UNITED STATES SECURITIES AND
EXCHANGE COMMISSION
Washington, DC
20549
Form 10-K
| |
|
|
|
þ
|
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the fiscal
year ended December 31, 2009
|
|
o
|
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the transition
period to
|
Commission File Number
001-33744
TRANS1 INC.
(Exact name of Registrant as
specified in its charter)
| |
|
|
DELAWARE
(State or other jurisdiction
of
incorporation or organization)
|
|
33-0909022
(I.R.S. employer
identification no.)
|
301 GOVERNMENT CENTER DRIVE, WILMINGTON, NC 28403
(Address of principal
executive office) (Zip code)
(910) 332-1700
(Registrant’s telephone
number, including area code)
Securities registered pursuant to Section 12(b) of the
Act
| |
|
|
|
Title of Each Class:
|
|
Name of Each Exchange on Which Registered:
|
|
|
|
Common Stock, par value $0.0001 per share
|
|
The NASDAQ Stock Market LLC
|
Securities registered pursuant to Section 12(g) of the
Act:
None
Indicate by check mark if the registrant is a well-known
seasoned issuer, as defined in Rule 405 of the Securities
Act. YES o NO þ
Indicate by check mark if the registrant is not required to file
reports pursuant to Section 13 or Section 15(d) of the
Act. YES o NO þ
Indicate by check mark whether the registrant (1) has filed
all reports required to be filed by Section 13 or 15(d) of
the Securities Exchange Act of 1934 during the preceding
12 months (or for such shorter period that the registrant
was required to file such reports), and (2) has been
subject to such filing requirements for the past
90 days. YES þ NO o
Indicate by check mark whether the registrant has submitted
electronically and posted on its corporate Web site, if any,
every Interactive Data File required to be submitted and posted
pursuant to Rule 405 of Regulations S-T
(§ 232.405 of this chapter) during the preceding
12 months (or for such shorter period that the registrant
was required to submit and post such
files). YES o NO o
Indicate by check mark if disclosure of delinquent filers
pursuant to Item 405 of
Regulation S-K
is not contained herein, and will not be contained, to the best
of registrant’s knowledge, in definitive proxy or
information statements incorporated by reference in
Part III of this
Form 10-K
or any amendment to this
Form 10-K. o
Indicate by check mark whether the registrant is a large
accelerated filer, an accelerated filer, a non-accelerated
filer, or a smaller reporting company. See the definitions of
“large accelerated filer,” “accelerated
filer” and “smaller reporting company” in Rule
12b-2 of the Exchange Act. (Check one):
| |
|
|
|
|
|
|
|
Large accelerated
filer o
|
|
Accelerated
filer þ
|
|
Non-accelerated
filer o
(Do not check if a smaller
reporting company)
|
|
Smaller Reporting
company o
|
Indicate by check mark whether the registrant is a shell company
(as defined in
Rule 12b-2
of the Exchange
Act). YES o NO þ
As of June 30, 2009, the last business day of our most
recently completed second fiscal quarter, the aggregate market
value of the voting stock held by non-affiliates was
approximately $107.6 million, based on the number of shares
held by non-affiliates of the registrant and based on the
reported last sale price of common stock on June 30, 2009.
This calculation does not reflect a determination that persons
are affiliates for any other purposes.
The number of shares of the registrant’s common stock
outstanding as of March 8, 2010 was 20,656,293 shares.
DOCUMENTS
INCORPORATED BY REFERENCE
Portions of the Registrant’s Definitive Proxy Statement to
be filed with the Securities and Exchange Commission within
120 days after the close of the fiscal year covered by this
annual report, relating to the Registrant’s annual meeting
of stockholders, are incorporated by reference into
Part III of this
Form 10-K.
With the exception of the portions of the Proxy Statement
specifically incorporated herein by reference, the Proxy
Statement is not deemed to be filed as part of this
Form 10-K.
TRANS1
INC.
FORM 10-K
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2009
TABLE OF CONTENTS
2
Cautionary
Note Regarding Forward-Looking Statements
In addition to historical financial information, this report
contains forward-looking statements within the meaning of
Section 27A of the Securities Act of 1933 and
Section 21E of the Securities Exchange Act of 1934 that
concern matters that involve risks and uncertainties that could
cause actual results to differ materially from those projected
in the forward-looking statements. All statements other than
statements of historical fact contained in this report,
including statements regarding future events, our future
financial performance, business strategy and plans and
objectives of management for future operations, are
forward-looking statements. We have attempted to identify
forward-looking statements by terminology including
“anticipates,” “believes,” “can,”
“continue,” “could,” “estimates,”
“expects,” “intends,” “may,”
“plans,” “potential,” “predicts,”
“should” or “will” or the negative of these
terms or other comparable terminology. Although we do not make
forward-looking statements unless we believe we have a
reasonable basis for doing so, we cannot guarantee their
accuracy. Such forward-looking statements are subject to risks,
uncertainties and other factors that could cause actual results
and the timing of certain events to differ materially from
future results expressed or implied by such forward-looking
statements. Readers are urged to carefully review and consider
the various disclosures made by us, which attempt to advise
interested parties of the risks, uncertainties, and other
factors that affect our business, operating results, financial
condition and stock price, including without limitation the
disclosures made under the captions “Management’s
Discussion and Analysis of Financial Condition and Results of
Operations” and “Risk Factors” in this report and
in the consolidated financial statements and notes thereto
included elsewhere in this report. Furthermore, such
forward-looking statements speak only as of the date of this
report. We expressly disclaim any intent or obligation to update
any forward-looking statements after the date hereof to conform
such statements to actual results or to changes in our opinions
or expectations.
References in this report to “TranS1”, “we”,
“our”, “us”, or the “Company”
refer to TranS1 Inc.
3
Overview
We are a medical device company focused on designing, developing
and marketing products that implement our proprietary minimally
invasive surgical approach to treat degenerative disc disease
and instability affecting the lower lumbar region of the spine.
Using this TranS1 pre-sacral approach, a surgeon can access
discs in the lower lumbar region of the spine through a 1.5 cm
incision adjacent to the tailbone and can perform an entire
fusion procedure through a small tube that provides direct
access to the intervertebral space. We developed our TranS1
pre-sacral approach to allow spine surgeons to access and treat
intervertebral spaces without compromising important surrounding
soft tissue. We believe this approach enables fusion procedures
to be performed with low complication rates, low blood loss,
short hospital stays, fast recovery times and reduced pain. We
have developed and currently market two single-level fusion
products, the
AxiaLIF®
and the AxiaLIF
360°tm
and a two-level fusion product, the AxiaLIF
2Ltm,
which include the Vectre and
Avatartm
posterior fixation systems for lumbar fixation supplemental to
AxiaLIF fusion.
Our AxiaLIF product received 510(k) clearance from the
U.S. Food and Drug Administration, or FDA, in December 2004
and a CE mark in March 2005 and was commercially launched in the
United States in January 2005. Our AxiaLIF 360° product
received FDA 510(k) clearance in September 2005 and a CE mark in
March 2006 and was commercially launched in the United States in
July 2006. We received a CE mark for our AxiaLIF 2L product in
the third quarter of 2005 and began commercialization in the
European market in the fourth quarter of 2006. We received
510(k) clearance for our AxiaLIF 2L from the FDA and began
marketing this product in the United States in April 2008. In
November 2009, we commenced the limited market release of our
next generation Vectre facet screw system. Our AxiaLIF 2L+
product received FDA 510(k) clearance, and we began marketing
the product, in January 2010. In January 2010, we entered into a
partnership agreement with Life Spine, Inc. to distribute
Avatar, a minimally invasive pedicle screw system. As of
December 31, 2009, over 8,600 fusion procedures have been
performed globally using our AxiaLIF products. At
December 31, 2009, we sold our AxiaLIF products through 55
direct sales personnel and 19 independent sales agents in the
United States and 4 direct sales personnel and 14 independent
distributors internationally. For the year ended
December 31, 2009, our revenues were $29.8 million and
our net loss was $23.2 million.
Lower back pain affects over six million people annually in the
United States and is a leading cause of healthcare expenditures
globally. Our currently marketed products address the lower
lumbar spine fusion market. We believe the introduction of
minimally invasive spine procedures, such as ours, will attract
more back pain patients, and attract them earlier, to a
definitive surgical solution, thereby increasing the rate at
which the market grows.
Spine
Anatomy
The human spine is the core of the human skeleton and provides
important structural support while remaining flexible to allow
movement. It consists of 33 separate interlocking bones called
vertebrae that are connected by soft tissue and provide
stability while facilitating motion. Vertebrae are paired into
motion segments that move by means of two facet joints and one
disc. The facet joints provide stability and enable the spine to
bend and twist while the discs absorb pressures and shocks to
the vertebrae. Nerves are contained in the spinal column and run
through the foramen openings to the rest of the body.
The vertebrae are categorized into five regions: cervical,
thoracic, lumbar, sacral and coccyx. The lumbar region, which is
at the bottom of the spine and consists of five vertebrae, is
capable of limited movement, and primarily functions as support
for the body’s weight. The sacrum consists of five fused
vertebrae labeled S1 through S5 directly below the lumbar region
and that provide attachment for the hipbones as well as
protection to organs in the pelvic area. The coccyx, also known
as the tailbone, is at the end of the spine.
4
Medical
Conditions Affecting the Lower Lumbar Spine and Traditional
Treatment Alternatives
Degenerative disc disease is a common medical condition
affecting the lower lumbar spine and refers to the degeneration
of the disc from aging and repetitive stresses resulting in a
loss of flexibility, elasticity and shock-absorbing properties.
As degenerative disc disease progresses, the space between the
vertebrae narrows, which can pinch the nerves exiting the spine
and result in back pain, leg pain, numbness and loss of motor
function. This lower back pain can be overwhelming for patients
as the resulting pain can have significant physical,
psychological and financial implications.
Treatment alternatives for lower lumbar spine conditions range
from non-operative conservative therapies to highly invasive
surgical interventions. Conservative therapies are typically the
initial treatments selected by patients and physicians and they
include rest, bracing, physical therapy, chiropractics,
electrical stimulation and drugs. When conservative therapies
fail to provide adequate pain relief, surgical interventions,
including fusion procedures, may be used to address the pain. If
a patient’s disc degeneration has not progressed to a stage
requiring fusion, but has progressed beyond the stage where
conservative therapies provide pain relief, physicians may use
non-fusion surgical procedures. Non-fusion surgical procedures
utilize implants that are designed to restore disc height and
allow limited movement of the vertebrae similar to a healthy
spine in order to avoid increased pressures on adjacent
vertebrae. These procedures and implants include dynamic
stabilization devices, artificial discs and prosthetic disc
nucleuses.
Fusion procedures attempt to alleviate lower back pain by
removing problematic disc material and permanently joining
together two or more opposing vertebrae. This is done in a
manner that restores the appropriate space between the vertebrae
surrounding the degenerative disc and eliminates mobility of the
affected vertebrae. By restoring disc height and eliminating
motion, fusion attempts to prevent the pinching of the nerves
exiting the spine and thereby reducing pain.
Traditional fusion procedures typically involve an incision in
the skin, and cutting muscle or moving organs to gain access to
the spine. The degenerated disc is then removed, referred to as
a discectomy, and a rigid implant is inserted, such as a bone
graft or cage, to stabilize the diseased vertebrae. This process
is referred to as fixation. The bone graft or cage promotes the
growth of bone between the vertebrae. Surgeons often also affix
supplementary rods and screws along the spine to provide
additional stabilization while the vertebrae fuse together
during the six to eighteen months following surgery. The primary
surgical fusion procedures performed in the lower lumbar region
include: ALIF, PLIF, TLIF and XLIF.
Anterior Lumbar Interbody Fusion, or ALIF. To
perform an ALIF procedure, surgeons access the spine through an
incision on the patient’s abdomen which provides them with
optimal access to the vertebral space for performing a
discectomy and inserting bone grafts for fusion. Supporting soft
tissue and nerves are manipulated or removed to accommodate the
anterior access required by the ALIF procedure. Surgeons
commonly perform ALIF procedures in conjunction with a general
or vascular surgeon because critical vasculature and organs must
be retracted to gain access to the spine. The assistance of a
second surgeon can reduce the economics for the spine surgeon
and increase the difficulty in scheduling the surgery.
Complications associated with ALIF procedures include vascular
damage to the vena cava and aorta.
Posterior Lumbar Interbody Fusion, or PLIF. To
perform a PLIF procedure, surgeons access the spine through an
incision on the center of the patient’s back. The surgeon
then navigates through muscles and nerves to gain access to the
spine. Once at the spine, the surgeon removes bone from the
lamina to gain access to the affected disc space where a
discectomy is performed and a bone graft is placed. When
compared to ALIF procedures, PLIF procedures are generally
considered easier to perform and can achieve better nerve root
decompression in certain cases. However, the anatomy of the
spine prevents surgeons from removing the entire degenerated
disc and obtaining optimal access for insertion of an implant or
bone graft. Complications associated with PLIF procedures
include nerve damage, soft tissue damage and implant migration.
Transforaminal Lumbar Interbody Fusion, or
TLIF. TLIF procedures are performed in a similar
manner to PLIF procedures, except the surgeon accesses the spine
through a small incision slightly to the left or right of the
center of the patient’s back. After reaching the spine, the
surgeon removes a portion of the facet joint and navigates
through the foramen which provides better visualization and disc
removal capabilities than PLIF.
5
Complications associated with TLIF procedures are similar to
those found in PLIF procedures including nerve damage, soft
tissue damage and implant migration.
Extreme Lateral Interbody Fusion, or XLIF. To
perform an XLIF procedure, the surgeon accesses the spine from
the patient’s side through one or two small incisions. The
XLIF procedure is not appropriate for fusions in the L5/S1
segment because the pelvis interferes with access. While the
XLIF procedure damages less patient tissue than other fusion
procedures, we believe that there is a significant physician
learning curve.
360° Lumbar Fusion Procedure. Currently,
the most common and structurally rigid lumbar fusion surgery is
referred to as a 360° fusion and requires a second surgical
procedure immediately following an ALIF, PLIF, TLIF or XLIF
procedure. The additional procedure involves the permanent
placement of screws and rods in the back to provide additional
support while the vertebrae fuse together during the six to
eighteen months following surgery.
Limitations
of Traditional Lower Lumbar Spine Procedures
While traditional and minimally invasive fusion procedures can
be effective at treating lower back pain, common drawbacks of
these procedures include:
|
|
|
| |
•
|
Disruption to Soft Tissue and Support
Structures. Current lumbar fusion procedures
require creating a pathway from the skin to the degenerative
disc that is large enough to allow direct visualization and work
by the surgeon. It is common to cut through healthy muscle or
move critical organs, arteries, nerves and soft tissue, which
can lead to bleeding, scarring, nerve damage and bowel
disruption.
|
| |
| |
•
|
Significant Blood Loss. As a result of
undergoing current lumbar fusion procedures, patients typically
experience significant blood loss of between 100 and 1,400 cc of
blood. As a result, it is common for patients to use the
hospital’s blood supply or donate units of their own blood
before a lumbar fusion procedure to replenish any significant
blood loss.
|
| |
| |
•
|
Lengthy Operative Procedure Times. We believe
it is common for current lumbar fusion procedures to take
between 90 minutes and 4 hours to complete. With some
procedures a second surgeon may be required. Long procedure
times increase the risks of complications and blood loss. Also,
hospital and physician resources are consumed for lengthy
periods of time, which can reduce productivity and increase
costs.
|
| |
| |
•
|
Lengthy Patient Hospital Stays. Patients
remain in the hospital for an average of three days following a
lumbar fusion procedure which consumes hospital and physician
resources.
|
| |
| |
•
|
Significant Patient Recovery Time. Patients
require three to six months to recover and rehabilitate after
undergoing a lumbar fusion procedure before resuming normal
activities.
|
| |
| |
•
|
Unresolved Patient Pain. Patients may continue
to experience lower back pain after undergoing lumbar fusion
procedures even though x-rays show successful fusion has been
achieved through the growth of new bone. We believe this may be
caused by muscle dissection, implant irritation and scar tissue
that develops around the site of the surgery.
|
Our
Solution
We believe we have developed the least invasive approach for
surgeons to perform fusion and motion preserving surgeries in
the L4/L5/S1 region without the drawbacks associated with
current lumbar fusion procedures. We refer to this unique
proprietary approach as our pre-sacral approach. We have
developed and are marketing three fusion products that are
delivered using our pre-sacral approach: AxiaLIF, AxiaLIF 360
and AxiaLIF 2L.
To access the spine using our pre-sacral approach, the surgeon
creates a 1.5 cm incision adjacent to the tailbone while the
patient is lying on their stomach. The surgeon then navigates a
blunt dissecting tool a short distance along the sacrum using
imaging technologies to a spine access point near the junction
of the S1/S2 vertebral bodies. As the dissecting tool is
advanced it moves soft tissue structures, including the bowel,
to the
6
side. From this access point, a guide pin is inserted through
the bone into the disc of the lowest lumbar motion segment known
as the L5/S1 disc. A tubular dissector is inserted over the
guide pin to create a tissue-protecting working channel between
the surgical access site and the L5/S1 disc where the entire
fusion operation is then performed. This protected working
channel provides access to the interior of the disc for removal
of disc material with rotating cutters and brushes, introduction
of bone graft material with special instrumentation and
insertion of our AxiaLIF implant. The implant immediately
provides fixation and restores disc height. The AxiaLIF 2L
procedure uses the same approach to provide access for our
procedure to both the L5/S1 and L4/L5 discs. The AxiaLIF
360° procedure supplements the AxiaLIF implant with
fixation for the back of the spine. In our AxiaLIF 360°
procedure, a second incision is made approximately eight inches
above the first incision, and two facet screws are implanted.
We believe our pre-sacral approach and its associated products
provide the following benefits for patients, providers and
payors:
|
|
|
| |
•
|
Least Invasive Approach Minimizes
Complications. Our AxiaLIF products are delivered
using our unique pre-sacral approach, which we believe is the
least invasive solution for delivering fusion and
motion-preserving products to the L4/L5/S1 region. Procedures
performed utilizing our pre-sacral approach have been documented
to have favorable clinical safety profiles with complication
rates of approximately 1%.
|
| |
| |
•
|
Spinal Stability. We believe our approach is
the only spinal fusion that does not violate or cut through any
muscles or ligaments that control the stability of the spine.
|
| |
| |
•
|
Short Learning Curve. We believe that the ease
of use of our pre-sacral approach enables reduced physician
training as compared to alternative lower lumbar spine
procedures.
|
| |
| |
•
|
Low Blood Loss. We believe the least invasive
nature of our pre-sacral approach results in the average patient
losing approximately 25 to 125 cc of blood during an AxiaLIF,
AxiaLIF 2L or AxiaLIF 360° procedure, which is much lower
than current techniques and correlates to reduced pain and
faster recoveries. Given the associated low blood loss, patients
generally do not need to donate blood prior to undergoing
procedures that utilize our pre-sacral approach.
|
| |
| |
•
|
Short Patient Hospital Stays. Patients
typically stay only one night in the hospital after receiving a
procedure performed using our pre-sacral approach. In a small
percentage of cases, patients are able to return home the same
day.
|
| |
| |
•
|
Reduction in Patient Pain. We believe our
pre-sacral approach is effective at reducing lower back pain
because surrounding soft tissue is not violated which prevents
the creation of scar tissue, a leading cause of pain.
|
Our
Strategy
Our goal is to become a global leader in the treatment of
conditions affecting the L4/L5/S1 region of the lumbar spine
utilizing our pre-sacral approach and associated
instrumentation. To achieve this goal, we are pursuing the
following strategies:
|
|
|
| |
•
|
Establish our Pre-Sacral Approach as a Standard of Care for
Lower Lumbar Spine Surgery. We believe patients
commonly avoid back surgery due to its invasive nature and other
drawbacks associated with current surgical treatment options. We
expend significant resources promoting our pre-sacral approach
as the least invasive approach to lower back surgery and we
believe the advantages of our technique will enable our AxiaLIF
products to become a standard of care for the lower lumbar
region of the spine.
|
| |
| |
•
|
Focus our Sales and Marketing Infrastructure to Drive Surgeon
Adoption. We intend to continue expending
significant resources targeting spine surgeons through our sales
and marketing efforts in the United States and internationally
in order to drive the adoption of our pre-sacral approach. We
believe the ease of use, short hospital stays and reduction in
patient pain will be compelling reasons for surgeon adoption of
our technologies.
|
7
|
|
|
| |
•
|
Opportunistically Pursue Acquisitions of Complementary
Businesses and Technologies. In addition to
building our internal product development efforts, we intend to
selectively license or acquire complementary products and
technologies that we believe will enable us to leverage our
growing distribution platform.
|
Products
Our products include surgical instruments for creating a safe
and reproducible access route to the L4/L5/S1 vertebrae, fusion
implants, as well as supplemental stabilization products. We
believe our AxiaLIF implants and instruments, combined with
facet screws, provide surgeons with the tools necessary to
perform a 360° lumbar fusion in the least invasive manner
available. We sell these products to our customers in procedure
kits that include all the instruments and implants needed to
complete a lumbar fusion.
AxiaLIF Lumbar Fusion Implants. Our AxiaLIF
implant is a threaded titanium rod, called our 3D Axial Rod,
that comes in varying lengths to enable one-level L5/S1
fusions and two-level L4/L5/S1 fusions. As it is implanted,
its proprietary thread design separates the vertebrae to restore
disc height. The increased disc height relieves pressure on the
nerve, while our 3D Axial Rod provides immediate rigid fixation.
AxiaLIF 360° Implants. Our proprietary
AxiaLIF 360° implants consist of our 3D Axial Rod plus our
titanium facet screws for supplemental posterior fixation. The
two AxiaLIF 360° facet screws are implanted through a
single 1.5 cm incision in the patient’s back using our
proprietary delivery system.
TranS1 Access and Disc Preparation
Instruments. Our pre-sacral approach requires the
use of a sterile set of surgical instruments that are used to
create a safe and reproducible working channel and to prepare
the disc and vertebrae for our implant. The instrumentation
contained in the set includes stainless steel navigation tools
and tubular dissectors to create the working channel, as well as
nitinol cutters and brushes to cut and remove the degenerated
disc material and prepare the disc space for our implant and the
bone graft material.
AVATAR Pedicle Screw System. In January 2010,
we entered into a partnership agreement with Life Spine, Inc. to
distribute Avatar, a minimally invasive pedicle screw system.
Avatar can be used with our implants to provide supplemental
posterior fixation. Avatar offers cannulated pedicle screws
inserted over a guidewire to reduce muscle and tissue trauma.
Extended tabs integrated to the screws combined with a variety
of rod insertion mechanisms provide secure implantation of the
rod while minimizing tissue dissection. In-situ reduction,
compression, and distraction are achieved simply and effectively
with intuitive instrumentation.
Product
Pipeline
We have re-prioritized our product development efforts and are
pursuing products that enhance our existing product line and
those that that have a shorter pathway to regulatory clearance
and commercialization. Due to an uncertain regulatory pathway,
we have decided to put our Percutaneous Nucleus Replacement, or
PNR, project on hold in the U.S., which also impacts our Partial
Disc Replacement project, which was an enhancement of the PNR.
In the future, we believe our product offerings will be expanded
to address additional clinical applications in the surgical
treatment of conditions affecting the lower lumbar spine. Such
applications would require FDA 510(k) clearance or PMA approval,
most likely supported by safety and efficacy data from clinical
trials. We also have an active program aimed at developing tools
that will lower complications for our procedures.
Sales and
Marketing
Our sales and marketing effort primarily targets industry
leaders and high volume spine surgeons. We also market our
products at various industry conferences and through industry
organized surgical training courses. In addition, we intend to
develop and implement marketing programs targeted at potential
patients, which we believe will accelerate the demand for our
products.
In 2009, no customer accounted for 10% or more of revenues.
8
In the United States, we market and sell our products through a
combination of direct sales representatives and independent
sales agents that have allocated representatives to solicit our
products. At December 31, 2009, our U.S. sales team
included territory managers, direct sales representatives, case
coverage specialists and independent sales agents covering
specific geographic regions. We select our sales representatives
and independent sales agents based on their expertise in spine
surgery medical device sales, reputation within the surgeon
community and sales coverage. Our sales representatives receive
a base salary and a percentage of the net sales that they
generate. In January 2010, as part of our on-going effort to
best address current market opportunities, we reduced our direct
sales representatives from 55 to 45. We have taken our most
successful reps and given them more territory to grow while
ensuring that all territories are appropriately covered. Our
overriding goal is to make each of our sales reps profitable as
quickly as possible. The sales management structure remains
unchanged. The independent sales agents are compensated based on
a percentage of the net sales that they generate. We have
agreements with our independent sales agents that provide them
with an exclusive right to sell our products in their
territories, which are generally terminable upon
90 days’ written notice.
Outside of the United States, we utilize third-party
distributors and our own direct sales representatives and
agents, with support from our vice president of international
sales and our U.S. and international sales and marketing
staff, to support the commercialization of our products. Through
December 31, 2009, the majority of our international sales
have been in Europe. In 2008, we hired direct sales
representatives in Germany, and in 2009 we began direct sales
through our own sales representatives and agents in Germany,
Switzerland, Netherlands and Belgium. In 2009, 69% of our
international revenues were through third-party distributors and
31% were through our direct sales efforts.
We intend to continue to hire sales and marketing personnel as
appropriate to enable us to support the commercialization of our
products.
Surgeon
Training
We devote significant resources to training and educating
surgeons on the specialized skills involved in the proper use of
our instruments and implants. We believe that the most effective
way to introduce and build market demand for our products is by
training spine surgeons in the use of our products. We
accomplish our training objectives primarily through cadaver and
surrogate models and live case observations with surgeons
experienced in our pre-sacral approach. We supplement this
training with online didactic tutorials. After this training,
surgeons are generally able to perform unsupervised surgeries
using our pre-sacral approach. As of December 31, 2009, we
had trained over 1,150 U.S. spine surgeons and 200 surgeons
outside of the U.S. in the use of our single-level product.
Of the U.S. surgeons trained on our pre-sacral approach,
approximately 380 have performed a procedure in the
12 months ended December 31, 2009 using our pre-sacral
approach. In addition, we have trained over 250
U.S. surgeons in the use of our AxiaLIF 2L product. We
believe we have the necessary capacity to train a sufficient
number of surgeons to meet our current goals.
Third-Party
Reimbursement
In the United States, healthcare providers generally rely on
third-party payors, principally private insurers and
governmental payors such as Medicare and Medicaid, to cover and
reimburse all or part of the cost of a spine fusion surgery in
which our medical device is used. Surgeons are reimbursed for
performing the surgical procedure, while hospitals are
reimbursed for the cost of the device, all patient care related
to the fusion procedure and the overhead associated with
maintaining the facility.
Most payors follow Medicare’s Diagnosis-Related Group, or
DRG, based payment system for reimbursing facilities. Under this
model, hospitals are paid a set amount to cover the costs
associated with a fusion patient, including the cost of the
device used in the procedure. The most commonly associated DRGs
for spinal fusion are
453/454/455
(“Combined Anterior/Posterior Spinal Fusion with major
complications and comorbidities (MCC), with complications and
comorbidities (CC) or without MCC/CC”) and
459/460
(“Spinal Fusion Except Cervical with or without MCC”).
Private payors typically use Medicare DRGs as a benchmark when
setting their own reimbursement rates for facilities.
9
Surgeons use the American Medical Association’s Current
Procedural Terminology, or CPT, system to bill payors for the
AxiaLIF procedure. CPT codes describe the services and
procedures provided for patients to third-party payors so that
physicians may be reimbursed. Effective January 1, 2009,
the AMA implemented a Category III code which may describe
the work involved in treating some AxiaLIF patients. Unlike
Category I CPT codes, Category III codes do not have a set
value which physicians use as a benchmark for setting their fee.
Additionally, some payors view Category III codes as
“investigational” or “experimental” and may
not reimburse them. However, AxiaLIF adoption continues to grow
and unlike many new or novel procedures, AxiaLIF is an access
variation on the current standard of care (spinal fusion) and
surgeons should code appropriately for the work they perform
based on the unique clinical decision making, time, risk, and
diagnosis of each patient.
As the breadth and depth of peer-reviewed clinical research
regarding AxiaLIF continues to grow, we will continue to
diligently work to ensure that patients have continued access to
AxiaLIF should their doctors determine this is best clinical
solution for their condition.
Internationally, reimbursement and healthcare payment systems
vary substantially from country to country and include
single-payor, government-managed systems as well as systems in
which private payors and government-managed systems exist
side-by-side.
Our ability to achieve market acceptance or significant sales
volume in international markets we enter will be dependent in
large part on the availability of reimbursement for procedures
performed using our products under the healthcare payment
systems in such markets. A small number of countries may require
us to gather additional clinical data before recognizing
coverage and reimbursement for our products. It is our intent to
complete the requisite clinical studies and obtain coverage and
reimbursement approval in countries where it makes economic and
strategic sense to do so.
We believe that the overall escalating cost of medical products
and services has led to, and will continue to lead to, increased
pressures on the healthcare industry to reduce the costs of
products and services. We cannot assure you that government or
private third-party payors will cover and reimburse the
procedures using our products in whole or in part in the future
or that payment rates will be adequate. In addition, it is
possible that future legislation, regulation, or reimbursement
policies of third-party payors will adversely affect the demand
for our procedures and products or our ability to sell them on a
profitable basis. The unavailability or inadequacy of
third-party payor coverage or reimbursement could have a
material adverse effect on our business, operating results and
financial condition.
Competition
The medical device industry is highly competitive, subject to
rapid technological change and significantly affected by new
product introductions and market activities of other
participants. Our currently marketed products are, and any
future products we commercialize will be, subject to intense
competition. Our competitors include providers of conservative,
non-operative therapies for lower lumbar spine conditions, as
well as a number of major medical device companies that have
developed or plan to develop products for minimally invasive
spine surgery in each of our current and future product
categories. We believe that the principal competitive factors in
our markets include:
|
|
|
| |
•
|
improved outcomes for medical conditions affecting the lower
lumbar spine;
|
| |
| |
•
|
acceptance by spine surgeons;
|
| |
| |
•
|
ease of use and reliability;
|
| |
| |
•
|
product price and qualification for reimbursement;
|
| |
| |
•
|
technical leadership and superiority;
|
| |
| |
•
|
effective marketing and distribution; and
|
| |
| |
•
|
speed to market.
|
We are aware of several companies that compete or are developing
technologies in our current and future product areas. As a
result, we expect competition to remain intense. We believe that
our most significant
10
competitors are Medtronic Sofamor Danek, Johnson &
Johnson DePuy Spine, Stryker Spine, NuVasive, Zimmer Spine,
Synthes, Orthofix International, Globus Medical, Alphatec Spine
and others, many of which have substantially greater sales and
financial resources than we do. In addition, these companies may
have more established distribution networks, entrenched
relationships with physicians, and greater experience in
launching, marketing, distributing and selling products.
Our ability to compete successfully will depend on our ability
to develop proprietary products that reach the market in a
timely manner, receive adequate reimbursement and are safer,
less invasive and less expensive than alternatives available for
the same purpose. Because of the size of the potential market,
we anticipate that companies will dedicate significant resources
to developing competing products.
Research
and Development
As of December 31, 2009, our research and development team
was comprised of 11 employees who have extensive experience
in developing products to treat medical conditions affecting the
lower lumbar spine. These employees work closely with our
clinical advisors and spine surgeon customers to design and
enhance our products and approach. Our R&D spending was
$6.4 million, $4.1 million and $3.9 million for
the years ending December 31, 2009, 2008 and 2007,
respectively. Since inception, we have devoted significant
resources to develop and enhance our AxiaLIF product kits
utilizing the pre-sacral approach.
Manufacturing
and Supply
We rely on third parties to manufacture all of our products and
their components, except for our nitinol nucleus cutter blades
and nucleus cutter sheaths, which we manufacture at our
facilities in Wilmington, North Carolina. Our outsourcing
partners are manufacturers that meet FDA, International
Organization for Standardization, or ISO, and other internal
quality standards. We believe these manufacturing relationships
allow us to work with suppliers who have the best specific
competencies while we minimize our capital investment, control
costs and shorten cycle times, all of which we believe allows us
to compete with larger-volume manufacturers of spine surgery
products.
All of our products and components are assembled, packaged,
labeled and sterilized at third-party facilities in the United
States under our existing contracts requiring compliance with
Good Manufacturing Processes, or GMPs. Following receipt of
products or components from our third-party manufacturers, we
inspect, warehouse and ship the products and components at our
facilities in Wilmington or at a third-party distribution
facility in Memphis, Tennessee. We reserve the exclusive right
to inspect and assure conformance of each product and component
to our specifications. In addition, FDA or other regulatory
authorities may inspect our facilities and those of our
suppliers to ensure compliance with quality system regulations.
The majority of our instruments and implants are produced by
third-party manufacturers on precision, high-speed machine shop
equipment. However, certain of our products, components,
materials used to manufacture such products and components, and
manufacturing operations are produced, performed or supplied by
third-party specialty vendors due to their proprietary or
non-conventional nature. For example, the blades for our nucleus
cutter are made from a metal called nitinol, which is converted
into strip form by three manufacturers in the United States
known to us. We have sourced nitinol strip from two of these
vendors. The nitinol strip is then further converted for us into
cutter blanks by a scalpel blade specialty vendor. Other vendors
are available to manufacture the cutter blanks, as we may deem
desirable or necessary. We convert the cutter blanks into cutter
blades at our facilities. Our tissue extractor product is
produced for us by a supplier that specializes in wire forming
and coiling specifically for the medical device industry. A
limited number of similar vendors exist that could be used to
produce the tissue extractor product, and we believe we could
replace this supplier on reasonable terms without substantial
delay, if necessary.
We are currently working with our third-party manufacturers to
plan for our manufacturing requirements as we increase our
commercialization efforts. In most cases, we have redundant
manufacturing capability with multiple vendors and enjoy the
significant capacity this arrangement provides to us. We may
consider manufacturing certain products or product components
internally, if and when demand or quality requirements
11
make it appropriate to do so. We believe the manufacturing
capacity available to us is sufficient to meet our demands into
the foreseeable future.
Patents
and Proprietary Technology
We rely on a combination of patent, trademark, copyright, trade
secret and other intellectual property laws, nondisclosure
agreements and other measures to protect our intellectual
property rights. We believe that in order to have a competitive
advantage, we must develop and maintain the proprietary aspects
of our technologies. We require our employees, consultants and
advisors to execute confidentiality agreements in connection
with their employment, consulting or advisory relationships with
us. We also require our employees, consultants and advisors who
we expect to work on our products to agree to disclose and
assign to us all inventions conceived during the work day, using
our property or which relate to our business. We cannot provide
any assurance that employees and consultants will abide by the
confidentiality or assignment terms of these agreements. Despite
any measures taken to protect our intellectual property,
unauthorized parties may attempt to copy aspects of our products
or to obtain and use information that we regard as proprietary.
Patents
As of December 31, 2009, we had 22 issued United States
patents, 46 pending patent applications in the United States, 2
issued European patents and 96 foreign patent applications as
counterparts of U.S. cases. The issued and pending patents
cover, among other things:
|
|
|
| |
•
|
our method for performing trans-sacral procedures in the spine,
including diagnostic or therapeutic procedures, and trans-sacral
introduction of instrumentation or implants;
|
| |
| |
•
|
apparatus for conducting these procedures including access, disc
preparation and implantation including the current TranS1
instruments individually and in kit form; and
|
| |
| |
•
|
implants for fusion and motion preservation in the spine.
|
Our issued patents begin to expire in 2021 assuming timely
payment of all maintenance fees. We have multiple patents
covering unique aspects and improvements for many of our methods
and products. We do not believe that the expiration of any
single patent is likely to significantly affect our business,
operating results or prospects.
Trademarks
We own four trademark registrations in the United States and
seven trademark registrations in the European Union. We also own
eight pending trademark applications in the United States, two
pending trademark applications in the European Union and eight
pending trademark applications in Canada.
Government
Regulation
Our products are medical devices subject to extensive regulation
by the FDA and other U.S. federal and state regulatory
bodies and comparable authorities in other countries. To ensure
that medical products distributed domestically and
internationally are safe and effective for their intended use,
FDA and comparable authorities in other countries have imposed
regulations that govern, among other things, the following
activities that we or our partners perform and will continue to
perform:
|
|
|
| |
•
|
product design and development;
|
| |
| |
•
|
registration and listing;
|
| |
| |
•
|
product testing (preclinical and clinical);
|
| |
| |
•
|
product manufacturing;
|
| |
| |
•
|
product labeling;
|
| |
| |
•
|
product storage;
|
12
|
|
|
| |
•
|
premarket clearance or approval;
|
| |
| |
•
|
advertising and promotion;
|
| |
| |
•
|
product marketing, sales and distribution; and
|
| |
| |
•
|
post-market surveillance, including reporting deaths or serious
injuries related to products and certain product malfunctions.
|
FDA’s
Premarket Clearance and Approval Requirements
Unless an exemption applies, each medical device we wish to
commercially distribute in the United States will require either
prior 510(k) clearance or prior premarket approval from the FDA.
The FDA classifies medical devices into one of three classes.
Devices deemed to pose lower risk are placed in either
class I or II, which in many cases requires the
manufacturer to submit to the FDA a premarket notification or
510(k) submission requesting permission for commercial
distribution. This process is known as requesting 510(k)
clearance. Some low risk devices are exempt from this
requirement. Devices deemed by the FDA to pose the greatest
risk, such as many life-sustaining, life-supporting or
implantable devices, or devices deemed not substantially
equivalent to a legally marketable device, are placed in
class III, requiring a PMA. Our current commercial products
are class II devices marketed under FDA 510(k) premarket
clearance. Both premarket clearance and PMA applications are
subject to the payment of user fees, paid at the time of
submission for FDA review.
510(k)
Clearance Pathway
To obtain 510(k) clearance, we must submit a premarket
notification demonstrating that the proposed device is
substantially equivalent to a legally marketable device not
requiring a PMA. Although statutorily mandated to clear or deny
a 510(k) premarket notification within 90 days of
submission of the application, FDA’s 510(k) clearance
pathway usually takes from three to twelve months, based on
requests for additional information by FDA, but it can take
significantly longer. Additional information can include
clinical data to make a determination regarding substantial
equivalence.
After a device receives 510(k) clearance, any modification that
could significantly affect its safety or effectiveness, or that
would constitute a new or major change in its intended use, will
require a new 510(k) clearance or, depending on the
modification, require a PMA. The FDA requires each manufacturer
to determine whether the proposed change requires submission of
a 510(k), or a PMA, but the FDA can review any such decision and
can disagree with a manufacturer’s determination. If the
FDA disagrees with a manufacturer’s determination, the FDA
can require the manufacturer to cease marketing
and/or
recall the modified device until 510(k) clearance or a PMA is
obtained. If the FDA requires us to seek 510(k) clearance or a
PMA for any modifications to a previously cleared product, we
may be required to cease marketing or recall the modified device
until we obtain this clearance or approval. Also, in these
circumstances, we may be subject to significant regulatory fines
or penalties. We have made and plan to continue to make
additional product enhancements to our AxiaLIF, AxiaLIF 2L
(including AxiaLIF 2L+) and AxiaLIF 360° (including Vectre)
products that we believe do not require new 510(k) clearances.
Premarket
Approval Pathway
A PMA application must be submitted if the device is not exempt
and cannot be cleared through the 510(k) process. The PMA
application process is generally more costly and time consuming
than the 510(k) process. A PMA application must be supported by
extensive data including, but not limited to, technical,
preclinical, clinical trials, manufacturing and labeling to
demonstrate to the FDA’s satisfaction the safety and
effectiveness of the device for its intended use.
After a PMA application is sufficiently complete, the FDA will
accept the application and begin an in-depth review of the
submitted information. By statute, the FDA has 180 days to
review the “accepted application”, although,
generally, review of the application can take between one and
three years, but it may take significantly longer. During this
review period, the FDA may request additional information or
13
clarification of information already provided. Also during the
review period, an advisory panel of experts from outside the FDA
may be convened to review and evaluate the application and
provide recommendations to the FDA as to the approvability of
the device. In addition, the FDA will conduct a preapproval
inspection of the manufacturing facility to ensure compliance
with quality system regulations. New PMA applications or PMA
application supplements are required prior to marketing for
product modifications that affect the safety and efficacy of the
device. PMA supplements often require submission of the same
type of information as a PMA application, except that the
supplement is limited to information needed to support any
changes from the device covered by the original PMA application,
and may not require as extensive clinical data or the convening
of an advisory panel. None of our products are currently
approved under a PMA but devices in development may
require it.
Clinical
Trials
Clinical trials are almost always required to support a PMA
application and are sometimes required for a 510(k) premarket
notification. In the U.S., these trials require submission of an
application for an investigational device exemption, or IDE. The
IDE application must be supported by appropriate data, such as
animal and laboratory testing results, showing that it is safe
to test the device in humans and that the testing protocol is
scientifically sound. The IDE application must be approved in
advance by the FDA for a specified number of patients, unless
the product is deemed a non-significant risk device and eligible
for more abbreviated IDE requirements. Clinical trials for a
significant risk device may begin once the IDE application is
approved by the FDA and the appropriate institutional review
boards at the clinical trial sites. Future clinical trials of
our motion preservation designs will require that we obtain an
IDE from the FDA prior to commencing clinical trials and that
the trial be conducted under the oversight of an institutional
review board at the clinical trial site. Our clinical trials
must be conducted in accordance with FDA regulations and federal
and state regulations concerning human subject protection,
including informed consent and healthcare privacy and financial
disclosure by clinical investigators. A clinical trial may be
suspended by FDA or the investigational review board at any time
for various reasons, including a belief that the risks to the
study participants outweigh the benefits of participation in the
study. Even if a study is completed, the results of our clinical
testing may not demonstrate the safety and efficacy of the
device, or may be equivocal or otherwise not be sufficient to
obtain clearance or approval of one of our products. Similarly,
in Europe the clinical study must be approved by the local
ethics committee and in some cases, including studies of
high-risk devices, by the Competent Authority in the applicable
country.
Pervasive
and Continuing FDA Regulation
After a device is placed on the market, numerous FDA and other
regulatory requirements continue to apply. These include:
|
|
|
| |
•
|
quality system regulation, which requires manufacturers,
including third-party contract manufacturers, to follow
stringent design, testing, control, documentation, and other
quality assurance controls, during all aspects of the
manufacturing process;
|
| |
| |
•
|
establishment registration and listing;
|
| |
| |
•
|
labeling regulations, and FDA prohibitions against the promotion
of products for uncleared or unapproved “off-label”
uses;
|
| |
| |
•
|
medical device reporting obligations, which require that
manufacturers submit reports to the FDA if information
reasonably suggests their device (i) may have caused or
contributed to a death or serious injury, or
(ii) malfunctioned and the device or a similar company
device would likely cause or contribute to a death or serious
injury if the malfunction were to recur; and
|
| |
| |
•
|
other post-market surveillance requirements, which apply when
necessary to protect the public health or to provide additional
safety and effectiveness data for the device.
|
14
We must register and list with FDA as medical device
manufacturers and must obtain all necessary state permits or
licenses to operate our business. As manufacturers, we are
subject to announced and unannounced inspections by FDA to
determine our compliance with quality system regulation and
other regulations. We have not yet been inspected by the FDA. We
believe that we are in substantial compliance with quality
system regulation and other regulations.
Failure to comply with applicable regulatory requirements can
result in enforcement action by the FDA, which may include,
among other things, any of the following sanctions:
|
|
|
| |
•
|
warning letters, fines, injunctions, consent decrees and civil
penalties;
|
| |
| |
•
|
repair, replacement, refunds, recall or seizure of our products;
|
| |
| |
•
|
operating restrictions, partial suspension or total shutdown of
production;
|
| |
| |
•
|
refusing our request for 510(k) clearance or premarket approval
of new products, new intended uses or other modifications to
existing products;
|
| |
| |
•
|
withdrawing or suspending premarket approvals that are already
granted; and
|
| |
| |
•
|
criminal prosecution.
|
We are subject to announced and unannounced inspections by the
FDA and these inspections may include the manufacturing
facilities of our subcontractors.
Fraud
and Abuse
We may directly or indirectly be subject to various federal and
state laws pertaining to healthcare fraud and abuse, including
anti-kickback laws. In particular, the federal healthcare
program anti-kickback statute prohibits persons from knowingly
and willfully soliciting, offering, receiving or providing
remuneration, directly or indirectly, in exchange for or to
induce either the referral of an individual, or the furnishing,
arranging for or recommending a good or service, for which
payment may be made in whole or part under federal healthcare
programs, such as the Medicare and Medicaid programs. The
anti-kickback statute is broad and prohibits many arrangements
and practices that are lawful in businesses outside of the
healthcare industry. In implementing the statute, the Office of
Inspector General, or OIG, has issued a series of regulations,
known as the “safe harbors,” which began in July 1991.
These safe harbors set forth provisions that, if all their
applicable requirements are met, will assure healthcare
providers and other parties that they will not be prosecuted
under the anti-kickback statute. The failure of a transaction or
arrangement to fit precisely within one or more safe harbors
does not necessarily mean that it is illegal or that prosecution
will be pursued. However, conduct and business arrangements that
do not fully satisfy all requirements of an applicable safe
harbor may result in increased scrutiny by government
enforcement authorities such as the OIG. Penalties for
violations of the federal anti-kickback statute include criminal
penalties and civil sanctions such as fines, imprisonment and
possible exclusion from Medicare, Medicaid and other federal
healthcare programs.
The federal False Claims Act prohibits persons from knowingly
filing or causing to be filed a false claim to, or the knowing
use of false statements to obtain payment from, the federal
government. Suits filed under the False Claims Act, known as
“qui tam” actions, can be brought by any individual on
behalf of the government. These individuals, sometimes known as
“relators” or, more commonly, as
“whistleblowers”, may share in any amounts paid by the
entity to the government in fines or settlement. The number of
filings of qui tam actions has increased significantly in recent
years, causing more healthcare companies to have to defend a
False Claim action. If an entity is determined to have violated
the federal False Claims Act, it may be required to pay up to
three times the actual damages sustained by the government, plus
civil penalties of between $5,500 and $11,000 for each separate
false claim. Various states have also enacted similar laws
modeled after the federal False Claims Act which apply to items
and services reimbursed under Medicaid and other state programs,
or, in several states, apply regardless of the payor.
The Health Insurance Portability and Accountability Act of 1996,
or HIPAA, created two new federal crimes: healthcare fraud and
false statements relating to healthcare matters. The healthcare
fraud statute
15
prohibits knowingly and willfully executing a scheme to defraud
any healthcare benefit program, including private payors. A
violation of this statute is a felony and may result in fines,
imprisonment or exclusion from government sponsored programs.
The false statements statute prohibits knowingly and willfully
falsifying, concealing or covering up a material fact or making
any materially false, fictitious or fraudulent statement in
connection with the delivery of or payment for healthcare
benefits, items, or services. A violation of this statute is a
felony and may result in fines or imprisonment.
We are also subject to certain state laws that are analogous to
each of the federal laws summarized above, such as anti-kickback
and false claims laws that may apply to items or services
reimbursed by any third-party payor, including commercial
insurers, and state laws governing the privacy of certain health
information, many of which differ from each other in significant
ways and often are not preempted by HIPAA, thus complicating
compliance efforts.
While we have adopted comprehensive compliance programs to
attempt to comply with these laws and regulations, if any of our
operations are found to have violated or be in violation of any
of the laws described above and other applicable state and
federal fraud and abuse laws, we may be subject to penalties,
among them being civil and criminal penalties, damages, fines,
exclusion from government healthcare programs, and the
curtailment or restructuring of our operations.
International
International sales of medical devices are subject to foreign
government regulations, which vary substantially from country to
country. The time required to obtain approval by a foreign
country may be longer or shorter than that required for FDA
clearance or approval, and the requirements may differ.
The European Union, which consists of 27 of the major countries
in Europe, has adopted numerous directives and standards
regulating the design, manufacture, clinical trials, labeling,
and adverse event reporting for medical devices. Other
countries, such as Switzerland, have voluntarily adopted laws
and regulations that mirror those of the European Union with
respect to medical devices. Devices that comply with the
requirements of a relevant directive will be entitled to bear CE
conformity marking and, accordingly, can be commercially
distributed throughout the member states of the European Union,
and other countries that comply with or mirror these directives.
The method of assessing conformity varies depending on the type
and class of the product, but normally involves a combination of
self-assessment by the manufacturer and a third-party assessment
by a “Notified Body,” an independent and neutral
institution appointed to conduct conformity assessments. This
third-party assessment consists of audits of the
manufacturer’s quality system. An assessment by a Notified
Body in one country within the European Union is required for
each product in order for a manufacturer to commercially
distribute the product throughout the European Union. Compliance
with voluntary harmonizing standards ISO 9001 and ISO 13845
issued by the ISO establishes the presumption of conformity with
the essential requirements for a CE mark. In August 2004, our
quality system was initially certified by Intertek ETL-Semko, a
Notified Body, under the European Union Medical Device Directive
to be in compliance with ISO standards 9001:2000 and ISO
13485:2003. The system was recertified in September 2007 and is
due for further recertification in September of 2010.
Employees
As of December 31, 2009, we had 148 employees, most of
whom were full-time employees, with 95 employees in
U.S. sales, marketing, customer service and training,
8 employees in international sales, marketing and training,
9 employees in manufacturing, 11 employees in research
and development, 15 employees in general and administrative
and 10 employees in clinical, regulatory and quality
assurance. We believe that our future success will depend in
part on our continued ability to attract, hire and retain
qualified personnel. None of our employees are represented by a
labor union, and we believe our employee relations are good.
16
General
Information
We were incorporated in Delaware in May 2000 under the name
“aXiaMed, Inc.” and changed our name to “TranS1
Inc.” in February 2003. Our principal executive office is
located at 301 Government Center Drive, Wilmington, North
Carolina 28403 and our telephone number is
(910) 332-1700.
Our website is located at www.trans1.com. The information
on, or that can be accessed through, our website is not
incorporated by reference into this report and should not be
considered to be a part of this report.
We make our annual reports on
Form 10-K,
quarterly reports on
Form 10-Q,
current reports on
Form 8-K
and amendments to these reports available on our website, at
www.trans1.com, free of charge as soon as practicable
after filing with the U.S. Securities and Exchange
Commission, or SEC.
All such reports are also available free of charge via EDGAR
through the SEC website at www.sec.gov. In addition, the public
may read and copy materials filed by us with the SEC at the
SEC’s public reference room located at 100 F St., NE,
Washington, D.C., 20549. Information regarding operation of
the SEC’s public reference room can be obtained by calling
the SEC at
1-800-SEC-0330.
Investing in our common stock involves a high degree of risk.
You should carefully consider the following risk factors, as
well as the other information contained in this report, before
deciding whether to invest in shares of our common stock. If any
of the following risks actually occur, our business, financial
condition, operating results and prospects would suffer. In that
case, the trading price of our common stock would likely decline
and you might lose all or part of your investment in our common
stock. The risks described below are not the only ones we face.
Additional risks that we currently do not know about or that we
currently believe to be immaterial may also impair our
operations and business results.
Risks
Related to Our Business
To be
commercially successful, spine surgeons must accept that our
products are a safe and effective alternative to existing
surgical treatments of certain spine disorders.
Our revenue is derived entirely from sales of our AxiaLIF
products and related surgical instruments. We expect that sales
of our AxiaLIF products will continue to account for a
substantial portion of our revenues for the foreseeable future.
We believe spine surgeons may not widely adopt our products
unless they determine, based on experience, long-term clinical
data and published peer reviewed journal articles, that our
products provide a safe and effective alternative to
conventional procedures used to treat certain spine disorders.
Spine surgeons may be slow to adopt our technology for the
following reasons, among others:
|
|
|
| |
•
|
lack of long-term clinical data supporting additional patient
benefits;
|
| |
| |
•
|
lack of experience with our products;
|
| |
| |
•
|
lack of evidence supporting cost savings of our procedure over
existing surgical alternatives;
|
| |
| |
•
|
perceived liability risks generally associated with the use of
new products and procedures;
|
| |
| |
•
|
training time required to use a new product; and
|
| |
| |
•
|
availability of adequate coverage and reimbursement for
hospitals and surgeons.
|
If we are unable to effectively demonstrate to spine surgeons
the benefits of our products as compared to existing surgical
treatments of spine disorders and our products fail to achieve
market acceptance, our future revenues will be adversely
impacted. In addition, we believe recommendations and support of
our products by influential spine surgeons are essential for
market acceptance and adoption. If we do not receive support
from these spine surgeons or do not have favorable long-term
clinical data, spine surgeons may not use our products and our
future revenues will be harmed and our stock price would likely
decline.
17
The
efficacy of our products is not yet supported by long-term
clinical data and may therefore prove to be less effective than
initially thought.
We obtained 510(k) clearance to manufacture, market and sell all
of our currently U.S. marketed products from the FDA. The
FDA’s 510(k) clearance process is less costly and rigorous
than the PMA process and requires less supporting clinical data.
As a result, we currently lack the breadth of published
long-term clinical data supporting the efficacy of our AxiaLIF
and AxiaLIF 360° products and the benefits they offer that
might have been generated in connection with the PMA process. In
addition, we may determine from post-market experience that
certain patient characteristics, such as age or preexisting
medical conditions, could affect fusion rates, which could lead
to misleading or contradictory data on the efficacy of our
products. For these reasons, spine surgeons may be slow to adopt
our products. Also, we may not be able to generate the
comparative data that our competitors have or are generating and
we may be subject to greater regulatory and product liability
risks. Further, any long-term safety or efficacy data we
generate may not be consistent with our existing data and may
demonstrate less favorable safety or efficacy. These results
could reduce demand for our products, significantly reduce our
ability to achieve expected revenues and could prevent us from
becoming profitable. Moreover, if future results and experience
indicate that our products cause unexpected or serious
complications or other unforeseen negative effects, we could be
subject to significant legal and regulatory liability and harm
to our business reputation.
The
demand for our products and the prices which customers and
patients are willing to pay for our products depend upon the
ability of our customers to obtain adequate third-party coverage
and reimbursement for their purchases of our
products.
Sales of our products depend in part on the availability of
adequate coverage and reimbursement from governmental and
private payors. In the United States, healthcare providers that
purchase our products generally rely on third-party payors,
principally Medicare, Medicaid and private health insurance
plans, to pay for all or a portion of the costs and fees
associated with the AxiaLIF procedure. Medicare coverage and
reimbursement policies are developed by the Centers for Medicare
and Medicaid Services, or CMS, the federal agency responsible
for administering the Medicare program, and its contractors. In
carrying out its responsibilities, the Agency must adhere to
relevant terms of the Social Security Act, and federal
regulations to ensure that it is complying with applicable
Medicare law, regulations and guidance. For a wide variety of
reasons, including the fact that the law permits coverage and
reimbursement for “medically necessary” procedures
approved by the Food and Drug Administration, AxiaLIF has strong
justification for coverage under the Medicare Program. However,
even when a product meets some of the criteria typically relied
upon to receive reimbursement under the Medicare Program, it
still must be “described adequately” under an American
Medical Association Current Procedural Terminology, or CPT code,
(typically used to describe services performed by physicians) or
included within a Diagnosis-Related Group payment (broad payment
categories used to reimburse hospitals).
As a result of some uncertainty regarding the CPT descriptions
used to describe the AxiaLIF surgery, some physicians have found
it more difficult to get reimbursed for the procedure. Moreover,
since many commercial payors rely upon Medicare policies as a
framework to establish commercial payment, some of the
uncertainty experienced by providers in receiving reimbursement
may not only negatively impact Medicare, and Medicaid
utilization, but commercial purchases as well. Accordingly, any
delays in obtaining, or an inability to clarify the
reimbursement policies for procedures using our products could
significantly affect the acceptance of our products and have a
material adverse effect on our business. If uncertainty remains,
or the reimbursement amount for a procedure declines, physicians
may revert to other fusion surgeries where reimbursement is
higher or more certain. Additionally, third-party payors
continue to review their coverage policies carefully for
existing and new therapies and can, without notice, deny
coverage for treatments that include the use of our products.
Our business would be negatively impacted to the extent any such
changes reduce reimbursement for our products.
With respect to coverage and reimbursement outside of the United
States, reimbursement systems in international markets vary
significantly by country, and by region within some countries,
and reimbursement approvals must be obtained on a
country-by-country
basis and can take up to 18 months, or longer. Many
18
international markets have government-managed healthcare systems
that govern reimbursement for new devices and procedures. In
most markets, there are private insurance systems as well as
government-managed systems. Additionally, some foreign
reimbursement systems provide for limited payments in a given
period and therefore result in extended payment periods.
Reimbursement in international markets may require us to
undertake country-specific reimbursement activities, including
additional clinical studies, which could be time consuming,
expensive and may not yield acceptable reimbursement rates.
Furthermore, healthcare costs have risen significantly over the
past decade. There have been and may continue to be proposals by
legislators, regulators and third-party payors to contain these
costs. These cost-control methods include managed care models,
prospective payment systems, capitated rates, group purchasing,
redesign of benefits, pre-authorizations or second opinions
prior to major surgery, encouragement of healthier lifestyles
and exploration of more cost-effective methods of delivering
healthcare. Healthcare providers may also attempt to control
costs by authorizing fewer elective surgical procedures or by
requiring the use of the least expensive devices possible. These
cost-control methods also potentially limit the amount which
healthcare providers may be willing to pay for medical devices.
In addition, in the United States, no uniform policy of coverage
and reimbursement for medical technology exists among all
payors. Therefore, coverage of and reimbursement for medical
technology can differ significantly from payor to payor. The
continuing efforts of third-party payors, whether governmental
or commercial, whether inside the United States or outside, to
contain or reduce these costs, combined with closer scrutiny of
such costs, could restrict our customers’ ability to obtain
adequate coverage and reimbursement from these third-party
payors. The cost containment measures that healthcare providers
are instituting both in the United States and internationally
could harm our business by adversely affecting the demand for
our products or the price at which we can sell our products.
Our
future growth depends on increasing physician awareness of our
pre-sacral approach and our related products for appropriate
treatment, intervention and referral.
We target our sales and education efforts to spine surgeons.
However, the initial point of contact for many patients may be
primary care physicians who commonly treat patients experiencing
lower lumbar spine pain. We believe that we must educate
physicians to change their screening and referral practices. If
we do not educate referring physicians about lower lumbar spine
conditions in general, and the existence of the pre-sacral
approach and our related products in particular, they may not
refer patients who are candidates for the procedures utilizing
our pre-sacral approach to spine surgeons, and those patients
may go untreated or receive conservative, non-operative
therapies. If we are not successful in educating physicians
about screening for lower lumbar spine conditions or about
referral opportunities, our ability to increase our revenue may
be impaired.
We
have incurred losses since inception and we expect to incur
increasing losses for the foreseeable future. We may never
achieve or sustain profitability.
We were incorporated in May 2000 and began commercial sales of
our products in early 2005. We have incurred net losses since
our inception and through December 31, 2009, we had an
accumulated deficit of $71.1 million. To date, we have
financed our operations primarily through sales of our equity
securities and have devoted substantially all of our resources
to research and development of our products and the commercial
launch of our AxiaLIF products. We expect our expenses to
increase significantly in connection with our additional
clinical trials and research and development activities, as well
as to support the expansion of our sales and marketing efforts.
As a result, we expect to continue to incur significant
operating losses for the foreseeable future. These losses will
continue to have an adverse effect on our stockholders’
equity and we may never achieve or sustain profitability.
19
We are
in a highly competitive market segment, which is subject to
rapid technological change. If our competitors are better able
to develop and market products that are safer, more effective,
less costly or otherwise more attractive than any products that
we may develop, our ability to generate revenue will be reduced
or eliminated.
The market for treatment of spine disorders is highly
competitive and subject to rapid and profound technological
change. Our success depends, in part, upon our ability to
maintain a competitive position in the development of
technologies and products for use in the treatment of spine
disorders. We face competition from both established and
development stage companies. Many of the companies developing or
marketing competing products are publicly traded or are
divisions of publicly-traded companies, and these companies
enjoy several competitive advantages, including:
|
|
|
| |
•
|
greater financial and human resources for product development,
sales and marketing and patent litigation;
|
| |
| |
•
|
significantly greater name recognition;
|
| |
| |
•
|
established relationships with spine surgeons, customers and
third-party payors;
|
| |
| |
•
|
additional lines of products, and the ability to offer rebates
or bundle products to offer greater discounts or incentives to
gain a competitive advantage;
|
| |
| |
•
|
established sales and marketing, and distribution
networks; and
|
| |
| |
•
|
greater experience in conducting research and development,
manufacturing, clinical trials, preparing regulatory submissions
and obtaining regulatory clearance or approval for products and
marketing approved products.
|
Our competitors may develop and patent processes or products
earlier than us, obtain regulatory clearance or approvals for
competing products more rapidly than us, and develop more
effective or less expensive products or technologies that render
our technology or products obsolete or non-competitive. We also
compete with our competitors in recruiting and retaining
qualified scientific and management personnel, establishing
clinical trial sites and patient enrollment in clinical trials,
as well as in acquiring technologies and technology licenses
complementary to our products or advantageous to our business.
If our competitors are more successful than us in these matters,
our business may be harmed.
Our
failure to continue building effective sales and marketing
capabilities for our products could significantly impair our
ability to increase sales of our products.
We commercially launched our AxiaLIF single-level product in
2005, our AxiaLIF 360° product in 2006, our AxiaLIF 2L
product in 2008 and our AxiaLIF 2L+ product in 2010, and we have
limited experience marketing and selling our products. We
utilize a hybrid model of independent sales agents and direct
sales representatives for product sales in the United States and
rely on third-party distributors, direct sales representatives
and agents for international sales. As of December 31,
2009, we employed 55 direct sales representatives in the United
States and 4 direct sales representatives in Europe. In January
2010, we reduced our U.S. force to 45 representatives to
better address current market opportunities. We have limited
experience managing a direct sales force, which can be an
expensive and time consuming process. If we are unable to
efficiently manage those individuals, our sales will suffer. We
also rely on marketing arrangements with independent sales
agents in the United States and independent distributors in
Europe, in particular their sales and service expertise and
relationships with the customers in the marketplace. We do not
control, nor monitor the marketing practices of our independent
sales agents or distributors and they may not be successful in
implementing our marketing plans or complying with applicable
laws regarding marketing practices. Independent distributors and
sales agents may terminate their relationship with us, or devote
insufficient sales efforts to our products. Our failure to
maintain our existing relationships with our independent sales
agents or distributors, or our failure to recruit and retain
additional skilled independent sales distributors and sales
agents or directly-employed sales professionals, could have an
adverse effect on our operations.
20
Our
future success depends on our ability to develop, receive
regulatory clearance or approval, and introduce new products or
product enhancements that will be accepted by the market in a
timely manner.
It is important to our business that we continue to build a more
complete product offering for treatment of spine disorders. As
such, our success will depend in part on our ability to develop
and introduce new products and enhancements to our existing
products to keep pace with the rapidly changing spine market.
However, we may not be able to successfully develop and obtain
regulatory clearance or approval for product enhancements, or
new products or our future products, or these products may not
be accepted by spine surgeons or the payors who financially
support many of the procedures performed with our products.
The success of any new product offering or enhancement to an
existing product will depend on several factors, including our
ability to:
|
|
|
| |
•
|
properly identify and anticipate spine surgeon and patient needs;
|
| |
| |
•
|
develop and introduce new products or product enhancements in a
timely manner;
|
| |
| |
•
|
avoid infringing upon the intellectual property rights of third
parties;
|
| |
| |
•
|
demonstrate, if required, the safety and efficacy of new
products with data from preclinical studies and clinical trials;
|
| |
| |
•
|
obtain the necessary regulatory clearances or approvals for new
products or product enhancements;
|
| |
| |
•
|
be fully FDA-compliant with marketing of new devices or modified
products;
|
| |
| |
•
|
provide adequate training to potential users of our products;
|
| |
| |
•
|
receive adequate coverage and reimbursement for procedures
performed with our products; and
|
| |
| |
•
|
develop an effective and FDA-compliant, dedicated marketing and
distribution network.
|
If we do not develop new products or product enhancements in
time to meet market demand or if there is insufficient demand
for these products or enhancements, our results of operations
will suffer.
If
clinical trials of our current or future product candidates do
not produce results necessary to support regulatory clearance or
approval in the United States or elsewhere, we will be unable to
commercialize these products.
We have several product candidates in our development pipeline
which might require a PMA from the FDA. A PMA application must
be supported by extensive information including, technical data,
preclinical and clinical trial data, and manufacturing and
labeling information to demonstrate to the FDA’s
satisfaction the safety and effectiveness of the device for its
intended use. As a result, to receive regulatory approval for
our products requiring PMA approval, we must conduct, at our own
expense, adequate and well controlled clinical trials to
demonstrate efficacy and safety in humans for their intended
uses. Clinical testing is expensive, typically takes many years
and has an uncertain outcome. The initiation and completion of
any of these studies may be prevented, delayed or halted for
numerous reasons, including, but not limited to, the following:
|
|
|
| |
•
|
the FDA, institutional review boards or other regulatory
authorities do not approve a clinical study protocol, force us
to modify a previously approved protocol, or place a clinical
study on hold;
|
| |
| |
•
|
patients do not enroll in, or enroll at the expected rate, or
complete a clinical study;
|
| |
| |
•
|
patients or investigators do not comply with study protocols;
|
| |
| |
•
|
patients do not return for post-treatment
follow-up at
the expected rate;
|
| |
| |
•
|
patients experience serious or unexpected adverse side effects
for a variety of reasons that may or may not be related to our
products such as the advanced stage of co-morbidities that may
exist at the time of treatment, causing a clinical study to be
put on hold;
|
| |
| |
•
|
sites participating in an ongoing clinical study may withdraw,
requiring us to engage new sites;
|
21
|
|
|
| |
•
|
difficulties or delays associated with bringing additional
clinical sites on-line;
|
| |
| |
•
|
third-party clinical investigators decline to participate in our
clinical studies, do not perform the clinical studies on the
anticipated schedule or consistent with the investigator
agreement, clinical study protocol, good clinical practices, and
other FDA and Institutional Review Board requirements;
|
| |
| |
•
|
third-party organizations do not perform data collection and
analysis in a timely or accurate manner;
|
| |
| |
•
|
regulatory inspections of our clinical studies require us to
undertake corrective action or suspend or terminate our clinical
studies;
|
| |
| |
•
|
changes in U.S. federal, state, or foreign governmental
statutes, regulations or policies;
|
| |
| |
•
|
interim results are inconclusive or unfavorable as to immediate
and long-term safety or efficacy; or
|
| |
| |
•
|
the study design is inadequate to demonstrate safety and
efficacy.
|
Clinical failure can occur at any stage of the testing. Our
clinical trials may produce negative or inconclusive results,
and we may decide, or regulators may require us, to conduct
additional clinical
and/or
non-clinical testing in addition to those we have planned. Our
failure to adequately demonstrate the efficacy and safety of any
of our devices would prevent receipt of regulatory clearance or
approval and, ultimately, the commercialization of that device.
Our
international operations subject us to certain operating risks,
which could adversely impact our net revenues, results of
operations and financial condition.
Sales of our products outside the United States represented 5.9%
of our revenue in 2009. Through December 31, 2009, we have
sold our products in the following countries outside of the
United States: United Kingdom, Italy, Austria, Australia,
Germany, Switzerland, Turkey, the Netherlands, Belgium, Israel,
Denmark, Spain, Greece, Hong Kong, Czech Republic, Slovenia,
Japan and Singapore. The sale and shipment of our products
across international borders, as well as the purchase of
components and products from international sources, subject us
to extensive U.S. and foreign governmental trade, import
and export, and custom regulations and laws. Compliance with
these regulations is costly and exposes us to penalties for
non-compliance. Other laws and regulations that can
significantly impact us include various anti-bribery laws,
including the U.S. Foreign Corrupt Practices Act and
anti-boycott laws. Any failure to comply with applicable legal
and regulatory obligations could impact us in a variety of ways
that include, but are not limited to, significant criminal,
civil and administrative penalties, including imprisonment of
individuals, fines and penalties, denial of export privileges,
seizure of shipments, restrictions on certain business
activities, and exclusion or debarment from government
contracting. Also, the failure to comply with applicable legal
and regulatory obligations could result in the disruption of our
shipping and sales activities.
In addition, many of the countries in which we sell our products
are, to some degree, subject to political, economic or social
instability. Our international operations expose us and our
distributors to risks inherent in operating in foreign
jurisdictions. These risks include:
|
|
|
| |
•
|
the imposition of additional U.S. and foreign governmental
controls or regulations;
|
| |
| |
•
|
the imposition of costly and lengthy new export licensing
requirements;
|
| |
| |
•
|
the imposition of U.S. or international sanctions against a
country, company, person or entity with whom we do business that
would restrict or prohibit continued business with the
sanctioned country, company, person or entity;
|
| |
| |
•
|
economic instability;
|
| |
| |
•
|
a shortage of high-quality sales people and distributors;
|
| |
| |
•
|
changes in third-party reimbursement policies that may require
some of the patients who receive our products to directly absorb
medical costs or that may necessitate the reduction of the
selling prices of our products;
|
22
|
|
|
| |
•
|
changes in duties and tariffs, license obligations and other
non-tariff barriers to trade;
|
| |
| |
•
|
the imposition of new trade restrictions;
|
| |
| |
•
|
the imposition of restrictions on the activities of foreign
agents, representatives and distributors;
|
| |
| |
•
|
scrutiny of foreign tax authorities which could result in
significant fines, penalties and additional taxes being imposed
on us;
|
| |
| |
•
|
pricing pressure that we may experience internationally;
|
| |
| |
•
|
laws and business practices favoring local companies;
|
| |
| |
•
|
longer payment cycles;
|
| |
| |
•
|
difficulties in maintaining consistency with our internal
guidelines;
|
| |
| |
•
|
difficulties in enforcing agreements and collecting receivables
through certain foreign legal systems; and
|
| |
| |
•
|
difficulties in enforcing or defending intellectual property
rights.
|
Any of these factors may adversely impact our operations. Our
international sales are predominately in Europe. In Europe,
healthcare regulation and reimbursement for medical devices vary
significantly from country to country. This changing environment
could adversely affect our ability to sell our products in some
European countries, which could negatively affect our results of
operations.
The
use, misuse or off-label use of our products may harm our image
in the marketplace or result in injuries that lead to product
liability suits, which could be costly to our business or result
in FDA sanctions if we are deemed to have engaged in such
promotion.
Our currently marketed products have been cleared by the
FDA’s 510(k) clearance process for use under specific
circumstances for the treatment of certain lower lumbar spine
conditions. We cannot, however, prevent a physician from using
our products or procedure outside of those indications cleared
for use, known as off-label use. There may be increased risk of
injury if physicians attempt to use our products off-label. We
train our sales force not to promote our products for off-label
uses. Furthermore, the use of our products for indications other
than those indications for which our products have been cleared
by the FDA may not effectively treat such conditions, which
could harm our reputation in the marketplace among physicians
and patients. Physicians may also misuse our products or use
improper techniques if they are not adequately trained,
potentially leading to injury and an increased risk of product
liability. If our products are misused or used with improper
technique, we may become subject to costly litigation by our
customers or their patients. Product liability claims could
divert management’s attention from our core business, be
expensive to defend and result in sizable damage awards against
us that may not be covered by insurance. If we are deemed by FDA
to have engaged in the promotion of any our products for
off-label use, we could be subject to FDA prohibitions on the
sale or marketing of our products or significant fines and
penalties, and the imposition of these sanctions could also
affect our reputation and position within the industry. Any of
these events could harm our business and results of operations
and cause our stock to decline.
We
purchase some of the key components of our products from single
suppliers. The loss of these suppliers could prevent or delay
shipments of our products or delay our clinical trials or
otherwise adversely affect our business.
Some of the key components of our products and related services
are currently purchased from only single suppliers. We do not
have long-term contracts with the third-party suppliers of our
product components. If necessary or desirable, we could source
our product components and related services from other
suppliers. However, establishing additional or replacement
suppliers for these components, and obtaining any additional
regulatory clearances or approvals, if necessary, that may
result from adding or replacing suppliers, will take a
substantial amount of time and could result in increased costs
and impair our ability to produce our products, which would
adversely impact our business, operating results and prospects.
In addition, some of our products,
23
which we acquire from third parties, are highly technical and
are required to meet exacting specifications, and any quality
control problems that we experience with respect to the products
supplied by third-party vendors could adversely and materially
affect our reputation, our attempts to complete our clinical
trials or commercialization of our products. We may also have
difficulty obtaining similar components from other suppliers
that are acceptable to the FDA or foreign regulatory
authorities, and the failure of our suppliers to comply with
strictly enforced regulatory requirements could expose us to
regulatory action including, warning letters, product recalls,
termination of distribution, product seizures or civil
penalties, among others. Furthermore, since some of these
suppliers may be located outside of the United States, we are
subject to foreign export laws and U.S. import and customs
statutes and regulations, which complicate and could delay
shipments of components to us.
If we experience any delay or deficiency in the quality of
products supplied to us by third-party suppliers, or if we have
to switch to replacement suppliers, we may face additional
regulatory delays and the manufacture and delivery of our
products would be interrupted for an extended period of time,
which would adversely affect our business, operating results and
prospects. In addition, we may be required to obtain prior
regulatory clearance or approval from the FDA or foreign
regulatory authorities to use different suppliers or components.
As a result, regulatory clearance or approval of our products
may not be received on a timely basis, or at all, and our
business, operating results and prospects would be harmed.
We
depend on our officers and other key employees, and if we are
not able to retain and motivate them or recruit additional
qualified personnel, our business will suffer.
We are highly dependent on our officers and other key employees.
Due to the specialized knowledge each of our officers and other
key employees possesses with respect to the treatment of spine
disorders and our operations, the loss of service of any of our
officers and other key employees could delay or prevent the
successful completion of our clinical trials, the growth of
revenue from existing products and the commercialization of our
new products. Each of our officers and key employees may
terminate his or her employment without notice and without cause
or good reason.
If we
fail to properly manage our anticipated growth, our business
could suffer.
The anticipated growth of our business could place a significant
strain on our managerial, operational and financial resources
and systems. To execute our anticipated growth successfully, we
must attract and retain qualified personnel and manage and train
them effectively. We must also review our internal business
processes and capabilities and internal controls to create the
scalability that a growing business demands. We will be
dependent on our personnel and third parties to accomplish this,
as well as to effectively market our products to an increasing
number of spine surgeons. We will also depend on our personnel
to develop next generation technologies.
Further, our anticipated growth will place additional strain on
our suppliers and manufacturers, resulting in increased need for
us to carefully monitor quality assurance. Any failure by us to
manage our growth effectively could have an adverse effect on
our ability to achieve our development and commercialization
goals.
We expect to expand our operations and grow our research and
development, product development, clinical, regulatory,
operations, sales and marketing and administrative functions.
Our growth will require hiring a significant number of qualified
clinical, scientific, regulatory, quality, commercial and
administrative personnel. Recruiting, motivating and retaining
such personnel will be critical to our success. There is intense
competition from other companies and research and academic
institutions for qualified personnel in the areas of our
activities. In addition, our operations are located in a
geographic region which historically does not have a large
number of medical device companies and it may be difficult to
convince qualified personnel to relocate to our area. If we fail
to identify, attract, retain and motivate these highly skilled
personnel, we may be unable to continue our development and
commercialization activities.
24
If we
need additional funding, we may be unable to raise capital when
needed, which would force us to delay, reduce, eliminate or
abandon our commercialization efforts or product development
programs.
We may need to raise substantial additional capital to:
|
|
|
| |
•
|
expand the commercialization of our products;
|
| |
| |
•
|
fund our operations and clinical trials;
|
| |
| |
•
|
continue our research and development;
|
| |
| |
•
|
defend, in litigation or otherwise, any claims that we infringe
third-party patents or other intellectual property rights;
|
| |
| |
•
|
address FDA or other governmental, legal/enforcement actions and
remediate underlying problems;
|
| |
| |
•
|
commercialize our new products, if any such products receive
regulatory clearance or approval for commercial sale; and
|
| |
| |
•
|
acquire companies and in-license products or intellectual
property.
|
We believe that our existing cash and cash equivalent balances
and cash receipts generated from sales of our products, will be
sufficient to meet our anticipated cash requirements for at
least the next two years. However, our future funding
requirements will depend on many factors, including:
|
|
|
| |
•
|
market acceptance of our products;
|
| |
| |
•
|
availability of adequate coverage and reimbursement for
hospitals and surgeons;
|
| |
| |
•
|
the scope, rate of progress and cost of our clinical trials;
|
| |
| |
•
|
the cost of our research and development activities;
|
| |
| |
•
|
the cost of filing and prosecuting patent applications and
defending and enforcing our patent and other intellectual
property rights;
|
| |
| |
•
|
the cost of defending, in litigation or otherwise, any claims
that we infringe third-party patent or other intellectual
property rights;
|
| |
| |
•
|
the cost and timing of additional regulatory clearances or
approvals;
|
| |
| |
•
|
the cost and timing of establishing additional sales, marketing
and distribution capabilities;
|
| |
| |
•
|
the effect of competing technological and market
developments; and
|
| |
| |
•
|
the extent to which we acquire or invest in businesses, products
and technologies, although we currently have no commitments or
agreements relating to any of these types of transactions.
|
If we raise additional funds by issuing equity securities, our
stockholders may experience dilution. Debt financing, if
available, may involve covenants restricting our operations or
our ability to incur additional debt. Any debt financing or
additional equity that we raise may contain terms that are not
favorable to us or our stockholders. If we raise additional
funds through collaboration and licensing arrangements with
third parties, it may be necessary to relinquish some rights to
our technologies or our products, or grant licenses on terms
that are not favorable to us. As a result of the recent and
continuing economic uncertainty, it has been difficult for
companies, particularly small cap medical device companies, to
obtain equity or debt financing. If we are unable to raise
adequate funds, we may have to liquidate some or all of our
assets, or delay, reduce the scope of or eliminate some or all
of our development programs.
If we do not have, or are not able to obtain, sufficient funds,
we may have to delay development or commercialization of our
products or license to third parties the rights to commercialize
products or technologies that we would otherwise seek to
commercialize. We also may have to reduce marketing, customer
support or other resources devoted to our products or cease
operations. Any of these factors could harm our operating
results.
25
If we
choose to acquire new businesses, products or technologies, we
may experience difficulty in the identification or integration
of any such acquisition, and our business may
suffer.
Our success depends on our ability to continually enhance and
broaden our product offerings in response to changing customer
demands, competitive pressures and technologies. Accordingly, we
may in the future pursue the acquisition of complementary
businesses, products or technologies instead of developing them
ourselves. We do not know if we will be able to identify or
complete any acquisitions, or whether we will be able to
successfully integrate any acquired business, product or
technology or retain key employees. Integrating any business,
product or technology we acquire could be expensive and time
consuming, The diversion of our management’s attention, and
any delays or difficulties encountered in connection with any of
our acquisitions, could result in the disruption of our ongoing
business or inconsistencies in standards, controls, procedures
and policies that could negatively affect our ability to
maintain relationships with customers, suppliers, employees and
others with whom we have business dealings. If we are unable to
integrate any acquired businesses, products or technologies
effectively, our business will suffer. In addition, any
amortization or charges resulting from acquisitions could harm
our operating results.
In addition, other companies, including those with substantially
greater resources than ours, may compete with us for the
acquisition of complementary businesses, products or
technologies, resulting in the possibility that we devote
resources to potential acquisitions or arrangements that are
never completed. If we do engage in any such acquisition, we
will incur a variety of costs, and we may never realize the
anticipated benefits of the acquisition in light of those costs.
If we fail to realize the expected benefits from acquisitions we
may consummate in the future, whether as a result of
unidentified risks, integration difficulties, regulatory
setbacks or other events, our business, consolidated results of
operations and financial condition could be adversely affected.
Furthermore, any strategic acquisition may require us to obtain
additional debt or equity financing, resulting in increased debt
obligations or dilution of ownership to our existing
stockholders, as applicable. Therefore, we may not be able to
finance acquisitions on terms satisfactory to us, if at all.
Consolidation
in the healthcare industry could lead to demands for price
concessions or to the exclusion of some suppliers from certain
of our markets, which could have an adverse effect on our
business, financial condition or results of
operations.
Because healthcare costs have risen significantly over the past
decade, numerous initiatives and reforms initiated by
legislators, regulators and third-party payors to curb these
costs have resulted in a consolidation trend in the healthcare
industry to create new companies with greater market power,
including hospitals. As the healthcare industry consolidates,
competition to provide products and services to industry
participants has become and will continue to become more
intense. This in turn has resulted and will likely continue to
result in greater pricing pressures and the exclusion of certain
suppliers from important market segments as group purchasing
organizations, independent delivery networks and large single
accounts continue to use their market power to consolidate
purchasing decisions for some of our customers. We expect that
market demand, government regulation, third-party reimbursement
policies and societal pressures will continue to change the
worldwide healthcare industry, resulting in further business
consolidations and alliances among our customers, which may
reduce competition, exert further downward pressure on the
prices of our products and may adversely impact our business,
financial condition or results of operations.
We
face the risk of product liability or other claims and may not
be able to obtain sufficient insurance coverage, if at
all.
Our business exposes us to the risk of product liability claims
that is inherent in the testing, manufacturing and marketing of
implantable medical devices. We may be subject to product
liability claims if our products cause, or merely appear to have
caused, an injury or death. Claims may be made by patients,
consumers or healthcare providers. Although we have product
liability and clinical trial liability insurance that we believe
is appropriate for our current level of operations, this
insurance is subject to deductibles and coverage limitations.
Our current product liability insurance may not continue to be
available to us on
26
acceptable terms, if at all, and, if available, the coverages
may not be adequate to protect us against any future product
liability claims. If we are unable to obtain insurance at
acceptable cost or on acceptable terms with adequate coverage or
otherwise protect against potential product liability claims, we
could be exposed to significant financial and other liabilities,
which may harm our business. A product liability claim, product
recall or other claim with respect to uninsured liabilities or
for amounts in excess of insured liabilities could have a
material adverse effect on our business, operating results and
prospects.
We may be subject to claims against us even if the apparent
injury is due to the actions of others. For example, we rely on
the expertise of spine surgeons, nurses and other associated
medical personnel to perform the medical procedure and related
processes for our products. If these medical personnel are not
properly trained or are negligent in their provision of care,
the therapeutic effect of our products may be diminished or the
patient may suffer critical injury, which may subject us to
liability. In addition, an injury that is caused by the
activities of our suppliers may be the basis for a claim against
us.
In addition, medical malpractice carriers are withdrawing
coverage in certain regions or substantially increasing
premiums. In the event we become a defendant in a product
liability suit in which the treating surgeon or hospital does
not have adequate malpractice insurance, the likelihood of
liability being imposed on us could increase.
These liabilities could prevent, delay or otherwise adversely
interfere with our product commercialization efforts, and result
in judgments, fines, damages and other financial liabilities
which have adverse effects on our business, operating results
and prospects. Defending a suit, regardless of merit, could be
costly, could divert management’s attention from our
business and might result in adverse publicity, which could
result in the withdrawal of, or inability to recruit, clinical
trial patient participants or result in reduced acceptance of
our products in the market. In addition to adversely impacting
our business and prospects, such adverse publicity could
materially adversely affect our stock price.
If our
independent contract manufacturers fail to timely deliver to us
sufficient quantities of some of our products and components in
a timely manner, our operations may be harmed.
Our reliance on independent contract manufacturers to
manufacture most of our products and components involves several
risks, including:
|
|
|
| |
•
|
inadequate capacity of the manufacturer’s facilities;
|
| |
| |
•
|
financial difficulties experienced by manufacturers due to the
current economic recession;
|
| |
| |
•
|
interruptions in access to certain process technologies; and
|
| |
| |
•
|
reduced control over product availability, quality, delivery
schedules, manufacturing yields and costs.
|
Shortages of raw materials, production capacity or financial
constraints, or delays by our contract manufacturers could
negatively affect our ability to meet our production obligations
and result in increased prices for affected parts. Any such
reduction, constraint or delay may result in delays in shipments
of our products or increases in the prices of components, either
of which could have a material adverse effect on our business.
We do not have supply agreements with all of our current
contract manufacturers and we often utilize purchase orders,
which are subject to acceptance by the supplier. Failure to
accept purchase orders could result in an inability to obtain
adequate supply of our product or components in a timely manner
or on commercially reasonable terms.
An unanticipated loss of any of our contract manufacturers could
cause delays in our ability to deliver our products while we
identify and qualify a replacement manufacturer, which delays
could negatively impact our revenues.
The liquidity of our customers and suppliers may also be
affected by the current economic and financial downturn. Our
suppliers may experience credit or liquidity problems which
could negatively affect sources of our products and components.
27
We
operate at a single location. Any disruption in this facility or
any inability to ship a sufficient number of our products to
meet demand could adversely affect our business and results of
operations.
We operate at a single location in Wilmington, North Carolina.
Our facility may be affected by man-made or natural disasters,
such as a hurricane. While we currently rely on third parties to
manufacture, assemble, package, label and sterilize our products
and components, and to warehouse and ship a portion of our
products, we might also be forced to rely on third parties to
inspect, warehouse or ship all our products and components in
the event our facilities were affected by a disaster. Our
facility, if damaged or destroyed, could be difficult to replace
and could require substantial lead-time to repair or replace. In
the case of a device with a PMA approval, we might be required
to obtain prior FDA, or notified body, approval of an alternate
facility, which could delay or prevent our marketing of the
affected product until this supplemental approval is obtained.
Although we believe we possess adequate insurance for damage to
our property and the disruption of our business from casualties,
this insurance may not be sufficient to cover all of our
potential losses and may not continue to be available to us on
acceptable terms, or at all.
Current
challenges in the commercial and credit environment may
adversely affect our business and financial
condition.
Unpredictable changes in economic conditions, including
recession, inflation, increased government intervention, or
other changes, may adversely affect our general business
strategy. Our ability to generate cash flows from operations or
enter into financing arrangements on acceptable terms could be
adversely affected if there is a material decline in the demand
for our products or in the solvency of our customers,
deterioration in our key financial ratios or credit ratings, or
other significantly unfavorable changes in conditions. While
these conditions and the current economic uncertainty have not
meaningfully impaired our ability to access credit markets or
meaningfully adversely affected our operations to date,
continuing volatility in the global financial markets could
increase borrowing costs or affect our ability to access the
capital markets. Current or worsening economic conditions may
also adversely affect the business of our customers, including
their ability to pay for our products and services, and the
amount spent on healthcare generally. This could result in a
decrease in the demand for our products and services, longer
sales cycles, slower adoption of new technologies and increased
price competition.
Our
future operating results are difficult to predict and may vary
significantly from quarter to quarter, which may negatively
impact our stock price in the future.
We have only commercially distributed our products since 2005.
Given this limited history, it is difficult to predict future
revenues derived from sales of our products. Because of this and
the uncertain effects of the following factors, our quarterly
revenues and results of operations may fluctuate in the future
due to, among others, the following reasons:
|
|
|
| |
•
|
market acceptance of our products;
|
| |
| |
•
|
availability of adequate coverage and reimbursement for
hospitals and surgeons;
|
| |
| |
•
|
the conduct and results of clinical trials;
|
| |
| |
•
|
the timing and expense of obtaining future regulatory approvals;
|
| |
| |
•
|
fluctuations in our expenses associated with expanding our
operations;
|
| |
| |
•
|
the introduction of new products by our competitors; and
|
| |
| |
•
|
changes in our pricing policies or in the pricing policies of
our competitors or suppliers.
|
Because of these and possibly other factors, it is possible that
in some future period our operating results will not meet
investor expectations or those of public market analysts.
Any unanticipated change in revenues or operating results is
likely to cause our stock price to fluctuate since such changes
reflect new information available to investors and analysts. New
information may cause investors and analysts to re-evaluate our
stock, which could cause a decline in the trading price of our
stock.
28
Risks
Related to Regulatory Environment
If we
fail to maintain regulatory approvals and clearances, or are
unable to obtain, or experience significant delays in obtaining,
FDA clearances or approvals for our future products or product
modifications, our ability to commercially distribute and market
these products could suffer.
Our products are subject to rigorous regulation by the FDA and
numerous other federal, state and foreign governmental
authorities. The process of obtaining regulatory clearances or
approvals to market a medical device can be costly and time
consuming, and we may not be able to obtain these clearances or
approvals on a timely basis, if at all. In particular, the FDA
permits commercial distribution of most new medical devices only
after the device has received clearance under
Section 510(k) of the Federal Food, Drug and Cosmetic Act,
or is the subject of an approved PMA. The FDA will clear
marketing of a non-exempt lower risk medical device through the
510(k) process if the manufacturer demonstrates that the new
product is substantially equivalent to other legally marketed
products not requiring PMA approval. High risk devices deemed to
pose the greatest risk, such as life-sustaining,
life-supporting, or implantable devices, or devices not deemed
substantially equivalent to a legally marketed device, require a
PMA. The PMA process is more costly, lengthy and uncertain than
the 510(k) clearance process. A PMA application must be
supported by extensive data, including, but not limited to,
technical, preclinical, clinical trial, manufacturing and
labeling data, to demonstrate to the FDA’s satisfaction the
safety and efficacy of the device for its intended use. Our
currently commercialized products have been cleared through the
510(k) process. However, we may need to submit a PMA for future
products we develop.
Certain of the FDA’s policies and procedures are under
review by new leadership and it is uncertain whether any changes
arising from such review could adversely affect our products and
business.
Our failure to comply with U.S. federal, state and foreign
governmental regulations could lead to the imposition of
injunctions, suspensions or loss of regulatory clearance or
approvals, product recalls, termination of distribution, product
seizures or civil penalties, among other things. In the most
extreme cases, criminal sanctions or closure of our
manufacturing facility are possible.
Foreign governmental authorities that regulate the manufacture
and sale of medical devices have become increasingly stringent
and, to the extent we market and sell our products
internationally, we may be subject to rigorous international
regulation in the future. In these circumstances, we would rely
significantly on our foreign independent distributors to comply
with the varying regulations, and any failures on their part
could result in restrictions on the sale of our products in
foreign countries.
Modifications
to our marketed products may require new 510(k) clearances or
PMA approvals, or may require us to cease marketing or recall
the modified products until clearances or approvals are
obtained.
Any modification to our currently marketed 510(k)-cleared
devices that could significantly affect its safety or efficacy,
or that would constitute a change in its intended use, requires
a new 510(k) clearance or, possibly, a PMA. The FDA requires
every manufacturer to make this determination in the first
instance, but the FDA may review the manufacturer’s
decision. The FDA may not agree with our decisions regarding
whether new clearances or approvals are necessary. If the FDA
requires us to seek 510(k) clearance or a PMA for any
modification to a previously cleared product, we may be required
to cease marketing and distributing, or to recall the modified
product until we obtain such clearance or approval, and we may
be subject to significant regulatory fines or penalties.
Further, our products could be subject to recall if the FDA
determines, for any reason, that our products are not safe or
effective because they are in violation of the FDCA. Any recall
or FDA requirement that we seek additional approvals or
clearances could result in significant delays, fines, increased
costs associated with modification of a product, loss of revenue
and potential operating restrictions imposed by the FDA.
29
Clinical
trials necessary to support a PMA application will be expensive
and will require the enrollment of large numbers of patients,
and suitable patients may be difficult to identify and recruit.
Delays or failures in our clinical trials will prevent us from
commercializing any modified or new products and will adversely
affect our business, operating results and
prospects.
Initiating and completing clinical trials necessary to support a
PMA application and additional safety and efficacy data beyond
that typically required for 510(k) clearances for possible
future product candidates, will be time consuming and expensive
and the outcomes uncertain. Moreover, the results of early
clinical trials are not necessarily predictive of future
results, and any product we advance into clinical trials may not
have favorable results in later clinical trials.
Conducting successful clinical studies may require the
enrollment of large numbers of patients, and suitable patients
may be difficult to identify and recruit. Patient enrollment in
clinical trials and completion of patient participation and
follow-up
depends on many factors, including the size of the patient
population, the nature of the trial protocol, the attractiveness
of, or the discomforts and risks associated with, the treatments
received by enrolled subjects, the availability of appropriate
clinical trial investigators, support staff, and proximity of
patients to clinical sites. For example, patients may be
discouraged from enrolling in our clinical trials if the trial
protocol requires them to undergo extensive post-treatment
procedures or
follow-up to
assess the safety and effectiveness of our products or if they
determine that the treatments received under the trial protocols
are not attractive or involve unacceptable risks or discomforts.
Patients may also not participate in our clinical trials if they
choose to participate in contemporaneous clinical trials of
competitive products. In addition, patients participating in
clinical trials may die before completion of the trial or suffer
adverse medical events unrelated to investigational products.
Development of sufficient and appropriate clinical protocols and
data to demonstrate safety and efficacy are required and we may
not adequately develop such protocols to support clearance and
approval. Further, the FDA may require us to submit data on a
greater number of patients than we originally anticipated
and/or for a
longer
follow-up
period or change the data collection requirements or data
analysis applicable to our clinical trials. Delays in patient
enrollment or failure of patients to continue to participate in
a clinical trial may cause an increase in costs and delays in
the clearance or approval and attempted commercialization of our
products or result in the failure of the clinical trial. In
addition, despite considerable time and expense invested in our
clinical trials, FDA may not consider our data adequate to
demonstrate safety and efficacy. Such increased costs and delays
or failures could adversely affect our business, operating
results and prospects.
If the
third parties on which we rely to conduct our clinical trials
and to assist us with pre-clinical development do not perform as
contractually required or expected, we may not be able to obtain
regulatory clearance or approval for or commercialize our
products.
We do not have the ability to independently conduct our
pre-clinical and clinical trials for our products and we must
rely on third parties, such as contract research organizations,
medical institutions, clinical investigators and contract
laboratories to conduct such trials. If these third parties do
not successfully perform their contractual duties or regulatory
obligations or meet expected deadlines, if these third parties
need to be replaced, or if the quality or accuracy of the data
they obtain is compromised due to the failure to adhere to our
clinical protocols or regulatory requirements or for other
reasons, our pre-clinical development activities or clinical
trials may be extended, delayed, suspended or terminated, and we
may not be able to obtain regulatory clearance or approval for,
or successfully commercialize, our products on a timely basis,
if at all, and our business, operating results and prospects may
be adversely affected. Furthermore, our third-party clinical
trial investigators may be delayed in conducting our clinical
trials for reasons outside of their control.
Even
if our products are approved by regulatory authorities, if we or
our suppliers fail to comply with ongoing FDA or other foreign
regulatory authority requirements, or if we experience
unanticipated problems with our products, these products could
be subject to restrictions or withdrawal from the
market.
Any product for which we obtain clearance or approval, and the
manufacturing processes, reporting requirements, post-approval
clinical data and labeling and promotional activities for such
product, will be subject
30
to continued regulatory review, oversight and periodic
inspections by the FDA and other domestic and foreign regulatory
bodies. In particular, we and our suppliers are required to
comply with the Quality System Regulations, or QSR, and MDD
regulations, which may include ISO standards, for the
manufacture of our products and other regulations which cover
the methods and documentation of the design, testing,
production, control, quality assurance, labeling, packaging,
storage and shipping of any product for which we obtain
clearance or approval. Regulatory bodies enforce the QSR and ISO
regulations through inspections. The failure by us or one of our
suppliers to comply with applicable statutes and regulations
administered by the FDA and other regulatory bodies, or the
failure to timely and adequately respond to any adverse
inspectional observations or product safety issues, could result
in, among other things, any of the following enforcement actions:
|
|
|
| |
•
|
warning letters or untitled letters;
|
| |
| |
•
|
fines and civil penalties;
|
| |
| |
•
|
unanticipated expenditures to address or defend such actions;
|
| |
| |
•
|
delays in clearing or approving, or refusal to clear or approve,
our products;
|
| |
| |
•
|
withdrawal or suspension of approval of our products or those of
our third-party suppliers by the FDA or other regulatory bodies;
|
| |
| |
•
|
product recall or seizure;
|
| |
| |
•
|
orders for physician notification or device repair, replacement
or refund;
|
| |
| |
•
|
interruption of production;
|
| |
| |
•
|
operating restrictions;
|
| |
| |
•
|
injunctions; and
|
| |
| |
•
|
criminal prosecution.
|
If any of these actions were to occur it would harm our
reputation and cause our product sales to suffer and may prevent
us from generating revenue. Furthermore, our key component
suppliers may not currently be or may not continue to be in
compliance with all applicable regulatory requirements which
could result in our failure to produce our products on a timely
basis and in the required quantities, if at all.
Even if regulatory clearance or approval of a product is
granted, such clearance or approval may be subject to
limitations on the intended uses for which the product may be
marketed and reduce our potential to successfully commercialize
the product and generate revenue from the product. If the FDA
determines that our promotional materials, labeling, training or
other marketing or educational activities constitute promotion
of an unapproved use, it could request that we cease or modify
our training educational, labeling or promotional materials or
subject us to regulatory enforcement actions. It is also
possible that other federal, state or foreign enforcement
authorities might take action if they consider our training
educational, labeling or other promotional materials to
constitute promotion of an unapproved use, which could result in
significant fines or penalties under other statutory
authorities, such as laws prohibiting false claims for
reimbursement.
In addition, we may be required to conduct costly post-market
testing and surveillance to monitor the safety or effectiveness
of our products, and we must comply with medical device
reporting requirements, including the reporting of adverse
events and certain malfunctions related to our products. Later
discovery of previously unknown problems with our products,
including unanticipated adverse events or adverse events of
unanticipated severity or frequency, manufacturing problems, or
failure to comply with regulatory requirements such as the QSR
or GMP, may result in changes to labeling, restrictions on such
products or manufacturing processes, withdrawal of the products
from the market, voluntary or mandatory recalls, a requirement
to repair, replace or refund the cost of any medical device we
manufacture or distribute, fines, suspension of regulatory
approvals, product seizures, injunctions or the imposition of
civil or criminal penalties which would adversely affect our
business, operating results and prospects.
31
We may
be subject to or otherwise affected by federal and state
healthcare laws, including fraud and abuse and health
information privacy and security laws, and could face
substantial penalties if we are unable to fully comply with such
laws.
Although we do not provide healthcare services, submit claims
for third-party reimbursement, or receive payments directly from
Medicare, Medicaid, or other third-party payors for our products
or the procedures in which our products are used, healthcare
regulation by federal and state governments could significantly
impact our business. Healthcare fraud and abuse and health
information privacy and security laws potentially applicable to
our operations include:
|
|
|
| |
•
|
the federal Anti-Kickback Law, which constrains our marketing
practices and those of our independent sales agents and
distributors, educational programs, pricing policies, and
relationships with healthcare providers, by prohibiting, among
other things, soliciting, receiving, offering or providing
remuneration, intended to induce the purchase or recommendation
of an item or service reimbursable under a federal healthcare
program (such as the Medicare or Medicaid programs);
|
| |
| |
•
|
federal false claims laws which prohibit, among other things,
knowingly presenting, or causing to be presented, claims for
payment from Medicare, Medicaid, or other third-party payors
that are false or fraudulent;
|
| |
| |
•
|
the federal Health Insurance Portability and Accountability Act
of 1996, or HIPAA, and its implementing regulations, which
created federal criminal laws that prohibit executing a scheme
to defraud any healthcare benefit program or making false
statements relating to healthcare matters and which also imposes
certain regulatory and contractual requirements regarding the
privacy, security and transmission of individually identifiable
health information; and
|
| |
| |
•
|
state laws analogous to each of the above federal laws, such as
anti-kickback and false claims laws that may apply to items or
services reimbursed by any third-party payor, including
commercial insurers, and state laws governing the privacy of
certain health information, many of which differ from each other
in significant ways and often are not preempted by HIPAA, thus
complicating compliance efforts.
|
We have adopted comprehensive compliance programs to attempt to
comply with these regulations. However, because of the breadth
of these laws and regulations and the sometimes subjective
nature of their application, it is possible that some of our
business activities could be subject to challenge under one or
more of such laws. If our past or present operations, or those
of our independent sales agents and distributors, are found to
be in violation of any of such laws or any other governmental
regulations that may apply to us, we may be subject to
penalties, including civil and criminal penalties, damages,
fines, exclusion from federal healthcare programs
and/or the
curtailment or restructuring of our operations. Similarly, if
the healthcare providers or entities with whom we do business
are found to be non-compliant with applicable laws, they may be
subject to sanctions, which could also have a negative impact on
us. Any penalties, damages, fines, curtailment or restructuring
of our operations could adversely affect our ability to operate
our business and our financial results. The risk of our being
found in violation of these laws is increased by the fact that
many of them have not been fully interpreted by the regulatory
authorities or the courts, and their provisions are open to a
variety of interpretations. Any action against us for violation
of these laws, even if we successfully defend against them,
could cause us to incur significant legal expenses and divert
our management’s attention from the operation of our
business.
Risks
Related to Our Intellectual Property
Our
ability to protect our intellectual property and proprietary
technology through patents and other means is
uncertain.
Our success depends significantly on our ability to protect our
proprietary rights to the procedures created with, and the
technologies used in, our products. We rely on patent
protection, as well as a combination of copyright, trade secret
and trademark laws, and nondisclosure, confidentiality and other
contractual restrictions to protect our proprietary technology.
However, these legal means afford only limited protection and
may not adequately protect our rights or permit us to gain or
keep any competitive advantage. For example, our
32
pending United States and foreign patent applications may not be
approved, may not issue as patents in a form that will be
advantageous to us, or may issue and be subsequently
successfully challenged by others and invalidated. In addition,
our pending patent applications include claims to material
aspects of our products and procedures that are not currently
protected by issued patents. Both the patent application process
and the process of managing patent disputes can be time
consuming and expensive. The patents we own may not be of
sufficient scope or strength to provide us with any meaningful
protection or commercial advantage, and competitors may be able
to design around our patents or develop products which provide
outcomes which are comparable to ours. Although we have taken
steps to protect our intellectual property and proprietary
technology, including entering into confidentiality agreements
and intellectual property assignment agreements with our
officers, employees, consultants and advisors, such agreements
may not be enforceable or may not provide meaningful protection
for our trade secrets or other proprietary information in the
event of unauthorized use or disclosure or other breaches of
such agreements. Furthermore, the laws of some foreign countries
do not protect our intellectual property rights to the same
extent as do the laws of the United States.
We rely on our trademarks, trade names, and brand names to
distinguish our products from the products of our competitors,
and have registered or applied to register many of these
trademarks. However, our trademark applications may not be
approved. Third parties may also oppose our trademark
applications, or otherwise challenge our use of the trademarks.
In the event that our trademarks are successfully challenged, we
could be forced to rebrand our products, which could result in
loss of brand recognition, and could require us to devote
resources to advertising and marketing new brands. Further, our
competitors may infringe our trademarks, or we may not have
adequate resources to enforce our trademarks.
In the event a competitor infringes upon our patent or other
intellectual property rights, enforcing those rights may be
costly, difficult and time consuming. Even if successful,
litigation to enforce our intellectual property rights or to
defend our patents against challenge could be expensive and time
consuming and could divert our management’s attention. We
may not have sufficient resources to enforce our intellectual
property rights or to defend our patents or other intellectual
property rights against a challenge.
Any
lawsuit, whether initiated by us to enforce our intellectual
property rights or by a third party against us alleging
infringement, may cause us to expend significant financial and
other resources, and may divert our attention from our business
and adversely affect our business, operating results and
prospects.
The medical device industry is characterized by the existence of
a large number of patents and frequent litigation based on
allegations of patent infringement. Patent litigation can
involve complex factual and legal questions and its outcome is
uncertain. Any claim relating to infringement of patents that is
successfully asserted against us may require us to pay
substantial damages. Even if we were to prevail, any litigation
could be costly and time-consuming and would divert the
attention of our management and key personnel from our business
operations. Our success will also depend in part on our not
infringing patents issued to others, including our competitors
and potential competitors. If our products are found to infringe
the patents of others, our development, manufacture and sale of
such products could be severely restricted or prohibited. In
addition, our competitors may independently develop similar
technologies. Because of the importance of our patent portfolio
and unpatented proprietary technology to our business, we may
lose market share to our competitors if we fail to protect our
patent rights.
As the number of entrants into our market increases, the
possibility of a patent infringement claim against us grows. Our
products and methods may be covered by patents held by our
competitors. Some of our competitors have considerable resources
available to them to engage in this type of litigation. We, on
the other hand, are an early stage company with comparatively
few resources available to us to engage in costly and protracted
litigation. Because some patent applications are maintained in
secrecy for a period of time after they are filed, there is a
risk that we could adopt a technology without knowledge of a
pending patent application, which technology would infringe a
third-party patent once that patent is issued. In addition, our
competitors may assert that future products we may market
infringe their patents.
A patent infringement suit or other infringement or
misappropriation claim brought against us or any of our
strategic partners or licensees may force us or any of our
strategic partners or licensees to stop or delay
33
developing, manufacturing or selling potential products that are
claimed to infringe a third party’s intellectual property,
unless that party grants us or any strategic partners or
licensees rights to use its intellectual property. In such
cases, we may be required to obtain licenses to patents or
proprietary rights of others in order to continue to
commercialize our products. However, we may not be able to
obtain any licenses required under any patents or proprietary
rights of third parties on acceptable terms, or at all. Even if
our strategic partners or licensees or we were able to obtain
rights to the third party’s intellectual property, these
rights may be non-exclusive, thereby giving our competitors
access to the same intellectual property. Ultimately, we may be
unable to commercialize some of our potential products or may
have to cease some of our business operations as a result of
patent infringement claims, which could severely harm our
business.
In any infringement lawsuit, a third party could seek to enjoin,
or prevent, us from commercializing our existing or future
products,
and/or may
seek damages from us, and any such lawsuit would likely be
expensive for us to defend against. A court may determine that
patents held by third parties are valid and infringed by us and
we may be required to:
|
|
|
| |
•
|
pay damages, including, but not limited to, treble damages and
attorneys’ fees, which may be substantial;
|
| |
| |
•
|
cease the development, manufacture, use and sale of products
that infringe the patent rights of others, through a
court-imposed sanction called an injunction;
|
| |
| |
•
|
expend significant resources to redesign our technology so that
it does not infringe others’ patent rights, or develop or
acquire non-infringing intellectual property, which may not be
possible;
|
| |
| |
•
|
discontinue manufacturing or other processes incorporating
infringing technology; or
|
| |
| |
•
|
obtain licenses to the infringed intellectual property, which
may not be available to us on acceptable terms, or at all.
|
Any development or acquisition of non-infringing products or
technology or licenses could require the expenditure of
substantial time and other resources and could have a material
adverse effect on our business and financial results. If we are
required to, but cannot, obtain a license to valid patent rights
held by a third party, we would likely be prevented from
commercializing the relevant product. We believe that it is
unlikely that we would be able to obtain a license to any
necessary patent rights controlled by companies against which we
would, directly or indirectly, compete. If we need to redesign
products to avoid third-party patents, we may suffer significant
regulatory delays associated with conducting additional studies
or submitting technical, manufacturing or other information
related to the redesigned product and, ultimately, in obtaining
regulatory approval.
Risks
Related to our Common Stock
A sale
of a substantial number of shares of our common stock may cause
the price of our common stock to decline.
If our stockholders sell substantial amounts of our common stock
in the public market, including shares issued upon the exercise
of options, the market price of our common stock could decline.
At December 31, 2009, we had 20,648,447 shares of
common stock outstanding. All of these shares are freely
tradable, without restriction, in the public market, of which
7,484,856 shares are held by directors, executive officers
and other affiliates and are subject to volume limitations under
Rule 144 under the Securities Act. In addition, the
2,216,026 shares of our common stock that are subject to
outstanding options as of December 31, 2009 will be
eligible for sale in the public market to the extent permitted
by the provisions of the various vesting agreements and
Rules 144 and 701 under the Securities Act. If these
additional shares are sold, or it is perceived they will be
sold, the trading price of our common stock could decline. These
sales also might make it more difficult for us to sell equity or
equity-related securities in the future at a time and price that
we deem reasonable or appropriate.
34
Our
directors, officers and principal stockholders have significant
voting power and may take actions that may not be in the best
interests of our other stockholders.
At December 31, 2009, our officers, directors and principal
stockholders, each holding more than 5% of our common stock,
collectively controlled approximately 56% of our outstanding
common stock. As a result, these stockholders, if they act
together, are able to control the management and affairs of our
company and most matters requiring stockholder approval,
including the election of directors and approval of significant
corporate transactions. This concentration of ownership may have
the effect of delaying or preventing a change in control and
might adversely affect the market price of our common stock.
This concentration of ownership may not be in the best interests
of our other stockholders.
If
securities or industry analysts do not publish research or
reports about our business, if they change their recommendations
regarding our stock adversely or if our operating results do not
meet their expectations, our stock price and trading volume
could decline.
The trading market for our stock may be influenced by the
research and reports that industry or securities analysts
publish about us or our business. If one or more of these
analysts cease coverage of us or fail to publish reports on us
regularly, we could lose visibility in the financial markets,
which in turn could cause our stock price or trading volume to
decline. Moreover, if one or more of the analysts who cover us
downgrade our stock or if our operating results do not meet
their expectations, our stock price could decline.
Trading
in our stock over the last twelve months has been limited, so
investors may not be able to sell as much stock as they want at
prevailing prices.
The average daily trading volume in our common stock for the
year ended December 31, 2009 was approximately
76,000 shares. If limited trading in our stock continues,
it may be difficult for investors to sell their shares in the
public market at any given time at prevailing prices. Moreover,
the market price for shares of our common stock may be made more
volatile because of the relatively low volume of trading in our
common stock. When trading volume is low, significant price
movement can be caused by the trading in a relatively small
number of shares. Volatility in our common stock could cause
stockholders to incur substantial losses.
Volatility
in the stock price of other companies may contribute to
volatility in our stock price.
The Nasdaq Global Market, particularly in recent years, has
experienced significant volatility with respect to medical
technology, pharmaceutical, biotechnology and other life science
company stocks. The volatility of medical technology,
pharmaceutical, biotechnology and other life science company
stocks often does not relate to the operating performance of the
companies represented by the stock. Further, there has been
particular volatility in the market price of securities of early
stage life science companies. These broad market and industry
factors may seriously harm the market price of our common stock,
regardless of our operating performance. In the past, following
periods of volatility in the market price of a company’s
securities, securities class action litigation has often been
instituted. A securities class action suit against us could
result in substantial costs, potential liabilities and the
diversion of management’s attention and resources.
Our
amended and restated certificate of incorporation, amended and
restated bylaws and Delaware law contain provisions that could
discourage a takeover.
Anti-takeover provisions of our amended and restated certificate
of incorporation, amended and restated bylaws and Delaware law
may have the effect of deterring or delaying attempts by our
stockholders to remove or replace management, engage in proxy
contests and effect changes in control. The provisions of our
charter documents include:
|
|
|
| |
•
|
a classified board so that only one of the three classes of
directors on our board of directors is elected each year;
|
| |
| |
•
|
procedures for advance notification of stockholder director
nominations and proposals;
|
35
|
|
|
| |
•
|
the ability of our board of directors to amend our bylaws
without stockholder approval;
|
| |
| |
•
|
a supermajority stockholder vote requirement for amending
certain provisions of our amended and restated certificate of
incorporation and our amended and restated bylaws; and
|
| |
| |
•
|
the ability of our board of directors to issue up to
5,000,000 shares of preferred stock without stockholder
approval upon the terms and conditions and with the rights,
privileges and preferences as our board of directors may
determine.
|
In addition, as a Delaware corporation, we are subject to
Delaware law, including Section 203 of the Delaware General
Corporation Law, or DGCL. In general, Section 203 prohibits
a Delaware corporation from engaging in any business combination
with any interested stockholder for a period of three years
following the date that the stockholder became an interested
stockholder unless certain specific requirements are met as set
forth in Section 203. These provisions, alone or together,
could have the effect of deterring or delaying changes in
incumbent management, proxy contests or changes in control.
We do
not anticipate declaring any cash dividends on our common
stock.
We have never declared or paid cash dividends on our common
stock and do not plan to pay any cash dividends in the near
future. Our current policy is to retain all funds and any
earnings for use in the operation and expansion of our business.
If we do not pay dividends, our stock may be less valuable to
you because a return on your investment will only occur if our
stock price appreciates.
|
|
|
Item 1B.
|
Unresolved
Staff Comments.
|
None.
At December 31, 2009, we leased approximately
30,000 square feet of space in a single-user building
located in an industrial park in Wilmington, North Carolina. Of
that amount, approximately 19,800 square feet were used for
manufacturing and warehousing, approximately 8,400 square
feet were used for office space and approximately
1,800 square feet were used for research and development
activities. This lease was terminated in February 2010. In
February 2010, we moved into a new facility of approximately
30,000 square feet, also located in Wilmington, North
Carolina. Of that amount, approximately 5,000 square feet
are used for manufacturing and warehousing, approximately
18,000 square feet are used for office space and
approximately 7,000 square feet are used for research and
development activities. This lease expires in December 2019. We
believe that our current facility will be sufficient to meet our
needs through that time.
|
|
|
Item 3.
|
Legal
Proceedings.
|
We are not currently party to any material legal proceedings. We
may be subject to various claims and legal actions arising in
the ordinary course of business from time to time.
|
|
|
Item 4.
|
Submission
of Matters to a Vote of Security Holders.
|
No matter was submitted to a vote of our security holders during
the quarter ended December 31, 2009.
36
PART II
|
|
|
Item 5.
|
Market
for the Registrant’s Common Equity, Related Stockholder
Matters and Issuer Purchases of Equity Securities
|
Price of
Common Stock
Our common stock is traded on the NASDAQ Global Market under the
symbol “TSON.” The following table sets forth the high
and low sales prices of our common stock as quoted on the NASDAQ
Global Market for the periods indicated.
| |
|
|
|
|
|
|
|
|
|
|
|
Price Range
|
|
|
|
High
|
|
Low
|
|
|
|
Fiscal 2009:
|
|
|
|
|
|
|
|
|
|
Fourth quarter
|
|
$
|
5.00
|
|
|
$
|
3.26
|
|
|
Third quarter
|
|
|
6.83
|
|
|
|
4.08
|
|
|
Second quarter
|
|
|
8.74
|
|
|
|
5.65
|
|
|
First quarter
|
|
|
7.66
|
|
|
|
4.55
|
|
|
Fiscal 2008:
|
|
|
|
|
|
|
|
|
|
Fourth quarter
|
|
$
|
9.54
|
|
|
$
|
5.82
|
|
|
Third quarter
|
|
|
14.25
|
|
|
|
7.45
|
|
|
Second quarter
|
|
|
15.74
|
|
|
|
9.93
|
|
|
First quarter
|
|
|
17.24
|
|
|
|
8.96
|
|
The closing price for our common stock as reported by the NASDAQ
Global Market on March 8, 2010 was $3.93 per share.
As of March 8, 2010, we had approximately 40 stockholders
of record based upon the records of our transfer agent, which
does not include beneficial owners of our common stock whose
shares are held in the names of various securities brokers,
dealers and registered clearing agencies.
Uses of
Proceeds from Sale of Registered Securities
On October 22, 2007, we completed our initial public
offering of 6,325,000 shares of common stock (inclusive of
825,000 shares sold to the underwriters upon exercise of
their over-allotment option) at the initial public offering
price of $15.00 per share. We effected the offering through a
Registration Statement on
Form S-1
(Registration
No. 333-144802),
which was declared effective by the SEC on October 16,
2007, and through a Registration Statement on
Form S-1
filed pursuant to Rule 462(b) under the Securities Act
(Registration
No. 333-146753),
which became effective upon filing on October 17, 2007
pursuant to Rule 462(b) (collectively, the
“Registration Statement”). The offering commenced on
October 17, 2007 and terminated on October 22, 2007
after all of the 6,325,000 shares of common stock
registered under the Registration Statement were sold. Our
initial public offering resulted in aggregate proceeds to us of
approximately $86.7 million, net of underwriting discounts
and commissions of approximately $6.6 million and offering
expenses of approximately $1.6 million. Lehman Brothers
Inc. and Piper Jaffray & Co. acted as joint
book-running managers for the offering with Cowen and Company,
LLC and Wachovia Capital Markets, LLC acting as co-managers.
No offering expenses were paid directly or indirectly to any of
our directors or officers (or their associates) or person owning
ten percent or more of any class of our equity securities or to
any other affiliates. All offering expenses were paid directly
to others.
As of December 31, 2009, we had used all of the net
proceeds for sales, marketing, general administrative and
research and development activities.
37
Dividend
Policy
We have never declared or paid any cash dividends on our capital
stock. We currently intend to retain future earnings, if any,
for development of our business and do not anticipate that we
will declare or pay cash dividends on our capital stock in the
foreseeable future.
Securities
Authorized For Issuance Under Equity Compensation
Plans
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(c)
|
|
|
|
|
|
|
|
|
|
|
Number of Securities
|
|
|
|
|
(a)
|
|
|
|
|
|
Remaining Available
|
|
|
|
|
Number of
|
|
|
(b)
|
|
|
for Future Issuance
|
|
|
|
|
Securities to be
|
|
|
Weighted-Average
|
|
|
Under Equity
|
|
|
|
|
Issued Upon Exercise
|
|
|
Exercise Price of
|
|
|
Compensation Plans
|
|
|
|
|
of Outstanding
|
|
|
Outstanding
|
|
|
(Excluding Securities
|
|
|
|
|
Options, Warrants
|
|
|
Options, Warrants
|
|
|
Reflected in Column
|
|
|
Plan Category
|
|
and Rights
|
|
|
and Rights
|
|
|
(a)
|
|
|
|
|
Equity compensation plans approved by security holders
|
|
|
2,216,026
|
|
|
$
|
6.96
|
|
|
|
1,032,914
|
|
|
Equity compensation plans not approved by security holders
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
2,216,026
|
|
|
$
|
6.96
|
|
|
|
1,032,914
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
38
Stock
Price Performance Graph
The following graph compares the cumulative total stockholder
return on our common stock from October 17, 2007 (the date
our common stock began trading on the NASDAQ Global Market)
through December 31, 2009 to that of the cumulative return
over such period for (i) The NASDAQ Stock Market Composite
Index, and (ii) NASDAQ Medical Equipment Index. Total
stockholder return assumes $100.00 invested at the beginning of
the period in our common stock and in each of the comparative
indices. The graph further assumes that such amount was
initially invested in our common stock at the closing market
price on the first day of trading, and that any dividends have
been reinvested. We have not paid any dividends on our common
stock. The stock price performance on the following graph is not
necessarily indicative of future stock price performance.
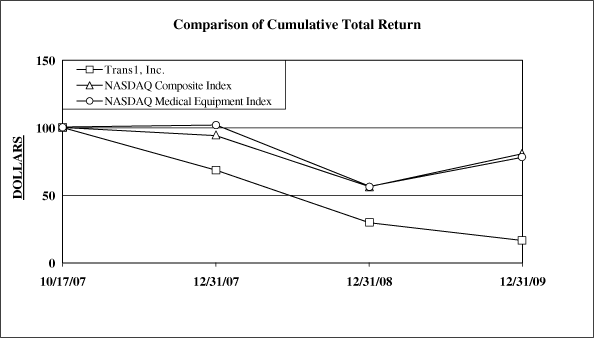
The material in the above performance graph does not
constitute soliciting material and should not be deemed filed or
incorporated by reference into any other Company filing, whether
under the Securities Act of 1933, as amended, or the Securities
Exchange Act of 1934, as amended, whether made on, before or
after the date of this report and irrespective of any general
incorporation language in such filing, except to the extent we
specifically incorporate this performance graph by reference
therein.
39
|
|
|
Item 6.
|
Selected
Financial Data.
|
The selected financial data has been derived from our audited
consolidated financial statements. You should read the following
financial information together with the information under
“Management’s Discussion and Analysis of Financial
Condition and Results of Operations” and our consolidated
financial statements and the notes included elsewhere in this
report.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
2006
|
|
|
2005
|
|
|
|
|
(In thousands, except share data)
|
|
|
|
|
Consolidated Statements of Operations Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenue
|
|
$
|
29,807
|
|
|
$
|
25,304
|
|
|
$
|
16,473
|
|
|
$
|
5,812
|
|
|
$
|
1,469
|
|
|
Cost of revenue
|
|
|
5,687
|
|
|
|
4,315
|
|
|
|
3,042
|
|
|
|
1,580
|
|
|
|
386
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gross profit
|
|
|
24,120
|
|
|
|
20,989
|
|
|
|
13,431
|
|
|
|
4,232
|
|
|
|
1,083
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating expenses
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
|
6,439
|
|
|
|
4,081
|
|
|
|
3,885
|
|
|
|
3,816
|
|
|
|
2,100
|
|
|
Sales and marketing
|
|
|
34,098
|
|
|
|
29,375
|
|
|
|
15,706
|
|
|
|
9,288
|
|
|
|
2,635
|
|
|
General and administrative
|
|
|
7,184
|
|
|
|
7,116
|
|
|
|
3,801
|
|
|
|
1,596
|
|
|
|
1,637
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
47,721
|
|
|
|
40,572
|
|
|
|
23,392
|
|
|
|
14,700
|
|
|
|
6,372
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating loss
|
|
|
(23,601
|
)
|
|
|
(19,583
|
)
|
|
|
(9,961
|
)
|
|
|
(10,468
|
)
|
|
|
(5,289
|
)
|
|
Interest income
|
|
|
405
|
|
|
|
2,548
|
|
|
|
1,384
|
|
|
|
858
|
|
|
|
264
|
|
|
Other income (expense)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
131
|
|
|
|
(17
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(23,196
|
)
|
|
$
|
(17,035
|
)
|
|
$
|
(8,577
|
)
|
|
$
|
(9,479
|
)
|
|
$
|
(5,042
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per common share - basic and diluted
|
|
$
|
(1.13
|
)
|
|
$
|
(0.84
|
)
|
|
$
|
(1.46
|
)
|
|
$
|
(3.91
|
)
|
|
$
|
(2.25
|
)
|
|
Weighted average common shares outstanding — basic and
diluted
|
|
|
20,603,600
|
|
|
|
20,288,711
|
|
|
|
5,872,008
|
|
|
|
2,423,223
|
|
|
|
2,243,018
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
2009
|
|
2008
|
|
2007
|
|
2006
|
|
2005
|
|
|
|
Consolidated Balance Sheet Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash, cash equivalents and short-term investments
|
|
$
|
55,251
|
|
|
$
|
77,266
|
|
|
$
|
93,921
|
|
|
$
|
14,962
|
|
|
$
|
25,994
|
|
|
Working capital
|
|
|
63,467
|
|
|
|
84,174
|
|
|
|
98,351
|
|
|
|
17,448
|
|
|
|
26,696
|
|
|
Total assets
|
|
|
68,991
|
|
|
|
90,491
|
|
|
|
102,856
|
|
|
|
20,004
|
|
|
|
28,013
|
|
|
Preferred stock
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
40,089
|
|
|
|
40,089
|
|
|
Common stock
|
|
|
2
|
|
|
|
2
|
|
|
|
2
|
|
|
|
—
|
|
|
|
—
|
|
|
Additional paid in capital
|
|
|
136,402
|
|
|
|
133,507
|
|
|
|
130,325
|
|
|
|
820
|
|
|
|
219
|
|
|
Total stockholders’ equity/(deficit)
|
|
|
65,280
|
|
|
|
85,586
|
|
|
|
99,439
|
|
|
|
(21,529
|
)
|
|
|
(12,654
|
)
|
40
|
|
|
Item 7.
|
Management’s
Discussion and Analysis of Financial Condition and Results of
Operations
|
The following discussion of our financial condition and results
of operations should be read in conjunction with our
consolidated financial statements and the related notes to our
consolidated financial statements included in this report. The
following discussion contains forward-looking statements. See
“Cautionary Note Regarding Forward-Looking Statements”
on page 1 of this report.
Overview
We are a medical device company focused on designing, developing
and marketing products that implement our proprietary minimally
invasive surgical approach to treat degenerative disc disease
and instability affecting the lower lumbar region of the spine.
Using this pre-sacral approach, a surgeon can access discs in
the lower lumbar region of the spine through a 1.5 cm incision
adjacent to the tailbone and can perform an entire fusion
procedure through a small tube that provides direct access to
the intervertebral space. We developed our pre-sacral approach
to allow spine surgeons to access and treat intervertebral
spaces without compromising important surrounding soft tissue.
We believe this approach enables fusion procedures to be
performed with low complication rates, low blood loss, short
hospital stays, fast recovery times and reduced pain. We have
developed and currently market in the United States and Europe
two single-level fusion products, AxiaLIF and AxiaLIF 360°,
and a two-level fusion product, the AxiaLIF 2L, which include
the Vectre and Avatar posterior fixation systems for lumbar
fixation supplemental to AxiaLIF fusion. All of our AxiaLIF
products are delivered using our pre-sacral approach.
From our incorporation in 2000 through 2004, we devoted
substantially all of our resources to research and development
and start-up
activities, consisting primarily of product design and
development, clinical trials, manufacturing, recruiting
qualified personnel and raising capital. We received FDA 510(k)
clearance for our AxiaLIF product in the fourth quarter of 2004,
and commercially introduced our AxiaLIF product in the United
States in the first quarter of 2005. We received FDA 510(k)
clearance for our AxiaLIF 360° product in the United States
in the third quarter of 2005 and began commercialization in the
United States in the third quarter of 2006. We received a CE
mark to market AxiaLIF in the European market in the first
quarter of 2005 and began commercialization in the first quarter
of 2006. For AxiaLIF 360°, we received a CE mark in the
first quarter of 2006. We received a CE mark for our AxiaLIF 2L
product in the third quarter of 2006 and began commercialization
in the European market in the fourth quarter of 2006. We
received 510(k) clearance for the AxiaLIF 2L from the FDA and
began marketing this product in the United States in the second
quarter of 2008. In November 2009, we commenced the limited
market release of our next generation Vectre facet screw system.
Our AxiaLIF 2L+ product received FDA 510 (k) clearance, and
we began marketing the product, in January 2010. In January
2010, we also entered into a partnership agreement with Life
Spine, Inc to distribute Avatar, a minimally invasive pedicle
screw system. We currently sell our products through a direct
sales force, independent sales agents and international
distributors.
We rely on third parties to manufacture most of our products and
their components. We believe these manufacturing relationships
allow us to work with suppliers who have the best specific
competencies while we minimize our capital investment, control
costs and shorten cycle times, all of which allows us to compete
with larger volume manufacturers of spine surgery products.
Since inception, we have been unprofitable. As of
December 31, 2009, we had an accumulated deficit of
$71.1 million.
We expect to continue to invest in maintaining our sales and
marketing infrastructure for our products in order to gain wider
acceptance for them. We also expect to continue to invest in
research and development and related clinical trials, and
increase general and administrative expenses as we grow. As a
result, we will need to generate significant revenue in order to
achieve profitability.
41
Financial
Operations
Revenue
We generate revenue from the sales of our procedure kits and
implants used in our AxiaLIF fusion procedure for the treatment
of degenerative disc disease and instability. Our revenue is
generated by our direct sales force, independent sales agents
and independent distributors. Our sales representatives or
independent sales agents hand deliver the procedure kit to the
customer on the day of the surgery or several days prior to the
surgery. The sales representative or independent agent is then
responsible for reporting the delivery of the procedure kit, and
the date of the operation to the corporate office for proper
revenue recognition. We recognize revenue upon the confirmation
that the procedure kit has been used in a surgical procedure.
The other sales method is for sales to distributors outside the
United States. These distributors order multiple procedure kits
at one time to have on hand. These transactions require the
customer to send in a purchase order before shipment will be
made to the customer. We determine revenue recognition on a case
by case basis dependent upon the terms and conditions of each
individual distributor agreement. Under the distributor
agreements currently in place, a distributor only has the right
of return for defective products and, accordingly, revenue is
recognized upon shipment of our products to our independent
distributors. Although we intend to continue to expand our
international sales and marketing efforts, we expect that a
substantial amount of our revenues will be generated in the
United States in future periods.
Cost
of Revenue
Cost of revenue consists primarily of material and overhead
costs related to our products. Cost of revenue also includes
facilities-related costs, such as rent, utilities and
depreciation.
Research
and Development
Research and development expenses consist primarily of personnel
costs, including stock-based compensation expense, within our
product development, regulatory and clinical functions and the
costs of clinical studies and product development projects. In
future periods, we expect research and development expenses to
grow as we continue to invest in basic research, clinical
trials, product development and in our intellectual property.
Sales
and Marketing
Sales and marketing expenses consist of personnel costs,
including stock-based compensation expense, sales commissions
paid to our direct sales representatives and independent sales
agents, and costs associated with physician training programs,
promotional activities, and participation in medical
conferences. In future periods, we expect sales and marketing
expenses to increase as we expand our sales and marketing
efforts.
General
and Administrative
General and administrative expenses consist of personnel costs,
including stock-based compensation, related to the executive,
finance, business development and information technology and
human resource functions, as well as professional service fees,
legal fees, accounting fees, insurance costs and general
corporate expenses. We expect general and administrative
expenses to increase as we grow our business.
Interest
Income
Interest income is primarily composed of interest earned on our
cash, cash equivalents and
available-for-sale
securities.
42
Results
of Operations
Comparison
of the Years Ended December 31, 2009, 2008 and
2007
Revenue. Revenue increased to
$29.8 million in 2009 from $25.3 million in 2008 and
$16.5 million in 2007. The $4.5 million increase in
revenue from 2008 to 2009, and the $8.8 million increase in
revenue from 2007 to 2008, was primarily attributable to an
increase in the number of products sold, which we believe
resulted from the continued market acceptance of our AxiaLIF,
AxiaLIF 360°, and AxiaLIF 2L products. None of this
increase was attributable to price increases. Beginning in the
second quarter of 2009, our revenue was negatively impacted by
lower than expected case volume as a result of concerns and
uncertainty in the marketplace surrounding physician
reimbursement for our AxiaLIF procedure. We are addressing these
issues with increased education and support resources for our
current and prospective surgeon users. These issues have more of
an impact on our AxiaLIF 360° procedure where ours is the
only procedure being performed. In cases where our procedure is
being performed in conjunction with other techniques or in
complex cases, we have seen a less-pronounced impact on cases.
Domestically, sales of our AxiaLIF 360° product decreased
to $7.8 million in 2009 from $8.5 million in 2008,
which was an increase from $6.8 million in 2007. Sales of
our AxiaLIF 2L product, which began commercialization in the
United States in the second quarter of 2008 and which has a
higher selling price than our other products, increased to
$8.0 million in 2009 from $3.3 million in 2008. As a
result, average selling prices in the United States increased to
approximately $10,500 in 2009 from approximately $9,850 in 2008
and $9,250 in 2007. In 2009, 2008 and 2007, we recorded 2,578,
2,280 and 1,591 domestic AxiaLIF cases, respectively. This
included 780, 852 and 677 AxiaLIF 360° cases in 2009, 2008
and 2007, respectively, and 596 and 229 AxiaLIF 2L cases in 2009
and 2008, respectively. Additionally, in 2009, 2008 and 2007, we
generated $953,000, $868,000 and $359,000, respectively, in
revenues from stand alone sales of our facet screw system.
Revenue generated outside the United States decreased to
$1.8 million in 2009 from $2.0 million in 2008, which
was an increase from $1.4 million in 2007. In 2009, 2008
and 2007, initial stocking shipments to new distributors were
$122,000, $382,000 and $438,000, respectively. In 2009, we began
direct sales through our own sales representatives and agents in
Europe and generated revenue of $543,000. In 2009, 2008 and
2007, 94%, 92% and 92%, respectively, of our revenues were
generated in the United States.
Cost of Revenue. Cost of revenue increased to
$5.7 million in 2009 from $4.3 million in 2008 and
$3.0 million in 2007. The $1.4 million increase from
2008 to 2009 and the $1.3 million increase from 2007 to
2008 were primarily the result of higher material and overhead
costs associated with increased sales volumes of our AxiaLIF,
AxiaLIF 360° and AxiaLIF 2L products. As a percentage of
revenue, cost of revenue was 19.1% in 2009, 17.1% in 2008 and
18.5% in 2007. The increase in cost of revenue as a percentage
of revenue from 2008 to 2009 was primarily related to specific
inventory reserves of $0.4 million taken in 2009 for excess
inventory and the under-absorption of manufacturing overhead of
$0.2 million as we reduced inventory purchases in response
to lower than anticipated business volume. The decrease in cost
of revenue as a percentage of revenue from 2007 to 2008 was
primarily attributable to increased efficiencies associated with
higher production and sales volumes.
Research and Development. Research and
development expenses increased to $6.4 million in 2009 from
$4.1 million in 2008 and $3.9 million in 2007. The
$2.3 million increase in expense from 2008 to 2009 was
primarily the result of increased research and development and
clinical project-related spending. The $0.2 million
increase in expense in 2008 compared to 2007 was primarily the
result of increases in personnel related costs, including
stock-based compensation expense, of $0.4 million,
partially offset by reductions in project related research and
development and clinical trial costs of $0.2 million.
Sales and Marketing. Sales and marketing
expenses increased to $34.1 million in 2009 from
$29.4 million in 2008 and $15.7 million in 2007. The
increase in expense from 2008 to 2009 of $4.7 million was
primarily attributable to increased personnel related costs,
including commissions and stock-based compensation expense, of
$3.6 million, as we continued to build out our sales and
marketing organization in order to continue to drive global
market acceptance of our AxiaLIF products, increased training
costs of $0.6 million and increased promotional activities
of $0.5 million. The increase in expenses from 2007 to 2008
of $13.7 million was primarily attributable to increased
personnel related costs, including commissions and stock-
43
based compensation expense, of $7.2 million, increased
travel costs of $2.7 million related to our expanded sales
force, increased training costs of $1.7 million and
increased tradeshow and promotional activities of
$1.2 million. In January 2010, we reduced our
U.S. sales force to better address current market
opportunities.
General and Administrative. General and
administrative expenses increased to $7.2 million in 2009
from $7.1 million in 2008 and $3.8 million in 2007.
The increase in expenses from 2008 to 2009 of $0.1 million
was primarily attributable to increased personnel related costs,
including stock-based compensation expenses, of
$0.5 million, partially offset by a decrease in consulting
expenses, of $0.4 million. The increase in expenses from
2007 to 2008 of $3.3 million was primarily attributable to
increased personnel related costs, including stock-based
compensation expenses, of $1.0 million, increased
professional fees of $1.5 million related to our operating
as a public company, including accounting, legal and board of
director expenses, increased directors and officers insurance
expense of $0.3 million and higher franchise taxes of
$0.2 million.
Other and Interest Income (Expense). Other and
interest income decreased to $0.4 million in 2009 from
$2.5 million in 2008 and $1.4 million in 2007. The
decrease of $2.1 million in other and interest income from
2008 to 2009 was primarily the result of historically low
interest rates, the conservative and liquid nature of the
investment portfolio and lower investment balances. The increase
$1.1 million from 2007 to 2008 was primarily due to
interest income on higher average cash and investment balances
from the net proceeds to the Company of $86.7 million from
our October 2007 IPO.
Liquidity
and Capital Resources
Sources
of Liquidity
Since our inception in 2000, we have incurred significant losses
and, as of December 31, 2009, we had an accumulated deficit
of $71.1 million. We have not yet achieved profitability,
and anticipate that we will continue to incur losses in the near
term. We expect that research and development, sales and
marketing and general and administrative expenses will continue
to grow and, as a result, we will need to generate significant
revenues to achieve profitability. To date, our operations have
been funded primarily with proceeds from the sale of preferred
stock and, most recently, the net proceeds from our October 2007
initial public offering. Gross proceeds from our preferred stock
sales totaled $40.5 million to date, and the net proceeds
from our October 2007 initial public offering were approximately
$86.7 million.
As of December 31, 2009, we did not have any outstanding
debt financing arrangements, we had working capital of
$63.5 million and our primary source of liquidity was
$55.3 million in cash, cash equivalents and short-term
investments. We currently invest our cash and cash equivalents
primarily in money market treasury funds and our short-term
investments primarily in U.S. agency backed debt
instruments.
Cash, cash equivalents and short-term investments
decreased from $77.3 million at December 31, 2008 to
$55.3 million at December 31, 2009. The decrease of
$22.0 million was primarily the result of net cash used in
operating activities of $20.8 million and purchases of
property and equipment of $1.3 million.
Cash, cash equivalents and short-term investments
decreased from $93.9 million at December 31, 2007 to
$77.3 million at December 31, 2008. The decrease of
$16.6 million was primarily the result of net cash used in
operating activities of $15.7 million and purchases of
property and equipment of $1.1 million.
Cash
Flows
Net Cash Used in Operating Activities. Net
cash used in operating activities was $20.8 million in
2009, $15.7 million in 2008 and $7.3 million in 2007.
For each of these periods, net cash used in operating activities
was attributable primarily to net losses after adjustment for
non-cash items, such as depreciation, stock-based compensation
expense, inventory reserves and increases in working capital
requirements to support the increased market acceptance of our
AxiaLIF products. The increase in working capital requirements
for 2009 was driven by the growth in inventories, and for 2008
and 2007 by the growth in inventories and related payables,
along with smaller changes in all years in prepaid assets and
accrued liabilities due to the timing of activities in those
accounts.
44
Net Cash Provided by (Used In) Investing
Activities. Net cash provided by investing
activities was $8.0 million in 2009. Net cash used in
investing activities was $7.1 million in 2008 and
$19.8 million in 2007. For each of these periods, this
amount reflected purchases or sales and maturities of
investments and purchases of property and equipment, primarily
for research and development, information technology,
manufacturing operations and capital improvements to our
facilities.
Net Cash Provided by Financing Activities. Net
cash provided by financing activities in 2009 and 2008 was
$0.1 million and $0.2 million, respectively,
representing proceeds from the issuance of shares of our common
stock upon the exercise of stock options. Net cash provided by
financing activities in 2007 was $86.8 million, which
represented the net proceeds to the Company of our October 2007
initial public offering of 6,325,000 shares of our common
stock, resulting in net proceeds to us, after deducting
underwriting discounts, commissions and offering expenses, of
approximately $86.7 million and $0.1 million in
proceeds from the issuance of shares of our common stock upon
the exercise of stock option.
Operating
Capital and Capital Expenditure Requirements
We believe that our existing cash and cash equivalents, together
with cash received from sales of our products, will be
sufficient to meet our cash needs for at least the next two
years. We intend to spend substantial sums on sales and
marketing initiatives to support the ongoing commercialization
of our products and on research and development activities,
including product development, regulatory and compliance,
clinical studies in support of our currently marketed products
and future product offerings, and the enhancement and protection
of our intellectual property. We may need to obtain additional
financing to pursue our business strategy, to respond to new
competitive pressures or to take advantage of opportunities that
may arise. The sale of additional equity or convertible debt
securities could result in dilution to our stockholders. If
additional funds are raised through the issuance of debt
securities, these securities could have rights senior to those
associated with our common stock and could contain covenants
that would restrict our operations. Any additional financing may
not be available in amounts or on terms acceptable to us, if at
all. If we are unable to obtain this additional financing, we
may be required to reduce the scope of our planned product
development and marketing efforts.
Contractual
Obligations
The following table discloses information about our contractual
obligations by the year in which payments are due as of
December 31, 2009:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Payments Due by Year
|
|
|
|
|
|
Less Than
|
|
|
|
|
|
After 5
|
|
Contractual Obligations
|
|
Total
|
|
1 Year
|
|
1-3 Years
|
|
3-5 Years
|
|
Years
|
|
|
|
( In thousands)
|
|
|
|
Operating leases(1)
|
|
$
|
4,128
|
|
|
$
|
374
|
|
|
$
|
695
|
|
|
$
|
735
|
|
|
$
|
2,324
|
|
|
|
|
|
(1) |
|
We rent office space under an operating lease which expires in
2019. |
Off-Balance
Sheet Arrangements
As of December 31, 2009, we did not have any outstanding
debt or available debt financing arrangements or off-balance
sheet liabilities.
Critical
Accounting Policies and Estimates
Our discussion and analysis of our financial condition and
results of operations is based upon our consolidated financial
statements, which have been prepared in accordance with
accounting principles generally accepted in the United States.
The preparation of financial statements requires management to
make estimates and judgments that affect the reported amounts of
assets and liabilities, revenue and expenses, and disclosures of
contingent assets and liabilities at the date of the financial
statements. On an on-going basis, we evaluate our estimates,
including those related to revenue recognition, accounts
receivable, inventories, income taxes and stock-based
compensation. We use authoritative pronouncements, historical
experience and other
45
assumptions as the basis for making estimates. Actual results
could differ from those estimates under different assumptions or
conditions.
We believe the following critical accounting policies affect our
more significant judgments and estimates used in the preparation
of our consolidated financial statements.
Revenue Recognition. We recognize revenue
based on the following criteria: (i) persuasive evidence
that an arrangement exists with the customer; (ii) the
delivery of the products
and/or
services has occurred (title has transferred); (iii) the
selling price has been fixed for the products or services
delivered; and (iv) the collection is reasonably assured.
Revenue is generated from the sale of our implants and procedure
kits, which consist of disposable instruments. We have two
distinct sale methods. The first method is when procedure kits
are sold directly to hospitals or surgical centers. Our sales
representatives or independent sales agents hand deliver the
procedure kit to the customer on the day of the surgery or
several days prior to the surgery. The sales representative or
independent agent is then responsible for reporting the delivery
of the procedure kit, and the date of the operation to our
corporate office for proper revenue recognition. We recognize
revenue upon the confirmation that the procedure kit has been
used in a surgical procedure. The other sales method is for
sales to distributors outside the United States. These
distributors order multiple procedure kits at one time to have
on hand. These transactions require the customer to send in a
purchase order before shipment will be made to the customer. We
determine revenue recognition on a case by case basis dependent
upon the terms and conditions of each individual distributor
agreement. Under the distributor agreements currently in place,
a distributor only has the right of return for defective
products and, accordingly, revenue is recognized upon shipment.
Accounts Receivable and Allowances. We monitor
collections and payments from our customers and maintain an
allowance for doubtful accounts based upon our historical
experience and any specific customer collection issues that we
have identified. While our credit losses have historically been
within our expectations and the allowance established, we may
not continue to experience the same credit loss rates that we
have in the past. We make estimates on the collectability of
customer accounts based primarily on analysis of historical
trends and experience and changes in customers’ financial
condition. Management uses its best judgment, based on the best
available facts and circumstances, and records a reserve against
the amounts due to reduce the receivable to the amount that is
expected to be collected.
These reserves are reevaluated and adjusted as additional
information is received that impacts the amount reserved.
Inventory. We state our inventories at the
lower of cost or market, computed on a standard cost basis,
which approximates actual cost on a
first-in,
first-out basis and market being determined as the lower of
replacement cost or net realizable value. Costs are monitored on
an annual basis and updated as necessary to reflect changes in
supplier costs and the rate of our overhead absorption is
adjusted based on projections of our manufacturing department
costs and production plan. Inventory reserves are established
when conditions indicate that the selling price could be less
than cost due to obsolescence, usage, or we deem we hold
excessive levels of inventory based on market demand.
Accounting for Income Taxes. Significant
management judgment is required in determining our provision for
income taxes, our deferred tax assets and liabilities and any
valuation allowance recorded against our net deferred tax
assets. We have recorded a full valuation allowance on our net
deferred tax assets as of December 31, 2009 due to
uncertainties related to our ability to utilize our deferred tax
assets in the foreseeable future.
Stock-Based Compensation. Effective
January 1, 2006, we adopted Accounting Standards
Codification, (“ASC”) 718 (formerly Statement of
Financial Accounting Standards No. 123R
(“SFAS 123R”), Share-Based Payment) using
the prospective transition method, which requires the
measurement and recognition of compensation expense for all
share-based payment awards granted, modified and settled to our
employees and directors after January 1, 2006. The fair
value of stock options was estimated using a Black-Scholes
option pricing model. This model requires the input of
subjective assumptions, including expected stock price
volatility, expected life and estimated forfeitures of each
award. The fair value of equity-based awards is
46
amortized over the vesting period of the award, and we have
elected to use the straight-line method of amortization. Due to
the limited amount of historical data available to us,
particularly with respect to stock-price volatility, employee
exercise patterns and forfeitures, actual results could differ
materially from our expectations.
Recent
Accounting Pronouncements
New
Accounting Standards
In February 2008, the Financial Accounting Standards Board
(“FASB”) issued ASC 820 (formerly Staff Position
No. FAS 157-2,
“Fair Value Measurements”), which delayed the
effective date of ASC 820 for non-financial assets and
liabilities to fiscal years beginning after November 15,
2008. We adopted ASC 820 for our non-financial assets and
non-financial liabilities on January 1, 2009, and it did
not have a material impact on our consolidated financial
statements.
In June 2009, the FASB issued the FASB Accounting Standards
Codification (“ASC”). The Codification has become the
single source for all authoritative GAAP recognized by the FASB
to be applied for financial statements issued for periods ending
after September 15, 2009. We applied the Codification to
our Annual Report on
Form 10-K
for the period ending December 31, 2009. The Codification
does not change GAAP and did not have an effect on our
consolidated financial position or results of operations.
In May 2009, the FASB issued ASC 855 (formerly
SFAS No. 165, “Subsequent Events”), which
establishes general standards of accounting for and disclosures
of events that occur after the balance sheet date but before the
financial statements are issued or are available to be issued.
ASC 855 provides guidance on the period after the balance sheet
date during which management of a reporting entity should
evaluate events or transactions that may occur for potential
recognition or disclosure in the financial statements, the
circumstances under which an entity should recognize events or
transactions occurring after the balance sheet date in its
financial statements and the disclosures that an entity should
make about events or transactions that occurred after the
balance sheet date. We adopted ASC 855 during the second quarter
of 2009, and its application did not have a material impact on
our consolidated financial statements.
No other recently issued, but not yet effective, accounting
standards are believed to have a material impact on us.
|
|
|
Item 7A.
|
Quantitative
and Qualitative Disclosures About Market Risk
|
Our exposure to interest rate risk at December 31, 2009 is
related to our investment portfolio. We invest our excess cash
primarily in money market funds and debt instruments of the
U.S. government and its agencies. Due to the short-term
nature of these investments, we have assessed that there is no
material exposure to interest rate risk arising from our
investments. Thus, a hypothetical 100 basis point adverse
move in interest rates along the entire interest rate yield
curve would not materially affect the fair market value of our
interest-sensitive financial investments. Declines in interest
rates over time will, however, reduce our investment income,
while increases in interest rates over time will increase our
interest expense. Historically, and as of December 31,
2009, we have not used derivative instruments or engaged in
hedging activities.
Although substantially all of our sales and purchases are
denominated in U.S. dollars, future fluctuations in the
value of the U.S. dollar may affect the competitiveness of
our products outside the United States. We do not believe,
however, that we currently have significant direct foreign
currency exchange rate risk and have not hedged exposures
denominated in foreign currencies.
47
|
|
|
Item 8.
|
Consolidated
Financial Statements and Supplementary Data.
|
The consolidated financial statements and supplementary data
required by this item are set forth under Item 15.
|
|
|
Item 9.
|
Changes
in and Disagreements with Accountants on Accounting and
Financial Disclosure.
|
None.
|
|
|
Item 9A.
|
Controls
and Procedures
|
Evaluation
of Disclosure Controls and Procedures
Our management, with the participation of our principal
executive officer and principal financial officer, evaluated the
effectiveness of our disclosure controls and procedures (as such
term is defined in
Rules 13a-15(e)
and
15d-15(e)
under the Securities Exchange Act of 1934, as amended, or the
Exchange Act) as of December 31, 2009. We maintain
disclosure controls and procedures that are designed to provide
a reasonable assurance level that information required to be
disclosed in our reports filed or submitted under the Exchange
Act is recorded, processed, summarized and reported within the
time periods specified in the SEC’s rules and forms and
that such information is accumulated and communicated to our
management, including our principal executive officer and
principal financial officer, as appropriate, to allow for timely
decisions regarding required disclosure. Our management
recognizes that any controls and procedures, no matter how well
designed and operated, can provide only reasonable assurance of
achieving their objectives and management necessarily applies
its judgment in evaluating the cost-benefit relationship of
possible controls and procedures. Based on the evaluation of our
disclosure controls and procedures as of December 31, 2009,
our principal executive officer and principal financial officer
concluded that, as of such date, our disclosure controls and
procedures were effective at the reasonable assurance level.
Internal
Control Over Financial Reporting
Management’s annual report on internal control over
financial reporting. Management’s report on
our internal control over financial reporting is included on
page 64 hereof. The report of our independent registered
public accounting firm related to their assessment of the
effectiveness of internal control over financial reporting is
included on page 65 hereof.
Changes in Internal Control over Financial
Reporting. There has been no change in our
internal control over financial reporting (as such term is
defined in
Rules 13a-15(f)
and
15d-15(f)
under the Exchange Act) during our most recent fiscal quarter
that has materially affected, or is reasonably likely to
materially affect, our internal control over financial reporting.
|
|
|
Item 9B.
|
Other
Information.
|
None.
PART III
|
|
|
Item 10.
|
Directors,
Executive Officers and Corporate Governance.
|
The information required by this Item is incorporated herein by
reference to our definitive Proxy Statement, which will be filed
within 120 days of December 31, 2009, and delivered to
stockholders in connection with our annual meeting of
stockholders.
|
|
|
Item 11.
|
Executive
Compensation.
|
The information required by this Item is incorporated herein by
reference to our definitive Proxy Statement, which will be filed
within 120 days of December 31, 2009, and delivered to
stockholders in connection with our annual meeting of
stockholders.
48
The material incorporated herein by reference to the material
under the caption “Compensation Committee Report” in
the Proxy Statement shall be deemed furnished, and not filed, in
this report and shall not be deemed incorporated by reference
into any filing under the Securities Act of 1933, as amended, or
the Securities Exchange Act of 1934, as amended, as a result of
this furnishing, except to the extent that we specifically
incorporate it by reference.
|
|
|
Item 12.
|
Security
Ownership of Certain Beneficial Owners and Management and
Related Stockholder Matters.
|
With the exception of the information regarding securities
authorized for issuance under our equity compensation plans,
which is set forth in Item 5 of this report under the
heading “Securities Authorized For Issuance under Equity
Compensation Plans” and incorporated herein by reference,
the information required by this Item is incorporated herein by
reference to our definitive Proxy Statement, which will be filed
within 120 days of December 31, 2009, and delivered to
stockholders in connection with our annual meeting of
stockholders.
|
|
|
Item 13.
|
Certain
Relationships and Related Transactions, and Director
Independence.
|
The information required by this Item is incorporated herein by
reference to our definitive Proxy Statement, which will be filed
within 120 days of December 31, 2009, and delivered to
stockholders in connection with our annual meeting of
stockholders.
|
|
|
Item 14.
|
Principal
Accounting Fees and Services.
|
The information required by this Item is incorporated herein by
reference to our definitive Proxy Statement, which will be filed
within 120 days of December 31, 2009, and delivered to
stockholders in connection with our annual meeting of
stockholders.
PART IV
|
|
|
Item 15.
|
Exhibits,
Financial Statement Schedules.
|
(a) Financial Statements
(1) Index to Financial Statements
| |
|
|
|
|
|
|
|
Page
|
|
|
|
Management Report on Internal Control Over Financial Reporting
|
|
|
52
|
|
|
Report of Independent Registered Public Accounting Firm
|
|
|
53
|
|
|
Consolidated Balance Sheets
|
|
|
54
|
|
|
Consolidated Statements of Operations
|
|
|
55
|
|
|
Consolidated Statements of Stockholders’ Equity (Deficit)
|
|
|
56
|
|
|
Consolidated Statements of Cash Flows
|
|
|
57
|
|
|
Notes to Consolidated Financial Statements
|
|
|
58
|
|
|
|
|
|
|
|
|
(2) Financial Statement Schedule:
Schedule II — Valuation Accounts
|
|
|
71
|
|
All other financial statement schedules have been omitted
because they are not applicable, not required or the information
required is shown in the financial statements or the notes
thereto.
The exhibits filed with this Annual Report on
Form 10-K
are listed in the Exhibit Index immediately following the
financial statement schedules, which Exhibit Index is
incorporated herein by reference.
49
SIGNATURES
Pursuant to the requirements of Section 13(a) or 15(d) of
the Securities Exchange Act of 1934, the registrant has duly
caused this report to be signed on its behalf by the undersigned
thereunto duly authorized.
TranS1 Inc.
Richard Randall
President and Chief Executive Officer
Date: March 12, 2010
POWER OF
ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose
signature appears below constitutes and appoints Richard Randall
and Michael Luetkemeyer, jointly and severally, his
attorneys-in-fact, each with the power of substitution, for him
in any and all capacities, to sign any amendments to this Report
on
Form 10-K,
and to file the same, with exhibits thereto and other documents
in connection therewith with the Securities and Exchange
Commission, hereby ratifying and confirming all that each of
said attorneys-in-fact, or his substitute or substitutes may do
or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of
1934, this report has been signed below by the following persons
on behalf of the registrant and in the capacities and on the
dates indicated.
| |
|
|
|
|
|
|
|
Signature
|
|
Title
|
|
Date
|
|
|
|
|
|
|
|
|
|
/s/ Richard
Randall
Richard
Randall
|
|
President and Chief Executive Officer (Principal Executive
Officer)
|
|
March 12, 2010
|
|
|
|
|
|
|
|
/s/ Michael
Luetkemeyer
Michael
Luetkemeyer
|
|
Chief Financial Officer
(Principal Financial and Accounting Officer)
|
|
March 12, 2010
|
|
|
|
|
|
|
|
/s/ Michael
Carusi
Michael
Carusi
|
|
Director
|
|
March 12, 2010
|
|
|
|
|
|
|
|
/s/ Mitchell
Dann
Mitchell
Dann
|
|
Director
|
|
March 12, 2010
|
|
|
|
|
|
|
|
/s/ Paul
LaViolette
Paul
LaViolette
|
|
Director
|
|
March 12, 2010
|
|
|
|
|
|
|
|
/s/ Jonathan
Osgood
Jonathan
Osgood
|
|
Director
|
|
March 12, 2010
|
|
|
|
|
|
|
|
/s/ James
Shapiro
James
Shapiro
|
|
Director
|
|
March 12, 2010
|
|
|
|
|
|
|
|
/s/ Joseph
Slattery
Joseph
Slattery
|
|
Director
|
|
March 12, 2010
|
50
Management
Report on Internal Control Over Financial Reporting
The management of TranS1 is responsible for establishing and
maintaining adequate internal control over financial reporting.
TranS1’s internal control system was designed to provide
reasonable assurance to the Company’s management and board
of directors regarding the preparation and fair presentation of
published financial statements.
Because of its inherent limitations, internal control over
financial reporting may not prevent or detect misstatements.
Also, projections of any evaluation of effectiveness to future
periods are subject to the risk that controls may become
inadequate because of changes in conditions, or that the degree
of compliance with the policies or procedures may deteriorate.
TranS1’s management assessed the effectiveness of the
Company’s internal control over financial reporting as of
December 31, 2009. In making this assessment, it used the
criteria set forth in Internal Control — Integrated
Framework issued by the Committee of Sponsoring
Organizations of the Treadway Commission. Based on
management’s assessment, we concluded that, as of
December 31, 2009, our internal control over financial
reporting was effective based on those criteria.
The effectiveness of the Company’s internal control over
financial reporting as of December 31, 2009 has been
audited by PricewaterhouseCoopers LLP, an independent registered
public accounting firm, as stated in their report which appears
herein.
52
Report of
Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of TranS1 Inc.
In our opinion, the accompanying consolidated financial
statements for 2009 and the financial statements for 2008 listed
in the index appearing under Item 15(a)(1) present fairly,
in all material respects, the financial position of TranS1 Inc.
and its subsidiaries at December 31, 2009 and 2008, and the
results of their operations and their cash flows for each of the
three years in the period ended December 31, 2009 in
conformity with accounting principles generally accepted in the
United States of America. In addition, in our opinion, the
financial statement schedule listed in the index appearing under
Item 15(a)(2) presents fairly, in all material respects,
the information set forth therein when read in conjunction with
the related consolidated financial statements. Also in our
opinion, the Company maintained, in all material respects,
effective internal control over financial reporting as of
December 31, 2009, based on criteria established in
Internal Control - Integrated Framework issued by the Committee
of Sponsoring Organizations of the Treadway Commission (COSO).
The Company’s management is responsible for these financial
statements and financial statement schedule, for maintaining
effective internal control over financial reporting and for its
assessment of the effectiveness of internal control over
financial reporting, included in the accompanying
Management’s Report on Internal Control over Financial
Reporting. Our responsibility is to express opinions on these
financial statements, on the financial statement schedule and on
the Company’s internal control over financial reporting
based on our integrated audits which were integrated audits in
2009 and 2008. We conducted our audits in accordance with the
standards of the Public Company Accounting Oversight Board
(United States). Those standards require that we plan and
perform the audits to obtain reasonable assurance about whether
the financial statements are free of material misstatement and
whether effective internal control over financial reporting was
maintained in all material respects. Our audits of the financial
statements included examining, on a test basis, evidence
supporting the amounts and disclosures in the financial
statements, assessing the accounting principles used and
significant estimates made by management, and evaluating the
overall financial statement presentation. Our audit of internal
control over financial reporting included obtaining an
understanding of internal control over financial reporting,
assessing the risk that a material weakness exists, and testing
and evaluating the design and operating effectiveness of
internal control based on the assessed risk. Our audits also
included performing such other procedures as we considered
necessary in the circumstances. We believe that our audits
provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a
process designed to provide reasonable assurance regarding the
reliability of financial reporting and the preparation of
financial statements for external purposes in accordance with
generally accepted accounting principles. A company’s
internal control over financial reporting includes those
policies and procedures that (i) pertain to the maintenance
of records that, in reasonable detail, accurately and fairly
reflect the transactions and dispositions of the assets of the
company; (ii) provide reasonable assurance that
transactions are recorded as necessary to permit preparation of
financial statements in accordance with generally accepted
accounting principles, and that receipts and expenditures of the
company are being made only in accordance with authorizations of
management and directors of the company; and (iii) provide
reasonable assurance regarding prevention or timely detection of
unauthorized acquisition, use, or disposition of the
company’s assets that could have a material effect on the
financial statements.
Because of its inherent limitations, internal control over
financial reporting may not prevent or detect misstatements.
Also, projections of any evaluation of effectiveness to future
periods are subject to the risk that controls may become
inadequate because of changes in conditions, or that the degree
of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers
LLP
Raleigh, NC
March 12, 2010
53
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands, except share amounts)
|
|
|
|
|
ASSETS
|
|
Current assets:
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
29,298
|
|
|
$
|
42,051
|
|
|
Short-term investments
|
|
|
25,953
|
|
|
|
35,215
|
|
|
Accounts receivable, net
|
|
|
3,926
|
|
|
|
4,812
|
|
|
Inventory
|
|
|
7,325
|
|
|
|
6,369
|
|
|
Prepaid expenses and other assets
|
|
|
676
|
|
|
|
632
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current assets
|
|
|
67,178
|
|
|
|
89,079
|
|
|
Property and equipment, net
|
|
|
1,813
|
|
|
|
1,412
|
|
|
|
|
|
|
|
|
|
|
|
|
Total assets
|
|
$
|
68,991
|
|
|
$
|
90,491
|
|
|
|
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY
|
|
Current liabilities:
|
|
|
|
|
|
|
|
|
|
Accounts payable
|
|
$
|
2,442
|
|
|
$
|
2,896
|
|
|
Accrued expenses
|
|
|
1,269
|
|
|
|
2,009
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current liabilities
|
|
|
3,711
|
|
|
|
4,905
|
|
|
|
|
|
|
|
|
|
|
|
|
Commitments (Note 5)
|
|
|
|
|
|
|
|
|
|
Stockholders’ equity:
|
|
|
|
|
|
|
|
|
|
Common stock, $0.0001 par value; 75,000,000 shares
authorized, 20,648,447 and 20,538,333 shares issued and
outstanding at December 31, 2009 and 2008,respectively
|
|
|
2
|
|
|
|
2
|
|
|
Preferred stock, $0.0001 par value; 5,000,000 shares
authorized, none issued and outstanding at December 31,
2009 and 2008.
|
|
|
—
|
|
|
|
—
|
|
|
Additional paid-in capital
|
|
|
136,402
|
|
|
|
133,507
|
|
|
Accumulated other comprehensive income
|
|
|
(5
|
)
|
|
|
—
|
|
|
Accumulated deficit
|
|
|
(71,119
|
)
|
|
|
(47,923
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Total stockholders’ equity
|
|
|
65,280
|
|
|
|
85,586
|
|
|
|
|
|
|
|
|
|
|
|
|
Total liabilities and stockholders’ equity
|
|
$
|
68,991
|
|
|
$
|
90,491
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of these
consolidated financial statements.
54
TranS1
Inc.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
(In thousands, except per share amounts)
|
|
|
|
|
Revenue
|
|
$
|
29,807
|
|
|
$
|
25,304
|
|
|
$
|
16,473
|
|
|
Cost of revenue
|
|
|
5,687
|
|
|
|
4,315
|
|
|
|
3,042
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gross profit
|
|
|
24,120
|
|
|
|
20,989
|
|
|
|
13,431
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
|
6,439
|
|
|
|
4,081
|
|
|
|
3,885
|
|
|
Sales and marketing
|
|
|
34,098
|
|
|
|
29,375
|
|
|
|
15,706
|
|
|
General and administrative
|
|
|
7,184
|
|
|
|
7,116
|
|
|
|
3,801
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
47,721
|
|
|
|
40,572
|
|
|
|
23,392
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating loss
|
|
|
(23,601
|
)
|
|
|
(19,583
|
)
|
|
|
(9,961
|
)
|
|
Interest income
|
|
|
405
|
|
|
|
2,548
|
|
|
|
1,384
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(23,196
|
)
|
|
$
|
(17,035
|
)
|
|
$
|
(8,577
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per common share — basic and diluted
|
|
$
|
(1.13
|
)
|
|
$
|
(0.84
|
)
|
|
$
|
(1.46
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average common shares outstanding — basic and
diluted
|
|
|
20,604
|
|
|
|
20,289
|
|
|
|
5,872
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of these
consolidated financial statements.
55
TranS1
Inc.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
|
|
|
Total
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
|
|
|
Other
|
|
|
|
|
|
Stockholders’
|
|
|
|
|
Common Stock
|
|
|
Paid-In
|
|
|
Notes
|
|
|
Comprehensive
|
|
|
Accumulated
|
|
|
Equity
|
|
|
|
|
Number
|
|
|
Amount
|
|
|
Capital
|
|
|
Receivable
|
|
|
Income
|
|
|
Deficit
|
|
|
(Deficit)
|
|
|
|
|
(In thousands, except share data)
|
|
|
|
|
Balance at December 31, 2006
|
|
|
2,435,484
|
|
|
$
|
—
|
|
|
$
|
820
|
|
|
$
|
(38
|
)
|
|
$
|
—
|
|
|
$
|
(22,311
|
)
|
|
$
|
(21,529
|
)
|
|
Issuance of common stock from exercised options
|
|
|
290,292
|
|
|
|
|
|
|
|
123
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
123
|
|
|
Issuance of common stock from initial public offering
|
|
|
6,325,000
|
|
|
|
1
|
|
|
|
86,658
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
86,659
|
|
|
Conversion of preferred stock to common stock
|
|
|
10,793,165
|
|
|
|
1
|
|
|
|
40,088
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
40,089
|
|
|
Stock based compensation
|
|
|
|
|
|
|
|
|
|
|
2,636
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2,636
|
|
|
Repayment of note receivable
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
38
|
|
|
|
|
|
|
|
|
|
|
|
38
|
|
|
Net loss
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(8,577
|
)
|
|
|
(8,577
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2007
|
|
|
19,843,941
|
|
|
|
2
|
|
|
|
130,325
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(30,888
|
)
|
|
|
99,439
|
|
|
Issuance of common stock from exercised options
|
|
|
694,392
|
|
|
|
|
|
|
|
203
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
203
|
|
|
Stock based compensation
|
|
|
|
|
|
|
|
|
|
|
2,979
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2,979
|
|
|
Net loss
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(17,035
|
)
|
|
|
(17,035
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2008
|
|
|
20,538,333
|
|
|
|
2
|
|
|
|
133,507
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(47,923
|
)
|
|
|
85,586
|
|
|
Issuance of common stock from exercised options
|
|
|
110,114
|
|
|
|
|
|
|
|
96
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
96
|
|
|
Stock based compensation
|
|
|
|
|
|
|
|
|
|
|
2,799
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2,799
|
|
|
Other comprehensive income
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(5
|
)
|
|
|
|
|
|
|
(5
|
)
|
|
Net loss
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(23,196
|
)
|
|
|
(23,196
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2009
|
|
|
20,648,447
|
|
|
$
|
2
|
|
|
$
|
136,402
|
|
|
$
|
—
|
|
|
$
|
(5
|
)
|
|
$
|
(71,119
|
)
|
|
$
|
65,280
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of these
consolidated financial statements.
56
TranS1
Inc.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
(In thousands)
|
|
|
|
|
Cash flows from operating activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(23,196
|
)
|
|
$
|
(17,035
|
)
|
|
$
|
(8,577
|
)
|
|
Adjustments to reconcile net loss to net cash used in operating
activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation
|
|
|
909
|
|
|
|
804
|
|
|
|
540
|
|
|
Stock-based compensation
|
|
|
2,799
|
|
|
|
2,979
|
|
|
|
2,636
|
|
|
Allowance for excess and obsolete inventory
|
|
|
505
|
|
|
|
400
|
|
|
|
306
|
|
|
Provision for bad debts
|
|
|
80
|
|
|
|
101
|
|
|
|
95
|
|
|
Changes in operating assets and liabilities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(Increase) decrease in accounts receivable
|
|
|
806
|
|
|
|
(1,688
|
)
|
|
|
(1,700
|
)
|
|
Increase in inventory
|
|
|
(1,461
|
)
|
|
|
(2,744
|
)
|
|
|
(2,251
|
)
|
|
Increase in prepaid expenses
|
|
|
(44
|
)
|
|
|
(35
|
)
|
|
|
(367
|
)
|
|
Increase (decrease) in accounts payable
|
|
|
(454
|
)
|
|
|
1,265
|
|
|
|
788
|
|
|
Increase (decrease) in accrued liabilities
|
|
|
(745
|
)
|
|
|
223
|
|
|
|
1,185
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in operating activities
|
|
|
(20,801
|
)
|
|
|
(15,730
|
)
|
|
|
(7,345
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash flows from investing activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Purchase of property and equipment
|
|
|
(1,310
|
)
|
|
|
(1,128
|
)
|
|
|
(516
|
)
|
|
Purchases of investments
|
|
|
(50,872
|
)
|
|
|
(55,761
|
)
|
|
|
(30,687
|
)
|
|
Sales of investments
|
|
|
60,134
|
|
|
|
49,791
|
|
|
|
11,370
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by (used in) investing activities
|
|
|
7,952
|
|
|
|
(7,098
|
)
|
|
|
(19,833
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash flows from financing activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from issuance of common stock
|
|
|
96
|
|
|
|
203
|
|
|
|
86,820
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by financing activities
|
|
|
96
|
|
|
|
203
|
|
|
|
86,820
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net increase (decrease) in cash and cash equivalents
|
|
|
(12,753
|
)
|
|
|
(22,625
|
)
|
|
|
59,642
|
|
|
Cash and cash equivalents, beginning of period
|
|
|
42,051
|
|
|
|
64,676
|
|
|
|
5,034
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents, end of period
|
|
$
|
29,298
|
|
|
$
|
42,051
|
|
|
$
|
64,676
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental disclosure of noncash financing activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Conversion of preferred stock to common stock
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
40,089
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of these
consolidated financial statements.
57
TranS1
Inc.
TranS1 Inc., a Delaware corporation (the “Company”),
was incorporated in May 2000 and is headquartered in Wilmington,
North Carolina. The Company is a medical device company focused
on designing, developing and marketing products that implement
its minimally invasive surgical approach to treat degenerative
disc disease and instability affecting the lower lumbar region
of the spine and operates in one business segment. The Company
has developed and currently markets two single-level fusion
products, the
AxiaLIF®
and the AxiaLIF
360°tm
and a two-level fusion product, the AxiaLIF
2Ltm.
All of the Company’s products are delivered using its
pre-sacral approach. The AxiaLIF product was commercially
released in January 2005, the AxiaLIF 360° product was
commercially released in July 2006 and the AxiaLIF 2L product
was commercially released in Europe in the fourth quarter of
2006. The Company received 510(k) clearance for the AxiaLIF 2L
from the FDA and began marketing this product in the United
States in the second quarter of 2008. The Company generates
revenue from the sale of implants and procedure kits. The
Company sells its products directly to hospitals and surgical
centers in the United States and in certain European countries
and to independent distributors outside the United States.
The Company is subject to a number of risks similar to other
companies in the medical device industry. These risks include
rapid technological change, uncertainty of market acceptance of
our products, uncertainty of regulatory approval, competition
from substitute products and larger companies, the need to
obtain additional financing, compliance with government
regulation, protection of proprietary technology, product
liability, and the dependence on key individuals.
|
|
|
2.
|
Summary
of Significant Accounting Policies
|
Basis
of Presentation
The Company has prepared the accompanying consolidated financial
statements in conformity with accounting principles generally
accepted in the United States of America. The Company’s
fiscal year ends on December 31. On October 5, 2007,
the Company’s Board of Directors approved an amendment to
the Company’s existing certificate of incorporation
effecting a 0.9-for-1 reverse stock split. All share and per
share information in the accompanying consolidated financial
statements and notes to the consolidated financial statements
has been retroactively restated to reflect the effect of the
stock split.
On October 22, 2007, all of the outstanding preferred
shares were converted into 10,793,165 common shares and the
Company completed its initial public offering of
6,325,000 shares of common stock, at an offering price of
$15.00 per share. The net proceeds of this offering, after
deducting the underwriting discounts, commissions and offering
expenses, were approximately $86.7 million.
In the second quarter of 2009, the Company revised the
classification of patent-related legal costs from research and
development expense to general and administrative expense as
such costs typically would be excluded from research and
development costs as defined by Accounting Standards
Codification 730 (formerly Statement of Financial Accounting
Standards No. 2, “Accounting for Research and
Development Costs”). Amounts related to prior periods are
not considered material to the financial statements taken as a
whole, but were revised for purposes of comparability. Such
amounts for the years ended December 31, 2008 and 2007 were
$940,000 and $900,000, respectively. The revision did not affect
previously reported total operating expenses, net loss, loss per
share, assets, liabilities, stockholders’ equity or cash
flows.
Principles
of Consolidation:
These consolidated financial statements include the accounts of
the Company and its subsidiaries. All intercompany transactions
and accounts have been eliminated in consolidation.
58
TranS1
Inc.
Notes to
Consolidated Financial
Statements — (Continued)
Use of
Estimates
The consolidated financial statements have been prepared in
conformity with accounting principles generally accepted in the
United States of America. These principles require management to
make estimates and assumptions that affect the reported amounts
of assets and liabilities and disclosure of contingent assets
and liabilities at the date of the consolidated financial
statements and the reported amounts of revenues and expenses
during the reporting period. Actual results could differ from
those estimates. The principal estimates relate specifically to
accounts receivable reserves, inventory reserves, stock-based
compensation, accrued expenses and income tax valuations.
Cash
and Cash Equivalents
The Company considers all highly liquid investments with an
original maturity of three months or less at the date of
purchase to be cash equivalents. Cash and cash equivalents
include money market funds and commercial paper. Cash
equivalents are carried at fair market value. Related unrealized
gains and losses were insignificant as of December 31, 2009
and 2008.
Investments
All marketable investments are classified as
available-for-sale
and therefore are carried at fair market value. Related
unrealized gains and losses were insignificant as of
December 31, 2009 and 2008. Realized gains and losses on
the sale of all such investments are reported in earnings and
computed using the specific identification cost method and were
insignificant for the years ended December 31, 2009 and
2008. All of the Company’s investments as of
December 31, 2009 have maturities of one year or less. The
following table presents the components of the Company’s
available-for-sale
investments:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Short-term investments:
|
|
|
|
|
|
|
|
|
|
U.S. government securities
|
|
$
|
25,953
|
|
|
$
|
9,035
|
|
|
Commercial paper
|
|
|
—
|
|
|
|
4,978
|
|
|
Corporate debt securities
|
|
|
—
|
|
|
|
18,972
|
|
|
Certificate of deposit
|
|
|
—
|
|
|
|
2,230
|
|
|
|
|
|
|
|
|
|
|
|
|
Total short-term investments
|
|
$
|
25,953
|
|
|
$
|
35,215
|
|
|
|
|
|
|
|
|
|
|
|
Fair
Value of Financial Instruments
The carrying values of cash equivalents, short-term investments,
accounts receivable, and accounts payable at December 31,
2009 and 2008 approximated their fair values due to the
short-term nature of these items.
At December 31, 2009, the Company holds certain assets that
are required to be measured at fair value on a recurring basis.
These assets include available for sale securities classified as
cash equivalents and short-term investments. Accounting
Standards Codification (“ASC”)
820-10
(formerly Statement of Financial Accounting Standards
No. 157 (SFAS 157), “Fair Value
Measurements”), requires the valuation of investments using
a three tiered approach, which requires that fair value
measurements be classified and disclosed in one of three tiers.
These tiers are: Level 1, defined as quoted prices in
active markets for identical assets or liabilities;
Level 2, defined as valuations based on observable inputs
other than those included in Level 1, such as quoted prices
for similar assets and liabilities in active markets, or other
inputs that are observable or can be corroborated by observable
input data; and Level 3, defined as valuations based on
unobservable inputs
59
TranS1
Inc.
Notes to
Consolidated Financial
Statements — (Continued)
reflecting the Company’s own assumptions, consistent with
reasonably available assumptions made by other market
participants.
At December 31, 2009, all available for sale securities are
classified as Level 1 assets with a fair value of
$54.9 million, which included money market funds of
$28.9 million and short-term investments of
$26.0 million. At December 31, 2008, all available for
sale securities are classified as Level 1 assets with a
fair value of $76.8 million, which included money market
funds of $41.6 million and short-term investments of
$35.2 million. The Company had no Level 2 or
Level 3 assets or liabilities at December 31, 2009 or
2008.
Accounts
Receivable
Accounts receivable are presented net of an allowance for
uncollectible accounts. Estimates on the collectability of
customer accounts are based primarily on an analysis of
historical trends and experience and changes in customers’
financial condition. The following table presents the components
of accounts receivable:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Gross accounts receivable
|
|
$
|
4,119
|
|
|
$
|
5,005
|
|
|
Allowance for uncollectible accounts
|
|
|
(193
|
)
|
|
|
(193
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Total accounts receivable, net
|
|
$
|
3,926
|
|
|
$
|
4,812
|
|
|
|
|
|
|
|
|
|
|
|
Concentration
of Credit Risk and Significant Customers
Financial instruments which potentially subject the Company to
concentrations of credit risk consist principally of cash and
cash equivalents.
The Company places cash deposits with a federally insured
financial institution, in amounts which at times exceed the
federally insured limit, which was $250,000 at December 31,
2009 and 2008. The total amount of deposits in excess of
federally insured limits was $71,000 and $1,914,000 at
December 31, 2009 and 2008, respectively.
In 2009, 2008 and 2007 no customer accounted for 10% or more of
revenues. As of December 31, 2009 and 2008, no single
customer accounted for 10% or more of the accounts receivable
balance.
Inventories
Inventories consist of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Raw materials
|
|
$
|
220
|
|
|
$
|
437
|
|
|
Work-in-process
|
|
|
3,993
|
|
|
|
3,334
|
|
|
Finished goods
|
|
|
3,112
|
|
|
|
2,598
|
|
|
|
|
|
|
|
|
|
|
|
|
Total inventories
|
|
$
|
7,325
|
|
|
$
|
6,369
|
|
|
|
|
|
|
|
|
|
|
|
Inventories are stated at the lower of cost or market, computed
on a standard cost basis, which approximates actual cost on a
first-in,
first-out basis and market being determined as the lower of
replacement cost or net realizable value. Inventory reserves are
established when conditions indicate that the selling price
could be less than cost due to obsolescence or the Company
determines that it holds excessive levels of inventory based on
market demand.
60
TranS1
Inc.
Notes to
Consolidated Financial
Statements — (Continued)
Property
and Equipment
Property and equipment are recorded at cost and depreciated over
their estimated useful lives using the straight-line method.
Leasehold improvements are amortized over the shorter of the
lease term or the estimated useful life of the related asset.
Maintenance and repairs are charged to expense as incurred. Upon
retirement or sale, the cost of assets disposed of and the
related accumulated depreciation and amortization are removed
from the accounts and any resulting gain or loss is credited or
charged to income.
The estimated useful lives are:
| |
|
|
|
Furniture and fixtures
|
|
5-10 years
|
|
Equipment
|
|
3-5 years
|
|
Other depreciable assets
|
|
2-10 years
|
|
Leasehold improvements
|
|
Lesser of estimated useful life or lease term
|
Revenue
Recognition
Revenue is recognized based on the following criteria:
(i) persuasive evidence that an arrangement exists with the
customer; (ii) the delivery of the products
and/or
services has occurred (title has transferred); (iii) the
selling price has been fixed for the products or services
delivered; and (iv) the collection is reasonably assured.
Revenue is generated from the sale of implants and procedure
kits, which consist of disposable instruments. The Company has
two distinct sale methods. The first method is when procedure
kits are sold directly to hospitals or surgical centers. The
Company’s sales representatives or independent sales agents
hand deliver the procedure kit to the customer on the day of the
surgery or several days prior to the surgery. The sales
representative or independent agent is then responsible for
reporting the delivery of the procedure kit, and the date of the
operation to the corporate office for proper revenue
recognition. The Company recognizes revenue upon the
confirmation that the procedure kit has been used in a surgical
procedure. The other sales method is for sales to distributors
outside the United States. These distributors order multiple
procedure kits at one time to have on hand. These transactions
require the customer to send in a purchase order before shipment
will be made to the customer. The Company determines revenue
recognition on a case by case basis dependent upon the terms and
conditions of each individual distributor agreement. Under the
distributor agreements currently in place, a distributor only
has the right of return for defective products and, accordingly,
revenue is recognized upon shipment.
Shipping
and Handling Costs
Shipping and handling costs in the United States are expensed as
incurred and are included in the cost of revenue. These costs
are not reimbursed by the Company’s customers. Shipping
costs to distributors outside the United States are either paid
directly by the distributor to the freight carrier or charged to
the distributor and reimbursed to the Company.
Sales
and Marketing Expenses
Sales and marketing expenses consist of personnel costs,
including stock-based compensation expense, sales commissions
paid to the Company’s direct sales representatives and
independent sales agents, and costs associated with physician
training programs, promotional activities, and participation in
medical conferences. All costs of advertising and promotional
activities are expensed as incurred. Advertising expenses were
$1.8 million, $1.7 million and $0.7 million for
the years ending December 31, 2009, 2008 and 2007,
respectively.
61
TranS1
Inc.
Notes to
Consolidated Financial
Statements — (Continued)
Research
and Development Expenses
Research and development expenses consist primarily of personnel
costs, including stock-based compensation expense, within the
Company’s product development, regulatory and clinical
functions and the costs of clinical studies and product
development projects. Research and development expenses are
expensed as incurred.
Patent
Costs
Costs associated with the submission of a patent application are
expensed as incurred given the uncertainty of the patents
resulting in probable future economic benefits to the Company.
Income
Taxes
The Company accounts for income taxes using the liability method
which requires the recognition of deferred tax assets or
liabilities for the temporary differences between financial
reporting and tax bases of the Company’s assets and
liabilities and for tax carryforwards at enacted statutory tax
rates in effect for the years in which the differences are
expected to reverse. The effect on deferred taxes of a change in
tax rates is recognized in the period that includes the
enactment date. In addition, valuation allowances are
established when necessary to reduce deferred tax assets to the
amounts expected to be realized.
Stock-Based
Compensation
The Company accounts for stock-based compensation under the fair
value provisions of ASC 718, (formerly SFAS 123R,
Share-Based Payment). ASC 718 requires the recognition of
compensation expense, using a fair-value-based method, for costs
related to all share-based payments including stock options. ASC
718 requires companies to estimate the fair value of share-based
payment awards on the date of grant using an option-pricing
model. The Company adopted ASC 718 using the prospective
transition method, which requires that for nonpublic entities
that used the minimum value method for either pro forma or
financial statements recognition purposes, ASC 718 shall be
applied to option grants or modifications to existing options
after the required effective date. For options granted prior to
the new ASC 718 effective date and for which the requisite
service period has not been performed as of January 1,
2006, the Company has continued to apply the intrinsic value
provisions of Accounting Principles Board Opinion No. 25
(APB 25), Accounting for Stock Issued to Employees, on
the remaining unvested awards. All options granted after
January 1, 2006 are expensed on a straight-line basis over
the vesting period.
The Company accounts for stock-based compensation arrangements
with nonemployees in accordance with ASC
505-50
(formerly the Emerging Issues Task Force Abstract
No. 96-18,
Accounting for Equity Instruments That Are Issued to Other
Than Employees for Acquiring, or in Conjunction with Selling
Goods or Services). The Company records the expense of such
services based on the estimated fair value of the equity
instrument using the Black-Scholes pricing model. The value of
the equity instruments is charged to earnings over the term of
the service agreement.
Net
Loss Per Common Share
Basic net loss per common share (“Basic EPS”) is
computed by dividing net loss available to common stockholders
by the weighted average number of common shares outstanding.
Diluted net loss available to common stockholders per common
share (“Diluted EPS”) is computed by dividing net loss
available to common stockholders by the weighted average number
of common shares and dilutive potential common share equivalents
then outstanding. The Company’s potential dilutive common
shares, which consist of shares issuable upon the exercise of
stock options and conversion of convertible preferred stock,
have not been included in the computation of diluted net loss
per share for all periods as the result would be anti-dilutive.
62
TranS1
Inc.
Notes to
Consolidated Financial
Statements — (Continued)
The following table sets forth the potential shares of common
stock that are not included in the calculation of diluted net
loss per share as the result would be anti-dilutive as of the
end of each period presented:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
2009
|
|
2008
|
|
2007
|
|
|
|
Weighted average stock options outstanding
|
|
|
2,110,689
|
|
|
|
1,915,687
|
|
|
|
1,928,495
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Segment
and Geographic Reporting
The Company applies ASC 280 (formerly SFAS No. 131,
“Disclosures about Segments of an Enterprise and Related
Information”). ASC 280 establishes standards for the
reporting by business enterprises of information about operating
segments, products and services, geographic areas, and major
customers. The Company has determined that it did not have any
separately reportable segments as of December 31, 2009,
2008 or 2007.
Revenue by geographic area was:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
(In thousands)
|
|
|
|
|
United States
|
|
$
|
28,045
|
|
|
$
|
23,322
|
|
|
$
|
15,085
|
|
|
Europe
|
|
|
1,602
|
|
|
|
1,854
|
|
|
|
1,183
|
|
|
Other
|
|
|
160
|
|
|
|
128
|
|
|
|
205
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
29,807
|
|
|
$
|
25,304
|
|
|
$
|
16,473
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Long-lived assets are primarily located in the United States.
Recently
Issued Accounting Standards
In February 2008, the FASB issued ASC 820 (formerly Staff
Position
No. FAS 157-2,
“Fair Value Measurements”), which delayed the
effective date of ASC 820 for non-financial assets and
liabilities to fiscal years beginning after November 15,
2008. The company adopted ASC 820 for its non-financial assets
and non-financial liabilities on January 1, 2009, and it
did not have a material impact on the Company’s
consolidated financial statements.
In June 2009, the FASB issued the FASB Accounting Standards
Codification (“ASC”). The Codification has become the
single source for all authoritative GAAP recognized by the FASB
to be applied for financial statements issued for periods ending
after September 15, 2009. The Company applied the
Codification to its Annual Report on
Form 10-K
for the period ending December 31, 2009. The Codification
does not change GAAP and did not have an effect on the
Company’s consolidated financial position or results of
operations.
In May 2009, the FASB issued ASC 855 (formerly
SFAS No. 165, “Subsequent Events”), which
establishes general standards of accounting for and disclosures
of events that occur after the balance sheet date but before the
financial statements are issued or are available to be issued.
ASC 855 provides guidance on the period after the balance sheet
date during which management of a reporting entity should
evaluate events or transactions that may occur for potential
recognition or disclosure in the financial statements, the
circumstances under which an entity should recognize events or
transactions occurring after the balance sheet date in its
financial statements and the disclosures that an entity should
make about events or transactions that occurred after the
balance sheet date. The Company adopted ASC 855 during the
second quarter of 2009, and its application did not have a
material impact on its consolidated financial statements.
63
TranS1
Inc.
Notes to
Consolidated Financial
Statements — (Continued)
No other recently issued, but not yet effective, accounting
standards are believed to have a material impact on the Company.
|
|
|
3.
|
Property
and Equipment
|
Property and equipment consist of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Furniture and fixtures
|
|
$
|
210
|
|
|
$
|
186
|
|
|
Equipment
|
|
|
2,620
|
|
|
|
2,061
|
|
|
Computer software
|
|
|
410
|
|
|
|
415
|
|
|
Leasehold improvements
|
|
|
380
|
|
|
|
380
|
|
|
Tools
|
|
|
73
|
|
|
|
73
|
|
|
Construction in process
|
|
|
588
|
|
|
|
14
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4,281
|
|
|
|
3,129
|
|
|
Less: accumulated depreciation and amortization
|
|
|
(2,468
|
)
|
|
|
(1,717
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
1,813
|
|
|
$
|
1,412
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization expense for the years ended
December 31, 2009, 2008 and 2007 was $909,000, $804,000,
and $540,000, respectively.
Accrued expenses consist of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Commissions
|
|
$
|
597
|
|
|
$
|
1,178
|
|
|
Vacation
|
|
|
186
|
|
|
|
163
|
|
|
Franchise taxes
|
|
|
122
|
|
|
|
121
|
|
|
Consulting
|
|
|
120
|
|
|
|
66
|
|
|
Legal and professional fees
|
|
|
90
|
|
|
|
69
|
|
|
Travel & entertainment
|
|
|
50
|
|
|
|
50
|
|
|
Bonus
|
|
|
10
|
|
|
|
270
|
|
|
Clinical
|
|
|
10
|
|
|
|
20
|
|
|
Other
|
|
|
84
|
|
|
|
72
|
|
|
|
|
|
|
|
|
|
|
|
|
Total accrued expenses
|
|
$
|
1,269
|
|
|
$
|
2,009
|
|
|
|
|
|
|
|
|
|
|
|
64
TranS1
Inc.
Notes to
Consolidated Financial
Statements — (Continued)
The Company rents office space under the terms of an operating
lease, which expires in 2019.
Future minimum lease payments under operating lease obligations
at December 31, 2009 are as follows (in thousands):
| |
|
|
|
|
|
Year ending December 31,
|
|
|
|
|
|
2010
|
|
$
|
374
|
|
|
2011
|
|
|
332
|
|
|
2012
|
|
|
363
|
|
|
2013
|
|
|
363
|
|
|
2014 and after
|
|
|
2,696
|
|
|
|
|
|
|
|
|
|
|
$
|
4,128
|
|
|
|
|
|
|
|
Rent expense related to operating leases for the years ended
December 31, 2009, 2008 and 2007 was $204,000, $157,000 and
$152,000, respectively.
At December 31, 2009 and 2008, the Company’s Amended
and Restated Certificate of Incorporation, which was adopted in
connection with the Company’s initial public offering,
authorized up to 80,000,000 shares of capital stock, of
which 75,000,000 shares were designated as common stock
with a par value of $0.0001 and up to 5,000,000 shares were
designated as preferred stock with a par value of $0.0001. At
December 31, 2009 and 2008, there were 20,648,447 and
20,538,333 shares of common stock issued and outstanding,
respectively, and there were no shares of preferred stock issued
and outstanding.
In 2009, the Company issued 110,114 shares of the common
stock to employees and consultants for $96,000 upon the exercise
of stock options. In 2008, the Company issued
694,392 shares of common stock to employees and consultants
for $203,000 upon the exercise of stock options. In 2007, the
Company issued 290,292 shares of common stock to employees
and consultants for $123,000 upon the exercise of stock options.
|
|
|
7.
|
Stock
Incentive Plans and Stock-Based Compensation
|
2007
Employee Stock Purchase Plan
The Company’s board of directors adopted the 2007 Employee
Stock Purchase Plan (the “ESPP”) in July 2007. The
ESPP became effective upon the completion of the Company’s
initial public offering. A total of 250,000 shares of
common stock are available for sale. In addition, the ESPP
provides for annual increases in the number of shares available
for issuance under the ESPP on the first day of each fiscal year
beginning in 2008, equal to the lesser of (i) 2.0% of the
outstanding shares of common stock on the first day of such
fiscal year or (ii) an amount determined by the
administrator of the ESPP. The Company’s Compensation
Committee administers the ESPP.
Shares shall be offered pursuant to the ESPP in six-month
periods commencing on the first trading day on or after June 1
and December 1 of each year, or on such other date as the
administrator may determine.
Our ESPP permits participants to purchase common stock through
payroll deductions of up to 10% of their eligible compensation,
which includes a participant’s base straight time gross
earnings, certain commissions, overtime and shift premium, but
exclusive of payments for incentive compensation, bonuses and
other compensation. A participant may purchase a maximum of
2,500 shares during a six-month purchase period. Amounts
deducted and accumulated by the participant are used to purchase
shares of the Company’s common
65
TranS1
Inc.
Notes to
Consolidated Financial
Statements — (Continued)
stock at the end of each six-month purchase period. The purchase
price of the shares will be 95% of the fair market value of
Company common stock on the exercise date. Participants may end
their participation at any time during an offering period, and
will be paid their accrued payroll deductions that have not yet
been used to purchase shares of common stock. Participation ends
automatically upon termination of employment with the Company.
Pursuant to ASC 718, the Company is not required to recognize
compensation expense in connection with purchases under the ESPP.
During fiscal years ended December 31, 2009, 2008 and 2007,
no shares were issued to participants under the ESPP.
2000
and 2007 Stock Incentive Plans
The Company established the TranS1 Inc. Stock Incentive Plan in
2000, (as amended, the “2000 Plan”) and the 2007 Stock
Incentive Plan (the “2007 Plan”) in October 2007
(collectively, the “Plans”) . Under the 2000 Plan and
the 2007 Plan, the Company may grant options to employees,
directors or service providers and contractors for a maximum of
3,159,108 and 1,400,000 shares, respectively, of the
Company’s common stock. Options granted under the Plans may
be incentive stock options or non-qualified stock options.
Non-qualified stock options may be granted to service providers
and incentive stock options may be granted only to employees.
The exercise periods may not exceed ten years for options.
However, in the case of an incentive stock option granted to an
optionee who, at the time of the option grant owns stock
representing more than 10% of the outstanding shares, the term
of the option shall be five years from the date of the grant.
The exercise price of incentive stock options cannot be less
than 100% of the fair market value per share of the
Company’s common stock on the grant date. The exercise
price of a nonqualified option under the 2000 Plan and the 2007
Plan shall not be less than 85% and 100%, respectively, of the
fair market value per share on the date the option is granted.
If an optionee owns more than 10% of the outstanding shares, the
exercise price cannot be less than 110% of the fair market value
of the stock on the date of the grant. Options granted under the
Plans generally vest over periods ranging from three to four
years.
The following table summarizes the activity of the
Company’s 2000 Plan and 2007 Plan, including the number of
shares under options (“Number”) and the weighted
average exercise price (“Price”):
| |
|
|
|
|
|
|
|
|
|
|
|
Number
|
|
|
Price
|
|
|
|
|
Outstanding as of December 31, 2008
|
|
|
2,247,733
|
|
|
$
|
7.12
|
|
|
Options granted
|
|
|
495,500
|
|
|
|
6.53
|
|
|
Options exercised
|
|
|
(110,114
|
)
|
|
|
0.91
|
|
|
Options forfeited
|
|
|
(417,093
|
)
|
|
|
8.88
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding as of December 31, 2009
|
|
|
2,216,026
|
|
|
$
|
6.96
|
|
|
|
|
|
|
|
|
|
|
|
The following table summarizes information about the
Company’s stock options at December 31, 2009:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted
|
|
|
Weighted
|
|
|
|
|
|
|
|
|
|
|
Average
|
|
|
Average
|
|
|
Number of
|
|
|
|
|
Number
|
|
|
Contractual
|
|
|
Exercise
|
|
|
Options
|
|
|
Range of Exercise Prices
|
|
Outstanding
|
|
|
Life (Years)
|
|
|
Price
|
|
|
Exercisable
|
|
|
|
|
$ 0.11 - $ 2.00
|
|
|
569,756
|
|
|
|
5.5
|
|
|
$
|
0.81
|
|
|
|
539,862
|
|
|
$ 2.01 - $ 6.00
|
|
|
689,770
|
|
|
|
8.1
|
|
|
|
5.69
|
|
|
|
255,923
|
|
|
$ 6.01 - $10.00
|
|
|
385,375
|
|
|
|
8.4
|
|
|
|
8.78
|
|
|
|
174,175
|
|
|
$12.00 - $13.00
|
|
|
380,084
|
|
|
|
7.9
|
|
|
|
12.39
|
|
|
|
186,875
|
|
|
$14.00 - $20.00
|
|
|
191,041
|
|
|
|
7.7
|
|
|
|
15.36
|
|
|
|
129,241
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2,216,026
|
|
|
|
7.4
|
|
|
|
6.96
|
|
|
|
1,286,076
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
66
TranS1
Inc.
Notes to
Consolidated Financial
Statements — (Continued)
The aggregate intrinsic value of outstanding stock options at
December 31, 2009 was $1.8 million. At
December 31, 2009, exercisable stock options had an
aggregate intrinsic value of $1.7 million, a weighted
average contractual life of 6.8 years and a weighted
average exercise price of $5.88.
Stock-Based
Compensation for Non-employees
During 2009, the Company issued options to purchase
2,500 shares of common stock with an exercise price of
$6.00 per share to consultants. These options vest over
3 years and expire 10 years from the date of issuance.
During 2008, the Company issued options to purchase
35,000 shares of common stock with an average exercise
price of $12.43 per share to consultants. These options vest
over a range of 1 to 3 years and expire 10 years from
the date of issuance. During 2007, the Company issued options to
purchase 50,207 shares of common stock with an average
exercise price of $7.52 per share to consultants. These options
vest over a range of zero to three years and expire
10 years from the date of issuance. The Company determined
the estimated fair value of the options issued to the
consultants using the Black-Scholes pricing model. The Company
used the following assumptions in the Black-Scholes pricing
model for 2009 grants: 60% volatility, 0% dividend yield, 2.65%
risk-free rate and a 6 year expected legal life. The
Company used the following assumptions in the Black-Scholes
pricing model for 2008 grants: 54% volatility, 0% dividend
yield, 1.54% risk-free rate and 6 year expected life. The
Company used the following assumptions in the Black-Scholes
pricing model for 2007 grants: 45% volatility, 0% dividend
yield, 4.92% risk-free rate and 6 year expected life.
Stock-based compensation expense charged to operations on
options granted to non-employees for years ended
December 31, 2009, 2008 and 2007 was $8,000, $118,000 and
$1.1 million, respectively. As of December 31, 2009,
there was $3,000 of total unrecognized compensation costs
related to non-vested stock option awards which is expected to
be recognized over a weighted-average period of 0.5 years.
Employee
Stock-Based Compensation
Under ASC 718, compensation cost for employee stock-based awards
is based on the estimated grant-date fair value and is
recognized over the vesting period of the applicable award on a
straight-line basis. For the period from January 1, 2006 to
December 31, 2009, the Company issued employee stock-based
awards in the form of stock options. The Company recorded
stock-based compensation expense of $2.8 million,
$2.9 million and $1.5 million for the years ended
December 31, 2009, 2008 and 2007, respectively. The
weighted average estimated fair value of the employee stock
options granted for the years ended December 31, 2009, 2008
and 2007 was $6.53, $11.77 and $10.34 per share, respectively.
The aggregate intrinsic value of stock options (the amount by
which the market price of the stock on the date of exercise
exceeded the exercise price of the option) exercised for the
years ended December 31, 2009, 2008 and 2007, was
$0.6 million, $5.4 million, and $3.8 million,
respectively.
The Company uses the Black-Scholes pricing model to determine
the fair value of stock options. The determination of the fair
value of stock-based payment awards on the date of grant is
affected by our stock price as well as assumptions regarding a
number of complex and subjective variables. These variables
include our expected stock price volatility over the term of the
awards, actual and projected employee stock option exercise
behaviors, risk-free interest rates and expected dividends.
Prior to the Company’s initial public offering, the Board
of Directors, with the assistance of management, performed
contemporaneous fair value analyses to determine the fair value
of the common stock at the time of the stock option grants. The
estimated grant-date fair values of the employee stock options
were calculated using the Black-Scholes valuation model, based
on the following assumptions for the years ended
December 31, 2009, 2008 and 2007:
Expected Life. The expected life of six years
is based on the “simplified” method described in the
SEC Staff Accounting Bulletin No. 110, which provides
guidance regarding the application of ASC 718.
Volatility. Through October 17, 2007, the
Company was a private entity with no historical data regarding
the volatility of its common stock. Accordingly, the expected
volatility used for 2007 was based
67
TranS1
Inc.
Notes to
Consolidated Financial
Statements — (Continued)
on volatility of similar entities, referred to as
“guideline” companies. In 2009 and 2008, the expected
volatility was based on a weighted average of the actual
volatility of the Company for the current year and the guideline
companies for prior periods. In evaluating similarity, the
Company considered factors such as industry, stage of life cycle
and size. The Company utilized an expected volatility range of
54% to 58% for 2009, 45% to 54% for 2008 and 45% for 2007.
Risk-Free Interest Rate. The risk-free rate is
based on U.S. Treasury zero-coupon issues with remaining
terms similar to the expected term on the options. The risk-free
rates were:
| |
|
|
|
Option Grant Year
|
|
Risk-Free Rate Range
|
|
|
|
2009
|
|
1.79% to 2.52%
|
|
2008
|
|
2.68% to 3.21%
|
|
2007
|
|
4.58% to 4.92%
|
Dividend Yield. The Company has never declared
or paid any cash dividends and does not plan to pay cash
dividends in the foreseeable future, and, therefore, used an
expected dividend yield of zero in the valuation model.
Forfeitures. ASC 718 also requires the Company
to estimate forfeitures at the time of grant, and revise those
estimates in subsequent periods if actual forfeitures differ
from those estimates. The Company uses historical data to
estimate pre-vesting option forfeitures and record stock-based
compensation expense only for those awards that are expected to
vest. All stock-based payment awards are amortized on a
straight-line basis over the requisite service periods of the
awards, which are generally the vesting periods.
As of December 31, 2009, there was $4.4 million of
total unrecognized compensation costs related to non-vested
employee stock option awards granted after January 1, 2006,
which is expected to be recognized over a weighted-average
period of 2.8 years.
No provision for federal or state income taxes has been recorded
as the Company has incurred net operating losses since inception.
Significant components of the Company’s deferred tax assets
and liabilities consist of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Deferred tax assets
|
|
|
|
|
|
|
|
|
|
Domestic net operating loss carryforwards
|
|
$
|
20,707
|
|
|
$
|
14,350
|
|
|
Inventory
|
|
|
272
|
|
|
|
200
|
|
|
Fixed assets
|
|
|
532
|
|
|
|
242
|
|
|
Other
|
|
|
638
|
|
|
|
485
|
|
|
Research and development credit
|
|
|
1,031
|
|
|
|
716
|
|
|
|
|
|
|
|
|
|
|
|
|
Total deferred tax assets
|
|
|
23,180
|
|
|
|
15,993
|
|
|
Valuation allowance for deferred assets
|
|
|
(23,180
|
)
|
|
|
(15,993
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Deferred tax assets
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
Deferred tax liabilities
|
|
|
|
|
|
|
|
|
|
Fixed assets
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
Total deferred tax liabilities
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
Net deferred tax assets (liabilities)
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
68
TranS1
Inc.
Notes to
Consolidated Financial
Statements — (Continued)
The Company provided a full valuation allowance against its net
deferred tax assets since realization of these benefits could
not be reasonably assured. The increase in valuation allowance
resulted primarily from the additional net operating loss
carryforward generated.
As of December 31, 2009, the Company had federal and state
net operating loss carryforwards of approximately
$54.7 million and $51.7 million, respectively. These
net operating loss carryforwards begin to expire in 2021 and
2016 for federal and state tax purposes, respectively.
Additionally, as of December 31, 2009, the Company had
research credit carryforwards of $1.1 million for federal
tax purposes. These credit carryforwards begin to expire in
2021. The utilization of the federal net operating loss
carryforwards may be subject to limitations under the rules
regarding a change in stock ownership as determined by the
Internal Revenue Code.
A reconciliation of differences between the U.S. federal
income tax rate and the Company’s effective tax rate for
the years ended December 31 is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
|
|
|
% of
|
|
|
|
|
|
% of
|
|
|
|
|
|
% of
|
|
|
|
|
Amount
|
|
|
Net Loss
|
|
|
Amount
|
|
|
Net Loss
|
|
|
Amount
|
|
|
Net Loss
|
|
|
|
|
(In thousands)
|
|
|
|
|
Tax at statutory rate
|
|
$
|
(8,119
|
)
|
|
|
35.0
|
%
|
|
$
|
(5,962
|
)
|
|
|
35.0
|
%
|
|
$
|
(3,002
|
)
|
|
|
35.0
|
%
|
|
State taxes
|
|
|
(59
|
)
|
|
|
0.3
|
%
|
|
|
(447
|
)
|
|
|
2.6
|
%
|
|
|
(394
|
)
|
|
|
4.6
|
%
|
|
Non deductible items
|
|
|
1,129
|
|
|
|
(4.9
|
)%
|
|
|
1,168
|
|
|
|
(6.9
|
)%
|
|
|
998
|
|
|
|
(11.6
|
)%
|
|
Other
|
|
|
215
|
|
|
|
(0.9
|
)%
|
|
|
(470
|
)
|
|
|
2.8
|
%
|
|
|
53
|
|
|
|
(0.6
|
)%
|
|
R&D credits
|
|
|
(486
|
)
|
|
|
2.1
|
%
|
|
|
(148
|
)
|
|
|
0.9
|
%
|
|
|
(115
|
)
|
|
|
1.3
|
%
|
|
Change in valuation allowance
|
|
|
7,320
|
|
|
|
(31.6
|
)%
|
|
|
5,859
|
|
|
|
(34.4
|
)%
|
|
|
2,460
|
|
|
|
(28.7
|
)%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
—
|
|
|
|
0.0
|
%
|
|
$
|
—
|
|
|
|
0.0
|
%
|
|
$
|
—
|
|
|
|
0.0
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of January 1, 2007, the Company adopted the provisions
of ASC 740 (formerly FASB Interpretation No. 48 or
FIN 48, “Accounting for Uncertainty in Income
Taxes — an interpretation of FASB Statement
No. 109”), which clarifies the accounting for
uncertainty in tax positions. As of that date, the Company had
$1.1 million of unrecognized tax benefits related to the
adoption of ASC 740. This would be recorded as a component of
income tax expense once the valuation allowance is released. For
the year ended December 31, 2009, the Company increased its
unrecognized tax benefits by $13,000. The change was recorded as
a reduction to the respective deferred tax asset which was
reflected as an increase in the valuation allowance. As of
December 31, 2009, the Company had $0.9 million of
unrecognized tax benefits which, if recognized, would be
recorded as a component of income tax expense. The
Company’s policy is to record estimated interest and
penalties related to the underpayment of income taxes as a
component of its income tax provision. As of December 31,
2007, 2008 and 2009, the Company had no accrued interest or tax
penalties recorded. A reconciliation of the beginning and ending
uncertain tax positions is as follows (in thousands):
| |
|
|
|
|
|
Balance at December 31, 2007
|
|
$
|
1,114
|
|
|
Gross decreases related to current period tax positions
|
|
|
(227
|
)
|
|
|
|
|
|
|
|
Balance at December 31, 2008
|
|
|
887
|
|
|
Gross increases related to current period tax positions
|
|
|
13
|
|
|
|
|
|
|
|
|
Balance at December 31, 2009
|
|
$
|
900
|
|
|
|
|
|
|
|
In many cases, uncertain tax positions are related to tax years
that remain subject to examination by the relevant tax
authorities. Given the losses accumulated to date, periods open
for examination are 2002 to 2009 for the primary taxing
jurisdictions of the United States and North Carolina. The
Company currently does not expect a significant change in the
FIN 48 liability in the next 12 months.
69
TranS1
Inc.
Notes to
Consolidated Financial
Statements — (Continued)
The following table presents the components of other
comprehensive loss:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
Net loss
|
|
$
|
(23,196
|
)
|
|
$
|
(17,035
|
)
|
|
$
|
(8,577
|
)
|
|
Other comprehensive income (loss):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Translation adjustments
|
|
|
(5
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total comprehensive loss
|
|
$
|
(23,201
|
)
|
|
$
|
(17,035
|
)
|
|
$
|
(8,577
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.
|
Quarterly
Data (Unaudited)
|
The following unaudited quarterly financial data, in the opinion
of management, reflects all adjustments, consisting of normal
recurring adjustments, necessary for a fair presentation of
results for the periods presented (in thousands, except per
share data):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, 2009
|
|
|
|
First
|
|
Second
|
|
Third
|
|
Fourth
|
|
|
|
Quarter
|
|
Quarter
|
|
Quarter
|
|
Quarter
|
|
|
|
Total revenues
|
|
$
|
8,678
|
|
|
$
|
7,938
|
|
|
$
|
6,912
|
|
|
$
|
6,279
|
|
|
Gross profit
|
|
|
7,136
|
|
|
|
6,423
|
|
|
|
5,546
|
|
|
|
5,015
|
|
|
Total operating expenses
|
|
|
12,517
|
|
|
|
13,293
|
|
|
|
11,171
|
|
|
|
10,740
|
|
|
Net loss
|
|
$
|
(5,164
|
)
|
|
$
|
(6,759
|
)
|
|
$
|
(5,571
|
)
|
|
$
|
(5,702
|
)
|
|
Basic and diluted net loss per common share
|
|
$
|
(0.25
|
)
|
|
$
|
(0.33
|
)
|
|
$
|
(0.27
|
)
|
|
$
|
(0.28
|
)
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, 2008
|
|
|
|
First
|
|
Second
|
|
Third
|
|
Fourth
|
|
|
|
Quarter
|
|
Quarter
|
|
Quarter
|
|
Quarter
|
|
|
|
Total revenues
|
|
$
|
5,978
|
|
|
$
|
5,951
|
|
|
$
|
6,021
|
|
|
$
|
7,354
|
|
|
Gross profit
|
|
|
4,940
|
|
|
|
4,814
|
|
|
|
5,010
|
|
|
|
6,225
|
|
|
Total operating expenses
|
|
|
8,319
|
|
|
|
10,801
|
|
|
|
10,363
|
|
|
|
11,089
|
|
|
Net loss
|
|
$
|
(2,439
|
)
|
|
$
|
(5,299
|
)
|
|
$
|
(4,764
|
)
|
|
$
|
(4,533
|
)
|
|
Basic and diluted net loss per common share
|
|
$
|
(0.12
|
)
|
|
$
|
(0.26
|
)
|
|
$
|
(0.23
|
)
|
|
$
|
(0.22
|
)
|
70
TRANS1
INC.
SCHEDULE II
VALUATION
AND QUALIFYING ACCOUNTS
FOR THE
YEARS ENDED DECEMBER 31, 2009, 2008 AND 2007
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at
|
|
|
|
|
|
Balance at
|
|
|
|
Beginning of
|
|
|
|
|
|
End of
|
|
|
|
Period
|
|
Additions(1)
|
|
Deductions(2)
|
|
Period
|
|
|
|
(In thousands)
|
|
|
|
Accounts Receivable Reserve:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year ended December 31, 2009
|
|
$
|
193
|
|
|
$
|
80
|
|
|
$
|
80
|
|
|
$
|
193
|
|
|
Year ended December 31, 2008
|
|
|
110
|
|
|
|
101
|
|
|
|
18
|
|
|
|
193
|
|
|
Year ended December 31, 2007
|
|
|
29
|
|
|
|
95
|
|
|
|
14
|
|
|
|
110
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at
|
|
|
|
|
|
Balance at
|
|
|
|
Beginning of
|
|
|
|
|
|
End of
|
|
|
|
Period
|
|
Additions(3)
|
|
Deductions(4)
|
|
Period
|
|
|
|
(In thousands)
|
|
|
|
Inventory Reserve:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year ended December 31, 2009
|
|
$
|
397
|
|
|
$
|
505
|
|
|
$
|
321
|
|
|
$
|
581
|
|
|
Year ended December 31, 2008
|
|
|
295
|
|
|
|
400
|
|
|
|
298
|
|
|
|
397
|
|
|
Year ended December 31, 2007
|
|
|
57
|
|
|
|
306
|
|
|
|
68
|
|
|
|
295
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at
|
|
|
|
|
|
Balance at
|
|
|
|
Beginning of
|
|
|
|
|
|
End of
|
|
|
|
Period
|
|
Additions
|
|
Deductions
|
|
Period
|
|
|
|
(In thousands)
|
|
|
|
Valuation Allowance for Deferred Tax Assets:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year ended December 31, 2009
|
|
$
|
15,993
|
|
|
$
|
7,559
|
|
|
$
|
372
|
|
|
$
|
23,180
|
|
|
Year ended December 31, 2008
|
|
|
9,524
|
|
|
|
6,469
|
|
|
|
—
|
|
|
|
15,993
|
|
|
Year ended December 31, 2007
|
|
|
8,121
|
|
|
|
2,460
|
|
|
|
1,057
|
|
|
|
9,524
|
|
|
|
|
|
(1) |
|
Amount represents customer balances deemed uncollectible. |
| |
|
(2) |
|
Uncollectible accounts written-off. |
| |
|
(3) |
|
Amount represents excess and obsolete reserve recorded to cost
of sales. |
| |
|
(4) |
|
Excess and obsolete inventory written-off against reserve. |
71
EXHIBIT INDEX
| |
|
|
|
|
Exhibit
|
|
|
|
No.
|
|
Description
|
|
|
|
|
3
|
.1
|
|
Amended and Restated Certificate of Incorporation of TranS1 Inc.
(incorporated by reference to Exhibit 3.2 to the
Registration Statement on
Form S-1,
as amended (File
No. 333-144802),
and as declared effective on October 16, 2007).
|
|
|
3
|
.2
|
|
Amended and Restated Bylaws of TranS1 Inc. (incorporated by
reference to Exhibit 3.4 to the Registration Statement on
Form S-1,
as amended (File
No. 333-144802),
and as declared effective on October 16, 2007).
|
|
|
4
|
.1
|
|
Specimen common stock certificate (incorporated by reference to
Exhibit 4.1 to the Registration Statement on
Form S-1,
as amended (File
No. 333-144802),
and as declared effective on October 16, 2007).
|
|
|
10
|
.1
|
|
Amended and Restated 2000 Stock Incentive Plan (incorporated by
reference to Exhibit 10.1 to the Registration Statement on
Form S-1,
as amended (File
No. 333-144802),
and as declared effective on October 16, 2007).*
|
|
|
10
|
.2
|
|
Form of Stock Option Agreement under Amended and Restated 2000
Stock Incentive Plan (incorporated by reference to
Exhibit 10.2 to the Registration Statement on
Form S-1,
as amended (File
No. 333-144802),
and as declared effective on October 16, 2007).*
|
|
|
10
|
.3
|
|
2007 Stock Incentive Plan, as amended (incorporated by reference
to Appendix A to the Definitive Proxy Statement on
Schedule 14A, as filed with the Commission on
April 30, 2009.*
|
|
|
10
|
.4
|
|
Form of Stock Option Agreement under 2007 Stock Incentive Plan
(incorporated by reference to Exhibit 10.4 to the
Registration Statement on
Form S-1,
as amended (File
No. 333-144802),
and as declared effective on October 16, 2007).*
|
|
|
10
|
.5
|
|
2007 Employee Stock Purchase Plan (incorporated by reference to
Exhibit 10.5 to the Registration Statement on
Form S-1,
as amended (File
No. 333-144802),
and as declared effective on October 16, 2007).*
|
|
|
10
|
.6
|
|
Lease, dated July 30, 2009, between TranS1 Inc. and Market
Place Group, LLC (incorporated by reference to Exhibit 10.1
of TranS1’s Current Report on
Form 8-K
filed with the Commission on March 11, 2010).
|
|
|
10
|
.7
|
|
Form of Indemnification Agreement (incorporated by reference to
Exhibit 10.7 to the Registration Statement on
Form S-1,
as amended (File
No. 333-144802),
and as declared effective on October 16, 2007).
|
|
|
10
|
.7.1
|
|
Schedule of Parties to Indemnification Agreement.**
|
|
|
10
|
.8
|
|
Offer Letter, dated December 11, 2009, between TranS1 Inc.
and Kenneth Reali (incorporated by reference to
Exhibit 10.1 of TranS1’s Current Report on
Form 8-K
filed with the Commission on January 12, 2010).*
|
|
|
10
|
.9
|
|
Separation Agreement, dated February 23, 2010, between
TranS1 Inc. and Michael Luetkemeyer (incorporated by reference
to Exhibit 10.1 of TranS1’s Current Report on
Form 8-K
filed with the Commission on February 25, 2010).*
|
|
|
23
|
.1
|
|
Consent of Independent Registered Public Accounting Firm.**
|
|
|
24
|
.1
|
|
Power of Attorney (included in the signature page).
|
|
|
31
|
.1
|
|
Certification of Chief Executive Officer Pursuant to
Rule 13a-14(a)
/ 15d-14(a) of the Securities Exchange Act of 1934.**
|
|
|
31
|
.2
|
|
Certification of Chief Financial Officer Pursuant to
Rule 13a-14(a)
/ 15d-14(a) of the Securities Exchange Act of 1934.**
|
|
|
32
|
.1
|
|
Certification of Chief Executive Officer Pursuant to
Rule 13a-14(b)
/ 15d-14(b) of the Securities Exchange Act of 1934 and
18 U.S.C. Section 1350.**
|
|
|
32
|
.2
|
|
Certification of Chief Financial Officer Pursuant to
Rule 13a-14(b)
/ 15d-14(b) of the Securities Exchange Act of 1934 and
18 U.S.C. Section 1350.**
|
|
|
|
|
* |
|
These exhibits are identified as management contracts or
compensatory plans or arrangements of the Registrant pursuant to
Item 15(a)(3) of
Form 10-K. |
72