Attached files
| file | filename |
|---|---|
| EX-21.1 - EX-21.1 - ALBIREO PHARMA, INC. | y80991exv21w1.htm |
| EX-23.1 - EX-23.1 - ALBIREO PHARMA, INC. | y80991exv23w1.htm |
| EX-10.20 - EX-10.20 - ALBIREO PHARMA, INC. | y80991exv10w20.htm |
| EX-32.01 - EX-32.01 - ALBIREO PHARMA, INC. | y80991exv32w01.htm |
| EX-10.22 - EX-10.22 - ALBIREO PHARMA, INC. | y80991exv10w22.htm |
| EX-31.02 - EX-31.02 - ALBIREO PHARMA, INC. | y80991exv31w02.htm |
| EX-10.19 - EX-10.19 - ALBIREO PHARMA, INC. | y80991exv10w19.htm |
| EX-31.01 - EX-31.01 - ALBIREO PHARMA, INC. | y80991exv31w01.htm |
Table of Contents
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
(Mark One)
| þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended September 30, 2009
or
| o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission File Number 001-33451
BIODEL INC.
(Exact Name of Registrant as Specified in Its Charter)
| Delaware | 90-0136863 | |
| (State or Other Jurisdiction of | (I.R.S. Employer | |
| Incorporation or Organization) | Identification No.) | |
| 100 Saw Mill Road | ||
| Danbury, CT | 06810 | |
| (Address of Principal Executive Offices) | (Zip Code) |
Registrant’s telephone number, including area code
(203) 796-5000
Securities registered pursuant to Section 12(b) of the Act:
(203) 796-5000
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class | Name of Each Exchange on Which Registered | |
| Common Stock, par value $0.01 per share | The NASDAQ Global Market |
Securities registered pursuant to Section 12(g) of the Act:
None
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in
Rule 405 of the Securities Act. Yes o No þ
Indicate by check mark if the registrant is not required to file reports pursuant to
Section 13 or Section 15(d) of the Act. Yes o No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed
by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or
for such shorter period that the registrant was required to file such reports), and (2) has been
subject to such filing requirements for the past 90 days. Yes þ No o
Indicate by check mark whether the registrant has submitted electronically and posted on its
corporate Web site, if any, every Interactive Data File required to be submitted and posted
pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months
(or for such shorter period that the registrant was required to submit and post such files). Yes
o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of
Regulation S-K (§ 229.405) is not contained herein, and will not be contained, to the best of
registrant’s knowledge, in definitive proxy or information statements incorporated by reference in
Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated
filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large
accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the
Exchange Act. (Check one):
| Large accelerated filer o | Accelerated filer þ | Non-accelerated filer o | Smaller reporting company o | |||
| (Do not check if a smaller reporting company) |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b2 of
the Exchange Act). Yes o No þ
The aggregate market value of the common stock of the registrant held by non-affiliates was
$60.6 million based on the last sales price at which the common stock was last sold on the NASDAQ
Global Market on March 31, 2009.
The number of shares outstanding of the registrant’s common stock, as of November 30, 2009 was
23,883,612.
Portions of the registrant’s definitive Proxy Statement, or the 2010 Proxy Statement, which
will be filed with the Securities and Exchange Commission not later than 120 days after
September 30, 2009, for its 2010 Annual Meeting of Stockholders are incorporated by reference into
Part III of this Report. With the exception of the portions of the 2010 Proxy Statement expressly
incorporated into this Annual Report on Form 10-K by reference, such document shall not be deemed
filed as part of this Annual Report on Form 10-K.
BIODEL INC.
INDEX TO REPORT ON FORM 10-K
INDEX TO REPORT ON FORM 10-K
| Page | ||||||||
| PART I |
||||||||
| 2 | ||||||||
| 15 | ||||||||
| 28 | ||||||||
| 28 | ||||||||
| 28 | ||||||||
| 28 | ||||||||
| PART II |
||||||||
| 29 | ||||||||
| 30 | ||||||||
| 31 | ||||||||
| 41 | ||||||||
| 41 | ||||||||
| 41 | ||||||||
| 41 | ||||||||
| 44 | ||||||||
| PART III |
||||||||
| 45 | ||||||||
| 45 | ||||||||
| 45 | ||||||||
| 45 | ||||||||
| 45 | ||||||||
| PART IV |
||||||||
| 45 | ||||||||
| EX-10.19 | ||||||||
| EX-10.20 | ||||||||
| EX-10.22 | ||||||||
| EX-21.1 | ||||||||
| EX-23.1 | ||||||||
| EX-31.01 | ||||||||
| EX-31.02 | ||||||||
| EX-32.01 | ||||||||
i
Table of Contents
FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements within the meaning of the
Private Securities Litigation Reform Act of 1995, that involve substantial risks and uncertainties.
All statements, other than statements of historical facts, included in this Annual Report on
Form 10-K regarding our strategy, future operations, future financial position, future revenues,
projected costs, prospects, plans and objectives of management are forward-looking statements. The
words “anticipates,” “believes,” “could,” “estimates,” “expects,” “intends,” “may,” “plans,”
“potential,” “predicts,” “projects,” “should,” “will,” “would” and similar expressions are intended
to identify forward-looking statements, although not all forward-looking statements contain these
identifying words.
Our forward-looking statements in this Annual Report on Form 10-K are subject to a number of
known and unknown risks and uncertainties that could cause actual results, performance or
achievements to differ materially from those described or implied in the forward-looking
statements, including:
| • | our ability to secure approval by the U.S. Food and Drug Administration, or FDA, for our product candidates under Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act, or FFDCA; | ||
| • | our ability to file our planned New Drug Application, or NDA, for VIAject® in December 2009 and, once filed, the length of time that will elapse before our NDA is fully reviewed by the FDA; | ||
| • | our ability to secure approval by the FDA for VIAject® without conducting additional pivotal clinical trials; | ||
| • | our ability to market, commercialize and achieve market acceptance for VIAject®; | ||
| • | the progress, timing or success of our product candidates, particularly VIAject®, and that of our research, development and clinical programs, including any resulting data analyses; | ||
| • | our ability to enter into collaboration arrangements for the commercialization of our product candidates and the success or failure of any such collaborations into which we enter, or our ability to commercialize our product candidates ourselves; | ||
| • | our ability to enforce our patent for VIAject® and our ability to secure additional patents for VIAject® and for our other product candidates; | ||
| • | our ability to protect our intellectual property and operate our business without infringing upon the intellectual property rights of others; | ||
| • | the degree of clinical utility of our products; | ||
| • | the ability of our major suppliers, including suppliers of insulin, to produce our product or products in our final dosage form; | ||
| • | our commercialization, marketing and manufacturing capabilities and strategies; and | ||
| • | our ability to accurately estimate anticipated operating losses, future revenues, capital requirements and our needs for additional financing. |
We may not actually achieve the plans, intentions or expectations disclosed in our
forward-looking statements, and you should not place undue reliance on our forward-looking
statements. Actual results or events could differ materially from the plans, intentions and
expectations disclosed in the forward-looking statements we make. We have included important
factors in the cautionary statements included in this Annual Report, particularly in Item 1A of
this Annual Report, and in our other public filings with the Securities and Exchange Commission
that could cause actual results or events to differ materially from the forward-looking statements
that we make.
You should read this Annual Report and the documents that we have filed as exhibits to the
Annual Report completely and with the understanding that our actual future results may be
materially different from what we expect. It is routine for internal projections and expectations
to change as the year, or each quarter in the year, progresses, and therefore it should be clearly
understood that the internal projections and beliefs upon which we base our expectations are made
as of the date of this Annual Report on Form 10-K and may change prior to the end of each quarter
or the year. While we may elect to update forward-looking statements at some point in the future,
we do not undertake any obligation to update any forward-looking statements whether as a result of
new information, future events or otherwise.
1
Table of Contents
PART I
ITEM 1: BUSINESS
Overview
We are a specialty biopharmaceutical company focused on the development and commercialization
of innovative treatments for diabetes, which may be safer, more effective and more convenient for
patients. We develop our product candidates by applying our proprietary formulation technologies to
existing drugs in order to improve their therapeutic profiles. Our initial development efforts are
focused on peptide hormones. Our most advanced product candidate, VIAject®, has been studied in two
pivotal Phase 3 clinical trials for the treatment of patients with Type 1 and Type 2 diabetes.
Diabetes is a disease characterized by abnormally high levels of blood glucose and inadequate
levels of insulin. Glucose is a simple sugar used by all the cells of the body to produce energy
and support life. Humans need a minimum level of glucose in their blood at all times to stay alive.
Insulin is a peptide hormone naturally secreted by the pancreas to regulate the body’s management
of glucose. When a healthy individual begins a meal, the pancreas releases a natural spike of
insulin called the first-phase insulin release, which is critical to the body’s overall control of
glucose. Virtually all patients with diabetes lack the first-phase insulin release. All patients
with Type 1 diabetes must treat themselves with mealtime insulin injections to compensate for the
lack of natural pancreatic first phase insulin release. As the disease progresses, patients with
Type 2 diabetes also require mealtime insulin. However, none of the currently marketed mealtime
insulin products adequately mimics the first-phase insulin release. As a result, patients using
insulin typically have inadequate levels of insulin in their systems at the start of a meal and too
much insulin in their systems between meals. This, in turn, results in the lack of adequate glucose
control associated with diabetes. The long-term adverse effects of elevated glucose levels include
blindness, loss of kidney function, nerve damage and loss of sensation and poor circulation in the
periphery, which in some severe cases, may lead to amputations.
Advances in insulin technology in the 1990s led to the development of new molecules, referred
to as rapid-acting insulin analogs, which are similar to insulin, but are absorbed into the blood
more rapidly.
VIAject® is our proprietary injectable formulation of recombinant human insulin designed to be
absorbed into the blood faster than the currently marketed rapid-acting insulin analogs. We have
completed two pivotal Phase 3 clinical trials of VIAject®, one in patients with Type 1 diabetes and
the other in patients with Type 2 diabetes. In both clinical trials we compared VIAject® to
Humulin® R, a form of recombinant human insulin, to determine if VIAject® is not inferior to
Humulin® R in the management of blood glucose levels, as measured by the mean change in patients’
glycosylated hemoglobin, or HbA1c, levels from baseline. Patients in both clinical trials were
treated for a period of six months. HbA1c is a measure of a patient’s average blood glucose level
over a period of approximately three months.
In March 2009, we announced our plan to submit an NDA to the FDA by the end of 2009 to market
VIAject® for the treatment of diabetes. We expect that the NDA will be submitted under section
505(b)(2) of the FFDCA and be based upon results from multiple pharmacokinetic and pharmacodynamic
studies as well as our two completed Phase 3 studies of VIAject® in patients with Type 1 and Type 2
diabetes. We intend to seek approval for a 100 IU/cc liquid formulation of VIAject® that is
bioequivalent to the two-part 25 IU/cc lyophilized powder formulation of VIAject® that was used in
our pivotal Phase 3 clinical trials.
In October 2009 we executed a letter of intent to purchase a disposable insulin pen designed
by Wockhardt Ltd. for use with VIAject®. We intend to submit this pen to the FDA for review at a
later date after completing certain modifications that we believe will improve its commercial
performance.
In 2010 we plan to conduct additional clinical trials designed to generate additional data to
enhance VIAject’s potential commercial success.
In addition to VIAject®, we are developing VIAtab™, a sublingual, or below the tongue, tablet
formulation of insulin. We have tested one formulation of VIAtab™ in a Phase 1 trial in patients
with Type 1 diabetes and are developing additional formulations for further clinical testing. We
believe that VIAtab™ has the potential to rapidly deliver insulin, while sparing patients from the
unpleasant aspects of injection therapy. We are developing VIAtab™ as a potential treatment for
patients with Type 2 diabetes who are in the early stages of their disease.
We have developed our product candidates utilizing our proprietary VIAdel™ technology which
allows us to study the interaction between peptide hormones and small molecules. We use our
technology to reformulate existing peptide drugs with small molecule ingredients that are generally
regarded as safe by the FDA so as to improve their therapeutic profiles. We believe that this
approach to drug development will allow us to utilize Section 505(b)(2) of the FFDCA for FDA
approval of our product candidates. Section 505(b)(2) provides for a type of NDA that allows
expedited development of new formulations of chemical entities and biological compounds that have
already undergone extensive clinical trials and been approved by the FDA. Both the time and cost of
development of a new product can be substantially less under a Section 505(b)(2) NDA than under a
full NDA.
2
Table of Contents
Our Strategy
Our goal is to build a leading specialty biopharmaceutical company focused on the development
and commercialization of innovative treatments for diabetes, which may be safer, more effective and
more convenient for patients. To achieve our goal, we are pursuing the following strategies:
| • | Obtain Regulatory Approval for VIAject®. Our current focus is to seek regulatory approval for VIAject® in the major world markets starting with the United States. | ||
| • | Continue the Development of VIAject’s® commercial profile. We plan to conduct additional clinical trials of VIAject® designed to enhance the product candidate’s potential commercial differentiation compared to other leading meal-time insulin products. | ||
| • | Commercialize our Product Candidates Through Strategic Collaborations. We intend to fund our clinical trial programs through at least proof-of-concept by ourselves. However, because our product candidates target large primary care markets, we intend to maximize the commercial potential of our product candidates by selectively entering into strategic arrangements with leading pharmaceutical or biotechnology companies for the commercialization of our product candidates. Because we are focusing on therapeutic indications in large markets, we believe that these larger companies have the marketing, sales and financial resources to maximize the commercial potential of our products. | ||
| • | Focus on the Section 505(b)(2) Regulatory Approval Pathway. Using our VIAdel™ technology, we seek to reformulate existing drugs with ingredients that are generally regarded as safe by the FDA. We believe that this approach to drug development will allow us to use the abbreviated development pathway of Section 505(b)(2) of the FFDCA, which can result in substantially less time and cost in bringing a new drug to market. We intend to continue to focus our efforts on reformulating new product candidates for which we will be able to seek regulatory approval pursuant to Section 505(b)(2) NDAs. | ||
| • | Pursue Additional Product Candidates. In addition to our pivotal Phase 3 clinical trials of VIAject®, we have conducted a Phase 1 clinical trial of VIAtab™, our sublingual insulin product candidate. In 2010 we may allocate limited resources to developing our VIAtab™ formulation. |
Diabetes and the Insulin Market
Diabetes Overview
Glucose is a simple sugar used by all the cells of the body to produce energy and support
life. Humans need a minimum level of glucose in their blood at all times to stay alive. The primary
manner in which the body produces blood glucose is through the digestion of food. When a person is
not getting this glucose from food digestion, glucose is produced from stores and released by the
liver. The body’s glucose levels are regulated by insulin. Insulin is a peptide hormone that is
naturally secreted by the pancreas. Insulin helps glucose enter the body’s cells to provide a vital
source of energy.
When a healthy individual begins a meal, the pancreas releases a natural spike of insulin
called the first-phase insulin release. In addition to providing sufficient insulin to process the
glucose coming into the blood from digestion of the meal, the first-phase insulin release acts as a
signal to the liver to stop making glucose while digestion of the meal is taking place. Because the
liver is not producing glucose and there is sufficient additional insulin to process the glucose
from digestion, the blood glucose levels of healthy individuals remain relatively constant and
their blood glucose levels do not become too high.
Diabetes is a disease characterized by abnormally high levels of blood glucose and inadequate
levels of insulin. There are two major types of diabetes — Type 1 and Type 2. In Type 1 diabetes,
the body produces no insulin. In the early stages of Type 2 diabetes, although the pancreas does
produce insulin, the body loses its early phase insulin response to a meal. In addition, the body’s
cells do not respond as well as they should to a normal amount of insulin, a condition known as
insulin resistance. According to the Centers for Disease Control and Prevention, or CDC, Type 2
diabetes is the more prevalent form of the disease, affecting approximately 90% to 95% of all
people diagnosed with diabetes.
Even before any other symptoms are present, one of the first effects of Type 2 diabetes is the
loss of the meal-induced first-phase insulin release. In the absence of the first-phase insulin
release, the liver will not receive its signal to stop making glucose. As a result, the liver will
continue to produce glucose at a time when the body begins to produce new glucose through the
digestion of the meal. As a result, the blood glucose level of patients with diabetes rises too
high after eating, a condition known as hyperglycemia. Hyperglycemia causes glucose to attach
unnaturally to certain proteins in the blood, interfering with the proteins’ ability to perform
their normal function of maintaining the integrity of the small blood vessels. With hyperglycemia
occurring after each meal, the tiny blood vessels eventually break down and leak. The long-term
adverse effects of hyperglycemia include blindness, loss of kidney function, nerve damage and loss
of sensation and poor circulation in the periphery, potentially requiring amputation of the
extremities.
Between two and three hours after a meal, an untreated diabetic’s blood glucose becomes so
elevated that the pancreas receives a signal to secrete an inordinately large amount of insulin. In
a patient with early Type 2 diabetes, the pancreas can still respond and secretes this large amount
of insulin. However, this occurs at the time when digestion is almost over and blood glucose levels
should begin to fall. This inordinately large amount of insulin has two detrimental effects. First,
it puts an undue extreme demand on an already compromised pancreas, which may lead to its more
rapid deterioration and eventually render the pancreas unable to produce insulin. Second, too much
insulin after digestion leads to weight gain, which may further exacerbate the disease condition.
3
Table of Contents
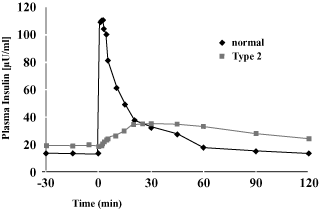
The figure below, which is derived from an article in the New England Journal of Medicine ,
illustrates the differences in the insulin release profiles of a healthy individual and a person in
the early stages of Type 2 diabetes. In response to an intravenous glucose injection, which
simulates eating a meal, the healthy individual produces the first-phase insulin release. In
contrast, the patient with Type 2 diabetes lacks the first-phase insulin release and releases the
insulin more slowly and over time. As a result, in the early stages of the disease, the Type
2 patient’s insulin level is too low at the initiation of a meal and too high after meal digestion.
First Phase Insulin Release
Current Treatments for Diabetes and their Limitations
Because patients with Type 1 diabetes produce no insulin, the primary treatment for Type 1
diabetes is daily intensive insulin therapy. The treatment of Type 2 diabetes typically starts with
management of diet and exercise. Although helpful in the short-run, treatment through diet and
exercise alone is not an effective long-term solution for the vast majority of patients with Type 2
diabetes. When diet and exercise are no longer sufficient, treatment commences with various
non-insulin oral medications. These oral medications act by increasing the amount of insulin
produced by the pancreas, by increasing the sensitivity of insulin-sensitive cells, by reducing the
glucose output of the liver or by some combination of these mechanisms. These treatments are
limited in their ability to manage the disease effectively and generally have significant side
effects, such as weight gain. Because of the limitations of non-insulin treatments, many patients
with Type 2 diabetes deteriorate over time and eventually require insulin therapy to support their
metabolism.
Insulin therapy has been used for more than 80 years to treat diabetes. This therapy usually
involves administering several injections of insulin each day. These injections consist of
administering a long-acting basal injection one or two times per day and an injection of a fast
acting insulin at mealtime. Although this treatment regimen is accepted as effective, it has
limitations. First, patients generally dislike injecting themselves with insulin due to the
inconvenience and pain of needles. As a result, patients tend not to comply adequately with the
prescribed treatment regimens and are often improperly medicated.
More importantly, even when properly administered, insulin injections do not replicate the
natural time-action profile of insulin. In particular, the natural spike of the first-phase insulin
release in a person without diabetes results in blood insulin levels rising within several minutes
of the entry into the blood of glucose from a meal. By contrast, injected insulin enters the blood
slowly, with peak insulin levels occurring within 80 to 100 minutes following the injection of
regular human insulin.
A potential solution is the injection of insulin directly into the vein of diabetic patients
immediately before eating a meal. In studies of intravenous injections of insulin, patients
exhibited better control of their blood glucose for 3 to 6 hours following the meal. However, for a
variety of medical reasons, intravenous injection of insulin before each meal is not a practical
therapy.
One of the key improvements in insulin treatments was the introduction in the 1990s and 2000s
of rapid-acting insulin analogs, such as Humalog®, NovoLog® and Apidra®. However, even with the
rapid-acting insulin analogs, peak insulin levels typically occur within 50 to 70 minutes following
the injection. Because the rapid-acting insulin analogs do not adequately mimic the first-phase
insulin release, diabetics using insulin therapy continue to have inadequate levels of insulin
present at the initiation of a meal and too much insulin present between meals. This lag in insulin
delivery can result in hyperglycemia early after meal onset. Furthermore, the excessive insulin
between meals may result in an abnormally low level of blood glucose known as hypoglycemia.
Hypoglycemia can result in loss of mental acuity, confusion, increased heart rate, hunger, sweating
and faintness. At very low glucose levels, hypoglycemia can result in loss of consciousness, coma
and even death. According to the American Diabetes Association, or ADA, insulin-using diabetic
patients have on average 1.3 serious hypoglycemic events per year, many of which require hospital
emergency room visits.
The Biodel Solution
Our two most advanced clinical programs are VIAject®, an injectable formulation of insulin,
and VIAtab™, a sublingual formulation of insulin. We believe these product candidates may change
the way Type 1 and Type 2 diabetic patients are treated by
4
Table of Contents
improving the efficacy, safety and ease-of-use of insulin. Based upon our preclinical and
clinical data, if approved, VIAject® may produce a profile of insulin levels in the blood that
approximates the natural first-phase insulin release normally seen in persons without diabetes
following a meal. In addition, VIAject® may be associated with medically important advantages with
regard to reduced hypoglycemia and reduced weight gain.
VIAject®
VIAject® is our proprietary formulation of injectable human insulin to be taken at mealtime.
VIAject® is designed to be absorbed into the blood faster than the currently marketed rapid-acting
insulin analogs. One of the key features of our formulation of insulin is that it allows the
insulin to disassociate, or separate, from the six molecule, or hexameric, form to the single
molecule, or monomeric, form and inhibits re-association to the hexameric form. We believe that by
favoring the monomeric form, VIAject® allows for more rapid delivery of insulin into the blood as
the human body requires insulin to be in the form of a single molecule before it can be absorbed
into the body to produce its desired biological effects.
Potential Advantages of VIAject® over Existing Insulin Treatments
We believe VIAject® offers a number of potential advantages over currently available
injectable insulin products, including the following:
| • | In our Phase 1 clinical trial in volunteers without diabetes, and in our Phase 1/2 clinical trials in patients with Type 1 diabetes, VIAject® reached the blood and exerted blood glucose lowering activity more rapidly than the rapid-acting insulin analog, Humalog®, and the regular human recombinant insulin, Humulin® R. Accordingly, we believe VIAject® more closely mimics the first-phase insulin release of healthy individuals at the beginning of a meal, which we believe reduces the risks of hyperglycemia and hypoglycemia. | ||
| • | In our analysis of the data from of our pivotal Phase 3 clinical trial in patients with Type 1 diabetes, we found that patients receiving VIAject®, when compared to those receiving Humulin® R, experienced fewer severe hypoglycemic events and did not gain weight, while those receiving Humulin® R did. In our analysis of the data from our pivotal Phase 3 clinical trial in patients with Type 2 diabetes, we found that patients receiving VIAject®, when compared to those receiving Humulin® R, experienced fewer mild and moderate, or non-severe, hypoglycemic events and gained less weight. Accordingly, we believe VIAject® may be a safer form of mealtime insulin therapy than is currently available to patients with Type 1 or Type 2 diabetes. |
Clinical Trials of VIAject®
Phase 1 and Phase 2 Clinical Trials. We have conducted Phase 1 and Phase 2 clinical trials
comparing the performance of VIAject® to Humalog®, the largest selling rapid-acting insulin analog
in the United States, and Humulin® R. In these trials, we observed that VIAject® produced a release
profile into the blood that more closely approximates the natural first-phase insulin release seen
in healthy individuals.
In 2005, we completed a Phase 1 clinical trial of VIAject®. This was a single center, open
label, five-way crossover study in healthy volunteers. All volunteers received insulin
subcutaneously. The study employed a “glucose clamp” procedure, in which glucose is automatically
infused into the volunteer’s blood so that his or her blood glucose will be maintained at a healthy
normal level. The effect of insulin is to lower blood glucose, thereby requiring an infusion of
glucose to maintain the normal glucose level. The rate at which glucose must be infused is called
the glucose infusion rate, or GIR. In this trial we found that VIAject® was faster than both
Humulin® R and Humalog® in the time to reach 50% of the maximum GIR, which provides evidence of the
insulin in VIAject® reaching the blood faster than that of Humulin® R and Humalog® . This faster
action for each dose of VIAject® was statistically significant as compared to both Humulin® R and
Humalog®.
In 2006, we completed a Phase 1/2 single-center, randomized, double blind, crossover, clinical
trial of VIAject® to compare the intra-subject variability of the timing and effect of repeated
doses of VIAject® to that of Humulin® R in fourteen patients with Type 1 diabetes. Repeated
administration of the same dose of both regular human insulin and rapid-acting insulin analogs are
known to produce variable blood insulin level results in the same patients. This is known as the
within-subject or intra-subject variability of insulin. In this trial we found that the
intra-subject variability of VIAject® was less than that of Humulin® R.
In 2007, we completed a Phase 2 clinical trial to examine VIAject® ’s ability to control blood
glucose after patients with Type 1 diabetes received a standardized meal. Patients received either
VIAject®, Humulin® R or Humalog® in conjunction with a standardized meal. Plasma insulin and blood
glucose levels were monitored throughout the study. We found that VIAject® statistically
significantly reduced maximal glucose after a standardized meal when compared to Humulin® R.
Maximal glucose was lower after patients were treated with VIAject®, as compared to Humalog®,
although this did not reach statistical significance. VIAject® was associated with less
hypoglycemia after a standardized meal when compared to Humulin® R. This finding was statistically
significant. While hypoglycemia was reduced for VIAject® compared to Humalog®, it did not reach
statistical significance.
Pivotal Phase 3 Clinical Trials. We completed our two pivotal Phase 3 clinical trials of
VIAject® in July 2008. Our pivotal Phase 3 clinical trials were open-label, parallel group,
randomized trials conducted at centers in the United States, Germany and India. The trials were
designed to compare the efficacy and safety of VIAject® to Humulin® R. One of the trials tested
VIAject® in patients
5
Table of Contents
with Type 1 diabetes and the other in patients with Type 2 diabetes. We enrolled more than
400 patients in each trial for a six month treatment period. Approximately one-half of the patients
in each trial were treated with VIAject® and the remainder with Humulin® R.
The primary objective of the trials was to determine if VIAject® is not inferior to Humulin® R
in the management of blood glucose levels, as measured by the mean change in patients’ glycosylated
hemoglobin, or HbA1c, levels from baseline to the end of the trial. HbA1c levels are a measure of
patients’ average blood glucose levels over a period of approximately 3 months. HbA1c is the FDA’s
preferred endpoint for diabetes trials. Predefined secondary endpoints in the trials included rates
of mild and moderate and severe hypoglycemic events, and changes in body weight.
In the Type 1 and Type 2 trials, HbA1c decreased comparably in the treatment groups, thereby
achieving the primary endpoint of non-inferiority. In the Type 1 trial, a statistically
significant interaction associated with HbA1c data from India was observed and efficacy results
from India were, therefore, not comparable to the results from the United States and Germany, which
together represented 77% of the treated patients participating in the Type 1 trial. These data were
not comparable to the rest of the data for several reasons including: (a) markedly increased HbA1c
levels both prior to and after study drug initiation, (b) a twofold higher rate of intra-subject
variability in HbA1c results and (c) markedly reduced reporting of hypoglycemic events. The
anomalies were observed in both the VIAject® and Humulin® R treatment groups. When we include all
data from India in our HbA1c analysis, we do not establish non-inferiority in the Type 1 trial.
However, we believe that including the data from India is not valid for determining
non-inferiority. When the statistically significant interaction associated with the HbA1c data
from India is taken into account, non-inferiority in the Type 1 trial is achieved.
In the Type 1 trial, patients receiving Humulin® R were almost twice as likely to have one or
more severe hypoglycemic events when compared to those receiving VIAject®. This result did not
achieve statistical significance due to the small number of patients experiencing one or more
severe hypoglycemic events (15 patients in the Humulin® R treatment group and 8 patients in the
VIAject® treatment group, for a total of twenty-three patients). Patients receiving VIAject® lost
0.1 pounds on average, while patients receiving Humulin® R gained 3.1 pounds on average, for a
difference of 3.2 pounds. This result was statistically significant.
In the Type 2 trial, the median number of non-severe hypoglycemic events in the Humulin® R arm
was twice as great as in the VIAject® arm. Patients treated with VIAject® gained less weight than
patients treated with Humulin® R. Patients receiving VIAject® gained 1.0 pounds on average, while
patients receiving Humulin® R gained 3.0 pounds on average, for a difference of 2.0 pounds. Both
the hypoglycemia and weight gain results were statistically significant. With regard to severe
hypoglycemic events, no meaningful comparison was possible due to the small number of events that
occurred in both the VIAject® and the Humulin® R treatment groups.
In both the Type 1 and Type 2 clinical trials, swelling, itching and redness were reported in
less than 5% of patients receiving VIAject® or Humulin® R. VIAject® was associated with injection
site discomfort, the prevalence of which decreased with time. Approximately 5% of the patients
treated with VIAject® dropped out of the two clinical trials primarily due to injection site
discomfort. No patients treated with Humulin® R dropped out of the two clinical trials due to
injection site discomfort.
Approximately 400 patients with Type 1 and Type 2 diabetes who completed the pivotal Phase 3
clinical trials elected to participate in a long term safety extension trial in which all patients
are treated with VIAject® as their mealtime insulin. Patients will complete the extension trial
upon the eighteenth month of patient treatment, subject to compassionate use exceptions. We intend
to include interim data from this extension trial in our NDA. We expect to complete dosing of all
patients in the extension trial in February 2010.
Bioequivalence and Tolerability Trials. The VIAject® formulation used in the pivotal Phase 3
clinical trials was a two vial presentation, with one vial containing lyophilized insulin and the
second vial containing 10cc of the proprietary VIAject® diluent, which upon reconstitution yields a
concentration of 25 IU/cc at a pH of 4. We have also developed two pre-mixed, liquid formulations
of VIAject® at concentrations of 100 IU/cc. One is at a pH of 4, and the other is at a pH of 7.
We have completed two trials studying the bioequivalence of our liquid formulations of
VIAject® to the lyophilized formulation that we used in our Phase 3 clinical trials. The first
bioequivalence trial was completed in December 2008 and compared the100 IU/cc liquid formulation at
a pH of 4 to the 25 IU/cc lyophilized formulation at a pH of 4. The second trial was completed in
October 2009 and compared the 100 IU/cc liquid formulation at a pH of 7 to the 25 IU/cc lyophilized
formulation at a pH of 4. Each trial showed the liquid formulation to be bioequivalent to the
lyophilized formulation. We expect our NDA for VIAject® will be based on the pH 7 100 IU/cc liquid
formulation, as we believe it may offer certain commercial advantages. We expect that the NDA will
include vials, for use with syringes and insulin pumps, and cartridges, for use in both disposable
and reusable pen injectors.
In October 2009 we completed a trial studying the tolerability of the pH 7 100 IU/cc liquid
formulation of VIAject® compared to both the VIAject® 25 IU/cc lyophilized formulation and
Humalog®. This was a double blind, randomized trial in which patients were injected with three
doses per day of one of the three insulin formulations. The trial was administered over three
days, with a different formulation each day. The primary endpoint of the trial was to show that
the liquid formulation of VIAject® that we intend to submit in our NDA presents less injection site
discomfort than the lyophilized formulation used in our Phase 3 clinical trials. Preliminary
results from the tolerability trial indicated that the liquid formulation of VIAject® presented a
statistically significant reduction in injection site discomfort when compared to the lyophilized
formulation. Additionally, a majority of patients stated that they experienced the same or less
discomfort with the liquid formulation of VIAject® than they did with their usual meal-time
insulin.
6
Table of Contents
Preliminary results also indicated, however, that a subset of patients experienced more
injection site discomfort with the liquid formulation of VIAject® than they did with Humalog®.
Regulatory Status. In March 2009, we announced our plan to submit an NDA to the FDA by the
end of 2009 to market VIAject® for the treatment of diabetes. After reviewing all of the data from
the two pivotal Phase 3 clinical trials with regulatory consultants and meeting with the FDA staff,
we decided to proceed with the submission of our NDA under section 505(b)(2) of the FFDCA. We
expect that the NDA will be based upon results from multiple pharmacokinetic and pharmacodynamic
studies as well as two completed Phase 3 studies of VIAject® in patients with Type 1 and Type 2
diabetes. We intend to seek approval for a pH 7 100 IU/cc liquid formulation of VIAject® that is
bioequivalent to the two-part 25 IU/cc lyophilized powder formulation of VIAject® that was used in
our pivotal Phase 3 clinical trials. We intend to submit the NDA in December 2009.
Additional Pipeline Opportunities
VIAtab™ is our formulation of recombinant human insulin designed to be taken orally via
sublingual administration. Unlike other oral insulin products under development that must be
swallowed, the sublingual delivery of VIAtab™ may avoid the destructive effects on insulin by the
stomach and liver. We are developing VIAtab™ as a potential treatment for patients with Type 2
diabetes in the early stages of their disease. We believe that VIAtab™ may be a suitable treatment
for these patients because of its potential rapid delivery and because it does not require
injections. In our preclinical in vitro and animal studies, we successfully delivered insulin by
sublingual administration. We have tested one formulation of VIAtab™ in a Phase 1 trial in patients
with Type 1 diabetes. In 2010 we may allocate limited resources to developing our VIAtab™
formulation.
Our VIAdel™ Technology
Peptide hormones, such as insulin, are valuable drugs used to treat a variety of important
human diseases. Peptide hormones are, in general, relatively unstable and poorly absorbed into the
blood from the gastrointestinal tract. As a result, they are typically given by subcutaneous
injection. Because peptide hormones are charged molecules, their absorption from injection sites is
inhibited and slowed. This is in contrast to their natural release into the blood, which is
typically in one or more very rapid, spike-like, secretions. Slowing of the rate of absorption
reduces the clinical efficacy of many peptide hormones, including insulin, parathyroid hormone and
calcitonin in particular.
Our VIAdel™ technology consists of several proprietary models that we have developed to study
the interaction of small molecules with peptide hormones and their effects on the stability,
apparent molecular size, complexed state, surface charge distribution and rate of absorption and
mechanisms of absorption of peptide hormones. These models have allowed us to develop proprietary
formulations designed to increase the rate of absorption and stability of these peptide hormones,
potentially allowing for improved efficacy by injection and for administration by non-invasive
routes, such as sublingual administration.
We use our VIAdel™ technology to develop proprietary formulations of small molecules which
form weak and reversible hydrogen bonds with their molecular cargo. By doing so, we believe that
our formulations mask the charge on peptides. As a consequence, many of the peptides in our
formulations face less resistance from cell membranes, which would generally repel them, thus
allowing them to pass through cell membranes into the blood more rapidly and in greater quantities
than other currently approved formulations of the same peptides. Our VIAdel™ technology is designed
to allow us to develop formulations that stabilize delicate peptides which can result in longer
shelf lives for our formulations. Furthermore, because we use our VIAdel™ technology to reformulate
existing peptide drugs with ingredients that are generally regarded as safe by the FDA and because
our reformulations do not drastically alter the structure of these peptides, we believe that our
VIAdel™ technology allows us to develop product candidates for which the Section 505(b)(2) approval
pathway is available.
Government Regulation
The FDA and other federal, state, local and foreign regulatory agencies impose substantial
requirements upon the clinical development, approval, labeling, manufacture, marketing and
distribution of drug products. These agencies regulate, among other things, research and
development activities and the testing, approval, manufacture, quality control, safety,
effectiveness, labeling, storage, record keeping, advertising and promotion of our product
candidates. The regulatory approval process is generally lengthy and expensive, with no guarantee
of a positive result. Moreover, failure to comply with applicable FDA or other requirements may
result in civil or criminal penalties, recall or seizure of products, injunctive relief including
partial or total suspension of production, or withdrawal of a product from the market.
United States Government Regulation
The FDA regulates, among other things, the research, manufacture, promotion and distribution
of drugs in the United States under the FFDCA and other statutes and implementing regulations. The
process required by the FDA before prescription drug product candidates may be marketed in the
United States generally involves the following:
| • | completion of extensive nonclinical laboratory tests, animal studies and formulation studies, all performed in accordance with the FDA’s Good Laboratory Practice, or GLP, regulations; | ||
| • | submission to the FDA of an IND which must become effective before human clinical trials may begin; |
7
Table of Contents
| • | for some products, performance of adequate and well-controlled human clinical trials in accordance with the FDA’s regulations, including Good Clinical Practices, to establish the safety and efficacy of the product candidate for each proposed indication; | ||
| • | submission to the FDA of an NDA; | ||
| • | satisfactory completion of an FDA preapproval inspection of the manufacturing facilities at which the product is produced to assess compliance with current Good Manufacturing Practice, or cGMP, regulations; and | ||
| • | FDA review and approval of the NDA prior to any commercial marketing, sale or shipment of the drug. |
The testing and approval process requires substantial time, effort and financial resources,
and we cannot be certain that any approvals for our product candidates will be granted on a timely
basis, if at all.
Nonclinical tests include laboratory evaluations of product chemistry, formulation and
stability, as well as studies to evaluate toxicity in animals and other animal studies. The results
of nonclinical tests, together with manufacturing information and analytical data, are submitted as
part of an IND to the FDA. Some nonclinical testing may continue even after an IND is submitted.
The IND also includes one or more protocols for the initial clinical trial or trials and an
investigator’s brochure. An IND automatically becomes effective 30 days after receipt by the FDA,
unless the FDA, within the 30-day time period, raises concerns or questions relating to the
proposed clinical trials as outlined in the IND and places the clinical trial on a clinical hold.
In such cases, the IND sponsor and the FDA must resolve any outstanding concerns or questions
before any clinical trials can begin. Clinical trial holds also may be imposed at any time before
or during studies due to safety concerns or non-compliance with regulatory requirements. An
independent institutional review board, or IRB, at each of the clinical centers proposing to
conduct the clinical trial must review and approve the plan for any clinical trial before it
commences at that center. An IRB considers, among other things, whether the risks to individuals
participating in the trials are minimized and are reasonable in relation to anticipated benefits.
The IRB also approves the consent form signed by the trial participants and must monitor the study
until completed.
Clinical Trials. Clinical trials involve the administration of the product candidate to
human subjects under the supervision of qualified medical investigators according to approved
protocols that detail the objectives of the study, dosing procedures, subject selection and
exclusion criteria, and the parameters to be used to monitor participant safety. Each protocol is
submitted to the FDA as part of the IND.
Human clinical trials are typically conducted in three sequential phases, but the phases may
overlap, or be combined.
| • | Phase 1 clinical trials typically involve the initial introduction of the product candidate into healthy human volunteers. In Phase 1 clinical trials, the product candidate is typically tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and pharmacodynamics. | ||
| • | Phase 2 clinical trials are conducted in a limited patient population to gather evidence about the efficacy of the product candidate for specific, targeted indications; to determine dosage tolerance and optimal dosage; and to identify possible adverse effects and safety risks. | ||
| • | Phase 3 clinical trials are undertaken to evaluate clinical efficacy and to test for safety in an expanded patient population at geographically dispersed clinical trial sites. The size of Phase 3 clinical trials depends upon clinical and statistical considerations for the product candidate and disease, but sometimes can include several thousand patients. Phase 3 clinical trials are intended to establish the overall risk-benefit ratio of the product candidate and provide an adequate basis for product labeling. |
Clinical testing must satisfy extensive FDA regulations. Reports detailing the results of the
clinical trials must be submitted at least annually to the FDA and safety reports must be submitted
for serious and unexpected adverse events. Success in early stage clinical trials does not assure
success in later stage clinical trials. The FDA, an IRB or we may suspend a clinical trial at any
time on various grounds, including a finding that the research subjects or patients are being
exposed to an unacceptable health risk.
New Drug Applications. Assuming successful completion of the required clinical trials, the
results of product development, nonclinical studies and clinical trials are submitted to the FDA as
part of an NDA. An NDA also must contain extensive manufacturing information, as well as proposed
labeling for the finished product. An NDA applicant must develop information about the chemistry
and physical characteristics of the drug and finalize a process for manufacturing the product in
accordance with cGMP. The manufacturing process must be capable of consistently producing quality
product within specifications approved by the FDA. The manufacturer must develop methods for
testing the quality, purity and potency of the final product. In addition, appropriate packaging
must be selected and tested and stability studies must be conducted to demonstrate that the product
does not undergo unacceptable deterioration over its shelf life. Prior to approval, the FDA will
conduct an inspection of the manufacturing facilities to assess compliance with cGMP.
The FDA reviews all NDAs submitted before it accepts them for filing. The FDA may request
additional information rather than accept an NDA for filing. In this event, the NDA must be
resubmitted with the additional information and is subject to review before the FDA accepts it for
filing. After an application is filed, the FDA may refer the NDA to an advisory committee for
review, evaluation and recommendation as to whether the application should be approved and under
what conditions. The FDA is not bound
8
Table of Contents
by the recommendation of an advisory committee, but it considers them carefully when making
decisions. The FDA may deny approval of an NDA if the applicable regulatory criteria are not
satisfied. Data obtained from clinical trials are not always conclusive and the FDA may interpret
data differently than we interpret the same data. The FDA may issue a complete response letter,
which may require additional clinical or other data or impose other conditions that must be met in
order to secure final approval of the NDA. If a product receives regulatory approval, the approval
may be significantly limited to specific diseases and dosages or the indications for use may
otherwise be limited, which could restrict the commercial value of the product. In addition, the
FDA may require us to conduct Phase 4 testing which involves clinical trials designed to further
assess a drug’s safety and effectiveness after NDA approval, and may require surveillance programs
to monitor the safety of approved products which have been commercialized. Once issued, the FDA may
withdraw product approval if ongoing regulatory requirements are not met or if safety or efficacy
questions are raised after the product reaches the market.
Section 505(b)(2) NDAs. There are two types of NDAs: the full NDA and the Section 505(b)(2)
NDA. We intend to file Section 505(b)(2) NDAs that might, if accepted by the FDA, save time and
expense in the development and testing of our product candidates. A full NDA is submitted under
Section 505(b)(1) of the FFDCA, and must contain full reports of investigations conducted by the
applicant to demonstrate the safety and effectiveness of the drug. A Section 505(b)(2) NDA may be
submitted for a drug for which one or more of the investigations relied upon by the applicant was
not conducted by or for the applicant and for which the applicant has no right of reference from
the person by or for whom the investigations were conducted. A Section 505(b)(2) NDA may be
submitted based in whole or in part on published literature or on the FDA’s finding of safety and
efficacy of one or more previously approved drugs, which are known as reference drugs. Thus, the
filing of a Section 505(b)(2) NDA may result in approval of a drug based on fewer clinical or
nonclinical studies than would be required under a full NDA. The number and size of studies that
need to be conducted by the sponsor depends on the amount and quality of data pertaining to the
reference drug that are publicly available, and on the similarity of and differences between the
applicant’s drug and the reference drug. In some cases, extensive, time-consuming, and costly
clinical and nonclinical studies may still be required for approval of a Section 505(b)(2) NDA.
Because we are developing new formulations of previously approved chemical entities, such as
insulin, our drug approval strategy is to submit Section 505(b)(2) NDAs to the FDA. We plan to
pursue similar routes for submitting applications for our product candidates in foreign
jurisdictions if available. The FDA may not agree that our product candidates are approvable as
Section 505(b)(2) NDAs. Insulin is a small protein molecule which is known to be associated with
significant intra- and interpatient variability of absorption and resulting glucose lowering
response. This makes it more difficult to demonstrate that two insulin substances are highly
similar than would be the case with many small molecule drugs. The availability of the
Section 505(b)(2) NDA pathway for insulin is even more controversial than for small molecule drugs,
and the FDA may not accept this pathway for our insulin drug candidates. There is no specific
guidance available for insulin Section 505(b)(2) NDAs, and no insulin product has been approved
under a Section 505(b)(2) NDA. If the FDA determines that Section 505(b)(2) NDAs are not
appropriate and that full NDAs are required for our product candidates, the time and financial
resources required to obtain FDA approval for our product candidates could substantially and
materially increase, and our products might be less likely to be approved. If the FDA requires full
NDAs for our product candidates, or requires more extensive testing and development for some other
reason, our ability to compete with alternative products that arrive on the market more quickly
than our product candidates would be adversely impacted.
Patent Protections. An applicant submitting a Section 505(b)(2) NDA must certify to the FDA
with respect to the patent status of the reference drug upon which the applicant relies in support
of approval of its drug. With respect to every patent listed in the FDA’s Orange Book, which is the
FDA’s list of approved drug products, as claiming the reference drug or an approved method of use
of the reference drug, the Section 505(b)(2) applicant must certify that: (1) there is no patent
information listed by the FDA for the reference drug; (2) the listed patent has expired; (3) the
listed patent has not expired, but will expire on a particular date; (4) the listed patent is
invalid, unenforceable, or will not be infringed by the manufacture, use, or sale of the product in
the Section 505(b)(2) NDA; or (5) if the patent is a use patent, that the applicant does not seek
approval for a use claimed by the patent. If the applicant files a certification to the effect of
clause (1), (2) or (5), FDA approval of the Section 505(b)(2) NDA may be made effective immediately
upon successful FDA review of the application, in the absence of marketing exclusivity delays,
which are discussed below. If the applicant files a certification to the effect of clause (3), the
Section 505(b)(2) NDA approval may not be made effective until the expiration of the relevant
patent and the expiration of any marketing exclusivity delays.
If the Section 505(b)(2) NDA applicant provides a certification to the effect of clause (4),
referred to as a paragraph IV certification, the applicant also must send notice of the
certification to the patent owner and the holder of the NDA for the reference drug. The filing of a
patent infringement lawsuit within 45 days of the receipt of the notification may prevent the FDA
from approving the Section 505(b)(2) NDA for 30 months from the date of the receipt of the
notification unless the court determines that a longer or shorter period is appropriate because
either party to the action failed to reasonably cooperate in expediting the action. However, the
FDA may approve the Section 505(b)(2) NDA before the 30 months have expired if a court decides that
the patent is invalid, unenforceable, or not infringed, or if a court enters a settlement order or
consent decree stating the patent is invalid or not infringed.
Notwithstanding the approval of many products by the FDA pursuant to Section 505(b)(2), over
the last few years certain brand-name pharmaceutical companies and others have objected to the
FDA’s interpretation of Section 505(b)(2). If the FDA’s interpretation of Section 505(b)(2) is
successfully challenged in court, the FDA may be required to change its interpretation of
Section 505(b)(2) which could delay or even prevent the FDA from approving any Section 505(b)(2)
NDA that we submit. The pharmaceutical industry is highly competitive, and it is not uncommon for a
manufacturer of an approved product to file a citizen petition with the FDA seeking to delay
approval of, or impose additional approval requirements for, pending competing products. If
successful, such
9
Table of Contents
petitions can significantly delay, or even prevent, the approval of the new product. Moreover,
even if the FDA ultimately denies such a petition, the FDA may substantially delay approval while
it considers and responds to the petition.
Marketing Exclusivity. Market exclusivity provisions under the FFDCA can delay the
submission or the approval of Section 505(b)(2) NDAs, thereby delaying a Section 505(b)(2) product
from entering the market. The FFDCA provides five-year marketing exclusivity to the first applicant
to gain approval of an NDA for a new chemical entity, or NCE, meaning that the FDA has not
previously approved any other drug containing the same active moiety. This exclusivity prohibits
the submission of a Section 505(b)(2) NDA for any drug product containing the active moiety during
the five-year exclusivity period. However, submission of a Section 505(b)(2) NDA that certifies
that a listed patent is invalid, unenforceable, or will not be infringed, as discussed above, is
permitted after four years, but if a patent infringement lawsuit is brought within 45 days after
such certification, FDA approval of the Section 505(b)(2) NDA may automatically be stayed until 7 1
/ 2 years after the NCE approval date. The FFDCA also provides three years of marketing
exclusivity for the approval of new and supplemental NDAs for product changes, including, among
other things, new indications, dosage forms, routes of administration or strengths of an existing
drug, or for a new use, if new clinical investigations, other than bioavailability studies, that
were conducted or sponsored by the applicant are deemed by FDA to be essential to the approval of
the application. Five-year and three-year exclusivity will not delay the submission or approval of
another full NDA; however, as discussed above, an applicant submitting a full NDA under
Section 505(b)(1) would be required to conduct or obtain a right of reference to all of the
preclinical and adequate and well-controlled clinical trials necessary to demonstrate safety and
effectiveness.
Other types of exclusivity in the United States include orphan drug exclusivity and pediatric
exclusivity. The FDA may grant orphan drug designation to a drug intended to treat a rare disease
or condition, which is generally a disease or condition that affects fewer than 200,000 individuals
in the United States, or more than 200,000 individuals in the United States and for which there is
no reasonable expectation that the cost of developing and making available in the United States a
drug for this type of disease or condition will be recovered from sales in the United States for
that drug. Seven-year orphan drug exclusivity is available to a product that has orphan drug
designation and that receives the first FDA approval for the indication for which the drug has such
designation. Orphan drug exclusivity prevents approval of another application for the same drug for
the same orphan indication, for a period of seven years, regardless of whether the application is a
full NDA or a Section 505(b)(2) NDA, except in limited circumstances, such as a showing of clinical
superiority to the product with orphan exclusivity. Pediatric exclusivity, if granted, provides an
additional six months to an existing exclusivity or statutory delay in approval resulting from a
patent certification. This six-month exclusivity, which runs from the end of other exclusivity
protection or patent delay, may be granted based on the voluntary completion of a pediatric study
in accordance with an FDA-issued “Written Request” for such a study.
Section 505(b)(2) NDAs are similar to full NDAs filed under Section 505(b)(1) in that they are
entitled to any of these forms of exclusivity if they meet the qualifying criteria. They also are
entitled to the patent protections described above, based on patents that are listed in the FDA’s
Orange Book in the same manner as patents claiming drugs and uses approved for NDAs submitted as
full NDAs.
Other Regulatory Requirements. Maintaining substantial compliance with appropriate federal,
state and local statutes and regulations requires the expenditure of substantial time and financial
resources. Drug manufacturers are required to register their establishments with the FDA and
certain state agencies, and after approval, the FDA and these state agencies conduct periodic
unannounced inspections to ensure continued compliance with ongoing regulatory requirements,
including cGMPs. In addition, after approval, some types of changes to the approved product, such
as adding new indications, manufacturing changes and additional labeling claims, are subject to
further FDA review and approval. The FDA may require post-approval testing and surveillance
programs to monitor safety and the effectiveness of approved products that have been
commercialized. Any drug products manufactured or distributed by us pursuant to FDA approvals are
subject to continuing regulation by the FDA, including:
| • | record-keeping requirements; | ||
| • | reporting of adverse experiences with the drug; | ||
| • | providing the FDA with updated safety and efficacy information; | ||
| • | reporting on advertisements and promotional labeling; | ||
| • | drug sampling and distribution requirements; and | ||
| • | complying with electronic record and signature requirements. |
In addition, the FDA strictly regulates labeling, advertising, promotion and other types of
information on products that are placed on the market. There are numerous regulations and policies
that govern various means for disseminating information to health-care professionals as well as
consumers, including to industry sponsored scientific and educational activities, information
provided to the media and information provided over the Internet. Drugs may be promoted only for
the approved indications and in accordance with the provisions of the approved label.
The FDA has very broad enforcement authority and the failure to comply with applicable
regulatory requirements can result in administrative or judicial sanctions being imposed on us or
on the manufacturers and distributors of our approved products, including warning letters, refusals
of government contracts, clinical holds, civil penalties, injunctions, restitution, and
disgorgement or profits,
10
Table of Contents
recall or seizure of products, total or partial suspension of production or distribution,
withdrawal of approvals, refusal to approve pending applications, and criminal prosecution
resulting in fines and incarceration. The FDA and other agencies actively enforce the laws and
regulations prohibiting the promotion of off-label uses, and a company that is found to have
improperly promoted off-label uses may be subject to significant liability. In addition, even after
regulatory approval is obtained, later discovery of previously unknown problems with a product may
result in restrictions on the product or even complete withdrawal of the product from the market.
New Legislation. On September 27, 2007, the President signed into law the Food and Drug
Administration Amendments Act of 2007, or FDAAA. This new legislation grants significant new powers
to the FDA, many of which are aimed at improving drug safety and assuring the safety of drug
products after approval. In particular, the new law authorizes the FDA to, among other things,
require post-approval studies and clinical trials, mandate changes to drug labeling to reflect new
safety information, and require risk evaluation and mitigation strategies for certain drugs,
including certain currently approved drugs. In addition, the new law significantly expands the
federal government’s clinical trial registry and results databank and creates new restrictions on
the advertising and promotion of drug products. Under the FDAAA, companies that violate these and
other provisions of the new law are subject to substantial civil monetary penalties.
The FDA has not yet implemented many of the provisions of the FDAAA, so we cannot predict the
impact of the new legislation on the pharmaceutical industry or our business. However, the
requirements and changes imposed by the FDAAA may make it more difficult, and more costly, to
obtain and maintain approval for new pharmaceutical products, or to produce, market and distribute
existing products. In addition, the FDA’s regulations, policies and guidance are often revised or
reinterpreted by the agency or the courts in ways that may significantly affect our business and
our products. It is impossible to predict whether additional legislative changes will be enacted,
or FDA regulations, guidance or interpretations changed, or what the impact of such changes, if
any, may be.
Regulations Outside the United States
In addition to regulations in the United States, we will be subject to a variety of
regulations in other jurisdictions governing clinical trials and commercial sales and distribution
of our products. Whether or not we obtain FDA approval for a product, we must obtain the necessary
approvals by the comparable regulatory authorities of countries outside the United States before we
can commence clinical trials or marketing of the product in those countries. The approval process
varies from country to country, and the time may be longer or shorter than that required for FDA
approval. The requirements governing the conduct of clinical trials, product licensing, pricing and
reimbursement also vary between jurisdictions.
To obtain regulatory approval of a drug under European Union regulatory systems, we may submit
applications for marketing authorizations either under a centralized or decentralized procedure.
The centralized procedure is compulsory for medicines produced by certain biotechnological
processes, new active substances indicated for the treatment of certain diseases such as AIDS,
cancer, neurodegenerative disorders and diabetes, and products designated as orphan medicinal
products, and optional for other new active substances and those products which constitute a
significant therapeutic, scientific or technical innovation. The procedure provides for the grant
of a single marketing authorization that is valid for all European Union member states, as well as
for Iceland, Liechtenstein, and Norway. The decentralized procedure provides for approval by one or
more other, or concerned, member states of an assessment of an application performed by one member
state, known as the reference member state. Under this procedure, an applicant submits an
application, or dossier, and related materials including a draft summary of product
characteristics, and draft labeling and package leaflet, to the reference member state and
concerned member states. The reference member state prepares a draft assessment and drafts of the
related materials within 120 days after receipt of a valid application. Within 90 days of receiving
the reference member state’s assessment report, each concerned member state must decide whether to
approve the assessment report and related materials. If a member state cannot approve the
assessment report and related materials on the grounds of potential serious risk to the public
health, the disputed points may eventually be referred to the European Commission, whose decision
is binding on all member states.
Competition
The pharmaceutical industry is characterized by intense competition and rapidly evolving
technology. For several decades, scientists have attempted to improve the bioavailability of
injected formulations and to devise alternative non-invasive delivery systems for the delivery of
macromolecules such as insulin. While we believe that product candidates using our VIAdel™
technology will be an improvement over existing products, our product candidates will compete
against many products with similar indications.
If approved, our primary competition for VIAject® will be rapid acting mealtime injectable
insulins such as Humalog® , which is marketed by Eli Lilly, NovoLog® , which is marketed by Novo
Nordisk, and Apidra® , which is marketed by Sanofi-Aventis.
In addition, other development stage rapid acting insulin formulations may be approved and
compete with VIAject®. Halozyme Therapeutics, Inc is developing two rapid-acting insulin products:
recombinant human insulin formulated with a recombinant human hyaluronidase enzyme, and a rapid
acting insulin analog formulated in combination with a recombinant human hyaluronidase enzyme. A
three month multidose crossover treatment study in patients with type 1 diabetes is currently
underway that compares a regular insulin / hyaluronidase combination with Humalog®. Results are
expected in the third quarter of 2010. Halozyme Therapeutics, Inc. has previously reported that,
in Phase 1 and Phase 2 clinical trials of Humulin® R and Humalog® in combination with a recombinant
human hyaluronidase enzyme, the combinations yielded pharmacokinetics and glucodynamics that better
mimicked physiologic mealtime insulin release and activity than Humulin® R or Humalog® alone.
11
Table of Contents
In addition, VIAject® may face competition from products employing non-invasive methods of
insulin delivery, such as oral insulin pills, which are currently in development, or others which
are in clinical development. Generex has developed an oral spray that is currently in Phase 3
development. The development of insulin formulations that are taken orally, or swallowed, face
problems because insulin is largely broken down in the digestive system and as a result much of the
insulin delivered orally does not enter the blood and the timing and amount of dosage that does is
variable and unpredictable.
MannKind’s pulmonary Technosphere™ technology is a New Chemical Entity which has been studied
in three Phase 3 clinical trials in patients with Type 1 and Type 2 diabetes. MannKind filed an NDA
for its product candidate in early 2009. Insulin administered as a nasal spray has been studied
extensively but does not appear to be a practical route for insulin administration because without
the addition of penetration enhancers, the bioavailability of the insulin is too low and too
variable. Nasally administered insulin using penetration enhancers has produced irritation and
destruction of the nasal passages with frequent use.
Intellectual Property and Proprietary Technology
Our technologies have been developed exclusively by our employees, without input from third
parties.
On October 9, 2007 the United States Patent and Trademark Office issued U.S. Patent
No. 7,279,457 encompassing VIAject® and VIAtab™. The patent will expire no earlier than January
2026.
On October 12, 2008 we reported that we received a notice of allowance from the European
Patent Office for patent claims encompassing VIAject® and VIAtab™. The European Patent granted as
EP 1 740 154 on June 17, 2009 and will have a term of 20 years from the international filing date
in the designated countries if all annuity fees are paid.
We have a policy of filing for patent protection on all our product candidates. Our patents
and patent applications consist of the following:
| • | one granted United States patent, one foreign patent, and several pending United States patent applications and corresponding foreign and international patent applications relating to our VIAject® and VIAtab™ technology; | ||
| • | one foreign patent and pending foreign patent applications relating to our technology for enhancing delivery of drugs in a form for absorption through the skin into the blood, a process known as transdermal drug delivery; | ||
| • | one foreign patent, two pending United States patent applications and corresponding foreign patent applications relating to sublingual and/or oral delivery devices that can be used to deliver the certain insulin based products; and | ||
| • | several United States patent applications and a corresponding international patent applications relating to other early stage product candidates. |
Our pending patent applications, those we may file in the future, or those we may license from
third parties, may not result in patents being issued.
The individual active and inactive ingredients in our VIAject® and VIAtab™ product candidates
have been known and used for many years and, therefore, are no longer subject to patent protection,
except in proprietary combinations. Accordingly, our patent and pending applications are directed
to the particular formulations of these ingredients in our products, and to their use. Although we
believe our formulations and their use are patented and provide a competitive advantage, our
patents may not prevent others from marketing formulations using the same active and inactive
ingredients in similar but different formulations.
We require our employees, consultants and members of our scientific advisory board to execute
confidentiality agreements upon the commencement of employment, consulting or collaborative
relationships with us. These agreements provide that all confidential information developed or made
known during the course of the relationship with us be kept confidential and not disclosed to third
parties except in specific circumstances. In the case of employees, the agreements provide that all
inventions resulting from work performed for us, utilizing our property or relating to our business
and conceived or completed by the individual during employment shall be our exclusive property to
the extent permitted by applicable law.
Manufacturing
We believe our laboratory in Danbury, Connecticut is equipped to meet the limited
manufacturing requirements of all of our product candidates through Phase 2 clinical trials. We
intend to manufacture our product candidates by contracting with third parties that operate
manufacturing facilities in accordance with cGMP. To date, we have relied on two commercial
manufacturers — Catalent Pharma Solutions (formerly known as Cardinal Health PTS, LLC) and
Hyaluron, Inc. — to manufacture our VIAject® product candidate. We believe that both manufacturers
comply with the relevant regulatory requirements. We have suspended plans to enlarge our own
laboratory or manufacturing facilities.
In September 2008, we received notice from Catalent Pharma Solutions that they intend to sell,
or possibly close, the facility where VIAject® has been manufactured. As a result, we now intend to
conduct the majority of our future manufacturing of VIAject®
12
Table of Contents
in the United Kingdom at a facility of a wholly owned subsidiary of Wockhardt U.K. Holdings,
Ltd. We intend to negotiate a commercial manufacturing agreement with the Wockhardt U.K. Holdings,
Ltd. subsidiary.
We have contracted with N.V. Organon (formerly known as Diosynth B.V.), a global producer of
insulin, to supply us with all of the insulin that we will need for the testing and manufacturing
of our product candidates. Our agreement with N.V. Organon will terminate in December 2011, and we
are discussing the possibility of extending the agreement. We believe that our current supplies of
insulin, together with the quantities of insulin called for under our existing supply agreement,
will be sufficient to allow us to complete our current and anticipated future clinical trials of
VIAject®. In addition, we believe that the available quantities under the agreement will be
sufficient to support our needs for approximately three years following the commercial launch of
VIAject®. We are seeking to qualify another insulin supplier to serve as additional or alternative
supplier.
Sales and Marketing
We currently have no sales and marketing capabilities and no distribution capabilities. Our
current strategy is to selectively enter into collaboration agreements with leading pharmaceutical
or biotechnology companies for the commercialization of our product candidates. In entering into
these collaboration agreements we may retain some commercial rights for certain product candidates
for which we receive marketing approvals in situations in which we believe it is possible to access
the market through a focused, specialized sales force. For example, we may focus on the pediatric
market because we believe VIAject® is particularly suited for the treatment of children with
diabetes, the number of pediatric endocrinologists is relatively few and we believe this patient
population is underserved.
In order to implement our strategy successfully, we may need to develop a specialized sales
and marketing organization with sufficient technical expertise. We do not expect to do so, however,
in fiscal year 2010.
Employees
At September 30, 2009 we had 54 full time-employees and several part-time consultants who
perform services for us on a regular basis. We consider our employee relations to be good.
Additional Information
Our website is www.biodel.com. We are not including the information contained on our
website as a part of, or incorporating it by reference into, this Annual Report on Form 10-K. We
make available free of charge on our website our Annual Reports on Form 10-K, Quarterly Reports on
Form 10-Q, Current Reports on Form 8-K and amendments to those reports filed or furnished pursuant
to Section 13(a) or 15(d) of the Exchange Act, as soon as reasonably practicable after we
electronically file such material with, or furnished it to, the Securities and Exchange Commission.
Our reports filed with the Securities and Exchange Commission are also available at the Securities
and Exchange Commission’s website at www.sec.gov.
Executive Officers of the Registrant
The following table sets forth our executive officers, their respective ages and positions as
of November 30, 2009:
| Name | Age | Position | ||
Dr. Solomon S. Steiner |
72 | Chairman, President and Chief Executive Officer | ||
Gerard Michel |
46 | Chief Financial Officer, Vice President, Corporate Development and Treasurer | ||
Erik Steiner |
43 | Vice President, Operations | ||
Dr. Alan Krasner |
46 | Chief Medical Officer | ||
Paul Bavier |
37 | General Counsel and Secretary |
Dr. Solomon S. Steiner co-founded our company and has served as our Chairman, President and
Chief Executive Officer since our inception in December 2003. In 1991, Dr. Steiner founded
Pharmaceutical Discovery Corporation, or PDC, a biopharmaceutical corporation. Dr. Steiner served
as PDC’s Chief Executive Officer and Chairman of the Board of Directors from its inception until
December 2001, when PDC was merged with two other companies to form MannKind Corporation. From
December 2001 to February 2003, Dr. Steiner served on MannKind’s Board of Directors and as a
Corporate Vice President and Chief Scientific Officer. In 1985, Dr. Steiner founded and was the
Chairman of the Board of Directors and President of Clinical Technologies Associates, Inc., or
CTAI, now known as Emisphere Technologies, Inc. Under his leadership CTAI went public in February
of 1989. Dr. Steiner is an inventor of Emisphere’s oral delivery system for peptides and
mucopolysaccharides. Dr. Steiner is currently an adjunct full professor at New York Medical College
and research full professor of psychiatry and neurology at New York University School of Medicine.
Dr. Steiner received a Ph.D. from New York University. Dr. Steiner is Erik Steiner’s father.
Mr. Gerard Michel joined our company in November 2007 as Chief Financial Officer, Vice
President of Corporate Development and Treasurer. From October 2003 to November 2007, Mr. Michel
served as Chief Financial Officer and from April 2006 to November 2007, Vice President, Corporate
Development of NPS Pharmaceuticals, a biopharmaceutical company. From June 1995 to July 2002,
Mr. Michel served as a Principal of the consulting firm Booz-Allen & Hamilton. Mr. Michel received
an MBA and B.S. from University of Rochester, and an M.S., Microbology from The University of
Rochester School of Medicine and Dentistry.
13
Table of Contents
Mr. Erik Steiner co-founded our company and has served as our Vice President, Operations since
our inception in December 2003. From February 2003 to December 2003, Mr. Steiner co-founded and
served as the Vice President, Operations of Steiner Ventures. From May 1999 to February 2003,
Mr. Steiner served as Head of Operations of Cabot McMullen Inc, a film and television production
company. Prior thereto, Mr. Steiner served as Administrative Director and Fiscal Administrator of
the New Jersey Public Interest Research Group. Mr. Steiner is Solomon Steiner’s son.
Dr. Alan Krasner joined our company in May 2008 as Chief Medical Officer. From 2002 to 2008,
Dr. Krasner served as Director of the Department of Clinical Research Metabolic Diseases at Pfizer
Global Research and Development where he was responsible for the design, execution, clinical
analysis, and reporting of multiple, global clinical trials supporting registration of late stage
drug candidates. Dr. Krasner currently serves as a consulting physician at the Joslin Diabetes and
Endocrinology Center of the Lawrence and Memorial Hospital in New London, Connecticut. Dr. Krasner
holds a B.S. from the Medical Education Honors Program at Northwestern University and a M.D. from
Northwestern University Medical School. He completed his residency at Johns Hopkins Hospital in
internal medicine and subsequently received a fellowship from Johns Hopkins Hospital in
endocrinology and metabolism.
Mr. Paul Bavier has served as our general counsel and secretary since December 2008. From
October 2007 to December 2008, Mr. Bavier served as our deputy general counsel. From November 2004
to October 2007, Mr. Bavier served as assistant general counsel at Gerber Scientific, Inc. Mr.
Bavier began his legal career as an associate in the corporate law department of Ropes & Gray in
Boston. He holds a B.A. from Middlebury College and a J. D. from the University of Michigan Law
School.
14
Table of Contents
PART II-OTHER INFORMATION
ITEM 1A. RISK FACTORS
Risks Related to Our Financial Position and Need for Additional Capital
We have incurred significant losses since our inception. We expect to incur losses for the
foreseeable future and may never achieve or maintain profitability.
Since our inception in December 2003, we have incurred significant operating losses. Our net
loss was approximately $43.3 million for the year ended September 30, 2009. As of September 30,
2009, we had a deficit accumulated during the development stage of approximately $126.5 million. We
have invested a significant portion of our efforts and financial resources in the development of
VIAject®, and our ability to generate near-term revenue depends on VIAject’s® success. We have not
completed development of VIAject® or any of our other product candidates. We expect to continue to
incur significant operating losses for at least the next several years as we:
| • | submit our NDA for VIAject® in December 2009; | ||
| • | continue our clinical trial extension program for the pivotal Phase 3 clinical trials of VIAject®, which is designed to run through February 2010; | ||
| • | conduct additional clinical trials of VIAject® to support our commercialization efforts and, potentially, FDA approval; | ||
| • | produce required validation batches of VIAject® vials, cartridges, and disposable pens to support our NDA for VIAject®; | ||
| • | purchase recombinant human insulin and other materials to build commercial supply inventory for VIAject®; and | ||
| • | conduct additional clinical development of our other product candidates. |
To become and remain profitable, we must succeed in developing and eventually commercializing
drugs with significant market potential. This will require us to be successful in a range of
challenging activities, including successfully completing preclinical testing and clinical trials
of our product candidates, obtaining regulatory approval for these product candidates and
manufacturing, marketing and selling those products for which we may obtain regulatory approval. We
may never succeed in these activities and may never generate revenues that are significant or large
enough to achieve profitability. Even if we do achieve profitability, we may not be able to sustain
or increase profitability on a quarterly or annual basis. Our failure to become and remain
profitable could depress the market price of our common stock and could impair our ability to raise
capital, expand our business or continue our operations. A decline in the market price of our
common stock could also cause you to lose all or a part of your investment.
We will need substantial additional funding and may be unable to raise capital when needed, which
would force us to delay, reduce or eliminate our product development programs or commercialization
efforts.
We are a development stage company with no commercial products. All of our product candidates
are still being developed, and all but VIAject® are in early stages of development. Our product
candidates will require significant additional clinical development, regulatory approvals and
related investment before they can be commercialized.
While we have suspended significant expenditures on our earlier stage product candidates
pending further development of our regulatory plans for VIAject®, we do not expect our research and
development expenses to decrease in 2010 as we continue our Phase 3 clinical trial extension
program for VIAject®, conduct new clinical trials of VIAject® to support our commercialization
efforts, produce validation batches, and purchase recombinant human insulin. In addition, subject
to obtaining regulatory approval of any of our product candidates, we expect to incur significant
commercialization expenses to produce commercial quantities of finished product; and we may incur
significant additional sales and marketing expenses depending on our role in commercializing any of
our products that obtain regulatory approval. If we commercialize VIAject® without a commercial
partner, we will need substantial additional funding and may be unable to raise capital when needed
or on attractive terms, which would force us to delay, reduce or eliminate our research and
development programs or commercialization efforts.
Based upon our current plans, we believe that our existing cash, cash equivalents and
marketable securities will enable us to fund our anticipated operating expenses and capital
expenditures at least through the second quarter of fiscal year 2011. We cannot assure you that our
plans will not change or that changed circumstances will not result in the depletion of our capital
resources more rapidly than we currently anticipate. Our future capital requirements will depend on
many factors, including:
| • | our ability to secure approval by the FDA for VIAject® under Section 505(b)(2) of the FFDCA; | ||
| • | our ability to file our NDA for VIAject® in December 2009 as planned and, once filed, the length of time that will elapse before our NDA is fully reviewed by the FDA; | ||
| • | the costs associated with preparing and submitting our Market Authorization Application, or MAA, for VIAject® to the European Medicines Agency, or EMEA; | ||
| • | our ability to market, commercialize and achieve market acceptance for product candidates, particularly VIAject®; |
15
Table of Contents
| • | our ability to secure approval by the FDA for VIAject® without conducting additional pivotal clinical trials; | ||
| • | the FDA’s findings regarding data anomalies observed in India in our Phase 3 clinical trial of VIAject® for patients with Type 1 diabetes and the impact of those findings on the timing of a regulatory approval; | ||
| • | the size, endpoints and duration of additional clinical trials of VIAject® in patients with Type 1 diabetes to support our commercialization efforts and, potentially, FDA approval; | ||
| • | the cost to fully develop the 100 IU/cc liquid formulation of VIAject®; | ||
| • | our ability to establish that the 100 IU/cc liquid formulation of VIAject® is well-tolerated in chronic use; | ||
| • | the cost to develop an insulin pen program for use with VIAject®; | ||
| • | the cost of purchasing recombinant human insulin and other materials to build commercial supply inventory for VIAject®, taking into account currency exchange rate fluctuations; | ||
| • | the costs of preparing, filing and prosecuting patent applications and maintaining, enforcing and defending intellectual property-related claims; | ||
| • | the cost associated with qualifying and obtaining regulatory approval of suppliers of insulin and manufacturers of our product candidates; | ||
| • | our ability to establish and maintain collaborations and the terms and success of the collaborations, including the timing and amount of payments that we might receive from potential strategic collaborators; and | ||
| • | the continued participation of patients in our VIAject® Phase 3 clinical trial extension program, which is designed to run through February 2010. |
Until such time, if ever, as we can generate substantial product revenues, we expect to
finance our cash needs through public or private equity offerings and debt financings, strategic
collaborations and licensing arrangements. If we raise additional funds by issuing additional
equity securities, our stockholders will experience dilution. Debt financing, if available, may
involve agreements that include covenants limiting or restricting our ability to take specific
actions, such as incurring additional debt, making capital expenditures or declaring dividends. Any
debt financing or additional equity that we raise may contain terms, such as liquidation and other
preferences, that are not favorable to us or our stockholders. If we raise additional funds through
collaboration, strategic alliance and licensing arrangements with third parties, it may be
necessary to relinquish valuable rights to our technologies or product candidates, future revenue
streams, research programs or product candidates or to grant licenses on terms that may not be
favorable to us.
Our short operating history may make it difficult for you to evaluate the success of our business
to date and to assess our future viability.
We commenced active operations in January 2004. Our operations to date have been limited to
organizing and staffing our company, developing and securing our technology and undertaking
preclinical studies and clinical trials of our product candidates. We have limited experience
completing large-scale, pivotal clinical trials and we have not yet demonstrated our ability to
successfully file an NDA, obtain regulatory approvals, manufacture a commercial scale product, or
arrange for a third party to do so on our behalf, or conduct sales and marketing activities
necessary for successful product commercialization. Consequently, any predictions you make about
our future success or viability may not be as accurate as they could be if we had a longer
operating history.
In addition, as a new business, we may encounter unforeseen expenses, difficulties,
complications, delays and other known and unknown factors. We will need to transition from a
company with a research focus to a company capable of supporting commercial activities. We may not
be successful in such a transition.
Risks Related to the Development and Commercialization of Our Product Candidates
We depend heavily on the success of our most advanced product candidate, VIAject®. The results
from our completed pivotal Phase 3 clinical trials of VIAject® may not be sufficient to file an
NDA for VIAject® or to obtain marketing approval from the FDA. If we are unable to commercialize
VIAject® or experience significant delays in doing so, our business will be materially harmed.
We have invested a significant portion of our efforts and financial resources in the
development of our most advanced product candidate, VIAject®. Our ability to generate significant
product revenues will depend heavily on the successful development and eventual commercialization
of this product candidate. The results from our completed pivotal Phase 3 clinical trials of
VIAject® may not be sufficient to file an NDA for VIAject® or obtain marketing approval from the
FDA. Due to data from India in our pivotal Phase 3 clinical trial for patients with Type 1
diabetes that we found to be anomalous when compared to data from the United States and Germany for
the same trial, the FDA could conclude we did not establish non-inferiority of VIAject® when
compared to Humulin® R in terms of blood glucose control. The FDA may require that we conduct
additional clinical trials with VIAject® before
considering or approving our marketing application. Even if it is determined that no
additional clinical trials will be required, we anticipate that VIAject® would not be commercially
available for at least the next 18 months, if at all.
16
Table of Contents
We may never reinitiate significant expenditures on our earlier stage product candidates.
We have suspended significant expenditures on the development of VIAtab™ and are unlikely to
reinitiate extensive development of our early stage product candidates unless we receive marketing
approval for VIAject® from the FDA. Even if VIAject® is approved by the FDA, we cannot guarantee
that we will have sufficient resources to allocate to earlier stage product candidates or that the
focus of our early stage product development program will not have changed.
The results of clinical trials do not ensure success in future clinical trials or commercial success.
We have completed and released the results of our two pivotal Phase 3 clinical trials of
VIAject®. Additionally, we have tested one formulation of VIAtab™ in Phase 1 clinical trials in
patients with Type 1 diabetes and are developing additional formulations for further clinical
testing. We have not completed the development of any products through commercialization. VIAject®
continues to be tested in our Phase 3 clinical trial extension program and we believe we will need
to conduct additional clinical trials to be successful in our commercialization efforts and,
potentially, in order to receive FDA approval. The outcomes of preclinical testing and clinical
trials may not be predictive of the success of later clinical trials. Furthermore, interim or
preliminary results of a clinical trial do not necessarily predict final results. We cannot assure
you that our additional clinical trials of VIAject® will ultimately be successful. New information
regarding the safety, efficacy and tolerability of VIAject® may arise that may be less favorable
than the data observed to date. In addition, we will need to conduct Phase 2 and Phase 3 clinical
trials of VIAtab™ in larger numbers of patients taking the drug for longer periods before we are
able to seek approvals to market and sell VIAtab™ from the FDA and similar regulatory authorities
outside the United States. If we are not successful in commercializing any of our product
candidates, or are significantly delayed in doing so, our business will be materially harmed. The
commercial success of our product candidates will depend on several factors, including the
following:
| • | successful completion of preclinical development and clinical trials; | ||
| • | our ability to identify and enroll patients who meet clinical trial eligibility criteria; | ||
| • | receipt of marketing approvals from the FDA and similar regulatory authorities outside the United States, including for the liquid formulation of VIAject®; | ||
| • | establishing that our liquid formulation of VIAject® is well-tolerated in chronic use; | ||
| • | establishing commercial manufacturing capabilities through arrangements with third-party manufacturers; | ||
| • | launching commercial sales of the products, whether alone or in collaboration with others; | ||
| • | competition from other products; and | ||
| • | a continued acceptable safety and tolerability profile of the products following approval. |
If the FDA does not approve our NDA, or if our clinical trials are delayed or do not produce
positive results, we may incur additional costs and ultimately be unable to commercialize our
product candidates.
Before obtaining regulatory approval for the sale of our product candidates, we must conduct,
at our own expense, extensive preclinical tests to demonstrate the safety of our product candidates
in animals and clinical trials to demonstrate the safety and efficacy of our product candidates in
humans. Preclinical and clinical testing is expensive, difficult to design and implement, can take
many years to complete and is uncertain as to outcome. A failure of one or more of our clinical
trials of VIAject® and VIAtab™ can occur at any stage of testing. We may experience numerous
unforeseen events during clinical trials of VIAject® and VIAtab™ or during the FDA’s review of our
planned NDA that could delay or prevent our ability to receive regulatory approval or commercialize
our product candidates, including:
| • | the FDA may require us to conduct additional clinical trials of VIAject® prior to accepting our NDA for filing or following its review of our NDA based on data from India in one of our pivotal Phase 3 clinical trials that we found to be anomalous when compared to data from the United States and Germany for the same clinical trial; | ||
| • | the number of patients required for our clinical trials may be larger than we anticipate, enrollment in our clinical trials may be slower than we currently anticipate, or participants may drop out of our clinical trials at a higher rate than we anticipate, any of which would result in significant delays; | ||
| • | our third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner; | ||
| • | we might have to suspend or terminate our clinical trials if the participants are being exposed to unacceptable health risks; | ||
| • | regulators or institutional review boards may require that we hold, suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements; | ||
| • | the cost of our clinical trials may be greater than we anticipate; |
17
Table of Contents
| • | the supply or quality of our product candidates or other materials necessary to conduct our clinical trials may be insufficient or inadequate; and | ||
| • | the effects of our product candidates may not be the desired effects or may include undesirable side effects or the product candidates may have other unexpected characteristics. |
If we are required to conduct additional clinical trials or other testing of our product
candidates beyond those that we currently contemplate, if we are unable to successfully complete
our clinical trials or other testing, if the results of these trials or tests are not positive or
are only modestly positive or if there are safety concerns, we may:
| • | be delayed in obtaining marketing approval for our product candidates; | ||
| • | not be able to obtain marketing approval; | ||
| • | obtain approval for indications that are not as broad as intended; or | ||
| • | have the product removed from the market after obtaining marketing approval. |
Our product development costs will also increase if we experience delays in testing or
approvals. We do not know whether any preclinical tests or clinical trials will begin as planned,
will need to be restructured or will be completed on schedule, if at all. Significant preclinical
or clinical trial delays also could shorten any periods during which we may have the exclusive
right to commercialize our product candidates or allow our competitors to bring products to market
before we do and impair our ability to commercialize our products or product candidates and may
harm our business and results of operations.
If our product candidates are found to cause undesirable side effects we may need to delay or
abandon our development and commercialization efforts.
Any undesirable side effects that might be caused by our product candidates could interrupt,
delay or halt clinical trials and could result in the denial of regulatory approval by the FDA or
other regulatory authorities for any or all targeted indications. In addition, if any of our
product candidates receive marketing approval and we or others later identify undesirable side
effects caused by the product, we could face one or more of the following:
| • | a change in the labeling statements or withdrawal of FDA or other regulatory approval of the product; | ||
| • | a change in the way the product is administered; or | ||
| • | the need to conduct additional clinical trials. |
Any of these events could prevent us from achieving or maintaining market acceptance of the
affected product or could substantially increase the costs and expenses of commercializing the
product, which in turn could delay or prevent us from generating significant revenues from its
sale.
In our analysis of our completed pivotal Phase 3 clinical trials we found that VIAject® was
associated with injection site discomfort, although the prevalence of discomfort decreased during
the course of the treatment. In addition, in an October 2009 tolerability trial of the liquid
formulation of VIAject® it was determined that a subset of patients experienced more injection site
discomfort with VIAject® than they did with Humalog®.
The commercial success of any product candidates that we may develop, including VIAject® and
VIAtab™ will depend upon the degree of market acceptance by physicians, patients, healthcare
payors and others in the medical community.
Any products that we bring to the market, including VIAject® and VIAtab™, if they receive
marketing approval, may not gain market acceptance by physicians, patients, healthcare payors and
others in the medical community. If these products do not achieve an adequate level of acceptance,
we may not generate significant product revenues and we may not become profitable. Physicians will
not recommend our product candidates until clinical data or other factors demonstrate the safety
and efficacy of our product candidates as compared to other treatments. Even if the clinical safety
and efficacy of our product candidates is established, physicians may elect not to recommend these
product candidates for a variety of reasons including the reimbursement policies of government and
third-party payors, the effectiveness of our competitors in marketing their products and, in the
case of VIAject®, the possibility that patients may experience more injection site discomfort than
they experience with competing products.
The degree of market acceptance of our product candidates, if approved for commercial sale,
will depend on a number of factors, including:
| • | the willingness and ability of patients and the healthcare community to adopt our technology; | ||
| • | the ability to manufacture our product candidates in sufficient quantities with acceptable quality and to offer our product candidates for sale at competitive prices; | ||
| • | the perception of patients and the healthcare community, including third-party payors, regarding the safety, efficacy and benefits of our product candidates compared to those of competing products or therapies; |
18
Table of Contents
| • | the convenience and ease of administration of our product candidates relative to existing treatment methods, such as our ability to gain regulatory approval for our liquid 100 IU/cc formulation of VIAject® and the degree to which injection site discomfort may be associated with this formulation; | ||
| • | the label and promotional claims allowed by the FDA, such as, in the case of VIAject®, claims relating to glycemic control, hypoglycemia, weight gain, injection site discomfort, expiry dating and required handling conditions; | ||
| • | the pricing and reimbursement of our product candidates relative to existing treatments; and | ||
| • | marketing and distribution support for our product candidates. |
If we fail to enter into strategic collaborations for the commercialization of our product
candidates or if our collaborations are unsuccessful, we may be required to establish our own
sales, marketing, manufacturing and distribution capabilities which will be expensive, require
additional capital we do not currently have, and could delay the commercialization of our product
candidates and have a material and adverse affect on our business.
A broad base of physicians, including primary care physicians, internists and
endocrinologists, treat patients with diabetes. A large sales force may be required to educate and
support these physicians. Therefore, our current strategy for developing, manufacturing and
commercializing our product candidates includes securing collaborations with leading pharmaceutical
and biotechnology companies for the commercialization of our product candidates. To date, we have
not entered into any collaborations with pharmaceutical or biotechnology companies. We face
significant competition in seeking appropriate collaborators. In addition, collaboration agreements
are complex and time-consuming to negotiate, document and implement. For all these reasons, it may
be difficult for us to find third parties that are willing to enter into collaborations on economic
terms that are favorable to us, or at all. If we do enter into any such collaboration, the
collaboration may not be successful. The success of our collaboration arrangements will depend
heavily on the efforts and activities of our collaborators. It is likely that our collaborators
will have significant discretion in determining the efforts and resources that they will apply to
these collaborations.
If we fail to enter into collaborations, or if our collaborations are unsuccessful, we may be
required to establish our own direct sales, marketing, manufacturing and distribution capabilities.
Establishing these capabilities can be time-consuming and expensive and we have little experience
in doing so. Because of our size, we would be at a disadvantage to our potential competitors to the
extent they collaborate with large pharmaceutical companies that have substantially more resources
than we do. As a result, we would not initially be able to field a sales force as large as our
competitors or provide the same degree of market research or marketing support. In addition, our
competitors would have a greater ability to devote research resources toward expansion of the
indications for their products. We cannot assure prospective investors that we will succeed in
entering into acceptable collaborations, that any such collaboration will be successful or, if not,
that we will successfully develop our own sales, marketing and distribution capabilities.
If we are unable to obtain adequate reimbursement from governments or third-party payors for any
products that we may develop or if we are unable to obtain acceptable prices for those products,
they may not be purchased or used and our revenues and prospects for profitability will suffer.
Our future revenues and profits will depend heavily upon the availability of adequate
reimbursement for the use of our approved product candidates from governmental and other
third-party payors, both in the United States and in other markets. Reimbursement by a third-party
payor may depend upon a number of factors, including the third-party payor’s determination that use
of a product is:
| • | a covered benefit under its health plan; | ||
| • | safe, effective and medically necessary; | ||
| • | appropriate for the specific patient; | ||
| • | cost-effective; and | ||
| • | neither experimental nor investigational. |
Obtaining reimbursement approval for a product from each government or other third-party payor
is a time-consuming and costly process that could require us to provide supporting scientific,
clinical and cost-effectiveness data for the use of our products to each payor. We may not be able
to provide data sufficient to gain acceptance with respect to reimbursement. Even when a payor
determines that a product is eligible for reimbursement, the payor may impose coverage limitations
that preclude payment for some uses that are approved by the FDA or comparable authorities. In
addition, eligibility for coverage does not imply that any product will be reimbursed in all cases
or at a rate that allows us to make a profit or even cover our costs. Interim payments for new
products, if applicable, may also not be sufficient to cover our costs and may not be made
permanent.
We are subject to pricing pressures and uncertainties regarding Medicare reimbursement and reform.
Reforms in Medicare added a prescription drug reimbursement benefit beginning in 2006 for all
Medicare beneficiaries. Although we cannot predict the full effects on our business of the
implementation of this legislation, it is possible that the new benefit, which will be managed by
private health insurers, pharmacy benefit managers, and other managed care organizations, will
result in decreased reimbursement for prescription drugs, which may further exacerbate
industry-wide pressure to reduce the prices charged for prescription drugs. This could harm our
ability to generate revenues.
19
Table of Contents
Governments outside the United States tend to impose strict price controls, which may adversely
affect our revenues, if any.
In some countries, particularly the countries of the European Union, the pricing of
prescription pharmaceuticals is subject to governmental control. In these countries, pricing
negotiations with governmental authorities can take considerable time after the receipt of
marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we
may be required to conduct a clinical trial that compares the cost-effectiveness of our product
candidate to other available therapies. If reimbursement of our products is unavailable or limited
in scope or amount, or if pricing is set at unsatisfactory levels, our business could be adversely
affected.
Legislation has been introduced in Congress that, if enacted, would permit more widespread
re-importation of drugs from foreign countries into the United States, which may include
re-importation from foreign countries where the drugs are sold at lower prices than in the United
States. Such legislation, or similar regulatory changes, could decrease the price we receive for
any approved products which, in turn, could adversely affect our operating results and our overall
financial condition.
Product liability lawsuits against us could cause us to incur substantial liabilities and to limit
commercialization of any products that we may develop.
We face an inherent risk of product liability exposure related to the testing of our product
candidates in human clinical trials and will face an even greater risk if we commercially sell any
products that we may develop. If we cannot successfully defend ourselves against claims that our
product candidates or products caused injuries, we will incur substantial liabilities. Regardless
of merit or eventual outcome, liability claims may result in:
| • | decreased demand for any product candidates or products that we may develop; | ||
| • | injury to our reputation; | ||
| • | withdrawal of clinical trial participants; | ||
| • | costs to defend the related litigation; | ||
| • | substantial monetary awards to trial participants or patients; | ||
| • | loss of revenue; and | ||
| • | the inability to commercialize any products that we may develop. |
We currently carry global liability insurance that we believe is sufficient to cover us from
potential damages arising from clinical trials of VIAject®. We also carry local insurance policies
per clinical trial of our product candidates. The amount of insurance that we currently hold may
not be adequate to cover all liabilities that we may incur. We intend to expand our insurance
coverage to include the sale of commercial products if we obtain marketing approval for any
products. Insurance coverage is increasingly expensive. We may not be able to maintain insurance
coverage at a reasonable cost. If losses from product liability claims exceed our liability
insurance coverage, we may ourselves incur substantial liabilities. If we are required to pay a
product liability claim, we may not have sufficient financial resources to complete development or
commercialization of any of our product candidates and, if so, our business and results of
operations would be harmed.
We face substantial competition in the development of our product candidates which may result in
others developing or commercializing products before or more successfully than we do.
We are engaged in segments of the pharmaceutical industry that are characterized by intense
competition and rapidly evolving technology. Many large pharmaceutical and biotechnology companies,
academic institutions, governmental agencies and other public and private research organizations
are pursuing the development of novel drugs that target endocrine disorders. We face, and expect to
continue to face, intense and increasing competition as new products enter the market and advanced
technologies become available. There are several approved injectable rapid-acting mealtime insulin
analogs currently on the market including Humalog® , marketed by Eli Lilly and Company, NovoLog® ,
marketed by Novo Nordisk A/S, and Apidra® , marketed by Sanofi-Aventis. These rapid-acting insulin
analogs provide improvement over regular forms of short-acting insulin, including faster
subcutaneous absorption, an earlier and greater insulin peak and more rapid post-peak decrease. In
addition, other development stage rapid-acting insulin formulations may be approved and compete
with VIAject®. Halozyme Therapeutics, Inc. has conducted a Phase 1 and Phase 2 clinical trial of
Humulin® R and Humalog® in combination with a recombinant human hyaluronidase enzyme and has
reported that in each case the combination yielded pharmacokinetics and glucodynamics that better
mimicked physiologic mealtime insulin release and activity than Humulin® R or Humalog® alone.
Generex Biotechnology Corporation has developed an oral spray that is currently in Phase 3
development. Several companies are also developing alternative insulin systems for diabetes,
including MannKind Corporation, which has submitted its NDA in early 2009. In addition, a number of
established pharmaceutical companies, including GlaxoSmithKline plc, and Bristol-Myers Squibb
Company, are developing proprietary technologies or have entered into arrangements with, or
acquired, companies with technologies for the treatment of diabetes.
Potential competitors also include academic institutions, government agencies and other public
and private research organizations that conduct research, seek patent protection and establish
collaborative arrangements for research, development, manufacturing and commercialization. Our
competitors may develop products that are more effective, safer, more convenient or less costly
than any that
20
Table of Contents
we are developing or that would render our product candidates obsolete or non-competitive. Our
competitors may also obtain FDA or other regulatory approval for their products more rapidly than
we may obtain approval for ours.
Many of our potential competitors have:
| • | significantly greater financial, technical and human resources than we have and may be better equipped to discover, develop, manufacture and commercialize product candidates; | ||
| • | more extensive experience in preclinical testing and clinical trials, obtaining regulatory approvals and manufacturing and marketing pharmaceutical products; | ||
| • | product candidates that have been approved or are in late-stage clinical development; or | ||
| • | collaborative arrangements in our target markets with leading companies and research institutions. |
Our product candidates may be rendered obsolete by technological change.
The rapid rate of scientific discoveries and technological changes could result in one or more
of our product candidates becoming obsolete or noncompetitive. For several decades, scientists have
attempted to improve the bioavailability of injected formulations and to devise alternative
non-invasive delivery systems for the delivery of drugs such as insulin. Our product candidates
will compete against many products with similar indications. In addition to the currently marketed
rapid-acting insulin analogs, our competitors are developing insulin formulations delivered by oral
pills, pulmonary devices and oral spray devices. Our future success will depend not only on our
ability to develop our product candidates, but also on our ability to maintain market acceptance
against emerging industry developments. We cannot assure present or prospective stockholders that
we will be able to do so.
Our business activities involve the storage and use of hazardous materials, which require
compliance with environmental and occupational safety laws regulating the use of such materials.
If we violate these laws, we could be subject to significant fines, liabilities or other adverse
consequences.
Our research and development work and manufacturing processes involve the controlled storage
and use of hazardous materials, including chemical and biological materials. Our operations also
produce hazardous waste products. We are subject to federal, state and local laws and regulations
governing the use, manufacture, storage, handling and disposal of these materials. Although we
believe that our safety procedures for handling and disposing of such materials and waste products
comply in all material respects with the standards prescribed by federal, state and local laws and
regulations, the risk of accidental contamination or injury from hazardous materials cannot be
completely eliminated. In the event of an accident or failure to comply with environmental laws, we
could be held liable for any damages that may result, and any such liability could fall outside the
coverage or exceed the limits of our insurance. In addition, we could be required to incur
significant costs to comply with environmental laws and regulations in the future or pay
substantial fines or penalties if we violate any of these laws or regulations. Finally, current or
future environmental laws and regulations may impair our research, development or production
efforts.
Risks Related to Our Dependence on Third Parties
Use of third parties to manufacture our product candidates may increase the risks that we will not
have sufficient quantities of our product candidates or such quantities at an acceptable cost, or
that our suppliers will not be able to manufacture our products in their final dosage form. In any
such case, clinical development and commercialization of our product candidates could be delayed,
prevented or impaired.
We do not currently own or operate manufacturing facilities for commercial production of our
product candidates. We have limited experience in drug manufacturing and we lack the resources and
the capabilities to manufacture any of our product candidates on a clinical or commercial scale.
Our current strategy is to outsource all manufacturing of our product candidates and products to
third parties. We also expect to rely upon third parties to produce materials required for the
commercial production of our product candidates if we succeed in obtaining necessary regulatory
approvals. We currently rely on one manufacturer to manufacture our VIAject® product candidate. We
intend to negotiate a commercial manufacturing agreement with this manufacturer but cannot
guarantee that we will reach agreement in a timely manner or on terms that are favorable to us.
There can be no assurance that this manufacturer will support our VIAject® or other
manufacturing programs in the future. The manufacture of pharmaceutical products requires
significant expertise and capital investment, including the development of advanced manufacturing
techniques, processes and quality controls.
Reliance on third-party manufacturers entails risks to which we would not be subject if we
manufactured product candidates or products ourselves, including:
| • | reliance on the third party for regulatory compliance and quality assurance; | ||
| • | the possible breach of the manufacturing agreement by the third party because of factors beyond our control; and | ||
| • | the possible refusal by the third party to support our manufacturing programs, based on its own business priorities, at a time that is costly or inconvenient for us. |
21
Table of Contents
Our manufacturers may not be able to comply with current good manufacturing practice, or cGMP,
regulations or other regulatory requirements or similar regulatory requirements outside the United
States. Our manufacturers are subject to unannounced inspections by the FDA, state regulators and
similar regulators outside the United States. Our failure, or the failure of our third-party
manufacturers, to comply with applicable regulations could result in sanctions being imposed on us,
including fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing
approval of our product candidates, delays, suspension or withdrawal of approvals, license
revocation, seizures or recalls of product candidates or products, operating restrictions and
criminal prosecutions, any of which could significantly and adversely affect supplies of our
product candidates.
Our product candidates and any products that we may develop may compete with other product
candidates and products for access to manufacturing facilities. There are a limited number of
manufacturers that operate under cGMP regulations and that are both capable of manufacturing for us
and willing to do so. If the third parties that we engage to manufacture product for our clinical
trials should cease to continue to do so for any reason, we likely would experience delays in
advancing these trials while we identify and qualify replacement suppliers and we may be unable to
obtain replacement supplies on terms that are favorable to us. In addition, if we are not able to
obtain adequate supplies of our product candidates or the drug substances used to manufacture them,
it will be more difficult for us to develop our product candidates and compete effectively.
Our current and anticipated future dependence upon others for the manufacture of our product
candidates may adversely affect our future profit margins and our ability to develop product
candidates and commercialize any products that receive regulatory approval on a timely and
competitive basis.
We rely on third parties to conduct our clinical trials and those third parties may not perform
satisfactorily, including failing to meet established deadlines for the completion of such trials.
We do not independently conduct clinical trials of our product candidates. We rely on third
parties, such as contract research organizations, clinical data management organizations, medical
institutions and clinical investigators, to enroll qualified patients and conduct our clinical
trials. Our reliance on these third parties for clinical development activities reduces our control
over these activities. We are responsible for ensuring that each of our clinical trials is
conducted in accordance with the general investigational plan and protocols for the trial.
Moreover, the FDA requires us to comply with standards, commonly referred to as Good Clinical
Practices, for conducting, recording, and reporting the results of clinical trials to assure that
data and reported results are credible and accurate and that the rights, integrity and
confidentiality of trial participants are protected. Our reliance on third parties that we do not
control does not relieve us of these responsibilities and requirements. Furthermore, these third
parties may also have relationships with other entities, some of which may be our competitors. If
these third parties do not successfully carry out their contractual duties, meet expected deadlines
or conduct our clinical trials in accordance with regulatory requirements or our stated protocols,
we will not be able to obtain, or may be delayed in obtaining, regulatory approvals for our product
candidates and will not be able to, or may be delayed in our efforts to, successfully commercialize
our product candidates.
If our suppliers, principally our sole insulin supplier, fail to deliver materials and provide
services needed for the production of VIAject® and VIAtab™ in a timely and sufficient manner, or
if they fail to comply with applicable regulations, clinical development or regulatory approval of
our product candidates or commercialization of our products could be delayed, producing additional
losses and depriving us of potential product revenue.
We need access to sufficient, reliable and affordable supplies of recombinant human insulin
and other materials for which we rely on various suppliers. We also must rely on those suppliers to
comply with relevant regulatory and other legal requirements, including the production of insulin
in accordance with cGMP. We can make no assurances that our suppliers, particularly our insulin
supplier, will comply with cGMP.
We have recently entered into a new agreement with our existing single insulin supplier from
which we obtain all of the insulin that we use for testing and manufacturing VIAject® and VIAtab™.
Our agreement with this insulin supplier will terminate in December 2011. We are discussing the
possibility of extending this supply agreement beyond 2011, but we cannot guarantee that this
effort will be successful.
We believe that our current supplies of insulin, together with the quantities of insulin
called for under our existing supply agreement, will be sufficient to allow us to complete our
current clinical trial extension program and anticipated future clinical trials of VIAject®. In
addition, we believe that the quantities available under the agreement will be sufficient to
support our needs for approximately three years following the commercial launch of VIAject®. We are
seeking to qualify other insulin suppliers to serve as additional or alternative suppliers if we
are unable or choose not to enter into a new commercial supply agreement with our existing
supplier. We cannot assure you that we will be able to qualify a new insulin supplier prior to
December 2011. Even if we do qualify a new supplier in a timely manner, the cost of switching or
adding additional suppliers may be significant, and we cannot assure you that we will be able to
enter into a commercial supply agreement with a new supplier on favorable terms. If we are unable
to procure sufficient quantities of insulin from our current or any future supplier, if supply of
recombinant human insulin and other materials otherwise becomes limited, or if our suppliers do not
meet relevant regulatory requirements, and if we were unable to obtain these materials in
sufficient amounts, in a timely manner and at reasonable prices, we could be delayed in the
manufacturing and future commercialization of VIAject® and VIAtab™, which would have a material
adverse effect on our business. We would incur substantial costs and manufacturing delays if our
suppliers are unable to provide us with products or services approved by the FDA or other
regulatory agencies.
22
Table of Contents
Risks Related to Our Intellectual Property
If we are unable to protect our intellectual property rights, our competitors may develop and
market similar or identical products that may reduce demand for our products, and we may be
prevented from establishing collaborative relationships on favorable terms.
The following factors are important to our success:
| • | receiving patent protection for our product candidates; | ||
| • | maintaining our trade secrets; | ||
| • | not infringing on the proprietary rights of others; and | ||
| • | preventing others from infringing our proprietary rights. |
We will be able to protect our proprietary rights from unauthorized use by third parties only
to the extent that our proprietary rights are covered by valid and enforceable patents or are
effectively maintained as trade secrets. We try to protect our proprietary position by filing U.S.
and foreign patent applications related to our proprietary technology, inventions and improvements
that are important to the development of our business. Because the patent position of
pharmaceutical companies involves complex legal and factual questions, the issuance, scope and
enforceability of patents cannot be predicted with certainty. Patents, if issued, may be
challenged, invalidated or circumvented. Thus, any patents that we own or license from others may
not provide any protection against competitors.
We have been granted one U.S. patent and one European patent in all of the designation
countries, several pending United States patent applications relating to our VIAject® and VIAtab™
technology and several pending U.S. foreign patent applications relating to our technology for
enhancing delivery of drugs. These pending patent applications, those we may file in the future, or
those we may license from third parties, may not result in patents being issued. If patents do not
issue with claims encompassing our products, our competitors may develop and market similar or
identical products that compete with ours. Even if patents are issued, they may not provide us with
proprietary protection or competitive advantages against competitors with similar technology.
Failure to obtain effective patent protection for our technology and products may reduce demand for
our products and prevent us from establishing collaborative relationships on favorable terms.
The active and inactive ingredients in our VIAject® and VIAtab™ product candidates have been
known and used for many years and, therefore, are no longer subject to patent protection.
Accordingly, our granted U.S. and foreign patents and pending patent applications are directed to
the particular formulations of these ingredients in our products, and their use. Although we
believe our formulations and their use are patentable and provide a competitive advantage, even if
issued, our patents may not prevent others from marketing formulations using the same active and
inactive ingredients in similar but different formulations.
We also rely on trade secrets, know-how and technology, which are not protected by patents, to
maintain our competitive position. We try to protect this information by entering into
confidentiality agreements with parties that have access to it, such as potential corporate
partners, collaborators, employees and consultants. Any of these parties may breach the agreements
and disclose our confidential information or our competitors may learn of the information in some
other way. Furthermore, others may independently develop similar technologies or duplicate any
technology that we have developed. If any trade secret, know-how or other technology not protected
by a patent were to be disclosed to or independently developed by a competitor, our business and
financial condition could be materially adversely affected.
The laws of many foreign countries do not protect intellectual property rights to the same
extent as do the laws of the United States.
We may become involved in lawsuits and administrative proceedings to protect, defend or enforce
our patents that would be expensive and time-consuming.
In order to protect or enforce our patent rights, we may initiate patent litigation against
third parties in the United States or in foreign countries. In addition, we may be subject to
certain opposition proceedings conducted in patent and trademark offices challenging the validity
of our patents and may become involved in future opposition proceedings challenging the patents of
others. The defense of intellectual property rights, including patent rights, through lawsuits,
interference or opposition proceedings, and other legal and administrative proceedings can be
costly and can divert our technical and management personnel from their normal responsibilities.
Such costs increase our operating losses and reduce our resources available for development
activities. An adverse determination of any litigation or defense proceedings could put one or more
of our patents at risk of being invalidated or interpreted narrowly and could put our patent
applications at risk of not issuing.
Furthermore, because of the substantial amount of discovery required in connection with
intellectual property litigation, there is a risk that some of our confidential information could
be compromised by disclosure during this type of litigation. For example, during the course of this
kind of litigation and despite protective orders entered by the court, confidential information may
be inadvertently disclosed in the form of documents or testimony in connection with discovery
requests, depositions or trial testimony. This disclosure could materially adversely affect our
business and financial results.
23
Table of Contents
Claims by other parties that we infringe or have misappropriated their proprietary technology may
result in liability for damages, royalties, or other payments, or stop our development and
commercialization efforts.
Competitors and other third parties may initiate patent litigation against us in the United
States or in foreign countries based on existing patents or patents that may be granted in the
future. Many of our competitors may have obtained patents covering products and processes generally
related to our products and processes, and they may assert these patents against us. Moreover,
there can be no assurance that these competitors have not sought or will not seek additional
patents that may cover aspects of our technology. As a result, there is a greater likelihood of a
patent dispute than would be expected if our competitors were pursuing unrelated technologies.
While we conduct patent searches to determine whether the technologies used in our products
infringe patents held by third parties, numerous patent applications are currently pending and may
be filed in the future for technologies generally related to our technologies, including many
patent applications that remain confidential after filing. Due to these factors and the inherent
uncertainty in conducting patent searches, there can be no guarantee that we will not violate
third-party patent rights that we have not yet identified.
There may be U.S. and foreign patents issued to third parties that relate to aspects of our
product candidates. There may also be patent applications filed by these or other parties in the
United States and various foreign jurisdictions that relate to some aspects of our product
candidates, which, if issued, could subject us to infringement actions. The owners or licensees of
these and other patents may file one or more infringement actions against us. In addition, a
competitor may claim misappropriation of a trade secret by an employee hired from that competitor.
Any such infringement or misappropriation action could cause us to incur substantial costs
defending the lawsuit and could distract our management from our business, even if the allegations
of infringement or misappropriation are unwarranted. A need to defend multiple actions or claims
could have a disproportionately greater impact. In addition, either in response to or in
anticipation of any such infringement or misappropriation claim, we may enter into commercial
agreements with the owners or licensees of these rights. The terms of these commercial agreements
may include substantial payments, including substantial royalty payments on revenues received by us
in connection with the commercialization of our products.
Payments under such agreements could increase our operating losses and reduce our resources
available for development activities. Furthermore, a party making this type of claim could secure a
judgment that requires us to pay substantial damages, which would increase our operating losses and
reduce our resources available for development activities. A judgment could also include an
injunction or other court order that could prevent us from making, using, selling, offering for
sale or importing our products or prevent our customers from using our products. If a court
determined or if we independently concluded that any of our products or manufacturing processes
violated third-party proprietary rights, our clinical trials could be delayed and there can be no
assurance that we would be able to reengineer the product or processes to avoid those rights, or to
obtain a license under those rights on commercially reasonable terms, if at all.
Risks Related to Regulatory Approval of Our Product Candidates
If we fail to submit our NDA for VIAject® as we intend, or if we are unable to obtain required
regulatory approvals, we will not be able to commercialize VIAject® as planned, or at all, and our
ability to generate revenue will be materially impaired.
We have invested a significant portion of our efforts and financial resources in the
development of VIAject®, and our ability to generate near-term revenue depends on VIAject’s®
success. All of our product candidates, including VIAject®, and the activities associated with
their development and commercialization, including their testing, manufacture, safety, efficacy,
recordkeeping, labeling, storage, approval, advertising, promotion, sale and distribution, are
subject to comprehensive regulation by the FDA and other regulatory agencies in the United States
and by comparable authorities in other countries. Failure to obtain regulatory approval for a
product candidate will prevent us from commercializing the product candidate. Securing FDA approval
requires the submission of an NDA containing extensive preclinical and clinical data and supporting
information for each therapeutic indication to establish the product candidate’s safety and
efficacy. Securing FDA approval also requires the submission of information about the product
manufacturing process to, and inspection of manufacturing facilities by, the FDA.
We have not yet submitted an NDA or received regulatory approval to market VIAject® or any of
our other product candidates in the United States or any other jurisdiction. We intend to submit
our NDA for VIAject® in December 2009. If we are delayed in filing our NDA, our ability to generate
revenue from the commercialization of VIAject® will be similarly delayed. Additionally, we cannot
guarantee that the FDA will accept our NDA as filed, review our NDA in a timely fashion or approve
our NDA. Many companies that have believed that their products performed satisfactorily in clinical
trials have nonetheless failed to obtain FDA approval for their products. Upon the FDA’s review of
our NDA, it may request that we conduct additional analyses of the data and, if it believes that
the data are not satisfactory, could request additional information from us, including data that
may necessitate additional clinical trials. While we announced positive results from our pivotal
Phase 3 clinical trial of VIAject®, data from patients with Type 1 diabetes in India were found to
be anomalous when compared to data from the United States and Germany for the same trial. As a
result, the FDA could conclude we did not establish non-inferiority of VIAject® when compared to
Humulin® R in terms of blood glucose control. Failure of the FDA to accept our NDA as filed, review
our NDA in a timely fashion, or approve our NDA will materially impair our ability to generate
product revenue and our business.
If the FDA does not believe that our product candidates satisfy the requirements for the Section
505(b)(2) approval procedure, the approval pathway will take longer and cost more than anticipated
and in either case may not be successful.
24
Table of Contents
We believe that VIAject® and VIAtab™ qualify for approval under Section 505(b)(2) of the
FFDCA. Because we are developing new formulations of previously approved chemical entities, such as
insulin, this may enable us to avoid having to submit certain types of data and studies that are
required in full NDAs and instead submit an NDA under Section 505(b)(2). The FDA may not agree that
our products are approvable under Section 505(b)(2). Insulin is a unique and complex drug that is
associated with more intra-and inter-patient variability than many small molecule drugs. The
availability of the Section 505(b)(2) pathway for insulin is even more controversial than for small
molecule drugs, and the FDA may not accept this pathway for our insulin product candidates. The FDA
has not published any guidance that specifically addresses an NDA for an insulin product candidate
under Section 505(b)(2). No other insulin product has yet been approved pursuant to an NDA under
Section 505(b)(2). If the FDA determines that NDAs under Section 505(b)(2) are not appropriate and
that full NDAs are required for our product candidates, the time and financial resources required
to obtain FDA approval for our product candidates could substantially and materially increase. This
would require us to obtain substantially more funding than previously anticipated which could
significantly dilute the ownership interests of our stockholders. Even with this investment, the
prospect for FDA approval may be significantly lower. If the FDA requires full NDAs for our product
candidates or requires more extensive testing and development for some other reason, our ability to
compete with alternative products that arrive on the market more quickly than our product
candidates would be adversely impacted.
Notwithstanding the approval of many products by the FDA under Section 505(b)(2) over the last
few years, certain brand-name pharmaceutical companies and others have objected to the FDA’s
interpretation of Section 505(b)(2). If the FDA’s interpretation of Section 505(b)(2) is
successfully challenged, the FDA may be required to change its interpretation of Section 505(b)(2)
which could delay or even prevent the FDA from approving any NDA under Section 505(b)(2) that we
submit. The pharmaceutical industry is highly competitive, and it is not uncommon for a
manufacturer of an approved product to file a citizen petition with the FDA seeking to delay
approval of, or impose additional approval requirements for, pending competing products. If
successful, such petitions can significantly delay, or even prevent, the approval of the new
product. However, even if the FDA ultimately denies such a petition, the FDA may substantially
delay approval while it considers and responds to the petition.
Moreover, even if VIAject® and VIAtab™ are approved under Section 505(b)(2), the approval may
be subject to limitations on the indicated uses for which the product may be marketed or to other
conditions of approval, or may contain requirements for costly post-marketing testing and
surveillance to monitor the safety or efficacy of the product.
Any product for which we obtain marketing approval could be subject to restrictions or withdrawal
from the market and we may be subject to penalties if we fail to comply with regulatory
requirements or if we experience unanticipated problems with our products, when and if any of them
are approved.
Any product for which we obtain marketing approval, along with the manufacturing processes,
post-approval clinical data, labeling, advertising and promotional activities for such product,
will be subject to continual requirements of and review by the FDA and comparable regulatory
authorities. These requirements include, in the case of FDA, submissions of safety and other
post-marketing information and reports, registration requirements, cGMP requirements relating to
quality control, quality assurance and corresponding maintenance of records and documents,
requirements regarding the distribution of samples to physicians and recordkeeping. Even if
regulatory approval of a product is granted, the approval may be subject to limitations on the
indicated uses for which the product may be marketed or to other conditions of approval, or may
contain requirements for costly post-marketing testing and surveillance to monitor the safety or
efficacy of the product. In addition, if any of our product candidates are approved, our product
labeling, advertising and promotion would be subject to regulatory requirements and continuing
regulatory review. The FDA strictly regulates the promotional claims that may be made about
prescription drug products. In particular, a drug may not be promoted in a misleading manner or for
uses that are not approved by the FDA as reflected in the product’s approved labeling. The FDA and
other agencies actively enforce the laws and regulations prohibiting misleading promotion and the
promotion of off-label uses, and a company that is found to have improperly promoted off-label uses
may be subject to significant liability.
Discovery after approval of previously unknown problems with our products, manufacturers or
manufacturing processes, or failure to comply with regulatory requirements, may result in actions
such as:
| • | restrictions on such products’ manufacturers or manufacturing processes; | ||
| • | restrictions on the marketing or distribution of a product; | ||
| • | warning letters; | ||
| • | withdrawal of the products from the market; | ||
| • | refusal to approve pending applications or supplements to approved applications that we submit; | ||
| • | recall of products; | ||
| • | fines, restitution or disgorgement of profits or revenue; | ||
| • | suspension or withdrawal of regulatory approvals; | ||
| • | refusal to permit the import or export of our products; | ||
| • | product seizure; |
25
Table of Contents
| • | injunctions; or | ||
| • | imposition of civil or criminal penalties. |
Legislation may make it more difficult and costly for us to obtain regulatory approval of our
product candidates and to produce, market and distribute our existing products.
On September 27, 2007, President Bush signed into law the Food and Drug Administration
Amendments Act of 2007, or the FDAAA. This new legislation grants significant new powers to the
FDA, many of which are aimed at improving drug safety and assuring the safety of drug products
after approval. Under the FDAAA, companies that violate the new law are subject to substantial
civil monetary penalties. While we expect the FDAAA to have a significant impact on the
pharmaceutical industry, the extent of the impact is not yet known. The new requirements and
changes imposed by the FDAAA may make it more difficult, and more costly, to obtain and maintain
approval of new pharmaceutical products and to produce, market and distribute existing products.
In addition, the FDA’s regulations, policies or guidance may change and new or additional
statutes or government regulations may be enacted that could prevent or delay regulatory approval
of our product candidates or further restrict or regulate post-approval activities. It is
impossible to predict whether additional legislative changes will be enacted, or FDA regulations,
guidance or interpretations implemented or modified, or what the impact of such changes, if any,
may be.
Failure to obtain regulatory approval in international jurisdictions would prevent us from
marketing our products abroad.
We intend to have our products marketed outside the United States. In order to market our
products in the European Union and many other jurisdictions, we must obtain separate regulatory
approvals and comply with numerous and varying regulatory requirements of other countries regarding
safety and efficacy and governing, among other things, clinical trials and commercial sales and
distribution of our products. The approval procedure varies among countries and can involve
additional testing. The time required to obtain approval may differ from that required to obtain
FDA approval. The regulatory approval process outside the United States may include all of the
risks associated with obtaining FDA approval, as well as additional risks. In addition, in many
countries outside the United States, it is required that the product be approved for reimbursement
before the product can be approved for sale in that country. We may not obtain approvals from
regulatory authorities outside the United States on a timely basis, if at all. Approval by the FDA
does not ensure approval by regulatory authorities in other countries or jurisdictions, and
approval by one regulatory authority outside the United States does not ensure approval by
regulatory authorities in other countries or jurisdictions or by the FDA. We may not be able to
file for regulatory approvals and may not receive necessary approvals to commercialize our products
in any market.
Reports of side effects or safety concerns in related technology fields or in other companies’
clinical trials could delay or prevent us from obtaining regulatory approval or negatively impact
public perception of our product candidates.
At present, there are a number of clinical trials being conducted by us and by other
pharmaceutical companies involving insulin or insulin delivery systems. The major safety concern
with patients taking insulin is the occurrence of hypoglycemic events. If we discover that our
product is associated with a significantly increased frequency of hypoglycemic or other adverse
events, or if other pharmaceutical companies announce that they observed frequent or significant
adverse events in their trials involving insulin or insulin delivery systems, we could encounter
delays in the commencement or completion of our clinical trials or difficulties in obtaining the
approval of our product candidates. In addition, the public perception of our products might be
adversely affected, which could harm our business and results of operations, even if the concern
relates to another company’s product.
Risks Related to Employee Matters and Managing Growth
Our future success depends on our ability to retain our chief executive officer and other key
executives and to attract, retain and motivate qualified personnel.
We are highly dependent on Dr. Solomon S. Steiner, our Chairman, President and Chief Executive
Officer, Gerard Michel, our Chief Financial Officer and Dr. Alan Krasner, our Chief Medical
Officer. Dr. Steiner is an inventor of our VIAdel™ technology. The loss of the services of any of
these persons might impede the achievement of our research, development and commercialization
objectives. With the exception of Dr. Steiner, we currently do not have employment agreements with
any other executive officers. Replacing key employees may be difficult and time-consuming because
of the limited number of individuals in our industry with the skills and experiences required to
develop, gain regulatory approval of and commercialize our product candidates successfully. Other
than a $1 million key person insurance policy on Dr. Steiner, we do not have key person life
insurance to cover the loss of any of our other employees.
Recruiting and retaining qualified scientific personnel, clinical personnel and sales and
marketing personnel will also be critical to our success. We may not be able to attract and retain
these personnel on acceptable terms, if at all, given the competition among numerous pharmaceutical
and biotechnology companies for similar personnel. We also experience competition for the hiring of
scientific and clinical personnel from other companies, universities and research institutions. In
addition, we rely on consultants and advisors, including scientific and clinical advisors, to
assist us in formulating our research and development and commercialization strategy. Our
consultants and advisors may be employed by employers other than us and may have commitments under
consulting or advisory contracts with other entities that may limit their availability to us.
We may expand our development, regulatory and sales and marketing capabilities, and as a result,
we may encounter difficulties in managing our growth, which could disrupt our operations.
26
Table of Contents
If our development and commercialization plans for VIAject® are successful, we may experience
significant growth in the number of our employees and the scope of our operations, particularly in
the areas of manufacturing, clinical trials management, and regulatory affairs. To manage our
anticipated future growth, we must continue to implement and improve our managerial, operational
and financial systems and continue to recruit and train additional qualified personnel. Due to our
limited financial resources we may not be able to effectively manage the expansion of our
operations or recruit and train additional qualified personnel. Any inability to manage growth
could delay the execution of our business plans or disrupt our operations.
Risks Related to Our Common Stock
Our executive officers, directors and principal stockholders have significant ability to control
all matters submitted to stockholders for approval.
Our executive officers, directors and principal stockholders, in the aggregate, beneficially
own shares representing approximately 39% of our outstanding capital stock. As a result, these
stockholders, if they act together, will be able to exercise a significant controlling influence
over matters requiring stockholder approval, including the election of directors and approval of
significant corporate transactions, such as mergers, consolidations and sales of all or
substantially all of our assets, and will have significant control over our management and
policies. The interests of this group of stockholders may not always coincide with our corporate
interests or the interests of other stockholders. This significant concentration of stock ownership
could also result in the entrenchment of our management and adversely affect the price of our
common stock.
Provisions in our corporate charter documents and under Delaware law could make an acquisition of
us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our
stockholders to replace or remove our current management.
Provisions in our corporate charter and bylaws may discourage, delay or prevent a merger,
acquisition or other change in control of us that stockholders may consider favorable, including
transactions in which you might otherwise receive a premium for your shares. These provisions could
also limit the price that investors might be willing to pay in the future for shares of our common
stock, thereby depressing the market price of our common stock. In addition, these provisions may
frustrate or prevent any attempts by our stockholders to replace or remove our current management
by making it more difficult for stockholders to replace members of our board of directors. Because
our board of directors is responsible for appointing the members of our management team, these
provisions could in turn affect any attempt by our stockholders to replace current members of our
management team.
Among others, these provisions:
| • | establish a classified board of directors such that not all members of the board are elected at one time; | ||
| • | allow the authorized number of our directors to be changed only by resolution of our board of directors; | ||
| • | limit the manner in which stockholders can remove directors from the board; | ||
| • | establish advance notice requirements for stockholder proposals that can be acted on at stockholder meetings and nominations to our board of directors; | ||
| • | require that stockholder actions must be effected at a duly called stockholder meeting and prohibit actions by our stockholders by written consent; | ||
| • | limit who may call stockholder meetings; | ||
| • | authorize our board of directors to issue preferred stock without stockholder approval, which could be used to institute a stockholder rights plan or “poison pill” that would work to dilute the stock ownership of a potential hostile acquirer, effectively preventing acquisitions that have not been approved by our board of directors; and | ||
| • | require the approval of the holders of at least 75% of the votes that all our stockholders would be entitled to cast to amend or repeal certain provisions of our charter or bylaws. |
In addition, because we are incorporated in Delaware, we are governed by the provisions of
Section 203 of the Delaware General Corporation Law, which generally prohibits a person who owns in
excess of 15% of our outstanding voting stock from merging or combining with us for a period of
three years after the date of the transaction in which the person acquired in excess of 15% of our
outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
If our stock price is volatile, purchasers of our common stock could incur substantial losses.
Our stock price may be volatile. The stock market in general and the market for biotechnology
companies in particular have experienced extreme volatility that has often been unrelated to the
operating performance of particular companies. The market price for our common stock may be
influenced by many factors, including:
| • | results of clinical trials of our product candidates or those of our competitors; | ||
| • | regulatory or legal developments in the United States and other countries; | ||
| • | variations in our financial results or those of companies that are perceived to be similar to us; |
27
Table of Contents
| • | developments or disputes concerning patents or other proprietary rights; | ||
| • | the recruitment or departure of key personnel; | ||
| • | changes in the structure of healthcare payment systems; | ||
| • | market conditions in the pharmaceutical and biotechnology sectors and issuance of new or changed securities analysts’ reports or recommendations; | ||
| • | general economic, industry and market conditions; and | ||
| • | the other factors described in this “Risk Factors” section. |
We have never paid any cash dividends on our capital stock and we do not anticipate paying any
cash dividends in the foreseeable future.
We have paid no cash dividends on our capital stock to date. We currently intend to retain our
future earnings, if any, to fund the development and growth of our business. In addition, the terms
of any future debt agreements may preclude us from paying dividends. As a result, we do not expect
to pay any cash dividends in the foreseeable future, and payment of cash dividends, if any, will
depend on our financial condition, results of operations, capital requirements and other factors
and will be at the discretion of our board of directors. Furthermore, we may in the future become
subject to contractual restrictions on, or prohibitions against, the payment of dividends. Capital
appreciation, if any, of our common stock will be investors’ sole source of gain for the
foreseeable future.
A significant portion of our total outstanding shares are restricted from immediate resale but may
be sold into the market in the near future. This could cause the market price of our common stock
to drop significantly, even if our business is doing well.
Sales of a substantial number of shares of our common stock in the public market could occur
at any time. These sales, or the perception in the market that the holders of a large number of
shares intend to sell shares, could reduce the market price of our common stock. As of November 30,
2009, we had approximately 24 million shares of common stock outstanding. Of these, approximately 9
million shares are able to be sold in accordance with the SEC’s Rule 144 and the remainder are
generally freely tradable without restriction under securities laws.
We incur substantial costs as a result of operating as a public company, and our management is
required to devote substantial time to comply with public company regulations.
We are subject to the reporting requirements of the Exchange Act, the Sarbanes-Oxley Act of
2002 as well as other federal and state laws. These requirements may place a strain on our people,
systems and resources. The Exchange Act requires that we file annual, quarterly and current reports
with respect to our business and financial condition. The Sarbanes-Oxley Act requires that we
maintain effective disclosure controls and procedures and internal controls over financial
reporting. In order to maintain and improve the effectiveness of our disclosure controls and
procedures and internal controls over financial reporting, significant resources and management
oversight will be required. This may divert management’s attention from other business concerns,
which could have a material adverse effect on our business, financial condition, results of
operations and cash flows.
ITEM 1B. UNRESOLVED STAFF COMMENTS
Not applicable.
ITEM 2. PROPERTIES
We lease approximately 29,300 square feet of office space and laboratory facilities in
Danbury, Connecticut from Mulvaney Properties LLC. Our corporate headquarters are located at 100
Saw Mill Road, Danbury, Connecticut, in approximately 19,500 square feet of rentable office space.
The lease for this office space expires July 31, 2014, subject to our right to renew the lease
under the same terms and conditions for an additional seven year term. Our laboratory facility is
located at 6 and 8 Christopher Columbus Avenue, Danbury, Connecticut, in approximately 7,200 and
2,600 square feet of rentable laboratory and office space. The leases for our facilities at 6 and 8
Christopher Columbus expire in January 2010. We expect to renew these leases before they expire.
Our laboratory facility is fully equipped to perform our current drug delivery and related research
and development activities, as well as to manufacture on a limited basis our own product line in
accordance with cGMP.
Mulvaney Properties LLC is controlled by a non-affiliated stockholder of ours.
ITEM 3. LEGAL PROCEEDINGS
We currently are not involved in any legal proceedings.
ITEM 4. SUBMISSION OF MATTERS TO VOTE OF SECURITY HOLDERS
None.
28
Table of Contents
PART II
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information
Since May 11, 2007, our common stock has traded on the NASDAQ Global Market under the symbol
“BIOD.”
The following table sets forth the high and low sale prices per share for our common stock for
each of the quarters in the period beginning October 1, 2008 through September 30, 2009, as
reported on the NASDAQ Global Market:
| Quarter Ended | High | Low | ||||||
December 31, 2008 |
$ | 4.99 | $ | 1.62 | ||||
March 31, 2009 |
$ | 6.00 | $ | 3.29 | ||||
June 30, 2009 |
$ | 6.02 | $ | 3.64 | ||||
September 30, 2009 |
$ | 5.48 | $ | 4.46 | ||||
The closing price of our common stock, as reported by the NASDAQ Global Market, was $3.99 on
December 8, 2009.
Holders
As of November 30, 2009, the number of holders of record of our common stock was 49.
Dividends
We have never paid or declared any cash dividends on our common stock. We currently intend to
retain earnings, if any, to finance the growth and development of our business. Payment of future
dividends, if any, will be at the discretion of our board of directors.
Equity Compensation Plan Information
Information relating to compensation plans under which our equity securities are authorized
for issuance is set forth under “Security Ownership of Certain Beneficial Owners and Management and
Related Stockholder Matters” in our definitive proxy statement for our 2010 Annual Meeting of
Stockholders.
Issuer Purchases of Equity Securities
We did not make any purchases of our shares of common stock in the fourth quarter of fiscal
2009, nor did any affiliated purchaser or anyone acting on behalf of us or an affiliated purchaser.
29
Table of Contents
ITEM 6. SELECTED FINANCIAL DATA
You should read the following selected financial data together with our financial statements
and the related notes which are included elsewhere in this Annual Report and the “Management’s
Discussion and Analysis of Financial Condition and Results of Operations” section of this Annual
Report. We have derived the statement of operations data set forth below for the three-year period
ended September 30, 2009 and the balance sheet data as of September 30, 2008 and 2009 set forth
below from our audited financial statements which are included in this Annual Report. We have
derived the statement of operations data set forth below for the years ended September 30, 2005,
2006 and 2007 and the balance sheet data as of September 30, 2005, 2006 and 2007 set forth below
from our audited financial statements, which are not included in this Annual Report. Our audited
financial statements include, in the opinion of our management, all adjustments, consisting of only
normal recurring accruals, necessary for a fair presentation of those statements. Historical
results for any prior or interim period are not necessarily indicative of results to be expected in
any future period or for a full fiscal year.
| Year Ended September 30, | ||||||||||||||||||||
| 2005 | 2006 | 2007 | 2008 | 2009 | ||||||||||||||||
| (In thousands, except share and per share amounts) | ||||||||||||||||||||
Statement of operations data: |
||||||||||||||||||||
Revenue |
$ | — | $ | — | $ | — | $ | — | $ | — | ||||||||||
Operating expenses: |
||||||||||||||||||||
Research and development |
2,666 | 5,987 | 15,939 | 32,554 | 32,325 | |||||||||||||||
General and administrative |
724 | 1,548 | 8,386 | 14,800 | 10,994 | |||||||||||||||
Total operating expenses |
3,390 | 7,535 | 24,325 | 47,354 | 43,319 | |||||||||||||||
Other (income) and expense: |
||||||||||||||||||||
Interest and other income |
(9 | ) | (182 | ) | (1,902 | ) | (3,010 | ) | (386 | ) | ||||||||||
Interest expense |
— | 78 | — | — | — | |||||||||||||||
Loss on settlement of debt |
— | 627 | — | — | — | |||||||||||||||
Operating loss before tax provision
(benefit) |
(3,381 | ) | (8,058 | ) | (22,423 | ) | (44,344 | ) | (42,933 | ) | ||||||||||
Tax provision (benefit) |
2 | 10 | 125 | (983 | ) | 337 | ||||||||||||||
Net loss |
(3,383 | ) | (8,068 | ) | (22,548 | ) | (43,361 | ) | (43,270 | ) | ||||||||||
Charge for accretion of beneficial
conversion rights |
— | (603 | ) | — | — | — | ||||||||||||||
Deemed dividend — warrants |
— | — | (4,457 | ) | — | — | ||||||||||||||
Net loss applicable to common
stockholders |
$ | (3,383 | ) | $ | (8,671 | ) | $ | (27,005 | ) | $ | (43,361 | ) | $ | (43,270 | ) | |||||
Net loss per share — basic and diluted |
$ | (0.56 | ) | $ | (1.05 | ) | $ | (1.76 | ) | $ | (1.94 | ) | $ | (1.82 | ) | |||||
Weighted average shares outstanding —
basic and diluted |
6,080,746 | 8,252,113 | 15,354,898 | 22,390,434 | 23,746,598 | |||||||||||||||
| As of September 30, | ||||||||||||||||||||
| 2005 | 2006 | 2007 | 2008 | 2009 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
Balance sheet data: |
||||||||||||||||||||
Cash, cash equivalents and marketable
investment securities |
$ | 368 | $ | 17,539 | $ | 80,022 | $ | 90,283 | $ | 54,640 | ||||||||||
Working capital (deficit) |
(98 | ) | 15,307 | 75,244 | 84,377 | 46,787 | ||||||||||||||
Total assets |
1,195 | 18,659 | 82,506 | 97,511 | 59,625 | |||||||||||||||
Deficit accumulated during the development stage |
(4,157 | ) | (12,828 | ) | (39,833 | ) | (83,194 | ) | (126,464 | ) | ||||||||||
Total stockholders’ equity |
654 | 16,348 | 77,223 | 88,487 | 50,538 | |||||||||||||||
30
Table of Contents
| ITEM 7. | MANAGEMENT DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
You should read the following discussion and analysis of our financial condition and results
of operations together with our financial statements and the related notes and other financial
information included elsewhere in this Form 10-K. Some of the information in this discussion and
analysis or set forth elsewhere in this Form 10-K, including our plans and strategies for our
business, includes forward-looking statements which involve risks and uncertainties. Please review
the “Forward-Looking Statements” and the “Risk Factors” sections of this Form 10-K for a discussion
of important factors that could cause actual results to materially differ from those anticipated or
implied by the forward-looking statements contained in the following discussion and analysis.
Overview
We are a specialty biopharmaceutical company focused on the development and commercialization
of innovative treatments for endocrine disorders such as diabetes, which may be safer, more
effective and more convenient for patients. We develop our product candidates by applying our
proprietary formulation technologies to existing drugs in order to improve their therapeutic
profiles. Our initial development efforts are focused on peptide hormones. Our most advanced
product candidate, VIAject®, has been studied in two pivotal Phase 3 clinical trials for the
treatment of patients with Type 1 and Type 2 diabetes. Earlier stage product candidates include
VIAtab™, a sublingual tablet formulation of insulin.
VIAject® is our proprietary injectable formulation of recombinant human insulin designed to be
absorbed into the blood faster than the currently marketed rapid-acting insulin analogs. We have
recently completed two pivotal Phase 3 clinical trials of VIAject®, one in patients with Type 1
diabetes and the other in patients with Type 2 diabetes. In both clinical trials we compared
VIAject® to Humulin® R, a form of recombinant human insulin. We believe VIAject® can improve the
management of blood glucose levels in patients with diabetes by more closely mimicking the natural
first-phase insulin release that healthy individuals experience at mealtime. Patients in both
clinical trials were treated for a period of six months.
In March 2009, we announced our plan to submit an NDA to the FDA by the end of 2009 to market
VIAject® for the treatment of diabetes. We expect that the NDA will be submitted under section
505(b)(2) of the FFDCA and be based upon results from multiple pharmacokinetic and pharmacodynamic
studies as well as our two completed Phase 3 studies of VIAject® in patients with Type 1 and Type 2
diabetes. We intend to seek approval for a 100 IU/cc liquid formulation of VIAject® that is
bioequivalent to the two-part 25 IU/cc lyophilized powder formulation of VIAject® that was used in
our pivotal Phase 3 clinical trials.
In October 2009 we executed a letter of intent to purchase a disposable insulin pen designed
by Wockhardt Ltd. for use with VIAject®. We intend to submit this pen to the FDA for review at a
later date after completing certain modifications that we believe will improve its commercial
performance.
In 2010 we plan to conduct additional clinical trials designed to generate additional data to
enhance VIAject’s potential commercial success.
We have developed all of our product candidates utilizing our proprietary VIAdel™ technology,
which allows us to study the interaction between peptide hormones and small molecules. We use our
technology to reformulate existing peptide drugs with small molecule ingredients that are generally
regarded as safe by the FDA to improve their therapeutic profiles.
We are a development stage company. We were incorporated in December 2003 and commenced active
operations in January 2004. To date, we have generated no revenues and have incurred significant
losses. We have financed our operations and internal growth through our initial public offering in
May 2007 and follow-on offering in February 2008 and prior to that, private placements of
convertible preferred stock and other securities. We have devoted substantially all of our efforts
to research and development activities, including clinical trials. Our net loss was $43.3 million
for the year ended September 30, 2009. As of September 30, 2009, we had a deficit accumulated
during the development stage of $126.5 million. The deficit accumulated during the development
stage is attributable primarily to our research and development activities and non-cash charges for
(1) accretion of beneficial conversion rights and (2) deemed dividend-warrants and stock-based
compensation. Research and development and general and administrative expenses, as a percentage of
net loss applicable to common stockholders, represent approximately 75% and 25%, respectively, of
the expenses that we have incurred since our inception. We expect to continue to generate
significant losses as we continue to develop our product candidates.
Financial Operations Overview
Revenues
To date, we have generated no revenues. We do not expect to begin generating any revenues
unless any of our product candidates receive marketing approval or if we receive payments in
connection with strategic collaborations that we may enter into for the commercialization of our
product candidates.
31
Table of Contents
Research and Development Expenses
Research and development expenses consist of the cost associated with our basic research
activities, as well as the costs associated with our drug development efforts, conducting
preclinical studies and clinical trials, manufacturing development efforts and activities related
to regulatory filings. Our research and development expenses consist of:
| • | external research and development expenses incurred under agreements with third-party contract research organizations and investigative sites, third-party manufacturing organizations and consultants; | ||
| • | employee-related expenses, which include salaries and benefits for the personnel involved in our preclinical and clinical drug development and manufacturing activities; and | ||
| • | facilities, depreciation and other allocated expenses, which include direct and allocated expenses for rent and maintenance of facilities, depreciation of leasehold improvements and equipment and laboratory and other supplies. |
While we have suspended significant expenditures on our earlier stage product candidates and
any enlargement of our laboratory facilities pending further development of our regulatory plans
for VIAject®, we expect to continue to incur significant operating losses for the next several
years as we:
| • | continue our clinical trial extension program for the pivotal Phase 3 clinical trials of VIAject®, which is designed to run through February 2010; | ||
| • | conduct additional clinical trials of VIAject® to support our commercialization efforts and, potentially, FDA approval; and | ||
| • | purchase recombinant human insulin and other materials to build commercial supply inventory for VIAject®. |
If we obtain regulatory approval for VIAject®, research and development expenses may increase
significantly as we prepare for the commercial launch of VIAject® and reinvigorate development work
on our early stage product candidates.
We have used our employee and infrastructure resources across multiple research projects,
including our drug development programs. To date, we have not tracked expenses related to our
product development activities on a program-by-program basis. Accordingly, we cannot reasonably
estimate the amount of research and development expenses that we incurred with respect to each of
our clinical and preclinical product candidates. However, substantially all of our research and
development expenses incurred to date are attributable to our VIAject® program.
The following table illustrates, for each period presented, our research and development costs
by nature of the cost.
| December 3, | ||||||||||||||||
| 2003 | ||||||||||||||||
| (Inception) to | ||||||||||||||||
| Year Ended September 30, | September 30, | |||||||||||||||
| 2007 | 2008 | 2009 | 2009 | |||||||||||||
| (In thousands) | ||||||||||||||||
Research and development expenses: |
||||||||||||||||
Pre-clinical expenses |
$ | 1,983 | $ | 4,230 | $ | 2,709 | $ | 12,253 | ||||||||
Manufacturing expenses |
2,141 | 6,728 | 11,674 | 22,061 | ||||||||||||
Clinical/regulatory expenses |
11,815 | 21,596 | 17,942 | 55,737 | ||||||||||||
Total |
$ | 15,939 | $ | 32,554 | $ | 32,325 | $ | 90,051 | ||||||||
The successful development of our product candidates is highly uncertain. At this time, we
cannot reasonably estimate or know the nature, specific timing and estimated costs of the efforts
that will be necessary to complete the remainder of the development of, or the period, if any, in
which material net cash inflows may commence from our product candidates. This is due to the
numerous risks and uncertainties associated with developing drugs, including the uncertainty of:
| • | our ability to secure approval by the FDA for our product candidates under Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act, or the FFDCA; | ||
| • | our ability to file our NDA for VIAject® in December 2009 as planned and, once filed, the length of time that will elapse before our NDA is fully reviewed by the FDA; | ||
| • | the costs associated with preparing and submitting our MAA for VIAject® to the EMEA; | ||
| • | our ability to market, commercialize and achieve market acceptance for product candidates, particularly VIAject®; | ||
| • | our ability to secure approval by the FDA for VIAject® without conducting additional pivotal clinical trials; |
32
Table of Contents
| • | the FDA’s findings regarding data anomalies observed in India in our Phase 3 clinical trial of VIAject® for patients with Type 1 diabetes and the impact of those findings on the timing of a regulatory approval; | ||
| • | the size, endpoints and duration of additional clinical trials of VIAject® to support our commercialization efforts and, potentially, FDA approval; | ||
| • | the cost to fully develop the 100 IU/cc liquid formulation of VIAject®; | ||
| • | our ability to establish that the 100 IU/cc liquid formulation of VIAject® is well-tolerated in chronic use; | ||
| • | the cost to develop an insulin pen program for use with VIAject®; | ||
| • | the costs of pre-commercialization activities, including increased insulin purchases; | ||
| • | the costs associated with qualifying and obtaining regulatory approval of suppliers of insulin and manufacturers of our product candidates; | ||
| • | the costs of preparing, filing and prosecuting patent applications and maintaining, enforcing and defending intellectual property-related claims; | ||
| • | the emergence of competing technologies and products and other adverse market developments; and | ||
| • | our ability to establish and maintain collaborations and the terms and success of the collaborations, including the timing and amount of payments that we might receive from potential strategic collaborators. |
A change in the outcome of any of these variables with respect to the development of VIAject®
or our other product candidates could mean a significant change in the costs and timing associated
with product development. For example, if the FDA or other regulatory authority were to require us
to conduct an additional pivotal Phase 3 clinical trial of VIAject®, we could be required to expend
significant additional financial resources and time on the completion of that clinical development
program.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and related expenses for
personnel, including stock-based compensation expenses, in our executive, legal, accounting,
finance and information technology functions. Other general and administrative expenses include
facility-related costs not otherwise allocated to research and development expense, travel
expenses, costs associated with industry conventions and professional fees, such as legal and
accounting fees and consulting costs.
We anticipate that our general and administrative expenses will not change significantly as we
focus our product development efforts on obtaining regulatory approval for VIAject®. Over the
longer term, however, these expenses could increase as we approach the commercial launch of
VIAject®:
Marketable Securities
In accordance with Accounting Standard Codification (ASC) Topic 320, Investments in Debt and
Equity Securities issued by the Financial Accounting Standard Board (“FASB”) in May 1993, our
marketable securities were classified as available-for-sale. In accordance with that standard,
these securities are reported at market value with unrealized gains and losses shown as a component
of accumulated other comprehensive income (loss). We regularly evaluate the performance of these
investments individually for impairment, taking into consideration the investment, volatility and
current returns. If a determination is made that a decline in fair value is other-than-temporary,
the related securities are written down to their estimated fair value. As of September 30, 2008 and
2009, the Company had $25.6 million and $0 marketable security investments, respectively.
Pre-Launch Inventory
Inventory costs associated with products that have not yet received regulatory approval are
capitalized if we believe there is probable future commercial use and future economic benefit. If
the probability of future commercial use and future economic benefit cannot be reasonably
determined, then costs associated with pre-launch inventory that has not yet received regulatory
approval are expensed as research and development expense during the period the costs are incurred.
For the year ended September 30, 2009, the Company expensed $6.5 million of costs associated with
the purchase of recombinant human insulin, as research and development expense after it passed
quality control inspection by the Company and transfer of title occurred. The Company plans on
submitting the NDA for VIAject® in December 2009. Until the Company can determine the probability
of VIAject® receiving regulatory approval, costs associated with the purchase of recombinant human
insulin will continue to be expensed as research and development.
Comprehensive Income (Loss)
We classify our marketable securities as available for sale, in accordance with ASC Topic 220,
Comprehensive Income, issued by the FASB in June 1997. Other Comprehensive Income include changes
in equity for unrealized holding gains (losses) on
marketable securities, which have arisen during the period. During the year ended September
30, 2009, marketable security investments were sold at a par value and the September 30, 2008 net
unrealized loss of $62 was fully recaptured.
33
Table of Contents
Interest Income
Interest income consists of interest earned on our cash and cash equivalents and marketable
securities, resulting primarily from the $125.6 million in net proceeds received from our initial
public offering in May 2007 and follow-on offering in February 2008. In November 2007, our board of
directors approved investment policy guidelines, the primary objectives of which are the
preservation of capital, the maintenance of liquidity and maintenance of appropriate fiduciary
control — subject to our business objectives and tax situation.
Due to the uncertainty in the credit and financial markets, along with our intention to file
an NDA in December 2009 and secure FDA approval of VIAject®, we have modified our investment
strategy and primarily invested in certain marketable securities, which consist primarily of
short-to-intermediate-term debt securities issued by the U.S. government, Treasury securities and
U.S. government agencies. The focus on preserving cash and investing in stable securities generated
lower returns during the year ended September 30, 2009. During the period from before we file our
NDA until the FDA finishes its review of our NDA, we intend to maintain this conservative strategy
until the credit and financial markets improve and become more stable.
Exercise of Warrants
In March 2007, we offered the holders of warrants to purchase an aggregate of 149,125 shares
of our Series B convertible preferred stock and an aggregate of 3,417,255 shares of our common
stock with an exercise price of $5.56 per share the opportunity to exercise such warrants at an
exercise price of $3.67, representing a 34% discount in the exercise price. Such holders exercised
all of such warrants on a combination of cashless and cash exercise basis. We issued an aggregate
of 2,636,907 shares of common stock and received aggregate cash proceeds of approximately $0.4
million in connection with such exercises.
As a result of the discounted exercise price, in the fiscal quarter ended March 31, 2007, we
recorded a deemed dividend charge of approximately $4.5 million for the warrants that were so
exercised.
As of September 30, 2009, we had warrants outstanding to purchase an aggregate of 118,815
shares of our common stock with an exercise price of $1.41 per share.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of our financial condition and results of operations
are based on our audited financial statements that have been prepared in accordance with accounting
principles generally accepted in the United States. The preparation of these financial statements
requires us to make estimates and assumptions that affect the reported amounts of assets and
liabilities and the disclosure of contingent assets and liabilities at the date of the financial
statements, as well as the reported expenses during the reporting periods. On an ongoing basis, we
evaluate our estimates and assumptions. We base our estimates on historical experience and on
various assumptions that we believe are reasonable under the circumstances, the results of which
form the basis for making judgments about the carrying values of assets and liabilities that are
not readily apparent from other sources. Actual results may differ from these estimates under
different assumptions or conditions.
While our significant accounting policies are more fully described in Note 2 to our financial
statements appearing at the end of this Form 10-K, we believe that the following accounting
policies, which we have discussed with our audit committee, are the most critical to aid you in
fully understanding and evaluating our financial condition and results of operations.
Preclinical Study and Clinical Trial Accruals
In preparing our financial statements, we must estimate accrued expenses pursuant to contracts
with multiple research institutions, clinical research organizations and contract manufacturers
that conduct and manage preclinical studies, clinical trials and manufacture product for these
trials on our behalf. This process involves communicating with relevant personnel to identify
services that have been performed on our behalf and estimating the level of services performed and
the associated costs incurred for services when we have not yet been invoiced for or otherwise
notified of the actual cost. We make estimates of our accrued expenses as of each balance sheet
date in our financial statements based on facts and circumstances known to us. The financial terms
of these agreements vary and may result in uneven payment flows. To date, we have not adjusted our
estimates at any balance sheet date in any material amount. Examples of preclinical study, clinical
trial and manufacturing expenses include the following:
| • | fees paid to contract research organizations in connection with preclinical and toxicology studies and clinical trials; | ||
| • | fees paid to investigative sites in connection with clinical trials; | ||
| • | fees paid to contract manufacturers in connection with the production of clinical trial materials; and | ||
| • | professional service fees. |
34
Table of Contents
Stock-Based Compensation
We recognize compensation costs related to share-based transactions, including employee stock
options, in the financial statements based on fair value. The fair value of the stock underlying
the options is a significant factor in determining credits or charges to operations appropriate for
the stock-based payments to both employees and non-employees.
We selected the Black-Scholes valuation model as the most appropriate valuation method for
stock option grants to employees, members of our board of directors and non-employees. The fair
value of these stock option grants is estimated as of their date of grant using the Black-Scholes
valuation model.
Because we lack sufficient company-specific historical and implied volatility information, we
based our estimate of expected volatility on the median historical volatility of a group of
publicly-traded companies that we believe are comparable to us based on the criteria set forth in
Accounting Standards Codification (ASC) Topic 718-10-55-37(c) and SAB Topic 14.D, particularly line
of business, stage of development, size and financial leverage. We will continue to consistently
apply this process using the same companies or, if those companies become no longer comparable,
other appropriately comparable companies until a sufficient amount of historical information
regarding the volatility of our share price becomes available. However, we will regularly review
these comparable companies, and may substitute more appropriate companies if facts and
circumstances warrant a change. We use the average of (1) the weighted average vesting period and
(2) the contractual life of the option, eight years, as the estimated term of the option. The risk
free rate of interest for periods within the contractual life of the stock option is based on the
yield of a U.S. Treasury strip on the date the award is granted with a maturity equal to the
expected term of the award. We estimate forfeitures based on actual forfeitures during our limited
history. Additionally, we have assumed that dividends will not be paid.
For options granted to non-employees and non-directors, primarily consultants serving on our
Scientific Advisory Board, we measure fair value of the equity instruments utilizing the
Black-Scholes valuation model, if that value is more reliably measurable than the fair value of the
consideration or service received. The fair value of these equity investments are periodically
revalued as the options vest and are recognized as expense over the related period of service or
the vesting period, whichever is longer. As of September 30, 2009, we issued to these non-employees
options to purchase an aggregate of 377,111 shares of our common stock. Because we must revalue
these options for accounting purposes each reporting period, the amount of the stock-based
compensation expense related to these non-employee options will increase or decrease, based on
changes in the price of our common stock. For the years ended September 30, 2009, 2008 and 2007,
the stock-based compensation expense related to these options was $0.1 million, $0.2 million, and
$0.7 million, respectively.
For the year ended September 30, 2009, the stock-based compensation expense was $5.1 million,
of which $1.7 million is reflected in research and development expenses and $3.4 million is
reflected in general and administrative expenses. For the year ended September 30, 2008, the
stock-based compensation expense was $6.7 million, of which $1.6 million is reflected in research
and development expenses and $5.1 million is reflected in general and administrative expenses. For
the year ended September 30, 2007, the stock-based compensation expense was $4.2 million, of which
$0.7 million is reflected in research and development expenses and $3.5 million is reflected in
general and administrative expenses.
Income Taxes
As part of the process of preparing our financial statements, we are required to estimate our
income taxes in each of the jurisdictions in which we operate. This process involves estimating our
actual current tax expense together with assessing temporary differences resulting from differing
treatments of items for tax and accounting purposes. These differences result in deferred tax
assets and liabilities. As of September 30, 2009, we had federal net operating loss carryforwards
of $100.9 million, Connecticut state net operating loss carryforwards of $100.4 million and federal
and local research and development tax credit carryovers of approximately $2.1 million, all of
which expire starting in 2024.
At September 30, 2009, we recorded a 100% valuation allowance against our net deferred tax
asset of approximately $43.2 million, as our management believes it is uncertain that it will be
fully realized. If we determine in the future that we will be able to realize all or a portion of
our net deferred tax asset, an adjustment to the deferred tax valuation allowance would increase
net income in the period in which we make such a determination.
35
Table of Contents
Results of Operations
Year Ended September 30, 2009 Compared to Year Ended September 30, 2008
Revenue. We did not recognize any revenue during the years ended September 30, 2009 or 2008.
Research and Development Expenses.
| Year Ended | ||||||||||||||||
| September 30, | Decrease | |||||||||||||||
| 2008 | 2009 | $ | % | |||||||||||||
| In thousands, except per share amounts | ||||||||||||||||
Research and Development |
$ | 32,554 | $ | 32,325 | $ | 229 | 0.7 | % | ||||||||
Percentage of net loss |
75.1 | % | 74.7 | % | ||||||||||||
Research and development expenses were $32.3 million for the year ended September 30, 2009, a
decrease of $0.2 million, or 0.7%, from $32.6 million for the year ended September 30, 2008. This
decrease was primarily attributable to a $5.8 million decrease in research and development costs
related to our recently completed pivotal Phase 3 clinical trials for VIAject® and a decrease of
$1.5 million in the costs of manufacturing clinical supplies. These decreases were offset by an
increase of $5.8 million for the purchase of recombinant human insulin during the fiscal year in
order to build commercial supply inventory for VIAject® and an increase of $0.9 million in
professional fees for the preparation of the filing of our planned NDA. Research and development
expenses for the year ended September 30, 2009 include $1.6 million in stock-based compensation
expense related to options granted to employees and $0.1 million in stock-based compensation
expense related to options granted to non-employees.
While we have suspended significant expenditures on our earlier stage product candidates
pending further development of our regulatory plans for VIAject®, we do not anticipate that our
research and development expenses will decrease materially, if at all, as we continue our Phase 3
clinical trial extension program for VIAject®, conduct new clinical trials of VIAject®, produce
validation batches, and purchase recombinant human insulin.
General and Administrative Expenses.
| Year Ended | ||||||||||||||||
| September 30, | Decrease | |||||||||||||||
| 2008 | 2009 | $ | % | |||||||||||||
| In thousands, except per share amounts | ||||||||||||||||
General and Administrative |
$ | 14,800 | $ | 10,994 | $ | 3,806 | 25.7 | % | ||||||||
Percentage of net loss |
33.1 | % | 25.5 | % | ||||||||||||
General and administrative expenses were $11.0 million for the year ended September 30, 2009,
a decrease of $3.8 million, or 25.7%, from $14.8 million for the year ended September 30, 2008.
This decrease is attributable to the following items: a decrease of $1.6 million in stock-based
compensation charges for the non-employee directors due to a change in vesting policy from
immediate vesting to vesting pro rata over one year; a decrease of $0.6 million in professional
fees, a decrease of $0.5 million in personnel expenses and a decrease of $0.6 million in travel
expenses were due to a one-time event that occurred in 2008. General and administrative expenses
for the year ended September 30, 2009 include $3.4 million in stock-based compensation expense
related to options granted to employees. Because we must revalue options granted to non-employees
for accounting purposes each reporting period, the amount of the stock-based compensation income
for the year ended September 30, 2009 was $13 thousand.
We do not expect our general and administrative expenses to increase significantly over the
next twelve months as we focus our product development efforts on obtaining regulatory approval for
VIAject®. Over the longer term, however, we anticipate these expenses will increase as we approach
the commercial launch of VIAject®.
36
Table of Contents
Interest and Other Income.
| Year Ended | ||||||||||||||||
| September 30, | Decrease | |||||||||||||||
| 2008 | 2009 | $ | % | |||||||||||||
| In thousands, except per share amounts | ||||||||||||||||
Interest and Other Income |
$ | 3,010 | $ | 386 | $ | 2,624 | 87.2 | % | ||||||||
Percentage of net loss |
6.8 | % | 0.9 | % | ||||||||||||
Interest and other income decreased to $0.4 million for the year ended September 30, 2009 from
$3.0 million for the year ended September 30, 2008. The decrease was due to shifting our
investments primarily into treasury securities. The focus on preserving cash and investing in
stable securities generated lower returns during the year ended September 30, 2009.
Interest Expense. For the years ended September 30, 2009 and 2008, we had no interest
expense.
Net Loss and Net Loss per Share.
| Year Ended | ||||||||||||||||
| September 30, | Decrease | |||||||||||||||
| 2008 | 2009 | $ | % | |||||||||||||
| In thousands, except per share amounts | ||||||||||||||||
Net loss |
$ | (43,361 | ) | $ | (43,270 | ) | $ | 91 | 0.2 | % | ||||||
Net loss per share |
$ | (1.94 | ) | $ | (1.82 | ) | ||||||||||
Net loss was $43.3 million, or $(1.82) per share, for the year ended September 30, 2009
compared to $43.4 million, or $(1.94) per share, for the year ended September 30, 2008. The
decrease in net loss was primarily attributable to the decreased expenses described above. While we
have suspended significant expenditures of our earlier stage product candidates in order to focus
our product developments efforts on obtaining regulatory approval for VIAject®, we expect our
losses to continue as we initiate additional clinical trials of VIAject® to support our
commercialization efforts and, potentially, FDA approval.
Year Ended September 30, 2008 Compared to Year Ended September 30, 2007
Revenue. We did not recognize any revenue during the years ended September 30, 2008 or 2007.
Research and Development Expenses.
| Year Ended | ||||||||||||||||
| September 30, | Increase | |||||||||||||||
| 2007 | 2008 | $ | % | |||||||||||||
| In thousands, except per share amounts | ||||||||||||||||
Research and Development |
$ | 15,939 | $ | 32,554 | $ | 16,615 | 104.2 | % | ||||||||
Percentage of net loss |
70.7 | % | 75.1 | % | ||||||||||||
Research and development expenses were $32.6 million for the year ended September 30, 2008, an
increase of $16.6 million, or 104.2%, from $15.9 million for the year ended September 30, 2007.
This increase was primarily attributable to increased research and development costs related to our recently
completed pivotal Phase 3 clinical trials for
VIAject®. Specific increases in research and development expenses included $7.8 million related to
increased clinical trial expenses in 2008; $3.3 million related to increased manufacturing expenses
in 2008 for the process development, scale-up and manufacture of commercial batches of VIAject® to
support our clinical trials and regulatory submissions; and $2.6 million related to increased
personnel costs, non-cash stock-based compensation expenses and consulting fees. Research and
development expenses for the year ended September 30, 2008 include $1.3 million in stock-based
compensation expense related to options granted to employees and $0.2 million in stock-based
compensation expense related to options granted to non-employees.
37
Table of Contents
General and Administrative Expenses.
| Year Ended | ||||||||||||||||
| September 30, | Increase | |||||||||||||||
| 2007 | 2008 | $ | % | |||||||||||||
| In thousands, except per share amounts | ||||||||||||||||
General and Administrative |
$ | 8,386 | $ | 14,800 | $ | 6,414 | 76.5 | % | ||||||||
Percentage of net loss |
37.2 | % | 33.1 | % | ||||||||||||
General and administrative expenses were $14.8 million for the year ended September 30, 2008,
an increase of $6.4 million, or 76.5%, from $8.4 million for the year ended September 30, 2007.
This increase was primarily attributable to a $4.5 million increase in personnel expense. The
balance of the increase was attributable to increases in insurance expenses, depreciation expenses
and higher legal and consulting fees associated with becoming a public company. General and
administrative expenses for the year ended September 30, 2008 include $5.1 million in stock-based
compensation expense related to options granted to employees. Because we must revalue options
granted to non-employees for accounting purposes each reporting period, the amount of the
stock-based compensation expense for the year ended September 30, 2008 was $2 thousand.
Interest and Other Income.
| Year Ended | ||||||||||||||||
| September 30, | Increase | |||||||||||||||
| 2007 | 2008 | $ | % | |||||||||||||
| In thousands, except per share amounts | ||||||||||||||||
Interest and Other Income |
$ | 1,902 | $ | 3,010 | $ | 1,108 | 58.3 | % | ||||||||
Percentage of net loss |
8.4 | % | 6.8 | % | ||||||||||||
Interest and other income increased to $3.0 million for the year ended September 30, 2008 from
$1.9 million for the year ended September 30, 2007. The increase was due to higher balances of cash
and cash equivalents in 2008, resulting primarily from the $125.6 million in net proceeds received
from our initial public offering in May 2007 and follow-on offering in February 2008.
Interest Expense. For the years ended September 30, 2008 and 2007, we had no interest
expense.
Deemed Dividend — Warrants. On March 20, 2007, we offered the holders of warrants to
purchase an aggregate of 78,183 shares of Series B convertible preferred stock and an aggregate of
2,558,724 shares of common stock with an exercise price of $5.56 per share the opportunity to
exercise such warrants at an exercise price of $3.67, representing a 34% discount in the exercise
price. Such holders exercised all such warrants on a combination of cashless and cash exercise
basis. We issued 2,636,907 shares of common stock and received aggregate cash proceeds of $0.4
million in connection with such exercises. As a result of the discounted exercise price, we
recorded a non-cash deemed dividend of approximately $4.5 million for the warrants that were
exercised in the year ended September 30, 2007. No equivalent charge to stockholders was incurred
in the year ended September 30, 2008.
Net Loss Applicable to Common Stockholders and Net Loss per Share.
| Year Ended | ||||||||||||||||
| September 30, | Increase | |||||||||||||||
| 2007 | 2008 | $ | % | |||||||||||||
| In thousands, except per share amounts | ||||||||||||||||
Net loss applicable to common stockholders |
$ | (27,005 | ) | $ | (43,361 | ) | $ | 16,356 | 60.6 | % | ||||||
Net loss per share |
$ | (1.76 | ) | $ | (1.94 | ) | ||||||||||
Net loss applicable to common stockholders was $43.4 million, or $(1.94) per share, for the
year ended September 30, 2008 compared to $27.0 million, or $(1.76) per share, for the year ended
September 30, 2007. The increase in net loss was primarily attributable to the increased expenses
described above.
38
Table of Contents
Liquidity and Capital Resources
Sources of Liquidity and Cash Flows
As a result of our significant research and development expenditures and the lack of any
approved products or other sources of revenue, we have not been profitable and have generated
significant operating losses since we were incorporated in 2003. We have funded our research and
development operations primarily through proceeds from our Series A convertible preferred stock
financing in 2005 and our mezzanine and Series B convertible preferred stock financings in 2006.
Through December 31, 2006, we had received aggregate gross proceeds of $26.6 million from these
sales. We received an aggregate of $125.6 million from our initial public offering in May 2007 and
follow-on offering in February 2008.
At September 30, 2009, we had cash and cash equivalents totaling approximately $54.6 million.
To date, we have invested our excess funds primarily in managed money funds with two major
financial institutions. All highly liquid investments with an original maturity of less than three
months at the date of purchase are categorized as cash equivalents. We plan to continue to invest
our cash and cash equivalents in accordance with our approved investment policy guidelines, which
set forth our policy to hold investment securities to maturity.
Net cash used in operating activities was $35.3 million for the year ended September 30, 2009,
$34.9 million for the year ended September 30, 2008 and $15.5 million for the year ended September
30, 2007. Net cash used in operating activities for the year ended September 30, 2009 primarily
reflects the net loss for the period, offset in part by stock-based compensation, depreciation and
amortization expenses and a decrease in income tax receivable, accounts payable, income tax payable
and accrued expenses. Net cash used in operations for the years ended September 30, 2008 and 2007
primarily reflects the net loss for the period, offset in part by depreciation, share-base
compensation and changes in accrued expenses, accounts payable and deferred compensation.
Net cash provided by (used in) investing activities was $25.0 million for the year ended
September 30, 2009, ($28.4) million for the year ended September 30, 2008 and ($1.4) million for
the year ended September 30, 2007. Net cash provided by investing activities for the year ended
September 30, 2009 primarily reflects sale of marketable securities partially offset by the
purchase of property and equipment. Net cash used in investing activities for the years ended
September 30, 2008 and 2007 primarily reflects the purchases of marketable securities, property and
equipment and leasehold improvement costs.
Net cash provided by financing activities was $0.2 million for the year ended September 30,
2009, $48.0 million for the year ended September 30, 2008 and $79.4 million for the year ended
September 30, 2007. Net cash provided by financing activities in 2009 primarily reflects the
proceeds from sale of stock through our employee stock purchase plan. Net cash provided by
financing activities in 2008 primarily reflects proceeds from our follow-on public offering. Net
cash provided by financing activities in 2007 primarily reflects proceeds from our initial public
offering.
In July 2008, we entered into a supply agreement with N.V. Organon, which will terminate in
December 2011, to purchase specified minimum quantities of recombinant human insulin. We anticipate
that our minimum purchase requirements for the next nine consecutive quarters will total as much as
$12 million depending on our regulatory plans for VIAject®.
On February 12, 2008, we completed a follow-on public offering of 3,260,000 shares of our
common stock at a price to the public of $15.50 per share and received net proceeds from this
offering, after deducting underwriting discounts and commissions and expenses, of $46.8 million.
Certain of our stockholders sold 550,000 shares in the offering. We did not receive any proceeds
from the sale of shares from the selling stockholders.
On May 16, 2007, we completed an initial public offering of 5,750,000 shares of our common
stock at a price to the public of $15.00 per share. The offering resulted in gross proceeds of
$86.3 million. We received net proceeds from the offering of approximately $78.8 million after
deducting underwriting discounts and commissions and additional offering expenses. The remaining
net proceeds were invested in cash, cash equivalents and short-term investments, in accordance with
our investment policy.
Funding Requirements
We believe that our existing cash and cash equivalents will be sufficient to fund our
anticipated operating expenses and capital expenditures at least through the second quarter of
fiscal year 2011. We have based this estimate upon assumptions that may prove to be wrong and we
could use our available capital resources sooner than we currently expect. For example, if the FDA
were to require us to conduct an additional pivotal Phase 3 clinical trial of VIAject®, our
existing capital resources may not be sufficient to complete that clinical development program.
Because of the numerous risks and uncertainties associated with the development and
commercialization of our product candidates, and to the extent that we may or may not enter into
collaborations with third parties to participate in their development and commercialization, we are
unable to estimate the amounts of increased capital outlays and operating expenditures associated
with our current anticipated clinical trials.
Our future capital requirements will depend on many factors, including:
| • | our ability to secure approval by the FDA for VIAject® under Section 505(b)(2) of the FFDCA; | ||
| • | our ability to file our NDA for VIAject® in December 2009 as planned and, once filed, the length of time that will elapse before our NDA is fully reviewed by the FDA; |
39
Table of Contents
| • | the costs associated with preparing and submitting our MAA for VIAject® to the EMEA; | ||
| • | our ability to market, commercialize and achieve market acceptance for product candidates, particularly VIAject®; | ||
| • | our ability to secure approval by the FDA for VIAject® without conducting additional pivotal clinical trials; | ||
| • | the FDA’s findings regarding data anomalies observed in India in our Phase 3 clinical trial of VIAject® for patients with Type 1 diabetes and the impact of those findings on the timing of a regulatory approval; | ||
| • | the size, endpoints and duration of additional clinical trials of VIAject® in patients with Type 1 diabetes to support our commercialization efforts and, potentially, FDA approval; | ||
| • | the cost to fully develop the 100 IU/cc liquid formulation of VIAject®; | ||
| • | our ability to establish that the 100 IU/cc liquid formulation of VIAject® is well-tolerated in chronic use; | ||
| • | the cost to develop an insulin pen program for use with VIAject®; | ||
| • | the cost of purchasing recombinant human insulin and other materials to build commercial supply inventory for VIAject®, taking into account currency exchange rate fluctuations; | ||
| • | the costs of preparing, filing and prosecuting patent applications and maintaining, enforcing and defending intellectual property-related claims; | ||
| • | the cost associated with qualifying and obtaining regulatory approval of suppliers of insulin and manufacturers of our product candidates; | ||
| • | our ability to establish and maintain collaborations and the terms and success of the collaborations, including the timing and amount of payments that we might receive from potential strategic collaborators; and | ||
| • | the continued participation of patients in our VIAject® Phase 3 clinical trial extension program, which is designed to run through February 2010. |
We do not anticipate generating product revenue for the next few years. In the absence of
additional funding, we expect our continuing operating losses to result in increases in our cash
used in operations over the next several years. To the extent our capital resources are
insufficient to meet our future capital requirements, we will need to finance our future cash needs
through public or private equity offerings, debt financings or corporate collaboration and
licensing arrangements. We do not currently have any commitments for future external funding.
Additional equity or debt financing or corporate collaboration and licensing arrangements may
not be available on acceptable terms, if at all. If adequate funds are not available, we may be
required to delay, reduce the scope of or eliminate some or all of our research and development
programs, reduce our planned commercialization efforts or obtain funds through arrangements with
collaborators or others that may require us to relinquish rights to certain drug candidates that we
might otherwise seek to develop or commercialize independently or enter into corporate
collaborations at a later stage of development. In addition, any future equity funding will dilute
the ownership of our equity investors.
40
Table of Contents
Off-Balance Sheet Arrangements
We have no off-balance sheet arrangements.
Contractual Obligations
The following table summarizes our significant contractual obligations and commercial
commitments as of September 30, 2009 (in thousands):
| Less Than | More Than | |||||||||||||||||||
| Total | 1 Year | 1-3 Years | 3-5 Years | 5 Years | ||||||||||||||||
Operating lease obligations |
$ | 2,563 | $ | 537 | $ | 1,561 | $ | 465 | $ | — | ||||||||||
Purchase commitments |
12,268 | 5,025 | 7,243 | — | — | |||||||||||||||
Total fixed contractual obligations |
$ | 14,831 | $ | 5,562 | $ | 8,804 | $ | 465 | $ | — | ||||||||||
Recent Accounting Pronouncements
Effective October 1, 2008, the Company adopted the provisions ASC Topic 820, Fair Value
Measurements and Disclosures, which substantially incorporates the FASB Statement of Financial
Accounting Standards (SFAS), No. 157, Fair Value Measurements. These provisions define fair value,
establish a framework for measuring fair value in generally accepted accounting principles, and
expand disclosures about fair value measurements. Portions of these provisions have been deferred
for one year for certain non-financial assets and liabilities. The adoption of these provisions did
not have a material effect on our financial statements.
During the fourth quarter of 2009, the Company adopted the FASB Accounting Standards Update
(“ASU”) No. 2009-01, “Amendments based on Statement of Financial Accounting Standards No. 168 —
The FASB Accounting Standards Codification and the Hierarchy of Generally Accepted Accounting
Principles” (the “Codification”). The Codification became the single source of authoritative GAAP
in the United States, and other than rules and interpretive releases issued by the United States
Securities and Exchange Commission (“SEC”). The Codification reorganized generally accepted
accounting principles, or GAAP, into a topical format that eliminates the previous GAAP hierarchy
and instead established two levels of guidance — authoritative and nonauthoritative. All
non-grandfathered, non-SEC accounting literature that was not included in the Codification became
nonauthoritative. The adoption of the Codification did not change previous GAAP, but rather
simplified user access to all authoritative literature related to a particular accounting topic in
one place. Accordingly, the adoption had no impact on the Company’s consolidated financial
position and results of operations. All prior references to previous GAAP in the Company’s
consolidated financial statements were updated for the new references under the Codification.
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
Our exposure to market risk is limited to our cash, cash equivalents and marketable
securities. We invest in high-quality financial instruments, as permitted by the terms of our
investment policy guidelines. Currently, our investments are primarily limited to highly liquid
money market investments. A portion of our investments may be subject to interest rate risk and
could fall in value if interest rates were to increase. The effective duration of our portfolio is
currently less than one year, which we believe limits interest rate and credit risk. We do not
hedge interest rate exposure.
Pursuant to our supply agreement with N.V. Organon, our purchases of insulin are denominated
in Euros. Most of our other transactions are denominated in United States dollars and do not
present a material exposure to fluctuations in currency exchange rates.
| ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
Refer to page F-1.
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURES |
Not applicable.
| ITEM 9A. | CONTROLS AND PROCEDURES |
Management’s Evaluation of Disclosure Controls and Procedures
We are required to maintain disclosure controls and procedures designed to ensure that
material information related to us is recorded, processed, summarized and reported within the time
periods specified in the SEC rules and forms. The term “disclosure controls and procedures,” as
defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, or
the
41
Table of Contents
Exchange Act, means controls and other procedures of a company that are designed to ensure that
information required to be disclosed by a company in the reports that it files or submits under the
Exchange Act is recorded, processed, summarized and reported, within the time periods specified in
the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls
and procedures designed to ensure that information required to be disclosed by a company in the
reports that it files or submits under the Exchange Act is accumulated and communicated to the
company’s management, including its principal executive and principal financial officers, as
appropriate to allow timely decisions regarding required disclosure. Management recognizes that any
controls and procedures, no matter how well designed and operated, can provide only reasonable
assurance of achieving their objectives and management necessarily applies its judgment in
evaluating the cost-benefit relationship of possible controls and procedures.
Our management, with the participation of our chief executive officer and chief financial
officer, evaluated the effectiveness of our disclosure controls and procedures as of September 30,
2009 and, based on this evaluation, our chief executive officer and chief financial officer have
concluded that, as of the end of the period covered by this report, our disclosure controls and
procedures were effective.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over
financial reporting for the company. Internal control over financial reporting is defined in Rule
13a-15(f) or 15d-15(f) promulgated under the Securities Exchange Act of 1934 as a process designed
by, or under the supervision of, the company’s principal executive and principal financial officers
and effected by the company’s board of directors, management and other personnel, to provide
reasonable assurance regarding the reliability of financial reporting and the preparation of
financial statements for external purposes in accordance with generally accepted accounting
principles and includes those policies and procedures that:
| • | pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of the company; | ||
| • | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and | ||
| • | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent
or detect misstatements. Projections of any evaluation of effectiveness to future periods are
subject to the risk that controls may become inadequate because of changes in conditions, or that
the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as
of September 30, 2009. In making this assessment, our management used the criteria set forth by the
Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal
Control-Integrated Framework.
Based on our assessment, management concluded that, as of September 30, 2009, our internal
control over financial reporting is effective based on those criteria.
Our independent registered public accountants have issued an audit report on our assessment of
our internal control over financial reporting. This report appears below.
42
Table of Contents
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders
Biodel Inc.
Danbury, Connecticut
Biodel Inc.
Danbury, Connecticut
We have audited Biodel Inc.’s (a development stage company) internal control over financial
reporting as of September 30, 2009, based on criteria established in Internal Control — Integrated
Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO
criteria). Biodel Inc.’s management is responsible for maintaining effective internal control over
financial reporting and for its assessment of the effectiveness of internal control over financial
reporting, included in the accompanying “Item 9A, Controls and Procedures, Management’s Annual
Report on Internal Control over Financial Reporting”. Our responsibility is to express an opinion
on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting
Oversight Board (United States). Those standards require that we plan and perform the audit to
obtain reasonable assurance about whether effective internal control over financial reporting was
maintained in all material respects. Our audit included obtaining an understanding of internal
control over financial reporting, assessing the risk that a material weakness exists, and testing
and evaluating the design and operating effectiveness of internal control based on the assessed
risk. Our audit also included performing such other procedures as we considered necessary in the
circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide
reasonable assurance regarding the reliability of financial reporting and the preparation of
financial statements for external purposes in accordance with generally accepted accounting
principles. A company’s internal control over financial reporting includes those policies and
procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately
and fairly reflect the transactions and dispositions of the assets of the company; (2) provide
reasonable assurance that transactions are recorded as necessary to permit preparation of financial
statements in accordance with generally accepted accounting principles, and that receipts and
expenditures of the company are being made only in accordance with authorizations of management and
directors of the company; and (3) provide reasonable assurance regarding prevention or timely
detection of unauthorized acquisition, use, or disposition of the company’s assets that could have
a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent
or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are
subject to the risk that controls may become inadequate because of changes in conditions, or that
the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Biodel Inc. maintained, in all material respects, effective internal control
over financial reporting as of September 30, 2009, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting
Oversight Board (United States), the balance sheets of Biodel Inc. as of September 30, 2009 and
2008, and the related statements of operations, stockholders’ equity, and cash flows for each of
the three years in the period ended September 30, 2009 and for the period from December 3, 2003 to
September 30, 2009 and our report dated December 14, 2009 expressed an unqualified opinion thereon.
/s/ BDO Seidman, LLP
New York, New York
December 14, 2009
New York, New York
December 14, 2009
Changes in Internal Control over Financial Reporting
No change in our internal control over financial reporting occurred during the fiscal quarter
ended September 30, 2009 that has materially affected, or is reasonably likely to materially
affect, our internal control over financial reporting.
43
Table of Contents
| ITEM 9B. | OTHER INFORMATION |
On December 14, 2009, the Company entered into indemnification agreements with certain of its
directors and executive officers. Each indemnification agreement is in the form currently in
effect between Company and its other directors and executive officers.
44
Table of Contents
PART III
Certain information required by Part III is omitted from this Annual Report on Form 10-K
because we will file a definitive proxy statement within 120 days after the end of our fiscal year
for our 2009 annual meeting of stockholders, or proxy statement, and the information included in
the proxy statement is incorporated herein by reference.
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
Certain information required by this Item is contained under the heading “Executive Officers
of the Registrant” in Part I of this Annual Report on Form 10-K. Other information required by this
Item will appear under the headings “Election of Directors”, “Section 16(a) Beneficial Ownership
Reporting Compliance” and “Corporate Governance” in our proxy statement, which sections are
incorporated herein by reference.
We have adopted a written code of business conduct and ethics that applies to our principal
executive officer, principal financial officer, and principal accounting officer or controller, or
persons performing similar functions. Our code of business conduct and ethics, which also applies
to our directors and all of our officers and employees, can be found on our website, which is
located at www.biodel.com. We intend to disclose any amendments to, or waivers from, our
code of business conduct and ethics that are required to be publicly disclosed pursuant to rules of
the Securities and Exchange Commission and the NASDAQ Global Market by filing such amendment or
waiver with the Securities and Exchange Commission and by posting it on our website.
| ITEM 11. | EXECUTIVE COMPENSATION |
The information required by this Item will appear under the heading “Executive Compensation”
including “Compensation Discussion and Analysis”, “Director Compensation”, “Compensation Committee
Interlocks and Insider Participation” and “Compensation Committee Report” in our proxy statement,
which sections are incorporated herein by reference.
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required by this Item will appear under the headings “Security Ownership of
Certain Beneficial Owners and Management” and “Securities Authorized for Issuance under Equity
Compensation Plans” in our proxy statement, which sections are incorporated herein by reference.
| ITEM 13. | CERTAIN RELATIONSHIP AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information required by this Item will appear under the headings “Certain Relationships and
Related Transactions” and “Corporate Governance” in our proxy statement, which sections are
incorporated herein by reference.
| ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The information required by this Item will appear under the heading “Auditors’ Fees” in our proxy
statement, which section is incorporated herein by reference.
PART IV
| ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES |
(1) Financial Statements: See Index to Financial Statements and Schedules.
(2) Financial Statement Schedules: Not applicable.
(3) Exhibits: The Exhibit Index annexed to this report is incorporated by reference.
45
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934,
the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto
duly authorized.
| BIODEL INC. |
||||
| By: | /s/ Solomon S. Steiner | |||
| Dr. Solomon S. Steiner | ||||
| President and Chief Executive Officer Date: December 14, 2009 |
||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been
signed below by the following persons on behalf of the registrant and in the capacities and on the
dates indicated.
| Signature | Title | Date | ||
/s/ Solomon S. Steiner
|
Chairman of the Board, President and Chief Executive Officer (Principal Executive Officer) | December 14, 2009 | ||
/s/ Gerard Michel
|
Chief Financial Officer, Vice President, Corporate Development and Treasurer (Principal Financial and Accounting Officer) | December 14, 2009 | ||
/s/ Barry Ginsberg
|
Director | December 14, 2009 | ||
/s/ David Kroin
|
Director | December 14, 2009 | ||
/s/ Ira W. Lieberman
|
Director | December 14, 2009 | ||
/s/ Daniel Lorber
|
Director | December 14, 2009 | ||
/s/ Brian J.G. Pereira
|
Director | December 14, 2009 | ||
/s/ Charles Sanders
|
Director | December 14, 2009 | ||
/s/ Scott A. Weisman
|
Director | December 14, 2009 |
46
Table of Contents
Exhibits Index
| Exhibit | ||
| Number | Description of Document | |
3.1
|
Registrant’s Second Amended and Restated Certificate of Incorporation (Incorporated by reference to the exhibits to the Registrant’s Registration Statement on Form S-1 (333-140504)). | |
3.2
|
Registrant’s Amended and Restated Bylaws (Incorporated by reference to the exhibits to the Registrant’s Registration Statement on Form S-1 (333-140504)). | |
4.1
|
Specimen Common Stock Certificate (Incorporated by reference to the exhibits to the Registrant’s Registration Statement on Form S-1 (333-140504)). | |
4.2
|
Form of Warrant issued to Scott Weisman and McGinn Smith Holdings LLC to Purchase Shares of Series A convertible preferred stock (Incorporated by reference to the exhibits to the Registrant’s Registration Statement on Form S-1 (333-140504)). | |
4.3
|
Form of Subscription and Rights Agreement by and among the Registrant and the holders of the Series A convertible preferred stock (Incorporated by reference to the exhibits to the Registrant’s Registration Statement on Form S-1 (333-140504)). | |
4.4
|
Amended and Restated Registration Rights Agreement, dated September 19, 2006, by and among the Registrant and other parties named therein (Incorporated by reference to the exhibits to the Registrant’s Registration Statement on Form S-1 (333-140504)). | |
10.1
|
Form of Indemnity Agreement entered into between the Registrant and each of Albert Cha, Robert Feldstein, David Kroin, Daniel Lorber, Ira Lieberman, Charles Sanders, Roderike Pohl, and Solomon Steiner, Paul Sekhri, Erik Steiner, Samuel Wertheimer, R. Timmis Ware, Andreas Pfützner, and Scott Weisman (Incorporated by reference to the exhibits to the Registrant’s Registration Statement on Form S-1 (333-140504)). | |
10.2
|
Amended and Restated 2004 Stock Incentive Plan (Incorporated by reference to the exhibits to the Registrant’s Registration Statement on Form S-1 (333-140504)). | |
10.3
|
2005 Employee Stock Purchase Plan (Incorporated by reference to the exhibits to the Registrant’s Registration Statement on Form S-1 (333-140504)). | |
10.4
|
2005 Non-Employee Directors’ Stock Option Plan (Incorporated by reference to the exhibits to the Registrant’s Registration Statement on Form S-1 (333-140504)). | |
10.5
|
Amended and Restated Employment Agreement, dated March 20, 2007, as amended November 20, 2007, between the Registrant and Solomon S. Steiner (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on November 20, 2007). | |
10.7
|
Amended and Restated Consulting Agreement entered into on November 13, 2007, effective June 5, 2007, between the Registrant and Dr. Andreas Pfützner (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on November 14, 2007). | |
10.8†
|
Manufacturing Agreement, dated December 20, 2005 between the Registrant and Cardinal Health — PTS, LLC (Incorporated by reference to the exhibits to the Registrant’s Registration Statement on Form S-1 (333-140504)). | |
10.9
|
Change of Control Agreement entered into between the Registrant and certain of its executive officers (Incorporated by reference to the exhibits to the Registrant’s Registration Statement on Form S-1 (333-140504)). | |
10.10
|
Executive Severance Agreement entered into between the Registrant and certain of its executive officers (Incorporated by reference to the exhibits to the Registrant’s Registration Statement on Form S-1 (333-140504)). | |
10.11
|
Lease Agreement, dated February 2, 2004, between the Registrant and Mulvaney Properties, LLC and amendment thereto dated September 29, 2006 (Incorporated by reference to the exhibits to the Registrant’s Registration Statement on Form S-1 (333-140504)). | |
10.12
|
Commercial Lease, dated July 23, 2007, by and between the Registrant and Mulvaney Properties LLC. (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on July 27, 2007). |
47
Table of Contents
| Exhibit | ||
| Number | Description of Document | |
10.13
|
Lease Amendment, dated October 1, 2007, between the Registrant and Mulvaney Properties LLC (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on October 4, 2007). | |
10.14
|
Amendment to Lease Agreement, dated February 2, 2004, as amended, by and between the Registrant and Mulvaney Properties LLC. (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on July 27, 2007). | |
10.15
|
Severance Agreement, dated November 14, 2007, by and between the Registrant and F. Scott Reding (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on November 14, 2007). | |
10.16
|
Offer Letter, dated November 12, 2007, by and between the Registrant and Gerard J. Michel. (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on November 14, 2007). | |
10.17
|
Form of Incentive Stock Option Agreement for 2004 Amended and Restated Stock Incentive Plan. (Incorporated by reference to the Registrant’s Annual Report on Form 10-K filed on December 21, 2007). | |
10.18
|
Form of Option Agreement for 2005 Non-Employee Directors’ Stock Option Plan. (Incorporated by reference to the Registrant’s Annual Report on Form 10-K filed on December 21, 2007). | |
10.19
|
Base salaries of Executive Officers of the Registrant. | |
10.20
|
Summary of the Registrant’s Non-Employee Director Compensation. | |
10.21†
|
Supply Agreement, dated July 7, 2008, between the Registrant and N.V. Organon (Incorporated by reference to the Registrant’s Quarterly Report on Form 10-Q filed on August 11, 2008). | |
10.22*
|
Letter Agreement, dated November 12, 2009, between the Registrant and N.V. Organon, amending the Supply Agreement, dated July 7, 2008, between the parties. | |
21.1
|
Subsidiaries of the Registrant. | |
23.1
|
Consent of BDO Seidman, LLP, Independent Registered Public Accounting Firm. | |
24.1
|
Powers of Attorney (included on signature page). | |
31.01
|
Chief Executive Officer — Certification pursuant to Rule 13a-14(a) or Rule 15d-14(a) of the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
31.02
|
Chief Financial Officer — Certification pursuant to Rule 13a-14(a) or Rule 15d-14(a) of the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
32.01
|
Chief Executive Officer and Chief Financial Officer — Certification pursuant to Rule 13a-14(b) or Rule 15d-14(b) of the Securities Exchange Act of 1934 and 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| † | Confidential treatment granted with respect to certain portions of this exhibit. Omitted portions have been filed separately with the Securities and Exchange Commission. | |
| * | Portions of this exhibit (indicated by asterisks) have been omitted pursuant to a request for confidential treatment and have been filed separately with the Securities and Exchange Commission. |
48
Table of Contents
BIODEL INC.
INDEX TO FINANCIAL STATEMENTS
| Page | ||||
| F-2 | ||||
| F-3 | ||||
| F-4 | ||||
| F-5 | ||||
| F-7 | ||||
| F-8 | ||||
F-1
Table of Contents
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders
Biodel Inc.
Danbury, Connecticut
Biodel Inc.
Danbury, Connecticut
We have audited the accompanying balance sheets of Biodel Inc. (a development stage company)
as of September 30, 2009 and 2008 and the related statements of operations, stockholders’ equity
and cash flows for each of the three years in the period ended September 30, 2009 and for the
period from December 3, 2003 (inception) to September 30, 2009. These financial statements are the
responsibility of the Company’s management. Our responsibility is to express an opinion on these
financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting
Oversight Board (United States). Those standards require that we plan and perform the audit to
obtain reasonable assurance about whether the financial statements are free of material
misstatement. An audit also includes examining, on a test basis, evidence supporting the amounts
and disclosures in the financial statements, assessing the accounting principles used and
significant estimates made by management, as well as evaluating the overall financial statement
presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material
respects, the financial position of Biodel Inc. at September 30, 2009 and 2008, and the results of
its operations and its cash flows for each of the three years in the period ended September 30,
2009, and for the period from December 3, 2003 (inception) to September 30, 2009, in conformity
with accounting principles generally accepted in the United States.
We also have audited, in accordance with the standards of the Public Company Accounting
Oversight Board (United States), Biodel Inc.’s internal control over financial reporting as of
September 30, 2009, based on criteria established in Internal Control — Integrated Framework issued
by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) and our report dated
December 14, 2009 expressed an unqualified opinion thereon.
/s/ BDO Seidman, LLP
New York, New York
December 14, 2009
December 14, 2009
F-2
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Balance Sheets
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
| September 30, | ||||||||
| 2008 | 2009 | |||||||
ASSETS |
||||||||
Current: |
||||||||
Cash and cash equivalents |
$ | 64,731 | $ | 54,640 | ||||
Marketable securities, available for sale |
25,552 | — | ||||||
Taxes receivable |
1,988 | 752 | ||||||
Prepaid and other assets |
1,130 | 482 | ||||||
Total current assets |
93,401 | 55,874 | ||||||
Property and equipment, net |
3,931 | 3,695 | ||||||
Intellectual property, net |
59 | 56 | ||||||
Other assets |
120 | — | ||||||
Total assets |
$ | 97,511 | $ | 59,625 | ||||
LIABILITIES AND STOCKHOLDERS’ EQUITY |
||||||||
Current: |
||||||||
Accounts payable |
$ | 813 | $ | 1,007 | ||||
Accrued expenses: |
||||||||
Clinical trial expenses |
4,163 | 5,647 | ||||||
Payroll and related |
1,420 | 1,117 | ||||||
Accounting and legal fees |
509 | 325 | ||||||
Severance |
268 | 183 | ||||||
Other |
839 | 643 | ||||||
Income taxes payable |
1,012 | 165 | ||||||
Total current liabilities |
9,024 | 9,087 | ||||||
Commitments |
||||||||
Stockholders’ equity: |
||||||||
Preferred stock, $.01 par value; 50,000,000 shares authorized, none outstanding |
— | — | ||||||
Common stock, $.01 par value; 100,000,000 shares authorized; 23,698,558 and
23,803,672 issued and outstanding |
237 | 238 | ||||||
Additional paid-in capital |
171,506 | 176,764 | ||||||
Accumulated other comprehensive loss |
(62 | ) | — | |||||
Deficit accumulated during the development stage |
(83,194 | ) | (126,464 | ) | ||||
Total stockholders’ equity |
88,487 | 50,538 | ||||||
Total liabilities and stockholders’ equity |
$ | 97,511 | $ | 59,625 | ||||
See accompanying notes to financial statements.
F-3
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Statements of Operations
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
| December 3, | ||||||||||||||||
| 2003 | ||||||||||||||||
| (Inception) to | ||||||||||||||||
| Year Ended September 30, | September 30, | |||||||||||||||
| 2007 | 2008 | 2009 | 2009 | |||||||||||||
Revenue |
$ | — | $ | — | $ | — | $ | — | ||||||||
Operating expenses: |
||||||||||||||||
Research and development |
15,939 | 32,554 | 32,325 | 90,051 | ||||||||||||
General and administrative |
8,386 | 14,800 | 10,994 | 36,645 | ||||||||||||
Total operating expenses |
24,325 | 47,354 | 43,319 | 126,696 | ||||||||||||
Other (income) and expense: |
||||||||||||||||
Interest and other income |
(1,902 | ) | (3,010 | ) | (386 | ) | (5,489 | ) | ||||||||
Interest expense |
— | — | — | 78 | ||||||||||||
Loss on settlement of debt |
— | — | — | 627 | ||||||||||||
Operating loss before tax provision
(benefit) |
(22,423 | ) | (44,344 | ) | (42,933 | ) | (121,912 | ) | ||||||||
Tax provision (benefit) |
125 | (983 | ) | 337 | (508 | ) | ||||||||||
Net loss |
(22,548 | ) | (43,361 | ) | (43,270 | ) | (121,404 | ) | ||||||||
Charge for accretion of beneficial
conversion rights |
— | — | — | (603 | ) | |||||||||||
Deemed dividend — warrants |
(4,457 | ) | — | — | (4,457 | ) | ||||||||||
Net loss applicable to common
stockholders |
$ | (27,005 | ) | $ | (43,361 | ) | $ | (43,270 | ) | $ | (126,464 | ) | ||||
Net loss per share — basic and diluted |
$ | (1.76 | ) | $ | (1.94 | ) | $ | (1.82 | ) | |||||||
Weighted average shares outstanding —
basic and diluted |
15,354,898 | 22,390,434 | 23,746,598 | |||||||||||||
See accompanying notes to financial statements.
F-4
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Statements of Stockholders’ Equity
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
| Deficit | ||||||||||||||||||||||||||||||||||||||||
| Accumulated | Accumulated | |||||||||||||||||||||||||||||||||||||||
| Common Stock | Series A Preferred stock | Series B Preferred stock | Additional | Other | During the | Total | ||||||||||||||||||||||||||||||||||
| $01 Par Value | $.01 Par Value | $.01 Par Value | Paid in | Comprehensive | Development | Stockholders’ | ||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Capital | Income (loss) | Stage | Equity | |||||||||||||||||||||||||||||||
Shares issued to employees |
732,504 | $ | 7 | — | $ | — | — | $ | — | $ | (7 | ) | $ | $ | — | $ | — | |||||||||||||||||||||||
January 2004 Proceeds from sale of common stock |
4,581,240 | 46 | — | — | — | — | 1,308 | — | 1,354 | |||||||||||||||||||||||||||||||
Net loss |
— | — | — | — | — | — | — | (774 | ) | (774 | ) | |||||||||||||||||||||||||||||
Balance, September 30, 2004 |
5,313,744 | 53 | — | — | — | — | 1,301 | — | (774 | ) | 580 | |||||||||||||||||||||||||||||
Additional stockholder contributions |
— | — | — | — | — | — | 514 | — | 514 | |||||||||||||||||||||||||||||||
Share-based compensation |
— | — | — | — | — | — | 353 | — | 353 | |||||||||||||||||||||||||||||||
Shares issued to employees and directors for services |
42,656 | 1 | — | — | — | — | 60 | — | 61 | |||||||||||||||||||||||||||||||
July 2005 Private placement — Sale of Series A preferred stock, net of issuance costs of $379 |
— | — | 569,000 | 6 | — | — | 2,460 | — | 2,466 | |||||||||||||||||||||||||||||||
Founder’s compensation contributed to capital |
— | — | — | — | — | — | 63 | — | 63 | |||||||||||||||||||||||||||||||
Net loss |
— | — | — | — | — | — | — | — | (3,383 | ) | (3,383 | ) | ||||||||||||||||||||||||||||
Balance, September 30, 2005 |
5,356,400 | 54 | 569,000 | 6 | — | — | 4,751 | — | (4,157 | ) | 654 | |||||||||||||||||||||||||||||
Share-based compensation |
— | — | — | — | — | — | 1,132 | — | 1,132 | |||||||||||||||||||||||||||||||
July 2006 Private placement — Sale of Series B preferred stock, net of issuance costs of $1,795 |
— | — | — | — | 5,380,711 | 54 | 19,351 | — | 19,405 | |||||||||||||||||||||||||||||||
July 2006 — Series B preferred stock units issued July 2006 to settle debt |
— | — | — | — | 817,468 | 8 | 3,194 | — | 3,202 | |||||||||||||||||||||||||||||||
Shares issued to employees and directors for services |
4,030 | — | — | — | — | — | 23 | — | 23 | |||||||||||||||||||||||||||||||
Accretion of fair value of beneficial conversion charge |
— | — | — | — | — | — | 603 | (603 | ) | — | ||||||||||||||||||||||||||||||
Net loss |
— | — | — | — | — | — | — | — | (8,068 | ) | (8,068 | ) | ||||||||||||||||||||||||||||
Balance, September 30, 2006 |
5,360,430 | 54 | 569,000 | 6 | 6,198,179 | 62 | 29,054 | — | (12,828 | ) | 16,348 | |||||||||||||||||||||||||||||
May 2007 Proceeds from sale of common stock |
5,750,000 | 58 | — | — | — | — | 78,697 | — | 78,755 | |||||||||||||||||||||||||||||||
Conversion of preferred stock on May 16, 2007 |
6,407,008 | 64 | (569,000 | ) | (6 | ) | (6,198,179 | ) | (62 | ) | 4 | — | — | |||||||||||||||||||||||||||
Share-based compensation |
— | — | — | — | — | — | 4,224 | — | 4,224 | |||||||||||||||||||||||||||||||
Shares issued to employees, non-employees and directors for services |
2,949 | — | — | — | — | — | 16 | — | 16 | |||||||||||||||||||||||||||||||
Stock options exercised |
3,542 | — | — | — | — | — | 5 | — | 5 | |||||||||||||||||||||||||||||||
March 2007 Warrants exercised |
2,636,907 | 26 | — | — | — | — | 397 | — | 423 | |||||||||||||||||||||||||||||||
Deemed dividend — warrants |
— | — | — | — | — | — | 4,457 | (4,457 | ) | — | ||||||||||||||||||||||||||||||
Net loss |
— | — | — | — | — | — | — | (22,548 | ) | (22,548 | ) | |||||||||||||||||||||||||||||
Balance, September 30, 2007 |
20,160,836 | 202 | — | — | — | — | 116,854 | — | (39,833 | ) | 77,223 | |||||||||||||||||||||||||||||
Proceeds from sale of common stock |
3,260,000 | 32 | — | — | — | — | 46,785 | — | 46,817 | |||||||||||||||||||||||||||||||
Issuance of restricted stock |
9,714 | — | — | — | — | — | 172 | — | 172 | |||||||||||||||||||||||||||||||
Share-based compensation |
— | — | — | — | — | — | 6,503 | — | 6,503 | |||||||||||||||||||||||||||||||
Stock options exercised |
174,410 | 1 | — | — | — | — | 901 | — | 902 | |||||||||||||||||||||||||||||||
Warrants exercised |
79,210 | 1 | — | — | — | — | 111 | — | 112 | |||||||||||||||||||||||||||||||
Net unrealized (loss) on Marketable Securities |
— | — | — | — | — | — | — | (62 | ) | — | (62 | ) | ||||||||||||||||||||||||||||
Proceeds from sale of stock — ESPP |
14,388 | 1 | — | — | — | — | 180 | — | 181 | |||||||||||||||||||||||||||||||
Net loss |
— | — | — | — | — | — | — | (43,361 | ) | (43,361 | ) | |||||||||||||||||||||||||||||
Balance, September 30, 2008 |
23,698,558 | $ | 237 | — | $ | — | — | $ | — | $ | 171,506 | $ | (62 | ) | $ | (83,194 | ) | $ | 88,487 | |||||||||||||||||||||
See accompanying notes to financial statements.
F-5
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Statements of Stockholders’ Equity
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
| Deficit | ||||||||||||||||||||||||||||||||||||||||
| Accumulated | Accumulated | |||||||||||||||||||||||||||||||||||||||
| Common Stock | Series A Preferred stock | Series B Preferred stock | Additional | Other | During the | Total | ||||||||||||||||||||||||||||||||||
| $01 Par Value | $.01 Par Value | $.01 Par Value | Paid in | Comprehensive | Development | Stockholders’ | ||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Capital | Income (loss) | Stage | Equity | |||||||||||||||||||||||||||||||
Balance, September 30, 2008 |
23,698,558 | $ | 237 | — | $ | — | — | $ | — | $ | 171,506 | $ | (62 | ) | $ | (83,194 | ) | $ | 88,487 | |||||||||||||||||||||
Share-based compensation |
5,064 | 5,064 | ||||||||||||||||||||||||||||||||||||||
Stock options exercised |
17,661 | — | — | — | — | — | 25 | — | — | 25 | ||||||||||||||||||||||||||||||
Net unrealized gain on
Marketable Securities |
— | — | — | — | — | — | — | 62 | — | 62 | ||||||||||||||||||||||||||||||
Proceeds from sale of stock —
ESPP |
87,453 | 1 | — | — | — | — | 169 | — | — | 170 | ||||||||||||||||||||||||||||||
Net loss |
(43,270 | ) | (43,270 | ) | ||||||||||||||||||||||||||||||||||||
Balance, September 30, 2009 |
23,803,672 | $ | 238 | — | $ | — | — | $ | — | $ | 176,764 | $ | — | $ | (126,464 | ) | $ | 50,538 | ||||||||||||||||||||||
See accompanying notes to financial statements.
F-6
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Statements of Cash Flows
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
| December 3, 2003 | ||||||||||||||||
| (Inception) to | ||||||||||||||||
| September 30, | ||||||||||||||||
| 2007 | 2008 | 2009 | 2009 | |||||||||||||
Cash flows from operating activities: |
||||||||||||||||
Net loss |
$ | (22,548 | ) | $ | (43,361 | ) | $ | (43,270 | ) | $ | (121,404 | ) | ||||
Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||||||
Depreciation and amortization |
254 | 567 | 877 | 2,149 | ||||||||||||
Founder’s compensation contributed to capital |
— | — | — | 271 | ||||||||||||
Share-based compensation for employees and directors |
3,567 | 6,434 | 4,970 | 15,204 | ||||||||||||
Share-based compensation for non-employees |
657 | 241 | 94 | 2,325 | ||||||||||||
Loss on settlement of debt |
— | — | — | 627 | ||||||||||||
Write-off of capitalized patent expense |
— | 208 | — | 208 | ||||||||||||
Write-off of loan to related party |
— | — | — | 41 | ||||||||||||
(Increase) decrease in: |
||||||||||||||||
Prepaid expenses and other assets |
(430 | ) | (745 | ) | 768 | (482 | ) | |||||||||
Income taxes receivable |
— | (1,988 | ) | 1,236 | (752 | ) | ||||||||||
Increase (decrease) in: |
||||||||||||||||
Accounts payable |
830 | (1,374 | ) | 194 | 1,007 | |||||||||||
Income tax payable |
255 | 917 | (847 | ) | 165 | |||||||||||
Deferred compensation |
(500 | ) | — | — | — | |||||||||||
Accrued expenses and other liabilities |
2,406 | 4,198 | 715 | 8,133 | ||||||||||||
Total adjustments |
7,039 | 8,458 | 8,007 | 28,896 | ||||||||||||
Net cash used in operating activities |
(15,509 | ) | (34,903 | ) | (35,263 | ) | (92,508 | ) | ||||||||
Cash flows from investing activities: |
||||||||||||||||
Purchase of property and equipment |
(1,315 | ) | (2,769 | ) | (637 | ) | (5,809 | ) | ||||||||
Purchase of marketable securities |
— | (25,614 | ) | — | (25,614 | ) | ||||||||||
Sale of marketable securities |
— | — | 25,614 | 25,614 | ||||||||||||
Capitalized intellectual properties |
(66 | ) | (17 | ) | — | (298 | ) | |||||||||
Loan to related party |
— | — | — | (41 | ) | |||||||||||
Net cash provided by (used in) investing activities |
(1,381 | ) | (28,400 | ) | 24,977 | (6,148 | ) | |||||||||
Cash flows from financing activities: |
||||||||||||||||
Options exercised |
5 | 902 | 25 | 932 | ||||||||||||
Warrants exercised |
423 | 112 | — | 535 | ||||||||||||
Deferred public offering costs |
(1,268 | ) | — | — | (1,458 | ) | ||||||||||
Stockholder contribution |
— | — | — | 1,660 | ||||||||||||
Net proceeds from sale of Series A preferred stock |
— | — | — | 2,466 | ||||||||||||
Net proceeds from employee stock purchase plan |
— | 181 | 170 | 351 | ||||||||||||
Net proceeds from sale of common stock |
80,213 | 46,817 | — | 127,030 | ||||||||||||
Proceeds from bridge financing |
— | — | — | 2,575 | ||||||||||||
Net proceeds from sale of Series B preferred stock |
— | — | — | 19,205 | ||||||||||||
Net cash provided by financing activities |
79,373 | 48,012 | 194 | 153,295 | ||||||||||||
Net increase (decrease) in cash and cash equivalents |
62,483 | (15,291 | ) | (10,091 | ) | 54,640 | ||||||||||
Cash and cash equivalents, beginning of period |
17,539 | 80,022 | 64,731 | — | ||||||||||||
Cash and cash equivalents, end of period |
$ | 80,022 | $ | 64,731 | $ | 54,640 | $ | 54,640 | ||||||||
Supplemental disclosures of cash flow information: |
||||||||||||||||
Cash paid for interest and income taxes was: |
||||||||||||||||
Interest |
$ | — | $ | — | $ | — | $ | 9 | ||||||||
Income taxes |
44 | 88 | 111 | 246 | ||||||||||||
Non-cash financing and investing activities: |
||||||||||||||||
Settlement of debt with Series B preferred stock |
$ | — | $ | — | $ | — | $ | 3,202 | ||||||||
Accrued expenses settled with Series B preferred stock |
— | — | — | 150 | ||||||||||||
Deemed dividend — warrants |
4,457 | — | — | 4,457 | ||||||||||||
Accretion of fair value of beneficial charge on preferred stock |
— | — | — | 603 | ||||||||||||
Conversion of convertible preferred stock to common stock |
68 | — | — | 68 | ||||||||||||
See accompanying notes to financial statements.
F-7
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statement
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
1. Business and Basis of Presentation
Business
Biodel Inc. (“Biodel” or the “Company”, and formerly Global Positioning Group Ltd.) is a
development stage specialty pharmaceutical company located in Danbury, Connecticut. The Company was
incorporated in the State of Delaware on December 3, 2003 and commenced operations in January 2004.
The Company is focused on the development and commercialization of innovative treatments for
diabetes. The Company develops product candidates by applying its proprietary formulation
technologies to existing drugs in order to improve their therapeutic results. The Company’s initial
development efforts are focused on peptide hormones. The Company’s most advanced product candidate,
VIAject®, has been studied in two pivotal Phase 3 clinical trials for the treatment of patients
with Type 1 and Type 2 diabetes. Earlier stage product candidates include VIAtab™, a sublingual
tablet formulation of insulin. The Company has developed all of its product candidates utilizing
its proprietary VIAdel™ technology that allows the Company to study the interaction between peptide
hormones and small molecules.
Basis of Presentation
The Company is in the development stage as its primary activities since incorporation have
been establishing its facilities, recruiting personnel, conducting research and development,
business development, business and financial planning and raising capital.
On April 12, 2007, the Company effected a 0.7085 for one (0.7085:1) reverse stock split (see
Note 11). All references in these financial statements and accompanying notes to units of common
stock or per share amounts are reflective of the reverse split for all periods reported.
2. Summary of Significant Accounting Policies
Research and Development Costs
The Company is in the business of research and development and, therefore, research and
development costs include, but are not limited to, salaries and benefits, lab supplies, preclinical
fees, clinical trial and related clinical manufacturing costs, allocated overhead costs and
professional service providers. Research and development costs are expensed when incurred. Research
and development costs aggregated $15,939, $32,554, and $32,325 for the years ended September 30,
2007, 2008 and 2009, respectively.
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles
requires management to make estimates and assumptions that affect the reported amounts of assets
and liabilities and disclosure of contingent assets and liabilities at the date of the financial
statements and the reported amounts of revenues and expenses during the reporting period. On an
ongoing basis, the Company evaluates its estimates and assumptions including, but not limited to,
accruals, income taxes payable, and deferred tax assets. Actual results may differ from those
estimates.
Cash and Cash Equivalents
The Company considers currency on hand, demand deposits and all highly liquid investments with
an original maturity of three months or less at the date of purchase to be cash and cash
equivalents. At September 30, 2009, cash and cash equivalents of $54,640 are primarily held in
U.S. treasury denominated, money market accounts.
Marketable securities
The Company’s marketable securities were classified as available-for-sale and were reported at
market value with unrealized gains and losses shown as a component of accumulated other
comprehensive income (loss). The Company regularly evaluates the performance of these investments
individually for impairment, taking into consideration the investment, volatility and current
returns. If a determination is made that a decline in fair value is other-than-temporary, the
related fund is written down to its estimated fair value. At September 30, 2008, marketable
securities total $25,552. Due to the short-term need for funds and uncertainty in the credit and
financial market, the Company modified its investment strategy and primarily invested in money
market accounts comprised of treasury securities. At September 30, 2009, the Company had no
marketable security investments.
F-8
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statement — (Continued)
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
Fair Value of Financial Instruments
The carrying amounts of the Company’s financial instruments, which include cash and cash
equivalents, accounts payable, and accrued expenses approximate their fair values due to their
short maturities.
Pre-Launch Inventory
Inventory costs associated with products that have not yet received regulatory approval are
capitalized if the Company believes there is probable future commercial use and future economic
benefit. If the probability of future commercial use and future economic benefit cannot be
reasonably determined, then costs associated with pre-launch inventory that has not yet received
regulatory approval are expensed as research and development expense during the period the costs
are incurred. For the year ended September 30, 2009, the Company expensed $6.5 million of costs
associated with the purchase of recombinant human insulin, as research and development expense
after it passed quality control inspection by the Company and transfer of title occurred. The
Company plans on submitting the NDA application for VIAject® in December 2009. Until the Company
can determine the probability of VIAject® receiving regulatory approval, costs associated with the
purchase of recombinant human insulin will continue to be expensed as research and development (see
Note 7).
Intellectual Property
The intangible asset consists primarily of capitalized costs associated with VIAject® patents
and the purchase of two domain addresses. They are amortized using the straight-line method over
twenty years. If the Company determines that a patent will not result in future revenues, the cost
related to such patent will be expensed in full on the date of that determination. Amortization
expense for the years ended September 30, 2007, 2008 and 2009 was $13, $13 and $3, respectively.
Property and Equipment
Property and equipment are stated at cost, net of accumulated depreciation or amortization.
Major improvements are capitalized, while maintenance and repairs are expensed in the period the
cost is incurred. Property and equipment are depreciated over their estimated useful lives using
the straight-line method. Leasehold improvements are amortized using the straight-line method over
their estimated useful lives, or the remaining term of the lease, whichever is less. When assets
are retired or otherwise disposed of, the assets and related accumulated depreciation are removed
from the accounts and resulting gains or losses are included in other income (expense) in the
statement of operations. Estimated useful life for each asset category is as follows: Furniture and
fixtures — 7 years, Leasehold improvements — estimated useful life or remaining term of lease,
whichever is shorter, Laboratory equipment — 7 years, Manufacturing equipment 5 years, Device
development— 5 years, Facility equipment— 3 years and 7 years, Computer equipment — 5 years and
Computer software — 3 years.
Impairment of Long-Lived Assets
Whenever events or changes in circumstances indicate that the carrying amounts of a long-lived
asset may not be recoverable, the Company reviews these assets for impairment and determines
whether adjustments are needed to carrying values. There were no adjustments to the carrying value
of long-lived assets at September 30, 2008 and 2009.
Comprehensive Income (Loss)
The Company classifies its marketable securities as available for sale. Other Comprehensive
Income (Loss) include changes in equity for unrealized holding gains/(losses) on marketable
securities, which have arose during the period. During the year ended September 30, 2009,
marketable security investments were sold at par value and the September 30, 2008 unrealized loss
of $62 was fully recaptured.
Income Taxes
The Company uses the asset and liability method of accounting for deferred income taxes. The
provision for income taxes includes income taxes currently payable and those deferred as a result
of temporary differences between the financial statement and tax bases of assets and liabilities. A
valuation allowance is provided to reduce deferred tax assets to the amount of future tax benefit
when it is more likely than not that some portion of the deferred tax assets will not be realized.
Projected future taxable income and ongoing tax planning strategies are considered and evaluated
when assessing the need for a valuation allowance. Any increase or decrease in a valuation
allowance could have a material adverse or beneficial impact on the Company’s income tax provision
and net income or loss in the period which the determination is made.
F-9
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statements — (Continued)
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
The Company adopted the provisions of Accounting Standards Codification (ASC) Topic 740,
Income Taxes, with respect to uncertain tax positions (substantially incorporating the Financial
Accounting Standards Board (FASB) Interpretation No. 48, Accounting for Uncertainty in Income
Taxes, or FIN 48) effective October 1, 2007. These provisions clarify the accounting for
uncertainty in income taxes recognized in an enterprise’s financial statements. Recognition
thresholds and measurement attributes were prescribed for financial statement recognition and
measurement of a tax position taken or expected to be taken in a tax return. Guidance was also
provided on derecognition, classification, interest and penalties, accounting in interim periods,
disclosures and transition. The adoption had no resulting effect on our financial statements. See
Note 8 for additional information.
Concentration of Risks and Uncertainties
Financial instruments that potentially subject the Company to a concentration of credit risk
consist of cash and cash equivalents. The Company deposits excess cash with major financial
institutions in the United States. Balances may exceed the amount of insurance provided on such
deposits. The Company believes that its investment policy guideline for its excess cash maintains
safety and liquidity through its policies on credit requirements, diversification and investment
maturity.
The Company has experienced significant operating losses since inception. At September 30,
2009, the Company had a deficit accumulated during the development stage of $126,464. The Company
has generated no revenue to date. The Company has funded its operations to date principally from
the sale of securities. The Company expects to incur substantial additional operating losses for
the next several years and will need to obtain additional financing in order to complete the
clinical development of VIAject®, launch and commercialize VIAject®, if it receives regulatory
approval, and continue research and development programs. There can be no assurance that such
financing will be available or will be at terms acceptable to the Company.
The Company is currently developing its first product candidates and has no products that have
received regulatory approval. Any products developed by the Company will require approval from the
U.S. Food and Drug Administration (“FDA”) or foreign regulatory agencies prior to commercial sales.
There can be no assurance that the Company’s products will receive the necessary approvals. If the
Company is denied such approvals or such approvals are delayed, it would have a material adverse
effect on the Company’s future operating results.
To achieve profitable operations, the Company must successfully develop, test, manufacture and
market products, as well as secure the necessary regulatory approvals. There can be no assurance
that any such products can be developed successfully or manufactured at an acceptable cost and with
appropriate performance characteristics, or that such products will be successfully marketed. These
factors would have a material adverse effect on the Company’s future financial results.
Share-Based Compensation
The Company recognizes share-based compensation arising from compensatory share-based
transactions using the fair value at the grant date of the award. Determining the fair value of
share-based awards at the grant date requires judgment. The Company uses an option-pricing model
(the Black-Scholes valuation model) to assist in the calculation of fair value. Due to its limited
history, the Company uses the “calculated value method” which relies on comparable company
historical volatility and uses the average of (1) the weighted average vesting period and (2) the
contractual life of the option, or eight years, as the estimated term of the option. The Company
bases its estimates of expected volatility on the median historical volatility of a group of
publicly traded companies that it believes are comparable to the Company based on the line of
business, stage of development, size and financial leverage.
The risk-free rate of interest for periods within the contractual life of the stock option
award is based on the yield of U.S. Treasury strips on the date the award is granted with a
maturity equal to the expected term of the award. The Company estimates forfeitures based on actual
forfeitures during its limited history. Additionally, the Company has assumed that dividends will
not be paid.
For stock options granted to non-employees, the Company measures fair value of the equity
instruments utilizing the Black-Scholes valuation model, if that value is more reliably measurable
than the fair value of the consideration or service received. The fair value of these instruments
are periodically revalued as the options vest, and are recognized as expense over the related
period of service or vesting period, whichever is longer. The total cost expensed for options
granted to non-employees for the years ended September 30, 2007, 2008 and 2009 was $657, $241, and
$94 respectively.
The Company expenses ratably over the vesting period the cost of the stock options granted to
employees and directors. The total compensation cost expensed for the years ended September 30,
2007, 2008 and 2009 was $3,567, $6,434, and $4,970 respectively. At September 30, 2009, the total
compensation cost related to non-vested options not yet recognized was $9,203, which will be
recognized over the next three years assuming the employees complete their service period for
vesting of the options. The Black-Scholes valuation model assumptions are as follows and were
determined as discussed above:
F-10
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statements — (Continued)
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
| Year Ended September 30, | ||||||||||||
| 2007 | 2008 | 2009 | ||||||||||
Expected life (in years) |
5.25 | 5.25 | 5.25 | |||||||||
Expected volatility |
60 - 70 | % | 57 - 60 | % | 59 - 68 | % | ||||||
Expected dividend yield |
0 | % | 0 | % | 0 | % | ||||||
Risk-free interest rate |
4.23% - 4.96 | % | 2.36% - 4.42 | % | 1.00% - 3.19 | % | ||||||
Weighted-average grant date fair value |
$ | 11.09 | $ | 13.92 | $ | 2.69 | ||||||
Subsequent Events
Effective April 1, 2009, the Company adopted the provisions of ASC Topic 855, Subsequent
Events (substantially incorporating SFAS No. 165, Subsequent Events) which establishes general
standards of accounting for and disclosure of events that occur after the balance sheet date but
before financial statements are issued or are available to be issued. The provisions set forth:
(i) the period after the balance sheet date during which management of a reporting entity should
evaluate events or transactions that may occur for potential recognition or disclosure in the
financial statements; (ii) the circumstances under which an entity should recognize events or
transactions occurring after the balance sheet date in its financial statements; and (iii) the
disclosures that an entity should make about events or transactions that occurred after the balance
sheet date. The adoption of these provisions did not have an effect on the Company’s financial
position and results of operations. The Company evaluated all events or transactions that occurred
after September 30, 2009 through December 14, 2009, the date it issued these financial statements.
During the period, the Company did not have any material recognizable or unrecognizable subsequent
events.
Recent Accounting Pronouncements
In August 2009, the Financial Accounting Standards Board (“FASB”) issued ASU 2009-5 Fair Value
Measurements and Disclosures (Topic 820) Measuring Liabilities at Fair Value (“ASU 2009-5”). ASU
2009-5 provides amendments to Subtopic 820-10, Fair Value Measurements and Disclosures-Overall, for
the fair value measurement of liabilities. ASU 2009-5 clarifies that in circumstances in which a
quoted price in an active market for the identical liability is not available, a reporting entity
is required to measure fair value. ASU 2009-5 is effective for the Company for interim and annual
periods ending after September 30, 2009. The Company does not expect the adoption of ASU 2009-5
to have a material impact on its consolidated results of operations or financial position.
Effective October 1, 2008, the Company adopted the provisions of ASC Topic 820, Fair Value
Measurements and Disclosures (substantially incorporating SFAS No. 157, Fair Value Measurements).
These provisions define fair value, establish a framework for measuring fair value in generally
accepted accounting principles, and expand disclosures about fair value measurements. The FASB has
agreed to defer for one year the effective date for certain provisions with respect to
non-financial assets and liabilities. The adoption of this accounting pronouncement did not have a
material effect on the Company’s financial statements.
During the fourth quarter of 2009, the Company adopted the Financial Accounting Standards
Board (“FASB”) Accounting Standards Update (“ASU”) No. 2009-01, “Amendments based on Statement of
Financial Accounting Standards No. 168 – The FASB Accounting Standards Codification and the
Hierarchy of Generally Accepted Accounting Principles” (the “Codification”). The Codification
became the single source of authoritative GAAP in the United States, and other than rules and
interpretive releases issued by the United States Securities and Exchange Commission (“SEC”). The
Codification reorganized GAAP into a topical format that eliminates the previous GAAP hierarchy and
instead established two levels of guidance – authoritative and nonauthoritative. All
non-grandfathered, non-SEC accounting literature that was not included in the Codification became
nonauthoritative. The adoption of the Codification did not change previous GAAP, but rather
simplified user access to all authoritative literature related to a particular accounting topic in
one place. Accordingly, the adoption had no impact on the Company’s consolidated financial
position and results of operations. All prior references to previous GAAP in the Company’s
consolidated financial statements were updated for the new references under the Codification.
F-11
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statements — (Continued)
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
3. Marketable Securities
The Company classifies marketable securities as available for sale. The Company determines the
appropriate classification of debt and equity securities at the time of purchase and re-evaluates
such designation as of each balance sheet date.
The Company invests in certain marketable securities, which consist primarily of
short-to-intermediate-term debt securities issued by the U.S. government, U.S. government agencies
and municipalities and investment grade corporate securities. The Company only invests in
marketable securities with active secondary or resale markets to ensure portfolio liquidity and the
ability to readily convert investments into cash to fund current operations, or satisfy other cash
requirements as needed. Due to the nature of the Company as a development stage company and its
funding needs at times being uncertain, the Company has classified all marketable securities as
available-for-sale. The unrealized gains and losses on these securities are included in accumulated
other comprehensive income as a separate component of stockholders’ equity. The
specific-identification method is used to determine the cost of a security sold or the amount
reclassified from accumulated other comprehensive income into earnings.
As of September 30, 2008, the Company conducted a periodic review to identify and evaluate
each investment that has an unrealized loss. Any unrealized loss identified as other-than-temporary
is recorded directly in the Statement of Operations.
As of September 30, 2008 the Company concluded the unrealized losses were temporary because
(1) the Company believes the market value is partially due to global and current economic
conditions which are at unprecedented levels and (2) the securities continue to be of high quality
and interest has been paid when due. The Company does not believe any unrealized losses represent
an other-than-temporary impairment based on its evaluation of available evidence as of
September 30, 2008. If in the future the Company determines that any decline is
other-than-temporary, the Company would have to recognize the loss in its Statement of Operations.
Unrealized gains and losses are recorded in accumulated other comprehensive income in the Company’s
Balance Sheet.
Marketable securities classified as available for sale are measured at fair value based on
quoted market prices. The amortized cost, gross unrealized gains and losses and fair value of
investment securities at September 30, 2008 are summarized below. As of September 30, 2009, the
Company had no marketable security investments.
| September 30, 2008 | ||||||||||||||||
| Amortized | Gross Unrealized | Fair | ||||||||||||||
| Cost | Gains | Losses | Value | |||||||||||||
Short term marketable securities |
||||||||||||||||
Commercial paper |
$ | 998 | $ | — | $ | (1 | ) | $ | 997 | |||||||
Certificates of deposits |
1,999 | 1 | — | 2,000 | ||||||||||||
US government agency securities |
10,170 | — | (54 | ) | 10,116 | |||||||||||
Corporate and agency bonds |
4,010 | — | (8 | ) | 4,002 | |||||||||||
Discount and bank notes |
8,437 | — | — | 8,437 | ||||||||||||
Total |
$ | 25,614 | $ | 1 | $ | (63 | ) | $ | 25,552 | |||||||
F-12
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statements — (Continued)
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
As of September 30, 2009, the Company classifies all the securities as current since all
investments are in U.S. Treasury based money market accounts with original maturities of three
months or less at the date of purchase.
4. Net Loss per Share
Basic and diluted net loss per share has been calculated by dividing net loss by the weighted
average number of common shares outstanding during the period. All potentially dilutive common
shares have been excluded from the calculation of weighted average common shares outstanding since
their inclusion would be antidilutive.
The amount of options and warrants excluded are as follows:
| Year Ended September 30, | ||||||||||||
| 2007 | 2008 | 2009 | ||||||||||
Common shares underlying warrants for Series A Preferred Stock
|
198,025 | 118,815 | 118,815 | |||||||||
Stock options
|
1,685,974 | 3,135,390 | 3,407,633 | |||||||||
5. Property and equipment
Property and equipment consists of the following:
| September 30, | ||||||||
| 2008 | 2009 | |||||||
Furniture and fixtures |
$ | 313 | $ | 318 | ||||
Leasehold improvements |
1,546 | 1,549 | ||||||
Construction-in-progress |
121 | 15 | ||||||
Laboratory equipment |
1,381 | 1,612 | ||||||
Manufacturing equipment |
144 | 372 | ||||||
Facility equipment |
50 | 50 | ||||||
Device development |
— | 157 | ||||||
Computer equipment and other |
1,150 | 1,270 | ||||||
Sub-Total |
4,705 | 5,343 | ||||||
Less: Accumulated depreciation and amortization |
774 | 1,648 | ||||||
Total |
$ | 3,931 | $ | 3,695 | ||||
Depreciation expense for the years ended September 30, 2007, 2008 and 2009 was $543, $554 and
$874, respectively.
6. Related Party Transactions
The following is a description of material transactions, other than compensation arrangements,
since the Company’s incorporation on December 3, 2003 to which the Company has been a party and in
which any of its directors, executive officers or persons who it knows held more than five percent
of any class of capital stock, including their immediate family members who had or will have a
direct or indirect material interest. The Company believes that the terms obtained or consideration
paid or received, as applicable, in connection with the transactions described below were
comparable to terms available or the amounts that would have been paid or received, as applicable,
in arm’s-length transactions.
Consulting and Clinical Research Services
Effective December 2008, Dr. Pfützner is no longer our Chief Medical Officer in Europe. Dr.
Pfützner has been retained as a consultant for ongoing clinical trials being conducted in Europe.
F-13
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statements — (Continued)
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
During the fiscal years ended September 30, 2007 and 2008, the Company paid approximately $926
and $2,710, respectively, in clinical related costs to the Institute for Clinical Research and
Development in Mainz, Germany where Andreas Pfützner, our former Chief Medical Officer in Europe,
served as its managing director. Dr. Pfützner and his spouse are majority shareholders of the
institute.
On April 1, 2005, the Company entered into a consulting agreement with Dr. Pfützner to provide
consulting services in connection with the research and development of the Company’s product
candidates. The consulting agreement was amended and restated effective June 5, 2007. The initial
term of the amended and restated agreement terminated on June 5, 2009 and automatically renews for
successive one-year terms until the agreement is terminated by either party on prior written notice
in accordance with the terms of the agreement. Under the agreement, Dr. Pfützner is entitled to
receive $2 for each full business day devoted to the performance of his services in addition to a
non-refundable payment of $150 per annum for the two-year period commencing June 5, 2007.
Dr. Pfützner is bound by non-competition and non-solicitation covenants that prohibit him from
competing with the Company during the term of the agreement and for one year after termination of
the agreement.
Issuance of Series A Convertible Preferred Stock
Between March and July 2005, the Company issued and sold an aggregate of 35,000 shares of its
Series A convertible preferred stock (see Note 9) to two executive officers and one director.
McGinnSmith & Company, Inc. (“MSI”) served as placement agent in connection with the offering
of the Series A convertible preferred stock pursuant to a letter agreement (the “Letter
Agreement”), for which MSI received $280 (excluding $15 reimbursement for expenses) and warrants to
purchase 55,900 shares of Series A convertible preferred stock at $5.00 per share. The fair value
of the warrants was $121 and was computed using the Black-Scholes valuation model using the
following assumptions: term of 7 years; volatility rate of 90%; risk free rate of 3.65% and a
dividend yield of 0.0%, which was treated as cost of raising capital. A member of the Board of
Directors of the Company was a managing director of MSI until May 2007.
In July 2005, Steiner Ventures LLC, (“SV”), an entity controlled by Dr. Solomon S. Steiner,
Chairman and Chief Executive Officer, entered into a subscription agreement with the Company to
purchase 60,000 shares of the Series A convertible preferred stock at a price of $5.00 per share
which could be accepted by the Company at any time until July 2006. At a meeting of the Board of
Directors held on October 24, 2005, the Board of Directors approved, with the agreement of SV, the
amendment of that subscription agreement into a subscription to purchase 12 Units in the Bridge
Financing (see Note 9) for $300. The Company accepted this subscription and SV purchased the Units.
Since all securities contemplated to be issued pursuant to the SV subscription agreement were
to be issued at fair value, no value was ascribed to the subscription agreement or amendment.
Bridge Financing
Between February and May 2006, the Company completed a Bridge Financing (see Note 9). Four
executive officers and one director purchased an aggregate of 23 units, or $575, as part of the
financing. These units were subsequently settled with 182,540 shares of Series B convertible
preferred stock (see Note 9) and warrants to purchase 98,275 shares of common stock.
In connection with the sales of units in the Bridge Financing, the Company paid MSI an
aggregate commission of $70 and issued to MSI additional warrants to purchase 22,222 shares of
Series B convertible preferred stock and a warrant to purchase 11,963 shares of common stock. The
fair value of the warrants was $22 as computed using the Black-Scholes valuation model using the
following assumptions: term of 3.5 years; volatility rate of 50%; risk free rate of 5.05% and a
dividend yield of 0.0%.
Issuance of Series B Convertible Preferred Stock
On July 19, 2006, the Company issued and sold 38,071 shares of Series B convertible preferred
stock (see Note 9) and a warrant to purchase 20,496 shares of common stock to its Chief Executive
Officer in exchange for a $150 bonus that was earned by him during the calendar year ended
December 31, 2005 but voluntarily deferred. At September 30, 2005, the Company accrued $113 of the
bonus and the balance of $37 was expensed in fiscal 2006. The full amount of the accrued bonus was
exchanged for Series B convertible preferred stock on July 19, 2006.
In connection with the issuance of the Series B convertible preferred stock, the Company
retained MSI to serve as placement agent pursuant to an amendment to the Letter Agreement. MSI was
paid (a) an aggregate commission of $350 from the sale of the Series B convertible preferred stock,
(b) a warrant to purchase 126,903 shares of Series B convertible preferred stock and (c) a warrant
to purchase 68,322 shares of common stock. On July 19, 2006, the Company also sold and issued to a
director 12,690 shares of Series B convertible preferred stock and a warrant to purchase
6,832 shares of common stock. At the completion of the Series B
F-14
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statements — (Continued)
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
preferred stock financing, the lead investor remitted the monies for its investment in the Series B
Round net of offering-related expenses incurred by the investor group for which the Company was
responsible. Total offering expenses were approximately $2,000, of which $1,470 was commissions for
the placement of the offering. A director of the Company had arranged to pay for an investment in
the Series B preferred stock financing (the “Investment”) utilizing a portion of commissions due.
Since the monies due for the commission were not received by the Company, the purchase price of the
Investment could not be deducted from the monies received. The fair values of the warrants for
common stock were $126 and $13 and were computed using the Black-Scholes valuation model using the
following assumptions: term of 3.5 years; volatility rate of 50%; risk free rate of 5.05% and a
dividend yield of 0.0%. The fair value of the warrants for preferred stock was $167 and was
computed using the Black-Scholes valuation model using the following assumptions: term of
3.5 years; volatility rate of 50%; risk free rate of 4.70% and a dividend yield of 0.0%. These
amounts were treated as cost of raising capital.
Deferred Compensation
On December 15, 2005, the Board of Directors authorized a bonus to be paid to SV, if the
Chairman and Chief Executive Officer directed the completion of a successful financing in excess of
$10,000. Pursuant to that board resolution, the Company owed SV $250 because of the issuance of the
Series B convertible preferred stock during the year ended September 30, 2006 but payment was
deferred by Dr. Steiner. The Company recorded compensation expense for this bonus and had reflected
the balance as due to related party at September 30, 2006. The balance was paid in July 2007.
Separately, Dr. Steiner voluntarily deferred his calendar year compensation of $250. The
Company recorded compensation expense for this salary and had reflected the balance as deferred
compensation at September 30, 2006. The balance was paid in July 2007.
7. Commitments
Change of Control Agreements and Severance Agreements
In June 2008, the Company entered into change of control agreements and severance agreements
with two of its executive officers.
Pursuant to the terms of the change of control agreement with its executive officers, they are
each entitled to the following upon termination of employment with the Company occurring within two
years of a change of control, unless such termination is by the executive officer for other than
good reason or by the Company for cause (each as defined in the agreement):
| • | annual base salary earned through the termination date; | ||
| • | in the event the executive officer satisfied the performance criteria for an annual bonus prior to termination, a portion of the annual bonus based on the number of days worked during the year; | ||
| • | if the performance criteria were not fully satisfied, but the board of directors determines that the criteria could have been satisfied had the executive officer remained employed for the full fiscal year, an amount equal to the average of the annual bonus paid to the executive officer over the last three fiscal years, portioned based on the number of days worked during the year (the “Average Annual Bonus”); | ||
| • | any compensation previously deferred by the executive officer and any accrued paid time-off; | ||
| • | annual base salary for a period of 18 months following the date of termination; | ||
| • | health insurance and, under certain circumstances, life, disability and other insurance benefits for a period of 18 months or until the executive officer qualifies for similar benefits from another employer; | ||
| • | 150% of the Average Annual Bonus (paid in addition to the bonus described immediately above); |
F-15
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statements — (Continued)
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
| • | acceleration of all outstanding options; and | ||
| • | extension of the exercisability of options. |
Under the change of control agreement, if the Company terminates the executive officer’s
employment for cause or the executive officer terminates his employment with the Company without
good reason, then the executive officer is not entitled to severance payments or other benefits.
Pursuant to the Company’s severance agreement with its officers each executive officer is
entitled to the following upon termination of employment with the Company, unless such termination
is by the executive officer for other than good reason or by the Company for cause:
| • | annual base salary earned through the termination date; | ||
| • | in the event the executive officer satisfied the performance criteria for an annual bonus prior to termination, a portion of the annual bonus based on the number of days worked during the year; | ||
| • | if the performance criteria were not fully satisfied, but the board of directors determines that the criteria could have been satisfied had the executive officer remained employed for the full fiscal year, the Average Annual Bonus; | ||
| • | any compensation previously deferred by the executive officer and any accrued paid time-off; | ||
| • | annual base salary for a period of 18 months following the date of termination; | ||
| • | health insurance for a period of 18 months or until the executive officer qualifies for similar benefits from another employer; | ||
| • | 150% of the Average Annual Bonus (paid in addition to the bonus described immediately above); | ||
| • | acceleration of all outstanding options; and | ||
| • | extension of the exercisability of the options. |
The aggregate amount of base salary for both executives is $615. Bonuses for the executives
are at the discretion of, and awarded by the Board of Directors.
Former General Counsel Severance Agreement
Effective January 1, 2009, R. Timmis Ware, the Company’s former General Counsel and Secretary,
resigned from all his positions with the Company. Pursuant to the Company’s severance agreement
with Mr. Ware, Mr. Ware will receive a bonus, continuation of salary and certain benefits until
June 30, 2010. Furthermore, the agreement permits for the acceleration of the vesting of options to
purchase 170,445 shares of common stock at exercise prices between $1.41 through $18.16 that remain
exercisable through the original expiration date. The charge of approximately $277 for the lump sum
payment, salary and benefit continuation for eighteen months and option acceleration modification
charge of approximately $100 were recorded. As of September 30, 2009 the Company has paid $138 of
the $277 obligation, which leaves of $139 as a short term obligation.
Former Chief Financial Officer Severance Agreement
On November 13, 2007, F. Scott Reding, the Company’s former Chief Financial Officer, Chief
Accounting Officer and Treasurer, resigned from all his positions with the Company. In connection
with Mr. Reding’s resignation, the Company and Mr. Reding entered into a severance agreement that
established the terms of Mr. Reding’s separation of employment. The charge of $482 for the lump sum
payment, salary and benefit continuation for two years were recorded in the year ended
September 30, 2008. The charge of $482 includes lump sum payment and payment for the continuation
of salary and certain benefits for two years. As of September 30, 2009, the Company has paid $451
of the $482 obligation, which leaves $31 as a short term obligation.
Leases
As of September 30, 2009, the Company leases three facilities in Danbury, Connecticut with
Mulvaney Properties, LLC, which is controlled by a non-affiliated stockholder of the Company.
The Company entered into its first lease for laboratory space in February 2004, which was
subsequently renewed in September 2006. The Company expects to renew this lease prior to its
expiry in January 2010. This lease provides for annual basic lease payments of $64, plus operating
expenses.
F-16
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statements — (Continued)
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
In July 2007 the Company entered into a second lease for its corporate office, which was
subsequently amended in October 2007. The October 2007 amendment increased the term from a five
year to a seven year term beginning August 1, 2007 until July 31, 2014. The renewal option was also
amended from a five year to a seven year term. This lease provides for annual basic lease payments
of $357, plus operating expenses.
In December 2008, the Company entered into a third lease agreement for additional office space
adjacent to its laboratory space. The lease is for fourteen months beginning December 1, 2008 and
ending on January 30, 2010. The Company has agreed to use the leased premises only for offices,
laboratories, research, development and light manufacturing. The Company expects to renew this
lease prior to its expiry in January 2010. This lease provides for annual basic lease payments of
$29, plus operating expenses.
Lease expense for the years ended September 30, 2007, 2008 and 2009 was $195, $383 and $591,
respectively.
Minimum lease payments under these agreements as of September 30, 2009, as well as equipment leases
subsequently entered into, are as follows:
| Years Ending September 30, | ||||
2010 |
537 | |||
2011 |
502 | |||
2012 |
519 | |||
2013 |
540 | |||
2014 |
465 | |||
Total |
$ | 2,563 | ||
Purchase Commitments
The Company contracted with N.V. Organon, a global producer of insulin, to supply the Company
with all of the insulin that the Company will need for testing and manufacturing of the Company’s
product candidates. As subsequently amended in November 2009, the agreement with N.V. Organon will
terminate in December 2011. As of September 30, 2009, the Company had purchase commitments of
approximately $12,268 associated with the signing of a renewed contract with Organon.
| Years Ending September 30, | ||||
2010 |
$ | 5,025 | ||
2011 |
7,243 | |||
Total |
$ | 12,268 | ||
8. Income Taxes
Effective October 1, 2007, the Company adopted the provisions of FIN48, which was incorporated
into the Codification within ASC Topic 740, Income Taxes, with respect to uncertain tax positions.
The Company did not recognize any increase or decrease in the liability for unrecognized tax
benefits related to tax positions taken in prior periods as a result of the adoption, therefore,
there was no corresponding adjustment to retained earnings.
F-17
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statements — (Continued)
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
The Company did not have any liabilities for unrecognized tax positions as of October 1, 2007
(adoption date). During the year ended September 30, 2009, the Company performed a review on the
research and development activities that occurred in the state of Connecticut that supports our
Connecticut research and development credits and refunds. The results of that study decreased
income tax receivable and the corresponding reserve relating to an anticipated tax refund from the
state of Connecticut for research and development activities. For the year-ended September 30,
2009, this resulted in a 1% increase to the Company’s effective tax rate. The reserve does not
include interest or penalties based on the nature of the liability. The Company plans to treat any
future interest or penalties as operating expense.
The following table summarized the activity related to the Company’s tax liabilities:
| Year Ended September 30, | ||||||||||||
| 2007 | 2008 | 2009 | ||||||||||
Balance, beginning of year |
$ | — | $ | 75 | $ | 988 | ||||||
Increase related to current year tax position |
75 | 913 | 6 | |||||||||
Increase related to prior year’s tax position |
— | — | 107 | |||||||||
Decrease related to prior year’s tax position |
— | — | (913 | ) | ||||||||
Balance, at end of year |
$ | 75 | $ | 988 | $ | 188 | ||||||
The Company files U.S. federal and state tax returns and has determined that its major tax
jurisdictions are the United States and Connecticut. The tax years through 2008 remain open due to
net operating loss carryovers and subject to examination by the appropriate governmental agencies
in the United States and Connecticut.
The provision (benefit) for income taxes is as follows:
| Year Ended September 30, | ||||||||||||
| 2007 | 2008 | 2009 | ||||||||||
Current expense |
||||||||||||
Federal |
$ | — | $ | — | $ | — | ||||||
State |
125 | (983 | ) | 337 | ||||||||
Deferred expense |
— | — | — | |||||||||
Actual tax provision (benefit) |
$ | 125 | $ | (983 | ) | $ | 337 | |||||
At September 30, 2009, the Company had available federal net operating loss carryforwards of
approximately $100,900 which expire commencing in fiscal 2024 through 2029 and $100,400 of state
net operating loss carryforwards, which expire commencing in fiscal 2024 through 2029. The Company
also has federal and state research and development credit carryovers of approximately $2,100,
which expire commencing in fiscal 2024.
F-18
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statement
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
The major components of deferred tax assets and valuation allowances and deferred tax
liabilities at September 30, 2008 and 2009 are as follows:
| September 30, | ||||||||
| 2008 | 2009 | |||||||
Deferred Tax Assets |
||||||||
Net operating losses |
$ | 26,889 | $ | 39,959 | ||||
Research and development credit |
1,652 | 2,149 | ||||||
Depreciation of fixed assets |
121 | 284 | ||||||
Other |
222 | 802 | ||||||
Total deferred tax asset |
28,884 | 43,195 | ||||||
Valuation Allowance |
(28,763 | ) | (43,195 | ) | ||||
Net Deferred Tax Assets |
121 | — | ||||||
Deferred Tax Liabilities |
||||||||
Other |
(121 | ) | — | |||||
Total deferred tax liabilities |
(121 | ) | — | |||||
Net Deferred Tax Asset (liabilities) |
$ | — | $ | — | ||||
The Company files its tax returns on a fiscal year basis. For the years ended September 30,
2007, 2008 and 2009, the Company only had to pay state taxes.
During the year ended September 30, 2009, the Company performed a book-to-tax reconciliation
that adjusted the deferred tax assets and valuation allowance by approximately $300. As the Company
has not yet achieved profitable operations, management does not believe that it is more likely than
not that the tax benefits as of September 30, 2009 will be realized and therefore has recorded a
valuation allowance against its deferred tax asset.
The following reconciles the amount of tax expense at the federal statutory rate and taxes on
loss as reflected in operations:
| Year Ended | ||||||||||||
| September 30, | ||||||||||||
| 2007 | 2008 | 2009 | ||||||||||
Federal statutory rate |
34.00 | % | 34.00 | % | 34.00 | % | ||||||
Federal taxes at statutory rate |
$ | (9,080 | ) | $ | (15,077 | ) | $ | (14,597 | ) | |||
Tax expense on permanent differences |
2,958 | 2,293 | 1,741 | |||||||||
Tax benefit on research and business credits |
(186 | ) | (325 | ) | (325 | ) | ||||||
State taxes, net of federal tax effect |
33 | 61 | 43 | |||||||||
State benefit, net operating loss |
(1,877 | ) | (2,784 | ) | (1,249 | ) | ||||||
Valuation allowance increase |
8,498 | 14,972 | 14,432 | |||||||||
Connecticut research and development refund |
— | (1,988 | ) | (40 | ) | |||||||
Reserve for uncertain tax positions |
— | 913 | 6 | |||||||||
Other |
(221 | ) | 952 | 326 | ||||||||
Actual tax provision |
$ | 125 | $ | (983 | ) | $ | 337 | |||||
F-19
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statement
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
9. Stockholders’ Equity
Common Stock
The Company’s authorized common stock consists of 100,000,000 shares of a single class of
common stock, having a par value of $0.01 per share. The holders of the common stock are entitled
to one vote for each share and have no cumulative voting rights or preemptive rights.
As of September 30, 2009, the Company had warrants outstanding to purchase an aggregate of
118,815 shares of its common stock with an exercise price of $1.41 per share.
On February 12, 2008, the Company completed a follow-on public offering of 3,260,000 shares of
its common stock at a price to the public of $15.50 per share. The Company received net proceeds
from this offering, after deducting underwriting discounts and commissions and expenses, of
$46,817. Certain of the Company’s stockholders sold 550,000 shares in the offering. The Company did
not receive any proceeds from the sale of shares from the selling stockholders.
On May 16, 2007, the Company completed an initial public offering of 5,750,000 shares of its
common stock at a price to the public of $15.00 per share. The offering resulted in gross proceeds
of $86,300. The Company received net proceeds from the offering of approximately $78,800 after
deducting underwriting discounts and commissions and additional offering expenses. The completion
of the initial public offering resulted in the conversion of the Company’s Series A and B
convertible preferred stock. A total of 6,407,008 shares of common stock were issued upon the
conversion of the preferred stock.
Preferred Stock
The Company is authorized to issue up to 50,000,000 shares of preferred stock, having a par
value of $0.01 per share. The Company’s preferred stock may be issued in one or more series, the
terms of which may be determined at the time of issuance by the Company’s Board of Directors,
without further action by stockholders, and may include voting rights (including the right to vote
as a series on particular matters), preferences as to dividends and liquidation and conversion,
redemption rights and sinking fund provisions. The issuance of preferred stock could reduce the
rights, including voting rights, of the holders of common stock and, therefore, could reduce the
value of the common stock. In particular, specific rights granted to holders of preferred stock
could be used to restrict the Company’s ability to merge with or sell the Company’s assets to a
third party, thereby preserving control of the Company by existing management.
F-20
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statements — (Continued)
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
Series A Convertible Preferred Stock
The Company authorized 1,050,000 shares of Series A convertible preferred stock with certain
rights and privileges, of which 569,000 and 0 shares were issued and outstanding as of
September 30, 2006 and 2007, respectively. In July 2005, the Company completed a private placement
of 569,000 shares of its Series A convertible preferred stock and received proceeds of $2,845. Fees
incurred as part of the private placement totaled $379.
In connection with the Series A convertible preferred stock issuance, the Company entered into a
registration rights agreement with the purchasers of its stock, which provided, among other things,
for liquidated damages if the Company were initially unable to register and obtain an effective
registration of the securities within the allotted time. The stockholders could not demand
registration until one hundred and eighty (180) days after the Company had effected a qualified
initial public offering. The penalties were (i) one and three quarters (13/4 %) percent of the
aggregate number of shares of underlying common stock for each month, or part thereof, after a
ninety (90) day period that a registration statement was not filed with the SEC or (ii) one (1%)
percent of the aggregate number of shares of underlying common stock for each month if the forgoing
filed registration statement was not declared effective by the SEC within one hundred and twenty
(120) days.
Each share of Series A convertible preferred stock was automatically convertible into a number
of shares of common stock equal to the quotient of $3.54 divided by $1.00 immediately subsequent to
the date of the initial public offering.
As part of the compensation agreement, the placement agent received 279,500 Series A Warrants.
Each warrant consists of the right to purchase one share of fully paid and non-assessable common
stock for a period of seven years which expires on July 12, 2012. The exercise price of each
warrant is $1.00 per share. The exercise price may be paid in cash or by tendering common stock.
The warrants are transferable and provide for anti-dilution protection. The Company evaluated the
warrants to ascertain if they should be recorded as equity instruments, or if they contained
features which require them to be recorded as derivative liabilities, and concluded they should be
classified as equity instruments on the balance sheet.
As a result of the conversion option, the Company considered the features contained in the
Series A convertible preferred stock to ascertain whether the shares contained a beneficial
conversion feature. The Company determined that the issuance of the Series A convertible preferred
stock did not result in a beneficial conversion feature.
Series B Convertible Preferred Stock
The Company authorized 6,500,000 shares of Series B convertible preferred stock (“Series B
Preferred Stock”) of which 6,198,179 and 0 shares were issued and outstanding as of September 30,
2006 and 2007, respectively. In July 2006, the Company completed a private placement of
5,380,711 shares of its Series B preferred stock and received gross proceeds of $21,200 as part of
the private placement, fees incurred totaled $1,795. Additionally in July 2006, 817,468 shares of
Series B preferred stock and 440,105 common stock warrants were issued to repay the Company’s
Bridge Financing units.
Each share of Series B convertible preferred stock was automatically convertible into a number
of shares of common stock equal to the quotient of $3.94 divided by $1.00 immediately subsequent to
the date of the initial public offering.
As part of the compensation agreement relating to the Series B Preferred Stock transaction,
the placement agent received 126,903 Agent Series B Preferred Warrants and 68,322 common stock
warrants. Each such warrant consisted of the right to purchase one share of Series B Preferred
Stock for a period of seven years which expires on July 19, 2013. The exercise price of each
warrant was $5.56 per share. The exercise price was payable in cash or by tendering common stock.
In the event the Company issued common stock or rights to purchase common stock below the then
conversion price, then the price per share at which the Series B preferred stock was to be
converted would be reduced to the weighted average of the existing conversion price per share and
the price per share of the newly-issued stock or rights.
Also, as part of the compensation agreement relating to the bridge financing transaction, the
placement agent received an aggregate of 22,222 Series B Preferred warrants and 11,963 common stock
warrants. Each warrant consisted of the right to purchase one share of fully paid and
non-assessable common stock for a period of seven years which expires on July 19, 2012. The
exercise price of each warrant was $5.56 per share. The exercise price was payable in cash or by
tendering common stock. In the event the Company issued common stock or rights to purchase common
stock below the then conversion price, then the price per share at which the Series B preferred
stock was to be converted would be reduced to the weighted average of the existing conversion price
per share and the price per share of the newly-issued stock or rights.
F-21
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statement
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
The Company evaluated all the warrants to ascertain if they should be recorded as equity
instruments, or if they contained features which require them to be recorded as derivative
liabilities and concluded they should be classified as equity on the balance sheet.
As a result of the conversion option, the Company considered the features contained in the
Series B convertible preferred stock to ascertain whether the shares contained a beneficial
conversion feature and determined that the issuance of the Series B convertible preferred stock
resulted in a beneficial conversion feature in the amount of $603.
The completion of the Company’s initial public offering in May 2007 resulted in the conversion
of 6,407,008 shares of the Company’s Series A and B convertible preferred stock.
Shares Reserved for Future Issuance
As of September 30, 2009, the Company reserved shares of common stock for future issuance as
follows:
2004 stock incentive plan |
4,700,000 | |||
2005 employee stock purchase plan |
1,500,000 | |||
2005 Non-employee directors’ stock option plan |
500,000 | |||
Exercise of warrants issued to placement agent |
118,815 | |||
Total |
6,818,815 | |||
2004 Stock Incentive Plan, as amended
The Company established the 2004 Stock Incentive Plan on October 1, 2004 (the “Plan”) and as
amended in March 2007. The Plan provides for the granting of shares of common stock or securities
convertible into or exercisable for shares of common stock, including stock options (“Incentive
Stock Options”) to directors, employees, consultants and advisors of or to the Company. Incentive
Stock Options can be awarded only to persons who are employees of the Company at the time of the
grant. Stock options are exercisable at the conclusion of the vesting period. Employees can
exercise their vested shares up to 90 days after termination of services. A total of 4,700,000
options to purchase the equivalent number of shares of common stock may be issued pursuant to the
Stock Incentive Plan. No awards may be granted under the plan after October 1, 2014.
The Plan is be administered by either the Board of Directors of the Company or a Committee
thereof, which determines the terms and conditions of the awards granted under the Plan, including
the recipient of the award, the nature of the award, the exercise price of the award, the number of
shares subject to the award and the exercisability thereof.
Non-employee directors are not entitled to receive awards other than the non-qualified stock
options the plan directs be issued to non-employee directors.
2005 Employee Stock Purchase Plan
The Company’s 2005 Employee Stock Purchase Plan, or the Purchase Plan, was adopted by its
Board of Directors and approved by its stockholders on March 20, 2007. The Purchase Plan became
effective upon the closing of the Company’s initial public offering. The Purchase Plan is intended
to qualify as an employee stock purchase plan within the meaning of Section 423 of the Code.
Under the Purchase Plan, eligible employees may contribute up to 15% of their eligible
earnings for the period of that offering withheld for the purchase of common stock under the
Purchase Plan. The employee’s purchase price is equal to the lower of: 85% of the fair market value
per share on the start date of the offering period in which the employee is enrolled or 85% of the
fair market value per share on the semi-annual purchase date. The Purchase Plan imposes a
limitation upon a participant’s right to acquire common stock if immediately after the purchase,
the employee would own 5% or more of the total combined voting power or value of the Company’s
common stock or of any of its affiliates not eligible to participate in the Purchase Plan. The
Purchase Plan provides for an automatic rollover when the purchase price for a new offering period
is lower than previously established purchase price(s). The Purchase Plan also provides for a
one-time election that allows an employee the opportunity to enroll into a new offering period when
the new offering is higher than their current offering price. This election must be made within
30 days from the start of a new offering period. Offering periods are twenty-seven months in
length. The compensation cost in connection with the plan for the years ended September 30, 2008
and 2009 was $49 and $233, respectively.
F-22
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statement
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
An aggregate of 1,500,000 shares of common stock are reserved for issuance pursuant to
purchase rights to be granted to the Company’s eligible employees under the Purchase Plan. The
Purchase Plan shares are replenished annually on the first day of each fiscal year by virtue of an
evergreen provision. The provision allows for share replenishment equal to the lesser of 1% of the
total number of shares outstanding on that date or 100,000 shares. As of September 30, 2009, a
total of 1,318,220 shares were reserved and available for issuance under this plan. As of
September 30, 2009, the Company issued 101,841 shares under the Purchase Plan.
2005 Non-Employee Directors’ Stock Option Plan
The Company’s 2005 Non-Employee Directors’ Stock Option Plan, or the Directors’ Plan, was
adopted by its Board of Directors and approved by its stockholders on March 20, 2007. The
Directors’ Plan became effective upon the closing of the Company’s initial public offering. An
aggregate of 500,000 shares of common stock are reserved for issuance under the Directors’ Plan.
Upon the effective date of the registration statement in connection with the Company’s initial
public offering, each of its non-employee directors automatically received an initial option to
purchase 25,000 shares of common stock. Each non-employee director who is first elected or
appointed to the Company’s Board of Directors after the closing of the Company’s initial public
offering will receive an initial option to purchase 25,000 shares of common stock on the date of
his or her election or appointment. In addition, each non-employee director receives an option to
purchase 20,000 shares of common stock on an annual basis. Effective March 3, 2009, these shares
vest prorata over one year. However, in the event a non-employee director has not served since the
date of the preceding annual meeting of stockholders, that director will receive an annual grant
that has been reduced pro rata for each full quarter prior to the date of grant during which such
person did not serve as a non-employee director.
The fair value per share is being recognized as compensation expense over the applicable
vesting period. The fair value per share for award granted as of December 31, 2008 through
September 30, 2009 were calculated using the Black-Scholes valuation model.
The fair value of the common stock for the grants from December 23, 2004 through November 1,
2006 was determined using a retrospective valuation. The fair value of the common stock for the
grants during December 2006 and subsequently were determined contemporaneously with the grants.
F-23
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statements — (Continued)
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
The following table summarizes the stock option activity through September 30, 2009:
| Weighted Average |
Aggregate | |||||||||||
| Number | Exercise Price | Intrinsic Value | ||||||||||
Balance, September 30, 2004 |
— | $ | — | |||||||||
Granted |
385,432 | 1.41 | $ | 1,526 | ||||||||
Outstanding balance, September 30, 2005 |
385,432 | 1.41 | $ | 1,526 | ||||||||
Granted |
461,602 | 5.65 | ||||||||||
Forfeited, expired |
60,222 | 3.40 | ||||||||||
Outstanding balance, September 30, 2006 |
786,812 | 3.23 | $ | 1,287 | ||||||||
Granted |
955,842 | 13.96 | ||||||||||
Exercised |
3,542 | 1.41 | $ | 14 | ||||||||
Forfeited, expired |
53,138 | 5.65 | ||||||||||
Outstanding balance, September 30, 2007 |
1,685,974 | 6.80 | $ | 1,273 | ||||||||
Granted |
1,727,397 | 16.88 | ||||||||||
Exercised |
174,410 | 5.18 | 33 | |||||||||
Forfeited, expired |
103,571 | 11.04 | ||||||||||
Outstanding balance, September 30, 2008 |
3,135,390 | $ | 13.92 | $ | 832 | |||||||
Granted |
611,500 | 2.69 | $ | 1,682 | ||||||||
Exercised |
17,661 | 1.41 | $ | 70 | ||||||||
Forfeited, expired |
321,596 | 13.88 | ||||||||||
Outstanding balance, September 30, 2009 |
3,407,633 | $ | 11.81 | $ | 2,514 | |||||||
Exercisable shares, September 30, 2009 |
1,818,056 | $ | 10.66 | $ | 1,464 | |||||||
10. Employee Benefit Plan
Effective January 1, 2006, the Company established a 401(k) plan covering substantially all
employees. Employees may contribute up to 100% of their salary per year (subject to maximum limit
prescribed by federal tax law). The Company may elect to make a discretionary contribution or match
a discretionary percentage of employee contributions. As of September 30, 2009, the Company had not
elected to make any contributions to the plan.
F-24
Table of Contents
Biodel Inc.
(A Development Stage Company)
(A Development Stage Company)
Notes to Financial Statements — (Continued)
(In thousands, except share and per share amounts)
(In thousands, except share and per share amounts)
11. Reverse Split
On April 12, 2007, the Company completed a 0.7085 for one (0.7085:1) reverse stock split
(“Reverse Split”) rounding all fractional shares down to the next full share. Stockholders received
cash in lieu of fractional shares. After the Reverse Split, there were 8,003,828 shares of common
stock outstanding. The Reverse Split did not reduce the number of authorized shares of common
stock, alter the par value or modify the voting rights or other terms thereof. As a result of the
Reverse Split, the conversion prices and/or the numbers of shares issuable upon the exercise of any
outstanding options and warrants to purchase common stock were proportionally adjusted pursuant to
the respective anti-dilution terms of the 2004 Stock Incentive Plan and the respective warrant
agreements. All references in these financial statements and accompanying notes to units of common
stock or per share amounts are reflective of the Reverse Split for all periods reported.
12. Summary Selected Quarterly Financial Data (Unaudited)
The following table sets forth certain unaudited consolidated quarterly statement of
operations data for the eight quarters ended September 30, 2009. This information is unaudited, but
in the opinion of management, it has been prepared substantially on the same basis as the audited
consolidated financial statements and all necessary adjustments, consisting only of normal
recurring adjustments, have been included in the amounts stated below to state fairly the unaudited
consolidated quarterly results of operations. The results of operations for any quarter are not
necessarily indicative of the results of operations for any future period.
Quarter Ended
(in thousands, except share and per share amounts)
(in thousands, except share and per share amounts)
| December 31, | March 31, | June 30, | September 30, | |||||||||||||
| 2008 | 2009 | 2009 | 2009 | |||||||||||||
Revenue |
$ | — | $ | — | $ | — | $ | — | ||||||||
Net loss |
$ | (10,024 | ) | $ | (11,629 | ) | $ | (11,148 | ) | $ | (10,469 | ) | ||||
Basic and
diluted net loss
per common share |
$ | (0.42 | ) | $ | (0.49 | ) | $ | (0.47 | ) | $ | (0.44 | ) | ||||
Weighted
average common
shares basic and
diluted |
23,706,148 | 23,717,800 | 23,759,675 | 23,802,286 | ||||||||||||
Quarter Ended
(in thousands, except share and per share amounts)
(in thousands, except share and per share amounts)
| December 31, | March 31, | June 30, | September 30, | |||||||||||||
| 2007 | 2008 | 2008 | 2008 | |||||||||||||
Revenue |
$ | — | $ | — | $ | — | $ | — | ||||||||
Net loss |
$ | (11,025 | ) | $ | (9,557 | ) | $ | (10,114 | ) | $ | (12,665 | ) | ||||
Basic and
diluted net loss
per common share |
$ | (0.55 | ) | $ | (0.43 | ) | $ | (0.43 | ) | $ | (0.53 | ) | ||||
Weighted
average common
shares basic and
diluted |
20,198,829 | 22,045,400 | 23,653,956 | 23,674,362 | ||||||||||||
13. Subsequent Event
In November 2009, the Company amended its insulin supply agreement with N.V. Organon so that
the term of the agreement extends through December 2011. The remaining minimum quantities of
insulin that the Company is obligated to purchase have been reduced slightly and redistributed over
the remaining term of the agreement.
F-25