balance sheets at December 31, 2009 and 2008 (in thousands):
| Basis of Fair Value Measurements | ||||||||||||
| Balance at December 31, 2009 |
Quoted Prices in Active Markets for Identical Items (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) | |||||||||
| Cash equivalents |
$ | 7,566 | $ | 7,566 | $ | — | $ | — | ||||
| Basis of Fair Value Measurements | ||||||||||||
| Balance at December 31, 2008 |
Quoted Prices in Active Markets for Identical Items (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) | |||||||||
| Cash equivalents |
$ | 9,882 | $ | 9,882 | $ | — | $ | — | ||||
| Asset-backed securities |
571 | — | 571 | — | ||||||||
| Total |
$ | 10,453 | $ | 9,882 | $ | 571 | $ | — | ||||
65
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
Investment securities are exposed to various risks, such as interest rate, market and credit. Due to the level of risk associated with certain investment securities and the level of uncertainty related to changes in the value of investment securities, it is possible that changes in these risk factors in the near term could have an adverse impact on our results of operations or stockholders’ equity.
In addition to the preceding disclosures on assets recorded at fair value in our balance sheets, ASC 820-10 also requires disclosure of the fair value of certain other financial instruments for which it is practicable to estimate fair value, whether or not such fair values are recognized in the balance sheets. At December 31, 2009 and 2008, the carrying amounts reported in the balance sheets for accounts receivable, accounts payable and accrued expenses approximate fair value because of the short-term nature of these items.
Effective April 1, 2009, we adopted ASC 820-10-65-4 (formerly FSP FAS No. 157-4, Determining Fair Value When the Volume and Level of Activity for the Asset or Liability Have Significantly Decreased and Identifying Transactions That Are Not Orderly). ASC 820-10-65-4 applies to all fair value measurements and provides additional clarification on estimating fair value when the market activity for an asset has declined significantly. ASC 820-10-65-4 also requires an entity to disclose a change in valuation technique and related inputs to the valuation calculation and to quantify its effects, if practicable. ASC 820-10-65-4 is effective for financial statements issued for fiscal years and interim periods ending after June 15, 2009. The adoption of ASC 820-10-65-4 did not have a material impact on our financial position or results of operations.
Effective April 1, 2009, we adopted ASC 820-10-50 (formerly FSP FAS No. 107-1 and Accounting Principles Board 28-1, Interim Disclosures about Fair Value of Financial Instruments). ASC 820-10-50 requires disclosures about the fair value of financial instruments in interim as well as in annual financial statements. ASC 820-10-50 is effective for financial statements issued for fiscal years and interim periods ending after June 15, 2009. The adoption of ASC 820-10-50 did not have a material impact on our financial position or results of operations.
Effective July 1, 2009, we adopted ASU No. 2009-05, Measuring Liabilities at Fair Value (ASU 2009-05). This update provides amendments to ASC 820-10, Fair Value Measurements and Disclosures (formerly SFAS No. 157, Fair Value Measurements), for the fair value measurement of liabilities. ASU 2009-05 is effective for financial statements issued for fiscal years and interim periods ending after August 28, 2009. The adoption of ASU 2009-05 did not have an impact on our financial position or results of operations; however, could have an impact in future periods.
Effective December 31, 2009 we adopted Accounting Standards Update (ASU) 2010-06, Improving Disclosures About Fair Value Measurements which adds new requirements for disclosures about transfers into and out of Levels 1 and 2 and separate disclosures about purchases, sales, issuances, and settlements relating to Level 3 measurements. It also clarifies existing fair value disclosures about the level of disaggregation and about inputs and valuation techniques used to measure fair value. The adoption of ASU 2010-06 did not have a material impact on our financial position or results of operations.
Impairments
Effective April 1, 2009, we adopted ASC 320-10-65, Investments—Debt and Equity Securities (formerly FSP FAS No. 115-2 and FAS 124-2, Recognition and Presentation of Other-than-Temporary Impairments), which amended the other-than-temporary impairment guidance for debt securities to make the guidance more operational and to improve the presentation and
66
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
disclosure in the financial statements. Under ASC 320-10-65, an other-than-temporary impairment must be recognized through earnings if an investor has the intent to sell the debt security or if it is more likely than not that the investor will be required to sell the debt security before recovery of its amortized cost basis. However, even if an investor does not expect to sell a debt security, it must evaluate expected cash flows to be received and determine if a credit loss has occurred. In the event of a credit loss, only the amount associated with the credit loss is recognized in income. The amount of loss relating to other factors is recorded in accumulated other comprehensive income. ASC 320-10-65 also requires additional disclosures regarding the calculation of credit losses and the factors considered in reaching a conclusion that an investment is not other-than-temporarily impaired. ASC 320-10-65 was effective for financial statements issued for fiscal years and interim periods ending after June 15, 2009. The adoption of the ASC 320-10-65 did not have a material impact on our financial position or results of operations.
For available-for-sale debt securities with unrealized losses, management performs an analysis in accordance with ASC 320-10-35, Investments—Debt and Equity Securities—Subsequent Measurement (formerly FSP FAS No.115-1, The Meaning of Other-than-Temporary Impairment and its Application to Certain Investments) to assess whether we intend to sell or whether we would more likely than not be required to sell the security before the expected recovery of the amortized cost basis. Where we intend to sell a security, or may be required to do so, the security’s decline in fair value is deemed to be other-than-temporary and the full amount of the unrealized loss is recorded within earnings as an impairment loss.
Regardless of our intent to sell a security, we perform additional analysis on all securities with unrealized losses to evaluate losses associated with the creditworthiness of the security. Credit losses are identified where we do not expect to receive cash flows sufficient to recover the amortized cost basis of a security.
No impairment losses were recognized through earnings related to available-for-sale securities during the years ended December 31, 2009, 2008 or 2007.
For the years ended December 31, 2009, 2008 and 2007, we recognized in other comprehensive income (loss) of $43,000, $(5,000), and $(25,000), respectively, in gains (charges) associated with the temporary impairment of available-for-sale securities primarily related to investments in mortgage and asset-backed securities.
NOTE 4 PROPERTY AND EQUIPMENT
Property and equipment consist of the following:
| December 31, (in thousands) |
2009 | 2008 | ||||||
| Leasehold improvements |
$ | 1,338 | $ | 1,338 | ||||
| Furniture and equipment |
3,295 | 3,310 | ||||||
| Total property and equipment |
4,633 | 4,648 | ||||||
| Accumulated depreciation |
(4,123 | ) | (3,767 | ) | ||||
| Property and equipment, net |
$ | 510 | $ | 881 | ||||
67
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
Depreciation expense amounted to $356,000, $412,000 and $359,000 for the years ended December 31, 2009, 2008 and 2007, respectively.
NOTE 5 ACCRUED EXPENSES
Accrued expenses consist of the following:
| December 31, (in thousands) |
2009 | 2008 | ||||
| Professional fees |
$ | 241 | $ | 264 | ||
| Accrued salaries |
93 | 481 | ||||
| Accrued bonus |
275 | 240 | ||||
| Project costs |
417 | 1,048 | ||||
| Other |
54 | 189 | ||||
| Total |
$ | 1,080 | $ | 2,222 | ||
NOTE 6 COMMITMENTS AND CONTINGENCIES
Our lease for office, warehouse and laboratory space expires in 2011. We also lease certain office equipment under operating lease arrangements, which expire in 2013. Our future minimum lease payments under these non-cancelable operating leases for facilities and equipment are as follows (in thousands):
| Year Ending December 31, |
Minimum Payments | ||
| 2010 |
$ | 568 | |
| 2011 |
137 | ||
| 2012 |
10 | ||
| 2013 |
4 | ||
| 2014 |
— | ||
| Total |
$ | 719 | |
Total rental expense for facilities and equipment was $568,000, $567,000 and $550,000 for the years ended December 31, 2009, 2008 and 2007, respectively.
As part of the sale of our cosmeceutical and toiletry business to RP Scherer Corporation in July 2000, we guaranteed a minimum gross profit percentage on RP Scherer’s sales of products to Ortho Neutrogena and Dermik (see Note 9).
As permitted under Delaware law and in accordance with our bylaws, we indemnify our officers and directors for certain events or occurrences while the officer or director is or was serving at our request in such capacity. The term of the indemnification period is for the officer’s or director’s lifetime. The maximum amount of potential future indemnification is unlimited; however,
68
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
we have a director or officer insurance policy that limits our exposure and may enable us to recover a portion of any future payments. We believe the fair value of these indemnification agreements is minimal. Accordingly, we have not recorded any liabilities for these agreements as of December 31, 2009.
In the normal course of business, we provide indemnifications of varying scope under our agreements with other companies, typically our clinical research organizations, investigators, clinical sites, suppliers and others. Pursuant to these agreements, we generally indemnify, hold harmless, and agree to reimburse the indemnified parties for losses suffered or incurred by the indemnified parties in connection with use or testing of our products or product candidates or with any U.S. patent or any copyright or other intellectual property infringement claims by any third party with respect to our products. The term of these indemnification agreements is generally perpetual. The potential future payments we could be required to make under these indemnification agreements is unlimited. Historically, costs related to these indemnification provisions have been immaterial. We also maintain various liability insurance policies that limit our exposure. As a result, we believe the fair value of these indemnification agreements is minimal. Accordingly, we have not recorded any liabilities for these agreements as of December 31, 2009.
NOTE 7 STOCKHOLDERS’ EQUITY
Amendments to Articles of Incorporation
On July 29, 2009, we amended our certificate of incorporation to increase the number of shares of authorized common stock to 100,000,000, par value $.01 per share. Prior to the amendment, the number of shares of authorized common stock was 50,000,000, par value $.01 per share. The certificate of amendment was approved by a majority of our stockholders on May 27, 2009.
Private Placement
In October 2009, in a private placement, we sold 7,954,543 shares of our common stock at $0.88 per share and warrants to purchase 3,977,270 shares of our common stock, exercisable through January 7, 2015, at $0.88 per share (Private Placement). The purchasers paid $0.125 per underlying share for the warrants. Additionally the purchasers have the right to purchase up to an additional 5,165,286 shares at $0.97 per share prior to May 14, 2010 and paid $0.125 per underlying share for the right to purchase such additional shares. Total proceeds were approximately $7.9 million, after deducting costs associated with the issuance. We are required to prepare and file Form S-3 registration statements, as permissible under SEC rules and regulations, beginning within 30 days of October 22, 2009, with the SEC for the purpose of registering for resale of the securities sold in this Private Placement. We filed a Form S-3 covering 7,532,617 shares on November 6, 2009, which was declared effective by the SEC on November 17, 2009. However, if we fail to meet future filing or effectiveness deadlines for any additional registration statements required or fail to keep any registration statements continuously effective, we may be obligated to pay to the holders of the shares and warrants liquidated damages in the amount of 1% per month of the purchase price for the shares and warrants, up to a maximum cap of 8%. In addition, at the closing of the Private Placement, Baker Brothers Investments was given the right to designate a representative to the Company’s Board of Directors. In February 2010, Stephen R. Davis was appointed to the Company’s Board of Directors as the Baker Brothers Investments designee. In the event Baker Brothers Investments exercises its right to purchase its designated additional shares under the terms of the Private Placement, Baker Brothers Investments shall have the right to designate an additional representative to the Company’s board of directors.
69
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
Public Offering
In June 2007, we sold 24,393,939 shares of our common stock in a public offering at a price of $1.65 per share, for net proceeds of approximately $37.2 million after deducting underwriting fees and costs associated with the offering.
Shareholders’ Rights Plan
On December 18, 2006, we entered into a Preferred Shares Rights Agreement. As part of this agreement, preferred stock purchase rights (the rights) were distributed to stockholders of record as of January 2, 2007 (and to each person who acquires our common stock after that date unless determined otherwise by the board of directors) at the rate of one right for each share of common stock held. The rights become exercisable only upon the acquisition, or the acquisition of the right to acquire, by a person or group of affiliated or associated persons, of 20% or more of the outstanding shares of our common stock. In connection with the Private Placement in October 2009, we amended our Preferred Shareholders Rights Agreement to permit Tang Capital Partners and Baker Brothers Investments, both purchasers under the Private Placement, to each beneficially own up to 34% and 30%, respectively, of our outstanding common stock. Once exercisable, each right entitles the holder to purchase, at a price of $44.00, one one-thousandth of a share of Series A Participating Preferred Stock. For a limited period of time following the announcement of any such acquisition or offer, the rights are redeemable by us at a price of $0.01 per right. If the rights are not redeemed or exchanged, each right will then entitle the holder to receive, upon exercise of such right, a number of shares of our common stock having a then current value equal to two times the purchase price of such right. Similarly, if the rights are not redeemed or exchanged, and following the acquisition of 20% (34% for Tang Capital Partners, LP and 30% for Baker Brothers Investments) or more of the outstanding shares of our common stock by a person or group of affiliated or associated persons: (i) we consolidate with or merge into another entity; (ii) another entity consolidates with or merges into us; or (iii) we sell or otherwise transfer 50% or more of our consolidated assets or earning power, each right will then entitle the holder to receive, upon exercise of such right, a number of shares of common stock of the acquiring company having a then current value equal to two times the purchase price. For a limited period of time after the exercisability of the rights, each right, at the discretion of the Board of Directors, may be exercised for such number of shares of common stock determined in accordance with the rights agreement. We have initially reserved 200,000 shares of preferred stock pursuant to the exercise of these rights. These rights expire on December 31, 2016.
Stock-Based Compensation Plans
We have two types of stock-based compensation plans, which consist of a stock purchase plan and three stock option plans.
In 1997, our stockholders approved our 1997 Employee Stock Purchase Plan (the “Purchase Plan”). In December 2007 and May 2009, our stockholders authorized increases in the number of shares reserved for issuance under the Purchase Plan by 100,000 and 200,000 shares, respectively, for a total of 500,000 shares reserved at December 31, 2009. Under the terms of the Purchase Plan, employees can elect to have up to a maximum of 10% of their base earnings withheld to purchase our common stock. The purchase price of the stock is 85% of the lower of the closing prices for our common stock on: (i) the first trading day in the enrollment period, as defined in the Purchase Plan, in which the purchase is made, or (ii) the purchase date. The length of the enrollment period may not exceed a maximum of six months. Our compensation committee modified the Purchase Plan such that beginning May 2008, the length of all offering periods was decreased from 24 months to six months.
70
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
Enrollment dates are the first business day of May and November and the first enrollment date was April 30, 1997. Approximately 39% of eligible employees participated in the Purchase Plan in 2009. Under the Purchase Plan, we issued 99,294, 75,787 and 22,860 shares in 2009, 2008 and 2007, respectively. The weighted-average fair value per share of purchase rights granted during 2009, 2008 and 2007 was $0.30, $0.34 and $1.82, respectively. The weighted-average exercise price per share of the purchase rights exercised during 2009, 2008 and 2007 was $0.42, $0.67 and $2.52, respectively. We had 158,079, 57,373 and 133,160 shares reserved for issuance under the Purchase Plan at December 31, 2009, 2008 and 2007, respectively.
We have three stock option plans currently from which we can grant options and restricted stock awards to employees, officers, directors and consultants. In December 2007, the stockholders approved our 2007 Equity Incentive Plan (the “2007 Plan”). We are authorized to issue up to 3,000,000 shares under the 2007 Plan. We also grant stock options and restricted stock awards under the 2002 Stock Incentive Plan (the “2002 Plan”) and the Non-Qualified Stock Plan (the “NQ Plan”). We are authorized to issue up to 425,000 shares under the 2002 Plan, 100,000 of which were approved by stockholders in May 2006, and 2,062,500 shares under the NQ Plan, a plan that has not undergone stockholder approval and can only be utilized to grant stock options and restricted stock awards as inducements to attract new employees, to which 1,000,000 shares were added by the Board of Directors in September 2007, and an additional 1,000,000 shares were added in July 2008. The options to purchase our common stock are granted with an exercise price which equals fair market value of the underlying common stock on the grant dates, and expire no later than ten years from the date of grant. The options are exercisable in accordance with vesting schedules that generally provide for them to be fully vested and exercisable four years after the date of grant. Any shares that are issuable upon exercise of options granted that expire or become un-exercisable for any reason without having been exercised in full are available for future grant and issuance under the same stock option plan.
As mentioned in Note 1, we adopted ASC 718-20 on January 1, 2006. Accordingly, we record stock-based compensation expense based on the grant date or purchase date fair value of stock options and purchase rights issued to employees in conjunction with our stock option plans or the Purchase Plan. We also record compensation expense for stock options issued to non-employees and restricted stock awards to employees and directors.
The fair value of each employee and director grant of options to purchase common stock and purchase rights under the Purchase Plan is estimated on the date of the grant using the Black-Scholes option-pricing model assuming no dividends and the following weighted-average assumptions:
| 2009 | 2008 | 2007 | ||||
| Expected term (years): |
||||||
| Stock options |
6.00 | 6.00 | 6.25 | |||
| Employee Stock Purchase Plan |
.50 | 1.00 | 1.25 | |||
| Risk-free interest rate: |
||||||
| Stock options |
1.8% | 3.3% | 4.3% | |||
| Employee Stock Purchase Plan |
0.5% | 2.3% | 3.9% | |||
| Volatility: |
||||||
| Stock options |
205% | 219% | 240% | |||
| Employee Stock Purchase Plan |
129% | 78% | 57% |
71
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
The expected term is based on historical data since 2008; prior to 2008, the expected term of options granted was based on the simplified method provided in Staff Accounting Bulletin No. 107 for “plain vanilla options.” The expected term for the Purchase Plan is based on the weighted-average purchase period of the Purchase Plan. The expected volatility is based on our historical stock prices and the estimated forfeiture rate of the options is based on historical data.
The Black-Scholes option valuation model requires the input of highly subjective assumptions, including the expected life of the award and stock price volatility. The assumptions listed above represent management’s best estimates, but these estimates involve inherent uncertainties and the application of management judgment. As a result, if other assumptions had been used, our recorded stock-based compensation expense could have been materially different.
Stock-based compensation expenses recorded for awards granted under the stock option plans and the Purchase Plan, net of estimated forfeitures, was as follows:
| For the Years Ended December 31, |
2009 | 2008 | 2007 | ||||||
| Research and development |
$ | 260,000 | $ | 442,000 | $ | 117,000 | |||
| General and administrative |
877,000 | 670,000 | 231,000 | ||||||
| Total |
$ | 1,137,000 | $ | 1,112,000 | $ | 348,000 | |||
No tax benefit was recognized related to stock-based compensation expense since we have incurred operating losses and we have established a full valuation allowance to offset all the potential tax benefits associated with our deferred tax assets.
72
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
The following table summarizes option activity for the years ended December 31, 2009, 2008 and 2007:
| 2009 | 2008 | 2007 | |||||||||||||||||||||
| Shares | Weighted- Average Exercise Price |
Weighted- Average Remaining Contractual Term (Years) |
Aggregate Intrinsic Value as of December 31, 2009 |
Shares | Weighted- Average Exercise Price |
Shares | Weighted- Average Exercise Price | ||||||||||||||||
| Outstanding at beginning of year |
2,701,073 | $ | 2.38 | 550,383 | $ | 8.57 | 532,308 | $ | 10.68 | ||||||||||||||
| Granted |
991,500 | .67 | 2,599,300 | 1.29 | 93,757 | 3.58 | |||||||||||||||||
| Exercised |
(27,651 | ) | 0.71 | (1,708 | ) | 1.37 | — | — | |||||||||||||||
| Expired or Forfeited |
(572,505 | ) | 2.02 | (446,902 | ) | 3.67 | (75,682 | ) | 16.45 | ||||||||||||||
| Outstanding at end of year |
3,092,417 | 1.91 | 7.90 | $ | 538,748 | 2,701,073 | 2.38 | 550,383 | 8.57 | ||||||||||||||
| Options exercisable at year end |
1,316,567 | 3.11 | 6.85 | $ | 78,251 | 707,338 | 416,599 | ||||||||||||||||
| Options vested or Expected to vest |
2,919,164 | 7.85 | 1.97 | $ | 493,226 | ||||||||||||||||||
| Shares available for future grant at year end |
1,959,758 | 2,789,796 | 4,064,444 | ||||||||||||||||||||
| Weighted-average fair value of stock options granted during the year |
$ | 0.67 | $ | 1.28 | $ | 3.57 | |||||||||||||||||
As of December 31, 2009 there was approximately $1,987,000 of total unrecognized compensation expense related to unvested stock options. This expense is expected to be recognized over a weighted-average period of 2.87 years. Cash received from option exercises for the years ended December 31, 2009, 2008 and 2007 was $20,000, $2,000 and $0, respectively. The total intrinsic value of options exercised in the year ended December 31, 2009 was $7,000, and none in December 31, 2008 and 2007.
73
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
The following table summarizes information about stock options outstanding at December 31, 2009:
| OPTIONS OUTSTANDING | OPTIONS EXERCISABLE | |||||||||||
| Range of Exercise Prices |
Number Outstanding |
Weighted- Average Remaining Contractual Life (Years) |
Weighted- Average Exercise Price |
Number Exercisable |
Weighted- Average Exercise Price | |||||||
| $0.61-$0.71 |
736,512 | 9.09 | $ | 0.66 | 79,169 | $ | 0.71 | |||||
| $1.19 |
1,400,000 | 8.52 | 1.19 | 495,833 | 1.19 | |||||||
| $1.24-$4.28 |
623,257 | 7.56 | 1.53 | 415,546 | 1.63 | |||||||
| $4.60-$13.37 |
327,648 | 3.36 | 8.35 | 321,019 | 8.42 | |||||||
| $14.00 |
5,000 | 0.58 | 14.00 | 5,000 | 14.00 | |||||||
| $0.61-$14.00 |
3,092,417 | 7.90 | $ | 1.91 | 1,316,567 | $ | 3.11 | |||||
As of December 31, 2009, we had a total of 140,000 shares of unvested restricted stock awards granted to employees and directors. The compensation cost that has been expensed in the statements of operations for the restricted stock awards issued to employees and directors and stock issued in lieu of fees was $375,000, $83,000 and $99,000 for 2009, 2008 and 2007, respectively.
The following table summarizes unvested restricted stock awards activity for the year ended December 31, 2009:
| Shares | Weighted-Average Grant Date Fair Value | |||||
| Outstanding at beginning of year |
72,750 | $ | 2.9893 | |||
| Awarded |
399,949 | 0.9842 | ||||
| Released |
(332,699 | ) | 1.4160 | |||
| Outstanding at end of year |
140,000 | $ | 1.0000 | |||
In November 2009, our Board of Directors extended the vesting period for certain restricted stock awards granted in May 2009 from December 31, 2009 to January 2, 2010. The modification of the restricted stock awards did not result in any additional compensation expense.
74
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
Common Stock Reserved for Issuance
As of December 31, 2009, the Company had reserved shares of common stock for issuance as follows:
| Issuance upon exercise of outstanding stock options |
3,092,417 | |
| Issuance of future grants under stock option plans |
1,959,758 | |
| Issuance upon exercise of warrants |
3,977,270 | |
| Issuance upon exercise of right to purchase additional shares under the Private Placement |
5,165,286 | |
| Total |
14,194,731 | |
NOTE 8 NET INCOME (LOSS) PER SHARE
The following options, unvested restricted stock awards and warrants were outstanding as of December 31, 2009, 2008 and 2007, but were not included in the computation of diluted net loss per share since inclusion of these potentially dilutive securities would have been anti-dilutive for the periods presented (in thousands):
| For the years ended December 31, |
2009 | 2008 | 2007 | |||
| Number of options outstanding |
3,092 | 2,701 | 550 | |||
| Number of unvested restricted stock awards outstanding |
140 | 73 | 34 | |||
| Number of warrants outstanding |
3,977 | — | — |
NOTE 9 DISCONTINUED OPERATIONS
We completed the sale of certain assets of our Analytical Standards division as well as certain technology rights for our topical pharmaceutical and cosmeceutical product lines and other assets (cosmeceutical and toiletry business) in February 2003 and July 2000, respectively.
The Analytical Standards division and cosmeceutical and toiletry business are reported as discontinued operations for all periods presented in the accompanying Statements of Operations.
Income (loss) from discontinued operations represents primarily the gain (loss) attributable to changes in estimates of our cosmeceutical and toiletry business that was sold to RP Scherer on July 25, 2000, as follows (in thousands):
| For the years ended December 31, |
2009 | 2008 | 2007 | ||||||||
| Cosmeceutical and Toiletry Business: |
|||||||||||
| Change in estimates for guarantees |
$ | 68 | $ | (200 | ) | $ | (342 | ) | |||
There was no revenue relating to discontinued operations for the years ended December 31, 2009, 2008 and 2007.
75
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
The following table sets forth our basic and diluted income (loss) per common share from discontinued operations for the years ended December 31, 2009, 2008 and 2007:
| For the years ended December 31, |
2009 | 2008 | 2007 | ||||||||
| Basic and diluted income (loss) per common share from discontinued operations |
$ | — | $ | (0.01 | ) | $ | (0.02 | ) | |||
The gain on disposition of discontinued operations recorded in 2007 of $20,000 relates to the gain on the sale of our Analytical Standards division as measured by royalty receipts in excess of guaranteed minimums.
As of December 31, 2009 and 2008, accrued disposition cost of $553,000 and $621,000, respectively, represents accruals for gross profit guarantees related to discontinued operations.
The cash used in discontinued operations of $169,000 in 2007 relates to payments made for the Gross Profit Guaranty, partially offset by the royalties received from GFS, from sales of Analytical Standards products. The cash provided by discontinued operations of $19,000 in 2008 relates to the royalties received from GFS from sales of Analytical Standards products, partially offset by severance payments made to former employees who were terminated as a result of the sale of the Analytical Standards division.
Analytical Standards Division
On February 13, 2003, we completed the sale of our Analytical Standards division to GFS. In this transaction, we received $2.1 million at closing and were entitled to receive royalties on sales of Analytical Standards products for a period of five years following the sale at rates ranging from 5% to 15%.
Cosmeceutical and Toiletry Business
On July 25, 2000, we completed the sale of certain technology rights for our cosmeceutical and toiletry business to RP Scherer Corporation, a subsidiary of Cardinal Health, Inc.
Under the terms of the agreement with RP Scherer, we guaranteed a minimum gross profit percentage on RP Scherer’s combined sales of products to Ortho Neutrogena (Ortho) and Dermik (Gross Profit Guaranty). The guaranty period initially commenced on July 1, 2000 and was to end on the earlier of July 1, 2010 or the end of two consecutive guaranty periods where the combined gross profit on sales to Ortho and Dermik equals or exceeds the guaranteed gross profit (the “two period test”). The Gross Profit Guaranty expense totaled $944,000 for the first seven guaranty years and in those years profits did not meet the two period test. Effective March 2007, in conjunction with a sale of assets by RP Scherer’s successor company to an Amcol International subsidiary (Amcol), a new agreement was signed between us and Amcol to provide continuity of product supply to Ortho and Dermik. This new agreement potentially extends the gross profit guaranty period an additional three years to July 1, 2013, unless it is terminated earlier with the two period test. Amcol has indicated that its costs differ from those it charged historically to the RP Scherer successor company to produce the products. We have requested documentation of the actual costs, but have accrued at the amount Amcol represents it is currently owed. As there is no minimum amount of Gross Profit Guaranty due, no accrual for the guaranty is estimable for future years. A liability of $553,000 related to the current amount due under gross profit guarantees is recorded in accrued disposition costs as of December 31, 2009.
76
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
NOTE 10 DEFINED CONTRIBUTION PLAN
We have a defined contribution plan (401k) covering substantially all of our employees. In the past three calendar years, we made matching cash contributions equal to 50% of each participant’s contribution during the plan year up to a maximum amount equal to the lesser of 3% of each participant’s annual compensation or $7,350, $6,900, and $6,750 for 2009, 2008, and 2007, respectively, and such amounts were recorded as expense in the corresponding years. We may also contribute additional discretionary amounts to the defined contribution plan as we may determine. For the years ended December 31, 2009, 2008 and 2007, we contributed to the plan approximately $76,000, $104,000 and $84,000, respectively. No discretionary contributions have been made to the plan since its inception.
NOTE 11 INCOME TAXES
There was an income tax benefit of $122,000 in 2009 from the carryback of the net operating loss under the Worker, Homeownership, and Business Assistance Act of 2009, which among other things suspends the 90% limitation on utilization of net operating loss for alternative minimum tax purposes. There was no provision for 2008 because we have incurred operating losses. Deferred income taxes reflect the net tax effects of net operating loss and tax credit carryovers and temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of our deferred tax assets are as follows (in thousands):
| December 31, |
2009 | 2008 | ||||||
| Deferred Tax Assets: |
||||||||
| Net operating loss carryforwards |
$ | 22,600 | $ | 16,300 | ||||
| Research credits |
3,000 | 2,600 | ||||||
| Capitalized research expenses |
0 | 100 | ||||||
| Other |
1,330 | 2,130 | ||||||
| Total deferred tax assets |
26,930 | 21,130 | ||||||
| Valuation allowance |
(26,930 | ) | (21,130 | ) | ||||
| Net deferred tax assets |
$ | — | $ | — | ||||
Realization of our deferred tax assets is dependent upon our future taxable income, if any, the timing and amount of which are uncertain. Accordingly, our deferred tax assets have been fully offset by a valuation allowance. The valuation allowance increased by $5.8 million, increased by $10.3 million, and decreased by $15.4 million during 2009, 2008 and 2007, respectively.
As of December 31, 2009, we had federal and California net operating loss carryforwards of $57.8 million and $48.0 million, respectively, and federal and California research and development tax credit carryforwards of $1.13 million and $2.8 million, respectively. Of the carryforwards, federal and California net operating loss carryforwards of $7.7 million and $3.4 million, respectively, are subject to annual limitations and will be available from 2010 through 2026, as a result of federal ownership change limitations. The federal and state net operating losses and the federal research and development credit carryforwards expire at various dates beginning in the years 2010 through 2029, if not utilized. The state research credits have no expiration date.
77
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
Federal and state laws limit the use of net operating loss and tax credit carryforwards in certain situations where changes occur in the stock ownership of a company. We recently conducted an analysis of our stock ownership under Internal Revenue Code Section 382 and have reported our deferred tax assets related to net operating loss and research credit carryforwards after recognizing change of control limitations in 2007. The limitation of our federal and state carryforwards associated with previous net operating loss and research credit carryforwards, and the associated reduction in our deferred tax assets, was offset by a reduction in our valuation allowance. Utilization of our remaining net operating loss and research and development credit carryforwards may still be subject to substantial annual limitations due to ownership change limitations after December 31, 2009. Such an annual limitation could result in the expiration of the net operating loss and research and development credit carryforwards available as of December 31, 2009 before utilization.
We adopted the provisions of ASC 740-10-50, Accounting for Uncertainty in Income Tax Provisions, as of January 1, 2007, which resulted in the reversal of certain fully reserved deferred tax assets totaling $1.02 million and the related valuation allowance. A reconciliation of the beginning and ending amounts of unrecognized tax benefits is as follows (in thousands):
| December 31, |
2009 | 2008 | |||||
| Unrecognized tax benefit: |
|||||||
| At the beginning of the period |
$ | 120 | $ | 1,022 | |||
| Gross increases – tax positions in the current period |
— | — | |||||
| Gross decreases – tax positions in the current period |
— | (902 | ) | ||||
| At the end of the period |
$ | 120 | $ | 120 | |||
The unrecognized tax benefit, if recognized in full, would result in adjustments to deferred taxes and the related valuation allowance. We do not currently anticipate any significant changes to the unrecognized tax benefits in 2010. Our policy is to recognize interest and penalties accrued on any unrecognized tax benefits as a component of tax expense. To date, we have not used the unrecognized tax benefits to reduce any of our past tax obligations. As a result, we had no accrual for the payment of interest and penalties related to the unrecognized tax benefits. As of December 31, 2009, our tax returns were subject to future examination in the U.S. federal and state tax jurisdictions for the tax years 1995 through 2009, due to net operating losses and research credits that are being carried forward.
NOTE 12 SIGNIFICANT AGREEMENTS
Paul Royalty Fund
On January 18, 2006, we sold our rights to royalties on sales of Retin-A Micro and Carac, effective October 1, 2005, to an affiliate of the Paul Royalty Fund for up to $30 million. Proceeds of $25 million were received upon the closing of the transaction and used primarily to fund the Phase 3 pivotal trial of APF530, our drug candidate for the prevention of both acute and delayed CINV. The remaining $5 million was to be received upon the achievement of certain milestones over the successive four years. Upon attainment of one milestone in 2007, an additional $2.5 million was received. The final $2.5 million was received in January 2010 subsequent to the balance sheet date as described in Note 13. No additional payments are due to us.
78
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
RHEI Pharmaceuticals, Inc.
On October 1, 2006, we entered into an agreement with RHEI Pharmaceuticals, Inc. (RHEI) in which we granted them an exclusive license to develop and market APF530 in Greater China. We received a license fee on the signing of the contract, which was recorded as deferred revenue on the balance sheet, and were due additional milestone payments upon the achievement of certain regulatory events.
Following the announcement of acceptance for filing of an NDA for our APF530 product candidate (APF530) by the FDA on July 20, 2009, RHEI became contractually obligated to pay us a milestone payment. RHEI did not make such milestone payment in the time required under the terms of our agreement and we provided RHEI with notice of its cure period. RHEI remained in default of this payment and, as a result, we elected to terminate the agreement for cause on September 29, 2009. No material termination penalties apply to us for the termination of the agreement.
Revenue of $1.0 million, previously deferred in conjunction with the RHEI agreement was recognized in 2009 as a result of the termination and is included in contract revenue.
Merial
In September 2009, we entered into a world-wide license and development agreement with Merial, a world leading animal health company, for a long acting pain management product for companion animals. The license and development agreement follows a successful proof-of-concept agreement. Under the terms of the new agreement, we received an upfront license fee and will receive development funding and potential milestone payments, and royalties following commercialization.
Under the license and development agreement, we are obligated to perform reimbursable development services and provide any improvements related to the licensed technology during the six-year development period. We are recognizing the upfront license fee ratably over the development period, and will recognize revenue from the development services when the services are rendered. Any milestone payments will be recognized when receipt of the payments is probable.
We recognized $228,000 in revenue from Merial for services rendered in 2009.
79
Table of Contents
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2009, 2008 AND 2007
NOTE 13 SUBSEQUENT EVENTS
In January 2010, we received a $2.5 million milestone from an affiliate of Paul Capital Healthcare. The payment represents a milestone payment that became due to us in January 2010 under an agreement that we entered into effective October 1, 2005 to sell our royalty rights to Retin-A Micro® and Carac®. As the milestone occurred after December 31, 2009, it is not reflected in our financial statements as of that date. We will record revenue related to milestones upon achievement.
NOTE 14 QUARTERLY RESULTS OF OPERATIONS (UNAUDITED)
The following table presents summarized unaudited results of operations for each of our quarters in the years ended December 31, 2009 and 2008.
Quarterly Results of Operations
(in thousands, except per share data)
(unaudited)
| Year Ended December 31, 2009 |
First Quarter |
Second Quarter |
Third Quarter |
Fourth Quarter |
||||||||||||
| Total revenue |
$ | 8 | $ | 14 | $ | 1,117 | $ | 122 | ||||||||
| Operating expenses |
2,977 | 3,974 | 2,330 | 2,222 | ||||||||||||
| Interest and other, net |
9 | 19 | (1 | ) | (3 | ) | ||||||||||
| Loss from continuing operations |
(2,960 | ) | (3,941 | ) | (1,214 | ) | (2,103 | ) | ||||||||
| Discontinued operations |
— | — | — | 68 | ||||||||||||
| Net loss |
(2,960 | ) | (3,941 | ) | (1,214 | ) | (1,913 | ) | ||||||||
| Basic and diluted loss per common share: |
||||||||||||||||
| Loss from continuing operations |
(0.10 | ) | (0.13 | ) | (0.04 | ) | (0.06 | ) | ||||||||
| Net loss |
(0.10 | ) | (0.13 | ) | (0.04 | ) | (0.05 | ) | ||||||||
| Year Ended December 31, 2008 |
First Quarter |
Second Quarter |
Third Quarter |
Fourth Quarter |
||||||||||||
| Total revenue |
$ | 133 | $ | 152 | $ | 64 | $ | 20 | ||||||||
| Operating expenses |
7,220 | 6,401 | 6,341 | 3,852 | ||||||||||||
| Interest and other, net |
283 | 159 | 112 | (34 | ) | |||||||||||
| Loss from continuing operations |
(6,804 | ) | (6,090 | ) | (6,165 | ) | (3,866 | ) | ||||||||
| Discontinued operations |
(40 | ) | (40 | ) | (40 | ) | (80 | ) | ||||||||
| Net loss |
(6,844 | ) | (6,130 | ) | (6,205 | ) | (3,946 | ) | ||||||||
| Basic and diluted loss per common share: |
||||||||||||||||
| Loss from continuing operations |
(0.22 | ) | (0.20 | ) | (0.20 | ) | (0.13 | ) | ||||||||
| Net loss |
(0.22 | ) | (0.20 | ) | (0.20 | ) | (0.13 | ) | ||||||||
80
Table of Contents
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A (T). CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Based on an evaluation as of the end of the period covered by this report, our Chief Executive Officer and Chief Financial Officer have concluded that our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) were effective as of the end of the period covered by this report to ensure that information that we are required to disclose in reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in SEC rules and forms.
Our disclosure controls and procedures are designed to provide reasonable assurance of achieving their objectives, and our Chief Executive Officer and Chief Financial Officer have concluded that these controls and procedures are effective at the “reasonable assurance” level. We believe that a control system, no matter how well designed and operated, cannot provide absolute assurance that the objectives of the control system are met, and no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within a company have been detected.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
There are inherent limitations in the effectiveness of any system of internal control, including the possibility of human error and the circumvention or overriding of controls. Accordingly, even effective internal controls can provide only reasonable assurances with respect to financial statement preparation. Further, because of changes in conditions, the effectiveness of internal control may vary over time.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2009. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations (COSO) of the Treadway Commission in Internal Control—Integrated Framework. Based on our assessment using the COSO criteria, management concluded that, as of December 31, 2009, our internal control over financial reporting is effective.
This annual report does not include an attestation report of our independent registered public accounting firm regarding internal control over financial reporting. Our internal control over financial reporting was not subject to attestation by our independent registered public accounting firm pursuant to temporary rules of the SEC that permit us to provide only management’s report in this annual report.
81
Table of Contents
Changes in Internal Controls Over Financial Reporting
There were no changes in our internal controls over financial reporting that occurred during our most recent fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
None.
82
Table of Contents
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
We have incorporated by reference the information set forth under the captions “Election of Directors”, “Executive Officers”, “Corporate Governance” and “Compliance with Section 16(a) of the Securities Exchange Act” of our Proxy Statement (the “Proxy Statement”) for the 2010 annual meeting of shareholders.
Code of Ethics
We have adopted a Code of Ethics that applies to all of our directors, officers and employees. The Code of Ethics is posted on our website at http://www.appharma.com under the caption “Investor Relations/ Corporate Governance”. If we make any substantive amendments to the code of ethics or grant any waiver, including implicit waiver, from a provision of the code of ethics to our principal executive officer, principal financial officer or principal accounting officer, we will disclose the nature of such amendment or waiver on our website or in a current report on Form 8-K that will be publicly filed.
ITEM 11. EXECUTIVE COMPENSATION
We have incorporated by reference the information set forth under the captions “Executive Compensation” and “Director Compensation” of the Proxy Statement.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
We have incorporated by reference the information set forth under the captions “Common Stock Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information” of the Proxy Statement.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS AND DIRECTOR INDEPENDENCE
We have incorporated by reference the information set forth under the captions “Related Party Transactions” and “Corporate Governance” of the Proxy Statement.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
We have incorporated by reference the information set forth under the captions “Report of the Audit Committee”, “Ratification of Independent Registered Public Accountants” and “Auditors Fees and Services” of the Proxy Statement.
83
Table of Contents
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
| (a) | 1. Financial Statements |
| The financial statements and supplementary data set forth in Part II of the Annual Report on Form 10-K are included herein. |
| 2. Financial Statement Schedules |
| Schedule II Valuation Accounts |
| All other schedules have been omitted because the information is not required or is not so material as to require submission of the schedule, or because the information is included in the financial statements or the notes thereto. |
| 3. Exhibits |
| See | Exhibit Index beginning on page 87. |
84
Table of Contents
Pursuant to the requirement of Section 13 or 15 (d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
A.P. PHARMA, INC.
| By: |
/s/ Ronald J. Prentki | |
| Ronald J. Prentki | ||
| President, Chief Executive Officer and Director | ||
| Date: March 12, 2010 |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENT, that each person whose signature appears below constitutes and appoints Ronald J. Prentki and John B. Whelan, jointly and severally, his or her attorneys-in-fact, with the power of substitution, for him or her in any and all capacities, to sign any amendments to this Annual Report on Form 10-K and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that said attorneys-in-fact, or his substitute or substitutes, may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ Ronald J. Prentki Ronald J. Prentki |
President, Chief Executive Officer (Principal Executive Officer) and Director |
March 12, 2010 | ||
| /s/ John B. Whelan John B. Whelan |
Vice-President and Chief Financial Officer (Principal Financial and Accounting Officer) |
March 12, 2010 | ||
| /s/ Paul Goddard Paul Goddard |
Chairman of the Board of Directors |
March 12, 2010 | ||
| /s/ Toby Rosenblatt Toby Rosenblatt |
Director |
March 12, 2010 | ||
| /s/ Kevin C. Tang Kevin C. Tang |
Director |
March 12, 2010 | ||
| /s/ Gregory Turnbull Gregory Turnbull |
Director |
March 12, 2010 | ||
| /s/ Robert Zerbe Robert Zerbe |
Director |
March 12, 2010 | ||
| /s/ Stephen R. Davis Stephen R. Davis |
Director |
March 12, 2010 | ||
85
Table of Contents
SCHEDULE II
VALUATION AND QUALIFYING ACCOUNTS (in thousands)
| Beginning Balance |
Additions Charged to Cost and Expense |
Deductions, Write-Offs and Recoveries |
Ending Balance | |||||||||
| DECEMBER 31, 2009 |
||||||||||||
| Note receivable, allowance for doubtful note |
$ | 394 | $ | — | $ | (394) | $ | — | ||||
| DECEMBER 31, 2008 |
||||||||||||
| Note receivable, allowance for doubtful note |
$ | 394 | $ | — | $ | — | $ | 394 | ||||
| DECEMBER 31, 2007 |
||||||||||||
| Note receivable, allowance for doubtful note |
$ | 394 | $ | — | $ | — | $ | 394 | ||||
86
Table of Contents
FORM 10-K ANNUAL REPORT
2.1 – Copy of Asset Purchase Agreement between Registrant and RP Scherer South, Inc. dated June 21, 2000.(1)
3-A – Copy of Registrant’s Certificate of Amendment of Certificate of Incorporation.(2)
3-B – Copy of Registrant’s Bylaws.(3)
3-C – Copy of Registrant’s Certificate of Designation.(4)
4-A – Copy of Registrant’s Preferred Shares Rights Agreement.(5)
4-B – Copy of Registrant’s Form of Rights Certificate.(6)
4-C – First Amendment to Registrant’s Preferred Shares Rights Agreement.(7)
4-D – Copy of Specimen Common Stock Certificate.(8)
10-C – Registrant’s 1992 Stock Plan dated August 11, 1992.(9)*
10-D – Registrant’s 1997 Employee Stock Purchase Plan, as amended to date.(10)*
10-E – Lease Agreement between Registrant and Metropolitan Life Insurance Company for lease of Registrant’s executive offices in Redwood City dated as of November 17, 1997.(11)
10-F – Registrant’s 2002 Equity Incentive Plan dated June 13, 2002.(12)*
10-G – Agreement between Registrant and RHEI Pharmaceuticals, Inc. (RHEI) granting exclusive license to RHEI to develop and sell APF530 in Greater China dated October 1, 2006.(13)
10-H – Royalty Interest Agreement between Registrant and Paul Royalty Fund dated January 18, 2006.(14)
10-I – Amended and Restated Retention and Non-Competition Agreement between the Registrant and Michael O’Connell effective August 23, 2007.(15)*
10-J – Management Retention Agreement between the Registrant and Dr. John Barr dated as of November 8, 2007.(16)*
10-K – Registrant’s 2007 Equity Incentive Plan.(17)*
10-L – Form of 2007 Equity Incentive Plan Stock Option Agreement.(18)*
10-M – Form of 2007 Equity Incentive Plan Restricted Stock Unit Agreement.(19)*
10-N – Agreement with Johnson & Johnson dated April 14, 1992.(20)
10-O – Form of 2007 Equity Incentive Plan Restricted Stock Award Agreement.(10)*
10-P – Form of 2002 Equity Incentive Plan Stock Option Agreement.(10)*
10-Q – Form of 2002 Equity Incentive Plan Restricted Stock Agreement.(10)*
10-R – Amendment to the Registrant’s Non-Qualified Plan.(21)*
10-S – Form of Indemnification Agreement.(10)*
10-T – Registrant’s Non-Qualified Plan dated June 13, 2002.(22)*
10-U – Employment Letter Agreement with Ronald Prentki, President and Chief Executive Officer dated July 3, 2008.(23)*
10-V – Amendment to Employment Letter Agreement with Ronald Prentki, President and Chief Executive Officer dated December 30, 2008.(24)
10-W – Amendment to Management Retention Agreement between the Registrant and Dr. John Barr dated December 23, 2008.(24)
10-X – Employment Letter Agreement with John B. Whelan, Chief Financial Officer dated as of February 9, 2008. (24)
10-Y – Development and License Agreement dated as of September 11, 2009, between the Registrant and Merial Limited.(25)
10-Z – Securities Purchase Agreement, dated as of October 19, 2009, by and among the Registrant and the purchasers listed therein.(26)
87
Table of Contents
10-AA – Registration Rights Agreement, dated as of October 22, 2009, by and among the Registrant and the purchasers listed therein.(27)
10-AB – Form of Warrant to Purchase Shares of Common Stock.(28)
10-AC – Second Amendment to Preferred Shares Rights Agreement, dated as of October 20, 2009, by and between the Registrant and Computershare Trust Company N.A.(29)
23.1 – Consent of Independent Registered Public Accounting Firm.
31.1 – Certification of Chief Executive Officer pursuant to Rules 13A-15(e) Promulgated under the Securities Exchange Act of 1934 as amended.
31.2 – Certification of Chief Financial Officer pursuant to Rules 13A-15(e) Promulgated under the Securities Exchange Act of 1934 as amended.
32 – Certification of Chief Executive Officer and Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.
| (1) | Filed as an Exhibit with corresponding Exhibit No. to Registrant’s Form 8-K filed August 9, 2000 (file No. 000-16109), and incorporated herein by reference. |
| (2) | Filed as Exhibit 3.1 to Registrant’s Form 10-Q filed August 4, 2009, and incorporated herein by reference. |
| (3) | Filed as an Exhibit with corresponding Exhibit No. to Registrant’s Registration Statement on Form S-1 (Registration No. 33-15429) and incorporated herein by reference. |
| (4) | Filed as Exhibit 3.C to Registrant’s Form 8-K filed December 19, 2006, and incorporated herein by reference. |
| (5) | Filed as Exhibit 4.A to Registrant’s Form 8-K filed December 19, 2006, and incorporated herein by reference. |
| (6) | Filed as Exhibit 4.B to Registrant’s Form 8-K filed December 19, 2006, and incorporated herein by reference. |
| (7) | Filed as Exhibit 4.1 to Registrant’s Form 8-K filed October 7, 2008, and incorporated herein by reference. |
| (8) | Filed as Exhibit 4.1 to Registrant’s Registration Statement on Form S-3 (Registration No. 333-162968) filed November 6, 2009, and incorporated herein by reference. |
| (9) | Filed as Exhibit No. 28.1 to Registrant’s Registration Statement on Form S-8 (Registration No. 33-50640), and incorporated herein by reference. |
| (10) | Filed as an Exhibit with corresponding Exhibit No. to Registrant’s Annual Report on Form 10-K for the year ended December 31, 2007, and incorporated herein by reference. |
| (11) | Filed as an Exhibit with corresponding Exhibit No. to Registrant’s Annual Report on Form 10-K for the year ended December 31, 1997, and incorporated herein by reference. |
| (12) | Filed as Exhibit No. 99.1 to Registrant’s Registration Statement on Form S-8 (Registration No. 333-90428), and incorporated herein by reference. |
| (13) | Filed as Exhibit 10.AA to Registrant’s Form 10-Q filed November 7, 2006, and incorporated herein by reference. |
| (14) | Filed as Exhibit 10-Y to Registrant’s Form 10-Q filed May 15, 2006, and incorporated herein by reference. |
| (15) | Filed as Exhibit 10.14 to the Registrant’s Form 10-Q filed November 14, 2007 and incorporated herein by reference. |
| (16) | Filed as Exhibit 10.15 to the Registrant’s Form 10-Q filed November 14, 2007 and incorporated herein by reference. |
| (17) | Filed as Exhibit No 4.1 to Registrant’s Registration Statement on Form S-8 (Registration No. 333-148660) and incorporated herein by reference. |
| (18) | Filed as Exhibit no. 4.3 to Registrant’s Registration Statement on Form S-8 (Registration No 333-148660) and incorporated herein by reference. |
88
Table of Contents
| (19) | Filed as Exhibit No 4.4 to Registrant’s Registration Statement on Form S-8 (Registration No. 333-148660), and incorporated herein by reference. |
| (20) | Filed as an Exhibit with corresponding Exhibit No. to Registrant’s Annual Report on Form 10-K for the year ended December 31, 1992, and incorporated herein by reference. |
| (21) | Filed as Exhibit 10.16 to the Registrant’s Form 10-Q dated November 14, 2007 and incorporated herein by reference. |
| (22) | Filed as Exhibit No. 99.2 to Registrant’s Registration Statement on Form S-8 (Registration No. 333-90428), and incorporated herein by reference. |
| (23) | Filed as an Exhibit with corresponding Exhibit No. to the Registrant’s Form 10-Q filed August 14, 2008, and incorporated herein by reference. |
| (24) | Filed as an Exhibit with corresponding Exhibit No. to Registrant’s Annual Report on Form 10-K filed March 30, 2009, and incorporated herein by reference. |
| (25) | Filed as Exhibit 10.1 to the Registrant’s Form 10-Q filed November 16, 2009 and incorporated herein by reference. |
| (26) | Filed as Exhibit 10.1 to the Registrant’s Form 8-K filed on October 22, 2009 and incorporated herein by reference. |
| (27) | Filed as Exhibit 10.2 to the Registrant’s Form 8-K filed on October 22, 2009 and incorporated herein by reference. |
| (28) | Filed as Exhibit 10.3 to the Registrant’s Form 8-K filed on October 22, 2009 and incorporated herein by reference. |
| (29) | Filed as Exhibit 10.4 to the Registrant’s Form 8-K filed on October 22, 2009 and incorporated herein by reference. |
| * | Management contract or compensatory plans. |
89