Attached files
| file | filename |
|---|---|
| EX-32.1 - EXHIBIT 32.1 - NANOSPHERE INC | c97489exv32w1.htm |
| EX-23.1 - EXHIBIT 23.1 - NANOSPHERE INC | c97489exv23w1.htm |
| EX-32.2 - EXHIBIT 32.2 - NANOSPHERE INC | c97489exv32w2.htm |
| EX-31.1 - EXHIBIT 31.1 - NANOSPHERE INC | c97489exv31w1.htm |
| EX-31.2 - EXHIBIT 31.2 - NANOSPHERE INC | c97489exv31w2.htm |
Table of Contents
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
(Mark One)
| þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2009
OR
| o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number 001-33775
Nanosphere, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 36-4339870 | |
| (State or other jurisdiction of | (I.R.S. Employer Identification No.) | |
| incorporation or organization) | ||
| 4088 Commercial Avenue | Northbrook, Illinois 60062 | |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code: (847) 400-9000
Securities registered pursuant to Section 12(b) of the Act:
| (Title of Each Class) | (Name of Each Exchange on Which Registered) | |
| Common Stock, par value $0.01 | NASDAQ Global Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark whether the registrant is a well-known seasoned issuer, as defined in
Rule 405 of the Securities Act. o Yes þ No
Indicate by check mark whether the registrant is not required to file reports pursuant to
Section 13 or Section 15(d) of the Act. o Yes þ No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed
by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or
for such shorter period that the registrant was required to file such reports), and (2) has been
subject to such filing requirements for the past 90 days. Yes þ No o
Indicate by check mark whether the registrant has submitted electronically and posted on its
corporate Web site, if any, every Interactive Data File required to be submitted and posted
pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months
(or for such shorter period that the registrant was required to submit and post such
files). Yes o No o (the Registrant is not yet required to submit Interactive Data)
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation
S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in
definitive proxy or information statements incorporated by reference in Part III of this Form 10-K
or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated
filer, a non-accelerated filer or a smaller reporting company. See definitions of “large
accelerated filer”, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the
Exchange Act. (Check one):
| Large accelerated filer o | Accelerated filer o | Non-accelerated filer þ | Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of
the Exchange Act). Yes o No þ
As of June 30, 2009, the last business day of the registrant’s most recently completed second
fiscal quarter, the aggregate market value of the common stock held by non-affiliates of the
registrant was approximately $58,030,023 based on the closing sale price for the registrant’s
common stock on the NASDAQ Global Market on that date of $4.91 per share. This number is provided
only for the purpose of this report on Form 10-K and does not represent an admission by either the
registrant or any such person as to the status of such person.
As of March 4, 2010, there were 28,425,871 outstanding shares of common stock. The common
stock is listed on the NASDAQ Global Market (trading symbol “NSPH”).
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive proxy statement for fiscal year ended December 31,
2009 to be issued in conjunction with the registrant’s annual meeting of shareholders expected to
be held on May 25, 2010 are incorporated by reference in Part III of this Form 10-K. The
definitive proxy statement will be filed by the registrant with the SEC not later than 120 days
from the end of the registrant’s fiscal year ended December 31, 2009. Except as specifically
incorporated herein by reference, the above mentioned Proxy Statement is not deemed filed as part
of this report.
NANOSPHERE, INC.
INDEX
INDEX
| Page | ||||||||
| 1 | ||||||||
| 15 | ||||||||
| 26 | ||||||||
| 26 | ||||||||
| 26 | ||||||||
| 26 | ||||||||
| 27 | ||||||||
| 29 | ||||||||
| 30 | ||||||||
| 39 | ||||||||
| 40 | ||||||||
| 59 | ||||||||
| 59 | ||||||||
| 60 | ||||||||
| 61 | ||||||||
| 61 | ||||||||
| 61 | ||||||||
| 61 | ||||||||
| 61 | ||||||||
| 62 | ||||||||
| 65 | ||||||||
| Exhibit 23.1 | ||||||||
| Exhibit 31.1 | ||||||||
| Exhibit 31.2 | ||||||||
| Exhibit 32.1 | ||||||||
| Exhibit 32.2 | ||||||||
Table of Contents
Forward-Looking Statements
This Annual Report on Form 10-K contains forward-looking statements about us and our industry
that involve substantial risks and uncertainties. All statements, other than statements of
historical facts, included in this Annual Report on Form 10-K regarding our strategy, future
operations, future financial position, future net sales, projected expenses, prospects and plans
and objectives of management are forward-looking statements. These statements involve known and
unknown risks, uncertainties and other factors that may cause our actual results, levels of
activity, performance or achievement to be materially different from those expressed or implied by
the forward-looking statements.
In some cases, you can identify forward-looking statements by terms such as “anticipate,”
“believe,” “estimate,” “expect,” “intend,” “may,” “might,” “plan,” “project,” “will,” “would,”
“should,” “could,” “can,” “predict,” “potential,” “continue,” “objective,” or the negative of these
terms, and similar expressions intended to identify forward-looking statements. However, not all
forward-looking statements contain these identifying words. These forward-looking statements
reflect our current views about future events and are based on assumptions and subject to risks and
uncertainties. Given these uncertainties, you should not place undue reliance on these
forward-looking statements. Actual events or results could differ materially from those expressed
or implied by these forward-looking statements as a result of various factors.
These forward-looking statements represent our estimates and assumptions only as of the date
of this Annual Report on Form 10-K. Unless required by U.S. federal securities laws, we do not
intend to update any of these forward-looking statements to reflect circumstances or events that
occur after the statement is made or to conform these statements to actual results. The following
discussion should be read in conjunction with the consolidated financial statements and notes
thereto appearing elsewhere in this Annual Report on Form 10-K. Our actual results may differ
materially from those anticipated in these forward-looking statements as a result of various
factors, including those set forth under “Item 1A — Risk Factors” and elsewhere in this Annual
Report on Form 10-K.
PART I.
| Item 1. | Business. |
References herein to “we,” “us,” “our” or “the Company” refer to Nanosphere, Inc. unless the
context specifically requires otherwise.
Overview
We develop, manufacture and market an advanced molecular diagnostics platform, the Verigene
System, that enables simple, low cost and highly sensitive genomic and protein testing on a single
platform. Our proprietary nanoparticle technology simplifies molecular diagnostic testing, provides
the ability to run multiple tests simultaneously on the same sample and has the potential to run a
broad menu of tests on a single platform. We have developed or are currently developing diagnostic
tests for markers which reveal the existence of a variety of medical conditions including
cardiovascular, respiratory, cancer, autoimmune, neurodegenerative and other infectious diseases,
as well as for pharmacogenomics.
Pharmacogenomics is an emerging subset of human genetic testing that correlates gene variation
with a drug’s efficacy or toxicity. These tests play a key role in the advancement of personalized
medicine where drug therapies and dosing are guided by each patient’s genetic makeup. There is a
growing demand on laboratories to implement molecular diagnostic testing, but the cost and
complexity of existing technologies and the need for specialized personnel and facilities have
limited the number of laboratories with these capabilities.
The Verigene System is differentiated by its ease of use, rapid turnaround times and ability
to support a broad test menu. It also offers lower cost for laboratories already performing
molecular diagnostic testing and allows a broader range of laboratories, including those operated
by local hospitals, to perform these tests. Our ability to detect proteins, which can be 100 times
more sensitive than current technologies for certain targets, may enable earlier detection of and
intervention in diseases associated with known biomarkers as well as the introduction of tests for
new biomarkers that exist in concentrations too low to be detected by current technologies. We
are focused on the clinical diagnostics market and may seek opportunities either directly or
through partnerships to commercialize our technologies in other markets.
1
Table of Contents
We received 510(k) clearance from the United States Food and Drug Administration (“FDA”) for
commercial sale of the Verigene System in the second half of 2007. 510(k) clearance is granted by
the FDA if the submitted information establishes that the proposed device is “substantially
equivalent” to a legally marketed Class I or Class II medical device or a pre-amendment Class III
medical device for which the FDA has not sought pre-market approval. We have also received 510(k)
clearance to use the Verigene System for four diagnostic tests. The first test is a warfarin
metabolism assay, which is a pharmacogenomic test to determine the existence of certain genetic
information believed to affect the metabolism of warfarin-based drugs, including Coumadin, the
most-prescribed oral anticoagulant in North America and Europe. The second test is a
hyper-coagulation assay, one of the highest volume human genetic tests currently performed, to
determine an individual’s risk, based upon genetic information, for the development of blood clots,
which can lead to stroke, pulmonary embolism and deep vein thrombosis.
The third test is our respiratory panel which detects the presence of influenza A and B as
well as respiratory syncitial virus (“RSV”) A and B. Influenza is commonly known as the seasonal
flu and RSV is a respiratory virus that infects the lungs and breathing passages. RSV is the most
common cause of bronchitis and pneumonia in children under the age of one year and has become a
significant concern for older adults. Our respiratory panel provides physicians with a highly
accurate, fast determination of which virus is present which helps guide the most appropriate
treatment therapy. Most of the respiratory tests currently on the market take days to generate a
result, because they depend on culturing, or do not provide a reliable result, because they are
rapid tests which lack specificity.
On October 9, 2009, we received 510(k) clearance of our second generation respiratory panel
and the Verigene SP System. The Verigene SP instrument automates sample preparation, which should
significantly reduce laboratory technician time required to perform this molecular test. We also
believe that our respiratory assay on the Verigene SP offers a simple to use molecular test for
diagnosing respiratory infections and the flu, while providing improved specificity over currently
available rapid tests. On November 24, 2009, we received
clearance for a product insert change for this
assay confirming that the novel H1N1 virus is detected as a positive Influenza A when using our
respiratory assay and the Verigene SP. However, it is uncertain whether the ability to use this
assay to detect H1N1 virus will be material to the Company based on the reduction in the number of
recently reported H1N1 cases.
The fourth test is our cystic fibrosis test that enables molecular laboratories to perform
prenatal screening and diagnostic confirmations through identification of the number of copies of
each of the 23 most common gene mutations recognized by the American College of Obstetricians and
Gynecologists as markers for cystic fibrosis.
We have elected to delay the launch of our 501(k) cleared cystic fibrosis test until such time
as it may receive 510(k) clearance for use on the Verigene SP. In addition, we plan to submit
additional FDA applications for each of our previously 510(k) cleared assays to allow their use on
the new Verigene SP. We have engaged in dialogue with the FDA to
identify any additional clinical studies that we need to receive 510(k) clearance
to run these assays on the Verigene SP.
In the first quarter of 2009, we filed a de novo 510(k) submission for a hereditary
hemochromatosis (“HFE”) genetic test. We re-submitted an amended 510(k) submission in October 2009
to supply additional data requested by the FDA. Mutations in the HFE gene are associated with
hemochromatosis, which is the leading cause of iron overload disease, a systematic iron build up
that can eventually adversely affect the heart, liver, pancreas, joints and pituitary gland.
Untreated, hemochromatosis can be fatal. Once detected, hemochromatosis is easily treated.
Approximately one in every 250 people of European descent has the disease and one in eight is a
carrier of at least one of the recessive gene mutations. This is a de novo 510(k) because there are
currently no FDA-cleared tests on the market to detect these mutations of the HFE gene. We have
received additional feedback from the FDA indicating that further testing would be necessary in
order to proceed with this application. This assay was submitted for clearance on the original
Verigene System and the FDA requires separate testing and a new application to clear this assay on
the Verigene SP. Therefore, we plan to complete all necessary testing
on the Verigene SP to submit a 510(k) application for it.
The first ultra-sensitive protein test we plan to commercialize is for cardiac troponin I
(“cTnI”), which is the gold standard biomarker for diagnosis of the occurrence of myocardial
infarction or heart attack and identification of patients at risk for
acute coronary syndromes at risk for subsequent cardiovascular events. We
submitted a 510(k) application to the FDA in the fourth quarter of 2009. We have received initial
feedback from the FDA regarding our 510(k) submission indicating that significant further studies
will be required to obtain clearance for our cTnI assay. We believe that some of the additional
requirements suggested by the FDA may be addressed by results from our ongoing FAST-TRAC clinical
study, which is designed to support and further demonstrate the clinical utility of ultra-sensitive
cTnI
measurements as a diagnostic tool for use in the management of both acute and chronic cardiac
disease. We plan to complete the FAST-TRAC clinical studies along with any other studies necessary
to respond to the FDA’s requests in 2010.
2
Table of Contents
We are currently conducting multi-site trials for our cytochrome P450 2C19 assay that detects
genetic mutations associated with deficiencies to metabolize clopidogrel, more commonly known by
the trade name Plavix. Clopidogrel inhibits platelet function and is a standard treatment to
reduce the risk of thrombolytic events for patients undergoing percutaneous coronary intervention
(“PCI”) procedures. Clopidogrel metabolism is affected by the Cytochrome P-450 family of genes. Up
to 30-50% of the patient population possess variations in these genes and do not properly
metabolize this drug, thus increasing the risk of adverse events. Our 2C19 assay is designed to
identify patients possessing these variations so that alternative
therapeutic approaches can be prescribed to
effectively reduce clotting post-PCI.
In addition, we currently have research and development efforts underway on additional
genetic, infectious disease and protein tests. Our test development pipeline includes a blood
infection screening assay, an ultra-sensitive prostate-specific antigen (“PSA”)
test for early diagnosis of recurrent prostate cancer and a multiplexed protein-based
connective-tissue panel for the detection of rheumatoid arthritis, lupus and other
related diseases. We are also investigating new biomarkers where our ultra-sensitive
protein detection technology may enable earlier detection of a broad range of
diseases, such as cancer. We are in the process of assessing the commercial
viability of these applications and the various paths to regulatory approval that may be available.
Our technology is broadly applicable beyond the clinical diagnostic market in both research
and industrial applications. The Verigene System is presently used in research laboratories
supporting collaborations and independent research in areas including
cancer, infectious disease and Alzheimer’s disease. We have developed and delivered a biosecurity platform for the detection of
various bioterrorism agents to the Technical Support Working Group, an agency affiliated with the
U.S. Department of Defense.
As of December 31, 2009, our patent portfolio is comprised, on a worldwide basis, of 132
issued patents and 97 pending patent applications which we own directly or for which we are the
exclusive licensee. Some of these patents and patent applications derive from a common parent
patent application or are foreign counterpart patent applications and relate to similar or
identical technological claims. The issued patents cover approximately 12 different technological
claims and the pending patent applications cover approximately 4 additional technological claims.
Many of our issued and pending patents were exclusively licensed from the International
Institute for Nanotechnology at Northwestern University (“Northwestern”) in May 2000 and they
generally cover our core technology, including nanotechnology based biodiagnostics and biobarcode
technology. Our issued patents expire between 2017 and 2025. We believe our patent portfolio
provides protection against other companies offering products employing the same technologies and
methods as we have patented. While we believe our patent portfolio establishes a proprietary
position, there are many competitive products utilizing other technologies that do not infringe on
our patents.
In addition, as of December 31, 2009, we have non-exclusive licenses for at least 44 U.S.
patents that cover 12 different technological claims from various third parties. Most of these
license agreements require us to pay the licensor royalty fees that typically expire upon the
patent expiration dates which range from 2010 to 2025. These license agreements are nonexclusive
and do not create a proprietary position. The expiration of these non-exclusive licenses will
result in the termination of certain royalty payments by us to the licensors.
Our Products
The Verigene System is a bench-top molecular diagnostics workstation that is a universal
platform for genomic and protein testing. While many systems currently available on the market
provide a diagnostic result for one test or a few tests within a specific market niche, the
Verigene System provides for multiple tests to be performed on a single platform including both
genomic and protein assays.
The Verigene System is comprised of a microfluidics processor, a touchscreen reader and
disposable test cartridges. The microfluidics processor interacts with and manipulates various
functional components of the test cartridge, accomplishing a number of necessary steps including
target binding to the nucleic acid or protein array, nanoparticle probe hybridization, intermediate
washes and signal amplification. The reader houses the optical detection module that illuminates
the test slide and automated spot recognition software that analyzes the resulting signal
intensities and provides the test results. The reader also serves as the control station for the
Verigene System and features a simple and intuitive touchscreen interface that allows users to
track samples and test cartridges, initiate and monitor test processing, analyze results and
generate reports. The reader is web-enabled to allow remote access to results and reports.
3
Table of Contents
To perform a test, the operator adds a prepared sample to a designated port in the test
cartridge, enters sample identification and test cartridge information into the reader using the
touchscreen keyboard or via the barcode wand, and inserts the test cartridge into the processor.
The processor assimilates information received from the reader and matches it to the inserted test
cartridge and initiates the specified test protocol. Once the assay process is complete the test
array is introduced into the reader for image analysis and result reporting.
During 2009, we received 510(k) clearance from the FDA for the Verigene SP which handles the
same processing steps, including DNA extraction, as the original Verigene system as well as sample
preparation. This additional capability further automates and simplifies the testing process by
extending the system workflow from sample to result. The Verigene System is also modular so that
customers can attach a number of processors to each reader depending upon their laboratory test
volume. We are also developing a fully automated instrument with high throughput and sample
preparation for larger hospital based laboratories. By basing future generations of our instrument
platform on existing design elements, each new generation of development will process assays
developed for previous generations.
Our Applications
We are commercializing or have in development several genomic and protein assays including the
following:
| Assay | Condition Detected | Status | ||
Genomic Tests |
||||
Hyper-coagulation
|
Genetic mutation that could indicate increased risk of blood clots, stroke and pulmonary embolism | Verigene FDA clearance received October 2007 | ||
Warfarin Metabolism
|
Genetic mutation important in proper initial dosing of leading anticoagulant | Verigene FDA clearance received September 2007 | ||
Cystic Fibrosis
|
Cystic fibrosis gene mutation; prenatal carrier screening & diagnosis confirmation | Verigene FDA clearance received July 2009 | ||
Respiratory Virus Panel
|
Respiratory illness | Verigene FDA clearance received May 2009; Verigene SP FDA clearance received October 2009 | ||
HFE — hemochromatosis
|
Hemochromatosis, iron overload disease | Amended 510(k) filed with the FDA in October 2009 | ||
CYP450 2C19
|
Clopidigrel metabolism | In clinical trials | ||
Blood Infection Panel
|
Blood infection | In development | ||
Protein Tests |
||||
Cardiac Troponin I
|
Cardiovascular disease; risk stratification; diagnosis of heart attack | Filed with the FDA in the fourth quarter of 2009 | ||
Connective
Tissue Disease
|
Rheumatoid arthritis, lupus, psoriatic arthritis, spondyloarthropathies, scleroderma and Sjogren’s syndrome | In development | ||
Prostate Specific Antigen
|
Post-surgical recurrence of prostate cancer | In development |
4
Table of Contents
We are also continuing to research the expansion of our test menu in areas including, cancer,
autoimmune, allergy, neurodegenerative, cardiovascular, infectious diseases and pharmacogenomics.
Genomic Assays
Hyper-coagulation. Currently available technologies for this test are limited by contamination
issues associated with the polymerase chain reaction, or PCR, genomic test as well as its cost
resulting from a complex and costly work flow and large number of process steps. Our
hyper-coagulation panel consists of a multiplex of three genetic markers. This test enables the
direct detection of mutations associated with a pre-disposition to blood clots on a much simpler
platform than current alternatives. Our hyper-coagulation assay received 510(k) clearance for use
on the original Verigene System in October 2007.
Warfarin Metabolism. Our warfarin metabolism assay received 510(k) clearance for use on the
original Verigene System in September 2007. Our assay is the first FDA-cleared genetic diagnostic
test to assess warfarin metabolism. Our warfarin metabolism panel detects three genetic markers
that play a critical role in metabolizing warfarin and determining an individual’s sensitivity to
the drug. Through detection of these genetic markers, doctors are able to determine the appropriate
initial warfarin dosage level in a safer and more efficient manner than current methods. Most of
the other assays available today require PCR prior to running the assay, which contributes
significantly to the cost and complexity of testing.
Cystic Fibrosis. Our cystic fibrosis assay received 510(k) clearance for use on the original
Verigene System in July 2009. This assay provides a multiplex panel for the detection of mutations
in the CFTR gene that contribute to a higher risk of cystic fibrosis. Currently available
technologies for this test typically require up-front PCR and post-PCR multiplexing technologies,
which involve
complex and costly work flow which require more testing duration and hands-on lab technician
time as well as additional process steps. Our direct detection method enables detection of each of
the 23 CFTR gene mutations related to cystic fibrosis recognized by the American College of
Obstetricians and Gynecologists.
Respiratory Virus Panel. On May 1, 2009, we received 510(k) clearance for the commercial sale
of our respiratory panel for use on the original Verigene System which detects the presence of
influenza A and B as well as RSV A and B. Influenza is commonly known as the seasonal flu and RSV
is a respiratory virus that infects the lungs and breathing passages. RSV is the most common cause
of bronchitis and pneumonia in children under 12 months and has become a significant concern for
older adults. Our respiratory panel provides physicians with a highly accurate, fast determination
of which virus is present which helps guide the most appropriate therapy. On October 9,
2009, we received 510(k) clearance of our second generation respiratory panel for use on the
Verigene SP. On November 24, 2009, we received clearance for a label change for this assay
confirming that the novel H1N1 virus is detected as a positive
Influenza A when using clinical isolates running our
respiratory assay on the Verigene SP.
HFE. A de novo 510(k) for our HFE assay for use on the original Verigene System was submitted
to the FDA in February 2009 and we submitted an amended 510(k) in October 2009. We have received
additional feedback from the FDA indicating that further testing would be necessary in order to
proceed with this application. This assay was submitted for clearance on the original Verigene
System and the FDA requires separate testing and a new application to clear this assay on the
Verigene SP. Therefore, we plan to complete all necessary testing on
the Verigene SP to submit a
510(k) application for it. Mutations in the HFE gene are
associated with hemochromatosis, which is the leading cause of iron overload disease. Over time,
hemochromatosis causes iron build up and eventually can cause diseases in the heart, liver,
pancreas, joints and pituitary gland. Untreated, hemochromatosis can be fatal. Once detected,
hemochromatosis is easily treated. Approximately one in every 250 people of European descent has
the disease and one in eight is a carrier of at least one of the recessive gene mutations. There
are currently no FDA cleared tests on the market.
Cytochrome P450 2C19. Our 2C19 assay is for the detection of mutations that lead to deficiency
in the metabolism of clopidogrel, more commonly known by the trade name Plavix. Clopidogrel
inhibits platelet function and is a standard treatment to reduce the risk of thrombolytic events
for patients undergoing percutaneous coronary intervention (“PCI”) procedures. Clopidogrel is
metabolized through the Cytochrome P-450 family of genes. Up to 30-50% of the patient population
possess variations in these genes and do not properly metabolize this drug, thus increasing the
risk of adverse events. Therapeutic approaches can be prescribed to
effectively reduce clotting post-PCI.
Blood Stream Infection. We are developing three blood stream infection panels for the earlier
determination of specific bacterium present within patients with blood stream infections.
Currently under development are gram positive, gram negative and fungal panels. These assays are
designed to enable physicians to pinpoint bacterial strains infecting patients and thus prescribe
the most appropriate antibiotic regimen within the first 24 hours rather than after several days.
We believe that this early detection will allow patients to avoid unnecessary treatments that often
include serious side effects.
5
Table of Contents
Protein Assays
Our initial assay development efforts are focused on the earlier detection of disease through
application of our ultra-sensitive protein detection technology to existing biomarkers. The
following assays are in development:
Cardiac Troponin I. The first ultra-sensitive protein test we plan to commercialize is for
cardiac troponin I (“cTnI”), which is the gold standard biomarker for diagnosis of the occurrence
of myocardial infarction or heart attack and identification of patients at risk for acute coronary
syndrome. We submitted a 510(k) application to the FDA in the fourth quarter of 2009. We have
received initial feedback from the FDA regarding our 510(k) submission indicating that significant
further studies will be required to obtain clearance for our cTnI assay. We believe that some of
the additional requirements suggested by the FDA may be addressed by results from our ongoing
FAST-TRAC clinical study, which is designed to support and further demonstrate the clinical utility
of ultra-sensitive cTnI
measurements as a diagnostic tool for use in the management of both acute and chronic cardiac
disease. We plan to complete the FAST-TRAC clinical studies along with any other studies necessary
to respond to the FDA’s requests in 2010.
This assay is for the detection of cTnI in patients suspected of having cardiovascular
disease. Although there are various diagnostic tests used to detect acute myocardial infarction,
both the American College of Cardiology and the American Heart Association issued guidelines
asserting that testing for cardiac troponin is the definitive laboratory standard for the
diagnosis of myocardial infarction. Troponin assays are not only more sensitive but also more
specific than tests for other existing cardiovascular disease biomarkers.
Troponin I is a protein that is found in cardiac muscle and is released when the heart is
injured, for instance during a myocardial infarction. Cardiac troponin blood diagnostic tests are
used to diagnose a heart attack and evaluate mild to severe heart injury in patients experiencing
heart/chest discomfort. However, limitations of current detection levels often result in the
failure to accurately diagnose all cases of cardiovascular disease. Each year, millions of patients
enter emergency rooms with chest pain, many of whom are discharged from hospital emergency
departments after current technologies fail to diagnose acute myocardial infarctions. Tens of
thousands of these patients are subsequently readmitted to the hospital or die from cardiac arrest
within a few weeks or months after the initial visit.
Initial clinical studies have demonstrated our ability to reliably identify a rise in cardiac
troponin significantly below the limits of detection of assays currently on the market, at levels
where the assay can diagnose acute myocardial infarctions earlier and detect precursor cardiac
events including unstable angina, cardiac necrosis and ischemia. Furthermore, this level of
sensitivity could also lead to more accurate risk stratification of people with early stages of
cardiovascular disease such as angina. We believe that a new definition of “normal” will be defined
at a lower troponin concentration level that currently marketed tests cannot detect. Our FAST-TRAC
trial is designed to test new diagnostic attributes of the assay, including its predictive value.
Prostate Specific Antigen — Recurrent Prostate Cancer. This assay is designed to detect a
recurrence of prostate cancer following prostate removal, a standard treatment for prostate cancer.
Prostate specific antigen, or PSA, is a protein produced by the cells of the prostate gland and may
be found in an increased amount in those with prostate cancer. Despite regular post-surgery PSA
testing, most cases of recurrence are not detected for several years because the rising PSA levels
in these patients does not reach the limit of detection using current technologies. We expect our
ultra-sensitive PSA detection assay will diagnose recurrence much more quickly. Our research data
suggests that patients without recurrence do not have rising PSA levels within the first six months
post prostatectomy. Therefore, we believe our assay will enable more immediate treatment for
patients with recurrence and avoidance of unnecessary treatments and life altering side effects for
patients without recurrence.
6
Table of Contents
Connective-tissue. We are developing a multiplexed protein test for the detection of
rheumatoid arthritis, lupus and other connective tissue diseases. This assay combines the
advantage of testing for multiple proteins on a single cartridge with higher sensitivity that
enables earlier detection of disease. Additionally, we have novel,
proprietary biomarkers in research. We believe these assays will provide rheumatologists an
economical and easy to use solution for diagnosing patients with connective tissue diseases.
Biomarker Validation. We are also applying our ultra-sensitive protein detection methods to
the development of tests for both established protein biomarkers as well as the validation of novel
protein targets that may lead to earlier detection of medical conditions including in the areas of
autoimmune diseases, allergies, cancers and neurodegenerative disorders.
Our Technology
We believe our technology will drive greater usage of ultra-sensitive and multiplexed protein
and genomic diagnostics in routine clinical laboratories, much as enzyme-linked immunosorbent
assay, or ELISA, accelerated the use of protein testing in the 1970s and 1980s and PCR catalyzed
the emergence of nucleic acid diagnostics in the 1990s.
Our Gold Nanoparticle Molecular Probes
At the core of our technology are gold nanoparticles which offer a unique set of physical
properties that can be exploited in the detection of biological molecules. In 1998, Dr. Mirkin and
Dr. Letsinger at Northwestern developed a novel process to prepare stable probes by covalently
attaching oligonucleotides to gold nanoparticles. This method, protected by patents, is exclusively
assigned to or owned by us. We have refined the synthesis methods to enable highly reproducible
production of nanoparticle probes with diameters in the 13-50 nanometer range required for highly
sensitive biomedical analysis. Subsequently, we have also developed methods for attaching
antibodies to gold nanoparticles, thereby producing highly stable probes for ultra-sensitive
detection of proteins.
The properties of nanoparticle probes can be tailored by controlling the size of the
particles, the density of recognition-oligomers or antibodies on the nanoparticles, the use of
diluent oligonucleotides, the use of spacer oligonucleotides and the salt concentration. Combined,
the optimization of these properties enables us to deliver superior analytical performance
characteristics versus other methods, for example:
| • | High Signal-to-Noise Ratio. Our nanoparticle probes deliver significantly stronger signals than the fluorescent probes, or fluorophores, used in most diagnostic platforms today. Nanoparticles are typically 10-100 nm in diameter and therefore significantly larger than conventional fluorophores. This size difference enables nanoparticles to produce up to 10,000 times more signal via light scattering than a fluorophore. A single nanoparticle can be detected with simple optical instrumentation with very high sensitivity, thus eliminating the need to employ our amplification techniques. | |
| • | Orders of Magnitude Greater Sensitivity and Lower Detection Limits. The sensitivity and limits of detection of our technology are further enhanced by a silver-staining step, which effectively amplifies the signal from each nanoparticle bound to a target molecule. In this process, silver is coated onto the gold nanoparticle surface, producing larger particles with enhanced optical properties. Whereas the leading technologies today can detect molecules at the picomolar range (10-12), our technology is capable of up to a million times higher sensitivity at the attomolar (10-18) range, enabling the unprecedented analysis of rarely expressed genes or low abundance proteins for early disease detection and diagnosis. | |
| • | Unparalleled Specificity. A key property of the oligonucleotide-linked gold nanoparticle is an extremely sharp melting curve. The melting curve is the temperature range during which the capture oligonucleotide dissociates with the complementary target oligonucleotide in the sample. Our nanoparticles exhibit dissociation transitions of less than one degree in Celsius temperature, whereas most alternative products are based on polymerase chain reaction, or PCR, which exhibits melt transitions typically in the 15-30 degree range. The narrow band of temperature in which binding and dissociation occurs, creates a significantly higher signal to noise ratio resulting in greater specificity. These qualities eliminate errors caused by mismatched nucleotide pairs, thereby allowing genomic targets differing by a single nucleotide (base pair) to be distinguished with unprecedented selectivity. Sharp melting curves are a proprietary feature of our nanoparticles and our patent portfolio includes issued patents protecting the methods and product performance related to melt transition curves. |
7
Table of Contents
| • | High Count Multiplexing. Our core technology enables high count multiplexing, or simultaneous multiple target identification in a single sample, using a simple low-density microarray. A sample and probe mixture is introduced simultaneously into a single self-contained reaction chamber pre-printed with multiple reaction spots, each containing capture strand oligonucleotides or proteins that are complementary to a specific target molecule of interest. By utilizing the sharp melt transition of the nanoparticle probes, multiple targets can be discretely identified in a single sample. This methodology eliminates the need for complex and costly means of physically isolating individual target molecules. | |
| • | Detection of Genomic and Protein Molecules Simultaneously. We are able to synthesize our gold nanoparticle probes for the simultaneous multiplexed detection of both protein and genomic targets in the same assay. | |
| • | Superior Reaction Kinetics. The sharp melt transition curves in our gold nanoparticles increase binding affinity thereby leading to improved assay kinetics and efficiency. | |
| • | Long-Term Stability. The high density of oligonucleotides per nanoparticle, serves both as a protective and recognition layer on the nanoparticle surface and ensures the long-term stability of our nanoparticles. We have patented approaches using localized salt and buffer concentrations that deliver long-shelf life for our technology and reagent set. |
Assay Format
Our silver-enhanced gold nanoparticles and related optical detection technology are used for
diagnostic assays which detect genomic and proteomic targets captured onto microarrays as shown in
the “Schematic of Microarray Based Detection Using
Nanoparticle Probes” below. The microarray format enables high count multiplexing of assay
targets, facilitating the development of a broad menu of tests, including for complex diseases
where multiple targets must be evaluated to provide a diagnosis, in a simple, scalable format.
Two probe types can be used in a single assay. Oligonucleotide probes are used for genomic
assays and antibodies for protein assays. One probe, complementary to a specific site on the target
molecule, is attached to a surface such as a glass slide and the other probe, complementary to a
different site on the target molecule, is attached to the surface of gold nanoparticles. In the
presence of the target molecule of interest, the probes and target form a three dimensional,
cross-linked aggregate. After silver coating the gold nanoparticles, light scatter is measured on
the surface of the microarray slide. The silver-enhanced gold nanoparticle probes located on the
slide surface scatter light in proportion to the concentration of the target in the sample, which
is detected through optical imaging and translated into clinical results via our proprietary
software algorithms.
8
Table of Contents
Schematic of Microarray Based Detection Using Nanoparticle Probes
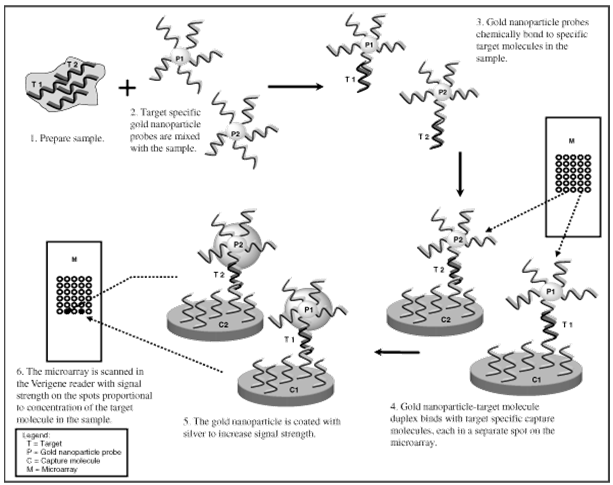
The above graphic depicts a genomic or proteomic assay utilizing a molecule attached to a gold
nanoparticle. In the case of a genomic assay, the molecule represents an oligonucleotide. In the
case of a proteomic assay, the molecule represents an antibody.
Research and Development
Our research and development efforts are focused on:
| • | Expanding and Enhancing the Capabilities of Our Instrument
Platform. Design elements and components of our current instrument
platform, the Verigene System, will serve as the foundation for future
generation development. The Verigene SP incorporates sample preparation
into our system. By adding this step, labs can now process a raw sample
material, in most cases whole blood, in a single step. This feature is
critical for analyzing infectious diseases and will further simplify the
processing of clinical samples from swab, cerebrospinal fluid and serum. We are also developing a fully automated instrument with increased high throughput and sample preparation for both infectious disease and human genetic tests for larger hospital based laboratories. By basing future generations of our instrument platform on existing design elements, each new generation of development will process assays developed for previous generations. |
| • | Developing Additional Genomic and Protein Assays. We are in various phases of developing and commercializing new assays for detecting protein biomarkers, infectious diseases and human genetic markers. Currently, we are researching additional human genetic, infectious disease and ultra-sensitive protein assays. |
9
Table of Contents
| • | Validating and Commercializing New Biomarkers. We have a dedicated team of protein scientists and assay developers who conduct assay development to support feasibility testing and new protein biomarker validation. This team is collaborating with clinical researchers in academic and private settings to apply our ultra-sensitive protein detection technology to the researchers’ efforts to create diagnostic methods with greater clinical sensitivity and specificity. We are also applying our ultra-sensitivity methods to the development of established protein biomarkers that may lead to earlier detection of medical conditions including cancer, neurodegenerative disorders including Alzheimer’s disease, sepsis and mad cow disease, as well as for blood screening and veterinary applications. |
| • | Enhancing Performance of Established Product Systems and Developing New Applications. We have entered into a license agreement with Northwestern which provides us with an exclusive license to certain patents and patent applications related to the application of nanotechnology to biodiagnostics and to biobarcode technology. This license covers all discoveries from the International Institute for Nanotechnology at Northwestern in the field of biodiagnostics through January 1, 2013. Nanosphere also has the right of first negotiation for an exclusive license on inventions after such date. Our research team utilizes the research and patents developed at Northwestern to develop diagnostic applications including additional genomic and protein testing assays for use in the Verigene System. |
Employees
As of December 31, 2009, we had 105 full-time employees. Of these employees, 46 were in
research and development, 31 were in manufacturing (in support of both product sales and the
research and development function), 15 were in sales and marketing and 13 were in general and
administrative functions. We have never had a work stoppage and none of our employees are covered
by collective bargaining agreements or represented by a labor union. We believe our employee
relations are good.
Government Grants and Contracts
We have received grants over the last five years that have allowed for the evaluation and
development of new technologies and also allowed for development of market specific diagnostic
products.
We have benefited from Small Business Innovation Research grants to prove feasibility of gold
nanoparticle based detection technology as well as evaluate potential new technologies and medical
diagnostic applications.
We have received government contracts for the development of automated biological agent
detection systems using nanoparticle probes that are capable of rapidly detecting biological
warfare agents and biological toxins. These products have potential applications for both
government contractors and civilian first responders. Since inception, we have recorded revenue of
approximately $9 million under these grants and contracts.
Manufacturing
We assemble and package all our finished products at our corporate headquarters in Northbrook,
Illinois. Our manufacturing facility occupies approximately 12,000 square feet of the 40,945 square
feet which we lease at our Northbrook facility. There, we manufacture our proprietary nanoparticle
probes, assay reagents, test cartridges and Verigene System instrumentation. We outsource much of
the disposable component molding. Reagent manufacturing and cartridge filling is performed under
the current Good Manufacturing Practice — Quality System Regulation as required by the FDA for the
manufacture of in vitro diagnostic products. These regulations carefully control the manufacture,
testing and release of diagnostics products as well as raw material receipt and control.
We have controlled methods for the consistent manufacturing of our proprietary nanoparticles
and production oligonucleotides at very high purity (greater than 95%). We also manufacture at our
Northbrook facility a proprietary linker to ensure stable bonding of the oligonucleotide to the
gold nanoparticle.
All quality control tests are validated to ensure product quality measurements are accurate.
Manufacturing of the Verigene System, including test cartridges, is tightly controlled with the use
of manufacturing batch records. These records control which product is produced and ensure that
each batch of product is manufactured consistently and according to the intended design.
10
Table of Contents
We plan to continue to manufacture components that we determine are highly proprietary or
difficult to produce consistently while outsourcing commodity components. As we continue to
execute on our sales and market plans, we have ramped-up our manufacturing operations to meet
demand. We are likely to establish additional outsourcing partnerships as we manufacture additional
products. While we believe our current facilities and expansion rights are adequate to meet our
manufacturing needs for at least the next three years, we may need to lease additional space. Our
recently revised facilities lease includes a right of first offer on additional available space in
our building. While we do not need to expand our facilities to meet anticipated demand for 2010,
we will likely require expanded facilities to meet anticipated demand beyond 2010.
Sales and Marketing
As a part of our business strategy, we have a direct sales and marketing organization to
support the sales of the Verigene System and its initial menu of tests in the United States. This
organization comprises geographically dispersed sales representatives and clinical support
specialists as well as a centralized staff of market and product managers. We believe that the
primary market for our diagnostic applications will be hospital-based laboratories and academic
research institutions in the United States. A customer may purchase the Verigene System, lease it
from a third party or enter into a reagent rental agreement. Our reagent rental agreements include
customer commitments to purchase a certain minimum volume of cartridges over the term of the
agreement. As part of these agreements, a portion of the charge for each cartridge is a rental fee
for use of the equipment.
Our sales and marketing organization provides customer service related to order fulfillment,
technical service and product support, and distribution logistics.
We believe that the primary international customers for our diagnostic applications will be
hospital-based laboratories and academic research institutions. We have obtained CE Mark approval
for sale of the Verigene System in European Union countries and will do so for each assay we plan
to market in Europe. Outside the United States, we anticipate initiating sales through marketing
partners and distributors. A distribution strategy is being developed for each relevant
international market. We expect to supplement marketing partnerships with specialists who will
train our partners’ sales forces and provide technical support.
Competition
We primarily face competition in the nucleic acid based testing market from companies that
provide PCR-based technologies. We believe that, over time, the Verigene System will compete with
these companies primarily on the following factors: (1) cost effectiveness; (2) ease of use;
(3) multiplex capability; (4) range of tests offered; (5) immediacy of results; and
(6) reliability.
We also face competition in the protein detection market from companies that provide mass
spectrometry systems. Although mass spectrometry systems offer high sensitivity, they are extremely
costly, require significant time and effort by sophisticated staff and cannot detect many complex,
disease-causing proteins. Due to these significant limitations we consider mass spectrometry
systems to be a lower competitive threat within commercial protein diagnostics laboratories.
The protein detection market also includes companies that provide ELISA-based testing systems.
We believe that our technology, which is at least 100 times more sensitive than ELISA-based
technologies provides a significant advantage because it can detect proteins at lower
concentrations equating to earlier detection of disease. This sensitivity will create new value for
existing biomarkers and allow the discovery of novel biomarkers for the treatment and monitoring of
disease where none exist today.
Regulation by the United States Food and Drug Administration
In the United States, the FDA regulates the sale and distribution, in interstate commerce, of
medical devices, including in vitro diagnostic test kits. Pursuant to the federal Food, Drug, and
Cosmetic Act, the FDA regulates the preclinical and clinical design, testing, manufacture,
labeling, distribution and promotion of medical devices. We will not be able to commence marketing
or commercial sales in the United States of new medical devices under development that fall within
the FDA’s jurisdiction until we receive clearance from the FDA.
11
Table of Contents
In the United States, medical devices are classified into one of three classes (i.e., Class I,
II or III) on the basis of the controls deemed necessary by the FDA to reasonably ensure their
safety and effectiveness. Class I devices are subject to general controls (e.g., establishment
registration, medical device listing, labeling regulations, possible premarket notification,
possible adherence to current Good Manufacturing Practice). However, most Class I devices are
exempt from premarket notification (510(k) clearance) and adherence to current Good Manufacturing
Practice. Class II devices are subject to general and special controls (e.g., special labeling
requirements, mandatory performance standards, premarket notification (510(k) clearance) often with
guidance from an FDA special control guideline, adherence to current Good Manufacturing Practice,
possible post-market surveillance). Generally, Class III devices are subject to general and special
controls and must receive premarket approval, or PMA, by the FDA to ensure their safety and
effectiveness (e.g., new devices for which insufficient information exists to assure safety and
effectiveness through general and special controls; often such devices are life-sustaining,
life-supporting and implantable). Many devices that have been approved by way of premarket approval
are required to perform post-market surveillance.
510(k) Clearance
The FDA will grant 510(k) clearance if the submitted information establishes that the proposed
device is “substantially equivalent” to a legally marketed Class I or Class II medical device or a
pre-amendment Class III medical device for which the FDA has not sought PMA. The FDA has recently
been requiring more rigorous demonstration of substantial equivalence than in the past. In some
cases, such as may be the case with our HFE and Cardiac Troponin I 510(k) submissions, the FDA may
require additional clinical data than it would have required in the past. The FDA may
determine that a proposed device is not substantially equivalent to a legally marketed device or
that additional information is needed before a substantial equivalence determination can be made. A
“not substantially equivalent” determination, or a request for additional information, could
prevent or delay the market introduction of new products that fall into this category. For any
devices that are cleared through the 510(k) process, modifications or enhancements that could
significantly affect safety or effectiveness, or constitute a major change in the intended use of
the device, require new 510(k) submissions and clearances.
Premarket Approval
A PMA application must be filed if a proposed device is a new device not substantially
equivalent to a legally marketed Class I or Class II device, or if it is a pre-amendment Class III
device for which the FDA has sought PMA. A PMA application must be supported by valid scientific
evidence to demonstrate the safety and effectiveness of the device, typically including the results
of clinical investigations, bench tests, and laboratory and animal studies. The PMA application
must also contain a complete description of the device and its components and a detailed
description of the method, facilities and controls used to manufacture the device. In addition, the
submission must include the proposed labeling, advertising literature and any training materials.
The PMA process can
be expensive, uncertain and lengthy, and a number of devices for which FDA approval has been
sought by other companies have never been approved for marketing.
Upon receipt of a PMA application, the FDA makes a threshold determination as to whether the
application is sufficiently complete to permit a substantive review. If the FDA determines that the
PMA application is complete, the FDA will accept the application for filing. Once the submission is
accepted, the FDA begins an in-depth review of the PMA. The FDA’s review of a PMA application
generally takes one to three years from the date the application is accepted, but may take
significantly longer. The review time is often extended by the FDA asking for more information or
clarification of information already provided in the submission. During the review period, an
advisory committee, typically a panel of clinicians, will likely be convened to review and evaluate
the application and provide recommendations to the FDA as to whether the device should be approved.
The FDA is not bound by the recommendation of the advisory panel. Toward the end of the PMA review
process, the FDA generally will conduct an inspection of the manufacturer’s facilities to ensure
that the facilities are in compliance with applicable current Good Manufacturing Practices
requirements.
If FDA evaluations of both the PMA application and the manufacturing facilities are favorable,
the FDA may issue either an approval letter or an approvable letter, which usually contains a
number of conditions that must be met in order to secure final approval of the PMA. When and if
those conditions have been fulfilled to the satisfaction of the FDA, the agency will issue a
premarket approval letter, authorizing commercial marketing of the device for certain indications.
If the FDA’s evaluation of the PMA application or manufacturing facilities is not favorable, the
FDA will deny approval of the PMA application or issue a non-approvable letter. The FDA may
determine that additional clinical investigations are necessary, in which case the PMA may be
delayed for one or more years while additional clinical investigations are conducted and submitted
in an amendment to the PMA.
Modifications to a device that is the subject of an approved PMA, including its labeling or
manufacturing process, may require approval by the FDA of PMA supplements or new PMAs. Supplements
to an approved PMA often require the submission of the same type of information required for an
initial PMA, except that the supplement is generally limited to that information needed to support
the proposed change from the product covered by the original PMA.
12
Table of Contents
Clinical Investigations
Before we can submit a medical device for 510(k) clearance, we may have to perform a short
(i.e., months) method comparison study at clinical sites to ensure that end-users can perform the
test successfully. This is a study in a clinical environment, but is not usually considered a
clinical trial. Alternatively, when we submit a PMA, we generally must conduct a longer (i.e.,
years) clinical trial of the device which supports the clinical utility of the device and how the
device will be used.
Although clinical investigations of most devices are subject to the investigational device
exemption, or IDE requirements, clinical investigations of in vitro diagnostic tests, including our
products and products under development, are exempt from the IDE requirements. Thus, our tests do
not require the FDA’s prior approval, provided the testing is non-invasive, does not require an
invasive sampling procedure that presents a significant risk, does not intentionally introduce
energy into the subject, and is not used as a diagnostic procedure without confirmation by another
medically established test or procedure. In addition, our tests must be labeled “for research use
only” or “for investigational use only,” and distribution controls must be established to assure
that our tests distributed for research, method comparisons or clinical trials are used only for
those purposes.
Obtaining FDA Clearance for Our Products
We received 510(k) clearance from the FDA for commercial sale of the Verigene System in the
second half of 2007. We have also received 510(k) clearance to use the Verigene System for four
diagnostic tests. The first test is a warfarin metabolism assay, which is a pharmacogenomic test
to determine the existence of certain genetic information believed to affect the metabolism of
warfarin based drugs, including Coumadin, the most-prescribed oral anticoagulant in North America
and Europe. The second test is a hyper-coagulation assay, one of the highest volume human genetic
tests currently performed, to determine an individual’s risk, based upon genetic information, for
the development of blood clots, which can lead to stroke, pulmonary embolism and deep vein
thrombosis.
The third test is our respiratory panel which detects the presence of influenza A and B as
well as respiratory syncitial virus (“RSV”) A and B. Influenza is commonly known as the seasonal
flu and RSV is a respiratory virus that infects the lungs and breathing passages. RSV is the most
common cause of bronchitis and pneumonia in children under the age of one year and has become a
significant concern for older adults. Our respiratory panel provides physicians with a highly
accurate, fast determination of which virus is present which helps guide the most appropriate
treatment therapy. Most of the respiratory tests currently on the market take
days to generate a result, because they depend on culturing, or do not provide a reliable
result, because they are rapid tests which lack specificity. The fourth test is our cystic
fibrosis test that enables molecular laboratories to perform prenatal screening and diagnostic
confirmations through identification of the number of copies of each of the 23 most common gene
mutations recognized by the American College of Obstetricians and Gynecologists as markers for
cystic fibrosis.
Most of our tests have special control guidances for 510(k) clearance. Some of our future
tests may be Class III devices. We also plan to conduct method comparison studies or clinical
trials of our products currently under development, which we intend to distribute in the United
States. Our future developments may not be exempt from IDE requirements and may require us to
obtain approval from the FDA through the PMA process rather than 510(k) clearance. In addition, any
failure to maintain compliance with the IDE exemption requirements could result in, among other
things, the loss of the IDE exemption or the imposition of other restrictions on the distribution
of our products.
Regulation After FDA Approval or Clearance
Any devices we manufacture or distribute pursuant to clearance or approval by the FDA are
subject to pervasive and continuing regulation by the FDA and certain state agencies. We are
required to adhere to applicable regulations setting forth detailed current Good Manufacturing
Practices requirements, which include testing, control and documentation requirements.
Non-compliance with these standards can result in, among other things, fines, injunctions, civil
penalties, recalls or seizures of products, total or partial suspension of production, failure of
the government to grant 510(k) clearance PMA for devices, withdrawal of marketing approvals and
criminal prosecutions. We have designed and implemented our manufacturing facilities under the
current Good Manufacturing Practices requirements. Our manufacturing facility has been inspected by
the FDA and will continue to be periodically audited by the FDA.
Because we are a manufacturer of medical devices, we must also comply with medical device
reporting requirements by reporting to the FDA any incident in which our product may have caused or
contributed to a death or serious injury. We must also report any incident in which our product
malfunctioned if that malfunction would likely cause or contribute to a death or serious injury if
it were to recur. Labeling and promotional activities are subject to scrutiny by the FDA and, in
certain circumstances, by the Federal Trade Commission. Current FDA enforcement policy prohibits
the marketing of approved medical devices for unapproved uses.
13
Table of Contents
We are also subject to numerous federal, state and local laws relating to such matters as safe
working conditions, manufacturing practices, environmental protection, fire hazard control and
disposal of hazardous or potentially hazardous substances. We have numerous policies and procedures
in place to ensure compliance with these laws and to minimize the risk of occupational exposure to
hazardous materials. In addition, we do not expect the operations of our products to produce
significant quantities of hazardous or toxic waste that would require extraordinary disposal
practices. Although the costs to comply with these applicable laws and regulations have not been
material, we cannot predict the impact on our business of new or amended laws or regulations, or
any changes in the way existing and future laws and regulations are interpreted or enforced.
Moreover, as we develop toxin and pathogen detection products for the food and agriculture markets,
we may be subject to the regulations of various food safety organizations, including the United
States Department of Agriculture.
Export of Our Products
Export of products subject to the 510(k) notification requirements, but not yet cleared to
market, are permitted with FDA authorization provided certain requirements are met. Unapproved
products subject to the PMA requirements must be approved by the FDA for export. To obtain FDA
export approval, we must meet certain requirements, including, with some exceptions, documentation
demonstrating that the product is approved for import into the country to which it is to be
exported and, in some instances, safety data for the devices.
Clinical Laboratory Improvement Amendments of 1988
The use of our products is also affected by the Clinical Laboratory Improvement Amendments of
1988, or CLIA, and related federal and state regulations, which provide for regulation of
laboratory testing. These regulations mandate that clinical laboratories must be certified by the
federal government, by a federally-approved accreditation agency or by a state that has been deemed
exempt from the regulation’s requirements. Moreover, these laboratories must meet quality
assurance, quality control and personnel standards, and they must undergo proficiency testing and
inspections. The CLIA standards applicable to clinical laboratories are based on the complexity of
the method of testing performed by the laboratory, which range from “waived” to “moderately
complex” to “highly complex.” We expect that most of our products will be categorized as either
“moderately complex” or “highly complex.”
Foreign Government Regulation
We anticipate that our products will be introduced in foreign markets in the future. We have
obtained CE Mark approval for sale of the Verigene System in European Union countries and will do
so for any assay we plan to launch in Europe. The regulatory review process varies from country to
country, and many countries also impose product standards, packaging requirements, labeling
requirements and import restrictions on devices. Each country has its own tariff regulations,
duties and tax requirements.
Other
Information
Copies of the
Company’s Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on
Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d)
of the Securities Exchange Act of 1934 are available free of charge through the investor relations
section of the Company’s website (www.nanosphere.us) as soon as reasonably practicable after the
Company electronically files the material with, or furnishes it to, the Securities and Exchange
Commission.
14
Table of Contents
| Item 1A. | Risk Factors. |
Our results from operations may be affected by the risk factors set forth below. All
investors should consider the following risk factors before deciding to purchase securities of the
Company. If any of the following events actually occur, our business, operating results, prospects
or financial condition could be materially and adversely affected. This could cause the trading
price of our common stock to decline and you may lose all or part of your investment. The risks
described below are not the only ones that we face. Additional risks not presently known to us or
that we currently deem immaterial may also significantly impair our business operations and could
result in a complete loss of your investment.
Risks Related to Our Business
We have a history of losses and we may never achieve or maintain profitability.
We have a limited operating history and have incurred significant losses in each fiscal year
since our inception, including net losses attributable to common stock of $33.9 million,
$37.0 million and $59.3 million in the years ended December 31, 2009, 2008 and 2007, respectively.
As of December 31, 2009, we had an accumulated deficit of approximately $239.3 million. Our losses
resulted principally from costs incurred in our research and development programs and from our
general and administrative expenses. In recent years, we have incurred significant costs in
connection with the development of the Verigene System and its test menu. We expect our research
and development expense levels to remain high for the foreseeable future as we seek to enhance our
existing product and develop new products. These losses, among other things, have had and will
continue to have an adverse effect on our working capital, total assets and stockholders’ equity.
Because of the numerous risks and uncertainties associated with our product development and
commercialization efforts, we are unable to predict when we will become profitable, and we may
never become profitable. If we fail to achieve profitability in the future, the market price of our
common stock could decline.
Our financial results depend on commercial acceptance of the Verigene System, its array of tests,
and the development of additional tests.
Our future depends on the success of the Verigene System, which depends primarily on its
acceptance by hospitals, research institutions, and independent diagnostic laboratories as a
reliable, accurate and cost-effective replacement for traditional molecular diagnostic measurement
methods. Many hospitals and laboratories already use expensive molecular diagnostic testing
instruments in their laboratories and may be reluctant to change their current procedures for
performing such analyses.
The Verigene System currently does not process a sufficiently broad menu of tests for some
hospitals and laboratories to consider adopting it. Although we continue to develop additional
tests to respond to hospitals’ and laboratories’ needs, we cannot guarantee that we will be able to
develop enough additional tests quickly enough or in a manner that is cost-effective or at all. The
development of new or enhanced products is a complex and uncertain process requiring the accurate
anticipation of technological and market trends, as well as precise technological execution. We are
currently not able to estimate when or if we will be able to develop, commercialize or sell
additional tests or enhance existing products. If we are unable to increase sales of the Verigene
System and its tests or to successfully develop and commercialize other products or tests, our
revenues and our ability to achieve profitability would be impaired.
The regulatory approval process is expensive, time consuming and uncertain and the failure to
obtain such approvals will prevent us from commercializing our future products.
Our products are subject to approval or clearance by the FDA or foreign governmental entities
prior to their marketing for commercial use. The 510(k) clearance and premarket approval processes
as well as the foreign approvals required to initiate sales outside the United States can be
expensive, time consuming and uncertain. It may take as long as eighteen months or longer from
submission to obtain 510(k) clearance, and from one to three years from submission to obtain
premarket approval; however, it may take longer, and 510(k) clearance or premarket approval may
never be obtained. Delays in receipt of, or failure to obtain, clearances or approvals for future
products, including tests that are currently in development, would result in delayed, or no,
realization of revenues from such products and in substantial additional costs which could decrease
our profitability. We have limited experience in filing FDA applications for 510(k) clearance and
premarket approval. There are no assurances that we will obtain any required clearance or approval.
Any such failure, or any material delay in obtaining the clearance or approval, could harm our
business, financial condition and results of operations.
15
Table of Contents
We and our customers are subject to various governmental regulations, and we may incur significant
expenses to comply with, and experience delays in our product commercialization as a result of,
these regulations.
The products we develop, manufacture and market are subject to regulation by the FDA and
numerous other federal, state and foreign governmental authorities. We generally are prohibited
from marketing our products in the United States unless we obtain either 510(k) clearance or
premarket approval from the FDA.
In addition, we are required to continue to comply with applicable FDA and other regulatory
requirements once we have obtained clearance or approval for a product. These requirements include
the Quality System Regulation, labeling requirements, the FDA’s general prohibition against
promoting products for unapproved or “off-label” uses and adverse event reporting regulations.
Failure to comply with applicable FDA product regulatory requirements could result in warning
letters, fines, injunctions, civil penalties, repairs, replacements, refunds, recalls or seizures
of products, total or partial suspension of production, the FDA’s refusal to grant future
pre-market clearances or approvals, withdrawals or suspensions of current product applications and
criminal prosecution. Any of these actions, in combination or alone, could prevent us from selling
our products and would likely harm our business.
Our manufacturing facilities are subject to periodic regulatory inspections by the FDA and
other federal and state regulatory agencies. The use of our diagnostic products by our customers is
also affected by the Clinical Laboratory Improvement Amendments of 1988, or CLIA, and related
federal and state regulations that provide for regulation of laboratory testing. CLIA is intended
to ensure the quality and reliability of clinical laboratories in the United States by mandating
specific standards in the areas of personnel qualifications, administration, participation in
proficiency testing, patient test management, quality and inspections. Current or future CLIA
requirements or the promulgation of additional regulations affecting laboratory testing may prevent
some laboratories from using some or all of our diagnostic products.
The FDA and foreign governmental regulators have made, and may continue to make, changes in
approval requirements and processes. We cannot predict what these changes will be, how or when they
will occur or what effect they will have on the regulation of our products. Any new regulations,
including regulations specifically related to nanotechnology, may impose additional costs or
lengthen review times of our products. Delays in receipt of or failure to receive regulatory
approvals or clearances for our new products would have a material adverse effect on our business,
financial condition and results of operations.
If third-party payors do not reimburse our customers for the use of our clinical diagnostic
products or if they reduce reimbursement levels, our ability to sell our products will be harmed.
We intend to sell our products primarily to hospital-based laboratories and academic research
institutions, substantially all of which receive reimbursement for the health care services they
provide to their patients from third-party payors, such as Medicare, Medicaid and other domestic
and international government programs, private insurance plans and managed care programs. Most of
these third-party payors may deny reimbursement if they determine that a medical product was not
used in accordance with cost-effective treatment methods, as determined by the third-party payor,
or was used for an unapproved indication. Third-party payors also may refuse to reimburse for
procedures and devices deemed to be experimental.
In the United States, the American Medical Association assigns specific Current Procedural
Terminology, or CPT, codes, which are necessary for reimbursement of diagnostic tests. Once the CPT
code is established, the Centers for Medicare and Medicaid Services establish reimbursement payment
levels and coverage rules under Medicaid and Medicare, and private payors establish rates and
coverage rules independently. Although the tests performed by our assays in development have
previously assigned CPT Codes, we cannot guarantee that our assays are covered by such CPT codes
and are therefore approved for reimbursement by Medicare and Medicaid as well as most third-party
payors. Additionally, certain of our future products may not be approved for reimbursement.
Third-party payors may choose to reimburse our customers on a per test basis, rather than on the
basis of the number of results given by the test. This may result in reference laboratories, public
health institutions and hospitals electing to use separate tests to screen for each disease so that
they can receive reimbursement for each test they conduct. In that event, these entities likely
would purchase separate tests for each disease, rather than products that multiplex.
Third-party payors are increasingly attempting to contain health care costs by limiting both
coverage and the level of reimbursement for medical products and services. Increasingly, Medicare,
Medicaid and other third-party payors are challenging the prices charged for medical services,
including clinical diagnostic tests. Levels of reimbursement may decrease in the future, and future
legislation, regulation or reimbursement policies of third-party payors may adversely affect the
demand for and price levels of our products. If our customers are not reimbursed for our products,
they may reduce or discontinue purchases of our products, which would cause our revenues to
decline.
16
Table of Contents
We may fail to receive positive clinical results from the diagnostic tests currently in development
that require clinical trials, and even if we receive positive clinical results, we may still fail
to receive the necessary clearances or approvals to market our products.
We are investing in the research and development of new products to expand the menu of testing
options for the Verigene System. In order to commercialize our products, we are required to
undertake time consuming and costly development activities, sometimes including clinical trials for
which the outcome is uncertain. Products that appear promising during early development and
preclinical studies may, nonetheless, fail to demonstrate the results needed to support regulatory
approval. Even if we receive positive clinical results, we may still fail to obtain the necessary
FDA clearance and approvals.
Our operating results may be variable and unpredictable.
The sales cycles for our products may be lengthy, which will make it difficult for us to
accurately forecast revenues in a given period, and may cause revenues and operating results to
vary significantly from period to period. In addition to its length, the sales cycle associated
with our products is subject to a number of significant risks, including the budgetary constraints
of our customers, their inventory management practices and possibly internal acceptance reviews,
all of which are beyond our control. Sales of our products will also involve the purchasing
decisions of large, medium and small hospitals and laboratories which can require many levels of
pre-approvals, further lengthening sales time. As a result, we may expend considerable resources on
unsuccessful sales efforts or we may not be able to complete transactions on the scheduled
anticipated.
If we do not achieve significant product revenue, we may not be able to meet our cash requirements
without obtaining additional capital from external sources, and if we are unable to do so, we may
have to curtail or cease operations.
We expect capital outlays and operating expenditures to increase over the next few years as we
expand our infrastructure, commercialization, manufacturing, and research and development
activities. We anticipate that our current cash and cash equivalents, which include the net
proceeds of our initial and secondary public offerings, will be sufficient to meet our estimated
needs for approximately two years. However, we operate in a market that makes our prospects
difficult to evaluate, and we may need additional financing to execute on our current or future
business strategies. The amount of additional capital we may need to raise depends on many factors,
including:
| • | the level of research and development investment required to maintain and improve our technology; | ||
| • | the amount and growth rate, if any, of our revenues; | ||
| • | changes in product development plans needed to address any difficulties in manufacturing or commercializing the Verigene System and enhancements to our system; | ||
| • | the costs of filing, prosecuting, defending and enforcing patent claims and other intellectual property rights; | ||
| • | competing technological and market developments; | ||
| • | our need or decision to acquire or license complementary technologies or acquire complementary businesses; | ||
| • | the expansion of our sales force; and | ||
| • | changes in regulatory policies, practices or laws that affect our operations, including clearance to market our products. |
We cannot be certain that additional capital will be available when and as needed or that our
actual cash requirements will not be greater than anticipated. If we require additional capital at
a time when investment in diagnostics companies or in the marketplace in general is limited due to
the then prevailing market or other conditions, we may not be able to raise such funds at the time
that we desire or any time thereafter. In addition, if we raise additional funds through the
issuance of common stock or convertible securities, the percentage ownership of our stockholders
could be significantly diluted, and these newly issued securities may have rights,
preferences or privileges senior to those of existing stockholders. If we obtain additional
debt financing, a substantial portion of our operating cash flow may be dedicated to the payment of
principal and interest on such indebtedness, and the terms of the debt securities issued could
impose significant restrictions on our operations. If we raise additional funds through
collaborations and licensing arrangements, we might be required to relinquish significant rights to
our technologies or products, or grant licenses on terms that are not favorable to us.
17
Table of Contents
The adverse capital and credit market conditions could affect our liquidity.
Adverse capital and credit market conditions could affect our ability to meet liquidity needs,
as well as our access to capital and cost of capital. The capital and credit markets have been
experiencing extreme volatility and disruption for more than 12 months. In recent months, the
volatility and disruption have reached unprecedented levels and the markets have exerted downward
pressure on availability of liquidity and credit capacity for certain issuers. For example,
recently credit spreads have widened considerably. Our results of operations, financial condition,
cash flows and capital position could be materially adversely affected by continued disruptions in
the capital and credit markets.
If our products do not perform as expected or the reliability of the technology on which our
products are based is questioned, we could experience lost revenue, delayed or reduced market
acceptance of our products, increased costs and damage to our reputation.
Our success depends on the market’s confidence that we can provide reliable, high-quality
diagnostics systems. We believe that customers in our target markets are likely to be particularly
sensitive to product defects and errors.
Our reputation and the public image of our products or technologies may be impaired if our
products fail to perform as expected or our products are perceived as difficult to use. Our
products are complex and may develop or contain undetected defects or errors. Any defects or errors
could lead to the filing of product liability claims, which could be costly and time-consuming to
defend and result in substantial damages. If we experience a sustained material defect or error,
this could result in loss or delay of revenues, delayed market acceptance, damaged reputation,
diversion of development resources, legal claims, increased insurance costs or increased service
and warranty costs, any of which could materially harm our business. We cannot assure you that our
product liability insurance would protect our assets from the financial impact of defending a
product liability claim. A product liability claim could have a serious adverse effect on our
business, financial condition and results of operations.
We rely on third-party license agreements for patents and other technology related to our products,
and the termination of these agreements could delay or prevent us from being able to commercialize
our products.
As of December 31, 2009, our patent portfolio is comprised, on a worldwide basis, of 132
issued patents and 97 pending patent applications which we own directly or for which we are the
exclusive licensee. Some of these patents and patent applications derive from a common parent
patent application or are foreign counterpart patent applications and relate to similar or
identical technological claims. The issued patents cover approximately 12 different technological
claims and the pending patent applications cover approximately 4 additional technological claims.
Many of our issued and pending patents were exclusively licensed from the International
Institute for Nanotechnology at Northwestern University (“Northwestern”) in May 2000 and they
generally cover our core technology, including nanotechnology based biodiagnostics and biobarcode
technology. Our issued patents expire between 2017 and 2025. Our patent portfolio provides
protection against other companies offering products employing the same technologies and methods as
we have patented. While we believe our patent portfolio establishes a proprietary position, there
are many competitive products utilizing other technologies that do not infringe on our patents.
In addition, we have non-exclusive licenses for 44 patents that cover 12 different
technological claims from various third parties. Most of these license agreements require us to pay
the licensor royalty fees that typically expire upon the patent expiration dates which range from
2009 to 2025. These license agreements are nonexclusive and do not create a proprietary position.
The expiration of these non-exclusive licenses will result in the termination of certain royalty
payments by us to the licensors.
If we are unable to obtain, maintain and enforce intellectual property protection covering our
products, others may be able to make, use, or sell our products, which could adversely affect our
ability to compete in the market.
Our success is dependent in part on obtaining, maintaining and enforcing intellectual property
rights, including patents. If we are unable to obtain, maintain and enforce intellectual property
legal protection covering our products, others may be able to make, use or sell products that are
substantially identical to ours without incurring the sizeable discovery, development and licensing
costs that we have incurred, which would adversely affect our ability to compete in the market.
18
Table of Contents
We seek to obtain and maintain patents and other intellectual property rights to restrict the
ability of others to market products that compete with our products. Currently, our patent
portfolio is comprised, on a worldwide basis, of 132 issued patents and 97 pending patent
applications which, in either case, we own directly or for which we are the exclusive licensee.
However, patents may not be issued from any pending or future patent applications owned by or
licensed to us, and moreover, issued patents owned or licensed to us now or in the future may be
found by a court to be invalid or otherwise unenforceable. Also, even if our patents are determined
by a court to be valid and enforceable, they may not be sufficiently broad to prevent others from
marketing products similar to ours or designing around our patents, despite our patent rights, nor
provide us with freedom to operate unimpeded by the patent rights of others.
Furthermore, we cannot be certain that we were the first to make the invention claimed in our
United States issued patents or pending patent applications, or that we were the first to file for
protection of the inventions claimed in our foreign issued patents or pending patent applications.
We may become subject to interference proceedings conducted in the patent and trademark offices of
various countries to determine our entitlement to patents, and these proceedings may conclude that
other patents or patent applications have priority over our patents or patent applications. It is
also possible that a competitor may successfully challenge our patents through various proceedings
and those challenges may result in the elimination or narrowing of our patents, and therefore
reduce our patent protection. Accordingly, rights under any of our issued patents, patent
applications or future patents may not provide us with commercially meaningful protection for our
products or afford us a commercial advantage against our competitors or their competitive products
or processes.
We have a number of foreign patents and applications. However, the laws of some foreign
jurisdictions do not protect intellectual property rights to the same extent as laws in the United
States, and many companies have encountered significant difficulties in protecting and defending
such rights in foreign jurisdictions. If we encounter such difficulties or we are otherwise
precluded from effectively protecting our intellectual property rights in foreign jurisdictions,
our business prospects could be substantially harmed.
We may initiate litigation to enforce our patent rights, which may prompt our adversaries in
such litigation to challenge the validity, scope or enforceability of our patents. Patent
litigation is complex and often difficult and expensive, and would consume the time of our
management and other significant resources. In addition, the outcome of patent litigation is
uncertain. If a court decides that our patents are not valid, not enforceable or of a limited
scope, we may not have the right to stop others from using the subject matter covered by those
patents.
We also rely on trade secret protection to protect our interests in proprietary know-how and
for processes for which patents are difficult to obtain or enforce. We may not be able to protect
our trade secrets adequately. In addition, we rely on non-disclosure and confidentiality agreements
with our employees, consultants and other parties to protect, in part, our trade secrets and other
proprietary technology. These agreements may be breached and we may not have adequate remedies for
any breach. Moreover, others may independently develop equivalent proprietary information, and
third parties may otherwise gain access to our trade secrets and proprietary knowledge. Any
disclosure of confidential data into the public domain or to third parties could allow our
competitors to learn our trade secrets and use the information in competition against us.
Our products could infringe patent rights of others, which may require costly litigation and, if we
are not successful, could cause us to pay substantial damages or limit our ability to commercialize
our products.
Our commercial success depends on our ability to operate without infringing the patents and
other proprietary rights of third parties. We are aware of third party patents that may relate to
our products and technology. There may also be other patents that relate to our products and
technology of which we are not aware. We may unintentionally infringe upon valid patent rights of
third parties. Although we are currently not involved in any material litigation involving patents,
a third party patent holder could assert a claim of patent infringement against us in the future.
Alternatively, we may initiate litigation against the third party patent holder to request that a
court declare that we are not infringing the third party’s patent and/or that the third party’s
patent is invalid or unenforceable. If a claim of infringement is asserted against us and is
successful, and therefore we are found to infringe, we could be required to pay damages for
infringement, including treble damages if it is determined that we knew or became aware of such a
patent and we failed to exercise due care in determining whether or not we infringed the patent. If
we have supplied infringing products to third parties or have licensed third parties to
manufacture, use or market infringing products, we may be obligated to indemnify these third
parties for damages they may be required to pay to the patent holder and for any losses they may
sustain. We can also be prevented from selling
or commercializing any of our products that use the infringing technology in the future,
unless we obtain a license from such third party. A license may not be available from such third
party on commercially reasonable terms, or may not be available at all. Any modification to include
a non-infringing technology may not be possible or if possible may be difficult or time-consuming
to develop, and require revalidation, which could delay our ability to commercialize our products.
19
Table of Contents
Any infringement action asserted against us, even if we are ultimately successful in defending
against such action, would likely delay the regulatory approval process of our products, harm our
competitive position, be expensive and require the time and attention of our key management and
technical personnel.
We have limited experience in sales and marketing and may be unable to successfully commercialize
our Verigene System, or it may be difficult to build brand loyalty.
We have limited marketing, sales and distribution experience and capabilities. Our ability to
achieve profitability depends on attracting customers for the Verigene System and building brand
loyalty. To successfully perform sales, marketing, distribution and customer support functions
ourselves, we will face a number of risks, including:
| • | our ability to attract and retain the skilled support team, marketing staff and sales force necessary to commercialize and gain market acceptance for our technology and our products; | ||
| • | the ability of our sales and marketing team to identify and penetrate the potential customer base including hospitals, research institutions, and independent diagnostic laboratories; | ||
| • | the time and cost of establishing a support team, marketing staff and sales force; and | ||
| • | the difficulty of establishing brand recognition and loyalty for our products. |
In addition, we may seek to enlist one or more third parties to assist with sales,
distribution and customer support globally or in certain regions of the world. If we do seek to
enter into such arrangements, we may not be successful in attracting desirable sales and
distribution partners, or we may not be able to enter into such arrangements on favorable terms. If
our sales and marketing efforts, or those of any third-party sales and distribution partners, are
not successful, our technologies and products may not gain market acceptance, which would
materially impact our business operations.
We may be unsuccessful in our long-term goal of expanding our product offerings outside the United
States.
To the extent we begin to offer our products broadly outside the United States, we expect that
we will be dependent on third-party distribution relationships. Distributors may not commit the
necessary resources to market and sell our products to the level of our expectations. If
distributors do not perform adequately, or we are unable to locate distributors in particular
geographic areas, our ability to realize long-term international revenue growth would be materially
adversely affected.
Additionally, our products may require regulatory clearances and approvals from jurisdictions
outside the United States. These products may not be sold in these jurisdictions until the required
clearances and approvals are obtained. We cannot assure you that we will be able to obtain these
clearances or approvals on a timely basis, or at all.
Manufacturing risks and inefficiencies may adversely affect our ability to produce products.
We must manufacture or engage third parties to manufacture components of our products in
sufficient quantities and on a timely basis, while maintaining product quality and acceptable
manufacturing costs and complying with regulatory requirements. In determining the required
quantities of our products and the manufacturing schedule, we must make significant judgments and
estimates based on historical experience, inventory levels, current market trends and other related
factors. Because of the inherent nature of estimates, there could be significant differences
between our estimates and the actual amounts of products we require. Additionally, some of the
components of the Verigene System are custom-made by only a few outside vendors, and we do not have
long-term supply contracts for the materials or components supplied by any of our vendors. If we
are unable to obtain from one or more of these vendors the needed materials or components that meet
our specifications on commercially reasonable terms, or at all, we may not be able to meet the
demand for our products. We have not arranged for alternate suppliers, and it may be difficult to
find alternate suppliers in a timely manner and on terms acceptable to us.
We manufacture in one facility. If there were to be a significant disruption in our ability
to use this facility, it would take significant time to setup and validate an alternative
manufacturing facility. Disruptions due to lack of power, flooding, fire and environmental
controls could adversely impact our ability to manufacture. In addition, we have been steadily
increasing manufacturing capacity to meet demand for our products. A disruption of our
manufacturing operations resulting from scale-up related challenges such as obtaining sufficient
raw materials, hiring of qualified factory personnel, installation and efficient operation of new
equipment, and management of our quality controls could cause us to cease, delay, or limit our
manufacturing operations and consequently adversely impact our business, our results of operations
and our financial condition.
20
Table of Contents
We may experience unforeseen technical complications in the processes we use to develop,
manufacture, customize or receive orders for our products. These complications could materially
delay or limit the use of products we attempt to commercialize, substantially increase the
anticipated cost of our products or prevent us from implementing our processes at appropriate
quality and scale levels, thereby causing our business to suffer. In addition, our manufacturing
operations use highly technical processes involving unique, proprietary techniques that our
manufacturing personnel must continuously monitor and update, especially as we develop more
products. In order to be profitable, we must manufacture greater quantities of products than we
have to date and we must do this more efficiently than we have in the past. We may not be able to
do so.
We will need to develop manufacturing capacity by ourselves or with third parties.
We will need to either continue to build internal manufacturing capacity or contract with one
or more manufacturing partners, or both. We currently use a combination of outsourced and internal
manufacturing activities. We may encounter difficulties in manufacturing our products and, due to
the complexity of our technology and our manufacturing process, we cannot be sure we fully
understand all of the factors that affect our manufacturing processes or product performance. We
may not be able to build manufacturing capacity internally or find one or more suitable
manufacturing partners, or both, to meet the volume and quality requirements necessary to be
successful in the market. If our products do not consistently meet our customers’ performance
expectations, we may be unable to generate sufficient revenues to become profitable. Significant
additional resources, implementation of additional manufacturing equipment and changes in our
manufacturing processes and organization may be required for the scale-up of each new product prior
to commercialization or to meet increasing customer demand once commercialization begins, and this
work may not be successfully or efficiently completed. Any delay in establishing or inability to
expand our manufacturing capacity could delay our ability to develop or sell our products, which
would result in lost revenue and seriously harm our business, financial condition and results of
operations.
Our business and future operating results may be adversely affected by events outside of our
control.
We develop and manufacture the Verigene System and assays in our facility located in
Northbrook, Illinois. This facility and the manufacturing equipment we use would be costly to
replace and could require substantial lead time to repair or replace. Our business and operating
results may be harmed due to interruption of our manufacturing by events outside of our control,
including earthquakes, tornadoes and fires. Other possible disruptions may include power loss and
telecommunications failures. In the event of a disruption, we may lose customers and we may be
unable to regain those customers thereafter. Our insurance may not be sufficient to cover all of
our potential losses and may not continue to be available to us on acceptable terms, or at all.
We face intense competition from established and new companies in the molecular diagnostics field.
We compete with companies that design, manufacture and market already existing and new
molecular diagnostics systems, and single target or low count multiplexing systems and assays are
abundant. We anticipate that we will face increased competition in the future as new companies
enter the market with new technologies and our competitors improve their current products. One or
more of our competitors may offer technology superior to ours and render our technology obsolete or
uneconomical. If a competitor were able to deliver a testing application that offers simplicity
and ease of use, high count multiplexing and high throughput and fast turnaround, our ability to
successfully market our products would be materially adversely affected. Most of our current
competitors, as well as many of our potential competitors, have greater name recognition, more
substantial intellectual property portfolios, longer operating histories, significantly greater
resources to invest in new technologies and more substantial experience in new product development,
regulatory expertise, manufacturing capabilities and the distribution channels to deliver products
to customers. If we are not able to compete successfully, we may not generate sufficient revenue to
become profitable.
Our success may depend upon how we and our competitors anticipate and adapt to market conditions.
The markets for our products are characterized by rapidly changing technology, evolving
industry standards, changes in customer needs, emerging competition and new product introductions.
The success of our products will depend on our ability to continue to increase their performance
and decrease their price. New technologies, techniques or products could emerge with similar or
better price-performance than our system and could exert pricing pressures on our products. It is
critical to our success for us to anticipate changes in technology and customer requirements and to
successfully introduce enhanced and competitive technology to meet our customers’ and prospective
customers’ needs on a timely basis. We may not be able to maintain our technological advantages
over emerging technologies in the future and we will need to respond to technological innovation in
a rapidly changing industry. If we fail to keep pace with emerging technologies our system will
become uncompetitive, our market share will decline and our business, revenue, financial condition
and operating results could suffer materially.
21
Table of Contents
We may not be able to manage our anticipated growth, and we may experience constraints or
inefficiencies caused by unanticipated acceleration and deceleration of customer demand.
Demand for our respiratory products is directly proportionate to the size and duration of
influenza and other respiratory illnesses. Unanticipated acceleration and deceleration of customer
demand for our products may result in constraints or inefficiencies related to our manufacturing,
sales force, implementation resources and administrative infrastructure. Such constraints or
inefficiencies may adversely affect us as a result of delays, lost potential product sales or loss
of current or potential customers due to their dissatisfaction. Similarly, over-expansion or
investments in anticipation of growth that does not materialize, or develops more slowly than we
expect, could harm our financial results and result in overcapacity.
To manage our anticipated future growth effectively, we must enhance our manufacturing
capabilities and operations, information technology infrastructure, and financial and accounting
systems and controls. Organizational growth and scale-up of operations could strain our existing
managerial, operational, financial and other resources. Our growth could require significant
capital expenditures and may divert financial resources from other projects, such as the
development of new products or enhancements of existing products. If our management is unable to
effectively manage our growth, our expenses may increase more than expected, our revenue could grow
more slowly than expected and we may not be able to achieve our research and development and
commercialization goals. Our failure to manage our anticipated growth effectively could have a
material adverse effect on our business, operating results or financial condition.
We use hazardous chemicals, biological materials, and infectious diseases in our business. Any
claims relating to improper handling, storage or disposal of these materials could be time
consuming and costly.
Our research and development and manufacturing processes involve the controlled use of
hazardous materials, including chemicals, biological materials and infectious diseases. Our
operations produce hazardous waste products. We cannot eliminate the risk of accidental
contamination or discharge and any resultant injury from these materials. We may be sued for any
injury or contamination that results from our use or the use by third parties of these materials,
and our liability may exceed our insurance coverage and our total assets. Federal, state and local
laws and regulations govern the use, manufacture, storage, handling and disposal of these hazardous
materials and specified waste products, as well as the discharge of pollutants into the environment
and human health and safety matters. Compliance with environmental laws and regulations may be
expensive, and may impair our research, development and production efforts. If we fail to comply
with these requirements, we could incur substantial costs, including civil or criminal fines and
penalties, clean-up costs, or capital expenditures for control equipment or operational changes
necessary to achieve and maintain compliance. In addition, we cannot predict the impact on our
business of new or amended environmental laws or regulations, or any changes in the way existing
and future laws and regulations are interpreted and enforced.
If we are unable to recruit and retain key executives and scientists, we may be unable to achieve
our goals.
Our performance is substantially dependent on the performance of our senior management and key
scientific and technical personnel. The loss of the services of any member of our senior management
or our scientific or technical staff could divert management’s attention to transition matters and
identification of suitable replacements, if any, and have a material adverse effect on our
business, operating results and financial condition. Each of our executive officers and other key
employees could terminate his or her relationship with us at any time. We do not maintain key man
life insurance on any of our employees.
In addition, our product development and marketing efforts could be delayed or curtailed if we
are unable to attract, train and retain highly skilled employees and scientific advisors,
particularly our management team, senior scientists and engineers and sales and marketing
personnel. To expand our research, product development and sales efforts we need additional people
skilled in areas such as protein science, information services, manufacturing, sales, marketing and
technical support. Because of the complex and technical
nature of our system and the dynamic market in which we compete, any failure to attract and
retain a sufficient number of qualified employees could materially harm our ability to develop and
commercialize our technology. We may not be successful in hiring or retaining qualified personnel
and our failure to do so could have a material adverse effect on our business, financial condition
and results of operations.
22
Table of Contents
Healthcare reform and restrictions on reimbursement may adversely affect our profitability.
In the United States, healthcare providers that purchase our products and other diagnostic
products generally rely on third-party payors to reimburse all or part of the cost of the
procedure. In international markets, reimbursement and healthcare payment systems vary
significantly by country, and include both government-sponsored healthcare and private insurance.
Third-party payors can affect the pricing or the relative attractiveness of our products by
regulating the maximum amount of reimbursement provided by such payors for laboratory testing
services. Lower-than-expected or decreases in reimbursement amounts for tests performed using our
products may decrease amounts physicians and other practitioners are able to charge patients, which
in turn may adversely affect the willingness of physicians and other practitioners to purchase our
products at prices we target, or at all. If we were not able to sell our products at target prices,
then we will suffer a decrease in expected profitability that would likely adversely affect our
business, financial condition and results of operations.
Risks Related to Our Common Stock
The market price of our common stock may be volatile and fluctuate significantly, which could
result in substantial losses for investors and subject us to securities class action litigation.
Market prices of diagnostics companies have been volatile. Among the factors that may cause
the market price of our common stock to fluctuate are the risks described in this “Risk Factors”
section and other factors, including:
| • | fluctuations in our quarterly operating results or the operating results of our competitors; | |
| • | changes in estimates of our financial results or recommendations by securities analysts; | |
| • | variance in our financial performance from the expectations of securities analysts; | |
| • | changes in the estimation of the future size and growth rate of our markets; | |
| • | changes in accounting principles or changes in interpretations of existing principles, which could affect our financial results; | |
| • | failure of our products to achieve or maintain market acceptance or commercial success; | |
| • | conditions and trends in the markets we serve; | |
| • | changes in general economic, industry and market conditions; | |
| • | success of competitive products and services; | |
| • | changes in market valuations or earnings of our competitors; | |
| • | changes in our pricing policies or the pricing policies of our competitors; | |
| • | announcements of significant new products, contracts, acquisitions or strategic alliances by us or our competitors; | |
| • | changes in legislation or regulatory policies, practices, or actions; | |
| • | the commencement or outcome of litigation involving our company, our general industry or both; | |
| • | recruitment or departure of key personnel; | |
| • | changes in our capital structure, such as future issuances of securities or the incurrence of additional debt; | |
| • | actual or expected sales of our common stock by our stockholders; and | |
| • | the trading volume of our common stock. |
23
Table of Contents
In addition, the stock market in general, the NASDAQ Global Market and the market for
diagnostics companies in particular, may experience a loss of investor confidence. Such loss of
investor confidence may result in extreme price and volume fluctuations in our common stock that
are unrelated or disproportionate to the operating performance of our business, financial condition
or results of operations. These broad market and industry factors may materially harm the market
price of our common stock and expose us to securities class action litigation. Such litigation,
even if unsuccessful, could be costly to defend and divert management’s attention and resources,
which could further materially harm our financial condition and results of operations.
If equity research analysts do not publish research or reports about our business or if they issue
unfavorable commentary or downgrade our common stock, the price of our common stock could decline.
The liquidity of the trading market for our common stock may be affected in part by the
research and reports that equity research analysts publish about us and our business. We do not
control the opinions of these analysts. The price of our stock could decline if one or more equity
analysts downgrade our stock or if those analysts issue other unfavorable commentary or cease
publishing reports about us or our business.
Certain provisions of our corporate governing documents could make an acquisition of our company
more difficult.
Certain provisions of our organizational documents could discourage potential acquisition
proposals, delay or prevent a change in control of us or limit the price that investors may be
willing to pay in the future for shares of our common stock. For example, our amended and restated
certificate of incorporation and amended and restated by-laws:
| • | authorize the issuance of preferred stock that can be created and issued by our board of directors without prior stockholder approval, commonly referred to as “blank check” preferred stock, with rights senior to those of our common stock; | |
| • | limit the persons who can call special stockholder meetings; | |
| • | provide that a majority vote of our stockholders is required to amend our amended and restated certificate of incorporation and amended and restated by-laws; | |
| • | establish advance notice requirements to nominate persons for election to our board of directors or to propose matters that can be acted on by stockholders at stockholder meetings; | |
| • | not provide for cumulative voting in the election of directors; and | |
| • | provide for the filling of vacancies on our board of directors by action of a majority of the directors and not by the stockholders. |
These and other provisions in our organizational documents could allow our board of directors
to affect your rights as a stockholder in a number of ways, including making it more difficult for
stockholders to replace members of the board of directors. Because our board of directors is
responsible for approving the appointment of members of our management team, these provisions could
in turn affect any attempt to replace the current management team. These provisions could also
limit the price that investors would be willing to pay in the future for shares of our common
stock.
Our amended and restated articles of incorporation provide that Section 203 of the Delaware
General Corporation Law, an anti-takeover law, will not apply to us. Section 203 generally
prohibits an interested stockholder from engaging in certain types of business combinations with a
Delaware corporation for three years after becoming an interested stockholder. An “interested
stockholder” is a person who, together with affiliates and associates, owns 15% or more of the
corporation.
24
Table of Contents
Our 2007 Long-Term Incentive Plan includes an automatic share replenishment, or “evergreen,”
provision that, unless our board of directors takes action to the contrary, will automatically
increase the number of shares of our common stock reserved for issuance under this plan each year.
Issuances of awards under this Plan would cause further dilution to existing stockholders.
In March 2007 our board of directors adopted and our shareholders approved our 2007 Long-Term
Incentive Plan (the “2007 Plan”). The 2007 Plan authorizes the grant of stock options, share
appreciation rights, restricted shares, restricted share units, unrestricted shares, incentive
stock options, deferred share units and performance awards. The total awards originally authorized
under the 2007 Plan was 4,106,009 shares, plus up to an additional 773,591 shares of common stock
that will become available in the event that awards made under our 2000 Equity Incentive Plan
expire, are forfeited or cancelled, plus an annual increase in the number of shares pursuant to the
evergreen provision equal to the least of: 900,000 shares of common stock; 4.0% of our outstanding
shares of common stock as of fiscal year end; and an amount determined by the board of directors.
At December 31, 2009, there were 28,422,461 outstanding shares of our common stock. In
addition, there were outstanding options to purchase 4,338,695 shares of our common stock
(including 620,732 shares authorized pursuant to the evergreen provision of the Plan), of which
3,843,095 were in-the-money based on our December 31, 2009 closing stock price. Pursuant to the
evergreen provision, an additional 900,000 shares of our common stock were authorized for issuance
under the 2007 Plan as of January 1, 2010. Collectively, the outstanding shares as of December 31,
2009, the in-the-money options and warrants as of December 31, 2009 and the additional shares
authorized on January 1, 2010 pursuant to the evergreen provision were 33,165,556 (the “Adjusted
Outstanding Shares”).
On January 1, 2011 and January 1, 2012, a maximum of 900,000 additional shares per year may be
authorized under our 2007 Plan as a result of this evergreen provision. If the maximum number of
shares under the evergreen provision were to be authorized and issued, the future shares issued
under the evergreen provision would result in an approximate 5% increase in the Adjusted
Outstanding Shares as of December 31, 2009.
The evergreen provision of the 2007 Plan will increase the likelihood that we will not request
existing stockholders to authorize additional shares for issuance under the 2007 Plan or a new
plan. However, other factors, such as a material increase in the number of our award-eligible
employees, or competitive conditions to attract or keep valuable employees, may affect the
likelihood of our requesting stockholders to authorize additional shares under this plan or a new
plan. The issuance, perception that issuance may occur, or exercise of these options may have a
dilutive impact on other stockholders and could have a material negative effect on the market price
of our common stock.
We do not currently intend to pay dividends on our capital stock and, consequently, your ability to
achieve a return on your investment will depend on appreciation in the price of our common stock.
We have never declared or paid any cash dividends on our common stock, and we currently intend
to invest our future earnings, if any, to fund the development and growth of our business.
Therefore, we do not anticipate declaring or paying cash dividends on our common stock in the
foreseeable future. The payment of dividends will be at the discretion of our board of directors
and will depend on our results of operations, capital requirements, financial condition, future
prospects, contractual arrangements, restrictions imposed by applicable law, any limitations on
payments of dividends present in our current and future debt agreements, and other factors our
board of directors may deem relevant. If we do not pay dividends, your ability to achieve a return
on your investment in our company will depend on any future appreciation in the market price of our
common stock. There is no guarantee that shares of our common stock will appreciate in value or
even maintain the price at which our stockholders have purchased their shares.
We will continue to incur costs and demands upon management as a result of complying with the laws
and regulations affecting public companies, which may adversely affect our operating results and
failure to achieve and maintain effective internal controls in accordance with Section 404 of the
Sarbanes-Oxley Act could cause investors to lose confidence in our operating results and in the
accuracy of our financial reports and could have a material adverse effect on our business and on
the price of our common stock.
As a public company, we are required, pursuant to Section 404 of the Sarbanes-Oxley Act of
2002, to furnish a report by management on, among other things, the effectiveness of our internal
control over financial reporting for our 2009 fiscal year. Management is responsible for
implementing controls and other procedures designed to ensure that information required to be
disclosed by us in the reports that we file with the SEC is disclosed accurately and is recorded,
processed, summarized and reported within the time periods specified in the SEC’s rules and forms.
While we have implemented the internal controls that we feel are necessary to comply with Section
404 of the Sarbanes Oxley Act, these controls may become inadequate because of changes in
conditions or the degree of compliance with these policies or procedures may deteriorate.
25
Table of Contents
Furthermore, as a public company, changing laws, regulations and standards relating to
corporate governance and public disclosure, including regulations implemented by the Securities and
Exchange Commission and the NASDAQ may increase legal and financial compliance costs and make some
activities more time consuming. These laws, regulations and standards are subject to varying
interpretations and, as a result, their application in practice may evolve over time as new
guidance is provided by regulatory and governing bodies. We intend to invest resources to comply
with evolving laws, regulations and standards, and this investment may result in increased general
and administrative expenses and a diversion of management’s time and attention from
revenue-generating activities to compliance activities. If notwithstanding our efforts to comply
with new laws, regulations and standards, we fail to comply, regulatory authorities may initiate
legal proceedings against us and our business may be harmed.
Failure to comply with these rules might also make it more difficult for us to obtain certain
types of insurance, including director and officer liability insurance, and we might be forced to
accept reduced policy limits and coverage and/or incur substantially higher costs to obtain the
same or similar coverage. The impact of these events could also make it more difficult for us to
attract and retain qualified persons to serve on our board of directors, on committees of our board
of directors, or as executive officers.
Concentration of ownership among some of our stockholders, including directors and management may
limit your ability to influence corporate matters.
As
of March 4, 2010, approximately 64% of our common stock including the exercise of all
outstanding warrants and exercisable options to purchase our common stock will be beneficially held
by our directors, our executive officers, and greater than five percent stockholders and their
respective affiliates. Lurie Investment Fund, L.L.C., Lurie Investments, Inc., AOQ Trust,
Alfa-Tech, L.L.C., and their respective affiliates, own 29% of our common stock, and Bain Capital
Venture Fund 2005, L.P. and their respective affiliates own 7% of our common stock. Consequently, a
small number of our stockholders may be able to substantially influence our management and affairs.
If they choose to act together, they would be able to influence most matters requiring approval by
our stockholders, including the election of directors, any merger, consolidation or sale of all or
substantially all of our assets and any other transaction. The concentration of ownership may also
delay or prevent a change in control of us even if such changes might otherwise be beneficial to
our stockholders. In addition the significant concentration of share ownership may adversely affect
the trading price of our common stock because investors often perceive disadvantages in owning
shares in companies with controlling stockholders.
| Item 1B. | Unresolved Staff Comments. |
None.
| Item 2. | Properties. |
Our executive, research and development and manufacturing functions are all located at a
40,945 square foot leased facility in Northbrook, Illinois. The lease for our Northbrook facility
expires in May 2014. Our recently revised facilities lease includes the right of first offer on
additional available space in our building. While we do not need to expand our facilities to meet
anticipated demand for 2010, we will likely require expanded facilities to meet anticipated demand
beyond 2010.
We do not own any real property.
| Item 3. | Legal Proceedings. |
We are from time to time subject to various claims and legal actions during the ordinary
course of our business. We believe that there are currently no claims or legal actions that would,
in management’s judgment based on information currently available, have a material adverse effect
on our results of operations or financial condition.
In July 2009, the Company was named as a defendant in a lawsuit filed by Eppendorf AG alleging
patent infringement. The Company intends to vigorously defend this case as it believes that the
claims lack merit. The Company believes that the resolution to these proceedings will not have a
material adverse effect on the Company’s financial position, liquidity or future operations. The
Company has not recorded any reserves related to this case.
| Item 4. | (Removed and Reserved). |
26
Table of Contents
PART II
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters, and Issuer Purchases of Equity Securities. |
Market Information
Our common stock has been traded on the NASDAQ Global Market since November 1, 2007 under the
symbol “NSPH”. The following table sets forth the high and low sale prices for our common stock for
each quarter of our two most recent fiscal years, as reported on the NASDAQ Global Market for the
period indicated.
| High | Low | |||||||
Fiscal year ended December 31, 2009 |
||||||||
First Quarter |
$ | 6.26 | $ | 2.71 | ||||
Second Quarter |
5.58 | 2.95 | ||||||
Third Quarter |
8.61 | 3.94 | ||||||
Fourth Quarter |
7.85 | 5.60 | ||||||
Fiscal year ended December 31, 2008 |
||||||||
First Quarter |
$ | 14.61 | $ | 6.78 | ||||
Second Quarter |
10.32 | 6.62 | ||||||
Third Quarter |
12.00 | 7.10 | ||||||
Fourth Quarter |
8.50 | 3.03 | ||||||
Stockholders
The last reported sale price of common stock on March 4, 2010 as reported on the NASDAQ Global
Market was $3.20. As of March 4, 2010, there were 185 holders of record of our common stock.
Dividend Policy
We have never declared or paid any cash dividends on our common stock and do not expect to pay
any dividends for the foreseeable future. We currently intend to retain any future earnings to fund
the operation, development and expansion of our business. Any future determination to pay dividends
will be at the sole discretion of our board of directors and will depend upon a number of factors,
including our results of operations, capital requirements, financial condition, future prospects,
contractual arrangements, restrictions imposed by applicable law, any limitations on payments of
dividends present in our current and future debt arrangements, and other factors our board of
directors may deem relevant.
27
Table of Contents
Stock Performance Graph
The following graph shows a comparison of cumulative total stockholder returns for our common
stock, the NASDAQ Composite Index and the NASDAQ Biotechnology Index. The graph assumes the
investment of $100 on November 1, 2007, and the reinvestment of all dividends. The performance
shown is not necessarily indicative of future performance.
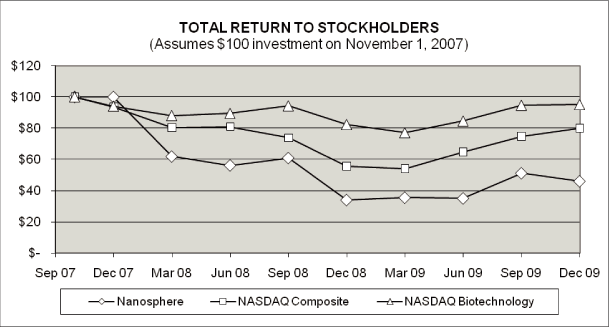
| Investment Return Analysis | Nov 07 | Dec 07 | Mar 08 | Jun 08 | Sep 08 | Dec 08 | Mar 09 | Jun 09 | Sep 09 | Dec 09 | ||||||||||||||||||||||||||||||
Nanosphere |
$ | 100.00 | $ | 99.93 | $ | 61.86 | $ | 56.14 | $ | 60.93 | $ | 34.00 | $ | 35.50 | $ | 35.07 | $ | 51.14 | $ | 46.00 | ||||||||||||||||||||
NASDAQ Composite |
$ | 100.00 | $ | 93.55 | $ | 80.39 | $ | 80.88 | $ | 73.79 | $ | 55.63 | $ | 53.92 | $ | 64.73 | $ | 74.86 | $ | 80.04 | ||||||||||||||||||||
NASDAQ Biotechnology |
$ | 100.00 | $ | 94.07 | $ | 87.98 | $ | 89.39 | $ | 94.20 | $ | 82.19 | $ | 76.93 | $ | 84.45 | $ | 94.59 | $ | 95.04 | ||||||||||||||||||||
The information contained in the graph above shall not be deemed to be “soliciting material” or to
be “filed” with the SEC, nor shall such information be incorporated by reference into any future
filing under the Securities Act or the Exchange Act, or subject to Regulation 14A or 14C
promulgated under the Exchange Act, other than as provided in Item 402 of the SEC’s Regulation S-K,
or to the liabilities of Section 18 of the Exchange Act, except to the extent that Nanosphere
specifically requests that the information be treated as soliciting material or specifically
incorporates it by reference in such filing.
Equity Compensation Plan Information
The following table provides information as of December 31, 2009 with respect to shares of the
Company’s common stock that may be issued under the 2007 Plan, which is the Company’s only existing
equity compensation plan under which grants can be made. Stockholders approved the Company’s 2007
Plan on March 27, 2007.
Equity Compensation Plan Information
| Number of securities | ||||||||||||
| remaining available for | ||||||||||||
| future issuance under | ||||||||||||
| Number of securities to be | equity compensation plans | |||||||||||
| issued upon exercise of | Weighted Average exercise | (excluding securities | ||||||||||
| outstanding awards | price of outstanding awards | reflected in column(a)) | ||||||||||
| Plan Category | (a) | (b) | (c) | |||||||||
Equity compensation plans
approved by
stockholders |
4,338,695 | $ | 5.75 | 268,416 | ||||||||
Equity compensation plans not
approved by stockholders |
— | — | — | |||||||||
Total |
4,338,695 | $ | 5.75 | 268,416 | ||||||||
| Pursuant to the evergreen provision, an additional 900,000 shares of common stock were authorized for issuance under the 2007 Plan as of January 1, 2010. | ||
28
Table of Contents
| Item 6. | Selected Financial Data. |
The following selected financial data should be read in conjunction with, and is qualified by
reference to, our financial statements and related notes and “Item 7. Management’s Discussion and
Analysis of Financial Condition and Results of Operations” appearing elsewhere in this report.
| As of December 31, | ||||||||||||||||||||
| Statements of Operations Data: | 2009 | 2008 | 2007 | 2006 | 2005 | |||||||||||||||
Total revenue |
$ | 2,214,103 | $ | 1,366,732 | $ | 1,167,364 | $ | 1,138,011 | $ | 1,914,517 | ||||||||||
Research and development expense |
18,607,921 | 23,675,138 | 21,445,994 | 18,185,564 | 13,717,654 | |||||||||||||||
Sales, general and administrative expense |
14,471,973 | 13,615,322 | 13,443,304 | 5,415,525 | 4,502,970 | |||||||||||||||
Net loss |
(33,948,653 | ) | (37,041,996 | ) | (53,199,184 | ) | (24,269,918 | ) | (16,408,082 | ) | ||||||||||
Net loss attributable to common stock |
(33,948,653 | ) | (37,041,996 | ) | (59,284,411 | ) | (46,421,053 | ) | (19,306,869 | ) | ||||||||||
Net loss per common share: |
||||||||||||||||||||
basic and diluted |
(1.46 | ) | (1.67 | ) | (14.18 | ) | (53.63 | ) | (31.57 | ) | ||||||||||
Weighted average number of common shares: |
||||||||||||||||||||
basic and diluted (1)(2) |
23,301,764 | 22,213,164 | 4,180,979 | 865,559 | 611,496 | |||||||||||||||
| As of December 31, | ||||||||||||||||||||
| Balance Sheet Data: | 2009 | 2008 | 2007 | 2006 | 2005 | |||||||||||||||
Cash and cash equivalents (1)(2) |
$ | 76,689,279 | $ | 75,356,960 | $ | 114,312,573 | $ | 29,112,429 | $ | 3,641,338 | ||||||||||
Working capital (1)(2) |
71,152,601 | 69,027,249 | 107,684,875 | 27,332,463 | (2,642,582 | ) | ||||||||||||||
Total assets (1)(2) |
88,668,954 | 86,895,910 | 125,963,862 | 37,968,243 | 9,015,260 | |||||||||||||||
Long-term debt |
— | 3,352,137 | 7,462,237 | 58,802 | — | |||||||||||||||
Convertible preferred stock (2) |
— | — | — | 108,868,040 | 51,143,984 | |||||||||||||||
Stockholders’ equity (deficit) (1)(2) |
79,081,998 | 74,541,108 | 109,199,722 | (108,307,662 | ) | (62,292,544 | ) | |||||||||||||
| (1) | In October 2009, we completed our underwritten public offering of 5,405,000 shares of common stock at $7.00 per share. We received approximately $35.4 million of net proceeds from the offering. | |
| (2) | In November 2007, we completed our initial public offering of 8,050,000 shares of common stock at $14.00 per share. We received approximately $102 million of net proceeds from the offering. All shares of convertible preferred stock were converted to common stock upon the closing of the initial public offering. |
29
Table of Contents
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations. |
The following discussion and analysis is based primarily on the financial statements of
Nanosphere, Inc. for the years presented and should be read together with the notes thereto
contained in this annual report on Form 10-K. Terms employed herein as defined terms, but without
definition, have the meanings set forth in the notes to the financial statements (see “Item 8.
Financial Statements and Supplementary Data”).
Business Overview
We develop, manufacture and market an advanced molecular diagnostics platform, the Verigene
System, that enables simple, low cost and highly sensitive genomic and protein testing on a single
platform. Our proprietary nanoparticle technology simplifies molecular diagnostic testing, provides
the ability to run multiple tests simultaneously on the same sample and has the potential to run a
broad menu of tests on a single platform. We have developed or are currently developing diagnostic
tests for markers which reveal the existence of a variety of medical conditions including
cardiovascular, respiratory, cancer, autoimmune, neurodegenerative and other infectious diseases,
as well as for pharmacogenomics.
Pharmacogenomics is an emerging subset of human genetic testing that correlates gene variation
with a drug’s efficacy or toxicity. These tests play a key role in the advancement of personalized
medicine where drug therapies and dosing are guided by each patient’s genetic makeup. There is a
growing demand on laboratories to implement molecular diagnostic testing, but the cost and
complexity of existing technologies and the need for specialized personnel and facilities have
limited the number of laboratories with these capabilities.
The Verigene System is differentiated by its ease of use, rapid turnaround times and ability
to support a broad test menu. It also offers lower cost for laboratories already performing
molecular diagnostic testing and allows a broader range of laboratories, including those operated
by local hospitals, to perform these tests. Our ability to detect proteins, which can be 100 times
more sensitive than current technologies for certain targets, may enable earlier detection of and
intervention in diseases associated with known biomarkers as well as the introduction of tests for
new biomarkers that exist in concentrations too low to be detected by current technologies. We are
focused on the clinical diagnostics market and may seek opportunities either directly or through
partnerships to commercialize our technologies in other markets.
We received 510(k) clearance from the United States Food and Drug Administration (“FDA”) for
commercial sale of the Verigene System in the second half of 2007. 510(k) clearance is granted by
the FDA if the submitted information establishes that the proposed device is “substantially
equivalent” to a legally marketed Class I or Class II medical device or a pre-amendment Class III
medical device for which the FDA has not sought pre-market approval. We have also received 510(k)
clearance to use the Verigene System for four diagnostic tests. The first test is a warfarin
metabolism assay, which is a pharmacogenomic test to determine the existence of certain genetic
information believed to affect the metabolism of warfarin-based drugs, including Coumadin, the
most-prescribed oral anticoagulant in North America and Europe. The second test is a
hyper-coagulation assay, one of the highest volume human genetic tests currently performed, to
determine an individual’s risk, based upon genetic information, for the development of blood clots,
which can lead to stroke, pulmonary embolism and deep vein thrombosis.
The third test is our respiratory panel which detects the presence of influenza A and B as
well as respiratory syncitial virus (“RSV”) A and B. Influenza is commonly known as the seasonal
flu and RSV is a respiratory virus that infects the lungs and breathing passages. RSV is the most
common cause of bronchitis and pneumonia in children under the age of one year and has become a
significant concern for older adults. Our respiratory panel provides physicians with a highly
accurate, fast determination of which virus is present which helps guide the most appropriate
treatment therapy. Most of the respiratory tests currently on the market take days to generate a
result, because they depend on culturing, or do not provide a reliable result, because they are
rapid tests which lack specificity.
On October 9, 2009, we received 510(k) clearance of our second generation respiratory panel
and the Verigene SP System. The Verigene SP instrument automates sample preparation, which should
significantly reduce laboratory technician time required to perform this molecular test. We also
believe that our respiratory assay on the Verigene SP offers a simple to use molecular test for
diagnosing respiratory infections and the flu, while providing improved specificity over currently
available rapid tests. On November 24, 2009, we received clearance for a label change for this
assay confirming that the novel H1N1 virus is detected as a positive Influenza A when using our
respiratory assay and the Verigene SP. However, it is uncertain whether the ability to use this
assay to detect H1N1 virus will be material to the Company based on the reduction in the number of
recently reported H1N1 cases.
30
Table of Contents
The fourth test is our cystic fibrosis test that enables molecular laboratories to perform
prenatal screening and diagnostic confirmations through identification of the number of copies of
each of the 23 most common gene mutations recognized by the American College of Obstetricians and
Gynecologists as markers for cystic fibrosis.
We have elected to delay the launch of our 501(k) cleared cystic fibrosis test until such time
as it may receive 510(k) clearance for use on the Verigene SP. In addition, we plan to submit
additional FDA applications for each of our previously 510(k) cleared assays to allow their use on
the new Verigene SP. We have engaged in dialogue with the FDA to identify the tests that we need
to receive 510(k) clearance to run these assays on the Verigene SP.
In the first quarter of 2009, we filed a de novo 510(k) submission for a hereditary
hemochromatosis (“HFE”) genetic test. We re-submitted an amended 510(k) submission in October 2009
to supply additional data requested by the FDA. Mutations in the HFE gene are associated with
hemochromatosis, which is the leading cause of iron overload disease, a systematic iron build up
that can eventually adversely affect the heart, liver, pancreas, joints and pituitary gland.
Untreated, hemochromatosis can be fatal. Once detected, hemochromatosis is easily treated.
Approximately one in every 250 people of European descent has the disease and one in eight is a
carrier of at least one of the recessive gene mutations. This is a de novo 510(k) because there are
currently no FDA-cleared tests on the market to detect these mutations of the HFE gene. We have
received additional feedback from the FDA indicating that further testing would be necessary in
order to proceed with this application. This assay was submitted for clearance on the original
Verigene System and the FDA requires separate testing and a new application to clear this assay on
the Verigene SP. Therefore, we plan to complete all necessary testing on the Verigene SP and submit
a new application for clearance of this assay on the Verigene SP.
The first ultra-sensitive protein test we plan to commercialize is for cardiac troponin I
(“cTnI”), which is the gold standard biomarker for diagnosis of the occurrence of myocardial
infarction or heart attack and identification of patients at risk for acute coronary syndrome. We
submitted a 510(k) application to the FDA in the fourth quarter of 2009. We have received initial
feedback from the FDA regarding our 510(k) submission indicating that significant further studies
will be required to obtain clearance for our cTnI assay. We believe that some of the additional
requirements suggested by the FDA may be addressed by results from our ongoing FAST-TRAC clinical
study, which is designed to support and further demonstrate the clinical utility of ultra-sensitive
cTnI measurements as a diagnostic tool for use in the management of both acute and chronic cardiac
disease. We plan to complete the FAST-TRAC clinical studies along with any other studies necessary
to respond to the FDA’s requests in 2010.
We are currently conducting multi-site trials for our cytochrome P450 2C19 assay that detects
genetic mutations associated with deficiencies to metabolize clopidogrel, more commonly known by
the trade name Plavix. Clopidogrel inhibits platelet function and is a standard treatment to
reduce the risk of thrombolytic events for patients undergoing percutaneous coronary intervention
(“PCI”) procedures. Clopidogrel metabolism is affected by the Cytochrome P-450 family of genes. Up
to 30-50% of the patient population possess variations in these genes and do not properly
metabolize this drug, thus increasing the risk of adverse events. Our 2C19 assay is designed to
identify patients possessing these variations so that alternative therapies can be prescribed to
effectively reduce clotting post-PCI.
In addition, we currently have research and development efforts underway on additional
genetic, infectious disease and protein tests. Our test development pipeline includes a blood
infection screening assay, a human papillomavirus (“HPV”) assay for cervical cancer screening, an
ultra-sensitive prostate-specific antigen (“PSA”) test for early diagnosis of recurrent prostate
cancer and a multiplexed protein-based connective-tissue panel for the detection of rheumatoid
arthritis, lupus and other related diseases. We are also investigating new biomarkers where our
ultra-sensitive protein detection technology may enable earlier detection of a broad range of
diseases. We are in the process of assessing the commercial viability of these applications and
the various paths to regulatory approval that may be available.
Our technology is broadly applicable beyond the clinical diagnostic market in both research
and industrial applications. The Verigene System is presently used in research laboratories
supporting collaborations and independent research in areas including ovarian cancer, mad cow
disease and HIV. We are currently working with the FDA on a joint research program to develop an
H5N1 avian flu assay. We have developed and delivered a biosecurity platform for the detection of
various bioterrorism agents to the Technical Support Working Group, an agency affiliated with the
U.S. Department of Defense.
As of December 31, 2009, our patent portfolio is comprised, on a worldwide basis, of 132
issued patents and 97 pending patent applications which we own directly or for which we are the
exclusive licensee. Some of these patents and patent applications derive from a common parent
patent application or are foreign counterpart patent applications and relate to similar or
identical technological claims. The issued patents cover approximately 12 different technological
claims and the pending patent applications cover approximately 4 additional technological claims.
31
Table of Contents
Many of our issued and pending patents were exclusively licensed from the International
Institute for Nanotechnology at Northwestern University (“Northwestern”) in May 2000 and they
generally cover our core technology, including nanotechnology based biodiagnostics and biobarcode
technology. Our issued patents expire between 2017 and 2025. We believe our patent portfolio
provides protection against other companies offering products employing the same technologies and
methods as we have patented. While we believe our patent portfolio establishes a proprietary
position, there are many competitive products utilizing other technologies that do not infringe on
our patents.
In addition, as of December 31, 2009, we have non-exclusive licenses for at least 44 U.S.
patents that cover 12 different technological claims from various third parties. Most of these
license agreements require us to pay the licensor royalty fees that typically expire upon the
patent expiration dates which range from 2010 to 2025. These license agreements are nonexclusive
and do not create a proprietary position. The expiration of these non-exclusive licenses will
result in the termination of certain royalty payments by us to the licensors.
Since inception we have incurred net losses each year, and we expect to continue to incur
losses for the foreseeable future. Our net losses attributable to common stock were approximately
$34 million for fiscal 2009. As of December 31, 2009, we had an accumulated deficit of
approximately $239 million. Our operations to date have been funded principally through capital
contributions from investors in two underwritten direct public offerings of common stock, and prior
thereto in our convertible preferred stock, which was converted to common stock in 2007, and our
debt borrowings.
In November 2007, we completed our initial public offering of 8,050,000 shares of common stock
at $14.00 per share. We received approximately $102 million of net proceeds from our initial
public offering. In October 2009, we completed a public offering of 5,405,000 shares of our common
stock at $7.00 per share. We received approximately $35.4 million of net proceeds from this
offering.
Financial Operations Overview
Revenue
Product sales revenue is derived from the sale or lease of the Verigene System, including
cartridges and related products sold to research laboratories and hospitals. Grant and contract
revenue consists of funds received under contracts and government grants, including funds for the
reimbursement of certain research and development expenses. Our market efforts are primarily
focused on driving product sales rather than grants and contracts. However, the Company recently
completed development of certain custom pharmacogenetic assays to be used in conjunction with the
clinical trials associated with new therapeutic drugs for a major pharmaceutical company. We will
continue to be opportunistic with regard to future contract and grant opportunities.
Cost of Sales
Cost of sales represents the cost of materials, direct labor and other manufacturing overhead
costs incurred to produce Verigene cartridges and instruments, as well as royalties on product
sales, amortization of purchased intellectual property relevant to products available for sale and
depreciation of instrument leases and rentals. Costs associated with custom assay development
contracts also include labor associated with assay development, validation and testing.
Research and Development Expenses
Research and development expenses primarily include all costs incurred during the development
of the Verigene System and assays, and the expenses associated with fulfilling our development
obligations related to the United States government contracts and grants. Such expenses include
salaries and benefits for research and development personnel, consulting services, materials,
patent-related costs and other expenses. We expense all research and development costs in the
periods in which they are incurred. We expect research and development expenses to grow modestly as
we continue to develop future generations of the Verigene System, and additional genomic and
protein tests.
32
Table of Contents
Sales, General and Administrative Expenses
Sales, general and administrative expenses principally include compensation for employees in
our sales, customer service, marketing, management and administrative functions. We also include
professional services, facilities, technology, communications and administrative expenses in sales,
general and administrative. The professional services costs primarily consist of legal and
accounting costs. We expect sales and marketing expenses will increase as additional sales and
customer support are needed to drive and support customer growth.
Interest Income
Interest income principally includes interest earned on our excess cash balances. Such
balances are primarily invested in money market and bank checking accounts at major financial
institutions. We anticipate that interest income will continue to decline as capital reserves are
consumed by operating losses and working capital. Recent declines in interest rates will also
contribute to reduced interest income for the foreseeable future.
Interest Expense
Interest expense includes the interest charges related to our debt borrowings, including
non-cash amortization of debt discount and issuance costs.
Fiscal 2009 Compared to 2008
Revenues
Revenues were $2.2 million for fiscal 2009 as compared to $1.4 million for fiscal 2008.
Product sales increased slightly to $1.1 million for fiscal 2009 as compared to $1.0 million for
fiscal 2008. The change in product sales was driven by a 47% increase in consumables revenue and a
50% reduction in system sales. Fiscal 2009 revenues also included $1.0 million of service revenue
related primarily to the assay development contracts with a major pharmaceutical company described
earlier. Revenues from government grants were $0.3 million for fiscal 2008.
Cost of Sales
For fiscal 2009, cost of sales was $2.2 million, as compared to $1.5 million for fiscal 2008.
The $0.7 million increase in cost of sales for fiscal 2009 resulted from the costs associated with
assay development revenue from our commercial contracts, the increase in product sales and the
fixed amortization of license fees. We have initiated several product cost reduction programs,
which include initiatives to reduce the cost of materials and labor. We expect per unit material
costs to decline as a result of process automation, additional multi-cavity molds and volume
absorption of overhead. Per unit labor costs will be reduced through additional cartridge assembly
automation, increased production lot sizes and higher overall volume. We expect to reduce cost per
unit substantially as we implement these programs and as volume increases.
Research and Development Expenses
Research and development expenses decreased from $23.7 million in fiscal 2008 to $18.6 million
in fiscal 2009. The $5.1 million decrease in research and development expenses for fiscal 2009
consists primarily of $2.0 million in staffing, $1.5 million of materials spending and $1.0 million
in reduced facilities and depreciation expense. The staffing reduction was driven by a more narrow
focus on core research and development projects. The materials reduction was primarily related to
the completion of the up-front investment in Verigene SP development activities.
Sales, General and Administrative Expenses
Sales, general and administrative expenses increased from $13.6 million for fiscal 2008 to
$14.5 million for fiscal 2009. The increase in sales, general and administrative expenses for
fiscal 2009 consists primarily of a $1.4 million increase in clinical trial expenses associated
with the FAST-TRAC Troponin I study intended to create market differentiation for this assay,
partially offset by a decrease in spending due to the completion of the initial implementation of
our Sarbanes-Oxley compliance program.
33
Table of Contents
Interest Expense
Interest expense was $1.3 million for fiscal 2009 as compared to $2.1 million for fiscal 2008.
The decrease in interest expense for fiscal 2009 resulted from increased principal payments and
lower interest, in accordance with our loan and security agreements.
Interest Income
Interest income was $0.4 million and $2.5 million for fiscal 2009 and 2008, respectively. The
decrease in interest income during fiscal 2009 resulted from a lower average cash balance during
this period as compared to fiscal 2008. In addition, interest rates declined significantly for
fiscal 2009 as compared to fiscal 2008.
Fiscal 2008 Compared to 2007
Revenues
Revenues were $1.4 million for fiscal 2008, as compared to $1.2 million for fiscal 2007.
Product revenues increased to $1.0 million for fiscal 2008 from $0.1 million for fiscal 2007 due to
sales and rentals of the Verigene System following clearance from the FDA during 2007. Revenues
from contracts and government grants were $0.3 million for fiscal 2008 and $1.1 million for fiscal
2007.
Cost of Sales
For fiscal 2008, cost of sales was $1.5 million, as compared to $0.1 million for fiscal 2007.
Cost of sales increased during 2008 due to the increase in Verigene System sales as well as a $0.3
million increase in amortization expense related to upfront license fees. Approximately 63% of the
2008 license fee amortization expense was associated with the license of a patent which expired in
May 2009.
Research and Development Expenses
Research and development expenses increased to $23.7 million in fiscal 2008, from
$21.4 million in fiscal 2007. The $2.3 million increase in research and development expenses for
2008 versus 2007 consisted primarily of $1.7 million in increased staffing and non-cash stock
compensation expenses and $0.6 million in increased depreciation expense. Research and development
staffing increases were primarily focused on development of our respiratory virus panel and
Troponin I assays as well as Verigene SP. Depreciation expense increased primarily due to
investments in leasehold improvements for lab space.
Sales, General and Administrative Expenses
Sales, general and administrative expenses remained relatively flat at $13.6 million for
fiscal 2008 versus $13.4 million for fiscal 2007. Sales and marketing spending increased by $0.8
million due to personnel and marketing expenses associated with our initial product launches.
General and administrative expenses increased by approximately $1.5 million for public company
expenses such as insurance, legal and accounting expenses. These increases were offset by a $1.5
million decrease in payroll expense due primarily to a bonus paid to our chief executive officer in
2007. Additional expense reductions totaling $0.6 million related to franchise taxes, which were
higher in 2007 due to our initial public offering, stock compensation expense, which was higher in
2007 due to shorter than normal vesting periods of certain stock options granted in 2007 and
facilities maintenance expenses.
Change in Fair Value of Convertible Derivative Liability
Our Series C-2 and Series D Convertible Preferred Stock contained conversion features which
were embedded derivatives and therefore required bifurcation and accounting at fair value separate
and distinct from the convertible preferred stock. Changes in the fair value of the conversion
liability were recognized in earnings. For fiscal 2007, the fair value of the conversion liability
increased, resulting in a $14.9 million non-cash charge to earnings. The increase in fair value
that resulted in the non-cash charge to earnings was due to the increased valuation of the Company.
Upon the closing of our initial public offering, our preferred stock was converted into common
stock and accordingly, we stopped incurring charges for this embedded derivative and the liability
converted into additional paid-in capital.
34
Table of Contents
Interest Expense
Interest expense remained relatively flat at $2.1 million for fiscal 2008 as compared to $2.0
million for fiscal 2007. In February 2007, the Company borrowed $12.5 million under two loan and
security agreements with Venture Lending & Leasing IV, Inc., and Venture Lending & Leasing V, Inc.
Interest Income
Interest income was $2.5 million and $1.9 million for fiscal 2008 and 2007, respectively. The
increase in interest income in 2008 resulted from the increase in cash from the initial public
offering, which was significantly offset by the reduction in interest rates.
Liquidity and Capital Resources
From our inception in December 1999 through December 31, 2009, we have received net proceeds
of $103.9 million from the sale of convertible preferred stock and issuance of notes payable that
were exchanged for convertible preferred stock, $102.2 million from our November 2007 initial
public offering, $35.4 million from our October 2009 underwritten public offering, $12.5 million
from our debt borrowings, and $9.2 million from government grant revenue. We have devoted
substantially all of these funds to research and development and sales, general and administrative
expenses. Since our inception, we have generated minimal revenues from the sale of the Verigene
System, including cartridges and related products, to our initial clinical customers, research
laboratories and government agencies. We also incurred significant losses and, as of December 31,
2009, we had an accumulated deficit of approximately $239.3 million. While we are currently in the
commercialization stage of operations, we have not yet achieved profitability and anticipate that
we will continue to incur net losses for the foreseeable future.
Because we recently began to commercialize our products, we do not anticipate achieving
positive operating cash flow before 2013. During this period we expect to increase spending on
additional manufacturing scale-up, research and development costs to expand our assay menu and to
develop a fully automated instrument with increased throughput, and continue to grow sales and
marketing personnel. Achievement of positive cash flow from operations will depend upon revenue
resulting from adoption of our initial products and expansion of the number of our FDA-cleared
tests. Demand for our respiratory products is directly proportional to the size and duration of
influenza and other respiratory illnesses. Any unanticipated acceleration or deceleration of
customer demand for our products relative to projections will have a material effect on our cash
flows. We anticipate that our current cash and cash equivalents will be sufficient to cover our
operating and investing activities as well as our debt interest and principal obligations for
approximately two years.
A customer may purchase the Verigene System, lease it from a third party or enter into a
reagent rental agreement. Our reagent rental agreements include customer commitments to purchase a
certain minimum volume of cartridges over the term of the agreement. As part of these agreements, a
portion of the charge for each cartridge is a rental fee for use of the equipment. To date, our
aggregate investment in systems rented to customers has not been material. However, we may need to
increase investment in such systems to support future product placements under reagent rental
agreements. We have established a relationship with a third party financing company to provide our
customers with lease financing for their Verigene equipment. This arrangement will help mitigate
the demand on our capital resources by allowing us to recover the cost of such systems immediately,
instead of over three to five years.
As of December 31, 2009, we had $76.7 million in cash and cash equivalents, compared to
$75.4 million at December 31, 2008. Cash used in operations decreased to $27.9 million for 2009 as
compared to $32.0 million in 2008 due to a decrease in research and development expenses during
2009.
Net cash used in investing activities decreased to $1.8 million for the year ended December
31, 2009 from $2.7 million for the year ended December 31, 2008. Investments in property and
equipment decreased $1.0 million during 2009 due to the significant spending in 2008 to scale-up
manufacturing for the commencement of product commercialization activities.
Net cash provided by financing activities was $31.1 million for the year ended December 31,
2009, compared to cash used by financing activities of $4.2 million for the year ended December 31,
2008. In October 2009, we completed our public offering of 5,405,000 shares of common stock at
$7.00 per share. We received approximately $35.4 million of net proceeds from this offering. In
2008 cash was used to repay our long-term debt, whereas in 2007, we received net proceeds of
approximately $102 million from our initial public offering of 8,050,000 shares of common stock and
we borrowed $12.5 million. In February 2007, we entered into two loan and security agreements with
Venture Lending & Leasing IV, Inc. and Venture Lending & Leasing V, Inc. Pursuant to these loan
agreements, we are required to pay interest and a minimal amount of principal, for the initial
twelve month period, which ended
in February 2008, followed by a thirty month period within which the note principal will be
amortized. Interest was paid during the initial twelve month period at a fixed annual interest rate
of 12.5% and will be paid during the following thirty month period at a fixed annual interest rate
of 10.0%.
35
Table of Contents
While we anticipate that our capital resources will be sufficient to meet our estimated needs
for approximately two years, we may need to increase our capital outlays and operating expenditures
over the next several years as we expand our product offering, drive product adoption, further
scale-up manufacturing and implement product cost savings. The amount of additional capital we may
need to raise depends on many factors, including:
| • | the level of research and development investment required to maintain and improve our technology; |
| • | the amount and growth rate of our revenues; |
| • | changes in product development plans needed to address any difficulties in manufacturing or commercializing the Verigene System and enhancements to our system; |
| • | the costs of filing, prosecuting, defending and enforcing patent claims and other intellectual property rights; |
| • | competing technological and market developments; |
| • | our need or decision to acquire or license complementary technologies or acquire complementary businesses; and |
| • | changes in regulatory policies or laws that affect our operations. |
We cannot be certain that additional capital will be available when and as needed or that our
actual cash requirements will not be greater than anticipated. If we require additional capital at
a time when investment in diagnostics companies or in the marketplace in general is limited due to
the then prevailing market or other conditions, we may not be able to raise such funds at the time
that we desire or any time thereafter. In addition, if we raise additional funds through the
issuance of equity or convertible debt securities, the percentage ownership of our stockholders
could be significantly diluted, and these newly-issued securities may have rights, preferences or
privileges senior to those of existing stockholders. If we obtain additional debt financing, a
substantial portion of our operating cash flow may be dedicated to the payment of principal and
interest on such indebtedness, and the terms of the debt securities issued could impose significant
restrictions on our operations. If we raise additional funds through collaborations and licensing
arrangements, we might be required to relinquish significant rights to our technologies or
products, or grant licenses on terms that are not favorable to us.
Income Taxes
Since inception, we have incurred operating losses and, accordingly, have not recorded a
provision for income taxes for any of the periods presented. As of December 31, 2009, we had net
operating loss carryforwards for federal and state income tax purposes of $157 million. The Company
also has federal research and development tax credit carryforwards of $8 million which will begin
to expire in 2020. Section 382 of the Internal Revenue Code subjects the utilization of net
operating loss and credit carryforwards to an annual limitation that is applicable if the Company
experiences an ownership change. The Company believes its public offerings and/or prior equity
investments may have triggered an ownership change as defined by the Internal Revenue Code.
However, the Company has yet to perform the computations under Section 382 which would determine
the amount of annual limitation on its utilization of its net operating loss and tax credit
carryforwards. The annual limitation may result in the expiration of the Company’s net operating
loss and tax credit carryforwards before they can be used.
36
Table of Contents
Contractual Obligations and Commitments
As of December 31, 2009, the annual amounts of future minimum payments under certain of our
contractual obligations were:
| Payments Due by Period | ||||||||||||||||||||
| Contractual Obligations | Total | Less than 1 Year | 1-3 Years | 3-5 Years | More than 5 Years | |||||||||||||||
Long-term debt obligations |
$ | 3,917,293 | $ | 3,917,293 | $ | — | $ | — | $ | — | ||||||||||
Interest on long-term debt obligations |
195,107 | 195,107 | — | — | — | |||||||||||||||
Operating lease |
1,972,488 | 465,595 | 865,564 | 641,329 | — | |||||||||||||||
Obligations under license agreements |
1,783,750 | 173,750 | 365,000 | 425,000 | 820,000 | |||||||||||||||
Total |
$ | 7,868,638 | $ | 4,751,745 | $ | 1,230,564 | $ | 1,066,329 | $ | 820,000 | ||||||||||
On August 28, 2009, the Company executed a lease renewal which commences on June 1, 2010 and
ends on May 31, 2014. Base rent on the lease renewal will range from $34,957 per month for the
first twelve months of the lease to $38,026 per month for the final twelve months of the lease
renewal. The Company will also pay as additional rent its proportionate share of real estate taxes
and operating expenses, which include all costs of operating, maintaining, replacing and repairing
the leased facility.
License Agreements
We have entered into several nonexclusive license agreements with various companies covering
certain technologies which are embedded in the Company’s diagnostic instruments and diagnostic test
products. Since inception, we have paid aggregate initial license fees of $2.4 million for these
licenses, and have agreed to pay a percentage of net sales as royalties, in percentage amounts
ranging from less than 1% to 12%. Certain of the license agreements have minimum annual royalty
payments, and such minimum payments are as shown above. These licenses expire at various times,
corresponding to the subject patents expirations, which currently range from 2010 to 2025.
We have entered into a license agreement with Northwestern which provides us with an exclusive
license to certain patents and patent applications related to the application of nanotechnology to
biodiagnostics and to biobarcode technology. This license covers all discoveries from the
International Institute for Nanotechnology at Northwestern in the field of biodiagnostics through
January 1, 2013. Nanosphere also has the right of first negotiation for an exclusive license on
inventions after such date. Our research team utilizes the research and patents developed at
Northwestern to develop diagnostic applications including additional genomic and protein testing
assays for use in the Verigene System.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet financing or unconsolidated special-purpose entities.
Critical Accounting Policies and Significant Judgments and Estimates
This discussion and analysis of our financial condition and results of operations is based on
our financial statements, which have been prepared in conformity with accounting principles
generally accepted in the United States of America. The preparation of these financial statements
requires management to make estimates and judgments that affect the reported amounts of assets,
liabilities and expenses and the disclosure of contingent assets and liabilities at the date of the
financial statements, as well as revenues and expenses during the reporting periods. We evaluate
our estimates and judgments on an ongoing basis. We base our estimates on historical experience and
on various other factors we believe are reasonable under the circumstances, the results of which
form the basis for making judgments about the carrying value of assets and liabilities that are not
readily apparent from other sources. Actual results could therefore differ materially from those
estimates under different assumptions or conditions.
Our significant accounting policies are described in Note 2 to our consolidated financial
statements included elsewhere in this Annual Report on Form 10-K. We believe the following critical
accounting policies reflect our more significant estimates and assumptions used in the preparation
of our financial statements.
Revenue Recognition
We recognize revenue under grants and contracts and for reimbursement of related research and
development expenses at the time the relevant expenses are incurred. For product sales, revenue is
recognized when persuasive evidence of an arrangement exists, title and risk of loss is transferred
to customers, the price to the buyer is fixed or determinable, and collectability is reasonably
assured.
37
Table of Contents
Verigene System instrument units are sold outright to customers or leased to customers
pursuant to operating leases. We recognize revenue from sales of the Verigene System, including
cartridges and related products, when the risks and rewards of ownership are transferred to the
customer. Revenue for Verigene System instrument units leased under operating lease arrangements is
recognized on an installment basis over the life of the lease while the cost of the leased
equipment is carried on the Company’s balance sheet and fully amortized over the life of lease
arrangements.
Stock-Based Compensation Expense
We have granted share-based compensation consisting of restricted stock and common stock
options issued to employees, consultants and founders. Compensation expense is recognized based on
the fair value of the stock-based awards granted utilizing various assumptions regarding the
underlying attributes of the options and our common stock. The estimated fair value of options
granted, net of forfeitures expected to occur during the vesting period, is determined using the
Black-Scholes option-pricing model and then amortized as compensation expense on a straight-line
basis over the vesting period of the options. All of the stock options granted prior to November
2007 have exercise prices at or above the estimated fair value of the common stock on the date of
grant, as determined by our board of directors prior to our initial public offering in November
2007, who used their knowledge of us and our affairs along with third-party valuation assessments,
to determine the fair value of our common stock. For option grants after our initial public
offering, we use the fair value of our common stock as determined by the closing price of our
common stock on NASDAQ on the date of grant. In addition to the grant date fair value of our
common stock, the Black-Scholes model requires inputs for risk-free interest rate, dividend yield,
volatility and expected lives of the options. Due to the Company’s limited period of trading
activity as a public company from 2007 through the third quarter of 2009, the expected volatility
of option grants prior to the fourth quarter of 2009 was based on historical data from various peer
public companies with similar product portfolios. The expected volatility for option awards granted
in the fourth quarter of 2009 was based on the Company’s actual historical volatility. The expected
life of options that vest ratably over four years of service is derived from the average of the
vesting period and the term of the option following the guidance in SEC Staff Accounting Bulletins
No. 107 and 110. The Company estimates the expected life of options with accelerated vesting terms
giving consideration to the dates that the Company expects to achieve key milestones under the
option agreements and the term of the option. The risk-free rate for periods within the contractual
life of the option is based on the U.S. Treasury yield curve in effect at the time of the grant.
Recent Accounting Pronouncements
Effective January 1, 2008, the Company adopted new accounting guidance on fair value
measurements. The new guidance clarifies the definition of fair value, prescribes methods for
measuring fair value, establishes a fair value hierarchy based on the inputs used to measure fair
value and expands disclosures about the use of fair value measurements. It was effective for
certain financial assets and liabilities beginning January 1, 2008. The Company adopted the
provisions of the new accounting guidance related to nonfinancial assets and nonfinancial
liabilities effective January 1, 2009, and such adoption had no material impact on the Company’s
financial statements.
In April 2008, the FASB issued new accounting guidance on the determination of the useful life
of intangible assets. The new guidance amends the factors that should be considered in developing
renewal or extension assumptions used to determine the useful life of a recognized intangible
asset. The Company adopted this new guidance on January 1, 2009 which resulted in no financial
impact.
In May 2009, the FASB issued new accounting and disclosure guidance for events that occur
subsequent to the balance sheet date but before the date that financial statements are issued or
are available to be issued. The Company’s management has evaluated its subsequent events for
disclosure in this annual report on Form 10-K through the date on which the financial statements
were issued.
In June 2009, the FASB issued authoritative guidance which replaced the previous hierarchy of
generally accepted accounting principles (“GAAP”) and establishes the FASB Codification as the
single source of authoritative U.S. GAAP recognized by the FASB to be applied by nongovernmental
entities. SEC rules and interpretive releases are also sources of authoritative GAAP for SEC
registrants. This guidance modifies the GAAP hierarchy to include only two levels of GAAP:
authoritative and nonauthoritative. This guidance was effective for the Company as of September 30,
2009. The adoption of this guidance did not impact the Company’s financial statements since the
FASB Codification is not intended to change or alter existing GAAP.
38
Table of Contents
In October 2009, the FASB issued authoritative guidance that amends existing guidance for
identifying separate deliverables in a revenue-generating transaction where multiple deliverables
exist, and provides guidance for allocating and recognizing revenue based
on those separate deliverables. The guidance is expected to result in more
multiple-deliverable arrangements being separable than under current guidance. This guidance is
effective for the Company beginning on January 1, 2011 and is required to be applied prospectively
to new or significantly modified revenue arrangements. The Company is currently assessing the
impact this guidance may have on its financial statements.
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk. |
Our exposure to market risk is currently confined to our cash and cash equivalents. We have
not used derivative financial instruments for speculation or trading purposes. The primary
objective of our investment activities is to preserve our capital for the purpose of funding
operations while at the same time maximizing the income we receive from our investments without
significantly increasing risk. To achieve these objectives, our investment policy allows us to
maintain a portfolio of cash equivalents and short-term investments through a variety of
securities, including commercial paper, money market funds and corporate debt securities. Our cash
and cash equivalents through December 31, 2009 included amounts in bank checking and liquid money
market accounts. As a result, we believe we have minimal interest rate risk; however, a one
percentage point decrease (or increase) in the average interest rate on our portfolio, if such a
decrease were possible, would have reduced (or increased) interest income for the year ended
December 31, 2009 by $655,930.
39
Table of Contents
| Item 8. | Financial Statements and Supplementary Data. |
The following financial statements and the related notes thereto, of Nanosphere, Inc. and the
Report of Independent Registered Public Accounting Firm, Deloitte & Touche LLP, are filed as a part
of this Form 10-K.
| Page | ||||
| 41 | ||||
| 43 | ||||
| 44 | ||||
| 45 | ||||
| 46 | ||||
| 47-58 | ||||
40
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of Nanosphere, Inc.
Northbrook, Illinois
Northbrook, Illinois
We have audited the accompanying balance sheets of Nanosphere, Inc. (the “Company”) as of December
31, 2009 and 2008, and the related statements of operations, stockholders’ equity (deficit), and
cash flows for each of the three years in the period ended December 31, 2009. These financial
statements are the responsibility of the Company’s management. Our responsibility is to express an
opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight
Board (United States). Those standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are free of material misstatement. An
audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the
financial statements. An audit also includes assessing the accounting principles used and
significant estimates made by management, as well as evaluating the overall financial statement
presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, such financial statements present fairly, in all material respects, the financial
position of the Company as of December 31, 2009 and 2008, and the results of its operations and its
cash flows for each of the three years in the period ended December 31, 2009, in conformity with
accounting principles generally accepted in the United States of America.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight
Board (United States), the Company’s internal control over financial reporting as of December 31,
2009, based on the criteria established in Internal Control—Integrated Framework issued by the
Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 11,
2010, expressed an unqualified opinion on the Company’s internal control over financial reporting.
/s/ DELOITTE & TOUCHE LLP
Chicago, Illinois
March 11, 2010
March 11, 2010
41
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of Nanosphere, Inc.
Northbrook, Illinois
Northbrook, Illinois
We have audited the internal control over financial reporting of Nanosphere, Inc. (the “Company”)
as of December 31, 2009, based on criteria established in Internal Control — Integrated Framework
issued by the Committee of Sponsoring Organizations of the Treadway Commission. The Company’s
management is responsible for maintaining effective internal control over financial reporting and
for its assessment of the effectiveness of internal control over financial reporting, included in
the accompanying Management’s Report on Internal Control over Financial Reporting. Our
responsibility is to express an opinion on the Company’s internal control over financial reporting
based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight
Board (United States). Those standards require that we plan and perform the audit to obtain
reasonable assurance about whether effective internal control over financial reporting was
maintained in all material respects. Our audit included obtaining an understanding of internal
control over financial reporting, assessing the risk that a material weakness exists, testing and
evaluating the design and operating effectiveness of internal control based on the assessed risk,
and performing such other procedures as we considered necessary in the circumstances.
We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed by, or under the
supervision of, the company’s principal executive and principal financial officers, or persons
performing similar functions, and effected by the company’s board of directors, management, and
other personnel to provide reasonable assurance regarding the reliability of financial reporting
and the preparation of financial statements for external purposes in accordance with generally
accepted accounting principles. A company’s internal control over financial reporting includes
those policies and procedures that (1) pertain to the maintenance of records that, in reasonable
detail, accurately and fairly reflect the transactions and dispositions of the assets of the
company; (2) provide reasonable assurance that transactions are recorded as necessary to permit
preparation of financial statements in accordance with generally accepted accounting principles,
and that receipts and expenditures of the company are being made only in accordance with
authorizations of management and directors of the company; and (3) provide reasonable assurance
regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the
company’s assets that could have a material effect on the financial statements.
Because of the inherent limitations of internal control over financial reporting, including the
possibility of collusion or improper management override of controls, material misstatements due to
error or fraud may not be prevented or detected on a timely basis. Also, projections of any
evaluation of the effectiveness of the internal control over financial reporting to future periods
are subject to the risk that the controls may become inadequate because of changes in conditions,
or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, the Company maintained, in all material respects, effective internal control over
financial reporting as of December 31, 2009, based on the criteria established in Internal Control
— Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway
Commission.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight
Board (United States), the financial statements as of and for the year ended December 31, 2009, of
the Company and our report dated March 11, 2010 expressed an unqualified opinion on those financial
statements.
/s/ DELOITTE & TOUCHE LLP
Chicago, Illinois
March 11, 2010
March 11, 2010
42
Table of Contents
Nanosphere, Inc.
Balance Sheets
| As of December 31, | ||||||||
| 2009 | 2008 | |||||||
CURRENT ASSETS: |
||||||||
Cash and cash equivalents |
$ | 76,689,279 | $ | 75,356,960 | ||||
Accounts receivable |
735,047 | 385,908 | ||||||
Inventories |
2,940,574 | 1,727,518 | ||||||
Other current assets |
374,657 | 559,528 | ||||||
Total current assets |
80,739,557 | 78,029,914 | ||||||
PROPERTY AND EQUIPMENT — Net |
6,144,555 | 7,294,224 | ||||||
INTANGIBLE ASSETS — Net of accumulated amortization |
1,709,842 | 1,443,582 | ||||||
OTHER ASSETS |
75,000 | 128,190 | ||||||
TOTAL |
$ | 88,668,954 | $ | 86,895,910 | ||||
CURRENT LIABILITIES: |
||||||||
Accounts payable |
$ | 2,509,405 | $ | 1,813,777 | ||||
Accrued compensation |
955,130 | 829,292 | ||||||
Other current liabilities |
2,303,631 | 1,541,385 | ||||||
Long-term debt — current portion |
3,818,790 | 4,818,211 | ||||||
Total current liabilities |
9,586,956 | 9,002,665 | ||||||
LONG-TERM LIABILITIES: |
||||||||
Long-term debt — noncurrent portion |
— | 3,352,137 | ||||||
Total liabilities |
9,586,956 | 12,354,802 | ||||||
COMMITMENTS AND CONTINGENCIES |
||||||||
STOCKHOLDERS’ EQUITY: |
||||||||
Common stock, $0.01 par value; 100,000,000 shares
authorized; 28,422,461 shares and 22,228,696
shares issued and outstanding as of December 31,
2009 and 2008, respectively |
284,225 | 222,287 | ||||||
Preferred stock, $0.01 par value; 10,000,000
shares authorized; no shares issued |
— | — | ||||||
Additional paid-in capital |
312,660,430 | 274,232,825 | ||||||
Warrants to acquire common stock |
5,423,771 | 5,423,771 | ||||||
Accumulated deficit |
(239,286,428 | ) | (205,337,775 | ) | ||||
Total stockholders’ equity |
79,081,998 | 74,541,108 | ||||||
TOTAL |
$ | 88,668,954 | $ | 86,895,910 | ||||
See notes to financial statements.
43
Table of Contents
Nanosphere, Inc.
Statements of Operations
| Years Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
REVENUE: |
||||||||||||
Grant and contract revenue |
$ | 1,089,890 | $ | 346,070 | $ | 1,056,874 | ||||||
Product sales |
1,124,213 | 1,020,662 | 110,490 | |||||||||
Total revenue |
2,214,103 | 1,366,732 | 1,167,364 | |||||||||
COSTS AND EXPENSES: |
||||||||||||
Cost of sales |
2,175,281 | 1,464,066 | 86,349 | |||||||||
Research and development |
18,607,921 | 23,675,138 | 21,445,994 | |||||||||
Sales, general, and administrative |
14,471,973 | 13,615,322 | 13,443,304 | |||||||||
Total costs and expenses |
35,255,175 | 38,754,526 | 34,975,647 | |||||||||
Loss from operations |
(33,041,072 | ) | (37,387,794 | ) | (33,808,283 | ) | ||||||
OTHER INCOME (EXPENSE): |
||||||||||||
Change in fair value of convertible derivative liability |
— | — | (14,860,901 | ) | ||||||||
Change in fair value of preferred stock warrants |
— | — | (4,414,207 | ) | ||||||||
Foreign exchange loss |
(4,207 | ) | (23,691 | ) | (52,362 | ) | ||||||
Interest expense |
(1,257,445 | ) | (2,080,629 | ) | (1,977,619 | ) | ||||||
Interest income |
354,071 | 2,450,118 | 1,914,188 | |||||||||
Total other income (expense) |
(907,581 | ) | 345,798 | (19,390,901 | ) | |||||||
NET LOSS |
(33,948,653 | ) | (37,041,996 | ) | (53,199,184 | ) | ||||||
Accumulated convertible preferred stock dividends |
— | — | (5,476,287 | ) | ||||||||
Convertible preferred stock redemption value adjustment |
— | — | (608,940 | ) | ||||||||
NET LOSS ATTRIBUTABLE TO COMMON STOCK |
$ | (33,948,653 | ) | $ | (37,041,996 | ) | $ | (59,284,411 | ) | |||
Net loss per common share — basic and diluted |
$ | (1.46 | ) | $ | (1.67 | ) | $ | (14.18 | ) | |||
Weighted average number of common shares outstanding —
basic and diluted |
23,301,764 | 22,213,164 | 4,180,979 | |||||||||
See notes to financial statements.
44
Table of Contents
Nanosphere, Inc.
Statements of Stockholders’ Equity (Deficit)
| Note | ||||||||||||||||||||||||||||
| Warrants | Receivable | |||||||||||||||||||||||||||
| Additional | To Acquire | From Chief | ||||||||||||||||||||||||||
| Common Stock | Paid-In | Common | Executive | Accumulated | ||||||||||||||||||||||||
| Shares | Par Value | Capital | Stock | Officer | Deficit | Total | ||||||||||||||||||||||
BALANCE — January 1, 2007 |
932,646 | $ | 92,580 | $ | 2,051,126 | $ | — | $ | (1,440,000 | ) | $ | (109,011,368 | ) | $ | (108,307,662 | ) | ||||||||||||
Share-based compensation related to stock options |
1,669,006 | 1,669,006 | ||||||||||||||||||||||||||
Exercise of stock options on common stock |
16,150 | 162 | 119,863 | 120,025 | ||||||||||||||||||||||||
Issuance of common stock from initial public
offering, net of offering expenses |
8,050,000 | 80,500 | 102,092,417 | 102,172,917 | ||||||||||||||||||||||||
Conversion of preferred stock to common stock |
13,048,119 | 130,481 | 116,151,384 | 116,281,865 | ||||||||||||||||||||||||
Conversion of convertible derivative liability
to additional paid-in capital |
47,554,882 | 47,554,882 | ||||||||||||||||||||||||||
Conversion of warrant liability to equity |
5,424,551 | 5,424,551 | ||||||||||||||||||||||||||
Exercise of warrants |
145,090 | 1,451 | 2,036,358 | 2,037,809 | ||||||||||||||||||||||||
Interest received on note receivable from chief
executive officer |
90,740 | 90,740 | ||||||||||||||||||||||||||
Proceeds from note receivable from chief
executive officer |
1,440,000 | 1,440,000 | ||||||||||||||||||||||||||
6% dividends, earned on Series C-2 and Series D
Convertible Preferred Stock |
(5,476,287 | ) | (5,476,287 | ) | ||||||||||||||||||||||||
Convertible preferred stock redemption value
adjustment |
(608,940 | ) | (608,940 | ) | ||||||||||||||||||||||||
Net loss |
(53,199,184 | ) | (53,199,184 | ) | ||||||||||||||||||||||||
BALANCE — December 31, 2007 |
22,192,005 | 305,174 | 271,765,776 | 5,424,551 | — | (168,295,779 | ) | 109,199,722 | ||||||||||||||||||||
Share-based compensation related to stock options |
2,217,858 | 2,217,858 | ||||||||||||||||||||||||||
Exercise of stock options on common stock |
36,491 | 365 | 163,845 | 164,210 | ||||||||||||||||||||||||
Exercise of warrants |
200 | 2 | 2,966 | (780 | ) | 2,188 | ||||||||||||||||||||||
Other |
(83,254 | ) | 82,380 | (874 | ) | |||||||||||||||||||||||
Net loss |
(37,041,996 | ) | (37,041,996 | ) | ||||||||||||||||||||||||
BALANCE — December 31, 2008 |
22,228,696 | 222,287 | 274,232,825 | 5,423,771 | — | (205,337,775 | ) | 74,541,108 | ||||||||||||||||||||
Share-based compensation |
672,500 | 6,725 | 2,527,352 | 2,534,077 | ||||||||||||||||||||||||
Exercise of stock options on common stock |
116,265 | 1,163 | 520,969 | 522,132 | ||||||||||||||||||||||||
Issuance of common stock from public offering,
net of offering expenses |
5,405,000 | 54,050 | 35,379,284 | 35,433,334 | ||||||||||||||||||||||||
Net loss |
(33,948,653 | ) | (33,948,653 | ) | ||||||||||||||||||||||||
BALANCE — December 31, 2009 |
28,422,461 | $ | 284,225 | $ | 312,660,430 | $ | 5,423,771 | $ | — | $ | (239,286,428 | ) | $ | 79,081,998 | ||||||||||||||
See notes to financial statements.
45
Table of Contents
Nanosphere, Inc.
Statements of Cash Flows
| Years Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
CASH FLOWS FROM OPERATING ACTIVITIES: |
||||||||||||
Net loss |
$ | (33,948,653 | ) | $ | (37,041,996 | ) | $ | (53,199,184 | ) | |||
Adjustments to reconcile net loss to net cash used in operating
activities: |
||||||||||||
Depreciation and amortization |
3,067,063 | 3,170,265 | 1,785,972 | |||||||||
Amortization of financing costs and accretion of debt discount |
499,386 | 762,277 | 671,480 | |||||||||
Loss from write-off of intangible assets |
— | 245,134 | — | |||||||||
Loss from disposal of fixed assets |
2,758 | 326,291 | — | |||||||||
Share-based compensation |
2,534,077 | 2,217,858 | 1,669,006 | |||||||||
Change in fair value of preferred stock warrants |
— | — | 4,414,207 | |||||||||
Change in fair value of convertible derivative liability |
— | — | 14,860,901 | |||||||||
Changes in operating assets and liabilities: |
||||||||||||
Accounts receivable |
(349,139 | ) | (285,109 | ) | (55,983 | ) | ||||||
Inventories |
(1,334,763 | ) | (1,078,965 | ) | (1,018,155 | ) | ||||||
Other current assets |
205,328 | 110,874 | (207,544 | ) | ||||||||
Other assets |
— | 2,630 | 10,830 | |||||||||
Accounts payable |
518,640 | (208,760 | ) | 712,225 | ||||||||
Accrued and other current liabilities |
856,137 | (195,047 | ) | 1,038,759 | ||||||||
Net cash used in operating activities |
(27,949,166 | ) | (31,974,548 | ) | (29,317,486 | ) | ||||||
CASH FLOWS FROM INVESTING ACTIVITIES: |
||||||||||||
Purchases of property and equipment |
(1,326,542 | ) | (2,332,267 | ) | (3,157,478 | ) | ||||||
Investments in intangible assets |
(507,794 | ) | (400,732 | ) | (399,911 | ) | ||||||
Net cash used in investing activities |
(1,834,336 | ) | (2,732,999 | ) | (3,557,389 | ) | ||||||
CASH FLOWS FROM FINANCING ACTIVITIES: |
||||||||||||
Proceeds from issuance of debt |
— | — | 12,500,000 | |||||||||
Repayment of long-term debt |
(4,818,211 | ) | (3,597,823 | ) | (166,673 | ) | ||||||
Payments on capital lease obligation |
(21,434 | ) | (37,036 | ) | (32,776 | ) | ||||||
Deferred financing fees |
— | — | (114,565 | ) | ||||||||
Proceeds from principal and interest on CEO note receivable |
— | — | 1,530,740 | |||||||||
Proceeds from the issuance of common stock, net of offering
expenses |
35,433,334 | (778,731 | ) | 102,951,648 | ||||||||
Proceeds from warrant redemptions |
— | 2,188 | 1,286,620 | |||||||||
Proceeds from stock option exercises |
522,132 | 164,210 | 120,025 | |||||||||
Other |
— | (874 | ) | — | ||||||||
Net cash (used in) provided by financing activities |
31,115,821 | (4,248,066 | ) | 118,075,019 | ||||||||
NET INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS |
1,332,319 | (38,955,613 | ) | 85,200,144 | ||||||||
CASH AND CASH EQUIVALENTS — Beginning of year |
75,356,960 | 114,312,573 | 29,112,429 | |||||||||
CASH AND CASH EQUIVALENTS — End of year |
$ | 76,689,279 | $ | 75,356,960 | $ | 114,312,573 | ||||||
NONCASH INVESTING AND FINANCING ACTIVITIES: |
||||||||||||
Capital expenditures included in accounts payable |
$ | 205,677 | $ | — | $ | 201,441 | ||||||
License costs capitalized and included in accounts payable |
— | — | 26,233 | |||||||||
License costs capitalized and included in other current liabilities |
278,381 | 225,000 | 347,500 | |||||||||
6% dividends earned on Series C-2 and Series D Convertible
Preferred Stock |
— | — | 5,476,287 | |||||||||
Conversion of Preferred Stock, including accumulated dividends on
Series C-2 and Series D Convertible Preferred Stock into common
stock |
— | — | 116,281,865 | |||||||||
Conversion of convertible derivative liability to additional
paid-in capital |
— | — | 47,554,882 | |||||||||
Conversion of preferred stock warrant liability to warrants to
acquire common stock |
— | — | 5,424,551 | |||||||||
Equity offering transaction costs included in accrued financing
costs |
— | — | 778,731 | |||||||||
Reclassification of inventory to equipment at customers |
251,727 | 1,254,451 | — | |||||||||
See notes to financial statements.
46
Table of Contents
Nanosphere, Inc.
Notes to Financial Statements
As of December 31, 2009 and 2008, and
For the years ended December 31, 2009, 2008 and 2007
As of December 31, 2009 and 2008, and
For the years ended December 31, 2009, 2008 and 2007
1. Description of Business
Nanosphere, Inc. (the “Company”) develops, manufactures and markets an advanced molecular
diagnostics platform, the Verigene System, that enables simple, low cost, and highly sensitive
genomic and protein testing on a single platform.
Basis of Presentation — The accompanying financial statements have been prepared on a
going-concern basis, which contemplates the realization of assets and the satisfaction of
liabilities in the normal course of business. The Company has incurred net losses attributable to
common stock of $239.3 million since inception, and has funded those losses primarily through the
sale and issuance of equity securities and secondarily through the issuance of debt. While the Company is no longer in the development stage and the focus of the
Company’s business activities has turned towards commercialization of its products, because of the
numerous risks and uncertainties associated with its product development and commercialization
efforts, the Company is unable to predict when it will become profitable, and the Company may
never become profitable. Capital outlays and operating expenditures may increase over the next
few years as the Company expands its infrastructure, commercialization, manufacturing, and research
and development activities. The Company operates in a market that makes its prospects difficult
to evaluate, and the Company may need additional financing in the future to execute on its current
or future business strategies.
2. Summary of Significant Accounting Policies
Cash and Cash Equivalents — The Company considers all highly liquid investments with a
maturity of three months or less, at date of purchase, to be cash equivalents. The majority of
these funds are held in interest-bearing money market and bank checking accounts. Interest income
is recorded on the accrual basis as earned.
Receivables — Accounts receivable consists of amounts due to the Company for sales of the
Verigene system as well as amounts due under various contracts and government grants. An allowance
for doubtful accounts is not recorded because the Company has no history of uncollectible
receivables and there are no specifically identified uncollectible accounts.
Inventories — Inventories are carried at the lower of cost or market, using the first-in,
first-out method. Certain finished goods inventory is ultimately leased rather than sold, and upon
the lease date is transferred to Property and equipment and subsequently depreciated to Cost of
sales over the period indicated below.
Property and Equipment — Property and equipment are recorded at cost and depreciated using the
straight-line method over the assets’ estimated useful lives, which are:
Equipment with customers |
3-5 years | |||
Computers and office equipment |
3 years | |||
Engineering and laboratory equipment, including tooling |
3-5 years | |||
Furniture and fixtures |
7 years | |||
Manufacturing equipment |
5-7 years | |||
The economic life of the Company’s equipment with customers is based on the original term of
the lease, which is typically three years. The Company believes that this is representative of the
period during which the instrument is expected to be economically usable.
Assets classified as leasehold improvements are amortized over the shorter of their estimated
useful lives or the lease term using the straight-line method. Maintenance and repair costs are
expensed as incurred.
Intangible Assets — Intangible assets are stated at cost less accumulated amortization and
consist of purchased intellectual property. Purchased intellectual property represents licenses and
is associated with patents owned by third-parties for technologies which are embedded in the
Company’s diagnostic instruments and diagnostic test products that the Company licensed in
anticipation of sales of such products. Amortization of purchased intellectual property begins upon
the Company obtaining FDA clearance to sell
products containing the licensed technology and is calculated using the straight-line method
over the remaining expected lives of the licensed technology, which range from 0.75 to 14.75 years.
Such amortization of upfront license fees is classified in Cost of sales on the statement of
operations.
47
Table of Contents
Nanosphere, Inc.
Notes to Financial Statements — (Continued)
Deferred Financing Costs — Deferred financing costs of $114,565 incurred in connection with
the Company’s issuance of debt are amortized over the life of the debt using the effective interest
rate method with amortization of such costs being charged to interest expense. Such costs, net of
accumulated amortization, are classified in Other Assets on the balance sheet.
Impairment of Long-Lived Assets — The Company assesses the recoverability of long-lived
assets, including intangible assets, by periodically evaluating the carrying value of such assets
whenever events or changes in circumstances indicate that the carrying amount of such assets may
not be recoverable. If impairment is indicated, the Company will value the asset at its estimated
fair value.
Use of Estimates — The preparation of financial statements in conformity with accounting
principles generally accepted in the United States of America requires management to make estimates
and assumptions that affect the amounts reported in the financial statements and the notes thereto.
The Company’s significant estimates included in the preparation of the financial statements are
related to inventories, property and equipment, intangible assets and share-based compensation.
Actual results could differ from those estimates.
Revenue Recognition — The Company recognizes revenue from product sales and contract
arrangements. The Company recognizes revenue from product sales when there is persuasive evidence
that an arrangement exists, delivery has occurred, the price is fixed or determinable and
collectability is reasonably assured. Verigene System instrument units are sold outright to
customers or leased to customers pursuant to operating leases. The Company recognizes revenue from
sales of the Verigene System, including cartridges and related products, when the risks and rewards
of ownership are transferred to the customer. Revenue for Verigene System instrument units sold
under operating lease arrangements is recognized on an installment basis over the life of the lease
while the cost of the leased equipment is carried on the Company’s balance sheet in Property and
equipment and depreciated over its estimated useful life to Cost of sales.
Shipping and handling costs are expensed as incurred and included in Cost of sales. In those
cases where the Company bills shipping and handling costs to customers, the amounts billed are
classified as revenue.
Grant and government sponsored research revenue and contract revenue related to research and
development services are recognized as the related services are performed based on the performance
requirements of the relevant contract. Under such agreements, the Company is required to perform
specific research and development activities and is compensated either based on the costs or costs
plus a mark-up associated with each specific contract over the term of the agreement or when
certain milestones are achieved.
Research and Development Costs — Research and development costs are expensed as incurred.
Income Taxes — The Company accounts for income taxes, including uncertain tax positions, under
the provisions of Financial Accounting Standards Board (“FASB”) Accounting Standards Codification
(“ASC”) Topic 740 “Accounting for Income Taxes”. This Topic requires recognition of deferred tax
assets and liabilities for the expected future tax consequences of events that have been included
in the financial statements or tax returns. Under this method, deferred tax assets and liabilities
are determined based on the differences between the financial reporting and tax bases of assets and
liabilities using enacted tax rates in effect for the year in which the differences are expected to
reverse. An allowance is provided to reduce net deferred tax assets to the amount management
believes will, more likely than not, be recovered.
Share-Based Compensation — The Company recognizes share-based compensation expense related to
restricted stock and common stock options issued to employees, consultants and directors. ASC Topic
718 “Stock Compensation” provides for recognition of compensation expense based on the fair value
of the stock-based compensation utilizing various assumptions regarding the underlying attributes
of the options and stock. The estimated fair value of options granted, net of forfeitures expected
to occur during the vesting period, is amortized as compensation expense on a straight-line basis
over the service period of the options.
Preferred Stock — Prior to the Company’s initial public offering in November 2007, the Company
had outstanding shares of preferred stock. The Company recognized changes in the redemption value
of its preferred stock immediately as they occurred and adjusted the carrying value of the
preferred stock to be equal to the redemption value of the preferred stock at the end of each
reporting period. Upon the closing of the initial public offering on November 6, 2007, all
preferred stock was converted to common stock.
48
Table of Contents
Nanosphere, Inc.
Notes to Financial Statements — (Continued)
Fair Value of Financial Instruments — The carrying amount of the Company’s financial
instruments, including cash and cash equivalents, accounts receivable and accounts payable
approximate their fair values. See Note 10 for information on the fair value of the Company’s
long-term debt.
Effective January 1, 2008, the Company adopted ASC Topic 820 “Fair Value Measurements and
Disclosures: Improving Disclosures about Fair Value Measurements,” which clarifies the definition
of fair value, prescribes methods for measuring fair value, establishes a fair value hierarchy
based on the inputs used to measure fair value and expands disclosures about the use of fair value
measurements. ASC Topic 820 and its applicable subtopics were effective for certain financial
assets and liabilities beginning January 1, 2008. The Company adopted the provisions of ASC Topic
820 related to nonfinancial assets and nonfinancial liabilities effective January 1, 2009, and such
adoption had no material impact on the Company’s financial statements.
New Accounting Standards — In May 2009, the FASB issued new accounting and disclosure guidance
for events that occur subsequent to the balance sheet date but before the date that financial
statements are issued or are available to be issued. The Company’s management has evaluated its
subsequent events for disclosure in this Form 10-K through the date on which the financial
statements were issued.
In October 2009, the FASB issued authoritative guidance that amends existing guidance for
identifying separate deliverables in a revenue-generating transaction where multiple deliverables
exist, and provides guidance for allocating and recognizing revenue based on those separate
deliverables. The guidance is expected to result in more multiple-deliverable arrangements being
separable than under current guidance. This guidance is effective for the Company beginning on
January 1, 2011 and is required to be applied prospectively to new or significantly modified
revenue arrangements. The Company is currently assessing the impact this guidance may have on its
financial statements.
Net Loss Per Common Share — Basic and diluted net loss per common share have been calculated
in accordance with ASC Topic 260, “Earnings Per Share”, for the years ended December 31, 2009, 2008
and 2007. As the Company had a net loss in each of the periods presented, basic and diluted net
loss per common share are the same.
The computation of basic net loss per common share for the year ended December 31, 2009
excluded 672,500 shares of restricted stock issued during 2009 (see Note 5). While these
restricted shares of stock are included in outstanding shares on the balance sheet at December 31,
2009, these restricted shares are excluded from basic net loss per common share in accordance with
ASC Topic 260 due to the forfeiture provisions associated with these shares.
The computations of diluted net loss per common share for the years ended December 31, 2009,
2008 and 2007 did not include the outstanding shares of restricted stock as well as the effects of
the following options to acquire common stock and common stock warrants as the inclusion of these
securities would have been antidilutive:
| Year ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
Restricted stock |
672,500 | — | — | |||||||||
Stock options |
4,338,695 | 3,362,721 | 3,141,530 | |||||||||
Common stock warrants |
1,300,119 | 1,300,119 | 1,300,319 | |||||||||
| 6,311,314 | 4,662,840 | 4,441,849 | ||||||||||
3. Intangible Assets
Intangible assets, consisting of purchased intellectual property, as of December 31, 2009 and
2008 comprise the following:
| December 31, 2009 | December 31, 2008 | |||||||||||||||||||||||
| Accumulated | Accumulated | |||||||||||||||||||||||
| Cost | Amortization | Net | Cost | Amortization | Net | |||||||||||||||||||
Intellectual property — licenses |
$ | 2,438,218 | $ | (728,376 | ) | $ | 1,709,842 | $ | 1,877,043 | $ | (433,461 | ) | $ | 1,443,582 | ||||||||||
49
Table of Contents
Nanosphere, Inc.
Notes to Financial Statements — (Continued)
Amortization expense for intangible assets amounted to $294,915, $343,217, and $90,244 for the
years ended December 31, 2009, 2008, and 2007, respectively. Estimated future amortization expense
is as follows:
| Years Ending December 31 | ||||
2010 |
$ | 447,744 | ||
2011 |
150,719 | |||
2012 |
139,810 | |||
2013 |
136,174 | |||
2014 |
136,174 | |||
Thereafter |
447,221 |
Licenses are amortized from the date of the U.S. Food and Drug Administration (the “FDA”)
clearance of products associated with the licensed technology and such amortization continues over
the remaining life of the license. The future amortization expense reflected above is based on
licenses related to products cleared by the FDA as of December 31, 2009. The amortization period
related to $0.3 million of licenses is not known as the diagnostic test products associated with
the licensed technology have not been cleared by the FDA and, accordingly, amortization has not
begun and no expense associated with the licenses is included in the table above. During the year
ended December 31, 2008, the Company wrote off capitalized license fees of $245,134 associated with
licenses which the Company did not plan to utilize in the Verigene System. There were no license
costs written off in the years ended December 31, 2009 or 2007.
4. Related Party Transactions
Robert Letsinger and Chad Mirkin, co-founders of the Company, provide contracted research and
development services to the Company, which are reimbursed based upon negotiated contract rates.
Such contracts automatically renew on an annual basis. The Company incurred expenses of $150,000
for these services in each of the years ended December 31, 2009, 2008 and 2007.
In March 2006, the Company issued to William Moffitt, the Company’s chief executive officer
and a director, 320,000 shares of restricted common stock at a price of $4.50 per share, for an
aggregate price of $1,440,000. The restrictions on the common stock lapsed in July 2007. In
connection with this sale of common stock, the Company received a full recourse, long-term
promissory note from William Moffitt for a total of $1,440,000, which note was secured by the
shares of common stock purchased. Interest on the promissory note accrued and was paid annually in
cash at an interest rate of 4.51%. Interest income on this note was $39,326 for the year ended
December 31, 2007. In 2007, $90,740 of interest was paid to the Company on the note and was
recorded as additional paid in capital. The note receivable due from the Company’s chief executive
officer was repaid in August 2007.
In August 2007, in accordance with terms of the Amended Bonus Agreement between the Company
and the chief executive officer, the Company paid a $2.3 million bonus payment to the chief
executive officer. $1.9 million of the expense for this bonus was recorded in 2007.
Brookside Capital Partners Fund, L.P., a 5% stockholder of the Company, purchased
892,857 shares of the Company’s common stock at $13.53 per share in the Company’s initial public
offering in November 2007.
AOQ Trust, a 5% stockholder of the Company, and LFT Partnership, an affiliate of AOQ Trust,
purchased 337,849 shares and 162,151 shares of the Company’s common stock, respectively, at $7.00
per share in the Company’s October 2009 underwritten public offering. Mark Slezak, a director of
the Company, is a trustee of AOQ Trust and the investment manager of LFT Partnership.
5. Equity Incentive Plans
The Company’s 2000 Equity Incentive Plan, as amended (the “2000 Plan”), permitted the grant of
options to employees, founders, and consultants for up to 1,600,000 shares of common stock. Option
awards are generally granted with an exercise price equal to the fair value of the Company’s common
stock at the date of grant; those option awards have various vesting structures and have 10 year
contractual terms. In connection with the approval of the 2007 Plan as defined below, the
Company terminated the 2000 Plan and therefore, the Company may not make any further awards of
options, share appreciations rights or restricted shares under the 2000 Plan.
50
Table of Contents
Nanosphere, Inc.
Notes to Financial Statements — (Continued)
In March 2007, the Company’s board of directors adopted and its shareholders approved the
Nanosphere 2007 Long-Term Incentive Plan (the “2007 Plan”). The 2007 Plan authorizes the grant of
stock options, share appreciation rights, restricted shares, restricted share units, unrestricted
shares, incentive stock options, deferred share units and performance awards. The total awards
originally authorized under the 2007 Plan was 4,106,009 shares, plus up to an additional 773,591
shares of common stock that will become available in the event that awards made under the 2000 Plan
expire, are forfeited or cancelled, plus an annual increase in the number of shares pursuant to the
evergreen provision equal to the least of: 900,000 shares of common stock; 4.0% of the Company’s
outstanding shares of common stock as of fiscal year end; and an amount determined by the board of
directors. Pursuant to the evergreen provision, an additional 900,000 shares of common stock
were authorized for issuance under the 2007 Plan as of January 1, 2010.
Certain options vest ratably over four years of service, while other options vest after seven
years of service but provide for accelerated vesting contingent upon the achievement of various
company-wide performance goals, such as decreasing time to market for new products and entering
into corporate collaborations (as defined in the option grant agreements). For these “accelerated
vesting” options, 20-25% of the granted option shares will vest upon the achievement of each of
four or five milestones as defined in the option grant agreements, with any remaining unvested
options vesting on the seven year anniversary of the option grant dates. Approximately 46% of the
options granted and outstanding contain “accelerated vesting” provisions.
The fair values of the Company’s option awards were estimated at the dates of grant using the
Black-Scholes option pricing model with the following assumptions:
| 2009 | 2008 | 2007 | ||||||||||
Expected dividend yield |
0 | % | 0 | % | 0 | % | ||||||
Expected volatility |
97 | % | 68 | % | 77 | % | ||||||
Risk free interest rate |
2.42 | % | 3.02 | % | 4.65 | % | ||||||
Weighted-average expected option life |
6.1 years | 6.4 years | 7.0 years | |||||||||
Estimated weighted-average fair value on the
date of grant based on the above assumptions |
$ | 4.59 | $ | 6.97 | $ | 3.38 | ||||||
Estimated forfeiture rate for unvested options |
4.4 | % | 4.6 | % | 4.6 | % | ||||||
Due to the Company’s limited period of trading activity as a public company from 2007 through
the third quarter of 2009, the expected volatility was based on historical data from various peer
public companies with similar product portfolios. The expected volatility for option awards granted
in the fourth quarter of 2009 was based on the Company’s actual historical volatility. The
risk-free rate is based on the U.S. Treasury yield curve in effect at the time of the grants for
periods consistent with the expected life of the option. The expected life of options that vest
ratably over four years of service is derived from the average of the vesting period and the term
of the option as defined in the Plans, following the guidance in Securities and Exchange Commission
(“SEC”) Staff Accounting Bulletin Nos. 107 and 110. The Company estimates the expected life of
options with accelerated vesting terms giving consideration to the dates that the Company expects
to achieve key milestones under the option agreements and the term of the option. Total
compensation cost associated with the option awards was $2,381,251, $2,217,858, and $1,669,006 in
2009, 2008, and 2007, respectively.
As of December 31, 2009, the total compensation cost not yet recognized related to the
nonvested awards is approximately $9.8 million, which amount is expected to be recognized over the
next four years, which is a weighted average term. Certain milestone events are deemed probable of
achievement prior to their seven year vesting term, and the acceleration of vesting resulting from
the achievement of such milestone events has been factored into the weighted average vesting term.
While the Company does not have a formally established policy, as a practice the Company has
delivered newly issued shares of its common stock upon the exercise of stock options.
51
Table of Contents
Nanosphere, Inc.
Notes to Financial Statements — (Continued)
A summary of option activity under the Plan as of December 31, 2009, and for the year then
ended is presented below:
| Weighted | ||||||||||||||||
| Weighted | Average | |||||||||||||||
| Average | Remaining | Aggregate | ||||||||||||||
| Number of | Exercise | Contractual | Intrinsic | |||||||||||||
| Options | Shares | Price | Term | Value of Options | ||||||||||||
Outstanding — January 1, 2009 |
3,362,721 | $ | 5.68 | |||||||||||||
Granted |
1,398,399 | $ | 5.92 | |||||||||||||
Exercised |
(116,265 | ) | $ | 4.49 | ||||||||||||
Expired |
(6,750 | ) | $ | 6.43 | ||||||||||||
Forfeited |
(299,410 | ) | $ | 6.22 | ||||||||||||
Outstanding — December 31, 2009 |
4,338,695 | $ | 5.75 | 7.86 | $ | 5,650,628 | ||||||||||
Exercisable — December 31, 2009 |
1,499,585 | $ | 5.82 | 6.69 | $ | 2,466,252 | ||||||||||
Vested and Expected to Vest — December 31, 2009 |
4,208,096 | $ | 5.75 | 7.84 | $ | 5,504,147 | ||||||||||
The intrinsic value of options exercised in 2009, 2008 and 2007 was $234,305, $205,385 and
$110,687, respectively.
Included in the number of options outstanding at December 31, 2009, are 1,982,056 options with
a weighted average exercise price of $5.30 per share and accelerated vesting provisions based on
the criteria mentioned above. During 2009, two of the five milestones as defined in the 2000 Plan
were achieved, and one of the four milestones as defined in the 2007 Plan was achieved. As a
result, 40% of the outstanding options under the 2000 Plan with accelerated vesting provisions were
vested as of December 31, 2009 and 25% of the outstanding options under the 2007 Plan with
accelerated vesting provisions were vested as of December 31, 2009. The total fair value of shares
vested during 2009, 2008, and 2007 was $3,068,055, $1,255,800, and $548,226, respectively.
On November 25, 2009, the Company granted 672,500 shares of restricted stock under the 2007
Plan, of which 50% vest in two, equal installments commencing on the first anniversary of the grant
date and are subject to forfeiture until vested, and 50% vest in four, equal annual installments
commencing on the first anniversary of the grant date and are subject to forfeiture until vested.
No shares were forfeited as of December 31, 2009. The weighted average grant-date fair value was
$6.06 per share. In 2009, the Company recognized $152,826 in compensation expense associated with
the restricted stock. As of December 31, 2009, the total compensation cost not yet recognized
related to the nonvested restricted stock awards is approximately $3.9 million, which amount is
expected to be recognized over the next two years, which is a weighted average term.
6. Income Taxes
Deferred tax assets consist primarily of net operating loss (“NOL”) carryforwards related to
U.S. federal and state taxes and research and development tax credits. Realization of future tax
benefits related to deferred tax assets is dependent on many factors, including the Company’s
ability to generate future taxable income. Due to the Company’s history of operating losses, the
Company has recorded a full valuation allowance against these assets.
NOL carryforwards of approximately $157 million for income tax purposes are available to
offset future taxable income. If not used, these carryforwards will expire in varying amounts from
2020 through 2029. The Company also has federal research and development tax credit carryforwards
of $8 million which will expire from 2020 through 2029. Section 382 of the Internal Revenue Code
subjects the utilization of net operating loss and credit carryforwards to an annual limitation
that is applicable if the Company experiences an ownership change. The Company believes its public
offerings and/or prior equity investments may have triggered an ownership change as defined by the
Internal Revenue Code. However, the Company has yet to perform the computations under Section 382
which would determine the amount of annual limitation on its utilization of its net operating loss
and tax credit carryforwards. The annual limitation may result in the expiration of the Company’s
net operating loss and tax credit carryforwards before they can be used.
52
Table of Contents
Nanosphere, Inc.
Notes to Financial Statements — (Continued)
The following is a summary of the components of the Company’s deferred tax assets and
liabilities as of December 31, 2009 and 2008:
| 2009 | 2008 | |||||||
Deferred tax assets: |
||||||||
Net operating losses |
$ | 60,851,659 | $ | 49,363,621 | ||||
Research and development credits |
8,130,439 | 6,974,400 | ||||||
Stock option compensation |
777,116 | 554,748 | ||||||
Amortization of intangible assets |
1,202,101 | 1,233,349 | ||||||
Other |
566,289 | 308,095 | ||||||
| 71,527,604 | 58,434,213 | |||||||
Valuation allowance |
(70,976,214 | ) | (57,995,579 | ) | ||||
Net deferred tax assets after valuation allowance |
551,390 | 438,634 | ||||||
Deferred tax liabilities: |
||||||||
Depreciation on property and equipment |
(551,390 | ) | (438,634 | ) | ||||
Deferred tax assets — net |
$ | — | $ | — | ||||
The reconciliation of the federal statutory rate to the Company’s effective tax rate of zero
percent for the years ended December 31, 2009, 2008 and 2007 is as follows:
| Years Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
Tax provision at the statutory federal rate |
34.0 | % | 34.0 | % | 34.0 | % | ||||||
State income taxes, net of federal income tax benefit |
4.8 | % | 4.8 | % | 4.8 | % | ||||||
Fair value adjustments to convertible derivative liability and preferred stock warrants |
— | — | (14.1) | % | ||||||||
Other |
(5.5 | )% | (5.8 | )% | 0.5 | % | ||||||
Valuation allowance |
(33.3 | )% | (33.0 | )% | (25.2 | )% | ||||||
| 0.0 | % | 0.0 | % | 0.0 | % | |||||||
As of December 31, 2009 and 2008, the Company had no liability recorded for unrecognized tax
benefits. The Company classifies penalties and interest expense related to income tax liabilities
as an income tax expense. There are no interest and penalties recognized in the statements of
operations for the years ended December 31, 2009, 2008 and 2007 or accrued on the balance sheets as
of December 31, 2009 and 2008.
The Company files tax returns in the U.S. and various states. All tax years since 1999 remain
open to examination by the major taxing jurisdictions to which the Company is subject. The Company
has not made any cash payments for income taxes since its inception.
7. License Agreements
In 2006, the Company entered into a license agreement with Northwestern University
(“Northwestern”), which provides the Company with an exclusive license to certain patents and
patent applications owned by Northwestern that are related to (1) nanotechnology, which technology
involves a particle where no single dimension is greater than 100 nanometers, or Nanotechnology,
and (2) biobarcode technology, which is analysis where oligonucleotides act as surrogate targets or
reporter molecules, or Biobarcode Technology. The license is limited to the “Biodiagnostics Field”
defined as qualitative or quantitative in vitro analysis, testing, measurement, or detection of
various biodiagnostics field subjects and target combinations.
The license agreement includes licenses to patents and patent applications based on existing
inventions and future inventions developed in the laboratory of Dr. Mirkin or Dr. Letsinger, by or
under their direct supervision, and conceived prior to January 1, 2013 that are Nanotechnology or
Biobarcode Technology referred to herein as Licensed Patents. The Company has an obligation to use
commercially reasonable efforts to bring the subject inventions of the Licensed Patents to market.
If the parties disagree as to whether they are meeting this diligence requirement, an arbitrator
may require the Company to comply with a timeline for cure or convert
the exclusive license to a
non-exclusive license; Northwestern does not have the right to revoke any license to the Licensed
Patents already granted to the Company.
53
Table of Contents
Nanosphere, Inc.
Notes to Financial Statements — (Continued)
The Company also has the first right to negotiate an exclusive license to inventions developed
in the laboratory of Dr. Mirkin or Dr. Letsinger, by or under their direct supervision, and
(1) conceived after January 1, 2013 that are Nanotechnology or Biobarcode Technology and (2) that
are not Nanotechnology or Biobarcode Technology, but otherwise within the Biodiagnostics Field,
conceived prior to January 1, 2013. Both (1) and (2) are herein referred to as Future Inventions.
If the parties cannot agree on the terms of the license for the Future Inventions, the parties
shall submit to arbitration to determine reasonable terms. For inventions conceived after
January 1, 2013 that are not Nanotechnology or Biobarcode Technology, but otherwise within the
Biodiagnostics Field, the Company has the right to negotiate a license if Northwestern offers such
inventions to third parties. If the Company has a license based on Future Inventions, Northwestern
has the right to terminate the license upon any material breach that the Company does not cure or
upon bankruptcy.
The Company has an obligation to pay Northwestern a royalty at a rate that is a percentage of
the gross profits of licensed products, subject to certain adjustments. The Company’s obligation
for payments to Northwestern pursuant to this agreement began on January 1, 2007, and obligations
and payments under this agreement have been insignificant through December 31, 2009.
The Company has entered into several nonexclusive license agreements with various companies
covering certain technologies which are embedded in the Company’s diagnostic instruments and
diagnostic test products. As of December 31, 2009, the Company has paid aggregate initial license
fees of $2.4 million for these licenses, and has agreed to pay a percentage of net sales as
royalties, in percentage amounts ranging from less than 1% to 12.0%. Royalties on net sales are
classified in Cost of sales. Certain of the license agreements have minimum annual royalty
payments, and such minimum payments are $173,750 in 2010, $178,750 in 2011, $186,250 in 2012,
$210,000 in 2013, $215,000 in 2014 and are $30,000 to $220,000 annually thereafter through the
dates the respective licenses terminate. These licenses expire at various times, corresponding to
the subject patents expirations, which currently range from 2010 to 2025.
8. Stockholders’ Equity
Common Stock
On October 16, 2007 (the “Effective Date”), each share of common stock, par value $0.01 per
share (the “Old Common Stock”), issued and outstanding immediately prior to the Effective Date, was
automatically reclassified as and converted into 1/25 of a share of common stock, par value $0.01
per share, of the Company. Additionally, in accordance with the Company’s Amended and Restated
Certificate of Incorporation, as amended, the conversion ratio of the Company’s convertible
preferred stock was also proportionately decreased.
On October 16, 2007, the majority of the shareholders of the Company’s Series C-2 and Series D
Convertible Preferred Stock, each series voting as a separate class, in accordance with the
Company’s Amended and Restated Certificate of Incorporation, as amended, approved the conversion of
all of the Company’s convertible preferred stock into shares of the Company’s common stock,
contingent upon the closing and effectiveness of the Company’s contemplated initial public offering
and the listing of the Company’s common stock on the NASDAQ Global Market, which occurred on
November 6, 2007.
On October 31, 2007, the Company’s registration statement for the initial public offering of
its common stock was declared effective by the SEC. On November 6, 2007, the Company closed on the
sale of 8,050,000 shares related to the initial public offering at $14.00 per share, less
underwriting discounts and commissions. Net proceeds from the initial public offering were
approximately $102 million, net of transaction expenses. Approximately $0.8 million of the
transaction expenses were paid in 2008.
On November 6, 2007, all of the Company’s then outstanding convertible preferred stock
converted into 13,048,119 shares of common stock upon the closing of the Company’s initial public
offering. The number of common shares issued included 755,758 shares of common stock issued in
connection with accrued and unpaid dividends on the convertible preferred stock through the closing
date. Further, included in the number of common shares issued upon conversion of the preferred
stock are convertible preferred shares issued immediately prior to the closing of the initial
public offering when approximately 99% of the outstanding warrants to purchase Series C and Series
C-2 Convertible Preferred Stock were exercised.
On November 6, 2007, the Company amended its bylaws and filed an amended certificate of
incorporation with the State of Delaware. As a result of these amendments, the number of
authorized common and preferred shares was reduced to 100 million and 10 million, respectively.
Further, the previously-issued series of convertible preferred stock were no longer authorized and
the number of shares of preferred stock outstanding was reduced to zero on this date.
54
Table of Contents
Nanosphere, Inc.
Notes to Financial Statements — (Continued)
The Company completed an underwritten public offering of 5,405,000 shares of common stock on
October 21, 2009 at a public offering price of $7.00 per share, less underwriting discounts and
commissions. Net proceeds from the public offering were approximately $35.4 million.
Registration Rights
Pursuant to an agreement between the Company and certain of its stockholders, the Company
has granted the following demand registration rights to Mr. Mark Slezak and Ms. Sheli Rosenberg,
who are members of the Company’s board of directors, AOQ Trust, Alfa-Tech, LLC, Lurie Investment
Fund, LLC, Lurie Investments, Inc. and their respective affiliates, and Bain Capital Venture Fund
2005, L.P., Brookside Capital Partners Fund, L.P., and their respective affiliates and other
stockholders. Mr. William P. Moffitt, III, the Company’s chief executive officer and a member of
the board of directors, and Dr. Chad Mirkin, a member of the board of directors, are parties to
this agreement, but do not have the right to demand registration. At any time after the earlier to
occur of (1) 120 days after the closing of an initial public offering, which occurred on
November 6, 2007, or (2) April 1, 2010:
| • | Long-Form Registrations. Stockholders holding at least 20% of the then outstanding shares of the Company’s common stock that are subject to the registration rights agreement, which are referred to as registrable securities, have the right to demand that the Company file a registration statement under the Securities Act on Form S-1 or any similar long-form registration covering their registrable securities. However, the Company is not obligated to file a long-form registration statement on more than three occasions upon the request of the stockholders. | ||
| • | Short-Form Registrations. Stockholders holding at least 10% of the then outstanding registrable securities have the right to demand that the Company file a registration statement on Form S-3 or any similar short-form registration covering their registrable securities, provided that such short-form registration is then available to the Company under applicable law. Such stockholders are entitled to request an unlimited number of short-form registrations. |
If the Company’s board of directors believes in its reasonable good faith that any demand
registration would require premature disclosure of a proposal or plan that the Company intends to
undertake, and such disclosure would have a material adverse effect on the Company, then it may
delay the registration once in any twelve month period for up to 90 days. Moreover, if the demand
registration is an underwritten offering, the Company may reduce the number of shares of
registrable securities to be registered upon the advice of the underwriters that such offering
exceeds the number of securities that can be sold in an orderly manner within an acceptable price
range. If shares of the Company’s common stock requested to be included in a registration must be
excluded pursuant to the underwriters’ advice, the Company will generally register a pro rata
portion of the shares requested to be registered.
Under the piggyback registration provisions, if the Company proposes to register any
securities under the Securities Act, other than pursuant to a demand registration, and the
registration form to be used may be used for the registration of registrable securities,
stockholders holding such registrable securities have the right to include their shares in the
registration statement. However, if the registration is an underwritten offering, the Company may
reduce the number of shares to be registered under the piggyback registration provisions upon the
advice of the underwriters that such offering exceeds the number of securities that can be sold in
an orderly manner within an acceptable price range. If shares of the Company’s common stock
requested to be included in a registration must be excluded pursuant to the underwriters’ advice,
the Company will generally register a pro rata portion of the shares requested to be registered
under the piggyback registration provisions. The piggyback registration rights granted under the
registration rights agreement have no expiration date.
Expenses of Registration. The Company will generally pay all registration expenses in
connection with the demand and piggyback registrations described above, including all registration
and filing fees, expenses and fees of compliance with securities laws, and fees and disbursements
of all counsel, independent certified public accountants, underwriters (excluding discounts and
commissions) and other persons retained by the Company. The Company will also pay the reasonable
fees and disbursements of one counsel chosen by the selling stockholders in each demand or
piggyback registration.
Transferability. The demand and piggyback registration rights described above are
generally transferable to any subsequent holder of registrable securities.
Convertible Preferred Stock
Prior to the completion of the Company’s IPO, the Company had outstanding four series of
convertible preferred stock, two of which paid dividends and also had a conversion feature that the
Company had determined was an embedded derivative instrument. The embedded derivative was
bifurcated from the convertible preferred stock and accounted for separately as a liability, the
convertible derivative liability. The conversion feature, when classified as a derivative
liability, was required to be initially recorded at fair value and to be marked to fair value at
the end of each reporting period, which resulted in a non-cash charge to other income or expense in
the statement of operations. Upon the conversion of the Series C-2 and Series D convertible
preferred stock to common stock during November 2007, the convertible derivative liability of $17.8
million was reclassified to additional paid-in capital.
55
Table of Contents
Nanosphere, Inc.
Notes to Financial Statements — (Continued)
Prior to the completion of the Company’s IPO, the Company also issued warrants to purchase
shares of convertible preferred stock in connection with certain of the convertible preferred stock
financings. These warrants were accounted for as liabilities initially at fair value and were
marked to fair value at the end of each reporting period up to the closing of the initial public
offering, which resulted in a non-cash charge to other income or expense in the statement of
operations.
With the exception of the Series D convertible preferred stock warrants, all convertible
preferred stock warrants were either exercised or expired upon the completion of the IPO. The
Series D convertible preferred stock warrants were converted to common stock warrants upon the
closing of the IPO. As of December 31, 2009 and 2008, there were outstanding warrants to acquire
shares of common stock of 1,300,119.
The expiration dates of the warrants outstanding at December 31, 2009 are as follows:
| Number of | Expiration | |||||||
| Series of Stock to which the Warrant is Exercisable | Warrants | Date | ||||||
Common — exercise price of $15.32 per share |
1,135,194 | April 2011 | ||||||
Common — exercise price of $8.75 per share |
164,925 | April 2013 | ||||||
The exercise price on the common stock warrants with an exercise price of $15.32 per share at
December 31, 2009 will increase to $17.50 per share in April 2010.
The fair values of the Company’s convertible preferred stock warrants were estimated as of
November 1, 2007 (the date of the initial public offering) using either a Black-Scholes or a
binominal pricing model, as appropriate given the terms of the warrant, with the following
weighted-average assumptions:
Expected dividend yield |
6 | % | ||
Expected volatility |
55 | % | ||
Risk free interest rate |
3.83 | % | ||
Weighted-average warrant term |
3.4 years | |||
Estimated weighted-average fair value |
$ | 0.17 | ||
9. Commitments and Contingencies
The Company’s existing operating lease for its office and laboratory space expires in May
2010. On August 28, 2009, the Company executed a lease renewal which commences in June 2010 and
ends in May 2014. Under the terms of the lease renewal, the Company has two successive three year
options to renew the lease, and the Company has the right of first refusal to lease additional
space within the facility.
Rent and operating expenses associated with the office and laboratory space were $472,751,
$781,900, and $680,559 in 2009, 2008, and 2007, respectively.
Annual future minimum obligations for operating leases as of December 31, 2009 are as follows:
| Operating | ||||
| Years Ending December 31 | Leases | |||
2010 |
$ | 465,595 | ||
2011 |
426,643 | |||
2012 |
438,921 | |||
2013 |
451,198 | |||
2014 |
190,131 | |||
Total minimum lease payments |
$ | 1,972,488 | ||
In July 2009, the Company was named as a defendant in a lawsuit filed by Eppendorf AG alleging
patent infringement. The Company intends to vigorously defend this case as it believes that the
claims lack merit. The Company believes that the resolution to
these proceedings will not have a material adverse effect on the Company’s financial position,
liquidity or future operations. The Company has not recorded any reserves related to this case.
56
Table of Contents
Nanosphere, Inc.
Notes to Financial Statements — (Continued)
10. Long-Term Debt
In February 2007, the Company entered into two loan and security agreements, with commitments
for debt financing with Venture Lending & Leasing IV, Inc., and Venture Lending & Leasing V, Inc.
The Company borrowed $12.5 million under these agreements in February 2007. Interest rates under
the agreements are 12.5% for the initial twelve month period and 10.0% during the following thirty
month period. Notes issued pursuant to this commitment are secured by a first security lien on all
of the Company’s assets including intellectual property. In connection with the execution of the
commitment and the initial note issuance under these agreements, the Company issued to the lenders
5,535,824 shares of the Company’s Series D Convertible Preferred Stock.
The $12.5 million of proceeds received were allocated to debt and the Series D Convertible
Preferred Stock based on their fair values at the borrowing date with $1.9 million allocated to
Series D Convertible Preferred Stock and the remaining $10.6 million allocated to debt. The
discount on the debt of $1.9 million resulted in an effective interest rate on the debt of 21% and
the discount will be amortized to interest expense over the term of the debt following the interest
method. Interest expense on this debt for the years ended December 31, 2009, 2008 and 2007 was
$1,224,039, $2,042,734 and $1,939,046, respectively, which includes $466,652, $729,544 and $642,838
of discount amortization, respectively. Cash interest paid on this debt was $806,639, $1,349,969
and $1,170,133 for the years ended December 31, 2009, 2008 and 2007, respectively. The fair value
of the debt at December 31, 2009 is $4.0 million, which was derived by applying an income approach
to determine the present value of forecast debt payments as required under the debt agreements. To
determine the appropriate yield by which to discount the future cash payments, the Company relied
upon observable inputs of yields for securities with comparable risk and return profiles, which are
Level 2 measurements under Codification Topic 820, “Fair Value Measurements and Disclosures”.
Aggregate principal payments on long-term debt in 2010 will be $3,917,293.
11. Supplemental Financial Information
| 2009 | 2008 | |||||||
Inventories: |
||||||||
Work-in-process component parts |
$ | 1,061,982 | $ | 1,068,840 | ||||
Finished goods |
1,878,592 | 658,678 | ||||||
Total |
$ | 2,940,574 | $ | 1,727,518 | ||||
| 2009 | 2008 | |||||||
Property and Equipment — Net: |
||||||||
Equipment with customers |
$ | 1,567,573 | $ | 1,582,580 | ||||
Computer equipment and software |
1,015,559 | 973,376 | ||||||
Laboratory equipment |
4,721,224 | 3,819,860 | ||||||
Furniture and fixtures |
269,047 | 269,047 | ||||||
Leasehold improvements |
2,869,258 | 2,787,273 | ||||||
Manufacturing equipment |
4,142,417 | 3,857,945 | ||||||
Office equipment |
67,068 | 67,068 | ||||||
Tooling |
1,377,386 | 1,159,222 | ||||||
Total property and equipment — at cost |
16,029,532 | 14,516,371 | ||||||
Less accumulated depreciation |
(9,884,977 | ) | (7,222,147 | ) | ||||
Property and Equipment — Net |
$ | 6,144,555 | $ | 7,294,224 | ||||
| 2009 | 2008 | |||||||
Other Current Liabilities: |
||||||||
Accrued clinical trial expenses |
$ | 939,509 | $ | 266,135 | ||||
All other |
1,364,122 | 1,275,250 | ||||||
Total |
$ | 2,303,631 | $ | 1,541,385 | ||||
57
Table of Contents
Nanosphere, Inc.
Notes to Financial Statements — (Continued)
12. Selected Quarterly Financial Data (Unaudited)
| 2009 Quarters | ||||||||||||||||
| First | Second | Third | Fourth | |||||||||||||
Total revenue |
$ | 255,225 | $ | 402,237 | $ | 729,017 | $ | 827,624 | ||||||||
Loss from operations |
$ | (7,982,063 | ) | $ | (8,131,319 | ) | $ | (7,996,289 | ) | $ | (8,931,401 | ) | ||||
Net loss |
$ | (8,193,473 | ) | $ | (8,384,143 | ) | $ | (8,242,361 | ) | $ | (9,128,676 | ) | ||||
Per share data: |
||||||||||||||||
Net loss per common
share — basic and
diluted |
$ | (0.37 | ) | $ | (0.38 | ) | $ | (0.37 | ) | $ | (0.34 | ) | ||||
| 2008 Quarters | ||||||||||||||||
| First | Second | Third | Fourth | |||||||||||||
Total revenue |
$ | 576,016 | $ | 194,715 | $ | 283,231 | $ | 312,770 | ||||||||
Loss from operations |
$ | (9,106,140 | ) | $ | (9,832,796 | ) | $ | (9,023,050 | ) | $ | (9,425,808 | ) | ||||
Net loss |
$ | (8,783,368 | ) | $ | (9,831,924 | ) | $ | (9,026,472 | ) | $ | (9,400,232 | ) | ||||
Per share data: |
||||||||||||||||
Net loss per common
share — basic and
diluted |
$ | (0.40 | ) | $ | (0.44 | ) | $ | (0.41 | ) | $ | (0.42 | ) | ||||
58
Table of Contents
| Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure. |
We have had no disagreements with our independent registered public accounting firm on any
matter of accounting principles or practices or financial statement disclosure.
| Item 9A. | Controls and Procedures. |
(a) Evaluation of Disclosure Controls and Procedures
Management of the Company, with the participation of the Chief Executive Officer and the Chief
Financial Officer, evaluated the effectiveness of the design and operation of the Company’s
disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) of the Securities
Exchange Act of 1934, as amended) as of December 31, 2009. The Company’s disclosure controls and
procedures are designed to ensure that information required to be disclosed by the Company in the
reports it files or submits under the Exchange Act is recorded, processed, summarized and reported
on a timely basis and that such information is accumulated and communicated to management,
including the Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely
decisions regarding disclosure. Based upon this evaluation, the Chief Executive Officer and the
Chief Financial Officer have concluded that the Company’s disclosure controls and procedures were
effective as of December 31, 2009.
(b) Management’s Report on Internal Control Over Financial Reporting
Internal control over financial reporting, as such term is defined in Rules 13a-15(f) and
15d-15(f) under the Exchange Act, is a process designed by, or under the supervision of, the
Company’s chief executive officer and chief financial officer, or persons performing similar
functions, and effected by the Company’s board of directors, management and other personnel, to
provide reasonable assurance regarding the reliability of financial reporting and the preparation
of financial statements for external purposes in accordance with generally accepted accounting
principles. The Company’s management, with the participation of the Company’s chief executive
officer and chief financial officer, has established and maintained policies and procedures
designed to maintain the adequacy of the Company’s internal control over financial reporting, and
includes those policies and procedures that:
| (1) | Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of the Company; | ||
| (2) | Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and | ||
| (3) | Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements. |
The Company’s management is responsible for establishing and maintaining adequate internal
control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f). The
Company’s management conducted an evaluation of the effectiveness of internal control over
financial reporting based on the framework in Internal Control — Integrated Framework issued by the
Committee of Sponsoring Organizations of the Treadway Commission. Based on its evaluation under the
framework in Internal Control — Integrated Framework, the Company’s management concluded that
internal control over financial reporting was effective as of December 31, 2009.
Because of its inherent limitations, internal control over financial reporting may not prevent
or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are
subject to the risk that controls may become inadequate because of changes in conditions, or that
the degree or compliance with the policies or procedures may deteriorate.
The Company’s independent registered public accounting firm, Deloitte & Touche LLP, has issued
an attestation report on the Company’s internal control over financial reporting as of December 31,
2009. Their report is included in this Form 10-K.
59
Table of Contents
(c) Changes in Internal Controls Over Financial Reporting
There have been no changes to the Company’s internal control over financial reporting during
the most recent fiscal quarter that have materially affected, or are reasonably likely to
materially affect, the Company’s internal control over financial reporting.
| Item 9B. | Other Information. |
None.
60
Table of Contents
PART III.
| Item 10. | Directors and Executive Officers of the Registrant. |
The information required by Item 10 will be set forth in the Company’s definitive proxy
statement for its annual meeting of shareholders expected to be held on May 25, 2010, and is
incorporated herein by reference.
| Item 11. | Executive Compensation. |
The information required by Item 11 will be set forth in the Company’s definitive proxy statement
for its annual meeting of shareholders expected to be held on May 25, 2010, and is incorporated
herein by reference.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters. |
The information required by Item 12 will be set forth in the Company’s definitive proxy
statement for its annual meeting of shareholders expected to be held on May 25, 2010, and is
incorporated herein by reference.
| Item 13. | Certain Relationships and Related Transactions, and Director Independence. |
The information required by Item 13 will be set forth in the Company’s definitive proxy statement
for its annual meeting of shareholders expected to be held on May 25, 2010, and is incorporated
herein by reference.
| Item 14. | Principal Accounting Fees and Services. |
The information required by Item 14 will be set forth in the Company’s definitive proxy
statement for its annual meeting of shareholders expected to be held on May 25, 2010, and is
incorporated herein by reference.
61
Table of Contents
PART IV.
| Item 15. | Exhibits, Financial Statement Schedules. |
(a)(1) Financial Statements
Reports of Independent Registered Public Accounting Firm |
||||
Balance Sheets as of December 31, 2009 and 2008 |
||||
Statements of Operations for the years ended December 31, 2009, 2008 and 2007 |
||||
Statements of Stockholders’ Equity (Deficit) for the years ended December 31, 2009, 2008 and 2007 |
||||
Statements of Cash Flows for the years ended December 31, 2009, 2008 and 2007 |
||||
Notes to Financial Statements |
(a)(2) Financial Statement Schedules
None
(a)(3) Exhibits required by Item 601 of Regulation S-K.
| Exhibit Number | Exhibit Description | |||
| 3.1 | Second Amended and Restated Certificate of Incorporation of Nanosphere, Inc. (3)
(Exhibit 3.1) |
|||
| 3.2 | Amended and Restated Bylaws of Nanosphere, Inc. (3) (Exhibit 3.2) |
|||
| 4.1 | Specimen of common stock certificate (4) (Exhibit 4.3) |
|||
| 10.1 | Nanosphere, Inc. 2000 Equity Incentive Plan (1) (Exhibit 10.1) |
|||
| 10.2 | Form of Nanosphere, Inc. 2000 Equity Incentive Plan Non-Qualified Stock Option Award
Agreement, as amended (1) (Exhibit 10.2) |
|||
| 10.3 | Form of Nanosphere, Inc. 2000 Equity Incentive Plan Option Award Agreement (1) (Exhibit
10.3) |
|||
| 10.4 | Nanosphere, Inc. 2007 Long-Term Incentive Plan, as amended and restated (4) (Exhibit 10.4) |
|||
| 10.5 | Form of Nanosphere, Inc. 2007 Long-Term Incentive Plan Incentive Stock Option Award
Agreement (Time Vested) (1) (Exhibit 10.5) |
|||
| 10.6 | Form of Nanosphere, Inc. 2007 Long-Term Incentive Plan Non-Qualified Stock Option Award
Agreement (Time Vested) (1) (Exhibit 10.6) |
|||
| 10.7 | Form of Nanosphere, Inc. 2007 Long-Term Incentive Plan Option Award Agreement
(Cliff-vested, performance-accelerated) (1) (Exhibit 10.7) |
|||
| 10.8 | Employment Agreement, dated as of July 19, 2004, by and between Nanosphere, Inc. and
William P. Moffitt III, as amended (1) (Exhibit 10.8) |
|||
| 10.9 | Restricted Stock Purchase Agreement, dated as of March 16, 2006, by and between
Nanosphere, Inc. and William P. Moffitt III (3) (Exhibit 10.9) |
|||
| 10.10 | Employment Agreement, dated January 2, 2001, by and between Nanosphere, Inc. and William
Cork (4) (Exhibit 10.10) |
|||
| 10.11 | Employment Agreement, dated May 13, 2005, by and between Nanosphere, Inc. and Gregory
Shipp (1) (Exhibit 10.11) |
|||
| 10.12 | Employment Agreement, dated September 5, 2005, by and between Nanosphere, Inc. and
Michael McGarrity (1) (Exhibit 10.12) |
|||
| 10.13 | Employment Agreement, dated April 25, 2007, by and between Nanosphere, Inc. and J. Roger
Moody, Jr. (1) (Exhibit 10.13) |
|||
| 10.14 | Employment Agreement, dated May 31, 2007, by and between Nanosphere, Inc. and Winton
Gibbons (1) (Exhibit 10.14) |
|||
62
Table of Contents
| Exhibit Number | Exhibit Description | |||
| 10.15 | Severance Agreement, dated as of June 4, 2007, by and between Nanosphere, Inc. and
Stephen Wasko (1) (Exhibit 10.15) |
|||
| 10.16 | License Agreement, dated as of January 1, 2006, by and between Northwestern University
and Nanosphere, Inc. (2)# (Exhibit 10.16) |
|||
| 10.17 | Non-Exclusive License Agreement, dated as of December 20, 2002, by and between
Nanosphere, Inc. and Abbott Laboratories (2)# (Exhibit 10.17) |
|||
| 10.18 | Lease with Motorola, Inc., dated as of March 24, 2003, as amended (1) (Exhibit 10.18) |
|||
| 10.19 | Loan and Security Agreement, dated as of February 7, 2007, by and between Nanosphere,
Inc. and Venture Lending & Leasing IV, Inc. (1) (Exhibit 10.19) |
|||
| 10.20 | Loan and Security Agreement, dated as of February 21, 2007, by and between Nanosphere,
Inc. and Venture Lending & Leasing V, Inc. (1) (Exhibit 10.20) |
|||
| 10.21 | Consulting and Non-Competition Agreement, dated as of October 31, 2002, by and between
Nanosphere, Inc. and Chad A. Mirkin, as amended (1) (Exhibit 10.21) |
|||
| 10.22 | Bonus Agreement, dated as of March 16, 2006, by and between Nanosphere, Inc. and William
P. Moffitt III, as amended (2) (Exhibit 10.22) |
|||
| 10.23 | Series D Preferred Stock and Warrant Purchase Agreement, dated as of April 12, 2006 (2)
(Exhibit 10.24) |
|||
| 10.24 | Note Purchase Agreement, dated as of March 15, 2006, by and between Nanosphere, Inc. and
Lurie Investment Fund, L.L.C. (2) (Exhibit 10.28) |
|||
| 10.25 | Form of Indemnification Agreement (3) (Exhibit 10.29) |
|||
| 10.26 | Non-Exclusive Financial Advisory Services Engagement Letter, dated as of August 8, 2007,
by and between Nanosphere, Inc. and Allen & Company LLC (4) (Exhibit 10.30) |
|||
| 10.27 | Amended and Restated Employment Agreement dated as of January 1, 2009, between
Nanosphere, Inc. and Mr. William P. Moffitt (5) (Exhibit 10.31) |
|||
| 10.28 | Second Amended and Restated Registration Rights Agreement, dated August 19, 2009 (6)
(Exhibit 10.1) |
|||
| 10.29 | Lease Agreement, dated August 28, 2009, between Nanosphere, Inc. and Northbrook
Commercial Properties, LLC (7) (Exhibit 10.1) |
|||
| 23.1 | Consent of Deloitte & Touche LLP* |
|||
| 31.1 | Certification of Chief Executive Officer pursuant to Exchange Act Rules 13a-14(a) and
15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 * |
|||
| 31.2 | Certification of Chief Financial Officer pursuant to Exchange Act Rules 13a-14(a) and
15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 * |
|||
| 32.1 | Certification of Chief Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted
pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 * |
|||
| 32.2 | Certification of Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted
pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 * |
|||
| * | Filed herewith | |
| # | Confidential treatment has been requested with respect to certain provisions of this agreement. Omitted portions have been filed separately with the SEC. | |
| (1) | Incorporated by reference from the Company’s Registration Statement on Form S-1 as filed with the Securities and Exchange Commission on August 13, 2007. The exhibit reference in parentheses indicates the corresponding exhibit number in such Registration Statement. | |
| (2) | Incorporated by reference from the Company’s Amendment No. 1 to Form S-1 as filed with the Securities and Exchange Commission on September 27, 2007. The exhibit reference in parentheses indicates the corresponding exhibit number in such Amendment. | |
| (3) | Incorporated by reference from the Company’s Amendment No. 2 to Form S-1 as filed with the Securities and Exchange Commission on October 17, 2007. The exhibit reference in parentheses indicates the corresponding exhibit number in such Amendment. |
63
Table of Contents
| (4) | Incorporated by reference from the Company’s Amendment No. 3 to Form S-1 as filed with the Securities and Exchange Commission on October 29, 2007. The exhibit reference in parentheses indicates the corresponding exhibit number in such Amendment. | |
| (5) | Incorporated by reference from the Company’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on March 5, 2009. The exhibit reference in parentheses indicates the corresponding exhibit number in such Current Report. | |
| (6) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q as filed with the Securities and Exchange Commission on November 5, 2009. The exhibit reference in parentheses indicates the corresponding exhibit number in such Quarterly Report. | |
| (7) | Incorporated by reference from the Company’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on September 1, 2009. The exhibit reference in parentheses indicates the corresponding exhibit number in such Current Report. |
64
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934,
the registrant has duly caused this report to be signed on its behalf by the undersigned,
thereunto duly authorized.
| NANOSPHERE, INC. |
||||
| By: | /s/ William P. Moffitt | |||
| William P. Moffitt | ||||
| President and Chief Executive Officer | ||||
Date: March 11, 2010
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been
signed below by the following persons on behalf of the Registrant and in the capacities and on
the dates indicated:
| Signature | Title(s) | Date | ||
/s/ William P. Moffitt
|
President, Chief Executive Officer, Director (principal executive officer) |
March 11, 2010 | ||
/s/ Roger Moody
|
Chief Financial Officer, Treasurer (principal financial officer and principal accounting officer) |
March 11, 2010 | ||
/s/ Mark Slezak
|
Chairman of the board of directors | March 11, 2010 | ||
/s/ Jeffrey R. Crisan
|
Director | March 11, 2010 | ||
/s/ André de Bruin
|
Director | March 11, 2010 | ||
/s/ Chad A. Mirkin
|
Director | March 11, 2010 | ||
/s/ James J. Nahirny
|
Director | March 11, 2010 | ||
/s/ Sheli Z. Rosenberg
|
Director | March 11, 2010 | ||
/s/ Lorin J. Randall
|
Director | March 11, 2010 |
65