Attached files
| file | filename |
|---|---|
| EX-31.1 - EX-31.1 - ANADYS PHARMACEUTICALS INC | a55326exv31w1.htm |
| EX-32.1 - EX-32.1 - ANADYS PHARMACEUTICALS INC | a55326exv32w1.htm |
| EX-23.1 - EX-23.1 - ANADYS PHARMACEUTICALS INC | a55326exv23w1.htm |
| EX-31.2 - EX-31.2 - ANADYS PHARMACEUTICALS INC | a55326exv31w2.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
SECURITIES AND EXCHANGE COMMISSION
Washington, D. C. 20549
FORM 10-K
(MARK ONE)
| þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2009
OR
| o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission File Number 0-50632
ANADYS PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 22-3193172 | |
| (State or other jurisdiction of | (I.R.S. Employer | |
| incorporation or organization) | Identification No.) | |
| 5871 Oberlin Drive, Suite 200, San Diego, California | 92121 | |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code: 858-530-3600
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Name of each exchange on which registered | |
| Common Stock, $0.001 par value | Nasdaq Global Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark whether the registrant is a well-known seasoned issuer as defined in
Rule 405 of the Securities Act. Yes o No þ
Indicate by check mark whether the registrant is not required to file reports pursuant to
Section 13 or Section 15(d) of the Act. Yes o
No þ
No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed
by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or
for such shorter period that the registrant was required to file such reports), and (2) has been
subject to such filing requirements for the past 90 days. Yes þ No o
Indicate by check mark whether the registrant has submitted electronically and posted on its
corporate Web site, if any, every Interactive Data File required to be submitted and posted
pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period
that the registrant was required to submit and post such files). Yes o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation
S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in
definitive proxy or information statements incorporated by reference in Part III of this Form 10-K
or any amendment to this Form 10-K. þ
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated
filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large
accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the
Exchange Act. (Check one):
| Large accelerated filer o | Accelerated filer þ | Non-accelerated filer o (Do not check if a smaller reporting company) |
Smaller Reporting Company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of
the Act). Yes o No þ
The aggregate market value of the common stock held by non-affiliates of the registrant
computed by reference to the closing price of the registrants common stock reported on the Nasdaq
Global Market as of the last business day of the registrant’s most recently completed second fiscal
quarter was approximately $66,821,443 as of such date.
As of February 17, 2010, the Registrant had outstanding 37,341,957 shares of common stock.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Company’s Proxy Statement to be filed with the Securities and Exchange
Commission in connection with the 2010 Annual Meeting of Stockholders are incorporated herein by
reference into Part III.
ANADYS PHARMACEUTICALS, INC.
ANNUAL REPORT ON FORM 10-K
ANNUAL REPORT ON FORM 10-K
Table of Contents
| Page | ||||||||
| PART I |
||||||||
| 3 | ||||||||
| 16 | ||||||||
| 32 | ||||||||
| 32 | ||||||||
| 32 | ||||||||
| 32 | ||||||||
| PART II |
||||||||
| 33 | ||||||||
| 35 | ||||||||
| 36 | ||||||||
| 42 | ||||||||
| 42 | ||||||||
| 42 | ||||||||
| 43 | ||||||||
| 45 | ||||||||
| PART III |
||||||||
| 46 | ||||||||
| 46 | ||||||||
| 46 | ||||||||
| 47 | ||||||||
| 47 | ||||||||
| PART IV |
||||||||
| 48 | ||||||||
| EX-23.1 | ||||||||
| EX-31.1 | ||||||||
| EX-31.2 | ||||||||
| EX-32.1 | ||||||||
Table of Contents
INFORMATION RELATED TO FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements. These forward-looking
statements involve a number of risks and uncertainties. Such forward-looking statements include
statements about our development plans and programs, clinical trials, strategies and objectives,
and other statements that are not historical facts, including statements which may be preceded by
the words “intend,” “will,” “plan,” “expect,” “anticipate,” “estimate,” “aim,” “seek,” “believe,”
“hope” or similar words. For such statements, we claim the protection of the Private Securities
Litigation Reform Act of 1995. Readers of this Annual Report on Form 10-K are cautioned not to
place undue reliance on these forward-looking statements, which speak only as of the date on which
they are made. We undertake no obligation to update publicly or revise any forward-looking
statements. Actual events or results may differ materially from our expectations. Important factors
that could cause actual results to differ materially from those stated or implied by our
forward-looking statements include, but are not limited to, the risk factors identified in our
periodic reports filed with the Securities and Exchange Commission (SEC), including, without
limitation, those discussed in “Item 1A. Risk Factors” and in “Item 7. Management’s Discussion and
Analysis of Financial Condition and Results of Operations” of this Annual Report on Form 10-K.
PART I
| Item 1. | Business |
Overview
Anadys Pharmaceuticals, Inc. is a biopharmaceutical company dedicated to improving patient
care by developing novel medicines for the treatment of hepatitis C. We believe hepatitis C
represents a large and significant unmet medical need. Our objective is to contribute to an
improved treatment outcome for patients with this serious disease.
We are currently focusing our efforts on the development of ANA598, a small-molecule,
non-nucleoside inhibitor of the NS5B polymerase for the treatment of hepatitis C. We also have
investigated ANA773, an oral, small-molecule inducer of endogenous interferons that acts via the
Toll-like receptor 7, or TLR7, pathway in a Phase I trial in hepatitis C.
Our expertise is based on two distinct scientific approaches to treating disease. With ANA598
we are focused on developing a direct antiviral, meaning a product candidate that acts by directly
interacting with, and blocking the function of, a component of the virus. We discovered ANA598
through an extensive structure-based drug design program that focused on parameters we feel are
critical for success in chronic viral diseases, including potency and sustained drug levels in
blood. With ANA773, the focus is to stimulate the patients’ own immune systems to block cells
infected with the hepatitis C virus from further producing more virus particles and amplifying the
infection. ANA773 stimulates the immune system through activating a key receptor on immune cells
known as TLR7. Our knowledge of TLR7 is buttressed by an extensive preclinical program exploring
the pharmacology of this receptor and by previous clinical experience with other molecules that act
via the TLR7 mechanism.
Activation of the TLR7 receptor may also allow the patient’s immune system to attack cancer
cells. Accordingly, we have also investigated ANA773 in a separate Phase I trial for the treatment
of patients with advanced cancer.
In June 2009, we initiated a strategic restructuring to focus our operations on the
development of ANA598, in particular the Phase II study of ANA598 in combination with pegylated
interferon and ribavirin. As part of the restructuring, we suspended further development of
ANA773. The strategic restructuring resulted in a reduction in our workforce of approximately 40%.
We retained the clinical development infrastructure required to conduct the Phase II study of
ANA598, key capabilities directed toward pharmaceutical development and next generation
non-nucleosides and a streamlined administrative staff.
Anadys retains all commercialization rights to both ANA598 and ANA773, which were discovered
at Anadys.
ANA598
ANA598 is a direct antiviral that blocks the hepatitis C virus’ (HCV) ability to multiply by
inhibiting the viral RNA polymerase. ANA598 belongs to a class of direct antivirals referred to as
non-nucleosides. We believe non-nucleosides will become an important component of future
combination regimens used to treat HCV infection, similar to the role played by non-nucleoside
inhibitors in HIV therapy. ANA598 has demonstrated positive antiviral response and a favorable
safety and tolerability profile in the ongoing Phase II combination trial, as described below.
ANA598 has also completed two long-term chronic toxicology studies (26 weeks duration in rats and
39 weeks duration in monkeys) with favorable results. Furthermore, ANA598 has demonstrated the
preclinical properties, including potency, pharmacokinetics and early safety, that we believe are
prerequisites for successful development in the HCV area.
3
Table of Contents
We are currently conducting a Phase II clinical trial of ANA598 in combination with pegylated
interferon-alpha and ribavirin, which is the current standard of care (SOC) for the treatment of
hepatitis C. The first dose cohort, receiving ANA598 at 200 mg twice daily (bid), initiated dosing
in September 2009 and has completed the 12-week ANA598 dosing portion of the trial. The second dose
cohort, receiving ANA598 at 400 mg bid, initiated dosing and fully enrolled in January 2010.
In the ongoing Phase II study, treatment-naïve (previously untreated) genotype 1 patients are
to receive ANA598 or placebo in combination with SOC for 12 weeks at dose levels of 200 mg bid or
400 mg bid, each with a loading dose of 800 mg bid on day one. After week 12, patients are to
continue receiving standard of care alone. Patients who achieve undetectable levels of virus at
weeks four and 12 will be randomized to stop all treatment at week 24 or 48. The primary endpoint
of the study is the proportion of patients who achieve undetectable levels of virus at week 12
(defined as complete Early Virological Response, or cEVR).
Additional endpoints include safety and tolerability as well as the proportion of patients with
undetectable levels of virus at week four (defined as Rapid Virological
Response, or RVR). Patients will be followed for 24 weeks after stopping therapy to
determine the rate of Sustained Virological Response, or SVR. Approximately
90 patients were to be and have been enrolled in this study – with approximately 30 patients
receiving ANA598 and 15 receiving placebo at each dose level. The study is being conducted at a
number of clinical sites in the United States.
In December 2009, we reported positive preliminary antiviral response and safety results at
week four from the first dose cohort (200 mg bid) and in February 2010, we reported positive
preliminary antiviral response and safety results at week 12 for this dose cohort. 56% of patients
receiving ANA598 at 200 mg bid in combination with SOC achieved undetectable levels of virus
(<15 IU/ml) at week 4, referred to as RVR, and 73% of patients receiving ANA598 at 200 mg bid in
combination with SOC achieved undetectable levels of virus (<15 IU/ml) at week 12, referred to
as cEVR. We are currently dosing patients at 400 mg bid and expect to analyze and release four week
data from this dose cohort prior to the end of the first quarter of 2010 and expect to analyze and
release 12-week data from this dose cohort during the second quarter of 2010.
We are in the early stages of Phase II drug development with ANA598. Substantial further
investment will be necessary in order to progress ANA598 beyond the events referenced above and
through additional clinical testing before we will be able to seek regulatory approval.
ANA773
ANA773 is a novel, oral inducer of endogenous interferons that acts via the TLR7 pathway that
we have investigated as a treatment for both HCV and cancer. Both the prodrug and its active
substance were discovered, designed and synthesized by Anadys scientists. Pharmacology studies have
shown that ANA773 can elicit desired immune responses and that components of the response can be
modulated by both dose and schedule of administration. We have also shown in our Phase I HCV
clinical study that
ANA773 can stimulate the immune system at a tolerated dose. However, we have elected to
suspend further development of ANA773 in order to focus our resources on ANA598. Currently, we are
not actively pursuing the development of ANA773 for either indication, except to allow currently
enrolled patients in the oncology trial described below to continue to receive ANA773 until disease
progression is observed. Further development of these programs will be contingent on obtaining
additional resources allocated to such programs or entering into a collaborative or licensing
arrangement around ANA773 with a third party.
The activities we have completed to date with ANA773 are part of early drug development. In
order for us to develop ANA773 further, we will require substantial additional funding or support
from a collaborator or licensee. We do not have any plans to further the development of ANA773 at
this time. If continued, substantial further investment would be necessary in order to progress
ANA773 beyond the events referenced above and through additional clinical testing before regulatory
approval could be sought.
ANA773 for HCV
Because interferon-alpha is the foundation of the current standard of care for hepatitis C and
the current development of direct antivirals is based on the addition of such direct antivirals to
interferon-based regimens, we believe that an oral agent that stimulates interferon production with
improved tolerability could potentially displace interferon from future regimens that contain
direct antivirals. We believe that ANA773 may offer the opportunity to be such an oral interferon
replacement.
4
Table of Contents
In 2009 we concluded a Phase I clinical trial of ANA773 in HCV patients. In the final cohort
of the trial, in which patients received 2000 mg of ANA773 every other day over 10 days, the mean
maximal decline in viral load was 1.3 log10, compared to a mean maximal decline of 0.3
log10 in patients who received placebo. Five of the eight patients who received 2000 mg
ANA773 experienced a maximal decline of greater than 1 log, while none of the eight patients who
received placebo experienced a decline of greater than 1 log. The mean end-of-treatment decline was
0.6 log10 in patients who received 2000 mg ANA773 compared to 0.1 log10 in
patients who received placebo. ANA773 was well-tolerated in patients throughout the course of the
study and there were no serious adverse events reported.
ANA773 for Cancer
We have also investigated ANA773 for the treatment of cancer in a Phase I clinical trial that
we initiated in 2008. We plan for currently enrolled patients to continue to receive ANA773 until
disease progression is observed and to conclude the trial once all patients reach this point.
Through our experience with the development of ANA773 for HCV, we have shown that ANA773 can
stimulate the human immune system at a tolerated dose and we continue to believe that ANA773 holds
promise as a therapy for cancer.
TLR7 agonists are of particular interest because there is precedent for their use in cancer
and small molecule ligands for this receptor have been identified. Topical imiquimod
(AldaraÒ) is approved for the treatment of basal cell carcinoma in the United States (U.S.),
and has demonstrated activity against other tumor types including melanoma and chronic lymphocytic
leukemia. Immunotherapy is potentially synergistic with other treatment modalities and new
approaches to manipulate conventional cancer therapies to work in concert with the immune system
are being explored.
Industry Background
HCV
Based on available market data, we estimate that the global HCV market in 2009 was between $2
and 3 billion. Due to significant global prevalence and substantial unmet medical need, improving
the treatment of chronic HCV infection remains an important priority for the medical
community and the pharmaceutical industry. Many patients with chronic HCV infection do not receive
the current standard of care due to concerns about adverse events or have incomplete response to
the current standard of care. If
untreated or inadequately treated, chronic HCV infection can result in significant liver
damage (cirrhosis), liver transplantation and liver cancer.
The World Health Organization (WHO) estimates that 170 million persons globally are
chronically infected with HCV and 3 to 4 million people are newly infected each year. Cirrhosis
develops in about 10% to 20% of people with chronic infection, and liver cancer develops in 1% to
5% of people with chronic infection over a period of 20 to 30 years. It is estimated that more
than 3 million people are chronically infected with HCV in the U.S. and that only about 100,000 of
these patients are currently under treatment. The National Institutes of Health estimates that HCV
results in 10,000 to 12,000 deaths in the U.S. annually and the Center for Disease Control and
Prevention estimates that the number of deaths could increase to nearly 40,000 by 2010. HCV also
exacerbates the severity of underlying liver disease when it coexists with other hepatic
conditions. In particular, liver disease progresses more rapidly among persons with alcoholic liver
disease and HIV infection.
There is currently no vaccine available to prevent infection with HCV. The current standard of
care for treatment of chronic HCV infection is a combination of pegylated interferon-alpha and
ribavirin. Interferon-alpha is administrated by injection and results in abnormally high levels of
this cytokine circulating systemically throughout the body. Therapy with interferon-alpha causes a
number of side effects in many patients, including depression, drops in blood cell count and
flu-like symptoms, sometimes experienced during the entire year-long primary course of therapy that
is standard for treatment of patients infected with genotype 1 HCV, the most difficult patient
group to treat. These side effects may make patients feel worse than foregoing treatment, which
reduces their motivation to initiate or continue HCV therapy. Many patients take additional drugs
to treat these side effects, further increasing the cost and the risk of additional side effects to
the patient. As a result, poor compliance with the HCV course of therapy may decrease the patient
response rate.
In addition to the side effects, current therapies do not provide sustained elimination of the
virus, called “sustained virologic response” (SVR), for a large proportion of chronically infected
patients. For example, in clinical trials, approximately 50 percent of the genotype 1 patients,
which represent the largest portion of HCV patients in the U.S., Europe and Japan, do not achieve
sustained virologic response six months after the end of the treatment. Due to the lack of
alternative treatments, patients without a sustained virologic response have no other treatment
option but to undergo a second 48-week course of interferon-alpha-based therapy with a different
brand of interferon-alpha. This second course of therapy subjects the relapsed patient to a similar
risk of side effects as the previous course of therapy and offers the benefit of SVR in only a
small fraction of patients who complete the 48 week treatment.
5
Table of Contents
In response to the limitations of existing treatments for HCV infection, direct antiviral
therapies (including protease, polymerase and NS5a inhibitors) have emerged as a potential addition
to or alternative to the current standard of care. Unlike interferons, which work by stimulating
the immune system’s response to viral infection, HCV direct antivirals directly target the virus by
inhibiting the protease, polymerase or NS5a region of the virus. Accordingly, direct antivirals
have the potential to significantly improve treatment outcomes, when added to the standard of care
in difficult-to-treat patients, including patients infected with HCV genotype 1. The addition of
direct antivirals to the standard of care could also lead to shorter treatment duration, which
could increase patient compliance. While direct antivirals will likely initially be used in
combination with pegylated interferon-alpha and ribavirin, it may be possible eventually to replace
one or both components of the current treatment regimen with a combination of oral therapies
directed at HCV, including protease, polymerase and NS5a inhibitors.
Quantification of viral concentration (viral load) in the blood is an accepted surrogate of
clinical effect in viral diseases. New treatments are evaluated on the ability to decrease or
eliminate detectable viral particles in blood. With viral load as an accepted surrogate, proof of
concept in the treatment of viral diseases can be obtained in Phase I human clinical trials. We
believe this early proof of concept results in a higher probability of success post Phase I than
the probability of success associated with drug development in many other therapeutic areas.
Cancer
Cancer remains a disease with significant unmet medical need. Each year, an estimated 12
million people worldwide are diagnosed with cancer and more than half will eventually die from
their disease. According to the American Cancer Society, the number of new cancer cases in the
United States is projected at 1.5 million for 2009, and approximately one out of every two men, and
one out of every three women, will develop cancer during his or her lifetime. Cancer accounts for
nearly one-quarter of all deaths in the United States, exceeded only by heart disease.
Cancer Treatment Today
Current treatments for cancer include surgery, chemotherapy, and radiation, as well as small
molecules, antibodies, hormone therapy, and other targeted agents. Surgical and radiation
treatments are limited in their effectiveness because they treat the tumor at a specific site, may
not remove all the cancer cells, and are not effective if the cancer has spread beyond its initial
site. Chemotherapy can treat the cancer at multiple sites, but causes severe side effects because
it destroys healthy cells and tissues as well as cancer cells. In many cases, chemotherapy can only
reduce tumors in size and not eliminate them completely, resulting in disease recurrence. Targeted
molecular therapies, including antibody and small molecule therapies, have shown promise, but
typically are most effective for only subsets of the patient population. Furthermore, all drug
therapies, both new and old, have been vulnerable to the emergence of tumor resistance and disease
recurrence.
Several clinical observations support the importance of tumor immune surveillance in humans.
The increased risk of tumor development in immunosuppressed patients, cases of spontaneous tumor
regression and the presence of tumor-reactive T cells and B cells correlating with improved
prognosis all point to a role for the immune system in controlling tumor growth. Immunotherapy has
had success in treating certain tumors and this approach remains of interest for improving cancer
treatment options. Immunotherapy is potentially synergistic with other treatment modalities and new
approaches to manipulate conventional cancer therapies to work in concert with the immune system
will be explored. Rational combinations and sequences of therapy, coupled with treatment strategies
based on emerging understanding of immunobiology, may result in therapies that control and/or
eradicate established cancer.
Toll-Like Receptors
Toll-like receptors — or TLRs — are a relatively new scientific discovery, though their
origins date back hundreds of millions of years. TLRs evolved as a way to protect organisms against
pathogens such as viruses and bacteria. This defense mechanism has proven so effective that it is
an integral part of the human immune system today and a promising target for innovative new
medicines.
In 1997, the first human TLR was cloned. To date, scientists have discovered 10 TLRs in
humans, each recognizing generic molecular patterns associated with a variety of invading
pathogens.
6
Table of Contents
TLR Agonists in Viral Diseases
Certain TLRs are responsible for fighting bacterial and fungal infections; others respond
specifically to viral infections.
Unlike adaptive immunity, which enables the immune system to remember and fight specific
infections that it has encountered before, innate immunity is the ability to recognize foreign
invaders upon their very first meeting. This function is regulated in part by TLRs, a family of
proteins that serve as a first line of defense in the body.
Once a TLR recognizes a particular pathogen, it launches a dual assault. First, it triggers
the body’s innate immunity, initiating an inflammatory response to fight the invader that includes
induction of interferon, a natural disease fighter that is the basis for many approved products. It
then alerts and educates the body’s adaptive immune system so that it will recognize the pathogen
in the future. If TLRs fail, the body is left vulnerable to infection.
TLR Agonists in Cancer
As key regulators of both innate and adaptive immune responses, TLRs have been shown in
research studies to affect several diseases, including cancer. Clinical studies have demonstrated
that activation of TLR7 is effective in treating certain cancers that appear on the skin.
Specifically, topical imiquimod (Aldara®) is approved for the treatment of superficial basal cell
carcinoma. Unfortunately, however, imiquimod is poorly tolerated when administered orally, limiting
its utility for broader indications requiring systemic exposure.
Additional justification for the investigation of TLR7 agonists for the treatment of cancer
comes from the many studies conducted with TLR9 agonists. TLR7 and TLR9 agonists share common
signaling pathways, partially overlap in cell-type expression, and have comparable direct and
indirect activities as immunostimulants. A large body of data exists from animal models and human
studies suggesting the potential utility of appropriately modified natural agonists of TLR9 either
in monotherapy or combination therapy for the treatment of cancer. TLR7 and TLR9 agonists are,
however, administered differently to patients: TLR7 agonists can be administered orally, while TLR9
agonists are thus far only injectable.
Our Strategy
The key elements of our strategy include the following:
| • | Advance the Development of ANA598 in HCV. We are developing ANA598, a non-nucleoside
inhibitor of the HCV NS5B polymerase. During 2010 we intend to: |
| o | Obtain 4-week data from the 400 mg bid cohort in the Phase II clinical trial in HCV infected patients, | ||
| o | Obtain 12-week data from the 400 mg bid cohort in the Phase II clinical trial in HCV infected patients, and | ||
| o | Obtain initial SVR data from patients able to stop SOC at week 24. |
| • | Pursue the development of novel, high quality product candidates in major disease areas.
We select our product candidates based on demonstrated properties that suggest the
potential to change treatment paradigms and become important products in time. Our strategy
is to couple high quality candidates with a disciplined investment approach, pursuing time-
and cost-efficient paths to obtaining clinical data. During 2010 we intend to identify a
non-nucleoside inhibitor of the HCV NS5B polymerase as a pre-clinical candidate that could
be suitable as a next generation agent to follow ANA598. |
||
| § | Opportunistically Explore Strategic Alliances around our product candidates. We intend
to explore potential strategic alliances and other transactions around ANA598 and ANA773. |
We currently have no ongoing collaborations.
7
Table of Contents
Our Development Programs
ANA598 for HCV
ANA598 is a direct antiviral that blocks the hepatitis C virus’ ability to multiply by
inhibiting the viral RNA polymerase. ANA598 belongs to a class of direct antivirals referred to as
non-nucleosides. We believe non-nucleosides will become an important component of future
combination regimens used to treat HCV infection, similar to the role played by non-nucleoside
inhibitors in HIV therapy. In 2007 we selected ANA598 as a development candidate. This selection
represented the culmination of a comprehensive structure-based drug design program directed towards
the viral RNA polymerase. ANA598 has demonstrated positive antiviral response and a favorable
safety and tolerability profile in the ongoing Phase II combination trial, as described below.
ANA598 has also completed two long-term chronic toxicology
studies (26 weeks duration in rats and 39 weeks duration in monkeys) with favorable results.
Furthermore, ANA598 has demonstrated the preclinical properties, including potency,
pharmacokinetics and early safety, that we believe are prerequisites for successful development in
the HCV area.
We believe that non-nucleoside NS5B polymerase inhibitors offer an exciting potential new way
to target treating HCV infection, as part of combination regimens which may include other direct
antivirals (such as protease inhibitors, nucleoside polymerase inhibitors and/or NS5a inhibitors)
and/or immunomodulators (such as pegylated interferon). We believe that polymerase inhibitors have
the potential to be equally important components of future regimens as protease inhibitors, which
is another class of HCV direct antivirals currently in clinical development by a number of
companies, including Vertex (with Mitsubishi and Johnson & Johnson) and Merck. Historically, it has
been challenging to identify non-nucleoside polymerase inhibitors that display both potency and
sustained drug levels in blood. With ANA598, we believe we have created a product candidate that
has the potential to overcome this challenge. We believe that we have the opportunity to be
competitive in the effort to develop non-nucleoside polymerase inhibitors for the treatment of HCV,
since, to our knowledge the number of non-nucleosides in development is smaller than the number of
potentially attractive combinations that can be formed with attractive protease inhibitors and
nucleoside polymerase inhibitors in development. We believe that the future evolution of HCV
therapy will likely include protease inhibitors, nucleosides and non-nucleosides used in various
combinations. Therefore, we view ANA598 as complementary to, rather than competitive with, protease
inhibitors and nucleosides that are currently in development as HCV therapies.
Preclinical evaluation of ANA598 required for initiation of clinical investigation was
completed in the first quarter of 2008, leading to submission of an Investigational New Drug (IND)
to the Food and Drug Administration (FDA), subsequent allowance of the IND by the FDA and
initiation of clinical investigation in the second quarter of 2008. In December 2008, we announced
that the FDA granted fast track designation to ANA598 for the treatment of chronic HCV infection.
We have completed three Phase I studies of ANA598 that have demonstrated potent antiviral activity
and good tolerability. We are currently conducting a Phase II study of ANA598 in combination with
current standard of care.
In the ongoing Phase II study, treatment-naïve genotype 1 patients are to receive ANA598 or
placebo in combination with SOC for 12 weeks at dose levels of 200 mg bid or 400 mg bid, each with
a loading dose of 800 mg bid on day one. After week 12, patients are to continue receiving SOC
alone. Patients who achieve undetectable levels of virus at weeks four and 12 will be randomized to
stop all treatment at week 24 or 48. The primary endpoint of the study is the proportion of
patients who achieve undetectable levels of virus at week 12 (defined as cEVR). Additional
endpoints include safety and tolerability as well as the proportion of patients with undetectable
levels of virus at week four (defined as RVR). Patients will be followed for 24 weeks after
stopping therapy to determine the rate of SVR. Approximately 90 patients were to be and have been
enrolled in this study – with approximately 30 patients receiving ANA598 and 15 receiving placebo
at each dose level. The study is being conducted at a number of clinical sites in the United
States.
In December 2009, we reported positive initial antiviral response and safety results from the
200 mg bid dose cohort based on a planned interim analysis of data at four weeks. In the group
receiving ANA598 added to SOC, there was a steady increase in the percentage of patients with
undetectable levels of virus from week one through week four, with 56% of patients achieving
undetectable levels of virus (defined as <15 IU/ml) at week four, referred to as RVR, compared to
20% of patients receiving placebo plus SOC achieving an RVR. In February 2010, we reported positive
preliminary antiviral response and safety results at 12 weeks from the 200 mg bid dose cohort. 73%
of patients receiving ANA598 at 200 mg bid in combination with SOC achieved undetectable levels of
virus (<15 IU/ml) at week 12, referred to as cEVR, compared to 71% of patients receiving placebo
plus SOC achieving a cEVR. Based on historical reports of cEVR rates for SOC control arms in larger
HCV trials, which generally range from 45% to 60%, the 71% seen in our 200 mg bid dose cohort
appears to be anomalously high and may be due to the small number of patients in the control arm.
Also, the 200 mg bid 12 week data showed the ability for ANA598 to significantly accelerate the
rate of patients achieving undetectable levels of virus when added to current treatment. In
clinical trials with other agents, accelerating the rate of achieving undetectable levels of virus
is associated with an increased chance of achieving a sustained viral response, or SVR. Although we
cannot
predict whether this will be the case with ANA598, we hope to show the same benefit at the
conclusion of this Phase II trial. Further, no patient in the 200 mg bid dose cohort experienced
viral rebound (defined as an increase in viral load of greater than 1 log10 from a prior
measurement) while receiving ANA598.
8
Table of Contents
In the ongoing Phase II study, ANA598 at 200 mg bid demonstrated a favorable safety and
tolerability profile through 12 weeks, although definitive conclusions regarding safety and
tolerability cannot be made until additional results in more patients and potentially over longer
duration are known. There were no serious adverse events reported and the profile of adverse events
reported was as expected for patients receiving SOC alone, with comparable rates observed between
the ANA598 and control arms. The incidence of rash was comparable between groups and consistent
with historical reports of rash rates due to interferon and ribavirin. In the ANA598 arm, 41% of
patients (12/29) developed a rash while 33% (5/15) of patients in the control group developed a
rash. Eleven of the twelve instances of rash in the ANA598 arm were mild. One patient in the ANA598
arm who had achieved undetectable levels of virus by week four developed a grade 3 rash after week
seven which began resolving rapidly upon stopping all study medication. In this instance, the rash
was characterized as grade 3 due to the extent of body surface covered by the rash, which was
maculopapular in nature (red spots, some raised). In accordance with the study design, this patient
resumed interferon/ribavirin alone, and continued in the study and maintained undetectable levels
of virus through week 12. The five instances of rash in the control arm were mild.
We are currently dosing patients at 400 mg bid and expect to analyze and release four week
data from this dose cohort prior to the end of the first quarter of 2010 and expect to analyze and
release 12-week data from this dose cohort during the second quarter of 2010. Based on preliminary
pharmacokinetic data from the 200 mg bid dose cohort, which show ANA598 levels in the patients’
blood through four weeks, it appears that the 800 mg loading dose given twice on day one has had
the desired effect of rapidly dropping viral load and minimizing the difference in response between
1a genotype and 1b genotype patients. Because this loading dose is also being administered to
patients in the 400 mg bid cohort, we expect to see a similar early response in patients receiving
ANA598 in that cohort. From the data seen to date, it is also apparent that patients in the 200 mg
bid cohort achieved undetectable levels of virus after four weeks of dosing even when the level of
ANA598 in their blood was among the lowest levels observed across patients. Accordingly, it may be
that, for a substantial number of patients, the antiviral response will plateau at 200 mg bid when
administered with an effective loading dose, and that we may not see an increase in antiviral
response in the 400 mg bid cohort. If this turns out to be the case and if 200 mg bid is identified
as the optimal dose to take forward in future clinical trials, we believe that the favorable
profile we have seen at the low dose of 200 mg bid will facilitate combining ANA598 with other
direct antivirals.
In January and April 2009, we announced data from the ANA598 Phase I study in which ANA598 was
dosed as monotherapy over three days in HCV patients at twice-daily doses of 200 mg, 400 mg or 800
mg. The data from the study demonstrated potent antiviral activity and good tolerability of ANA598
as a single agent at all dose levels. The median viral load reduction over three days ranged from
2.4 to 2.9 log10 in the three dose groups studied. No patient at any dose level showed evidence of
viral rebound while on ANA598 and there were no serious adverse events reported. Also in April
2009, we reported results from a 14-day study of ANA598 in healthy volunteers. ANA598 was generally
well-tolerated in all cohorts in the healthy volunteer study with no serious adverse events. Three
instances of mild-to-moderate rash were observed at the higher dose levels. Pharmacokinetic results
from this trial confirmed the plasma half-life of ANA598 of approximately 24 hours, and
demonstrated that steady-state levels of ANA598 in plasma are reached after six to seven days of
dosing.
During 2009 we also completed two long-term chronic toxicology studies of ANA598 (26 weeks
duration in rats and 39 weeks duration in monkeys) with favorable results. The completed toxicology
studies confirm the favorable toxicology profile of ANA598 and support dosing durations of as long
as one year if desirable in future clinical studies.
In 2009, we presented in vitro data showing that combinations of ANA598 with interferon-alpha,
the protease inhibitor telaprevir and the nucleoside polymerase inhibitor PSI-6130 appear to be
synergistic. Synergistic means that the actual combined effect of the two agents is greater than
would be predicted from
simply adding the effects of each agent alone. These studies also show that ANA598 retained
activity in vitro against mutants known to confer resistance to other classes of direct antivirals,
including protease inhibitors, nucleoside inhibitors and non-nucleosides that, through virtue of
binding at a different site than ANA598, display a resistance profile distinct from that of ANA598.
We also showed that genotypic mutations resistant to ANA598 appear to be fully susceptible to
interferon-alpha, telaprevir and PSI-6130. Previously, we have also presented data demonstrating
synergy between ANA598 and immunoregulatory proteins termed “cytokines” induced by ANA773, Anadys’
TLR7 agonist oral prodrug which has also been investigated for hepatitis C.
ANA773 for HCV
Because interferon-alpha is the foundation of the current standard of care for hepatitis C and
the current development of direct antivirals is based on the addition of such direct antivirals to
interferon-based regimens, we believe that an oral agent that stimulates interferon production with
improved tolerability could potentially displace interferon from future regimens that contain
direct antivirals. We believe that ANA773 may offer the opportunity to be such an oral interferon
replacement.
9
Table of Contents
In 2009 we concluded a Phase I clinical trial of ANA773 in HCV patients. In the first three
cohorts of the patient portion of this trial, HCV patients received oral ANA773 or placebo at every
other day over 28 days, at doses of 800 mg, 1200 mg or 1600 mg, with six subjects receiving ANA773
and two receiving placebo in each cohort. At these doses, data showed an encouraging trend toward
viral load reduction. We then amended the protocol to provide for a higher dose. In this final
cohort of the trial, in which patients received 2000 mg of ANA773 every other day over 10 days, the
mean maximal decline in viral load was 1.3 log10, compared to a mean maximal decline of 0.3 log10
in patients who received placebo. Five of the eight patients who received 2000 mg ANA773
experienced a maximal decline of greater than 1 log, while none of the eight patients who received
placebo experienced a decline of greater than 1 log. The mean end-of-treatment decline was 0.6
log10 in patients who received 2000 mg ANA773 compared to 0.1 log10 in patients who
received placebo. ANA773 was well-tolerated in patients throughout the course of the study and
there were no serious adverse events reported.
Results from pre-clinical pharmacology studies showed that ANA773 elicited desired immune
responses and that the profile of response could be modulated by both dose and schedule of
administration. Results of 13-week GLP toxicology studies showed that with every-other-day dosing
of ANA773, immune stimulation of a magnitude believed to confer therapeutic potential could be
achieved without adverse toxicology findings. The immune stimulation observed with every-other-day
dosing of ANA773 in monkeys included induction of interferon-alpha and interferon dependent
responses at levels that were sustained over 13 weeks of dosing.
ANA773 for Cancer
We have also investigated ANA773 as a potential treatment for cancer. ANA773 stimulates the
body’s immune system through activation of the TLR7 receptor. Through our experience with the
development of ANA773 for HCV we have shown that ANA773 can stimulate the human immune system at a
tolerated dose.
The pharmacologic consequences of TLR7 activation are broad and include induction of cytokines
such as interferon-alpha as well as activation of immune effector cell populations known as natural
killer (NK) cells and cytotoxic T lymphocytes (CTLs). The cytokine induction and cellular
activation mechanisms both offer the potential for direct control of tumor cell growth.
Furthermore, there is evidence to support the concept that activation of NK and CTL cells may be
beneficial in enhancing the effect of existing cancer therapies, including monoclonal antibodies
and certain chemotherapies.
There is a degree of precedent to support the use of the TLR7 mechanism in cancer. One
marketed product that works by stimulating the immune system via the TLR7 mechanism is imiquimod
(Aldara®). While the FDA-approved indications for imiquimod are limited to topical treatment of
skin diseases, such
as superficial basal cell carcinoma, actinic keratosis and warts induced by the human
papillomavirus, there are several reports of imiquimod demonstrating activity against skin
metastases from solid tumors, such as breast cancer. However, to date no one has successfully
developed an oral TLR7 agonist for cancer. We believe the combination of our prodrug approach,
described below, and our understanding of TLR7 pharmacology provides us an opportunity to utilize
the TLR7 mechanism to systemically treat a broad spectrum of cancers, including solid tumors and B
cell diseases, with oral administration of ANA773.
ANA773 is a prodrug of an active TLR7 agonist we believe may confer benefit in cancer
treatment. As a prodrug, ANA773 itself does not activate the TLR7 receptor. Rather, the body’s
metabolic processes transform ANA773 to an active form after absorption from the digestive tract,
resulting in the active TLR7 agonist circulating in the blood. Both ANA773 and the active agent it
delivers were designed and synthesized by our scientists. The use of a prodrug provides for
efficient delivery of the active agent to the bloodstream and avoids undesirable effects of an
active TLR7 agent in the digestive tract prior to absorption. We have shown in multiple preclinical
studies that oral delivery of ANA773 produced the desired blood concentrations of the active agent
and provided immune stimulation. In our clinical investigations, we have administered ANA773
orally.
We have reported the activity of ANA773 and its active form at multiple scientific
conferences. In October 2007, we presented data showing that activation of TLR7 in vivo by the
active form of ANA773 leads to the expected cellular responses, including activation of NK cells
and CTLs. Earlier in 2007, we presented data from an in vitro study demonstrating that ANA773 and
its active metabolite stimulate secretion of interferon alpha and enhance direct tumor cell killing
by NK cells. In addition to enhancing direct NK cell killing, the active metabolite of ANA773 also
enhanced the ability of rituximab, an antibody against CD20, to trigger immune-mediated cell
killing of transformed B cells. We have also presented data from in vivo preclinical studies
showing that the schedule of administration had a significant effect on the profile of immune
stimulation induced by ANA773. Alternating dosing with periods of no dosing led to more robust NK
cell activation and more stable levels of interferon-alpha induction, compared to chronic daily
administration. We anticipate that different dosing schedules may be required in different tumor
settings if ANA773 is pursued in the future as a therapy for cancer.
10
Table of Contents
Manufacturing and Supply
All of our manufacturing is out-sourced to third parties, with control by our internal
managers. We rely on third-party manufacturers to produce sufficient quantities of ANA598 and
ANA773 for use in clinical trials. We intend to continue this practice for any future clinical
trials and large-scale commercialization of ANA598 and/or ANA773. Both ANA598 and ANA773 are
small-molecule drugs. Historically, these drugs have been simpler and less expensive to manufacture
than biologic drugs.
Intellectual Property
Our policy is to pursue patents and to otherwise endeavor to protect our technology,
inventions and improvements that are commercially important to the development of our business. We
also rely upon trade secrets that may be important to the development of our business.
Our success will depend in large part on our ability to:
| • | Obtain and maintain patent and other proprietary protection for the technology, inventions and improvements we consider important to our business; | ||
| • | Defend and enforce our patents; | ||
| • | Preserve the confidentiality of our trade secrets; and | ||
| • | Operate without infringing the patents and proprietary rights of third parties. |
We hold issued patents and pending patent applications in the United States and in foreign
countries we deem appropriate, covering intellectual property developed as part of our research and
development programs. Our intellectual property holdings as of December 31, 2009, include, but are
not limited to, the United States and foreign patents and patent applications described below.
In our HCV non-nucleoside polymerase program, we hold two issued United States patents related
to our ANA598 program (patent numbers 7,462,611 and 7,582,754) which expire in 2027, one issued
patent in Malta expiring in 2028 and 81 pending United States and/or foreign patent applications
(in Australia, Brazil, Canada, China, the European Patent Convention, India, Japan, Mexico, Taiwan
and certain other foreign jurisdictions) covering ANA598 and/or other non-nucleoside NS5B
polymerase inhibitor compounds and the manufacture, pharmaceutical compositions, and methods of use
of these compounds.
In our ANA773 program, we hold one issued United States patent (patent number 7,560,544)
expiring in 2026, two issued patents in Malta with expiration dates of 2025 and 2027, and 75
pending United States and/or foreign patent applications (in Australia, Brazil, Canada, China, the
European Patent Convention, India, Japan, Mexico, Taiwan and certain other foreign jurisdictions)
covering ANA773 and related compounds and prodrugs, and the manufacture, pharmaceutical
compositions, and methods of use of these compounds.
We also hold one United States patent (patent number 7,576,068) expiring in 2026, one issued
patent in Morocco expiring in 2024, one issued patent in Georgia expiring in 2024 and 14 pending
United States and/or foreign patent applications (in Australia, Brazil, Canada, China, the European
Patent Convention, India, Japan, Mexico, Taiwan and certain other foreign jurisdictions) that
relate to methods of use of certain TLR7 agonists and TLR7 agonist prodrugs. In addition, we hold
patents and patent applications in the United States and foreign countries covering composition of
matter and methods of use of certain other TLR7 agonists and TLR7 agonist prodrugs, with patent
expiration dates beginning in 2022.
We intend to continue using our scientific expertise to pursue and file patent applications on
new developments with respect to uses, methods and compositions of matter in order to enhance our
intellectual property position in our areas of therapeutic focus.
We intend to aggressively prosecute our patent applications and enforce and defend our patents
and otherwise protect our proprietary technology. Although we believe our rights under patents and
patent applications provide a competitive advantage, the patent positions of pharmaceutical and
biotechnology companies are highly uncertain and involve complex legal and factual questions. We
may not be able to develop patentable products or processes, and may not be able to obtain patents
from pending applications. Even if patent claims are allowed, the claims may not issue, or in the
event of issuance, may not be sufficient to protect the technology owned by or licensed to us. Any
patents or patent rights that we obtain may be circumvented, challenged or invalidated by our
competitors.
11
Table of Contents
We also rely on trade secrets and proprietary know-how, especially when we do not believe that
patent protection is appropriate or can be obtained. Our practice is to require our employees,
consultants, outside scientific collaborators, sponsored researchers and other advisors to execute
confidentiality agreements upon the commencement of employment or other relationships with us.
These agreements generally provide that all confidential information developed by or made known to
the individual during the course of the individual’s relationship with us is to be kept
confidential and not disclosed to third parties. In the case of employees, the agreements generally
provide that all discoveries, developments, inventions and other intellectual property conceived or
reduced to practice by the individual while employed by us will be our exclusive property. In the
case of advisors and consultants, the agreements generally provide that all discoveries,
developments, inventions, and other intellectual property conceived or reduced to practice by the
individual as a result of performance of services for us and not resulting from research related to
work supported by another entity with which the individual is party to a confidentiality agreement,
shall be our exclusive property. These agreements may not effectively prevent disclosure of
confidential information nor result in the effective assignment to us of intellectual property, and
may not provide an adequate remedy to us in the event of unauthorized disclosure of confidential
information or other breaches of the agreements.
Competition
The biotechnology and pharmaceutical industries are very competitive and subject to rapid and
significant technological change. Our product candidates, if approved for sale, will compete with
existing therapies. In addition, a number of companies are pursuing the development of
pharmaceuticals that target the same diseases and medical conditions that we are targeting. We
believe that a significant number of drugs are currently under development and may become available
in the future for the treatment of HCV and cancer. Due to the level of focus on developing
treatments for these indications, ongoing research efforts are intense and new treatments are being
sought out and developed by our competitors. Some of these products use therapeutic approaches that
may compete directly or indirectly with ANA598 or ANA773. In addition, less expensive generic forms
of currently marketed drugs could lead to additional competition upon patent expiration or
invalidations.
HCV
Treating HCV with Interferon-based Therapies
Current standard treatments for HCV include an interferon-based product combined with
ribavirin. Although interferons result in antiviral effects, they are injectable products and cause
numerous side effects. Next generation interferon-based products, so-called pegylated interferons,
were developed to provide an improved dosing regimen and are approved as once-per-week injected
products. Currently approved therapies for the treatment of HCV infection include Peg-Intron
(pegylated interferon-alpha-2b) and Intron-A (interferon-alpha-2b), which are marketed by Merck,
Pegasys (pegylated interferon-alpha-2a) and Roferon-A (interferon-alpha-2a), which are marketed by
Roche and several branded and generic versions of ribavirin.
Many patients experience unpleasant side effects when receiving interferon-based products,
including flu-like symptoms such as fatigue, pyrexia, myalgia, cough, headache, and rigors,
psychiatric reactions, such as depression, irritability and anxiety, as well as neutropenia and
thyroid dysfunction. Due to the nature of HCV infection, patients may not show any symptoms from
the HCV itself when they initiate therapy. Ironically, harsh side effects often make patients feel
sicker than the disease itself. As a result, physicians often delay treatment of HCV-infected
patients until tests of liver function demonstrate initial liver degeneration due to the infection.
In clinical studies, harsh side effects have caused discontinuation of treatment in approximately
10 to 20 percent of patients. These side effects also require additional drug therapies, which
increase the cost to the patient. Further, the optimal dose, treatment length and response rates to
interferon and ribavirin therapy vary considerably based on HCV genotype and mode of therapy, i.e.,
monotherapy or combination therapy.
Direct Antivirals in Development for Treating HCV
In response to the limitations of existing treatments for HCV infection, the development of
direct antiviral therapies (including protease, polymerase and NS5a inhibitors) has emerged as a
potential addition to or alternative to the standard treatment. Unlike interferons, which work by
stimulating the immune system’s response to viral infection, HCV direct antivirals directly target
the virus by inhibiting the protease, polymerase or NS5a region of the virus. Accordingly, direct
antivirals may significantly improve treatment outcomes, when added to the standard of care in
difficult-to-treat patients, including patients infected with HCV genotype 1, relative to treatment
with the standard of care alone. The addition of direct antivirals to the standard of care could
also lead to shorter treatment duration, which could increase patient compliance. While direct
antivirals will likely initially be used in combination with pegylated interferon-alpha and
ribavirin, it may be possible eventually to replace one or both components of the current treatment
regimen with a combination of oral therapies directed at HCV, including both protease and
polymerase inhibitors.
12
Table of Contents
ANA598 belongs to a class of direct antivirals known as non-nucleoside polymerase inhibitors.
If approved, ANA598 would likely be used in combination with the current standard of care and/or
other
direct antiviral agents such as protease inhibitors, other polymerase inhibitors, or NS5a
inhibitors. Although any product currently approved or approved in the future for the treatment of
HCV infection could potentially decrease or eliminate the commercial opportunity of ANA598, we
expect that in a combination setting a non-nucleoside polymerase inhibitor would be complementary
with a protease inhibitor, a nucleoside polymerase inhibitor and an NS5a inhibitor. We believe that
other non-nucleoside polymerase inhibitors would likely be the most direct competitors of ANA598,
but depending on the resistance profiles of the compounds, it is possible that even two
non-nucleoside polymerase inhibitors could be complementary. To our knowledge, other non-nucleoside
polymerase inhibitor programs are currently under clinical evaluation by Pfizer, Gilead, Merck,
Abbott, Boehringer Ingleheim and Vertex. Further, a number of companies have non-nucleoside
polymerase inhibitor research programs.
Additional compounds in late stage clinical trials for HCV that may be complementary to or
competitive with ANA598 include Zalbin/Joulferon (albinterferon alfa-2b), in development by Human
Genome Sciences and Novartis, telaprevir, in development by Vertex Pharmaceuticals, Janssen
Pharmaceutica (Johnson & Johnson) and Mitsubishi Tanabe Pharma, boceprevir, narlaprevir and MK-7009 in development by
Merck, ITMN-191, in development by Intermune and Roche, TMC-435350, in development by Tibotec and
Medivir, BI-201335, in development by Boehringer Ingelheim, and RG7128 in development by Roche and
Pharmasset.
Immunological Agents in Development for Treating HCV
Due to the side effects and poor treatment response to interferon therapy discussed above,
there are currently a number of agents in development that could potentially replace today’s
pegylated interferons. ANA773 is an oral prodrug of a TLR7 agonist that we have evaluated for the
treatment of HCV. There are a number of agents in clinical development that could potentially
compete with ANA773 as new agents for the treatment of HCV, including, Zalbin/Joulferon
(albinterferon alfa-2b), in development by Novartis and Human Genome Sciences, and Locteron, in
development by Biolex Therapeutics, both of which are longer-acting versions of interferon alfa.
Also, in development as potential improvements to existing interferons are PEG-interferon lambda,
in development by Zymogenetics and Bristol Myers-Squibb, and omega interferon in development by
Intarcia Therapeutics. IMO-2055, a TLR9 agonist in development by Idera, and SD-101, a TLR9 agonist
in development by Dynavax Technologies are also being studied in early stage clinical trials in HCV
patients.
Cancer
Current treatments for cancer include surgery, chemotherapy, and radiation, as well as small
molecules, antibodies, hormone therapy, and other targeted agents. Surgical and radiation
treatments are limited in their effectiveness because they treat the tumor at a specific site, may
not remove all the cancer cells, and are not effective if the cancer has spread beyond its initial
site. Chemotherapy can treat the cancer at multiple sites, but causes severe side effects because
it destroys healthy cells and tissues as well as cancer cells. In many cases, chemotherapy can only
reduce tumors in size and not eliminate them completely, resulting in disease recurrence. Targeted
molecular therapies, including antibody and small molecule therapies, have shown promise, but
typically are most effective for only subsets of the patient population. Furthermore, all drug
therapies, both new and old, have been vulnerable to the emergence of tumor resistance and disease
recurrence.
ANA773 is a prodrug of a TLR7 agonist that we have evaluated for oncology indications. Any product
currently approved or approved in the future for the treatment of cancer could decrease or
eliminate the commercial opportunity of ANA773. Programs that most directly compete with ANA773 at
this time are several TLR9 agonists under evaluation for oncology indications, including IMO-2055,
in development by Idera and Merck KGaA, and a cancer program in development by Dynavax.
Competitive Risks
We are in the early stages of a Phase II study of ANA598, which was initiated following
completion of three short term Phase I studies, and we have only conducted short term Phase I studies
of ANA773.
Therefore, it is difficult to predict the efficacy, safety and tolerability that these product
candidates will demonstrate in longer term trials, alone or in combination with other agents. It is
also difficult to predict how these product candidates will interact with other product candidates
in development or on the market, until we perform combination studies. Further, it is difficult to
predict whether our product candidates will cause any toxicity issues, potential side effects, or
other negative consequences associated with their long-term use. During the course of future
clinical trials, we may discover that these product candidates are less effective, require
unacceptable dosing regimens, or have a similar side effect profile as the profile associated with
current therapies or future competitors. This may result in our product candidates being less
advantageous or less desirable from a patient and treating physician perspective as compared to
current therapies for HCV or cancer.
13
Table of Contents
We face competition from pharmaceutical and biotechnology companies both in the U.S. and
abroad. Our competitors may utilize discovery technologies and techniques or partner with
collaborators in order to develop products more rapidly or successfully than we or our future
collaborators are able to do. Many of our competitors, particularly large pharmaceutical companies,
have substantially greater financial, technical and human resources than we do and far more
experience in the discovery and development of product candidates and the commercialization of
potential products. In addition, academic institutions, government agencies, and other public and
private organizations conducting research may seek patent protection with respect to potentially
competitive products or technologies. These organizations may also establish exclusive
collaborative or licensing relationships with our competitors.
We believe that our ability to compete depends, in part, upon our ability to create, maintain
and license scientifically advanced technology. Further, we need to attract and retain qualified
personnel, obtain patent protection or otherwise develop proprietary technology or processes and
secure sufficient capital resources for the substantial time period between technological
conception and commercial sales of products based upon our technology.
We expect that competition among HCV and cancer therapies approved for sale will be based on
various factors, including improved product efficacy, safety and tolerability, ease of
administration (e.g., oral vs. intravenous administration), availability, price, reimbursement
status and patent position. Potential competitors may develop treatments for HCV or cancer that are
more effective and/or safer or more convenient than our product candidates or that would make our
technology and product candidates obsolete or non-competitive.
Government Regulations
We are subject to regulation by the FDA and comparable regulatory agencies in foreign
countries with respect to the development and commercialization of products and services resulting
from our drug discovery activities. These agencies and other federal, state and local entities
regulate research and development activities and the testing, manufacture, quality control, safety,
efficacy, labeling, storage, record keeping, advertising and promotion of these products and
services.
As an initial step in the drug approval process of pharmaceuticals, an applicant typically
conducts preclinical laboratory and animal studies of the product candidate. Following these
studies, the applicant will submit an IND (or equivalent) application to the FDA (or comparable
foreign regulatory agency). Once the IND becomes effective, the applicant can commence clinical
studies of the product candidate in humans to determine safety, tolerability and efficacy.
Following clinical studies, the marketing of a new drug requires the filing of a New Drug
Application (NDA) with the FDA and its subsequent approval (similar requirements exist within
foreign agencies). The process required by the FDA and comparable agencies before a pharmaceutical
or biologic device may be marketed in the U.S. or in any other country generally requires many
years and substantial effort and financial resources, and approval from the FDA may not be received
in a timely manner, if at all. The time required to satisfy FDA requirements or similar
requirements of foreign regulatory agencies may vary substantially based upon the type, complexity
and novelty of the product or the targeted disease. Even if a product receives regulatory approval,
later discovery of previously unknown problems with a product may result in restrictions on the
product or even complete withdrawal of the product from the market.
Under the FDA’s regulations, the clinical testing program required for marketing approval of a
new drug typically involves three sequential phases, which may overlap.
| • | Phase I: Studies are conducted on normal, healthy human volunteers or patients to determine safety, dosage tolerance, absorption, metabolism, distribution and excretion. If possible, Phase I studies may also be designed to gain early evidence of effectiveness. | ||
| • | Phase II: Studies are conducted on small groups of patients afflicted with a specific disease to gain preliminary evidence of efficacy, to determine the common short-term side effects and risks associated with the substance being tested and to determine dosage tolerance and optimal dosage. | ||
| • | Phase III: Involves large-scale studies conducted on disease-afflicted patients to provide statistical evidence of efficacy and safety and to provide an adequate basis for physician labeling. |
14
Table of Contents
Frequent reports are required in each phase, and, if unwarranted hazards to subjects are
found, the FDA may request modification or discontinuance of clinical testing until further
preclinical testing is conducted. Additional testing (Phase IV) may be conducted after FDA approval
for marketing is granted and could be designed to evaluate alternative utilizations of drug
products prior to their being marketed for such additional utilizations as well as to test for
complications resulting from long-term exposure not revealed in earlier clinical testing.
Environmental and Safety Matters
Certain of our development activities involve the controlled use of biological, hazardous and
radioactive materials and waste. We are also subject to numerous federal, state and local
environmental and safety laws and regulations, including those governing the use, manufacture,
storage, handling and disposal of hazardous materials and waste products. The cost of compliance
with and any violation of these regulations could have a material adverse effect on our business
and results of operations. Although we believe that our safety procedures for handling and
disposing of these materials comply with the standards prescribed by state and federal regulations,
we cannot assure investors that accidental contamination or injury from these materials will not
occur.
To date, compliance with laws and regulations relating to the protection of the environment
has not had a material effect on our capital expenditures or our competitive position. However, we
are not able to predict the extent of government regulation, and the cost and effect thereof on our
results of operations, which might result from any legislative or administrative action pertaining
to environmental or safety matters. In the event of contamination or injury, we could be held
liable for substantial damages or penalized with fines in an amount exceeding our resources, and
our clinical trials could be suspended. In addition, we may have to incur significant costs to
comply with future environmental laws and regulations.
Research and Development Expenses
Research and development expenses consist primarily of costs associated with the discovery,
preclinical and clinical development of our product candidates. Research and development expenses
are the primary source of our expenses and totaled $19.5 million, $26.0 million and $28.2 million
for the years ended December 31, 2009, 2008 and 2007, respectively.
Employees
As of March 1, 2010, we had 28 regular, full-time employees and six other employees, including
22 in research and development, and the balance in general and administrative positions, with 18 of
our employees holding Ph.D., M.D. or other advanced degrees. None of our employees is represented
by a labor union, and we consider our employee relations to be good.
Executive Officers of the Registrant
The following table sets forth information regarding our executive officers as of March 1,
2010:
| Name | Age | Position | ||||
Steve Worland, Ph.D.
|
52 | President and Chief Executive Officer | ||||
James L. Freddo, M.D.
|
55 | Senior Vice President, Drug Development and Chief Medical Officer | ||||
Elizabeth E. Reed, J.D.
|
39 | Senior Vice President, Legal Affairs and General Counsel | ||||
Peter T. Slover, CPA
|
35 | Vice President, Finance and Operations | ||||
Steve Worland, Ph.D. was appointed President and Chief Executive Officer and a member of the
Board of Directors in 2007. Dr. Worland joined Anadys in 2001 as Chief Scientific Officer and
served as President, Pharmaceuticals prior to being named CEO. Prior to joining Anadys, Dr. Worland
was Vice President, Head of Antiviral Research at Agouron Pharmaceuticals, a Pfizer Company. Dr.
Worland was at Agouron from 1988 through the acquisition of Agouron by Warner-Lambert in 1999,
where he held various positions and responsibilities that culminated with him assuming global
responsibility for anti-infective strategy as Vice President for Warner-Lambert. At Agouron,
Warner-Lambert and Pfizer, Dr. Worland led teams responsible for discovery and clinical development
in the areas of HIV, HCV and respiratory infections. Dr. Worland was a National Institutes of
Health Postdoctoral Fellow in Molecular Biology at Harvard University from 1985 to 1988. He
received his B.S. with highest honors in Biological Chemistry from the University of Michigan and
his Ph.D. in Chemistry from the University of California, Berkeley.
15
Table of Contents
James L. Freddo, M.D. joined us in July 2006 as Chief Medical Officer and was named Senior
Vice President, Drug Development and Chief Medical Officer in July 2008. Prior to joining Anadys,
Dr. Freddo was Vice President, Clinical Site Head and Development Site Head, Pfizer Global Research
and Development, La Jolla. Previously at Pfizer, he was Executive Director, Site Therapeutic Area
Leader, Clinical Development, Oncology. While at Pfizer, Dr. Freddo led the team responsible for
the registration of Sutent® (sunitinib malate), a drug approved by the FDA in January 2006 for
treating advanced kidney cancer and gastrointestinal stromal tumors. Prior to Pfizer, Dr. Freddo
held a variety of senior management positions at Wyeth-Ayerst Research from December 1996 until
June 2002, including Senior Director, Oncology, Senior Director, Infectious Diseases, and Senior
Director, Transplantation Immunology. Dr. Freddo currently serves as a member of the Board of
Directors of InfuSystem Holdings, Inc., a healthcare services company. He holds a B.S. degree in
Medical Technology from the State University of New York at Stony Brook, and a M.D. degree from the
University of North Carolina, where he also completed his fellowship training.
Elizabeth E. Reed, J.D joined us in October 2001 and was named Senior Vice President, Legal
Affairs and General Counsel in August 2009. Ms. Reed has also served as our Corporate Secretary
since January 2002. Previously, Ms. Reed served as our Vice President, Legal Affairs from December
2006 to August 2009, as our Senior Director, Legal Affairs from December 2002 to December 2006 and
as our Director of Legal Affairs from October 2001 to December 2002. Prior to joining us, Ms. Reed
was associated with the law firms of Cooley Godward LLP and Brobeck, Phleger & Harrison LLP. Ms.
Reed is a member of the State Bar of California and received her B.S. in Business Administration
with an emphasis in finance from the Haas School of Business at the University of California,
Berkeley and holds a J.D., cum laude, from Harvard Law School.
Peter T. Slover joined us in April 2004 and was named Vice President, Finance and Operations
in July 2009. Mr. Slover joined us as Manager of Financial Reporting and served in this position
through December 2005. From January 2006 to July 2006, Mr. Slover served as the Company’s Senior
Manager, Financial Reporting and Internal Controls, from August 2006 to December 2006 as our
Associate Controller, from January 2007 to December 2008 as our Controller and from January 2009 to
July 2009 as our Senior Director, Finance and Corporate Controller. Prior to joining the Company,
Mr. Slover began his career as an auditor at KPMG LLP where he spent seven years in public
accounting. Mr. Slover is a licensed Certified Public Accountant in the State of California. Mr.
Slover received a B.S. degree in Business Administration from Shippensburg University.
Company Website
We file annual, quarterly, current reports, proxy statements and other information with the
Securities and Exchange Commission. Our primary website can be found at
http://www.anadyspharma.com. We make available free of charge at this website (under the
“Investors — SEC Filings” caption) all of our reports filed or furnished pursuant to Section 13(a)
or 15(d) of the Securities Exchange Act of 1934, including our Annual Report on Form 10-K, our
Quarterly Reports on Form 10-Q and our Current Reports on Form 8-K and amendments to those reports.
These reports are made available on the website as soon as reasonably practicable after their
filing with, or furnishing to, the Securities and Exchange Commission. Furthermore, we also make
available on our website free of charge, and in print to any shareholder who requests it, the
Committee Charters for our Audit, Compensation, and Corporate Governance and Nominating Committees,
as well as the Code of Business Conduct and Ethics that applies to all directors, officers and
employees of the Company. Amendments to these documents or waivers related to the Code of Business
Conduct and Ethics will be made available on our website as soon as reasonably practicable after
their execution.
The Company was incorporated in Delaware in September 1992 as ScripTech Pharmaceuticals, Inc.,
and in 1994 we changed our name to Scriptgen Pharmaceuticals, Inc. In May 2000, following the
addition of a substantially new management team and the infusion of new capital, product candidates
and technologies, we changed our name to Anadys Pharmaceuticals, Inc.
| Item 1A. | Risk Factors |
You should consider carefully the following information about the risks described below,
together with the other information contained in this Annual Report and in our other public filings
before making any investment decisions regarding our stock. If any of the following risks actually
occurs, our business, financial condition, results of operations and future growth prospects would
likely be materially and adversely affected. In these circumstances, the market price of our common
stock would likely decline, and you may lose all or part of the money you paid to buy our common
stock.
Risks Related to Our Business
Any significant set-back regarding, or the failure of, ANA598 will have a large negative
impact on our business and stock price.
Currently, we are actively pursuing only the development of ANA598. As a result, our
development portfolio entails a highly concentrated risk of failure. If the timing or results of
clinical trials and non clinical studies of ANA598 do not meet our, your, analysts’ or others’
expectations, the market price of our common stock could decline significantly. Any significant
set-back regarding, or the failure of, ANA598 will have a significant negative impact on us and our
stock price.
16
Table of Contents
We may be unable to enter into future strategic transactions, and in particular
transactions around ANA598 or ANA773, on terms acceptable to us, or at all.
Our near and long-term viability will depend in part on our ability to successfully
establish strategic transactions with pharmaceutical and biotechnology companies. Since we do not
currently possess the resources necessary to independently fully develop and commercialize ANA598
or ANA773, we either will need to develop or acquire these resources on our own, which will require
substantial funding, time and effort, or will need to enter into collaborative agreements to assist
in the development and commercialization of these potential products. If we fail to establish
collaborations or licensing arrangements on acceptable terms, we may need to delay or terminate one
or more of our programs. Even if we successfully establish new collaborations, these relationships
may never result in the successful development or commercialization of any product candidates or
the generation of sales or royalty revenue.
More specifically, we elected to initiate the Phase II study of ANA598 prior to further
exploring interest in a possible transaction around ANA598. In addition, we plan to seek
outlicensing opportunities as a way
to potentially continue advancing the development of ANA773. There is no guarantee that we will
enter into a future transaction around ANA598 or ANA773 on favorable terms, or at all, or that
discussions will initiate or progress on our desired timelines. Completing transactions of this
nature is difficult and time-consuming. Potentially interested parties may decline to re-engage or
may terminate discussions based upon their assessment of our competitive, financial, regulatory or
intellectual property position or for any other reason. Furthermore, we may choose to defer
consummating a transaction relating to ANA598 until additional data is obtained. If we do not
actively pursue a transaction around ANA598 until we have additional data, we and our stockholders
will bear the risk that ANA598 will fail prior to any future transaction.
We will need additional funding and may be unable to raise capital when needed, which
would force us to delay, reduce or eliminate our development programs.
Our December 31, 2009 cash, cash equivalents and marketable securities balance was $20.5
million. We believe that this balance will be sufficient to satisfy our anticipated operational
cash needs for at least the next 12 months. However, we will need to seek additional funding in
order to conduct future development activities. There is no guarantee that additional funding will
be available to us on acceptable terms, or at all. If funds are not available, we may be required
to delay, reduce the scope of or eliminate one or more of our development programs.
In addition, we will need to raise additional capital if we choose to conduct certain
activities, including:
| • | fund our development programs; | ||
| • | acquire rights to products or product candidates, technologies or businesses; | ||
| • | establish and maintain manufacturing, sales and marketing operations; and | ||
| • | commercialize our product candidates, if any, that receive regulatory approval. |
Our future funding requirements will depend on, and could increase significantly as a
result of many factors, including:
| • | the progress of our clinical trials; | ||
| • | the progress of our nonclinical development activities; | ||
| • | our ability to establish and maintain strategic alliances; | ||
| • | the costs involved in enforcing or defending patent claims and other intellectual property rights; | ||
| • | the pace and timing of development activities conducted under joint development arrangements we may establish; |
17
Table of Contents
| • | the cost and timing of regulatory approvals; | ||
| • | the costs of establishing or expanding manufacturing, sales and distribution capabilities; | ||
| • | the costs related to development and manufacture of pre-clinical, clinical and validation lots for regulatory and commercialization of drug supply; | ||
| • | the commercialization of ANA598, ANA773 and any additional products; and | ||
| • | the extent to which we acquire or invest in other products technologies and businesses. |
We do not anticipate that we will generate significant revenues from operations for at
least several years, if ever. Until we can generate significant revenues from operations, we expect
to satisfy our future cash needs through public or private equity offerings, debt financings,
strategic alliances or other transactions, project financing and grant funding, as well as through
interest income earned on cash balances. We cannot be certain that additional funding will be
available to us on acceptable terms, or at all. If funds are not available, we may be required to
delay, reduce the scope of or eliminate one or more of our research or development programs or our
commercialization efforts.
Raising additional funds by issuing securities or through debt or project financing or
strategic alliances and licensing arrangements may cause dilution to existing stockholders,
restrict our operations or require us to relinquish proprietary rights.
We may seek to raise additional funds through public or private equity offerings, debt
financings, project financings or strategic alliances and licensing arrangements. We cannot be
certain that additional funding will be available on acceptable terms, or at all. To the extent
that we raise additional capital by issuing equity securities, our stockholders’ ownership will be
diluted. Other financing activities may also have an equity component, which also may lead to
dilution. Any debt or project financing we enter into may involve covenants that restrict our
operations. These restrictive covenants may include limitations on borrowing, specific restrictions
on the use of our assets as well as prohibitions on our ability to create liens, pay dividends,
redeem capital stocks or make investments. In addition, if we raise additional funds through
strategic alliances and licensing arrangements, it may be necessary to relinquish potentially
valuable rights to our potential products or proprietary technologies, or grant licenses on terms
that are not favorable to us. For example, we might be forced to relinquish all or a portion of our
sales and marketing rights with respect to potential products or license intellectual property that
enables licensees to develop competing products.
We are at an early stage of development, and we may never attain product sales.
Our existing organizational structure was formed in May 2000. Since then, most of our
resources have been dedicated to the development of our proprietary drug discovery technologies,
research and development and preclinical and early-stage clinical testing of compounds. Our current
product candidates are at only the very early stages of clinical trials. ANA598, ANA773 and any
other compounds that we may develop, may never be approved for commercial sales. These compounds
will require extensive and costly development, preclinical testing and clinical trials prior to
seeking regulatory approval for commercial sales. The time required to attain product sales and
profitability is lengthy and highly uncertain, and we cannot assure you that we will be able to
achieve or maintain product sales.
We expect our net losses to continue for at least several years, and we are unable to
predict the extent of future losses and when we will become profitable in our business operations,
if ever.
We have incurred net losses since our incorporation in 1992, and through December 31,
2009 we have an accumulated deficit of $283.3 million. Our losses are attributable in large part to
the significant research and development costs required to identify and validate potential product
candidates and conduct preclinical studies and clinical trials. To date, we have generated limited
revenues, consisting of one-time or limited payments associated with past collaborations or grants,
and we do not anticipate generating product revenues for at least several years, if ever. We would
need to increase our operating expenses over at least the next several years in order to fund the
development costs of our product candidates and further our development activities. As a result,
over time, we expect to continue to incur significant and increasing operating losses for the
foreseeable future. Because of the numerous risks and uncertainties associated with our research
and product development efforts, we are unable to predict the extent of any future losses or when
we will become profitable in our business operations, if ever. Even if we do achieve profitability
in our business operations, we may not be able to sustain or increase such profitability on an
ongoing basis.
18
Table of Contents
The technologies on which we rely are unproven and may not result in the development of
commercially viable products.
Our current product candidates, ANA598 and ANA773, were selected based on the presumption
that intervention at their respective targets, HCV polymerase and TLR7, offers a therapeutic
benefit. There can be no assurance that intervention at either target will offer sufficient benefit
and acceptable toxicity to warrant continued development and approval. ANA773 relies on the biology
of a specific receptor, or protein, named Toll-Like Receptor-7, or TLR7. However, the interaction
between small molecules and TLR7 represents a relatively new mechanism of action for the treatment
of disease, including HCV and cancer, and there is no guarantee that an acceptable balance between
therapeutic benefit and risk will be achieved with TLR7 agonists in HCV infected patients or in
cancer patients. For example, in June 2006 we suspended dosing of ANA975, a TLR7 agonist prodrug,
in our then on-going ANA975 clinical trial due to information from 13-week toxicology studies in
animals that showed intense immune stimulation. We subsequently conducted additional pre-clinical
studies and were unable to identify an acceptable balance between therapeutic benefit and risk
using a daily dosing schedule over 13-weeks. Accordingly, we subsequently discontinued the
development of ANA975 as a therapy for HCV infection. The science underlying ANA598 is also new and
unproven, as no products acting at the HCV polymerase have been approved for marketing. ANA598 and
ANA773 are at only the early stage of clinical investigation. The process of successfully
discovering product candidates is expensive, time-consuming and unpredictable, and the historical
rate of failure for drug candidates is extremely high. If our approaches to drug discovery and
development are not successful, we will not be able to establish or maintain a clinical development
portfolio or generate product revenue.
Because the results of preclinical studies and early clinical trials are not necessarily
predictive of future results, we can provide no assurances that ANA598 or ANA773 will have
favorable results in future clinical trials, or receive regulatory approval.
Positive results from preclinical studies or early clinical trials should not be relied
upon as evidence that later or larger-scale clinical trials will succeed. There is typically an
extremely high rate of attrition from the failure of drug candidates proceeding through clinical
trials. There is no guarantee that viral load declines seen in early patient trials will be
replicated in future trials of longer duration and/or larger patient populations. For example,
short-term viral load data from our ANA598 Phase Ib study may not translate into long-term benefit
due to the potential emergence of resistant variants or other factors. In particular, there is a
perception by some within the scientific community that the administration of non-nucleosides is
more likely to result in resistance than the administration of other types of direct acting
antivirals in development for HCV (including protease inhibitors and nucleoside polymerase
inhibitors) due to the lower genetic barrier of resistance to non-nucleosides relative to these
other types of compounds; however, we and others within the scientific community believe that this
concern is not likely to be a significant issue when non-nucleosides are dosed in combination with
other agents. However, the outcome of this debate will not be known until data from longer term
trials of non-nucleosides dosed in combination with other agents become available. Similarly, there
is no guarantee that favorable safety and tolerability seen in short term studies will be
replicated in studies of longer duration and/or in larger subject populations. For example, in a
14 day healthy volunteer study conducted in 2009, three of the 24 subjects who received ANA598
discontinued from the study due to the onset of a skin rash during the study. Furthermore, if
future toxicology studies have unexpected results, the clinical development of the compound at
issue could be suspended, delayed and/or terminated. If ANA598, or any other product candidate,
fails to demonstrate sufficient safety and efficacy in any clinical trial or shows unexpected
findings in future toxicology studies, we would experience potentially significant delays in, or be
required to abandon, development of ANA598. If we delay or abandon our development efforts related
to ANA598, we may not be able to generate sufficient revenues to become profitable, and our
reputation in the industry and in the investment community would likely be significantly damaged,
each of which would cause our stock price to decrease significantly.
We intend to develop ANA598 as a component of combination treatments, which presents
additional challenges to the drug development process.
We are developing ANA598 as a potential component of future combination treatments. We
may face additional challenges with this approach, as opposed to developing product candidates for
monotherapy. For example, any negative properties of our product candidates may be exacerbated when
combined with
other agents and/or have unexpected effects in humans. Furthermore, the optimal development of our
product candidates may entail explorations of combinations with other agents, which could require
us to establish agreements or alliances with other companies or third parties. There is no
guarantee that we will be able to enter into such alliances or agreements on terms that we view as
favorable, or at all. An important element of development for an agent such as ANA598 will be to
test the agent in combination with one or more other direct antivirals. In order for us to pursue
this development strategy, we will need to engage the interest of other biopharmaceutical or
pharmaceutical companies, since we do not have another direct antiviral to combine with ANA598. Our
ability to engage this interest will be impacted by other companies’ internal HCV portfolio
dynamics, with such dynamics influencing the companies’ perceived need to combine with an agent
such as ANA598. For those companies
19
Table of Contents
that have a perceived need to combine with an agent such as
ANA598, we will be dependent on their perception of the profile of ANA598 and their desire to test
ANA598 in combination with their agents. If they do not view the profile of ANA598 as favorably as
we do, or if they establish other criteria for combination that we have not yet satisfied with
ANA598, we could experience difficulties or delays in pursuing such combination trials. If we are
unable to optimize the development of ANA598, our business prospects could be harmed, causing our
stock price to suffer.
There is no guarantee that in future studies of ANA598, in which ANA598 may be dosed for
longer duration in combination with other agents, that we will be able to identify safe and
tolerable doses that result in clinical benefit, as measured by clearance of virus and durability
of that clearance.
Although we are currently conducting a Phase II study in which ANA598 is being dosed for
12 weeks with current standard of care, it is presently unknown what duration of dosing of ANA598
will be most appropriate when used as a component of combination therapy, and further studies of
longer duration may need to be explored. In addition, although we have presented in vitro data
showing that combinations of ANA598 with current standard of care and with certain direct antiviral
agents appear to be synergistic, these results may not be replicated in clinical trials. Also, it
is possible that ANA598 will not be additive or synergistic with other potential components of
future treatment regimens. Furthermore, it is possible that tolerability will be worse over longer
durations of treatment than was seen for the same dose at a shorter duration of treatment. For
example, in a 14 day healthy volunteer study conducted in 2009, three of the 24 subjects who
received ANA598 discontinued from the study due to the onset of a skin rash characterized as mild
to moderate with itching during the study, at comparable dose levels that were
well tolerated over three days in patients. Similarly, if the tolerability of doses of ANA598
required for long-term treatment as part of future combinations is unacceptable or unfavorable
relative to competitive product candidates, then the prospects for developing ANA598 as a treatment
for chronic hepatitis C will be diminished, causing our stock price to decrease significantly.
The FDA could impose additional requirements on the development of ANA598 which could
result in unexpected cost increases and/or delays to our development timelines.
The development of ANA598 in the United States is subject to ongoing regulation by the FDA.
There is no guarantee that the FDA will not impose additional requirements on our development
program for ANA598, including requirements associated with patient enrollment, manufacturing
processes of our clinical trials materials or other development activities related to ANA598, which
could result in increased costs to us or a delay in our desired timelines.
Fast track designation does not guarantee approval, or expedited approval, of ANA598 and
there is no guarantee that ANA598 will maintain fast track designation.
In December 2008, we announced that the FDA granted fast track designation to ANA598 for
the treatment of chronic HCV infection. Under the FDA Modernization Act of 1997, fast track
designation is designed to facilitate the development and expedite the review of new drugs that are
intended to treat serious or life-threatening conditions. Compounds selected must demonstrate the
potential to address an unmet medical need for such a condition. Mechanisms intended to facilitate
development include opportunities for frequent dialogue with FDA reviewers and for timely review of
submitted protocols.
However, the designation does not guarantee approval or expedited approval of any application for
the product. Furthermore, the FDA may revoke fast track designation from a product candidate at any
time if it determines that the criteria are no longer met.
During 2009 we completed a Phase I clinical trial of ANA773 for HCV and intend to explore
partnership opportunities as a way to potentially continue the development of ANA773. There is no
guarantee that we will be able to obtain a partnership around ANA773 or that the development of the
program will be continued.
During 2009 we completed a Phase I trial of ANA773 in HCV infected patients. While we are
encouraged by the data obtained to date, particularly at the higher doses at which we were able to
confirm a dose response for ANA773 in HCV infected patients, further schedule exploration will
likely be advisable before proceeding into larger scale studies. In connection with the
restructuring we implemented in the middle of 2009 and our desire to focus our resources on our
ANA598 program, we have decided to suspend further development of ANA773 while we look for a
partner to potentially fund further development of the program. There is no guarantee that we will
be able to obtain a partnership around ANA773 or that the development of the program will be able
to be continued. If the program is continued, if we or a licensee are unable to achieve viral load
reduction at levels comparable to injectable interferon but with a cleaner side effect profile, the
prospects for developing ANA773 as a competitive HCV product will be diminished. Furthermore, the
Phase I clinical trial was conducted in the Netherlands and not under a U.S. investigational new
drug
20
Table of Contents
application, or IND. If, in the future, we or a licensee want to proceed with the development
of ANA773 for HCV in the United States, approval from the FDA under a U.S. IND will be required.
There is no assurance that the FDA will agree that ANA773 should be tested as an investigational
treatment for HCV. Currently, there is no evidence that a TLR7 agonist can confer long-term benefit
as a therapy for HCV at an acceptable safety risk, and there is no assurance that the FDA will view
the data from our Phase I study in the Netherlands as sufficiently compelling to allow clinical
investigation. If the FDA does not view the data from our Phase I study in the Netherlands as
sufficiently compelling, it may not allow studies under a U.S. IND, in which case development and
commercialization of ANA773 for HCV in the United States would be precluded.
In 2007 we terminated our ANA975 development program due to challenges seen in animal
toxicology studies. To the extent that the ANA975 toxicology observations are mechanism related,
our ANA773 programs for cancer and hepatitis C and ability to out-license this product candidate
could be negatively impacted, causing our stock price to decline.
ANA975 is an oral prodrug of isatoribine, a TLR7 agonist. In 2007 we discontinued the
development of ANA975 as a treatment for HCV infection due to intense immune stimulation in
animals. To the extent that any of the ANA975 toxicology observations are mechanism related, rather
than compound specific, we, or a potential future collaborator, will need to determine whether the
level of immune stimulation induced by TLR7 agonists can be modulated to achieve a potential
therapeutic benefit with an acceptable safety profile. Although results from our ANA773 13-week
animal toxicology study indicated that with every-other-day dosing of ANA773, immune stimulation of
a magnitude believed to confer therapeutic potential can be achieved without adverse toxicology
findings, there is no guarantee that this favorable toxicology profile will persist in future
toxicology studies of longer duration, or that we will not see adverse safety findings in humans.
If we are unable to modulate the immunomodulatory effect with a dose and schedule that provides
therapeutic benefit without causing unacceptable adverse events, then the future development of
ANA773 may not be viable or attractive to a potential licensee, which could materially and
adversely affect our business and cause our stock price to decline.
Delays in the commencement of clinical testing of our current and potential product
candidates could result in increased costs to us and delay our ability to generate revenues.
Our potential drug products will require additional nonclinical testing and extensive
clinical trials prior to submission of any regulatory application for commercial sales. Previously,
we have conducted only early-stage clinical trials on our own. As a result, we have very limited
experience conducting clinical trials. In part because of this limited experience, we cannot be
certain that planned clinical trials will begin
or be completed on time, if at all. Delays in the commencement of clinical testing could
significantly increase our product development costs and delay product commercialization. In
addition, many of the factors that may cause, or lead to, a delay in the commencement of clinical
trials may also ultimately lead to denial of regulatory approval of a product candidate.
The commencement of clinical trials can be delayed for a variety of reasons, including
delays in:
| • | demonstrating sufficient safety and efficacy to obtain regulatory approval to commence a clinical trial; | ||
| • | reaching agreement on acceptable terms with prospective contract research organizations and trial sites; | ||
| • | manufacturing sufficient quantities or producing drug meeting our quality standards for a product candidate; | ||
| • | obtaining approval of an IND application or proposed trial design from the FDA; and | ||
| • | obtaining institutional review board approval to conduct a clinical trial at a prospective site. |
In addition, the commencement of clinical trials may be delayed due to insufficient
patient enrollment, which is a function of many factors, including the size and nature of the
patient population, the nature of the protocol, the proximity of patients to clinical sites, the
availability of effective treatments for the relevant disease, the number of other products under
development competing for the same patients in trials and the eligibility criteria for the clinical
trial.
Delays in the completion of, or the termination of, clinical testing of our current and
potential product candidates could result in increased costs to us and delay or prevent us from
generating revenues.
Once a clinical trial has begun, it may be delayed, suspended or terminated by us,
potential future collaborators, the FDA, or other regulatory authorities due to a number of
factors, including:
21
Table of Contents
| • | ongoing discussions with the FDA or other regulatory authorities regarding the scope or design of our clinical trials; | ||
| • | failure to conduct clinical trials in accordance with regulatory requirements; | ||
| • | lower than anticipated enrollment or retention rate of patients in clinical trials; | ||
| • | inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities resulting in the imposition of a clinical hold; | ||
| • | lack of adequate funding to continue clinical trials; | ||
| • | negative results of clinical trials; | ||
| • | negative or potentially problematic results of ongoing and concurrent non-clinical toxicology studies; | ||
| • | requests by the FDA for supplemental information on, or clarification of, the results of clinical trials conducted in other countries; | ||
| • | insufficient supply or deficient quality of drug candidates or other materials necessary for the conduct of our clinical trials; or | ||
| • | serious adverse events or other undesirable drug-related side effects experienced by participants. |
Many of the factors that may lead to a delay, suspension or termination of clinical
testing of a current or potential product candidate may also ultimately lead to denial of
regulatory approval of a current or potential product candidate. If we experience delays in the
completion of, or termination of, clinical testing, our financial results and the commercial
prospects for our product candidates may be harmed, and our ability to generate product revenues
will be delayed.
Even if we successfully complete clinical trials of ANA598 or any future product
candidate, there are no assurances that we will be able to submit, or obtain FDA approval of, a new
drug application.
There can be no assurance that if our clinical trials of ANA598 or any other potential
product candidate are successfully completed, we will be able to submit a new drug application, or
NDA, to the FDA or that any NDA we submit will be approved by the FDA in a timely manner, if at
all. If we are unable to submit a NDA with respect to ANA598 or any future product candidate, or if
any NDA we submit is not approved by the FDA, we will be unable to commercialize that product in
the United States. The FDA can and does reject NDAs and may require additional clinical trials,
even when drug candidates performed well or achieved favorable results in large-scale Phase III
clinical trials. If we fail to commercialize ANA598 or any future product candidate, we may be
unable to generate sufficient revenues to attain profitability, and our reputation in the industry
and in the investment community would likely be damaged, each of which would cause our stock price
to decrease.
If we successfully develop products but those products do not achieve and maintain market
acceptance, our business will not be profitable.
Even if ANA598 or any future product candidates are approved for commercial sale by the
FDA or other regulatory authorities, the degree of market acceptance of any approved product
candidate by physicians, healthcare professionals and third-party payors and our profitability and
growth will depend on a number of factors, including:
| • | our ability to provide acceptable evidence of safety and efficacy; | ||
| • | relative convenience and ease of administration; | ||
| • | the prevalence and severity of any adverse side effects; | ||
| • | availability of alternative treatments; | ||
| • | pricing and cost effectiveness; |
22
Table of Contents
| • | effectiveness of our or our collaborators’ sales and marketing strategy; | ||
| • | our ability to obtain sufficient third-party insurance coverage or reimbursement; and | ||
| • | our ability to establish or maintain an attractive price for ANA598 when used in combination with other agents. |
If ANA598 or any future product candidate does not provide additional clinical benefit when
included within a treatment regimen, that product likely will not be accepted favorably by the
market. Similarly, if ANA773 does not provide additional clinical benefit when included within a
treatment regimen, that product will likewise not be accepted favorably by the market. If any
products we or our collaborators may develop do not achieve market acceptance, then we will not
generate sufficient revenue to achieve or maintain profitability.
In addition, even if our products achieve market acceptance, we may not be able to maintain
that market acceptance over time if:
| • | new products or technologies are introduced that are more favorably received than our products, are more cost effective or render our products obsolete; or | ||
| • | complications, such as long-term toxicities and viral resistance, arise with respect to use of our products. |
We depend on outside parties to conduct our clinical trials, which may result in costs and
delays that prevent us from obtaining regulatory approval or successfully commercializing product
candidates.
We engage clinical investigators and medical institutions to enroll patients in planned
clinical trials and contract research organizations to perform data collection and analysis and
other aspects of our preclinical studies and clinical trials. As a result, we depend on these
clinical investigators, medical institutions and contract research organizations to properly
perform the studies and trials. If these parties do not successfully carry out their contractual
duties or obligations or meet expected deadlines, or if the quality or accuracy of the clinical
data they obtain is compromised due to the failure to adhere to our clinical protocols or for other
reasons, our clinical trials may be extended, delayed or terminated. We may not be able to enter
into replacement arrangements without undue delays or excessive expenditures. If there are delays
in testing or regulatory approvals as a result of the failure to perform by third-parties, our drug
development costs will increase and we may not be able to obtain regulatory approval for or
successfully commercialize our product candidates. In addition, we may not be able to maintain any
of these existing relationships, or establish new ones on acceptable terms, if at all.
We do not have internal manufacturing capabilities, and if we fail to develop and maintain
supply relationships with future collaborators or other outside manufacturers, we may be unable to
develop or commercialize any of our products.
Our ability to develop and commercialize products will depend in part on our ability to
manufacture, or arrange for collaborators or other parties to manufacture, our products at a
competitive cost, in accordance with regulatory requirements and in sufficient quantities for
clinical testing and eventual commercialization. Our current manufacturing agreements reflect a
much smaller scale than would be required for commercialization. If we are unable to enter into or
maintain commercial-scale manufacturing agreements with future collaborators or capable contract
manufacturers on acceptable terms the development and commercialization of our products could be
delayed, which would adversely affect our ability to generate revenues and would increase our
expenses.
If we are unable to establish sales and marketing capabilities or enter into agreements with
third parties to sell and market any products we may develop, we may not be able to generate
product revenue.
We do not currently have the capabilities for the sales, marketing and distribution of
pharmaceutical products. In order to commercialize any products, we would have to build our sales,
marketing, distribution, managerial and other non-technical capabilities or make arrangements with
third parties to perform these services. The establishment and development of our own sales force
to market any products we may develop in the United States will be expensive and time-consuming and
could delay any product launch, and we cannot be certain that we would be able to successfully
develop this capacity. If we are unable to establish our sales and marketing capability or any
other non-technical capabilities necessary to commercialize any product we may develop, we will
need to contract with third parties to market and sell any products we may develop in the United
States. We will also need to develop a plan to market and sell any products we may develop outside
the United States. If we are unable to establish adequate sales, marketing and distribution
capabilities, whether independently or with third parties, we may not be able to generate product
revenue and may not become profitable.
23
Table of Contents
If we are unable to retain key management and scientific staff, we may be unable to
successfully develop or commercialize our product candidates.
We are a small company and have approximately 30 employees. Our success depends on our
continued ability to retain and motivate highly qualified management and scientific personnel. In
particular, our programs depend on our ability to retain highly skilled clinical and preclinical
personnel in the field of HCV, as well as biologists and chemists.
We may not be able to retain qualified management and scientific personnel in the future due
to the intense competition for qualified personnel among biotechnology and pharmaceutical
businesses, particularly in the San Diego, California area. If we are not able to retain the
necessary personnel to accomplish our business objectives, we may experience constraints that will
impede significantly the achievement of our development objectives. In addition, all of our
employees are “at will” employees, which means that any employee may quit at any time and we may
terminate any employee at any time. Currently we do not have employment agreements with any
employees or members of senior management that provide any guarantee of continued employment by us.
We do not currently carry “key person” insurance covering members of senior management other than
Steve Worland, Ph.D., our President and Chief Executive Officer. The insurance covering Dr. Worland
is in the amount of $1.5 million. If we lose the services of Dr. Worland, or James L. Freddo, M.D.,
our Senior Vice President, Drug Development and Chief Medical Officer, or other members of our
senior management team or key personnel, we may not be able to find suitable replacements, and our
business may be harmed as a result.
Earthquake or wildfire damage to our facilities could delay our research and development
efforts and adversely affect our business.
Our headquarters and research and development facilities in San Diego, California, are located
in a seismic zone, and there is the possibility of an earthquake, which could be disruptive to our
operations and result in delays in our research and development efforts. In addition, San Diego has
experienced several severe wildfires during the past several years which have destroyed or damaged
many businesses and residences in the San Diego area. In the event of an earthquake or a severe
wildfire, if our facilities or the equipment in our facilities are significantly damaged or
destroyed for any reason, or we are otherwise required to shut down our operations, we may not be
able to rebuild or relocate our facility or replace any damaged equipment, or otherwise recommence
our business operations, in a timely manner and our business, financial condition and results of
operations could be materially and adversely affected.
Our securities available-for-sale held in the form of marketable securities are subject to
market, interest and credit risk that may reduce their value.
A portion of our securities available-for-sale is invested in marketable securities. Our cash
position may be adversely affected by changes in the value of these securities. In particular, the
value of these holdings may be adversely affected by increases in interest rates, downgrades by
rating agencies on the issuers of corporate bonds included in the portfolio and by other factors
which may result in other than temporary declines in value of the investments. Each of these events
may cause us to record charges to reduce the carrying value of our investment portfolio and may
adversely affect our cash position.
Risks Related to Our Industry
Because our product candidates and development and collaboration efforts depend on our
intellectual property rights, adverse events affecting our intellectual property rights will harm
our ability to commercialize products.
Our commercial success depends on obtaining and maintaining patent protection and trade secret
protection of our product candidates, proprietary technologies and their uses, as well as
successfully defending these patents against third-party challenges. We will only be able to
protect our product candidates, proprietary technologies and their uses from unauthorized use by
third parties to the extent that valid and enforceable patents or effectively-protected trade
secrets cover them.
Due to evolving legal standards relating to the patentability, validity and enforceability of
patents covering pharmaceutical inventions and the scope of claims made under these patents, our
ability to obtain and enforce patents is uncertain and involves complex legal and factual
questions. Accordingly, rights under any issued patents may not provide us with sufficient
protection for ANA598 or ANA773 or provide sufficient protection to afford us a commercial
advantage against competitive products or processes. In addition, we cannot guarantee that any
patents will issue from any pending or future patent applications owned by or licensed to us.
24
Table of Contents
Even with respect to patents that have issued or will issue, we cannot guarantee that the
claims of these patents are, or will be valid, enforceable or will provide us with any significant
protection against competitive products or otherwise be commercially valuable to us. For example:
| • | we might not have been the first to make, conceive, or reduce to practice the inventions covered by all or any of our pending patent applications and issued patents; | ||
| • | we might not have been the first to file patent applications for these inventions; | ||
| • | others may independently develop similar or alternative technologies or duplicate any of our technologies; | ||
| • | it is possible that none of our pending patent applications will result in issued patents; | ||
| • | our issued or acquired patents may not provide a basis for commercially viable products, may not provide us with any competitive advantages, or may be challenged by third parties; | ||
| • | our issued patents may not be valid or enforceable; | ||
| • | we may not develop additional proprietary technologies that are patentable; or | ||
| • | the patents of others may have an adverse effect on our business. |
Patent applications in the United States are maintained in confidence for up to 18 months or
longer after their filing. Consequently, we cannot be certain that we were the first to invent, or
the first to file patent applications on our product candidates. In the event that a third party
has also filed a U.S. patent application relating to our product candidates or a similar invention,
we may have to participate in interference proceedings declared by the U.S. Patent Office to
determine priority of invention in the United States. The costs of these proceedings could be
substantial and it is possible that our efforts would be unsuccessful, resulting in a material
adverse effect on our U.S. patent position. Furthermore, we may not have identified all U.S. and
foreign patents or published applications that affect our business either by blocking our ability
to commercialize our drugs or by covering similar technologies that affect our drug market.
In addition, some countries, including many in Europe, do not grant patent claims directed to
methods of treating humans, and in these countries patent protection may not be available at all to
protect our drug candidates. Even if patents issue, we cannot guarantee that the claims of those
patents will be valid and enforceable or provide us with any significant protection against
competitive products, or otherwise be commercially valuable to us. We may be particularly affected
by this because we expect that ANA598, if approved, will be marketed in foreign countries with high
incidences of HCV infection.
Other companies may obtain patents and/or regulatory approvals to use the same drugs to treat
diseases other than HCV or cancer. As a result, we may not be able to enforce our patents
effectively because we may not be able to prevent healthcare providers from prescribing,
administering or using another company’s product that contains the same active substance as our
products when treating patients infected with HCV or who have cancer.
If we fail to obtain and maintain patent protection and trade secret protection of ANA598 or
ANA773, proprietary technologies and their uses, the competition we face would increase, reducing
our potential revenues and adversely affecting our ability to attain or maintain profitability.
If we are sued for infringing intellectual property rights of others, it will be costly and
time-consuming, and an unfavorable outcome in that litigation would have a material adverse effect
on our business.
Our commercial success also depends upon our ability to develop, manufacture, market and sell
our product candidates and use our proprietary technologies without infringing the proprietary
rights of third parties. We may be exposed to future litigation by third parties based on claims
that our product candidates, technologies or activities infringe the intellectual property rights
of others. Numerous U.S. and foreign issued patents and pending patent applications owned by others
exist in HCV and cancer. These could materially affect our ability to develop our drug candidates
or sell our products. Because patent applications can take many years to issue, there may be
currently pending applications, unknown to us, which may later result in issued patents that our
product candidates
25
Table of Contents
or technologies may infringe. There also may be existing patents, of which we are not aware, that
our product candidates or technologies may inadvertently infringe. Further, there may be issued
patents and pending patent applications in fields relevant to our business, of which we may become
aware from time to time, that we believe we do not infringe or that we believe are invalid or
relate to immaterial portions of our overall drug discovery and development efforts. We cannot
assure you that third parties holding any of these patents or patent applications will not assert
infringement claims against us for damages or seeking to enjoin our activities. We also cannot
assure you that, in the event of litigation, we will be able to successfully assert any belief we
may have as to non-infringement, invalidity or immateriality, or that any infringement claims will
be resolved in our favor.
There is a substantial amount of litigation involving patent and other intellectual property
rights in the biotechnology and biopharmaceutical industries generally. Any litigation or claims
against us, with or without merit, may cause us to incur substantial costs, could place a
significant strain on our financial resources, divert the attention of management from our core
business and harm our reputation. In addition, intellectual property litigation or claims could
result in substantial damages and force us to do one or more of the following if a court decides
that we infringe on another party’s patent or other intellectual property rights:
| • | cease selling, incorporating or using any of our product candidates or technologies that incorporate the challenged intellectual property; | ||
| • | obtain a license from the holder of the infringed intellectual property right, which license may be costly or may not be available on reasonable terms, it at all; or | ||
| • | redesign our processes or technologies so that they do not infringe, which could be costly and time consuming and may not be possible. |
If we find during clinical evaluation that our drug candidates for the treatment of HCV or
cancer should be used in combination with a product covered by a patent held by another company or
institution, and that a labeling instruction is required in product packaging recommending that
combination, we could be accused of, or held liable for, inducing infringement of the third-party
patents covering the product recommended for co-administration with our product. In that case, we
may be required to obtain a license from the other company or institution to use the required or
desired package labeling, which may not be available on reasonable terms, or at all.
If we fail to obtain any required licenses or make any necessary changes to our technologies,
we may be unable to develop or commercialize some or all of our product candidates.
We may be involved in lawsuits or proceedings to protect or enforce our patent rights, trade
secrets or know-how, which could be expensive and time-consuming.
The defense and prosecution of intellectual property suits and related legal and
administrative proceedings can be both costly and time-consuming. Litigation and interference
proceedings could result in substantial expense to us and significant diversion of effort by our
technical and management personnel. Further, the outcome of patent litigation is subject to
uncertainties that cannot be adequately quantified in advance, including the demeanor and
credibility of witnesses and the identity of the adverse party. This is especially true in
biotechnology related patent cases that may turn on the testimony of experts as to technical facts
upon which experts may reasonably disagree and which may be difficult to comprehend by a judge or
jury. An adverse determination in an interference proceeding or litigation with respect to ANA598
or ANA773, to which we may become a party could subject us to significant liabilities to third
parties or require us to seek licenses from third parties. If required, the necessary licenses may
not be available on acceptable terms, or at all. Adverse determinations in a judicial or
administrative proceeding or failure to obtain necessary licenses could prevent us from
commercializing ANA598 or ANA773, which could have a material and adverse effect on our results of
operations.
Furthermore, because of the substantial amount of pre-trial document and witness discovery
required in connection with intellectual property litigation, there is risk that some of our
confidential information could be compromised by disclosure during this type of litigation. In
addition, during the course of this kind of litigation, there could be public announcements of the
results of hearings, motions or other interim proceedings or developments. If securities analysts
or investors perceive these results to be negative, it could have a substantial adverse effect on
the trading price of our common stock.
26
Table of Contents
Confidentiality agreements with employees and others may not adequately prevent disclosure of
trade secrets and other proprietary information and may not adequately protect our intellectual
property.
We also rely on trade secrets to protect our technology, especially where we do not believe
patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. In
order to protect our proprietary technology and processes, we also rely in part on confidentiality
and intellectual property assignment agreements with our corporate partners, employees,
consultants, outside scientific collaborators and sponsored researchers and other advisors. These
agreements may not effectively prevent disclosure of confidential information nor result in the
effective assignment to us of intellectual property, and may not provide an adequate remedy in the
event of unauthorized disclosure of confidential information or other breaches of the agreements.
In addition, others may independently discover our trade secrets and proprietary information, and
in such case we could not assert any trade secret rights against such party. Enforcing a claim that
a party illegally obtained and is using our trade secrets is difficult, expensive and
time-consuming, and the outcome is unpredictable. In addition, courts outside the United States may
be less willing to protect trade secrets. Costly and time-consuming litigation could be necessary
to seek to enforce and determine the scope of our proprietary rights, and failure to obtain or
maintain trade secret protection could adversely affect our competitive business position.
Many competitors have significantly more resources and experience, which may harm our
commercial opportunity.
The biotechnology and pharmaceutical industries are subject to intense competition and rapid
and significant technological change. We have many potential competitors, including major drug and
chemical companies, specialized biotechnology firms, academic institutions, government agencies and
private and public research institutions. Many of our competitors have significantly greater
financial resources, experience and expertise in:
| • | research and development; | ||
| • | preclinical testing; | ||
| • | clinical trials; | ||
| • | regulatory approvals; | ||
| • | manufacturing; and | ||
| • | sales and marketing of approved products. |
Smaller or early-stage companies and research institutions may also prove to be significant
competitors, particularly through collaborative arrangements with large and established
pharmaceutical or other companies. We will also face competition from these parties in recruiting
and retaining qualified scientific and management personnel, establishing clinical trial sites and
patient registration for clinical trials, and acquiring and in-licensing technologies and products
complementary to our programs or potentially advantageous to our business. If any of our
competitors succeed in obtaining approval from the FDA or other regulatory authorities for their
products sooner than we do or for products that are more effective or less costly than ours, our
commercial opportunity could be significantly reduced.
If our competitors develop treatments for HCV or cancer that are approved faster, marketed
better or demonstrated to be more effective than ANA598, ANA773, or any other products that we may
develop, our commercial opportunity will be reduced or eliminated.
We believe that a significant number of drugs are currently under development and may become
available in the future for the treatment of HCV and certain cancers. Potential competitors may
develop treatments for HCV or certain cancers that are more effective or less costly than our
product candidates or that would make our product candidates obsolete or non-competitive. Some of
these products may use therapeutic approaches that compete directly with ANA598 or ANA773. In
addition, less expensive generic forms of currently marketed drugs could lead to additional
competition upon patent expiration or invalidations.
ANA598, a non-nucleoside polymerase inhibitor, was selected as a development candidate in June
2007. If approved, ANA598 would likely be used in combination with the current standard of care
and/or other direct antiviral agents such as protease inhibitors and polymerase inhibitors. Any
product currently approved or approved in the future for the treatment of HCV infection could
decrease or eliminate the commercial opportunity of ANA598. Other non-nucleoside inhibitors would
likely be the most direct competitors for ANA598. To our knowledge, non-nucleoside polymerase
inhibitor programs are currently under clinical evaluation by Pfizer, Gilead, Merck, Abbott,
Boehringer Ingelheim and Vertex. Further, a number of companies have non-nucleoside polymerase
inhibitor research and pre-clinical development programs.
27
Table of Contents
Other potential competitors are products currently approved for the treatment of HCV
infection: Peg- Intron (pegylated interferon-alpha-2b), Rebetol (ribavirin), and Intron-A
(interferon-alpha-2b), which are marketed by Merck, Pegasys (pegylated interferon-alpha-2a),
Copegus (ribavirin USP), and Roferon- A (interferon-alpha-2a), which are marketed by Roche.
Additional compounds in late state clinical trials for HCV include Zalbin/Joulferon (albinterferon
alfa-2b), in development by Human Genome Sciences and Novartis, telaprevir, in development by
Vertex Pharmaceuticals, Janssen Pharmaceutica (Johnson & Johnson) and Mitsubishi Tanabe Pharma, boceprevir, MK-7009 and
narlaprevir, in development by Merck, ITMN-191, in development by Intermune and Roche, TMC-435350,
in development by Johnson & Johnson (Tibotec) and Medivir, BI-201335 in development by Boehringer
Ingelheim, and RG7128 in development by Roche and Pharmasset.
ANA773 is also subject to competition in the treatment of HCV from all of the HCV products and
compounds in development listed above as potential competitors of ANA598 and most specifically from
the products and development candidates that act as an immunomodulator or have an immunomodulatory
component, including Peg-Intron (pegylated interferon-alpha-2b), Rebetol (ribavirin), Intron-A
(interferonalpha- 2b), Pegasys (pegylated interferon-alpha-2a), Copegus (ribavirin USP), and
Roferon-A (interferon-alpha- 2a), each of which are products currently approved for the treatment
of HCV. IMO-2055, a TLR9 agonist in development by Idera, is also being studied in early stage
clinical trials in HCV patients. Other agents in development as potential replacements to pegylated
interferon-alfa include Zalbin/Joulferon (albinterferon alfa-2b), in development by Human Genome
Sciences and Novartis and Locteron, in development by Biolex Therapeutics, both of which are
longer-acting versions of interferon alfa. Also, in development as potential improvements to
existing interferons are PEG-interferon lambda, in development by Zymogenetics and Bristol
Myers-Squibb, and omega interferon in development by Intarcia Therapeutics.
Any product currently approved or approved in the future for the treatment of cancer could
decrease or eliminate the commercial opportunity of ANA773 in the oncology markets. Programs that
most directly compete with the ANA773 oncology program at this time are other TLR agonists under
evaluation for oncology indications, IMO-2055, in development by Idera and Merck KGaA and a cancer
program in development by Dynavax.
If we cannot establish pricing of our product candidates acceptable to the government,
insurance companies, managed care organizations and other payors, any product sales will be
severely hindered.
The continuing efforts of the government, insurance companies, managed care organizations and
other payors of health care costs to contain or reduce costs of health care may adversely affect:
| • | our ability to set a price we believe is fair for any products we or our collaborators may develop; | ||
| • | our ability to generate adequate revenues and gross margins; and | ||
| • | the availability of capital. |
In certain foreign markets, the pricing of prescription pharmaceuticals is subject to
government control. In the United States, given recent federal and state government initiatives
directed at lowering the total cost of health care, the U.S. Congress and state legislatures will
likely continue to focus on health care reform, the cost of prescription pharmaceuticals and on the
reform of the Medicare and Medicaid systems. The trend toward managed health care in the United
States, which could significantly influence the purchase of health care services and products, as
well as legislative proposals to reform health care, control pharmaceutical prices or reduce
government insurance programs, may result in lower prices for our product candidates. While we
cannot predict whether any legislative or regulatory proposals affecting our business will be
adopted, the announcement or adoption of these proposals could have a material and adverse effect
on our potential revenues and gross margins.
If we cannot arrange for reimbursement policies favorable to our product candidates, their
sales will be severely hindered.
Our ability to commercialize ANA598 or any other product candidate successfully will depend in
part on the extent to which governmental authorities, private health insurers and other
organizations establish appropriate reimbursement levels for the cost of ANA598 or any other
product and related treatments. Third party payors are increasingly challenging the prices charged
for medical products and services, including treatments for HCV. Also, the trend toward managed
health care in the United States as well as legislative proposals to reform health care, control
pharmaceutical prices or reduce government insurance programs, may also result in exclusion of our
product candidates from reimbursement programs. The cost containment measures that health care
payors and providers are instituting and the effect of any health care reform could materially and
adversely affect our ability to earn product revenue and generate significant profits and could
impact our ability to raise capital.
28
Table of Contents
Product liability claims may damage our reputation and, if insurance proves inadequate, the
product liability claims may harm our results of operations.
We face an inherent risk of product liability exposure for claimed injuries related to the
testing of our product candidates in human clinical trials, and will face an even greater risk if
we or our potential future collaborators sell our product candidates commercially. If we cannot
successfully defend ourselves against product liability claims, we will incur substantial
liabilities. Regardless of merit or eventual outcome, product liability claims may result in:
| • | decreased demand for our product candidates; | ||
| • | injury to our reputation; | ||
| • | withdrawal of clinical trial participants; | ||
| • | the inability to establish new collaborations with potential collaborators; | ||
| • | substantial costs of related litigation; | ||
| • | substantial monetary awards to patients; and | ||
| • | the inability to commercialize our product candidates. |
We currently have product liability insurance that covers our clinical trials and plan to
increase and expand this coverage as we commence larger scale trials. We also intend to expand our
insurance coverage to include the sale of commercial products if marketing approval is obtained for
any of our product candidates. However, insurance coverage is increasingly expensive. We may not be
able to maintain insurance coverage at a reasonable cost and we may not be able to obtain insurance
coverage that will be adequate to satisfy any liability that may arise.
Any claims relating to our improper handling, storage or disposal of biological, hazardous and
radioactive materials could be time-consuming and costly.
Our research and development involves the controlled use of hazardous materials, including
chemicals that cause cancer, volatile solvents, including ethylacetate and acetonitrile,
radioactive materials and biological materials including plasma from patients infected with HCV or
other infectious diseases that have the potential to transmit disease. Our operations also produce
hazardous waste products. We are subject to federal, state and local laws and regulations governing
the use, manufacture, storage, handling and disposal of these materials and waste products. If we
fail to comply with these laws and regulations or with the conditions attached to our operating
licenses, the licenses could be revoked, and we could be subjected to criminal sanctions and
substantial liability or required to suspend or modify our operations. Although we believe that our
safety procedures for handling and disposing of these materials comply with legally prescribed
standards, we cannot completely eliminate the risk of accidental contamination or injury from these
materials. In the event of contamination or injury, we could be held liable for damages or
penalized with fines in an amount exceeding our resources, and our clinical trials could be
suspended. In addition, we may have to incur significant costs to comply with future environmental
laws and regulations.
Our business and operations would suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems are vulnerable
to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and
telecommunication and electrical failures. Any system failure, accident or security breach that
causes interruptions in our operations could result in a material disruption of our development
programs. To the extent that any disruption or security breach results in a loss or damage to our
data or applications, or inappropriate disclosure of confidential or proprietary information, we
may incur liability as a result, our development programs may be adversely affected and the further
development of our product candidates may be delayed. In addition, we may incur additional costs to
remedy the damages caused by these disruptions or security breaches.
29
Table of Contents
Risks Related to Our Common Stock
Future sales of our common stock may cause our stock price to decline.
Our current stockholders hold a substantial number of shares of our common stock that they are
able to sell in the public market. Significant portions of these shares are held by a small number
of stockholders. Sales by our current stockholders of a substantial number of shares or the
expectation that such sale may occur, could significantly reduce the market price of our common
stock.
Our stock price may be volatile.
The market price of our common stock may fluctuate significantly in response to a number of
factors, most of which we cannot control, including:
| • | changes in the regulatory status of our product candidates, including the status and results of our clinical trials of ANA598 and ANA773; | ||
| • | significant contracts, new technologies, acquisitions, commercial relationships, joint ventures or capital commitments; | ||
| • | disputes or other developments relating to proprietary rights, including patents, trade secrets, litigation matters, and our ability to patent or otherwise protect our product candidates and technologies; | ||
| • | conditions or trends in the pharmaceutical and biotechnology industries; | ||
| • | fluctuations in stock market prices and trading volumes of similar companies, of our competitors or of the markets generally; | ||
| • | variations in our quarterly operating results; | ||
| • | changes in securities analysts’ estimates of our financial performance; | ||
| • | failure to meet or exceed securities analysts’ or investors’ expectations of our quarterly financial results, clinical results or our achievement of milestones; | ||
| • | sales of large blocks of our common stock, or the expectation that such sales may occur, including sales by our executive officers, directors and significant stockholders; | ||
| • | additions or departures of key personnel; | ||
| • | discussion of our business, products, financial performance, prospects or our stock price by the financial and scientific press and online investor communities such as chat rooms; | ||
| • | regulatory developments in the United States and foreign countries; | ||
| • | economic and political factors, including wars, terrorism and political unrest; and | ||
| • | technological advances by our competitors. |
Our quarterly results may fluctuate significantly, resulting in fluctuations in our stock
price.
We expect our results of operations to be subject to quarterly fluctuations. The level of our
revenues, if any, and results of operations at any given time, will be based primarily on the
following factors:
| • | the status of development of ANA598, ANA773 and our other product candidates, including results of preclinical studies and clinical trials and changes in regulatory status; |
30
Table of Contents
| • | our execution of collaborative, licensing or other arrangements, including arrangements involving ANA773, and the timing and accounting treatment of payments we make or receive under these arrangements; | ||
| • | whether or not we achieve specified research or commercialization milestones under any agreement that we enter into with collaborators and the timely payment by commercial collaborators of any amounts payable to us; | ||
| • | variations in the level of expenses related to our product candidates or potential product candidates during any given period; and | ||
| • | the effect of competing technological and market developments. |
These factors, some of which are not within our control, may cause the price of our stock to
fluctuate substantially. In particular, if our quarterly operating results fail to meet or exceed
the expectations of securities analysts or investors, our stock price could drop suddenly and
significantly. We believe that quarterly comparisons of our financial results are not necessarily
meaningful and should not be relied upon as an indication of our future performance.
Our largest stockholders may take actions that are contrary to your interests, including
selling their stock.
A small number of our stockholders hold a significant amount of our outstanding stock. These
stockholders may support competing transactions and have interests that are different from yours.
In addition, the average number of shares of our stock that trade each day is generally low. As a
result, sales of a large number of shares of our stock by these large stockholders or other
stockholders within a short period of time could adversely affect our stock price.
Anti-takeover provisions in our organizational documents and Delaware law may discourage or
prevent a change in control, even if an acquisition would be beneficial to our stockholders, which
could affect our stock price adversely and prevent attempts by our stockholders to replace or
remove our current management.
Our amended and restated certificate of incorporation and amended and restated bylaws contain
provisions that may delay or prevent a change in control, discourage bids at a premium over the
market price of our common stock and adversely affect the market price of our common stock and the
voting and other rights of the holders of our common stock. These provisions include:
| • | dividing our board of directors into three classes serving staggered three-year terms; | ||
| • | prohibiting our stockholders from calling a special meeting of stockholders; | ||
| • | permitting the issuance of additional shares of our common stock or preferred stock without stockholder approval; | ||
| • | prohibiting our stockholders from making certain changes to our amended and restated certificate of incorporation or amended and restated bylaws except with 66 2/3% stockholder approval; and | ||
| • | requiring advance notice for raising matters of business or making nominations at stockholders’ meetings. |
We are also subject to provisions of the Delaware corporation law that, in general, prohibit
any business combination with a beneficial owner of 15% or more of our common stock for five years
unless the holder’s acquisition of our stock was approved in advance by our board of directors.
Although we believe these provisions collectively provide for an opportunity to receive higher bids
by requiring potential acquirers to negotiate with our board of directors, they would apply even if
the offer may be considered beneficial by some stockholders. In addition, these provisions may
frustrate or prevent any attempts by our stockholders to replace or remove our current management
by making it more difficult for stockholders to replace members of our board of directors, which is
responsible for appointing the members of our management.
We have never paid cash dividends on our capital stock and we do not anticipate paying
dividends in the foreseeable future.
We have paid no cash dividends on any of our classes of capital stock to date, and we
currently intend to retain our future earnings, if any, to fund the development and growth of our
business. In addition, the terms of any future debt or credit facility may preclude us from paying
any dividends. As a result, capital appreciation, if any, of our common stock will be your sole
source of potential gain for the foreseeable future.
31
Table of Contents
Item 1B. Unresolved Staff Comments
Not Applicable.
Item 2. Properties
Our headquarters and research and development facility is located in approximately 14,000
square feet of office and laboratory space in San Diego, California. We occupy this facility under
a lease, which expires on January 31, 2011.
Item 3. Legal Proceedings
We are currently not a party to any material legal proceedings.
Item 4.
Reserved
Reserved.
32
Table of Contents
Part II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchase of
Equity Securities
Market Information
Our common stock is traded on the Nasdaq Global Market under the symbol ANDS. The following
table sets forth the high and low sales prices for our common stock for the periods indicated, as
reported on the Nasdaq Global Market.
| 2009 | High | Low | ||||||
First Quarter |
$ | 8.43 | $ | 1.61 | ||||
Second Quarter |
6.90 | 1.68 | ||||||
Third Quarter |
3.32 | 1.44 | ||||||
Fourth Quarter |
3.02 | 1.79 | ||||||
| 2008 | High | Low | ||||||
First Quarter |
$ | 1.77 | $ | 1.36 | ||||
Second Quarter |
3.13 | 1.50 | ||||||
Third Quarter |
2.98 | 2.10 | ||||||
Fourth Quarter |
2.70 | 1.50 | ||||||
Holders
As of February 17, 2010, there were approximately 5,400 holders of our common stock.
Dividend Policy
We have never declared or paid any cash dividends on our capital stock. We currently intend to
retain future earnings, if any, for development of our business and therefore do not anticipate
that we will declare or pay cash dividends on our capital stock in the foreseeable future.
33
Table of Contents
Performance Measurement Comparison (1)
The following graph shows a comparison of the five year total cumulative returns of an
investment of $100 in cash on December 31, 2004 in (i) our common stock (ii) the Nasdaq Composite
Index and (iii) the Nasdaq Biotechnology Index. All values assume reinvestment of the full amount
of all dividends (to date, we have not declared any dividends).
Comparison of cumulative total return on investment since December 31, 2004:
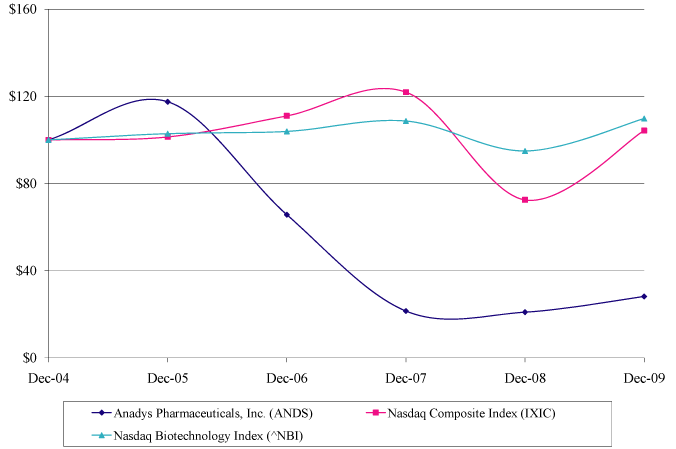
| December 31, 2004 | December 31, 2005 | December 31, 2006 | December 31, 2007 | December 31, 2008 | December 31, 2009 | |||||||||||||||||||||||||||
Anadys Pharmaceuticals, Inc. |
$ | 100.00 | $ | 117.49 | $ | 65.69 | $ | 21.50 | $ | 20.96 | $ | 28.17 | ||||||||||||||||||||
NASDAQ Composite Index |
100.00 | 101.37 | 111.03 | 121.92 | 72.49 | 104.31 | ||||||||||||||||||||||||||
NASDAQ Biotechnology Index |
100.00 | 102.84 | 103.89 | 108.65 | 94.93 | 109.77 | ||||||||||||||||||||||||||
| (1) | This section is not “soliciting material,” is not deemed “filed” with the SEC and is not to be incorporated by reference in any filing of the Company under the 1933 Act or the 1934 Act whether made before or after the date hereof and irrespective of any general incorporation language in any such filing. |
34
Table of Contents
Item 6. Selected Financial Data
The selected financial data set forth below with respect to our consolidated statements of
operations for each of the three years in the period ended December 31, 2009 and, with respect to
our consolidated balance sheets, at December 31, 2009 and 2008 are derived from our consolidated
financial statements that have been audited by Ernst & Young LLP, an independent registered public
accounting firm, which are included elsewhere in this Annual Report on Form 10-K. The statement of
operations data for the years ended December 31, 2006 and 2005 and the balance sheet data as of
December 31, 2007, 2006 and 2005 are derived from our audited consolidated financial statements
that are not included in this Annual Report on Form 10-K. The information set forth below is not
necessarily indicative of the results of future operations and should be read in conjunction with
Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and
the consolidated financial statements and notes thereto appearing elsewhere in this Annual Report
on Form 10-K.
| For the years ended December 31, | ||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||
| (In thousands, except net loss per share) | ||||||||||||||||||||
Consolidated Statements of Operations Data: |
||||||||||||||||||||
Revenues |
$ | — | $ | — | $ | 24,118 | $ | 5,420 | $ | 4,887 | ||||||||||
Operating expenses: |
||||||||||||||||||||
Research and development(1) |
19,494 | 25,993 | 28,192 | 25,419 | 20,901 | |||||||||||||||
General and administrative(1) |
8,243 | 8,109 | 8,692 | 11,308 | 7,705 | |||||||||||||||
Total operating expenses(1) |
27,737 | 34,102 | 36,884 | 36,727 | 28,606 | |||||||||||||||
Loss from operations |
(27,737 | ) | (34,102 | ) | (12,766 | ) | (31,307 | ) | (23,719 | ) | ||||||||||
Other income (expense): |
||||||||||||||||||||
Interest income |
478 | 1,482 | 3,611 | 4,727 | 2,103 | |||||||||||||||
Interest expense |
— | — | — | (69 | ) | (189 | ) | |||||||||||||
Loss from valuation of common stock
warrant liability |
(151 | ) | — | — | — | — | ||||||||||||||
Other, net |
132 | 218 | (17 | ) | (111 | ) | (118 | ) | ||||||||||||
Total other income (expense), net |
459 | 1,700 | 3,594 | 4,547 | 1,796 | |||||||||||||||
Net loss |
$ | (27,278 | ) | $ | (32,402 | ) | $ | (9,172 | ) | $ | (26,760 | ) | $ | (21,923 | ) | |||||
Net loss per share, basic and diluted: |
$ | (0.81 | ) | $ | (1.13 | ) | $ | (0.32 | ) | $ | (0.94 | ) | $ | (0.89 | ) | |||||
Shares used in calculating net loss per
share, basic and diluted: |
33,775 | 28,750 | 28,646 | 28,512 | 24,756 | |||||||||||||||
| (1) | As a result of the adoption of ASC 718 on January 1, 2006, there is a lack of comparability in our research and development expenses and our general and administrative expenses for the periods presented prior to January 1, 2006. Please reference Note 10 in our consolidated financial statements for additional information related to the impact of ASC 718 on our research and development expenses and our general and administrative expenses. |
| As of December 31, | ||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
Consolidated Balance Sheet Data: |
||||||||||||||||||||
Cash, cash equivalents and securities available-for-sale |
$ | 20,490 | $ | 27,936 | $ | 56,495 | $ | 82,149 | $ | 104,851 | ||||||||||
Working capital |
13,769 | 24,325 | 52,084 | 75,054 | 98,682 | |||||||||||||||
Total assets |
21,735 | 31,674 | 61,526 | 89,401 | 116,976 | |||||||||||||||
Long-term debt, net of current portion |
— | — | — | — | 682 | |||||||||||||||
Accumulated deficit |
(283,332 | ) | (256,054 | ) | (223,652 | ) | (214,480 | ) | (187,720 | ) | ||||||||||
Total stockholders’ equity |
14,429 | 25,825 | 55,679 | 60,325 | 78,936 | |||||||||||||||
35
Table of Contents
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
MANAGEMENT’S DISCUSSION AND ANALYSIS OF
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
This discussion and analysis should be read in conjunction with our financial statements and
notes thereto included in this Annual Report on Form 10-K (this Annual Report). Operating results
are not necessarily indicative of results that may occur in future periods.
This Annual Report contains forward-looking statements. These forward-looking statements
involve a number of risks and uncertainties. Such forward-looking statements include statements
about our development plans and programs, clinical trials, strategies, objectives, and other
statements that are not historical facts, including statements which may be preceded by the words
“intend,” “will,” “plan,” “expect,” “anticipate,” “estimate,” “aim,” “seek,” “believe,” “hope” or
similar words. For such statements, we claim the protection of the Private Securities Litigation
Reform Act of 1995. Readers of this Annual Report are cautioned not to place undue reliance on
these forward-looking statements, which speak only as of the date on which they are made. We
undertake no obligation to update publicly or revise any forward-looking statements. Actual events
or results may differ materially from our expectations. Important factors that could cause actual
results to differ materially from those stated or implied by our forward-looking statements
include, but are not limited to, the risk factors identified in our periodic reports filed with the
Securities and Exchange Commission (SEC), including those set forth in “Item 1A. Risk Factors” in
this Annual Report.
Overview
Anadys Pharmaceuticals, Inc. is a biopharmaceutical company dedicated to improving patient
care by developing novel medicines for the treatment of hepatitis C. We believe hepatitis C
represents a large and significant unmet medical need. Our objective is to contribute to an
improved treatment outcome for patients with this serious disease. We are currently focusing our
efforts on the development of ANA598, a small-molecule, non-nucleoside inhibitor of the NS5B
polymerase for the treatment of hepatitis C. We are currently conducting a Phase II study of ANA598
in combination with pegylated interferon alpha and ribavirin, which is the current standard of care
(SOC) for the treatment of hepatitis C. This study is being conducted in patients infected with
hepatitis C virus (HCV).
On June 3, 2009, we initiated a strategic restructuring to focus our operations on the
development of ANA598, in particular the Phase II study of ANA598 in combination with interferon
and ribavirin. The strategic restructuring resulted in a reduction in our workforce of
approximately 40%, with most of the employees having departed by the end of June 2009 and several
others departing at later dates prior to the end of 2009. We incurred a cash charge of
approximately $1.3 million for the year ended December 31, 2009, which is included in operating
expenses, for cash severance, benefits and outplacement services in connection with the workforce
reduction. In addition, we incurred a noncash charge of $0.4 million associated with the
modification of stock options for individuals included in the strategic restructuring.
We also have investigated ANA773, an oral, small-molecule inducer of endogenous interferons
that acts via the Toll-like receptor 7, or TLR7, pathway in a Phase I trial in hepatitis C. In 2009
we concluded a Phase I clinical trial of ANA773 in HCV patients.
We have also investigated ANA773 for the treatment of cancer. In 2009 we elected to stop
enrollment of new patients in the ongoing Phase I oncology trial to focus our resources on ANA598.
We plan for currently enrolled patients to continue to receive ANA773 until disease progression is
observed and to conclude the trial once all patients reach this point.
We have incurred significant operating losses since our inception and, as of December 31,
2009, our accumulated deficit was $283.3 million. We expect to incur substantial losses for at
least the next several years as we:
| • | continue the development of ANA598 for the treatment of HCV; | ||
| • | develop methods for and scale-up manufacturing of ANA598 for clinical trials and potential commercialization; | ||
| • | commercialize any product candidates that receive regulatory approval; and | ||
| • | potentially in-license technology and acquire or invest in businesses, products or technologies that are synergistic with our own. |
36
Table of Contents
Research and Development
During 2009 and 2008, research and development expenses consisted primarily of costs
associated with clinical development of the Company’s product candidates. During 2007, research and
development expenses consisted primarily of costs associated with the discovery, preclinical and
clinical development of our product candidates. Research and development expenses may
include external costs such as fees paid to clinical research organizations, clinical trial
investigators, contract research organizations, drug substance and drug product manufacturers and
consultants. Research and development expenses may also include internal costs such as
compensation, supplies, materials, an allocated portion of facilities costs, an allocated portion
of information systems support personnel and depreciation.
At this time, due to the risks inherent in the clinical trial process and given the
early-stage of development of our product candidates, we are unable to estimate with any certainty
the costs we will incur in the continued development of our product candidates for
commercialization. Clinical development timelines, likelihood of success and total costs vary
widely. However, we expect our research and development costs to be substantial and to increase as
we advance our product candidates through clinical development.
The following summarizes our research and development expenses for the years ended December
31, 2009, 2008 and 2007 (in thousands):
| For the years ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
ANA598 |
$ | 10,355 | $ | 11,044 | $ | 8,390 | ||||||
ANA773 |
3,103 | 8,177 | 6,136 | |||||||||
ANA380 |
— | — | 700 | |||||||||
ANA975, net of reimbursement |
— | 117 | 560 | |||||||||
Discovery stage programs |
— | — | 1,936 | |||||||||
Infrastructure, support personnel and other |
4,052 | 5,384 | 7,194 | |||||||||
Severance related to reduction in force |
630 | — | 813 | |||||||||
Non-cash employee and non-employee share-based compensation |
1,354 | 1,271 | 2,463 | |||||||||
Total research and development expense |
$ | 19,494 | $ | 25,993 | $ | 28,192 | ||||||
Effective July 1, 2008, we began allocating costs for ANA773 between our HCV and oncology
program. For the year ended December 31, 2009, ANA773 HCV related costs were $2.3 million and
ANA773 oncology related costs were $0.8 million. For the six months ended December 31, 2008, ANA773
HCV related costs were $3.1 million and ANA773 oncology related costs were $0.9 million.
General and Administrative
General and administrative expenses consist primarily of salaries and benefits for executive,
finance, investor relations, business development, human resources and legal personnel. In
addition, general and administrative expenses include insurance costs, professional services and an
allocated portion of facilities costs and information systems support personnel.
Critical Accounting Policies
Our discussion and analysis of our financial condition and results of operations are based on
our consolidated financial statements, which have been prepared in accordance with United States
generally accepted accounting principles (GAAP). The preparation of these consolidated financial
statements requires us to make estimates and judgments that affect the reported amounts of assets,
liabilities and expenses and related disclosure of contingent assets and liabilities. We review our
estimates on an on-going basis and make adjustments to the consolidated financials statements as
considered necessary. We base our estimates on historical experience and on various other
assumptions that we believe to be reasonable under the circumstances, the results of which form the
basis for making judgments about the carrying values of assets and liabilities. Actual results may
differ from these estimates under different assumptions or conditions. While all of our significant
accounting policies are described in Note 1 to our consolidated financial statements included in
this Annual Report, we believe the following accounting policies involve the judgments and
estimates used in the preparation of our consolidated financial statements:
37
Table of Contents
Drug Development Costs. Drug development costs include costs associated with the development
of our product candidates including non-clinical activities, toxicology studies, manufacturing of
non-clinical and clinical materials and clinical trials. We review and accrue drug development
costs based on work performed. We estimate work performed utilizing factors such as subject
enrollment, estimated timeline for completion of studies and other factors. These costs and
estimates vary based on the type, scope and length of non-clinical and clinical studies as well as
other factors. Drug development cost accruals are subject to revisions as studies, projects and
trials progress to completion. Expense is adjusted for revisions in the period in which the facts
that give rise to the revision become known.
Common Stock Warrant Liability. We account for common stock warrants which may potentially be
settled with cash as a liability. The common stock warrants have been recorded at their fair value
at issuance and will continue to be recorded at fair value each subsequent balance sheet date until
such time that they are exercised or are otherwise modified to remove the provisions that require
this treatment, at which time the warrants will be adjusted to fair value and reclassified from
liabilities to stockholders’ equity. Any change in value between reporting periods will be recorded
as other income (expense) at each reporting date. The fair value of the warrants is estimated using
the Black-Scholes pricing model.
Share-Based Compensation. Share-based compensation cost is estimated at the grant date based
on the award’s fair-value as calculated by a Black-Scholes pricing model and the portion that is
expected to vest is recognized as expense evenly over the requisite service period. The
determination of the fair value of share-based payment awards on the date of grant using an
option-pricing model is affected by our stock price as well as assumptions regarding a number of
complex and subjective variables. These variables include, but are not limited to, our expected
stock price volatility over the term of the awards, the risk-free interest rate, the expected term
of the awards and expected forfeitures. If any of the assumptions used in the model change
significantly, share-based compensation expense may differ materially in the future from that
recorded in the current period.
Results of Operations
Comparison of the Years Ended December 31, 2009, 2008 and 2007
Revenue. During 2009 and 2008, we did not recognize any revenue. We recorded revenue of $24.1
million for the year ended December 31, 2007. The $24.1 million decrease from 2007 to 2008 was
primarily attributed to the recognition in 2007 of previously deferred revenue upon the termination
of our License and Co-Development Agreement with Novartis.
Research and Development Expenses. Research and development expenses were $19.5 million,
$26.0 million and $28.2 million for the years ended December 31, 2009, 2008 and 2007, respectively.
The $6.5 million decrease from 2008 to 2009 was primarily due to $6.4 million in cost savings
associated with our strategic restructuring initiated in June 2009 of which, $5.1 million was due
to reduced ANA773 development costs and $1.3 million of which was due to reduced infrastructure and support personnel costs.
These decreases were partially offset by severance costs of $0.6 million. During 2009, we incurred
the following external development costs associated with our ANA598 program: $1.2 million
associated with our completed Phase Ib clinical trial in HCV patients, $1.0 million associated with
our completed long-term chronic toxicology studies of ANA598, $0.8 million associated with our
completed 14-day healthy volunteer study and $2.0 million for our on-going Phase II clinical trial
in HCV patients. During 2009, we incurred the following external development costs associated with
our ANA773 program: $1.4 million associated with our completed Phase I clinical trial for HCV and
$0.3 million associated with our on-going Phase I clinical trial for oncology. As we have elected
to suspend the development of ANA773 for HCV and oncology, we do not anticipate incurring
significant costs related to this program in future periods. Our non-cash share-based compensation
expense associated with share-based payments granted to our research and development employees was
$1.4 million for the year ended December 31, 2009 compared to $1.3 million for the year ended
December 31, 2008. Included in our non-cash share-based compensation expense for the year-ended
December 31, 2009 is $0.3 million associated with the modification of stock options for individuals
included in our strategic restructuring.
The $2.2 million decrease in research and development expenses from 2007 to 2008 was primarily
due to cost savings derived from our completed 2007 strategic restructuring which included the
halting of early stage discovery efforts and the termination of our collaborations with both
Novartis and LG Life Sciences. These decreases were partially offset by an increase in development
costs for ANA598 and ANA773. In 2008, we incurred development costs associated with our ANA598
program related to our completed Phase Ia clinical trial in healthy volunteers, our on-going Phase
Ib clinical trial in HCV patients, the initiation of long-term chronic toxicology studies of ANA598
and the manufacturing of clinical and non-clinical materials. 2008 development costs for ANA773
include costs associated with the manufacturing of non-clinical and clinical materials, our
completed 13-week GLP toxicology studies, our then on-going Phase I clinical trial for HCV and our
then on-going Phase I clinical trial for oncology. Our non-cash share-based compensation expense
associated with share-based payments granted to our research and development employees was $1.3
million
and $2.5 million for the years ended December 31, 2008 and 2007, respectively. The decrease in
our non-cash share-based compensation expense was primarily associated with the cancellation of
outstanding stock options for personnel included in our 2007
workforce reduction, as well as a
reduction in the weighted average fair value assigned to stock options granted in 2008 compared to
2007.
38
Table of Contents
General and Administrative Expenses. General and administrative expenses were $8.2 million,
$8.1 million and $8.7 million for the years ended December 31, 2009, 2008 and 2007, respectively.
The $0.1 million increase from 2008 to 2009 was primarily attributable to severance costs of $0.7
million, which were partially offset by a reduction in allocated facility costs associated with the
relocation of our corporate headquarters to a smaller facility. The $0.6 million decrease from 2007
to 2008 was primarily the result of cost savings derived from our completed 2007 strategic
restructuring. Non-cash share-based compensation expense associated with share-based payments
granted to our general and administrative employees and non-employee directors for the years ended
December 31, 2009, 2008 and 2007 was $1.4 million, $1.5 million and $1.7 million, respectively.
Included in our non-cash share-based compensation expense for the year-ended December 31, 2009 is
$0.1 million associated with the modification of stock options for individuals included in our 2009
strategic restructuring.
Interest Income. Interest income was $0.5 million, $1.5 million and $3.6 million for the years
ended December 31, 2009, 2008 and 2007, respectively. The $1.0 million decrease in our interest
income from 2008 to 2009 was the result of a lower average cash, cash equivalents and securities
available-for-sale balance and lower interest rates during 2009 compared to 2008. Our average
balance of cash, cash equivalents and securities available-for-sale, which were invested in
interest bearing securities, was $22.9 million in 2009 compared to $40.6 million in 2008. The
decrease in our average cash balance from 2008 to 2009 was driven by our use of cash, cash
equivalents and securities to fund our on-going operations partially offset by proceeds received
from the equity financing during June 2009. The $2.1 million decrease in our interest income from
2007 to 2008 was the result of a lower average cash, cash equivalents and securities
available-for-sale balance and lower interest rates during 2008 compared to 2007. Our average
balance of cash, cash equivalents and securities available-for-sale, which were invested in
interest bearing securities, was $40.6 million in 2008 compared to $67.7 million in 2007. The
decrease in our average cash balance from 2007 to 2008 was driven by our use of cash, cash
equivalents and securities to fund our on-going operations.
Common Stock Warrant Liability. Non-operating expense associated with the increase in common
stock warrant liability was $0.2 million for the year ended December 31, 2009. This represents the
increase in the fair value of the warrants from June 3, 2009 to December 31, 2009. The fair value
was calculated using the Black Scholes pricing model and is remeasured at each reporting period.
Potential future increases in our stock price will result in losses being recognized in our
statement of operations in future periods. Conversely, potential future declines in our stock price
will result in gains being recognized in our statement of operations in future periods.
Liquidity and Capital Resources
Overview
Our December 31, 2009 cash, cash equivalents and marketable securities balance was $20.5
million. Our cash, cash equivalents and available-for sale securities decreased by $7.4 million
from December 31, 2008 to December 31, 2009. The decrease in cash, cash equivalents and securities
available-for-sale is the result of year-to-date cash utilization to fund our operations, partially
offset by net proceeds of $16.0 million received from our completed equity financing during June
2009. We believe that our existing cash, cash equivalents and securities available-for-sale will be
sufficient to meet our projected operating requirements for at least the next twelve months.
On June 3, 2009, the Company sold 8.4 million shares of common stock and warrants to purchase
2.9 million shares of common stock to institutional investors for net proceeds of approximately
$16.0 million. The proceeds from this equity financing are being utilized to fund operating
activities and to continue to advance ANA598 for the treatment of HCV.
Excluding the net proceeds from our equity financing completed during June 2009, we used $23.4
million in cash to fund operations during the year ended December 31, 2009 compared to $28.6
million during the year ended December 31, 2008. The decease in our operating cash burn can be
attributed to the following factors: our strategic restructuring, initiated in June 2009, to focus
our operations on the development of ANA598, our decision to suspend the development of ANA773 for
HCV and oncology and the relocation of our corporate headquarters to a smaller facility in July
2009. We anticipate the strategic restructuring completed in 2009 will generate annual cash expense savings of
approximately $4.0 million to $5.0 million. We anticipate the relocation of our operations to
a smaller building to achieve an annual facility expense reduction of approximately $1.8
million.
39
Table of Contents
Future Cash Requirements
Over time we expect our development expenses to be substantial and to increase as we continue
the advancement of our development programs. The lengthy process of completing clinical trials and
seeking regulatory approval for our product candidates requires the expenditure of substantial
resources. Any failure by us or delay in completing clinical trials, or in obtaining regulatory
approvals, could cause our research and development expenses to increase and, in turn, have a
material adverse effect on our results of operations.
Our future capital uses and requirements depend on numerous forward-looking factors. These
factors may include but are not limited to the following:
| • | the progress of our clinical trials; | ||
| • | the progress of our nonclinical development activities; | ||
| • | our ability to establish and maintain strategic alliances; | ||
| • | the costs involved in enforcing or defending patent claims and other intellectual property rights; | ||
| • | the costs and timing of regulatory approvals; | ||
| • | the costs of establishing or expanding manufacturing, sales and distribution capabilities; | ||
| • | the costs related to development and manufacture of non-clinical, clinical and validation lots for regulatory and commercialization of drug supply; | ||
| • | the success of the commercialization of ANA598, ANA773 or any other product candidates we may develop; and | ||
| • | the extent to which we acquire or invest in other products, technologies and businesses. |
Investment Portfolio
As of December 31, 2009, we have $20.1 million of securities available-for-sale
consisting of money market funds, commercial paper, municipal bonds, U.S. treasury notes, U.S.
government sponsored enterprise securities and corporate debt securities with maturities that range
from one day to 12 months with an overall average months to maturity of 5.1 months. We have the
ability to liquidate these marketable securities without restriction or penalty.
As of December 31, 2009, we performed a review of all of the securities in our portfolio with
an unrealized loss position, to determine if any other-than-temporary impairments were required to
be recorded. Factors considered in our assessment included but were not limited to the following:
our ability and intent to hold the security until maturity; the number of months until the
security’s maturity, the number of quarters that each security was in an unrealized loss position,
ratings assigned to each security by independent rating agencies, the magnitude of the unrealized
loss compared to the face value of the security and other market conditions. No
other-than-temporary impairments were identified as of December 31, 2009 related to securities
currently in our portfolio. We also noted that none of the securities as of December 31, 2009 have
been in an unrealized loss position for greater than one year. As of December 31, 2009 we do not
own any asset-backed securities or auction rate securities.
Cash Flows from Operating Activities and Investing Activities
Our consolidated statements of cash flows are summarized as follows (in thousands):
| For the years ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
Net cash used in operating activities |
$ | (24,229 | ) | $ | (28,288 | ) | $ | (25,658 | ) | |||
Cash provided by (used in) investing activities |
||||||||||||
Purchase of securities available-for-sale |
$ | (24,657 | ) | $ | (8,806 | ) | $ | (15,131 | ) | |||
Proceeds from sale of securities available-for-sale |
26,484 | 12,463 | 13,170 | |||||||||
Purchase of property and equipment |
(88 | ) | (213 | ) | (356 | ) | ||||||
Proceeds from disposal of property and equipment |
111 | 392 | — | |||||||||
Net cash provided by (used in) investing activities |
$ | 1,850 | $ | 3,836 | $ | (2,317 | ) | |||||
40
Table of Contents
We expect to continue to utilize cash and marketable securities to fund our operating
activities as we continue to advance our wholly owned product candidate ANA598. We are not
currently party to any development collaborations and therefore cash to fund future operations will
most likely have to be obtained from one of the following sources: our current investment
portfolio, the sale of equity securities, new strategic alliance agreements or other transactions,
project financing or debt financing.
Cash Flows from Financing Activities
Our consolidated statements of cash flows are summarized as follows (in thousands):
| For the years ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
Cash provided by financing activities |
||||||||||||
Proceeds from exercise of stock options and employee stock purchase plan |
$ | 385 | $ | 259 | $ | 257 | ||||||
Proceeds from equity financing, net of issuance costs |
16,015 | — | — | |||||||||
Net cash provided by financing activities |
$ | 16,400 | $ | 259 | $ | 257 | ||||||
On June 3, 2009, the Company sold 8.4 million shares of common stock and warrants to purchase
2.9 million shares of common stock to institutional investors for net proceeds of approximately
$16.0 million. The proceeds from this equity financing are being utilized to fund operating
activities and to advance ANA598 for the treatment of HCV.
Aggregate Contractual Obligations
The following summarizes our contractual obligations as of December 31, 2009 (in thousands):
| Less | ||||||||||||||||||||
| than 1 | 2011 to | 2013 to | ||||||||||||||||||
| Contractual Obligations | Total | Year | 2012 | 2014 | Thereafter | |||||||||||||||
Operating leases |
$ | 388 | $ | 358 | $ | 30 | $ | — | $ | — | ||||||||||
Minimum royalty commitment |
700 | 100 | 200 | 200 | 200 | |||||||||||||||
| $ | 1,088 | $ | 458 | $ | 230 | $ | 200 | $ | 200 | |||||||||||
We also enter into agreements with clinical sites and contract research organizations that
conduct our clinical trials. We generally make payments to these entities based upon the number of
subjects enrolled and the length of their participation in the trials. To date, the majority of our
clinical costs have been related to the costs of subjects entering our clinical trials as well as
the manufacturing of compounds to be used in our clinical trials. Costs associated with clinical
trials will continue to vary as the trials go through their natural phases of enrollment and
follow-up. The costs will also be influenced by the pace of the development activities, timing of
the development activities and regulatory requirements associated with the conduct of our clinical
trials. At this time, due to the risks inherent in the clinical trial process and given the early
stage of development of our product development programs, we are unable to estimate with any
certainty the total costs we will incur in the continued development of our product candidates for
potential commercialization. Due to these same factors, we are unable to determine the anticipated
completion dates for our current product development programs. Clinical development timelines,
probability of success and development costs vary widely. As we continue our development programs,
we anticipate that we will make determinations as to how much funding to direct to each program on
an ongoing basis in response to the scientific and clinical success of each product candidate, as
well as an ongoing assessment of the product candidate’s commercial potential. In addition, we
cannot forecast with any degree of certainty whether any of our product candidates will be subject
to future partnering, when such arrangements will be secured, if at all, and to what degree such
arrangements would affect our development plans and capital requirements. As a result, we cannot be
certain when, or if, and to what extent we will receive cash inflows from the commercialization of
our product candidates.
Fair Value Inputs
Fair value is a market-based measurement that is determined based on assumptions that market
participants would use in pricing an asset or liability. See Notes 2 and 3 to the audited
consolidated financial statements, which are included elsewhere in this Annual Report.
We value our marketable securities by using quoted market prices, broker or dealer quotations
or alternative pricing sources with reasonable levels of price transparency. The types of
securities valued based on quoted market prices in active markets include money market securities.
We do not adjust the quoted price for such securities. The types of instruments valued based on
quoted prices in markets that are not active, broker or dealer quotations, or alternative pricing
sources with reasonable levels of price transparency
41
Table of Contents
include commercial paper, municipal bonds, U.S. treasury notes, U.S. government
sponsored enterprise securities and corporate debt securities. The price for each security at the
measurement date is sourced from an independent pricing vendor. Periodically, management assesses
the reasonableness of these sourced prices by comparing them to the prices provided by our
portfolio managers to derive the fair value of these financial instruments. Historically, we have
not experienced significant deviation between the prices from the independent pricing vendor and
our portfolio managers. Management assesses the inputs of the pricing in order to categorize the
financial instruments into the appropriate hierarchy levels. The fair value of the common stock
warrants, which may potentially be settled with cash and are therefore treated as a liability, is
estimated using the Black-Scholes pricing model.
Off-Balance Sheet Arrangements
As of December 31, 2009, 2008 and 2007, we did not have any relationships with unconsolidated
entities or financial partnerships, such as entities often referred to as structured finance or
special purpose entities, which would have been established for the purpose of facilitating
off-balance sheet arrangements or other contractually narrow or limited purposes. In addition, we
do not engage in trading activities involving non-exchange traded contracts. As such, we are not
materially exposed to any financing, liquidity, market or credit risk that could arise if we had
engaged in these relationships. We do not have relationships or transactions with persons or
entities that derive benefits from their non-independent relationship with us or our related
parties.
Item 7A. Quantitative and Qualitative Disclosure About Market Risk
Our primary exposure to market risk is interest income sensitivity, which is affected by
changes in the general level of U.S. interest rates, particularly because the majority of our
investments are in short-term marketable securities. Due to the nature of our short-term
investments, we believe that we are not subject to any material market risk exposure. We do not
have any foreign currency or other derivative financial instruments.
Item 8. Financial Statements and Supplementary Data
The consolidated financial statements and related financial information required to be filed
are indexed on page F-1 of this Annual Report on Form 10-K and are incorporated herein.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
Not Applicable.
42
Table of Contents
Item 9A. Controls and Procedures
Management’s Report on Internal Control Over Financial Reporting
Evaluation of Disclosure Controls and Procedures: Our President and Chief Executive Officer
and Vice President, Finance and Operations performed an evaluation of the effectiveness of our
disclosure controls and procedures (as defined in Rules 13a-l5(e) and l5d-15(e) of the Securities
Exchange Act of 1934) as of the end of the period covered by this Annual Report. Based on that
evaluation, our Chief Executive Officer and Vice President, Finance and Operations concluded that
our disclosure controls and procedures were effective as of December 31, 2009 in providing them
with material information related to the Company in a timely manner, as required to be disclosed in
the reports the Company files under the Exchange Act.
Management’s Annual Report on Internal Control over Financial Reporting: Our management is
responsible for establishing and maintaining adequate internal control over financial reporting, as
such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f).
Under the supervision and with the participation of our management, including our President
and Chief Executive Officer and Vice President, Finance and Operations, we conducted an evaluation
of the effectiveness of our internal control over financial reporting based on the framework in
Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the
Treadway Commission. Based on our evaluation under the framework in Internal Control — Integrated
Framework, our management concluded that our internal control over financial reporting was
effective as of December 31, 2009.
The effectiveness of our internal control over financial reporting as of December 31, 2009 has
been audited by Ernst & Young LLP, an independent registered public accounting firm.
Changes in Internal Control Over Financial Reporting: There was no significant change in our
internal control over financial reporting that occurred during our most recent fiscal quarter that
has materially affected, or is reasonably likely to materially affect, our internal control over
financial reporting.
43
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of Anadys Pharmaceuticals, Inc.
We have audited Anadys Pharmaceuticals, Inc.’s internal control over financial reporting as of
December 31, 2009, based on criteria established in Internal Control-Integrated Framework issued by
the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Anadys
Pharmaceuticals, Inc.’s management is responsible for maintaining effective internal control over
financial reporting, and for its assessment of the effectiveness of internal control over financial
reporting included in the accompanying Management’s Report on Internal Control Over Financial
Reporting. Our responsibility is to express an opinion on the company’s internal control over
financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting
Oversight Board (United States). Those standards require that we plan and perform the audit to
obtain reasonable assurance about whether effective internal control over financial reporting was
maintained in all material respects. Our audit included obtaining an understanding of internal
control over financial reporting, assessing the risk that a material weakness exists, testing and
evaluating the design and operating effectiveness of internal control based on the assessed risk,
and performing such other procedures as we considered necessary in the circumstances. We believe
that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide
reasonable assurance regarding the reliability of financial reporting and the preparation of
financial statements for external purposes in accordance with generally accepted accounting
principles. A company’s internal control over financial reporting includes those policies and
procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately
and fairly reflect the transactions and dispositions of the assets of the company; (2) provide
reasonable assurance that transactions are recorded as necessary to permit preparation of financial
statements in accordance with generally accepted accounting principles, and that receipts and
expenditures of the company are being made only in accordance with authorizations of management and
directors of the company; and (3) provide reasonable assurance regarding prevention or timely
detection of unauthorized acquisition, use, or disposition of the company’s assets that could have
a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent
or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are
subject to the risk that controls may become inadequate because of changes in conditions, or that
the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Anadys Pharmaceuticals, Inc. maintained, in all material respects, effective
internal control over financial reporting as of December 31, 2009, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting
Oversight Board (United States), the consolidated balance sheets of Anadys Pharmaceuticals, Inc. as
of December 31, 2009 and 2008, and the related consolidated statements of operations, stockholders’
equity and cash flows for each of the three years in the period ended December 31, 2009 of Anadys
Pharmaceuticals, Inc. and our report dated March 1, 2010 expressed an unqualified opinion
thereon.
| /s/ Ernst & Young LLP | ||||
San Diego, California
March 1, 2010
March 1, 2010
44
Table of Contents
Table of Contents
Part III
Certain information required by Part III of Form 10-K is omitted from this report because we
expect to file a definitive proxy statement for our 2010 Annual Meeting of Stockholders (the Proxy
Statement) within 120 days after the end of our fiscal year pursuant to Regulation 14A promulgated
under the Securities Exchange Act of 1934, as amended, and the information included in the Proxy
Statement is incorporated herein by reference to the extent provided below.
| Item 10. | Directors, Executive Officers and Corporate Governance |
The information required by Item 10 of Form 10-K is incorporated by reference to the
information under the headings “Election of Directors,” “Section 16(a) Beneficial Ownership
Reporting Compliance,” “Audit Committee” and “Shareholder Communications with the Board of
Directors” in our Proxy Statement.
Certain information required by Item 10 of Form 10-K regarding our executive officers is set
forth in Item 1 of Part I of this Annual Report under the caption “Executive Officers of the
Registrant.”
We have adopted a Code of Business Conduct and Ethics, which applies to all our directors,
officers and employees, including our President and Chief Executive Officer and Vice President,
Finance and Operations and all of our finance team. The Code of Business Conduct and Ethics is
posted on our website, http://www.anadyspharma.com (under the “Investors — Corporate
Governance” caption). In addition, we will provide to any person without charge, upon request,
addressed to the Corporate Secretary at Anadys Pharmaceuticals, Inc., 5871 Oberlin Drive, Suite
200, San Diego, CA 92121, a copy of our Code of Business Conduct and Ethics. We intend to satisfy
the disclosure requirement regarding any amendment to, or waiver of, a provision of the Code of
Business Conduct and Ethics for our President and Chief Executive Officer and Vice President,
Finance and Operations or persons performing similar functions, by posting such information on our
website.
| Item 11. | Executive Compensation |
The information required by Item 11 of Form 10-K is incorporated by reference to the
information under the heading “Compensation of Executive Officers” and “Compensation of Directors”
in our Proxy Statement.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The following table summarizes our outstanding securities and securities available for future
issuance under our equity compensation plans as of December 31, 2009. Security holders of the
Company have approved the 2002 Equity Incentive Plan, 2004 Equity Incentive Plan (2004 Plan), 2004
Non-Employee Directors’ Stock Option Plan and 2004 Employee Stock Purchase Plan.
In connection with the hiring of certain executive officers during 2006, the Compensation
Committee of our Board of Directors approved inducement grants of non-qualified stock options.
These option awards were granted without security holder approval pursuant to NASDAQ Marketplace
Rule 4350(i)(1)(A)(iv). Although these options were granted outside the 2004 Plan, they are subject
to substantially identical terms and conditions as those contained in the 2004 Plan.
| (c) | ||||||||||||
| Number of securities | ||||||||||||
| remaining available for | ||||||||||||
| (a) | (b) | future issuance under equity | ||||||||||
| Number of securities to | Weighted-average | compensation plans | ||||||||||
| be issued upon exercise | exercise price of | (excluding securities | ||||||||||
| Plan Category | of outstanding options | outstanding options | reflected in column (a)) | |||||||||
Equity
compensation plans
approved by
security holders |
6,817,036 | $ | 3.60 | 1,977,430 | ||||||||
Equity compensation
plans not approved
by security holders |
342,975 | $ | 3.00 | — | ||||||||
Total |
7,160,011 | 1,977,430 | ||||||||||
46
Table of Contents
The additional information required by Item 12 of Form 10-K related to security ownership of
certain beneficial owners and management is incorporated herein by reference to the information
under the heading “Security Ownership of Certain Beneficial Owners and Management” in our Proxy
Statement.
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
The information required by Item 13 of Form 10-K related to transactions with related persons,
promoters and certain control persons, if any, is incorporated herein by reference to the
information under the heading “Certain Transactions” in our Proxy Statement. The information
required by Item 13 of Form 10-K relating to director independence is incorporated herein by
reference to the information under the heading “Election of Directors” in our Proxy Statement.
| Item 14. | Principal Accounting Fees and Services |
The information required by Item 14 of Form 10-K is incorporated herein by reference to the
information under the heading “Ratification of Selection of Independent Registered Accounting Firm”
in our Proxy Statement.
47
Table of Contents
Part IV
Item 15. Exhibits and Financial Statement Schedules
(a) The following financial statements, financial statements schedules and exhibits are filed
as part of this report or incorporated herein by reference:
| (1) | Financial Statements. See index to consolidated financial statements on page F-1. | ||
| (2) | Financial Statement Schedules. All financial statements schedules for which provision is made in Regulation S-X are omitted because they are not required under the related instructions, are inapplicable, or the required information is given in the financial statements, including the notes thereto. | ||
| (3) | Exhibits. |
| Exhibit | ||||
| Number | Exhibit Description | Incorporated by Reference or Attached Hereto | ||
3.1
|
Form of Amended and Restated Certificate of Incorporation of the Registrant | Incorporated by reference to the Registrant’s Quarterly Report on Form 10-Q (SEC File No. 000-50632) filed on May 14, 2004. | ||
3.2
|
Amended and Restated Bylaws of the Registrant | Incorporated by reference to the Registrant’s Current Report on Form 8-K (SEC File No. 000-50632) filed on December 5, 2007. | ||
4.1
|
Form of Specimen Common Stock Certificate | Incorporated by reference to the Registrant’s Registration Statement on Form S-1 (SEC File No. 333-110528) filed on March 18, 2004. | ||
4.2
|
Form of Warrant | Incorporated by reference to the Registrant’s Current Report on Form 8-K (SEC File No. 000-50632) filed on June 4, 2009. | ||
10.1#
|
2002 Equity Incentive Plan | Incorporated by reference to Exhibit 10.3 in the Registrant’s Registration Statement on Form S-1 (SEC File No. 333-110528) filed on November 14, 2003. | ||
10.2#
|
Form of Stock Option Agreement under 2002 Equity Incentive Plan | Incorporated by reference to Exhibit 10.4 in the Registrant’s Registration Statement on Form S-1 (SEC File No. 333-110528) filed on November 14, 2003. | ||
10.3#
|
2004 Equity Incentive Plan | Incorporated by reference to Exhibit 10.5 in the Registrant’s Registration Statement on Form S-1 (SEC File No. 333-110528) filed on March 18, 2004. | ||
10.4#
|
Form of Stock Option Agreement under 2004 Equity Incentive Plan | Incorporated by reference to Exhibit 10.6 in the Registrant’s Registration Statement on Form S-1 (SEC File No. 333-110528) filed on March 18, 2004. | ||
10.5#
|
Form of Amendment to Stock Option Agreement Under 2004 Equity Incentive Plan, applicable to Non-Employee Director grants | Incorporated by reference to Exhibit 10.5 in the Registrant’s Annual Report on Form 10-K (SEC File No. 000-50632) filed on March 3, 2009. | ||
10.6#
|
2004 Employee Stock Purchase Plan | Incorporated by reference to Exhibit 10.7 in the Registrant’s Registration Statement on Form S-1 (SEC File No. 333-110528) filed on March 18, 2004. | ||
10.7#
|
Form of Offering Document under the 2004 Employee Stock Purchase Plan | Incorporated by reference to Exhibit 10.8 in the Registrant’s Registration Statement on Form S-1 (SEC File No. 333-110528) filed on March 18, 2004. | ||
10.8#
|
Form of Indemnification Agreement by and between the Registrant and each of its directors and officers | Incorporated by reference to Exhibit 10.11 in the Registrant’s Registration Statement on Form S-1 (SEC File No. 333-110528) filed on November 14, 2003. |
48
Table of Contents
| Exhibit | ||||
| Number | Exhibit Description | Incorporated by Reference or Attached Hereto | ||
10.9#
|
Form of Stock Option Agreement Under 2004 Non-Employee Directors’ Stock Option Plan | Incorporated by reference to Exhibit 10.10 in the Registrant’s Registration Statement on Form S-1 (SEC File No. 333-110528) filed on March 18, 2004. | ||
10.10#
|
Terms of Employment dated February 1, 2001 by and between the Registrant and Steve Worland, Ph.D. | Incorporated by reference to Exhibit 10.27 in the Registrant’s Registration Statement on Form S-1 (SEC File No. 333-110528) filed on November 14, 2003. | ||
10.11#
|
Terms of Employment dated October 2, 2001 by and between the Registrant and Elizabeth E. Reed | Incorporated by reference to Exhibit 10.30 in the Registrant’s Registration Statement on Form S-1 (SEC File No. 333-110528) filed on March 18, 2004. | ||
10.12#
|
Form of Inducement Stock Option Agreement | Incorporated by reference to Exhibit 10.42 in the Registrant’s Current Report on Form 8-K (SEC File No. 000-50632) filed on September 25, 2006. | ||
10.13#
|
Terms of Employment dated September 11, 2006 by and between the Registrant and James T. Glover | Incorporated by reference to Exhibit 10.43 in the Registrant’s Current Report on Form 8-K (SEC File No. 000-50632) filed on September 25, 2006. | ||
10.14#
|
Amended and Restated Severance and Change in Control Agreement dated March 3, 2008 by and between the Registrant and Stephen T. Worland, Ph.D. | Incorporated by reference to Exhibit 10.16 in the Registrant’s Annual Report on Form 10-K (SEC File No. 000-50632) filed on March 5, 2008. | ||
10.15#
|
Amended and Restated Severance and Change in Control Agreement dated March 3, 2008 by and between the Registrant and James L. Freddo, M.D. | Incorporated by reference to Exhibit 10.18 in the Registrant’s Annual Report on Form 10-K (SEC File No. 000-50632) filed on March 5, 2008. | ||
10.16#
|
Amended and Restated Severance and Change in Control Agreement dated March 3, 2008 by and between the Registrant and Elizabeth E. Reed | Incorporated by reference to Exhibit 10.19 in the Registrant’s Annual Report on Form 10-K (SEC File No. 000-50632) filed on March 5, 2008. | ||
10.17#
|
Terms of Employment dated June 21, 2006 by and between the registrant and James L. Freddo, M.D. | Incorporated by reference to Exhibit 10.21 in the Registrant’s Annual Report on Form 10-K (SEC File No. 000-50632) filed on March 5, 2008. | ||
10.18#
|
Terms of Employment dated March 6, 2001 by and between the Registrant and Mary-Yaroshevsky-Glanville | Incorporated by reference to Exhibit 10.22 in the Registrant’s Annual Report on Form 10-K (SEC File No. 000-50632) filed on March 5, 2008. | ||
10.19#
|
Amended and Restated 2004 Non-Employee Directors’ Stock Option Plan | Incorporated by reference to Exhibit 10.24 in the Registrant’s Quarterly Report on Form 10-Q (SEC File No. 000-50632) filed on May 1, 2008. | ||
10.20
|
Sub-lease agreement dated June 18, 2009 by and between the Registrant and Phenomix Corporation | Incorporated by reference to Exhibit 10.24 in the Registrant’s Quarterly Report on Form 10-Q (SEC File No. 000-50632) filed on July 31, 2009. | ||
10.21#
|
Severance Agreement and General Release dated June 30, 2009 by and between the Registrant and James T. Glover | Incorporated by reference to Exhibit 10.25 in the Registrant’s Quarterly Report on Form 10-Q (SEC File No. 000-50632) filed on July 31, 2009. | ||
10.22#
|
Terms of Employment dated July 1, 2009 for Peter T. Slover | Incorporated by reference to Exhibit 10.26 in the Registrant’s Quarterly Report on Form 10-Q (SEC File No. 000-50632) filed on July 31, 2009. | ||
10.23#
|
Severance and Change in Control Agreement dated July 1, 2009 by and between the Registrant and Peter T. Slover | Incorporated by reference to Exhibit 10.27 in the Registrant’s Quarterly Report on Form 10-Q (SEC File No. 000-50632) filed on July 31, 2009. | ||
10.24#
|
Severance Agreement and General Release dated September 30, 2009 by and between the Registrant and Mary Yaroshevsky-Glanville | Incorporated by reference to Exhibit 10.28 in the Registrant’s Quarterly Report on Form 10-Q (SEC File No. 000-50632) filed on October 30, 2009. |
49
Table of Contents
| Exhibit | ||||
| Number | Exhibit Description | Incorporated by Reference or Attached Hereto | ||
10.25#
|
Anadys Pharmaceuticals, Inc. Executive Officer Bonus Plan | Incorporated by reference to Exhibit 10.29 in the Registrant’s Quarterly Report on Form 10-Q (SEC File No. 000-50632) filed on October 30, 2009. | ||
21.1
|
List of Subsidiaries of the Registrant | Incorporated by reference to Exhibit 21.1 in the Registrant’s Registration Statement on Form S-1 (SEC File No. 333-110528) filed on November 14, 2003. | ||
23.1
|
Consent of Independent Registered Public Accounting Firm | Attached Hereto. | ||
31.1
|
Certification of President and Chief Executive Officer pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under the Securities Act of 1934, as amended | Attached Hereto. | ||
31.2
|
Certification of Vice President, Finance and Operations pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under the Securities Act of 1934, as amended | Attached Hereto. | ||
32.1
|
Certifications of President and Chief Executive Officer and Vice President, Finance and Operations pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | Attached Hereto. |
| # | Indicates management contract or compensatory plan. |
50
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Act of 1934, as amended,
the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto
duly authorized, in the City of San Diego, State of California, on the 1st day of March,
2010.
| ANADYS PHARMACEUTICALS, INC. |
||||
| By: | /s/ STEPHEN T. WORLAND, PH.D. | |||
| Stephen T. Worland, Ph.D. | ||||
| President and Chief Executive Officer | ||||
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes
and appoints Stephen T. Worland, Ph.D. and Peter T. Slover, and each of them, as his true and
lawful attorneys-in-fact and agents, with full power of substitution and resubstitution, for him
and in his name, place, and stead, in any and all capacities, to sign any and all amendments to
this Annual Report on Form 10-K, and any other documents in connection therewith, and to file the
same, with all exhibits thereto, with the Securities and Exchange Commission, granting unto said
attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and
every act and thing requisite and necessary to be done in connection therewith, as fully to all
intents and purposes as he might or could do in person, hereby ratifying and confirming all that
said attorneys-in-fact and agents, or any of them or their or his substitute or substituted, may
lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act of 1934, as amended, this report has been
signed by the following persons on behalf of the Registrant and in the capacities and on the dates
indicated.
| Signature | Title | Date | ||
/s/ STEPHEN T. WORLAND, PH.D.
|
President, Chief Executive Officer and Director | March 1, 2010 | ||
Stephen T. Worland, Ph.D.
|
(Principal Executive Officer) | |||
/s/ PETER T. SLOVER
|
Vice President, Finance and Operations | March 1, 2010 | ||
Peter T. Slover
|
(Principal Financial and Accounting Officer) | |||
/s/ GEORGE A. SCANGOS, PH.D.
|
Chairman of the Board | March 1, 2010 | ||
George A. Scangos, Ph.D. |
||||
/s/ MARK G. FOLETTA
|
Director | March 1, 2010 | ||
Mark G. Foletta |
||||
/s/ MARIOS FOTIADIS
|
Director | March 1, 2010 | ||
Marios Fotiadis |
||||
/s/ STEVEN H. HOLTZMAN
|
Director | March 1, 2010 | ||
Steven H. Holtzman |
||||
/s/ STELIOS PAPADOPOULOS, PH.D.
|
Director | March 1, 2010 | ||
Stelios Papadopoulos, Ph.D. |
||||
/s/ KLEANTHIS G. XANTHOPOULOS, PH.D.
|
Director | March 1, 2010 | ||
Kleanthis G. Xanthopoulos, Ph.D. |
51
Table of Contents
ANADYS PHARMACEUTICALS, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| Page | ||
| F-2 | ||
| F-3 | ||
| F-4 | ||
| F-5 | ||
| F-6 | ||
| F-7 |
F-1
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of Anadys Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of Anadys Pharmaceuticals, Inc. as of
December 31, 2009 and 2008, and the related consolidated statements of operations, stockholders’
equity, and cash flows for each of the three years in the period ended December 31, 2009. These
financial statements are the responsibility of the Company’s management. Our responsibility is to
express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight
Board (United States). Those standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are free of material misstatement. An
audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the
financial statements. An audit also includes assessing the accounting principles used and
significant estimates made by management, as well as evaluating the overall financial statement
presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all
material respects, the consolidated financial position of Anadys Pharmaceuticals, Inc. at December
31, 2009 and 2008, and the consolidated results of its operations and its cash flows for each of
the three years in the period ended December 31, 2009, in conformity with U.S. generally accepted
accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight
Board (United States), Anadys Pharmaceuticals, Inc.’s internal control over financial reporting as
of December 31, 2009, based on criteria established in Internal Control-Integrated Framework issued
by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March
1, 2010 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
San Diego, California
March 1, 2010
March 1, 2010
F-2
Table of Contents
ANADYS PHARMACEUTICALS, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except share data)
(In thousands, except share data)
| December 31, | December 31, | |||||||
| 2009 | 2008 | |||||||
Assets |
||||||||
Current assets: |
||||||||
Cash and cash equivalents |
$ | 4,497 | $ | 10,476 | ||||
Securities available-for-sale |
15,993 | 17,460 | ||||||
Prepaid expenses and other current assets |
559 | 2,202 | ||||||
Total current assets |
21,049 | 30,138 | ||||||
Property and equipment, net |
626 | 1,476 | ||||||
Other assets |
60 | 60 | ||||||
Total assets |
$ | 21,735 | $ | 31,674 | ||||
Liabilities and Stockholders’ Equity |
||||||||
Current liabilities: |
||||||||
Accounts payable |
$ | 740 | $ | 558 | ||||
Accrued expenses |
2,643 | 4,823 | ||||||
Common stock warrant liability |
3,897 | — | ||||||
Current portion of deferred rent |
— | 348 | ||||||
Other current liabilities |
— | 84 | ||||||
Total current liabilities |
7,280 | 5,813 | ||||||
Other long-term liabilities |
26 | 36 | ||||||
Commitments and contingencies |
||||||||
Stockholders’ equity: |
||||||||
Preferred stock, $0.001 par value; 10,000,000 shares authorized at December 31, 2009 and
December 31, 2008; no shares issued and outstanding at December 31, 2009 and
December 31, 2008 |
— | — | ||||||
Common stock, $0.001 par value; 90,000,000 shares authorized at December 31, 2009 and
December 31, 2008; 37,341,957 and 28,816,763 shares issued and outstanding at
December 31, 2009 and December 31, 2008, respectively |
37 | 29 | ||||||
Additional paid-in capital |
297,687 | 282,297 | ||||||
Accumulated other comprehensive gain (loss) |
37 | (447 | ) | |||||
Accumulated deficit |
(283,332 | ) | (256,054 | ) | ||||
Total stockholders’ equity |
14,429 | 25,825 | ||||||
Total liabilities and stockholders’ equity |
$ | 21,735 | $ | 31,674 | ||||
See accompanying notes to consolidated financial statements.
F-3
Table of Contents
ANADYS PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except net loss per share)
(In thousands, except net loss per share)
| For the Years Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
Revenues: |
||||||||||||
Collaborative agreements |
$ | — | $ | — | $ | 24,118 | ||||||
Total revenues |
— | — | 24,118 | |||||||||
Operating Expenses: |
||||||||||||
Research and development |
19,494 | 25,993 | 28,192 | |||||||||
General and administrative |
8,243 | 8,109 | 8,692 | |||||||||
Total operating expenses |
27,737 | 34,102 | 36,884 | |||||||||
Loss from operations |
(27,737 | ) | (34,102 | ) | (12,766 | ) | ||||||
Other income (expense): |
||||||||||||
Interest income |
478 | 1,482 | 3,611 | |||||||||
Loss from valuation of common stock warrant liability |
(151 | ) | — | — | ||||||||
Other, net |
132 | 218 | (17 | ) | ||||||||
Total other income (expense), net |
459 | 1,700 | 3,594 | |||||||||
Net loss |
$ | (27,278 | ) | $ | (32,402 | ) | $ | (9,172 | ) | |||
Net loss per share, basic and diluted |
$ | (0.81 | ) | $ | (1.13 | ) | $ | (0.32 | ) | |||
Shares used in calculating net loss per share, basic and diluted |
33,775 | 28,750 | 28,646 | |||||||||
See accompanying notes to consolidated financial statements.
F-4
Table of Contents
ANADYS PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(In thousands, except share data)
(In thousands, except share data)
| Accumulated | ||||||||||||||||||||||||||||||||
| other | ||||||||||||||||||||||||||||||||
| Additional | comprehensive | Total | ||||||||||||||||||||||||||||||
| Preferred stock | Common stock | paid-in | gain | Accumulated | stockholders’ | |||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | capital | (loss) | deficit | equity | |||||||||||||||||||||||||
Balance at December 31, 2006 |
— | $ | — | 28,596,198 | $ | 29 | $ | 274,798 | $ | (22 | ) | $ | (214,480 | ) | $ | 60,325 | ||||||||||||||||
Issuance of common stock pursuant to the exercise of stock options and
warrants |
— | — | 30,192 | — | 89 | — | — | 89 | ||||||||||||||||||||||||
Issuance of common stock pursuant to the employee stock purchase plan |
— | — | 70,558 | — | 168 | — | — | 168 | ||||||||||||||||||||||||
Compensation related to stock options and warrants issued to non-employees |
— | — | — | — | 101 | — | — | 101 | ||||||||||||||||||||||||
Share-based compensation expense including forfeitures |
— | — | — | — | 4,065 | — | — | 4,065 | ||||||||||||||||||||||||
Comprehensive loss: |
||||||||||||||||||||||||||||||||
Unrealized gain on short-term investments |
— | — | — | — | — | 103 | — | 103 | ||||||||||||||||||||||||
Net loss |
— | — | — | — | — | — | (9,172 | ) | (9,172 | ) | ||||||||||||||||||||||
Comprehensive loss |
— | — | — | — | — | — | — | (9,069 | ) | |||||||||||||||||||||||
Balance at December 31, 2007 |
— | $ | — | 28,696,948 | $ | 29 | $ | 279,221 | $ | 81 | $ | (223,652 | ) | $ | 55,679 | |||||||||||||||||
Issuance of common stock pursuant to the exercise of stock options and
warrants |
— | — | 36,567 | — | 106 | — | — | 106 | ||||||||||||||||||||||||
Issuance of common stock pursuant to the employee stock purchase plan |
— | — | 83,248 | — | 153 | — | — | 153 | ||||||||||||||||||||||||
Compensation related to stock options and warrants issued to non-employees |
— | — | — | — | 66 | — | — | 66 | ||||||||||||||||||||||||
Share-based compensation expense including forfeitures |
— | — | — | — | 2,751 | — | — | 2,751 | ||||||||||||||||||||||||
Comprehensive loss: |
||||||||||||||||||||||||||||||||
Unrealized loss on short-term investments |
— | — | — | — | — | (528 | ) | — | (528 | ) | ||||||||||||||||||||||
Net loss |
— | — | — | — | — | — | (32,402 | ) | (32,402 | ) | ||||||||||||||||||||||
Comprehensive loss |
— | — | — | — | — | — | — | (32,930 | ) | |||||||||||||||||||||||
Balance at December 31, 2008 |
— | $ | — | 28,816,763 | $ | 29 | $ | 282,297 | $ | (447 | ) | $ | (256,054 | ) | $ | 25,825 | ||||||||||||||||
Issuance of common stock pursuant to the exercise of stock options |
— | — | 84,465 | — | 232 | — | — | 232 | ||||||||||||||||||||||||
Issuance of common stock pursuant to the employee stock purchase plan |
— | — | 82,729 | — | 153 | — | — | 153 | ||||||||||||||||||||||||
Issuance of common stock associated with equity financing, net of issuance
costs |
— | — | 8,358,000 | 8 | 16,007 | — | — | 16,015 | ||||||||||||||||||||||||
Fair value of common stock warrants issued in connection with equity
financing |
— | — | — | — | (3,746 | ) | — | — | (3,746 | ) | ||||||||||||||||||||||
Compensation related to stock options and warrants issued to non-employees |
— | — | — | — | 125 | — | — | 125 | ||||||||||||||||||||||||
Share-based compensation expense including forfeitures |
— | — | — | — | 2,619 | — | — | 2,619 | ||||||||||||||||||||||||
Comprehensive loss: |
||||||||||||||||||||||||||||||||
Unrealized gain on short-term investments |
— | — | — | — | — | 484 | — | 484 | ||||||||||||||||||||||||
Net loss |
— | — | — | — | — | — | (27,278 | ) | (27,278 | ) | ||||||||||||||||||||||
Comprehensive loss |
— | — | — | — | — | — | — | (26,794 | ) | |||||||||||||||||||||||
Balance at December 31, 2009 |
— | $ | — | 37,341,957 | $ | 37 | $ | 297,687 | $ | 37 | $ | (283,332 | ) | $ | 14,429 | |||||||||||||||||
See accompanying notes to consolidated financial statements.
F-5
Table of Contents
ANADYS PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
(In thousands)
| For the years ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
Cash Flows from Operating Activities: |
||||||||||||
Net loss |
$ | (27,278 | ) | $ | (32,402 | ) | $ | (9,172 | ) | |||
Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
Depreciation and amortization |
853 | 1,141 | 1,431 | |||||||||
Share-based compensation |
2,619 | 2,751 | 4,065 | |||||||||
Amortization of premium/discount on securities available-for-sale |
155 | 157 | — | |||||||||
(Gain) loss on the sale of available-for-sale securities |
(31 | ) | 24 | — | ||||||||
Compensation related to stock option issuances to non-employees |
96 | 16 | 52 | |||||||||
Loss on valuation of common stock warrant liability |
151 | — | — | |||||||||
Rent expense related to warrants issued in connection with operating
lease of the Company’s former facility |
29 | 50 | 49 | |||||||||
(Gain) loss from disposal of property and equipment |
(26 | ) | (149 | ) | 27 | |||||||
Changes in operating assets and liabilities: |
||||||||||||
Accounts receivable |
— | — | 1,175 | |||||||||
Prepaid expenses and other current assets |
1,643 | (1,198 | ) | (63 | ) | |||||||
Other assets |
— | 1,320 | 7 | |||||||||
Accounts payable |
182 | (526 | ) | 290 | ||||||||
Accrued expenses |
(2,180 | ) | 1,057 | 449 | ||||||||
Deferred rent |
(348 | ) | (584 | ) | (466 | ) | ||||||
Deferred revenue |
— | — | (23,567 | ) | ||||||||
Other liabilities |
(94 | ) | 55 | 65 | ||||||||
Net cash used in operating activities |
(24,229 | ) | (28,288 | ) | (25,658 | ) | ||||||
Cash Flows from Investing Activities: |
||||||||||||
Purchase of securities available-for-sale |
(24,657 | ) | (8,806 | ) | (15,131 | ) | ||||||
Proceeds from sale and maturity of securities available-for-sale |
26,484 | 12,463 | 13,170 | |||||||||
Purchase of property and equipment |
(88 | ) | (213 | ) | (356 | ) | ||||||
Proceeds from the sale of property and equipment |
111 | 392 | — | |||||||||
Net cash provided by (used in) investing activities |
1,850 | 3,836 | (2,317 | ) | ||||||||
Cash Flows from Financing Activities: |
||||||||||||
Proceeds from exercise of stock options and employee stock purchase plan |
385 | 259 | 257 | |||||||||
Proceeds from equity financing, net of issuance costs |
16,015 | — | — | |||||||||
Net cash provided by financing activities |
16,400 | 259 | 257 | |||||||||
Net decrease in cash and cash equivalents |
(5,979 | ) | (24,193 | ) | (27,718 | ) | ||||||
Cash and cash equivalents at beginning of year |
10,476 | 34,669 | 62,387 | |||||||||
Cash and cash equivalents at end of year |
$ | 4,497 | $ | 10,476 | $ | 34,669 | ||||||
Supplemental Disclosure of Non-Cash Investing and Financing Activities: |
||||||||||||
Fair value of common stock warrant liability |
$ | 3,746 | $ | — | $ | — | ||||||
Unrealized gain (loss) on securities available-for-sale |
$ | 484 | $ | (528 | ) | $ | 103 | |||||
See accompanying notes to consolidated financial statements.
F-6
Table of Contents
ANADYS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Organization and Summary of Significant Accounting Policies
Organization and Business
Anadys Pharmaceuticals, Inc. (Anadys or the Company) is a biopharmaceutical company dedicated
to improving patient care by developing novel medicines for the treatment of hepatitis C. The
Company believes hepatitis C represents a large and significant unmet medical need. The Company’s
objective is to contribute to an improved treatment outcome for patients with this serious disease.
The Company is currently focusing its efforts on the development of ANA598, a small-molecule,
non-nucleoside inhibitor of the NS5B polymerase for the treatment of hepatitis C. The Company has
also investigated ANA773, an oral, small-molecule inducer of endogenous interferons that acts via
the Toll-like receptor 7, or TLR7, pathway in a Phase I trial in hepatitis C and in a separate
Phase I trial for treatment of patients with advanced cancer.
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of the Company and its
wholly owned subsidiaries, Anadys Pharmaceuticals Europe GmbH and Anadys Development Limited. All
significant intercompany accounts and transactions have been eliminated. In 2003, the Company
discontinued its Anadys Pharmaceuticals Europe GmbH operations and intends to dissolve that entity.
Anadys Development Limited was established in 2005 to serve as a legal representative of the
Company for conducting clinical trials in Europe. As of and for the year ended December 31, 2009,
neither Anadys Pharmaceuticals Europe GmbH nor Anadys Development Limited had active operations.
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting
principles (GAAP) in the United States requires management to make estimates and assumptions that
affect the amounts reported in the consolidated financial statements and accompanying notes. Actual
amounts could differ from those estimates.
Cash and Cash Equivalents
Cash and cash equivalents are comprised of highly liquid investments with an original maturity
of less than three months when purchased and are readily convertible without prior notice or
penalty to known amounts of cash.
Securities Available-for-Sale
Investments with an original maturity of more than three months when purchased have been
classified by management as securities available-for-sale. Such investments are carried at fair
value, with unrealized gains and losses included as a component of accumulated other comprehensive
gain (loss) in stockholders’ equity. Realized gains and losses and declines in value judged to be
other-than-temporary (of which there have been none to date) on available-for-sale securities are
included in interest income. The cost of securities sold is based on the specific-identification
method. The Company views its available-for-sale securities as available for use in current
operations. Accordingly, the Company has classified all investments as short-term, even though the
stated maturity date may be one year or more beyond the current balance sheet date.
Fair Value of Financial Instruments
The carrying amount of cash, cash equivalents, securities available-for-sale, accounts payable
and accrued expenses are considered to be representative of their respective fair value because of
the short-term nature of those items.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to a significant concentration of
credit risk consist primarily of cash, cash equivalents and securities available-for-sale. The
Company maintains deposits in federally insured financial institutions in excess of federally
insured limits. Management, however, believes the Company is not exposed to significant credit risk
due to the financial position of the depository institutions in which those deposits are held.
Additionally, the Company has established guidelines regarding diversification of its investments
and their maturities, which are designed to maintain safety and liquidity.
F-7
Table of Contents
ANADYS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
During 2007, the Company derived a majority of its revenues from a License and Co-Development
Agreement with Novartis International Pharmaceutical Ltd. (Novartis) which was terminated during
2007.
Property and Equipment
Property and equipment are stated at cost and depreciated over the estimated useful lives of
the assets (ranging from three to five years) using the straight-line method. Leasehold
improvements are amortized over the estimated useful life of the asset or the lease term, whichever
is shorter.
Impairment of Long-Lived Assets
If indicators of impairment exist, the Company assesses the recoverability of the affected
long-lived assets by determining whether the carrying value of such assets can be recovered through
undiscounted future operating cash flows. If impairment is indicated, the Company measures the
amount of such impairment by comparing the fair value of the asset to the carrying value of the
asset and records the impairment as a reduction in the carrying value of the related asset and a
charge to operating results. The Company has not recognized any impairment loss for the period
ended December 31, 2009.
Research and Development
During 2009 and 2008, research and development expenses consisted primarily of costs
associated with clinical development of the Company’s product candidates. During 2007, research and
development expenses consisted primarily of costs associated with the discovery, preclinical and
clinical development of the Company’s product candidates. Research and development expenses may
include external costs such as fees paid to clinical research organizations, clinical trial
investigators, contract research organizations, drug substance and drug product manufacturers and
consultants. Research and development expenses may also include internal costs such as
compensation, supplies, materials, an allocated portion of facilities costs, an allocated portion
of information systems support personnel and depreciation.
Under the Company’s former License and Co-Development Agreement with Novartis, which was
terminated during 2007, reimbursements of development costs for ANA975 from Novartis were recorded
as an offset to research and development expense.
Accumulated Other Comprehensive Gain (Loss)
All components of comprehensive gain (loss), including net income (loss), are reported in the
financial statements in the period in which they are recognized. Comprehensive income (loss) is
defined as the change in equity during a period from transactions and other events and
circumstances from non-owner sources. Net income (loss) and other comprehensive income (loss),
including unrealized gains and losses on investments and foreign currency translation adjustments,
are reported, net of their related tax effect, to arrive at comprehensive income (loss).
Deferred Rent
Rent expense is recorded on a straight-line basis over the term of the lease. The difference
between rent expense recorded and amounts paid under the lease agreement is recorded as deferred
rent in the accompanying consolidated balance sheet. During 2004, the Company entered into a
sub-lease agreement to lease the Company’s former corporate headquarters and research and
development facility located in San Diego, California. This lease expired on August 1, 2009. In
accordance with the sub-lease agreement, the Company was allocated a $1.6 million tenant
improvement allowance as an incentive to move into the facility. The Company recorded this
incentive as an increase to both property and equipment and deferred rent and these amounts were
being amortized on a straight-line basis over the life of the lease of 62 months. As of December
31, 2008, the Company had $0.2 million of unamortized deferred rent associated with the lease
incentive. As of December 31, 2009 there was no unamortized deferred rent associated with the lease
incentive. On June 18, 2009, the Company entered into a new sublease agreement to lease its current
corporate headquarters and research and development facility located in San Diego, California.
There is no deferred rent associated with this lease.
Share-Based Compensation
Share-based compensation expense for options granted to employees and non-employee directors
is estimated at the grant date based on the award’s fair value as calculated using a Black-Scholes
pricing model and the portion that is ultimately expected to vest is recognized as expense on a
straight-line basis over the requisite service period. The Company accounts for compensation
expense for options granted to non-employees based on the fair value of the options issued using
the Black-Scholes pricing model and is periodically re-measured as the underlying options vest. The
Company records share-based compensation as components of either research and development expense
or general and administrative expense.
F-8
Table of Contents
ANADYS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Net Loss Per Share
Basic loss per share (EPS) is calculated by dividing the net loss by the weighted-average
number of common shares outstanding for the period, without consideration for common stock
equivalents. Diluted EPS is computed by dividing the net loss by the weighted-average number of
common stock equivalents outstanding for the period determined using the treasury-stock method. For
purposes of this calculation, common stock subject to repurchase by the Company, preferred stock,
options and warrants are considered to be common stock equivalents and are only included in the
calculation of diluted earnings per share when their effect is dilutive.
Common stock equivalents from stock options and warrants of approximately 10.1 million, 6.6
million and 5.6 million were excluded from the calculation of net loss per share for the years
ended December 31, 2009, 2008 and 2007, respectively, because the effect would be antidilutive.
Subsequent Events
The Company evaluated all events or transactions that occurred after the balance sheet date of
December 31, 2009 through the date it issued these financial statements. No
subsequent events were identified requiring additional disclosure in the notes to these financial
statements.
Adoption of Recent Accounting Pronouncements
In June 2009, the Financial Accounting Standards Board (FASB) created the Accounting Standards
Codification (ASC), which is codified as ASC 105. Effective September 15, 2009, the Company adopted
the provisions of ASC 105. The ASC was established as the single source of authoritative accounting
principles recognized by the FASB to be applied by nongovernmental entities in preparation of
financial statements in conformity with U.S. GAAP. The adoption of ASC 105 did not have a material
impact on the Company’s consolidated financial statements.
Effective June 15, 2009, the Company adopted the provisions of ASC 855, which establishes
general standards of accounting for and disclosure of events that occur after the balance sheet
date but before financial statements are issued. The adoption of ASC 855 did not have a material
impact on the Company’s consolidated financial statements.
Effective June 15, 2009, the Company adopted the provisions of ASC 825, which extends the
disclosure requirements regarding the fair value of financial instruments to interim financial
statements of publicly traded companies. The adoption of ASC 825 did not have a material effect on
the Company’s consolidated financial statements.
Effective June 15, 2009, the Company adopted the provisions of ASC 320, which extends the
disclosure requirements regarding debt and equity securities to interim financial statements of
publicly traded companies as well as provides new disclosure requirements. The adoption of ASC 320
did not have a material effect on the Company’s consolidated financial statements.
In December 2007, the FASB ratified the consensus reached by the Emerging Issues Task Force
(EITF) on ASC 808. ASC 808 requires collaborators to present the results of activities for which
they act as the principal on a gross basis and report any payments received from (made to) other
collaborators based on other applicable U.S. GAAP or, in the absence of other applicable U.S. GAAP,
based on analogy to authoritative accounting literature or a reasonable, rational, and consistently
applied accounting policy election. Further, ASC 808 clarified that the determination of whether
transactions within a collaborative arrangement are part of a vendor-customer (or analogous)
relationship subject to ASC 605. ASC 808 was effective for us beginning on January 1, 2009. The
adoption of ASC 808 did not have a material effect on the Company’s consolidated financial
statements.
F-9
Table of Contents
ANADYS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
2. Securities Available-for-Sale
Securities available-for-sale consisted of the following as of December 31, 2009 and 2008,
respectively (in thousands):
| December 31, 2009 | ||||||||||||||||
| Amortized | Unrealized | Market | ||||||||||||||
| Cost | Gain | Loss | Value | |||||||||||||
Commercial paper |
$ | 2,199 | $ | — | $ | — | $ | 2,199 | ||||||||
Municipal bonds |
1,026 | — | — | 1,026 | ||||||||||||
U.S. treasury notes |
2,048 | — | (1 | ) | 2,047 | |||||||||||
U.S. government sponsored enterprise securities |
9,127 | 8 | (5 | ) | 9,130 | |||||||||||
Corporate debt securities |
1,556 | 35 | — | 1,591 | ||||||||||||
| $ | 15,956 | $ | 43 | $ | (6 | ) | $ | 15,993 | ||||||||
| December 31, 2008 | ||||||||||||||||
| Amortized | Unrealized | Market | ||||||||||||||
| Cost | Gain | Loss | Value | |||||||||||||
U.S. Government sponsored enterprise securities |
$ | 6,604 | $ | 72 | $ | — | $ | 6,676 | ||||||||
Corporate debt securities |
11,304 | 64 | (584 | ) | 10,784 | |||||||||||
| $ | 17,908 | $ | 136 | $ | (584 | ) | $ | 17,460 | ||||||||
The amortized cost and estimated fair value of the Company securities available-for-sale by
contractual maturity as of December 31, 2009 and 2008 are shown below (in thousands):
| December 31, 2009 | ||||||||||||||||
| Amortized | Unrealized | Market | ||||||||||||||
| Cost | Gain | Loss | Value | |||||||||||||
Within one year |
$ | 15,956 | $ | 43 | $ | (6 | ) | $ | 15,993 | |||||||
After one year |
— | — | — | — | ||||||||||||
| $ | 15,956 | $ | 43 | $ | (6 | ) | $ | 15,993 | ||||||||
| December 31, 2008 | ||||||||||||||||
| Amortized | Unrealized | Market | ||||||||||||||
| Cost | Gain | Loss | Value | |||||||||||||
Within one year |
$ | 15,641 | $ | 106 | $ | (580 | ) | $ | 15,167 | |||||||
After one year through two years |
2,267 | 30 | (4 | ) | 2,293 | |||||||||||
| $ | 17,908 | $ | 136 | $ | (584 | ) | $ | 17,460 | ||||||||
Included in the Company’s securities portfolio as of December 31, 2008 was an American General
Finance Corporation, a subsidiary of American International Group, Inc. (AIG), corporate bond which
had an unrealized loss position of $0.6 million. This security matured on August 15, 2009 at par
value.
As of December 31, 2009, the Company performed a review of all of the securities in its
portfolio with an unrealized loss position to determine if any other-than-temporary impairments
were required to be recorded. Factors considered in the Company’s assessment included, but were not
limited to the following: the Company’s ability and intent to hold the security until maturity; the
number of months until the security’s maturity, the number of quarters that each security has been
in an unrealized loss position, ratings assigned to each security by independent rating agencies,
the magnitude of the unrealized loss compared to the face value of the security and other market
conditions. No other-than-temporary impairments were identified as of December 31, 2009 related to
securities currently in the Company’s portfolio. The Company also noted that none of the securities
as of December 31, 2009 have been in an unrealized loss position for greater than one year.
3. Fair Value Measurements
As of December 31, 2009, the Company has $20.1 million of marketable securities consisting of
money market funds, commercial paper, municipal bonds, U.S. treasury notes, U.S. government
sponsored enterprise securities and corporate debt securities with maturities that range from one
day to 12 months with an overall average time to maturity of 5.1 months. The Company has the
ability to liquidate these investments without restriction or penalty.
F-10
Table of Contents
ANADYS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Valuation techniques are based on observable and unobservable inputs. Observable inputs
reflect readily obtainable data from independent sources, while unobservable inputs reflect the
Company’s market assumptions. These inputs are classified into the following hierarchy:
Level 1 Inputs — Quoted prices for identical instruments in active markets.
Level 2 Inputs — Quoted prices for similar instruments in active markets; and quoted prices
for identical or similar instruments in markets that are not active.
Level 3 Inputs — Unobservable inputs based on the Company’s assessment that market
participants would use in pricing the instruments.
The Company determines fair value for marketable securities with Level 1 inputs through quoted
market prices. The Company determines fair value for marketable securities with Level 2 inputs
through broker or dealer quotations or alternative pricing sources with reasonable levels of price
transparency. The Company determines fair value for the common stock warrants with Level 3 inputs
through a Black-Scholes pricing model.
The following table presents the Company’s assets and liabilities that are measured at fair
value on a recurring basis as of December 31, 2009 (in thousands):
| Fair Value Measurements at Reporting Date Using | ||||||||||||||||
| Quoted | ||||||||||||||||
| Prices in | ||||||||||||||||
| Active | Significant | |||||||||||||||
| Markets for | Other | Significant | ||||||||||||||
| Identical | Observable | Unobservable | ||||||||||||||
| December 31, | Assets | Inputs | Inputs | |||||||||||||
| 2009 | (Level 1) | (Level 2) | (Level 3) | |||||||||||||
Description |
||||||||||||||||
Assets: |
||||||||||||||||
Money market funds |
$ | 2,101 | $ | 2,101 | $ | — | $ | — | ||||||||
Commercial paper |
3,199 | — | 3,199 | — | ||||||||||||
Municipal bonds |
1,026 | — | 1,026 | — | ||||||||||||
U.S. treasury notes |
2,047 | — | 2,047 | — | ||||||||||||
U.S. government sponsored enterprise securities |
9,130 | — | 9,130 | — | ||||||||||||
Corporate debt securities |
2,599 | — | 2,599 | — | ||||||||||||
Total financial assets |
$ | 20,102 | $ | 2,101 | $ | 18,001 | $ | — | ||||||||
Liabilities: |
||||||||||||||||
Common stock warrants |
$ | 3,897 | $ | — | $ | — | $ | 3,897 | ||||||||
Total financial liabilities |
$ | 3,897 | $ | — | $ | — | $ | 3,897 | ||||||||
As of December 31, 2009, the Company has a $3.9 million common stock warrant liability. This
liability is associated with warrants to purchase 2.9 million shares of common stock, issued in
connection with the Company’s common stock offering, which closed in June 2009. See additional information
related to the forms of these warrants at Note 5.
The Company initially assessed the fair value of the warrants on June 3, 2009, the date of
their issuance, at $3.7 million. The Company reassesses the fair value of the warrants at each
reporting date utilizing a Black-Scholes pricing model. As of June 3, 2009, inputs used in the
Black Scholes pricing model included a dividend yield of 0%, expected volatility of 88.79%,
risk-free interest rate of 2.54% and expected life of approximately five years. As of December 31,
2009, inputs used in the Black-Scholes pricing model included a dividend yield of 0%, expected
volatility of 93.82%, risk-free interest rate of 2.69% and expected life of approximately 4.4
years. As a result of this calculation, the Company recorded a loss of $0.2 million for the year
ended December 31, 2009. The loss is reflected in the Company’s consolidated statement of
operations as a component of other income (expense), net.
F-11
Table of Contents
ANADYS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The following table is a roll forward of the fair value of the common stock warrants, as to
which fair value is determined by Level 3 inputs (in thousands):
| As of December 31, | ||||
| 2009 | ||||
Description |
||||
Beginning balance |
$ | — | ||
Purchases, issuances, and settlements |
3,746 | |||
Realized loss included in net loss |
151 | |||
Ending balance |
$ | 3,897 | ||
4. Property and Equipment
Property and equipment consist of the following (in thousands):
| As of December 31, | ||||||||
| 2009 | 2008 | |||||||
Furniture and fixtures |
$ | 41 | $ | 69 | ||||
Equipment |
3,914 | 5,412 | ||||||
Computers and software |
1,399 | 1,798 | ||||||
Leasehold improvements |
30 | 1,833 | ||||||
| 5,384 | 9,112 | |||||||
Less accumulated depreciation and amortization |
(4,758 | ) | (7,636 | ) | ||||
| $ | 626 | $ | 1,476 | |||||
Depreciation and amortization expense relating to property and equipment for the years ended
December 31, 2009, 2008 and 2007 was $0.9 million, $1.1 million and $1.4 million, respectively.
5. Equity Financing
On June 3, 2009, the Company sold 8.4 million shares of common stock and warrants to purchase
2.9 million shares of common stock to institutional investors for gross proceeds of approximately
$17.5 million. The shares of common stock and the warrants were
sold in units, with each unit consisting of one share of common stock and a warrant to purchase
0.35 of a share of common stock, at a purchase price of $2.09375 per unit. Each warrant has an
exercise price of $2.75 per share, is exercisable 6 months after issuance and will expire five
years from the date of issuance. The Company agreed to pay the Placement Agent, Cowen & Company,
LLC, a fee equal to 6% of the gross proceeds from the offering of common stock and common stock
warrants in the offering, and to reimburse it for legal and other expenses, not to exceed $100,000.
The net proceeds related to this transaction were approximately $16.0 million.
The warrants included in this transaction contain a “fundamental change” provision, which may
in certain circumstances allow the common stock warrants to be redeemed for cash at an amount equal
to the Black-Scholes Value, as defined in the warrants. In addition, the warrants include a
“failure to timely deliver shares” provision, which may require the Company to pay cash to the
warrant holder in certain circumstances as defined in the warrants. Accordingly, the common stock
warrants are recorded as a liability and then marked to market each period through earnings in
other income (expense). See a discussion on the fair value of the common stock warrants at Note 3.
6. Restructuring
On June 3, 2009, the Company initiated a strategic restructuring to focus its operations on
the development of ANA598, in particular a Phase II study of ANA598 in combination with interferon
and ribavirin. The strategic restructuring resulted in a reduction in the Company’s workforce of
approximately 40%, with most of the employees having departed by the end of June 2009 and several
others departing at later dates prior to the end of 2009. The Company incurred a cash charge of
$1.3 million for the year ended December 31, 2009, which is included in operating expenses, for
cash severance, benefits and outplacement services in connection with the workforce reduction. In
addition, the Company incurred a noncash charge of $0.4 million associated with the modification of
stock options for individuals included in the strategic restructuring.
F-12
Table of Contents
ANADYS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The following is a rollforward of the Company’s restructuring liability (in thousands):
| As of December 31, | ||||
| 2009 | ||||
Description |
||||
Beginning balance |
$ | — | ||
Additions |
1,283 | |||
Severance payments |
(1,231 | ) | ||
Ending balance |
$ | 52 | ||
The Company expects that the restructuring liability of $0.1 million at December 31, 2009 to be
utilized during 2010.
7. Other Balance Sheet Captions
| As of December 31, | ||||||||
| 2009 | 2008 | |||||||
Prepaid expenses and other current assets consist of the following (in thousands): |
||||||||
Tenant deposit on former facility |
$ | — | $ | 1,260 | ||||
Prepaid insurance |
208 | 193 | ||||||
Interest receivable |
138 | 278 | ||||||
Other prepaid expenses |
213 | 471 | ||||||
| $ | 559 | $ | 2,202 | |||||
The tenant deposit on the Company’s former facility was collected in September 2009.
| As of December 31, | ||||||||
| 2009 | 2008 | |||||||
Other assets consist of the following (in thousands): |
||||||||
Note receivable |
$ | 30 | $ | 60 | ||||
Tenant deposit on current facility |
30 | — | ||||||
| $ | 60 | $ | 60 | |||||
| As of December 31, | ||||||||
| 2009 | 2008 | |||||||
Accrued expenses consist of the following (in thousands): |
||||||||
Accrued personnel costs |
$ | 320 | $ | 344 | ||||
Accrued employee bonus |
988 | 1,347 | ||||||
Accrued drug development |
508 | 2,110 | ||||||
Accrued legal and patent costs |
243 | 198 | ||||||
Accrued facility costs |
20 | 124 | ||||||
Accrued severance costs |
52 | — | ||||||
Other accrued expenses |
512 | 700 | ||||||
| $ | 2,643 | $ | 4,823 | |||||
8. Commitments and Contingencies
On June 18, 2009, the Company entered into a Sublease Agreement with Phenomix Corporation for
the lease of approximately 14,000 square feet of office and laboratory space in which the Company
is conducting its ongoing operations. Each month during the approximate 19.5 month term of the
lease which commenced on July 9, 2009, the Company is required to remit base rent of $0.03 million.
The lease also provides for additional payments, including a $0.03 million security deposit, common
area maintenance charges, taxes, maintenance and utilities. This leased space, located in San
Diego, California, replaced the Company’s former headquarters and research and development facility
in which the Company occupied approximately 40,000 square feet under a lease that expired on August
1, 2009. Gross rent expense for the years ended December 31, 2009, 2008 and 2007 was approximately
$1.4 million, $2.1 million and $2.0 million, respectively.
Future minimum lease payments under equipment and facility leases are as follows as of
December 31, 2009 (in thousands):
2010 |
$ | 358 | ||
2011 |
30 | |||
| $ | 388 | |||
F-13
Table of Contents
ANADYS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
9. Collaboration and License Agreements
Novartis International Pharmaceutical Ltd.
During 2007, the Company and Novartis decided to discontinue the development of ANA975. As a
result, during 2007 the Company recognized $21.0 million of previously deferred revenue. No revenue
was recognized under the Novartis collaboration in 2008 or 2009.
During the years ended December 31, 2008, and 2007, the Company recorded $0.05 million and
$0.5 million as offsets to research and development expense, which represent Novartis’ share of
ANA975 expenses incurred by the Company. The Company did not record an offset to research and
development expense during the year ended December 31, 2009.
10. Stockholders’ Equity
Warrants
As of December 31, 2009, the Company had outstanding warrants to purchase 2.9 million shares
of common stock with an average exercise price of $2.78. These warrants expire at various times
between December 17, 2012 and June 3, 2014. See additional information related to the warrants at
Note 5.
Stock Options
In 2002, the Company adopted the 2002 Equity Incentive Plan (the 2002 Plan). In connection
with the adoption of the 2002 Plan, the Company’s 1994 Stock Option Plan and 1998 Equity Incentive
Plan (collectively, the “Prior Plans”) were amended and restated into the 2002 Plan. All options
that were previously granted under the Prior Plans became governed by the 2002 Plan and the Prior
Plans no longer existed as individual plans. The 2002 Plan provided for the issuance of incentive
stock options to officers and other employees of the Company and non-qualified stock options,
awards of stock and direct stock purchase opportunities to directors, officers, employees and
consultants of the Company.
During March 2004 upon the effectiveness of the Company’s initial public offering (IPO), the
2004 Equity Incentive Plan (the 2004 Plan) was adopted. The initial share reserve under the 2004
Plan was equal to the number of shares of common stock reserved under the 2002 Plan that remained
available for future stock awards upon the effectiveness of the IPO. Options granted under the 2002
Plan continue to be governed by the provisions of the 2002 Plan. On October 30, 2009, the Company
registered an additional
1,000,000 shares for issuance under the 2004 Plan in accordance with the provisions of the
2004 Plan. The total number of shares which remain available for grant under the 2004 Plan is
609,295 shares at December 31, 2009. The options are exercisable at various dates and will expire
no more than ten years from their date of grant, or in the case of certain non-qualified options,
ten years from the date of grant. The exercise price of each option shall be determined by the
Board of Directors although generally options have an exercise price equal to the fair market value
of the Company’s stock on the date of the option grant. In the case of incentive stock options, the
exercise price shall not be less than 100% of the fair market value of the Company’s common stock
at the time the option is granted. For holders of more than 10% of the Company’s total combined
voting power of all classes of stock, incentive stock options may not be granted at less than 110%
of the fair market value of the Company’s common stock at the date of grant and for a term not to
exceed five years.
Upon the effectiveness of the initial public offering, the 2004 Non-Employee Directors’ Stock
Option Plan (the NEDSOP Plan) was adopted. On October 30, 2009, the Company registered an
additional 186,549 shares for issuance under the NEDSOP Plan in accordance with the provisions of
the NEDSOP Plan. The total number of shares which remain available for grant under the NEDSOP Plan
is 325,512 shares at December 31, 2009. The options are exercisable at various dates and will
expire no more than ten years from their date of grant. The exercise price of each option shall be
determined by the Board of Directors although generally options have an exercise price equal to the
fair market value of the Company’s stock on the date of the option grant.
In connection with the hiring of certain executive officers during 2006, the Compensation
Committee of the Company’s Board of Directors approved inducement grants of non-qualified stock
options to purchase shares of Anadys’ Common Stock. The total number of shares which remain
outstanding under the inducement grants is 342,975 shares at December 31, 2009. These option awards
were granted without stockholder approval pursuant to NASDAQ Marketplace Rule 4350(i)(1)(A)(iv).
Although these options were granted outside the 2004 Plan, they are subject to substantially
identical terms and conditions as those contained in the 2004 Plan.
F-14
Table of Contents
ANADYS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The following table summarizes information about stock options outstanding under the 2002
Plan, 2004 Plan, the NEDSOP Plan and inducement grants as of December 31, 2009:
| Options Outstanding | Options Exercisable | |||||||||||||||||||||||
| Weighted | Weighted | Weighted | ||||||||||||||||||||||
| Range of | Average | Average | Average | |||||||||||||||||||||
| Exercise | Number | Remaining | Exercise | Number | Exercise | |||||||||||||||||||
| Price | Outstanding | Contractual Life | Price | Exercisable | Price | |||||||||||||||||||
| $ | 1.48-$2.24 | 1,826,018 | 8.24 | $ | 2.00 | 561,712 | $ | 1.93 | ||||||||||||||||
| $ | 2.25-$2.42 | 1,814,223 | 8.09 | $ | 2.36 | 757,453 | $ | 2.30 | ||||||||||||||||
| $ | 2.68-$3.00 | 1,508,828 | 4.13 | $ | 2.92 | 1,401,613 | $ | 2.93 | ||||||||||||||||
| $ | 3.46-$8.16 | 1,797,514 | 5.26 | $ | 5.98 | 1,678,639 | $ | 6.09 | ||||||||||||||||
| $ | 8.37-$15.61 | 213,428 | 4.70 | $ | 11.48 | 211,868 | $ | 11.45 | ||||||||||||||||
| 7,160,011 | 4,611,285 | |||||||||||||||||||||||
A summary of the Company’s stock option activity and related information is as follows:
| Weighted | Weighted-Average | Aggregate Intrinsic | ||||||||||||||
| Options | Average | Remaining | Value | |||||||||||||
| Outstanding | Exercise Price | Contractual Term | (in thousands) | |||||||||||||
Balance at December 31, 2006 |
5,047,180 | $ | 5.77 | |||||||||||||
Granted |
2,005,875 | 2.59 | ||||||||||||||
Exercised |
(30,192 | ) | 2.95 | |||||||||||||
Cancelled |
(1,475,036 | ) | 6.52 | |||||||||||||
Balance at December 31, 2007 |
5,547,827 | $ | 4.44 | |||||||||||||
Granted |
1,508,493 | 2.07 | ||||||||||||||
Exercised |
(36,567 | ) | 2.90 | |||||||||||||
Cancelled |
(474,387 | ) | 5.47 | |||||||||||||
Balance at December 31, 2008 |
6,545,366 | $ | 3.83 | |||||||||||||
Granted |
1,359,736 | 2.34 | ||||||||||||||
Exercised |
(84,465 | ) | 2.74 | |||||||||||||
Cancelled |
(660,626 | ) | 4.34 | |||||||||||||
Balance at December 31, 2009 |
7,160,011 | $ | 3.57 | 6.48 | $ | 364 | ||||||||||
Exercisable at December 31, 2009 |
4,611,285 | $ | 4.25 | 5.06 | $ | 144 | ||||||||||
The total intrinsic value of options exercised during the year ended December 31, 2009 was
$0.2 million determined as of the date of exercise. There was no material intrinsic value for
options exercised during the year ended December 31, 2008. The total intrinsic value of options
exercised during the year ended December 31, 2007 was $0.05 million determined as of the date of
exercise. The Company settles employee stock option exercises with newly issued common shares.
The Company granted 15,000 stock options to non-employees for the year ended December 31,
2009. The Company did not grant any stock options to non-employees for the years ended December 31,
2008 and 2007. Compensation expense related to non-employee stock option grants was $0.1 million,
$0.01 million and $0.05 million for the years ended December 31, 2009, 2008 and 2007, respectively.
Share-Based Compensation
The Company is required to record share-based compensation as components of either research
and development expense or general and administrative expense. The Company has reported the
following amounts of share-based compensation expense in the consolidated Statements of Operations
(in thousands, except per share data):
| For the years ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
Research and development expense |
$ | 1,354 | $ | 1,271 | $ | 2,463 | ||||||
General and administrative expense |
1,361 | 1,492 | 1,650 | |||||||||
Total share-based compensation expense |
$ | 2,715 | $ | 2,763 | $ | 4,113 | ||||||
Net share-based compensation expense, per common share
basic and diluted |
$ | 0.08 | $ | 0.10 | $ | 0.14 | ||||||
F-15
Table of Contents
ANADYS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Included in the research and development expense and general and administrative expense for
the year ended December 31, 2009 is $0.3 million and $0.1 million, respectively, of share-based
compensation expense related to the modification of stock options for employees included in the
June 2009 reduction in workforce.
As of December 31, 2009, there was an additional $3.1 million of total unrecognized
compensation cost related to unvested share-based awards granted under the Company’s stock option
plans. This unrecognized compensation cost is expected to be recognized over a weighted-average
period of 2.54 years.
The fair value of options granted to employees and directors was estimated at the date of
grant using a Black-Scholes pricing model with the weighted-average assumptions stated below.
| For the years ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
Risk-free interest rate |
2.20 | % | 2.45 | % | 4.20 | % | ||||||
Dividend yield |
0 | % | 0 | % | 0 | % | ||||||
Volatility factors of the expected market price of the Company’s common stock |
80 | % | 71 | % | 70 | % | ||||||
Weighted-average expected life of option (years) |
5.94 | 5.76 | 5.60 | |||||||||
The estimated weighted-average fair value of stock options granted during 2009, 2008 and 2007
was $1.62, $1.31 and $1.67, respectively.
Dividend Yield—The Company has never declared or paid dividends on common stock and has no
plans to do so in the foreseeable future.
Expected Volatility—Volatility is a measure of the amount by which a financial variable such
as a share price has fluctuated or is expected to fluctuate during a period. The Company considered
the historical volatility from its IPO through the dates of grants, in combination with the
historical volatility of peer companies and business and economic considerations in order to
estimate the expected volatility, due to the Company’s short history as a public company.
Risk-Free Interest Rate—This is the U.S. Treasury rate for the week of each option grant
during the quarter having a term that most closely resembles the expected life of the option.
Expected Life of the Option Term—This is the period of time that the options granted are
expected to remain unexercised. Options granted during the year have a maximum contractual term of
ten years. The Company estimates the expected life of the option term based on actual past behavior
for similar options with further consideration given to the class of employees to whom the options
were granted.
Forfeitures are estimated at the time of grant and revised, if necessary, in subsequent
periods if actual forfeitures differ from those estimates. The Company assesses the forfeiture rate
on an annual basis and revises the rate when deemed necessary.
Employee Stock Purchase Plan
Under the Company’s 2004 Employee Stock Purchase Plan (Purchase Plan), employees may purchase
common stock every six months (up to but not exceeding 12% of each employee’s wages) over the
offering period at 85% of the fair market value of the common stock at certain specified dates. The
offering period may not exceed 24 months. This purchase discount is significant enough to be
considered compensatory. As a result, the Company recorded $0.2 million, $0.2 million and $0.04
million in share-based compensation related to the Purchase Plan for the years ended December 31,
2009, 2008 and 2007, respectively.
F-16
Table of Contents
ANADYS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For the years ended December 31, 2009, 2008 and 2007, 82,729 shares, 83,248 shares and 70,558
shares of common stock were issued under the Purchase Plan, respectively. The weighted-average fair
value of employee stock Purchase Plan purchases was $1.85, $1.84 and $2.38 per share for 2009, 2008
and 2007, respectively.
Shares Reserved for Issuance
Shares of common stock reserved for future issuance as of December 31, 2009 are as follows:
| December 31, 2009 | ||||
Warrants |
2,944,234 | |||
Employee Stock Purchase Plan |
1,041,054 | |||
Stock options under the Company’s Plans: |
||||
Granted and outstanding |
7,160,011 | |||
Reserved for future issuance |
936,376 | |||
| 8,096,387 | ||||
11. Income Taxes
As of December 31, 2009 and 2008, the Company has not recorded any uncertain tax benefits.
Significant components of the Company’s deferred tax assets are shown below. A valuation
allowance of $91.0 million and $80.5 million has been established to offset the deferred tax
assets, as realization of such assets has not met the more likely than not threshold, as of
December 31, 2009 and 2008, respectively.
| As of December 31, | ||||||||
| 2009 | 2008 | |||||||
| (in thousands) | ||||||||
Deferred tax assets: |
||||||||
Net operating loss carryforwards |
$ | 52,972 | $ | 46,681 | ||||
Research and development credits |
5,574 | 4,663 | ||||||
Non-qualified stock options |
4,914 | 3,844 | ||||||
Capitalized research and development expense |
27,415 | 25,058 | ||||||
Accruals |
98 | 248 | ||||||
Other |
33 | 30 | ||||||
Total deferred tax assets |
91,006 | 80,524 | ||||||
Valuation allowance for deferred tax assets |
(90,991 | ) | (80,524 | ) | ||||
Net deferred taxes |
$ | 15 | $ | — | ||||
Deferred tax liabilities: |
||||||||
Unrealized gain (loss) on securities available-for-sale |
(15 | ) | — | |||||
Total deferred tax liabilities |
(15 | ) | — | |||||
Net deferred taxes |
$ | — | $ | — | ||||
As of December 31, 2009 the Company had federal and state tax net operating loss (NOL)
carryforwards of approximately $133.5 million and $109.5 million, respectively. Approximately $4.1
million of the federal loss carryforwards will begin expiring in 2010 and approximately $5.8
million of the state loss carryforwards will begin expiring in 2011, unless previously utilized.
The Company also has federal and state research tax credit (R&D credit) carryforwards of
approximately $2.2 million and $5.2 million respectively. The federal research credits will begin
expiring in 2027 unless previously utilized. The state research credits do not expire.
Utilization of the NOL and R&D credit carryforwards may be subject to a substantial annual
limitation under Section 382 of the Internal Revenue Code of 1986, and similar state provisions due
to ownership change limitations that have occurred previously or that could occur in the future.
These ownership changes may limit the amount of NOL and R&D credit carryforwards and other deferred
tax assets that can be utilized to offset future taxable income and tax, respectively. In general,
an ownership change, as defined by Section 382, results from transactions increasing ownership of
certain shareholders or public groups in the stock of the corporation by
F-17
Table of Contents
ANADYS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
more than 50 percentage
points over a three-year period. During 2007, the Company completed an analysis of Section 382 and has determined that, as of
December 31, 2009 and 2008, approximately $14.8 million and $16.0 million, respectively, of the deferred
tax assets related to NOL and credit carryforwards will expire unused and, accordingly, the Company has removed
such assets from its deferred tax assets with a corresponding reduction to its valuation allowance. During 2009, the Company
completed an initial analysis of Section 382 and, based on this analysis, the Company believes that an ownership change, as defined
in Section 382, has occurred during 2009 and, therefore, its NOL and R&D credit carryforwards and other deferred tax assets
will be subject to annual limitations in future periods. As of the date of the filing of this Annual Report on Form 10-K, a final
determination of the annual limitations has not been determined and, therefore, the Company’s deferred tax assets related to
its NOLs and R&D credit carryforwards have not been reduced for the limitations associated with the change in ownership
which occurred during 2009.
There may be additional limitations imposed on the Company’s ability to
fully utilize its remaining deferred tax assets due to future ownership changes. Any amounts that
are determined by the Company to expire prior to their utilization due to such limitations will be
removed from deferred tax assets with a corresponding reduction of the valuation allowance. In
addition, future changes in the unrecognized tax benefit will have no impact on the effective tax
rate due to the existence of the valuation allowance.
The Company’s practice is to recognize interest and/or penalties related to income tax matters
in income tax expense. As of December 31, 2009, the Company did not record any interest or
penalties.
The provision for income taxes on earnings subject to income taxes differs from the
statutory federal rate at December 31, 2009, 2008 and 2007, due to the following (in thousands):
| 2009 | 2008 | 2007 | ||||||||||
Federal income taxes at 35% |
$ | (9,547 | ) | $ | (11,341 | ) | $ | (3,210 | ) | |||
State income taxes, net of federal benefit |
(1,566 | ) | (1,748 | ) | (383 | ) | ||||||
Tax effect on non-deductible expenses and credits |
(980 | ) | (567 | ) | (792 | ) | ||||||
Increase in valuation allowance |
10,467 | 12,940 | (11,033 | ) | ||||||||
Expiration of net operating loss carryforwards |
1,426 | 703 | — | |||||||||
Other |
200 | 13 | 2 | |||||||||
Adjustment for Section 382 limitation |
— | — | 15,416 | |||||||||
| $ | — | $ | — | $ | — | |||||||
The tax years 1993 to 2009 remain open to examination by the major taxing jurisdictions to
which the Company is subject, as tax authorities may have the right to examine prior periods where
net operating losses or tax credits were generated and carried forward.
12. Savings Plan
The Company has a retirement savings plan for all employees, subject to certain age
requirements, pursuant to Section 401(k) of the Internal Revenue Code. The Company matches 25% of
employee contributions up to 6% of eligible compensation. Employer contributions were $0.1 million
for each of the years ended December 31, 2009, 2008 and 2007.
13. Unaudited Quarterly Results of Operations
The following quarterly financial data, in the opinion of management, reflects all
adjustments, consisting of normal recurring adjustments, necessary for a fair presentation of
results for the periods presented.
| First | Second | Third | Fourth | |||||||||||||
| Fiscal year 2009 | Quarter | Quarter | Quarter | Quarter | ||||||||||||
| (in thousands, except net loss per share) | ||||||||||||||||
Revenues |
$ | — | $ | — | $ | — | $ | — | ||||||||
Net loss |
(8,759 | ) | (6,530 | ) | (7,732 | ) | (4,257 | ) | ||||||||
Net loss per share, basic and diluted |
(0.30 | ) | (0.21 | ) | (0.21 | ) | (0.11 | ) | ||||||||
| First | Second | Third | Fourth | |||||||||||||
| Fiscal year 2008 | Quarter | Quarter | Quarter | Quarter | ||||||||||||
| (in thousands, except net loss per share) | ||||||||||||||||
Revenues |
$ | — | $ | — | $ | — | $ | — | ||||||||
Net loss |
(7,444 | ) | (7,092 | ) | (9,349 | ) | (8,517 | ) | ||||||||
Net loss per share, basic and diluted |
(0.26 | ) | (0.25 | ) | (0.32 | ) | (0.30 | ) | ||||||||
F-18