UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FOR ANNUAL AND TRANSITION REPORTS PURSUANT TO SECTIONS 13 OR 15(d)
OF THE SECURITIES EXCHANGE ACT OF 1934
(Mark One)
| |
|
|
| þ |
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2009
OR
| |
|
|
| o |
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from
to
001-33357
(Commission file number)
PROTALIX BIOTHERAPEUTICS, INC.
(Exact name of registrant as specified in its charter)
| |
|
|
| |
|
|
| Florida
|
|
65-0643773 |
|
|
|
|
State or other jurisdiction
of incorporation or organization
|
|
(I.R.S. Employer
Identification No.) |
| |
|
|
2 Snunit Street
Science Park
POB 455
Carmiel, Israel
|
|
20100 |
|
|
|
|
| (Address of principal executive offices)
|
|
(Zip Code) |
972-4-988-9488
Registrant’s telephone number, including area code
Securities registered pursuant to Section 12(b) of the Act:
| |
|
|
| Title of each class
|
|
Name of each exchange on which registered |
| Common stock, par value $0.001 per share
|
|
NYSE AMEX |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in
Rule 405 of the Securities Act. Yes o No þ
Indicate by check mark if the registrant is not required to file reports pursuant to
Section 13 or Section 15(d) of the Act. Yes o No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed
by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or
for such shorter period that the registrant was required to file such reports), and (2) has been
subject to such filing requirements for the past 90 days. Yes þ No o
Indicate by check mark whether the registrant has submitted electronically and posted on its
corporate Web site, if any, every Interactive Data File required to be submitted and posted
pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period
that the registrant was required to submit and post such files). Yes o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation
S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in
definitive proxy or information statements incorporated by reference in Part III of this Form 10-K
or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large
accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions
of “large accelerated filer,” “accelerated filer” and “smaller reporting
company” in Rule 12b-2 of the
Exchange Act. (Check one):
| |
|
|
|
|
|
|
| Large accelerated filer o
|
|
Accelerated filer þ
|
|
Non-accelerated filer o
|
|
Smaller reporting company o |
| |
|
(Do not check if a smaller reporting company)
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of
the Exchange Act). Yes o No þ
The aggregate market value of the voting stock held by non-affiliates of the Registrant, as of
June 30, 2009 was approximately $162 million (based upon the closing price for shares of the
Registrant’s common stock as reported by the NYSE Amex) as of June 30, 2009 of $4.52). Shares of
common stock held by each officer, director and holder of 5% or more of the outstanding common
stock have been excluded in that such persons may be deemed to be affiliates. This determination
of affiliate status is not necessarily a conclusive determination for other purposes.
On February 15, 2010, approximately 80,745,296 shares of the Registrant’s common stock, par
value $0.001 per share, were outstanding.
FORM 10-K
TABLE OF CONTENTS
i
PART I
Except where the context otherwise requires, the terms, “we,” “us,” “our” or “the Company,” refer
to the business of Protalix BioTherapeutics, Inc. and its consolidated subsidiaries, and “Protalix”
or “Protalix Ltd.” refers to the business of Protalix Ltd., our wholly-owned subsidiary and sole
operating unit.
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
The statements set forth under the captions “Business,” “Management’s Discussion and Analysis of
Financial Condition and Results of Operations” and “Risk Factors,” and other statements included
elsewhere in this Annual Report on Form 10-K, which are not historical, constitute “forward-looking
statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, and
Section 21E of the Securities Exchange Act of 1934, as amended, including statements regarding
expectations, beliefs, intentions or strategies for the future. When used in this report, the
terms “anticipate,” “believe,” “estimate,” “expect” and “intend” and words or phrases of similar
import, as they relate to our or our subsidiaries or our management, are intended to identify
forward-looking statements. We intend that all forward-looking statements be subject to the
safe-harbor provisions of the Private Securities Litigation Reform Act of 1995. These
forward-looking statements are only predictions and reflect our views as of the date they are made
with respect to future events and financial performance, and we undertake no obligation to update
any forward-looking statement to reflect events or circumstances after the date on which the
statement is made or to reflect the occurrence of unanticipated events, except as may be required
under applicable law. Forward-looking statements are subject to many risks and uncertainties that
could cause our actual results to differ materially from any future results expressed or implied by
the forward-looking statements.
Examples of the risks and uncertainties include, but are not limited to, the following:
| |
• |
|
the inherent risks and uncertainties in developing drug platforms and products of
the type we are developing; |
| |
| |
• |
|
delays in our preparation and filing of applications for regulatory approval; |
| |
| |
• |
|
delays in the approval or the potential rejection of any applications we file with
the U.S. Food and Drug Administration, or the FDA, or other regulatory authorities; |
| |
| |
• |
|
any lack of progress of our research and development (including the results of
clinical trials we are conducting); |
| |
| |
• |
|
obtaining on a timely basis sufficient patient enrollment in our clinical trials; |
| |
| |
• |
|
the impact of development of competing therapies and/or technologies by other
companies; |
| |
| |
• |
|
our ability to obtain additional financing required to fund our research programs; |
| |
| |
• |
|
the risk that we will not be able to develop a successful sales and marketing
organization in a timely manner, if at all; |
| |
| |
• |
|
our ability to establish and maintain strategic license, collaboration and
distribution arrangements and to manage our relationship with Pfizer Inc., Teva Ltd. or
with any other collaborator, distributor or partner; |
| |
| |
• |
|
potential product liability risks and risks of securing adequate levels of product
liability and clinical trial insurance coverage; |
| |
| |
• |
|
the availability of reimbursement to patients from health care payors for any of our
product candidates, if approved; |
| |
| |
• |
|
the possibility of infringing a third party’s patents or other intellectual property
rights; |
| |
| |
• |
|
the uncertainty of obtaining patents covering our products and processes and in
successfully enforcing our intellectual property rights against third parties; |
| |
| |
• |
|
the possible disruption of our operations due to terrorist activities and armed
conflict, including as a result of the disruption of the operations of regulatory
authorities, our subsidiaries, our manufacturing facilities and our customers,
suppliers, distributors, collaborative partners, licensees and clinical trial sites;
and |
| |
| |
• |
|
other risks and uncertainties detailed in Section 1A of this Annual Report on Form
10-K. |
In addition, companies in the pharmaceutical and biotechnology industries have suffered significant
setbacks in advanced or late-stage clinical trials, even after obtaining promising earlier trial
results or preliminary findings for such clinical trials. Even if favorable testing data is
generated by clinical trials of drug products, the FDA may not accept or approve an NDA filed by a
pharmaceutical or biotechnology company for such drug product. These and other risks and
uncertainties are detailed under the heading “Risk Factors” in this Annual Report on Form 10-K and
are described from time to time in the reports we file with the Securities and Exchange Commission.
We undertake no obligation to update, and we do not have a policy of updating or revising, these
forward-looking statements.
Item 1. Business
We are a biopharmaceutical company focused on the development and commercialization of recombinant
therapeutic proteins based on our proprietary ProCellExTM protein expression system, or
ProCellEx. Using our ProCellEx system, we are developing a pipeline of proprietary and biosimilar
or “generic” versions of recombinant therapeutic proteins based on our plant cell-based expression
technology that target large, established pharmaceutical markets and that rely upon known
biological mechanisms of action. Our initial commercial focus has been on complex therapeutic
proteins, including proteins for the treatment of genetic disorders, such as Gaucher disease and
Fabry disease. We believe our ProCellEx protein expression system will enable us to develop
proprietary recombinant proteins that are therapeutically equivalent or superior to existing
recombinant proteins currently marketed for the same indications. Because we are primarily
targeting biologically equivalent versions of highly active, well-tolerated and commercially
successful therapeutic proteins, we believe our development process is associated with relatively
less risk compared to other biopharmaceutical development processes for completely novel
therapeutic proteins.
Our lead product development candidate is prGCD (designated this year as taliglucerase alfa) for
the treatment of Gaucher disease, which we are developing using our ProCellEx protein expression
system. Gaucher disease is a rare and serious lysosomal storage disorder with severe and
debilitating symptoms. Taliglucerase alfa is our proprietary recombinant form of
glucocerebrosidase (GCD), an enzyme naturally found in human cells that is mutated or deficient in
patients with Gaucher disease. In July 2007, we reached an agreement with the U.S. Food and Drug
Administration, or the FDA, on the final design of our pivotal phase III clinical trial of
taliglucerase alfa, through the FDA’s special protocol assessment (SPA) process. The phase III
clinical trial was completed in September 2009 and, on October 15, 2009, we announced positive
top-line results from the trial. On December 9, 2009, we filed a New Drug Application (NDA) for
taliglucerase alfa. In January 2010, the FDA requested additional data regarding the Chemistry,
Manufacturing and Controls (CMC) section of the NDA. No additional clinical or preclinical
information was requested. The FDA’s request focused primarily on the validation of the
manufacturing process for our upgraded manufacturing facility. A validation plan for our
manufacturing process of taliglucerase alfa has already been established and reviewed by the FDA.
We are working diligently to provide the requested data to the FDA and anticipate submitting the
requested data during the second quarter of 2010. In addition, we expect to submit similar
applications with other comparable regulatory agencies in other countries during 2010.
In addition to our recently completed pivotal phase III clinical trial, during the third quarter of
2008, we initiated a double-blind, follow-on extension study as part of this trial. We also
initiated a home care treatment program for patients enrolled in the extension study and in
December 2008, we initiated a clinical study evaluating the safety and efficacy of switching
Gaucher patients currently treated under the current standard of care to treatment with
taliglucerase alfa. The current standard of care for Gaucher patients is enzyme replacement
therapy with CerezymeTM which is produced by Genzyme Corporation and currently the only
approved enzyme replacement therapy for Gaucher disease. Enzyme replacement therapy is a medical
treatment in which recombinant enzymes are infused into patients in whom the enzyme is lacking or
dysfunctional. The switch-over study is not a prerequisite for marketing approval from the FDA of
taliglucerase alfa. In December 2009 we filed a proposed pediatric investigation plan to the
Pediatric Committee of the European Medicines Agency, or the EMEA.
In July 2009, following a request by the FDA, we submitted an expanded access program treatment
protocol in order to address an expected shortage of the current enzyme replacement therapy
approved for Gaucher disease. The treatment protocol was approved by the FDA in August 2009. We
are also providing taliglucerase alfa to patients in the European Union under a compassionate use
protocol. In August 2009, we received Fast Track Designation for taliglucerase alfa and in
September 2009, the FDA’s Office of Orphan Product Development granted taliglucerase alfa Orphan
Drug Status. The fast track mechanism was created to facilitate the development and approval of
new drugs intended for the treatment of life-threatening conditions for which there are no
effective treatments and which demonstrate the potential to address unmet medical needs for the
conditions. The fast track process includes the scheduling of meetings to seek FDA input into
development plans, the option of submitting an NDA serially in sections rather than submitting all
components simultaneously, the option to request evaluation of studies using surrogate endpoints,
and the potential for a priority review. The fast track designation may be withdrawn by the FDA at
any time. The fast track designation does not guarantee that we will qualify for or be able to
take advantage of the expedited review procedures and does not increase the likelihood that
taliglucerase alfa will receive regulatory approval.
The Orphan Drug designation for taliglucerase alfa for the treatment of Gaucher Disease provides
special status to taliglucerase alfa provided that it meets certain criteria. As a result of the
orphan designation, we are qualified for the tax credit and marketing incentives provided under the
Orphan Drug Act of 1983. A marketing application for a prescription drug product that has been
designated as a drug for a rare disease or condition is not subject to a prescription drug user fee
unless the application includes an indication for other than a rare disease or condition.
2
On November 30, 2009, Protalix Ltd. and Pfizer Inc., or Pfizer, entered into an exclusive license
and supply agreement pursuant to which Pfizer was granted an exclusive, worldwide license to
develop and commercialize taliglucerase alfa. Under the terms and conditions of the Pfizer
agreement, Protalix Ltd. retained the right to commercialize taliglucerase alfa in Israel. In
connection with the execution of the Pfizer agreement, Pfizer made an upfront payment to Protalix
Ltd. of $60.0 million in connection with the execution of the agreement and subsequently paid to
Protalix Ltd. an additional $5.0 million upon our filing of a proposed pediatric investigation plan
to the Pediatric Committee of the EMEA. Protalix Ltd. is also eligible to receive potential
milestone payments of up to $50.0 million, in the aggregate, for the successful achievement of
other regulatory-related milestones and to payments equal to 40% of the net profits earned by
Pfizer on sales of taliglucerase alfa. In calculating the net profits, there are certain agreed
upon limits on the amounts that may be deducted from gross sales for certain expenses and costs of
goods sold. Protalix Ltd. retained the manufacturing rights to taliglucerase alfa and Pfizer and
Protalix Ltd. have agreed to a specific allocation of the responsibilities for the continued
development efforts for taliglucerase alfa.
Although Gaucher disease is a relatively rare disease, it represents a large commercial market due
to the severity of the symptoms and the chronic nature of the disease. The annual worldwide sales
of Cerezyme were approximately $793 million in 2009, compared with $1.2 billion for the previous
year, according to public reports by Genzyme. According to Genzyme, it suffered a temporary
interruption in production of Cerezyme in 2009 associated with the remediation of a contamination
in one of its manufacturing facilities, and, as a result, shipments of Cerezyme were limited during
the second half of 2009. Taliglucerase alfa is a plant cell expressed version of the GCD enzyme,
developed through our ProCellEx protein expression system. Taliglucerase alfa has an amino acid,
glycan and three-dimensional structure that is very similar to its naturally-produced counterpart
as well as to Cerezyme, which is a mammalian cell expressed version of the same protein. We
believe taliglucerase alfa may prove more cost-effective than the currently marketed alternative
due to the cost benefits of expression through our ProCellEx protein expression system. In
addition, based on our laboratory testing, preclinical and clinical results, we believe that
taliglucerase alfa may have the potential for increased potency and efficacy in certain parameters
compared to the existing enzyme replacement therapy for Gaucher disease.
In addition to taliglucerase alfa, we are developing an innovative product pipeline using our
ProCellEx protein expression system. Our product pipeline currently includes, among other
candidates, therapeutic protein candidates for the treatment of Fabry disease, a rare, genetic
lysosomal disorder in humans, an acetylcholinesterase enzyme-based therapy for biodefense and
intoxication treatments, antiTNF, a plant cell expressed recombinant fusion protein made from the
soluble form of the human TNF receptor (TNFR), fused to the Fc component of a human antibody IgG1
domain and additional undisclosed therapeutic proteins, most of which are currently being evaluated
in animal studies. During the fourth quarter of 2009, we filed an IND (investigational new drug
application) with the FDA for our acetylcholinesterase enzyme-based therapy for biodefense
applications after successfully completing pre-clinical studies for this indication. We expect to
initiate a phase I clinical study for our acetylcholinesterase enzyme-based therapy during the
first quarter of 2010. We plan to file an investigational new drug application (IND) with the FDA
with respect to an additional product candidate during 2010.
In September 2009, we announced preliminary preclinical data regarding pr-antiTNF, our proprietary
product candidate for the treatment of certain immune diseases such as rheumatoid arthritis,
juvenile idiopathic arthritis, ankylosing, spondylitis, psoriatic arthritis and plaque psoriasis.
Our pr-antiTNF product candidate has an amino acid sequence that is similar to Enbrel™, which is
one of the treatments for patients of those diseases. We believe that we may be able to reduce the
development risks and time to market for our product candidates as our product candidates are based
on well-understood proteins with known biological mechanisms of action. Except for the rights to
commercialize taliglucerase alfa worldwide (other than Israel) which we licensed to Pfizer, we hold
the worldwide commercialization rights to our proprietary development candidates, and we intend to
establish an internal, commercial infrastructure and targeted sales force to market taliglucerase
alfa in Israel and our other products, if approved, in North America, the European Union and in
other significant markets, including Israel. In addition we are continuously evaluating potential
strategic marketing partnerships.
Our ProCellEx protein expression system consists of a comprehensive set of technologies and
capabilities for the development of recombinant proteins, including advanced genetic engineering
technology and plant cell-based protein expression methods. Through our ProCellEx protein
expression system, we can develop highly complex recombinant therapeutic proteins all the way to
the scale-up of a purified product produced in compliance with current good manufacturing
practices, or cGMP. We believe that our plant cell-based expression technology will enable us, in
certain cases, to develop and commercialize recombinant proteins without infringing upon the
method-based patents or other intellectual property rights of third parties. The major elements of
our ProCellEx system are patent protected in most major countries. Moreover, we expect to enjoy
method-based patent protection for the proteins we develop using our proprietary ProCellEx protein
expression technology, although there can be no assurance that any such patents will be granted.
In some cases, we may be able to obtain patent protection for the compositions of the proteins
themselves. We have filed for United States and international composition of matter patents for
taliglucerase alfa.
3
Our ProCellEx protein expression system is built on flexible custom-designed bioreactors made of
polyethylene and optimized for the development of complex proteins in plant cell cultures. These
bioreactors entail low initial capital investment, are rapidly scalable at a low cost and require
less hands-on maintenance between cycles, compared to the highly complex, expensive, stainless
steel bioreactors typically used in mammalian cell-based production systems. As a result, through
our ProCellEx protein expression system, we believe that we can develop recombinant therapeutic
proteins yielding substantial cost advantages, accelerated development and other competitive
benefits as compared to mammalian cell-based protein expression systems.
We have successfully demonstrated the feasibility of our ProCellEx system through clinical and pre
clinical studies performed by us to date including the positive efficacy and safety data in our
phase III study for taliglucerase alfa, pre clinical results in well-known models in our enzyme for
Fabry disease and pr-antiTNF, and extensive animal studies for our acetylcholinesterase enzyme, and
by expressing, on an exploratory, research scale, many additional complex therapeutic proteins
belonging to different drug classes, such as enzymes, hormones, monoclonal antibodies, cytokines
and vaccines. The therapeutic proteins we have expressed to date in research models have produced
the intended composition and similar biological activity compared to their respective
human-equivalent proteins. Moreover, several of such proteins demonstrated advantageous biological
activity when compared to the biotherapeutics currently available in the market to treat the
applicable disease or disorder. We believe that the clinical success of taliglucerase alfa
represents a strong proof-of-concept for our ProCellEx protein expression system and plant
cell-based protein expression technology. We also believe that the significant benefits of our
ProCellEx protein expression system, if further substantiated in clinical trials and
commercialization of our product candidates, have the potential to transform the industry standard
for the development of complex therapeutic proteins.
Our goal is to become a leading fully integrated biopharmaceutical company focused on the
development and commercialization of proprietary and biosimilar or generic versions of recombinant
therapeutic proteins. To that end, we are leveraging our ProCellEx protein expression system to
develop a pipeline of proprietary and biosimilar versions of recombinant therapeutic proteins. In
addition to the product candidates that we are developing internally, we have entered into
agreements for additional compounds with academic institutions, including a licensing agreement
with the technology transfer arm of Israel’s Weizmann Institute of Science and an agreement with
the technology transfer arm of the Hebrew University of Jerusalem. In addition, we are
collaborating with other pharmaceutical companies to develop therapeutic proteins that can benefit
from the significant cost, intellectual property and other competitive advantages of our ProCellEx
protein expression system. We entered into an agreement with Teva Pharmaceutical Industries Ltd.
in September 2006 under which we have agreed to collaborate on the research and development of two
proteins to be identified by Teva and us, using our ProCellEx protein expression system. The
agreement also identifies additional matters for collaboration between Teva and us. Subsequently,
two proteins were identified to be researched and developed under the agreement but in 2009, both
of the projects were terminated for commercial reasons. We also continuously review and consider additional development
and commercialization alliances with other pharmaceutical companies and academic institutions.
Industry Overview
Recombinant proteins have revolutionized the treatment of a variety of diseases and disorders.
Recombinant proteins are forms of human proteins that are produced, or expressed, using a
mammalian, plant, bacterial or yeast cell as a production engine. In the early 1970s, a number of
key scientific breakthroughs, including, among others, the demonstration of genetic engineering and
genetic sequencing techniques, as well as the synthesis of genes, led to the advancement of
recombinant protein technology.
As a result, the market for pharmaceutical therapeutics has undergone a transformation as
recombinant proteins and other biologic products have become an increasingly significant portion of
the global drug market and the focus of research worldwide. Based upon data from the Biotechnology
Industry Organization, an organization that provides information, advocacy and business support to
the biotechnology industry, since the introduction in 1982 of recombinant human insulin, the
world’s first genetically engineered pharmaceutical product, over 254 biotechnology drugs have been
approved for over 392 indications. According to Datamonitor, a provider of business information to
the pharmaceutical and other industries, the overall global biologics market size is expected to
grow to $105.2 billion in 2010, from $56.1 billion in 2004, representing a compounded annual growth
rate (CAGR) of 11.1%.
Mammalian cell-based systems are the current industry standard for expression of recombinant
therapeutic glycoproteins (complex proteins that contain sugar residues), including catalytic
enzymes and monoclonal antibodies. Mammalian cell-based systems were first introduced in the late
1980s and are currently used to produce many of the biotechnology industry’s largest and most
successful therapeutic proteins, including Epogen®, Neupogen®, Cerezyme,
Rituxan®, Enbrel®, Neulasta® and Herceptin®. Mammalian
cell-based expression technology is based on the introduction of a human gene encoding for a
4
specific therapeutic protein into the genome of a mammalian cell. The cells most often used in
connection with mammalian cell-based protein expression are Chinese hamster ovary (CHO) cells.
Mammalian cell-based expression systems have become the dominant system for the expression of
recombinant proteins due to their capacity for sophisticated, proper protein folding (which is
necessary for proteins to carry out their intended biological activity), assembly and
post-expression modification, such as glycosilation (the addition of sugar residues to a protein
which is necessary to enable specific biological activity by the protein). While bacterial and
yeast cell-based expression systems were the first protein expression systems developed by the
biotechnology industry and remain cost-effective compared to mammalian cell-based production
methodologies, proteins expressed in bacterial and yeast cell-based systems lack the capacity for
sophisticated protein folding, assembly and post-expression modifications, which are key factors of
mammalian cell-based systems. Accordingly, such systems cannot be used to produce glycoproteins or
other complex proteins and, therefore, bacterial and yeast cell-based systems are limited to the
expression of the most basic, simple proteins, such as insulin and growth hormones. Due to their
significant advantages, mammalian cell-based expression systems can produce proteins with superior
quality and efficacy compared to proteins expressed in bacteria and yeast cell-based systems. As a
result, the majority of currently approved therapeutic proteins, as well as those under
development, are produced in mammalian cell-based systems.
Despite the utility and widespread use of mammalian cell-based systems, they are subject to a
number of disadvantages. CHO cells and other mammalian cells are highly sensitive and can only be
grown under near perfect conditions, requiring highly complex, expensive, stainless steel
bioreactors which tightly regulate the required temperature, pH and oxygen levels. As a result,
such bioreactor systems are very costly and complicated to operate. CHO cells and other mammalian
cells are also susceptible to viral infections, including human viruses, and several cases of viral
contamination have occurred recently. The FDA and other regulatory
authorities require viral inactivation and other rigorous and detailed procedures for mammalian
cell-based manufacturing processes in order to address these potential hazards, thereby increasing
the cost and time demands of such expression systems. Furthermore, the current FDA and other
procedures only ensure screening for scientifically identified, known viruses. Accordingly,
compliance with current FDA and other procedures does not fully guarantee that patients are
protected against transmission of unknown or new potentially fatal viruses that may infect
mammalian cells. In addition, mammalian cell-based expression systems require large quantities of
sophisticated and expensive growth medium to accelerate the expression process.
Several companies and research institutions have explored alternatives to mammalian cell-based
production technologies that overcome some of these disadvantages, focusing primarily on the
expression of human proteins in genetically-modified organisms, or GMOs, such as transgenic
field-grown, whole plants and transgenic animals. However, these alternate techniques may be
restricted by regulatory and environmental risks regarding contamination of agricultural crops and
by the difficulty in applying cGMP standards of the pharmaceutical industry to these expression
technologies.
ProCellEx: Our Proprietary Protein Expression System
ProCellEx is our proprietary production system that we have developed based on our plant cell
culture technology for the development, expression and manufacture of recombinant proteins.
Our expression system consists of a comprehensive set of capabilities and proprietary
technologies, including advanced genetic engineering and plant cell culture technology, which
enables us to produce complex, proprietary and biologically equivalent proteins for a variety
of human diseases. Our protein expression system facilitates the creation and selection of
high expressing, genetically stable cell lines capable of expressing recombinant proteins. The
entire protein expression process, from initial nucleotide cloning to large-scale production of
the protein product, occurs under cGMP-compliant, controlled processes. Our plant cell culture
technology uses plant cells, such as carrot and tobacco cells, which undergo advanced genetic
engineering and are grown on an industrial scale in a flexible bioreactor system. Cell growth,
from scale up through large-scale production, takes place in flexible, sterile, polyethylene
bioreactors which are confined to a clean-room environment. Our bioreactors are well-suited
for plant cell growth using a simple, inexpensive, chemically-defined growth medium as a
catalyst for growth. The reactors are custom-designed and optimized for plant cell cultures,
easy to use, entail low initial capital investment, are rapidly scalable at a low cost and
require less hands-on maintenance between cycles. Our protein expression system does not
involve mammalian or animal components or transgenic field-grown, whole plants at any point in
the production process.
Our ProCellEx system is capable of producing proteins with an amino acid structure practically
equivalent to that of the desired human protein, and with a very similar, although not
identical, glycan, or sugar, structure. Our internal research and external laboratory studies
have demonstrated that ProCellEx is capable of producing recombinant proteins that exhibit a
glycan and amino acid structure similar to their naturally-produced human counterparts. In
collaboration with Israel’s Weizmann Institute of Science, we have demonstrated that the
three-dimensional structure of a protein expressed
5
in our proprietary plant cell-based expression system retains the same three-dimensional
structure as exhibited by the mammalian cell-based expressed version of the same protein. In
addition, proteins produced by our ProCellEx system maintain the biological activity that
characterize that of the naturally-produced proteins. Based on these results, we believe that
proteins developed using our ProCellEx protein expression system have the intended composition
and correct biological activity of their human equivalent proteins.
Competitive Advantages of Our ProCellEx Protein Expression System
We believe that our ProCellEx protein expression system, including our advanced genetic
engineering technology and plant cell-based protein expression methods, affords us a number of
significant advantages over mammalian, bacterial, yeast and transgenic cell-based expression
technologies, including the following:
Ability to Penetrate Certain Patent-Protected Markets. We seek to develop recombinant proteins
that we believe we can produce and commercialize without infringing upon the method-based patents
or other intellectual property rights of third parties. In several cases, a marketed
biotherapeutic protein is not itself subject to patent protection and is available for use in the
public domain; however, the process of expressing the protein product in mammalian or bacterial
cell systems is protected by method-based patents. Using our plant cell-based protein expression
technology, we are able to express an equivalent protein without infringing upon these method-based
patents. Moreover, we expect to enjoy method-based patent protection for the proteins we develop
using our proprietary ProCellEx protein expression technology, although there can be no assurance
that any such patents will be granted. In some cases, we may be able to obtain patent protection
for the compositions of the proteins themselves. We have filed for United States and international
composition of matter patents for taliglucerase alfa.
Significantly Lower Capital and Production Costs. Plant cells have a number of dynamic qualities
that make them well-suited for the production of therapeutic proteins. Plant cells grow rapidly
under a variety of conditions and are not as sensitive to temperature, pH and oxygen levels as
mammalian cells. Our ProCellEx protein expression system, therefore, requires significantly less
upfront capital expenditures as it does not use highly complex, expensive, stainless steel
bioreactors typically used in mammalian cell-based production systems to maintain very specific
temperature, pH and oxygen levels. Instead, we use simple polyethylene bioreactors that are able
to be maintained at the room temperature of the clean-room in which they are placed. This system
also reduces ongoing production and monitoring costs typically incurred by companies using
mammalian cell-based expression technologies. Furthermore, while mammalian cell-based systems
require very costly growth media at various stages of the production process to achieve target
yields of their proteins, plant cells require only simple and much less expensive solutions based
on sugar, water and microelements at infrequent intervals to achieve target yields. We believe
that these factors will potentially result in lower capital and production costs for the commercial
scale production of proteins by our ProCellEx system thereby providing us with a competitive
advantage over competing protein expression technologies.
Elimination of the Risk of Viral Transmission or Infection by Mammalian Components. By nature,
plant cells do not carry the risk of infection by human or other animal viruses. As a result, the
risk of contamination of our products under development and the potential risk of viral
transmission from our products under development to future patients, whether from known or unknown
viruses, is eliminated. Because our product candidates do not bear the risk of viral transmission,
we are not required by the FDA or other regulatory authorities to perform the constant monitoring
procedures for mammalian viruses during the protein expression process that mammalian cell-based
manufacturers are required to undertake. In addition, the production process of our ProCellEx
protein expression system is void of any mammalian components which are susceptible to the
transmission of prions, such as those related to bovine spongiform encephalopathy (commonly known
as “mad-cow disease”). These factors further reduce the risks and operating costs of our ProCellEx
system compared to mammalian cell-based expression systems.
More Effective and Potent End Product Relative to Mammalian Based Systems. Our ProCellEx protein
expression system produces enzymes which have uniform glycosilation patterns and therefore do not
require the lengthy and expensive post-expression modifications that are required for certain
proteins produced by mammalian cell-based systems, including the proteins for the treatment of
Gaucher disease. Such post-expression modifications in mammalian cell-produced proteins are made
in order to expose the terminal mannose sugar residues, which are structures on a protein that are
key elements in allowing the produced protein to bind to a target cell and subsequently be taken
into the target cell for therapeutic benefit. In the production of Cerezyme, exposing these
terminal mannose sugar residues involves a multitude of highly technical steps which add time and
cost to the production process. In addition, these steps do not guarantee the exposure of all of
the required terminal mannose sugar residues, resulting in potentially lower effective yields and
inconsistency in potency from batch to batch. Our ProCellEx protein expression system, by
contrast, produces taliglucerase alfa in a “ready to use” form that does not require additional
glycosilation or other modifications to make taliglucerase alfa suitable for use in enzyme
6
replacement therapy for Gaucher disease. We believe this quality increases the potency and
consistency of the expressed proteins, thereby further increasing the cost advantages of our
ProCellEx protein expression system over competing protein expression methodologies.
Broad Range of Expression Capabilities. Unlike bacterial and yeast cell-based systems, which are
unable to produce complex proteins, our ProCellEx protein expression system is able to produce a
broad array of complex glycosilated proteins. We have successfully demonstrated the feasibility of
our ProCellEx system by producing, on an exploratory, research scale, a variety of therapeutic
proteins belonging to different classes of recombinant drugs, such as enzymes, hormones, monoclonal
antibodies, cytokines and vaccines. We have demonstrated that the recombinant proteins we have
expressed to date have the intended composition and correct biological activity of their
human-equivalent protein, with several of such proteins demonstrating advantageous biological
activity compared to the currently available biotherapeutics. In specific cases, we have been
successful in expressing proteins that have not been successfully expressed in other production
systems.
Our Strategy
Our goal is to become a leading fully integrated biopharmaceutical company focused on the
development and commercialization of proprietary and biosimilar recombinant therapeutic
proteins. To achieve our goal, we intend to:
Facilitate the successful development and commercialization of taliglucerase alfa by Pfizer. We
intend to work with our licensee, Pfizer, to develop and commercialize taliglucerase alfa. We have
begun collaborating with Pfizer to facilitate the transition of certain of our taliglucerase alfa
assets to Pfizer’s organization. We are cooperating with Pfizer with respect to our Expanded
Access protocol for taliglucerase alfa in order to facilitate the participation of additional
physicians in additional sites in the protocol. Pfizer is promoting the protocol to new clinical
sites and is recruiting additional patients. We have also begun to facilitate relationships
between Pfizer and the Gaucher community and third-party payors. We intend to actively participate
and provide our expertise in Pfizer’s development and commercialization efforts with respect to
taligluerase alfa.
Obtain Regulatory Approval for Taliglucerase Alfa for the Treatment of Gaucher Disease. We
completed successfully our pivotal phase III clinical trial of taliglucerase alfa in September 2009
and announced the positive top-line study results in October 2009 and full study results in
February 2010. We filed a New Drug Application (NDA) to the FDA in December 2009 and in January
2010, the FDA requested additional data regarding the Chemistry, Manufacturing and Controls (CMC)
section of the NDA. No additional clinical or preclinical information was requested. The FDA’s
request focused primarily on the validation of the manufacturing process for our upgraded
manufacturing facility. A validation plan for our manufacturing process of taliglucerase alfa has
already been established and reviewed by the FDA. We are working diligently to provide the
requested data to the FDA and anticipate submitting the requested data during the second quarter of
2010. We expect to submit similar applications with other comparable regulatory agencies in other
countries during 2010. On February 1, 2010, we held a pre-Marketing Approval Application (MAA)
meeting with the EMEA. Our phase III clinical trial was conducted in selected leading medical
centers worldwide in North America, South America, Israel, Europe and South Africa. In the third
quarter of 2008, we initiated a double blind, follow-on extension study as part of the phase III
clinical trial in which patients that successfully completed treatment in the trial were given the
opportunity to continue to be treated with taliglucerase alfa at the same dose that they received
in the trial. We are compiling additional information relating to the long term safety and
efficacy of taliglucerase alfa through the follow-on study. In addition, in the fourth quarter of
2008 we announced the enrollment of the first patient in a worldwide, multi-center, open-label,
switch-over trial to assess the safety and efficacy of taliglucerase alfa. The switch-over trial,
which is not a pre requisite for marketing approval from the FDA, was designed to include 15
patients with Gaucher disease that are currently undergoing enzyme replacement therapy with
imiglucerase (Cerezyme). Due to the shortage in 2009 of the currently available enzyme replacement
therapy for Gaucher disease, after fully enrolling the 15 patients we extended the trial to include
up to 30 patients in total. In December 2009, we filed a proposed pediatric investigation plan to
the Pediatric Committee of the EMEA. We believe that taliglucerase alfa may have cost, efficacy
and potency advantages over the currently available enzyme replacement therapy for Gaucher disease
and we intend to pursue post-marketing studies to confirm these advantages. Although Gaucher
disease is a relatively rare disease, it represents a substantial commercial market due to the
severity of the symptoms and the chronic nature of the disease. We believe that the approval of
taliglucerase alfa as a treatment for Gaucher disease, if at all, with its potentially longer
acting profile and more cost-effective development process, may lead to an increase in the number
of patients who will be able to have access to and afford such treatment, thereby expanding the
size of the market for Gaucher disease treatments.
Develop a Pipeline of Innovative or Biosimilar Versions of Recombinant Therapeutic Proteins. We
are leveraging our ProCellEx protein expression system to develop a pipeline of innovative or
biosimilar versions of recombinant proteins, with an emphasis on therapeutic treatments with large
market opportunities. We select additional therapeutic candidates for
7
development through in-house testing, licensing agreements with academic institutions and
collaborations with pharmaceutical partners. We have currently identified several product
candidates that are mainly oriented towards the specialty disease and therapeutic market segments,
including treatments for Fabry disease and an acetylcholinesterase enzyme based therapy for
biodefense and intoxication treatments. We have also identified several other product candidates
that are chemical equivalents of approved therapeutic products that will no longer be patent
protected within the next couple of years, such as pr-antiTNF, our proprietary product candidate
for the treatment of certain immune diseases such as rheumatoid arthritis. We believe our
cost-effective technology will be an important asset for the commercialization of such drug
candidates. We believe that the clinical and regulatory pathway for many of our pipeline product
programs candidates is already established, and that this may reduce the risks and costs associated
with our clinical development programs. Furthermore, established markets already exist for the
development of most of our current product candidates. We plan to apply the manufacturing,
clinical and regulatory experience we have gained from the development of our lead product
candidate to advance a number of our preclinical product candidates into clinical trials over the
next few years.
Collaborate with Third Party Pharmaceutical Suppliers and build a Targeted Sales and Marketing
Infrastructure. We have licensed to Pfizer the right to commercialize taliglucerase worldwide,
except in Israel. We plan to establish our own, internal sales and marketing capabilities for
taliglucerase in Israel, and, for our other product candidates, in North America, the European
Union and in other significant markets, including Israel. We believe that the focus of our current
clinical pipeline on relatively rare genetic disorders with small patient populations and a highly
concentrated group of physicians focused on treating patients with such disorders may facilitate
our creation of a targeted internal sales force. In addition we are continuously evaluating
potential strategic marketing partnerships with respect to our other product candidates.
Establish Development and Commercialization Alliances with Corporate Partners. We believe that our
technology and know-how has broad applicability to many classes of proteins and can be used to
develop and potentially enhance numerous existing marketed protein therapeutics. We intend to
leverage our technology and know-how by pursuing development and commercialization alliances with
corporate partners for specific products and territories in order to enable us to optimize our
resources and effectively penetrate a wider range of target diseases and therapeutic markets. In
November 2009, we entered into a license and supply agreement with Pfizer for the development and
commercialization of taliglucerase alfa. We entered into an agreement with Teva in September 2006
for the development of two proteins. In 2009, programs relating to two proteins to be developed
under the agreement were terminated for commercial reasons. Last, we are in various stages of discussions with a number
of multinational pharmaceutical companies regarding additional collaboration agreements.
Acquire or In-License New Technologies, Products or Companies. We continuously seek attractive
product candidates and innovative technologies to in-license or acquire. We intend to focus on
product candidates that would be synergistic with our ProCellEx protein expression system and
expertise and that represent large potential market opportunities. We believe that by pursuing
selective acquisitions of technologies in businesses that complement our own, we will be able to
enhance our competitiveness and strengthen our market position.
Leverage Strength and Experience of Our Management Team and Board of Directors. Our management
team has extensive experience in the biotechnology and pharmaceutical industry. The Chairman of
our Board of Directors, Mr. Eli Hurvitz, is an experienced pharmaceutical industry veteran and the
current Chairman of the Board and former President and Chief Executive Officer of Teva. In
February 2008, we appointed Professor Roger D. Kornberg, a renowned biochemist and laureate of the
Nobel Prize in Chemistry, to our Board of Directors. We will continue to leverage their experience
and established track record as well as their relationships across the biotechnology and
pharmaceutical industries.
Our Pipeline Drug Candidates
Our Lead Product Candidate, Taliglucerase Alfa
Taliglucerase alfa, our lead proprietary product candidate, is a plant cell expressed
recombinant glucocerebrosidase enzyme (GCD) for the treatment of Gaucher disease. In July
2007, we reached an agreement with the FDA on the final design of our pivotal phase III
clinical trial of taliglucerase alfa through the FDA’s special protocol assessment (SPA)
process. We successfully completed our phase III pivotal clinical trial of taliglucerase alfa
in September 2009 and announced positive top line results of the clinical trial in October 2009
and full study results in February 2010. We submitted a New Drug Application (NDA) to the FDA
in December 2009. In January 2010, the FDA requested additional data regarding the Chemistry,
Manufacturing and Controls (CMC) section of the NDA. No additional clinical or preclinical
information was requested. The FDA’s request focused primarily on the validation of the
manufacturing process for our upgraded manufacturing facility. A validation plan for our
manufacturing process of taliglucerase alfa has already been established and reviewed by the
FDA. We are working diligently to provide the requested data to the FDA and anticipate
submitting the requested data during the second quarter of 2010. In addition, we expect to
submit
8
similar applications with other comparable regulatory agencies in other countries during 2010.
During the third quarter of 2008, we initiated a double blind, follow-on extension study as
part of the phase III clinical trial in which patients that successfully completed treatment in
the trial were given the opportunity to continue to be treated with taliglucerase alfa at the
same dose that they received in the trial. We are compiling additional information relating to
the long term safety and efficacy of taliglucerase alfa through the follow-on study. In
addition, in the fourth quarter of 2008 we announced the enrollment of the first patient in a
worldwide, multi-center, open-label, switch-over trial which has been reviewed by the FDA and
is designed to assess the safety and efficacy of taliglucerase alfa. The switch-over trial,
which is not a pre requisite for approval, is designed to include 15 patients with Gaucher
disease that are currently undergoing enzyme replacement therapy with imiglucerase (Cerezyme).
Due to the shortage of the currently available enzyme replacement therapy for Gaucher disease,
after fully enrolling the 15 patients we extended the trial to include up to 30 patients in
total. In addition, in December 2009, we filed a proposed pediatric investigation plan to the
Pediatric Committee of the EMEA. In clinical trials in healthy subjects and in vivo primate
studies, taliglucerase alfa has demonstrated an increased half-life and prolonged presence of
the enzyme in the blood serum of the subjects as compared to Cerezyme, the only enzyme
replacement therapy currently marketed to treat Gaucher disease.
We believe that taliglucerase alfa, if approved, has the potential to offer patients and
healthcare payors a more effective and cost efficient treatment of Gaucher disease because of
the following features:
Increased Glycan Efficacy and Consistency. We believe that our ProCellEx protein expression system
produces recombinant proteins that exhibit consistent enzymatic activity from batch to batch. This
results in a highly active product that may achieve a desired therapeutic effect more effectively
than the activity demonstrated in proteins produced through mammalian cell-based expression systems
due to its greater glycan efficacy and consistency. This quality increases the effective
consistency in potency and further increases the cost advantages from using our plant cell-based
expression technology compared to competing protein expression methodologies.
Longer Half-Life. The data generated in preclinical and human clinical trials relating to the
half-life of taliglucerase alfa in the subjects’ blood serum after infusion showed that the
half-life of taliglucerase alfa is significantly longer than that of Cerezyme when measured and
compared to publicly available data on Cerezyme.
Cost-Effective. Taliglucerase alfa is potentially less expensive to produce as the manufacturing
process does not require the large initial set-up investments involved in mammalian cell-based
protein production, the extensive ongoing costs associated with growth media and monitoring
throughout the production process nor any of the post-expression modification costs in order to
modify the glycosilation of the proteins produced through the mammalian cell-based methodologies.
As such, we believe that taliglucerase alfa’s potential advantages may lead taliglucerase alfa to
become a highly efficacious and cost-effective treatment alternative for Gaucher disease patients.
In addition, we are developing a new method for delivering active recombinant proteins systemically
through oral administration of transgenic plant cells expressing biotherapeutic proteins. If
proven effective, we intend to apply this breakthrough technology to taliglucerase alfa before we
apply it to any other product candidates. If proven effective, our experimental oral taliglucerase
alfa would be the first protein to be administered orally rather than through intravenous therapy.
We are developing our oral taliglucerase alfa product candidate to be used in enzyme replacement
therapy, not as a small molecule. This differentiates our oral product candidate from other early
clinical stage, experimental, small molecule, oral drugs which are being developed for the
treatment of Gaucher disease by Amicus Therapeutics, Inc., or Amicus Therapeutics, and Genzyme.
Small molecule based treatments for Gaucher disease, such as Zavesca, have different mechanisms of
action than those associated with enzyme replacement therapy, and may be associated with a number
of side effects. We have filed patent applications with respect to this new protein delivery
mechanism in other countries with commercially significant markets. Currently, we are the
exclusive owners of all rights to this technology.
Gaucher Disease Background
Gaucher disease, a hereditary, genetic disorder with severe and debilitating symptoms, is the
most prevalent lysosomal storage disorder in humans. Lysosomal storage disorders are metabolic
disorders in which a lysosomal enzyme, a protein that degrades cellular substrates in the
lysosomes of cells, is mutated or deficient. Lysosomes are small membrane-bound cellular
structures within cells that contain enzymes necessary for intracellular digestion. Gaucher
disease is caused by mutations or deficiencies in the gene encoding GCD, a lysosomal enzyme
that catalyzes the degradation of the fatty substrate, glucosylceramide (GlcCer). The normal
degradation products of GlcCer are glucose and ceramide, which are easily excreted by the cells
through normal biological processes. Patients with Gaucher disease lack or otherwise have
dysfunctional GCD and, accordingly, are not able to break down GlcCer. The absence of an
active GCD enzyme leads to the accumulation of GlcCer in lysosomes of certain white blood cells
called macrophages. Macrophages affected by the
9
disease become highly enlarged due to the accumulation of GlcCer and are referred to as
“Gaucher cells.” Gaucher cells accumulate in the spleen, liver, lungs, bone marrow and brain.
Signs and symptoms of Gaucher disease may include enlarged liver and spleen, abnormally low
levels of red blood cells and platelets and skeletal complications. In some cases, the patient
may suffer an impairment of the central nervous system.
Current Treatments for Gaucher Disease
The standard of care for Gaucher disease is enzyme replacement therapy using recombinant GCD to
replace the mutated or deficient natural GCD enzyme. The latest studies estimate that there
are approximately 10,000 patients suffering from Gaucher disease worldwide. Enzyme replacement
therapy is a medical treatment in which recombinant enzymes are injected into patients in whom
the enzyme is lacking or dysfunctional. Cerezyme, an enzyme replacement therapy commercialized
by Genzyme Corporation, is the only recombinant GCD currently available on the market and
approved worldwide for the treatment of Gaucher disease. According to public reports issued by
Genzyme, Cerezyme had annual sales of approximately $793 million in 2009, compared to $1.2
billion in 2008. According to Genzyme it suffered a temporary interruption in production of
Cerezyme in 2009 associated with the remediation of a contamination in one of its manufacturing
facilities, and, as a result, shipments of Cerezyme were limited during the second half of
2009. Cerezyme is produced through a mammalian cell-based protein expression process in CHO
cells. There are no known severe side effects to the use of Cerezyme and its approved use over
the past decade suggests that it is an effective treatment of Gaucher disease. However,
Cerezyme is subject to the limitations of most mammalian cell-based therapeutic proteins,
including lengthy and costly production processes and contamination risks. As enzyme
replacement therapy does not cure the genetic disorder, but rather provides an external source
for transfusion of the missing or mutated enzyme, Gaucher disease patients generally receive
the treatment over their entire lifetime. The current average annual cost for enzyme
replacement therapy for an adult Gaucher disease patient in the United States is in excess of
$200,000.
The only other approved drug for the treatment of Gaucher disease at this time is Zavesca
(miglustat), marketed by Actelion Ltd. Zavesca has been approved by the FDA for use in the
United States as an oral treatment. However, it has many side effects and the FDA has approved
it only for administration to those patients who cannot be treated through enzyme replacement
therapy, and, accordingly, have no other treatment alternative. As a result, Zavesca’s use has
been extremely limited. Actelion has reported sales of Zavesca of approximately CHF 53.1
million (approximately $51.6 million) in 2009.
Taliglucerase Alfa Development Program
We believe the clinical development path for taliglucerase alfa will be similar to that
followed by the existing enzyme replacement therapy currently on the market. The primary
efficacy endpoint for our pivotal phase III study was the reduction in size of spleen and the
secondary endpoints for our pivotal phase III study included increase in platelet and
hemoglobin counts and reduction in liver size, all of which are generally well-established and
accepted by regulatory agencies and specifically agreed to by the FDA in the special protocol
assessment (SPA) of the final design of our pivotal phase III clinical trial for taliglucerase
alfa. We met all of the endpoints in our phase III trial which was successfully completed in
September 2009. See “—Phase III Clinical Trial.” The primary end point for our switch-over
study, which is not a prerequisite for approval, is non deterioration in the patient’s clinical
condition as measured through significant, well established end points such as platelet and
hemoglobin counts and spleen and liver size.
Laboratory Testing and Preclinical Studies of Taliglucerase Alfa
We conducted several in vitro tests and in vivo preclinical studies of taliglucerase alfa. Our
preclinical rodent and primate trials generated extensive toxicological and safety data that
demonstrated no adverse effects, even with very high doses of taliglucerase alfa being
administered via intravenous infusions. In short term repeat dose studies in rodents and
primates and nine month repeat dose studies in primates, no toxicity was observed at dosage
levels of up to 10 times the current dose recommended for GCD in clinical use. Furthermore, no
neutralizing antibodies were detected in any of the primates treated in the studies. The
presence of neutralizing antibodies would have implied a likelihood of the host rejecting the
therapeutic enzyme or reacting to it in a less efficient manner.
Our laboratory and preclinical data demonstrate that taliglucerase alfa has the potential to be
an efficacious enzyme replacement therapy for the treatment of Gaucher disease. Data produced
from these preliminary development studies show that, relative to Cerezyme, taliglucerase alfa
has:
10
| • |
|
an equivalent to superior level of enzymatic activity (see Figure 1); |
| |
| • |
|
enhanced uptake based on observed GlcCer substrate degradation (see Figure 2); and |
| |
| • |
|
a prolonged half-life (see Figure 3). |
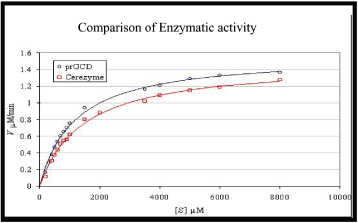
As shown in Figure 1, we compared the enzymatic activity of taliglucerase alfa and Cerezyme
using an in vitro assay where increasing amounts of GlcCer substrate (S), provided in
millimolar, were degraded by a fixed amount of taliglucerase alfa and Cerezyme, measured in
milligrams. Enzymatic activity was measured by the rate of degradation of GlcCer into glucose
and ceramide (its normal degradation products), measured by millimoles of product produced per
minute per fixed amount of enzyme. In the study assays performed, one demonstrated that
taliglucerase alfa had enzymatic activity that was equivalent to Cerezyme; the other studies
demonstrated superior activity by taliglucerase alfa. Figure 1 demonstrates that the enzymatic
activity of taliglucerase alfa was superior to Cerezyme.
Figure 1: Taliglucerase Alfa and Cerezyme Enzymatic Activity
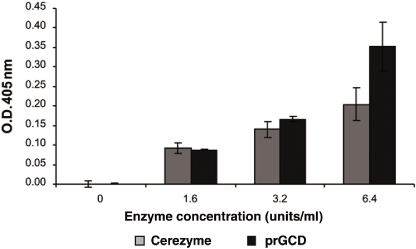
As shown in Figure 2, we compared the uptake of increasing amounts of Cerezyme and
taliglucerase alfa into the target cell, using an ex vivo mouse macrophage cell model.
Cellular uptake was measured in cell lysates, solutions containing the contents of burst cells,
by comparing enzymatic activity at various enzyme concentrations of Cerezyme and taliglucerase
alfa based on the amount of GlcCer substrate degradation into glucose and ceramide, measured in
a microplate absorbance reader, a flat plate with multiple “wells” used as small test tubes, at
an optical density of 405 nanometers. The results in Figure 2 demonstrate that the uptake into
the macrophage cells of taliglucerase alfa was greater than the uptake of Cerezyme at higher
enzyme concentrations, as measured by the resulting enzymatic activity in the cells. We
believe that the ability of the plant cells to directly generate the required terminal mannose
structures for efficient glycosilation of taliglucerase alfa, results in the enhanced uptake of
taliglucerase alfa into the Gaucher cells. In contrast, Cerezyme requires post-expression and
purification modifications to expose the terminal mannose structures, which modification
process can yield enzymes with less consistent glycosilation patterns and could reduce cellular
uptake of Cerezyme.
11
Figure 2: Taliglucerase Alfa and Cerezyme Cellular Uptake
Furthermore, the data generated in preclinical trials relating to pharmacokinetic parameters,
specifically the half-life of enzyme in the subjects’ blood serum after infusion, showed that
the half-life of taliglucerase alfa is significantly longer than that of Cerezyme based upon
data disclosed publicly by Genzyme. We believe the extended half-life of taliglucerase alfa
relative to Cerezyme is attributable to the different glycoside profile, thereby resulting in
the enhanced uptake of taliglucerase alfa into the Gaucher cells.
Figure 3: Taliglucerase Alfa and Cerezyme Half-Life Data
| |
|
|
|
|
| |
|
Taliglucerase Alfa |
|
Cerezyme |
| |
Primates
|
|
~13.0-20.0 minutes
|
|
~ 6.8-8.0 minutes (1) |
Humans
|
|
~10.5-14.5 minutes
|
|
~3.6-10.4 minutes (2) |
|
|
|
| (1) |
|
Source: Cerezyme NDA – PharmTox review |
| |
| (2) |
|
Source: Cerezyme labeling approved by FDA for package insert |
Prior to submitting the NDA for taliglucerase alfa, we conducted further, standard preclinical
studies, mainly reproductive studies, of taliglucerase alfa, which further support the safety
profile of taliglucerase alfa.
Phase I Clinical Trial
We completed a phase I clinical trial of taliglucerase alfa in June 2006 which was performed
under an FDA Investigational New Drug (IND) approval. The phase I clinical trial was a
single-center, non-randomized, open label, dose ranging study designed to evaluate the safety
and pharmacokinetics of taliglucerase alfa in healthy subjects. The trial was conducted on
healthy subjects over a four-week period in which subjects received three single escalating
doses of taliglucerase alfa administered as intravenous infusions.
All doses administered to subjects in the phase I clinical trial, including the highest dose,
which was the same dosage currently suggested with respect to the treatment by Cerezyme,
demonstrated a strong safety profile. The data from our phase I clinical trial showed that
taliglucerase alfa was safe and well tolerated at all doses. See Figure 4.
12
Figure 4: Adverse Events presented by: Dose Group, Severity and Relation to Study
Treatment (Incidents; Subjects (% of Subjects))
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Relation between Event to Drug |
|
15 U/kg |
|
30 U/kg |
|
60 U/kg |
|
Placebo |
|
Events Severity |
|
Total |
Unrelated to drug (1)
|
|
0; 0 (0%)
|
|
0; 0 (0%)
|
|
2; 1 (17%)
|
|
0; 0 (0%)
|
|
Moderate
|
|
|
2 |
|
Remotely related to drug (2)
|
|
4; 2 (33%)
|
|
1; 1 (17%)
|
|
2; 1 (17%)
|
|
1; 1 (17%)
|
|
Mild
|
|
|
8 |
|
Possibly related to drug (3)
|
|
0; 0 (0%)
|
|
0; 0 (0%)
|
|
0; 0 (0%)
|
|
0; 0 (0%)
|
|
|
|
|
0 |
|
Probably related to drug (4)
|
|
0; 0 (0%)
|
|
0; 0 (0%)
|
|
0; 0 (0%)
|
|
0; 0 (0%)
|
|
|
|
|
0 |
|
Related to drug (5)
|
|
0; 0 (0%)
|
|
0; 0 (0%)
|
|
0; 0 (0%)
|
|
0; 0 (0%)
|
|
|
|
|
0 |
|
|
|
|
| (1) |
|
The event is clearly related to other factors, such as a subject’s clinical state,
therapeutic interventions or concomitant medications. |
| |
| (2) |
|
The event was most likely produced by other factors, such as a subject’s clinical
state, therapeutic interventions or concomitant medications, and does not follow a known
response pattern to the study drug. |
| |
| (3) |
|
The event has a reasonable temporal relationship to the study drug administration and
follows a known response pattern to the study drug. However, a potential alternate etiology
may be responsible for the event. The effect of drug withdrawal is unclear. Rechallenge
information is unclear or lacking. |
| |
| (4) |
|
The event follows a reasonable temporal sequence from the time of drug administration,
and follows a known response pattern to the study drug and cannot be reasonably explained by
other factors. There is a reasonable response to withdrawal of the drug. Rechallenge
information is not available or advisable. |
| |
| (5) |
|
The event follows a temporal sequence from the time of drug administration and follows
a known response pattern to the study drug. The event either occurs immediately following
the study drug administration, improves on stopping the drug or reappears on repeated
exposure. |
There were no serious adverse events and no subjects withdrew from the trial or discontinued
treatment due to an adverse event.
In addition, as illustrated in Figure 3 above, the half-life of taliglucerase alfa was found to
be significantly longer than that of Cerezyme, based upon data disclosed publicly by Genzyme,
which was consistent with our preclinical data.
Further, no neutralizing antibodies or adverse immunological responses were detected in any of
the subjects treated in the phase I clinical trial. The presence of neutralizing antibodies
would imply that the human body may reject the therapeutic enzyme.
Phase III Clinical Trial
After the conclusion of the phase I clinical trial and discussions with the FDA, we applied to
commence a pivotal phase III clinical trial of taliglucerase alfa without the requirement to
first complete a phase II clinical trial. In April 2007, we received approval from the FDA to
initiate a pivotal phase III clinical trial. We submitted to the FDA a request for a special
protocol assessment (SPA) of the final design of our pivotal phase III clinical trial for
taliglucerase alfa. In July 2007, we reached an agreement with the FDA on the design that we
submitted in the SPA request and in the third quarter of 2007 we initiated enrollment and
treatment of naive patients in the phase III clinical trial. In accordance with the terms of
the SPA, the phase III clinical trial was a multi-center, world-wide, randomized, double-blind,
parallel group, dose-ranging study to assess the safety and efficacy of taliglucerase alfa in
31 treatment-naive patients suffering from Gaucher disease. In the trial, patients were
selected randomly for one of two dosing arms (60 U/kg or 30 U/kg) and received intravenous
infusions of taliglucerase alfa once every two weeks for a nine-month period. The primary
endpoint of the study was a 20% mean reduction from baseline in spleen volume after nine
months, as measured by MRI. Major secondary endpoints were an increase in hemoglobin, decrease
in liver volume and increase in platelet count. The trial enrolled patients at 11 centers
throughout Europe, Israel, North America, South America and South Africa. We commenced
enrollment and treatment of patients in our phase III clinical trial in the third quarter of
2007 and completed enrollment in the fourth quarter of 2008. During the third quarter of 2008,
we initiated a double blind, follow-on
13
extension study as part of our phase III clinical trial in which patients that successfully
completed treatment in the trial were given the opportunity to continue to be treated with
taliglucerase alfa at the same dose that they received in the trial. We are compiling
additional information relating to the long term safety and efficacy of taliglucerase alfa
through the follow-on study. In addition, in the fourth quarter of 2008, we announced the
enrollment of the first patient in a worldwide, multi-center, open-label, switch-over trial to
assess the safety and efficacy of taliglucerase alfa. The switch-over trial, which is not a
pre requisite for approval, was originally designed to include 15 patients with Gaucher disease
that are currently undergoing enzyme replacement therapy with imiglucerase (Cerezyme). Due to
the shortage of Cerezyme in 2009, after fully enrolling the 15 patients we extended the trial
to include up to 30 patients in total. We successfully completed the phase III clinical trial
in September 2009.
Phase III Clinical Trial Results
We reported positive top line results of our phase III clinical trial of taliglucerase alfa in
October 2009 and full study results in February 2010. In the clinical trial, taliglucerase alfa
significantly reduced mean spleen volume after nine months compared with baseline in both treatment
groups. The 60 U/kg group demonstrated a statistically significant mean reduction in spleen volume
of 38.0% (p<0.0001) and the 30 U/kg group demonstrated a significant mean reduction in spleen
volume of 26.9% (p<0.0001). In addition, the primary endpoint was achieved in both treatment
groups after only six months of therapy.
Statistically significant improvements were also observed for the secondary endpoints after nine
months when compared to baseline for the 60 U/kg dose. Patients demonstrated a mean increase in
hemoglobin of 2.2 g/dL or 22.2% (p<0.0001), a mean decrease in liver volume of 11.1%
(p<0.0001) and a mean elevation in platelet count of 41,494 ml or 72.1% (p=0.0031). For
patients in the 30 U/kg dose, statistically significant improvements after nine months compared
with baselines were observed for hemoglobin level (increased 1.6 g/dL or 14.8%; p=0.0010) and liver
size (decreased 10.48%; p=0.0041); a nominal elevation in platelet count was also seen (11,427 ml
or 13.7%; p=0.0460).
Thirty patients in the trial had Chitotriosidase measurements, a biomarker for clinical symptoms of
Gaucher disease. In these patients, Chitotriosidase decreased from baseline in both the 30U/kg and
60U/kg groups by 47.3% and 58.4%, respectively.
The safety analysis for both treatment groups showed that taliglucerase alfa was well tolerated and
no serious or severe adverse events were reported. Two patients in the trial developed antibodies
to taliglucerase alfa and no patients developed neutralizing antibodies. In addition, two patients
experienced hypersensitivity reactions to taliglucerase alfa. No anti-taliglucerase antibodies
were detected in these patients and both reactions were treated in the physicians’ clinic and
reversed.
Most adverse events were considered unrelated to taliglucerase alfa. The most frequent mild to
moderate adverse event was headache. Other mild to moderate adverse events included dizziness,
muscle spasm, chest discomfort, nausea, skin irritation and arthalgia.
Other Drug Candidates in Our Pipeline
We are developing other recombinant therapeutic proteins to be expressed by our ProCellEx protein
expression system, with an emphasis on treatments for which there are large, established
pharmaceutical markets and where our proprietary protein expression system enables us to develop
and commercialize recombinant proteins that are patent-protected and therapeutically equivalent or
superior to the existing treatments. We select additional therapeutic candidates for development
by testing candidates in-house and through collaborations with academic partners. We have
identified several product candidates oriented towards specialty disease and therapeutic market
segments, including treatments for Fabry disease. We are also conducting initial research to
evaluate potential programs in the fields of monoclonal antibodies, cytokines and vaccines. We
filed an investigational new drug application (IND) with the FDA for our acetylcholinesterase
enzyme (AChE) during the last quarter of 2009. In addition, we plan to file an IND with the FDA
with respect to our Fabry project during the second half of 2010. Last, we are developing a new
method for delivering active recombinant proteins systemically through oral administration of
transgenic plant cells expressing such biotherapeutic proteins.
PRX-102
We are developing a proprietary alpha Galactosidase enzyme, currently titled PRX-102, which is a
therapeutic enzyme for the treatment of Fabry disease, a rare genetic lysosomal storage disorder in
humans, the symptoms of which involve the accumulation of lipids in the cells of the kidneys, heart
and other organs. These symptoms may lead to kidney failure and increased risk of heart attack and
stroke. Fabry disease affects more than 8,000 people globally. We believe that the treatment of
Fabry disease is a specialty clinical niche with the potential for high growth. Currently there
are two drugs available on the market to treat Fabry disease. Fabrazyme, made by Genzyme, was
approved for the treatment of Fabry disease in the European Union in 2001 and the United States in
2003. Genzyme reported $431 million in worldwide sales of
14
Fabrazyme in 2009, compared to $494 million in 2008. According to Genzyme, it suffered a temporary
interruption in production of Fabrazyme in 2009 associated with the remediation of a contamination
in one of its manufacturing facilities, and, as a result, shipments of Fabrazyme were limited
during the second half of 2009. The other approved drug for the treatment of Fabry disease in the
European Union is Replagal, which is sold by Shire plc. Shire reported $194 million in sales of
Replagal in 2009. According to public reports by Shire, it filed a BLA with the FDA for Replagal
in the United States in December 2009.
We are currently in the animal evaluation testing phase of the development of PRX-102, which tests
are based on a well established mouse model for Fabry disease. Initial pre clinical trial results
demonstrate the successful reduction of the GB3 level of mice suffering from Fabry disease when
compared to the control. These results are similar to the results of treatment with Fabrazyme. We
expect to file an IND with the FDA for PRX-102 in the second half of 2010 following the completion
of additional animal studies. As was the case in our development of taliglucerase alfa, our
development of PRX-102 involves the expression by our proprietary protein expression system of a
naturally occurring enzyme to be used in enzyme replacement therapy for the treatment of Fabry
disease. Based on our experience with taliglucerase alfa and the experience of other companies
developing enzyme replacement therapies for Fabry disease, we have reason to believe that, if
favorable data is accumulated in preclinical and phase I clinical trials, the FDA may allow us to
proceed directly with a pivotal phase III clinical trial without the need to complete a phase II
clinical trial. However, there can be no assurance that we will initiate phase I clinical trials
and if we do, that such trials will result in favorable data. In addition, there can be no
assurance that the FDA will allow us to proceed directly with a phase III clinical trial after
completion of a phase I clinical trial.
Acetylcholinesterase
In August 2007, we entered into an agreement with the Yissum Research and Development Company and
the Boyce Thompson Institute, Inc. pursuant to which we are developing a proprietary plant
cell-based acetylcholinesterase (AChE) and its molecular variants for the use in several
therapeutic and prophylactic indications, as well as in a biodefense program and an
organophosphate-based pesticide treatment program. Pursuant to the terms of the agreement, we have
received an exclusive, worldwide right and license to certain technology, including patents and
certain patent applications relating to AChE for the therapeutic and prophylactic indications as
well as an exclusive license not limited to such indications with respect to certain of those
patents and patent applications. In consideration for those licenses, we have agreed to make
certain regulatory milestone payments, a sales-based milestone payment, a license maintenance fee
and a royalty on net sales of any products developed with the licensed technology.
In January 2008, we expanded the scope of our acetylcholinesterase program with Yissum after we
achieved proof of concept results in an animal study conducted as part of the program. In our
animal study, the plant cell expressed form of the acetylcholinesterase protein demonstrated full
protection from organophosphate poisoning, stimulating the capacity of the plant cell expressed
acetylcholinesterase protein to treat nerve gas and pesticide poisoning. Under our agreement with
Yissum, we intend to conduct a collaborative research program in the laboratory of Professor
Hermona Soreq, a world leader in the field of acetylcholinesterase research and Dean of the Faculty
of Science at the Hebrew University.
To date, our in vitro experiments have shown that the acetylcholinesterase enzyme expressed in our
ProCellEx protein expression system demonstrates promising biological activity on biochemical and
cellular levels. In addition, early animal studies demonstrated that the acetylcholinesterase
expressed in our ProCellEx protein expression system was able to successfully treat animals exposed
to the nerve gas agent analogs, both when injected with our acetylcholinesterase product candidate
immediately before exposure or when injected after exposure. In December 2009, we submitted an IND
application for our acetylcholinesterase enzyme with the FDA, and we intend to initiate a clinical
study immediately after our IND is accepted, if at all.
We are currently in discussions with different U.S. civil and military organizations regarding
certain grants for which our acetylcholinesterase program is eligible. We anticipate applying for
specific grants during 2010 to support the further development of our acetylcholinesterase program.
pr-antiTNF
In September 2009, we announced preliminary preclinical data regarding an antiTNF (Tumor, Necrosis
Factor) protein that we are expressing through our proprietary ProCellEx system. We have designed
this antiNF as pr-antiNF. pr-antiNF is a candidate for the treatment of certain autoimmune
diseases such as rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing, spondylitis,
psoriatic arthritis and plaque psoriasis. Amgen Inc. has reported total sales of Enbrel of $3.5
billion for 2009 and Wyeth Pharmaceuticals has reported total sales of Enbrel of $1.4 billion for
the six months ended June 30, 2009. Wyeth was acquired by Pfizer
in 2009.
15
pr-antiTNF is a plant cell-expressed recombinant fusion protein made from the soluble form of the
human TNF receptor (TNFR), fused to the Fc component of a human antibody domain. pr-antiTNF has an
identical amino acid sequence to Enbrel and our in vitro and preclinical animal studies have
demonstrated that pr-antiTNF exhibits similar activity to Enbrel. Specifically, pr-antiTNF binds
TNF thereby inhibiting it from binding to cellular surface TNF receptors and protects L929 cells
from TNF-induced apoptosis in a dose-dependent manner. In a proof-of-concept in vivo study using
an established arthritis animal model, pr-antiTNF, when injected in mice, significantly improved
the clinical arthritis parameters associated with this accepted arthritis mouse model, including
joint inflammation, swelling and tissue degradation. We intend to conduct additional animal
studies to collect additional data to form a basis for a discussion with the FDA to explore the
regulatory pathway for our antiTNF program. Patents for the Enbrel start to expire as early as
2012, and we expect to use our cost effective manufacturing platform to facilitate entry into this
market upon approval of our pr-antiTNF product, if at all.
Commercialization Agreement
On November 30, 2009, Protalix Ltd. and Pfizer entered into a license and supply agreement pursuant
to which Pfizer was granted an exclusive, worldwide license to develop and commercialize
taliglucerase alfa. Under the terms and conditions of the Pfizer agreement, Protalix Ltd. retained
the right to commercialize taliglucerase alfa in Israel. In connection with the execution of the
Pfizer agreement, Pfizer made an upfront payment to Protalix Ltd. of $60.0 million in connection
with the execution of the agreement and subsequently paid to Protalix Ltd. an additional $5.0
million upon our filing of a proposed pediatric investigation plan to the Pediatric Committee of
the EMEA. Protalix Ltd. is also eligible to receive potential milestone payments of up to $50.0
million, in the aggregate, for the successful achievement of other regulatory-related milestones
and to payments equal to 40% of the net profits earned by Pfizer on sales of taliglucerase alfa.
In calculating the net profits, there are certain agreed upon limits on the amounts that may be
deducted from gross sales for certain expenses and costs of goods sold. Protalix Ltd. retained the
manufacturing rights to taliglucerase alfa and Pfizer and Protalix Ltd. have agreed to a specific
allocation of the responsibilities for the continued development efforts for taliglucerase alfa.
Protalix Ltd. will manufacture all of the taliglucerase alfa needed for all purposes under the
agreement and Pfizer will purchase the taliglucerase alfa from Protalix Ltd., subject to certain
terms and conditions. The Pfizer agreement also provides for reimbursement by Pfizer of certain
costs to be incurred by Protalix Ltd.
In connection with the payments made under the Pfizer agreement, Protalix Ltd. has accrued a
sublicense fee equal to $1.6 million payable to the academic institution from who it licensed
certain technology relating to taligluceraze alfa. Future milestone payments will be subject
to a 2.5% royalty, and all of the royalty payments we receive under the agreement will be
subject to a 0.75% royalty payable to the same institution until 2016, when a patent related to
taliglucerase alfa licensed to us will expire. We are also required to pay a royalty equal to
3% of the revenues we record from Pfizer under the Pfizer agreement to the OCS.
We will be subject to a withholding tax on the U.S. revenue source portion of the payments made to
us for our share of Pfizer’s in net profits under the Pfizer agreement. Currently, the withholding
tax rate is 15%.
Strategic Collaborations
Teva Pharmaceutical Industries
In September 2006, we entered into a Collaboration and Licensing Agreement with Teva for the
development and manufacture of two proteins, to be identified by Teva and us using our ProCellEx
protein expression system. The agreement also identifies additional matters for collaboration
between Teva and us. Subsequently, two proteins were identified to be researched and developed
under the agreement but in 2009, both of the projects were terminated for commercial reasons. These proteins were not
part of our current product development pipeline. Pursuant to the agreement, we have agreed to
collaborate on the research and development of the two proteins utilizing our ProCellEx protein
expression system. If the research and preclinical development efforts for either protein are
successful and if Teva elects to pursue clinical trials for the development of either protein
through our ProCellEx protein expression system, we have agreed to grant to Teva an exclusive
license to commercialize the products developed based on the protein in return for royalty and
milestone payments payable upon the achievement of certain pre-defined goals. We will retain
certain exclusive manufacturing rights with respect to the active pharmaceutical ingredient of the
proteins following the first commercial sale of a licensed product under the agreement and other
rights. See “Risk Factors—Our strategy, in many cases, is to enter into collaboration agreements
with third parties to leverage our ProCellEx system to develop product candidates. If we fail to
enter into these agreements or if we or the third parties do not perform under such agreements or
terminate or elect to discontinue the collaboration, it could have a material adverse affect on our
revenues.
16
Weizmann Institute of Science
In March 2006, we entered into a Research and License Agreement with the Yeda Research and
Development Company Limited, the technology transfer arm of the Weizmann Institute of Science,
pursuant to which Yeda is using its technology to design a next generation of GCD for the treatment
of Gaucher disease that can be expressed using our ProCellEx protein expression system and that may
have certain benefits over the first generation treatments used today. The technology licensed
from Yeda provides a methodology for the rational design of an improved drug for the treatment of
Gaucher disease by enzyme replacement therapy, based on the three-dimensional crystal structure of
GCD that was solved by scientists from the Weizmann Institute of Science. In consideration for
Yeda’s research, we agreed to pay a fixed research budget amount. Yeda has granted us a license to
use their technology and discoveries for the development, production and sale of enzymatically
active mutations of GCD and derivatives thereof for the treatment of Gaucher disease. We are
responsible for commercializing the products developed under the license. Under the agreement, we
are obligated to pay certain minimum royalty amounts and varying fixed royalty amounts on net sales
of products developed using the licensed technology for the treatment of Gaucher disease and other
indications as well as for sublicensing revenues. Accordingly, we will have certain payment
obligations to Yeda even if we were to fail to generate any revenue from the licensed technology.
See “Risk Factors—If we cannot meet requirements under our license agreements, we could lose the
rights to our products, which could have a material adverse effect on our business.”
Intellectual Property
We maintain a proactive intellectual property strategy which includes patent filings in multiple
jurisdictions, including the United States and other commercially significant markets. We hold 14
granted patents and 84 patent applications currently pending with respect to various compositions,
methods of production and methods of use relating to our ProCellEx protein expression system and
our proprietary product pipeline. Of such patent applications, two were filed internationally
during the first quarter of 2010. We also have one joint patent with a third party and hold
licensed rights to seven patents and five patent applications.
Our competitive position and future success depend in part on our ability, and that of our
licensees, to obtain and leverage the intellectual property covering our product candidates,
know-how, methods, processes and other technologies, to protect our trade secrets, to prevent
others from using our intellectual property and to operate without infringing the intellectual
property of third parties. We seek to protect our competitive position by filing United States,
European Union, Israeli and other foreign patent applications covering our technology, including
both new technology and improvements to existing technology. Our patent strategy includes
obtaining patents, where possible, on methods of production, compositions of matter and methods of
use. We also rely on know-how, continuing technological innovation, licensing and partnership
opportunities to develop and maintain our competitive position. Lastly, we monitor third parties
for activities that may infringe our intellectual property, as well as the progression of third
party patent applications that may cover our product candidates or expression methods and thus,
potentially, interfere with the development of our business. We are aware, for example, of United
States patents, and corresponding international counterparts of such patents, owned by third
parties that contain claims covering methods of producing GCD. We do not believe that, if any
claim of infringement were to be asserted against us based upon such patents, taliglucerase alfa
would be found to infringe any valid claim under such patents. However, there can be no assurance
that a court would find in our favor or that, if we choose or are required to seek a license to any
one or more of such patents, a license would be available to us on acceptable terms or at all.
Our patent portfolio consists of several patent families (consisting of patents and/or patent
applications) covering our technology, protein expression methodologies and system and product
candidates. We have been issued, and hold licensed rights to, patents in the United States, the
European Union, Israel, Canada, the Czech Republic, Hungary, Japan, Poland, Mexico, Hong Kong and
India that cover our ProCellEx protein expression system, including the methods that we use for
culturing and harvesting plant cells and/or tissues in consecutive cycles. Another patent family
in our patent portfolio contains patent applications relating to the production of glycosilated
proteins in our plant culture platform, particularly proteins having a terminal mannose
glycosilation, including taliglucerase alfa. An additional patent family contains patent
applications relating to a system and method for production of antibodies in a plant cell culture,
and antibodies produced in such a system. In addition, our patent portfolio includes a patent
family for a new method for delivering active recombinant proteins systemically through oral
administration of transgenic plant cells and a patent family related to saccharides containing
protein conjugates. Lastly, our patent portfolio includes a patent that we co-own and that covers
human glycoprotein hormone and chain splice variants, including isolated nucleic acids encoding
these variants. More specifically, this patent portfolio covers a new splice variant of human FSH.
In April 2004, we entered into a Collaborative Research Agreement with Icon Genetics AG (which was
subsequently acquired by Bayer Corporation), or Icon, regarding an option to license Icon’s
amplification technology for utilization in the
17
expression of our products under development in order to improve our yield. In connection with
such option, we entered into a license agreement with Icon in April 2005, pursuant to which we
received an exclusive worldwide license to develop, test, use and commercialize Icon’s technology
to express certain proteins in our ProCellEx protein expression system. In addition, we are
entitled to a non-exclusive worldwide license to make and have made other proteins expressed by
using Icon’s technology in our technology. In consideration for the licenses, we are obligated to
pay to Icon development milestone payments and royalties. See “Risk Factors—If we fail to
adequately protect or enforce our intellectual property rights or secure rights to third party
patents, the value of our intellectual property rights would diminish and our business, competitive
position and results of operations would suffer.”
Manufacturing
We are obligated to manufacture all of the taliglucerase alfa drug product needed under the Pfizer
agreement, subject to certain terms and conditions. Our drug product candidates, including
taliglucerase alfa, must be manufactured in a sterile environment and in compliance with cGMPs set
by the FDA and other relevant foreign regulatory authorities. We use our current facility, which
has approximately 20,000 sq/ft of clean rooms built according to industry standards, to develop,
process and manufacture taliglucerase alfa and other recombinant proteins. We have completed the
final upgrade of the manufacturing space within our facility to ensure that the manufacturing space
will be able to comply with the good laboratory, clinical and manufacturing practices required by
the FDA and other comparable regulatory authorities for production of pharmaceutical products on a
commercial scale. We intend to use our current manufacturing space to produce all of the
taliglucerase alfa we need in the near future, included the taliglucerase alfa to be purchased by
Pfizer. Current capacity of our facility can serve approximately 20% of the Gaucher disease
patients that are currently under treatment. We intend to expand our current facility in order to
reach a capacity of approximately 50% of the Gaucher disease patients that are currently under
treatment and to house the laboratory space necessary for further development of other product
candidates in our pipeline. Total expected cost for such expansion is estimated to be
approximately $20.0 million and the process is expected to be completed during 2011.
We have entered into a contract with Teva pursuant to which Teva is performing the final filling
and freeze drying steps for taliglucerase alfa. According to our agreement with Pfizer, Pfizer
will be responsible for the fill and finish activities for taliglucerase alfa.
Our current facility in Israel has been granted “Approved Enterprise” status, and we have elected
to participate in the alternative benefits program. Our facility is located in a Zone A location,
and, therefore, our income from the Approved Enterprise will be tax exempt in Israel for a period
of 10 years, commencing with the year in which we first generate taxable income from the relevant
Approved Enterprise. To remain eligible for these tax benefits, we must continue to meet certain
conditions, and if we increase our activities outside of Israel, for example, by future
acquisitions, such increased activities generally may not be eligible for inclusion in Israeli tax
benefit programs. In addition, our technology is subject to certain restrictions with respect to
the transfer of technology and manufacturing rights. “See Risk Factors—The manufacture of our
products is an exacting and complex process, and if we or one of our materials suppliers encounter
problems manufacturing our products, it will have a material adverse effect on our business and
results of operations.”
Raw Materials and Suppliers
We believe that the raw materials that we require throughout the manufacturing process of our
current and potential drug product candidates are widely available from numerous suppliers and are
generally considered to be generic industrial biological supplies. We do not rely on a single or
unique supplier for any materials relating to the current production of any biotherapeutic proteins
in our pipeline.
Development and regulatory approval of our pharmaceutical products are dependent upon our ability
to procure active ingredients and certain packaging materials from sources approved by the FDA and
other regulatory authorities. Since the FDA and other regulatory approval processes require
manufacturers to specify their proposed suppliers of active ingredients and certain packaging
materials in their applications, FDA approval of a supplemental application to use a new supplier
in connection with any drug candidate or approved product, if any, would be required if active
ingredients or such packaging materials were no longer available from the specified supplier, which
could result in manufacturing delays. From time to time, we intend to identify alternative
FDA-approved suppliers to ensure the continued supply of necessary raw materials.
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly evolving technology
and significant competition. Competition from numerous existing companies and others entering the
fields in which we operate is intense and expected to increase. Most of these companies have
substantially greater research and development, manufacturing,
18
marketing, financial, technological personnel and managerial resources than we do. In addition,
many specialized biotechnology companies have formed collaborations with large, established
companies to support research, development and commercialization of products that may be
competitive with our current and future product candidates and technologies. Acquisitions of
competing companies by large pharmaceutical or biotechnology companies could enhance such
competitors’ financial, marketing and other resources. Academic institutions, governmental
agencies and other public and private research organizations are also conducting research
activities and seeking patent protection and may commercialize competitive products or technologies
on their own or through collaborations with pharmaceutical and biotechnology companies.
We specifically face competition from companies with approved treatments of Gaucher disease,
including Genzyme and to a much lesser extent, Actelion. Shire is currently developing a
gene-activated enzyme expressed in human cancer cells to treat Gaucher disease. Shire has
submitted marketing applications for its enzyme replacement therapy treatment for Gaucher
disease. According to public reports by Shire, its application is being reviewed by the FDA
under Priority Review with a PDUFA date of February 28, 2010 and the EMEA’s Committee for
Medicinal Products for Human Use has granted accelerated review for Shire’s Marketing
Authorization Application. In addition, we are aware of other early clinical stage,
experimental, small molecule, oral drugs which are being developed for the treatment of Gaucher
disease by Amicus Therapeutics, which according to public filings by Amicus Therapeutics has
been suspended, and Genzyme. We also face competition from companies with approved enzyme
treatments of Fabry disease, including Genzyme and Shire, and we are aware of other early stage
drugs which are being developed for the treatment of Fabry disease, including a drug being
developed by Amicus Therapeutics.
We also face competition from companies that are developing other platforms for the expression of
recombinant therapeutic pharmaceuticals. We are aware of companies that are developing alternative
technologies to develop and produce therapeutic protein in anticipation of the expiration of
certain patent claims covering marketed proteins. Competitors developing alternative expression
technologies include Crucell N.V., Shire and GlycoFi, Inc. (which was acquired by Merck & Co.
Inc.). Other companies are developing alternate plant-based technologies, include Biolex, Inc.,
Chlorogen, Inc., Greenovation Biotech GmbH, and Symbiosys, none of which are cell-based. Rather,
such companies base their product development on transgenic plants or whole plants.
Several biogeneric companies are pursuing the opportunity to develop and commercialize follow-on
versions of other currently marketed biologic products, including growth factors, hormones,
enzymes, cytokines and monoclonal antibodies, which are areas that interest us. These companies
include, among others, Novartis AG/Sandoz Pharmaceuticals, BioGeneriX AG, Stada Arzneimittel AG,
BioPartners GmbH and Teva.
Key differentiating elements affecting the success of our product candidates are likely to be their
potency and efficacy profiles, as well as their cost-effectiveness as compared to other existing
therapies. See “Risk Factors—Developments by competitors may render our products or technologies
obsolete or non-competitive which would have a material adverse effect on our business and results
of operations.”
Scientific Advisory Board
Members of our scientific advisory board, who are experts in the fields of plant molecular and cell
biology as well as Gaucher disease and various hematological and genetic disorders, consult with
our management within their professional areas of expertise; exchange strategic and business
development ideas with our management; attend scientific, medical and business meetings with our
management, such as meetings with the FDA and comparable foreign regulatory authorities, meetings
with strategic or potential strategic partners and other meetings relevant to their areas of
expertise; and attend meetings of our scientific advisory board. We expect our scientific advisory
board to convene at least twice annually, and we frequently consult with the individual members of
our Scientific Advisory Board. Our scientific advisory board currently includes the following
people:
19
| |
|
|
| Name |
|
Affiliation |
Professor Aaron Ciechanover, M.D., D.Sc. |
|
Laureate of the Nobel Prize in
Chemistry
Distinguished research Professor at the
Cancer and Vascular Biology Research Center
of the Rappaport Research Institute and
Faculty of Medicine at the Technion |
|
|
|
|
|
American Academy of Arts and Sciences, Member |
|
|
|
Professor Gad Galili, Ph.D.
|
|
Chairman of the Department of Plant
Sciences, The Weizmann Institute of Science,
Rehovot, Israel |
|
|
|
Professor Ari Zimran, M.D.
|
|
Director of the Gaucher Clinic, Shaare Zedek
Medical Center, Jerusalem, Israel |
|
|
|
|
|
Associate Professor of Medicine, Hebrew
University-Hadassah Medical School,
Jerusalem, Israel |
Government Regulation
The testing, manufacture, distribution, advertising and marketing of drug products are subject to
extensive regulation by federal, state and local governmental authorities in the United States,
including the FDA, and by similar authorities in other countries. Any product that we develop must
receive all relevant regulatory approvals or clearances, as the case may be, before it may be
marketed in a particular country.
The regulatory process, which includes overseeing preclinical studies and clinical trials of each
pharmaceutical compound to establish its safety and efficacy and confirmation by the FDA that good
laboratory, clinical and manufacturing practices were maintained during testing and manufacturing,
can take many years, requires the expenditure of substantial resources and gives larger companies
with greater financial resources a competitive advantage over us. Delays or terminations of
clinical trials that we undertake would likely impair our development of product candidates.
Delays or terminations could result from a number of factors, including stringent enrollment
criteria, slow rate of enrollment, size of patient population, having to compete with other
clinical trials for eligible patients, geographical considerations and others.
The FDA review process can be lengthy and unpredictable, and we may encounter delays or rejections
of our applications when submitted. Generally, in order to gain FDA approval, we must first
conduct preclinical studies in a laboratory and in animal models to obtain preliminary information
on a compound and to identify any potential safety problems. The results of these studies are
submitted as part of an IND application that the FDA must review before human clinical trials of an
investigational drug can commence. Clinical trials may be terminated by the clinical trial site,
sponsor or the FDA if toxicities appear that are either worse than expected or unexpected.
Clinical trials are normally performed in three sequential phases and generally take two to five
years, or longer, to complete. Phase I consists of testing the drug product in a small number of
humans, normally healthy volunteers, to determine preliminary safety and tolerable dose range.
Phase II usually involves studies in a limited patient population to evaluate the effectiveness of
the drug product in humans having the disease or medical condition for which the product is
indicated, determine dosage tolerance and optimal dosage and identify possible common adverse
effects and safety risks. Phase III consists of additional controlled testing at multiple clinical
sites to establish clinical safety and effectiveness in an expanded patient population of
geographically dispersed test sites to evaluate the overall benefit-risk relationship for
administering the product and to provide an adequate basis for product labeling. Phase IV clinical
trials may be conducted after approval to gain additional experience from the treatment of patients
in the intended therapeutic indication.
After completion of clinical trials of a new drug product, FDA and foreign regulatory authority
marketing approval must be obtained. Assuming that the clinical data support the product’s safety
and effectiveness for its intended use, a New Drug Application (NDA) is submitted to the FDA for
its review. Generally, it takes one to three years to obtain approval. If questions arise during
the FDA review process, approval may take a significantly longer period of time. The testing and
approval processes require substantial time and effort and approval on a timely basis, if at all,
or the approval that we receive may be for a narrower indication than we had originally sought,
potentially undermining the commercial viability of the product. Even if regulatory approvals are
obtained, approved products are subject to continual review and holders of an approved product are
required, for example, to report certain adverse reactions and production problems, if any, to the
FDA, and to comply with certain requirements concerning advertising and promotional labeling for
the product. Also, quality
20
control and manufacturing procedures relating to a product must continue to conform to cGMP after
approval, and the FDA periodically inspects manufacturing facilities to assess compliance with
cGMP. Accordingly, manufacturers must continue to expend time, money and effort in the area of
production and quality control to comply with cGMP and other aspects of regulatory compliance. The
later discovery of previously unknown problems or failure to comply with the applicable regulatory
requirements with respect to any product may result in restrictions on the marketing of the product
or withdrawal of the product from the market as well as possible civil or criminal sanctions. See
also “—International Regulation.”
Under the Orphan Drug Act of 1983, the FDA may grant orphan drug designation to drugs and
biological products intended to treat a rare disease or condition, which is generally a disease or
condition that affects fewer than 200,000 individuals in the United States. In September 2009, we
received orphan drug designation for taliglucerase alfa for the treatment of Gaucher disease. The
FDA grants orphan drug designation to drugs that may provide a significant therapeutic advantage
over existing treatments and target conditions affecting 200,000 or fewer U.S. patients per year.
Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory
review and approval process. Among the other benefits of orphan drug designation are possible
funding and tax savings to support clinical trials and for other financial incentives and a waiver
of the marketing application user fee and most likely priority review.
The FDA has a fast track program that is intended to expedite or facilitate the process for
reviewing new drugs and biological products that meet certain criteria. Specifically, new drugs
and biological products are eligible for fast track designation if they are intended to treat a
serious or life-threatening condition and demonstrate the potential to address unmet medical needs
for the condition. Fast track designation applies to the combination of the product and the
specific indication for which it is being studied. For a fast track product, the FDA may consider
for review on a rolling basis sections of the NDA before the complete application is submitted, if
the sponsor provides a schedule for the submission of the sections of the NDA, the FDA agrees to
accept sections of the NDA as they become available and determines that the schedule is acceptable,
and the sponsor pays any required user fees upon submission of the first section of the NDA. We
used the rolling submission option for our NDA for our lead product candidate, taliglucerase alfa,
which we completed in December 2009.
None of our products under development has been approved for marketing in the United States or
elsewhere. We may not be able to obtain regulatory approval for any of our products under
development in a timely manner, if at all. Failure to obtain requisite governmental approvals or
failure to obtain approvals of the scope requested will delay or preclude us, or our licensees or
marketing partners, from marketing our products, or limit the commercial use of our products, and
thereby would have a material adverse effect on our business, financial condition and results of
operations. See “Risk Factors—We may not obtain the necessary U.S. or worldwide regulatory
approvals to commercialize our drug candidates in a timely manner, if at all, which would have a
material adverse effect on our business and results of operations.”
The United States federal government regulates healthcare through various agencies, including but
not limited to the following: (i) the FDA, which administers the Federal Food, Drug, and Cosmetic
Act (FDCA), as well as other relevant laws; (ii) the Center for Medicare & Medicaid Services (CMS),
which administers the Medicare and Medicaid programs; (iii) the Office of Inspector General (OIG)
which enforces various laws aimed at curtailing fraudulent or abusive practices, including by way
of example, the Anti-Kickback Law, the Anti-Physician Referral Law, commonly referred to as Stark,
the Anti-Inducement Law, the Civil Money Penalty Law and the laws that authorize the OIG to exclude
healthcare providers and others from participating in federal healthcare programs; and (iv) the
Office of Civil Rights, which administers the privacy aspects of the Health Insurance Portability
and Accountability Act of 1996 (HIPAA). All of the aforementioned are agencies within the
Department of Health and Human Services (HHS). Healthcare is also provided or regulated, as the
case may be, by the Department of Defense through its TriCare program, the Department of Veterans
Affairs, especially through the Veterans Health Care Act of 1992, the Public Health Service within
HHS under Public Health Service Act § 340B (42 U.S.C. § 256b), the Department of Justice through
the Federal False Claims Act and various criminal statutes, and state governments under the
Medicaid and other state sponsored or funded programs and their internal laws regulating all
healthcare activities. Many states also have anti-kickback and anti-physician referral laws that
are similar to the federal laws, but may be applicable in situations where federal laws do not
apply.
Medicare is the federal healthcare program for those who are (i) over 65 years of age, (ii)
disabled, (iii) suffering from end-stage renal disease or (iv) suffering from Lou Gehrig’s disease.
Medicare consists of part A, which covers inpatient costs, part B, which covers services by
physicians and laboratories, durable medical equipment and certain drugs, primarily those
administered by physicians, and part D, which provides drug coverage for most prescription drugs
other than those covered under part B. Medicare also offers a managed care option under part C.
Medicare is administered by CMS. In contrast, Medicaid is a state-federal healthcare program for
the poor and is administered by the states pursuant to an agreement with the Secretary of Health
and Human Services. Most state Medicaid programs cover most outpatient prescription drugs.
21
International Regulation
We are subject to regulations and product registration requirements in many foreign countries in
which we may sell our products, including in the areas of product standards, packaging
requirements, labeling requirements, import and export restrictions and tariff regulations, duties
and tax requirements. The time required to obtain clearance required by foreign countries may be
longer or shorter than that required for FDA clearance, and requirements for licensing a product in
a foreign country may differ significantly from FDA requirements.
Pharmaceutical products may not be imported into, or manufactured or marketed in, the State of
Israel absent drug registration. The three basic criteria for the registration of pharmaceuticals
in Israel is quality, safety and efficacy of the pharmaceutical product and the Israeli Ministry of
Health requires pharmaceutical companies to conform to international developments and standards.
Regulatory requirements are constantly changing in accordance with scientific advances as well as
social and ethical values.
The relevant legislation of the European Union requires that medicinal products, including generic
versions of previously approved products, and new strengths, dosage forms and formulations, of
previously approved products, shall have a marketing authorization before they are placed on the
market in the European Union. Authorizations are granted after the assessment of quality, safety
and efficacy by the respective health authorities. In order to obtain an authorization, an
application must be made to the competent authority of the member state concerned or in a
centralized procedure to the EMEA. Besides various formal requirements, the application must
contain the results of pharmaceutical (physico-chemical, biological or microbiological) tests, of
preclinical (toxicological and pharmacological) tests as well as of clinical trials. All of these
tests must have been conducted in accordance with relevant European Union regulations and must
allow the reviewer to evaluate the quality, safety and efficacy of the medicinal product. On
January 2010, the Committee for Orphan Medicinal Products (COMP) of the EMEA recommended that the
European Commission grant orphan drug designation to taliglucerase alfa. Orphan drug designation
in the European Union is granted to medicinal products intended for the diagnosis, prevention and
treatment of life-threatening diseases and very serious conditions that affect not more than five
in 10,000 people in the European Union. Orphan drug designation is generally given to medicinal
products that treat conditions for which no current therapy exists or are expected to bring a
significant benefit to patients over existing therapies. If granted by the European Commission,
orphan drug designation will provide us a centralized procedure for obtaining marketing
authorization for taliglucerase alfa, with a single marketing authorization valid throughout all EU
Member States. We may also be eligible for a number of additional incentives including protocol
assistance, reduction in registration fees and eligibility for grants and initiatives supporting
research and development related to the orphan drug designation.
Israeli Government Programs
The following is a summary of the current principal Israeli tax laws applicable to us and Protalix
Ltd., and of the Israeli Government programs from which Protalix Ltd. benefits. Some parts of this
discussion are based on new tax legislation that has not been subject to judicial or administrative
interpretation. Therefore, the views expressed in the discussion may not be accepted by the tax
authorities in question. The discussion should not be construed as legal or professional tax
advice and does not cover all possible tax considerations.
General Corporate Tax Structure in Israel
Generally, Israeli companies are subject to corporate tax at the rate of 29% on taxable income and
are subject to real capital gains tax at a rate of 25% on capital gains (other than gains derived
from the sale of listed securities that are taxed at the prevailing corporate tax rates) derived
after January 1, 2003. The corporate tax rate was reduced in June 2004, from 36% to 35% for the
2004 tax year, 34% for the 2005 tax year, 31% for the 2006 tax year, 29% for the 2007 tax year, 27%
for the 2008 tax year, 26% for the 2009 tax year and 25% for the 2010 tax year and thereafter.
Additional, gradual corporate tax reductions were adopted in 2008, as follows: 24% for the 2011
tax year; 23% for the 2012 tax year; 22% for the 2013 tax year; 21% for the 2014 tax year; 20% for
the 2015 tax year; and 18% thereafter. As discussed below, the corporate tax rate may be less for
income derived from an Approved Enterprise. In addition to the corporate taxes in Israel, we are
subject to a withholding tax on the U.S. revenue source portion of the payments made to us for our
share of Pfizer’s net profits under the Pfizer agreement. The withholding tax rate is 15%. See
“Business — Commercialization Agreement.”
Law for the Encouragement of Capital Investments, 1959
The Law for the Encouragement of Capital Investments, 1959, known as the Investment Law, provides
certain incentives for capital investments in a production facility (or other eligible assets).
Generally, an investment program that is implemented in accordance with the provisions of the
Investment Law, referred to as an “Approved Enterprise,” is entitled to benefits.
22
These benefits may include cash grants from the Israeli government and tax benefits, based upon,
among other things, the location of the facility in which the investment is made and specific
elections made by the grantee.
The Investment Law was significantly amended effective in April 2005. Protalix Ltd. will continue
to enjoy the tax benefits under the pre-revision provisions of the Investment Law. If any new
benefits are granted to Protalix Ltd. in the future, Protalix Ltd. will be subject to the
provisions of the amended Investment Law with respect to these new benefits. Therefore, the
following discussion is a summary of the Investment Law prior to its amendment as well as the
relevant changes contained in the new legislation.
Under the Investment Law prior to its amendment, a company that wished to receive benefits had to
receive approval from the “Investment Center” of the Israeli Ministry of Industry, Trade and Labor,
or the Investment Center. Each certificate of approval for an Approved Enterprise relates to a
specific investment program in the Approved Enterprise, delineated both by the financial scope of
the investment and by the physical characteristics of the facility or the asset, e.g., the
equipment to be purchased and utilized pursuant to the program.
An Approved Enterprise may elect to forego any entitlement to the grants otherwise available under
the Investment Law and, instead, participate in an alternative benefits program under which the
undistributed income from the Approved Enterprise is fully exempt from corporate tax for a defined
period of time. Under the alternative package of benefits, a company’s undistributed income
derived from an Approved Enterprise will be exempt from corporate tax for a period of between two
and 10 years from the first year of taxable income, depending upon the geographic location within
Israel of the Approved Enterprise. Upon expiration of the exemption period, the Approved
Enterprise is eligible for the reduced tax rates otherwise applicable under the Investment Law for
any remainder of the otherwise applicable benefits period (up to an aggregate benefits period of
either seven or 10 years, depending on the location of the company or its definition as a foreign
investors’ company). If a company has more than one Approved Enterprise program or if only a
portion of its capital investments are approved, its effective tax rate is the result of a weighted
combination of the applicable rates. The tax benefits from any certificate of approval relate only
to taxable profits attributable to the specific Approved Enterprise. Income from activity that is
derived from different Approved Enterprises does not enjoy these tax benefits.
A company that has an Approved Enterprise program is eligible for further tax benefits if it
qualifies as a foreign investors’ company. A foreign investors’ company eligible for benefits is
essentially a company in which more than 25% of the share capital (in terms of shares, rights to
profit, voting and appointment of directors) is owned (measured by both share capital and combined
share and loan capital) by non-Israeli residents. A company that qualifies as a foreign investors’
company and has an Approved Enterprise program is eligible for tax benefits for a 10-year benefit
period and may enjoy a reduced corporate tax rate of 10% to 25%, depending on the amount of the
company’s shares held by non-Israeli shareholders.
If a company that has an Approved Enterprise program is a wholly owned subsidiary of another
company, then the percentage of foreign investments is determined based on the percentage of
foreign investment in the parent company. The tax rates and related levels of foreign investments
are set forth in the following table:
| |
|
|
| Percent of Foreign |
|
Rate of Reduced |
| Ownership |
|
Tax |
| 0-49%
|
|
25% |
| 49-74%
|
|
20% |
| 74-90%
|
|
15% |
| 90-100%
|
|
10% |
Our original facility in Israel has been granted “Approved Enterprise” status, and it has elected
to participate in the alternative benefits program. Under the terms of its Approved Enterprise
program, the facility is located in a top priority location, or “Zone A,” and, therefore, the
income from that Approved Enterprise will be tax exempt in Israel for a period of 10 years,
commencing with the year in which taxable income is first generated from the relevant Approved
Enterprise. The current benefits program may not continue to be available and Protalix Ltd. may
not continue to qualify for its benefits.
A company that has elected to participate in the alternative benefits program and that subsequently
pays a dividend out of the income derived from the Approved Enterprise during the tax exemption
period will be subject to corporate tax in respect of the amount distributed at the rate that would
have been applicable had the company not elected the alternative benefits program (generally 10% to
25%, depending on the extent to which non-Israeli shareholders hold such company’s shares). If the
dividend is distributed within 12 years after the commencement of the benefits period (or, in the
case of a foreign
investor’s company, without time limitation), the dividend recipient is taxed at the reduced
withholding tax rate of 15%
23
applicable to dividends from approved enterprises, or at the lower rate
under an applicable tax treaty. After this period, the withholding tax rate is 25%, or at the
lower rate under an applicable tax treaty. In the case of a company with a foreign investment
level (as defined by the Investment Law) of 25% or more, the 12-year limitation on reduced
withholding tax on dividends does not apply. The company must withhold this tax at its source,
regardless of whether the dividend is converted into foreign currency.
The Investment Law also provides that an Approved Enterprise is entitled to accelerated
depreciation on its property and equipment that are included in an approved investment program.
This benefit is an incentive granted by the Israeli government regardless of whether the
alternative benefits program is elected.
The benefits available to an Approved Enterprise are conditioned upon terms stipulated in the
Investment Law and regulations and the criteria set forth in the applicable certificate of
approval. If Protalix Ltd. does not fulfill these conditions in whole or in part, the benefits can
be canceled and Protalix Ltd. may be required to refund the received benefits, linked to the
Israeli consumer price index with the addition of interest or alternatively with an additional
penalty payment. We believe that Protalix Ltd. currently operates in compliance with all
applicable conditions and criteria, but there can be no assurance that Protalix Ltd. will continue
to do so. Furthermore, there can be no assurance that any Approved Enterprise status granted to
Protalix Ltd.’s facilities will entitle Protalix Ltd. to the same benefits to which it is currently
entitled.
Pursuant to the March 2005 amendment to the Investment Law, the approval of the Investment Center
is required only for Approved Enterprises that receive cash grants. Approved Enterprises that do
not receive benefits in the form of governmental cash grants, but only tax benefits, are no longer
required to obtain this approval. Instead, these Approved Enterprises are required to make certain
investments as specified in the Investment Law.
The amended Investment Law specifies certain conditions for an Approved Enterprise to be entitled
to benefits. These conditions include:
| • |
|
the Approved Enterprise’s revenues from any single country or a separate customs
territory may not exceed 75% of the Approved Enterprise’s total revenues; or |
| |
| • |
|
at least 25% of the Approved Enterprise’s revenues during the benefits period must be
derived from sales into a single country or a separate customs territory with a population
of at least 12 million. |
There can be no assurance that Protalix Ltd. will comply with the above conditions in the future or
that Protalix Ltd. will be entitled to any additional benefits under the Investment Law. In
addition, it is possible that Protalix Ltd. may not be able to operate in a way that maximizes
utilization of the benefits under the Investment Law.
From time to time, the Israeli Government has discussed reducing the benefits available to
companies under the Investment Law. The termination or substantial reduction of any of the
benefits available under the Investment Law could materially impact the cost of our future
investments.
Encouragement of Industrial Research and Development Law, 1984
In the past, Protalix Ltd. received grants from the Office of the Chief Scientist of the Israeli
Ministry of Industry, Trade and Labor, the OCS, for the financing of a portion of its research and
development expenditures in Israel. As of December 31, 2009, the OCS approved grants in respect of
Protalix Ltd.’s continuing operations totaling approximately $16.7 million, measured from
inception. Protalix Ltd. is required to repay up to 100% of grants actually received (plus
interest at the LIBOR rate applied to the grants received on or after January 1, 1999) to the OCS
through payments of royalties at a rate of 3% to 6% of the revenues generated from an OCS-funded
project, depending on the period in which revenues were generated. As of December 31, 2009,
Protalix Ltd. had not paid royalties and Protalix Ltd.’s contingent liability to the OCS with
respect to grants received was approximately $14.8 million, after the accrual of a $1.9 million
royalty payment to the OCS in connection with the $65.0 million we received in 2009 from Pfizer.
Under the Israeli Law for the Encouragement of Industrial Research and Development, 1984 and
related regulations, the Research Law, recipients of grants from the OCS are prohibited from
manufacturing products developed using these grants outside of the State of Israel without special
approvals, although the Research Law does enable companies to seek prior approval for conducting
manufacturing activities outside of Israel without being subject to increased royalties. If
Protalix Ltd. receives approval to manufacture the products developed with government grants
outside of Israel, it will be required to pay an increased total amount of royalties (possibly up
to 300% of the grant amounts plus interest), depending on the manufacturing volume that is
performed outside of Israel, as well as at a possibly increased royalty rate.
24
Additionally, under the Research Law, Protalix Ltd. is prohibited from transferring the OCS
financed technologies and related intellectual property rights outside of the State of Israel
except under limited circumstances and only with the approval of the Research Committee of the OCS.
Protalix Ltd. may not receive the required approvals for any proposed transfer and, if received,
Protalix Ltd. may be required to pay the OCS a portion of the consideration that it receives upon
any sale of such technology by a non-Israeli entity. The scope of the support received, the
royalties that Protalix Ltd. has already paid to the OCS, the amount of time that has elapsed
between the date on which the know-how was transferred and the date on which the OCS grants were
received and the sale price and the form of transaction will be taken into account in order to
calculate the amount of the payment to the OCS. Approval of the transfer of technology to
residents of the State of Israel is required, and may be granted in specific circumstances only if
the recipient abides by the provisions of applicable laws, including the restrictions on the
transfer of know-how and the obligation to pay royalties. No assurances can be made that approval
to any such transfer, if requested, will be granted.
In March 2005, an amendment to the Research Law was enacted. One of the main modifications
included in the amendment was an authorization of the Research Committee to allow the transfer
outside of Israel of know-how derived from an approved program and the related manufacturing
rights. In general, the Research Committee may approve transfer of know-how in limited
circumstances as follows:
| • |
|
in the event of a sale of the know-how itself to a non affiliated third party, provided
that upon such sale the owner of the know-how pays to the OCS an amount, in cash, as set
forth in the Research Law. In addition, the amendment provides that if the purchaser of
the know-how gives the selling Israeli company the right to exploit the know-how by way of
an exclusive, irrevocable and unlimited license, the research committee may approve such
transfer in special cases without requiring a cash payment. |
| |
| • |
|
in the event of a sale of the company which is the owner of know-how, pursuant to which
the company ceases to be an Israeli company, provided that upon such sale, the owner of the
know-how makes a cash payment to the OCS as set forth in the Research Law. |
| |
| • |
|
in the event of an exchange of know-how such that in exchange for the transfer of
know-how outside of Israel, the recipient of the know-how transfers other know-how to the
company in Israel in a manner in which the OCS is convinced that the Israeli economy
realizes a greater, overall benefit from the exchange of know-how. |
Another provision in the amendment concerns the transfer of manufacturing rights. The research
committee may, in special cases, approve the transfer of manufacture or of manufacturing rights of
a product developed within the framework of the approved program or which results therefrom,
outside of Israel.
The State of Israel does not own intellectual property rights in technology developed with OCS
funding and there is no restriction on the export of products manufactured using technology
developed with OCS funding. The technology is, however, subject to transfer of technology and
manufacturing rights restrictions as described above. For a description of such restrictions,
please see “Risk Factors—Risks Relating to Our Operations in Israel.” OCS approval is not required
for the export of any products resulting from the research or development or for the licensing of
any technology in the ordinary course of business.
Special Provisions Relating to Taxation under Inflationary Conditions
Protalix Ltd. is taxed in Israel under the Income Tax Law (Inflationary Adjustments), 1985,
generally referred to as the Inflationary Adjustments Law. The Inflationary Adjustments Law is
highly complex, and represents an attempt to overcome the problems presented to a traditional tax
system by an economy undergoing rapid inflation. The provisions that are material to us are
summarized below:
| • |
|
Where a company’s equity, as calculated under the Inflationary Adjustments Law, exceeds
the depreciated cost of its fixed assets (as defined in the Inflationary Adjustments Law),
a deduction from taxable income is permitted equal to this excess multiplied by the
applicable annual rate of inflation. The maximum deduction permitted under this provision
in any single tax year is 70% of taxable income. The unused portion linked to the Israeli
consumer price index, may be carried forward. |
| • |
|
Where a company’s depreciated cost of fixed assets exceeds its equity, the excess
multiplied by the applicable annual rate of inflation is added to taxable income. |
| • |
|
Subject to specified limitations, depreciation deductions carryforwards on fixed assets
and losses are adjusted for inflation based on the change in the consumer price index. |
25
Under the Inflationary Adjustments Law, results for tax purposes are measured in real terms, in
accordance with changes in the Israeli consumer price index. The difference between the change in
the Israeli consumer price index and the exchange rate of Israeli currency in relation to the U.S.
dollar may in future periods cause significant differences between taxable income and the income
measured in dollars as reflected in our consolidated financial statements.
Law for the Encouragement of Industry (Taxes), 1969
We believe that Protalix Ltd. currently qualifies as an “Industrial Company” within the meaning of
the Law for the Encouragement of Industry (Taxes), 1969, or the Industry Encouragement Law. The
Industry Encouragement Law defines “Industrial Company” as a company resident in Israel that
derives 90% or more of its income in any tax year (other than specified kinds of passive income
such as capital gains, interest and dividends) from an “Industrial Enterprise” that it owns. An
“Industrial Enterprise” is defined as an enterprise whose major activity in a given tax year is
industrial production.
The following corporate tax benefits, among others, are available to Industrial Companies:
| • |
|
amortization of the cost of purchased know-how and patents over an eight-year period for
tax purposes; |
| • |
|
accelerated depreciation rates on equipment and buildings; |
| • |
|
under specified conditions, an election to file consolidated tax returns with other
related Israeli Industrial Companies; and |
| • |
|
expenses related to a public offering are deductible in equal amounts over three years. |
Eligibility for the benefits under the Industry Encouragement Law is not subject to receipt of
prior approval from any governmental authority. It is possible that Protalix Ltd. may fail to
qualify or may not continue to qualify as an “Industrial Company” or that the benefits described
above will not be available in the future.
Tax Benefits for Research and Development
Under specified conditions, Israeli tax laws allow a tax deduction by a company for research and
development expenditures, including capital expenditures, for the year in which such expenditures
are incurred. These expenditures must relate to scientific research and development projects and
must be approved by the OCS. Furthermore, the research and development projects must be for the
promotion of the company and carried out by or on behalf of the company seeking such tax deduction.
However, the amount of such deductible expenditures is reduced by the sum of any funds received
through government grants for the finance of such scientific research and development projects.
Expenditures not so approved are deductible over a three-year period.
Employees
As of December 31, 2009, we had 182 employees, of whom 29 have an M.D. or a Ph.D. in their
respective scientific fields. We believe that our relations with these employees are good. We
intend to continue to hire additional employees in research and development, manufacturing and
administration in order to meet our operating plans. We believe that our success will greatly
depend on our ability to identify, attract and retain capable employees. The Israeli Ministry of
Labor and Welfare is authorized to make certain industry-wide collective bargaining agreements that
apply to types of industries or employees including ours (“Expansion Orders”). These agreements
affect matters such as cost of living adjustments to salaries, length of working hours and week,
recuperation, travel expenses, and pension rights. Otherwise, our employees are not represented by
a labor union or represented under a collective bargaining agreement. See “Risk Factors—We depend
upon key employees and consultants in a competitive market for skilled personnel. If we are unable
to attract and retain key personnel, it could adversely affect our ability to develop and market
our products.”
Company Background
Our principal business address is set forth below. Our executive offices and our main research
manufacturing facility are located at that address. Our telephone number is +972-4-988-9488. From
May 2001 through December 31, 2006, our company had no operations. We were originally formed as
Embassy Acquisition Corp., a Florida corporation, in November 2005 and changed our name to
Orthodontix, Inc., in April, 1992. On December 31, 2006, we acquired, through a merger with our
wholly-owned subsidiary, Protalix Acquisition Co. Ltd., all of the outstanding shares of Protalix
Ltd., in exchange for shares of our common stock. As a result, Protalix Ltd. is now our
wholly-owned subsidiary. In connection with the merger, we completed a one-for-ten reverse stock
split and on February 26, 2007, we changed our name to Protalix
26
BioTherapeutics, Inc. Unless otherwise indicated, all share numbers in this annual report on Form
10-K give effect to such reverse stock split.
Our wholly-owned subsidiary and sole operating unit, Protalix Ltd., is an Israeli corporation and
was originally incorporated in Israel as Metabogal Ltd. on December 27, 1993. During 1999,
Protalix Ltd. changed its focus from plant secondary metabolites to the expression of recombinant
therapeutic proteins in plant cells, and in April 2004 changed its name to Protalix Ltd.
ProCellExTM is our trademark. Each of the other trademarks, trade names or service
marks appearing in this Annual Report on Form 10-K belongs to its respective holder.
Available Information
Our corporate website is www.protalix.com. We make available on our website, free of charge, our
Securities and Exchange Commission, or the Commission, filings, including our Annual Report on Form
10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and any amendments to these
reports, as soon as reasonably practicable after we electronically file these documents with, or
furnish them to, the Commission. Additionally, from time to time, we provide notifications of
material news including press releases and conferences on our website. Webcasts of presentations
made by our company at certain conferences may also be available on our website, to the extent the
webcasts are available. The content of our website is not intended to be incorporated by reference
into this report or in any other report or document we file and any references to these websites
are intended to be inactive textual references only.
Our website also includes printable versions of our Code of Business Conduct and Ethics and the
charters for each of the Audit, Compensation and Nominating Committees of our Board of Directors.
Each of these documents is also available in print to any shareholder who requests a copy by
addressing a request to:
Protalix BioTherapeutics, Inc.
2 Snunit Street
Science Park
POB 455
Carmiel 20100, Israel
Attn: Mr. Yossi Maimon, Chief Financial Officer
27
Item 1A. Risk Factors
You should carefully consider the risks described below together with the other information
included in this Annual Report on Form 10-K. Our business, financial condition or results of
operations could be adversely affected by any of these risks. If any of these risks occur, the
value of our common stock could decline.
Risks Related to Our Business
We currently have no significant product revenues and will need to raise additional capital to
operate our business, which may not be available on favorable terms, or at all, and which will have
a dilutive effect on our shareholders.
To date, we have generated no significant revenues from product sales and only minimal revenues
from research and development services and other fees. For the years ended December 31, 2009,
2008 and 2007, we had net losses of $31.4 million, $22.4 million and $32.1 million,
respectively, primarily as a result of expenses incurred through a combination of research and
development activities and expenses supporting those activities, which includes share-based
compensation expense. Drug development and commercialization is very capital intensive. Until
we receive approval from the FDA and other regulatory authorities for our drug candidates, we
cannot sell our drugs and will not have product revenues, except for the upfront payment we
received in connection with the Pfizer agreement and certain regulatory-related milestone
payments in connection with the agreement which we expect to earn prior to any sales of
taliglucerase alfa. Therefore, for the foreseeable future, we will have to fund all of our
operations and capital expenditures from our cash on hand, potential regulatory-related
milestone payments under the Pfizer agreement, other licensing fees and grants and the net
proceeds of any equity or debt offerings. Over the next 12 months, we expect to spend a minimum
of approximately $30.0 million building an internal sales and marketing force for the sale of
taliglucerase alfa in Israel, expanding our manufacturing capacity and on preclinical and
clinical development for our products candidates. Based on our current plans and capital
resources, we believe that our cash and cash equivalents together with the regulatory milestones
payments we anticipate receiving from Pfizer will be sufficient to enable us to meet our planned
operating needs for the foreseeable future. However, changes may occur that could consume our
existing capital at a faster rate than projected, including, among others, changes in the
progress of our research and development efforts, the cost and timing of regulatory approvals
and the costs of protecting our intellectual property rights. We may seek additional financing
to implement and fund product development, preclinical studies and clinical trials for the drugs
in our pipeline, as well as additional drug candidates and other research and development
projects. If we are unable to secure additional financing in the future on acceptable terms, or
at all, we may be unable to commence or complete planned preclinical and clinical trials or
obtain approval of our drug candidates from the FDA and other regulatory authorities. In
addition, we may be forced to reduce or discontinue product development or product licensing,
reduce or forego sales and marketing efforts and other commercialization activities or forego
attractive business opportunities in order to improve our liquidity and to enable us to continue
operations which would have a material adverse effect on our business and results of operations.
Any additional sources of financing will likely involve the issuance of our equity securities,
which will have a dilutive effect on our shareholders.
We are not currently profitable and may never become profitable which would have a material adverse
effect on our business and results of operations and could negatively impact the value of our
common stock.
We expect to incur substantial losses for the foreseeable future and may never become
profitable. We also expect to continue to incur significant operating and capital expenditures,
and we anticipate that our expenses will increase substantially in the foreseeable future as we:
| |
• |
|
continue to undertake preclinical development and clinical trials for our current and
new drug candidates; |
| |
| |
• |
|
seek regulatory approvals for our drug candidates; |
| |
| |
• |
|
implement additional internal systems and infrastructure; |
| |
| |
• |
|
seek to license-in additional technologies to develop; and |
| |
| |
• |
|
hire additional personnel. |
We also expect to continue to experience negative cash flow for the foreseeable future as we
fund our operating losses and capital expenditures. As a result, we will need to generate
significant revenues in order to achieve and maintain profitability. We may not be able to
generate these revenues or achieve profitability in the future. Any failure to achieve
28
or maintain profitability would have a material adverse effect on our business and results of
operations and could negatively impact the value of our common stock.
We have a limited operating history which may limit the ability of investors to make an informed
investment decision.
We are a clinical stage biopharmaceutical company. To date, we have not commercialized any of
our drug candidates or received any FDA or other approval to market any drug. The successful
commercialization of our drug candidates will require us to perform a variety of functions,
including:
| |
• |
|
continuing to undertake preclinical development and clinical trials; |
| |
| |
• |
|
participating in regulatory approval processes; |
| |
| |
• |
|
formulating and manufacturing products; and |
| |
| |
• |
|
conducting sales and marketing activities. |
Our operations have been limited to organizing and staffing our company, acquiring, developing
and securing our proprietary technology and undertaking, through third parties, preclinical
trials and clinical trials of our principal drug candidates. To date, we have commenced a phase
III clinical trial in connection with only one drug candidate, taliglucerase alfa, which trial
was completed in August 2009, and we have not commenced the preclinical trial phase of
development under Good Laboratory Practice (GLP) standards for any of our other drug candidates.
These operations provide a limited basis for investors to assess our ability to commercialize
our drug candidates and whether to invest in us.
Our ProCellEx protein expression system is based on our proprietary plant cell-based expression
technology which has a limited history and any material problems with the system, which may be
unforeseen, may have a material adverse effect on our business and results of operations.
Our ProCellEx protein expression system is based on our proprietary plant cell-based expression
technology. Our business is dependent upon the successful development and approval of our
product candidates produced through our protein expression system. Our ProCellEx protein
expression system is novel and is still in the early stages of development and optimization,
and, accordingly, is subject to certain risks. Mammalian cell-based protein expression systems
have been used in connection with recombinant therapeutic protein expression for more than 20
years and are the subject of a wealth of data; in contrast, there is not a significant amount of
data generated regarding plant cell-based protein expression and, accordingly, plant cell-based
protein expression systems may be subject to unknown risks. In addition, the protein
glycosilation pattern created by our protein expression system is not identical to the natural
human glycosilation pattern and its long term effect on human patients is still unknown.
Lastly, as our protein expression system is a new technology, we cannot always rely on existing
equipment; rather, there is a need to design custom-made equipment and to generate specific
growth media for the plant cells, which may not be available at favorable prices, if at all.
Any material problems with the technology underlying our plant cell-based protein expression
system may have a material adverse effect on our business and results of operations.
We currently depend heavily on the success of taliglucerase alfa, our lead product candidate. Any
failure to commercialize taliglucerase alfa, or the experience of significant delays in doing so,
will have a material adverse effect on our business, results of operations and financial condition.
We have invested a significant portion of our efforts and financial resources in the development
of taliglucerase alfa. Our ability to generate product revenue, depends heavily on the
successful development and commercialization of taliglucerase alfa. In November 2009, we
granted to Pfizer an exclusive worldwide license to develop and commercialize taliglucerase alfa
except in Israel. We retained such rights in Israel. The successful commercialization of
taliglucerase alfa will depend on several factors, including the following:
| |
• |
|
successful completion of our ongoing studies of taliglucerase alfa; |
| |
| |
• |
|
obtaining marketing approvals from the FDA and other foreign regulatory authorities; |
| |
| |
• |
|
maintaining the cGMP compliance of our manufacturing facility or establishing
manufacturing arrangements with third parties; |
| |
| |
• |
|
the successful audit of our facilities by the FDA and other foreign regulatory
authorities;
|
29
| |
• |
|
Pfizer’s efforts under the license and supply agreement we entered into in November
2009; |
| |
| |
• |
|
our development of a successful sales and marketing organization for taliglucerase in
Israel; |
| |
| |
• |
|
the availability of reimbursement to patients from healthcare payors for our drug
products, if approved; |
| |
| |
• |
|
a continued acceptable safety and efficacy profile of our product candidates
following approval; and |
| |
| |
• |
|
other risks described in these Risk Factors. |
Any failure to commercialize taliglucerase alfa or the experience of significant delays in doing
so will have a material adverse effect on our business, results of operations and financial
condition.
Our strategy, in many cases, is to enter into collaboration agreements with third parties to
leverage our ProCellEx system to develop product candidates. If we fail to enter into these
agreements or if we or the third parties do not perform under such agreements or terminate or elect
to discontinue the collaboration, it could have a material adverse affect on our revenues.
Our strategy, in many cases, is to enter into arrangements with pharmaceutical companies to
leverage our ProCellEx system to develop additional product candidates. Under these
arrangements, we may grant to our partners rights to license and commercialize pharmaceutical
products developed under the applicable agreements. Our partners may control key decisions
relating to the development of the products and we may depend on our partners’ expertise and
dedication of sufficient resources to develop and commercialize our product candidates. The
rights of our partners limit our flexibility in considering alternatives for the
commercialization of our product candidates. To date, we have entered into a license and supply
agreement with Pfizer relating to the development and commercialization of taliglucerase alfa
and an agreement with Teva Pharmaceutical Industries Ltd., which relates to the development by
us of two proteins, and the licensing by Teva of such proteins in consideration for royalties
and milestone payments. Subsequently, two proteins were identified to be researched and
developed under the agreement but in 2009, both of the projects were terminated for commercial reasons. We may not
identify any additional proteins to be developed in a collaboration between us and Teva under
the agreement, which may have a material adverse effect on our business, results of operations
and financial condition. If we or any of our partners breach or terminate the agreements that
make up such arrangements, our partners otherwise fail to conduct their obligations under such
arrangements in a timely manner, there is a dispute about their obligations or if either party
terminates the applicable agreement or elects not to continue the arrangement, we may not enjoy
the benefits of the agreements or receive a sufficient amount of royalty or milestone payments
from them, if any.
All of our product candidates other than taliglucerase alfa are in pre clinical or research stages.
If we are unable to develop and commercialize our other product candidates, our business will be
adversely affected.
A key element of our strategy is to develop and commercialize a portfolio of new products in
addition to taliglucerase alfa. We are seeking to do so through our internal research programs and
strategic collaborations for the development of new products. Research programs to identify new
product candidates require substantial technical, financial and human resources, whether or not any
product candidates are ultimately identified. Our research programs may initially show promise in
identifying potential product candidates, yet fail to yield product candidates for clinical
development for many reasons, including the following:
| |
• |
|
the research methodology used may not be successful in identifying potential product
candidates; |
| |
| |
• |
|
competitors may develop alternatives that render our product candidates obsolete; |
| |
| |
• |
|
a product candidate may on further study be shown to have harmful side effects or
other characteristics that indicate it is unlikely to be effective or otherwise does not
meet applicable regulatory approval; |
| |
| |
• |
|
a product candidate is not capable of being produced in commercial quantities at an
acceptable cost, or at all; or |
| |
| |
• |
|
a product candidate may not be accepted by patients, the medical community or
third-party payors. |
Any failure to develop or commercialize any of our other product candidates may have a material
adverse effect on our business, results of operations and financial condition.
30
We may not obtain the necessary U.S. or worldwide regulatory approvals to commercialize our drug
candidates in a timely manner, if at all, which would have a material adverse effect on our
business and results of operations.
We will need FDA approval to commercialize our drug candidates in the United States and
approvals from foreign regulators to commercialize our drug candidates elsewhere. In order to
obtain FDA approval of any of our drug candidates, we must submit to the FDA a New Drug
Application, an NDA, or a Biologic License Application, a BLA, demonstrating that the drug
candidate is safe for humans and effective for its intended use. This demonstration requires
significant research and animal tests, which are referred to as preclinical studies, as well as
human tests, which are referred to as clinical trials. Satisfaction of the FDA’s regulatory
requirements typically takes many years, and depends upon the type, complexity and novelty of
the drug candidate and requires substantial resources for research, development and testing. In
December 2009 we completed the filing of an NDA for taliglucerase alfa for the treatment of
Gaucher disease. Our research and clinical efforts may not result in drugs that the FDA
considers safe for humans and effective for indicated uses which would have a material adverse
effect on our business and results of operations. After clinical trials are completed for any
drug candidate, if at all, the FDA has substantial discretion in the drug approval process of
the drug candidate and may require us to conduct additional clinical testing or to perform
post-marketing studies which would cause us to incur additional costs. Incurring such costs
could have a material adverse effect on our business and results of operations.
The approval process for any drug candidate may also be delayed by changes in government
regulation, future legislation or administrative action or changes in FDA policy that occur
prior to or during its regulatory review of such drug candidate. Delays in obtaining regulatory
approvals with respect to any drug candidate may:
| |
• |
|
delay commercialization of, and our ability to derive product revenues from, such
drug candidate; |
| |
| |
• |
|
delay the regulatory-related milestone payments we anticipate receiving from Pfizer; |
| |
| |
• |
|
require us to perform costly procedures with respect to such drug candidate; or |
| |
| |
• |
|
otherwise diminish any competitive advantages that we may have with respect to such
drug candidate. |
Even if we comply with all FDA requests, the FDA may ultimately reject the NDA we filed for
taliglucerase alfa or one or more of the NDAs we file in the future, if any, or we might not
obtain regulatory clearance in a timely manner for taliglucerase alfa or any of our other
product candidates. Companies in the pharmaceutical and biotechnology industries have suffered
significant setbacks in advanced or late-stage clinical trials, even after obtaining promising
earlier trial results or in preliminary findings for such clinical trials. Further, even if
favorable testing data is generated by clinical trials of drug products, the FDA may not accept
or approve an NDA filed by a pharmaceutical or biotechnology company for such drug product.
Failure to obtain FDA approval of any of our drug candidates in a timely manner, if at all, will
severely undermine our business and results of operation by reducing our potential marketable
products and our ability to generate corresponding product revenues.
The “fast track” designation for the development program of taliglucerase alfa for the treatment of
Gaucher disease may not lead to a faster development or regulatory review or approval process.
If a human medicine is intended for the treatment of a serious or life-threatening condition and
the medicine demonstrates the potential to address unmet medical needs for this condition, the
sponsor of an IND may apply for FDA “fast track” designation for a particular indication.
Marketing applications submitted by sponsors of product candidates in fast track development may
qualify for expedited FDA review under the policies and procedures offered by the FDA, but the
fast track designation does not assure any such qualification. Although the FDA has granted
fast track designation for taliglucerase alfa for the treatment of Gaucher disease, we may not
experience a faster development process, review or approval compared to applications considered
for approval under conventional FDA procedures. In addition, the FDA may withdraw the fast
track designation at any time. If the FDA withdraws the fast track designation of taliglucerase
alfa, the approval process for taliglucerase alfa may be delayed. In addition, the fast track
designation does not guarantee that taliglucerase alfa will qualify for or be able to take
advantage of the expedited review procedures and does not increase the likelihood that
taliglucerase alfa will receive regulatory approval for the treatment of Gaucher disease.
Clinical trials are very expensive, time-consuming and difficult to design and implement and may
result in unforeseen costs which may have a material adverse effect on our business and results of
operations.
Human clinical trials are very expensive and difficult to design and implement, in part because
they are subject to rigorous regulatory requirements. The clinical trial process is also
time-consuming. Other than taliglucerase alfa, our drug candidates
31
are in early stages of preclinical studies or research stages. Other, ongoing clinical trials of
taliglucerase alfa, and anticipated clinical trial of our other potential drug candidates which
have not yet been initiated, will take at least several years to complete. Preliminary and initial
results from a clinical trial do not necessarily predict final results, and failure can occur at
any stage of the trials. We may encounter problems that cause us to abandon or repeat preclinical
studies or clinical trials. Companies in the pharmaceutical and biotechnology industries have
suffered significant setbacks in advanced clinical trials, even after obtaining promising results
in earlier trials. Data obtained from tests are susceptible to varying interpretations which may
delay, limit or prevent regulatory approval. Failure or delay in the commencement or completion of
our clinical trials may be caused by several factors, including:
| • |
|
unforeseen safety issues; |
| |
| • |
|
determination of dosing issues; |
| |
| • |
|
lack of effectiveness during clinical trials; |
| |
| • |
|
slower than expected rates of patient recruitment; |
| |
| • |
|
inability to monitor patients adequately during or after treatment; |
| |
| • |
|
inability or unwillingness of medical investigators and institutional review boards to
follow our clinical protocols; and |
| |
| • |
|
lack of sufficient funding to finance the clinical trials. |
Any failure or delay in commencement or completion of any clinical trials may have a material
adverse effect on our business and results of operations. In addition, we or the FDA or other
regulatory authorities may suspend any clinical trial at any time if it appears that we are
exposing participants in the trial to unacceptable safety or health risks or if the FDA or such
other regulatory authorities, as applicable, find deficiencies in our IND submissions or the
conduct of the trial. Any suspension of clinical trial may have a material adverse effect on our
business, financial condition and results of operations.
If the results of our clinical trials do not support our claims relating to any drug candidate or
if serious side effects are identified, the completion of development of such drug candidate may be
significantly delayed or we may be forced to abandon development altogether, which will
significantly impair our ability to generate product revenues.
The results of our clinical trials with respect to any drug candidate might not support our
claims of safety or efficacy, the effects of our drug candidates may not be the desired effects
or may include undesirable side effects or the drug candidates may have other unexpected
characteristics. Further, success in preclinical testing and early clinical trials does not
ensure that later clinical trials will be successful, and the results of later clinical trials
may not replicate the results of prior clinical trials and preclinical testing. The clinical
trial process may fail to demonstrate that our drug candidates are safe for humans and effective
for indicated uses. In addition, our clinical trials may involve a specific and small patient
population. Results of early clinical trials conducted on a small patient population may not be
indicative of future results. Adverse or inconclusive results may cause us to abandon a drug
candidate and may delay development of other drug candidates. Any delay in, or termination of,
our clinical trials will delay the filing of our NDAs with the FDA and, ultimately,
significantly impair our ability to commercialize our drug candidates and generate product
revenues which would have a material adverse effect on our business, financial condition and
results of operations.
We may find it difficult to enroll patients in our clinical trials, which could cause significant
delays in the completion of such trials or may cause us to abandon one or more clinical trials.
Most of the diseases or disorders that our product candidates are intended to treat are
relatively rare and we expect only a subset of the patients with these diseases to be eligible
for our clinical trials. Given that each of our product candidates other than taliglucerase
alfa is in the early stages of preclinical or research stages, we may not be able to initiate
clinical trials for each or all of our product candidates if we are unable to locate a
sufficient number of eligible subjects to participate in the clinical trials required by the FDA
and/or other foreign regulatory authorities. The requirements of our clinical trials generally
mandate that a patient cannot be involved in another clinical trial for the same indication. We
are aware that our competitors have ongoing clinical trials for products that are competitive
with our product candidates and subjects who would otherwise be eligible for our clinical trials
may be involved in such testing, rendering them unavailable for testing of our product
candidates. Our inability to enroll a sufficient number of patients for any of our current or
future clinical trials would result in significant delays or may require us to abandon one or
more clinical trials altogether, which would have a material adverse effect on our business.
32
If physicians, patients, third party payors and others in the medical community do not accept and
use our drugs, our ability to generate revenue from sales of our products under development will be
materially impaired.
Even if the FDA or other foreign regulatory authorities approve any of our drug candidates for
commercialization, physicians and patients, and other healthcare providers, may not accept and
use such candidates. Future acceptance and use of our products will depend upon a number of
factors including:
| |
• |
|
perceptions by physicians, patients, third party payors and others in the medical
community, about the safety and effectiveness of our drug candidates; |
| |
| |
• |
|
the willingness of the target patient population to try new therapies and of
physicians to prescribe these therapies; |
| |
| |
• |
|
the prevalence and severity of any side effects, including any limitations or
warnings contained in our products’ approved labeling; |
| |
| |
• |
|
pharmacological benefit of our products relative to competing products and products
under development; |
| |
| |
• |
|
the efficacy and potential advantages relative to competing products and products
under development; |
| |
| |
• |
|
relative convenience and ease of administration; |
| |
| |
• |
|
effectiveness of education, marketing and distribution efforts by us and our
licensees and distributors, if any; |
| |
| |
• |
|
publicity concerning our products or competing products and treatments; |
| |
| |
• |
|
reimbursement of our products by third party payors; and |
| |
| |
• |
|
the price for our products and competing products. |
Because we expect sales of our current drug candidates, if approved, to generate substantially
all of our product revenues for the foreseeable future, the failure of any of these drugs to
find market acceptance would have a material adverse effect on our business and revenues from
sales of our products would be materially impaired.
Because our clinical trials depend upon third-party researchers, the results of our clinical trials
and such research activities are subject to delays and other risks which are, to a certain extent,
beyond our control, which could impair our clinical development programs and our competitive
position.
We depend upon independent investigators and collaborators, such as universities and medical
institutions, to conduct our preclinical and clinical trials. These collaborators are not our
employees, and we cannot control the amount or timing of resources that they devote to our
clinical development programs. The investigators may not assign as great a priority to our
clinical development programs or pursue them as diligently as we would if we were undertaking
such programs directly. If outside collaborators fail to devote sufficient time and resources
to our clinical development programs, or if their performance is substandard, the approval of
our FDA and other applications, if any, and our introduction of new drugs, if any, may be
delayed which could impair our clinical development programs and would have a material adverse
effect on our business and results of operations. The collaborators may also have relationships
with other commercial entities, some of whom may compete with us. If our collaborators also
assist our competitors, our competitive position could be harmed.
The manufacture of our products is an exacting and complex process, and if we or one of our
materials suppliers encounter problems manufacturing our products, it will have a material adverse
effect on our business and results of operations.
The FDA and foreign regulators require manufacturers to register manufacturing facilities. The
FDA and foreign regulators also inspect these facilities to confirm compliance with cGMP or
similar requirements that the FDA or foreign regulators establish. We or our materials
suppliers may face manufacturing or quality control problems causing product production and
shipment delays or a situation where we or the supplier may not be able to maintain compliance
with the FDA’s cGMP requirements, or those of foreign regulators, necessary to continue
manufacturing our drug candidates. Any failure to comply with cGMP requirements or other FDA or
foreign regulatory requirements could adversely affect our clinical research activities and our
ability to market and develop our products. Our current facility has not been audited by the
FDA or other foreign regulatory authorities but must be audited in connection with the NDA we
submitted for
33
taliglucerase alfa. There can be no assurance that we will be able to comply with FDA or
foreign regulatory manufacturing requirements for our current facility or any future facility
that we may establish, which would have a material adverse effect on our business.
We rely on third parties for final processing of taliglucerase alfa, which exposes us to a number
of risks that may delay development, regulatory approval and commercialization of our product
candidates or result in higher product costs.
We have no experience in the final filling and freeze drying steps of the drug manufacturing
process. According to our license and supply agreement with Pfizer, Pfizer will be responsible
for the fill and finish activities for taliglucerase alfa. Upon our receipt of marketing
approval from the FDA or other regulatory authorities for taliglucerase alfa, if at all, we will
rely primarily on Pfizer and/or other third-party contractors to perform the final manufacturing
steps for taliglucerase alfa on a commercial scale. We may be unable to identify manufacturers
and replacement manufacturers on acceptable terms or at all because the number of potential
manufacturers is limited and the FDA and other regulatory authorities, as applicable, must
approve any replacement manufacturer, including us, and we or any such third party manufacturer
might be unable to formulate and manufacture our drug products in the volume and of the quality
required to meet our clinical and commercial needs. If we engage any contract manufacturers,
such manufacturers may not perform as agreed or may not remain in the contract manufacturing
business for the time required to supply our clinical or commercial needs. Each of these risks
could delay our clinical trials, the approval, if any, of taliglucerase alfa and our other
potential drug candidates by the FDA or other regulatory authorities, or the commercialization
of taliglucerase alfa and our other drug candidates or could result in higher product costs or
otherwise deprive us of potential product revenues.
We have no experience selling, marketing or distributing products and no internal capability to do
so.
We currently have no sales, marketing or distribution capabilities and no experience in building a
sales force and distribution capabilities. To be able to commercialize taliglucerase alfa upon
approval, if at all, in Israel, and to commercialize any of our other product candidates, we must
either develop internal sales, marketing and distribution capabilities, which will be expensive and
time consuming, or make arrangements with third parties to perform these services. In November
2009, we granted to Pfizer an exclusive, worldwide right to develop and commercialize taliglucerase
alfa, but retained such rights in Israel. If we decide to market any of our products directly, we
must commit significant financial and managerial resources to develop a marketing and sales force
with technical expertise and with supporting distribution capabilities. Factors that may inhibit
our efforts to commercialize our products directly and without strategic partners include:
| |
• |
|
our inability to recruit and retain adequate numbers of effective sales and
marketing personnel; |
| |
| |
• |
|
the inability of sales personnel to obtain access to or persuade adequate numbers of
physicians to prescribe our products; |
| |
| |
• |
|
the lack of complementary products to be offered by sales personnel, which may put us
at a competitive disadvantage relative to companies with more extensive product
lines; and |
| |
| |
• |
|
unforeseen costs and expenses associated with creating and sustaining an independent
sales and marketing organization. |
We may not be successful in recruiting the sales and marketing personnel necessary to sell any of
our products upon approval, if at all, and even if we do build a sales force, it may not be
successful in marketing our products, which would have a material adverse effect on our business,
financial condition and results of operations.
If the market opportunities for our current product candidates are smaller than we believe they
are, our revenues may be adversely affected and our business may suffer.
The primary focus of our current clinical pipeline is on relatively rare disorders with small
patient populations, in particular Gaucher disease and Fabry disease. Currently, most reported
estimates of the prevalence of these diseases are based on studies of small subsets of the
population of specific geographic areas, which are then extrapolated to estimate the prevalence
of the diseases in the broader world population. As new studies are performed, the estimated
prevalence of these diseases may change. There can be no assurance that the prevalence of
Gaucher disease or Fabry disease in the study populations, particularly in these newer studies,
accurately reflect the prevalence of these diseases in the broader world population. If the
market opportunities for our current product candidates are smaller than we believe they are,
our revenues may be adversely affected and our business may suffer.
34
We may enter into distribution arrangements and marketing alliances for certain products and any
failure to successfully identify and implement these arrangements on favorable terms, if at all,
may impair our ability to commercialize our product candidates.
While we intend to build a sales force to market taliglucerase alfa in Israel and other product
candidates worldwide, we do not anticipate having the resources in the foreseeable future to
develop global sales and marketing capabilities for all of the products we develop, if any. We
may pursue arrangements regarding the sales and marketing and distribution of one or more of our
product candidates, such as our license and supply agreement with Pfizer, and our future
revenues may depend, in part, on our ability to enter into and maintain arrangements with other
companies having sales, marketing and distribution capabilities and the ability of such
companies to successfully market and sell any such products. Any failure to enter into such
arrangements and marketing alliances on favorable terms, if at all, could delay or impair our
ability to commercialize our product candidates and could increase our costs of
commercialization. Any use of distribution arrangements and marketing alliances to
commercialize our product candidates will subject us to a number of risks, including the
following:
| |
• |
|
we may be required to relinquish important rights to our products or product
candidates; |
| |
| |
• |
|
we may not be able to control the amount and timing of resources that our
distributors or collaborators may devote to the commercialization of our product
candidates; |
| |
| |
• |
|
our distributors or collaborators may experience financial difficulties; |
| |
| |
• |
|
our distributors or collaborators may not devote sufficient time to the marketing and
sales of our products; and |
| |
| |
• |
|
business combinations or significant changes in a collaborator’s business strategy
may adversely affect a collaborator’s willingness or ability to complete its obligations
under any arrangement. |
We may need to enter into additional co-promotion arrangements with third parties where our own
sales force is neither well situated nor large enough to achieve maximum penetration in the
market. We may not be successful in entering into any co-promotion arrangements, and the terms
of any co-promotion arrangements we enter into may not be favorable to us.
Developments by competitors may render our products or technologies obsolete or non-competitive
which would have a material adverse effect on our business and results of operations.
We compete against fully integrated pharmaceutical companies and smaller companies that are
collaborating with larger pharmaceutical companies, academic institutions, government agencies
and other public and private research organizations. Our drug candidates will have to compete
with existing therapies and therapies under development by our competitors. In addition, our
commercial opportunities may be reduced or eliminated if our competitors develop and market
products that are less expensive, more effective or safer than our drug products. Other
companies have drug candidates in various stages of preclinical or clinical development to treat
diseases for which we are also seeking to develop drug products. Some of these potential
competing drugs are further advanced in development than our drug candidates and may be
commercialized earlier. Even if we are successful in developing effective drugs, our products
may not compete successfully with products produced by our competitors.
We specifically face competition from companies with approved treatments of Gaucher disease,
including Genzyme and to a much lesser extent, Actelion. In addition, we are aware of other
early stage, experimental, small molecule, oral drugs which are being developed for the
treatment of Gaucher disease by Amicus Therapeutics, a trial which Amicus Therapeutics reports
has been suspended, and Genzyme. Shire plc is currently developing a gene-activated enzyme
expressed in human cancer cells to treat Gaucher disease. Shire has submitted marketing
applications in the United States, the European Union and Canada for its enzyme replacement
therapy treatment for Gaucher disease. According to public reports by Shire, its application
is being reviewed by the FDA under Priority Review with a PDUFA date of February 28, 2010 and
the EMEA’s Committee for Medicinal Products for Human Use has granted accelerated review for
Shire’s Marketing Authorization Application. We also face competition from companies with
approved treatments of Fabry disease, including Genzyme and Shire, and we are aware of other
early stage drugs which are being developed for the treatment of Fabry disease, including a
drug being developed by Amicus Therapeutics.
We also face competition from companies that are developing other platforms for the expression of
recombinant therapeutic pharmaceuticals. We are aware of companies that are developing alternative
technologies to develop and produce therapeutic proteins in anticipation of the expiration of
certain patent claims covering marketed proteins. Competitors developing
35
alternative expression technologies include Crucell N.V., Shire and GlycoFi Inc. (which was
acquired by Merck). Other companies are developing alternate plant-based technologies, include
Biolex, Inc., Chlorogen, Inc., Greenovation Biotech GmbH and Dow Agroscience.
Several biogeneric companies are pursuing the opportunity to develop and commercialize follow-on
versions of other currently marketed biologic products, including growth factors, hormones,
enzymes, cytokines and monoclonal antibodies, which are areas that interest us. These companies
include, among others, Novartis AG/Sandoz Pharmaceuticals, BioGeneriX AG, Stada Arzneimittel AG,
BioPartners GmbH and Teva.
Most of our competitors, either alone or together with their collaborative partners, operate larger
research and development programs, staff and facilities and have substantially greater financial
resources than we do, as well as significantly greater experience in:
| |
• |
|
developing drugs; |
| |
| |
• |
|
undertaking preclinical testing and human clinical trials; |
| |
| |
• |
|
obtaining FDA and other regulatory approvals of drugs; |
| |
| |
• |
|
formulating and manufacturing drugs; and |
| |
| |
• |
|
launching, marketing and selling drugs. |
These organizations also compete with us to attract qualified personnel, acquisitions and joint
ventures candidates and for other collaborations. Activities of our competitors may impose
unanticipated costs on our business which would have a material adverse effect on our business,
financial condition and results of operations.
If we fail to adequately protect or enforce our intellectual property rights or secure rights to
third party patents, the value of our intellectual property rights would diminish and our business,
competitive position and results of operations would suffer.
As of December 31, 2009, we had 84 pending patent applications and held licensed rights to
five pending patent applications. However, the filing of a patent application does not mean that
we will be issued a patent, or that any patent eventually issued will be as broad as requested
in the patent application or sufficient to protect our technology. Any modification required to
a current patent application may delay the approval of such patent application which would have
a material adverse effect on our business and results of operations. In addition, there are a
number of factors that could cause our patents, if granted, to become invalid or unenforceable
or that could cause our patent applications to not be granted, including known or unknown prior
art, deficiencies in the patent application or the lack of originality of the technology.
Our competitive position and future revenues will depend in part on our ability and the ability
of our licensors and collaborators to obtain and maintain patent protection for our products,
methods, processes and other technologies, to preserve our trade secrets, to prevent third
parties from infringing on our proprietary rights and to operate without infringing the
proprietary rights of third parties. We have filed U.S. and international patent applications
for process patents, as well as composition of matter patents, for taliglucerase alfa. However,
we cannot predict:
| |
• |
|
the degree and range of protection any patents will afford us against competitors and
those who infringe upon our patents, including whether third parties will find ways to
invalidate or otherwise circumvent our licensed patents; |
| |
| |
• |
|
if and when patents will issue; |
| |
| |
• |
|
whether or not others will obtain patents claiming aspects similar to those covered
by our licensed patents and patent applications; or |
| |
| |
• |
|
whether we will need to initiate litigation or administrative proceedings, which may
be costly, and whether we win or lose. |
As of December 31, 2009, we hold, or have license rights to, 22 patents. If patent rights
covering our products are not sufficiently broad, they may not provide us with sufficient
proprietary protection or competitive advantages against competitors with similar products and
technologies. Furthermore, if the U.S. Patent and Trademark Office or foreign
36
patent offices issue patents to us or our licensors, others may challenge the patents or
circumvent the patents, or the patent office or the courts may invalidate the patents. Thus,
any patents we own or license from or to third parties may not provide any protection against
our competitors and those who infringe upon our patents.
Furthermore, the life of our patents is limited. The patents we hold relating to our ProCellEx
protein expression system will expire in 2016. If patents issue from other currently pending
patent applications, those patents will expire between 2023 and 2028.
We rely on confidentiality agreements that could be breached and may be difficult to enforce which
could have a material adverse effect on our business and competitive position.
Our policy is to enter agreements relating to the non-disclosure of confidential information
with third parties, including our contractors, consultants, advisors and research collaborators,
as well as agreements that purport to require the disclosure and assignment to us of the rights
to the ideas, developments, discoveries and inventions of our employees and consultants while we
employ them. However, these agreements can be difficult and costly to enforce. Moreover, to
the extent that our contractors, consultants, advisors and research collaborators apply or
independently develop intellectual property in connection with any of our projects, disputes may
arise as to the proprietary rights to the intellectual property. If a dispute arises, a court
may determine that the right belongs to a third party, and enforcement of our rights can be
costly and unpredictable. In addition, we rely on trade secrets and proprietary know-how that
we seek to protect in part by confidentiality agreements with our employees, contractors,
consultants, advisors or others. Despite the protective measures we employ, we still face the
risk that:
| |
• |
|
these agreements may be breached; |
| |
| |
• |
|
these agreements may not provide adequate remedies for the applicable type of breach;
or |
| |
| |
• |
|
our trade secrets or proprietary know-how will otherwise become known. |
Any breach of our confidentiality agreements or our failure to effectively enforce such
agreements would have a material adverse effect on our business and competitive position.
If we infringe the rights of third parties we could be prevented from selling products, forced to
pay damages and required to defend against litigation which could result in substantial costs and
may have a material adverse effect on our business and results of operations.
We have not received to date any claims of infringement by any third parties. However, as our
drug candidates progress into clinical trials and commercialization, if at all, our public
profile and that of our drug candidates may be raised and generate such claims. Defending
against such claims, and occurrence of a judgment adverse to us, could result in unanticipated
costs and may have a material adverse effect on our business and competitive position. If our
products, methods, processes and other technologies infringe the proprietary rights of other
parties, we may incur substantial costs and we may have to:
| |
• |
|
obtain licenses, which may not be available on commercially reasonable terms, if at
all; |
| |
| |
• |
|
redesign our products or processes to avoid infringement; |
| |
| |
• |
|
stop using the subject matter claimed in the patents held by others, which could
cause us to lose the use of one or more of our drug candidates; |
| |
| |
• |
|
defend litigation or administrative proceedings that may be costly whether we win or
lose, and which could result in a substantial diversion of management resources; or |
| |
| |
• |
|
pay damages. |
Any costs incurred in connection with such events or the inability to sell our products may have
a material adverse effect on our business, financial condition and results of operations.
37
If we cannot meet requirements under our license agreements, we could lose the rights to our
products, which could have a material adverse effect on our business.
We depend on licensing agreements with third parties to maintain the intellectual property
rights to certain of our products under development. Presently, we have licensed rights from
the Yeda Research and Development Company Limited, the technology transfer arm of the Weizman
Institute of Science, which allow us to use their technology and discoveries for the
development, production and sale of enzymatically active mutations of GCD and derivatives
thereof for the treatment of Gaucher disease. In addition, pursuant to our agreement with the
Yissum Research and Development Company, or Yissum, the technology transfer arm of the Hebrew
University of Jerusalem, Israel, and the Boyce Thompson Institute for Plant Research, at Cornell
University, we have received an exclusive worldwide right and license to certain technology,
including patents and additional patent applications relating to acetylcholinesterase (AChE),
for all therapeutic and prophylactic indications as well as an exclusive license not limited to
such indications with respect to certain of these patents and patent applications. Under the
agreement with Yissum, we intend to develop a proprietary plant cell-based acetylcholinestrase
(AChE) and its molecular variants for the use in several therapeutic and prophylactic
indications, including a biodefense program. Our license agreements require us to make payments
and satisfy performance obligations in order to maintain our rights under these agreements. All
of these agreements last either throughout the life of the patents that are the subject of the
agreements, or with respect to other licensed technology, for a number of years after the first
commercial sale of the relevant product.
In addition, we are responsible for the cost of filing and prosecuting certain patent
applications and maintaining certain issued patents licensed to us. If we do not meet our
obligations under our license agreements in a timely manner, we could lose the rights to our
proprietary technology which could have a material adverse effect on our business.
If we in-license drug candidates, we may delay or otherwise adversely affect the development of our
existing drug candidates, which may negatively impact our business, results of operations and
financial condition.
In addition to our own internally developed drug candidates, we proactively seek opportunities
to in-license and advance other drug candidates that are strategic and have value-creating
potential to take advantage of our development know-how and technology. If we in-license any
additional drug candidates, our capital requirements may increase significantly. In addition,
in-licensing additional drug candidates may place a strain on the time of our existing
personnel, which may delay or otherwise adversely affect the development of our existing drug
candidates or cause us to re-prioritize our drug pipeline if we do not have the necessary
capital resources to develop all of our drug candidates, which may delay the development of our
drug candidates and negatively impact our business, results of operations and financial
condition.
If we are unable to successfully manage our growth, there could be a material adverse impact on our
business, results of operations and financial condition.
We have grown rapidly and expect to continue to grow. We expect to hire more employees,
particularly in the areas of drug development, manufacturing, regulatory affairs and sales and
marketing, and increase our facilities and corporate infrastructure, further increasing the size
of our organization and related expenses. To manage our anticipated future growth, we must
continue to implement and improve our managerial, operational and financial systems, expand our
facilities and continue to recruit and train additional qualified personnel. Due to our limited
resources, we may not be able to effectively manage the expansion of our operations or recruit
and train additional qualified personnel. The expansion of our operations may lead to
significant costs and may divert our management and business development resources. Any
inability on the part of our management to manage growth could delay the execution of our
business plans or disrupt our operations. If we are unable to manage our growth effectively, we
may not use our resources in an efficient manner, which may delay the development of our drug
candidates and negatively impact our business, results of operations and financial condition.
If we acquire companies, products or technologies, we may face integration risks and costs
associated with those acquisitions that could negatively impact our business, results from
operations and financial condition.
If we are presented with appropriate opportunities, we may acquire or make investments in
complementary companies, products or technologies. We may not realize the anticipated benefit
of any acquisition or investment. If we acquire companies or technologies, we will face risks,
uncertainties and disruptions associated with the integration process, including difficulties in
the integration of the operations of an acquired company, integration of acquired technology
with our products, diversion of our management’s attention from other business concerns, the
potential loss of key employees or customers of the acquired business and impairment charges if
future acquisitions are not as successful as we originally anticipate. In addition, our
operating results may suffer because of acquisition-related costs or amortization expenses or
charges relating to acquired intangible assets. Any failure to successfully integrate other
companies, products or
38
technologies that we may acquire may have a material adverse effect on our business and results
of operations. Furthermore, we may have to incur debt or issue equity securities to pay for any
additional future acquisitions or investments, the issuance of which could be dilutive to our
existing shareholders.
We depend upon key employees and consultants in a competitive market for skilled personnel. If we
are unable to attract and retain key personnel, it could adversely affect our ability to develop
and market our products.
We are highly dependent upon the principal members of our management team, especially our
President and Chief Executive Officer, Dr. David Aviezer, Ph.D., as well as our directors,
including Eli Hurvitz, the Chairman of our Board of Directors, our scientific advisory board
members, consultants and collaborating scientists. Many of these people have been involved with
us for many years and have played integral roles in our progress, and we believe that they will
continue to provide value to us. A loss of any of these personnel may have a material adverse
effect on aspects of our business and clinical development and regulatory programs. We have
employment agreements with Dr. Aviezer and four other officers that may be terminated by us or
the applicable officer at any time with varying notice periods of 60 to 90 days. Although these
employment agreements generally include non-competition covenants and provide for severance
payments that are contingent upon the applicable employee’s refraining from competition with us,
the applicable noncompetition provisions can be difficult and costly to monitor and enforce.
The loss of any of these persons’ services would adversely affect our ability to develop and
market our products and obtain necessary regulatory approvals. Further, we do not maintain
key-man life insurance.
We also depend in part on the continued service of our key scientific personnel and our ability
to identify, hire and retain additional personnel, including marketing and sales staff. We
experience intense competition for qualified personnel, and the existence of non-competition
agreements between prospective employees and their former employers may prevent us from hiring
those individuals or subject us to suit from their former employers. While we attempt to
provide competitive compensation packages to attract and retain key personnel, many of our
competitors are likely to have greater resources and more experience than we have, making it
difficult for us to compete successfully for key personnel.
Our collaborations with outside scientists and consultants may be subject to restriction and
change.
We work with chemists, biologists and other scientists at academic and other institutions, and
consultants who assist us in our research, development, regulatory and commercial efforts,
including the members of our scientific advisory board. These scientists and consultants have
provided, and we expect that they will continue to provide, valuable advice on our programs.
These scientists and consultants are not our employees, may have other commitments that would
limit their future availability to us and typically will not enter into non-compete agreements
with us. If a conflict of interest arises between their work for us and their work for another
entity, we may lose their services. In addition, we will be unable to prevent them from
establishing competing businesses or developing competing products. For example, if a key
scientist acting as a principal investigator in any of our clinical trials identifies a
potential product or compound that is more scientifically interesting to his or her professional
interests, his or her availability to remain involved in our clinical trials could be restricted
or eliminated.
Under current U.S. and Israeli law, we may not be able to enforce employees’ covenants not to
compete and therefore may be unable to prevent our competitors from benefiting from the expertise
of some of our former employees.
We have entered into non-competition agreements with all of our employees. These agreements
prohibit our employees, if they cease working for us, from competing directly with us or working
for our competitors for a limited period. Under current U.S. and Israeli law, we may be unable
to enforce these agreements against most of our employees and it may be difficult for us to
restrict our competitors from gaining the expertise our former employees gained while working
for us. If we cannot enforce our employees’ non-compete agreements, we may be unable to prevent
our competitors from benefiting from the expertise of our former employees.
If product liability claims are brought against us, it may result in reduced demand for our
products or damages that exceed our insurance coverage.
The clinical testing, marketing and use of our products exposes us to product liability claims
in the event that the use or misuse of those products causes injury, disease or results in
adverse effects. Use of our products in clinical trials, as well as commercial sale, could
result in product liability claims. We presently carry clinical trial liability insurance with
coverages of up to $5.0 million per occurrence and $5.0 million in the aggregate, an amount we
consider reasonable and customary. However, this insurance coverage includes various
deductibles, limitations and exclusions from coverage, and in any event might not fully cover
any potential claims. We may need to obtain additional clinical trial liability coverage prior
to initiating additional clinical trials. We expect to obtain product liability insurance
coverage before
39
commercialization of our proposed products; however, such insurance is expensive and insurance
companies may not issue this type of insurance when we need it. We may not be able to obtain
adequate insurance in the future at an acceptable cost. Any product liability claim, even one
that was not in excess of our insurance coverage or one that is meritless and/or unsuccessful,
could adversely affect our cash available for other purposes, such as research and development,
which could have a material adverse effect on our business, financial condition and results of
operations. Product liability claims may result in reduced demand for our products, if
approved, which would have a material adverse effect on our business, financial condition and
results of operations. In addition, the existence of a product liability claim could affect the
market price of our common stock.
Reimbursement may not be available for our product candidates, which could diminish our sales or
affect our ability to sell our products profitably.
Market acceptance and sales of our product candidates will depend on worldwide reimbursement
policies. Government authorities and third-party payors, such as private health insurers and
health maintenance organizations, decide which drugs they will pay for and establish
reimbursement levels. We cannot be sure that reimbursement will be available for any of our
product candidates, if approved for marketing and sale. Obtaining reimbursement approval for an
approved product from every government or other third party payor is a time consuming and costly
process that could require us to provide supporting scientific, clinical and cost-effectiveness
data for the use of our products, if and when approved, to every payor. We may not be able to
provide data sufficient to gain acceptance with respect to reimbursement or we might need to
conduct post-marketing studies in order to demonstrate the cost-effectiveness of any approved
products, if any, to such payors’ satisfaction. Such studies might require us to commit a
significant amount of management time and financial and other resources. Even if a payor
determines that an approved product is eligible for reimbursement, the payor may impose coverage
limitations that preclude payment for some uses that are approved by the FDA or other regulatory
authorities. In addition, there is a risk that full reimbursement may not be available for high
priced products. Moreover, eligibility for coverage does not imply that any approved product
will be reimbursed in all cases or at a rate that allows us to make a profit or even cover our
costs. Also, we cannot be sure that reimbursement amounts will not reduce the demand for, or
the price of, our product candidates. Except with respect to taliglucerase alfa, we have not
commenced efforts to have our product candidates reimbursed by government or third-party payors.
If reimbursement is not available or is available only to limited levels, the sales of our
products, if approved may be diminished or we may not be able to sell such products profitably.
Reforms in the healthcare industry and the uncertainty associated with pharmaceutical pricing,
reimbursement and related matters could adversely affect the marketing, pricing and demand for our
products, if approved.
Increasing expenditures for healthcare have been the subject of considerable public attention in
the United States. Both private and government entities are seeking ways to reduce or contain
healthcare costs. Numerous proposals that would effect changes in the U.S. healthcare system
have been introduced or proposed in the U.S. Congress and in some state legislatures within the
United States, including reductions in the pricing of prescription products and changes in the
levels at which consumers and healthcare providers are reimbursed for purchases of
pharmaceutical products. For example, the Medicare Prescription Drug Improvement, and
Modernization Act of 2003 and the proposed rules thereunder impose new requirements for the
distribution and pricing of prescription drugs that began in 2006, which could reduce
reimbursement of prescription drugs for healthcare providers and insurers. Although we cannot
predict the full effect on our business of the implementation of this legislation, it is
possible that the new Medicare prescription drug benefit, which will be managed by private
health insurers and other managed care organizations, will result in additional government
reimbursement for prescription drugs, which may make some prescription drugs more affordable but
may further exacerbate industry-wide pressure to reduce prescription drug prices. We believe
that legislation that reduces reimbursement for our product candidates could adversely impact
how much or under what circumstances healthcare providers will prescribe or administer our
products, if approved. This could materially and adversely impact our business by reducing our
ability to generate revenue, raise capital, obtain additional collaborators and market our
products, if approved. In addition, we believe the increasing emphasis on managed care in the
United States has and will continue to put pressure on the price and usage of pharmaceutical
products, which may adversely impact product sales, upon approval, if at all.
Governments outside the United States tend to impose strict price controls and reimbursement
approval policies, which may adversely affect our prospects for generating revenue.
In some countries, particularly European Union countries, the pricing of prescription
pharmaceuticals is subject to governmental control. In these countries, pricing negotiations
with governmental authorities can take considerable time (six to 12 months or longer) after the
receipt of marketing approval for a product. To obtain reimbursement or pricing
40
approval in some countries with respect to any product candidate that achieves regulatory
approval, we may be required to conduct a clinical trial that compares the cost-effectiveness of
our product candidate to other available therapies. If reimbursement of our products upon
approval, if at all, is unavailable or limited in scope or amount, or if pricing is set at
unsatisfactory levels, our prospects for generating revenue, if any, could be adversely affected
which would have a material adverse effect on our business and results of operations. Further,
if we achieve regulatory approval of any product, we must successfully negotiate product pricing
for such product in individual countries. As a result, the pricing of our products, if
approved, in different countries may vary widely, thus creating the potential for third-party
trade in our products in an attempt to exploit price differences between countries. This
third-party trade of our products could undermine our sales in markets with higher prices.
Risks Relating to Our Operations in Israel
Potential political, economic and military instability in the State of Israel, where the majority
of our senior management and our research and development facilities are located, may adversely
affect our results of operations.
Our executive office and operations are located in the State of Israel. Accordingly, political,
economic and military conditions in Israel directly affect our business. Since the State of
Israel was established in 1948, a number of armed conflicts have occurred between Israel and its
Arab neighbors. Any hostilities involving Israel or the interruption or curtailment of trade
between Israel and its present trading partners, or a significant downturn in the economic or
financial condition of Israel, could affect adversely our operations. Since October 2000 there
have been increasing occurrences of terrorist violence. Ongoing and revived hostilities or
other Israeli political or economic factors could harm our operations and product development
and cause our revenues to decrease. Furthermore, several countries, principally those in the
Middle East, still restrict business with Israel and Israeli companies. These restrictive laws
and policies may limit seriously our ability to sell our products in these countries.
Although Israel has entered into various agreements with Egypt, Jordan and the Palestinian
Authority, there have been times since October 2000 when Israel has experienced an increase in
unrest and terrorist activity. The establishment in 2006 of a government in the Palestinian
Authority by representatives of the Hamas militant group has created additional unrest and
uncertainty in the region. In mid-2006, there was a war between Israel and the Hezbollah in
Lebanon, resulting in thousands of rockets being fired from Lebanon up to 50 miles into Israel.
Our current facilities are located in northern Israel, are in range of rockets that were fired
from Lebanon into Israel during the war and suffered minimal damages during one of the rocket
attacks. If our facilities are damaged as a result of hostile action, our operations may be
materially adversely affected.
Our operations may be disrupted by the obligations of our personnel to perform military service
which could have a material adverse effect on our business.
Many of our male employees in Israel, including members of senior management, are obligated to
perform up to one month (in some cases more) of annual military reserve duty until they reach
the age of 45 and, in the event of a military conflict, could be called to active duty. Our
operations could be disrupted by the absence of a significant number of our employees related to
military service or the absence for extended periods of military service of one or more of our
key employees. A disruption could have a material adverse effect on our business.
Because a certain portion of our expenses is incurred in New Israeli Shekels, or NIS, our results
of operations may be seriously harmed by currency fluctuations and inflation.
We report our financial statements in U.S. dollars, our functional currency, but we pay a
meaningful portion of our expenses in NIS. As a result, we are exposed to risk to the extent
that the inflation rate in Israel exceeds the rate of devaluation of the NIS in relation to the
U.S. dollar or if the timing of these devaluations lags behind inflation in Israel. In that
event, the U.S. dollar cost of our operations in Israel will increase and our U.S.
dollar-measured results of operations will be adversely affected. To the extent that the value
of the NIS increases against the dollar, our expenses on a dollar cost basis increase. Our
operations also could be adversely affected if we are unable to guard against currency
fluctuations in the future. To date, we have not engaged in hedging transactions. In the
future, we may enter into currency hedging transactions to decrease the risk of financial
exposure from fluctuations in the exchange rate of the U.S. dollar against the NIS. These
measures, however, may not adequately protect us from material adverse effects.
41
The tax benefits available to us require that we meet several conditions and may be terminated or
reduced in the future, which would increase our taxes and would have a material adverse effect on
our business and results of operations.
We are able to take advantage of tax exemptions and reductions resulting from the “Approved
Enterprise” status of our facilities in Israel. To remain eligible for these tax benefits, we
must continue to meet certain conditions, including making specified investments in property and
equipment, and financing at least 30% of such investments with share capital. If we fail to
meet these conditions in the future, the tax benefits would be canceled and we may be required
to refund any tax benefits we already have enjoyed. These tax benefits are subject to
investment policy by the Investment Center and may not be continued in the future at their
current levels or at any level. In recent years the Israeli government has reduced the benefits
available and has indicated that it may further reduce or eliminate some of these benefits in
the future. The termination or reduction of these tax benefits or our inability to qualify for
additional “Approved Enterprise” approvals may increase our tax expenses in the future, which
would reduce our expected profits and adversely affect our business and results of operations.
Additionally, if we increase our activities outside of Israel, for example, by future
acquisitions, such increased activities generally may not be eligible for inclusion in Israeli
tax benefit programs.
The Israeli government grants we have received for certain research and development expenditures
restrict our ability to manufacture products and transfer technologies outside of Israel and
require us to satisfy specified conditions. If we fail to satisfy these conditions, we may be
required to refund grants previously received together with interest and penalties which could have
a material adverse effect on our business and results of operations.
Our research and development efforts have been financed, in part, through grants that we have
received from the OCS. We, therefore, must comply with the requirements of the Israeli Law for
the Encouragement of Industrial Research and Development, 1984, and related regulations, or the
Research Law.
Under the Research Law, the discretionary approval of an OCS committee is required for any
transfer of technology developed with OCS funding. OCS approval is not required for the export
of any products resulting from the research or development, or for the licensing of the
technology in the ordinary course of business. We may not receive the required approvals for
any proposed transfer. Such approvals, if granted, may be subject to the following additional
restrictions:
| |
• |
|
we may be required to pay the OCS a portion of the consideration we receive upon any
sale of such technology to an entity that is not Israeli. The scope of the support
received, the royalties that were paid by us, the amount of time that elapses between
the date on which the know-how is transferred and the date on which the grants were
received, as well as the sale price, will be taken into account in order to calculate
the amount of the payment; and |
| |
| |
• |
|
the transfer of manufacturing rights could be conditioned upon an increase in the
royalty rate and payment of increased aggregate royalties (up to 300% of the amount of
the grant plus interest, depending on the percentage of the manufacturing that is
foreign). |
These restrictions may impair our ability to sell our technology assets or to outsource
manufacturing outside of Israel. We have no current intention to manufacture or transfer
technologies out of Israel. The restrictions will continue to apply even after we have repaid
the full amount of royalties payable for the grants. If we fail to satisfy these conditions, we
may be required to refund grants previously received together with interest and penalties which
could have a material adverse effect on our business and results of operations.
Investors may have difficulties enforcing a U.S. judgment, including judgments based upon the civil
liability provisions of the U.S. federal securities laws against us, our executive officers and
most of our directors or asserting U.S. securities laws claims in Israel.
Most of our directors and officers are not residents of the United States and most of their
assets and our assets are located outside the United States. Service of process upon our
non-U.S. resident directors and officers and enforcement of judgments obtained in the United
States against us, some of our directors and executive officers may be difficult to obtain
within the United States. We have been informed by our legal counsel in Israel that investors
may find it difficult to assert claims under U.S. securities laws in original actions instituted
in Israel or obtain a judgment based on the civil liability provisions of U.S. federal
securities laws against us, our officers and our directors. Israeli courts may refuse to hear a
claim based on a violation of U.S. securities laws against us or our officers and directors
because Israel is not the most appropriate forum to bring such a claim. In addition, even if an
Israeli court agrees to hear a claim, it may determine that Israeli law and not U.S. law is
applicable to the claim. If U.S. law is found to be applicable, the content of applicable U.S.
law must be proved as a fact which can be a time-consuming and costly process. Certain matters
of procedure will also be governed by Israeli law. There is little binding case law in Israel
addressing the matters described above.
42
Israeli courts might not enforce judgments rendered outside Israel which may make it difficult
to collect on judgments rendered against us. Subject to certain time limitations, an Israeli
court may declare a foreign civil judgment enforceable only if it finds that:
| |
• |
|
the judgment was rendered by a court which was, according to the laws of the state of
the court, competent to render the judgment; |
| |
| |
• |
|
the judgment may no longer be appealed; |
| |
| |
• |
|
the obligation imposed by the judgment is enforceable according to the rules relating
to the enforceability of judgments in Israel and the substance of the judgment is not
contrary to public policy; and |
| |
| |
• |
|
the judgment is executory in the state in which it was given. |
Even if these conditions are satisfied, an Israeli court will not enforce a foreign judgment if
it was given in a state whose laws do not provide for the enforcement of judgments of Israeli
courts (subject to exceptional cases) or if its enforcement is likely to prejudice the
sovereignty or security of the State of Israel. An Israeli court also will not declare a
foreign judgment enforceable if:
| |
• |
|
the judgment was obtained by fraud; |
| |
| |
• |
|
there is a finding of lack of due process; |
| |
| |
• |
|
the judgment was rendered by a court not competent to render it according to the laws
of private international law in Israel; |
| |
| |
• |
|
the judgment is at variance with another judgment that was given in the same matter
between the same parties and that is still valid; or |
| |
| |
• |
|
at the time the action was brought in the foreign court, a suit in the same matter
and between the same parties was pending before a court or tribunal in Israel. |
Risks Related to Investing in Our Common Stock
The market price of our common stock may fluctuate significantly.
The market price of our common stock may fluctuate significantly in response to numerous
factors, some of which are beyond our control, such as:
| |
• |
|
the announcement of new products or product enhancements by us or our competitors; |
| |
| |
• |
|
developments concerning intellectual property rights and regulatory approvals; |
| |
| |
• |
|
variations in our and our competitors’ results of operations; |
| |
| |
• |
|
results of our ongoing studies regarding our lead product candidate taliglucerase
alfa, or communications from the FDA or other regulatory authorities regarding our NDA
for taliglucerase alfa or similar filings; |
| |
| |
• |
|
changes in earnings estimates or recommendations by securities analysts, if our
common stock is covered by analysts; |
| |
| |
• |
|
developments in the biotechnology industry; and |
| |
| |
• |
|
general market conditions and other factors, including factors unrelated to our
operating performance. |
Further, stock markets in general, and the market for biotechnology companies in particular,
have recently experienced price and volume fluctuations. Continued market fluctuations could
result in extreme volatility in the price of our common stock, which could cause a decline in
the value of our common stock. Price volatility of our common stock may be worse if the trading
volume of our common stock is low. We have not paid, and do not expect to pay, any cash
dividends on our common stock as any earnings generated from future operations will be used to
finance our operations.
43
As a result, investors will not realize any income from an investment in our common stock until
and unless their shares are sold at a profit.
All liabilities of our company have survived the merger and there may be undisclosed liabilities
that could harm our revenues, business, prospects, financial condition and results of operations.
Protalix Ltd. and its counsel conducted due diligence on us that was customary and appropriate
for the reverse merger transaction consummated on December 31, 2006. However, the due diligence
process may not have revealed all our material liabilities then existing or that could be
asserted in the future against us relating to our activities before the consummation of the
merger. Any such potential liabilities survive the merger and could harm our revenues,
business, prospects, financial condition and results of operations.
Future sales of our common stock could reduce our stock price.
The market price of our common stock could drop significantly if our existing shareholders sell a
large number of shares of our common stock or are perceived by the market as intending to sell
them. All of the shares sold in our public offering in October 2007 were freely tradable without
restriction or further registration under the federal securities laws, unless purchased by our
“affiliates” as that term is defined in Rule 144 under the Securities Act. In addition, all of the
outstanding shares of our common stock are freely tradable without restriction or further
registration under the federal securities laws, unless owned by our affiliates. At December 31,
2009, there were options issued and outstanding to purchase 6,805,421 shares of our common stock
with a weighted average exercise price of $2.89 per share. Also at December 31, 2009, there were
1,433,623 shares of common stock remaining available for future for issuance in connection with
future grants of incentives under our 2006 Stock Incentive Plan.
Directors, executive officers, principal shareholders and affiliated entities own a significant
percentage of our capital stock, and they may make decisions that an investor may not consider to
be in the best interests of our shareholders.
Our directors, executive officers, principal shareholders and affiliated entities beneficially
own, in the aggregate, approximately 38% of our outstanding common stock. As a result, if some
or all of them acted together, they would have the ability to exert substantial influence over
the election of our Board of Directors and the outcome of issues requiring approval by our
shareholders. This concentration of ownership may have the effect of delaying or preventing a
change in control of our company that may be favored by other shareholders. This could prevent
the consummation of transactions favorable to other shareholders, such as a transaction in which
shareholders might otherwise receive a premium for their shares over current market prices.
Failure to maintain effective internal controls in accordance with Section 404 of the
Sarbanes-Oxley Act could have a material adverse effect on our business and operating results. In
addition, current and potential shareholders could lose confidence in our financial reporting,
which could have a material adverse effect on the price of our common stock.
Effective internal controls are necessary for us to provide reliable financial reports and
effectively prevent fraud. If we cannot provide reliable financial reports or prevent fraud,
our results of operation could be harmed.
Section 404 of the Sarbanes-Oxley Act of 2002 requires annual management assessments of the
effectiveness of our internal controls over financial reporting and a report by our independent
registered public accounting firm addressing these assessments. We continuously monitor our
existing internal controls over financial reporting systems to confirm that they are compliant
with Section 404, and we may identify deficiencies that we may not be able to remediate in time
to meet the deadlines imposed by the Sarbanes-Oxley Act. This process may divert internal
resources and will take a significant amount of time and effort to complete.
If, at any time, it is determined that we are not in compliance with Section 404, we may be
required to implement new internal control procedures and reevaluate our financial reporting.
We may experience higher than anticipated operating expenses as well as increased independent
auditor fees during the implementation of these changes and thereafter. Further, we may need to
hire additional qualified personnel. If we fail to maintain the adequacy of our internal
controls, as such standards are modified, supplemented or amended from time to time, we may not
be able to conclude on an ongoing basis that we have effective internal controls over financial
reporting in accordance with Section 404 of the Sarbanes-Oxley Act, which could result in our
being unable to obtain an unqualified report on internal controls from our independent auditors.
Failure to maintain an effective internal control environment could also cause investors to
lose confidence in our reported financial information, which could have a material adverse
effect on the price of our common stock.
44
Compliance with changing regulation of corporate governance and public disclosure may result in
additional expenses, divert management’s attention from operating our business which could have a
material adverse effect on our business.
There have been other changing laws, regulations and standards relating to corporate governance
and public disclosure in addition to the Sarbanes-Oxley Act, as well as new regulations
promulgated by the Commission and rules promulgated by the national securities exchanges,
including the NYSE Amex and the NASDAQ. These new or changed laws, regulations and standards
are subject to varying interpretations in many cases due to their lack of specificity, and as a
result, their application in practice may evolve over time as new guidance is provided by
regulatory and governing bodies, which could result in continuing uncertainty regarding
compliance matters and higher costs necessitated by ongoing revisions to disclosure and
governance practices. As a result, our efforts to comply with evolving laws, regulations and
standards are likely to continue to result in increased general and administrative expenses and
a diversion of management time and attention from revenue-generating activities to compliance
activities. Our board members, Chief Executive Officer and Chief Financial Officer could face
an increased risk of personal liability in connection with the performance of their duties. As
a result, we may have difficulty attracting and retaining qualified board members and executive
officers, which could have a material adverse effect on our business. If our efforts to comply
with new or changed laws, regulations and standards differ from the activities intended by
regulatory or governing bodies, we may incur additional expenses to comply with standards set by
regulatory authorities or governing bodies which would have a material adverse effect on our
business and results of operations.
We are a holding company with no operations of our own.
We are a holding company with no operations of our own. Accordingly, our ability to conduct our
operations, service any debt that we may incur in the future and pay dividends, if any, is
dependent upon the earnings from the business conducted by Protalix Ltd. The distribution of those
earnings or advances or other distributions of funds by our subsidiary to us, as well as our
receipt of such funds, are contingent upon the earnings of our subsidiary and are subject to
various business considerations and U.S. and Israeli law. If Protalix Ltd. is unable to make
sufficient distributions or advances to us, or if there are limitations on our ability to receive
such distributions or advances, we may not have the cash resources necessary to conduct our
corporate operations which would have a material adverse effect on our business and results of
operations.
The issuance of preferred stock or additional shares of common stock could adversely affect the
rights of the holders of shares of our common stock.
Our Board of Directors is authorized to issue up to 100,000,000 shares of preferred stock without
any further action on the part of our shareholders. Our Board of Directors has the authority to
fix and determine the voting rights, rights of redemption and other rights and preferences of
preferred stock. Currently, we have no shares of preferred stock outstanding.
Our Board of Directors may, at any time, authorize the issuance of a series of preferred stock that
would grant to holders the preferred right to our assets upon liquidation, the right to receive
dividend payments before dividends are distributed to the holders of common stock and the right to
the redemption of the shares, together with a premium, before the redemption of our common stock,
which may have a material adverse effect on the rights of the holders of our common stock. In
addition, our Board of Directors, without further shareholder approval, may, at any time, issue
large blocks of preferred stock. In addition, the ability of our Board of Directors to issue
shares of preferred stock without any further action on the part of our shareholders may impede a
takeover of our company and may prevent a transaction that is favorable to our shareholders.
Item 1B. Unresolved Staff Comments
None.
Item 2. Properties
Our manufacturing facility and executive offices are located in Carmiel, Israel. The facilities
currently contain approximately 15,000 sq/ft of manufacturing space and additional 40,000 sq/ft of
laboratory, warehouse and office space and are leased at a rate of approximately $63,000 per month.
Our facilities are equipped with the requisite laboratory services required to conduct our
business, and we believe that the existing facilities are adequate to meet our needs for the
foreseeable future. We have leased the facility through 2017, subject to three options exercisable
by us to extend the term for a five-year period, for an aggregate of 15 additional years. Upon the
exercise of each option to extend the term of the lease, if any, the then current base rent shall
be increased by 10%. We also lease an office in Ramat Gan, Israel, for approximately $1,700 per
month.
45
Item 3. Legal Proceedings
We are not involved in any material legal proceedings.
Item 4. Submission of Matters to a Vote of Security Holders
At our Annual Meeting of Shareholders held on November 9, 2009, the following matters were voted on
by our shareholders: (i) the election of nine directors; and (ii) the approval of the appointment
of Kesselman & Kesselman, Certified Public Accountant (Isr.), A member of PricewaterhouseCoopers
International Limited, as our independent registered public accounting firm for the fiscal year
ended December 31, 2009. The results of such shareholder votes are as follows:
(i) Election of Directors
| |
|
|
|
|
|
|
|
|
| |
|
For |
|
Withheld |
Eli Hurvitz |
|
|
46,816,221 |
|
|
|
260,960 |
|
David Aviezer, Ph.D., MBA |
|
|
47,064,250 |
|
|
|
12,931 |
|
Yoseph Shaaltiel, Ph.D. |
|
|
46,858,832 |
|
|
|
218,349 |
|
Alfred Akirov |
|
|
46,941,083 |
|
|
|
136,098 |
|
Amos Bar Shalev |
|
|
46,565,514 |
|
|
|
511,667 |
|
Zeev Bronfeld |
|
|
45,584,634 |
|
|
|
1,492,547 |
|
Yodfat Harel Gross |
|
|
46,662,025 |
|
|
|
415,156 |
|
Roger D. Kornberg, Ph.D. |
|
|
46,859,840 |
|
|
|
217,341 |
|
Eyal Sheratzky |
|
|
47,061,035 |
|
|
|
16,146 |
|
(ii) Approval of Kesselman & Kesselman, Certified Public Accountant (Isr.), A member of
PricewaterhouseCoopers International Limited, as our independent registered public accounting firm
for the fiscal year ended December 31, 2009.
| |
|
|
|
|
For
|
|
Against
|
|
Abstain |
|
|
|
|
|
|
| 46,945,171
|
|
129,176
|
|
2,834 |
46
PART II
|
|
|
| Item 5. |
|
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer
Purchases of Equity Securities |
Our common stock is traded on the NYSE Amex under the symbol PLX. The following table sets forth
the quarterly sales price ranges of our common stock for the periods indicated, as reported by the
NYSE Amex.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
2009 |
|
2008 |
| Quarter Ended |
|
High |
|
Low |
|
High |
|
Low |
March 31 |
|
$ |
2.99 |
|
|
$ |
1.74 |
|
|
$ |
3.59 |
|
|
$ |
2.60 |
|
June 30 |
|
$ |
5.29 |
|
|
$ |
2.00 |
|
|
$ |
3.70 |
|
|
$ |
2.56 |
|
September 30 |
|
$ |
8.49 |
|
|
$ |
4.49 |
|
|
$ |
3.06 |
|
|
$ |
2.08 |
|
December 31 |
|
$ |
12.50 |
|
|
$ |
6.37 |
|
|
$ |
2.17 |
|
|
$ |
0.96 |
|
These quotations reflect prices between dealers and do not include retain mark-ups, mark-downs and
commissions and may not necessarily represent actual transactions.
There were
approximately 40 holders of record of our common stock at February 15, 2010. A
substantially greater number of holders of our common stock are “street name” or beneficial
holders, whose shares are held of record by banks, brokers and other financial institutions. To
date, we have not declared or paid any cash dividends on our common stock. We do not anticipate
paying any dividends on our common stock in the foreseeable future.
STOCK PERFORMANCE GRAPH
The following graph compares the cumulative total shareholder return data for our common stock from
December 31, 2004 through December 31, 2009 to the cumulative return over such time period of (i)
The AMEX Composite Index and (ii) The AMEX Biotechnology Index. The graph assumes an investment of
$100 on December 31, 2004 in each of our common stock, and the stocks comprising the AMEX Composite
Index and the stocks comprising the AMEX Biotechnology Index, including dividend reinvestment, if
any.
The stock price performance shown on the graph below represents historical price performance and is
not necessarily indicative of any future stock price performance. Specifically, during the period
from December 31, 2004 through December 31, 2006, our company did not have any operations and our
common stock was quoted on the OTC® Bulletin Board. The historical performance of our
common stock prior to January 2, 2007, represents the performance of our company prior to the
merger on December 31, 2006, and, therefore, is not indicative of the performance of our common
stock after the merger or the performance of our common stock after it was listed for trade on the
NYSE AMEX on March 12, 2007.
47
COMPARISON OF 5 YEAR CUMULATIVE TOTAL RETURN*
Among Protalix BioTherapeutics, Inc., The NYSE Amex Composite Index
and The NYSE Arca Biotechnology Index
Notwithstanding anything to the contrary set forth in any of our previous filings under the
Securities Act of 1933, as amended, or the Exchange Act, which might incorporate future filings
made by us under those statutes, this Stock Performance Graph will not be incorporated by reference
into any of those prior filings, nor will such report or graph be incorporated by reference into
any future filings made by us under those Acts.
Use of Proceeds
The effective date of our first registration statement, filed on Form S-3 under the Securities Act
of 1933, which was accompanied by a registration statement on Form S-3 filed pursuant to Rule
462(b) under the Securities Act (Nos. 333-144801 and 333-146919), relating to a public offering of
our common stock, was September 26, 2007 and the offering date was October 25, 2007. The sole
book-running manager of the offering was UBS Investment Bank and CIBC World Markets (now
Oppenheimer & Co., Inc.) served as the co-manager. In the offering we sold 10,000,000 shares of
common stock at a price per share of $5.00. Our aggregate net proceeds (after underwriting
discounts and expenses) amounted to approximately $46 million. The offering closed on October 30,
2007.
The amount of the underwriting discount paid by us was $3.5 million and the expenses of the
offering, not including the underwriting discount, were approximately $810,000.
Between October 30, 2007 and December 31, 2009, we have used approximately $43.7 million of the net
proceeds to fund our operating activities, including activities related to the development of our
clinical and preclinical product candidates and for working capital, capital expenditures and other
general corporate purposes. During the year ended December 31, 2009, our research and development
expenses comprised approximately 78% of our operating expenses. We have deposited the net proceeds
of the offering in accordance with our investment policy in short-term bank-deposits. There has
been no material change in our planned use of proceeds from our public offering as described in our
registration statement.
48
Item 6. Selected Financial Data
The selected consolidated financial data below should be read in conjunction with “Management’s
Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated
financial statements and the related notes included elsewhere in this Annual Report on Form 10-K.
The selected consolidated statements of operations data for the years ended December 31, 2009, 2008
and 2007 and for the period from December 27, 1993 through December 31, 2009 and the selected
consolidated balance sheet data as of December 31, 2009 and 2008, are derived from the audited
consolidated financial statements included elsewhere in this Annual Report. The selected
consolidated statements of operations data for the year ended December 31, 2007 and the selected
consolidated balance sheet data as of December 31, 2007 have been adjusted to reflect the
restatement of our financial results. The statement of operations data for the years ended
December 31, 2005 and 2006 and the balance sheet data as of December 31, 2005, 2006 and 2007 are
derived from audited financial statements not included in this Annual Report. The historical
results presented below are not necessarily indicative of future results.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year Ended December 31, |
|
| |
|
2005 |
|
|
2006 |
|
|
2007 |
|
|
2008 |
|
|
2009 |
|
| |
|
|
|
|
|
(in thousands, except share and per share amounts) |
|
|
|
|
|
Consolidated Statement of Operations Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenues |
|
$ |
150 |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
$ |
388 |
|
Cost of revenues |
|
|
35 |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
1,625 |
|
Gross profit (loss) |
|
|
115 |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
(1,237 |
) |
Research and development expenses, net |
|
|
3,773 |
|
|
$ |
5,246 |
|
|
$ |
13,570 |
|
|
$ |
17,401 |
|
|
$ |
23,588 |
|
General and administrative expenses |
|
|
2,131 |
|
|
|
4,525 |
|
|
|
20,594 |
|
|
|
6,770 |
|
|
|
7,144 |
|
Finance expense (income) |
|
|
(43 |
) |
|
|
(344 |
) |
|
|
(2,080 |
) |
|
|
(1,757 |
) |
|
|
(529 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss before change in accounting principle |
|
$ |
5,746 |
|
|
$ |
9,427 |
|
|
$ |
32,084 |
|
|
$ |
22,414 |
|
|
$ |
31,440 |
|
Cumulative effect of change in accounting
principle |
|
|
— |
|
|
|
(37 |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
5,746 |
|
|
$ |
9,390 |
|
|
$ |
32,084 |
|
|
$ |
22,414 |
|
|
$ |
31,440 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per share of common stock, basic and
diluted (1) |
|
$ |
0.31 |
|
|
$ |
0.32 |
|
|
$ |
0.48 |
|
|
$ |
0.30 |
|
|
$ |
0.41 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average number of shares of common
stock used in computing net loss per share of
common stock (2) |
|
|
18,801,527 |
|
|
|
29,300,987 |
|
|
|
67,187,329 |
|
|
|
75,890,633,344 |
|
|
|
76,942,840 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Consolidated Balance Sheet Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
4,741 |
|
|
$ |
15,378 |
|
|
$ |
61,813 |
|
|
$ |
42,596 |
|
|
$ |
81,266 |
|
Other assets |
|
|
2,484 |
|
|
|
11,610 |
|
|
|
6,324 |
|
|
|
8,215 |
|
|
|
17,405 |
|
Total assets |
|
|
7,225 |
|
|
|
26,988 |
|
|
|
68,137 |
|
|
|
50,811 |
|
|
|
98,671 |
|
Current liabilities |
|
|
845 |
|
|
|
2,268 |
|
|
|
3,762 |
|
|
|
5,527 |
|
|
|
21,530, |
|
Liabilities |
|
|
1,130 |
|
|
|
2,704 |
|
|
|
4,452 |
|
|
|
6,464 |
|
|
|
82,788 |
|
Shareholders’ equity |
|
|
6,095 |
|
|
|
24,284 |
|
|
|
63,685 |
|
|
|
44,347 |
|
|
|
15,838 |
|
|
|
|
| * |
|
Represents less than $1. |
| |
| (1) |
|
Reflects the retroactive effects of the impact of our merger with Protalix Ltd. and the
resulting exchange of shares of common stock for the ordinary shares of Protalix Ltd. at an
exchange ratio of approximately 61.08 shares of our common stock per ordinary share of
Protalix Ltd. for all periods presented. |
| |
| (2) |
|
In connection with the merger, we completed a one-for-ten reverse stock split, therefore all
share numbers presented in this Annual Report on Form 10-K give retroactive effect to the
reverse stock split, as applicable. |
49
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis of our financial condition and results of
operations together with our consolidated financial statements and the related notes included
elsewhere in this Annual Report on Form 10-K. Some of the information contained in this discussion
and analysis, particularly with respect to our plans and strategy for our business and related
financing, includes forward-looking statements that involve risks and uncertainties. You should
read “Risk Factors” in Item 1A of this Annual Report for a discussion of important factors that
could cause actual results to differ materially from the results described in or implied by the
forward-looking statements contained in the following discussion and analysis.
Overview
We are a biopharmaceutical company focused on the development and commercialization of recombinant
therapeutic proteins based on our proprietary ProCellExTM protein expression system, or
ProCellEx. Using our ProCellEx system, we are developing a pipeline of proprietary and biosimilar
or “generic” versions of recombinant therapeutic proteins based on our plant cell-based expression
technology that target large, established pharmaceutical markets and that rely upon known
biological mechanisms of action. Our initial commercial focus has been on complex therapeutic
proteins, including proteins for the treatment of genetic disorders, such as Gaucher disease and
Fabry disease. We believe our ProCellEx protein expression system will enable us to develop
proprietary recombinant proteins that are therapeutically equivalent or superior to existing
recombinant proteins currently marketed for the same indications. Because we are primarily
targeting biologically equivalent versions of highly active, well-tolerated and commercially
successful therapeutic proteins, we believe our development process is associated with relatively
less risk compared to other biopharmaceutical development processes for completely novel
therapeutic proteins.
Our lead product development candidate is prGCD (taliglucerase alfa) for the treatment of Gaucher
disease, which we are developing using our ProCellEx protein expression system. Gaucher disease is
a rare and serious lysosomal storage disorder with severe and debilitating symptoms. Taliglucerase
alfa is our proprietary recombinant form of glucocerebrosidase (GCD), an enzyme naturally found in
human cells that is mutated or deficient in patients with Gaucher disease. In July 2007, we
reached an agreement with the U.S. Food and Drug Administration, or the FDA, on the final design of
our pivotal phase III clinical trial of taliglucerase alfa, through the FDA’s special protocol
assessment (SPA) process. The phase III clinical trial was completed in September 2009 and, on
October 15, 2009, we announced positive top-line results from the trial. On December 9, 2009, we
filed our New Drug Application (NDA) for taliglucerase alfa, and in January 2010 the FDA requested
additional data regarding the Chemistry, Manufacturing and Controls (CMC) section of our NDA. No
additional clinical or preclinical information was requested. The request focused primarily on the
validation of the manufacturing process in our upgraded manufacturing facility. A validation plan
for our manufacturing process of taliglucerase alfa has already been established and reviewed by
the FDA. We are working diligently to provide the requested data to the FDA and anticipate
submitting the requested data during the second quarter of 2010. In addition, we expect to submit
similar applications with other comparable regulatory agencies in other countries during 2010.
In addition to our recently completed phase III clinical trial, during the third quarter of 2008,
we initiated a double-blind, follow-on extension study as part of the trial. We also initiated a
home care treatment program for patients enrolled in the extension study and in December 2008, we
initiated a clinical study evaluating the safety and efficacy of switching Gaucher patients
currently treated under the current standard of care to treatment with taliglucerase alfa. The
current standard of care for Gaucher patients is enzyme replacement therapy with
CerezymeTM which is produced by Genzyme Corporation and currently the only approved
enzyme replacement therapy for Gaucher disease. Enzyme replacement therapy is a medical treatment
in which recombinant enzymes are injected into patients in whom the enzyme is lacking or
dysfunctional. The switch-over study is not a prerequisite for approval of taliglucerase alfa. In
December 2009 we filed a proposed pediatric investigation plan to the Pediatric Committee of the
EMEA.
On November 30, 2009, Protalix Ltd. and Pfizer entered into an exclusive license and supply
agreement pursuant to which Pfizer was granted an exclusive, worldwide license to develop and
commercialize taliglucerase alfa. Under the terms and conditions of the Pfizer agreement, Protalix
Ltd. retained the right to commercialize taliglucerase alfa in Israel. In connection with the
execution of the Pfizer agreement, Pfizer made an upfront payment to Protalix Ltd. of $60.0 million
in connection with the execution of the agreement and subsequently paid to Protalix Ltd. an
additional $5.0 million upon our filing of a proposed pediatric investigation plan to the Pediatric
Committee of the EMEA. Protalix Ltd. is also eligible to receive potential milestone payments
exceeding $50.0 million for the successful achievement of other developmental milestones and to
royalties equal to 40% of the net profits earned on Pfizer’s sales of taliglucerase alfa. Pfizer
and Protalix Ltd. have agreed to a specific allocation of the responsibilities for the continued
development efforts for taliglucerase alfa.
50
Our business is conducted by our wholly-owned subsidiary, Protalix Ltd., which we acquired through
a reverse merger transaction effective December 31, 2006. The merger transaction was treated as a
recapitalization for accounting purposes and, as such, the results of operations discussed below
are those of Protalix Ltd. Prior to the merger transaction, we had not conducted any operations
for several years. Protalix Ltd. was originally incorporated in Israel in December 1993. Since
its inception in December 1993, Protalix Ltd. has generated significant losses in connection with
its research and development, including the clinical development of taliglucerase alfa. Since we
do not generate significant revenue from any of our product candidates, we expect to continue to
generate losses in connection with the continued clinical development of taliglucerase alfa and the
research and development activities relating to our technology and other drug candidates. Such
research and development activities are budgeted to expand over time and will require further
resources if we are to be successful. As a result, we believe that our operating losses are likely
to be substantial over the next several years. We will need to obtain additional funds for the
commercialization of our lead product, taliglucerase alfa, and to further develop the research and
clinical development of our other programs.
Critical Accounting Policies
Our significant accounting policies are more fully described in Note 1 to our consolidated
financial statements appearing at the end of this Annual Report. We believe that the accounting
policies below are critical for one to fully understand and evaluate our financial condition and
results of operations.
The discussion and analysis of our financial condition and results of operations is based on our
financial statements, which we prepared in accordance with U.S. generally accepted accounting
principles. The preparation of these financial statements requires us to make estimates and
assumptions that affect the reported amounts of assets and liabilities and the disclosure of
contingent assets and liabilities at the date of the financial statements, as well as the reported
revenues and expenses during the reporting periods. On an ongoing basis, we evaluate such
estimates and judgments, including those described in greater detail below. We base our estimates
on historical experience and on various other factors that we believe are reasonable under the
circumstances, the results of which form the basis for making judgments about the carrying value of
assets and liabilities that are not readily apparent from other sources. Actual results may differ
from these estimates under different assumptions or conditions.
Functional Currency
The currency of the primary economic environment in which our operations are conducted is the
dollar. As we have no significant source of revenues, we considered the currency of the primary
economic environment to be the currency in which we expend cash. Most of our expenses and capital
expenditures are incurred in dollars, and a significant source of our financing has been provided
in U.S. dollars.
Revenues
We have applied guidance regarding Accounting for Revenue Arrangements with Multiple Deliverables
in connection with our receipt of revenues. In accordance with the guidance, we determined that
the various deliverables due from us under our license and supply agreement with Pfizer represent
separate units of accounting for revenue recognition purposes. The initial, non-refundable upfront
license fee payment of $60.0 million together with the first $5.0 million milestone will be
recognized on a straight line basis as revenue over the estimated relationship period. We have
estimated that the performance period under the agreement with Pfizer will be 14 years based on our
last relevant patent to expire.
Our deliverables under the agreement with Pfizer primarily include an exclusive license to
taliglucerase alfa as an enzyme replacement therapy for the treatment of Gaucher disease, the
manufacture of taliglucerase alfa, the performance of certain research and development services as
required under the agreement and participation in a joint steering committee. The $60.0 million
up-front payment and the subsequent $5.0 million milestone payment were recorded as deferred
revenue. These amounts will be amortized over the performance period at a rate of approximately
$1.1 million per fiscal quarter, with $388,000 recorded as revenues in 2009.
Research and Development Expense
We expect our research and development expense to remain our primary expense in the near future as
we continue to develop our product candidates. Research and development expense consists of:
| |
• |
|
internal costs associated with research and development activities; |
| |
| |
• |
|
payments made to third party contract research organizations, investigative sites and
consultants; |
| |
| |
• |
|
manufacturing development costs;
|
51
| |
• |
|
personnel-related expenses, including salaries, benefits, travel, and related costs for
the personnel involved in research and development; |
| |
| |
• |
|
activities relating to the advancement of product candidates through preclinical studies
and clinical trials; and |
| |
| |
• |
|
facilities and other allocated expenses, which include direct and allocated expenses for
rent and maintenance of facilities, as well as laboratory and other supplies. |
The following table identifies our current major research and development projects:
| |
|
|
|
|
| Project |
|
Status |
|
Expected Near Term Milestone |
| |
taliglucerase alfa for the
treatment of Gaucher disease
|
|
NDA Filed
|
|
PDUFA date |
|
|
|
|
|
PRX 102 — alpha Galactosidase enzyme
|
|
Pre Clinical
|
|
Pre IND meeting with FDA |
|
|
|
|
|
Acetylcholinesterase
|
|
Pre Clinical
|
|
IND approval and initiation of
phase I clinical trial |
|
|
|
|
|
pr-antiTNF
|
|
Research
|
|
Additional animal studies |
All of our projects, other than our recently completed phase III clinical trial of taliglucerase
alfa, are in the pre clinical or research phase with relatively immaterial costs. Most of our
research and development costs were incurred in connection with our phase III clinical trial of
taliglucerase alfa. Our internal resources, employees and infrastructure are not tied to any
individual research project and are typically deployed across all of our projects. We currently do
not record and maintain research and development costs per project.
The costs and expenses of our projects are partially funded by grants we have received from the
OCS. Each grant is deducted from the related research and development expenses as the costs are
incurred. For additional information regarding the grant process, see “Business—Israeli Government
Programs—Encouragement of Industrial Research and Development Law, 1984” in Item 1 of this Annual
Report. There can be no assurance that we will continue to receive grants from the OCS in amounts
sufficient for our operations, if at all.
At this time, due to the inherently unpredictable nature of preclinical and clinical development
processes and given the early stage of our preclinical product development programs, we are unable
to estimate with any certainty the costs we will incur in the continued development of the product
candidates in our pipeline for potential commercialization. Clinical development timelines, the
probability of success and development costs can differ materially from expectations. While we are
currently focused on advancing each of our product development programs, our future research and
development expenses will depend on the clinical success of each product candidate, as well as
ongoing assessments of each product candidate’s commercial potential. In addition, we cannot
forecast with any degree of certainty which product candidates may be subject to future
collaborations, when such arrangements will be secured, if at all, and to what degree such
arrangements would affect our development plans and capital requirements. See “Risk Factors—All of
our product candidates other than taliglucerase alfa are in research stages. If we are unable to
develop and commercialize our other product candidates, our business will be adversely affected”
and “—We may not obtain the necessary U.S. or worldwide regulatory approvals to commercialize our
drug candidates in a timely manner, if at all, which would have a material adverse effect on our
business and results of operations.”
We expect our research and development expenses to increase in the future as we continue the
advancement of our clinical trials and preclinical product development programs. The lengthy
process of completing clinical trials and seeking regulatory approval for our product candidates
requires expenditure of substantial resources. Any failure or delay in completing clinical trials,
or in obtaining regulatory approvals, could cause a delay in generating product revenue and cause
our research and development expense to increase and, in turn, have a material adverse effect on
our operations. We filed a New Drug Application, or NDA, for taliglucerase alfa with the FDA in
the last quarter of 2009. Because of the factors set forth above, we are not able to estimate with
any certainty when we would recognize any net cash inflows from our projects. See “Risk
Factors—Clinical trials are very expensive, time-consuming and difficult to design and implement
and may result in unforeseen costs which may have a material adverse effect on our business and
results of operations.”
52
General and Administrative Expense
General and administrative expense consists primarily of salaries and other related costs,
including share-based compensation expense, for persons serving as our executive, finance,
accounting and administration functions. Other general and administrative expense includes
facility-related costs not otherwise included in research and development expense, costs associated
with industry and trade shows and professional fees for legal and accounting services. We expect
that our general and administrative expenses will increase as we add additional personnel and
continue to comply with the reporting and other obligations applicable to public companies in the
United States.
Financial Expense and Income
Financial Expense and Income consists of the following:
| |
• |
|
interest earned on our cash and cash equivalents; |
| |
| |
• |
|
interest expense on short term bank credit and loan; and |
| |
| |
• |
|
expense or income resulting from fluctuations of the New Israeli Shekel (NIS), in which
a portion of our assets and liabilities are denominated, against the United States Dollar
and other foreign currencies. |
Share-Based Compensation
The discussion below regarding share-based compensation relates to share-based compensation paid by
Protalix Ltd., our wholly-owned subsidiary.
In accordance with the guidance, we record the benefit of any grant to a non-employee and remeasure
the benefit in any future vesting period for the unvested portion of the grants, as applicable. In
addition, we use the straight-line accounting method for recording the benefit of the entire grant,
unlike the graded method we use to record grants made to employees.
We measure share-based compensation cost for all share-based awards at the fair value on the grant
date and recognition of share-based compensation over the service period for awards that we expect
will vest. The fair value of stock options is determined based on the number of shares granted and
the price of our ordinary shares, and calculated based on the Black-Scholes valuation model. We
recognize such value as expense over the service period, net of estimated forfeitures, using the
accelerated method.
For purposes of determining the fair value of the options and shares of restricted common stock
granted to employees and non-employees during the fiscal year ended December 31, 2008, including
shares held by non employees that vested during such period, our management used the fair value of
our common stock which was the closing sale price of our common stock on the NYSE Amex on the date
of calculation.
The guidance allows companies to estimate the expected term of the option rather than simply using
the contractual term of an option. Because of lack of data on past option exercises by employees,
the expected term of the options could not be based on historic exercise patterns. Accordingly, we
adopted the simplified method, according to which companies may calculate the expected term as the
average between the vesting date and the expiration date, assuming the option was granted as a
“plain vanilla” option.
In performing the valuation, we assumed an expected 0% dividend yield in the previous years and in
the next years. We do not have a dividend policy and given the lack of profitability, dividends
are not expected in the foreseeable future, if at all. The guidance stipulates a number of factors
that should be considered when estimating the expected volatility, including the implied volatility
of traded options, historical volatility and the period that the shares of the company are being
publicly traded. As we do not have any traded shares or options, the expected volatility figures
used in this valuation have been calculated by using the historical volatility of traded shares of
similar companies. In addition, we examined the standard deviation of shares of similar
biotechnology companies that engage in research and development, generally with no significant
revenues. We found that the standard deviation of the shares of comparable companies was in the
range of 40%-60% over periods of three to six years. The volatility used for each grant differed
based on its expected term. For the term of each grant of our options, the historical volatility
was calculated based upon the overall trading history of the common stock of comparable companies.
The risk-free interest rate in the table above has been based on the implied yield of U.S. federal
reserve zero–coupon government bonds. The remaining term of the bonds used for each valuation was
equal to the expected term of the grant. This methodology has been applied to all grants valued by
us. The guidance requires the use of a risk–free interest rate based on the implied yield
currently available on zero–coupon government issues of the country in whose currency the exercise
53
price is expressed, with a remaining term equal to the expected life of the option being valued.
This requirement has been applied for all grants valued as part of this report.
Results of Operations
Year Ended December 31, 2009 Compared to the Year Ended December 31, 2008
Revenues
We recorded revenue of $388,000 during the year ended December 31, 2009. The revenue represents
the pro rata amortization of the $60.0 million upfront payment and $5.0 million milestone payment
we received in connection with our license and supply agreement with Pfizer. The payments were
recorded as deferred revenue and the amounts will be amortized over the performance period,
estimated at approximately 14 years, at a rate of approximately $1.1 million per quarter. No
revenues were recorded during the year ended December 31, 2008.
Research and Development Expenses
Research and development expenses were $27.4 million for the year ended December 31, 2009, an
increase of $5.3 million, or 24%, from $22.1 million for the year ended December 31, 2008. The
increase resulted primarily from an increase of $1.2 million in development expenses related to
salaries for personnel involved in research and development, and $2.2 million in related
subcontractors and consultants expenses, mainly in connection with our phase III clinical trial of
taliglucerase alfa. The increase in research and development expenses was further increased by the
recognition of grants equal to $3.8 million from the OCS during 2009, a decrease of approximately
$912,000, or 19%, compared to the recognition of grants equal to $4.7 million during 2008.
We expect research and development expenses to continue to be our primary expense as we enter into
a more advanced stage of pre clinical trials for certain of our product candidates and into
clinical trials for our AChE program.
General and Administrative Expenses
General and administrative expenses were $7.1 million for the year ended December 31, 2009, an
increase of $374,000, or approximately 6.0%, from $6.8 million for the year ended December 31,
2008. The increase resulted primarily from a $543,000 increase in salaries expenses during 2009.
Financial Expenses and Income
Financial income was $529,000 for the year ended December 31, 2009, a decrease of $1.2 million, or
approximately 70.0%, compared to $1.8 million for the year ended December 31, 2008. The decrease
resulted primarily from a lower interest rate for deposits in 2009 which contributed to lower
financial income during 2009.
Year Ended December 31, 2008 Compared to the Year Ended December 31, 2007
Revenues
No revenues were recorded during the years ended December 31, 2008 or 2007.
Research and Development Expenses
Research and development expenses were $22.1 million for the year ended December 31, 2008, an
increase of $7.5 million, or 51%, from $14.6 million for the year ended December 31, 2007. The
increase resulted primarily from the increase of $1.8 million in development expenses related to
salaries for personnel involved in research and development and $2.1 million in related
subcontractors and consultants expenses, mainly in connection with our on-going phase III clinical
trial of taliglucerase alfa. The increase in research and development expenses was partially
offset by the recognition of grants equal to $4.7 million from the OCS during 2008, an increase of
approximately $3.6 million compared to the recognition of grants equal to $1.1 million during 2007.
We expect research and development expenses to continue to be our primary expense as we enter into
a more advanced stage of clinical trials for our product candidates, especially with respect to the
anticipated continued progress in our phase III clinical trial for taliglucerase alfa.
54
General and Administrative Expenses
General and administrative expenses were $6.8 million for the year ended December 31, 2008, a
decrease of $13.8 million, or approximately 67%, from $20.6 million for the year ended December 31,
2007. The decrease resulted primarily from a $15.0 million decrease in share-based compensation
during 2008.
Financial Expenses and Income
Financial income was $1.8 million for the year ended December 31, 2008, a decrease of $323,000, or
approximately 16%, compared to $2.1 million for the year ended December 31, 2007. The decrease
resulted primarily from a lower interest rate for deposits in 2008 and the devaluation of the NIS
against USD, both of which contributed to lower financial income during 2008.
Liquidity and Capital Resources
Sources of Liquidity
As a result of our significant research and development expenditures and the lack of any approved
products to generate product sales revenue, we have not been profitable and have generated
operating losses since our inception. To date, we have funded our operations primarily with
proceeds equal to $31.3 million from the sale of shares of our common stock and from sales of
convertible preferred and ordinary shares of Protalix Ltd., and an additional $14.2 million in
connection with the exercise of warrants issued in connection with the sale of such ordinary
shares, through December 31, 2008. In addition, on October 25, 2007, we generated gross proceeds
of $50 million in connection with an underwritten public offering of our common stock.
Furthermore, on November 30, 2009, we entered into an exclusive license and supply agreement with
Pfizer, pursuant to which Pfizer made an upfront payment to Protalix Ltd. of $60.0 million in
connection with the execution of the agreement and subsequently paid to Protalix Ltd. an additional
$5.0 million upon meeting a certain milestone. Protalix Ltd. is also eligible to receive potential
milestone payments of up to $50.0 million for the successful achievement of other
regulatory-related milestones. We are also entitled to payments equal to 40% of the net profits
earned by Pfizer on its sales of taliglucerase alfa, if any. In calculating net profits there are
certain agreed upon limits on the amounts that may be deducted from gross sales for certain
expenses and costs of goods sold. We believe that the funds currently available to us as are
sufficient to satisfy our capital needs for the foreseeable future.
The following table summarizes our past funding sources:
| |
|
|
|
|
|
|
|
|
|
|
|
|
| Security |
|
Year |
|
Number of Shares |
|
Amount(1) |
Ordinary Shares |
|
|
1996-2000 |
|
|
|
18,801,527 |
(2) |
|
$ |
1,100,000 |
|
Series A Convertible Preferred Shares |
|
|
2001 |
|
|
|
11,635,090 |
|
|
$ |
2,000,000 |
|
Series B Convertible Preferred Shares(3) |
|
|
2004-2005 |
|
|
|
7,225,357 |
|
|
$ |
4,500,000 |
|
Series C Convertible Preferred Shares(4) |
|
|
2005 |
|
|
|
5,513,422 |
|
|
$ |
7,700,000 |
|
Ordinary Shares(5) |
|
|
2006 |
|
|
|
10,637,686 |
|
|
$ |
16,000,000 |
|
Common Stock |
|
|
2007 |
|
|
|
10,000,000 |
|
|
$ |
50,000,000 |
|
|
|
|
| (1) |
|
Gross proceeds; does not include proceeds from warrant exercises. |
| |
| (2) |
|
Includes the issuance of ordinary shares to founders. |
| |
| (3) |
|
During 2005, 1,035,569 Series B Preferred Shares were converted on a 1:1 basis into Series C
Preferred Shares for no additional consideration. Also, in connection with such funding,
warrants to purchase 181,228 Series B Preferred Shares were issued for no additional
consideration with an aggregate exercise price of $100,000. As of the closing date of the
merger, 168,034 of such warrants were exercised for net proceeds equal to approximately
$96,000 and 13,194 of such warrants were forfeited. |
| |
| (4) |
|
In connection with such funding, warrants to purchase an additional 8,862,803 Series C
Preferred Shares were granted to the investors for no additional consideration with a total
exercise price equal to $9.0 million. As of the closing date of the merger, 5,296,279 of such
warrants were exercised for net proceeds equal to $8.7 million, 3,384,502 were assumed by our
company and 182,022 expired. |
| |
| (5) |
|
In connection with such funding, warrants to purchase 3,875,416 ordinary shares were issued
for no additional consideration with an aggregate exercise price equal to $5.3 million. These
warrants were exercised in full on January 31, 2007. |
55
Cash Flows
Net cash provided from operations was $44.5 million for the year ended December 31, 2009. The net
loss for 2009 of $31.4 million was reversed by $65.0 million received under the license and supply
agreement with Pfizer. Such reversal was partially offset by non-cash charges for share-based
compensation of $2.7 million, and depreciation of $2.0 million. Net cash used in investing
activities for 2009 was $6.2 million and consisted primarily of purchases of property and
equipment. Net cash provided from financing activities for 2009 was approximately $293,000 due to
option exercise.
Net cash used in operations was $16.0 million for the year ended December 31, 2008. The net loss
for 2008 of $22.4 million was mainly offset by non-cash charges for share-based compensation of
$3.1 million, and depreciation of $1.3 million. Net cash used in investing activities for 2008 was
$3.7 million and consisted primarily of purchases of property and equipment. Net cash used by
financing activities for 2008 was approximately $51,000 due to certain fundraising costs incurred
in 2008 in connection with the offering of 2007.
Future Funding Requirements
We expect to incur losses from operations for the foreseeable future. We expect to incur
increasing research and development expenses, including expenses related to the hiring of personnel
and the advancement of our additional pipeline of product candidate into the various clinical
trials. We expect that general and administrative expenses will also increase as we expand our
finance and administrative staff, add infrastructure, and incur additional costs related to our
preparation for the commercial phase for our lead product candidate, taliglucerase alfa. In
addition, we are working on the expansion of our manufacturing facility that would meet
approximately half of the market for our lead product candidate, which would increase our capital
expenditures significantly, the first phase of which has commenced in the first quarter of 2010 and
estimated to cost approximately $20.0 million in total.
We believe that our existing cash and cash equivalents and the regulatory milestone payments we
anticipate receiving from Pfizer will be sufficient to enable us to fund our operating expenses and
capital expenditure requirements for the foreseeable future. We have based this estimate on
assumptions that are subject to change and may prove to be wrong, and we may be required to use our
available capital resources sooner than we currently expect. Because of the numerous risks and
uncertainties associated with the development and commercialization of our product candidates, we
are unable to estimate the amounts of increased capital outlays and operating expenditures
associated with our current and anticipated clinical trials.
Our future capital requirements will depend on many factors, including the progress and results of
our clinical trials, costs of commercialization activities, including product marketing, sales and
distribution and whether these efforts will be performed internally or through some form of
collaboration with third parties, the duration and cost of discovery and preclinical development,
and laboratory testing and clinical trials for our product candidates, the timing and outcome of
regulatory review of our product candidates, the costs involved in preparing, filing, prosecuting,
maintaining, defending and enforcing patent claims and other intellectual property rights and the
number and development requirements of other product candidates that we pursue.
We may need to finance our future cash needs through public or private equity offerings, debt
financings, or additional corporate collaboration and licensing arrangements. We currently do not
have any commitments for future external funding, other than the potential regulatory-related
milestone payments from Pfizer. We may need to raise additional funds more quickly if one or more
of our assumptions prove to be incorrect or if we choose to expand our product development efforts
more rapidly than we presently anticipate. We may also decide to raise additional funds even
before we need them if the conditions for raising capital are favorable. The sale of additional
equity or debt securities will likely result in dilution to our shareholders. The incurrence of
indebtedness would result in increased fixed obligations and could also result in covenants that
would restrict our operations. Additional equity or debt financing, grants or corporate
collaboration and licensing arrangements may not be available on acceptable terms, if at all. If
adequate funds are not available, we may be required to delay, reduce the scope of or eliminate our
research and development programs, reduce our planned commercialization efforts or obtain funds
through arrangements with collaborators or others that may require us to relinquish rights to
certain product candidates that we might otherwise seek to develop or commercialize independently.
Effects of Inflation and Currency Fluctuations
Inflation generally affects us by increasing our cost of labor and clinical trial costs. We do not
believe that inflation has had a material effect on our results of operations during the years
ended December 31, 2007, 2008 or 2009.
56
Currency fluctuations could affect us by increased or decreased costs mainly for goods and services
acquired outside of Israel. We do not believe currency fluctuations have had a material effect on
our results of operations during the years ended December 31, 2007, 2008 or 2009.
Off-Balance Sheet Arrangements
We have no off-balance sheet arrangements as of December 31, 2008 and 2009. See Note 4 of the
consolidated financial statements for a full description of certain contingent royalty payments.
Recently Issued Accounting Pronouncements
In May 2009, the Financial Accounting Standards Board, or FASB, issued ASC Topic 855 “Subsequent
Events” (formerly SFAS No. 165, Subsequent Events). ASC 855 sets forth the period after the
balance sheet date during which management of a reporting entity should evaluate events or
transactions that may occur for potential recognition or disclosure in the financial statements,
the circumstances under which an entity should recognize events or transactions occurring after the
balance sheet date in its financial statements, and the disclosures that an entity should make
about events or transactions that occurred after the balance sheet date. ASC 855 iseffective for
interim or annual periods ending after June 15, 2009. We adopted the provisions of ASC 855 for the
quarter ended June 30, 2009. The adoption of ASC 855 did not have a material impact on our
condensed financial statements.
In June 2009, the FASB issued Accounting Standards Update, “Topic 105 — Generally Accepted
Accounting Principles” which amended ASC 105 “The “FASB Accounting Standards Codification” and the
Hierarchy of Generally Accepted Accounting Principles (formerly SFAS No. 168 “The FASB Accounting
Standards Codification and the Hierarchy of Generally Accepted Accounting Principles – A
Replacement of FASB Statement No. 162”). ASU 2009-1 establishes the FASB Accounting Standards
CodificationTM (Codification) as the single source of authoritative U.S. generally
accepted accounting principles (U.S. GAAP) recognized by the FASB to be applied by nongovernmental
entities. Rules and interpretive releases of the SEC under authority of federal securities laws
are also sources of authoritative U.S. GAAP for SEC registrants. ASU 2009-1 and the Codification
are effective for financial statements issued for interim and annual periods ending after September
15, 2009. The Codification supersedes all existing non-SEC accounting and reporting standards.
All other nongrandfathered non-SEC accounting literature not included in the Codification will
become nonauthoritative. The FASB will no longer issue new standards in the form of Statements,
FASB Staff Positions, or Emerging Issues Task Force Abstracts. Instead, the FASB will issue
Accounting Standards Updates, which will serve only to: (a) update the Codification; (b) provide
background information about the guidance; and (c) provide the bases for conclusions on the
change(s) in the Codification. The adoption of ASU 2009-1 did not have a material impact on our
financial statements.
Contractual Obligations
The following table summarizes our significant contractual obligations at December 31, 2009:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Total |
|
|
Less than 1 year |
|
|
1-3 years |
|
|
3-5 years |
|
|
More than 5 years |
|
Operating lease obligations |
|
$ |
4,695 |
|
|
$ |
1,136 |
|
|
$ |
2,038 |
|
|
$ |
1,521 |
|
|
|
— |
|
Purchase obligations (1) |
|
$ |
5,419 |
|
|
$ |
5,419 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Certain Clinical contract obligations |
|
$ |
3,693 |
|
|
$ |
2,954 |
|
|
$ |
739 |
|
|
|
— |
|
|
|
— |
|
Other long
term liabilities reflected on the balance sheet under GAAP |
|
$ |
1,209 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
$ |
1,209 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total |
|
$ |
15,016 |
|
|
$ |
9,510 |
|
|
$ |
2,777 |
|
|
$ |
1,521 |
|
|
$ |
1,209 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (1) |
|
Represents open purchase orders issued to certain suppliers and other vendors mainly in
connection with certain improvements to our manufacturing facility, that were outstanding as of
December 31, 2009. |
57
Selected Quarterly Financial Data (unaudited)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Three Months Ended |
|
| |
|
2008 |
|
|
2009 |
|
| |
|
(U.
S.
dollars in thousands) |
|
| |
|
March 31 |
|
|
June 30 |
|
|
Sept. 30 |
|
|
Dec. 31 |
|
|
March 31 |
|
|
June 30 |
|
|
Sept. 30 |
|
|
Dec. 31 |
|
Revenues |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
388 |
|
Net loss |
|
$ |
5,113 |
|
|
$ |
4,229 |
|
|
$ |
6,496 |
|
|
$ |
6,576 |
|
|
$ |
5,183 |
|
|
$ |
5,427 |
|
|
$ |
5,894 |
|
|
$ |
14,936 |
|
Net loss per share
of common stock,
basic and diluted |
|
$ |
0.07 |
|
|
$ |
0.06 |
|
|
$ |
0.09 |
|
|
$ |
0.09 |
|
|
$ |
0.07 |
|
|
$ |
0.07 |
|
|
$ |
0.08 |
|
|
$ |
0.19 |
|
58
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Currency Exchange Risk
The currency of the primary economic environment in which our operations are conducted is the
dollar. We are currently have no significant source of revenues; therefore we consider the
currency of the primary economic environment to be the currency in which we expend cash.
Approximately 50% of our expenses and capital expenditures are incurred in dollars, and a
significant source of our financing has been provided in U.S. dollars. Since the dollar is the
functional currency, monetary items maintained in currencies other than the dollar are remeasured
using the rate of exchange in effect at the balance sheet dates and non-monetary items are
remeasured at historical exchange rates. Revenue and expense items are remeasured at the average
rate of exchange in effect during the period in which they occur. Foreign currency translation
gains or losses are recognized in the statement of operations.
Approximately 35% of our costs, including salaries, expenses and office expenses, are incurred in
New Israeli Shekels, the NIS. Inflation in Israel may have the effect of increasing the U.S.
dollar cost of our operations in Israel. If the U.S. dollar declines in value in relation to the
NIS, it will become more expensive for us to fund our operations in Israel. A revaluation of 1% of
the NIS will affect our income before tax by less than 1%. The exchange rate of the U.S. dollar to
the NIS, based on exchange rates published by the Bank of Israel, was as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year Ended December 31, |
| |
|
2007 |
|
2008 |
|
2009 |
Average rate for period |
|
|
4.1081 |
|
|
|
3.5878 |
|
|
|
3.933 |
|
Rate at year-end |
|
|
3.8460 |
|
|
|
3.8020 |
|
|
|
3.775 |
|
To date, we have not engaged in hedging transactions. In the future, we may enter into currency
hedging transactions to decrease the risk of financial exposure from fluctuations in the exchange
rate of the U.S. dollar against the NIS. These measures, however, may not adequately protect us
from material adverse effects due to the impact of inflation in Israel.
Interest Rate Risk
Our exposure to market risk is confined to our cash and cash equivalents. We consider all short
term, highly liquid investments, which include short-term deposits with original maturities of
three months or less from the date of purchase, that are not restricted as to withdrawal or use and
are readily convertible to known amounts of cash, to be cash equivalents. The primary objective of
our investment activities is to preserve principal while maximizing the interest income we receive
from our investments, without increasing risk. We invest any cash balances primarily in bank
deposits and investment grade interest-bearing instruments. We are exposed to market risks
resulting from changes in interest rates. We do not use derivative financial instruments to limit
exposure to interest rate risk. Our interest gains may decline in the future as a result of
changes in the financial markets.
Item 8. Financial Statements and Supplementary Data
See the Index to Consolidated Financial Statements on Page F-1 attached hereto.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
We conducted an evaluation of the effectiveness of the design and operation of our disclosure
controls and procedures as of the end of the period covered by this Form 10-K. The controls
evaluation was conducted under the supervision and with the participation of management, including
our Chief Executive Officer and Chief Financial Officer. Disclosure controls and procedures are
controls and procedures designed to reasonably assure that information required to be disclosed in
our reports filed under the Exchange Act, such as this Form 10-K, is recorded, processed,
summarized and reported within the time periods specified in the Commission’s rules and forms.
Disclosure controls and procedures are also designed to reasonably assure that such information is
accumulated and communicated to our management, including the Chief Executive Officer and Chief
Financial Officer, as appropriate to allow timely decisions regarding required disclosure.
59
The evaluation of our disclosure controls and procedures included a review of the controls’
objectives and design, our implementation of the controls and their effect on the information
generated for use in this Form 10-K. In the course of the controls evaluation, we reviewed
identified data errors, control problems or acts of fraud, and sought to confirm that appropriate
corrective actions, including process improvements, were being undertaken. This type of evaluation
will be performed on a quarterly basis so that the conclusions of management, including the Chief
Executive Officer and Chief Financial Officer, concerning the effectiveness of the disclosure
controls and procedures can be reported in our periodic reports on Form 10-Q and Form 10-K. The
overall goals of these various evaluation activities are to monitor our disclosure controls and
procedures, and to modify them as necessary. Our intent is to maintain the disclosure controls and
procedures as dynamic systems that change as conditions warrant.
Based on the controls evaluation, our Chief Executive Officer and Chief Financial Officer have
concluded that, as of the end of the period covered by this Form 10-K, our disclosure controls and
procedures were effective to provide reasonable assurance that information required to be disclosed
in our Exchange Act reports is recorded, processed, summarized and reported within the time periods
specified by the Commission, and that material information related to our company and our
consolidated subsidiary is made known to management, including the Chief Executive Officer and
Chief Financial Officer, particularly during the period when our periodic reports are being
prepared.
Management Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over
financial reporting to provide reasonable assurance regarding the reliability of our financial
reporting and the preparation of financial statements for external purposes in accordance with U.S.
generally accepted accounting principles. Internal control over financial reporting includes those
policies and procedures that (i) pertain to the maintenance of records that in reasonable detail
accurately and fairly reflect the transactions and dispositions of our assets; (ii) provide
reasonable assurance that transactions are recorded as necessary to permit preparation of financial
statements in accordance with U.S. generally accepted accounting principles, and that receipts and
expenditures of our company are being made only in accordance with authorizations of management and
our directors; and (iii) provide reasonable assurance regarding prevention or timely detection of
unauthorized acquisition, use or disposition of our assets that could have a material effect on our
financial statements.
Management assessed our internal control over financial reporting as of December 31, 2009, the end
of our fiscal year. Management based its assessment on criteria established in Internal
Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway
Commission. Management’s assessment included evaluation of elements such as the design and
operating effectiveness of key financial reporting controls, process documentation, accounting
policies and our overall control environment.
Based on our assessment, management has concluded that our internal control over financial
reporting was effective as of the end of the fiscal year to provide reasonable assurance regarding
the reliability of financial reporting and the preparation of financial statements for external
reporting purposes in accordance with U.S. generally accepted accounting principles. We reviewed
the results of management’s assessment with the Audit Committee of our Board of Directors.
Our independent registered public accounting firm has audited management’s assessment of our
internal control over financial reporting, and issued an unqualified
opinion dated February 25,
2010 on such assessment and on our internal control over financial reporting, which opinion is
included herein.
Inherent Limitations on Effectiveness of Controls
Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect
that our disclosure controls and procedures or our internal control over financial reporting will
prevent or detect all error and all fraud. A control system, no matter how well designed and
operated, can provide only reasonable, not absolute, assurance that the control system’s objectives
will be met. The design of a control system must reflect the fact that there are resource
constraints, and the benefits of controls must be considered relative to their costs. Further,
because of the inherent limitations in all control systems, no evaluation of controls can provide
absolute assurance that misstatements due to error or fraud will not occur or that all control
issues and instances of fraud, if any, within a company have been detected. These inherent
limitations include the realities that judgments in decision-making can be faulty and that
breakdowns can occur because of simple error or mistake. Controls can also be circumvented by the
individual acts of some persons, by collusion of two or more people or by management override of
the controls. The design of any system of controls is based in part on certain assumptions about
the likelihood of future events, and there can be no assurance that any design will succeed in
achieving its stated goals under all potential future conditions. Projections of any evaluation of
controls effectiveness to future periods are subject to risks. Over
60
time, controls may become inadequate because of changes in conditions or deterioration in the
degree of compliance with policies or procedures.
Changes in internal controls
There were no changes in our internal control over financial reporting (as defined in Rules 13a-15f
and 15d-15f under the Exchange Act) that occurred during the period ended December 31, 2009 that
have materially affected, or that are reasonably likely to materially affect, our internal control
over financial reporting.
Item 9B. Other Information
None.
61
PART III
Item 10. Directors, Executive Officers and Corporate Governance
Our directors and executive officers, their ages and positions as of February 15, 2010, are as
follows:
| |
|
|
|
|
|
|
| Name |
|
Age |
|
Position |
Directors |
|
|
|
|
|
|
Eli Hurvitz |
|
|
77 |
|
|
Chairman of the Board |
David Aviezer, Ph.D., MBA |
|
|
45 |
|
|
Director, President and Chief Executive Officer |
Yoseph Shaaltiel, Ph.D. |
|
|
56 |
|
|
Director and Executive VP, Research and Development |
Alfred Akirov (1)(2)(3) |
|
|
69 |
|
|
Director |
Amos Bar Shalev (1)(2)(3) |
|
|
57 |
|
|
Director |
Zeev Bronfeld |
|
|
58 |
|
|
Director |
Yodfat Harel Gross (1)(2)(3) |
|
|
37 |
|
|
Director |
Roger D. Kornberg, Ph.D. |
|
|
62 |
|
|
Director |
Eyal Sheratzky |
|
|
41 |
|
|
Director |
Executive Officers |
|
|
|
|
|
|
Einat Brill Almon, Ph.D. |
|
|
50 |
|
|
Vice President, Product Development |
Yossi Maimon, CPA |
|
|
39 |
|
|
Vice President, Chief Financial Officer, Treasurer and Secretary |
Sandra L Lauterbach |
|
|
41 |
|
|
Vice President, Sales and Commercial Affairs |
|
|
|
| (1) |
|
Member of Nominating Committee |
| |
| (2) |
|
Member of Audit Committee |
| |
| (3) |
|
Member of Compensation Committee |
Eli Hurvitz. Mr. Hurvitz serves as Chairman of our Board of Directors and has served as a director
of Protalix Ltd. since 2005 and as our director since December 31, 2006. Mr. Hurvitz has served as
Chairman of the Board of Teva (TASE:TEVA; NASDAQ:TEVA) since April 2002. Previously, he served as
Teva’s President and Chief Executive Officer for over 25 years and has been involved with Teva in
various capacities for over 40 years. Mr. Hurvitz is an experienced veteran of the pharmaceutical
industry. In addition, he currently serves as Chairman of the Board of The Israel Democracy
Institute (IDI), Chairman of the Board of NeuroSurvival Technologies Ltd. (a private company) and a
director of Vishay Intertechnology. He served as Chairman of the Israel Export Institute from 1974
through 1977 and as the President of the Israel Manufacturers Association from 1981 through 1986.
He served as Chairman of the Board of Bank Leumi Ltd. from 1986 through 1987. He was a director of
Koor Industries Ltd. from 1997 through 2004 and a member of the Belfer Center for Science and
International Affairs at the John F. Kennedy School of Government at Harvard University from 2002
through 2005. He received his B.A. in Economics and Business Administration from the Hebrew
University of Jerusalem in 1957.
David Aviezer, Ph.D., MBA. Dr. Aviezer has served as Chief Executive Officer of Protalix Ltd.
since 2002 and its director since 2005 and as our director since December 31, 2006. On December
31, 2006, he became our President and Chief Executive Officer. Dr. Aviezer has over 15 years of
experience in biotechnology management, advancing products from early-stage research up to their
regulatory approval and commercialization. Prior to joining Protalix Ltd., from 1996 to 2002, he
served as General Manager of ProChon Biotech Ltd., an Israeli company focused on orthopedic
disorders. Previously, Dr. Aviezer was a visiting scientist at the Medical Research Division of
American Cyanamid, a subsidiary of Wyeth which was subsequently acquired by Pfizer (NYSE:PFE), in
New York. Since 1996, Dr. Aviezer has served as an Adjunct Lecturer at Bar Ian University. Dr.
Aviezer is the recipient of the Clore Foundation Award and the J.F. Kennedy Scientific Award. He
holds a Ph.D. in Molecular Biology and Biochemistry from the Weizmann Institute of Science and an
MBA from the Bar Ilan University Business School.
Yoseph Shaaltiel, Ph.D. Dr. Shaaltiel founded Protalix Ltd. in 1993 and has served as a member of
our Board of Directors and as our Vice President, Research and Development since December 31, 2006.
Prior to establishing Protalix Ltd., from 1988 to 1993, Dr. Shaaltiel was a Research Associate at
the MIGAL Technological Center. He also served as Deputy Head of the Biology Department of the
Biological and Chemical Center of the Israeli Defense Forces and as a Biochemist at Makor Chemicals
Ltd. Dr. Shaaltiel was a Postdoctoral Fellow at the University of California at Berkeley and at
Rutgers University in New Jersey. He has co-authored over 40 articles and abstracts on plant
biochemistry and holds seven patents. Dr. Shaaltiel received his Ph.D. in Plant Biochemistry from
the Weizmann Institute of Science, an M.Sc. in Biochemistry from the Hebrew University and a B.Sc.
in Biology from the Ben Gurion University.
62
Alfred Akirov. Mr. Akirov has served as our director since January 2008. Mr. Akirov is the
founder, chairman of the Board of Directors and chief executive officer of the Alrov Group (TASE:
ALRO), an Israeli publicly-traded company that is listed on the Tel Aviv Stock Exchange. Mr.
Akirov founded the Alrov Group in 1978 and it is currently one of Israel’s largest real-estate
companies. The Alrov Group holds 80% of the capital stock of Techno-Rov Holdings (1993) Ltd., one
of our shareholders. Mr. Akirov serves in different capacities, including chairman, chief
executive officer and director, for a number of private companies in the Alrov Group and Techno-Rov
portfolios. Mr. Akirov serves on the Executive Council and the Board of Governors of the Tel Aviv
University.
Amos Bar Shalev. Mr. Bar Shalev has served as our director since July 2008. Mr. Bar Shalev served
as a director of Protalix Ltd. from 2005 through January 31, 2008, and as our director from
December 31, 2006 through January 31, 2008. Mr. Bar Shalev was not nominated for reelection at our
annual meeting of shareholders on January 31, 2008. On July 14, 2008, our Board of Directors
appointed Mr. Bar Shalev to serve on the board. Mr. Bar Shalev brings to us extensive experience
in managing technology companies. Currently, Mr. Bar Shalev manages the Technorov portfolio.
Until 2004, he was the Managing Director of TDA Capital Partners, a management company of the TGF
(Templeton Tadiran) Fund. Prior to that, from 2004 through 2007, he was the President of Win Buyer
Ltd. From 2000 through 2007, Mr. Bar Shalev served the Director of Technorov Holdings (1993) Ltd.
and from 2004 through 2007 he served as the Director of Golden Wings Investment Company Ltd. He
has served on the board of directors of many companies, such as Golden Wings Investment Company
Ltd., Win Buyer Ltd. and Sun Light. He received his B.Sc. in Electrical Engineering from the
Technion, Israel in 1978 and M.B.A. from the Tel Aviv University in 1981. He holds the highest
award from the Israeli Air Force for technological achievements.
Zeev Bronfeld. Mr. Bronfeld has served as a director of Protalix Ltd. since 1996 and as our
director since December 31, 2006. Mr. Bronfeld brings to us vast experience in management and
value building of biotechnology companies. Mr. Bronfeld is an experienced businessman who is
involved in a number of biotechnology companies. He is a co-founder of Biocell Ltd. (TASE:BCEL),
an Israeli publicly traded holding company specializing in biotechnology companies and has served
as its Chief Executive Officer since 1986. Mr. Bronfeld currently serves as a director of Biocell
Ltd., D. Medical Industries Ltd. (TASE:DMDC), and Biomedix Incubator Ltd. (TASE:BMDX), all of which
are public companies traded on the Tel Aviv Stock Exchange. Mr. Bronfeld is also a director of
each of the following privately-held companies: Meitav Technological Incubator Ltd., Ecocycle
Israel Ltd., Contipi Ltd., Nilimedix Ltd., G-Sense Ltd., Sindolor Medical Ltd., L.N. Innovative
Technologies, A.T.I Ashkelon Industries Information Technologies Ltd., T.I.F. Ventures Ltd., MOFET
B’Yehuda – Industrial Research & Development in Judea Ltd., Incubator for Management of
Technological Entrepreneurship Misgav Ltd., A.Y.M.B. Holdings and Investments Ltd., Macrocure Ltd.,
Medx-set Ltd., Braintact Ltd., Active P Ltd., and Angio B Ltd. Mr. Bronfeld received a B.A. in
Economics from the Hebrew University in 1975.
Yodfat Harel Gross. Ms. Harel Gross has served as our director since June 2007. Since 2006, Ms.
Harel Gross has been a Managing Director of Tamares Capital Ltd., a private investment group with
interests in real estate, technology, manufacturing, leisure and media. At Tamares Capital, Ms.
Harel Gross serves as the Business Development Director and the head of the Israel office. Prior
to joining Tamares Capital, from 2004 to 2006, she was the Head of the Medical Desk of Orbotech,
Ltd. (NASDAQ:ORBK), a company providing high-tech inspection and imaging solutions for bare printed
circuit board (PCB), flat panel display (FPD) and PCB assembly manufacturing worldwide. Prior to
that, from 1994 to 2003, she was a Managing Director of Harel-Hertz Investment House Ltd., a
business investment company with offices in Tel Aviv, Israel and Tokyo, Japan. In 2002,
Harel-Hertz Investment House became the Israeli representative office for ITX Corporation, a
publicly-traded company in Japan. Ms. Harel Gross currently serves on the board of directors of
Tamares Capital, Tamares Hotels, Tamares Real Estate, Storewiz and Halman-Aldubi Provident Funds,
Ltd. Ms. Harel Gross holds a B.A. in Communication and Political Science from Bar Ilan University
and an executive M.B.A. from Bradford University, Great Britain. She has also completed programs
in Directors’ Studies and Advanced Advertising and Marketing at the Israel Management Center.
Roger D. Kornberg, Ph.D. Professor Kornberg has served as our director since February 2008. He
has served as a director of Teva since 2007. Professor Kornberg is a member of the U.S. National
Academy of Sciences and the Winzer Professor of Medicine in the Department of Structural Biology at
Stanford University, Stanford, California. He has been a member of the faculty of Stanford
University since 1972. Prior to that, he was a professor at Harvard Medical School. Professor
Kornberg is a renowned biochemist and in 2006 he was awarded the Nobel Prize in Chemistry in
recognition for his studies of the molecular basis of eukaryotic transcription, the process by
which DNA is copied to RNA. Professor Kornberg is also the recipient of several awards, including
the 2001 Welch Prize, the highest award granted in the field of chemistry in the United States, and
the 2002 Leopold Mayer Prize, the highest award granted in the field of biomedical sciences from
the French Academy of Sciences. He received his B.S. in Chemistry from Harvard University in 1967
and his Ph.D. in Chemistry from
63
Stanford University in 1972. He holds honorary degrees from universities in Europe and Israel,
including the Hebrew University in Jerusalem, where he currently is a visiting professor.
Eyal Sheratzky. Mr. Sheratzky has served as a director of Protalix Ltd. since 2005 and as our
director since December 31, 2006. Mr. Sheratzky has served as a director of Ituran Location &
Control (NASDAQ:ITRN), a publicly-traded company quoted on the Nasdaq, since 1995 and as a Co-Chief
Executive Officer since 2003. Prior to such date, he served as an alternate Chief Executive
Officer of Ituran from 2002 through 2003 and as Vice President of Business Development from 1999
through 2002. Mr. Sheratzky is the Chairman of the Board of Directors of Biocell and serves as a
director of Moked Ituran Ltd. and of Ituran’s subsidiaries. From 1994 to 1999 he served as the
Chief Executive Officer of Moked Services, Information and Investments Ltd. and as legal advisor to
several of Ituran’s affiliated companies. Mr. Sheratzky holds LL.B and LL.M degrees from Tel Aviv
University School of Law and an Executive M.B.A. degree from Kellogg University.
Einat Brill Almon, Ph.D. Dr. Almon joined Protalix Ltd. in December 2004 as its Vice President,
Product Development and became our Vice President, Product Development on December 31, 2006. Dr.
Almon has many years of experience in the management of life science projects and companies,
including biotechnology and agrobiotech, with direct experience in clinical, device and scientific
software development, as well as a strong background and work experience in Intellectual Property.
Prior to joining Protalix Ltd., from 2001 to 2004, she served as Director of R&D and IP of
Biogenics Ltd., a company that developed an autologous platform for tissue based protein drug
delivery. Biogenics, based in Israel, is a wholly-owned subsidiary of Medgenics Inc. Dr. Almon
has trained as a biotechnology patent agent at leading IP firms in Israel. Dr. Almon holds a Ph.D.
and an M.Sc. in molecular biology of cancer research from the Weizmann Institute of Science, a
B.Sc. from the Hebrew University and has carried out Post-Doctoral research at the Hebrew
University in the area of plant molecular biology.
Yossi Maimon, CPA. Mr. Maimon joined Protalix Ltd. on October 15, 2006 as its Chief Financial
Officer and became our Vice President and Chief Financial Officer on December 31, 2006. Prior to
joining Protalix, from 2002 to 2006, he served as the Chief Financial Officer of Colbar LifeScience
Ltd., a biomaterial company focusing on aesthetics, where he led all of the corporate finance
activities, fund raisings and legal aspects of Colbar including the sale of Colbar to Johnson and
Johnson. Mr. Maimon has a B.A. in accounting from the City University of New York and an M.B.A.
from Tel Aviv University, and he is a Certified Public Accountant in the United States (New York
State) and Israel.
Sandra L. Lauterbach. Ms. Lauterbach has served as our Vice President, Sales and Commercial
Affairs since December 18, 2009. Prior to joining our company, Ms. Lauterbach was the Vice
President of Marketing, Endocrinology of EMD Serono, Inc., from July 2008 through July 2009. Prior
to that, from August 2003 through July 2008, she served in a number of positions at Genzyme
Corporation, the last position being the Senior Director, Global Marketing for Fabrazyme. Ms.
Lauterbach holds a B.Sc. in Molecular Biology from the University of Wisconsin and an MBA from the
University of South Florida.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our directors, executive officers and holders of more
than 10% of our common stock to file with the Commission reports regarding their ownership and
changes in ownership of our equity securities. Dr. Shaaltiel, and Mr. Bar Shalev each filed a late
Form 4 in connection with certain purchases of our common stock in 2009. Otherwise, we believe
that all Section 16 filings requirements were met during 2009. In making this statement, we have
relied solely upon examination of the copies of Forms 3, 4 and 5 provided to us and the written
representations of our former and current directors, officers and 10% shareholders.
Audit Committee
We require that all Audit Committee members possess the required level of financial literacy and at
least one member of the Audit Committee meet the current standard of requisite financial management
expertise as required by the NYSE Amex and applicable rules and regulations of the SEC. Messrs.
Bar-Shalev and Akirov, and Ms. Harel Gross have been appointed by the Board of Directors to serve
on the Audit Committee until their respective successors have been duly elected.
Our Audit Committee operates under a formal charter that governs its duties and conduct.
All members of the Audit Committee are independent from our executive officers and management.
Our independent registered public accounting firm reports directly to the Audit Committee.
64
Our Audit Committee meets with management and representatives of our registered public accounting
firm prior to the filing of officers’ certifications with the Commission to receive information
concerning, among other things, effectiveness of the design or operation of our internal controls
over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act of 2002.
Our Audit Committee has adopted a Policy for Reporting Questionable Accounting and Auditing
Practices and Policy Prohibiting Retaliation against Reporting employees to enable confidential and
anonymous reporting of improper activities to the Audit Committee.
Messrs. Bar-Shalev and Akirov qualify as “audit committee financial experts” under the applicable
rules of the SEC. In making the determination as to these individuals’ status as audit committee
financial experts, our Board of Directors determined they have accounting and related financial
management expertise within the meaning of the aforementioned rules, as well as the listing
standards of the NYSE Amex.
Code of Business Conduct and Ethics
We have adopted a Code of Business Conduct and Ethics that includes provisions ranging from
restrictions on gifts to conflicts of interest. All of our employees and directors are bound by
this Code of Business Conduct and Ethics. Violations of our Code of Business Conduct and Ethics
may be reported to the Audit Committee.
The Code of Business Conduct and Ethics includes provisions applicable to all of our employees,
including senior financial officers and members of our Board of Directors and is posted on our
website (www.protalix.com). We intend to post amendments to or waivers from any such Code of
Business Conduct and Ethics.
Item 11. Executive Compensation
Compensation Discussion and Analysis
The primary goals of the Compensation Committee of our Board of Directors with respect to executive
compensation are to attract and retain the most talented and dedicated executives possible, to tie
annual and long-term cash and stock incentives to achievement of specified performance objectives,
and to align executives’ incentives with shareholder value creation. To achieve these goals, the
Compensation Committee intends to implement and maintain compensation plans that tie a portion of
executives’ overall compensation to key strategic goals such as developments in our clinical path,
the establishment of key strategic collaborations, the build-up of our pipeline and the
strengthening of our financial position. The Compensation Committee evaluates individual executive
performance with a goal of setting compensation at levels the committee believes are comparable
with executives in other companies of similar size and stage of development operating in the
biotechnology industry while taking into account our relative performance and our own strategic
goals.
Elements of Compensation
Executive compensation consists of following elements:
Base Salary. Base salaries for our executives are established based on the scope of their
responsibilities taking into account competitive market compensation paid by other companies for
similar positions. Generally, we believe that executive base salaries should be targeted near the
median of the range of salaries for executives in similar positions with similar responsibilities
at comparable companies. Base salaries are usually reviewed annually, and adjusted from time to
time to realign salaries with market levels after taking into account individual responsibilities,
performance and experience. The review for 2009 took place during the first quarter of 2010. The
base salaries of our Named Executive Officers are set forth in “Employment Arrangements.”
On February 25, 2010, our Board of Directors, acting upon the resolution of a majority of our
independent directors, resolved to increase for 2010 the monthly salaries of our Named Executive
Officers at further described below.
Annual Bonus. The Compensation Committee has the authority to award discretionary annual
bonuses to our executive officers. It has established a formal bonus plan for certain milestones
expected to occur during 2010. These awards are intended to compensate officers for achieving
financial, clinical and operational goals and for achieving individual annual performance
objectives. These objectives vary depending on the individual executive, but relate generally to
strategic factors such as developments in our clinical path, the execution of license agreement for
the commercialization of our lead product candidates, the establishment of key strategic
collaborations, the build-up of our pipeline and to financial factors such as raising capital.
65
For each year, the Compensation Committee will select, in its discretion, the executive
officers of our company or our subsidiary who are eligible to receive bonuses. Any bonus granted
by the Compensation Committee will generally be paid in the first quarter following completion of a
given year, unless such bonuses were specifically granted for a specific milestone achieved during
the year which was payable immediately after the achievement of the milestone. Similar to bonuses
paid in the past, the actual amount of discretionary bonus will be determined following a review of
each executive’s individual performance and contribution to our goals. The Compensation Committee
has not fixed a minimum or maximum payout for any officer’s annual discretionary bonus, unless
specified in an executive’s employment agreement.
Pursuant to each officer’s employment agreement, the executive officer is eligible for a
discretionary annual bonus. The Compensation Committee determined the discretionary annual bonus
to be paid to our executive officers, and the discretionary bonus to be awarded to certain officers
in 2010 for performance in 2009 and in 2008. The Compensation Committee elected to pay bonuses to
the executive officers for their performance in 2008 as bonuses were not paid in 2009 due to the
general market conditions and our cash balance at that time. The actual amount of the
discretionary bonus to be paid to each executive officer is determined following a review of the
executive’s individual performance and contribution to our strategic goals conducted during the
first quarter of each fiscal year. The Compensation Committee has not fixed a minimum or a maximum
amount for any officer’s annual discretionary bonus.
On February 25, 2010, our Board of Directors, acting upon the resolution of a majority of our
independent directors, awarded a total of approximately
$2.6 million in bonuses to our executive officers, out of which
approximately $1.0 million is to
be paid during the first quarter of 2010 for achievements for the years 2008 and 2009 including,
among others, the execution of the license and supply agreement for the commercialization of our
lead product candidate, the completion of our phase III clinical trial for our lead product
candidate and the upgrade of our manufacturing facility. The
remaining approximately $1.5 million is to be paid
upon, and subject to, the FDA’s approval of taliglucerase alfa
and upon the first shipment of taliglucerase alfa, if at all.
Options. Our 2006 Stock Option Plan authorizes us to grant options to purchase shares of
common stock to our employees, directors and consultants. Our Compensation Committee is the
administrator of the stock option plan. Stock option grants are generally made at the commencement
of employment and following a significant change in job responsibilities or to meet other special
retention or performance objectives. The Compensation Committee reviews and approves stock option
awards to executive officers based upon a review of competitive compensation data, its assessment
of individual performance, a review of each executive’s existing long-term incentives, and
retention considerations. The exercise price of stock options granted under the 2006 Stock
Incentive Plan must be equal to at least 100% of the fair market value of our common stock on the
date of grant; however, in certain circumstances, grants may be made at a lower price to Israeli
grantees who are residents of the State of Israel.
On February 25, 2010, our Board of Directors, acting upon the resolution of a majority of our
independent directors, granted stock options to our President and Chief Executive Officer, our
Executive Vice President, Research and Development, our Vice President, Product Development and our
Vice President and Chief Financial Officer. The number of shares of common stock underlying the
option grants was 250,000, 145,000, 130,000 and 130,000, respectively. The options have
an exercise price of $6.90 per share and will vest quarterly over three
years after the FDA’s approval of taliglucerase alfa, if at all. The grants of stock options to such
officers were in recognition their ongoing efforts in achieving our milestones regarding clinical
developments, research and development, financial developments and other factors during 2009 and 2010.
Severance and Change in Control Benefits. Pursuant to the employment agreements entered into
with each of our executive officers based in Israel, the executive officer is entitled to be
insured by Protalix Ltd. under a Manager’s Policy in lieu of severance. The intention of such
Manager’s Policies is to provide the Israel-based officers with severance protection of one month’s
salary for each year of employment. In addition, stock option agreements with each of our named
executive officers, as amended, provide that all of the outstanding options of each Named Executive
Officer are subject to accelerated vesting immediately upon a change in control of our company.
Other Compensation. Consistent with our compensation philosophy, we intend to continue to
maintain our current benefits for our executive officers; however, the Compensation Committee in
its discretion may revise, amend, or add to the officer’s executive benefits if it deems it
advisable. As an additional benefit to all of our Israel-based Named Executive Officers and for
most of our employees, we generally contribute to certain funds amounts equaling a total of
approximately 15% of their gross salaries for certain pension and other savings plans for the
benefit of the Named Executive Officers. In addition, in accordance with customary practice in
Israel, our Israel-based executives’ agreements require us to contribute towards their vocational
studies, and to provide annual recreational allowances, a company car and a company phone. We
believe these benefits are currently equivalent with median competitive levels for comparable
companies.
Executive Compensation. We refer to the “Summary Compensation Table” set forth in Section 11
of the Annual Report on Form 10-K for information regarding the compensation earned during the
fiscal year ended December 31, 2009 by:
66
our President and Chief Executive Officer; our Executive Vice President, Research and
Development; our Vice President, Product Development; and our Vice President and Chief Financial
Officer, who we refer to collectively as the “Named Executive Officers.” There are no other
executive officers for 2009 whose total compensation exceeded $100,000 during that fiscal year
other than the Named Executive Officers.
Compensation Committee Report
The above report of the Compensation Committee does not constitute soliciting material and shall
not be deemed filed or incorporated by reference into any other filing by us under the Securities
Act of 1933 or the Securities Exchange Act of 1934.
The Compensation Committee has reviewed and discussed the Compensation Discussion and Analysis set
forth below with our management. Based on this review and discussion, the Compensation Committee
has recommended to our Board of Directors that the Compensation Discussion and Analysis be included
in our Annual Report on Form 10–K and our annual proxy statement on Schedule 14A.
Respectfully submitted on February 25, 2010, by the members of the Compensation Committee of the
Board of Directors.
Yodfat Harel Gross
Alfred Akirov
Amos Bar Shalev
Summary Compensation Table
The following table sets forth a summary for the fiscal years ended December 31, 2009 and 2008
respectively, of the cash and non-cash compensation awarded, paid or accrued by us or Protalix Ltd.
to each of our President and Chief Executive Officer, our Executive Vice President, Research and
Development, our Vice President, Product Development and our Vice President and Chief Financial
Officer, who we refer to collectively as the “Named Executive Officers.” There were no restricted
stock awards, long-term incentive plan payouts or other compensation paid during fiscal years 2009
and 2008 by us or Protalix Ltd. to the Named Executive Officers, except as set forth below. All of
the Named Executive Officers are employees of our subsidiary, Protalix Ltd. All currency amounts
are expressed in U.S. dollars.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-Equity |
|
Nonqualified |
|
All Other |
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock |
|
Option |
|
Incentive Plan |
|
Deferred |
|
Compen- |
|
|
| Name and |
|
|
|
|
|
Salary |
|
Bonus |
|
Award(s) |
|
Award(s) |
|
Compensation |
|
Compensation |
|
sation |
|
Total |
| Principal Position |
|
Year |
|
($) |
|
($) |
|
($) |
|
($) |
|
($) |
|
Earnings ($) |
|
($)(1) |
|
($) |
David Aviezer, Ph.D., MBA |
|
|
2009 |
|
|
|
427,970 |
|
|
|
500,000 |
|
|
|
|
|
|
|
529,951 |
|
|
|
|
|
|
|
|
|
|
|
62,167 |
|
|
|
1,520,088 |
|
President and |
|
|
2008 |
|
|
|
486,305 |
|
|
|
— |
|
|
|
|
|
|
|
565,394 |
|
|
|
|
|
|
|
|
|
|
|
93,224 |
|
|
|
1,144,923 |
|
Chief Executive Officer |
|
|
2007 |
|
|
|
341,074 |
|
|
|
239,210 |
|
|
|
|
|
|
|
351,343 |
|
|
|
|
|
|
|
|
|
|
|
67,990 |
|
|
|
999,617 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Yoseph Shaaltiel, Ph.D. |
|
|
2009 |
|
|
|
196,271 |
|
|
|
160,000 |
|
|
|
|
|
|
|
225,336 |
|
|
|
|
|
|
|
|
|
|
|
37,981 |
|
|
|
619,588 |
|
Executive Vice President, |
|
|
2008 |
|
|
|
226,652 |
|
|
|
— |
|
|
|
|
|
|
|
163,328 |
|
|
|
|
|
|
|
|
|
|
|
54,704 |
|
|
|
444,684 |
|
Research and Development |
|
|
2007 |
|
|
|
117,297 |
|
|
|
50,000 |
|
|
|
|
|
|
|
2,420 |
|
|
|
|
|
|
|
|
|
|
|
47,339 |
|
|
|
277,056 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Einat Brill Almon, Ph.D. |
|
|
2009 |
|
|
|
172,210 |
|
|
|
160,000 |
|
|
|
|
|
|
|
203,388 |
|
|
|
|
|
|
|
|
|
|
|
36,927 |
|
|
|
652,525 |
|
Vice President, |
|
|
2008 |
|
|
|
195,559 |
|
|
|
28,932 |
|
|
|
|
|
|
|
253,862 |
|
|
|
|
|
|
|
|
|
|
|
51,223 |
|
|
|
529,576 |
|
Product Development |
|
|
2007 |
|
|
|
153,254 |
|
|
|
65,171 |
|
|
|
|
|
|
|
94,482 |
|
|
|
|
|
|
|
|
|
|
|
42,282 |
|
|
|
355,189 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Yossi Maimon, CPA |
|
|
2009 |
|
|
|
186,478 |
|
|
|
160,000 |
|
|
|
|
|
|
|
253,030 |
|
|
|
|
|
|
|
|
|
|
|
41,051 |
|
|
|
640,599 |
|
Vice President, |
|
|
2008 |
|
|
|
203,097 |
|
|
|
30,659 |
|
|
|
|
|
|
|
238,194 |
|
|
|
|
|
|
|
|
|
|
|
83,808 |
|
|
|
555,758 |
|
Chief Financial Officer |
|
|
2007 |
|
|
|
156,444 |
|
|
|
77,223 |
|
|
|
|
|
|
|
247,815 |
|
|
|
|
|
|
|
|
|
|
|
41,975 |
|
|
|
523,457 |
|
|
|
|
| (1) |
|
Includes employer contributions to pension and/or insurance plans and other miscellaneous
payments. |
67
The following table summarizes the grant of awards made to the Named Executive Officers during
2009 as of December 31, 2009.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| GRANTS OF PLAN-BASED AWARDS |
| |
|
Grant |
|
Estimated Future Payouts Under |
|
Estimated Future Payouts Under Equity |
|
|
|
|
|
|
|
|
|
|
| Name |
|
Date |
|
Non-Equity Incentive Plan Awards |
|
Incentive Plan Awards |
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
All |
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other |
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock Awards: |
|
All other |
|
|
|
|
|
Grant |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Number |
|
Option Awards: |
|
Exercise |
|
Date fair |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
of |
|
Number of |
|
or Base |
|
Value of |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Shares |
|
Securities |
|
Price of |
|
Stock and |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
of Stock |
|
Underlying |
|
Option |
|
Option |
| |
|
|
|
|
|
Threshold |
|
Target |
|
Maximum |
|
Threshold |
|
Target |
|
Maximum |
|
or Units |
|
Options (#) |
|
Awards |
|
Awards |
| |
|
|
|
|
|
($) |
|
($) |
|
($) |
|
($) |
|
($) |
|
($) |
|
(#) |
|
(1) |
|
($/Sh) (2) |
|
($)(3) |
| |
| (a) |
|
(b) |
|
(c) |
|
(d) |
|
(e) |
|
(f) |
|
(g) |
|
(h) |
|
(i) |
|
(j) |
|
(k) |
|
(l) |
| |
David Aviezer |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Yoseph Shaaltiel |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Einat Brill Almon |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Yossi Maimon |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (1) |
|
Represents outstanding options at December 31, 2009. |
| |
| (2) |
|
Represents the range of the exercise price of the stock options. |
| |
| (3) |
|
Represents the fair value as recorded on the grant date of the stock options. |
Outstanding Equity Awards at Fiscal Year-End
The following table sets forth information with respect to the Named Executive Officers concerning
equity awards as of December 31, 2009.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Option Awards |
|
Stock Awards |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Equity |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Equity |
|
Incentive |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Incentive |
|
Plan |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Plan |
|
Awards: |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Awards: |
|
Market |
| |
|
|
|
|
|
|
|
|
|
Equity |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Number |
|
or Payout |
| |
|
|
|
|
|
|
|
|
|
Incentive |
|
|
|
|
|
|
|
|
|
|
|
|
|
Market |
|
of |
|
Value |
| |
|
|
|
|
|
|
|
|
|
Plan |
|
|
|
|
|
|
|
|
|
|
|
|
|
Value of |
|
Unearned |
|
of |
| |
|
|
|
|
|
|
|
|
|
Awards: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Shares |
|
Shares, |
|
Unearned |
| |
|
Number |
|
Number |
|
Number |
|
|
|
|
|
|
|
|
|
Number |
|
or Units |
|
Units or |
|
Shares, |
| |
|
of Securities |
|
of Securities |
|
of Securities |
|
|
|
|
|
|
|
|
|
of Shares |
|
of Stock |
|
Other |
|
Units or |
| |
|
Underlying |
|
Underlying |
|
Underlying |
|
|
|
|
|
|
|
|
|
or Units |
|
That |
|
Rights |
|
Other |
| |
|
Unexercised |
|
Unexercised |
|
Unexercised |
|
Option |
|
|
|
of Stock |
|
Have |
|
That |
|
Rights |
| |
|
Options |
|
Options |
|
Unearned |
|
Exercise |
|
Option |
|
That |
|
Not |
|
Have Not |
|
That |
| |
|
Exercisable |
|
Unexercisable |
|
Options |
|
Price |
|
Expiration |
|
Have Not |
|
Vested |
|
Vested |
|
Have Not |
| Name |
|
(#) |
|
(#) |
|
(#) |
|
($) |
|
Date |
|
Vested (#) |
|
($) |
|
(#) |
|
Vested ($) |
David Aviezer |
|
|
326,267 |
|
|
|
— |
|
|
|
— |
|
|
|
0.120 |
|
|
|
8/1/2013 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
855,135 |
|
|
|
122,162 |
|
|
|
— |
|
|
|
0.972 |
|
|
|
9/10/2016 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
200,001 |
|
|
|
399,999 |
|
|
|
— |
|
|
|
5.00 |
|
|
|
2/7/2018 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
100,000 |
|
|
|
— |
|
|
|
— |
|
|
|
2.65 |
|
|
|
2/25/2019 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Yoseph Shaaltiel |
|
|
244,324 |
|
|
|
— |
|
|
|
— |
|
|
|
0.001 |
|
|
|
6/30/2011 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
87,910 |
|
|
|
175,818 |
|
|
|
— |
|
|
|
5.00 |
|
|
|
2/7/2018 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
50,000 |
|
|
|
— |
|
|
|
— |
|
|
|
2.65 |
|
|
|
2/25/2019 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Einat Brill Almon |
|
|
4,740 |
|
|
|
— |
|
|
|
— |
|
|
|
0.399 |
|
|
|
5/23/2016 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
158,811 |
|
|
|
73,297 |
|
|
|
— |
|
|
|
0.972 |
|
|
|
8/13/2016 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
103,756 |
|
|
|
207,516 |
|
|
|
|
|
|
|
5.00 |
|
|
|
2/7/2018 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
50,000 |
|
|
|
— |
|
|
|
— |
|
|
|
2.65 |
|
|
|
2/25/2019 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Yossi Maimon |
|
|
107,488 |
|
|
|
116,245 |
|
|
|
— |
|
|
|
0.972 |
|
|
|
9/19/2016 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
58,333 |
|
|
|
116,667 |
|
|
|
|
|
|
|
5.00 |
|
|
|
2/7/2018 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
50,000 |
|
|
|
— |
|
|
|
— |
|
|
|
2.65 |
|
|
|
2/25/2019 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
68
================================================================================
Option exercises during 2009 and vested stock awards for Named Executive Officers as of
December 31, 2009 were as follows:
OPTION EXERCISES AND STOCK VESTED
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Option Awards |
|
Stock Awards |
| |
| |
|
Number of Shares Acquired |
|
Value Received on |
|
Number of Shares Acquired |
|
Value Received on |
| Name |
|
on Exercise (#) |
|
Exercise($) |
|
on Vesting (#) |
|
Vesting($) |
| (a) |
|
(b) |
|
(c) |
|
(d) |
|
(e) |
| |
David Aviezer(1) |
|
|
469,238- |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Yossi Maimon(1) |
|
|
251,890 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Yoseph Shaaltiel |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Einat Brill Almon(1) |
|
|
185,000 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
| (1) |
|
Options were exercised through net exercise with no value received by our company in
connection with the exercise. |
Potential Payments upon Termination or Change-in-Control
Each of our Named Executive Officers is entitled to be insured by Protalix Ltd. under a Manager’s
Policy in lieu of severance upon termination. The intention of such Manager’s Policies is to
provide the Israel-based officers with severance protection of one month’s salary for each year of
employment. We do not provide any change in control benefits to our Named Executive Officers
except that their stock option agreements, as amended, provide that all of the outstanding options
of each Named Executive Officers are subject to accelerated vesting immediately upon a change in
control of our company as defined in our 2006 Stock Incentive Plan. If we had experienced a change
of control on December 31, 2009, the value of the acceleration of the stock options held by each of
Dr. Aviezer, Dr. Shaaltiel, Dr. Brill Almon and Mr. Maimon would be $1.3 million, $284,000,
$750,000 and $846,000, respectively. For a discussion regarding the change of control under our
2006 Stock Incentive Plan, see “—2006 Stock Incentive Plan.”
Employment Arrangements
David Aviezer, Ph.D., MBA. Dr. Aviezer originally served as Protalix Ltd.’s Chief Executive
Officer on a consultancy basis pursuant to a Consulting Services Agreement between Protalix Ltd.
and Agenda Biotechnology Ltd., a company wholly-owned by Dr. Aviezer. On September 11, 2006,
Protalix Ltd. entered into an employment agreement with Dr. Aviezer pursuant to which he agreed to
be employed as Protalix Ltd.’s President and Chief Executive Officer, which agreement supersedes
the Consultancy Services Agreement. Dr. Aviezer currently serves as our President and Chief
Executive Officer. Dr. Aviezer’s current monthly base
salary is NIS 148,000 (approximately
$39,200) and he is entitled to an annual bonus at the Board’s discretion. The monthly salary is
subject to cost of living adjustments from time to time. Dr. Aviezer is eligible to receive a
substantial bonus in the event of certain public offerings or acquisition transactions, which bonus
shall be at the discretion of the Board, and certain specified bonuses in the event Protalix
achieves certain specified milestones. In connection with the employment agreement, in addition to
other options already held by Dr. Aviezer granted to Dr. Aviezer options to purchase 16,000
ordinary shares of Protalix Ltd. at an exercise price equal to $59.40 per share, which we assumed
as options to purchase 977,297 shares of our common stock at $0.97 per share. Such options vest
quarterly retroactively from June 1, 2006, over a four-year period. In addition, in 2008 we
granted to Dr. Aviezer an option
to purchase 600,000 shares of our common stock at an exercise price equal to $5.00 per share. The
option vests variably over a five-year period that commenced on January 1, 2008. In 2009, we
granted Dr. Aviezer an option to purchase 100,000 shares of our common stock at an exercise price
equal to $2.65 per share. As of December 31, 2009, all of those options had fully vested. Dr.
Aviezer’s employment agreement is terminable by either party on 90 days’ written notice for any
reason and we may terminate the agreement for cause without notice. Dr. Aviezer is entitled to be
insured by Protalix Ltd. under a Manager’s Policy in lieu of severance, company contributions
towards vocational studies, annual recreational allowances, a company car and a company phone. Dr.
Aviezer is entitled to 24 working days of vacation. All stock options that have not vested as of
the date of termination shall be deemed to have expired.
Yoseph Shaaltiel, Ph.D. Dr. Shaaltiel founded Protalix Ltd. in 1993 and currently serves as our
Executive Vice President, Research and Development. Dr. Shaaltiel entered into an employment
agreement with Protalix Ltd. on September 1, 2001. Pursuant to the employment agreement, his
current monthly base salary is NIS 85,000 (approximately $22,500) per month. The employment
agreement is terminable by Protalix Ltd. on 90 days’ written notice for any reason and we may
terminate the agreement for cause without notice. In 2008 we granted to Dr. Shaaltiel an option to
purchase 263,728 shares of our common stock at an exercise price equal to $5.00 per share. The
option vests variably over a five-year period that commenced on January 1, 2008. In 2009, we
granted Dr. Shaaltiel an option to purchase 50,000 shares of our common stock at an exercise price
equal to $2.65 per share. As of December 31, 2009, all of those options had fully vested. Dr.
Shaaltiel is
69
entitled to be insured by Protalix Ltd. under a Manager’s Policy in lieu of severance,
company contributions towards vocational studies, annual recreational allowances, a company car and
a company phone. Dr. Shaaltiel is entitled to 24 working days of vacation.
Einat Brill Almon, Ph.D. Dr. Brill Almon joined Protalix Ltd. on December 19, 2004 as its Vice
President, Product Development, pursuant to an employment agreement effective on December 19, 2004
by and between Protalix Ltd. and Dr. Brill Almon, and currently serves as our Senior Vice
President, Product Development. Pursuant to the employment agreement, her current monthly base
salary is NIS 73,500 per month (approximately $19,500). She is also entitled to certain specified
bonuses in the event that Protalix achieves certain specified clinical development milestones
within specified timelines. In connection with the employment agreement, Protalix agreed to grant
to Dr. Brill Almon options to purchase 7,919 ordinary shares of Protalix Ltd. at exercise prices
equal to $24.36 and $59.40 per share, which we assumed as options to purchase 483,701 shares of our
common stock at $0.40 and $0.97 per share. The options vest over four years. In addition, in 2008
we granted to Dr. Almon an option to purchase 311,272 shares of our common stock at an exercise
price equal to $5.00 per share. The option vests variably over a five-year period that commenced
on January 1, 2008. In 2009, we granted to Dr. Brill Almon an option to purchase 50,000 shares of
our common stock at an exercise price equal to $2.65 per share. As of December 31, 2009, all of
those options had fully vested. The employment agreement is terminable by either party on 60 days’
written notice for any reason and we may terminate the agreement for cause without notice. Dr.
Brill Almon is entitled to be insured by Protalix Ltd. under a Manager’s Policy in lieu of
severance, company contributions towards vocational studies, annual recreational allowances, a
company car and a company phone at up to NIS 1,000 per month. Dr. Brill Almon is entitled to 22
working days of vacation. All stock options that have not vested as of the date of termination
shall be deemed to have expired.
Yossi Maimon, CPA. Mr. Maimon joined Protalix Ltd. as its Chief Financial Officer pursuant to an
employment agreement effective as of October 15, 2006 by and between Protalix Ltd. and Mr. Maimon
and currently serves as our Chief Financial Officer. Pursuant to the employment agreement, his
current monthly base salary is NIS 73,500 (approximately $19,500) and Mr. Maimon is entitled to an
annual discretionary bonus and additional discretionary bonuses in the event Protalix achieves
significant financial milestones, subject to the Board’s sole discretion. The monthly salary is
subject to cost of living adjustments from time to time. In connection with the employment
agreement, Protalix agreed to grant to Mr. Maimon options to purchase 10,150 ordinary shares of
Protalix Ltd. at an exercise price equal to $59.40 per share, which we assumed as options to
purchase 619,972 shares of our common stock at $0.97 per share. The first 25% of such options
shall vest on the first anniversary of the grant date and the remainder shall vest quarterly in 12
equal increments. In addition, in 2008 we granted to Mr. Maimon an option to purchase 175,000
shares of our common stock at an exercise price equal to $5.00 per share. The option vests
variably over a five-year period that commenced on January 1, 2008. In 2009, we granted to Mr.
Maimon an option to purchase 50,000 shares of our common stock at an exercise price equal to $2.65
per share. As of December 31, 2009, all of those options had fully vested. The employment
agreement is terminable by either party on 60 days’ written notice for any reason and we may
terminate the agreement for cause without notice. Mr. Maimon is entitled to be insured by Protalix
Ltd. under a Manager’s Policy in lieu of severance, company contributions towards vocational
studies, annual recreational allowances, a company car and a company phone. Mr. Maimon is entitled
to 24 working days of vacation. All stock options that have not vested as of the date of
termination shall be deemed to have expired.
Sandra L. Lauterbach. Ms. Lauterbach joined our company as our Vice President, Sales and
Commercial Affairs, pursuant to an employment agreement effective December 18, 2009. Pursuant to
the employment agreement, Ms. Lauterbach’s annual
base salary is $180,000 and we may elect to pay her an annual discretionary bonus in an amount and
based upon criteria determined by either the Compensation Committee of our Board of Directors, or
the entire Board of Directors, at their sole discretion. She is also entitled to certain health
care insurance benefits and contributions to retirement plans, and allowances for car and cell
phone expenses. In connection with the employment agreement, the Board of Directors granted to Ms.
Lauterbach stock options to purchase 160,000 shares of our common stock at an exercise price equal
to $6.81. The options vest over a period of four years, with 25% of the options vesting upon the
lapse of one year from the date of grant and the remainder of the options vesting on a quarterly
basis in 12 equal installments, commencing on the initial vesting date. The unvested portion of
the option will vest automatically upon a change of control of our company. The employment
agreement is terminable by either party on 60 days’ written notice for any reason and we may
terminate the agreement for cause without notice.
2006 Stock Incentive Plan
Our Board of Directors and a majority of our stockholders approved our 2006 Stock Incentive Plan on
December 14, 2006 and cancelled our 1998 stock option plan (no options were outstanding under the
1998 plan at that time). We have reserved 9,741,655 shares of our common stock for issuance, in
the aggregate, under the 2006 Stock Incentive Plan, subject to adjustment for a stock split or any
future stock dividend or other similar change in our common stock or our capital structure.
70
As of
February 25, 2010, options to acquire 230,000 of common stock remain available for grant under
our 2006 Stock Incentive Plan.
Our 2006 Stock Incentive Plan provides for the grant of stock options, restricted stock, restricted
stock units, stock appreciation rights and dividend equivalent rights, collectively referred to as
“awards.” Stock options granted under the 2006 Stock Incentive Plan may be either incentive stock
options under the provisions of Section 422 of the Internal Revenue Code, or non-qualified stock
options. Incentive stock options may be granted only to employees. Awards other than incentive
stock options may be granted to employees, directors and consultants. The 2006 Stock Incentive
Plan is also in compliance with the provisions of the Israeli Income Tax Ordinance New Version,
1961 (including as amended pursuant to Amendment 132 thereto) and is intended to enable us to grant
awards to grantees who are Israeli residents as follows: (i) awards to employees pursuant to
Section 102 of the Tax Ordinance (definition refers only to employees, office holders and directors
of our company or a related entity excluding those who are considered “Controlling Shareholders”
pursuant to the Tax Ordinance); and (ii) awards to non-employees pursuant to Section 3(I) of the
Tax Ordinance. In accordance with the terms and conditions imposed by the Tax Ordinance, grantees
who receive awards under the 2006 Stock Incentive Plan may be afforded certain tax benefits in
Israel as described below.
Our Board of Directors or the Compensation Committee, referred to as the “plan administrator,”
administers our 2006 Stock Incentive Plan, including selecting the grantees, determining the number
of shares to be subject to each award, determining the exercise or purchase price of each award,
and determining the vesting and exercise periods of each award.
The exercise price of stock options granted under the 2006 Stock Incentive Plan must be equal to at
least 100% of the fair market value of our common stock on the date of grant; however, in certain
circumstances, grants may be made at a lower price to Israeli grantees who are residents of the
State of Israel. If, however, incentive stock options are granted to an employee who owns stock
possessing more than 10% of the voting power of all classes of our stock or the stock of any parent
or subsidiary of our company, the exercise price of any incentive stock option granted must equal
at least 110% of the fair market value on the grant date and the maximum term of these incentive
stock options must not exceed five years. The maximum term of all other awards must not exceed 10
years. The plan administrator will determine the exercise or purchase price (if any) of all other
awards granted under the 2006 Stock Incentive Plan.
Under the 2006 Stock Incentive Plan, incentive stock options and options to Israeli grantees may
not be sold, pledged, assigned, hypothecated, transferred or disposed of in any manner other than
by will or by the laws of descent or distribution and may be exercised during the lifetime of the
participant only by the participant. Other awards shall be transferable by will or by the laws of
descent or distribution and to the extent and in the manner authorized by the plan administrator by
gift or pursuant to a domestic relations order to members of the participant’s immediate family.
The 2006 Stock Incentive Plan permits the designation of beneficiaries by holders of awards,
including incentive stock options.
If the service of a participant in the 2006 Stock Incentive Plan is terminated for any reason other
than cause, disability or death, the participant may exercise awards that were vested as of the
termination date for a period ending upon the earlier of 12 months or the expiration date of the
awards unless otherwise determined by the plan administrator.
In the event of a corporate transaction or a change of control, all awards will terminate unless
assumed by the successor corporation. Unless otherwise provided in a participant’s award
agreement, in the event of a corporate transaction for the portion of each award that is assumed or
replaced, then such award will automatically become fully vested and exercisable
immediately upon termination of a participant’s service if the participant is terminated by the
successor company or us without cause within 12 months after the corporate transaction. For the
portion of each award that is not assumed or replaced, such portion of the award will automatically
become fully vested and exercisable immediately prior to the effective date of the corporate
transaction so long as the participant’s service has not been terminated prior to such date.
In the event of a change in control, except as otherwise provided in a participant’s award
agreement, following a change in control (other than a change in control that also is a corporate
transaction) and upon the termination of a participant’s service without cause within 12 months
after a change in control, each award of such participant that is outstanding at such time will
automatically become fully vested and exercisable immediately upon the participant’s termination.
Under our 2006 Stock Incentive Plan, a corporate transaction is generally defined as:
• a merger or consolidation in which we are not the surviving entity, except for the principal
purpose of changing our company’s state of incorporation;
• the sale, transfer or other disposition of all or substantially all of our assets;
71
• the complete liquidation or dissolution of our company;
• any reverse merger in which we are the surviving entity but our shares of common stock
outstanding immediately prior to such merger are converted or exchanged by virtue of the merger
into other property, whether in the form of securities, cash or otherwise, or in which securities
possessing more than forty percent (40%) of the total combined voting power of our outstanding
securities are transferred to a person or persons different from those who held such securities
immediately prior to such merger; or
• acquisition in a single or series of related transactions by any person or related group of
persons of beneficial ownership of securities possessing more than fifty percent (50%) of the total
combined voting power of our outstanding securities but excluding any such transaction or series of
related transactions that the plan administrator determines not to be a corporate transaction
(provided however that the plan administrator shall have no discretion in connection with a
corporate transaction for the purchase of all or substantially all of our shares unless the
principal purpose of such transaction is changing our company’s state of incorporation).
Under our 2006 Stock Incentive Plan, a change of control is defined as:
• the direct or indirect acquisition by any person or related group of persons of beneficial
ownership of securities possessing more than fifty percent (50%) of the total combined voting power
of our outstanding securities pursuant to a tender or exchange offer made directly to our
shareholders and which a majority of the members of our board (who have generally been on our board
for at least 12 months) who are not affiliates or associates of the offeror do not recommend
shareholders accept the offer; or
• a change in the composition of our board over a period of 12 months or less, such that a
majority of our board members ceases, by reason of one or more contested elections for board
membership, to be comprised of individuals who were previously directors of our company.
Unless terminated sooner, the 2006 Stock Incentive Plan will automatically terminate in 2016. Our
Board of Directors has the authority to amend, suspend or terminate our 2006 Stock Incentive Plan.
No amendment, suspension or termination of the 2006 Stock Incentive Plan shall adversely affect any
rights under awards already granted to a participant. To the extent necessary to comply with
applicable provisions of federal securities laws, state corporate and securities laws, the Internal
Revenue Code, the rules of any applicable stock exchange or national market system, and the rules
of any non-U.S. jurisdiction applicable to awards granted to residents therein (including the Tax
Ordinance), we shall obtain shareholder approval of any such amendment to the 2006 Stock Incentive
Plan in such a manner and to such a degree as required.
Impact of Israeli Tax Law
The awards granted to employees pursuant to Section 102 of the Tax Ordinance under the 2006 Stock
Incentive Plan may be designated by us as approved options under the capital gains alternative, or
as approved options under the ordinary income tax alternative.
To qualify for these benefits, certain requirements must be met, including registration of the
options in the name of a trustee. Each option, and any shares of common stock acquired upon the
exercise of the option, must be held by the trustee for a period commencing on the date of grant
and deposit into trust with the trustee and ending 24 months thereafter.
Under the terms of the capital gains alternative, we may not deduct expenses pertaining to the
options for tax purposes.
Under the 2006 Stock Incentive Plan, we may also grant to employees options pursuant to Section
102(c) of the Tax Ordinance that are not required to be held in trust by a trustee. This
alternative, while facilitating immediate exercise of vested options and sale of the underlying
shares, will subject the optionee to the marginal income tax rate of up to 50% as well as payments
to the National Insurance Institute and health tax on the date of the sale of the shares or
options. Under the 2006 Stock Incentive Plan, we may also grant to non-employees options pursuant
to Section 3(I) of the Tax Ordinance. Under that section, the income tax on the benefit arising to
the optionee upon the exercise of options and the issuance of common stock is generally due at the
time of exercise of the options.
These options shall be further subject to the terms of the tax ruling that has been obtained by
Protalix Ltd. from the Israeli tax authorities in connection with the merger. Under the tax
ruling, the options issued by us in connection with the assumption of Section 102 options
previously issued by Protalix Ltd. under the capital gains alternative shall be issued to a
trustee, shall
72
be designated under the capital gains alternative and the issuance date of the
original options shall be deemed the issuance date for the assumed options for the calculation of
the respective holding period.
Compensation of Directors
The following table sets forth information with respect to compensation of our non-employee
directors during fiscal year 2009. The fees to our current directors were paid by Protalix Ltd.
Prior to the merger, Protalix Ltd. compensated only certain of its directors, which compensation
was limited to the granting of options under its employee stock option plan.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Fees |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Earned or |
|
|
|
|
|
|
|
|
|
Non-Equity |
|
Nonqualified |
|
All Other |
|
|
| |
|
Paid in |
|
Stock |
|
|
|
|
|
Incentive |
|
Deferred Compen- |
|
Compen- |
|
Total |
| Name |
|
Cash ($) |
|
Award ($) |
|
OptionAwards ($) |
|
Plan Compensation ($) |
|
sation Earnings ($) |
|
sation ($) |
|
($) |
Current Directors |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Eli Hurvitz(1) |
|
|
33,000 |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
33,000 |
|
Alfred Akirov |
|
|
33,000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Amos Bar Shalev |
|
|
33,000 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
33,000 |
|
Zeev Bronfeld |
|
|
33,000 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
33,000 |
|
Yodfat Harel Gross |
|
|
33,000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
33,000 |
|
Roger D. Kornberg |
|
|
33,000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
33,000 |
|
Eyal Sheratzky |
|
|
33,000 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
33,000 |
|
Sharon
Toussia-Cohen(2) |
|
|
11,000 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
11,000 |
|
|
|
|
| (1) |
|
Represents amounts paid to Pontifax Management Company, Ltd. pursuant to a management
consulting agreement. |
| |
| (2) |
|
Mr. Toussia-Cohen resigned from our Board of Directors on May 10, 2009. |
Our Board of Directors will review director compensation annually and adjust it according to
then current market conditions and corporate governance guidelines.
Compensation Committee Interlocks and Insider Participation
Our Compensation Committee currently consists of Messrs. Akirov and Bar Shalev and Ms. Harel Gross,
who were appointed to the Committee during 2009. In addition, until May 10, 2009, Mr.
Toussia-Cohen served on our Compensation Committee. No member of our Compensation Committee or any
executive officer of our company or of Protalix Ltd. has a
relationship that would constitute an interlocking relationship with executive officers or
directors of another entity. No Compensation Committee member is or was an officer or employee of
ours or of Protalix Ltd. Further, none of our executive officers serves on the board of directors
or compensation committee of any entity that has one or more executive officers serving as a member
of our Board of Directors or Compensation Committee.
73
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder
Matters
Security Ownership of Certain Beneficial Owners and Management
The following table sets forth information, as of February 15, 2010, regarding beneficial ownership
of our common stock:
| |
• |
|
each person who is known by us to own beneficially more than 5% of our common stock; |
| |
| |
• |
|
each director; |
| |
| |
• |
|
each of our executive officers; and |
| |
| |
• |
|
all of our directors and executive officers collectively. |
Unless otherwise noted, we believe that all persons named in the table have sole voting and
investment power with respect to all shares of our common stock beneficially owned by them. For
purposes of these tables, a person is deemed to be the beneficial owner of securities that can be
acquired by such person within 60 days from February 15, 2010 upon exercise of options,
warrants and convertible securities. Each beneficial owner’s percentage ownership is determined by
assuming that options, warrants and convertible securities that are held by such person (but not
those held by any other person) and that are exercisable within such 60 days from such date have
been exercised.
The address for all directors and officers is c/o Protalix BioTherapeutics, Inc., 2 Snunit Street,
Science Park, POB 455, Carmiel, Israel, 20100.
| |
|
|
|
|
|
|
|
|
| |
|
Amount and Nature of |
|
Percentage |
| Name and Address of Beneficial Owner |
|
Beneficial Ownership |
|
of Class |
Board of Directors and Executive Officers |
|
|
|
|
|
|
|
|
Eli Hurvitz |
|
|
455,240 |
|
|
|
* |
% |
David Aviezer, Ph.D., MBA |
|
|
1,609,155 |
(1) |
|
|
2.0 |
|
Yoseph Shaaltiel, Ph.D. |
|
|
1,573,828 |
(2) |
|
|
1.9 |
|
Alfred Akirov |
|
|
6,186,046 |
(3) |
|
|
7.7 |
|
Amos Bar Shalev |
|
|
— |
|
|
|
— |
|
Zeev Bronfeld |
|
|
14,466,319 |
(4) |
|
|
17.9 |
|
Yodfat Harel Gross |
|
|
— |
|
|
|
— |
|
Roger D. Kornberg. Ph.D. |
|
|
21,875 |
(5) |
|
|
* |
|
Eyal Sheratzky |
|
|
— |
|
|
|
— |
|
Einat Brill Almon, Ph.D. |
|
|
278,590 |
(6) |
|
|
* |
|
Yossi Maimon |
|
|
312,761 |
(7) |
|
|
* |
|
Sandra L. Lauterbach |
|
|
— |
(8) |
|
|
— |
|
All executive officers and directors as a group
(12 persons) |
|
|
24,903,814 |
(9) |
|
|
29.7 |
|
|
|
|
|
|
|
|
|
|
5% Holders |
|
|
|
|
|
|
|
|
Biocell Ltd. |
|
|
14,466,319 |
(10) |
|
|
17.9 |
|
Techno-Rov Holdings (1993) Ltd. |
|
|
6,186,046 |
(11) |
|
|
7.7 |
|
FMR LLC |
|
|
4,193,555 |
(12) |
|
|
5.4 |
|
Frost Gamma Investment Trust |
|
|
7,188,132 |
(13) |
|
|
8.9 |
|
|
|
|
| * |
|
less than 1%. |
| |
| (1) |
|
Consists of 1,609,155 shares of our common stock issuable upon exercise of outstanding
options within 60 days of February 15, 2010. Does not include 394,049 shares of common stock
issuable upon exercise of outstanding options that are not exercisable within 60 days of
February 15, 2010. |
| |
| (2) |
|
Consists of 1,363,754 shares of our common stock held by Dr. Shaaltiel and 210,074 shares of
our common stock issuable upon exercise of outstanding options within 60 days of February 15,
2010. Does not include 225,816 shares of common stock issuable upon exercise of outstanding
options that are not exercisable within 60 days of February 15, 2010. |
74
|
|
|
| (3) |
|
Consists of 6,186,046 shares of our common stock held by Techno-Rov Holdings (1993) Ltd. Mr.
Akirov is the Chief Executive Officer of Techno-Rov Holdings and has the power to control its
investment decisions. |
| |
| (4) |
|
Consists of 14,466,319 shares of our common stock held by Biocell Ltd. Mr. Bronfeld is a
director and Chief Executive Officer of Biocell. Mr. Bronfeld disclaims beneficial ownership
of these shares. |
| |
| (5) |
|
Consists of 21,875 shares of our common stock issuable upon exercise of outstanding options
within 60 days of February 15, 2010. Does not include 28,125 shares of common stock issuable
upon exercise of outstanding options that are not exercisable within 60 days of February 15,
2010. |
| |
| (6) |
|
Consists of 278,590 shares of our common stock issuable upon exercise of outstanding options
within 60 days of February 15, 2010. Does not include 319,530 shares of common stock issuable
upon exercise of outstanding options that are not exercisable within 60 days of February 15,
2010. |
| |
| (7) |
|
Consists of 312,761 shares of our common stock issuable upon exercise of outstanding options
within 60 days of February 15, 2010. Does not include 135,972 shares of common stock issuable
upon exercise of outstanding options that are not exercisable within 60 days of February 15,
2010. |
| |
| (8) |
|
Does not include 160,000 shares of common stock issuable upon exercise of outstanding options
that are not exercisable within 60 days of February 15, 2010. |
| |
| (9) |
|
Consists of 22,471,359 shares of our common stock and 2,432,455 shares of our common stock
issuable upon exercise of options within 60 days of February 15, 2010. Does not include
1,103,852 shares of common stock issuable upon exercise of outstanding options that are not
exercisable within 60 days of February 15, 2010. |
| |
| (10) |
|
The address is Moshe Aviv Tower, 7 Jabotinsky Street, Ramat Gan, Israel. Biocell Ltd.’s
investment and voting decisions are made collectively by its board of directors. |
| |
| (11) |
|
The address is Alrov Tower, 46 Rothschild Blvd., Tel Aviv, Israel. Mr. Akirov is the Chief
Executive Officer of Techno-Rov Holdings (1993) Ltd. and has the power to control its
investment decisions. |
| |
| (12) |
|
Based solely on a Schedule 13G filed by FMR LLC on February 12, 2010, reporting the above
stock ownership as of December 31, 2009. FMR LLC reports that it has the sole power to
dispose or to direct the disposition of 4,193,555 shares of common stock. The address is 82
Devonshire Street, Boston, MA 02109. |
| |
| (13) |
|
The address is 4400 Biscayne Blvd., Miami, Florida 33137. Frost Gamma, L.P. is the sole and
exclusive beneficiary of Frost Gamma Investments Trust. Dr. Phillip Frost is the sole limited
partner of Frost Gamma, L.P. The general partner of Frost Gamma, L.P. is Frost Gamma, Inc.
and the sole shareholder of Frost Gamma, Inc. is Frost-Nevada Corporation. Dr. Frost is also
the sole shareholder of Frost-Nevada Corporation. |
Equity Compensation Plan Information
The following table provides information as of December 31, 2009 with respect to the shares of our
common stock that may be issued under our existing equity compensation plan.
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
A |
|
|
B |
|
|
C |
|
| |
|
|
|
|
|
|
|
Number of Securities Remaining |
|
| |
|
Number of Securities |
|
|
|
|
|
Available for Future Issuance |
|
| |
|
to be Issued |
|
|
Weighted Average |
|
|
Under Equity Compensation Plans |
|
| |
|
Upon Exercise of |
|
|
Exercise Price of |
|
|
(Excluding Securities Reflected in |
|
| Plan Category |
|
Outstanding Options |
|
|
Outstanding Options |
|
|
Column A) |
|
Equity
Compensation Plans
Approved by
Shareholders |
|
|
6,173,555 |
|
|
$ |
2.13 |
|
|
|
1,433,623 |
|
Equity Compensation
Plans Not Approved
by Shareholders |
|
|
631,866 |
|
|
$ |
10.24 |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total |
|
|
6,805,421 |
|
|
$ |
2.89 |
|
|
|
1,433,623 |
|
|
|
|
|
|
|
|
|
|
|
Item 13. Certain Relationships and Related Transactions, and Director Independence
On March 17, 2005, Protalix Ltd. entered into a Management Services Agreement with Pontifax
Management Company, Ltd. in connection with the purchase of Protalix’s Series B Preferred Shares by
the Pontifax Funds. Pursuant to the Management Services Agreement, Mr. Hurvitz serves as a member
of the Board of Directors. Further, Protalix agreed not to designate a
75
permanent chairman of the
Board of Directors until Pontifax Management Company chose to nominate Mr. Hurvitz as the Chairman
of the Board in 2006. In consideration for Mr. Hurvitz’s services, Protalix is required to pay
Pontifax Management Company a fee equal to $3,000 per month plus required taxes on such payment.
In addition, in connection with the execution of the Management Services Agreement, Protalix issued
to Pontifax options to purchase a number of its Series B Preferred Shares equal to 3.5% of the then
outstanding share capital with an exercise price equal to the par value of the shares. Lastly,
upon the appointment of Mr. Hurvitz as Chairman of the Board of Directors, Protalix issued to
Pontifax additional warrants for Series B Preferred Shares equal to 3.76% of the then outstanding
share capital of Protalix. In connection with the merger, we assumed the Management Services
Agreement and all options granted under the Management Services Agreement have been converted into
options to purchase 3,384,502 shares of our common stock. Under the terms of the assumed
Management Services Agreement, we are obligated only to use our best efforts to nominate Mr.
Hurvitz for election to our Board of Directors, which remains subject to the review and approval of
the Nominating Committee of the Board of Directors and the entire Board of Directors, as
applicable. For 2010, the fee payable under this agreement will be $33,000, which is the same fee
payable to the other non-executive directors.
On September 14, 2006, Protalix Ltd. entered into a collaboration and licensing agreement with Teva
for the development and manufacture of two proteins using ProCellEx. Mr. Hurvitz, the Chairman of
our Board of Directors, is the Chairman of Teva’s Board of Directors, and Phillip Frost M.D., a
former director and a major shareholder of our company, is the Vice Chairman of Teva’s Board of
Directors and Professor Roger D. Kornberg, a member of our Board of Directors also serves as a
member of the board of directors of Teva. The agreement provides that we will collaborate with
Teva on the research and development of two proteins using ProCellEx. Subsequently, two proteins
were identified to be researched and developed under the agreement but in 2009, both of the
projects were terminated for commercial reasons. Protalix Ltd. has granted to Teva an exclusive license to commercialize
any products developed under the collaboration in return for royalty and milestone payments payable
upon the achievement of certain pre-defined goals. Protalix Ltd. will retain certain exclusive
manufacturing rights with respect to the active pharmaceutical ingredient of the proteins following
the first commercial sale of a licensed product under the agreement and other rights thereafter.
All related party transactions are reviewed and approved by the Audit Committee, as required by the
Audit Committee Charter.
Corporate Governance and Independent Directors
In compliance with the listing requirements of the NYSE Amex, we have a comprehensive plan of
corporate governance for the purpose of defining responsibilities, setting high standards of
professional and personal conduct and assuring compliance with such responsibilities and standards.
We currently regularly monitor developments in the area of corporate governance to ensure we are
in compliance with the standards and regulations required by the NYSE Amex. A summary of our
corporate governance measures follows.
Independent Directors
We believe a majority of the members of our Board of Directors are independent from management.
When making determinations from time to time regarding independence, the Board of Directors will
reference the listing standards adopted by the NYSE Amex as well as the independence standards set
forth in the Sarbanes-Oxley Act of 2002 and the rules and regulations promulgated by the Commission
under that Act. In particular, our Audit Committee periodically evaluates and reports to the Board
of Directors on the independence of each member of the Board. We anticipate our audit committee
will analyze whether a director is independent by evaluating, among other factors, the following:
| |
• |
|
Whether the member of the Board of Directors has any material relationship with us,
either directly, or as a partner, shareholder or officer of an organization that has a
relationship with us; |
| |
| |
• |
|
Whether the member of the Board of Directors is a current employee of our company or
any of our subsidiaries, or was an employee of our company or any of our subsidiaries
within three years preceding the date of determination; |
| |
| |
• |
|
Whether the member of the Board of Directors is, or in the three years preceding the
date of determination has been, affiliated with or employed by (i) a present internal
or external auditor of our company or any affiliate of
such auditor or (ii) any former internal or external auditor of our company or any
affiliate of such auditor, which performed services for us within three years preceding
the date of determination; |
76
| |
• |
|
Whether the member of the Board of Directors is, or in the three years preceding the
date of determination has been, part of an interlocking directorate, in which any of
our executive officers serve on the Compensation Committee of another company that
concurrently employs the member as an executive officer; |
| |
| |
• |
|
Whether the member of the Board of Directors receives any compensation from us,
other than fees or compensation for service as a member of the Board of Directors and
any committee of the Board of Directors and reimbursement for reasonable expenses
incurred in connection with such service and for reasonable educational expenses
associated with Board of Directors or committee membership matters; |
| |
| |
• |
|
Whether an immediate family member of the member of the Board of Directors is a
current executive officer of our company or was an executive officer of our company
within three years preceding the date of determination; |
| |
| |
• |
|
Whether an immediate family member of the member of the Board of Directors is, or in
the three years preceding the date of determination has been, affiliated with or
employed in a professional capacity by (i) a present internal or external auditor of
ours or any of our affiliates or (ii) any former internal or external auditor of our
company or any affiliate of ours which performed services for us within three years
preceding the date of determination; and |
| |
| |
• |
|
Whether an immediate family member of the member of the Board of Directors is, or in
the three years preceding the date of determination has been, part of an interlocking
directorate, in which any of our executive officers serve on the Compensation Committee
of another company that concurrently employs the immediate family member of the member
of the Board of Directors as an executive officer. |
The above list is not exhaustive and we anticipate that the Audit Committee will consider all other
factors which could assist it in its determination that a director will have no material
relationship with us that could compromise that director’s independence.
Under these standards, our Board of Directors has determined that Messrs. Akirov and Bar–Shalev and
Ms. Harel Gross are considered “independent” pursuant to the rules of the NYSE Amex and Section
10A(m)(3) of the Securities Exchange Act of 1934, as amended. In addition, our Board of Directors
has determined that at least two of these directors are able to read and understand fundamental
financial statements and have substantial business experience that results in their financial
sophistication, qualifying them for membership on any audit committee we form. Our Board of
Directors has also determined that Messrs. Akirov, Bar Shalev, Bronfeld and Sheratzky, Ms. Harel
Gross and Dr. Kornberg are “independent” pursuant to the rules of the NYSE Amex.
The position of chairman of the board is not held by our chief executive officer at this time. The
Board of Directors does not have a policy mandating the separation of these functions. However,
when Mr. Hurvitz joined our company in 2005, the Board of Directors at that time determined that it
was in our best interest that Mr. Hurvitz serve as the chairman of the board. This decision was
based on Mr. Hurvitz’s vast experience in the pharmaceutical industry in Israel and globally. Our
non-management directors hold formal meetings, separate from management, at least twice per year.
We have no formal policy regarding attendance by our directors at annual shareholders meetings,
although we encourage such attendance and anticipate most of our directors will attend these
meetings. Messrs. Hurvitz, Bronfeld, Bar Shalev, Sheratzky, Akirov, Aviezer and Shaaltiel, and Ms.
Harel Gross, attended our 2009 annual meeting of shareholders.
77
Item 14. Principal Accountant Fees and Services
The following table sets forth fees billed to us by our independent registered public accounting
firm during the fiscal years ended December 31, 2009 and 2008 for: (i) services rendered for the
audit of our annual financial statements and the review of our quarterly financial statements; (ii)
services by our independent registered public accounting firm that are reasonably related to the
performance of the audit or review of our financial statements and that are not reported as Audit
Fees; (iii) services rendered in connection with tax compliance, tax advice and tax planning; and
(iv) all other fees for services rendered.
| |
|
|
|
|
|
|
|
|
| |
|
Year ended December 31, |
| |
|
2009 |
|
2008 |
Audit Fees |
|
$ |
259,000 |
|
|
$ |
249,000 |
|
Audit Related Fees |
|
$ |
78,039 |
|
|
$ |
49,000 |
|
Tax Fees |
|
$ |
197,282 |
|
|
$ |
76,000 |
|
All Other Fees |
|
$ |
— |
|
|
$ |
— |
|
Policy on Audit Committee Pre-Approval of Audit and Permissible Non-Audit Services of Independent
Auditors
Prior to entering into the engagement letter with our independent registered accountants, our Audit
Committee approved the 2009 audit fees. For fiscal year 2010, our Audit Committee has approved
fees for certain services to be rendered by our independent registered accounting firm.
78
PART IV
Item 15. Exhibits and Financial Statement Schedules
The following documents are filed as part of this Annual Report on Form 10-K:
1. Financial Statements. The following Consolidated Financial Statements of Protalix
BioTherapeutics, Inc. are included in Item 8 of this Annual Report on Form 10-K:
| |
|
|
|
|
| |
|
Page |
|
Report of Independent Registered Public Accounting Firm |
|
|
F-2 |
|
|
|
|
|
|
Consolidated Balance Sheets as of December 31, 2008, and 2009 |
|
|
F-3 |
|
|
|
|
|
|
Consolidated Statements of Operations for the years ended
December 31, 2007, 2008, and 2009 |
|
|
F-4 |
|
|
|
|
|
|
Consolidated Statements of Changes in Shareholders’ Equity for the
years ended December 31, 2007, 2008, and 2009 |
|
|
F-5 |
|
|
|
|
|
|
Consolidated Statements of Cash Flows for the years ended December
31, 2007, 2008, and 2009 |
|
|
F-6 |
|
|
|
|
|
|
Notes to Consolidated Financial Statements |
|
|
F-8 |
|
2. Financial Statement Schedule. Financial statement schedules have been omitted since
they are either not required, are not applicable or the required information is shown in the
consolidated financial statements or related notes.
| |
|
|
|
|
| Exhibit |
|
|
|
|
| Number |
|
Exhibit Description |
|
Method of Filing |
3.1
|
|
Amended and Restated Articles of
Incorporation of the Company
|
|
Incorporated by
reference to our
Registration
Statement on Form
S-4 filed on March
26, 1998 |
|
|
|
|
|
3.2
|
|
Article of Amendment to Articles of
Incorporation dated June 9, 2006
|
|
Incorporated by
reference to our
Registration
Statement on Form
8-A filed on March
9, 2007 |
|
|
|
|
|
3.3
|
|
Article of Amendment to Articles of
Incorporation dated December 13, 2006
|
|
Incorporated by
reference to our
Registration
Statement on Form
8-A filed on March
9, 2007 |
|
|
|
|
|
3.4
|
|
Article of Amendment to Articles of
Incorporation dated December 26, 2006
|
|
Incorporated by
reference to our
Registration
Statement on Form
8-A filed on March
9, 2007 |
|
|
|
|
|
3.5
|
|
Article of Amendment to Articles of
Incorporation dated February 26, 2007
|
|
Incorporated by
reference to our
Registration
Statement on Form
8-A filed on March
9, 2007 |
|
|
|
|
|
3.6
|
|
Amended and Restated Bylaws of the Company
|
|
Incorporated by
reference to our
Quarterly Report on
Form 10-Q for the
quarter ended June
30, 2008, filed on
August 8, 2008 |
|
|
|
|
|
10.1
|
|
2006 Stock Incentive Plan
|
|
Incorporated by
reference to our
Amended Annual
Report on Form
10-K/A for the year
ended December 31,
2006, filed on
July 13, 2007 |
79
| |
|
|
|
|
| Exhibit |
|
|
|
|
| Number |
|
Exhibit Description |
|
Method of Filing |
10.2
|
|
Employment Agreement between Protalix
Ltd. and Yoseph Shaaltiel, dated as of
September 1, 2004
|
|
Incorporated by
reference to our
Current Report on
Form 8-K filed on
January 8, 2007 |
|
|
|
|
|
10.3
|
|
Employment Agreement between Protalix
Ltd. and Einat Almon, dated as of
December 19, 2004
|
|
Incorporated by
reference to our
Current Report on
Form 8-K filed on
January 8, 2007 |
|
|
|
|
|
10.4
|
|
Employment Agreement between Protalix
Ltd. and David Aviezer, dated as of
September 11, 2006
|
|
Incorporated by
reference to our
Current Report on
Form 8-K filed on
January 8, 2007 |
|
|
|
|
|
10.5
|
|
Employment Agreement between Protalix
Ltd. and Yossi Maimon, dated as of
October 15, 2006
|
|
Incorporated by
reference to our
Current Report on
Form 8-K filed on
January 8, 2007 |
|
|
|
|
|
10.6†
|
|
License Agreement entered into as of
April 12, 2005, by and between Icon
Genetics AG and Protalix Ltd.
|
|
Incorporated by
reference to our
Amended Current
Report on Form
8-K/A filed on
September 20, 2007 |
|
|
|
|
|
10.7†
|
|
Research and License Agreement between
Yeda Research and Development Company
Limited and Protalix Ltd. dated as of
March 15, 2006
|
|
Incorporated by
reference to our
Amended Current
Report on Form
8-K/A filed on
September 20, 2007 |
|
|
|
|
|
10.8†
|
|
Agreement between Teva Pharmaceutical
Industries Ltd. and Protalix Ltd., dated
September 14, 2006
|
|
Incorporated by
reference to our
Amended Current
Report on Form
8-K/A filed on
September 20, 2007 |
|
|
|
|
|
10.9
|
|
Lease Agreement between Protalix Ltd. and
Angel Science Park (99) Ltd., dated as of
October 28, 2003 as amended on April 18,
2005
|
|
Incorporated by
reference to our
Current Report on
Form 8-K filed on
January 8, 2007 |
|
|
|
|
|
10.10
|
|
Merger Agreement and Plan of
Reorganization made and entered into as
of August 21, 2006, by and among the
Company, Protalix Acquisition Co., Ltd.
and Protalix Ltd.
|
|
Incorporated by
reference to our
Current Report on
Form 8-K filed on
January 8, 2007 |
|
|
|
|
|
10.11
|
|
Stock Option Award Agreement grant by and
between the Company and Steven Rubin,
dated as of December 31, 2006
|
|
Incorporated by
reference to our
Annual Report on
Form 10-K for the
year ended December
31, 2007, filed on
March 30, 2007 |
|
|
|
|
|
10.12
|
|
First Amendment to the December 31, 2006
Stock Option Award Agreement by and
between the Company and Steven Rubin,
effective as of February 28, 2007
|
|
Incorporated by
reference to our
Annual Report on
Form 10-K for the
year ended December
31, 2008 filed on
March 30, 2007 |
|
|
|
|
|
10.13
|
|
Scientific Advisory Board Agreement dated
August 5, 2007 by and between the Company
and Aaron Ciechanover, M.D.
|
|
Incorporated by
reference to our
Current Report on
Form 8-K filed on
August 6, 2007 |
|
|
|
|
|
10.14†
|
|
Research and License Agreement made on
August 8, 2007, by and between Yissum
Research Development Company of
Jerusalem, the Boyce Thompson Institute
and Protalix Ltd.
|
|
Incorporated by
reference to our
Quarterly Report on
Form 10-Q for the
quarter ended
September 30, 2007,
filed on November
14, 2007 |
|
|
|
|
|
10.15
|
|
Unprotected Lease Agreement
|
|
Incorporated by
reference to our
Annual Report on
Form 10-K for the
year ended December
31, 2007, filed on
March 17, 2008. |
|
|
|
|
|
10.16†
|
|
Exclusive License and Supply Agreement
dated as of November 30, 2009 between
Protalix Ltd. and Pfizer Inc.
|
|
Filed herewith |
|
|
|
|
|
10.17
|
|
Employment Agreement by and between the
Company and Sandra Lauterbach dated as of
December 17, 2009
|
|
Incorporated by
reference to our
Current Report on
Form 8-K filed on
December 22, 2009 |
|
|
|
|
|
21.1
|
|
Subsidiaries
|
|
Filed herewith |
80
| |
|
|
|
|
| Exhibit |
|
|
|
|
| Number |
|
Exhibit Description |
|
Method of Filing |
23.1
|
|
Consent of Kesselman & Kesselman,
Certified Public Accountant (Isr.), A
member of PricewaterhouseCoopers
International Limited, independent
registered public accounting firm for the
Registrant
|
|
Filed herewith |
|
|
|
|
|
31.1
|
|
Certification of Chief Executive Officer
pursuant to Rule 13a-14(a) as adopted
pursuant to Section 302 of the
Sarbanes-Oxley Act of 2002
|
|
Filed herewith |
|
|
|
|
|
31.2
|
|
Certification of Chief Financial Officer
pursuant to Rule 13a-14(a) as adopted
pursuant to Section 302 of the
Sarbanes-Oxley Act of 2002
|
|
Filed herewith |
|
|
|
|
|
32.1
|
|
18 U.S.C. Section 1350, as adopted
pursuant to Section 906 of the
Sarbanes-Oxley Act of 2002, Certification
of Chief Executive Officer
|
|
Filed herewith |
|
|
|
|
|
32.2
|
|
18 U.S.C. Section 1350, as adopted
pursuant to Section 906 of the
Sarbanes-Oxley Act of 2002, Certification
of Chief Financial Officer
|
|
Filed herewith |
|
|
|
| † |
|
Portions of this exhibit were omitted and have been filed separately with the Secretary of the
Securities and Exchange Commission pursuant to the Registrant’s application requesting confidential
treatment under Rule 24b-2 of the Exchange Act. |
81
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as
amended, the registrant has duly caused this report to be signed on its behalf by the undersigned,
thereunto duly authorized, as of February 25, 2010.
| |
|
|
|
|
| |
PROTALIX BIOTHERAPEUTICS, INC.
|
|
| |
By: |
/s/ David Aviezer
|
|
| |
|
David Aviezer, Ph.D. |
|
| |
|
|
|
| |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes
and appoints David Aviezer, Ph.D. and Yossi Maimon, and each of them, as his true and lawful
attorneys-in-fact and agents, with full power of substitution and re-substitution, for him and in
his name, place and stead, in any and all capacities, to sign any and all amendments to this Report
on Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection
therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and
agents, and each of them, full power and authority to do and perform each and every act and thing
requisite and necessary to be done in connection therewith, as fully to all intents and purposes as
he might or could do in person, hereby ratifying and confirming that said attorneys-in-fact and
agents, or any of them, or their or his substitute or substitutes, may lawfully do or cause to be
done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been
signed below by the following persons on behalf of the Registrant and in the capacities and on the
dates indicated.
| |
|
|
|
|
| Signature |
|
Title |
|
Date |
| /s/ David Aviezer |
|
President, Chief Executive |
|
February 25, 2010 |
| |
|
|
|
|
| David Aviezer, Ph.D. |
|
Officer (Principal Executive Officer) and
Director |
|
|
| |
|
|
|
|
| /s/ Yossi Maimon |
|
Chief Financial Officer, |
|
February 25, 2010 |
| |
|
|
|
|
| Yossi Maimon |
|
Treasurer and Secretary (Principal
Financial and Accounting
Officer) |
|
|
| |
|
|
|
|
| /s/ Yoseph Shaaltiel |
|
Executive VP, Research and |
|
February 25, 2010 |
| |
|
|
|
|
| Yoseph Shaaltiel, Ph.D. |
|
Development and Director |
|
|
| |
|
|
|
|
| /s/ Alfred Akirov |
|
Director |
|
February 25, 2010 |
| |
|
|
|
|
| Alfred Akirov |
|
|
|
|
| |
|
|
|
|
| /s/ Amos Bar Shalev |
|
Director |
|
February 25, 2010 |
| |
|
|
|
|
| Amos Bar Shalev |
|
|
|
|
| |
|
|
|
|
| /s/ Zeev Bronfeld |
|
Director |
|
February 25, 2010 |
| |
|
|
|
|
| Zeev Bronfeld |
|
|
|
|
| |
|
|
|
|
| /s/ Yodfat Harel Gross |
|
Director |
|
February 25, 2010 |
| |
|
|
|
|
| Yodfat Harel Gross |
|
|
|
|
82
| |
|
|
|
|
| Signature |
|
Title |
|
Date |
| /s/ Eyal Sheratzky
|
|
Director
|
|
February 25, 2010 |
| |
|
|
|
|
| Eyal Sheratzky |
|
|
|
|
| |
|
|
|
|
| /s/ Roger D. Kornberg
|
|
Director
|
|
February 25, 2010 |
| |
|
|
|
|
| Roger D. Kornberg, Ph.D. |
|
|
|
|
83
PROTALIX BIOTHERAPEUTICS, INC.
CONSOLIDATED FINANCIAL STATEMENTS
TABLE OF CONTENTS
| |
|
|
|
|
| |
|
Page |
|
|
|
F-2 |
|
|
|
|
|
|
CONSOLIDATED FINANCIAL STATEMENTS |
|
|
|
|
|
|
|
|
|
|
|
|
F-3 |
|
|
|
|
|
|
|
|
|
F-4 |
|
|
|
|
|
|
|
|
|
F-5 |
|
|
|
|
|
|
|
|
|
F-6 |
|
|
|
|
|
|
|
|
|
F-8 |
|
The dollar amounts are stated in U.S. dollars ($)
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the shareholders of
PROTALIX BIOTHERAPEUTICS, INC.
In our opinion, the consolidated balance sheets and the related statements of operations, changes
in shareholders’ equity and cash flows present fairly, in all material respects, the financial
position of Protalix BioTherapeutics, Inc. and its subsidiaries at December 31, 2008 and 2009, and
the results of their operations and their cash flows for each of the three years in the period
ended December 31, 2009, in conformity with accounting principles generally accepted in the United
States of America. Also, in our opinion, the Company maintained, in all material respects,
effective internal control over financial reporting as of December 31, 2009, based on criteria
established in Internal Control — Integrated Framework issued by the Committee of Sponsoring
Organizations of the Treadway Commission (COSO). The Company’s management is responsible for these
financial statements, for maintaining effective internal control over financial reporting and for
its assessment of the effectiveness of internal control over financial reporting, included in the
accompanying “Management Report on Internal Control over Financial Reporting” appearing under Item
9A. Our responsibility is to express opinions on these financial statements and on the Company’s
internal control over financial reporting based on our integrated audits. We conducted our audits
in accordance with the standards of the Public Company Accounting Oversight Board (United States).
Those standards require that we plan and perform the audits to obtain reasonable assurance about
whether the financial statements are free of material misstatement and whether effective internal
control over financial reporting was maintained in all material respects. Our audits of the
financial statements included examining, on a test basis, evidence supporting the amounts and
disclosures in the financial statements, assessing the accounting principles used and significant
estimates made by management, and evaluating the overall financial statement presentation. Our
audit of internal control over financial reporting included obtaining an understanding of internal
control over financial reporting, assessing the risk that a material weakness exists, and testing
and evaluating the design and operating effectiveness of internal control based on the assessed
risk. Our audits also included performing such other procedures as we considered necessary in the
circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable
assurance regarding the reliability of financial reporting and the preparation of financial
statements for external purposes in accordance with generally accepted accounting principles. A
company’s internal control over financial reporting includes those policies and procedures that:
(i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect
the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance
that transactions are recorded as necessary to permit preparation of financial statements in
accordance with generally accepted accounting principles, and that receipts and expenditures of the
company are being made only in accordance with authorizations of management and directors of the
company; and (iii) provide reasonable assurance regarding prevention or timely detection of
unauthorized acquisition, use or disposition of the company’s assets that could have a material
effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or
detect misstatements. Also, projections of any evaluation of effectiveness to future periods are
subject to the risk that controls may become inadequate because of changes in conditions, or that
the degree of compliance with the policies or procedures may deteriorate.
| |
|
|
|
|
Tel-Aviv, Israel |
|
/s/ Kesselman & Kesselman |
February 25, 2010 |
|
Kesselman & Kesselman |
|
|
Certified Public Accountant (Isr.) |
|
|
A member of PricewaterhouseCoopers |
|
|
International Limited |
F-2
PROTALIX BIOTHERAPEUTICS, INC.
CONSOLIDATED BALANCE SHEETS
(U.S. dollars in thousands, except shares and per share amounts)
| |
|
|
|
|
|
|
|
|
| |
|
December 31, |
|
| |
|
2008 |
|
|
2009 |
|
ASSETS |
|
|
|
|
|
|
|
|
CURRENT ASSETS: |
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
42,596 |
|
|
$ |
81,266 |
|
Accounts receivable |
|
|
793 |
|
|
|
2,144 |
|
|
|
|
|
|
|
|
Total current
assets |
|
|
43,389 |
|
|
|
83,410 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
FUNDS IN RESPECT OF EMPLOYEE
RIGHTS UPON RETIREMENT |
|
|
581 |
|
|
|
724 |
|
|
|
|
|
|
|
|
PROPERTY AND EQUIPMENT, NET |
|
|
6,841 |
|
|
|
14,537 |
|
|
|
|
|
|
|
|
Total assets |
|
$ |
50,811 |
|
|
$ |
98,671 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LIABILITIES AND SHAREHOLDERS’ EQUITY |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CURRENT LIABILITIES: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accounts payable and accruals: |
|
|
|
|
|
|
|
|
Trade |
|
$ |
2,235 |
|
|
$ |
3,406 |
|
Other |
|
|
3,292 |
|
|
|
13,561 |
|
Deferred revenues |
|
|
— |
|
|
|
4,563 |
|
|
|
|
|
|
|
|
Total current
liabilities |
|
|
5,527 |
|
|
|
21,530 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LONG TERM LIABILITIES: |
|
|
|
|
|
|
|
|
Deferred revenues |
|
|
|
|
|
|
60,049 |
|
Liability for employee rights upon retirement |
|
|
937 |
|
|
|
1,209 |
|
|
|
|
|
|
|
|
Total long term
liabilities |
|
|
937 |
|
|
|
61,258 |
|
|
|
|
|
|
|
|
COMMITMENTS |
|
|
|
|
|
|
|
|
Total liabilities |
|
|
6,464 |
|
|
|
82,788 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SHAREHOLDERS’ EQUITY: |
|
|
|
|
|
|
|
|
Common Stock, $0.001 par value: |
|
|
|
|
|
|
|
|
Authorized — as of December 31, 2008 and 2009,
150,000,000 shares; issued and outstanding —
as of December 31, 2008 and 2009, 75,938,059
and 80,841,237 shares, respectively |
|
|
76 |
|
|
|
81 |
|
Additional paid-in capital |
|
|
119,281 |
|
|
|
122,252 |
|
Accumulated deficit |
|
|
(75,010 |
) |
|
|
(106,450 |
) |
|
|
|
|
|
|
|
Total shareholders’ equity |
|
|
44,347 |
|
|
|
15,883 |
|
|
|
|
|
|
|
|
Total liabilities and shareholders’ equity |
|
$ |
50,811 |
|
|
$ |
98,671 |
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of the consolidated financial statements.
F-3
PROTALIX BIOTHERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(U.S. dollars in thousands, except shares and per share amounts)
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year ended December 31, |
|
| |
|
2007 |
|
|
2008 |
|
|
2009 |
|
REVENUES |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
388 |
|
ROYALTIES EXPENSES |
|
|
— |
|
|
|
— |
|
|
|
3,575 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
3,187 |
|
|
|
|
|
|
|
|
|
|
|
|
|
RESEARCH AND DEVELOPMENT EXPENSES |
|
|
14,641 |
|
|
|
22,115 |
|
|
$ |
27,390 |
|
Less — grants |
|
|
(1,071 |
) |
|
|
(4,714 |
) |
|
|
(5,752 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
13,570 |
|
|
|
17,401 |
|
|
|
21,638 |
|
|
|
|
|
|
|
|
|
|
|
GENERAL AND ADMINISTRATIVE EXPENSES |
|
|
20,594 |
|
|
|
6,770 |
|
|
|
7,144 |
|
|
|
|
|
|
|
|
|
|
|
OPERATING LOSS |
|
|
34,164 |
|
|
|
24,171 |
|
|
|
31,969 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
FINANCIAL INCOME — NET |
|
|
(2,080 |
) |
|
|
(1,757 |
) |
|
|
(529 |
) |
|
|
|
|
|
|
|
|
|
|
NET LOSS FOR THE YEAR |
|
$ |
32,084 |
|
|
$ |
22,414 |
|
|
$ |
31,440 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per share of common stock — basic and diluted: |
|
$ |
0.48 |
|
|
$ |
0.30 |
|
|
$ |
0.41 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average number of shares of common stock used
in computing loss per share of common stock, basic and
diluted: |
|
|
67,187,329 |
|
|
|
75,890,633 |
|
|
|
76,942,840 |
|
|
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of the consolidated financial statements.
F-4
PROTALIX BIOTHERAPEUTICS, INC.
CONSOLIDATED STATEMENT OF CHANGES IN SHAREHOLDERS’ EQUITY
(U.S. dollars in thousands, except share data)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional |
|
|
|
|
|
|
|
| |
|
Common |
|
|
Common |
|
|
|
|
|
|
paid-in |
|
|
Accumulated |
|
|
|
|
| |
|
Stock |
|
|
Stock |
|
|
Warrants |
|
|
Capital |
|
|
deficit |
|
|
Total |
|
| |
|
Number of |
|
|
|
|
| |
|
shares |
|
|
Amount |
|
Balance at January 1, 2007 |
|
|
61,781,959 |
|
|
|
62 |
|
|
|
355 |
|
|
|
44,379 |
|
|
|
(20,512 |
) |
|
|
24,284 |
|
Changes during 2007: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Common Stock issued for cash (net of issuance costs
of $4,310) (see Note 5c) |
|
|
10,000,000 |
|
|
|
10 |
|
|
|
— |
|
|
|
45,680 |
|
|
|
— |
|
|
|
45,690 |
|
Share-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
20,437 |
|
|
|
— |
|
|
|
20,437 |
|
Exercise of options granted to employees |
|
|
110,064 |
|
|
|
* |
|
|
|
— |
|
|
|
14 |
|
|
|
— |
|
|
|
14 |
|
Exercise of warrants |
|
|
3,875,416 |
|
|
|
4 |
|
|
|
(355 |
) |
|
|
5,684 |
|
|
|
— |
|
|
|
5,333 |
|
Restricted Common Stock issued for future services (1) |
|
|
8,000 |
|
|
|
* |
|
|
|
— |
|
|
|
11 |
|
|
|
— |
|
|
|
11 |
|
Net Loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(32,084 |
) |
|
|
(32,084 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2007 |
|
|
75,775,439 |
|
|
|
76 |
|
|
|
— |
|
|
|
116,205 |
|
|
|
(52,596 |
) |
|
|
63,685 |
|
Changes during 2008: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Restricted Common Stock issued for future services (1) |
|
|
(5,333 |
) |
|
|
* |
|
|
|
— |
|
|
|
(3 |
) |
|
|
— |
|
|
|
(3 |
) |
Share-based compensation |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
3,074 |
|
|
|
— |
|
|
|
3,074 |
|
Exercise of options granted to employees (includes
net exercise) |
|
|
167,953 |
|
|
|
* |
|
|
|
— |
|
|
|
5 |
|
|
|
— |
|
|
|
5 |
|
Net Loss |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
|
(22,414 |
) |
|
|
(22,414 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2008 |
|
|
75,938,059 |
|
|
|
76 |
|
|
|
— |
|
|
|
119,281 |
|
|
|
(75,010 |
) |
|
|
44,347 |
|
Changes during 2009: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Share-based compensation |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
2,683 |
|
|
|
— |
|
|
|
2,683 |
|
Exercise of options granted to employees and
non-employees (includes net exercise) |
|
|
4,903,178 |
|
|
|
5 |
|
|
|
— |
|
|
|
288 |
|
|
|
— |
|
|
|
293 |
|
Net Loss |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
|
(31,440 |
) |
|
|
(31,440 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2009 |
|
|
80,841,237 |
|
|
|
81 |
|
|
|
— |
|
|
|
122,252 |
|
|
|
(106,450 |
) |
|
|
15,883 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| * |
|
Represents an amount less than $1. |
| |
| (1) |
|
The Company issued a total of 8,000 shares of restricted Common Stock in consideration for
services provided by, and to be provided by, a member of the Company’s Scientific Advisory
Board. Such services were terminated in October 2008 while the forfeiture provisions of the
restricted stock were still in effect. Accordingly, 5,333 shares of the restricted Common
Stock were forfeited. See Note 5d(2)(a)(1). |
The accompanying notes are an integral part of the consolidated financial statements.
F-5
PROTALIX BIOTHERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(U.S. dollars in thousands)
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year ended December 31, |
|
| |
|
2007 |
|
|
2008 |
|
|
2009 |
|
CASH FLOWS FROM OPERATING
ACTIVITIES: |
|
|
|
|
|
|
|
|
|
|
|
|
Net Loss |
|
$ |
(32,084 |
) |
|
$ |
(22,414 |
) |
|
$ |
(31,440 |
) |
Adjustments required to reconcile net loss to net cash
provided by (used in) operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
Share based compensation |
|
|
20,448 |
|
|
|
3,071 |
|
|
|
2,683 |
|
Depreciation and impairment of fixed assets |
|
|
759 |
|
|
|
1,301 |
|
|
|
1,990 |
|
Financial income net (mainly exchange differences) |
|
|
(806 |
) |
|
|
(270 |
) |
|
|
(166 |
) |
Changes in accrued liability for employee rights
upon retirement |
|
|
254 |
|
|
|
247 |
|
|
|
265 |
|
Loss (Gain) on amounts funded in respect of employee
rights upon retirement |
|
|
(57 |
) |
|
|
39 |
|
|
|
(81 |
) |
Loss (Gain) on sale of fixed assets |
|
|
(6 |
) |
|
|
|
|
|
|
29 |
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
|
|
|
Increase in deferred revenues (including non-current portion) |
|
|
|
|
|
|
|
|
|
|
64,612 |
|
Decrease (increase) in accounts receivable |
|
|
140 |
|
|
|
636 |
|
|
|
(1,224 |
) |
Increase in accounts payable and accruals |
|
|
903 |
|
|
|
1,375 |
|
|
|
7,784 |
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by (used in) operating activities |
|
$ |
(10,449 |
) |
|
$ |
(16,015 |
) |
|
$ |
44,452 |
|
|
|
|
|
|
|
|
|
|
|
CASH FLOWS FROM INVESTING
ACTIVITIES: |
|
|
|
|
|
|
|
|
|
|
|
|
Purchase of property and equipment |
|
$ |
(2,335 |
) |
|
$ |
(3,371 |
) |
|
$ |
(6,195 |
) |
Investment in restricted deposit |
|
|
— |
|
|
|
(175 |
) |
|
|
|
|
Proceeds from sale of property and equipment |
|
|
11 |
|
|
|
1 |
|
|
|
73 |
|
Amounts funded in respect of employee rights
upon retirement, net |
|
|
(114 |
) |
|
|
(156 |
) |
|
|
(52 |
) |
|
|
|
|
|
|
|
|
|
|
Net cash used in investing activities |
|
$ |
(2,438 |
) |
|
$ |
(3,701 |
) |
|
$ |
(6,174 |
) |
|
|
|
|
|
|
|
|
|
|
CASH FLOWS FROM FINANCING
ACTIVITIES: |
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of shares and warrants, net of issuance cost |
|
$ |
45,746 |
|
|
$ |
(56 |
) |
|
|
|
|
Exercise of options and warrants |
|
|
12,924 |
|
|
|
5 |
|
|
$ |
293 |
|
Merger with a wholly-owned subsidiary of the Company, net of
issuance cost |
|
|
(104 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by (used in) financing activities |
|
$ |
58,566 |
|
|
$ |
(51 |
) |
|
$ |
293 |
|
|
|
|
|
|
|
|
|
|
|
EFFECT OF EXCHANGE RATE CHANGES ON
CASH |
|
|
756 |
|
|
|
550 |
|
|
|
99 |
|
|
|
|
|
|
|
|
|
|
|
NET INCREASE (DECREASE) IN CASH AND
CASH EQUIVALENTS |
|
|
46,435 |
|
|
|
(19,217 |
) |
|
|
38,670 |
|
BALANCE OF CASH AND CASH
EQUIVALENTS AT BEGINNING OF YEAR |
|
|
15,378 |
|
|
|
61,813 |
|
|
|
42,596 |
|
|
|
|
|
|
|
|
|
|
|
BALANCE OF CASH AND CASH
EQUIVALENTS AT END OF YEAR |
|
$ |
61,813 |
|
|
$ |
42,596 |
|
|
$ |
81,266 |
|
|
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of the consolidated financial statements.
F-6
PROTALIX BIOTHERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(U.S. dollars in thousands)
(CONTINUED)
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year ended December 31, |
|
| |
|
2007 |
|
|
2008 |
|
|
2009 |
|
SUPPLEMENTARY INFORMATION ON
INVESTING AND FINANCING ACTIVITIES
NOT INVOLVING CASH FLOWS: |
|
|
|
|
|
|
|
|
|
|
|
|
Purchase of property and equipment |
|
$ |
666 |
|
|
$ |
932 |
|
|
$ |
4,525 |
|
|
|
|
|
|
|
|
|
|
|
Issuance cost not yet paid and accruals — other |
|
$ |
61 |
|
|
$ |
5 |
|
|
$ |
5 |
|
|
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of the consolidated financial statements.
F-7
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 1 — SIGNIFICANT ACCOUNTING POLICIES
| |
1. |
|
Operation |
| |
| |
|
|
Protalix BioTherapeutics, Inc. and its wholly-owned subsidiary, Protalix Ltd. (the
“Israeli Subsidiary” or “Protalix Ltd.,” and collectively with Protalix
BioTherapeutics, Inc., the “Company”), are biopharmaceutical companies focused on
the development and commercialization of recombinant therapeutic proteins based on
the Company’s proprietary ProCellExtm protein expression system
(“ProCellEx”). In September 2009, the Company formed another wholly-owned
subsidiary under the laws of the Netherlands in connection with the EMEA application
process in Europe. The Company’s lead product development candidate is
taliglucerase alfa for the treatment of Gaucher disease (the brand name for which is
UPLYSO™), which the Company is developing using its ProCellEx protein expression
system. |
| |
| |
|
|
In September 2009, the Company successfully completed its phase III pivotal trial of
taliglucerase alfa. In December 2009, the Company filed a New Drug Application
(“NDA”) submission with the U.S. Food and Drug Administration (“FDA”) for
taliglucerase alfa for the treatment of Gaucher disease. |
| |
| |
|
|
In addition to its phase III clinical trial, the Company initiated a clinical study
in December 2008 to evaluate the safety and efficacy of switching Gaucher patients
currently treated under the current standard of care to treatment with taliglucerase
alfa. This switchover-study is not a prerequisite for the marketing approval of
taliglucerase alfa. In August 2009, the Company received Fast Track Designation for
taliglucerase alfa, and in September 2009, the FDA’s Office of Orphan Product
Development granted taliglucerase alfa Orphan Drug Status. |
| |
| |
|
|
The Company has been in the development stage since its inception until November
2009 (see 2 below). |
| |
| |
|
|
On November 30, 2009, Protalix Ltd. and Pfizer Inc. (“Pfizer”) entered into an
Exclusive License and Supply Agreement (the “Pfizer Agreement”) pursuant to which
Protalix Ltd. granted Pfizer an exclusive, worldwide license to develop and
commercialize taliglucerase alfa, except in Israel. Under the terms and conditions
of the Pfizer Agreement, Protalix Ltd. retained the right to commercialize
taliglucerase alfa in Israel. See Note 9. |
| |
| |
|
|
Successful completion of the Company’s development program and its transition to
normal operations is dependent upon obtaining necessary regulatory approvals from
the FDA prior to selling its products within the United States, and foreign
regulatory approvals must be obtained to sell its products internationally. There
can be no assurance that the Company will receive regulatory approval of any of its
product candidates, and a substantial amount of time may pass before the Company
achieves a level of sales adequate to support the Company’s operations, if at all.
The Company will also incur substantial expenditures in connection with the
regulatory approval process for each of its product candidates during the
developmental period. Obtaining marketing approval will be directly dependent on
the Company’s ability to implement the necessary regulatory steps required to obtain
marketing approval in the United States and in other countries. The Company cannot
predict the outcome of these activities. |
F-8
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 1 — SIGNIFICANT ACCOUNTING POLICIES (continued):
| |
2. |
|
The Merger |
| |
| |
|
|
On December 31, 2006, Protalix BioTherapeutics, Inc. (formerly Orthodontix, Inc.)
consummated the acquisition of Protalix Ltd., a privately-held Israeli biotechnology
company incorporated on December 27, 1993, by the merger (the “Merger”) of its
wholly-owned subsidiary, Protalix Acquisition Co., Ltd., with Protalix Ltd. At and
as of the Merger, the former shareholders of Protalix Ltd. received a number of
shares of Common Stock equal to more than 99% of the outstanding shares of Common
Stock. As a result, Protalix Ltd. is now the Company’s wholly-owned subsidiary. As
of that date, for accounting purposes, the Merger was accounted for as a
recapitalization of Protalix Ltd. Accordingly, the historical financial statements
of the Company reflect the historical operations and financial statements of the
Subsidiary before the Merger. |
| |
| |
3. |
|
Subsequent Events |
| |
| |
|
|
The Company has evaluated events through February 25, 2010, the date of issuance of
the
financial statements. See Note 10. |
| |
b. |
|
Basis of presentation |
| |
| |
|
|
The Company’s financial statements have been prepared in accordance with generally
accepted accounting principles in the United States (“U.S. GAAP”). Prior to December
2009, the Company was a development stage company as defined under the guidance for
Development Stage Enterprises. Commencing November 2009, the Company ceased to
consider itself a development stage company. |
| |
| |
|
|
In June 2009, the Financial Accounting Standards Board (“FASB”) issued the FASB
Accounting Standards Codification (“Codification” or “ASC”). The Codification became
the single authoritative source for U.S. GAAP and changed the way in which the
accounting literature is organized. The Codification does not change U.S. GAAP and
accordingly its adoption did not have a material impact on the Company’s consolidated
financial statements. |
| |
| |
c. |
|
Use of estimates in the preparation of financial statements |
| |
| |
|
|
The preparation of financial statements in conformity with U.S. GAAP requires
management to make estimates and assumptions that affect the reported amounts of assets
and liabilities, the disclosure of contingent assets and liabilities at the date of the
financial statements and the reported amounts of revenues and expenses during the
reporting period. Actual results may differ from those estimates. |
| |
| |
d. |
|
Functional currency |
| |
| |
|
|
The dollar is the currency of the primary economic environment in which the operations
of the Company and its Subsidiaries are conducted. The Company’s revenues are derived
in dollars. Most of the Company’s expenses and capital expenditures are incurred in
dollars, and the major source of the Company’s financing to date has been provided in
dollars. |
| |
| |
|
|
Transactions and balances originally denominated in dollars are presented at their
original amounts. Balances in non-dollar currencies are translated into dollars using
historical and current exchange rates for non-monetary and monetary balances,
respectively. For non-dollar transactions and other items (stated below) reflected in
the statements of operations, the following exchange rates are used: (i) for
transactions — exchange rates at the transaction dates or average rates; and (ii) for
other items (derived from non-monetary balance sheet items such as depreciation and
amortization, etc.) — historical exchange rates. Currency transaction gains or losses
are carried to financial income or expenses, as appropriate. |
F-9
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 1 — SIGNIFICANT ACCOUNTING POLICIES (continued):
| |
e. |
|
Cash equivalents |
| |
| |
|
|
The Company considers all short-term, highly liquid investments, which include
short-term bank deposits with original maturities of three months or less from the date
of purchase, that are not restricted as to withdrawal or use and are readily
convertible to known amounts of cash, to be cash equivalents. |
| |
| |
f. |
|
Property and equipment |
| |
1. |
|
Property and equipment are stated at cost, net of accumulated
depreciation and amortization. |
| |
| |
2. |
|
The Company’s assets are depreciated by the straight-line method on
the basis of their estimated useful lives as follows: |
| |
|
|
|
|
| |
|
years |
Laboratory equipment |
|
|
5 |
|
Furniture |
|
|
10-15 |
|
Computer equipment |
|
|
3 |
|
Leasehold improvements are amortized by the straight-line method over the
expected lease term, which is shorter than the estimated useful life of the
improvements.
| |
g. |
|
Impairment in value of long-lived assets: |
| |
| |
|
|
The Company tests long-lived assets for impairment if an indication of impairment
exists. If the sum of expected future cash flows of definite life of long lived assets
(undiscounted and without interest charges) is less than the carrying amount of such
assets, the Company recognizes an impairment loss, and writes down the assets to their
estimated fair values, calculated based on expected future discounted cash flows. See
Note 2c. |
| |
| |
h. |
|
Income taxes |
| |
1. |
|
Deferred income taxes |
| |
| |
|
|
Deferred taxes are determined utilizing the assets and liabilities method based on
the estimated future tax effects of the differences between the financial accounting
and tax bases of assets and liabilities under the applicable tax laws. Deferred tax
balances are computed using the tax rates expected to be in effect when those
differences reverse. A valuation allowance in respect of deferred tax assets is
provided if, based upon the weight of available evidence, it is more likely than not
that some or all of the deferred tax assets will not be realized. The Company has
provided a full valuation allowance with respect to its deferred tax assets. |
| |
| |
|
|
The guidance prohibits the recognition of deferred tax liabilities or assets that
arise from differences between the financial reporting and tax bases of assets and
liabilities that are measured from the local currency into dollars using historical
exchange rates, and that result from changes in exchange rates or indexing for tax
purposes. Consequently, the above mentioned differences with respect to Protalix
Ltd. were not reflected in the computation of deferred tax assets and liabilities. |
| |
| |
2. |
|
Uncertainty in income taxes |
| |
| |
|
|
As of January 1, 2007, the Company adopted accounting guidance with respect to
accounting for uncertainty in income taxes. |
| |
| |
|
|
Tax benefits recognized in the financial statements are at least more likely than
not of being sustained, based on technical merits. The amount of benefits recorded
for these tax benefits is measured as the largest benefit more likely than not to be
sustained. |
F-10
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 1 — SIGNIFICANT ACCOUNTING POLICIES (continued):
| |
i. |
|
Revenue Recognition |
| |
| |
|
|
The Company earns revenue under collaboration agreements with third parties to develop
and produce drug candidates. The Company recognizes revenue and milestone payments in
accordance with guidance regarding revenue recognition and accounting for revenue
arrangements with multiple deliverables. Pursuant to this guidance, the Company
determines whether an arrangement involves multiple revenue-generating deliverables
that should be accounted for as a combined unit of accounting or separate units of
accounting for revenue recognition purposes. If it is determined that there are
multiple units of accounting, the consideration from the arrangement is allocated among
the separate units based on a relative fair value allocation. If the arrangement
represents a single unit of accounting, the revenue is recognized over the performance
obligation period. Non-refundable up-front license payments, where continuing
involvement is required of the Company, are deferred and recognized over the related
performance period. The Company estimates its performance period based on the specific
terms of each collaboration agreement and adjusts the performance periods, if
appropriate, based on the applicable facts and circumstances. |
| |
| |
j. |
|
Research and development costs |
| |
| |
|
|
Research and development costs are expensed as incurred and consist primarily of
personnel, subcontractors and consultants, facilities, equipment and supplies for
research and development activities. Grants received by the Israeli Subsidiary from
the Office of the Chief Scientist of Israel’s Ministry of Industry, Trade and Labor
(the “OCS”) and other research foundations are recognized when the grant becomes
receivable, provided there is reasonable assurance that the Company or the Subsidiary
will comply with the conditions attached to the grant and there is reasonable assurance
the grant will be received. The grant is deducted from the related research and
development expenses as the applicable costs are incurred. See Note 4(a). |
| |
| |
|
|
In connection with purchases of assets, amounts assigned to intangible assets to be
used in a particular research and development project that have no alternative future
use are charged to research and development costs at the purchase date. |
| |
| |
|
|
Nonrefundable advance payments for goods or services that will be used or rendered for
future research and development activities are deferred and amortized over the period
that the goods are delivered or the related services are performed, subject to an
assessment of recoverability. |
| |
| |
k. |
|
Comprehensive loss |
| |
| |
|
|
The Company has no other comprehensive loss components other than net loss for the
reported periods. |
| |
| |
l. |
|
Concentration of credit risks |
| |
| |
|
|
Financial instruments that potentially subject the Company to concentration of credit
risk consist principally of bank deposits. The Company deposits these instruments with
highly rated financial institutions, mainly in Israeli banks, and, as a matter of
policy, limits the amounts of credit exposure to any one financial institution. The
Company has not experienced any credit losses in these accounts and does not believe it
is exposed to any significant credit risk on these instruments. |
F-11
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 1 — SIGNIFICANT ACCOUNTING POLICIES (continued):
| |
m. |
|
Share-based compensation |
| |
| |
|
|
The Company accounts for employee’s share-based payment awards classified as equity
awards using the grant-date fair value method. The fair value of share-based payment
transactions is recognized as an expense over the requisite service period, net of
estimated forfeitures. The Company estimated forfeitures based on historical
experience and anticipated future conditions. |
| |
| |
|
|
The Company elected to recognize compensation cost for an award with only service
conditions that has a graded vesting schedule using the accelerated method based on the
multiple-option award approach. |
| |
| |
|
|
When stock options are granted as consideration for services provided by consultants
and other non-employees, the transaction is accounted for based on the fair value of
the consideration received or the fair value of the stock options issued, whichever is
more reliably measurable. The fair value of the options granted is measured on a final
basis at the end of the related service period and is recognized over the related
service period using the straight-line method. |
| |
| |
n. |
|
Net Loss per share |
| |
| |
|
|
Basic and diluted loss per share (“LPS”) are computed by dividing net loss by the
weighted average number of shares of Common Stock outstanding for each period. |
| |
| |
|
|
Shares of restricted Common Stock and the shares of Common Stock underlying outstanding
options of the Company were not included in the computation of diluted LPS because of
the anti-dilutive effect of doing so. |
| |
| |
|
|
Diluted LPS does not include options and restricted shares of Common Stock and warrants
of the Company in the amount of 11,959,795, 11,037,356 and 10,660,447 shares of Common
Stock for the years 2007, 2008 and 2009, respectively. |
| |
| |
o. |
|
Fair Value Measurement |
| |
| |
|
|
Fair value is defined as the price that would be received to sell an asset or paid to
transfer a liability (i.e., the “exit price”) in an orderly transaction between market
participants at the measurement date. |
| |
| |
p. |
|
Newly Issued and Recently Adopted Accounting Pronouncements |
| |
1. |
|
In May 2009, the FASB issued ASC Topic 855 “Subsequent Events”
(formerly SFAS No. 165, Subsequent Events). ASC 855 sets forth the period after
the balance sheet date during which management of a reporting entity should
evaluate events or transactions that may occur for potential recognition or
disclosure in the financial statements, the circumstances under which an entity
should recognize events or transactions occurring after the balance sheet date in
its financial statements, and the disclosures that an entity should make about
events or transactions that occurred after the balance sheet date. ASC 855 is
effective for interim or annual periods ending after June 15, 2009. The Company
adopted the provisions of ASC 855 for the quarter ended June 30, 2009. The
adoption of ASC 855 did not have a material impact on the Company’s condensed
financial statements. |
F-12
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 1 — SIGNIFICANT ACCOUNTING POLICIES (continued):
| |
2. |
|
In June 2009, the FASB issued Accounting Standards Update (“ASU”) No.
2009-1, “Topic 105 — Generally Accepted Accounting Principles” which amended ASC
105 “The “FASB Accounting Standards Codification” and the Hierarchy of Generally
Accepted Accounting Principles (formerly SFAS No. 168 “The FASB Accounting
Standards Codification and the Hierarchy of Generally Accepted Accounting
Principles — A Replacement of FASB Statement No. 162”). ASU 2009-1 establishes
the FASB Accounting Standards CodificationTM (Codification) as the
single source of authoritative U.S. GAAP recognized by the FASB to be applied by
nongovernmental entities. Rules and interpretive releases of the U.S. Securities
and Exchange Commission (the “SEC”) under authority of federal securities laws are
also sources of authoritative U.S. GAAP for SEC registrants. ASU 2009-1 and the
Codification are effective for financial statements issued for interim and annual
periods ending after September 15, 2009. The Codification supersedes all existing
non-SEC accounting and reporting standards. All other nongrandfathered non-SEC
accounting literature not included in the Codification will become
nonauthoritative. The FASB will no longer issue new standards in the form of
Statements, FASB Staff Positions, or Emerging Issues Task Force Abstracts.
Instead, the FASB will issue Accounting Standards Updates, which will serve only
to: (a) update the Codification; (b) provide background information about the
guidance; and (c) provide the bases for conclusions on the change(s) in the
Codification. The adoption of ASU 2009-1 did not have a material impact on the
Company’s financial statements. |
| |
| |
3. |
|
Multiple Deliverable Revenue Arrangements |
| |
| |
|
|
In October 2009, the FASB issued an Accounting Standards Update to ASC 605, ASU No.
2009-13, “Multiple Deliverable Revenue Arrangements” (“ASU 2009-13”). ASU 2009-13
provides guidance on whether multiple deliverables in a revenue arrangement exist,
how the arrangement should be separated, and how the consideration should be
allocated. Pursuant to ASU 2009-13, when vendor specific objective evidence or
third party evidence for deliverables in an arrangement cannot be determined, a best
estimate of the selling price is required to separate deliverables and allocate
arrangement consideration, using the relative selling price method. In addition,
the residual method of allocating arrangement consideration is no longer permitted
under ASU 2009-13. |
| |
| |
|
|
ASU 2009-13 is effective for revenue arrangements entered into or materially
modified in fiscal years beginning on or after June 15, 2010. The Company is
currently evaluating the potential impact of ASU 2009-13 on its consolidated
financial position, results of operations and cash flows. |
NOTE 2 — PROPERTY AND EQUIPMENT
| |
a. |
|
Composition of property and equipment grouped by major classifications, and
changes, is as follows: |
| |
|
|
|
|
|
|
|
|
| |
|
December 31, |
|
| |
|
2008 |
|
|
2009 |
|
Laboratory equipment |
|
$ |
5,898 |
|
|
$ |
10,008 |
|
Furniture and computer equipment |
|
|
699 |
|
|
|
944 |
|
Leasehold improvements |
|
|
3,058 |
|
|
|
5,665 |
|
Equipment under construction |
|
|
97 |
|
|
|
2,615 |
|
|
|
|
|
|
|
|
|
|
$ |
9,752 |
|
|
$ |
19,232 |
|
Less — accumulated depreciation
and amortization |
|
|
(2,911 |
) |
|
|
(4,695 |
) |
|
|
|
|
|
|
|
|
|
$ |
6,841 |
|
|
$ |
14,537 |
|
|
|
|
|
|
|
|
F-13
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 2 — PROPERTY AND EQUIPMENT (continued):
| |
b. |
|
Depreciation and amortization in respect of property and equipment totaled
$692, $1,301 and $1,990 for the years ended December 31, 2007, 2008 and 2009,
respectively. |
| |
| |
c. |
|
The Company tested the carrying value of certain long lived assets. As a
result, during the year ended December 31, 2007, the Company recorded a total
impairment of $67. See Notes 7c and 7d. The impaired long lived assets were mainly
laboratory equipment and leasehold improvements. |
NOTE 3 — LIABILITY FOR EMPLOYEE RIGHTS UPON RETIREMENT
The Israeli Subsidiary is required to make a severance payment upon dismissal of an
employee, or upon termination of employment in certain circumstances. The severance pay
liability to the employees (based upon length of service and the latest monthly salary — one
month’s salary for each year employed) is reflected by a balance sheet accrual under
“Liability for employee rights upon retirement.” The liability is recorded as if it were
payable at each balance sheet date on an undiscounted basis.
The liability is funded in part by the purchase of insurance policies or pension funds and
by the deposit of funds in dedicated deposits. The amounts funded are included in the
balance sheets under “Funds in respect of employee rights upon retirement.” These policies
are the Company’s assets. However, under labor agreements and subject to certain
limitations, any policy may be transferred to the ownership of the individual employee for
whose benefit the funds were deposited in the policy. In the years ended December 31, 2007,
2008 and 2009, the Company deposited $128, $161 and $79, respectively, with the insurance
companies in connection with its severance payment obligations.
In accordance with the current employment agreements with certain employees, the Company
makes regular deposits with certain insurance companies for accounts controlled by each
applicable employee in order to secure the employee’s rights upon retirement. The Company
is fully relieved from any severance pay liability with respect to each such employee after
it makes the payments on behalf of the employee. The liability accrued in respect of these
employees and the amounts funded, as of the respective agreement dates, are not reflected in
the balance sheets, as the amounts funded are not under the control and management of the
Company and the pension or severance pay risks have been irrevocably transferred to the
applicable insurance companies (the “Contribution Plans”).
The Company accounts for the severance pay obligations as contemplated by guidance regarding
“determination of vested benefit obligation for a defined benefit pension plan” and,
accordingly, records the obligations on a non-discounted basis as if they were payable at
each balance sheet date.
The amounts of severance pay expenses were $365, $563 and $488 for the years ended December
31, 2007, 2008 and 2009, respectively, of which $140, $319 and $335 in the years ended
December 31, 2007, 2008 and 2009, respectively, were in respect of a Contribution Plan.
Gain (loss) on amounts funded in respect of employee rights upon retirement totaled $57,
$(39) and $81 for the years ended December 31, 2007, 2008 and 2009, respectively.
The Company expects to contribute approximately $570 in 2010 to insurance companies in
connection with its severance liabilities for its 2010 operations, $420 of which will be
contributed to one or more Contribution Plans.
During the 10-year period following December 31, 2009, the Company expects to pay future
benefits to two employees upon their normal retirement age, which is anticipated to amount
to $64 and $23 during the years 2010 and 2012, respectively. These amounts were determined
based on each such employee’s current salary rates and the number of years of employment
that will be accumulated upon the retirement date of each such employee. This expectation
does not include additional amounts that might be paid to employees that will cease working
for the Company before their normal retirement age.
F-14
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 4 — COMMITMENTS
| |
1. |
|
The Company is obligated to pay royalties to the OCS on proceeds from
the sale of products developed from research and development activities that were
funded, partially, by grants from the OCS. At the time the grants were received,
successful development of the related projects was not assured. |
| |
| |
|
|
In the case of failure of a project that was partly financed as described above, the
Company is not obligated to pay any such royalties or repay funding received from the
OCS. |
| |
| |
|
|
Under the terms of the funding arrangements with the OCS, royalties of 3% to 6% are
payable on the sale of products developed from projects funded by the OCS, which
payments shall not exceed, in the aggregate, 100% of the amount of the grant received
(dollar linked), plus, commencing upon January 1, 2001, interest at annual rate based
on LIBOR. In addition, if the Company receives approval to manufacture the products
developed with government grants outside the State of Israel, it will be required to
pay an increased total amount of royalties (possibly up to 300% of the grant amounts
plus interest), depending on the manufacturing volume that is performed outside the
State of Israel, and, possibly, an increased royalty rate. |
| |
| |
|
|
The Company is obligated to pay the OCS royalties in respect of the amounts received
from Pfizer under the Pfizer Agreement. Royalty expenses are included in the
statement of operations as a component of royalties’ expenses and were approximately
$1,950 in the aggregate during the year ended December 31, 2009. |
| |
| |
|
|
At December 31, 2009, the maximum royalty amount payable by the Company under these
funding arrangements is approximately $13,941 (without interest, assuming 100% of the
funds are payable). |
| |
| |
2. |
|
The Company is a party to certain research and license agreements.
Under the agreements, the Company is obligated to pay royalties at varying rates
from its future revenues. As of December 31, 2009, royalty payments in the amount
of $1,625 have become payable under the agreements due to the execution of the
Pfizer Agreement and are included in the statement of operations as a component of
royalties’ expenses. |
| |
| |
|
|
Under each agreement, the Company is also obligated to pay milestone, licensing and
other payments to the counterparties of the agreement. The payments under the
agreements are for varying amounts and are subject to varying conditions. If all of
the contingencies with respect to milestone payments under the research and license
agreements are met, the aggregate milestone payments payable would be approximately
$1,050 and would be payable, if at all, as the Company’s projects progress over the
course of a number of years. A total of $70 of milestone payments have become
payable under the Company’s agreements for the year ended December 31, 2009 in
connection with the filing of the NDA for taliglucerase alfa with the FDA. |
| |
| |
|
|
None of the agreements has a fixed termination date. Subject to earlier termination
for other reasons, each agreement terminates after a certain number of years
following the first commercial sale of any licensed product under the agreement or
after a certain number of years without the initiation of commercial sales of any
product under the agreement. |
| |
b. |
|
Subcontracting Agreements |
| |
| |
|
|
The Company has entered into sub-contracting agreements with several clinical providers
in Israel, the United States and certain other countries in connection with its primary
product development process. As of December 31, 2009, total commitments under said
agreements were approximately $3,693. |
F-15
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 4 — COMMITMENTS (continued):
| |
c. |
|
Lease Agreements |
| |
| |
|
|
The Company is a party to a number of lease agreements for its facilities, the latest of
which expires in 2017. The Company has the option to extend certain of such agreements
on three occasions for additional five-year periods, for a total of 15 additional years.
Under the leases, the aggregate monthly rental payments are approximately $63. As of
December 31, 2008, the Company provided bank guarantees of approximately $226, in the
aggregate, to secure the fulfillment of its obligations under the lease agreements. The
future minimum lease payments required in each of the next five years under the
operating leases for such premises are approximately as follows: 2010 — $791, 2011 -
$798, 2012 — $798, 2013 — $798 and 2014 — $798. Lease expenses totaled $197, $220 and
$780 for the years ended December 31, 2007, 2008 and 2009, respectively. |
| |
| |
d. |
|
Vehicle Lease and Maintenance Agreements |
| |
| |
|
|
In July 2004, the Company entered into several three-year lease and maintenance
agreements for vehicles which are regularly amended as new vehicles are leased. The
current monthly lease fees aggregate approximately $31. The expected lease payments for
the years ending December 31, 2010, 2011 and 2012 are $346, $270 and $173, respectively. |
| |
| |
e. |
|
Teva Agreement |
| |
| |
|
|
On September 14, 2006, the Company entered into agreement (the “Teva Agreement”) with
Teva Pharmaceutical Industries Ltd. (“Teva”) under which the Company agreed to
collaborate on the research and development of two proteins to be identified by Teva and
the Company, using ProCellEx. The Teva Agreement also identifies additional matters for
collaboration between Teva and the Company. The Company granted to Teva an exclusive
license to commercialize any products developed under the collaboration in return for
royalty and milestone payments payable upon the achievement of certain pre-defined
goals. The Company will retain certain exclusive manufacturing rights with respect to
the active pharmaceutical ingredient of the proteins following the first commercial sale
of a licensed product under the agreement and other rights thereafter. Subsequently,
two proteins were identified to be researched and developed under the agreement but in
2009, both of the projects were terminated for commercial reasons. Eli Hurvitz, the Chairman of the Company’s
Board of Directors, is the Chairman of Teva’s Board of Directors, and Phillip Frost
M.D., a former director and a large, indirect shareholder of the Company, is the Vice
Chairman of Teva’s Board of Directors. |
| |
| |
f. |
|
Yissum Agreement |
| |
| |
|
|
On August 8, 2007, the Company signed an agreement with the Yissum Research and
Development Company, the technology transfer arm of the Hebrew University of Jerusalem,
Israel, and the Boyce Thompson Institute for Plant Research, at Cornell University,
Ithaca, New York, to develop a proprietary plant cell-based acetylcholinesterase (AChE)
and its molecular variants for the use in several therapeutic and prophylactic
indications, including a biodefense program and organophosphate-based pesticide
treatment. Pursuant to the agreement, the Company has received an exclusive worldwide
right and license to certain technology, including patents and additional patent
applications relating to AChE (the “Licensed Technology”), for all therapeutic and
prophylactic indications. In consideration for the license, the Company is required to
make certain milestone payments upon its achievement of clinical milestones and
royalties from sales derived from any drugs developed by it with the Licensed
Technology. The agreement does not terminate until either party to the agreement elect
to terminate the agreement, subject to certain terms and conditions set forth therein. |
F-16
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 5 — SHARE CAPITAL
| |
a. |
|
Rights of the Company’s Stock |
| |
1. |
|
Common Stock |
| |
| |
|
|
Each share of Common Stock is entitled to one vote. The holders of Common Stock are
also entitled to receive dividends whenever funds are legally available, when and if
declared by the Board of Directors. Since its inception, the Company has not
declared any dividends. |
| |
| |
2. |
|
Preferred Shares |
| |
| |
|
|
The preferred shares were authorized in the Company’s Restated Articles of
Incorporation on April 16, 1998. The rights and privileges of the preferred stock
may be established by the Company’s Board of Directors. The directors have not
designated any class of preferred stock and no shares of preferred stock have ever
been issued. |
| |
b. |
|
On January 31, 2007, warrants that were issued in connection with a share
purchase agreement dated August 2006, were exercised for 3,875,416 shares of Common
Stock for an aggregate exercise price of $5,333. |
| |
| |
c. |
|
In October 2007, the Company issued and sold 10,000,000 shares of Common Stock
in an underwritten public offering at a price equal to $5.00 per share. The net
proceeds to the Company were $45,690 (net of underwriting commissions and issuance
costs of $4,310). |
| |
| |
d. |
|
Stock based compensation |
| |
| |
|
|
On December 14, 2006, the Board of Directors adopted the Protalix BioTherapeutics, Inc.
2006 Stock Incentive Plan (the “Plan”). The grant of options to Israeli employees
under the Plan is subject to the terms stipulated by Sections 102 and 102A of the
Israeli Income Tax Ordinance. Each option grant is subject to the track chosen by the
Company, either Section 102 or Section 102A of the Israeli Income Tax Ordinance, and
pursuant to the terms thereof, the Company is not allowed to claim, as an expense for
tax purposes, the amounts credited to employees as a benefit, including amounts
recorded as salary benefits in the Company’s accounts, in respect of options granted to
employees under the Plan, with the exception of the work-income benefit component, if
any, determined on the grant date. For Israeli non-employees, the share option plan is
subject to Section 3(i) of the Israeli Income Tax Ordinance. |
| |
| |
|
|
Immediately prior to the closing of the Merger, options to purchase 88,001 ordinary
shares of Protalix Ltd. were outstanding under the Plan. Pursuant to the terms of the
Merger Agreement, the Company assumed all of the outstanding obligations under such
plan and, accordingly, the Company issued options to purchase 5,375,174 shares of
Common Stock, in the aggregate, in lieu of ordinary shares of Protalix Ltd., and has
reserved an additional 4,366,481 shares of Common Stock under the Plan for future
allocation under the Plan. As of December 31, 2009, 1,433,623 shares of Common Stock
remain available for grant under the Plan. |
| |
| |
|
|
For purposes of determining the fair value of the options and shares of restricted
Common Stock granted to employees and non-employees, the Company’s management uses the
fair value of the Common Stock. |
F-17
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 5 — SHARE CAPITAL (continued):
| |
|
|
During the years ended December 31, 2001 through 2009, the Company granted options
and shares of restricted Common Stock to certain employees and non-employees as
follows: |
| |
| |
1. |
|
Options granted to employees: |
a) Below is a table summarizing all of the option grants to employees from inception
through December 31, 2009:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Year of |
|
No. of options |
|
Exercise |
|
|
|
|
|
Fair value |
|
Exercise |
| Grant |
|
granted |
|
price range |
|
Vesting period |
|
at grant |
|
Period |
2001 |
|
|
244,324 |
|
|
|
0.001 |
|
|
immediate |
|
$ |
42 |
|
|
10 years |
2003 |
|
|
1,243,977 |
|
|
|
0.12 |
|
|
Approximately 610,017 immediate
and the remainder in 4 years |
|
$ |
389 |
|
|
10 years |
2005 |
|
|
1,182,591 |
|
|
|
0.12-0.40 |
|
|
4 years |
|
$ |
939 |
|
|
10 years |
2006* |
|
|
2,201,972 |
|
|
|
0.97 |
|
|
4 years |
|
$ |
1,963 |
|
|
10 years |
2007 |
|
|
204,351 |
|
|
|
4.33 |
|
|
4 years |
|
$ |
5,790 |
|
|
10 years |
2008 |
|
|
2,060,000 |
|
|
|
2.35-5.00 |
|
|
4 -5 years |
|
$ |
2,914 |
|
|
10 years |
2009** |
|
|
504,000 |
|
|
|
2.65 |
|
|
Upon achievement of certain milestones |
|
$ |
1,068 |
|
|
10 years |
2009 |
|
|
120,400 |
|
|
|
2.65 |
|
|
4 years |
|
$ |
212 |
|
|
10 years |
| |
|
|
|
7,761,615 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| * |
|
Includes options granted outside of an option plan. |
| |
| ** |
|
The milestone was achieved as of December 31,
2009 rendering the options fully vested. |
Set forth below are grants made by the Company in 2009 and grants made by the
Company prior to 2009 to certain related parties (such grants appear in the table
above):
| |
1. |
|
In May 2007, the Company’s Board of Directors approved
the grant of options to purchase 204,351 shares of Common Stock to a
newly-hired officer of the Company, at an exercise price equal to $4.33 per
share. The options vest over a four-year period and are exercisable for a
10-year period commencing on the date of grant. In May 2008, the officer’s
employment by the Company was terminated. As a result, 127,719 of the
options granted to the officer were forfeited. |
| |
| |
2. |
|
In February 2008, the Company’s Board of Directors
approved the grant of options to purchase 1,900,000 shares of Common Stock
to certain officers and employees of the Company with an exercise price
equal to $5.00 per share. The options vest variably over a period of up to
five years. The options are exercisable over a 10-year period commencing on
the date of grant. |
| |
| |
3. |
|
In October 2008, the Company’s Board of Directors
approved the grant of options to purchase 160,000 shares of Common Stock to
a new newly-hired officer of the Company of the Company with an exercise
price equal to $2.35 per share. The options vest over a of four-year period.
The options are exercisable over a 10-year period commencing on the date of
grant. |
| |
| |
4. |
|
In February 2008, the Company amended the stock option
agreements of certain executive officers. As amended, such stock option
agreements provide for the full acceleration of the vesting period of
unvested options held by such officers immediately upon a change of
control. The Company concluded that there was no incremental increase in the
value of the awards and therefore no accounting charges need to be recorded
in connection with such modification. |
F-18
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 5 — SHARE CAPITAL (continued):
| |
5. |
|
In February, 2009, the Company’s Board of Directors
approved the grant of options to purchase 624,400 shares of Common Stock to
certain officers and employees of the Company with an exercise price equal
to $2.65 per share. The options vest as follows: |
(i) 504,000 of the options vest immediately upon the achievement of certain
clinical and operational performance milestones, which milestones must be
achieved within one year of the date of grant or the options will be
forfeited. The options are exercisable over a 10-year period commencing on
the date of grant. The Company estimated the fair value of the options on
the date of grant using the Black-Scholes option-pricing model to be
approximately $1,068, based on the following weighted average assumptions:
dividend yield of 0% for all years; expected volatility of 75.3%; risk-free
interest rates of 2.95%; and expected life of 10 years. The vesting
conditions of these options were satisfied prior to December 31, 2009.
Accordingly, these options were fully vested at December 31, 2009 and the
Company recognized all of the expenses for these options during 2009.
(ii) 120,400 of the options vest as follows: 25% within one year from the
date of grant, with the remainder vesting in 12 equal quarterly tranches over
36 months. The options are exercisable over a 10-year period commencing on
the date of grant. The Company estimated the fair value of the options on
the date of grant using the Black-Scholes option-pricing model to be
approximately $212, based on the following weighted average assumptions:
dividend yield of 0% for all years; expected volatility of 75.3%; risk-free
interest rates of 1.84%; and expected life of six years. The Company’s
management assumed the simplified method to reflect the expected life
regarding these options. The Company continued to use the simplified method
in 2009 as the Company does not have sufficient historical exercise data to
provide a reasonable basis upon which to estimate expected term due to the
limited period of time its equity shares have been publicly traded.
b) The fair value of options granted during the years ended December 31, 2007, 2008
and 2009 were $5,790, $2,914 and $1,280, respectively. The fair value of each
option granted is estimated on the date of grant using the Black-Scholes
option-pricing model, with the following weighted average assumptions:
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
2007 |
|
2008 |
|
2009 |
Dividend yield |
|
|
0 |
% |
|
|
0 |
% |
|
|
0 |
% |
|
|
|
|
|
|
|
|
|
|
|
|
|
Expected volatility |
|
|
53 |
% |
|
|
63 |
% |
|
|
75 |
% |
|
|
|
|
|
|
|
|
|
|
|
|
|
Risk-free interest rate |
|
|
4.62 |
% |
|
|
2.99 |
% |
|
|
2.74 |
% |
|
|
|
|
|
|
|
|
|
|
|
|
|
Expected life — in years |
|
|
6.0 |
|
|
|
6.0 |
|
|
|
9.2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
The expected volatility is based on the historical volatility of the Common
Stock and those of comparable companies. The risk-free interest rate
assumption is based on observed interest rates appropriate for the expected
term of the stock options granted in dollar terms. The Company’s management
uses the contractual term or its expectations, as applicable (through 2008 — using the simplified method), of each
option as its expected life. The pre-vesting forfeiture rate of
approximately 7% is estimated based on pre-vesting forfeiture experience. |
| |
| |
|
|
The total unrecognized compensation cost of employee stock options at
December 31, 2009 is $782 (net of forfeiture rate), and it is expected to be
recognized over a weighted average period of 0.9 years. |
| |
| |
|
|
The total cash received from employees as a result of employee stock option
exercises for the years ended December 31, 2007, 2008 and 2009 was $14, $5
and $293, respectively. The Company did not realize any tax benefit in
connection with these exercises. |
F-19
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 5 — SHARE CAPITAL (continued):
| |
|
|
During 2009, the Company issued 1,049,737 shares of Common Stock in
connection with the exercise of 1,146,912 options by certain officers and
employees of the Company. The Company received cash proceeds equal to $293
in connection with such exercises as 642,050 of such options were exercised
on a “net-exercise” basis. |
| |
2. |
|
Options and shares of restricted Common Stock granted to consultants,
directors, and other service providers: |
a) Set forth below is a table summarizing all of the option and restricted stock
grants issued by the Company to consultants, directors and other service providers
from its inception through December 31, 2009:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
Fair value |
|
|
| Year of |
|
No. of options |
|
Exercise price |
|
Vesting |
|
at grant |
|
Exercise |
| Grant |
|
granted |
|
range |
|
period |
|
($) |
|
Period |
1999* |
|
|
384,811 |
|
|
$ |
0.1 |
|
|
3 years |
|
|
27 |
|
|
10 years |
2000* |
|
|
349,017 |
|
|
$ |
0.001 |
|
|
immediate |
|
|
35 |
|
|
5.5 years |
2001 |
|
|
837,727 |
|
|
$ |
0.17 |
|
|
mainly 2 years |
|
|
51 |
|
|
7 years |
2003 |
|
|
1,601,912 |
|
|
$ |
0.12 |
|
|
4 years |
|
|
498 |
|
|
10 years |
2005* |
|
|
2,315,890 |
|
|
$ |
0.001-$0.57 |
|
|
2-4 years |
|
|
2,466 |
|
|
10 years |
2006* |
|
|
4,629,516 |
|
|
$ |
0.001-$16.7 |
|
|
2.5 years |
|
|
2,237 |
|
|
10 years |
2007 |
|
|
16,000 |
|
|
$ |
0.001 |
|
|
4 years |
|
|
246 |
|
|
10 years |
2008 |
|
|
50,000 |
|
|
$ |
3.02 |
|
|
4 years |
|
|
109 |
|
|
10 years |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10,184,873 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| * |
|
Includes options granted outside of an option plan. |
Set forth below are grants made by the Company during the year ended December
31, 2009 and grants made by the Company prior to 2009 to certain related parties
(such grants appear in the table above):
| |
1. |
|
In May 2007, the Company’s Board of Directors approved
the grant of 8,000 shares of restricted Common Stock to a new member of its
Scientific Advisory Board. The shares vest as follows: 25% vest 12 months
after the grant date and the remaining 75% of the shares vest over three
years in 36 equal monthly installments. In October 2008, such services were
terminated and 5,333 the 8,000 shares of restricted Common Stock were
forfeited. |
| |
| |
2. |
|
In February 2008, the Company’s Board of Directors
approved the grant of options to purchase 50,000 shares of Common Stock to a
new director of the Company with an exercise price equal to $3.02 per share.
The options vest over a four-year period and are exercisable over a 10-year
period commencing on the date of grant. |
b) The fair value of options and shares of restricted Common Stock granted to
consultants and other non-employees during the years ended December 31, 2007, 2008
and 2009 were $246, $109 and $0 respectively. The fair value of each option granted
is estimated on the date of grant using the Black-Scholes option-pricing model, with
the following weighted average assumptions:
| |
|
|
|
|
| |
|
2008 |
Dividend yield |
|
|
0 |
% |
|
|
|
|
|
Expected volatility |
|
|
63 |
% |
|
|
|
|
|
Risk-free interest rate |
|
|
2.99 |
% |
|
|
|
|
|
Expected life — in years |
|
|
10 |
|
|
|
|
|
|
F-20
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 5 — SHARE CAPITAL (continued):
| |
|
|
The expected volatility is based on the historical volatility of the Company’s stock
and those of comparable companies. The risk-free interest rate assumption is based
on observed interest rates appropriate for the expected term of the stock options
granted in dollar terms. The Company’s management used the contractual terms as the
expected life in 2009. |
| |
| |
|
|
The total unrecognized compensation cost as of December 31, 2009, is $39, and it is
expected to be recognized over a weighted average period of 0.9 years. |
| |
| |
|
|
No cash was received from consultants as a result of consultant stock option
exercises for the years ended December 31, 2007, 2008 and 2009. The Company did not
realize any tax benefits in connection with these exercises. |
| |
| |
|
|
During the year ended December 31, 2009, the Company issued 3,853,441 shares of
Common Stock in connection with the exercise of 3,866,093 options by certain
consultants, directors, and other service providers of the Company. The Company did
not receive cash proceeds in connection with such exercises as all of such options
were exercised on a “net-exercise” basis. |
| |
e. |
|
A summary of share option plans, and related information, under all of the
Company’s equity incentive plans for the years ended December 31, 2007, 2008 and 2009
are as follows: |
| |
1. |
|
Options granted to employees: |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year ended December 31, |
|
| |
|
2007 |
|
|
2008 |
|
|
2009 |
|
| |
|
|
|
|
|
Weighted |
|
|
|
|
|
|
Weighted |
|
|
|
|
|
|
Weighted |
|
| |
|
Number |
|
|
average |
|
|
Number |
|
|
average |
|
|
Number |
|
|
average |
|
| |
|
of |
|
|
exercise |
|
|
of |
|
|
exercise |
|
|
of |
|
|
exercise |
|
| |
|
options |
|
|
price |
|
|
options |
|
|
price |
|
|
options |
|
|
price |
|
Outstanding at beginning
of year |
|
|
4,144,817 |
|
|
$ |
0.635 |
|
|
|
4,212,686 |
|
|
$ |
0.830 |
|
|
|
5,890,641 |
|
|
$ |
2.118 |
|
Changes during the year: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Granted |
|
|
204,351 |
|
|
|
4.330 |
|
|
|
2,060,000 |
|
|
|
4.794 |
|
|
|
624,400 |
|
|
|
2.650 |
|
Forfeited |
|
|
25,197 |
|
|
|
0.120 |
|
|
|
177,237 |
|
|
|
4.405 |
|
|
|
1,400 |
|
|
|
2.650 |
|
Expired |
|
|
1,221 |
|
|
|
0.970 |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
Exercised (*) |
|
|
110,064 |
|
|
|
0.131 |
|
|
|
204,808 |
|
|
|
0.565 |
|
|
|
1,146,912 |
|
|
|
0.733 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding at end of year |
|
|
4,212,686 |
|
|
$ |
0.830 |
|
|
|
5,890,641 |
|
|
$ |
2.118 |
|
|
|
5,366,729 |
|
|
$ |
2.476 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exercisable at end of year |
|
|
2,503,399 |
|
|
$ |
0.365 |
|
|
|
3,267,607 |
|
|
$ |
0.995 |
|
|
|
3,680,382 |
|
|
$ |
1.785 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (*) |
|
The total intrinsic value of options exercised during the years ended
December 31, 2007, 2008 and 2009, was $907, $450 and $7,258, respectively. |
F-21
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 5 — SHARE CAPITAL (continued):
| |
2. |
|
Options granted to consultants, directors, and other service
providers: |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year ended December 31, |
|
| |
|
2007 |
|
|
2008 |
|
|
2009 |
|
| |
|
|
|
|
|
Weighted |
|
|
|
|
|
|
Weighted |
|
|
|
|
|
|
Weighted |
|
| |
|
Number |
|
|
average |
|
|
Number |
|
|
average |
|
|
Number |
|
|
average |
|
| |
|
of |
|
|
exercise |
|
|
of |
|
|
exercise |
|
|
of |
|
|
exercise |
|
| |
|
options |
|
|
price |
|
|
options |
|
|
price |
|
|
options |
|
|
price |
|
Outstanding at beginning of
year |
|
|
7,572,035 |
|
|
$ |
5.996 |
|
|
|
5,254,785 |
|
|
$ |
1.250 |
|
|
|
5,304,785 |
|
|
$ |
1.285 |
|
Changes during the year: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Granted |
|
|
8,000 |
|
|
|
0.001 |
|
|
|
50,000 |
|
|
|
3.020 |
|
|
|
|
|
|
|
|
|
Forfeited |
|
|
2,325,250 |
|
|
|
16.700 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exercised (*) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3,866,093 |
|
|
|
0.016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding at end of year |
|
|
5,254,785 |
|
|
$ |
1.250 |
|
|
|
5,304,785 |
|
|
$ |
1.285 |
|
|
|
1,438,692 |
|
|
$ |
4.697 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exercisable at end of year |
|
|
4,341,917 |
|
|
$ |
0.023 |
|
|
|
5,133,189 |
|
|
$ |
0.902 |
|
|
|
1,407,234 |
|
|
$ |
4.674 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (*) |
|
The total intrinsic value of options exercised during the years ended December 31,
2007, 2008 and 2009, was $0, $0 and $41,281, respectively. |
| |
f. |
|
The following tables summarize information concerning outstanding and exercisable
options as of December 31, 2009: |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| December 31, 2009 |
| Options outstanding |
|
Options exercisable |
| |
|
Number of |
|
Weighted |
|
Number of |
|
Weighted |
| |
|
options |
|
average |
|
options |
|
average |
| |
|
outstanding |
|
remaining |
|
exercisable |
|
remaining |
| Exercise |
|
at end of |
|
contractual |
|
at end of |
|
contractual |
| prices |
|
year |
|
life |
|
year |
|
life |
$0.001 |
|
|
919,207 |
|
|
|
4.09 |
|
|
|
915,875 |
|
|
|
4.08 |
|
$0.120 |
|
|
1,107,926 |
|
|
|
4.32 |
|
|
|
1,107,926 |
|
|
|
4.32 |
|
$0.399 |
|
|
47,569 |
|
|
|
5.35 |
|
|
|
47,569 |
|
|
|
5.35 |
|
$0.972 |
|
|
1,666,365 |
|
|
|
6.51 |
|
|
|
1,312,287 |
|
|
|
6.49 |
|
$2.350 |
|
|
160,000 |
|
|
|
8.82 |
|
|
|
40,000 |
|
|
|
8.82 |
|
$2.650 |
|
|
623,000 |
|
|
|
9.15 |
|
|
|
504,000 |
|
|
|
9.15 |
|
$3.020 |
|
|
50,000 |
|
|
|
8.10 |
|
|
|
21,875 |
|
|
|
8.10 |
|
$5.000 |
|
|
1,843,812 |
|
|
|
8.10 |
|
|
|
750,542 |
|
|
|
8.10 |
|
$16.700 |
|
|
387,542 |
|
|
|
7.00 |
|
|
|
387,542 |
|
|
|
7.00 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6,805,421 |
|
|
|
|
|
|
|
5,087,616 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The aggregate intrinsic value of the total outstanding and of total vested and
exercisable options as of December 31, 2009, is $29,317 and $24,438, respectively.
| |
g. |
|
The following table illustrates the effect of share-based compensation on the
statement of operations: |
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year ended December 31, |
|
| |
|
2007 |
|
|
2008 |
|
|
2009 |
|
Research and development expenses |
|
$ |
3,587 |
|
|
$ |
1,226 |
|
|
$ |
1,489 |
|
General and administrative expenses |
|
|
16,861 |
|
|
|
1,845 |
|
|
|
1,194 |
|
|
|
|
|
|
|
|
|
|
|
|
|
$ |
20,448 |
|
|
$ |
3,071 |
|
|
$ |
2,683 |
|
|
|
|
|
|
|
|
|
|
|
F-22
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 6 — TAXES ON INCOME
| |
a. |
|
The Company |
| |
| |
|
|
Protalix BioTherapeutics, Inc. is taxed according to tax laws of the United States. The
income of Protalix BioTherapeutics, Inc. is taxed in the United States at the rate of up
to 39.4%. |
| |
| |
b. |
|
Protalix Ltd. |
| |
| |
|
|
The Israeli Subsidiary is taxed according to Israeli tax laws: |
| |
1. |
|
Measurement of results for tax purposes under the Income Tax
(Inflationary Adjustments) Law, 1985 (hereafter — the inflationary adjustments law) |
| |
| |
|
|
Pursuant to the Israel Income Tax Law (Adjustments for Inflation), 1985 (hereinafter
— the Adjustments Law), the results for tax purposes have been measured through 2007
on a real basis, based on changes in the Israel consumer price index. The
Subsidiary is taxed under this law. |
| |
| |
|
|
Under the Israel Income Tax Law (Adjustments for Inflation) (Amendment No. 20), 2008
(the “amendment”), the provisions of the Adjustments Law will cease to apply to the
Subsidiary in the 2008 tax year, and, therefore, commencing in 2008, the results of
the Israeli Subsidiary have been measured for tax purposes in nominal terms. The
amendment includes a number of transition provisions regarding the end of
application of the Adjustments Law, which applied to the Israeli Subsidiary through
the end of the 2007 tax year. |
| |
| |
2. |
|
Tax rates |
| |
| |
|
|
The income of the Israeli Subsidiary (other than income from “Approved Enterprises”
see 3 below is taxed in Israel at the regular rate. According to the provisions of
the Law for Amending the Israel Income Tax Ordinance, 2005 (“Amendment 147”) of
August 2005, corporate tax rates will be gradually lowered, resulting in the
corporate following tax rates for 2007 and thereafter: 2007 — 29%, 2008 — 27%,
2009 — 26% and for 2010 and thereafter — 25%. |
| |
| |
|
|
Capital gain for assets purchased since January 1, 2003 are subject to real capital
gain tax at 25% and exempted from inflationary capital gains tax. |
| |
| |
|
|
On July 23, 2009, the Israel Economic Efficiency Law (Legislation Amendments for
Applying the Economic Plan for 2009 and 2010), 2009 (the “2009 Amendment”), became
effective, stipulating, among other things, an additional gradual decrease in tax
rates in 2011 and thereafter, as follows: 2011 — 24%, 2012 — 23%, 2013 — 22%,
2014 — 21%, 2015 — 20% and 2016 and thereafter — 18%. |
| |
| |
|
|
In addition to the above decrease in corporate tax, the real capital gain tax was
reduced to be in line with corporate tax in the year of selling the asset. |
| |
| |
3. |
|
The Law for the Encouragement of Capital Investments, 1959
(hereinafter, the “Law”) |
| |
a. |
|
Reduced tax rates |
| |
| |
|
|
The Israeli Subsidiary has been granted “Approved Enterprise” status under the
Law for the Encouragement of Capital Investments, 1959. Income derived from the
Approved Enterprise during a period of 10 years from the year in which the
enterprise first realizes taxable income is tax exempt, provided that the
maximum period to which it is restricted by the law has not elapsed. |
| |
| |
|
|
The Israeli Subsidiary has an “Approved Enterprise” plan since 2004. The period
of benefits in respect of the main enterprise of the Company, which has not yet
commenced in 2009 expires in 2017. |
F-23
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 6 — TAXES ON INCOME (continued):
| |
|
|
If the Israeli Subsidiary subsequently pays a dividend out of income derived
from the “Approved Enterprise” during the tax exemption period, it will be
subject to tax on the amount distributed, including any company tax on these
amounts, at the rate which would have been applicable had such income not been
exempted. |
| |
| |
|
|
In addition to the corporate taxes in Israel, the Company is subject to a
withholding tax on the U.S. revenue source portion of the payments made to the
Company for its share of Pfizer’s net profits under the Pfizer Agreement. The
withholding tax rate is currently 15%. |
| |
b. |
|
Accelerated depreciation |
| |
| |
|
|
The Israeli Subsidiary is entitled to claim accelerated depreciation as provided by
Israeli law, commencing in the first year of operation of each asset, in respect of
buildings, machinery and equipment used by the Approved Enterprise. |
| |
| |
c. |
|
Conditions for entitlement to the benefits |
| |
| |
|
|
The entitlement to the above benefits is conditional upon the Israeli Subsidiary
fulfilling the conditions stipulated by the law, rules and regulations published
thereunder, and the instruments of approval for the specific investment in an
approved enterprise. In the event of any failure of the Israeli Subsidiary to
comply with these conditions, the benefits may be cancelled and the Subsidiary may
be required to refund the amount of the benefits, in whole or in part, with
interest. |
| |
| |
|
|
The Investment Center of Israeli Ministry of Industry, Trade and Labor (the
“Investment Center”) is currently reviewing the Israeli Subsidiary’s final
implementation report and, as a result, the Company has not yet received a final
implementation approval with respect to its “Approved Enterprise” from the
Investment Center. Additionally, given the Israeli Subsidiary’s significant amount
of net operating losses and the limitation mentioned above to the benefit period,
the Israeli Subsidiary cannot predict when it would be able to enjoy the tax
benefits described above, if at all. |
| |
| |
5. |
|
The Law for the Encouragement of Industry (Taxation), 1969: |
| |
| |
|
|
The Israeli Subsidiary is an “industrial company”, as defined under the Law for the
Encouragement of Industry (Taxation), 1969. As such, the Israeli Subsidiary is
entitled to claim depreciation at increased rates for equipment used in industrial
activity, as stipulated by regulations published under law, and has done so. |
| |
| |
|
|
Under the provisions of the Income Tax Regulations “Accelerated Depreciation in
respect of Equipment acquired during the Defined Period” (Temporary Orders),
industrial companies whose operations are mostly “eligible operations” are entitled
to claim accelerated depreciation at the rate of 50% on machinery and equipment
acquired from June 1, 2008 to May 31, 2009. The accelerated depreciation is to be
claimed over two years. For the year in which the equipment was acquired,
depreciation is recorded at the regular rate. In the second year and thereafter,
depreciation is recorded at a rate that would make the aggregate rate is 100%. |
| |
| |
|
|
Under the regulations, the Company is entitled to accelerated depreciation in 2009
in respect of machines and equipment purchased in 2008 and 2009. The effect of the
change in the rates of accelerated depreciation was included in deferred taxes
described above. |
F-24
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 6 — TAXES ON INCOME (continued):
| |
c. |
|
Tax losses carried forward to future years |
| |
| |
|
|
As of December 31, 2009, the Company had aggregate net operating loss (NOL)
carry-forwards equal to approximately $14,690 that are available to reduce future
taxable income as follows: |
| |
1. |
|
The Company |
| |
| |
|
|
The NOL carry-forward of the Company equal to approximately $5,911 may be restricted
under Section 382 of the Internal Revenue Code (“IRC”). IRC Section 382 applies
whenever a corporation with NOL experiences an ownership change. As a result of IRC
Section 382, the taxable income for any post change year that may be offset by a
pre-change NOL may not exceed the general IRC Section 382 limitation, which is the
fair market value of the pre-change entity multiplied by the IRC long-term tax
exempt rate. |
| |
| |
2. |
|
Protalix Ltd. |
| |
| |
|
|
At December 31, 2009, the Israeli Subsidiary had approximately $8,779 of NOL
carry-forwards that are available to reduce future taxable income with no limited
period of use. |
| |
d. |
|
Deferred income taxes: |
| |
| |
|
|
The components of the Company’s net deferred tax asset at December 31, 2008 and 2009
were as follows: |
| |
|
|
|
|
|
|
|
|
| |
|
December 31, |
|
| |
|
2008 |
|
|
2009 |
|
In respect of: |
|
|
|
|
|
|
|
|
R&D expenses |
|
$ |
2,728 |
|
|
|
|
|
Property and equipment |
|
|
7 |
|
|
$ |
(161 |
) |
Holiday and recreation pay |
|
|
162 |
|
|
|
200 |
|
Severance pay obligation |
|
|
89 |
|
|
|
87 |
|
Deferred revenues |
|
|
— |
|
|
|
8,345 |
|
Net operating loss carry forwards |
|
|
11,624 |
|
|
|
2,246 |
|
Valuation allowance |
|
|
(14,610 |
) |
|
|
(10,717 |
) |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
| |
e. |
|
Reconciliation of the theoretical tax expense to actual tax expense |
| |
|
|
The main reconciling items between the statutory tax rate of the Company and the
effective rate is the tax exemptions in connections with the Approved Enterprise and
the provision for full valuation allowance in respect of tax benefits from carry
forward tax losses due to the uncertainty of the realization of such tax benefits
(see above). |
| |
|
|
In accordance with the Income Tax Ordinance, as of December 31, 2009, all of
Protalix Ltd.’s tax assessments through tax year 2005 are considered final. |
| |
|
A summary of open tax years by major jurisdiction is presented below: |
| |
|
|
|
|
| Jurisdiction: |
|
Years: |
Israel |
|
|
2005-2009 |
|
United States (*) |
|
|
2002-2009 |
|
Netherlands |
|
|
2009 |
|
|
|
|
| (*) |
|
Includes federal, state and local (or similar provincial jurisdictions) tax
positions. |
F-25
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 7— SUPPLEMENTARY FINANCIAL STATEMENT INFORMATION (continued):
| |
|
|
|
|
|
|
|
|
| |
|
Year Ended |
|
| |
|
December 31, |
|
| |
|
2008 |
|
|
2009 |
|
a. Accounts receivable: |
|
|
|
|
|
|
|
|
Institutions |
|
$ |
223 |
|
|
$ |
740 |
|
State of Israel (see Note 4a ) |
|
|
166 |
|
|
|
832 |
|
Restricted deposit |
|
|
211 |
|
|
|
213 |
|
Prepaid expenses |
|
|
176 |
|
|
|
208 |
|
Sundry |
|
|
17 |
|
|
|
151 |
|
|
|
|
|
|
|
|
|
|
$ |
793 |
|
|
$ |
2,144 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
b. Accounts payable and accruals — other: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Payroll and related expenses |
|
$ |
766 |
|
|
$ |
2,731 |
|
Provision for vacation and recreation pay |
|
|
624 |
|
|
|
799 |
|
Accrued expenses |
|
|
970 |
|
|
|
1,931 |
|
Royalties payable |
|
|
|
|
|
|
3,575 |
|
Property and equipment supplier |
|
|
932 |
|
|
|
4,525 |
|
|
|
|
|
|
|
|
|
|
$ |
3,292 |
|
|
$ |
13,561 |
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year ended December 31, |
|
| |
|
2007 |
|
|
2008 |
|
|
2009 |
|
c. Research and development expenses — net: |
|
|
|
|
|
|
|
|
|
|
|
|
Payroll and related expenses |
|
$ |
7,510 |
|
|
$ |
9,296 |
|
|
$ |
10,479 |
|
Subcontractors and consultants |
|
|
3,141 |
|
|
|
5,289 |
|
|
|
7,469 |
|
Materials and consumables |
|
|
1,875 |
|
|
|
3,799 |
|
|
|
3,852 |
|
Rent, insurance and maintenance |
|
|
905 |
|
|
|
1,592 |
|
|
|
2,238 |
|
Patent registration and licensing |
|
|
331 |
|
|
|
182 |
|
|
|
387 |
|
Depreciation and impairment |
|
|
689 |
|
|
|
1,171 |
|
|
|
1,799 |
|
Other |
|
|
190 |
|
|
|
786 |
|
|
|
1,166 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
14,641 |
|
|
|
22,115 |
|
|
|
27,390 |
|
Less — grants (see Note 4a) |
|
|
1,071 |
|
|
|
4,714 |
|
|
|
5,752 |
|
|
|
|
|
|
|
|
|
|
|
|
|
$ |
13,570 |
|
|
$ |
17,401 |
|
|
$ |
21,638 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
d. General and administrative expense: |
|
|
|
|
|
|
|
|
|
|
|
|
Payroll and related expenses |
|
$ |
1,562 |
|
|
$ |
2,261 |
|
|
$ |
2,804 |
|
Management and consulting fees |
|
|
16,407 |
|
|
|
1,335 |
|
|
|
681 |
|
Rent, insurance and maintenance |
|
|
112 |
|
|
|
191 |
|
|
|
347 |
|
Professional fees |
|
|
1,702 |
|
|
|
1,362 |
|
|
|
2,274 |
|
Travel |
|
|
400 |
|
|
|
472 |
|
|
|
251 |
|
Depreciation |
|
|
69 |
|
|
|
130 |
|
|
|
191 |
|
Other |
|
|
342 |
|
|
|
1,019 |
|
|
|
596 |
|
|
|
|
|
|
|
|
|
|
|
|
|
$ |
20,594 |
|
|
$ |
6,770 |
|
|
$ |
7,144 |
|
|
|
|
|
|
|
|
|
|
|
F-26
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 7— SUPPLEMENTARY FINANCIAL STATEMENT INFORMATION (continued):
| |
e. |
|
Financial income: |
| |
| |
|
|
During the years 2007, 2008 and 2009 financial income includes transaction gain (loss)
rate in the amount of $50, ($280) and $67, respectively. |
NOTE 8 — RELATED PARTY TRANSACTIONS
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year ended December 31, |
|
| |
|
2007 |
|
|
2008 |
|
|
2009 |
|
a. Management and
consulting fees to the Chairman
of the Board |
|
$ |
36 |
|
|
$ |
33 |
|
|
$ |
33 |
|
|
|
|
|
|
|
|
|
|
|
b. Rent payments to a shareholder |
|
$ |
10 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
c. Compensation to the
non-executive directors (except
the Chairman of the Board) |
|
|
|
|
|
$ |
210 |
|
|
$ |
209 |
|
|
|
|
|
|
|
|
|
|
|
|
| |
d. |
|
With respect to options granted to the Company’s Chief Executive
Officer and to a shareholder, see Notes 5(d)(1) and (2). |
| |
| |
e. |
|
In March 2005, Protalix Ltd. entered into a management services
agreement with Pontifax Management Company, Ltd. in connection with an
investment in Protalix Ltd. by affiliates of Pontifax. The monthly
management fees under the management services agreement are $3. The
management services agreement shall be in full force as long as Mr.
Hurvitz serves as a member of the Company’s Board of Directors. In 2009,
the amount was set to $33. |
NOTE 9 -LICENSE AND SUPPLY AGREEMENT
| |
|
On November 30, 2009, Protalix Ltd. and Pfizer Inc entered into the Pfizer Agreement
pursuant to which Pfizer was granted an exclusive, worldwide license to develop and
commercialize taliglucerase alfa, except Israel. Under the terms and conditions of the
Pfizer Agreement, Protalix Ltd. retained the right to commercialize taliglucerase alfa in
Israel. Under the Pfizer Agreement, Pfizer made an upfront payment to Protalix Ltd. of
$60,000 in connection with the execution of the agreement and shortly thereafter paid to
Protalix Ltd. an additional $5,000 upon the Company’s filing of a proposed pediatric
investigation plan to the Pediatric Committee of the European Medicines Agency (EMEA).
Protalix Ltd. is also eligible to receive additional potential milestone payments totaling
up to $50,000 for the successful achievement of other regulatory milestones. Protalix Ltd.
is entitled to 40% of the profits earned on Pfizer’s sales of taliglucerase alfa. Such
profit will be calculated, among other things, while taking into account Protalix Ltd.’s
cost of goods sold and Pfizer’s commercial expenses with certain expenses capped or borne
soley by one party. |
| |
|
The Company has determined that the initial, non-refundable upfront license fee payment of
$60,000 together with the first $5,000 payment will be recognized on a straight line basis
as revenue over the estimated relationship period. The Company has estimated that its
relationship period will be approximately 14 years based on the Company’s last significant
patent to expire. |
| |
|
The Company’s deliverables under this collaboration include an exclusive license to
taliglucerase alfa as an enzyme replacement therapy for the treatment of Gaucher disease,
certain research and development services as required under the Pfizer Agreement for
taliglucerase alfa, manufacturing of taliglucerase alfa and optional participation in a
joint steering committee. |
| |
| |
|
In connection with the payments received under the Pfizer Agreement Protalix Ltd is
obligated to pay certain royalties. See Note 4a. |
F-27
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands)
NOTE 10 — SUBSEQUENT EVENTS
| |
a. |
|
On February 7, 2010, the Company’s Board of Directors approved the grant of
options to purchase 160,000 shares of Common Stock to a new executive officer of the
Company with an exercise price equal to $6.81 per share. The options vest over a
four-year period, with the first 25% to vest on the first anniversary of the date of
the grant and the remaining 75% in equal tranches on a quarterly basis for three years
thereafter. The options are exercisable over a 10-year period commencing on the date of
grant. The Company estimated the fair value of the options on the date of grant using
the Black-Scholes option-pricing model to be approximately $740, based on the following
weighted average assumptions: dividend yield of 0% for all years; expected volatility
of 76.02%; risk-free interest rates of 2.96%; and expected life of 6 years. |
| |
| |
b. |
|
During January and February 2010, the Company issued a total of 14,000 shares
of Common Stock in connection with the exercise of options to purchase 14,000 shares of
Common Stock by certain employees of the Company. The Company received aggregate cash
proceeds equal to approximately $2. |
| |
| |
c. |
|
In February 2010, the Company’s Board of Directors approved the grant of
options to purchase 1,043,623 shares of Common Stock, in the aggregate, to certain officers and employees of
the Company with an exercise price equal to $6.90 per share. The options vest quarterly over three years, commencing after the FDA’s approval of
taliglucerase alfa, if at all.
The options are exercisable over a 10-year period commencing on the date of grant. The
Company estimated the fair value of the options on the date of grant using the
Black-Scholes option-pricing model to be approximately $5.8 million, based on the
following weighted average assumptions: dividend yield of 0% for all years; expected
volatility of 75.74%; risk-free interest rates of 3.69%; and expected
life of 10
years. |
F-28